

Abstract
Drug metabolism and pharmacokinetics (DMPK) is an important branch of pharmaceutical sciences. The nature of ADME (absorption, distribution, metabolism, excretion) and PK (pharmacokinetics) inquiries during drug discovery and development has evolved in recent years from being largely descriptive to seeking a more quantitative and mechanistic understanding of the fate of drug candidates in biological systems. Tremendous progress has been made in the past decade, not only in the characterization of physiochemical properties of drugs that influence their ADME, target organ exposure, and toxicity, but also in the identification of design principles that can minimize drug-drug interaction (DDI) potentials and reduce the attritions. The importance of membrane transporters in drug disposition, efficacy, and safety, as well as the interplay with metabolic processes, has been increasingly recognized. Dramatic increases in investments on new modalities beyond traditional small and large molecule drugs, such as peptides, oligonucleotides, and antibody-drug conjugates, necessitated further innovations in bioanalytical and experimental tools for the characterization of their ADME properties. In this review, we highlight some of the most notable advances in the last decade, and provide future perspectives on potential major breakthroughs and innovations in the translation of DMPK science in various stages of drug discovery and development.
Key words: Drug discovery and development, New drug application, Biologics license application, Pharmacokinetics, ADME, New modalities, Model-informed drug development, Micro-physiological systems
Graphical abstract
This review highlights some of the most notable advances in the translation of DMPK science in drug discovery and development in the last decade and predicts future breakthroughs and innovations.
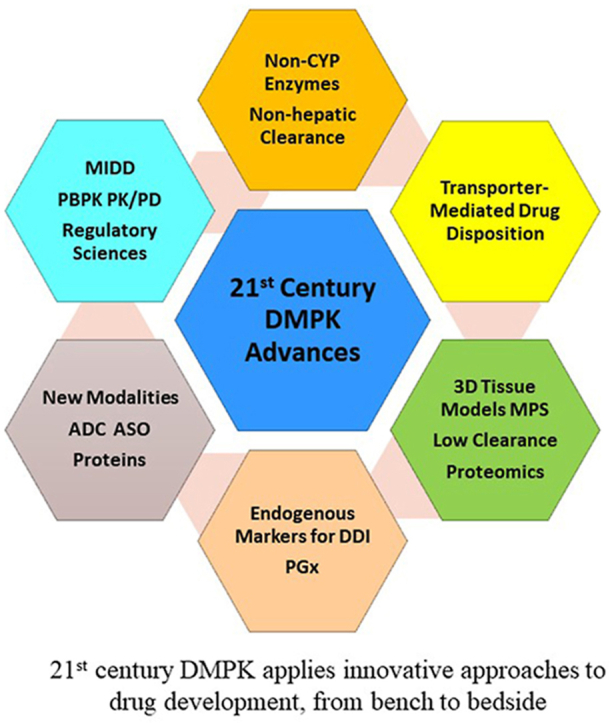
1. Introduction
Drug metabolism and pharmacokinetics (DMPK) is conventionally known as a scientific discipline that studies the availability of drugs or drug candidates for pharmacological processes and characterizes their entry (absorption) into the body, fate within the body (including distribution and biotransformation), and elimination from the body, overtime. The basic research into DMPK mechanisms has been the driving force for advancement in a number of scientific areas, such as the biochemistry, pharmacology, and genetics of drug-metabolizing enzymes (DMEs) and transporters, as well as their regulators1. The translation of the new knowledge and technical development in basic DMPK science, carried out by researchers from academia, pharmaceutical industry, and regulatory agencies, has played essential roles in the success of the pharmaceutical sciences field in developing new therapies for numerous human diseases and will remain so for the continued quest for discovering and developing better drugs in shorter time frames2.
The overall process of drug discovery and development can be divided into six stages: hit to lead, lead optimization, candidate selection, preclinical development, clinical development, registration and launch and post-marketing surveillance (Fig. 1). The processes of studying and characterizing ADME-PK properties have been well recognized as an integral discipline, which are indispensable and permeate all phases of the drug discovery and development pipeline (Fig. 1). Prior to 2000s, the focus of DMPK scientists in the pharmaceutical industry is primarily to provide a descriptive characterization for drug candidates in support of clinical trials and regulatory registration. Over the past decade, numerous high throughput tools have been adapted in pharmaceutical industry that enable a large volume of compounds entering the ADME testing funnels. However, the high throughput screening did not shorten the time of drug discovery from the bench to bedside; rather, the paradigm of DMPK inquiries has been dramatically shifted by the advances in related fields, such as pharmacogenetics, pharmacogenomics and the functional characterization of various drug transporters located in different organs, to focusing on gaining a more quantitative and mechanistic understanding of the fate of drug candidates in biological systems. Consequently, unprecedented insights can now be obtained on the molecular and mechanistic bases of the potential for drug–drug interactions (DDIs), interindividual variability of drug exposure, and asymmetric exposure in key organs either on or off the intended drug targets. As such, the discipline is well integrated into the holistic drug discovery paradigm to optimize ADME properties of molecules early and select drug candidates for entry into development.
Figure 1.

Overview of DMPK related activities in drug discovery and development. The overall process can be divided into six stages: hit to lead, lead optimization, candidate selection, preclinical development, clinical development and registration and launch. Key DMPK related activities at each of the six stages are listed in the text boxes separated by small molecule and biologics.
From a research and drug development viewpoint, here we highlight some of the key DMPK advancements that have been well-established during the past decades. A major focus is placed on advances in human clearance prediction for small-molecule drugs (Section 2), an area where tremendous progress has been made, not only in the characterization of the physiochemical properties of drugs that influence their ADME, target organ exposure, and toxicity, but also in the identification of design principles that can minimize DDI potentials and reduce the attritions caused by a lack of efficacy or poor safety profiles. Additional highlights are provided, which cover DMPK advances that enable development of new modalities (Section 3), model-informed drug development (MIDD) (Section 4), and advancement of regulatory sciences (Section 5). Notably, many ADME genes also play critical roles in common or rare human diseases and are well-acknowledged drug targets. However, the physiological functions of ADME genes will be beyond the scope of this review. Finally, perspectives on potential future breakthroughs and innovations in the translation of DMPK science in various stages of drug discovery and development are also discussed.
2. Major advances in human clearance prediction for small-molecule drugs
The selection of a drug candidate for further development is based on a balance among adequate target potency, optimized ADME-PK properties, and safety profiles, which ensure the proper dose and dose regimen with minimal DDI potential and adverse drug effects. For small molecules, the ability to accurately predict rates of drug clearance in humans is often considered critical in multiple stages during drug discovery and preclinical drug development (Fig. 1). Clearance prediction is particularly challenging for small-molecule drugs, which remain to be the dominant form of therapeutics in spite of recent surge in new modalities. It is a complicated process to characterize compound specificity in metabolism and multiplicity of various phase I and phase II biotransformation enzymes, as well as the involvement of drug transporters. In addition, drug tissue distribution, DDI liability and non-CYP elimination pathways are also important considerations for the accuracy of predictions. In this section, several topics will be reviewed to highlight key advances and remaining challenges. This includes clearance prediction for non-CYP enzymes (2.1), transporter-mediated drug disposition and elimination (2.2), new tools to quantitatively assess drug metabolizing enzyme and transporter activities (2.3), utility of endogenous biomarkers in DDI predictions (2.4), consideration of ADME pharmacogenetics in drug discovery and development (2.5), and non-hepatic clearance mechanisms (2.6).
2.1. Progress in clearance prediction for non-CYP enzymes
2.1.1. Overview
Prediction of human cytochrome P450 (CYP)-mediated drug clearance has become increasingly accurate due to the availability of high-quality reagents, such as human liver microsomes (HLM) and human hepatocytes (HHEP). Misprediction is typically the exception than the norm, although literature analyses of available data showed a general trend of under-prediction by several fold of in vivo clearance using liver microsomes or hepatocytes3. Our understanding of enzyme tissue distribution has been greatly enhanced, largely due to advances in protein quantification using proteomic approaches. The protein expression data are incorporated into physiologically based pharmacokinetic (PBPK) modeling, which allows more accurate and quantitative prediction of human PK and DDIs (See Section 4). Further developments will be needed to address CYP enzymes that are relatively less well-studied for human drug metabolism, such as CYP1A1, 2A6, 2J2, and CYP4 enzymes, as well as non-CYP drug-metabolizing enzymes (which are discussed in greater detail below), to better understand their roles in drug metabolism and disposition. For example, CYP4 enzymes, which hydroxylate the terminal carbon atom on an alkyl chain, metabolize a number of drugs, including ebastine, terfenadine and fingolimod4. These new developments will also include exploring in vitro to in vivo clearance extrapolation (IVIVE) and identifying selective substrates and inhibitors, in addition to enzyme distribution and comparative biochemistry studies.
CYPs are the major family of enzymes responsible for the metabolism of most small-molecule drugs; however, non-CYP enzymes also contribute significantly to clearance of many marketed drugs5. The major non-CYP drug metabolizing enzymes include UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), aldehyde oxidase (AO), carboxylesterases (CESs) and amidases, N-acetyltransferases (NATs), monoamine oxidases (MAOs), flavin-containing monooxygenases (FMOs), and glutathione-S-transferases (GSTs). Table 1 illustrates the contributions of various non-CYP enzymes toward the clearance of selected small-molecule drugs approved during 2011–2020.
Table 1.
Contributions of non-CYP enzymes to the clearance of drugs approved during 2011–2020 (https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm).
| Drug name | Drug class or mechanism | UGT | SULT1 | AO | CES1 | CES2 | MAO-A | NAT2 | FMO | Amidase | NDA # | Year |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Canagliflozin | Sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor | x | 204,042 | 2013 | ||||||||
| Fostamatinib | Tyrosine kinase inhibitor | x | 209,299 | 2018 | ||||||||
| Mirabegron | β3-Adrenergic agonist | x | 202,611 | 2012 | ||||||||
| Nintedanib | Respiratory agent | x | 205,832 | 2014 | ||||||||
| Safinamide | MAO-B inhibitor | x | 207,145 | 2017 | ||||||||
| Siponimod | Immunomodulator | x | 209,884 | 2019 | ||||||||
| Apixaban | Anticoagulant and antiplatelets cardiovascular drug | x | 202,155 | 2012 | ||||||||
| Opicapone | COMT inhibitor for Parkinson's disease | x | 209,510 | 2020 | ||||||||
| Crizotinib | Kinase inhibitors for cancer treatment | x | 202,570 | 2011 | ||||||||
| Idelalisib | Kinase inhibitors for cancer treatment | x | 206,545 | 2015 | ||||||||
| Edoxaban | Anticoagulant and antiplatelet | x | 206,316 | 2015 | ||||||||
| Sacubitril | Angiotensin II inhibitor | x | 207,620 | 2015 | ||||||||
| Selexipag | Vasodilator drug | x | 207,947 | 2015 | ||||||||
| Sofosbuvir | Antiviral | x | 205,834 | 2014 | ||||||||
| Tenofovir | Nucleoside reverse transcriptase inhibitor (NRTIs) for treatment of AIDS | x | x | 203,100 | 2012 | |||||||
| Telotristat etiprate | Antidiarrheals for gastrointestinal disorder | x | 208,794 | 2017 | ||||||||
| Safinamide | MAO-A substrate for Parkinson's disease | x | 207,145 | 2017 | ||||||||
| Retigabine | Anticonvulsant | x | 022,345 | 2011 | ||||||||
| Eravacycline | Anti-infective | x | 211,109 | 2018 | ||||||||
| Brivaracetam | Anticonvulsants | x | 205,836 | 2016 | ||||||||
| Eravacycline | Antibiotics | x | 211,109 | 2018 | ||||||||
| Safinamide | MAO-B inhibitors | x | 207,145 | 2017 |
x, enzymes contribute to metabolism of the corresponding drug.
Although the liver is still the major focus of drug metabolism research, the small intestine represents an important organ in pre-systemic drug metabolism6, which may affect the bioavailability of a drug and it may sometimes also affect pro-drug activation. Recently, good quality cryopreserved human intestinal mucosa samples from the duodenum, jejunum, and ileum have become commercially available. The activities and proteins of many CYP and non-CYP drug-metabolizing enzymes can be detected in these GI mucosa reagents, which provide useful tools for drug metabolism studies. Unfortunately, GI mucosa reagents for other preclinical species are not yet widely available, which limits the potential for inter-species comparisons and in vitro-in vivo correlation7,8. Metabolic enzymes are also expressed in other extrahepatic organs and may contribute to phase I and phase II metabolism. Similar to the characterization of liver and intestinal enzymes, the in vitro metabolic activities in these other organs can be characterized by incubation of extrahepatic samples with drugs of interest.
2.1.2. Clearance prediction for major non-CYP drug metabolizing enzymes
2.1.2.1. Uridine 5′-diphospho-glucuronosyltransferases (UGTs).
The UGTs are a family of enzymes that conjugate a glucuronic acid to drugs or drug metabolites containing oxygen, nitrogen, or sulfur, such as the hydroxyl, carboxyl, amino or thiol groups. The conjugation makes a drug (or metabolite) more polar and often more easily excreted into urine or bile. UGTs such as UGT1A1, UGT1A7, UGT1A9, and UGT2B7 are usually low-affinity and high-capacity enzymes that lack selective inhibitors and substrates9, 10, 11. Similar to studies on CYP enzymes, relative activity factor approaches obtained from recombinant UGTs and HLM with known substrates12 are usually applied to determine the fraction metabolized (fm) by UGTs (fm,UGT). Assays of UGT activities with multiple substrates in a cocktail were proposed to overcome potential enzyme selectivity of individual substrates13. A recent review discussed the optimal experimental conditions for UGT reaction phenotyping14. Species differences in UGT activities are well documented, with humans demonstrated to have higher UGT activities than rodents15,16. UGT clearance determined from in vitro assays (e.g., in hepatocyte suspension) tends to underpredict in vivo clearance. The prediction accuracy can be improved by using long-term hepatocyte co-culture systems17.
2.1.2.2. Sulfotransferases (SULTs)
Sulfation is an important pathway for detoxification and elimination of xenobiotics. SULTs (SULT1A1, SULT1A3, SULT1B1, SULT1E1, and SULT2A1) are cytosolic enzymes that transfer a sulfonate group from 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to a drug molecule to form a conjugated metabolite that is more polar and readily excreted in urine or bile. PAPS is a high-energy sulfate donor, which is generated by human PAPS synthases isoforms PAPSS1 and PAPSS2; these PAPS synthases are required for all human sulfation pathways18. These SULT enzymes are mostly expressed in the liver and intestine (except SULT1A3, which is mainly expressed in the intestine), and to a lesser extent in the lung and kidney19. In contrast to UGTs, SULTs are usually high-affinity but low-capacity enzymes. SULTs and UGTs share similar substrates and their contributions (fm,SULT and fm,UGT) to drug clearance are often dose-dependent. At low concentrations, SULTs typically have higher contributions than UGTs. As exemplified by the metabolism of acetaminophen9, UGTs usually have greater contributions at high concentrations, when SULTs are likely saturated. Thus, it is important to conduct in vitro studies using clinically relevant drug concentrations, to capture the contributions of the sulfation pathway correctly20. Using recombinant SULTs and the REF (relative expression factor) approach21, the individual contributions of the major SULTs towards the metabolism of a given drug can be elucidated.
2.1.2.3. Aldehyde oxidase (AO)
AO is a cytosolic molybdoflavoprotein enzyme involved in the oxidation of a wide variety of compounds, especially azaheterocyclics9,22. In addition, AO catalyzes the reduction of multiple functional groups, including nitrite, nitro groups, S-oxides, N-oxides, hydroxamate, isoxazoles and isothiazoles. AO occurs as a homodimer, and it requires flavin adenine dinucleotide, molybdenum cofactor and two 2Fe–2S clusters as cofactors9,23,24. In humans, only one functional AO gene, AOX1, is present, although several AOX1 orthologs are found in almost all other mammals25. AO is ubiquitously expressed, but with the greatest abundance in hepatic cytosol26.
Compounds with dominant metabolism by AO should be avoided in drug development pipeline because of the uncertainty in human clearance. Given the species differences in AO activities and poor in vitro–in vivo correlation, the prediction of human AO clearance has not been very successful using either human in vitro or animal in vivo data22. An inter-species correlation approach is challenged by the facts that rodents have low and variable AO expression, dogs have no AO expression, and monkeys, like humans, tend to have large inter-individual variations in AO expression. IVIVE using data from human hepatocytes or S9 fractions resulted in underprediction of in vivo clearance via AO26,27. A common approach to estimating human AO-mediated clearance is to test a group of known AO substrates with drug candidates and rank order the clearance using a few compounds with clinical data (e.g., zaleplon–low, zoniporide–medium, carbazeran–high) as “yardsticks”26. In clinic, compounds that show clearance slower than zaleplon can often survive, i.e., have a reasonable human PK property. A drug discovery decision tree for AO substrate was presented in a recent review22. An AO-humanized mouse model is available, which, though demonstrating human AO activity, has yet to be improved, so that it is able to predict human AO clearance quantitively28.
2.1.2.4. Carboxylesterases (CESs) and amidases
CESs are an important group of enzymes in the hydrolase family, which catalyze the hydrolysis of esters, amides, carbamates, thioesters and hydroxamic acids29,30. In humans, both CES1 and CES2 are expressed in the liver, but only CES2 is present in the intestine. Irinotecan is an example of an ester pro-drug, which is hydrolyzed by CES2 in the intestine to form the active metabolite SN-38 found in systemic circulation31. Large species differences have been observed for the CESs. Species differences and human CES tissue distribution were summarized in a recent review29. Many nonspecific hydrolase inhibitors inhibit both CES1 and CES2, such as bis(p-nitrophenyl)phosphate, diisopropylfluorophosphate, tetraisopropyl pyrophosphoramide, and paraoxon. Selective inhibitors of CES1 (e.g., digitonin)32 and CES2 (e.g., telmisartan and eserine)32,33 have been reported. However, their selectivity against CYPs and UGTs will need to be further evaluated in HHEP system33. The intestinal S9 fraction has been shown to be effective in the prediction of fraction of gut metabolism (Fg) mediated by CESs using various models34. Accurate prediction of CES-mediated clearance in humans remains to be challenging due to extra-hepatic contributions35.
Drug molecules containing amide bonds generally undergo slower hydrolysis as compared to esters. The amide bonds are hydrolyzed by amidases36, a class of enzymes that also encompass aminoacylases, peptide amidase and fatty acid amide hydrolases, which produces a carboxylic acid and ammonia. Amidases can have esterase activities and some amides can also be hydrolyzed by the liver microsomal carboxylesterases.
2.1.2.5. N-acetyltransferases (NATs)
NATs are cytosolic enzymes that catalyze the conjugation of aryl and alkyl amine compounds with an acetyl group using acetyl-CoA as a cofactor. NAT1 and NAT2 are two human N-acetyltransferases, which have 81% amino acid sequence identity37. NAT1 is ubiquitously expressed in many human tissues, with the highest mRNA expression in the intestine, colon and liver, whereas NAT2 is mainly expressed in the liver, with lower expression in the small intestine, colon and duodenum (The Human Protein Atlas https://www.proteinatlas.org/ENSG00000156006-NAT2/tissue). NAT2 is a polymorphic enzyme; 50%–55% Caucasians, <10% Asian and as high as 83% Egyptian are poor metabolizers37. NAT1 and NAT2 have overlapping, but different, substrate specificity. Caffeic acid, ferulic acid and gallic acid selectively inhibit NAT1, whereas scopuletin and curcumin selectively inhibit NAT2. Human recombinant NATs are commercially available for qualitative reaction phenotyping, to determine which NAT may be involved in the metabolism of a test compound. The pan-CYP inhibitor, 1-aminobenzotriazole, is a more potent inhibitor for NAT2 than NAT1 (IC50 158 μmol/L vs. > 1 mmol/L)38. A PBPK model was developed to predict oral clearance of isoniazid by NAT2 and the model was verified using different NAT2 acetylator status39.
2.1.2.6. Monoamine oxidases (MAOs)
MAOs are flavin-containing enzymes located in the outer mitochondrial membrane of mammalian cells in various tissues and are often involved in amine oxidation. MAO-A and MAO-B are two forms of MAOs present throughout the human body, including the brain. Serotonin is a MAO-A substrate, whereas β-phenylethylamine is a MAO-B substrate. By using selective inhibitors, e.g., clorgyline for MAO-A and deprenyl for MAO-B, the relative contributions of MAO-A and MAO-B can be elucidated40. Although MAOs are also present in HLM preparations, the mitochondria fraction isolated from the liver is most commonly used to study MAOs. Given the fact that mitochondrial fractions are not always available for human and preclinical species, liver microsome preparations can also be used to estimate the hepatic clearance mediated by MAOs. A semi-quantitative IVIVE study, performed using seven MAO substrates, showed that HLM slightly underpredict in vivo clearance41. Some MAO-B inhibitors are promising treatments for the astrogliosis in the human brain related to Parkinson's or Alzheimer's disease. Because of the CNS location of MAO-B, a peripheral biomarker is not readily available. A MAO-B tracer, (S)-(2-methylpyrid-5-yl)-6-[(3-18F-fluoro-2-hydroxy)propoxy]quinoline has been developed, which can be used for PET imaging. In a preclinical study, it showed a fast brain uptake, quick washout, and was replaceable by MAO-B inhibitors, such as lazabemide42.
2.1.2.7. Flavin-containing monooxygenases (FMOs)
FMOs are microsomal enzymes that co-exist with CYPs in the liver and they oxidize drugs containing soft nucleophiles such as nitrogen, sulfur, phosphorus, and selenium. Similar to CYPs, FMOs require NADPH for activity. To differentiate the potential contribution of FMOs from CYPs, FMO can be inactivated by heat (e.g., 50 °C for 1 min) in the absence of NADPH in liver microsomes, or it can be inhibited by a chemical inhibitor (e.g., 500 μmol/L methimazole)43. HLM and HHEP both showed a good IVIVE for FMO in a recent study using ten FMO substrates44. Ketoconazole, a CYP3A inhibitor commonly used in clinical DDI studies, until its withdrawal from clinical application due to hepatotoxicity, is metabolized by FMO10. FMO-mediated metabolism of ketoconazole is believed to lead to formation of reactive metabolites hydroxylamine and nitrone, which form protein adducts10.
