

Abstract
Background:
The collecting duct (CD) is a major site of both biosynthesis and action of prostaglandin E2 (PGE2) as highlighted by the predominant expression of cyclooxygenase-2 (COX-2) and some E-prostanoid (EP) subtypes at this nephron site. The purpose of this study was to determine the relevance and mechanism of CD COX-2/PGE2/EP1 signaling for the regulation of Na+ hemostasis during Na+ depletion.
Methods:
Mice with Aqp2Cre-driven deletion of COX-2 (COX-2fl/flAqp2Cre+) or the EP1 subtype (EP1fl/flAqp2Cre+) were generated and the Na+-wasting phenotype of these mice during low-salt (LS) intake was examined. EP subtypes responsible for PGE2-induced local renin response were analyzed in primary cultured mouse inner medullary CD cells.
Results:
Following 28-day LS intake, COX-2fl/flAqp2Cre+ mice exhibited a higher urinary Na+ excretion and lower cumulative Na+ balance, accompanied with suppressed intrarenal renin, angiotensin II, and aldosterone, expression of CYP11B2, and blunted expression of epithelial sodium channel (ENaC) subunits compared to floxed controls (COX-2fl/flAqp2Cre−), whereas no differences were observed for indices of systemic renin-angiotensin-aldosterone system (RAAS). In cultured CD cells, exposure to PGE2 stimulated release of soluble (pro)renin receptor, prorenin/renin and aldosterone and the stimulation was more sensitive to antagonism of EP1 as compared other EP subtypes. Subsequently, EP1fl/flAqp2Cre+ mice largely recapitulated Na+-wasting phenotype seen in COX-2fl/flAqp2Cre+ mice.
Conclusions:
The study for the first time reports that CD COX-2/EP1 pathway might play a key role in maintenance of Na+ homeostasis in the face of Na+ depletion, at least in part, through activation of intrarenal RAAS and ENaC.
Keywords: Cyclooxygenase-2, prostaglandin E2, EP1 receptors, intrarenal RAAS, ENaC, collecting duct
Introduction
A key function of the kidney is to retain Na+ in response to Na+ depletion so that urinary Na+ excretion is reduced to match restricted Na+ intake. The Na+ retaining action is indispensable for maintenance of plasma volume and blood pressure (BP). Although multiple mechanisms are responsible for eliciting renal Na+ retaining response, the activation of the renin-angiotensin-aldosterone-system (RAAS) plays an essential role[1]. The synthesis and secretion of renin from the juxtaglomerular (JG) cells of the kidney is the rate-limiting step of activation of the RAAS[2]. macula dense (MD)-dependent renin secretion is mainly dependent on enhancement of MD cyclooxygenase-2 (COX-2) expression that releases prostaglandin E2 (PGE2) acting in a paracrine manner to activate E-prostanoid receptor 4 (EP4) receptors in the JG cells[3–5]. In these cells, activation of EP4 receptors elevates intracellular cAMP that stimulates the release as well as expression of renin [6–9].
Apart from the systemic renin-angiotensin-system (RAS), the existence of intrarenal RAS has been demonstrated by a large body of experimental evidence[10]. In particular, renin is expressed in the collecting duct (CD) where its expression is positively regulated by angiotensin II (AngII), contrasting the negative regulation of JG renin by AngII[11, 12]. At functional level, CD renin represents an important driver of intrarenal RAS mediating the pathogenic role in hypertension development induced by infusion of pharmacological doses of AngII[13, 14]. BP is elevated by CD overexpression of renin[14] but decreased by CD-specific deletion of renin[13] during AngII infusion. In the AngII infusion model, enhancement of expression of renin along with (pro)renin receptor (PRR) depends on COX-2-derived PGE2 via EP4 receptors[11, 12]. However, a potential physiological function of prostaglandin (PG)-dependent activation of intrarenal RAS during Na+ deletion has not been tested by prior studies.
Within the kidney, the CD, composed of intercalated cells (ICs) and principal cells (PCs), is a major site for both production and action of PGE2[15, 16]. Indeed, COX-2 has recently been localized to the ICs where PGE2 is released and may act in a paracrine manner to modulate Na+ transport in the neighboring PCs of the CD[17]. PGE2 exerts its biologic action through G protein-coupled E-proteinoid (EP) receptors designated EP1, EP2, EP3, and EP4[18]. Among the 4 EP subtypes, the expression of EP1 and EP4 receptors are detected in the CD[18]. The present study attempted to examine the physiological function of CD COX-2 in regulation of Na+ homeostasis during Na+ depletion and further to identify EP1 receptors as the responsible EP subtype that controls intrarenal RAAS and epithelial sodium channel (ENaC).
Materials and Methods
The authors declare that all supporting data are available within the article (and its online-only Data Supplement)
Animals.
Aqp2Cre-driven deletion of COX-2 (COX-2fl/flAqp2Cre+) mice, Aqp2Cre-driven deletion of EP1 (EP1fl/flAqp2Cre+) mice, and floxed control (COX-2fl/flAqp2Cre− and EP1fl/flAqp2Cre−) mice (3-month old) were all given free access to tap water and fed the standard diet (Na+: 0.3% and K+: 1%). Mice were housed in a temperature- and humidity-controlled room with a 12:12-h light-dark cycle. All animal studies were conducted with the approval of the University of Utah Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Statistical analysis.
Data are summarized as means ± SEM. Statistical analysis was performed by using one-way analysis of variance (ANOVA) with the Bonferroni test for multiple comparisons, by repeated measures ANOVA for the interaction (time × genotype), by unpaired student t-test for two comparisons at either different time points or under different conditions/treatments using IBM SPSS 19 software. Difference was considered to be significant when the probability value was less than 0.05.
The rest of the Methods is available in the online-only Data Supplement.
Results
Verification of Aqp2Cre-driven COX-2 deletion.
COX-2fl/flAqp2Cre+ mice were generated by crossing COX-2-floxed mice[19] with Aquaporin 2 (Aqp2)-Cre mice[20]. Figure S1A illustrates the gene targeting strategy. COX-2fl/flAqp2Cre+ mice were born at the expected frequency, had normal growth and survival. PCR of DNA isolated from various organs from COX-2fl/flAqp2Cre+ mouse demonstrated strong recombination in the kidneys with minor recombination in other organs (Figure S1C). Renal protein expression of COX-2 was examined by immunofluorescence with co-labeling with ani-AQP2 antibody as a marker for PCs of the CD. Within the kidney, COX-2 was predominantly expressed in CD cells that were negative for AQP2 labeling, indicating ICs [17]. COX-2 labeling in ICs was reduced in COX-2fl/flAqp2Cre+ mice as compared with floxed controls (Figure S2A). Compared with COX-2fl/flAqp2Cre+ mice, COX-2fl/flAqp2Cre+ mice had 50% lower COX-2 mRNA expression in the renal cortex and medulla under normal condition, and completely blocked low Na+ (LS)-induced upregulation of COX-2 mRNA expression in the renal medulla but not renal cortex (Figure S2B). By immunoblotting analysis, there were no detectable differences in renal cortical or medullary COX-2 protein between the genotypes on a normal Na+ (NS) diet (Figure S2C). Aqp2Cre-driven COX-2 deletion completely abolished LS-stimulated COX-2 protein expression in the renal medulla but not renal cortex (Figure S2C). By ELISA, 24-h urinary PGE2 excretion was not different between the genotypes under basal condition. In contrast, the increase of urinary PGE2 excretion in response to 28-day LS intake was completely abolished by Aqp2Cre-driven COX-2 deletion (Figure S2D). We detected EP4 mRNA expression in the kidney regions by RT-qPCR. Under normal condition, Aqp2Cre-driven COX-2 deletion increased EP4 mRNA expression in both renal cortex and medulla (Figure S3). Interestingly, renal cortical EP4 mRNA was similarly elevated by LS intake in both genotypes whereas there was no further increase in renal medullary EP4 mRNA in response to LS intake in the null mice (Figure S3).
Aqp2Cre-driven COX-2 deletion induced Na+-wasting phenotype during LS intake.
As shown in Table S1, under basal condition, both COX-2fl/flAqp2Cre+ and COX-2fl/flAqp2Cre− mice had comparable food and water intake, body weight, urine volume, urinary excretion of Na+, K+, and Cl−, and plasma Na+, K+, and Cl− concentrations. Within days of LS intake, a sharp decrease in 24-h urinary Na+ excretion was similarly observed in both genotypes but with continued LS intake, the value became higher in COX-2fl/flAqp2Cre+ mice than COX-2fl/flAqp2Cre− mice (Figure 1A). As a result, cumulative Na+ balance was lowered in COX-2fl/flAqp2Cre+ mice than COX-2fl/flAqp2Cre− mice, confirming the Na+ wasting phenotype (Figure 1A). A small but statistically significant reduction of plasma Na+ concentration occurred in COX-2fl/flAqp2Cre+ mice following 28-day LS intake as compared with floxed control (Table S1). However, radiotelemetry detected no difference in BP between COX-2fl/flAqp2Cre+ mice and their floxed controls under either normal salt or LS conditions (Figure S4).
Figure 1.
