

Abstract
The currently circulating Omicron sub-variants are the SARS-CoV-2 strains with the highest number of known mutations. Herein, we found that human angiotensin-converting enzyme 2 (hACE2) binding affinity to the receptor-binding domains (RBDs) of the four early Omicron sub-variants (BA.1, BA.1.1, BA.2, and BA.3) follows the order BA.1.1 > BA.2 > BA.3 ≈ BA.1. The complex structures of hACE2 with RBDs of BA.1.1, BA.2, and BA.3 reveal that the higher hACE2 binding affinity of BA.2 than BA.1 is related to the absence of the G496S mutation in BA.2. The R346K mutation in BA.1.1 majorly affects the interaction network in the BA.1.1 RBD/hACE2 interface through long-range alterations and contributes to the higher hACE2 affinity of the BA.1.1 RBD than the BA.1 RBD. These results reveal the structural basis for the distinct hACE2 binding patterns among BA.1.1, BA.2, and BA.3 RBDs.
Keywords: SARS-CoV-2, Omicron, hACE2, RBDs
Graphical abstract
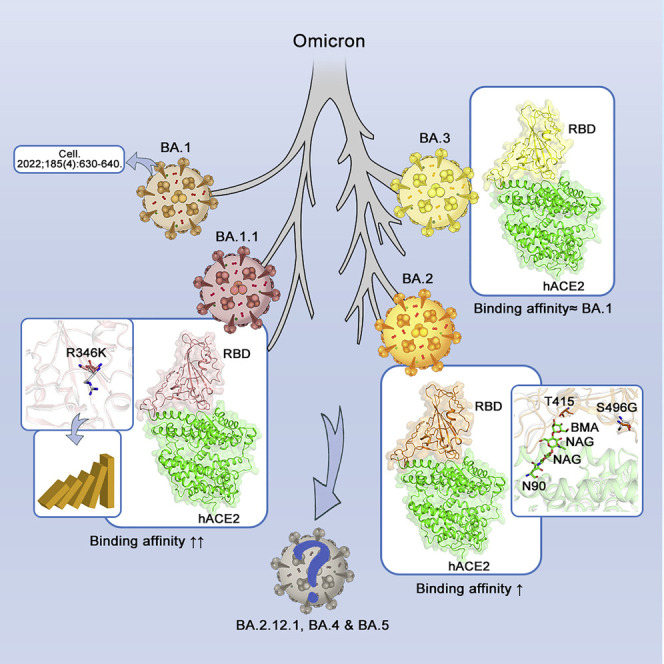
The biochemical and structural analysis of the human angiotensin-converting enzyme-2 (ACE2, the receptor for SARS-CoV-2 viral entry) and the receptor-binding domain (RBD) of four Omicron sub-variants—BA.1, BA.1.1, BA.2, and BA.3—helps to reveal the structural basis of differences in sub-variant binding affinities and the impact of RBD mutations.
Introduction
Omicron (B.1.1.529), which is the SARS-CoV-2 variant with most mutations, was defined as the fifth variant of concern (VOC) by the World Health Organization (WHO) on November 26, 2021 (Kandeel et al., 2022). The Omicron variants were recently divided into four sub-variants, i.e., BA.1, BA.1.1, BA.2, and BA.3 (PAHO and WHO, 2022). The BA.2 sub-variant was suggested to have a growth advantage over other circulating variants and is more transmissible than the BA.1 sub-variant. Statistical analysis shows that the effective reproduction number of BA.2 is 1.4-fold higher than that of BA.1 (Yamasoba et al., 2022). During the revision of this manuscript, some more Omicron sub-variants are emerging, e.g., BA.2.12.1, BA.4, BA.5, etc.
The growth advantage of BA.2 was thought to be related to its higher capacity for immune escape. However, the difference between the level of neutralizing antibodies against BA.1 and BA.2 in individuals who accepted vaccination or experienced SARS-CoV-2 infection was small, so it is difficult to explain the higher transmissibility of BA.2 than BA.1 (Bruel et al., 2022; Callaway, 2022; Iketani et al., 2022; Yu et al., 2022).
SARS-CoV-2 binding to its receptor, human angiotensin-converting enzyme 2 (hACE2), is a critical step for initiating the infection. The binding of SARS-CoV-2 depends on the interactions between hACE2 and the receptor-binding domain (RBD) in the spike (S) protein of the virus. There are 15–16 amino acid (aa) mutations in the RBD of Omicron sub-variants compared with the prototype (PT) virus (Lu et al., 2020b; Tan et al., 2020; Wei et al., 2020).
The RBD of the S protein is also the major target of neutralizing antibodies (Dai and Gao, 2021; Peng et al., 2021; Xu et al., 2021). Previously, we solved both the crystal and cryo-EM structures of the Omicron BA.1 RBD in complex with hACE2, revealing that although some mutations in the BA.1 RBD increase the affinity to hACE2, the others attenuate it. Consequently, the affinity of Omicron BA.1 RBD is similar to that of the PT strain RBD (Han et al., 2022). In this report, we evaluated the binding properties of BA.1.1, BA.2, and BA.3 RBDs to hACE2 and explored the structural basis for the difference in their binding affinities.
Results
Affinity of RBDs from the SARS-CoV-2 variants to hACE2 and entry efficiency of the pseudoviruses
Among the SARS-CoV-2 RBDs, there are 15 aa mutations in BA.1 and 16 in BA.1.1 with an extra mutation at R346K. BA.2 and BA.3 contain 16 and 15 aa mutations, correspondingly (Figure 1 A). As of April 28, 2022, there have been 75 Pango Omicron sub-variants. Among these sub-variants, BA.1.1 is the globally dominant Omicron sub-variant, accounting for approximately 26.62% (874,168) of the total Omicron genomes. The other top 10 sub-variants are BA.2.9, BA.2, BA.1.17.2, BA.1.15, BA.1.16, BA.1, BA.1.1.1, BA.2.10, and BA.1.14, accounting for approximately 10.55% (346,329), 8.83% (290,135), 6.50% (213,581), 6.34% (208,159), 4.80% (157,468), 4.59% (150,661), 2.73% (89,652), 2.46% (80,763), and 2.37% (77,890) of the total Omicron genomes, correspondingly (Figure 1B) (outbreak.info, 2022).
Figure 1.

Mutation distribution in VOC S proteins and SPR for VOC RBD binding to hACE2 and VOC pseudovirus entry assay
(A) Schematic diagram for mutation distribution in the S proteins from the five VOCs, including the four Omicron sub-variants (BA.1, BA.1.1, BA.2, and BA.3). The mutations unique to BA.1.1, BA.2, or BA.3 are indicated in blue. The mutations unique to BA.2 and BA.3 are indicated in green. The mutations present in BA.1 and BA.1.1 but absent in BA.2 and BA.3 are shown with dashed lines.