2.1.2.8. Glutathione S-transferases (GSTs)
GSTs are known to play important roles in cellular detoxification by conjugating reactive metabolites (or electrophiles generated by the body) to the reduced form of glutathione. In recent years, it is becoming popular to develop covalent modifiers that can reach high inhibition toward certain targets, such as the JAK345. Besides the concern of potential off-target toxicity, one challenge of developing covalent modifiers is the prediction of human clearance, as GSTs are expressed all over the body, i.e., hepatic (microsomal, cytosolic and mitochondrial) and extra-hepatic in many tissues. A recent study demonstrated that in vitro blood clearance can be used to represent extrahepatic metabolism by GSTs, with use of a correction factor. Together with the clearance determined in hepatocytes, the in vitro clearances can be successfully scaled up to in vivo human clearance for GST substrates after factoring in the hepatic blood flow rate for hepatic clearance and cardiac output for blood clearance45.
2.1.3. The challenges and in vitro tools to assess low clearance compounds
With the implementation of high throughput metabolic stability screening and rapid metabolite identification, medicinal chemists are able to design and create compounds that have very low clearance, in order to lower drug dose and reduce dosing frequency46,47. For certain indications, the extremely long half-life resulting from a very low clearance could be a “no-go” for the project. The difficulty associated with characterization of the low clearance in vitro has been identified as a major gap of DMPK science in the past decades, as these compounds often hit the lower limits of the standard HLM and HHEP stability assays48,49. Driven by the needs of drug discovery to measure intrinsic clearance beyond the standard metabolic stability assay limits, several approaches have been developed recently to address the challenges of low clearance. As summarized in “Tools to quantitatively assess in vitro clearance”, these include the hepatocyte relay method, long-term coculture systems, and measurement of metabolite formation rate.
2.1.3.1. Hepatocyte relay method
The hepatocyte relay method was developed, by prolonging the incubation time of test compounds with fresh-thawed hepatocytes using a relay approach, to extend the lower limit of a hepatocyte metabolic stability assay49, 50, 51. The method involves incubation of test compounds with hepatocytes for 4 h. At the end of incubation, the supernatant is transferred to freshly thawed hepatocytes of the same lot and incubated for another 4 h. The incubation can be repeated five times to achieve a cumulative incubation time of 20 h. The supernatant can be stored in a freezer before the next relay and different cell densities can be used to achieve desired lower limit of detection for determination of intrinsic clearance. The hepatocyte relay assay is an extension of the standard hepatocyte assay using the same lot of hepatocytes, which makes it easy for drug discovery teams to understand the data and its applications. Hepatocytes from preclinical species can be used as well in order to develop pre-clinical IVIVE50. The hepatocyte relay method has been applied not only to measuring intrinsic clearance, but also for metabolite identification and reaction phenotyping21,51. The characteristics of the method is summarized in Table 252, 53, 54, 55, 56, 57, 58.
Table 2.
Characteristics of low clearance methods.
| Methods | Hepatocyte relay | HepatoPac | Hurel |
|---|---|---|---|
| Hepatocyte density (million cells/mL) | Any, typically 0.5–2 | 0.0752 | 0.3053,54 |
| Incubation time | 4 h/relay, accumulatively 20 h for 5 relays | Up to 7 days without media change52 | 3 days53,54 |
| CLint, app lower limit (μL/min/million cells)a | 0.58 (0.5 million cells/mL) 0.14 (2 million cells/mL) |
0.49 (7-day incubation at 0.07 million cells/mL) | 0.27 (3-day incubation at 0.30 million cells/mL) |
| CLint, app, scaled lower limit (mL/min/kg)b | 1.5 (0.5 million cells/mL, 20 h) 0.36 (2 million cells/mL, 20 h) |
1.2 (7-day incubation at 0.07 million cells/mL) | 0.67 (3-day incubation at 0.30 million cells/mL) |
| Enzyme activity compared to suspension hepatocytes | Same as suspension hepatocytes | Higher UGT and AKR activities from mouse fibroblast Lower activities of CYP2D6, AO and FMO55 |
Higher UGT and AKR activities from mouse fibroblast Lower activities of CYP2D6, AO and FMO55 |
| Donors | Single or pooled | Single or pooled | Single or pooled |
| Preclinical species | Any | Any | Any |
| Coculture | None | Mouse fibroblasts | Proprietary non-parenchymal stromal cell line56 |
| Plate format | Standard | Micropatterned plates | Standard |
2.1.3.2. Micropatterned hepatocyte coculture system
Micro-patterned long-term coculture system with primary human hepatocytes and 3T3-J2 mouse embryonic fibroblasts is engineered to have extended cell viability and metabolic activity59. The hepatocyte cultures can maintain functional activity for up to 7 days without a medium change52. This co-culture system has been used to predict low clearance and profile metabolite structures with good success17,52,60,61. However, significant metabolism of compounds by aldo-keto reductases (AKRs) and UGTs in the fibroblasts of the co-culture was observed, so were reductions in the activities of hepatocyte CYP2D6, AO and FMO, compared to hepatocytes in suspension culture55. These events can complicate clearance prediction and metabolite identification55. The lower limits for CLint determination with a 7-day incubation, compared to the hepatocyte relay method, are summarized in Table 2.
2.1.3.3. Plated hepatocyte coculture system
The plated hepatocyte co-culture system with a proprietary supporting non-parenchymal stromal cell line enables long-term culture and maintain functional activities56. Test compounds can be incubated with the cells for 3 days without media change, which allow studies of low metabolic clearance53,54. Several studies showed that the co-culture system provided good prediction of in vivo clearance and human relevant metabolites for low clearance compounds53, 54, 55. Similar to the micropatterned hepatocyte co-culture system, significant metabolism of test compounds by AKR and UGT in the supporting cells of the co-culture and reductions in the activities of hepatocyte CYP2D6, AO and FMO were observed, which can complicate clearance prediction and metabolite identification55. The lower limits for CLint determination using the hepatocyte coculture system is found to be comparable with the hepatocyte relay method (Table 2).
2.1.3.4. Measurement of metabolite formation rate
Rates of metabolite formation can be used to estimate rates of depletion of parent compounds, as monitoring metabolite formation is a more sensitive approach than monitoring disappearance of the parent for low clearance compounds. When radiolabeled material is not available for clearance study, which is a typical case in the drug discovery phase, the metabolite formation rate approaches require: (1) all major primary metabolite standards are available for determination of the metabolite formation rate, (2) metabolite formation rates are measured at low substrate consumption (typically <20%) and show linearity with incubation time and protein concentration, (3) primary metabolites do not have rapid conversions to secondary metabolites, and (4) the total number of major metabolites is relatively small. The occurrence of multiple metabolites can introduce high variability due to additive errors from multiple rate measurements. Using this approach, intrinsic clearance was determined for several low clearance compounds, such as tolbutamide, S-warfarin and S-mephenytoin62.
2.2. Characterization of transporter-mediated drug disposition and elimination
Over the past two decades, more than 450 membrane transporters have been cloned in humans. They are classified into two superfamilies–the ATP-binding cassette (ABC) proteins and solute carrier (SLC) proteins63, 64, 65. Membrane transporters are expressed on the barrier surface of organs, such as the kidney, liver, brain and placenta, and can transport endogenous substances or xenobiotics and their metabolites either into (influx/uptake) or out of (efflux) cell membranes and organ barriers. Numerous clinical studies have demonstrated that modulators of transporter expression and/or activity can affect drug PK, pharmacodynamics (PD), and organ exposure of substrates. Transporter-mediated DDIs are well-documented in the literature66.
The ABC gene family encodes 49 members in the human genome, which belong to seven transporter subfamilies, ABCA through ABCG. The proteins can hydrolyze adenosine triphosphate (ATP) to generate energy for translocating vast and diverse substrates, including lipids, drugs, and metabolic products, from intracellular to extracellular spaces65,67,68. The most relevant ABC transporters to the efficacy and toxicity of drugs include the P-glycoprotein (P-gp/MDR1/ABCB1), the breast cancer resistance protein (BCRP/ABCG2), the multi-drug resistance proteins (MRPs/ABCCs), and the bile salt export protein BSEP/ABCB11)69.
The SLCs belong to a highly diverse superfamily containing 395 identified human transporters70. The most relevant SLC transporters to drug disposition and DDIs include organic anion transporting polypeptides (OATPs), organic anion and organic cation transporters (OATs and OCTs), and multidrug and toxin extrusion transporters (MATEs).
2.2.1. Transporter role in drug absorption
The gastrointestinal (GI) tract expresses many transporters that are found to significantly impact drug absorption. On the luminal membrane of enterocytes, uptake transporters like OATP2B1, peptide transporter (PEPT) 1, apical sodium-dependent bile acid transporter and monocarboxylate transporter 1 can uptake drugs from the gut lumen into enterocytes. Drugs with low permeability could have a boosted absorption if they are substrates of uptake transporters. Meanwhile, efflux transporters expressed on the luminal membrane, such as P-gp, MRP2, and BCRP, can efflux drugs back to the gut lumen66,71,72. A decreased fraction of oral absorption (fa) could occur with drugs transported by luminal efflux transporters. Inhibition of P-gp or BCRP transporters can increase oral bioavailability and decrease the active excretion in urine or bile, resulting in an increase of systemic exposure73. P-gp can be induced by PXR nuclear receptor activators, such as rifampin, leading to the substantial reduction of oral absorption of digoxin74, albeit the induction is less sensitive than CYP3A75. Transporters expressed on the basolateral membrane of enterocytes, such as organic cation transporter (OCT1) and MRP3, can transport intracellular drugs across the basolateral membrane to reach the blood stream. However, the clinical significance of these basolateral transporters in drug absorption is still not well characterized.
2.2.2. Transporter role in drug clearance
Transporter-mediated hepatic uptake can act as the ‘gatekeeper’ of a drug entering hepatocytes, where the transporters provide the access of substrates to intracellular metabolizing enzymes or additional efflux transporters for elimination into the bile76,77. OATP1B1/1B3, OAT2, sodium-taurocholate co-transporting polypeptide, and OCT1 are expressed in the liver and play a key role in hepatic drug disposition and elimination. For example, the OATP1B transporters are responsible for the uptake of a wide range of amphiphilic organic compounds into the liver. The OATP-mediated transport may be the rate-determining step for the overall clearance of OATP substrate drugs, and is thus important for drugs whose site of action is in the liver. When OATP transport is inhibited or reduced due to genetic polymorphisms, drug exposure in systemic circulation often increases, which supports the notion that OATPs play important roles in hepatic clearance and uptake-mediated drug disposition. Similarly, reduced OATP-mediated transport can result in higher plasma levels of endogenous substrates—such as tetradecanedioic acid (TDA), hexadecanedioic acid (HDA), and coproporphyrins I and III78. Because these substrates are in vivo endogenous biomarkers for OATP functional activities, monitoring their levels during first-in-human trials could elucidate the potential of OATP inhibition-mediated DDIs79, 80, 81. On the other hand, transporters expressed on the canalicular membrane of hepatocytes play an important role in biliary excretion of drugs and their metabolites. MRP2 contributes to the biliary excretion of endogenous chemicals, such as conjugated bilirubin, and many structurally diverse xenobiotics and their metabolites82, 83, 84, 85, 86. While the physiological role of BSEP is to secrete bile salts to the bile duct and maintain the normal bile flow, inhibition of BSEP has been found to associate with cholestatic liver injury87.
The measurement of intrinsic clearance (CLint) using in vitro liver-derived reagents has proved pivotal for the prediction of in vivo hepatic clearance88,89. With the term “CLint”, the well-stirred model is adapted, which refers to the assumption that no limitations or barriers within the liver exist. Widening disconnects of in vitro–in vivo clearance correlation or IVIVE have recently been documented for drugs that are substrates of drug transporters90. To incorporate transporter-mediated hepatic clearance, newer extrapolation approaches have been developed to avoid the systemic bias in clearance prediction. In recent years, an extended clearance model is often used to estimate the overall hepatic clearance91, 92, 93. While the extended clearance model can be useful to identify rate-determining processes of drug clearance, the limitations of the concept have been raised, regarding its adequacy in PK prediction when selecting compounds for drug development94.
Renal clearance is another important elimination pathway for many clinical drugs and their metabolites95,96. It consists of glomerular filtration and tubular secretion and reabsorption. Renal transporters, such as OAT1/3, OCT2, and MATE1/2-K, play important roles in the renal elimination of drugs. OAT1 and OAT3 expressed on the basolateral membrane of the proximal renal tubular cells are responsible for the uptake of anionic drugs from the blood stream into the proximal tubule cells for either metabolic processing or elimination into the urine80,97. OCT1, OCT2 and OCT3 mediate the facilitative diffusion of organic cations via an electrochemical gradient98. OCT1 is exclusively expressed on the basolateral membrane of hepatocytes, while OCT2 is expressed at the basolateral surface of the kidney proximal tubule cells99,100. OCT1 is a thiamine transporter; it regulates the exposure of thiamine and its active metabolite, thiamine pyrophosphate, in the liver, to change hepatic energy status and modulate activation of AMP-activated protein kinase. It is found to be a major hepatic transporter in metabolic function and play a critical role in hepatic steatosis101. MATE1 and MATE2-K are expressed on luminal membrane of renal proximal tubule cells; they act in an antipodal fashion to OCT2-mediated uptake, to efflux drugs into the urine. Inhibition of these renal transporters can reduce renal secretion of drugs, leading to an increase of plasma exposure102. A determination of whether OCT2-mediated uptake or MATE-mediated excretion was inhibited is critical for clearance prediction, as inhibition of drug efflux can result in drug accumulation in the proximal tubule cells, leading to renal toxicity103,104.
2.2.3. Transporter role in drug organ distribution
Transporter-mediated organ uptake is generally considered as a drug distribution process. In addition to influencing drug pharmacokinetics, transporter proteins are the main determinants of drug distribution. The spatial expression of transporters within or along the tissue or organ is another determinant to their net impact. In addition, the co-expression of different transporters acting in the same or opposing direction have overall relevance in their impact. For example, efflux transporters are expressed on the barriers of liver, kidney, brain endothelium, mammary tissue, and testis, and have particular importance in the distribution of drugs to organs that are protected by these tissue barriers. P-gp and BCRP can limit drug transport across the blood–brain, blood–testis and maternal–fetal barriers, and reduce oral absorption of drugs105. In the kidney, OAT1, OAT3 and OCT2 are expressed on the basolateral membrane of proximal tubular cells and work in concert with apically expressed MATE1 and MATE2-K transporters to determine the local drug concentration. Antivirals (e.g., acyclic nucleoside phosphonates), antibiotics (e.g., β-lactams), and chemotherapeutic agents (e.g., methotrexate and cisplatin) are substrates for these transporters, and can accumulate in the proximal tubule and cause direct cellular toxicity106, 107, 108. OAT-mediated cidofovir uptake leads to the accumulation in the proximal tubule cells that is the cause of nephrotoxicity. Administration of OAT inhibitor probenecid is prescribed to use clinically prior to cidofovir to reduce the accumulation and the incidence of renal adverse events109. In contrast, the nephrotoxicity of cisplatin is the result of its being a basal OCT2 and apical MATE substrate110,111. In mice or transfected cell lines, co-administration of MATE inhibitors ondansetron or vandetanib can cause cisplatin accumulation in the kidney, resulting in enhanced toxicity112,113.
2.3. New tools to quantitatively assess drug metabolizing enzyme and transporter activities
2.3.1. Three-dimensional (3D) in vitro human tissue models
Current drug ADMET studies of new molecule entities (NME) often rely on the use of two-dimensional (2D) cell culture systems, such as Caco-2 human colorectal adenocarcinoma cells, Madin–Darby canine kidney cells (MDCK) and plated hepatocytes. The 2D cell culture systems have proven to be convenient and effective models to evaluate permeability, transport, metabolism, and cytotoxicity of compounds. However, 2D cultures have several limitations, including a lack of tissue specific architectures, cell-to-cell interactions, and mechanic flow. There have been significant advances in the development of 3D cell culture systems as alternative models that may better recapitulate the in vivo physiology compared to static culture 2D models114,115. More specifically, spheroids, organoids, and organs-on-chips have been introduced as novel systems to assess ADMET properties116. For instance, the effects of dynamic flow conditions of a gut-on-chip culture system on the gene expression profiles were investigated117. The in vitro gene expression profiles under 3D culture conditions were similar to those of human in vivo duodenum, jejunum, ileum, and colon tissue samples, indicating that the dynamic gut-on-chip platform is a suitable model for studying human intestinal drug transporters, enzymes, and toxicological responses117. In addition, several other intestinal chip models were recently developed to study drug absorption, metabolism, and bioavailability118, 119, 120, 121. Hepatocyte spheroids, liver organoids, and liver-on-chips have been used to investigate drug metabolism and drug-induced toxicity. Sandwich cultured hepatocyte, in which a hepatocyte monolayer is cultured between two extracellular matrix layers, has been shown to maintain activities of drug metabolizing-enzymes and transporters. The sandwich cultured hepatocyte model is commonly used to study in vitro drug uptake, biliary excretion, and ADMET gene regulations122,123.
Complex chip-based tissue culture systems having a co-culture system with multiple cell types or organoids are even more attractive because tissue ultrastructure, enzyme and transporter activities, and multi-organ interactions are well preserved. Moreover, these complex systems show different advantages and limitations for characterizing ADMET properties of drug candidates124. For example, in a simplified liver-testis-on-chip model, liver spheroids grown in microfluidic conditions abundantly and stably expressed DMEs compared to those under other conditions125. By comparison, testosterone secreted by testicular organoids cultured without liver spheroids can reach higher levels. This suggests that the liver spheroids have metabolized the testosterone synthesized by testis organoids in the co-cultures, as naturally occurs in vivo. Furthermore, the addition of cyclophosphamide led to upregulation of specific CYP enzymes in liver spheroids, and loss of germ cells in testicular organoids, in the multi-organ-chip co-cultures, but not in testis-only culture, confirming multi-organ interactions125. Currently, various 3D culture systems, from simple spheroids to more complicated organoids, are being developed to improve cell- and organ-based assays, to more closely mimicking in vivo human drug metabolism and disposition, or susceptibility to adverse drug response124.
2.3.2. Proteomics-informed prediction of drug clearance to improve IVIVE
Protein quantification using proteomic approaches has made giant leaps in the past decade. Protein expression levels of major DMEs and transporters have been mapped out in the major tissues, specific populations and disease states, as well as in vitro systems126, 127, 128, 129, 130, 131, 132. This information helps to develop relative expression factor (REF) and inter-system extrapolation factor values for reaction phenotyping, cross-tissue translation of enzyme and transporter activities, and PBPK modeling.
During the past decade, liquid chromatography–tandem mass spectrometry (LC–MS/MS) based quantitative proteomics has been applied to measuring absolute and/or relative abundance of DMEs and transporters in ADME-related tissues/organs131,133, 134, 135. Compared to conventional immune-quantitative methods, such as Western blotting and enzyme-linked immunosorbent assays (ELISA), these state-of-the-art proteomic techniques select targeted peptides based on sequence information, separate and identify protein digests with multiple reaction monitoring. This allows quantitative measurement of multiple DMEs and transporters simultaneously with high selectivity and sensitivity. Quantitative proteomic techniques have been broadly applied to generate rich datasets on the abundance of ADME-related enzymes and transporters in various human tissues (e.g., liver, intestine, kidney, and brain) in healthy and special populations, and preclinical animal models132,136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146, 147. These data help to further understand the effects of intrinsic and extrinsic factors (e.g., age, disease, ethnicity, and genetics) on the expression of enzymes and transporters, and their impact on PK and efficacy of drugs when using PBPK modeling. The protein expression data are used as system-dependent parameters, under the assumption that the protein abundance data are correlated with the activity of functional proteins148,149. Quantitative proteomics is also a promising approach for IVIVE of transporter-mediated clearance. Using LC–MS/MS-based quantitative targeted proteomics, in vitro clearance by the transporter of interest measured in recombinant transporter systems can be extrapolated to in vivo using REF method, which is defined as relative abundance of a selected transporter measured in a relevant tissue (e.g., liver for hepatic transporters) to that in an in vitro system (e.g., transporter transfected cells)150. These REF values have been used as scaling factors to successfully predict hepatic clearance of rosuvastatin in rat and human and renal clearance of metformin in human150, 151, 152, 153, 154. Additional studies are needed to expand this approach to additional compounds with diversified structures. REF approach is also useful to estimate relative contributions of various transporters to overall elimination of drugs, which is an important parameter for DDI prediction155.
Despite the success of quantitative proteomics in ADME related areas, some challenges remain. For instance, there is high variability in ADME proteomic data generated in different laboratories, which is largely caused by differences in sample matrices used, and in protein extraction, digestion, and measurement methods156. Such variability highlights current limitations of these techniques, which may lead to high uncertainty when applying proteomic data for quantitative translation and modeling. Protocol optimization and harmonization, as well as adherence to best practices for quantitative proteomics, will help to address some of the issues157. Furthermore, establishing the relationship between protein abundance and functional activity of transporters and enzymes will ultimately improve the confidence of applying quantitative proteomic based approach to predict PK, DDIs and efficacy of drugs.