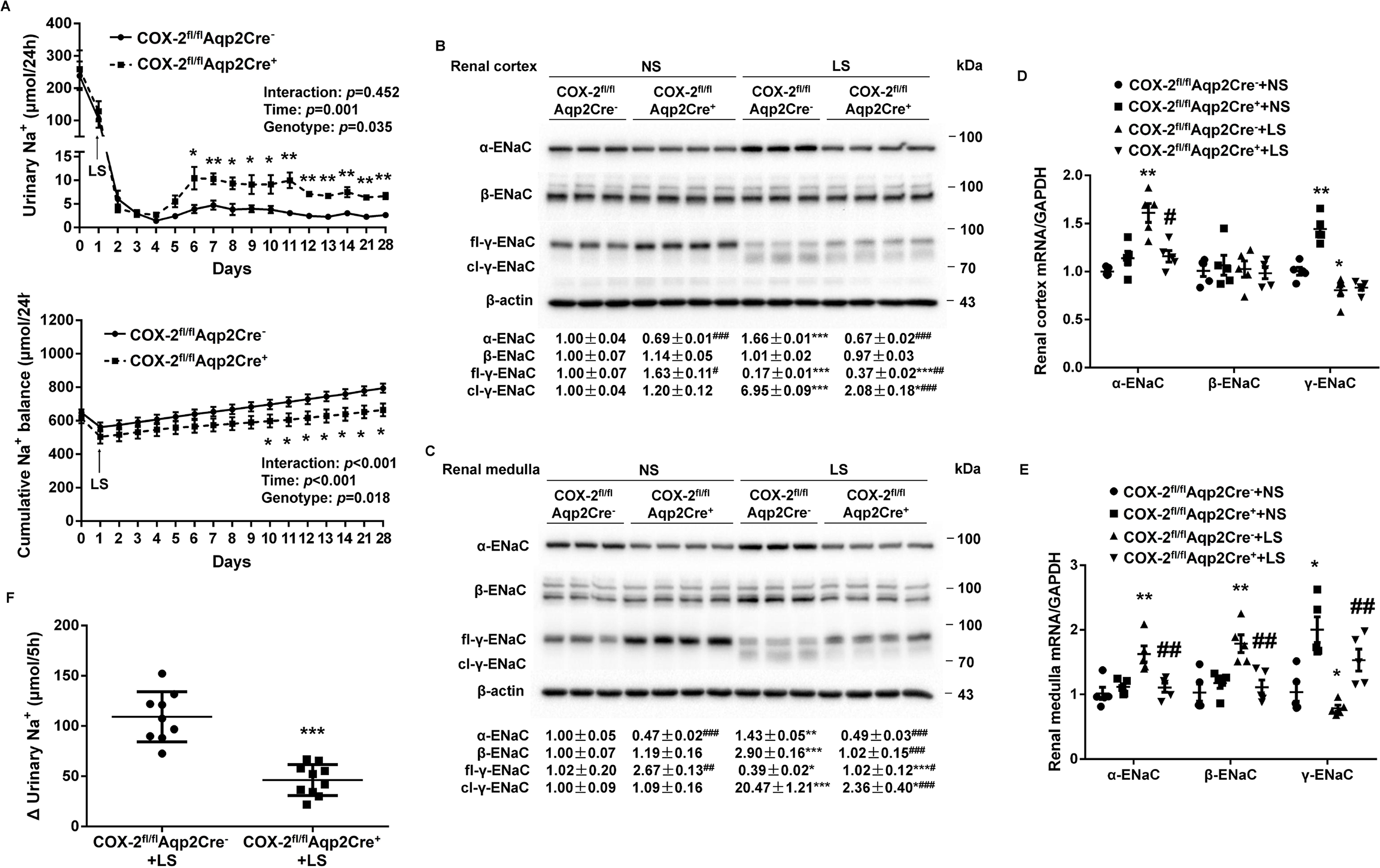
Analysis of urinary Na+ excretion, cumulative Na+ balance, and renal expression of epithelial sodium channel (ENaC) subunits in COX-2fl/flAqp2Cre− and COX-2fl/flAqp2Cre+ mice on a normal Na+ (NS) or low Na+ (LS) diet. (A) 24-h urinary Na+ excretion and cumulative Na+ balance in COX-2fl/flAqp2Cre− and COX-2fl/flAqp2Cre+ mice on a 4-wk NS or LS diet. N = 10 per group. Data are mean ± SEM. *p<0.05, **p<0.01 vs. COX-2fl/flAqp2Cre− at the corresponding time period (unpaired Student’s t test) (analysis of the interaction [time × genotype] by repeated-measures ANOVA). (B, C) Immunoblotting and densitometric analysis of ENaC subunits in the renal cortex (B) and medulla (C). γ-ENaC-probed membrane was stripped and re-probed with β-actin as an internal control for equal loading of the samples. N = 3–4 per group for statistical analysis. Data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 vs. NS; #p<0.05, ##p<0.01, ###p<0.001 vs. COX-2fl/flAqp2Cre− (ANOVA with the Bonferroni test). (D, E) RT-qPCR analysis of ENaC subunits mRNA in the renal cortex (D) and medulla (E) with GAPDH as an internal control. N = 5 per group. Data are mean ± SEM. *p<0.05, **p<0.01 vs. COX-2fl/flAqp2Cre−+NS; #p<0.05, ##p<0.01 vs. COX-2fl/flAqp2Cre−+LS (ANOVA with the Bonferroni test). (F) In vivo ENaC activity as reflected by rapid natriuretic responses to amiloride. LS-loaded COX-2fl/flAqp2Cre− and COX-2fl/flAqp2Cre+ mice were all subjected to a single dose of amiloride (10 mg/kg by intraperitoneal injection) or vehicle treatment, followed by 5-h urine collection, and shown was the change of 5-h urinary Na+ excretion, determined by the delta value of 5-h urinary Na+ excretion of vehicle and amiloride treatment. N = 9–10 per group. Data are mean ± SEM. ***p<0.001 vs. COX-2fl/flAqp2Cre−+LS (unpaired Student’s t test).
ENaC, a major Na+ transporter in the epithelial cells of the CD, is under the control of aldosterone (Aldo) during Na+ depletion. We therefore examined renal expression of the 3 subunits, α, β, and γ, in the two strains of mice on a NS or LS diet by immunoblotting analysis. On a NS diet, COX-2fl/flAqp2Cre+ mice had lower α-ENaC but higher full-length γ-ENaC (fl-γ-ENaC) protein expression in the renal cortex (Figure 1B) and medulla (Figure 1C) with unchanged β-ENaC expression in the two kidney regions (Figure 1B and 1C) as compared with COX-2fl/flAqp2Cre− mice. In response to LS intake, protein expression of ENaC subunits exhibited distinct responses with increases in both renal cortical and medullary α-ENaC expression and cleavage of γ-ENaC (cl-γ-ENaC) and in renal medullary but not renal cortical β-ENaC protein expression (Figure 1B and 1C). In contrast, these changes in all 3 subunits were attenuated in COX-2fl/flAqp2Cre+ mice (Figure 1B and 1C). By RT-qPCR, Aqp2Cre-driven COX-2 deletion significantly blocked LS-stimulated renal cortical α-ENaC mRNA expression (Figure 1D) and renal medullary α-ENaC and β-ENaC mRNA expression (Figure 1E), but remarkably increased γ-ENaC mRNA expression in both renal cortex and medulla under NS condition (Figure 1D and 1E). Consistently, amiloride-induced increases in urinary Na+ excretion were decreased in COX-2fl/flAqp2Cre+ mice as compared with COX-2fl/flAqp2Cre− mice under LS condition (Figure 1F). These results suggest that Aqp2Cre-driven COX-2 deletion resulted in attenuated ENaC activity in the face of Na+ depletion.
Aqp2Cre-driven COX-2 deletion blocked LS-induced activation of intrarenal RAS.
Activation of the RAS is a prerequisite for maintenance of Na+ homeostasis in the face of Na+ depletion. However, the relative importance of systemic versus intrarenal RAS and its relationship with PGs are only partially understood. Therefore, we comprehensively examined multiple parameters indicative of the two systems in COX-2fl/flAqp2Cre+ and COX-2fl/flAqp2Cre− mice on NS or LS intake. On a NS diet, there were no detectable differences in 24-h urinary prorenin/renin and AngII excretion, urinary renin activity (Figure 2A), plasma prorenin/renin and AngII concentrations (Figure S4A & C), or plasma renin activity (Figure S4B) between the genotypes. However, Aqp2Cre-driven COX-2 deletion significantly attenuated LS-stimulated 24-h urinary prorenin/renin excretion (13.08±2.94 vs. 54.80±7.48 ng/24h, p<0.05), urinary renin activity (0.43±0.10 vs. 1.54±0.33 ng/24h, p<0.01), and 24-h urinary AngII excretion (0.25±0.04 vs. 1.12±0.18 ng/24h, p<0.01) in COX-2fl/flAqp2Cre− mice (Figure 2A). In contrast, LS-stimulated increase of plasma prorenin/renin concentration (Figure S5A), plasma renin activity (Figure S5B), and plasma AngII concentration (Figure S5C) were unaffected by Aqp2Cre-driven COX-2 deletion. In agreement with these results, Aqp2Cre-driven COX-2 deletion remarkably inhibited LS-induced upregulation of renal expression of PRR and prorenin protein and renin mRNA expression in the renal medulla but not renal cortex (Figure 2C).
Figure 2.
