

Abstract
Hederacoside C (HSC) has attracted much attention as a novel modulator of inflammation, but its anti-inflammatory mechanism remains elusive. In the present study, we investigated how HSC attenuated intestinal inflammation in vivo and in vitro. HSC injection significantly alleviated TNBS-induced colitis by inhibiting pro-inflammatory cytokine production and colonic epithelial cell apoptosis, and partially restored colonic epithelial cell proliferation. The therapeutic effect of HSC injection was comparable to that of oral administration of mesalazine (200 mg·kg−1·d−1, i.g.). In LPS-stimulated human intestinal epithelial Caco-2 cells, pretreatment with HSC (0.1, 1, 10 μM) significantly inhibited activation of MAPK/NF-κB and its downstream signaling pathways. Pretreatment with HSC prevented LPS-induced TLR4 dimerization and MyD88 recruitment in vitro. Quantitative proteomic analysis revealed that HSC injection regulated 18 proteins in the colon samples, mainly clustered in neutrophil degranulation. Among them, S100A9 involved in the degranulation of neutrophils was one of the most significantly down-regulated proteins. HSC suppressed the expression of S100A9 and its downstream genes including TLR4, MAPK, and NF-κB axes in colon. In Caco-2 cells, recombinant S100A9 protein activated the MAPK/NF-κB signaling pathway and induced inflammation, which were ameliorated by pretreatment with HSC. Notably, HSC attenuated neutrophil recruitment and degranulation as well as S100A9 release in vitro and in vivo. In addition, HSC promoted the expression of tight junction proteins and repaired the epithelial barrier via inhibiting S100A9. Our results verify that HSC ameliorates colitis via restoring impaired intestinal barrier through moderating S100A9/MAPK and neutrophil recruitment inactivation, suggesting that HSC is a promising therapeutic candidate for colitis.
Keywords: hederacoside C, colitis, quantitative proteomics, S100A9, neutrophils, MAPK/NF-κB signaling
Introduction
Inflammatory bowel disease (IBD) has become a global disease with a rapidly rising incidence in developing countries [1]. This disease, including Crohn’s disease (CD) and ulcerative colitis (UC), is mainly characterized by chronic or relapsing intestinal inflammation and tissue damage [2]. The clinical symptoms of IBD are mainly abdominal pain, diarrhea, rectal bleeding, and weight loss [3].
The pathogenesis of UC is multifactorial and has not been completely clarified. It is currently accepted that alterations in the gut microbiota, genetic susceptibility, immunological regulation, and intestinal barrier are involved in the etiopathogenesis of UC [4–6]. Among them, epithelial barrier dysfunction is one of the main factors in the pathogenesis of UC which lead to intestinal bacteria and antigens penetrating into intestinal mucosa [7].
The intestinal epithelial barrier is composed of the mucous layer, epithelial cells, and intercellular tight junctions (TJs), which are responsible for regulating the permeability of mucosa and form the physical, protective, and host defense barrier against the harmful luminal microenvironment [8]. The ability of intestinal epithelial barrier is mediated by the formation of protein complex connections between adjacent cells in intestinal epithelia. These connections include tight junctions (TJ) and adherent junctions (AJ), which form the apical junction complex (AJC), as well as desmosomes, which are located in the basolateral membrane [9]. Tight junctions complexes are composed of junctional adhesion molecules, claudins, occludins, and zonula occludens (ZO), which seal neighboring cells together [10]. A growing body of evidence suggests that TJs are critical for maintaining barrier function during the shedding of intestinal epithelial cells, which occurs continuously from villus tips or colonic surfaces as a result of migration of the epithelial cells up the crypt-villus axis from stem cells at the base of the crypt [11]. The integrity of intestinal epithelial barrier is critical for the health of the gut, and epithelial barrier dysfunction is a hallmark of IBD.
Several pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interferon-γ, have been shown to increase TJ permeability and to induce apoptosis of intestinal epithelial cells. This leads to the loss of epithelial barrier function and induces epithelial damage [12]. Anti-inflammatory agents such as sulfasalazine, glucocorticoids, and nonsteroids are used for IBD. However, these drugs are less effective for some patients and frequently cause severe side effects, including opportunistic infections and malignancies [13]. Thus, it is essential to discover alternative effective drugs for IBD that can restore the impaired intestinal barrier function.