2.3.3. Genetically engineered in vitro and in vivo models
Genetically engineered cell or animal models have been increasingly used to identify ADME genes linked to specific drug metabolism or transport pathways. A well-characterized genetic model offers the possibility to directly validate the in vivo outcomes predicted from in vitro data and mathematical modeling. Over the past decades, various engineered models, generated through the use of genome editing approaches, such as zinc-finger nucleases (ZFN) and transcription activator-like effector nucleases (TALEN), became available for pharmaceutical research. For example, ZFN technology was applied to knock-out drug efflux transporter genes ABCB1, ABCC2 or ABCG2 in a subclone of Caco-2 cells158 and MDCK cells159. A Bsep knockout model, in which the Abcb11 gene is knocked out by ZFN technology in Sprague–Dawley rats, has become a useful tool to study bile acid disposition and compensatory gene regulation160. Recently, the powerful gene editing technology of clustered regularly interspaced short palindromic repeats associated protein 9 (CRISPR-Cas9) has been increasingly applied for studying ADME gene function in cell lines or in animals161,162, as exemplified by the Cyp2d knock-out/human CYP2D6 knock-in163, and Slco1b2 gene knockout rats164. The MDCK cell line with canine Abcb1 gene knockout is a valuable tool for permeability assessment and is routinely used in major pharmaceutical companies165. However, despite these successful examples, the application of gene editing technologies has just started in the field of ADME research, and there are only a few models available for routinely screening compounds.
2.4. Utility of endogenous biomarkers in DDI prediction
The last decade has witnessed significant efforts toward the discovery and development of endogenous biomarkers of the activities of DMEs and transporters. In particular, there have been significant progresses in the identification, characterization, and validation of endogenous biomarkers for assessing DDI potentials for hepatic OATP1B1 and OATP1B3. Conventional approaches to DDI studies, using probe drugs that are likely to be used concomitantly with investigational drugs to define DDI magnitudes, have certain limitations when utilized during drug development. For example, the efficacious dose of a drug candidate is a moving target at early drug development stages. Setting an efficacious dose can be even more complicated if a drug has several different indications, because each indication can have its own optimal dose. This makes it challenging to select a dose early for DDI studies. Safety issues of some drug probes and the various probe combinations can be of concern. A retrospective analysis of 107 clinical DDI studies pertaining to OATP1B inhibition demonstrated that the cutoff value for the static DDI prediction parameter (R-value) of >1.1 or 1.04, as recommended by FDA and EMA, resulted in 21% and 31% of false-positive predictions, respectively, and it may not be possible to accurately predict the DDI magnitudes for true-positives166. Although PBPK models have been applied to the translation of in vitro DME inhibition to in vivo DDI, the predictive performance of PBPK for transporter-mediated DDIs has not been well established, especially when the ADMET properties of an investigational drug are not fully understood at early drug discovery phases.
Such a dilemma could potentially be solved by using circulating and urinary endogenous biomarkers that are selective for individual or combinations of hepatic and renal transporters. Using endogenous biomarkers could circumvent the need to dose probe substrates and thus reduce pill burden in the trials. There are great potentials to utilize endogenous biomarkers as probe substrates to assess DDI risk in the clinic, especially when investigating DDIs in patients with organ impairment or other diseases167,168. In addition, the measurements of endogenous biomarkers enable evaluations of dose-dependent transporter inhibition in phase I clinical trials to determine in vivo inhibition parameters169. The biomarker data generated in phase I studies together with model-based predictions would complement regulatory decision tree-based approaches by minimizing false-positive predictions170,171. The biomarker approaches also support earlier risk assessment and optimization of drug development plan by properly determining the need for, and timing of, DDI studies. As a result, the investigation of endogenous clinical biomarkers of transporters, including identification and validation of these biomarkers, has increased exponentially during the last few years80,81,172,173.
To date, a number of laboratories have studied various structurally diverse, circulating endogenous compounds as transporter biomarkers, by using genome-wide association in humans (e.g., HDA and TDA) or monkeys [e.g., coproporphyrin I (CPI) and III (CPIII)], and/or human clinical DDI studies with known inhibitors (e.g., N1-methylnicotinamide)81. Several endogenous compounds have been identified as substrates for OATP1B1 and/or OATP1B3, including CPI, CPIII, HDA, TDA, unconjugated and conjugated bilirubin, glycochenodeoxycholate-3-sulfate (GCDCA3S), and glycochenodeoxycholate-3-glucuronide (Table 3). Among them, CPI, CPIII, and GCDCA3S have been proposed as promising endogenous biomarkers for OATP1B function81,173,174. These endogenous biomarkers, in particular CPI, displayed good sensitivity and dose-dependency in systemic exposure to rifampin, a known OATP1B inhibitor, dosed in the range of 150–600 mg in healthy subjects169,175,176. Recent popPK (population pharmacokinetics) modeling analysis confirmed the sensitivity of CPI to detect weak and moderate OATP1B inhibition177. The significantly increased plasma CPI, CPIII, and GCDCA3S concentrations in healthy subjects carrying the SLCO1B1 c.521C/C genotype, compared to those with the c.521T/T genotype, corroborates the role of OATP1B1 in the disposition of these endogenous biomarkers178,179.
Table 3.
Examples of ADME biomarkers that can be used to detect drug–drug interactions.
| Enzymes or transporters | Potential endogenous biomarkers |
|---|---|
| CYP3A4/5 |
|
| OATP1B1/1B3 |
|
| OAT1/OAT3 |
|
| OCT2/MATE1/2κ |
|
N1-Methylnicotinamide and creatinine are the most studied endogenous biomarkers for renal OCT2, MATE1 and MATE2-K174,180. The endogenous biomarkers of renal OAT1 and OAT3 are relatively under-investigated. Taurine and GCDCA-S have been identified as potential endogenous biomarkers of OAT1 and OAT3, respectively181. Recently, pyridoxic acid (PDA) and homovanillic acid were discovered and validated as promising plasma-based endogenous biomarkers of OAT1 and OAT3 in cynomolgus monkey and they were also evaluated in humans182,183. The selectivity of PDA and homovanillic acid as OAT1 and OAT3 biomarkers in human was confirmed by assessing substrate potential of the compounds towards major renal and hepatic transporters182,184. A human popPK model has been developed, and the analysis supports qualification of PDA as endogenous biomarkers of OAT1 and OAT3185.
Challenges remain with the use of endogenous biomarkers in support of clinical transporter inhibition risk assessment. The selectivity, sensitivity, biosynthesis, and ADME properties of an endogenous biomarker, and its ability to differentiate different transporter inhibitors, need to be characterized. In addition, investigational drugs may affect the synthesis of endogenous biomarkers and their transport to blood from the synthesis site(s) in the body. With that in mind, the OCT2 endogenous biomarkers have not been fully validated, since there is no selective inhibitor of OCT2 over MATE1 and MATE2-K. Endogenous biomarkers for assessing the inhibition of P-gp or BCRP have not been identified, likely due to the limited involvement of intestinal P-gp and BCRP in oral absorption of endogenous compounds. In addition, it is difficult to dissect the relative contributions of intestinal, hepatic, and renal P-gp and BCRP to altered systemic exposure of endogenous biomarkers.
Endogenous biomarkers are also needed for DMEs. A notable progress in this area in the last decade is the identification of 4β-hydroxycholesterol (4β-HC) as a CYP3A biomarker186, 187, 188, 189, 190. However, although CYP3A induction potential in humans may be predicted by measuring plasma 4β-HC, plasma 4β-HC level increases only ∼2–4-folds with a typical 2-week rifampicin treatment, and it has a relatively narrow dynamic range compared to oral midazolam (CYP3A probe drug) clearance191. Changes in plasma 4β-HC level primarily reflect induction or inhibition of hepatic but not intestinal CYP3A192. In addition, the long half-life of 4β-HC makes it more suitable for measuring baseline or steady-state hepatic CYP3A activity, than for measuring acute changes in CYP3A activity, such as those resulting from enzyme inhibition. With all the inherent limitations of 4β-HC, it is well recognized that it cannot replace the dedicated utility of midazolam for detecting CYP3A DDI. Conversely, 4β-HC may be used for the following clinical scenarios: 1) in multiple-dose studies, where an increase in 4β-HC may provide an early signal for strong hepatic CYP3A inducers; 2) in long-term treatment studies or in patient populations, where a midazolam DDI study is not feasible; and 3) in efficacy studies for chronic conditions, in which hepatic CYP3A activity is altered by disease193.
2.5. Consideration of ADME pharmacogenetics (PGx) during drug discovery and development
There has been increasing understanding of clinically relevant polymorphisms of ADME genes and their impact on the drug PK/PD characteristics. The observed heterogeneity in the drug PK/PD and clinical responses may be attributed to the underlying genetic variations in ADME genes that are important regulators of drug exposure. Many reviews on these topics have been written, focusing on CYPs194,195, conjugating enzymes such as UGTs196, carboxylesterases197, as well as transporters OATP1B1198 and BCRP199.
In parallel to scientific advances, regulatory agencies have also issued guidance on examining the role of ADME pharmacogenetics to explain PK variability and associated clinical outcomes, setting clear expectations on sponsors in this regard200,201. As of today, approximately 100 US FDA-approved drugs have PGx languages in the labeling, of which more than 60% are related to ADME genes (https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling). These include examples of some well-known and important drug-gene pairs, such as clopidogrel and CYP2C19, statins and OATP1B1, and irinotecan and UGT1A1.
In addition to the label impact, ADME genetics has been utilized in drug discovery and development for purposes of marketplace differentiation, clinical dosing adjustment, mechanistic understanding of PK/PD variability, and predictions of DDIs and clinical outcomes202,203. One key component in deciphering the impact of ADME genetics is to appropriately determine genotype and phenotype relationships during clinical genetic analyses. To this end, it is important to first select genetic variants of interest based on consistent selection criteria, some of which were discussed previously204, and then leverage preclinical data on fractional clearance (fCL) of a compound to predict metabolic or transport phenotypes based on genotypes. The predicted phenotype of a given genotype may differ for different compounds, as the fCL of the ADME properties of interest may vary202. Examples of consortium efforts on defining genotype and phenotype relationships can be found at the Pharmacogene Variation Consortium (http://www.pgrn.org/pharmvar.html) and the Clinical Pharmacogenetics Implementation Consortium (https://cpicpgx.org/). Finally, as demonstrated in literature205,206 and the recently approved eliglustat labeling, mechanistic modeling is anticipated to continue to facilitate the understanding of PK/PD variability, DDIs, and clinical outcomes, through integration of multiple genetic factors and clinical variables207.
2.6. Characterization of non-hepatic clearance mechanisms
2.6.1. Prediction of human renal clearance
Although renal clearance as a major clearance pathway only accounts for approximately 32% of the top 200 drugs prescribed in US in 2010208, an accurate prediction of renal clearance is important to the estimation of the total systemic clearance, doses, dose regimen, and DDIs. An observed renal clearance of a compound reflects the collective effects of multiple mechanisms, including glomerular filtration, active secretion, and passive and active reabsorption. When renal clearance is mainly driven by passive mechanisms, i.e., glomerular filtration and passive reabsorption, allometric scaling from animals to humans for renal clearance prediction generally works well. Allometric scaling using animal data is currently the recommended approach to predict human renal clearance that is predominantly mediated by passive mechanisms. A kidney PBPK model has recently been developed, which is able to successfully predict passive renal clearance (glomerular filtration and passive reabsorption) for compounds without active processes in human, dog and rat209. The model requires input parameters of passive permeability from monolayer cultured low-efflux MDCK cells210, plasma protein binding, and pKa209. The model also enables prediction of drug-related risks for crystalluria when solubility information is included209.
When active secretion and reabsorption are the dominant mechanisms for renal clearance, allometric scaling typically is no longer able to accurately predict human renal clearance, due to species differences in transporter expression, affinity, and selectivity. As stated above, transporters play important roles in active renal secretion and reabsorption of compounds. OAT1, OAT3 and OCT2 are the major drug transporters on the basolateral membrane, whereas P-gp, MATE1, MATE2-K, MRP2, and MRP4 are the predominant efflux transporters on the apical membrane of proximal tubules that facilitate active renal secretion211. PEPT1, PEPT2 and urate transporter 1 are the major uptake transporters on the apical membrane to enable active reabsorption from renal tubules211. The relative activity factor approach was developed to quantitatively predict active renal secretion by OAT1, OAT2 and OAT3212. A study conducted with a set of 36 diverse drugs indicated that the most accurate prediction was achieved through direct correlation between human and dog renal clearance after correcting for differences in plasma protein binding and kidney blood flow213. Various in silico models have been developed to predict renal clearance and fraction of renal secretion with good accuracy214,215.
2.6.2. Drug elimination from intestinal excretion: An under-investigated drug clearance mechanism
Biliary excretion, renal elimination via glomerular filtration or transporter-facilitated excretion, and metabolism are important clearance pathways for small-molecule drugs. However, the contribution of the intestine and the kidney to drug distribution can be more complex than the initial absorption and excretion. Direct intestinal excretion (IE) has not been widely recognized as a common elimination pathway because the parent drug found in feces in radioactive ADME studies is often treated as unabsorbed materials or biliary excretion of intact drug, and because of a lack of readily available techniques to directly measure IE. IE is responsible for transporter-mediated elimination of endogenous compounds, such as bilirubin and cholesterol, and efflux transporter substrates, such as digoxin and paclitaxel216, 217, 218, 219, 220. For a compound with a high apparent permeability and in vitro metabolic stability, such as apixaban, passive diffusion coupled with intestinal peristaltic movement, as well as intestinal blood flow, generates a significant level of IE221,222. P-gp or BCRP gene knockout, or even simultaneous inhibition of both P-gp and BCRP, only affected apixaban IE to a small extent (<40%)223. Therefore, passive permeation can be more important than efflux transporters to IE of apixaban, a highly permeable compound. Transporters BCRP and P-gp only slightly enhanced IE of apixaban when the drug was dosed orally. In comparison, nitrofurantoin and digoxin had low levels of IE because of their low apparent permeability. BCRP and P-gp gene knockout further reduced the already low level of IE for nitrofurantoin and digoxin222. Of note, IE of a highly soluble drug may be self-saturated when dosed orally.
The extent of IE also depends on other elimination pathways such as biotransformation, urinary elimination, and biliary excretion. For example, metabolism appeared to be more important for apixaban elimination in humans (25% of dose) than in rats and dogs (<15%; urinary excretion was the major elimination pathways of apixaban in rat but not in dog or human). Despite these variables that make it hard to determine the exact contributions of various elimination pathways, IE accounted for approximately >20%, >42%, and >27% of apixaban dose clearance in rats, dogs, and humans, respectively224. Finally, it is always important to distinguish the rate from the extent of IE. If the system is modulated via activated charcoal or BDC (bile duct cannulation) or transporter inhibition, it is important to examine the extent of change in an elimination pathway as well as the rate.
Several studies have provided evidence for regional IE through use of the Ussing chambers, everted intestinal sacs, perfused intestinal segments, and in vivo with activated charcoal treatment218,220,225, 226, 227, 228, 229. Several drugs were discovered to undergo extensive IE only after investigation using various in vitro and in vivo models, and in limited examples fecal elimination resulting from regional IE has been unambiguously demonstrated218,228. Measuring the amount of a drug in feces after intravenous (IV) administration (preferably with radiolabeled drugs) to BDC animals represents a direct method to assess IE. Effects of experimental treatment with activated charcoal on PK/elimination and intestinal recovery of a drug in BDC animals following IV administration would provide ultimate IE assessment230,231.
3. DMPK advances that enable development of new modalities
New disease targets are continuously discovered and new therapeutic modalities that represent a diverse array of biologics, such as antibodies, antibody–drug conjugates, therapeutic replacement enzymes, peptides, and small interfering RNAs, are rapidly entering drug development pipelines. The new modalities, which, with increasing complexity and diversity, have revolutionized the treatment of a wide range of diseases, such as cancer, neurodegenerative disorders, and autoimmune diseases, require more comprehensive preclinical DMPK tools. Here we highlight several DMPK advancements that enable the development of new modalities.
3.1. Antisense oligonucleotide drugs
As a new class of drugs, several antisense oligonucleotide (ASO) drugs, such as inotersen and nusinersen, have been approved232. ASOs are small (∼18–30 nucleotides), single-stranded, chemically modified oligonucleotides with complementary sequences to specific mRNA sites. The action of ASOs is accomplished through post-transcriptional modulation, via two distinct mechanisms. One mechanism, exemplified by inotersen, is through RNase H-mediated cleavage. The ASO drugs bind to their complementary mRNA transcripts; the resulting complex is cleaved by endogenous RNase H enzymes in the nuclei and cytosol, resulting in a reduction of mRNA levels and consequent reduction of the levels of target, pathogenic proteins. The other mechanism, exemplified by nusinersen, is via modulation of pre-mRNA splicing, to increase levels of functional mRNAs and its encoded proteins.
Compared to traditional, small-molecule drugs, ASOs have a much higher molecular weight (4–10 kDa), and, though lipophilic, they possess multiple negative charges. As expected, they have very low membrane permeability, negligible oral bioavailability, and poor blood–brain barrier penetration. Their routes of administration are either IV or subcutaneous (SC) injections, to target the liver or muscle, or direct injections into the site of action, such as intrathecal injection to target the spinal cord or intravitreal injection to target the eye. Due to the low permeability, they are delivered into cells via endocytosis. Once inside the cell, they are sequestered into endosomes and then further into lysosomes for degradation or exocytosis. A portion of the ASO molecules may escape the lysosome degradation and reach the cytosol and nucleus to interact with drug targets.
Following either IV or SC administration, plasma ASO concentrations decline rapidly from peak concentrations in a multiexponential fashion. This process is characterized as an initial rapid distribution phase, when an ASO drug distributes to tissues, such as the liver and kidney, in minutes to a few hours, followed by a slow terminal elimination phase with a terminal plasma half-life of up to 2–4 weeks. Following SC administration, ASOs are absorbed rapidly from the injection site and released into the systemic circulation. The time to reach peak plasma concentration is typically 3–4 h. Phosphorothioate ASO drugs have relatively high plasma protein binding, preventing them from rapid renal excretion.
ASOs are not metabolized by the major “drug metabolizing” enzymes, such as CYPs. Instead, they are metabolized by endonucleases and exonucleases. The metabolites are fragments of the parent oligonucleotides and are eliminated from the body via renal clearance. Therefore, ASOs are not involved in the typical drug metabolizing enzyme-mediated DDIs233, 234, 235. ASOs typically have no or minimal inhibition or induction of major DMEs and transporters at clinically relevant concentrations236.
3.2. Therapeutic protein drugs
The protein therapeutics field has grown significantly since the introduction of the first human protein therapeutic, i.e., human insulin derived from recombinant DNA, in 1982. As of May 1, there are about 18 investigational therapeutic proteins (TPs) in regulatory review either by the US or EU regulatory agencies in 2021. Of those, three TPs were granted first approvals in either the US or EU from January 1 to May 1, 2021237. It is estimated that the global protein therapeutics market will be approximately $315 billion by 2025238. TPs have several advantages over small molecule drugs, which currently dominate the pharmaceutical market. TPs are capable of performing highly specific and complex functions, which is challenging for small molecule drugs to achieve. The higher specificity of proteins also results in lesser off-target toxicity. Much of this growth can be attributed to the development of monoclonal antibody (mAb) therapeutics as new therapeutic targets are discovered (e.g., checkpoint blockades). In addition, beside the advances in bispecific antibodies and multifunctional antibody-based medicines (e.g., antibody–drug conjugates (ADCs), non-antibody-based proteins such as replacement and non-endogenous proteins are also being developed. The first nanobody, caplacizumab, that contains a single-domain antibody fragment was approved in 2019.
Compared to small molecules, the ADME process for proteins are much less well characterized. In general, the PK and disposition properties of mAbs and peptides are influenced by two broadly categorized phenomena: target-mediated drug disposition (TMDD) and nontarget-related clearance mechanisms239. The nontarget-mediated PK and disposition of either mAbs or peptides is complex and depends on protein structure and the intrinsic physiologic mechanisms. Physiochemical attributes, such as molecular weight, secondary and tertiary structures, charge- and hydrophobicity-related attributes, post-translational modifications (e.g., glycosylation, deamidation, and methylation), and thermal and catabolic stability, each plays a role in the clearance of mAbs and peptides to various degrees240,241. Proteins are administered parenterally and usually not available as oral drugs due to denaturation in the gut. Little is known about the mechanism of absorption of mAbs administered via SC or intramuscular injection, although uptake is recognized to be primarily via the lymphatic system. Absorption of mAbs from the SC route is slow, with peak concentrations occurring approximately 6–9 days after administration. Bioavailability of most mAbs ranges from 40% to 80%242. Large proteins are not able to efficiently penetrate the tissue to reach their target sites due to their size and hydrophilicity. The distribution of proteins into tissues is generally slow and via convective transport through pores on capillary walls as well as transcytosis from circulation to the extracellular space. Presence of target in peripheral tissue can significantly change the tissue distribution of proteins and lead to potential asymmetric distribution of proteins between plasma and tissues239.
The disposition of mAbs has been characterized using a platform PBPK model based on experimental mouse data. The model has been further applied to describe the plasma and tissue PK of nonspecific or antigen-specific mAbs in various preclinical species and humans243. A common mechanism of protein elimination includes filtration into urine, secretion into the bile and biotransformation (e.g., metabolism or catabolism). There is an inverse relationship between the rate of renal clearance (or elimination) of protein-based biologics and their size (or molecular weight). The general philosophy, based on a number of studies, suggests that molecules that are less than 50 kDa are removed from systemic circulation by renal filtration through the renal glomeruli. The large molecular weight of mAbs (∼150 kDa) clearly precludes these molecules from renal clearance mechanisms; however, for peptides, renal filtration can play a major role in their removal from systemic circulation. Secretion into the bile is an important pathway of elimination of IgA antibodies, but this route is not a significant contributor to the elimination of IgG antibodies. The majority of IgG elimination occurs via intracellular catabolism, following fluid-phase or receptor-mediated endocytosis. However, mAbs, as well as endogenous IgG/albumin, can be protected from degradation by tight binding to protective receptors [the neonatal Fc-receptor (FcRn)] at acidic pH in the early endosome. The IgG–FcRn complexes are not delivered to the lysosome for catabolism but rather are sorted to the cell surface for fusion with the cell membrane. IgG/albumin dissociates from the receptor at physiological pH and is rapidly released into extracellular fluid. As a result, these molecules exhibit relatively long terminal half-lives, usually weeks. IgG affinity for FcRn is species specific239,244. By taking advantage of the recycling by FcRn receptors, the fusion of a small therapeutic protein with the Fc binding moiety of mAbs or albumin can enhance its half-life.