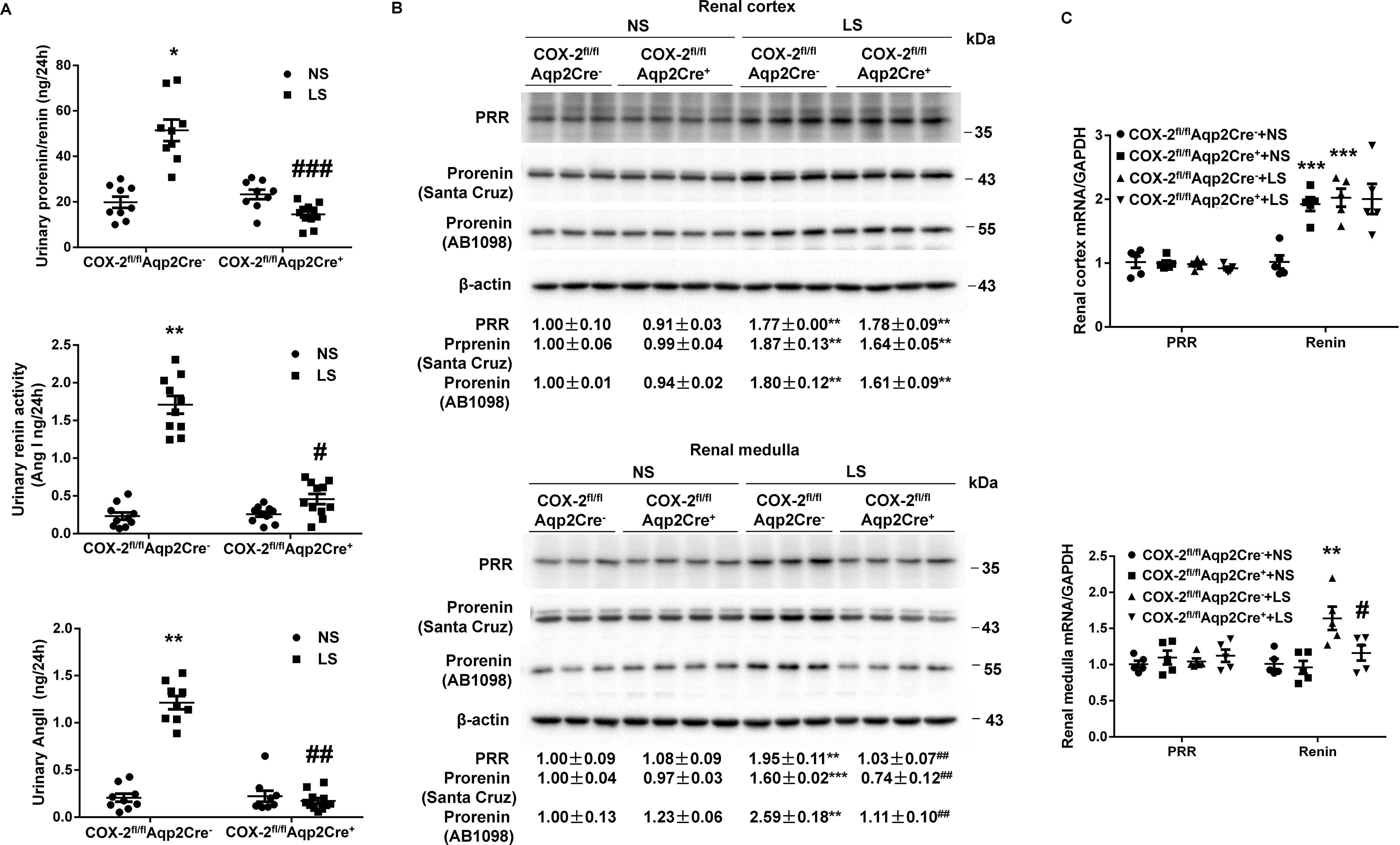
Responses of key components of the renin-angiotensin-system in urine and the kidney of COX-2fl/flAqp2Cre− and COX-2fl/flAqp2Cre+ mice during normal-Na+ (NS) and low Na+ (LS) intake. (A) The urine samples were assayed for total renin/prorenin, renin activity, and angiotensin II (Ang II). N=9–11 per group. Data are mean ± SEM. *p<0.05, **p<0.01 vs. NS; #p<0.05, ##p<0.01, ###p<0.001 vs. COX-2fl/flAqp2Cre− (ANOVA with the Bonferroni test). (B) Representative immunoblotting and densitometric analysis of (Pro)renin receptor (PRR) and prorenin expression in the renal cortex and medulla with β-actin as an internal control. Prorenin-probed membranes were re-probed with anti-β-actin antibody. N = 3–4 per group for statistical analysis. Data are mean ± SEM. **p<0.01, ***p<0.001 vs. NS; ##p<0.01 vs. COX-2fl/flAqp2Cre− (ANOVA with the Bonferroni test). (C) RT-qPCR analysis of PRR and renin mRNA in the renal cortex and medulla with GAPDH as an internal control. N = 5 per group. Data are mean ± SEM. **p<0.01, ***p<0.001 vs. COX-2fl/flAqp2Cre−+NS; #p<0.05 vs. COX-2fl/flAqp2Cre−+LS (ANOVA with the Bonferroni test).
Aqp2Cre-driven COX-2 deletion blocked LS-induced intrarenal Aldo biosynthesis.
Aldo derived from the adrenal glands through the activity of CYP11B2 (Cytochrome P450 Family 11 Subfamily B Member 2) is traditionally considered to act in an endocrine manner to modulate Na+ reabsorption in the Aldo-sensitive distal nephron, whereas the contribution of intrarenal generation of Aldo to this phenomenon has not been tested by any prior study. We examined status of Aldo biosynthesis in the kidney versus the adrenal glands in COX-2fl/flAqp2Cre+ mice and COX-2fl/flAqp2Cre− mice on NS or LS intake. We performed ELISA to measure free and total Aldo from plasma and urine, and tissues, and conducted immunoblotting and RT-qPCR to determined tissue CYP11B2 expression. The basal parameters including urinary and plasma Aldo as well as tissue Aldo were largely comparable between the genotypes except that renal medullary but not cortical Aldo content was reduced in COX-2fl/flAqp2Cre+ mice (Figure 3). LS intake significantly promoted urinary free and total Aldo excretion (Figure 3A) and renal cortical and medullary Aldo level (Figure 3B) in floxed mice, which were all abolished in COX-2fl/flAqp2Cre+ mice. In contrast, plasma Aldo concentration and adrenal Aldo level were responsive to LS intake in a similar extent between the genotypes (Figure S5D). Accordingly, we examined CYP11B2 mRNA and protein expression in the kidney regions and adrenal glands by RT-qPCR and immunoblotting analysis, respectively. Consistent with the Aldo data, CYP11B2 protein and mRNA expressions in renal cortex and medulla (Figure 3C & D) of floxed mice were consistently upregulated by LS intake, which was abolished in COX-2fl/flAqp2Cre+ mice. In contrast, LS-induced increases in CYP11B2 expression in adrenal glands remained unchanged in the null mice (Figure S5E & F). Additionally, we detected CYP11B1 mRNA expression in the kidney regions and adrenal gland by RT-qPCR. Under normal condition, Aqp2Cre-driven COX-2 deletion significantly increased CYP11B1 mRNA expression in renal cortex (Figure S6A) and medulla (Figure S6B) but not adrenal gland (Figure S6C). LS intake enhanced CYP11B1 mRNA expression in renal cortex (Figure S6A) and medulla (Figure S6B) but slightly decreased the levels of CYP11B1 mRNA in the adrenal gland (Figure S6C) in the floxed mice. The effect of LS on CYP11B1 mRNA expression was unaffected in the null mice (Figure S6).
Figure 3.

Responses of intrarenal aldosterone biosynthesis in COX-2fl/flAqp2Cre− and COX-2fl/flAqp2Cre+ mice during normal-Na+ (NS) and low-Na+ (LS) intake. (A, B) Aldosterone levels in the urine (N=9–11 per group) (A), and renal cortex and medulla (N=5 per group) (B) were determined by ELISA. Data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 vs. NS; #p<0.05, ##p<0.01, ###p<0.001 vs. COX-2fl/flAqp2Cre− (ANOVA with the Bonferroni test). (C) Representative immunoblotting and densitometric analysis of CYP11B2 (Cytochrome P450 Family 11 Subfamily B Member 2) in the renal cortex and medulla. CYP11B2-probed membranes were stripped and re-probed with anti-β-actin antibody. N = 3–4 per group for statistical analysis. Data are mean ± SEM. **p<0.01, ***p<0.001 vs. NS; ###p<0.001 vs. COX-2fl/flAqp2Cre− (ANOVA with the Bonferroni test). (D) RT-qPCR analysis of CYB11B2 mRNA in the renal cortex and medulla with GAPDH as an internal control. N=5 per group. Data are mean ± SEM. *p<0.05, ***p<0.001 vs. NS; ##p<0.01, ###p<0.001 vs. COX-2fl/flAqp2Cre− (ANOVA with the Bonferroni test).
EP subtypes in PGE2-stimulated prorenin/renin and Aldo release from cultured IMCD cells.
We examined the direct effect of PGE2, a major prostanoid in the kidney, on the release of soluble PRR (sPRR), prorenin/renin, and Aldo in primary cultured mouse inner medullary collecting duct (IMCD) cells[21] and further determined the EP subtype involved. EP1 and EP4 receptors are known to be the major EP subtypes expressed in the CD[18]. PGE2 treatment for 24 h significantly stimulated sPRR, prorenin/renin, and Aldo secretion (Figure S7). PRO20, a decoy inhibitor of PRR, almost completely abolished PGE2-induced prorenin/renin and Aldo release (Figure S7B & C). An EP1 antagonist SC-19220 abolished the effect of PGE2 on sPRR, prorenin/renin, and Aldo release (Figure S7A–C). Relatively, antagonism of EP3 or EP4 receptors was less effective in that they failed to suppress the release of prorenin/renin or Aldo despite the inhibitory effect on sPRR (Figure S7A–C). The involvement of the EP2 receptor was not examined in the present study mainly due to the lack of an effective antagonist for this EP subtype.
Verification of Aqp2Cre-driven EP1 deletion.
To generate EP1-floxed mice, a targeting construct[22] was made wherein exon 2 of the EP1 gene was flanked by two loxP sites (floxed) and electroporated into mouse embryonic stem cells. EP1-floxed mice were not different from wild-type (WT) mice and therefore served as normal controls. EP1 floxed mice were crossed with Aqp2-Cre mice and termed as EP1fl/flAqp2Cre+ mice. Figure S8A illustrates the targeting strategy. EP1fl/flAqp2Cre+ mice were born at the expected frequency, had normal growth and survival. PCR of DNA isolated from various organs from EP1fl/flAqp2Cre+ mice demonstrated strong recombination in the kidneys with minor recombination in brain and testis (Figure S8D). As compared with EP1fl/flAqp2Cre− mice, EP1fl/flAqp2Cre+ mice had 50% lower EP1 mRNA expression in the renal cortex and medulla (Figure S8E). Under normal condition, Aqp2Cre-driven EP1 deletion increased EP4 mRNA expression in renal cortex and medulla without affecting the stimulation effect of LS on EP4 mRNA expression in these regions (Figure S9).
Aqp2Cre-driven EP1 deletion induced Na+ wasting phenotype during LS intake.