Hederacoside C (HSC), a natural triterpenoidal glycoside mainly isolated from Pulsatilla chinensis (Bunge) Regel, was used for the treatment of cancers and inflammatory disorders in traditional Chinese medicine. Previous studies have demonstrated that it possesses anti-inflammatory, anti-oxidation, anti-microbial, anti-schistosomal and antispasmodic activity with low toxicity [14–16]. It reduces the expression of TNF-α, IL-1β, iNOS, and COX-2 in LPS-stimulated peritoneal macrophages via inhibiting interleukin-1 (IL-1) receptor-associated kinase 1 (IRAK1) [17]. It also prevents TNBS-induced colonic inflammation, including decreasing pro-inflammatory cytokine expression and NF-κB activation in mice. Although its chemotherapeutic effect on colitis has already been demonstrated, the molecular mechanism of action of HSC is unclear.
In the present work, we conducted a series of experiments to discover the molecular mechanism of HSC in vitro and in vivo. We first measured anti-inflammation effects of HSC on TLR4 dimerization or MyD88 recruitment induced by LPS stimuli using immunoprecipitation and immunoblotting techniques. We then searched for the differentially regulated proteins altered by HSC in colon samples treated by quantitative proteomic analyses and analyzed the biological processes that changed proteins participated in through the Metascape database. We further investigated the regulatory effect of HSC on S100A9 protein levels, and the downstream signal pathways of it. Finally, we commented on the potential function of HSC in the intestinal epithelial barrier through the S100A9-dependent pathway.
Materials and methods
Animals
Male Sprague–Dawley (SD) rats weighing 180 to 220 g were obtained from the Experimental Animal Center of Soochow University (Suzhou, China). All rats were housed in cages at temperature of 22 ± 2 °C in a 12-h light/dark cycle, and fed with standard diet and water ad libitum. All animal experiments were conducted in accordance with the procedure approved by the Ethical Review Committee for Laboratory Animal Welfare of Soochow University. All animal experiments were approved by the Ethical Review Committee for Laboratory Animal Welfare of Soochow University and were conducted in accordance with the principles established for the care and use of laboratory animals by the College of Pharmaceutical Sciences of Soochow University.
HSC administration
HSC (purity ≥ 99.0%) was obtained from P. chinensis in our laboratory as described in the previous work [16]. Drug administration to animals began 24 h after induction of colitis for 7 d. Mesalazine was purchased from Ethypharm Pharmaceutical Co., Ltd. (Shanghai, China). The mesalazine group was orally administered with mesalazine aqueous solution (200 mg/kg) by gavage once a day according to the reference [18]. Because pharmacokinetics of HSC was complex, the microorganism of colon could transform and metabolise HSC, we choose the injection of HSC [19, 20]. The HSC groups were intraperitoneally injected with HSC in physiological saline at doses of 0.625, 1.25, and 2.5 mg/kg, 2 times/day for 7 d.
TNBS-induced colitis
The rats were fasted for 24 h before experiment but allowed free access to water, and then anesthetized by intraperitoneal injection of 5% chloral hydrate. After anesthesia, TNBS (2,4,6-trinitrobenzene sulfonic acid, 80 mg/kg, #MB5547, Dalian Meilun Biotechnology Co., Ltd., Dalian, China) dissolved in equal volume of absolute ethanol was administered in the rectum through the catheter. Following the administration of TNBS, the animals were maintained in a head-down position for 2 min to prevent leakage. Rats in control group received 0.9% saline, and the rest of the procedure is the same as that of the model rats. Next, the animals were placed in separate cages with free access to food and water.
Disease activity index (DAI) score
DAI score was used to assess the clinical severity of colitis by scoring the daily body weight, stool consistency, and rectal bleeding. The loss of body weight was scored as follows: 0, no weight loss; 1, weight loss of 0 to 5%; 2, weight loss of 5% to 10%; 3, weight loss of 10% to 20%; and 4, weight loss >20%. Stool consistency was scored as follows: 0, normally formed pellets; 2, pasty and semiformal pellets; and 4, liquid stool. Rectal bleeding was scored as follows: 0, no blood from the rectum; 2, occult blood detected in rectum; and 4, gross bleeding from the rectum. The three scores were summed, resulting in a total clinical score ranging from 0 to 12.
Histopathological analysis of rectums
The colonic tissues were quickly dissected and rinsed with saline to remove fecal residues. Central portions of colonic tissue were fixed in 4% paraformaldehyde for histological assessments. Sections of rectums with a thickness of 5 μm were stained with hematoxylin and eosin (H&E) to visualize cell morphology using an optical microscope. Light microscopy and panoramic viewer camera system (CX31, Olympus Optical Co., Ltd., Tokyo, Japan) were used to examine, scan, and analyze the histopathological features of colon.