TMDD is common for TPs, especially mAbs, due to their relatively low systemic clearance and high target-binding affinity. Target-mediated elimination is saturable because of the finite expression level of the target. The rate of uptake and elimination of antibodies by target-mediated pathways is a function of dose and the expression level of the target, as well as a function of the kinetics of receptor internalization and intracellular catabolism. Target-mediated elimination happens following Fab binding, not only to the cell-surface receptor, but also to soluble targets, particularly multimeric substances with several repeated epitopes, leading to the formation of large complexes that may be rapidly eliminated by phagocytosis. A consequence of TMDD is non-linear PK, with high clearance observed at lower does and dose-dependent clearance decrease and half-life increase. IgG antibodies may also interact with Fc-γ-receptors (FcγR); the endocytosis and catabolism of IgG-FcγR complexes may contribute to the elimination of circulating antibodies. However, considering the relatively high affinity of IgG for FcγR and the high endogenous concentrations of IgG in plasma (∼65 μmol/L), it has been argued that FcγR-mediated elimination is unlikely to be important for monomeric IgG239,242.
PK from cynomolgus monkeys alone can be successfully scaled to humans within a linear range, using a simple allometry with fixed exponent within reasonable accuracy, i.e., within 2-fold of the observed values. The mean scaling exponents in the allometric equation for CL and Vss, against body weight, were calculated to be 0.85 and 1.00, respectively245. The non-linear PK profile can be predicted through the TMDD modeling approach if target expression and physiological parameters are available in patients or when Michaelis–Menten parameters (Km and Vmax) in monkeys and humans are assumed scalable246. The PK/PD models and concepts of TPs are generally similar to small molecules.
As TPs do not typically depend on metabolism or transport for clearance, there is less potential for concomitantly administered drugs to affect therapeutic protein disposition or elimination. However, evolving data indicate that TPs may affect the disposition of other drugs. Exogenously administered immunologic proteins, such as interferons and interleukins, may result in changes in the metabolic capacity of the liver that are similar to what has been observed during acute infection or inflammation. It has been demonstrated that cytokines can suppress gene transcription, resulting in downregulation of CYP enzymes and drug transporters247. FDA guidance248 suggests evaluating the DDI potential for TPs as perpetrators if they are proinflammatory cytokines or cytokine modulators, or when a therapeutic protein affects human physiological processes that can in turn alter the PK profiles of co-administered medications. Conversely, the therapeutic protein should be evaluated as a victim if co-administered medications compromise the function of FcRn, or when co-administered medications include an immunosuppressor and a therapeutic protein whose PK properties are affected by immunogenicity. If co-administered medications impact the TP's target or target-mediated disposition, the DDI potential of the TPs may be evaluated as perpetrators or as victims, depending on the role of the TPs in the DDI.
The translation of in vitro data or animal DDI data to humans has been limited. Some pre-clinical methods could provide mechanistic understanding of the DDI potential of TPs, which in some cases can be combined with PBPK models. Clinical studies of TPs should consider the suspected mechanism for the DDI when selecting the relevant study population and the interacting drugs to evaluate. The study design (parallel or crossover) should be informed by the suspected mechanism of the DDI and the PK characteristics of the drugs (e.g., the drug's half-life). Although PBPK modeling may be applied to TP–drug interactions to evaluate complex mechanisms and explore the potential for clinically significant interactions, this approach is still emerging and currently not comprehensive enough to substitute for in vivo studies.
3.3. Antibody–drug conjugates (ADCs)
ADCs are a class of complex molecules that compose of a monoclonal antibody (mAb) linked to a biologically active small molecule (payload) through a specialized chemical linker forming a conjugate. mAbs specifically bind to tumor associated antigens that are highly expressed in cancer cells, which allow for the discrimination between normal and cancer cells. Upon binding to the surface antigen, ADCs can be internalized by endocytosis and trafficked to endosomes and lysosomes, where lysosomal proteolysis catalyzes the linker to release the biologically active small molecule. While the antigen is selected to target cancer cells, the linker is designed to be stable in systemic circulation but rapidly cleaved in the lysosome. Once released from the mAb, it is thought that payload can freely diffuse through the lysosomal membrane to intact with intracellular targets249. The cytotoxic payloads are often substrates of efflux transporters. The efflux transporters that are also expressed on the lysosomal membrane can sequester the payload and cause multidrug resistance of an ADC250,251. The in vivo forms of ADC species can be very heterogeneous, including unconjugated payload, unconjugated antibody, and ADCs with varying levels of conjugation252.
This in vivo structural variation, in combination with the lack of specific agency guidance for their bioanalysis, makes it essential for sponsors to design a robust and relevant bioanalytical strategy for each ADC. It is critical for bioanalytical scientists to engage key stakeholders, such as clinical pharmacologists, clinical safety scientists, and regulatory specialists, to apply sound scientific principles to ensure a bioanalytical strategy that generates robust data that are meaningful for efficacy and safety endpoints. A stage-specific bioanalytical strategy may be needed for the bioanalysis of ADCs. For example, investigations of in vivo metabolism of an ADC may be needed in early-stage clinical studies. As clinical development progresses, the bioanalytical strategy may evolve once correlative relationships between different species of the ADC and various safety and efficacy signals are established.
From an analytical perspective, it is important to know the fundamentals of the assays, such as the exact analyte, the variability of the measurements, and the reliability of procedures, from sample collection, handling, to storage. The heterogenous in vivo species could also potentially interfere with each other's quantitation, due to issues such as continuous catalysis during sample handling and storage. Typically, the PK bioanalysis strategy of an ADC involves the measurements of the concentrations of ADCs, total antibodies (ADC plus free antibodies), and free warhead in circulation. The ADCs and the total antibodies are usually measured by ligand binding assays (LBA) (Fig. 1). They can also be measured by hybrid immunocapture–LC/MS methods, where a surrogate signature peptide can be used as a read out of total antibodies while the warhead can be used for the quantification of ADCs. Recent advances in analytical technologies, such as high-resolution mass spectrometers, have enabled direct quantification of intact macromolecules, including free antibodies and ADCs. Like other biotherapeutic modalities, ADCs can induce host immune responses that may affect the exposure, safety, and efficacy of the drug. Molecular structure factors specific to ADCs, such as the payload and hapten-like structures, may increase the risk of immunogenicity.
3.4. Advancement of bioanalytical methods to enable development of large-molecule drugs
3.4.1. Ligand-binding assays (LBA) for ASO drugs
LBA, specifically hybridization ELISA, has been the most used bioanalytical technology for ASOs. In this method, an ASO drug in the biologic matrix is captured with an immobilized oligonucleotide, which has a complementary sequence to one part of the ASO drug, and the ASO drug is further hybridized with another oligonucleotide which has a complementary sequence to the other part of the ASO drug to be detected. The advantages of the hybridization ELISA method include high sensitivity, with capability of quantitation of ASOs at single-digit ng/mL levels in biological fluids, and relatively high throughput. However, the major challenge of hybridization ELISA is its limited specificity, as hybridization probes may react with the truncated metabolites of the target ASOs. Chromatographic assays coupled to high-resolution mass spectrometers or MS/MS have been used as alternatives to hybridization ELISA for ASO bioanalysis. The main advantages of the MS-coupled assays are that there is no need for special reagents or enhanced specificity, and that the assay can distinguish truncated metabolites from the parent ASO drugs. However, the relatively low sensitivity of the LC–MS methods has limited their application in ASO drug discovery. Recently, a method combining the advantages of LBAs and LC–MS assays, called hybridization LC–MS/MS assay, was reported, which can achieve both high specificity and high sensitivity253.
3.4.2. Analytical methods for immunogenicity of ADCs
As is for other biotherapeutics, the immunogenicity assay strategy of an ADC should be developed based on its risk assessment, including the probability of immunogenicity against the ADC and its consequences254. However, there are potential consequences that are unique to ADCs. For example, formation of immune complexes containing anti-drug antibodies (ADAs) with the payload by immune cells may result in toxicity. In addition, prior exposure to therapeutics containing the same linker or small-molecule drug may further complicate the immunogenicity risks.
A standard three-tier strategy is often appropriate for ADC immunogenicity monitoring255. The decision to perform ADA domain mapping should be based on a thorough risk assessment. Typically, specificity is mapped against the mAb vs. linker/payload. Determination of domain specificity can be achieved by two approaches. In a competition strategy, ADA domain mapping is accomplished through a confirmatory competitive inhibition assay. For example, signals derived from ADA against the mAb can be ablated by addition of unlabeled mAb in the confirmatory assay. Alternatively, ADC components may be used as capture and detection reagents to detect the existence of ADA specific to that component.
4. Model-informed drug development (MIDD)
The concept of PBPK modelling is not new256; however, significant progresses in the utility of PBPK modelling have been made in the past 10 years, through its incorporation into drug discovery and development processes257, 258, 259, 260, 261, 262, 263. These advances were propelled by the scientific community, through efforts from three main sectors. Firstly, commercial PBPK platforms were developed by incorporating a large number of species-dependent physiological system parameters and drug-dependent physicochemical, in vitro, and in vivo ADME data; these included Simcyp (https://www.certara.com/software/simcyp-pbpk), GastroPlus (www.simulations-plus.com), PKSIM (www.systemsbiology.com/products/pk-sim.html), ADMEWORKS DDI Simulator (http://www.fqs.pl/chemistry_materials_life_science/products/ddi_simulator), CLOEPK (http://www.cyprotex.com/insilico/), and many others. Secondly, experienced modelers from pharmaceutical industry and academic institutions utilized these commercial PBPK platforms to address predictive PK-related questions and support the internal decision making in drug discovery and development. Thirdly, modelers from regulatory agencies reviewed the PBPK applications in drug development and provided guidance on the best PBPK practices.
4.1. Application of physiologically based pharmacokinetic (PBPK) modeling
DMPK scientists from pharmaceutical industry are not only concerned with whether certain PK behavior of a molecule can be predicted, but are also interested in providing an understanding of the confidence level for such predictions in general to enable the internal decision making for key milestones for drug discovery and development. Therefore, scientists from pharmaceutical companies formed a working group under the International Consortium for Innovation and Quality in Pharmaceutical Development, with a common goal of understanding and improving the prediction accuracy and confidence. Moderate to high confidence predictions have been achieved in the following areas: (1) preclinical and clinical PK prediction for CYP-mediated mechanisms, (2) DDI-related reversible CYP inhibition alone, (3) absorption prediction for high solubility and high permeability compounds [Biopharmaceutics Classification System (BCS) Class 1], (4) PK prediction for special populations related to PGx, and (5) PBPK-PD prediction and target organ distribution for small, passively permeable molecules. It is particularly notable that there has been an improved understanding of the PK prediction for specific populations (e.g., patients with renal or hepatic impairment). For examples, a retrospective analysis conducted by 19 pharmaceutical companies suggested 90% (n = 47/50 arms) and 70% (n = 43/56 arms) successful rate (predicted within twofold of observed AUC ratios) for mild renal and hepatic impairment PK prediction, respectively258. The confidence in predicting PK for moderate and severe hepatic impairment populations remains relatively low. An accurate reflection of the pathophysiological changes for the targeted patient population in the PBPK platform and the accurate characterization of the clearance pathway for a drug would contribute to the successful prediction. Based on these collective efforts, it was recommended that PBPK may be used to support mild renal impairment study waivers, a view that is not yet universally accepted, particularly by regulatory agencies264.
Another retrospective analysis conducted by 23 pharmaceutical companies suggests an 80% successful rate (24/30 drugs) for correctly capturing the likelihood of food effect, without any optimization of the absorption parameters. In addition, there is no clear correlation between the prediction confidence and BCS category and/or food effect type. However, an association is demonstrated with the key mechanism(s) driving the food effect. High confidence in PBPK prediction of food effects is typically observed for compounds where the mechanism of food effect was related to physiology, including changes in the gastrointestinal (GI) luminal fluids, fluid volume, motility, pH, ion pairing, and bile salts. Low confidence in prediction is associated with the food effects related to drug formulation interactions with the intestinal microenvironment, specifically with respect to salts and weak bases, such that the model and/or the biorelevant media used could not capture the dynamic effects of the drug on its microenvironment. Low confidence in modeling is also observed with the food effects related to fed-state hydrodynamics (e.g., GI fluid viscosity) and food–drug/micelle–drug interactions where standard in vitro assays are not able to characterize the food effect mechanistically.
Compared to the CYP-mediated clearance and DDI prediction, the confidence of the transporter translation is considered to be relatively low. Nevertheless, 25 case examples of transporter-mediated DDI simulation using PBPK models, which either successfully supported submissions to the regulatory agencies or played a crucial role in trial design/strategic internal decision making (between 2013 and 2018) is a testimony of the tremendous progress made in the science of PBPK modeling for transporter substrates and inhibitors263. The remaining challenges and opportunities are identified in the following areas: 1) poor IVIVE translation for transporter substrates with the general underprediction trend of in vitro kinetics parameters, 2) gaps in system data such as transporter protein abundance in vitro and in vivo, localization and intestinal/renal/hepatic/brain parameters, 3) incorporation of the transporter contributions to the Vss prediction, 4) poor IVIVE translation for transporter perpetrators, with a general trend of underprediction by in vitro inhibition Ki values. Future advancements in these areas will improve our confidence in the prediction of unbound tissue concentration, which will impact the accuracy in predicting efficacy and toxicity of drugs.
4.2. PK/PD considerations and dose selection for cell and oligonucleotide therapy
Applications of MIDD to cell therapy are still in the early stages, and rapidly evolving. MIDD approaches can be leveraged to address important PK/PD questions to support dose selection, with consideration of the complexities resulting from the unique cellular PD. For example, what's the optimal binding affinity and receptor expression density for a given target? How translatable are the in vitro and animal models? How should these data be used in making predictions about the first-in-human dose? Are there any intrinsic or extrinsic patient factors (e.g., tumor burden or co-administered therapy) that should inform dose regimen?
The first clinical cellular kinetic model for chimeric antigen receptor (CAR)-producing T-cells (CAR-T) was developed for tisagenlecleucel and was used for confirming that medications used to treat cytokine release syndrome (tocilizumab and low-dose corticosteroids) did not impact the rate of CAR-T expansion265,266. The authors reported that blood cellular kinetics usually exhibit a rapid expansion, beyond the infused cell dose, and then long-term persistence. The mathematical model (Fig. 2267) describes the exponential expansion, with the rate constant ρ up to time Tmax, followed by biphasic decline after Tmax with rate constants α (a rapid decline reflecting the apoptosis of most activated lymphocytes following the initial immune response) and β (a slower decline due to a gradual decrease of T cells that can persist in the body). The recently published work in the xenograph mouse model268 presented a more mechanistic modeling example by including tumor killing components, but further work remains to translate such models and insights into clinical development.
Figure 2.

Multi-phase kinetics of CAR-T cell and model structure. (a) A typical multi-phase kinetics of CAR-T cell: distribution, expansion, contraction and persistence phase; (b) model structure for CAR-T kinetics; (c) schematic diagram of a three-step workflow for modeling and analysis. ALL, acute lymphocytic leukemia; CAR-T, chimeric antigen receptor-T; CLL, chronic lymphocytic leukemia; Cmax, peak plasma concentration; CR/PD, complete response/progressive disease; DLBCL, diffuse large-B cell lymphoma; MM, multiple myeloma. Reproduced with permission from Ref. 267. Copyright © 2020 The authors and American Society for Clinical Pharmacology and Therapeutics.
Plasma PK for RNA oligonucleotides (ASO and siRNA) is typically characterized by a rapid distribution phase driven by uptake into major distribution organs (liver and kidney), and a slower terminal elimination phase269, 270, 271. Allometric scaling approaches can in general be used for inter-species scaling of plasma clearance across nucleotide sequences and between species. Liver: plasma concentration ratios of ∼5000-fold have been reported after the initial distribution phase for ASOs, and these numbers are consistent across species and between different ASOs269, 270, 271. The liver is the chosen site of action for a number of oligonucleotide therapies given their preferential liver uptake. Exposure (plasma or liver)–response relationship established in preclinical models can be used to inform human translation. The EC50 values estimated in mice extrapolated well to humans269,270.
5. Advancement of regulatory sciences
Scientific advances in DMEs and transporters strengthened our understanding of PK DDIs. Drug interaction potential is recognized as an important consideration in the evaluation of an NME and is an integral part of drug development and regulatory review prior to its market approval272, 273, 274, 275. Metabolism, transport, and drug-interaction information play a key role in benefit/risk assessment. Since the publication of the first DDI guidance in 1997, FDA has updated DDI guidance documents multiple times over the past two decades to incorporate updated knowledge and recommendations on DDI evaluation during drug development so that DDIs can be managed properly (Fig. 3105,276, 277, 278, 279, 280). FDA DDI guidance documents281 recommend an integrated approach that includes in vitro and in vivo studies and in silico tools (e.g., PBPK modeling) to help determine the need of in vivo DDI studies, study design, and inform labeling recommendations. There is an increase in PBPK modeling in drug development and regulatory submissions. PBPK models have been used in lieu of some in vivo DDI studies, for example, to evaluate the effects of moderate inhibitors or inducers on the PK of an NME and complex DDIs mediated by CYPs264,282, 283, 284, 285. PBPK analyses to evaluate transporter-mediated DDIs can be scientifically challenging, yet they still play a role in regulatory submissions.
Figure 3.

FDA DDI Guidance History (1997–2020). Major CYP enzymes recommended for routine assessment to identify potential metabolism-mediated interactions include: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A. Transporter-mediated DDI was first recommended in 2006 draft FDA DDI guidance, focusing on P-glycoprotein. Evolving knowledge on roles of transporters in DDI, safety and efficacy, and collaborative work led by the International Transporter Consortium has led to more transporters being studied105,276, 277, 278. To date, nine clinically important transporters are recommended in FDA DDI guidance documents for routine evaluations of DDI potential for investigational drugs, which include four efflux transporters (P-gp, BCRP, MATE1 and MATE2-K) and five uptake transporters (OATP1B1, OATP1B3, OAT1, OAT3, and OCT2)279,280. Basic and more mechanistic models and decision criteria have been developed and refined over the past decade. The international harmonization efforts on DDI evaluation are being pursued at the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). An ICH M12 guideline on DDI is being developed to provide a consistent approach in designing, conducting, and interpreting DDI studies during the development of a therapeutic product (https://www.ich.org/page/multidisciplinary-guidelines).
Potential DDIs resulting from effects of selected drugs on the NME and effects of the NME on other selected drugs should be explored during drug development to ensure an adequate assessment of an NME's safety and effectiveness. Drug withdrawal due to DDIs has rarely occurred since 2002, which may be attributed to potential DDIs being evaluated early so that proper mitigation strategies can be implemented prior to drug approval.
Besides enzyme or transporter-mediated DDIs, absorption-related DDI for drugs whose solubility is pH-dependent can be caused by pH changes in the gastrointestinal tract that are induced by acid reducing agents (e.g., proton pump inhibitors, H2 blockers, and antacids). pH-dependent DDI should be evaluated during drug development and a preliminary decision framework has been proposed to investigate this type of DDI with a draft guidance published by the FDA in 2020286. PBPK modeling efforts to predict pH-dependent DDI have been explored287,288.
The increased clinical use of therapeutic proteins raises concerns regarding their impact on DMEs or transporters and the potential for drug interactions. Generally, these interactions between therapeutic proteins and small molecule drugs may not be adequately detected by in vitro assessments289,290. Therefore, in vivo drug interaction studies may be indicated when a therapeutic protein is known to modulate enzymes or transporters (e.g., proinflammatory cytokines) or is designed to be administered with other drug product(s) as a combination regimen, especially if the co-administered drug has a narrow therapeutic range. Evaluation of DDIs for therapeutic proteins is still an evolving area. FDA published a separate draft guidance on DDI assessment for therapeutic proteins in 2020. Overviews of clinically reported therapeutic protein–drug interactions and the information included in the product package insert have recently been presented291, 292, 293.
6. Summary and future perspectives
The competitive landscape in drug discovery and commercialization to enhance drug access requires DMPK scientists to continue apply innovative approaches to advance drug candidates in ever accelerated timelines. As we reflect on some of the major ADME and PK advancements in the past decade, the future looks even brighter on the translation of new innovations and cutting-edge science and technologies for drug discovery and development. Here we list some of the areas where breakthroughs are needed and/or likely to occur in the next decade.
6.1. Clearance prediction
The confidence in human clearance prediction has increased greatly due to the high-quality reagents (e.g., HLM and HHEP) available and the increased understanding of clearance mechanisms, as well as availability of state-of-the-art modeling and simulation tools to integrate complex processes. Low-clearance methods have been developed and implemented to detect intrinsic clearance and identify metabolites of slow-turnover compounds. Much progress has been made with the prediction of transporter-mediated clearance and enzyme-transporter interplay, and new tools are continuously being developed to further mitigate challenges. Further enhancements in clearance prediction will likely come from breakthroughs in our ability to more accurately predict non-CYP mediated clearance, extra-hepatic clearance, transporter-mediated hepatic and renal clearance, enterohepatic recirculation, and biliary clearance. Significant innovation will be needed to develop new tools to address these gaps. Continuous optimization of 3D culture systems and increased utility of humanized animals294 are potential solutions for these challenging areas. Knowledge gaps underlying the IVIVE disconnect remain to be filled.
6.2. Protein quantification
Major technological advances in proteomics have enabled rapid quantification of DMEs and transporters in various human tissues and in vitro systems. These data are highly informative; they make it practical to bridge in vitro cDNA expressed systems to more physiologically relevant systems (e.g., HLM or HHEP), by using the REF approach. They also allow cross-tissue scaling of enzyme or transporter protein levels when using PBPK modeling. Currently, the variability of the proteomics approach is still high, with inconsistency observed between different methods or laboratories. Relationships between protein membrane expression and activity need to be established and verified further. As the field learns to standardize the approaches for protein quantification and validate the data against activity information, the quality of the data will improve. Overall, we expect that the proteomics protein quantification approaches will continue to have a high impact in the ADME and PK field in the next decade.