(B) Donut chart for the global composition of Omicron sub-variants (data were collected from GISAID on April 25th, 2022).
(C) The SPR curves for the VOC RBDs binding to hACE2.
(D) Pseudovirus entry assay for the five VOCs, including the four Omicron sub-variants (BA.1, BA.1.1, BA.2, and BA.3). The error bars indicate the standard deviations (SD) for three or four independent experiments.
See also Figure S1.
We assessed the binding affinities between hACE2 and RBDs from the four Omicron sub-variants (BA.1, BA.1.1, BA.2, and BA.3), together with the other earlier four VOCs (Alpha, Beta, Gamma, and Delta), and the PT virus (Lu et al., 2020a; Wei et al., 2020) by surface plasmon resonance (SPR) (Figure 1C). Our results showed that the K D values for the binding of the PT RBD to hACE2 is 24.4 nM, similar to what was previously reported (Han et al., 2022; Wang et al., 2020; Zhang et al., 2021), whereas those of Omicron sub-variants, BA.1, BA.1.1, BA.2, and BA.3, were 19.5, 5.9, 10.0, and 22.1 nM, correspondingly. Compared with BA.1 RBD, the K D values of BA.1.1 and BA.2 were 3.3- and 2.0-fold lower, respectively, indicating that their affinities to hACE2 were a bit higher than that of the PT RBD. However, the affinity of BA.3 to hACE2 was very close to BA.1. The K D values of the RBDs binding to hACE2 in Alpha, Beta, Gamma, and Delta variants were 6.7, 19.7, 16.0, and 25.1 nM, correspondingly, suggesting that only Alpha RBD showed a comparable affinity with BA.1.1. The increased affinity of the BA.1.1 and BA.2 compared with BA.1 is consistent with the high frequencies of these two sub-variants in GISAID. We further detected the binding capacity of the above RBDs to hACE2 expressed on the surface of BHK-21 cells by flow cytometry (Figure S1 ). These results suggested that all the RBDs tested could efficiently bind to hACE2 on the cell surface. In order to detect the entry efficiency of the Omicron sub-variants, we prepared the VSV-backbone pseudoviruses expressing the S proteins from the SARS-CoV-2 PT and the VOCs Alpha, Beta, Gamma, Delta, and the four Omicron sub-variants. The entry efficiency of the pseudoviruses into Vero cells was evaluated (Figure 1D). All Omicron sub-variants were able to enter the cells efficiently. The relative entry efficiencies of Omicron sub-variants are consistent with the recent research (Saito et al., 2022).
Figure S1.

Flow cytometry assay for VOC RBDs binding to hACE2-expressing BHK-21 cells, related to Figure 1
The BHK-21 transfected with hACE2 fused with eGFP were incubated with the indicated His-tagged RBDs and stained with the anti-His-APC antibody. Non-transfected, non-stained control: the cells were neither transfected with hACE2 nor stained with the RBDs and the anti-His-APC antibody. Transfected, non-stained control: the cells were transfected with hACE2 but not stained with the RBDs and the anti-His-APC antibody.
Structural comparison of hACE2 in complex with BA.1, BA.1.1, BA.2, and BA.3 RBDs
The crystal structures of BA.1.1, BA.2, and BA.3 RBDs in the complex with hACE2 have been solved at resolutions of 3.0, 2.9, and 2.7 Å, correspondingly, in this study (Table S1).
We performed an interface analysis of the three complex structures with the cutoff for the atom contact distance being 4.5 Å (the cutoff distance for hydrogen bonds and salt bridges being 3.5 Å) and made a comparison with the interaction interface in the BA.1 RBD/hACE2 structure we previously solved (Han et al., 2022). The results show that the same 21 hACE2 residues are involved in constituting the binding interface with 20 BA.1 RBD residues, 21 BA.1.1 RBD residues, 21 BA.2 RBD residues, and 19 BA.3 RBD residues in the four complexes, correspondingly. The interaction atom pairs and numbers of interface atom contacts, hydrogen bonds, and salt bridges are listed in Tables S2 and S3.
Among the BA.1 RBD/hACE2, BA.2 RBD/hACE2, and BA.3 RBD/hACE2 complexes, the BA.2 RBD/hACE2 complex interface has the largest total number of atom contacts and hydrogen bonds, which might correlate with the higher hACE2 affinity of the BA.2 RBD compared with BA.1 and BA.3. However, the BA.1.1 RBD with the highest hACE2 affinity forms a less total number of atom contacts and hydrogen bonds with hACE2 than BA.2 RBD, but it has two more salt bridges than BA.2 RBD (Figure S2 ; Table S3). Therefore, the descending order of BA.1.1, BA.2, BA.1, and BA.3 RBD affinity to hACE2 can be partly explained by the different number of total atomic interactions, hydrogen bonds, and salt bridges, although other factors including the strength of individual interactions, especially those of critical mutation sites also play a role. This is consistent with our previous report that the lower affinity of dog ACE2 (dACE2) to the SARS-CoV-2 RBD than hACE2 can be explained by fewer interface atom contacts, hydrogen bonds, and salt bridges of dACE2 with RBD (Zhang et al., 2021).
Figure S2.

Salt bridges formed between BA.1.1 RBD/hACE2 and BA.2 RBD/hACE2 interfaces, related to Figure 2
(A) BA.1.1 RBD R493 forms four salt bridges with hACE2 E35 and D38.
(B) BA.2 RBD R493 forms a salt bridge with hACE2 E35 and BA.2 RBD R498 forms a salt bridge with hACE2 D38.
The contact interfaces between hACE2 and SARS-CoV-2 or the Omicron sub-variant BA.1 RBD are distributed over two patches (Han et al., 2022; Wang et al., 2020). As Patch 1 in the BA.1 RBD/hACE2 complex, which we previously reported (Han et al., 2022), all Patch 1 of the three complexes solved here contains 5 hACE2 residues (S19, Q24, H34, E35, and Y83), which form hydrogen bonds or salt bridges with RBD residues, but Patch 1 of the BA.1.1 complex has an extra hACE2 D38, forming a hydrogen bond and a salt bridge with BA.1.1 RBD S496 and R493, correspondingly (Figures 2 A–2D, upper panels).
Figure 2.