As shown in Table S2, under baseline conditions, male 3-mo-old EP1fl/flAqp2Cre+ mice and EP1fl/flAqp2Cre− mice had comparable food and water intake, body weight, urine volume, urinary excretion of Na+, K+, and Cl−, and plasma Na+, K+, and Cl− concentrations. Following LS intake, EP1fl/flAqp2Cre+ mice had a similar reduction of 24-h urinary Na+ excretion within days but elevated values thereafter as compared with EP1fl/flAqp2Cre− mice (Figure 4A). As a result, EP1fl/flAqp2Cre+ mice exhibited a reduced cumulative Na+ balance (Figure 4A) during LS intake. A small but statistically significant reduction of plasma Na+ concentration occurred in EP1fl/flAqp2Cre+ mice following 28-day LS intake as compared with EP1fl/flAqp2Cre− mice (Table S2). However, radiotelemetry detected no difference in BP between EP1fl/flAqp2Cre+ and EP1fl/flAqp2Cre− mice and under either normal salt or LS conditions (Figure S10).
Figure 4.
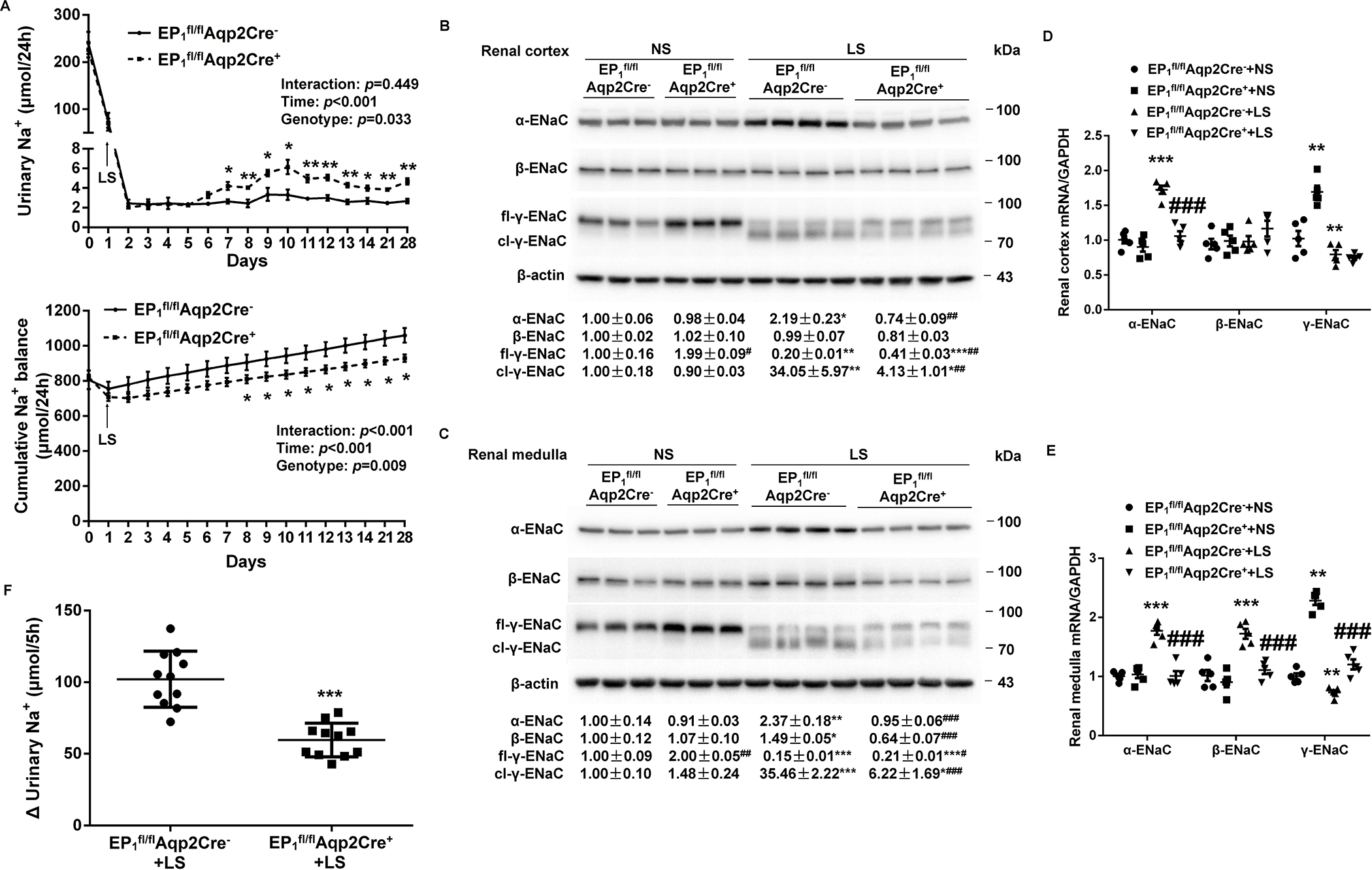
Analysis of urinary Na+ excretion, cumulative Na+ balance, and renal epithelial sodium channel (ENaC) expression and activity in EP1fl/flAqp2Cre− and EP1fl/flAqp2Cre+ mice on a normal-Na+ (NS) or low-Na+ (LS) diet. (A) 24-h urinary Na+ excretion and cumulative Na+ balance in EP1fl/flAqp2Cre− and EP1fl/flAqp2Cre+ mice fed NS or LS diets. N = 10 per group. Data are mean ± SEM. *p<0.05, **p<0.01 vs. EP1fl/flAqp2Cre− at the corresponding time period (unpaired Student’s t test) (analysis of the interaction [time × genotype] by repeated-measures ANOVA). (B, C) Immunoblotting and densitometric analysis of ENaC subunits in the renal cortex (B) and medulla (C) in NS and LS treated EP1fl/flAqp2Cre− and EP1fl/flAqp2Cre+ mice. γ-ENaC-probed membrane was stripped and re-probed with anti-β-actin antibody. N = 3–4 per group for statistical analysis. Data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 vs. NS; #p<0.05, ##p<0.01, ###p<0.001 vs. EP1fl/flAqp2Cre− (ANOVA with the Bonferroni test). (D, E) RT-qPCR analysis of ENaC subunits mRNA in the renal cortex (D) and medulla (E) with GAPDH as an internal control. N = 5 per group. Data are mean ± SEM. **p<0.01, ***p<0.001 vs. EP1fl/flAqp2Cre−+NS; ###p<0.001 vs. EP1fl/flAqp2Cre−+LS (ANOVA with the Bonferroni test). (F) In vivo ENaC activity as reflected by rapid natriuretic responses to amiloride. LS-loaded EP1fl/flAqp2Cre− and EP1fl/flAqp2Cre+ mice were all subjected to a single dose of amiloride (10 mg/kg by intraperitoneal injection) or vehicle treatment, followed by 5-h urine collection, and shown was the change of 5-h urinary Na+ excretion, determined by the delta value of 5-h urinary Na+ excretion of vehicle and amiloride treatment. N = 11 per group. Data are mean ± SEM. ***p<0.001 vs. EP1fl/flAqp2Cre−+LS (unpaired Student’s t test).
The renal expression of ENaC subunits in EP1fl/flAqp2Cre+ and EP1fl/flAqp2Cre− mice was examined using immunoblotting analysis. On a NS diet, EP1fl/flAqp2Cre+ mice had higher full-length γ-ENaC protein expression in the kidney cortex (Figure 4B) and medulla (Figure 4C), as compared with EP1fl/flAqp2Cre− mice but without differences in expression of α-ENaC and β-ENaC in either renal cortex (Figure 4B) or medulla (Figure 4C). LS intake in EP1fl/flAqp2Cre− mice induced ENaC activation as evidenced by increased α-ENaC protein expression and cleavage of γ-ENaC in both renal cortex and medulla, and β-ENaC protein expression selectively in renal medulla but not renal cortex. The LS-induced changes in the 3 subunits were all significantly blocked by Aqp2Cre-driven EP1 deletion (Figure 4B & 4C). By RT-qPCR, Aqp2Cre-driven EP1 deletion remarkably blocked LS-stimulated renal cortical α-ENaC mRNA expression (Figure 4D) and renal medullary α-ENaC and β-ENaC mRNA expression (Figure 4E), but increased γ-ENaC mRNA expression in both renal cortex and medulla under NS condition (Figure 4D and 4E). The in vivo ENaC activity as reflected by amiloride-induced increases in urinary Na+ excretion was significantly attenuated by Aqp2Cre-driven EP1 deletion during LS intake (Figure 4F).
Aqp2Cre-driven EP1 deletion blocked LS-induced activation of intrarenal RAS.
Aqp2Cre-driven EP1 deletion attenuated the basal levels of 24-h urinary renin activity and AngII excretion but not prorenin/renin content. Following LS intake, Aqp2Cre-driven EP1 deletion significantly blocked LS-stimulated 24-h urinary prorenin/renin excretion (10.03±2.64 vs. 40.32±6.93 ng/24h, p<0.01), urinary renin activity (0.33±0.06 vs. 1.37±0.17 ng/24h, p<0.01), and 24-h urinary AngII excretion (0.28±0.05 vs. 1.09±0.14 ng/24h, p<0.05) seen in EP1fl/flAqp2Cre− mice (Figure 5A). In contrast, Aqp2Cre-driven EP1 deletion didn’t affect the basal or LS-stimulated plasma prorenin/renin concentrations (Figure S11A), renin activity (Figure S11B), or AngII concentration (Figure S11C). The protein abundances of prorenin, PRR, and sPRR in the renal cortex and medulla were determined by immunoblotting analysis. RT-qPCR was performed to examine mRNA expression of renin and PRR in the two renal regions. Following LS intake, parallel increases were observed for protein abundances of prorenin and PRR/sPRR, and mRNA expression of renin and PRR in both renal cortex and medulla of EP1fl/flAqp2Cre− mice. Interestingly, Aqp2Cre-driven EP1 deletion selectively blocked LS-induced increases in protein abundances of prorenin and PRR/sPRR and mRNA expression of renin in the renal medulla but not renal cortex (Figure 5B & C). Of note, similar results were obtained with immunoblotting analysis of prorenin protein expression using two different antibodies against prorenin.
Figure 5.