6.3. Endogenous biomarker strategies
The biomarker field has matured significantly during the past decade. Biomarkers are widely applied in efficacy and safety studies in drug discovery and development. ADMET biomarkers have shown great promises in predicting DDI and exposure changes in disease populations, such as 4β-hydroxycholesterol for CYP3A4/5 and CPI for OATP1B. ADMET biomarkers have been used regularly in the pharmaceutical industry to aid internal decision making. As more and more validations are available between biomarker approaches and traditional studies, the ADMET biomarker strategies are more likely to gain acceptance by regulatory agencies in the future.
6.4. DDI prediction
Our ability to accurately predict DDI has improved tremendously in the past decade. This is largely due to in-depth mechanistic understanding of the kinetic parameters influencing DDI, and significant developments in PBPK modeling to integrate complex processes, such as DDI involving metabolites, autoinhibition/autoinduction, and enzyme-transporter interplay. The regulatory DDI guidelines have continuously evolved as our knowledge expands in ADMET science. PBPK modeling and simulations have provided highly valuable information to enable project progression without conducting certain clinical trials. Future enhancements of the PBPK modeling tools, along with the scientific and technological advancements, will enable more reliable DDI predictions for the more challenging processes, such as transporter-mediated DDI and complex interplay of reversible/time dependent inhibition and induction.
6.5. New modalities
As the pharmaceutical industry is putting more emphasis on new areas of peptides, oligonucleotides, highly-engineered proteins, antibody–drug conjugates, and RNA vaccines, new ADMET knowledge and enabling technologies are being developed to address the paradigm shift. Quantification of new modalities requires innovative approaches and creative thinking. Measurement of ADCs may mean detection of multiple species rather than a single molecule. ADMET of new modalities can be very different than those of small molecules. TMDD is the norm rather than the exception for therapeutic proteins compared to small molecules. CYP-mediated DDI is common for small molecules, but rare for ASO or therapeutic proteins, which may have quite different DDI mechanisms. Major breakthroughs are expected in this area to support development of new approaches and fill knowledge gaps in our understanding of the DMPK differences between the new modalities and small-molecule drugs.
6.6. Application of microphysiological systems
Although 3D cell culture systems are miniaturized organs that possess some key features of intact tissues, they are still over-simplified versions of the native tissues in many ways. Many challenges remain for widespread adoption of 3D cell culture systems in drug discovery and development. Current 3D culture systems are diverse in terms of complexity, architecture, size, morphology, and assay protocols. This leads to challenges in standardization with respect to culture and assay methods, phenotypes, and output data analysis. In addition, the predictive values of 3D cell cultures for ADMET need further validation with human in vivo data. Furthermore, the assays using 3D cell models are far less developed with respect to quantification, imaging, and analysis throughput, compared with established 2D cell culture models. Taken together, 3D cell cultures are promising tools in drug discovery and development, and have high potential to capture the in vivo functions of organs and tissues. Moreover, the development of screening-compatible 3D cell cultures would transform the drug discovery process, as it would enable obtaining more physiologically relevant pharmacokinetic and toxicity data early.
Acknowledgments
We would like to thank Dr. Ping Zhao and Dr. Donglu Zhang for providing scientific insights on this review article. Research in Xinxin Ding's laboratory was supported in part by grants from the National Institutes of Health (CA023074, CA092596, ES004940, ES006694, and ES020867, USA).
Author contributions
Wrote and reviewed the manuscript: Yurong Lai, Xiaoyan Chu, Li Di, Wei Gao, Yingying Guo, Xingrong Liu, Chuang Lu, Jialin Mao, Hong Shen, Huaping Tang, Cindy Q. Xia, Lei Zhang, and Xinxin Ding.
Conflicts of interest
The views expressed are those of authors and do not necessarily reflect FDA's views or policy. The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Yurong Lai, Email: Yurong.lai@gilead.com.
Xinxin Ding, Email: xding@pharmacy.arizona.edu.
References
- 1.Chung T.D.Y., Terry D.B., Smith L.H. In: Assay guidance manual. Markossian S., Grossman A., Brimacombe K., Arkin M., Auld D., Austin C.P., et al., editors. Eli Lilly & Company and the National Center for Advancing Translational Sciences; Bethesda (MD): 2004–; 2015. In vitro and in vivo assessment of ADME and PK properties during lead selection and lead optimization—guidelines, benchmarks and rules of thumb. [PubMed] [Google Scholar]
- 2.Li Y., Meng Q., Yang M., Liu D., Hou X., Tang L., et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm Sin B. 2019;9:1113–1144. doi: 10.1016/j.apsb.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood F.L., Houston J.B., Hallifax D. Clearance prediction methodology needs fundamental improvement: trends common to rat and human hepatocytes/microsomes and implications for experimental methodology. Drug Metab Dispos. 2017;45:1178–1188. doi: 10.1124/dmd.117.077040. [DOI] [PubMed] [Google Scholar]
- 4.Edson K.Z., Rettie A.E. CYP4 enzymes as potential drug targets: focus on enzyme multiplicity, inducers and inhibitors, and therapeutic modulation of 20-hydroxyeicosatetraenoic acid (20-HETE) synthase and fatty acid omega-hydroxylase activities. Curr Top Med Chem. 2013;13:1429–1440. doi: 10.2174/15680266113139990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saravanakumar A., Sadighi A., Ryu R., Akhlaghi F. Physicochemical properties, biotransformation, and transport pathways of established and newly approved medications: a systematic review of the top 200 most prescribed drugs vs. the FDA-approved drugs between 2005 and 2016. Clin Pharmacokinet. 2019;58:1281–1294. doi: 10.1007/s40262-019-00750-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie F., Ding X., Zhang Q.Y. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm Sin B. 2016;6:374–383. doi: 10.1016/j.apsb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li A.P., Ho M.D., Alam N., Mitchell W., Wong S., Yan Z., et al. Inter-individual and inter-regional variations in enteric drug metabolizing enzyme activities: results with cryopreserved human intestinal mucosal epithelia (CHIM) from the small intestines of 14 donors. Pharmacol Res Perspect. 2020;8:e00645. doi: 10.1002/prp2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H., Wolford C., Basit A., Li A.P., Fan P.W., Murray B.P., et al. Regional proteomic quantification of clinically relevant non-cytochrome P450 enzymes along the human small intestine. Drug Metab Dispos. 2020;48:528–536. doi: 10.1124/dmd.120.090738. [DOI] [PubMed] [Google Scholar]
- 9.Di L. The role of drug metabolizing enzymes in clearance. Expet Opin Drug Metabol Toxicol. 2014;10:379–393. doi: 10.1517/17425255.2014.876006. [DOI] [PubMed] [Google Scholar]
- 10.Foti R.S., Dalvie D.K. Cytochrome P450 and non-cytochrome P450 oxidative metabolism: contributions to the pharmacokinetics, safety, and efficacy of xenobiotics. Drug Metab Dispos. 2016;44:1229–1245. doi: 10.1124/dmd.116.071753. [DOI] [PubMed] [Google Scholar]
- 11.Yang G., Ge S., Singh R., Basu S., Shatzer K., Zen M., et al. Glucuronidation: driving factors and their impact on glucuronide disposition. Drug Metab Rev. 2017;49:105–138. doi: 10.1080/03602532.2017.1293682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohnert T., Patel A., Templeton I., Chen Y., Lu C., Lai G., et al. Evaluation of a new molecular entity as a victim of metabolic drug–drug interactions—an industry perspective. Drug Metab Dispos. 2016;44:1399–1423. doi: 10.1124/dmd.115.069096. [DOI] [PubMed] [Google Scholar]
- 13.Seo K.A., Kim H.J., Jeong E.S., Abdalla N., Choi C.S., Kim D.H., et al. In vitro assay of six UDP-glucuronosyltransferase isoforms in human liver microsomes, using cocktails of probe substrates and liquid chromatography–tandem mass spectrometry. Drug Metab Dispos. 2014;42:1803–1810. doi: 10.1124/dmd.114.058818. [DOI] [PubMed] [Google Scholar]
- 14.Miners J.O., Rowland A., Novak J.J., Lapham K., Goosen T.C. Evidence-based strategies for the characterisation of human drug and chemical glucuronidation in vitro and UDP-glucuronosyltransferase reaction phenotyping. Pharmacol Ther. 2021;218:107689. doi: 10.1016/j.pharmthera.2020.107689. [DOI] [PubMed] [Google Scholar]
- 15.Furukawa T., Naritomi Y., Tetsuka K., Nakamori F., Moriguchi H., Yamano K., et al. Species differences in intestinal glucuronidation activities between humans, rats, dogs and monkeys. Xenobiotica. 2014;44:205–216. doi: 10.3109/00498254.2013.828362. [DOI] [PubMed] [Google Scholar]
- 16.Kaivosaari S., Finel M., Koskinen M. N-Glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Xenobiotica. 2011;41:652–669. doi: 10.3109/00498254.2011.563327. [DOI] [PubMed] [Google Scholar]
- 17.Docci L., Klammers F., Ekiciler A., Molitor B., Umehara K., Walter I., et al. In vitro to in vivo extrapolation of metabolic clearance for UGT substrates using short-term suspension and long-term co-cultured human hepatocytes. AAPS J. 2020;22:131. doi: 10.1208/s12248-020-00482-9. [DOI] [PubMed] [Google Scholar]
- 18.Mueller J.W., Idkowiak J., Gesteira T.F., Vallet C., Hardman R., van den Boom J., et al. Human DHEA sulfation requires direct interaction between PAPS synthase 2 and DHEA sulfotransferase SULT2A1. J Biol Chem. 2018;293:9724–9735. doi: 10.1074/jbc.RA118.002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riches Z., Stanley E.L., Bloomer J.C., Coughtrie M.W. Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT "pie". Drug Metab Dispos. 2009;37:2255–2261. doi: 10.1124/dmd.109.028399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li A.P., Hartman N.R., Lu C., Collins J.M., Strong J.M. Effects of cytochrome P450 inducers on 17α-ethinyloestradiol (EE2) conjugation by primary human hepatocytes. Br J Clin Pharmacol. 1999;48:733–742. doi: 10.1046/j.1365-2125.1999.00081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di L. Reaction phenotyping to assess victim drug–drug interaction risks. Expet Opin Drug Discov. 2017;12:1105–1115. doi: 10.1080/17460441.2017.1367280. [DOI] [PubMed] [Google Scholar]
- 22.Dalvie D., Di L. Aldehyde oxidase and its role as a drug metabolizing enzyme. Pharmacol Ther. 2019;201:137–180. doi: 10.1016/j.pharmthera.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Garattini E., Terao M. The role of aldehyde oxidase in drug metabolism. Expet Opin Drug Metabol Toxicol. 2012;8:487–503. doi: 10.1517/17425255.2012.663352. [DOI] [PubMed] [Google Scholar]
- 24.Coelho C., Mahro M., Trincao J., Carvalho A.T., Ramos M.J., Terao M., et al. The first mammalian aldehyde oxidase crystal structure: insights into substrate specificity. J Biol Chem. 2012;287:40690–40702. doi: 10.1074/jbc.M112.390419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garattini E., Fratelli M., Terao M. Mammalian aldehyde oxidases: genetics, evolution and biochemistry. Cell Mol Life Sci. 2008;65:1019–1048. doi: 10.1007/s00018-007-7398-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zientek M., Jiang Y., Youdim K., Obach R.S. In vitro–in vivo correlation for intrinsic clearance for drugs metabolized by human aldehyde oxidase. Drug Metab Dispos. 2010;38:1322–1327. doi: 10.1124/dmd.110.033555. [DOI] [PubMed] [Google Scholar]
- 27.Akabane T., Gerst N., Masters J.N., Tamura K. A quantitative approach to hepatic clearance prediction of metabolism by aldehyde oxidase using custom pooled hepatocytes. Xenobiotica. 2012;42:863–871. doi: 10.3109/00498254.2012.670736. [DOI] [PubMed] [Google Scholar]
- 28.Uehara S., Yoneda N., Higuchi Y., Yamazaki H., Suemizu H. Human aldehyde oxidase 1-mediated carbazeran oxidation in chimeric TK-NOG mice transplanted with human hepatocytes. Drug Metab Dispos. 2020;48:580–586. doi: 10.1124/dmd.120.091090. [DOI] [PubMed] [Google Scholar]
- 29.Di L. The impact of carboxylesterases in drug metabolism and pharmacokinetics. Curr Drug Metabol. 2019;20:91–102. doi: 10.2174/1389200219666180821094502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hermant P., Bosc D., Piveteau C., Gealageas R., Lam B., Ronco C., et al. Controlling plasma stability of hydroxamic acids: a medchem toolbox. J Med Chem. 2017;60:9067–9089. doi: 10.1021/acs.jmedchem.7b01444. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed F., Vyas V., Cornfield A., Goodin S., Ravikumar T.S., Rubin E.H., et al. In vitro activation of irinotecan to SN-38 by human liver and intestine. Anticancer Res. 1999;19:2067–2071. [PubMed] [Google Scholar]
- 32.Shimizu M., Fukami T., Nakajima M., Yokoi T. Screening of specific inhibitors for human carboxylesterases or arylacetamide deacetylase. Drug Metab Dispos. 2014;42:1103–1109. doi: 10.1124/dmd.114.056994. [DOI] [PubMed] [Google Scholar]
- 33.Umehara K., Zollinger M., Kigondu E., Witschi M., Juif C., Huth F., et al. Esterase phenotyping in human liver in vitro: specificity of carboxylesterase inhibitors. Xenobiotica. 2016;46:862–867. doi: 10.3109/00498254.2015.1133867. [DOI] [PubMed] [Google Scholar]
- 34.Trapa P.E., Beaumont K., Atkinson K., Eng H., King-Ahmad A., Scott D.O., et al. In vitro–in vivo extrapolation of intestinal availability for carboxylesterase substrates using portal vein-cannulated monkey. J Pharmaceut Sci. 2017;106:898–905. doi: 10.1016/j.xphs.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Rautio J., Meanwell N.A., Di L., Hageman M.J. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 2018;17:559–587. doi: 10.1038/nrd.2018.46. [DOI] [PubMed] [Google Scholar]
- 36.Weber B.W., Kimani S.W., Varsani A., Cowan D.A., Hunter R., Venter G.A., et al. The mechanism of the amidases: mutating the glutamate adjacent to the catalytic triad inactivates the enzyme due to substrate mispositioning. J Biol Chem. 2013;288:28514–28523. doi: 10.1074/jbc.M113.503284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rawal J., Jones R., Payne A., Gardner I. Strategies to prevent N-acetyltransferase-mediated metabolism in a series of piperazine-containing pyrazalopyrimidine compounds. Xenobiotica. 2008;38:1219–1239. doi: 10.1080/00498250802334417. [DOI] [PubMed] [Google Scholar]
- 38.Sun Q., Harper T.W., Dierks E.A., Zhang L., Chang S., Rodrigues A.D., et al. 1-Aminobenzotriazole, a known cytochrome P450 inhibitor, is a substrate and inhibitor of N-acetyltransferase. Drug Metab Dispos. 2011;39:1674–1679. doi: 10.1124/dmd.111.039834. [DOI] [PubMed] [Google Scholar]
- 39.Cordes H., Thiel C., Aschmann H.E., Baier V., Blank L.M., Kuepfer L. A physiologically based pharmacokinetic model of isoniazid and its application in individualizing tuberculosis chemotherapy. Antimicrob Agents Chemother. 2016;60:6134–6145. doi: 10.1128/AAC.00508-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalgutkar A.S., Castagnoli N., Jr. Selective inhibitors of monoamine oxidase (MAO-A and MAO-B) as probes of its catalytic site and mechanism. Med Res Rev. 1995;15:325–388. doi: 10.1002/med.2610150406. [DOI] [PubMed] [Google Scholar]
- 41.Masuo Y., Nagamori S., Hasegawa A., Hayashi K., Isozumi N., Nakamichi N., et al. Utilization of liver microsomes to estimate hepatic intrinsic clearance of monoamine oxidase substrate drugs in humans. Pharm Res. 2017;34:1233–1243. doi: 10.1007/s11095-017-2140-4. [DOI] [PubMed] [Google Scholar]
- 42.Harada R., Hayakawa Y., Ezura M., Lerdsirisuk P., Du Y., Ishikawa Y., et al. 18F-SMBT-1: a selective and reversible PET tracer for monoamine oxidase-B imaging. J Nucl Med. 2021;62:253–258. doi: 10.2967/jnumed.120.244400. [DOI] [PubMed] [Google Scholar]
- 43.Cashman J.R., Zhang J. Human flavin-containing monooxygenases. Annu Rev Pharmacol Toxicol. 2006;46:65–100. doi: 10.1146/annurev.pharmtox.46.120604.141043. [DOI] [PubMed] [Google Scholar]
- 44.Jones B.C., Srivastava A., Colclough N., Wilson J., Reddy V.P., Amberntsson S., et al. An investigation into the prediction of in vivo clearance for a range of flavin-containing monooxygenase substrates. Drug Metab Dispos. 2017;45:1060–1067. doi: 10.1124/dmd.117.077396. [DOI] [PubMed] [Google Scholar]
- 45.Leung L., Yang X., Strelevitz T.J., Montgomery J., Brown M.F., Zientek M.A., et al. Clearance prediction of targeted covalent inhibitors by in vitro–in vivo extrapolation of hepatic and extrahepatic clearance mechanisms. Drug Metab Dispos. 2017;45:1–7. doi: 10.1124/dmd.116.072983. [DOI] [PubMed] [Google Scholar]
- 46.Rudewicz P.J., Yue Q., Shin Y. CRC Press; Boca Raton: 2009. High-throughput strategies for metabolite identification in drug discovery. [Google Scholar]
- 47.Di L., Kerns E.H. 2 edition. Elsevier/AP; Boston: 2016. Drug-like properties: concepts, structure design and methods: from ADME to toxicity optimization. [Google Scholar]
- 48.Di L., Obach R.S. Addressing the challenges of low clearance in drug research. AAPS J. 2015;17:352–357. doi: 10.1208/s12248-014-9691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di L., Trapa P., Obach R.S., Atkinson K., Bi Y.A., Wolford A.C., et al. A novel relay method for determining low-clearance values. Drug Metab Dispos. 2012;40:1860–1865. doi: 10.1124/dmd.112.046425. [DOI] [PubMed] [Google Scholar]
- 50.Di L., Atkinson K., Orozco C.C., Funk C., Zhang H., McDonald T.S., et al. In vitro–in vivo correlation for low-clearance compounds using hepatocyte relay method. Drug Metab Dispos. 2013;41:2018–2023. doi: 10.1124/dmd.113.053322. [DOI] [PubMed] [Google Scholar]
- 51.Yang X., Atkinson K., Di L. Novel cytochrome P450 reaction phenotyping for low-clearance compounds using the hepatocyte relay method. Drug Metab Dispos. 2016;44:460–465. doi: 10.1124/dmd.115.067876. [DOI] [PubMed] [Google Scholar]
- 52.Lin C., Shi J., Moore A., Khetani S.R. Prediction of drug clearance and drug–drug interactions in microscale cultures of human hepatocytes. Drug Metab Dispos. 2016;44:127–136. doi: 10.1124/dmd.115.066027. [DOI] [PubMed] [Google Scholar]
- 53.Bonn B., Svanberg P., Janefeldt A., Hultman I., Grime K. Determination of human hepatocyte intrinsic clearance for slowly metabolized compounds: comparison of a primary hepatocyte/stromal cell co-culture with plated primary hepatocytes and HepaRG. Drug Metab Dispos. 2016;44:527–533. doi: 10.1124/dmd.115.067769. [DOI] [PubMed] [Google Scholar]
- 54.Hultman I., Vedin C., Abrahamsson A., Winiwarter S., Darnell M. Use of HmuREL human coculture system for prediction of intrinsic clearance and metabolite formation for slowly metabolized compounds. Mol Pharm. 2016;13:2796–2807. doi: 10.1021/acs.molpharmaceut.6b00396. [DOI] [PubMed] [Google Scholar]
- 55.Kratochwil N.A., Meille C., Fowler S., Klammers F., Ekiciler A., Molitor B., et al. Metabolic profiling of human long-term liver models and hepatic clearance predictions from in vitro data using nonlinear mixed-effects modeling. AAPS J. 2017;19:534–550. doi: 10.1208/s12248-016-0019-7. [DOI] [PubMed] [Google Scholar]
- 56.Novik E.I., Dwyer J., Morelli J.K., Parekh A., Cho C., Pludwinski E., et al. Long-enduring primary hepatocyte-based co-cultures improve prediction of hepatotoxicity. Toxicol Appl Pharmacol. 2017;336:20–30. doi: 10.1016/j.taap.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 57.Keefer C., Chang G., Carlo A., Novak J.J., Banker M., Carey J., et al. Mechanistic insights on clearance and inhibition discordance between liver microsomes and hepatocytes when clearance in liver microsomes is higher than in hepatocytes. Eur J Pharmaceut Sci. 2020;155:105541. doi: 10.1016/j.ejps.2020.105541. [DOI] [PubMed] [Google Scholar]
- 58.Di L., Kerns E.H., Gao N., Li S.Q., Huang Y., Bourassa J.L., et al. Experimental design on single-time-point high-throughput microsomal stability assay. J Pharmaceut Sci. 2004;93:1537–1544. doi: 10.1002/jps.20076. [DOI] [PubMed] [Google Scholar]
- 59.Khetani S.R., Bhatia S.N. Microscale culture of human liver cells for drug development. Nat Biotechnol. 2008;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 60.Chan T.S., Yu H., Moore A., Khetani S.R., Tweedie D. Meeting the challenge of predicting hepatic clearance of compounds slowly metabolized by cytochrome P450 using a novel hepatocyte model, HepatoPac. Drug Metab Dispos. 2013;41:2024–2032. doi: 10.1124/dmd.113.053397. [DOI] [PubMed] [Google Scholar]