The complex structures of SARS-CoV-2 sub-variants BA1.1, BA.2, and BA.3 RBDs bound to hACE2
(A–D) The hydrogen bond (red) and salt bridge (yellow) networks of BA.1 (A), BA1.1 (B), BA.2 (C), and BA.3 (D) RBDs contact with hACE2. The complex structures are shown as a cartoon, and residues involved in hydrogen bond or salt bridge formation are shown as sticks. The RBDs of BA.1, BA1.1, BA.2, and BA.3 are shown as cyan, salmon, orange, and yellow, correspondingly. hACE2 is shown as green.
(E–H) The binding surface of hACE2 with the BA.1 (E), BA1.1 (F), BA.2 (G), and BA.3 (H) RBDs.
See also Figure S2.
Unlike the BA.1.1 complex, hACE2 D38 in the BA.1 RBD, BA.2 RBD, and BA.3 RBD complexes forms a hydrogen bond and a salt bridge with RBD Y449 and R498, correspondingly, in all the three complexes. In addition to hACE2 D38, Patch 2 of the BA.1.1 complex contains Y41, K353, and D355, whereas that of the three complexes contains hACE2 Y41, Q42, and K353. The hydrogen bonds network in Patch 2 of the BA.2 RBD complex is the most extensive, whereas that of the BA.1.1 RBD complex is the weakest (Figures 2A–2D, lower panels).
The binding interface residues of RBDs in the four complexes shown in (Figures 2E–2H) are evidently different. The two patches of the BA.1 and BA.1.1 RBDs are connected, whereas the boundaries of those in the BA.2 and BA.3 are distinguishable. Among all the mutations in the four RBDs, only the G496S is located in the binding interface. Notably, T415 of BA.2 RBD forms a hydrogen bond with hACE2 N90-glycan, which is discussed later (Figure 2G).
Correlation of the S496G substitution in the RBDs with hACE2 binding
The affinity of BA.2 RBD to hACE2 is a little bit higher, if any, than that of the BA.1/BA.3 RBDs. The difference between the BA.2 RBD and the BA.1 RBD is that BA.2 contains S371F, T376A, D405N, and R408S mutations, but BA.1 RBD does not, whereas BA.1 contains the G446S and G496S mutations, but BA.2 does not. Among these mutations, only the G496S mutation is in the RBD/hACE2 interface. In the BA.1 RBD/hACE2, BA.2 RBD/hACE2, and BA.3 RBD/hACE2 complexes, hACE2 D38 forms a salt bridge and hydrogen bond networks with RBD R498 and Y449. However, the side chains of these residues appear less compact in the BA.1 RBD/hACE2 complex with the G496S mutation, whereas in the BA.1.1 complex with the same mutation, this interaction network is disrupted (Figure 3 A). Therefore, we speculate that S496 may disturb the polar interaction network formed between hACE2 D38 and RBD R498/Y449.
Figure 3.

The G496S mutation in RBD and N90-glycan in hACE2 modulate the RBDs/hACE2 interaction
(A) The G496S mutation affects the local polar interaction networks at the RBD/hACE2 interfaces.
(B) The interaction between hACE2 N90-glycan may be related to the R408S mutations in the BA.2 RBD/hACE2 complex. The four panels from the leftmost one in (A) and (B) are BA.1 RBD/hACE2 (PDB: 7WBP), BA.1.1 RBD/hACE2, BA.2 RBD/hACE2, and BA.3 RBD/hACE2, correspondingly. The numbers on the dash lines are distances (Å) between the two atoms they link.
See also Figures S3–S6.
To verify the effects of G496S on receptor binding, we first evaluated the reverse mutation of S496G in BA.1.1 RBD on the ACE2 affinity. Our results indicate that the S496G site mutation induces an approximately 3.6-fold affinity increase, whereas the G496S mutation in BA.2 RBD induces an approximately 1.5-fold decrease in hACE2 binding affinity (Figure S3 ). Therefore, the SPR measurements support that G496S attenuates RBD binding.
Figure S3.

The SPR curves for the PT RBD, BA.1.1 RBD, BA.1.1 with the S496G mutation, BA.2 RBD, and BA.2 with the G496S mutation binding to hACE2, related to Figure 3
The mean and standard deviation of KD values representing three independent experiments are shown.
To further explore the mechanism for the reduced binding associated with the G496S mutation, we performed molecular dynamics (MDs) simulations and examined the effect of the G496S mutation on the free energy of BA.1 RBD binding. Our results indicate that the molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) free energy of the BA.1 RBD with G496 binding to hACE2 is 5.34 kcal/mol (ΔΔEBinding) lower than that with S496 (Table S4). Representative snapshots clustered from the MD trajectories suggested that the more compact intra-chain hydrogen bonding networks formed by the hydroxyl side chain of S496 within RBD reduce the inter-chain interactions between BA.1 RBD and ACE2 (Figure S4 ). For control, we also performed the simulations of S446 BA.1 versus G446 BA.1 binding, showing that ΔΔEBinding between S446 and G446 is only 1.15 kcal mol−1, which is much lower than the ΔΔEBinding between S496 and G496. These results also support that the G496S mutation attenuates the binding affinity of RBDs to hACE2.
Figure S4.

Representative snapshots clustered from MD trajectories of the (A and B) S496 BA.1 RBD-ACE2 system and the (C and D) G496 BA.1 RBD-ACE2 system, related to Figure 3
RBD domains are colored in cyan, and ACE2 is colored in yellow. H-bonds are displayed as black dotted lines.
Interestingly, the visibility of the electronic densities of the hACE2 N90-linked glycans was very different in the four complexes. The densities for the first three glycan residues (N-acetyl-β-d-glucosamine [NAG], NAG, and Beta-D-mannose [BMA]) of the hACE2 N90-linked glycan in the BA.2 RBD/hACE2 complex were sharp and clear, but only the densities of the first residues (NAG) of ACE2 N90-glycan in the BA.1 RBD/hACE2 and BA.1.1 RBD/hACE2 complexes and the first two residues (NAG and NAG) of ACE2 N90-glycan in the BA.3 RBD/hACE2 complex were visible (Figure 3B). More importantly, in the BA.2 RBD/hACE2 complex, the third residue (BMA) of the hACE2 N90-glycan chain forms a hydrogen bond with BA.2 T145, which stabilizes the glycan chain (Figures 3B and S5 ) and presumably increases the affinity of the BA.2 RBD to hACE2. The unique mutation of BA.2 is R408S. We hypothesized that the large side chain of R408 in RBDs of BA.1, BA.1.1, and BA.3 has steric clashes with the N90-glycan chain. With the R408S mutation in BA.2, the steric clashes decrease, and therefore, the N90-glycan has a higher probability to get close and bind to T415. The glycosylation of viral envelope proteins together with receptors has a wide range of functions, including regulating viral tropism (Tian et al., 2021). Here, we present a molecular insight into how glycosylation regulates virus receptor binding.