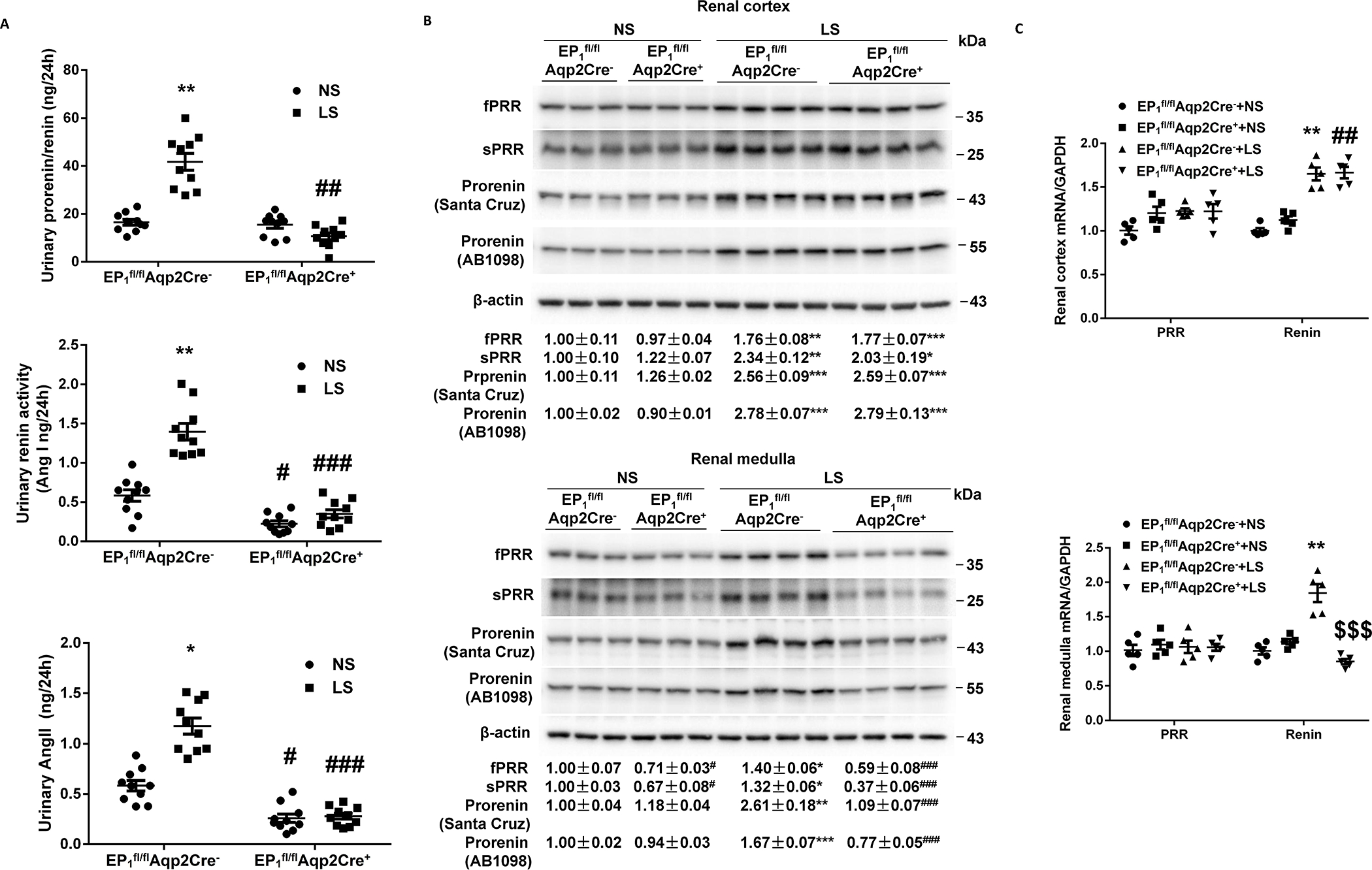
Responses of key components of the renin-angiotensin-system in urine and the kidney of EP1fl/flAqp2Cre− and EP1fl/flAqp2Cre+ mice during normal-Na+ (NS) and low-Na+ (LS) intake. (A) The urine samples were assayed for total renin/prorenin, renin activity, and angiotensin II (Ang II). N=10 per group. Data are mean ± SEM. *p<0.05, **p<0.01 vs. NS; #p<0.05, ##p<0.01, ###p<0.001 vs. EP1fl/flAqp2Cre− (ANOVA with the Bonferroni test). (B) Representative immunoblotting and densitometric analysis of (Pro)renin receptor (PRR) and prorenin expression in the renal cortex and medulla with β-actin as an internal control. Prorenin-probed membranes were probed with anti-β-actin antibody. N = 3–4 per group for statistical analysis. Data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 vs. NS; #p<0.05, ###p<0.001 vs. EP1fl/flAqp2Cre− (ANOVA with the Bonferroni test). (C) RT-qPCR analysis of PRR and renin mRNA in the renal cortex and medulla with GAPDH as an internal control. N = 5 per group. Data are mean ± SEM. **p<0.01 vs. EP1fl/flAqp2Cre−+NS; ##p<0.01 vs. EP1fl/flAqp2Cre++NS, $ $ $p<0.001 vs. EP1fl/flAqp2Cre−+LS (ANOVA with the Bonferroni test).
Aqp2Cre-driven EP1 deletion blocked LS-induced intrarenal Aldo biosynthesis.
We assessed intrarenal and adrenal Aldo biosynthesis in EP1fl/flAqp2Cre+ and EP1fl/flAqp2Cre− mice under NS or LS condition. On a NS diet, there were no detectable differences in 24-h urinary Aldo excretion, intrarenal Aldo level, plasma Aldo concentrations, and adrenal gland Aldo levels, renal and adrenal expression of CYP11B2 between the genotypes (Figure 6 & S8). In contrast, Aqp2Cre-driven EP1 deletion abolished LS-induced urinary free and total Aldo excretion (Figure 6A) and renal cortical and medullary Aldo content (Figure 6B) and CYP11B2 protein and mRNA expression (Figure 6C & D), whereas the increased plasma Aldo concentration and adrenal Aldo content (Figure S8D) and CYP11B2 expression (Figure S8E & F) were unaffected. Under normal condition, Aqp2Cre-driven EP1 deletion significantly increased CYP11B1 mRNA expression in renal cortex (Figure S12A) and medulla (Figure S12B) but not adrenal gland (Figure S12C). The effect of LS on CYP11B1 mRNA expression in both regions was unaffected in the mice with Aqp2Cre-driven EP1 deletion (Figure S12).
Figure 6.
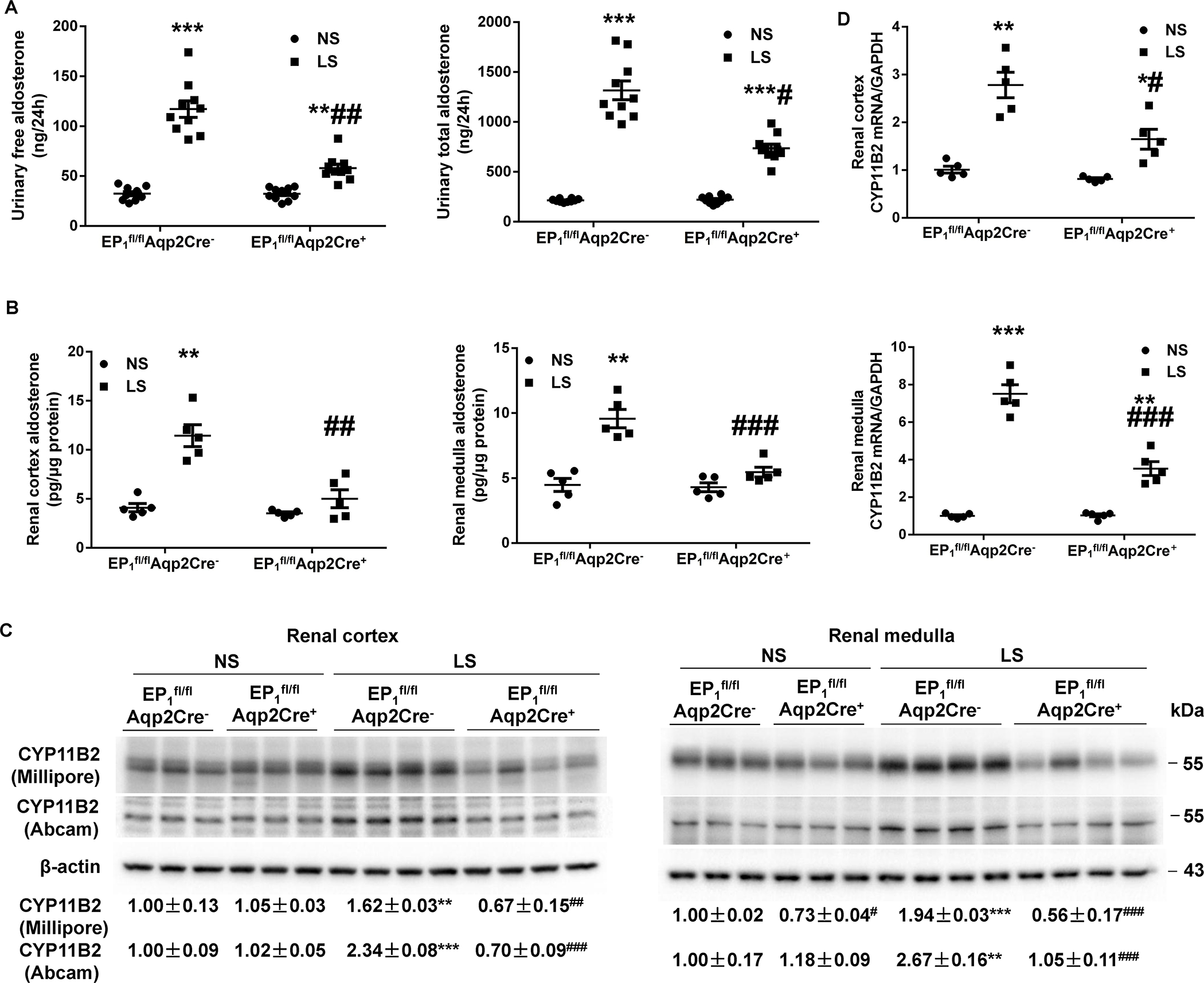
Responses of intrarenal aldosterone biosynthesis in EP1fl/flAqp2Cre− and EP1fl/flAqp2Cre+ mice during normal-Na+ (NS) and low-Na+ (LS) intake. (A, B) Aldosterone levels in the urine (N=10 per group) (A), and renal cortex and medulla (N=5 per group) (B) were determined by ELISA. Data are mean ± SEM. **p<0.01, ***p<0.001 vs. NS; #p<0.05, ##p<0.01, ###p<0.001 vs. EP1fl/flAqp2Cre− (ANOVA with the Bonferroni test). (C) Representative immunoblotting and densitometric analysis of CYP11B2 (Cytochrome P450 Family 11 Subfamily B Member 2) in the renal cortex and medulla. CYP11B2-probed membranes were stripped and re-probed with anti-β-actin antibody. N = 3–4 per group for statistical analysis. Data are mean ± SEM. **p<0.01, ***p<0.001 vs. NS; #p<0.05, ##p<0.01, ###p<0.001 vs. EP1fl/flAqp2Cre− (ANOVA with the Bonferroni test). (D) RT-qPCR analysis of CYP11B2 mRNA in the renal cortex and medulla with GAPDH as an internal control. N=5 per group. Data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 vs. NS; #p<0.05, ###p<0.001 vs. EP1fl/flAqp2Cre− (ANOVA with the Bonferroni test).