- 61.Wang W.W., Khetani S.R., Krzyzewski S., Duignan D.B., Obach R.S. Assessment of a micropatterned hepatocyte coculture system to generate major human excretory and circulating drug metabolites. Drug Metab Dispos. 2010;38:1900–1905. doi: 10.1124/dmd.110.034876. [DOI] [PubMed] [Google Scholar]
- 62.Brown H.S., Griffin M., Houston J.B. Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos. 2007;35:293–301. doi: 10.1124/dmd.106.011569. [DOI] [PubMed] [Google Scholar]
- 63.Muller J., Keiser M., Drozdzik M., Oswald S. Expression, regulation and function of intestinal drug transporters: an update. Biol Chem. 2017;398:175–192. doi: 10.1515/hsz-2016-0259. [DOI] [PubMed] [Google Scholar]
- 64.Colas C., Ung P.M., Schlessinger A. SLC transporters: structure, function, and drug discovery. Medchemcomm. 2016;7:1069–1081. doi: 10.1039/C6MD00005C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benadiba M., Maor Y. Importance of ABC transporters in drug development. Curr Pharmaceut Des. 2016;22:5817–5829. doi: 10.2174/1381612822666160810120359. [DOI] [PubMed] [Google Scholar]
- 66.International Transporter Consortium. Giacomini K.M., Huang S.M., Tweedie D.J., Benet L.Z., Brouwer K.L., et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jani M., Ambrus C., Magnan R., Jakab K.T., Beery E., Zolnerciks J.K., et al. Structure and function of BCRP, a broad specificity transporter of xenobiotics and endobiotics. Arch Toxicol. 2014;88:1205–1248. doi: 10.1007/s00204-014-1224-8. [DOI] [PubMed] [Google Scholar]
- 68.Lund M., Petersen T.S., Dalhoff K.P. Clinical implications of P-glycoprotein modulation in drug–drug interactions. Drugs. 2017;77:859–883. doi: 10.1007/s40265-017-0729-x. [DOI] [PubMed] [Google Scholar]
- 69.Konig J., Muller F., Fromm M.F. Transporters and drug–drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65:944–966. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 70.Colas C., Schlessinger A., Pajor A.M. Mapping functionally important residues in the Na+/dicarboxylate cotransporter, NaDC1. Biochemistry. 2017;56:4432–4441. doi: 10.1021/acs.biochem.7b00503. [DOI] [PubMed] [Google Scholar]
- 71.Estudante M., Morais J.G., Soveral G., Benet L.Z. Intestinal drug transporters: an overview. Adv Drug Deliv Rev. 2013;65:1340–1356. doi: 10.1016/j.addr.2012.09.042. [DOI] [PubMed] [Google Scholar]
- 72.Katsura T., Inui K. Intestinal absorption of drugs mediated by drug transporters: mechanisms and regulation. Drug Metabol Pharmacokinet. 2003;18:1–15. doi: 10.2133/dmpk.18.1. [DOI] [PubMed] [Google Scholar]
- 73.Kruijtzer C.M., Beijnen J.H., Rosing H., ten Bokkel Huinink W.W., Schot M., Jewell R.C., et al. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 74.Greiner B., Eichelbaum M., Fritz P., Kreichgauer H.P., von Richter O., Zundler J., et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zamek-Gliszczynski M.J., Patel M., Yang X., Lutz J.D., Chu X., Brouwer K.L.R., et al. Intestinal P-gp and putative hepatic OATP1B induction: international transporter consortium perspective on drug development implications. Clin Pharmacol Ther. 2021;109:55–64. doi: 10.1002/cpt.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mao Q., Lai Y., Wang J. Drug transporters in xenobiotic disposition and pharmacokinetic prediction. Drug Metab Dispos. 2018;46:561–566. doi: 10.1124/dmd.118.081356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Varma M.V., El-Kattan A.F. Transporter-enzyme interplay: deconvoluting effects of hepatic transporters and enzymes on drug disposition using static and dynamic mechanistic models. J Clin Pharmacol. 2016;56 Suppl 7:S99–S109. doi: 10.1002/jcph.695. [DOI] [PubMed] [Google Scholar]
- 78.Yee S.W., Giacomini M.M., Shen H., Humphreys W.G., Horng H., Brian W., et al. Organic anion transporter polypeptide 1B1 polymorphism modulates the extent of drug–drug interaction and associated biomarker levels in healthy volunteers. Clin Transl Sci. 2019;12:388–399. doi: 10.1111/cts.12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mori D., Kashihara Y., Yoshikado T., Kimura M., Hirota T., Matsuki S., et al. Effect of OATP1B1 genotypes on plasma concentrations of endogenous OATP1B1 substrates and drugs, and their association in healthy volunteers. Drug Metabol Pharmacokinet. 2019;34:78–86. doi: 10.1016/j.dmpk.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 80.Chu X., Chan G.H., Evers R. Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter-mediated drug–drug interactions. J Pharmaceut Sci. 2017;106:2357–2367. doi: 10.1016/j.xphs.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 81.Chu X., Liao M., Shen H., Yoshida K., Zur A.A., Arya V., et al. Clinical probes and endogenous biomarkers as substrates for transporter drug–drug interaction evaluation: perspectives from the international transporter consortium. Clin Pharmacol Ther. 2018;104:836–864. doi: 10.1002/cpt.1216. [DOI] [PubMed] [Google Scholar]
- 82.Paulusma C.C., van Geer M.A., Evers R., Heijn M., Ottenhoff R., Borst P., et al. Canalicular multispecific organic anion transporter/multidrug resistance protein 2 mediates low-affinity transport of reduced glutathione. Biochem J. 1999;338 Pt 2:393–401. [PMC free article] [PubMed] [Google Scholar]
- 83.Keppler D., Arias I.M. Hepatic canalicular membrane. Introduction: transport across the hepatocyte canalicular membrane. FASEB J. 1997;11:15–18. doi: 10.1096/fasebj.11.1.9034161. [DOI] [PubMed] [Google Scholar]
- 84.Ito K., Suzuki H., Hirohashi T., Kume K., Shimizu T., Sugiyama Y. Functional analysis of a canalicular multispecific organic anion transporter cloned from rat liver. J Biol Chem. 1998;273:1684–1688. doi: 10.1074/jbc.273.3.1684. [DOI] [PubMed] [Google Scholar]
- 85.Konig J., Nies A.T., Cui Y., Leier I., Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta. 1999;1461:377–394. doi: 10.1016/s0005-2736(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 86.Kusuhara H., Sugiyama Y. Role of transporters in the tissue-selective distribution and elimination of drugs: transporters in the liver, small intestine, brain and kidney. J Control Release. 2002;78:43–54. doi: 10.1016/s0168-3659(01)00480-1. [DOI] [PubMed] [Google Scholar]
- 87.Morgan R.E., Trauner M., van Staden C.J., Lee P.H., Ramachandran B., Eschenberg M., et al. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci. 2010;118:485–500. doi: 10.1093/toxsci/kfq269. [DOI] [PubMed] [Google Scholar]
- 88.Hallifax D., Rawden H.C., Hakooz N., Houston J.B. Prediction of metabolic clearance using cryopreserved human hepatocytes: kinetic characteristics for five benzodiazepines. Drug Metab Dispos. 2005;33:1852–1858. doi: 10.1124/dmd.105.005389. [DOI] [PubMed] [Google Scholar]
- 89.Davies M., Jones R.D.O., Grime K., Jansson-Lofmark R., Fretland A.J., Winiwarter S., et al. Improving the accuracy of predicted human pharmacokinetics: lessons learned from the AstraZeneca drug pipeline over two decades. Trends Pharmacol Sci. 2020;41:390–408. doi: 10.1016/j.tips.2020.03.004. [DOI] [PubMed] [Google Scholar]
- 90.Naritomi Y., Terashita S., Kimura S., Suzuki A., Kagayama A., Sugiyama Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab Dispos. 2001;29:1316–1324. [PubMed] [Google Scholar]
- 91.Patilea-Vrana G., Unadkat J.D. Transport vs. metabolism: what determines the pharmacokinetics and pharmacodynamics of drugs? Insights from the extended clearance model. Clin Pharmacol Ther. 2016;100:413–418. doi: 10.1002/cpt.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.El-Kattan A.F., Varma M.V.S. Navigating transporter sciences in pharmacokinetics characterization using the extended clearance classification system. Drug Metab Dispos. 2018;46:729–739. doi: 10.1124/dmd.117.080044. [DOI] [PubMed] [Google Scholar]
- 93.Watanabe T., Kusuhara H., Maeda K., Shitara Y., Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Therapeut. 2009;328:652–662. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- 94.Liang X., Lai Y. Overcoming the shortcomings of the extended-clearance concept: a framework for developing a physiologically-based pharmacokinetic (PBPK) model to select drug candidates involving transporter-mediated clearance. Expet Opin Drug Metabol Toxicol. 2021;17:869–886. doi: 10.1080/17425255.2021.1912012. [DOI] [PubMed] [Google Scholar]
- 95.Lee W., Kim R.B. Transporters and renal drug elimination. Annu Rev Pharmacol Toxicol. 2004;44:137–166. doi: 10.1146/annurev.pharmtox.44.101802.121856. [DOI] [PubMed] [Google Scholar]
- 96.Wright S.H., Dantzler W.H. Molecular and cellular physiology of renal organic cation and anion transport. Physiol Rev. 2004;84:987–1049. doi: 10.1152/physrev.00040.2003. [DOI] [PubMed] [Google Scholar]
- 97.Liu H.C., Goldenberg A., Chen Y., Lun C., Wu W., Bush K.T., et al. Molecular properties of drugs interacting with SLC22 transporters OAT1, OAT3, OCT1, and OCT2: a machine-learning approach. J Pharmacol Exp Therapeut. 2016;359:215–229. doi: 10.1124/jpet.116.232660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zamek-Gliszczynski M.J., Giacomini K.M., Zhang L. Emerging clinical importance of hepatic organic cation transporter 1 (OCT1) in drug pharmacokinetics, dynamics, pharmacogenetic variability, and drug interactions. Clin Pharmacol Ther. 2018;103:758–760. doi: 10.1002/cpt.941. [DOI] [PubMed] [Google Scholar]
- 99.Roth M., Obaidat A., Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165:1260–1287. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nies A.T., Koepsell H., Damme K., Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011:105–167. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 101.Chen L., Shu Y., Liang X., Chen E.C., Yee S.W., Zur A.A., et al. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc Natl Acad Sci U S A. 2014;111:9983–9988. doi: 10.1073/pnas.1314939111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yin J., Wang J. Renal drug transporters and their significance in drug–drug interactions. Acta Pharm Sin B. 2016;6:363–373. doi: 10.1016/j.apsb.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang S., Lovejoy K.S., Shima J.E., Lagpacan L.L., Shu Y., Lapuk A., et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yokoo S., Yonezawa A., Masuda S., Fukatsu A., Katsura T., Inui K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol. 2007;74:477–487. doi: 10.1016/j.bcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 105.Giacomini K.M., Huang S.M., Tweedie D.J., Benet L.Z., Brouwer K.L., Chu X., et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee W.K., Wolff N.A., Thevenod F. Organic cation transporters: physiology, toxicology and special focus on ethidium as a novel substrate. Curr Drug Metabol. 2009;10:617–631. doi: 10.2174/138920009789375360. [DOI] [PubMed] [Google Scholar]
- 107.Hagos Y., Wolff N.A. Assessment of the role of renal organic anion transporters in drug-induced nephrotoxicity. Toxins (Basel) 2010;2:2055–2082. doi: 10.3390/toxins2082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ciarimboli G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014;34:547–550. [PubMed] [Google Scholar]
- 109.Cundy K.C., Petty B.G., Flaherty J., Fisher P.E., Polis M.A., Wachsman M., et al. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 1995;39:1247–1252. doi: 10.1128/aac.39.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yonezawa A., Masuda S., Yokoo S., Katsura T., Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family) J Pharmacol Exp Therapeut. 2006;319:879–886. doi: 10.1124/jpet.106.110346. [DOI] [PubMed] [Google Scholar]
- 111.Filipski K.K., Loos W.J., Verweij J., Sparreboom A. Interaction of cisplatin with the human organic cation transporter 2. Clin Cancer Res. 2008;14:3875–3880. doi: 10.1158/1078-0432.CCR-07-4793. [DOI] [PubMed] [Google Scholar]
- 112.Li Q., Guo D., Dong Z., Zhang W., Zhang L., Huang S.M., et al. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs) Toxicol Appl Pharmacol. 2013;273:100–109. doi: 10.1016/j.taap.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen H., Yang Z., Zhao W., Zhang Y., Rodrigues A.D. Assessment of vandetanib as an inhibitor of various human renal transporters: inhibition of multidrug and toxin extrusion as a possible mechanism leading to decreased cisplatin and creatinine clearance. Drug Metab Dispos. 2013;41:2095–2103. doi: 10.1124/dmd.113.053215. [DOI] [PubMed] [Google Scholar]
- 114.Steinway S.N., Saleh J., Koo B.K., Delacour D., Kim D.H. Human microphysiological models of intestinal tissue and gut microbiome. Front Bioeng Biotechnol. 2020;8:725. doi: 10.3389/fbioe.2020.00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Calitz C., Hamman J.H., Fey S.J., Wrzesinski K., Gouws C. Recent advances in three-dimensional cell culturing to assess liver function and dysfunction: from a drug biotransformation and toxicity perspective. Toxicol Mech Methods. 2018;28:369–385. doi: 10.1080/15376516.2017.1422580. [DOI] [PubMed] [Google Scholar]
- 116.Fang Y., Eglen R.M. Three-dimensional cell cultures in drug discovery and development. SLAS Discov. 2017;22:456–472. doi: 10.1177/1087057117696795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kulthong K., Hooiveld G., Duivenvoorde L., Miro Estruch I., Marin V., van der Zande M., et al. Transcriptome comparisons of in vitro intestinal epithelia grown under static and microfluidic gut-on-chip conditions with in vivo human epithelia. Sci Rep. 2021;11:3234. doi: 10.1038/s41598-021-82853-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Santbergen M.J.C., van der Zande M., Gerssen A., Bouwmeester H., Nielen M.W.F. Dynamic in vitro intestinal barrier model coupled to chip-based liquid chromatography mass spectrometry for oral bioavailability studies. Anal Bioanal Chem. 2020;412:1111–1122. doi: 10.1007/s00216-019-02336-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Guo Y., Li Z., Su W., Wang L., Zhu Y., Qin J. A biomimetic human gut-on-a-chip for modeling drug metabolism in intestine. Artif Organs. 2018;42:1196–1205. doi: 10.1111/aor.13163. [DOI] [PubMed] [Google Scholar]
- 120.Kasendra M., Tovaglieri A., Sontheimer-Phelps A., Jalili-Firoozinezhad S., Bein A., Chalkiadaki A., et al. Development of a primary human small intestine-on-a-chip using biopsy-derived organoids. Sci Rep. 2018;8:2871. doi: 10.1038/s41598-018-21201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kasendra M., Luc R., Yin J., Manatakis D.V., Kulkarni G., Lucchesi C., et al. Duodenum intestine-chip for preclinical drug assessment in a human relevant model. Elife. 2020;9:1–23. doi: 10.7554/eLife.50135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Niu C., Smith B., Lai Y. Transporter gene regulation in sandwich cultured human hepatocytes through the activation of constitutive androstane receptor (CAR) or aryl hydrocarbon receptor (AhR) Front Pharmacol. 2020;11:620197. doi: 10.3389/fphar.2020.620197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bi Y.A., Kimoto E., Sevidal S., Jones H.M., Barton H.A., Kempshall S., et al. In vitro evaluation of hepatic transporter-mediated clinical drug–drug interactions: hepatocyte model optimization and retrospective investigation. Drug Metab Dispos. 2012;40:1085–1092. doi: 10.1124/dmd.111.043489. [DOI] [PubMed] [Google Scholar]
- 124.Wang H., Brown P.C., Chow E.C.Y., Ewart L., Ferguson S.S., Fitzpatrick S., et al. 3D cell culture models: drug pharmacokinetics, safety assessment, and regulatory consideration. Clin Transl Sci. 2021;14:1659–1680. doi: 10.1111/cts.13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Baert Y., Ruetschle I., Cools W., Oehme A., Lorenz A., Marx U., et al. A multi-organ-chip co-culture of liver and testis equivalents: a first step toward a systemic male reprotoxicity model. Hum Reprod. 2020;35:1029–1044. doi: 10.1093/humrep/deaa057. [DOI] [PubMed] [Google Scholar]
- 126.Li J., Zhu H.J. Liquid chromatography–tandem mass spectrometry (LC–MS/MS)-based proteomics of drug-metabolizing enzymes and transporters. Molecules. 2020;25:2718. doi: 10.3390/molecules25112718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Couto N., Al-Majdoub Z.M., Achour B., Wright P.C., Rostami-Hodjegan A., Barber J. Quantification of proteins involved in drug metabolism and disposition in the human liver using label-free global proteomics. Mol Pharm. 2019;16:632–647. doi: 10.1021/acs.molpharmaceut.8b00941. [DOI] [PubMed] [Google Scholar]
- 128.Khatri R., Fallon J.K., Rementer R.J.B., Kulick N.T., Lee C.R., Smith P.C. Targeted quantitative proteomic analysis of drug metabolizing enzymes and transporters by nano LC–MS/MS in the sandwich cultured human hepatocyte model. J Pharmacol Toxicol Methods. 2019;98:106590. doi: 10.1016/j.vascn.2019.106590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nakamura K., Hirayama-Kurogi M., Ito S., Kuno T., Yoneyama T., Obuchi W., et al. Large-scale multiplex absolute protein quantification of drug-metabolizing enzymes and transporters in human intestine, liver, and kidney microsomes by SWATH-MS: comparison with MRM/SRM and HR-MRM/PRM. Proteomics. 2016;16:2106–2117. doi: 10.1002/pmic.201500433. [DOI] [PubMed] [Google Scholar]
- 130.Ladumor M.K., Thakur A., Sharma S., Rachapally A., Mishra S., Bobe P., et al. A repository of protein abundance data of drug metabolizing enzymes and transporters for applications in physiologically based pharmacokinetic (PBPK) modelling and simulation. Sci Rep. 2019;9:9709. doi: 10.1038/s41598-019-45778-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bhatt D.K., Prasad B. Critical issues and optimized practices in quantification of protein abundance level to determine interindividual variability in DMET proteins by LC–MS/MS proteomics. Clin Pharmacol Ther. 2018;103:619–630. doi: 10.1002/cpt.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Couto N., Al-Majdoub Z.M., Gibson S., Davies P.J., Achour B., Harwood M.D., et al. Quantitative proteomics of clinically relevant drug-metabolizing enzymes and drug transporters and their intercorrelations in the human small intestine. Drug Metab Dispos. 2020;48:245–268. doi: 10.1124/dmd.119.089656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ohtsuki S., Uchida Y., Kubo Y., Terasaki T. Quantitative targeted absolute proteomics-based ADME research as a new path to drug discovery and development: methodology, advantages, strategy, and prospects. J Pharmaceut Sci. 2011;100:3547–3559. doi: 10.1002/jps.22612. [DOI] [PubMed] [Google Scholar]
- 134.Prasad B., Unadkat J.D. Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J. 2014;16:634–648. doi: 10.1208/s12248-014-9602-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xu M., Saxena N., Vrana M., Zhang H., Kumar V., Billington S., et al. Targeted LC–MS/MS proteomics-based strategy to characterize in vitro models used in drug metabolism and transport studies. Anal Chem. 2018;90:11873–11882. doi: 10.1021/acs.analchem.8b01913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Uchida Y., Zhang Z., Tachikawa M., Terasaki T. Quantitative targeted absolute proteomics of rat blood–cerebrospinal fluid barrier transporters: comparison with a human specimen. J Neurochem. 2015;134:1104–1115. doi: 10.1111/jnc.13147. [DOI] [PubMed] [Google Scholar]
- 137.Prasad B., Evers R., Gupta A., Hop C.E., Salphati L., Shukla S., et al. Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab Dispos. 2014;42:78–88. doi: 10.1124/dmd.113.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Al-Majdoub Z.M., Al Feteisi H., Achour B., Warwood S., Neuhoff S., Rostami-Hodjegan A., et al. Proteomic quantification of human blood–brain barrier SLC and ABC transporters in healthy individuals and dementia patients. Mol Pharm. 2019;16:1220–1233. doi: 10.1021/acs.molpharmaceut.8b01189. [DOI] [PubMed] [Google Scholar]
- 139.Billington S., Salphati L., Hop C., Chu X., Evers R., Burdette D., et al. Interindividual and regional variability in drug transporter abundance at the human blood–brain barrier measured by quantitative targeted proteomics. Clin Pharmacol Ther. 2019;106:228–237. doi: 10.1002/cpt.1373. [DOI] [PubMed] [Google Scholar]
- 140.Al-Majdoub Z.M., Achour B., Couto N., Howard M., Elmorsi Y., Scotcher D., et al. Mass spectrometry-based abundance atlas of ABC transporters in human liver, gut, kidney, brain and skin. FEBS Lett. 2020;594:4134–4150. doi: 10.1002/1873-3468.13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Uchida Y., Ohtsuki S., Katsukura Y., Ikeda C., Suzuki T., Kamiie J., et al. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J Neurochem. 2011;117:333–345. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 142.Prasad B., Johnson K., Billington S., Lee C., Chung G.W., Brown C.D., et al. Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab Dispos. 2016;44:1920–1924. doi: 10.1124/dmd.116.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang L., Prasad B., Salphati L., Chu X., Gupta A., Hop C.E., et al. Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab Dispos. 2015;43:367–374. doi: 10.1124/dmd.114.061580. [DOI] [PubMed] [Google Scholar]