Figure S5.

Electron densities of the hydrogen bond between BA.2 T415 and hACE2 N90-glycan, related to Figure 3
The local 2Fo-Fc electronic density map at 1.0 σ for hACE2 N90-glycan and BA.2 T415 is shown as mesh lines. The hydrogen bond is shown as the yellow dash line and its length is indicated.
To test the role of the R408S mutation on RBD/hACE2 interaction, we assessed the hACE2 affinity of BA.2 RBD with the S408R mutation, showing that the K D values of the BA.2 RBD (15.67 nM) and BA.2 RBD with the S408R mutation (16.37 nM) were also similar (Figure S6 ). These results suggest that the R408S mutation has very limited RBD binding to hACE2.
Figure S6.

The SPR curves for the PT RBD, BA.2, and BA.2 with the S408R mutation, related to Figure 3
The mean and standard deviation of KD values representing three independent experiments are shown.
Correlation of the enhanced binding of the BA.1.1 RBD to hACE2 compared with the BA.1 RBD with the long-distance “conduction” interactions induced by R346K
Our SPR data showed that the affinity of the BA.1.1 RBD to hACE2 is higher than the BA.1 RBD. Sequence alignment indicated that there is only one substitution residue (R346K) located far away from the receptor-binding motif (RBM) between BA.1 RBD and BA.1.1 RBD. Although this residue does not contact with hACE2 directly (Figure 4 A), the binding affinity of BA.1.1 RBD with hACE2 was enhanced compared with BA.1 RBD with hACE2 (Figure 1C). To explore the underlying molecular mechanism, we determined the structure of the BA.1.1 RBD/hACE2 complex.
Figure 4.

A single substitution far away from the binding motif of BA.1.1 RBD changes the interaction network with hACE2
(A–C) Structural comparison between BA.1 RBD/hACE2 complex and BA.1.1 RBD/hACE2 complex. The complex structures are shown as a cartoon and the key amino acids are shown as sticks. The key amino acids in BA.1 RBD and BA.1.1 RBD are colored by cyan and salmon, correspondingly. The key residues in the hACE2 are colored green.
(D and E) Pattern diagram of R346K substitution in BA.1.1 RBD changing the interaction network. (D) Schematic of the conformation of the key amino acids in BA.1 RBD.
(E) Schematic of the conformation change of the key amino acids after R346K substitution in BA.1.1 RBD.
In the BA.1 RBD/hACE2 complex, R346 “pushes” N450 and changes the conformation of the corresponding loop to hACE2. Then, Y449 on the same loop contacts with D38 of hACE2 (Figure 4, Figure 4A, 4B, and 4D). When R346 was substituted by K346, it points to the opposite side and does not influence N450, the corresponding loop springs back, and the Y449 no longer contacts with D38 of hACE2. D38 of hACE2 points to the other side and loses the interactions with Y449 and R498 (Figure 4, Figure 4A, 4C, and 4E) and contacts with R493 of BA.1.1 RBD instead. Consequently, BA.1.1 RBD R493 forms salt bridges with E35 and D38 of hACE2, and it also forms contacts with H34 of hACE2. In addition, BA.1.1 RBD S496 forms a hydrogen bond with hACE2 D38. Therefore, these residues form a hub that generates strong interface interactions (Figures 4C and 4E). In summary, although R346K is far away from the RBM region, it also enhances the binding affinity of BA.1.1 RBD with hACE2 through long-distance “conduction” interactions like the “butterfly effect” (Figure 4E).
Discussion
Since its first appearance, SARS-CoV-2 Omicron disseminated rapidly and became the dominant circulating variant globally. Although only four Omicron sub-variants (BA.1, BA.1.1, BA.2, and BA.3) were defined as of February 3, 2022 (PAHO and WHO, 2022), the Omicron family of viruses continues to grow in complexity. Over the past few months, several new Omicron sub-variants are gaining attention, including BA.2.12.1, which is spreading widely in the Northeast United States, and BA.4 and BA.5, which are growing rapidly to dominate South Africa (Haseltine, 2022; Maxmen, 2022). These sub-variants can evolve mutations to specifically evade humoral immunity (Cao et al., 2022). Previous studies showed that hACE2 affinities of Omicron (B.1.1.529 or BA.1) S proteins/RBDs are comparable with those of PT S proteins/RBDs (Han et al., 2022; Mannar et al., 2022) or higher than them (Cameroni et al., 2022; Cui et al., 2022; Lan et al., 2022; Yin et al., 2022). In this study, we showed that RBDs from other two Omicron sub-variants, BA.1.1 and BA.2, have higher hACE2 affinities than that of BA.1 and gave structural explanations for the difference in binding ability.
First, we propose that the absence of the G496S mutation is related to the enhanced affinity of BA.2 RBD. Here, we observed that S496 disturbs the local interaction networks in hACE2 D38, RBD R498, and Y449. Our SPR data and MD simulations of BA.1.1 RBD S496G mutants confirmed that the G496S mutation reduces RBD affinity. Our results are consistent with previous findings showing that G496S mutation attenuates the affinity of the RBD to hACE2 in deep mutational scanning studies (Starr et al., 2020; Haseltine, 2021).
An intriguing finding of this study is that hACE2 N90-glycan forms a hydrogen bond with BA.2 T415. The explicit binding of BA.2 T415 to hACE2 N90-glycan and the enhanced hACE2 affinity of BA.2 RBD led us to propose that N90-glycan/T415 interaction is favorable to the binding of BA.2 RBD to hACE2. However, Chan et al. reported that hACE2 N90-glycosylation inhibits PT RBD binding to hACE2 (Chan et al., 2020). Solving this paradox, we propose that the binding of hACE2 N90-glycan to BA.2 RBD T415 alleviates the inhibitory effect of N90-glycan on RBD/hACE2 binding. Because of the adjacency of the R408S mutation to T415, we hypothesized that the binding of hACE2 N90-glycan to T415 is related to the R408S mutation in BA.2. However, the SPR results showed that the R408S mutation in BA.1.1 RBD and the S408R reverse mutation in BA.2 RBD had little effect on the hACE2 affinity. Therefore, either the R408S mutation has indeed no effect on the glycan/T415 binding, or the effect is too weak to be detected by SPR, i.e., the limitation of the method used. Anyway, here we provide evidence showing that hACE2 glycan is involved in SARS-CoV-2 RBD binding.