Discussion
The purpose of our study was to determine the relevance and mechanism of CD COX-2/PGE2/EP1 signaling for the regulation of Na+ hemostasis during Na+ depletion. The experimental approach mainly involves the generation and characterization of two strains of mouse models with CD-specific deletion of COX-2 or the EP1 subtype in combination of in vitro analysis of EP subtypes responsible for PGE2-induced local renin response. Our findings demonstrate that Aqp2Cre-driven deletion of COX-2 or EP1 receptors induced Na+-wasting phenotype associated with suppressed renal expression of PRR, prorenin, and CYP11B2 expression, renal Aldo, and urinary excretion of prorenin/renin, AngII, and Aldo, all indicative of suppressed intrarenal RAAS. In contrast, LS-induced systemic RAAS remained unchanged in either strain of the null mice. In addition, the two strains of null mice also exhibited impairment of LS-induced activation of ENaC in the kidney.
The present study for the first time generated mice with Aqp2Cre-driven deletion of COX-2 by using Aqp2-Cre and examined the phenotype during Na+ depletion. While Aqp2 promoter was originally thought to be PC-specific[13, 23], increasing evidence shows that this promoter was active before the IC appears [24–27] supporting the conclusion that ICs derive from AQP2 expressing cells during nephrogenesis[28, 29]. Indeed, single-cell RNA-seq identified a small fraction of hybrid cells expressing AQP2 (PC marker) and AE1 or pendrin transcripts (IC marker) [30]. Therefore, Aqp2-Cre targets both PCs and ICs in the CDs [31, 32]. In the present study, we found that IC expression of COX-2 was reduced in COX-2fl/flAqp2Cre+ mice. As expected, urinary PGE2 excretion, a reliable index of renal PG synthesis, was elevated in floxed mice during Na+ depletion. To our surprise, LS-induced renal PGE2 synthesis traditionally considered as being attributable to MD COX-2[33, 34], was completely blocked by Aqp2Cre-driven COX-2 deletion. This finding supports CD rather than MD COX-2 as the predominant source of LS-simulated renal PGE2 synthesis. In agreement with this notion, COX-2fl/flAqp2Cre+ mice developed Na+-wasting phenotype over 28 days of LS intake as compared with floxed controls. It is interesting to note that the difference in urinary Na+ excretion between the genotypes didn’t become significant until day 6 of LS intake. This result may suggest that a distinct mechanism may contribute to the rapid decline of urinary Na+ excretion in response to LS intake within days. The nature of such mechanism remains elusive but could be related to alteration of renal hemodynamics as a result of enhanced vasoconstriction induced by AngII. We would like to propose an intriguing possibility that LS-induced rapid renal hemodynamic response may depend on MD COX-2-dependent activation of systemic RAAS. In this way, COX-2 in the MD and the CD may act in concert to coordinate Na+-retaining response within days and weeks. In support of this notion, we have previously reported that 7-day LS induced COX-2 expression in the MD but not in the renal medulla [5]. In vitro evidence demonstrates that low Cl− is responsible for induction of COX-2 expression and PGE2 release via rapid activation of MAP kinase[35]. In the present study, we found that 28-day LS stimulated renal medullary COX-2 expression. The cellular mechanism for the slow response of renal medullary COX-2 to LS remains elusive.
Within the renal medulla, COX-2 expression has been detected in inner medullary interstitial cells (RMICs) [15, 36], and ICs of CDs [16, 17]. Several previous studies from our group and others have shown that renal medullary COX-2 expression is induced in the RMICs by as short as 7-day HS intake as a mechanism of promoting natriuresis[36–40]. However, this notion is at odds with the current observation of LS upregulation of renal medullary COX-2 expression. While the exact reason for this discrepancy is unclear, the cellular localization of COX-2 induction during the two conditions appears different. In this regard, COX-2 expression is induced in RMICs by HS intake but in ICs by LS intake. It seems likely that RMIC-derived PGE2 may directly inhibit tubular ion transport to induce natriuresis[41] during HS intake whereas as shown by the present study IC-derived PGE2 activates intrarenal RAAS to elicit Na+ retaining response during LS intake.
The evidence from our previous study demonstrates that COX-2-dependent activation of renal PRR and intrarenal RAS mediate elevated BP induced by AngII infusion [11]. This conclusion is largely based on the inhibitory effects of pharmacological inhibition of COX-2 on renal PRR expression and renal medullary and urinary renin levels induced by AngII infusion [11]. The present study has extended this observation in the following aspects. First, the generation of Aqp2Cre-driven COX-2 deletion precisely mapped the action of COX-2 to the CD. Second, the present study for the first time examined the physiological role of CD COX-2 in regulation of Na+ balance during Na+ depletion. Third, besides local PRR and renin, renal Aldo synthesis is also under the control of COX-2-derived PGs, supporting existence of the intrarenal RAAS. These results support an important physiological role of COX-2-dependent intrarenal RAAS in regulation of Na+ homeostasis independently of the systemic RAAS. In this regard, COX-2/PGE2 stimulates PRR/prorenin signaling to directly increase CYP11B2 expression[21], or indirectly enhance CYP11B2 expression via activating intrarenal AngII/AT1R signaling[42], resulting in Aldo synthesis in the CD in response to LS intake.
The present study has defined a specific EP subtype, e.g. the EP1 subtype acting downstream of COX-2 in regulation of CD function during Na+ depletion. In cultured IMCD cells, antagonism of EP1 receptor completely abolished PGE2-induced sPRR, prorenin/renin, and Aldo secretion whereas antagonism of EP3 or EP4 subtype effectively suppressed PGE2-induced sPRR but failed to block the rise of prorenin/renin or Aldo levels, in agreement with the report by Gonzalez et al.[43, 44]. The potential mechanism may involve EP1-mediated activation of PKC/cAMP/CREB pathway[43]. Indeed, the EP1 subtype has been implicated in modulating water and Na+ transport in the CD[45, 46]. Given the broad roles of the EP1 in multiple tissue types, it is imperative to take a cell-specific strategy to address the function of this EP subtype in the CD. In the present study we for the first time employed homologous recombination strategy to generate mice with EP1 floxed allele. The floxed mice were indistinguishable from their WT controls. EP1-floxed mice were crossed with Aqp2-Cre mice to generate EP1fl/flAqp2Cre+ mice. The null mice developed Na+-wasting phenotype accompanied with suppressed intrarenal RAAS but unaltered systemic RAAS, almost completely analogous to that of COX-2fl/flAqp2Cre+ mice. The two strains of null mice also developed a similar extent of hyponatremia during LS intake. These results suggest that the EP1 contributes to the Na+-retaining action of CD COX-2 via intrarenal RAAS at least in the setting of Na+ depletion. Whether other EP subtypes are involved remains elusive. In particular, the EP4 subtype couples with COX-2 to regulate intrarenal PRR and renin during AngII-induced hypertension [11, 12]. Furthermore, PGE2 exhibited delayed stimulatory effect on ENaC and Na+ reabsorption by increasing intracellular cAMP levels via EP4 receptors in renal collecting duct cells [47]. Indeed, we found an increased expression of EP4 receptor in both mouse strains with Aqp2Cre-driven deletion of COX-2 or EP1 receptors. This result may suggest a compensatory stimulation of EP4-dependent pathways in the absence of EP1 receptors or COX-2. It seems reasonable to speculate a potential interaction between EP1 and EP4 subtypes in regulation of Na+ balance during Na+ depletion.
ENaC is a major Na+ transporter on the apical membrane of the CD, playing a key role in fine-tuning urinary Na+ excretion during Na+ depletion. In response to Na+ depletion or Aldo infusion in rats, renal ENaC is activated as highlighted by increased protein expression of α-ENaC and the cleavage of γ-ENaC without a significant change in β-ENaC protein expression[48, 49]. In the present study, Na+ depletion-induced increases in α-ENaC protein expression and the cleavage of γ-ENaC were similarly observed in the cortex and medulla of floxed mice undergoing Na+ depletion, and an increased β-ENaC protein expression was seen in the medulla but not the cortex. In contrast, the changes in the three subunits were all blocked by Aqp2Cre-driven COX-2 or EP1 deletion, suggesting an essential role of COX-2/EP1 pathway in determining the ENaC response to Na+ depletion. The reason of the differential regulation on β-ENaC expression in the kidney regions is unclear and may warrant future investigation. The activation of ENaC during Na+ depletion is traditionally attributed to Aldo released from adrenal glands, a key component of the systemic RAAS[8, 50]. Relatively, intrarenal generation of Aldo particularly as it is related to renal control of Na+ handling is much less understood. The present study takes advantage of CD-specific targeting of COX-2 and EP1 subtype to avoid confounding influence from non-CD cell types. Specifically, deletion of CD COX-2 or EP1 subtype effectively blocked LS-induced increases in renal and urinary Aldo and renal CYP11B2 levels without affecting circulating and adrenal levels of these parameters. Our in vitro data further showed antagonism of EP1 but not EP3 or EP4 abolished PGE2-stimulated Aldo secretion in cultured IMCD cells. These results provide strong evidence to suggest that activation of ENaC during Na+ depletion is under the control of COX-2/EP1-dependent intrarenal but not adrenal generation of Aldo. However, the human protein atlas and genecards showed that CYP11B2 expression was exclusively (https://www.proteinatlas.org/ENSG00000179142-CYP11B2/tissue) or predominantly (https://www.genecards.org/cgi-bin/carddisp.pl?gene=CYP11B2) expressed in human adrenal glands. While this data still needs to be verified, there appears to be differences in extra-adrenal CYP11B2 expression between humans and rodents. The functional role of CYP11B2 in the mouse kidney will be ideally addressed by generating renal-specific deletion of CYP11B2.