- 144.Li C.Y., Hosey-Cojocari C., Basit A., Unadkat J.D., Leeder J.S., Prasad B. Optimized renal transporter quantification by using aquaporin 1 and aquaporin 2 as anatomical markers: application in characterizing the ontogeny of renal transporters and its correlation with hepatic transporters in paired human samples. AAPS J. 2019;21:88. doi: 10.1208/s12248-019-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bhatt D.K., Mehrotra A., Gaedigk A., Chapa R., Basit A., Zhang H., et al. Age- and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin Pharmacol Ther. 2019;105:131–141. doi: 10.1002/cpt.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Billington S., Ray A.S., Salphati L., Xiao G., Chu X., Humphreys W.G., et al. Transporter expression in noncancerous and cancerous liver tissue from donors with hepatocellular carcinoma and chronic hepatitis C infection quantified by LC–MS/MS proteomics. Drug Metab Dispos. 2018;46:189–196. doi: 10.1124/dmd.117.077289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Prasad B., Gaedigk A., Vrana M., Gaedigk R., Leeder J.S., Salphati L., et al. Ontogeny of hepatic drug transporters as quantified by LC–MS/MS proteomics. Clin Pharmacol Ther. 2016;100:362–370. doi: 10.1002/cpt.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.El-Khateeb E., Vasilogianni A.M., Alrubia S., Al-Majdoub Z.M., Couto N., Howard M., et al. Quantitative mass spectrometry-based proteomics in the era of model-informed drug development: applications in translational pharmacology and recommendations for best practice. Pharmacol Ther. 2019;203:107397. doi: 10.1016/j.pharmthera.2019.107397. [DOI] [PubMed] [Google Scholar]
- 149.Al Feteisi H., Achour B., Rostami-Hodjegan A., Barber J. Translational value of liquid chromatography coupled with tandem mass spectrometry-based quantitative proteomics for in vitro–in vivo extrapolation of drug metabolism and transport and considerations in selecting appropriate techniques. Expet Opin Drug Metabol Toxicol. 2015;11:1357–1369. doi: 10.1517/17425255.2015.1055245. [DOI] [PubMed] [Google Scholar]
- 150.Kumar A.R., Prasad B., Bhatt D.K., Mathialagan S., Varma M.V.S., Unadkat J.D. In vivo-to-in vitro extrapolation of transporter-mediated renal clearance: relative expression factor versus relative activity factor approach. Drug Metab Dispos. 2020;49:470–478. doi: 10.1124/dmd.121.000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kumar V., Yin M., Ishida K., Salphati L., Hop C., Rowbottom C., et al. Prediction of transporter-mediated rosuvastatin hepatic uptake clearance and drug interaction in humans using proteomics-informed REF approach. Drug Metab Dispos. 2021;49:159–168. doi: 10.1124/dmd.120.000204. [DOI] [PubMed] [Google Scholar]
- 152.Sachar M., Kumar V., Gormsen L.C., Munk O.L., Unadkat J.D. Successful prediction of positron emission tomography-imaged metformin hepatic uptake clearance in humans using the quantitative proteomics-informed relative expression factor approach. Drug Metab Dispos. 2020;48:1210–1216. doi: 10.1124/dmd.120.000156. [DOI] [PubMed] [Google Scholar]
- 153.Wegler C., Prieto Garcia L., Klinting S., Robertsen I., Wisniewski J.R., Hjelmesaeth J., et al. Proteomics-informed prediction of rosuvastatin plasma profiles in patients with a wide range of body weight. Clin Pharmacol Ther. 2021;109:762–771. doi: 10.1002/cpt.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ishida K., Ullah M., Toth B., Juhasz V., Unadkat J.D. Successful prediction of in vivo hepatobiliary clearances and hepatic concentrations of rosuvastatin using sandwich-cultured rat hepatocytes, transporter-expressing cell lines, and quantitative proteomics. Drug Metab Dispos. 2018;46:66–74. doi: 10.1124/dmd.117.076539. [DOI] [PubMed] [Google Scholar]
- 155.Vildhede A., Karlgren M., Svedberg E.K., Wisniewski J.R., Lai Y., Noren A., et al. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug–drug interactions. Drug Metab Dispos. 2014;42:1210–1218. doi: 10.1124/dmd.113.056309. [DOI] [PubMed] [Google Scholar]
- 156.Wegler C., Gaugaz F.Z., Andersson T.B., Wisniewski J.R., Busch D., Groer C., et al. Variability in mass spectrometry-based quantification of clinically relevant drug transporters and drug metabolizing enzymes. Mol Pharm. 2017;14:3142–3151. doi: 10.1021/acs.molpharmaceut.7b00364. [DOI] [PubMed] [Google Scholar]
- 157.Prasad B., Achour B., Artursson P., Hop C., Lai Y., Smith P.C., et al. Toward a consensus on applying quantitative liquid chromatography–tandem mass spectrometry proteomics in translational pharmacology research: a white paper. Clin Pharmacol Ther. 2019;106:525–543. doi: 10.1002/cpt.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sampson K.E., Brinker A., Pratt J., Venkatraman N., Xiao Y., Blasberg J., et al. Zinc finger nuclease-mediated gene knockout results in loss of transport activity for P-glycoprotein, BCRP, and MRP2 in Caco-2 cells. Drug Metab Dispos. 2015;43:199–207. doi: 10.1124/dmd.114.057216. [DOI] [PubMed] [Google Scholar]
- 159.Gartzke D., Delzer J., Laplanche L., Uchida Y., Hoshi Y., Tachikawa M., et al. Genomic knockout of endogenous canine P-glycoprotein in wild-type, human P-glycoprotein and human BCRP transfected MDCKII cell lines by zinc finger nucleases. Pharm Res. 2015;32:2060–2071. doi: 10.1007/s11095-014-1599-5. [DOI] [PubMed] [Google Scholar]
- 160.Cheng Y., Chen S., Freeden C., Chen W., Zhang Y., Abraham P., et al. Bile salt homeostasis in normal and Bsep gene knockout rats with single and repeated doses of troglitazone. J Pharmacol Exp Therapeut. 2017;362:385–394. doi: 10.1124/jpet.117.242370. [DOI] [PubMed] [Google Scholar]
- 161.Lu J., Liu J., Guo Y., Zhang Y., Xu Y., Wang X. CRISPR-Cas9: a method for establishing rat models of drug metabolism and pharmacokinetics. Acta Pharm Sin B. 2021;11:2973–2982. doi: 10.1016/j.apsb.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Karlgren M., Simoff I., Keiser M., Oswald S., Artursson P. CRISPR-Cas9: a new addition to the drug metabolism and disposition tool box. Drug Metab Dispos. 2018;46:1776–1786. doi: 10.1124/dmd.118.082842. [DOI] [PubMed] [Google Scholar]
- 163.Yoshimi K., Kunihiro Y., Kaneko T., Nagahora H., Voigt B., Mashimo T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun. 2016;7:10431. doi: 10.1038/ncomms10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ma X., Shang X., Qin X., Lu J., Liu M., Wang X. Characterization of organic anion transporting polypeptide 1b2 knockout rats generated by CRISPR/Cas9: a novel model for drug transport and hyperbilirubinemia disease. Acta Pharm Sin B. 2020;10:850–860. doi: 10.1016/j.apsb.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen E.C., Broccatelli F., Plise E., Chen B., Liu L., Cheong J., et al. Evaluating the utility of canine Mdr1 knockout Madin-Darby canine kidney I cells in permeability screening and efflux substrate determination. Mol Pharm. 2018;15:5103–5113. doi: 10.1021/acs.molpharmaceut.8b00688. [DOI] [PubMed] [Google Scholar]
- 166.Vaidyanathan J., Yoshida K., Arya V., Zhang L. Comparing various in vitro prediction criteria to assess the potential of a new molecular entity to inhibit organic anion transporting polypeptide 1B1. J Clin Pharmacol. 2016;56 Suppl 7:S59–S72. doi: 10.1002/jcph.723. [DOI] [PubMed] [Google Scholar]
- 167.Mori D., Ishida H., Mizuno T., Kusumoto S., Kondo Y., Izumi S., et al. Alteration in the plasma concentrations of endogenous organic anion-transporting polypeptide 1B biomarkers in patients with non-small cell lung cancer treated with paclitaxel. Drug Metab Dispos. 2020;48:387–394. doi: 10.1124/dmd.119.089474. [DOI] [PubMed] [Google Scholar]
- 168.Tatosian D.A., Yee K.L., Zhang Z., Mostoller K., Paul E., Sutradhar S., et al. A microdose cocktail to evaluate drug interactions in patients with renal impairment. Clin Pharmacol Ther. 2021;109:403–415. doi: 10.1002/cpt.1998. [DOI] [PubMed] [Google Scholar]
- 169.Mori D., Kimoto E., Rago B., Kondo Y., King-Ahmad A., Ramanathan R., et al. Dose-dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin Pharmacol Ther. 2020;107:1004–1013. doi: 10.1002/cpt.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cheung K.W.K., Yoshida K., Cheeti S., Chen B., Morley R., Chan I.T., et al. GDC-0810 pharmacokinetics and transporter-mediated drug interaction evaluation with an endogenous biomarker in the first-in-human, dose escalation study. Drug Metab Dispos. 2019;47:966–973. doi: 10.1124/dmd.119.087924. [DOI] [PubMed] [Google Scholar]
- 171.Yoshida K., Guo C., Sane R. Quantitative prediction of OATP-mediated drug–drug interactions with model-based analysis of endogenous biomarker kinetics. CPT Pharmacometrics Syst Pharmacol. 2018;7:517–524. doi: 10.1002/psp4.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Muller F., Sharma A., Konig J., Fromm M.F. Biomarkers for in vivo assessment of transporter function. Pharmacol Rev. 2018;70:246–277. doi: 10.1124/pr.116.013326. [DOI] [PubMed] [Google Scholar]
- 173.Rodrigues A.D., Taskar K.S., Kusuhara H., Sugiyama Y. Endogenous probes for drug transporters: balancing vision with reality. Clin Pharmacol Ther. 2018;103:434–448. doi: 10.1002/cpt.749. [DOI] [PubMed] [Google Scholar]
- 174.Mochizuki T., Mizuno T., Maeda K., Kusuhara H. Current progress in identifying endogenous biomarker candidates for drug transporter phenotyping and their potential application to drug development. Drug Metabol Pharmacokinet. 2021;37:100358. doi: 10.1016/j.dmpk.2020.09.003. [DOI] [PubMed] [Google Scholar]
- 175.Lai Y., Mandlekar S., Shen H., Holenarsipur V.K., Langish R., Rajanna P., et al. Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug–drug interactions mediated by organic anion transporting polypeptide inhibition. J Pharmacol Exp Therapeut. 2016;358:397–404. doi: 10.1124/jpet.116.234914. [DOI] [PubMed] [Google Scholar]
- 176.Shen H., Christopher L., Lai Y., Gong J., Kandoussi H., Garonzik S., et al. Further studies to support the use of coproporphyrin I and III as novel clinical biomarkers for evaluating the potential for organic anion transporting polypeptide 1B1 and OATP1B3 inhibition. Drug Metab Dispos. 2018;46:1075–1082. doi: 10.1124/dmd.118.081125. [DOI] [PubMed] [Google Scholar]
- 177.Barnett S., Ogungbenro K., Menochet K., Shen H., Lai Y., Humphreys W.G., et al. Gaining mechanistic insight into coproporphyrin I as endogenous biomarker for OATP1B-mediated drug–drug interactions using population pharmacokinetic modeling and simulation. Clin Pharmacol Ther. 2018;104:564–574. doi: 10.1002/cpt.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Neuvonen M., Hirvensalo P., Tornio A., Rago B., West M., Lazzaro S., et al. Identification of glycochenodeoxycholate 3-O-glucuronide and glycodeoxycholate 3-O-glucuronide as highly sensitive and specific OATP1B1 biomarkers. Clin Pharmacol Ther. 2021;109:646–657. doi: 10.1002/cpt.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Suzuki Y., Sasamoto Y., Koyama T., Yoshijima C., Nakatochi M., Kubo M., et al. Substantially increased plasma coproporphyrin-I concentrations associated with OATP1B1∗15 allele in Japanese general population. Clin Transl Sci. 2021;14:382–388. doi: 10.1111/cts.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chu X., Bleasby K., Chan G.H., Nunes I., Evers R. The complexities of interpreting reversible elevated serum creatinine levels in drug development: does a correlation with inhibition of renal transporters exist?. Drug Metab Dispos. 2016;44:1498–1509. doi: 10.1124/dmd.115.067694. [DOI] [PubMed] [Google Scholar]
- 181.Tsuruya Y., Kato K., Sano Y., Imamura Y., Maeda K., Kumagai Y., et al. Investigation of endogenous compounds applicable to drug–drug interaction studies involving the renal organic anion transporters, OAT1 and OAT3, in humans. Drug Metab Dispos. 2016;44:1925–1933. doi: 10.1124/dmd.116.071472. [DOI] [PubMed] [Google Scholar]
- 182.Shen H., Nelson D.M., Oliveira R.V., Zhang Y., McNaney C.A., Gu X., et al. Discovery and validation of pyridoxic acid and homovanillic acid as novel endogenous plasma biomarkers of organic anion transporter (OAT) 1 and OAT3 in cynomolgus monkeys. Drug Metab Dispos. 2018;46:178–188. doi: 10.1124/dmd.117.077586. [DOI] [PubMed] [Google Scholar]
- 183.Shen H., Holenarsipur V.K., Mariappan T.T., Drexler D.M., Cantone J.L., Rajanna P., et al. Evidence for the validity of pyridoxic acid (PDA) as a plasma-based endogenous probe for OAT1 and OAT3 function in healthy subjects. J Pharmacol Exp Therapeut. 2019;368:136–145. doi: 10.1124/jpet.118.252643. [DOI] [PubMed] [Google Scholar]
- 184.Willemin M.E., Van Der Made T.K., Pijpers I., Dillen L., Kunze A., Jonkers S., et al. Clinical investigation on endogenous biomarkers to predict strong OAT-mediated drug–drug interactions. Clin Pharmacokinet. 2021;60:1187–1199. doi: 10.1007/s40262-021-01004-2. [DOI] [PubMed] [Google Scholar]
- 185.Ahmad A., Ogungbenro K., Kunze A., Jacobs F., Snoeys J., Rostami-Hodjegan A., et al. Population pharmacokinetic modeling and simulation to support qualification of pyridoxic acid as endogenous biomarker of OAT1/3 renal transporters. CPT Pharmacometrics Syst Pharmacol. 2021;10:467–477. doi: 10.1002/psp4.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Diczfalusy U., Miura J., Roh H.K., Mirghani R.A., Sayi J., Larsson H., et al. 4β-Hydroxycholesterol is a new endogenous CYP3A marker: relationship to CYP3A5 genotype, quinine 3-hydroxylation and sex in Koreans, Swedes and Tanzanians. Pharmacogenetics Genom. 2008;18:201–208. doi: 10.1097/FPC.0b013e3282f50ee9. [DOI] [PubMed] [Google Scholar]
- 187.Bodin K., Andersson U., Rystedt E., Ellis E., Norlin M., Pikuleva I., et al. Metabolism of 4β-hydroxycholesterol in humans. J Biol Chem. 2002;277:31534–31540. doi: 10.1074/jbc.M201712200. [DOI] [PubMed] [Google Scholar]
- 188.Kasichayanula S., Boulton D.W., Luo W.L., Rodrigues A.D., Yang Z., Goodenough A., et al. Validation of 4β-hydroxycholesterol and evaluation of other endogenous biomarkers for the assessment of CYP3A activity in healthy subjects. Br J Clin Pharmacol. 2014;78:1122–1134. doi: 10.1111/bcp.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Suzuki Y., Itoh H., Fujioka T., Sato F., Kawasaki K., Sato Y., et al. Association of plasma concentration of 4β-hydroxycholesterol with CYP3A5 polymorphism and plasma concentration of indoxyl sulfate in stable kidney transplant recipients. Drug Metab Dispos. 2014;42:105–110. doi: 10.1124/dmd.113.054171. [DOI] [PubMed] [Google Scholar]
- 190.Suzuki Y., Itoh H., Sato F., Kawasaki K., Sato Y., Fujioka T., et al. Significant increase in plasma 4β-hydroxycholesterol concentration in patients after kidney transplantation. J Lipid Res. 2013;54:2568–2572. doi: 10.1194/jlr.P040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bjorkhem-Bergman L., Backstrom T., Nylen H., Ronquist-Nii Y., Bredberg E., Andersson T.B., et al. Comparison of endogenous 4β-hydroxycholesterol with midazolam as markers for CYP3A4 induction by rifampicin. Drug Metab Dispos. 2013;41:1488–1493. doi: 10.1124/dmd.113.052316. [DOI] [PubMed] [Google Scholar]
- 192.Shin K.H., Choi M.H., Lim K.S., Yu K.S., Jang I.J., Cho J.Y. Evaluation of endogenous metabolic markers of hepatic CYP3A activity using metabolic profiling and midazolam clearance. Clin Pharmacol Ther. 2013;94:601–609. doi: 10.1038/clpt.2013.128. [DOI] [PubMed] [Google Scholar]
- 193.Mao J., Martin I., McLeod J., Nolan G., van Horn R., Vourvahis M., et al. Perspective: 4β-hydroxycholesterol as an emerging endogenous biomarker of hepatic CYP3A. Drug Metab Rev. 2017;49:18–34. doi: 10.1080/03602532.2016.1239630. [DOI] [PubMed] [Google Scholar]
- 194.Zanger U.M., Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 195.Tornio A., Backman J.T. Cytochrome P450 in pharmacogenetics: an update. Adv Pharmacol. 2018;83:3–32. doi: 10.1016/bs.apha.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 196.Stingl J.C., Bartels H., Viviani R., Lehmann M.L., Brockmoller J. Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: a quantitative systematic review. Pharmacol Ther. 2014;141:92–116. doi: 10.1016/j.pharmthera.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 197.Merali Z., Ross S., Pare G. The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect. Drug Metabol Drug Interact. 2014;29:143–151. doi: 10.1515/dmdi-2014-0009. [DOI] [PubMed] [Google Scholar]
- 198.Niemi M., Pasanen M.K., Neuvonen P.J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–181. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
- 199.Hira D., Terada T. BCRP/ABCG2 and high-alert medications: biochemical, pharmacokinetic, pharmacogenetic, and clinical implications. Biochem Pharmacol. 2018;147:201–210. doi: 10.1016/j.bcp.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 200.FDA . 2013. Clinical pharmacogenomics: premarket evaluation in early-phase clinical studies and recommendations for labeling.https://www.fda.gov/files/drugs/published/Clinical-Pharmacogenomics–Premarket-Evaluation-in-Early-Phase-Clinical-Studies-and-Recommendations-for-Labeling.pdf January. Accessed [February, 2022]. Available from: [Google Scholar]
- 201.EMA/CHMP . January 19, 2011. Guideline on the use of pharmacogenetic methodologies in the pharmacokinetic evaluation of medicinal products.https://www.ema.europa.eu/documents/scientific-guideline/guideline-use-pharmacogenetic-methodologies-pharmacokinetic-evaluation-medicinal-products_en.pdf Updated [August 1, 2012]. Accessed [February, 2022]. Available from: [Google Scholar]
- 202.Brian W., Tremaine L.M., Arefayene M., de Kanter R., Evers R., Guo Y., et al. Assessment of drug metabolism enzyme and transporter pharmacogenetics in drug discovery and early development: perspectives of the I-PWG. Pharmacogenomics. 2016;17:615–631. doi: 10.2217/pgs.16.9. [DOI] [PubMed] [Google Scholar]
- 203.Tremaine L., Brian W., DelMonte T., Francke S., Groenen P., Johnson K., et al. The role of ADME pharmacogenomics in early clinical trials: perspective of the Industry Pharmacogenomics Working Group (I-PWG) Pharmacogenomics. 2015;16:2055–2067. doi: 10.2217/pgs.15.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Giacomini K.M., Balimane P.V., Cho S.K., Eadon M., Edeki T., Hillgren K.M., et al. International transporter consortium commentary on clinically important transporter polymorphisms. Clin Pharmacol Ther. 2013;94:23–26. doi: 10.1038/clpt.2013.12. [DOI] [PubMed] [Google Scholar]
- 205.Guo Y., Lucksiri A., Dickinson G.L., Vuppalanchi R.K., Hilligoss J.K., Hall S.D. Quantitative prediction of CYP3A4- and CYP3A5-mediated drug interactions. Clin Pharmacol Ther. 2020;107:246–256. doi: 10.1002/cpt.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tsamandouras N., Dickinson G., Guo Y., Hall S., Rostami-Hodjegan A., Galetin A., et al. Identification of the effect of multiple polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid using a population-modeling approach. Clin Pharmacol Ther. 2014;96:90–100. doi: 10.1038/clpt.2014.55. [DOI] [PubMed] [Google Scholar]
- 207.Fowler S., Kletzl H., Finel M., Manevski N., Schmid P., Tuerck D., et al. A UGT2B10 splicing polymorphism common in african populations may greatly increase drug exposure. J Pharmacol Exp Therapeut. 2015;352:358–367. doi: 10.1124/jpet.114.220194. [DOI] [PubMed] [Google Scholar]
- 208.Morrissey K.M., Stocker S.L., Wittwer M.B., Xu L., Giacomini K.M. Renal transporters in drug development. Annu Rev Pharmacol Toxicol. 2013;53:503–529. doi: 10.1146/annurev-pharmtox-011112-140317. [DOI] [PubMed] [Google Scholar]
- 209.Li Z., Litchfield J., Tess D.A., Carlo A.A., Eng H., Keefer C., et al. A physiologically based in silico tool to assess the risk of drug-related crystalluria. J Med Chem. 2020;63:6489–6498. doi: 10.1021/acs.jmedchem.9b01995. [DOI] [PubMed] [Google Scholar]
- 210.Di L., Whitney-Pickett C., Umland J.P., Zhang H., Zhang X., Gebhard D.F., et al. Development of a new permeability assay using low-efflux MDCKII cells. J Pharmaceut Sci. 2011;100:4974–4985. doi: 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- 211.Zamek-Gliszczynski M.J., Taub M.E., Chothe P.P., Chu X., Giacomini K.M., Kim R.B., et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin Pharmacol Ther. 2018;104:890–899. doi: 10.1002/cpt.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mathialagan S., Piotrowski M.A., Tess D.A., Feng B., Litchfield J., Varma M.V. Quantitative prediction of human renal clearance and drug–drug interactions of organic anion transporter substrates using in vitro transport data: a relative activity factor approach. Drug Metab Dispos. 2017;45:409–417. doi: 10.1124/dmd.116.074294. [DOI] [PubMed] [Google Scholar]