Another interesting finding is that the R346K mutation induces major alterations at the RBD/hACE2 interface through long-distance conduction interactions. Mutations located outside of the binding interface affecting the affinity via long-range conduction effects have been reported previously (Joughin et al., 2005; Unal et al., 2013). Actually, the “push” configuration induced by the R346K mutation captured by crystallography may represent one of the dynamic states for BA.1.1. Collectively, the available evidence warns against the misleading concept suggesting that only interface residues determine affinity and explain the role of SARS-CoV-2 RBD mutations.
Limitations of the study
Receptor binding is the initial step for SARS-CoV-2 infection. In this study, we focused on the interaction between the RBDs of four Omicron sub-variants and hACE2. The transmissibility of a virus is affected by multiple factors of complex nature, such as viral stability, receptor-binding affinity, fusion efficiency, replication activity, host immune responses, etc. To provide a deeper understanding of how the Omicron variants are transmitted, these aspects need to be further investigated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
Bacterial strains | ||
| Escherichia coli strain DH5α | Vazyme | Cat# C502-02 |
| Escherichia coli strain BL21 (DE3) | Vazyme | Cat# C504-02 |
| Escherichia coli strain DH10Bac | Gibco | Cat# 10361012 |
|
Chemicals, antibodies and recombinant proteins | ||
| Polyethylenimine (PEI) | Polysciences | Cat# 24885-2 |
| Anti-His/APC | Miltenyi Biotec | Cat# 130-119-820; RRID:SCR_008984 |
| FuGENE 6 Transfection Reagent | Promega | Cat# E2691 |
| BaseMuncher endonuclease | Abcam | Cat# ab270049 |
| SARS-CoV-2 prototype RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_402119 |
| SARS-CoV-2 Alpha RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_683466 |
| SARS-CoV-2 Beta RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_678615 |
| SARS-CoV-2 Gamma RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_833172 |
| SARS-CoV-2 Delta RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_2020954 |
| SARS-CoV-2 Omicron BA.1 RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_6640916 |
| SARS-CoV-2 Omicron BA.1.1 RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_ 496747 |
| SARS-CoV-2 Omicron BA.2 RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_9845731 |
| SARS-CoV-2 Omicron BA.3 RBD protein with His-tag, spike residues 319-541 | This paper | accession number: EPI_ISL_7605713 |
| Human ACE2 protein, residues 19-615 | This paper | accession number: NP_001358344 |
|
Critical commercial assays | ||
| HisTrap HP 5 mL column | GE Healthcare | Cat# 17524802 |
| Superdex 200 Increase 10/300 GL | GE Healthcare | Cat# 28990944 |
| HiLoad 16/600 Superdex 200 pg | GE Healthcare | Cat# 28989335 |
| Series S Sensor Chip SA | GE Healthcare | Cat# 29104992 |
| Membrane concentrator | Millipore | Cat# UFC901096 |
| Membrane concentrator | Millipore | Cat# SLHP033RB |
| Deposited data | ||
| Omicron BA.1.1 RBD/hACE2 complex (X-RAD) | This paper | PDB: 7XAZ |
| Omicron BA.2 RBD/hACE2 complex (X-RAD) | This paper | PDB: 7XB0 |
| Omicron BA.3 RBD/hACE2 complex (X-RAD) | This paper | PDB: 7XB1 |
| Experimental models: Cell lines | ||
| HEK293F cells | Gibco | Cat# 11625-019 |
| HEK293T cells | ATCC | ATCC CRL-3216 |
| BHK-21 cells | ATCC | CCL-10 |
| Sf9 cells | Gibco | Cat# 11496015 |
| Recombinant DNA | ||
| pEGFP-N1 | MiaoLingPlasmid | Cat# P0133 |
| pCAGGS | MiaoLingPlasmid | Cat# P0165 |
| pFastBac1 | Invitrogen | Cat# 10359-016 |
| Software and algorithms | ||
| PyMOL software | Molecular Graphics System, Version 1.8 Schrö dinger | https://pymol.org/2/ |
| Graphpad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| BIAcore 8K Evaluation software | GE Healthcare | N/A |
| FlowJo V10 | FLOWJO | https://www.flowjo.com/solutions/flowjo/downloads |
| HKL2000 | HKL Research | https://www.hkl-xray.com/hkl-2000 |
| COOT | (Emsley and Cowtan, 2004) | http://www.mrc-lmb.cam.ac.uk/personal/peemsley/coot/ |
| Phenix | (Adams et al., 2010) | http://www.phenix-online.org/ |
| MolProbity | Duke Biochemistry | http://molprobity.biochem.duke.edu/index.php |
| GROMACS | (Abraham et al., 2015) | http://www.gromacs.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, George F. Gao (gaof@im.ac.cn).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cells
HEK293F suspension-cultured cells (Gibco, Cat#11625-019) were cultured at 37 °C in SMM 293-TII Expression Medium (Sino Biological, Cat# M293TII), and BHK-21 adherent cells (ATCC CCL-10) were cultured at 37 °C in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Method details
Gene cloning
The extracellular domain of hACE2 (residues 19-615, GenBank: NP_001358344) was cloned into the Baculovirus transfection vector pFastBac1 (Invitrogen) with the gp67 signal peptide sequence added to the N-terminus of the hACE2 gene and the Hexa-His tag sequence at the C-terminus. The coding sequences of RBD of SARS-CoV-2 (residue 319-541), RBDs of VOCs (residue 319-541) and hACE2 (residues 19-615, GenBank:NP_001358344) including the Hexa-His tag sequence at the C-terminus were inserted into the pCAGGS vector.
Protein expression and purification
The pFastBac1-hACE2 recombinant bacmids produced by transforming the plasmid into DH10Bac E.coli. were transfected into Sf9 cells using FuGENE 6 Transfection Reagent (Promega). The virus was amplified, and the High Five cells were used for protein expression. Supernatants were collected after 48 h-infection. The pCAGGS-hACE2 and pCAGGS-RBDs were expressed in HEK293F cells. Cell culture supernatants were collected after a 5-day infection.
The supernatants containing the proteins mentioned before were purified using His-Trap HP columns (GE Healthcare) and the SuperdexTM 200 Increase 10/300 GL column (GE Healthcare). Purified proteins were stored in a buffer containing 20 mM Tris-HCl and 150 mM NaCl (pH 8.0).
The hACE2 protein expressed via the Bac-to-Bac Baculovirus expression system was used for crystallization. The hACE2 protein produced by HEK293F cells was used for SPR assay. The RBD proteins produced by HEK293F cells were used for crystallization and SPR. The proteins for SPR assay were stored in PBST buffer (1.8 mM KH2PO4, 10 mM Na2HPO4 (pH 7.4), 137 mM NaCl, 2.7 mM KCl, and 0.005% (v/v) Tween 20).