We performed radiotelemetry experiments to measure BP in COX-2fl/flAqp2Cre+ and EP1fl/flAqp2Cre+ mice and their respective floxed controls during LS intake. To our surprise, no significant changes were observed in either null strain as compared with their floxed controls under basal condition or following LS intake. Likely, despite salt-wasting, the null strains were able to maintain normal BP, highlighting powerful compensatory mechanisms in the face of Na+ depletion. It’s well known that BP is controlled by multiple interdependent factors involving renal handling of Na+ and plasma volume, vascular reactivity, sympathetic nervous system, etc. In response to Na+ depletion, the kidney plays a key role in fine-tuning urinary Na+ excretion to match Na+ intake to reestablish Na+ balance. The current study contributes to identifying a local COX-2/EP1 pathway during renal handling Na+ balance. However, the renal mechanism is not the only determinant of BP at least during LS intake. Besides the renal mechanism, Na+ depletion also enhances vascular reactivity and stimulates synthetic nervous activity. For example, Kahlil et al. conducted myograph experiments to demonstrate that the reactivity and Ca2+ mobilization was elevated in aortic rings of rabbits on a LS diet[51]. Rocks et al. provided further evidence that LS-induced vascular reactivity might be due to the altered counter-regulatory effect of angiotensin-(1–7) on AngII[52]. Evidence is also available to suggest activation of synthetic nervous activity in response to LS intake. In this regard, Volpe et al. showed that plasma norepinephrine concentration increased significantly during 7 days of LS intake, returning to baseline after 30 days of the treatment[53]. Likely, activation of vascular and neural mechanisms may help maintain BP homeostasis in the KO mice following LS intake.
The present study has several limitations. For example, the specificity of AQP2-Cre-mediated DNA recombination is not limited to in the ICs. A future study using an IC-specific strategy such as the V-ATPase B1-Cre will be needed. Secondly, the measurement of AngII and aldosterone was solely dependent on ELISA assay. The method is limited in that the absolute values can vary significantly in different assays. Additionally, the antibody against AngII in the assay may recognize AngII metabolites and therefore AngII ELISA kit actually detects AngII plus its metabolites. Similarly, the Aldo ELISA kit is also known to have cross-reactivity with other steroid hormones, especially 3β, 5β-tetrahydroaldosterone and corticosterone.
Perspectives
Activation of the systemic RAAS through macula densa COX-2-dependent renin release from juxtaglomerular cells is thought to be essential for maintenance of Na+ balance, plasma volume, and BP during Na+ restriction[4, 5, 35, 54, 55]. On the other hand, the CD is an important site for production and action of PGE2 with COX-2 being found in the ICs. The present study has defined COX-2/PGE2/EP1 pathway in the CD in maintaining Na+ homeostasis in response to LS intake through modulating the intrarenal RAAS and expression of ENaC. This finding offers new perspectives on local mechanisms within the distal nephron for regulation of Na+ homeostasis.
Supplementary Material
Novelty and Relevance.
What Is New?
We for the first time demonstrate that a local mechanism within the collecting duct (CD) involving cyclooxygenase-2 (COX-2)-derived prostaglandin E2 (PGE2) via E-prostanoid receptor 1 (EP1) receptors might play a pivotal role in maintaining Na+ homeostasis in the face of Na+ depletion by activating intrarenal renin-angiotensin-aldosterone-system (RAAS) and epithelial sodium channel (ENaC).
What Is Relevant?
Na+ is a major ion in the extracellular fluid that determines plasma volume and blood pressure. Maintenance of Na+ balance in the face of Na+ depletion has been traditionally thought to be a major function of the systemic RAAS. The present study uncovered a novel role of intrarenal RAAS in the Na+ regulatory process.
Clinical/Pathophysiological Implications?
The present study comprehensively examined the role of CD COX-2 and EP1 in renal Na+ handling during 28-day low Na+ intake and the underlying mechanism. Na+-wasting phenotype was consistently observed in mice with Aqp2Cre-driven conditional deletion of COX-2 or EP1 in the CD, accompanied with attenuated responses of intrarenal RAAS and ENaC without affecting systemic RAAS. Overall, these results support existence of Na+-retaining COX-2/PGE2/EP1 pathway in the distal nephron.
Acknowledgments
We thank Dr. Harvey Herschman (University of California) for providing COX-2fl/fl mice.
Sources of Funding
This work was supported by National Institutes of Health Grants HL139689, DK104072, HL135851, VA Merit Review I01 BX004871 from the Department of Veterans Affairs, and Postdoctoral Fellowship Award 19POST34410068 from American Heart Association. T. Yang is Senior Research Career Scientist in Department of Veterans Affairs.
Nonstandard abbreviations
- Aldo
aldosterone
- AngII
angiotensin II
- BP
blood pressure
- CD
collecting duct
- COX-2
cyclooxygenase-2
- CYP11B2
Cytochrome P450 Family 11 Subfamily B Member 2
- ENaC
epithelial sodium channel
- EP
E-prostanoid
- IC
intercalated cell
- LS
low Na+
- NS
normal Na+
- PC
principal cell
- PGE2
prostaglandin E2
- PRR
(Pro)renin receptor
- RAAS
renin-angiotensin-aldosterone system
Footnotes
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007; 59: 251–87. 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 2.Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C. Physiology of kidney renin. Physiol Rev. 2010; 90: 607–73. 10.1152/physrev.00011.2009. [DOI] [PubMed] [Google Scholar]
- 3.Mann B, Hartner A, Jensen BL, Kammerl M, Kramer BK, Kurtz A. Furosemide stimulates macula densa cyclooxygenase-2 expression in rats. Kidney Int. 2001; 59: 62–8. 10.1046/j.1523-1755.2001.00466.x. [DOI] [PubMed] [Google Scholar]
- 4.Yang T, Endo Y, Huang YG, Smart A, Briggs JP, Schnermann J. Renin expression in COX-2-knockout mice on normal or low-salt diets. Am J Physiol Renal Physiol. 2000; 279: F819–25. 10.1152/ajprenal.2000.279.5.F819. [DOI] [PubMed] [Google Scholar]
- 5.Yang T, Singh I, Pham H, Sun D, Smart A, Schnermann JB, Briggs JP. Regulation of cyclooxygenase expression in the kidney by dietary salt intake. Am J Physiol. 1998; 274: F481–9. 10.1152/ajprenal.1998.274.3.F481. [DOI] [PubMed] [Google Scholar]
- 6.Nusing RM, Treude A, Weissenberger C, Jensen B, Bek M, Wagner C, Narumiya S, Seyberth HW. Dominant role of prostaglandin E2 EP4 receptor in furosemide-induced salt-losing tubulopathy: a model for hyperprostaglandin E syndrome/antenatal Bartter syndrome. J Am Soc Nephrol. 2005; 16: 2354–62. 10.1681/ASN.2004070556. [DOI] [PubMed] [Google Scholar]
- 7.Facemire CS, Nguyen M, Jania L, Beierwaltes WH, Kim HS, Koller BH, Coffman TM. A major role for the EP4 receptor in regulation of renin. Am J Physiol Renal Physiol. 2011; 301: F1035–41. 10.1152/ajprenal.00054.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pöschke A, Kern N, Maruyama T, Pavenstädt H, Narumiya S, Jensen BL, Nüsing RM. The PGE(2)-EP4 receptor is necessary for stimulation of the renin-angiotensin-aldosterone system in response to low dietary salt intake in vivo. Am J Physiol Renal Physiol. 2012; 303: F1435–1442. [DOI] [PubMed] [Google Scholar]
- 9.Friis UG, Stubbe J, Uhrenholt TR, Svenningsen P, Nusing RM, Skott O, Jensen BL. Prostaglandin E2 EP2 and EP4 receptor activation mediates cAMP-dependent hyperpolarization and exocytosis of renin in juxtaglomerular cells. Am J Physiol Renal Physiol. 2005; 289: F989–97. 10.1152/ajprenal.00201.2005. [DOI] [PubMed] [Google Scholar]
- 10.Yang T, Xu C. Physiology and Pathophysiology of the Intrarenal Renin-Angiotensin System: An Update. J Am Soc Nephrol. 2017; 28: 1040–1049. 10.1681/ASN.2016070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F, Lu X, Peng K, Zhou L, Li C, Wang W, Yu X, Kohan DE, Zhu SF, Yang T. COX-2 mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Am J Physiol Renal Physiol. 2014; 307: F25–32. 10.1152/ajprenal.00548.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang A, Yang T. Prostaglandin E-prostanoid4 receptor mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Hypertension. 2014; 64: 369–77. 10.1161/HYPERTENSIONAHA.114.03654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramkumar N, Stuart D, Rees S, Hoek AV, Sigmund CD, Kohan DE. Collecting duct-specific knockout of renin attenuates angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2014; 307: F931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramkumar N, Ying J, Stuart D, Kohan DE. Overexpression of Renin in the collecting duct causes elevated blood pressure. Am J Hypertens. 2013; 26: 965–72. [DOI] [PubMed] [Google Scholar]