- 213.Paine S.W., Menochet K., Denton R., McGinnity D.F., Riley R.J. Prediction of human renal clearance from preclinical species for a diverse set of drugs that exhibit both active secretion and net reabsorption. Drug Metab Dispos. 2011;39:1008–1013. doi: 10.1124/dmd.110.037267. [DOI] [PubMed] [Google Scholar]
- 214.Chen J., Yang H., Zhu L., Wu Z., Li W., Tang Y., et al. In silico prediction of human renal clearance of compounds using quantitative structure-pharmacokinetic relationship models. Chem Res Toxicol. 2020;33:640–650. doi: 10.1021/acs.chemrestox.9b00447. [DOI] [PubMed] [Google Scholar]
- 215.Doddareddy M.R., Cho Y.S., Koh H.Y., Kim D.H., Pae A.N. In silico renal clearance model using classical Volsurf approach. J Chem Inf Model. 2006;46:1312–1320. doi: 10.1021/ci0503309. [DOI] [PubMed] [Google Scholar]
- 216.Kotal P., Van der Veere C.N., Sinaasappel M., Elferink R.O., Vitek L., Brodanova M., et al. Intestinal excretion of unconjugated bilirubin in man and rats with inherited unconjugated hyperbilirubinemia. Pediatr Res. 1997;42:195–200. doi: 10.1203/00006450-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 217.van der Velde A.E., Vrins C.L., van den Oever K., Kunne C., Oude Elferink R.P., Kuipers F., et al. Direct intestinal cholesterol secretion contributes significantly to total fecal neutral sterol excretion in mice. Gastroenterology. 2007;133:967–975. doi: 10.1053/j.gastro.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 218.Mayer U., Wagenaar E., Beijnen J.H., Smit J.W., Meijer D.K., van Asperen J., et al. Substantial excretion of digoxin via the intestinal mucosa and prevention of long-term digoxin accumulation in the brain by the mdr 1a P-glycoprotein. Br J Pharmacol. 1996;119:1038–1044. doi: 10.1111/j.1476-5381.1996.tb15775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sparreboom A., van Asperen J., Mayer U., Schinkel A.H., Smit J.W., Meijer D.K., et al. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci U S A. 1997;94:2031–2035. doi: 10.1073/pnas.94.5.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gramatté T., Oertel R. Intestinal secretion of intravenous talinolol is inhibited by luminal R-verapamil. Clin Pharmacol Ther. 1999;66:239–245. doi: 10.1016/S0009-9236(99)70031-7. [DOI] [PubMed] [Google Scholar]
- 221.Zhang D., He K., Herbst J.J., Kolb J., Shou W., Wang L., et al. Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab Dispos. 2013;41:827–835. doi: 10.1124/dmd.112.050260. [DOI] [PubMed] [Google Scholar]
- 222.Zhang D., Wei C., Hop C., Wright M.R., Hu M., Lai Y., et al. Intestinal excretion, intestinal recirculation, and renal tubule reabsorption are underappreciated mechanisms that drive the distribution and pharmacokinetic behavior of small molecule drugs. J Med Chem. 2021;64:7045–7059. doi: 10.1021/acs.jmedchem.0c01720. [DOI] [PubMed] [Google Scholar]
- 223.Zhang D., Frost C.E., He K., Rodrigues A.D., Wang X., Wang L., et al. Investigating the enteroenteric recirculation of apixaban, a factor Xa inhibitor: administration of activated charcoal to bile duct-cannulated rats and dogs receiving an intravenous dose and use of drug transporter knockout rats. Drug Metab Dispos. 2013;41:906–915. doi: 10.1124/dmd.112.050575. [DOI] [PubMed] [Google Scholar]
- 224.Raghavan N., Frost C.E., Yu Z., He K., Zhang H., Humphreys W.G., et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37:74–81. doi: 10.1124/dmd.108.023143. [DOI] [PubMed] [Google Scholar]
- 225.Stass H., Kubitza D., Moller J.G., Delesen H. Influence of activated charcoal on the pharmacokinetics of moxifloxacin following intravenous and oral administration of a 400 mg single dose to healthy males. Br J Clin Pharmacol. 2005;59:536–541. doi: 10.1111/j.1365-2125.2005.02357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gramatte T., Oertel R., Terhaag B., Kirch W. Direct demonstration of small intestinal secretion and site-dependent absorption of the beta-blocker talinolol in humans. Clin Pharmacol Ther. 1996;59:541–549. doi: 10.1016/S0009-9236(96)90182-4. [DOI] [PubMed] [Google Scholar]
- 227.Ramon J., Ben-Haim M., Shabtai M., Rubinstein E. Transepithelial intestinal excretion of ciprofloxacin in humans. Clin Infect Dis. 2001;32:822–823. doi: 10.1086/319206. [DOI] [PubMed] [Google Scholar]
- 228.Garner C.E., Solon E., Lai C.M., Lin J., Luo G., Jones K., et al. Role of P-glycoprotein and the intestine in the excretion of DPC 333 [(2R)-2-{(3R)-3-amino-3-[4-(2-methylquinolin-4-ylmethoxy)phenyl]-2-oxopyrrolidin-1-yl}-N-hydroxy-4-methylpentanamide] in rodents. Drug Metab Dispos. 2008;36:1102–1110. doi: 10.1124/dmd.107.017038. [DOI] [PubMed] [Google Scholar]
- 229.Gnoth M.J., Buetehorn U., Muenster U., Schwarz T., Sandmann S. In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J Pharmacol Exp Therapeut. 2011;338:372–380. doi: 10.1124/jpet.111.180240. [DOI] [PubMed] [Google Scholar]
- 230.Wang X., Mondal S., Wang J., Tirucherai G., Zhang D., Boyd R.A., et al. Effect of activated charcoal on apixaban pharmacokinetics in healthy subjects. Am J Cardiovasc Drugs. 2014;14:147–154. doi: 10.1007/s40256-013-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wong P.C., Pinto D.J., Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis. 2011;31:478–492. doi: 10.1007/s11239-011-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Agrawal S., Jiang Z., Zhao Q., Shaw D., Cai Q., Roskey A., et al. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: in vitro and in vivo studies. Proc Natl Acad Sci U S A. 1997;94:2620–2625. doi: 10.1073/pnas.94.6.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yu A.M., Choi Y.H., Tu M.J. RNA drugs and RNA targets for small molecules: principles, progress, and challenges. Pharmacol Rev. 2020;72:862–898. doi: 10.1124/pr.120.019554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bennett C.F. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med. 2019;70:307–321. doi: 10.1146/annurev-med-041217-010829. [DOI] [PubMed] [Google Scholar]
- 235.Roberts T.C., Langer R., Wood M.J.A. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19:673–694. doi: 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Shemesh C.S., Yu R.Z., Warren M.S., Liu M., Jahic M., Nichols B., et al. Assessment of the drug interaction potential of unconjugated and GalNAc3-conjugated 2′-MOE-ASOs. Mol Ther Nucleic Acids. 2017;9:34–47. doi: 10.1016/j.omtn.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Society TA. Therapeutic monoclonal antibodies approved or in review in the EU or US. Accessed [May 1, 2021]. Available from: https://www.antibodysociety.org/antibody-therapeutics-product-data.
- 238.Lu R.M., Hwang Y.C., Liu I.J., Lee C.C., Tsai H.Z., Li H.J., et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wang W., Wang E.Q., Balthasar J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 240.Tibbitts J., Canter D., Graff R., Smith A., Khawli L.A. Key factors influencing ADME properties of therapeutic proteins: a need for ADME characterization in drug discovery and development. MAbs. 2016;8:229–245. doi: 10.1080/19420862.2015.1115937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Datta-Mannan A. Mechanisms influencing the pharmacokinetics and disposition of monoclonal antibodies and peptides. Drug Metab Dispos. 2019;47:1100–1110. doi: 10.1124/dmd.119.086488. [DOI] [PubMed] [Google Scholar]
- 242.Mould D.R., Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24:23–39. doi: 10.2165/11530560-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 243.Shah D.K., Betts A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39:67–86. doi: 10.1007/s10928-011-9232-2. [DOI] [PubMed] [Google Scholar]
- 244.Sand K.M., Bern M., Nilsen J., Noordzij H.T., Sandlie I., Andersen J.T. Unraveling the interaction between FcRn and albumin: opportunities for design of albumin-based therapeutics. Front Immunol. 2014;5:682. doi: 10.3389/fimmu.2014.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Deng R., Iyer S., Theil F.P., Mortensen D.L., Fielder P.J., Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned?. MAbs. 2011;3:61–66. doi: 10.4161/mabs.3.1.13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Dua P., Hawkins E., van der Graaf P.H. A tutorial on target-mediated drug disposition (TMDD) models. CPT Pharmacometrics Syst Pharmacol. 2015;4:324–337. doi: 10.1002/psp4.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Morgan E.T., Goralski K.B., Piquette-Miller M., Renton K.W., Robertson G.R., Chaluvadi M.R., et al. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab Dispos. 2008;36:205–216. doi: 10.1124/dmd.107.018747. [DOI] [PubMed] [Google Scholar]
- 248.FDA. Drug–drug interaction assessment for therapeutic proteins Published [August 2020]; Accessed [February, 2022]. Available from: https://www.fda.gov/media/140909/download.
- 249.Tumey L.N. An overview of the current ADC discovery landscape. Methods Mol Biol. 2020;2078:1–22. doi: 10.1007/978-1-4939-9929-3_1. [DOI] [PubMed] [Google Scholar]
- 250.Liu-Kreyche P., Shen H., Marino A.M., Iyer R.A., Humphreys W.G., Lai Y. Lysosomal P-gp-MDR1 confers drug resistance of brentuximab vedotin and its cytotoxic payload monomethyl auristatin E in tumor cells. Front Pharmacol. 2019;10:749. doi: 10.3389/fphar.2019.00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Przepiorka D., Deisseroth A., Kane R., Kaminskas E., Farrell A.T., Pazdur R. Gemtuzumab ozogamicin. J Clin Oncol. 2013;31:1699–1700. doi: 10.1200/JCO.2012.48.1887. [DOI] [PubMed] [Google Scholar]
- 252.Matsuda Y., Mendelsohn B.A. An overview of process development for antibody–drug conjugates produced by chemical conjugation technology. Expet Opin Biol Ther. 2021;21:963–975. doi: 10.1080/14712598.2021.1846714. [DOI] [PubMed] [Google Scholar]
- 253.Li P., Gong Y., Kim J., Liu X., Gilbert J., Kerns H.M., et al. Hybridization liquid chromatography–tandem mass spectrometry: an alternative bioanalytical method for antisense oligonucleotide quantitation in plasma and tissue samples. Anal Chem. 2020;92:10548–10559. doi: 10.1021/acs.analchem.0c01382. [DOI] [PubMed] [Google Scholar]
- 254.Hock M.B., Thudium K.E., Carrasco-Triguero M., Schwabe N.F. Immunogenicity of antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 2015;17:35–43. doi: 10.1208/s12248-014-9684-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Carrasco-Triguero M., Dere R.C., Milojic-Blair M., Saad O.M., Nazzal D., Hong K., et al. Immunogenicity of antibody–drug conjugates: observations across 8 molecules in 11 clinical trials. Bioanalysis. 2019;11:1555–1568. doi: 10.4155/bio-2018-0259. [DOI] [PubMed] [Google Scholar]
- 256.Teorell T. Kinetics of distribution of substances administered to the body. I. The extravascular modes of administration. Arch Int Pharmacodyn Ther. 1937;57:205–225. [Google Scholar]
- 257.Rowland M., Peck C., Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 258.Heimbach T., Chen Y., Chen J., Dixit V., Parrott N., Peters S.A., et al. Physiologically-based pharmacokinetic modeling in renal and hepatic impairment populations: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2021;110:297–310. doi: 10.1002/cpt.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Jones H.M., Chen Y., Gibson C., Heimbach T., Parrott N., Peters S.A., et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97:247–262. doi: 10.1002/cpt.37. [DOI] [PubMed] [Google Scholar]
- 260.Sinha V., Zhao P., Huang S.M., Zineh I. Physiologically based pharmacokinetic modeling: from regulatory science to regulatory policy. Clin Pharmacol Ther. 2014;95:478–480. doi: 10.1038/clpt.2014.46. [DOI] [PubMed] [Google Scholar]
- 261.Wagner C., Zhao P., Pan Y., Hsu V., Grillo J., Huang S.M., et al. Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst Pharmacol. 2015;4:226–230. doi: 10.1002/psp4.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Riedmaier A.E., DeMent K., Huckle J., Bransford P., Stillhart C., Lloyd R., et al. Use of physiologically based pharmacokinetic (PBPK) modeling for predicting drug–food interactions: an industry perspective. AAPS J. 2020;22:123. doi: 10.1208/s12248-020-00508-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Taskar K.S., Pilla Reddy V., Burt H., Posada M.M., Varma M., Zheng M., et al. Physiologically-based pharmacokinetic models for evaluating membrane transporter mediated drug–drug interactions: current capabilities, case studies, future opportunities, and recommendations. Clin Pharmacol Ther. 2020;107:1082–1115. doi: 10.1002/cpt.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Zhang X., Yang Y., Grimstein M., Fan J., Grillo J.A., Huang S.M., et al. Application of PBPK modeling and simulation for regulatory decision making and its impact on US prescribing information: an update on the 2018–2019 submissions to the US FDA's Office of Clinical Pharmacology. J Clin Pharmacol. 2020;60 Suppl 1:S160–S178. doi: 10.1002/jcph.1767. [DOI] [PubMed] [Google Scholar]
- 265.Mueller K.T., Waldron E., Grupp S.A., Levine J.E., Laetsch T.W., Pulsipher M.A., et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24:6175–6184. doi: 10.1158/1078-0432.CCR-18-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Stein A.M., Grupp S.A., Levine J.E., Laetsch T.W., Pulsipher M.A., Boyer M.W., et al. Tisagenlecleucel model-based cellular kinetic analysis of chimeric antigen receptor-T cells. CPT Pharmacometrics Syst Pharmacol. 2019;8:285–295. doi: 10.1002/psp4.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Liu C., Ayyar V.S., Zheng X., Chen W., Zheng S., Mody H., et al. Model-based cellular kinetic analysis of chimeric antigen receptor-T cells in humans. Clin Pharmacol Ther. 2021;109:716–727. doi: 10.1002/cpt.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Singh A.P., Zheng X., Lin-Schmidt X., Chen W., Carpenter T.J., Zong A., et al. Development of a quantitative relationship between CAR-affinity, antigen abundance, tumor cell depletion and CAR-T cell expansion using a multiscale systems PK–PD model. MAbs. 2020;12:1688616. doi: 10.1080/19420862.2019.1688616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Yu R.Z., Grundy J.S., Geary R.S. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expet Opin Drug Metabol Toxicol. 2013;9:169–182. doi: 10.1517/17425255.2013.737320. [DOI] [PubMed] [Google Scholar]
- 270.Yu R.Z., Kim T.W., Hong A., Watanabe T.A., Gaus H.J., Geary R.S. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab Dispos. 2007;35:460–468. doi: 10.1124/dmd.106.012401. [DOI] [PubMed] [Google Scholar]
- 271.Geary R.S. Antisense oligonucleotide pharmacokinetics and metabolism. Expet Opin Drug Metabol Toxicol. 2009;5:381–391. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- 272.Huang S.M., Strong J.M., Zhang L., Reynolds K.S., Nallani S., Temple R., et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48:662–670. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- 273.Zhang L., Reynolds K.S., Zhao P., Huang S.M. Drug interactions evaluation: an integrated part of risk assessment of therapeutics. Toxicol Appl Pharmacol. 2010;243:134–145. doi: 10.1016/j.taap.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 274.Rekic D., Reynolds K.S., Zhao P., Zhang L., Yoshida K., Sachar M., et al. Clinical drug–drug interaction evaluations to inform drug use and enable drug access. J Pharmaceut Sci. 2017;106:2214–2218. doi: 10.1016/j.xphs.2017.04.016. [DOI] [PubMed] [Google Scholar]
- 275.Yoshida K., Zhao P., Zhang L., Abernethy D.R., Rekic D., Reynolds K.S., et al. In vitro–in vivo extrapolation of metabolism- and transporter-mediated drug–drug interactions-overview of basic prediction methods. J Pharmaceut Sci. 2017;106:2209–2213. doi: 10.1016/j.xphs.2017.04.045. [DOI] [PubMed] [Google Scholar]
- 276.Huang S.M., Zhang L., Giacomini K.M. The International Transporter Consortium: a collaborative group of scientists from academia, industry, and the FDA. Clin Pharmacol Ther. 2010;87:32–36. doi: 10.1038/clpt.2009.236. [DOI] [PubMed] [Google Scholar]
- 277.Giacomini K.M., Huang S.M. Transporters in drug development and clinical pharmacology. Clin Pharmacol Ther. 2013;94:3–9. doi: 10.1038/clpt.2013.86. [DOI] [PubMed] [Google Scholar]
- 278.Giacomini K.M., Galetin A., Huang S.M. The International Transporter Consortium: summarizing advances in the role of transporters in drug development. Clin Pharmacol Ther. 2018;104:766–771. doi: 10.1002/cpt.1224. [DOI] [PubMed] [Google Scholar]
- 279.FDA. In vitro drug interaction studies—cytochrome P450 enzyme- and transporter-mediated drug interactions. Published [January 2020]. Accessed [February 2022]. Available form: https://www.fda.gov/media/134582/download.
- 280.FDA. Clinical drug interaction studies—cytochrome P450 enzyme- and transporter-mediated drug interactions. Published [January 2020]. Accessed [February 2022]. Available form: https://www.fda.gov/media/134581/download.
- 281.FDA. Drug interaction & labeling. Published [January 2020]. Accessed [February 2022]. Available from: https://www.fda.gov/drugs/development-resources/drug-interactions-labeling.
- 282.Zhao P., Zhang L., Grillo J.A., Liu Q., Bullock J.M., Moon Y.J., et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89:259–267. doi: 10.1038/clpt.2010.298. [DOI] [PubMed] [Google Scholar]
- 283.Zhao P., Rowland M., Huang S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin Pharmacol Ther. 2012;92:17–20. doi: 10.1038/clpt.2012.68. [DOI] [PubMed] [Google Scholar]
- 284.Wagner C., Pan Y., Hsu V., Sinha V., Zhao P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin Pharmacokinet. 2016;55:475–483. doi: 10.1007/s40262-015-0330-y. [DOI] [PubMed] [Google Scholar]
- 285.Wagner C., Pan Y., Hsu V., Grillo J.A., Zhang L., Reynolds K.S., et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54:117–127. doi: 10.1007/s40262-014-0188-4. [DOI] [PubMed] [Google Scholar]
- 286.Zhang L., Wu F., Lee S.C., Zhao H., Zhang L. pH-dependent drug–drug interactions for weak base drugs: potential implications for new drug development. Clin Pharmacol Ther. 2014;96:266–277. doi: 10.1038/clpt.2014.87. [DOI] [PubMed] [Google Scholar]
- 287.Dong Z., Li J., Wu F., Zhao P., Lee S.C., Zhang L., et al. Application of physiologically-based pharmacokinetic modeling to predict gastric pH-dependent drug–drug interactions for weak base drugs. CPT Pharmacometrics Syst Pharmacol. 2020;9:456–465. doi: 10.1002/psp4.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Mitra A., Parrott N., Miller N., Lloyd R., Tistaert C., Heimbach T., et al. Prediction of pH-dependent drug–drug interactions for basic drugs using physiologically based biopharmaceutics modeling: industry case studies. J Pharmaceut Sci. 2020;109:1380–1394. doi: 10.1016/j.xphs.2019.11.017. [DOI] [PubMed] [Google Scholar]
- 289.Evers R., Dallas S., Dickmann L.J., Fahmi O.A., Kenny J.R., Kraynov E., et al. Critical review of preclinical approaches to investigate cytochrome P450-mediated therapeutic protein drug–drug interactions and recommendations for best practices: a white paper. Drug Metab Dispos. 2013;41:1598–1609. doi: 10.1124/dmd.113.052225. [DOI] [PubMed] [Google Scholar]
- 290.Kenny J.R., Liu M.M., Chow A.T., Earp J.C., Evers R., Slatter J.G., et al. Therapeutic protein drug–drug interactions: navigating the knowledge gaps-highlights from the 2012 AAPS NBC Roundtable and IQ Consortium/FDA workshop. AAPS J. 2013;15:933–940. doi: 10.1208/s12248-013-9495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Huang S.M., Zhao H., Lee J.I., Reynolds K., Zhang L., Temple R., et al. Therapeutic protein–drug interactions and implications for drug development. Clin Pharmacol Ther. 2010;87:497–503. doi: 10.1038/clpt.2009.308. [DOI] [PubMed] [Google Scholar]
- 292.Lee J.I., Zhang L., Men A.Y., Kenna L.A., Huang S.M. CYP-mediated therapeutic protein–drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet. 2010;49:295–310. doi: 10.2165/11319980-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 293.Jing X., Ji P., Schrieber S.J., Fletcher E.P., Sahajwalla C. Update on therapeutic protein–drug interaction: information in labeling. Clin Pharmacokinet. 2020;59:25–36. doi: 10.1007/s40262-019-00810-z. [DOI] [PubMed] [Google Scholar]
- 294.Bissig K.D., Han W., Barzi M., Kovalchuk N., Ding L., Fan X., et al. P450-humanized and human liver chimeric mouse models for studying xenobiotic metabolism and toxicity. Drug Metab Dispos. 2018;46:1734–1744. doi: 10.1124/dmd.118.083303. [DOI] [PMC free article] [PubMed] [Google Scholar]