SPR assay
The biotinylated SARS-CoV-2 and VOCs RBD proteins were transferred into PBST buffer (1.8 mM KH2PO4, 10 mM Na2HPO4 (pH 7.4), 137 mM NaCl, 2.7 mM KCl, and 0.05% (v/v) Tween 20) and immobilized on SA chip. Serially diluted hACE2s were then flowed over the chip in PBST buffer. Binding affinities were measured using a BIAcore 8K (GE Healthcare) at 25 °C in the single-cycle mode. Binding kinetics were analyzed with Biacore Insight software (GE Healthcare) using a 1:1 Langmuir binding model.
KD values of SPR experiments were obtained with BIAcore 8K Evaluation Software (GE Healthcare), using a 1:1 binding model. The values indicate the mean ± SD of three independent experiments.
Complex preparation and Crystallization
Purified hACE2 were mixed and incubated with BA.1.1 RBD, BA.2 RBD and BA.3 RBD on ice for about 2 h. The three mixtures were then purified on HiLoad 16/600 Superdex 200PG column in a buffer containing 20 mM Tris (pH 8.0) and 50 mM NaCl. Purified complex proteins (BA.1.1, RBD-hACE2, BA.2 RBD-hACE2 and BA.3 RBD-hACE2) were concentrated to 7 ∼ 10 mg/mL for crystallization using a vapour-diffusion sitting-drop method. In general, 1 μL protein was mixed with 1 μL reservoir solutions, and the mixtures were sealed and equilibrated against 100 μL of reservoir solutions at 18°C. After 48h, high-resolution complex crystals of BA.2 RBD-hACE2 were obtained in 0.1 M ammonium acetate, 0.1 M Bis-Tris pH5.5, 17% w/v PEG10,000 and complex crystals of BA.3 RBD-hACE2 were grown in 0.1 M Imidazole pH7.0, 12% w/v Polyethylene glycol 20,000, whereas the complex crystals of BA.1.1 RBD-hACE2 were grown in 0.1 M Potassium chloride, 0.1 M Sodium HEPES pH7.5, 15% w/v PEG 6000.
X-ray diffraction data collection and structure determination
To collect the data, complex crystals were briefly soaked in the corresponding reservoir solutions containing 20% (v/v) glycerol for cryo-protection and then flash-cooling in liquid nitrogen. The diffraction data were collected at the Shanghai Synchrotron Radiation Facility (SSRF) BL02U1. The datasets were processed using HKL2000 software (Otwinowski and Minor, 1997). The structures of these three complexes were determined via the molecular replacement method using Phaser (Adams et al., 2010) with a search model (previously reported complex structure: SARS-CoV-2 RBD binding with hACE2 (PDB: 6LZG)). The atomic models were built using Coot (Emsley and Cowtan, 2004) and refined with phenix.refine (Adams et al., 2010). MolProbity tool (Williams et al., 2018) was used to assess the stereochemical qualities of the final models. Finally, all structures were generated using Pymol software (https://pymol.org/2/).
Flow cytometry assay
The plasmids containing hACE2 fused with eGFP were transfected into BHK-21 cells. Then stable cells were obtained by antibiotic screening. A mixture containing RBDs from SARS-CoV-2 PT (0.3 μg/mL) or VOCs (0.3μg/mL) was incubated with the hACE2-expressing BHK-21 stable cells at 4°C for 0.5 h. Subsequently, cells were washed with PBS thrice and stained with APC mouse anti-his secondary antibody for 0.5 h before being analyzed using BD FACS Canto FlowCytometer (BD Biosciences). BHK-21 cells were used as negative controls. The data of all samples were analyzed using FlowJo 10.8.1 (TreeStar Inc., Ashland, OR, USA).
All experiments were performed three times; one representative of each experiment is shown in the Figure S1.
Production and quantification of pseudoviruses
To obtain SARS-CoV-2 PT and VOC pseudoviruses, a replication-deficient vesicular stomatitis virus (VSV) vector backbone (VSV-ΔG-GFP) expressing the corresponding spike proteins were constructed as described protocols (Muik et al., 2021). Overall, 30 μg of spike protein expression plasmids were transfected into HEK293T cells. After 24h, The VSV-ΔG-GFP pseudoviruses were added to the transfected cell supernatant. After incubation for 1 h at 37°C, the inoculum was removed. Then, the cells were washed with PBS and cultured in DMEM containing both 10% FBS and anti-VSV-G antibody produced by I1Hybridoma ATCC® CRL2700™. The pseudoviruses were obtained 30 h post-infection. After being filtered by 0.45 μm filters (Millipore, Cat#SLHP033RB), the pseudoviruses were aliquoted and stored at −80 °C.
Prior to pseudovirus quantification, 0.5 U/μL BaseMuncher endonuclease (Abcam) was used to remove unpackaged RNA (37 °C for 1.5 h). Viral RNA was extracted using an RNA extraction kit (Bioer Technology) and quantified by quantitative RT–PCR (qPCR) using a 7500 fast real-time PCR system (Applied Biosystems). The primers and probes used to detect the L gene of the VSV virus are as described in the previous literature (Hole et al., 2010).
Pseudovirus infection assays
The numbers of the pseudovirus particles for SARS-CoV-2 and its variants were normalized by the VSV L gene qPCR, and then diluted to have the same number of viral particles per microliter via quantitative RT-PCR. Then, 100 μL of pseudovirus was added to each well of 96-well plates containing Vero cells. After 15 h, each whole well was scanned using a CQ1 confocal image cytometer (Yokogawa) and the total numbers of GFP-positive cells were determined using the software bundled with the instrument (Yokogawa). Each group included 6 biological replicates, and the analysis was repeated 3-4 times. Statistical analysis was performed using Graphpad Prism 8.