- 15.Zhang MZ, Wang S, Wang Y, Zhang Y, Ming Hao C, Harris RC. Renal Medullary Interstitial COX-2 (Cyclooxygenase-2) Is Essential in Preventing Salt-Sensitive Hypertension and Maintaining Renal Inner Medulla/Papilla Structural Integrity. Hypertension. 2018; 72: 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez AA, Luffman C, Bourgeois CR, Vio CP, Prieto MC. Angiotensin II-independent upregulation of cyclooxygenase-2 by activation of the (Pro)renin receptor in rat renal inner medullary cells. Hypertension. 2013; 61: 443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stegbauer J, Chen D, Herrera M, Sparks MA, Yang T, Königshausen E, Gurley SB, Coffman TM. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight. 2017; 2: e92720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breyer MD, Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol. 2000; 279: F12–23. 10.1152/ajprenal.2000.279.1.F12. [DOI] [PubMed] [Google Scholar]
- 19.Ishikawa TO, Herschman HR. Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis. 2006; 44: 143–9. 10.1002/gene.20192. [DOI] [PubMed] [Google Scholar]
- 20.Nelson RD, Stricklett P, Gustafson C, Stevens A, Ausiello D, Brown D, Kohan DE. Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. Am J Physiol. 1998; 275: C216–226. [DOI] [PubMed] [Google Scholar]
- 21.Xu C, Fang H, Zhou L, Lu A, Yang T. High potassium promotes mutual interaction between (pro)renin receptor and the local renin-angiotensin-aldosterone system in rat inner medullary collecting duct cells. American Journal Of Physiology-Cell Physiology. 2016; 311: C686–C695. 10.1152/ajpcell.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu S, Ying G, Wu Q, Capecchi MR. A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nat Protoc. 2008; 3: 1056–76. [DOI] [PubMed] [Google Scholar]
- 23.Ramkumar N, Stuart D, Mironova E, Abraham N, Gao Y, Wang S, Lakshmipathi J, Stockand JD, Kohan DE. Collecting duct principal, but not intercalated, cell prorenin receptor regulates renal sodium and water excretion. Am J Physiol Renal Physiol. 2018; 315: F607–F617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parreira KS, Debaix H, Cnops Y, Geffers L, Devuyst O. Expression patterns of the aquaporin gene family during renal development: influence of genetic variability. Pflugers Arch. 2009; 458: 745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jouret F, Auzanneau C, Debaix H, Wada GH, Pretto C, Marbaix E, Karet FE, Courtoy PJ, Devuyst O. Ubiquitous and kidney-specific subunits of vacuolar H+-ATPase are differentially expressed during nephrogenesis. J Am Soc Nephrol. 2005; 16: 3235–46. [DOI] [PubMed] [Google Scholar]
- 26.Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, Persson AE, Bergström G Gö, Enerbäck S. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest. 2004; 113: 1560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Al-Awqati Q. Segmental expression of Notch and Hairy genes in nephrogenesis. Am J Physiol Renal Physiol. 2005; 288: F939–52. [DOI] [PubMed] [Google Scholar]
- 28.Wu H, Chen L, Zhou Q, Zhang X, Berger S, Bi J, Lewis DE, Xia Y, Zhang W. Aqp2-expressing cells give rise to renal intercalated cells. J Am Soc Nephrol. 2013; 24: 243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trepiccione F, Soukaseum C, Iervolino A, Petrillo F, Zacchia M, Schutz G, Eladari D, Capasso G, Hadchouel J. A fate-mapping approach reveals the composite origin of the connecting tubule and alerts on “single-cell”-specific KO model of the distal nephron. Am J Physiol Renal Physiol. 2016; 311: F901–F906. [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, Brown D, Burg MB, Knepper MA. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A. 2017; 114: E9989–E9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L, Gao C, Zhang L, Zhang Y, Chen E, Zhang W. Highly tamoxifen-inducible principal cell-specific Cre mice with complete fidelity in cell specificity and no leakiness. Am J Physiol Renal Physiol. 2018; 314: F572–F583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang F, Lu X, Peng K, Fang H, Zhou L, Su J, Nau A, Yang KT, Ichihara A, Lu A, Zhou SF, Yang T. Antidiuretic Action of Collecting Duct (Pro)Renin Receptor Downstream of Vasopressin and PGE2 Receptor EP4. J Am Soc Nephrol. 2016; 27: 3022–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peti-Peterdi J, Komlosi P, Fuson AL, Guan Y, Schneider A, Qi Z, Redha R, Rosivall L, Breyer MD, Bell PD. Luminal NaCl delivery regulates basolateral PGE2 release from macula densa cells. J Clin Invest. 2003; 112: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest. 1994; 94: 2504–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang T, Park JM, Arend L, Huang Y, Topaloglu R, Pasumarthy A, Praetorius H, Spring K, Briggs JP, Schnermann J. Low chloride stimulation of prostaglandin E2 release and cyclooxygenase-2 expression in a mouse macula densa cell line. J Biol Chem. 2000; 275: 37922–9. [DOI] [PubMed] [Google Scholar]
- 36.Ye W, Zhang H, Hillas E, Kohan DE, Miller RL, Nelson RD, Honeggar M, Yang T. Expression and function of COX isoforms in renal medulla: evidence for regulation of salt sensitivity and blood pressure. Am J Physiol Renal Physiol. 2006; 290: F542–9. [DOI] [PubMed] [Google Scholar]
- 37.Yang T, Singh I, Pham H, Sun D, Smart A, Schnermann JB, Briggs JP. Regulation of cyclooxygenase expression in the kidney by dietary salt intake. Am J Physiol. 1998; 274: F481–9. [DOI] [PubMed] [Google Scholar]
- 38.Yu Y, Stubbe J, Ibrahim S, Song WL, Smyth EM, Funk CD, FitzGerald GA. Cyclooxygenase-2-dependent prostacyclin formation and blood pressure homeostasis: targeted exchange of cyclooxygenase isoforms in mice. Circ Res. 2010; 106: 337–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Zhao M, He W, Milne GL, Howard JR, Morrow J, Hébert RL, Breyer RM, Chen J, Hao CM. Increased dietary NaCl induces renal medullary PGE2 production and natriuresis via the EP2 receptor. Am J Physiol Renal Physiol. 2008; 295: F818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He W, Zhang M, Zhao M, Davis LS, Blackwell TS, Yull F, Breyer MD, Hao CM. Increased dietary sodium induces COX2 expression by activating NFκB in renal medullary interstitial cells. Pflugers Arch. 2014; 466: 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stokes JB. Effect of prostaglandin E2 on chloride transport across the rabbit thick ascending limb of Henle. Selective inhibitions of the medullary portion. J Clin Invest. 1978; 64: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue C, Siragy HM. Local renal aldosterone system and its regulation by salt, diabetes, and angiotensin II type 1 receptor. Hypertension. 2005; 46: 584–90. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez AA, Salinas-Parra N, Leach D, Navar LG, Prieto MC. PGE2 upregulates renin through E-prostanoid receptor 1 via PKC/cAMP/CREB pathway in M-1 cells. American Journal Of Physiology-Renal Physiology. 2017; 313: F1038–F1049. 10.1152/ajprenal.00194.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salinas-Parra N, Reyes-Martinez C, Prieto MC, Gonzalez AA. Prostaglandin E2 Induces Prorenin-Dependent Activation of (Pro)renin Receptor and Upregulation of Cyclooxygenase-2 in Collecting Duct Cells. Am J Med Sci. 2017; 354: 310–318. 10.1016/j.amjms.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breyer MD, Breyer RM. G protein-coupled prostanoid receptors and the kidney. Annu Rev Physiol. 2001; 63: 579–605. [DOI] [PubMed] [Google Scholar]
- 46.Olesen ET, Fenton RA. Is there a role for PGE2 in urinary concentration? J Am Soc Nephrol. 2013; 24: 169–78. [DOI] [PubMed] [Google Scholar]
- 47.Mansley MK, Niklas C, Nacken R, Mandery K, Glaeser H, Fromm MF, Korbmacher C, Bertog M. Prostaglandin E2 stimulates the epithelial sodium channel (ENaC) in cultured mouse cortical collecting duct cells in an autocrine manner. J Gen Physiol. 2020; 152: e201912525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest. 1999; 104: R19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frindt G, Ergonul Z, Palmer LG. Na channel expression and activity in the medullary collecting duct of rat kidney. Am J Physiol Renal Physiol. 2007; 292: F1190–6. [DOI] [PubMed] [Google Scholar]
- 50.Terker AS, Ellison DH. Renal mineralocorticoid receptor and electrolyte homeostasis. Am J Physiol Regul Integr Comp Physiol. 2015; 309: R1068–70. 10.1152/ajpregu.00135.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khalil RA, Crews JK, Carroll JF, Hall JE. Enhanced vascular reactivity and Ca2+ entry with low-salt diet: effect of obesity. Hypertension. 1999; 34: 882–8. [DOI] [PubMed] [Google Scholar]
- 52.Roks AJ, Nijholt J, van Buiten A, van Gilst WH, de Zeeuw D, Henning RH. Low sodium diet inhibits the local counter-regulator effect of angiotensin-(1–7) on angiotensin II. J Hypertens. 2004; 22: 2355–61. [DOI] [PubMed] [Google Scholar]
- 53.Volpe M, Müller FB, Trimarco B. Transient enhancement of sympathetic nervous system activity by long-term restriction of sodium intake. Circulation. 1985; 72: 47–52. [DOI] [PubMed] [Google Scholar]
- 54.Harris RC, Mckanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD. Cyclooxygenase-2 Is Associated with the Macula Densa Of Rat-Kidney And Increases with Salt Restriction. Journal Of Clinical Investigation. 1994; 94: 2504–2510. Doi 10.1172/Jci117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harding P, Carretero OA, Beierwaltes WH. Chronic cyclooxygenase-2 inhibition blunts low sodium-stimulated renin without changing renal haemodynamics. Journal Of Hypertension. 2000; 18: 1107–1113. Doi 10.1097/00004872-200018080-00016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.