Molecular dynamics (MD) simulations
The structure of the BA.1 RBD/hACE2 complex without the RBD G496S mutation (G496 BA.1 RBD/hACE2) was modeled based on the BA.1 RBD/hACE2 complex structure. After stripping N-acetyl-β-glucosaminide glycans and water molecules from the crystal structure and adding TIP3P water (Best et al., 2012; Jorgensen et al., 1983) and counter ions, both G496 RBD-ACE2 and S496 RBD-ACE2 systems were subjected to five parallel MD simulations with different random seeds. MD simulations were performed using a GPU cluster with a GROMACS package (version 2020.5) (Abraham et al., 2015) at the temperature of 300 K and with the time step of 2 fs. The CHARMM36 force field was applied. All bonds involving hydrogen atoms were constrained using the LINCS algorithm (Hess et al., 1997) and water molecules were restrained using the SETTLE algorithm (Miyamoto and Kollman, 1992). Atom-based truncation scheme was applied, with a non-bond cutoff of 12 Å, and with the force switching function initiated at 10 Å for van-der-Waals interactions. Long-range electrostatic interactions were calculated using the Particle Mesh Ewald method with fourth-order cubic interpolation and 1.6 Å grid spacing (Darden et al., 1993). The dynamics of each system was modeled with a step-by-step equilibration. Energy was minimized using the steepest descent algorithm with the maximum force less than 1000 kJ/mol/nm, 200 ps of number of particles, volume, and temperature (NVT) equilibration with 1000 kJ/mol·nm2 harmonic positional restraints on the heavy atoms of the protein, and three 200 ps steps of number of particles, pressure, and temperature (NPT) equilibration with 1000, 1000 and 500 kJ/mol·nm2 harmonic positional restraints on the protein backbone heavy atoms, correspondingly. Then each equilibrated system was subjected to 200 ns NPT MD production using a Velocity rescaling thermostat without any restraints on the protein atoms (Bussi et al., 2007).
Molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) calculations
For both the G496 RBD-ACE2 and S496 RBD-ACE2 systems, 100 snapshots were extracted from the 200 ns trajectories of each simulation. A total of 500 snapshots of each system was subjected to MM/PBSA calculations using the g_mmpbsa tool (Kumari et al., 2014). The entropy contribution was ignored and the solute dielectric constant was set to 2. The binding energy between RBD and ACE2 was calculated as ΔEBinding = ERBD/hACE2 – ERBD - EhACE2. Besides, the binding energy was decomposed on each residue.
Quantification and statistical analysis
Flow cytometry analysis
All experiments were performed three times independently; one representative of each experiment is shown in this article.
Binding affinity analysis
KD values of SPR experiments were obtained with BIAcore 8K Evaluation Software (GE Healthcare), using a 1:1 binding model. The values indicate the mean ± SD of three replicated experiments.
Pseudovirus infection assays
Each group contained 6 replicates. And the analysis was repeated 3-4 times.
Acknowledgments
We are particularly grateful to the Pathogen Microbiology and Immunology Public Technology Service Center for its support. We thank Hua Tao and Tong Zhao from the Institute of Microbiology, Chinese Academy of Sciences, for their technical support in the FACS assay. We acknowledge the staff of beamline BL02U1, BL10U2, and BL19U1 at the Shanghai Synchrotron Radiation Facility for assistance during the data collection. This work was supported by the National Key R&D Program of China (2021YFC2301401, 2020YFA0907102, 2020YFA0509202, and 2021YFC0863300), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29010202 and XDB37030204), the Queensland-Chinese Academy of Sciences Collaborative Science Fund 2020 (153211KYSB20200001), and the National Natural Science Foundation of China (grant 92169208).
Author contributions
N.H., J.Q., and G.F.G. initiated and designed the project. J.Q. and N.H. supervised and coordinated the experiments for crystal preparations, diffraction data collection, and structure determinations. G.F.G. supervised the structural analysis, SPR and FACS assays, and pseudoviruses transductions. L.L., H.L., Y.M., and W.L. purified the proteins and grew the crystals with the help of P.H., Q.W., Y.Z., Y.Z., and Q.W.; L.L. and H.L. performed the SPR analysis with the help of Z.F.; and Y.M. and W.L. conducted the flow cytometry assay. J.Q. collected the crystal structural data and solved the structures. D.L. and X.Z. performed the pseudotyped virus-related assays. L.W. collected and analyzed the sequence data from GISAID. K.L., X.Z., Y.S., N.H., J.Q., and G.F.G. wrote and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.06.023.
Supplemental information
Data and code availability
-
•
The crystal structures of Omicron BA.1.1 RBD/hACE2 complex (PDB: 7XAZ), Omicron BA.2 RBD/hACE2 complex (PDB: 7XB0), and Omicron BA.3 RBD/hACE2 complex (PDB: 7XB1) have been deposited in the Protein Data Bank (www.rcsb.org).
-
•
This study did not generate custom computer code.
-
•
Any additional information required to reanalyze the data reported in this work is available from the Lead Contact upon request.
References
- Abraham M.J., Murtolad T., Schulzbc R., Pálla S., Smithbc J.C., Hessa B., Lindahlad E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R.B., Zhu X., Shim J., Lopes P.E., Mittal J., Feig M., Mackerell A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel T., Hadjadj J., Maes P., Planas D., Seve A., Staropoli I., Guivel-Benhassine F., Porrot F., Bolland W.H., Nguyen Y., et al. Serum neutralization of SARS-CoV-2 Omicron sublineages BA.1 and BA.2 in patients receiving monoclonal antibodies. Nat. Med. 2022;28:1297–1302. doi: 10.1038/s41591-022-01792-5. [DOI] [PubMed] [Google Scholar]
- Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Callaway E. Why does the Omicron sub-variant spread faster than the original? Nature. 2022;602:556–557. doi: 10.1038/d41586-022-00471-2. [DOI] [PubMed] [Google Scholar]
- Cameroni E., Bowen J.E., Rosen L.E., Saliba C., Zepeda S.K., Culap K., Pinto D., VanBlargan L.A., De Marco A., di Iulio J., et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2022;602:664–670. doi: 10.1038/s41586-021-04386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Yisimayi A., Jian F., Song W., Xiao T., Wang L., Du S., Wang J., Li Q., Chen X., et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 2022 doi: 10.1038/s41586-022-04980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.K., Dorosky D., Sharma P., Abbasi S.A., Dye J.M., Kranz D.M., Herbert A.S., Procko E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science. 2020;369:1261–1265. doi: 10.1126/science.abc0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z., Liu P., Wang N., Wang L., Fan K., Zhu Q., Wang K., Chen R., Feng R., Jia Z., et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell. 2022;185 doi: 10.1016/j.cell.2022.01.019. 860.e13–871.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L., Gao G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021;21:73–82. doi: 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T., York D., Pedersen L. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Han P., Li L., Liu S., Wang Q., Zhang D., Xu Z., Han P., Li X., Peng Q., Su C., et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell. 2022;185 doi: 10.1016/j.cell.2022.01.001. 630.e10–640.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseltine W.A. 2021. How omicron evades natural immunity, vaccination, and monoclonal antibody treatments.https://www.forbes.com/sites/williamhaseltine/2021/12/17/how-omicron-evades-natural-immunity-vaccination-and-monoclonal-antibody-treatments/?sh=3c9a3f2460e0 [Google Scholar]
- Haseltine W.A. 2022. New members of the omicron family of viruses: BA.2.12.1, BA.4, and BA.5.https://www.forbes.com/sites/williamhaseltine/2022/04/20/new-members-of-the-omicron-family-of-viruses-ba2121-ba4-and-ba5/?sh=255752617ff8 [Google Scholar]
- Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- Hole K., Velazquez-Salinas L., Clavijo A. Improvement and optimization of a multiplex real-time reverse transcription polymerase chain reaction assay for the detection and typing of vesicular stomatitis virus. J. Vet. Diagn. Invest. 2010;22:428–433. doi: 10.1177/104063871002200315. [DOI] [PubMed] [Google Scholar]
- Iketani S., Liu L., Guo Y., Liu L., Chan J.F., Huang Y., Wang M., Luo Y., Yu J., Chu H., et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature. 2022;604:553–556. doi: 10.1038/s41586-022-04594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- Joughin B.A., Green D.F., Tidor B. Action-at-a-distance interactions enhance protein binding affinity. Protein Sci. 2005;14:1363–1369. doi: 10.1110/ps.041283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M., Mohamed M.E.M., Abd El-Lateef H.M., Venugopala K.N., El-Beltagi H.S. Omicron variant genome evolution and phylogenetics. J. Med. Virol. 2022;94:1627–1632. doi: 10.1002/jmv.27515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Open Source Drug Discovery Consortium. Lynn A. g_mmpbsa—a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Lan J., He X., Ren Y., Wang Z., Zhou H., Fan S., Zhu C., Liu D., Shao B., Liu T.Y., et al. Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction. Cell Res. 2022;32:593–595. doi: 10.1038/s41422-022-00644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Niu P., Zhao L., Wang H., Wang W., Tan W. Sequencing the complete genome of COVID-19 virus from clinical samples using the sanger method. China CDC Wkly. 2020;2:447–452. doi: 10.46234/ccdcw2020.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannar D., Saville J.W., Zhu X., Srivastava S.S., Berezuk A.M., Tuttle K.S., Marquez A.C., Sekirov I., Subramaniam S. SARS-CoV-2 Omicron variant: antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science. 2022;375:760–764. doi: 10.1126/science.abn7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxmen A. Why call it BA.2.12.1? A guide to the tangled Omicron family. Nature. 2022;606:446–447. doi: 10.1038/d41586-022-01466-9. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Kollman P.A. Settle: an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. doi: 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Muik A., Wallisch A.K., Sänger B., Swanson K.A., Mühl J., Chen W., Cai H., Maurus D., Sarkar R., Türeci Ö., et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. Science. 2021;371:1152–1153. doi: 10.1126/science.abg6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- outbreak.info . 2022. Omicron variant report.https://outbreak.info/situation-reports/omicron?loc=ZAF&loc=GBR&loc=USA&selected=ZAF [Google Scholar]
- PAHO. WHO . 2022. Sublineages of SARS-CoV-2 VOC omicron.https://www.paho.org/en/documents/sublineages-sars-cov-2-voc-omicron [Google Scholar]
- Peng R.C., Wu L.A., Wang Q.L., Qi J.X., Gao G.F. Cell entry by SARS-CoV-2. Trends Biochem. Sci. 2021;46:848–860. doi: 10.1016/j.tibs.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A., Irie T., Suzuki R., Maemura T., Nasser H., Uriu K., Kosugi Y., Shirakawa K., Sadamasu K., Kimura I., et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature. 2022;602:300–306. doi: 10.1038/s41586-021-04266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C., et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182 doi: 10.1016/j.cell.2020.08.012. 1295.e20–1310.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W., Zhao X., Ma X., Wang W., Niu P., Xu W., Gao G.F., Wu G. A novel coronavirus genome identified in a cluster of pneumonia cases — Wuhan, China 2019–2020. China CDC Wkly. 2020;2:61–62. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W., Li D., Zhang N., Bai G., Yuan K., Xiao H., Gao F., Chen Y., Wong C.C.L., Gao G.F. O-glycosylation pattern of the SARS-CoV-2 spike protein reveals an “O-Follow-N” rule. Cell Res. 2021;31:1123–1125. doi: 10.1038/s41422-021-00545-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal H., Jagannathan R., Bhatnagar A., Tirupula K., Desnoyer R., Karnik S.S. Long range effect of mutations on specific conformational changes in the extracellular loop 2 of angiotensin II type 1 receptor. J. Biol. Chem. 2013;288:540–551. doi: 10.1074/jbc.M112.392514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.Y., et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181 doi: 10.1016/j.cell.2020.03.045. 894–904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q., Wang Y., Ma J., Han J., Jiang M., Zhao L., Ye F., Song J., Liu B., Wu L., et al. Description of the first strain of 2019-nCoV, C-Tan-nCoV wuhan strain – National Pathogen Resource Center, China, 2020. China CDC Wkly. 2020;2:81–82. doi: 10.46234/ccdcw2020.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B., et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Dai L., Gao G.F. Humoral and cellular immunity and the safety of COVID-19 vaccines: a summary of data published by 21 May 2021. Int. Immunol. 2021;33:529–540. doi: 10.1093/intimm/dxab061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasoba D., Kimura I., Nasser H., Morioka Y., Nao N., Ito J., Uriu K., Tsuda M., Zahradnik J., Shirakawa K., et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell. 2022;185 doi: 10.1016/j.cell.2022.04.035. 2103.e19–2115.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W., Xu Y., Xu P., Cao X., Wu C., Gu C., He X., Wang X., Huang S., Yuan Q., et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science. 2022;375:1048–1053. doi: 10.1126/science.abn8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Collier A.Y., Rowe M., Mardas F., Ventura J.D., Wan H., Miller J., Powers O., Chung B., Siamatu M., et al. Neutralization of the SARS-CoV-2 omicron BA.1 and BA.2 Variants. N. Engl. J. Med. 2022;386:1579–1580. doi: 10.1056/NEJMc2201849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Zhang Y., Liu K., Li Y., Lu Q., Wang Q., Zhang Y., Wang L., Liao H., Zheng A., et al. The molecular basis for SARS-CoV-2 binding to dog ACE2. Nat. Commun. 2021;12:4195. doi: 10.1038/s41467-021-24326-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The crystal structures of Omicron BA.1.1 RBD/hACE2 complex (PDB: 7XAZ), Omicron BA.2 RBD/hACE2 complex (PDB: 7XB0), and Omicron BA.3 RBD/hACE2 complex (PDB: 7XB1) have been deposited in the Protein Data Bank (www.rcsb.org).
-
•
This study did not generate custom computer code.
-
•
Any additional information required to reanalyze the data reported in this work is available from the Lead Contact upon request.