5-Ethynyl-2′-Deoxyuridine (EdU) Assay
BeyoClick™ EdU-488 Kit (Beyotime, Haimen, China) was used to measure the cell proliferation in colon. Briefly, 4 h before rats were sacrificed, they were injected intraperitoneally with 5 mg/kg EdU solution and then the colon was collected and rinsed with saline. Colon samples were then processed using the BeyoClick™ EdU-488 kit according to the manufacturer’s instructions [21]. EdU-positive cells were detected with a confocal laser scanning microscope (LSM 710, ZEISS, Germany).
Apoptosis detection assay
To measure the apoptosis, a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay was performed by using an in situ Cell Death Detection Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer’s instructions. In this assay, colon samples were collected and stained with the kit. Fluorescein labels incorporated in nucleotides were detected by a laser scanning confocal microscope and the fluorescent cells were counted.
Cell culture and drug preparation
The human colon adenocarcinoma Caco-2 cell line was purchased from Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM medium (#11995065, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (#10270106, Gibco), 100 µg/mL streptomycin and 100 U/mL penicillin (#10378016, Gibco) in a humidified 5% CO2 atmosphere at 37 °C. LPS (#L6529, Sigma-Aldrich, St. Louis, MO, USA) or S100A9 (#ab95909, Abcam, Cambridge, MA, USA) was dissolved in PBS. Cells were pretreated with 0.1, 1, 10 µM HSC for 1 h, and then 1 µg/mL LPS or recombinant S100A9 protein was added into the cells. Samples were collected at the corresponding time.
Real-time polymerase chain reaction (RT-PCR)
According to the manufacturer’s instructions, total RNA was extracted with TRIzol reagent (Ambion, USA) and quantified by a NanoDrop 2000 spectrophotometer (Thermo Scientific). After quality check, mRNA was reverse-transcribed to cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). Advanced™ Universal SYBR® Green (Bio-Rad) was used for qPCR measurement. Standard curves were constructed with the CFX96™ real-time PCR detection system (Bio-Rad). The cycling conditions for the qPCR were as follows: 95 °C for 3 min, followed by 37 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. The relative expression levels of genes were calculated using the 2−ΔΔCt method based on the threshold cycle (Ct) value.
ELISA assays
Frozen colonic samples were homogenized mechanically in lysis buffer. Homogenized tissue samples were centrifuged at 12,000 × g at 4 °C for 15 min. Supernatants from homogenized tissue or cell culture were collected. Cytokine levels and myeloperoxidase in the supernatant were determined using ELISA kits according to the manufacturer’s instructions.
Western blotting analysis
Proteins were extracted with ice-cold RIPA buffer supplemented with protease inhibitor mixture (Thermo Fisher, USA) from cells and mouse colonic tissues. After separated by SDS-PAGE, the proteins were transferred to PVDF membranes. The membranes were blocked by incubation in 5% nonfat dry milk for 1 h at room temperature and incubated overnight with the primary antibodies overnight at 4 °C with gentle shaking. The primary antibodies used in this work were obtained from the following sources: anti-JNK (1:1000, #9252), anti-p-JNK (1:2000, #9255), anti-ERK1/2 (1:2000, #4695), anti-p-ERK1/2 (1:1000, #4370), anti-p65 (1:2000, #8242), anti-p-p65 (1:1000, #3033), anti-p38 (1:2000, #8960), anti-p-p38 (1:1000, #4511), anti-Bcl-2 (1:1000, #3498), anti-cleaved-caspase 3 (1:1000, #9664), anti-S100A9 (1:1000, #73425) and anti-caspase 3 (1:1000, #9662), anti-Bax (1:2000, #sc-20067), anti-p53 (1:1000, #sc-126), anti-TLR4 (1:1000, #sc-293072), anti-GAPDH (1:2000, #sc-365062) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Then the membranes were incubated with horseradish peroxidase (HRP) conjugated goat anti-rabbit or anti-mouse secondary antibodies (Beyotime, Shanghai, China) for 1 h at room temperature. The bound antibodies were exposed to HRP substrates (Millipore Corporation, Billerica, USA) and detected with the ChemiDoc XRS Imager (Bio-Rad, USA). Protein bands were quantified by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
TLR4 dimerization and MyD88 recruitment
HEK293T cells (2 × 106/10-cm dish) were transfected with HA-TLR4 (12 μg) and Flag-TLR4 (12 μg) plasmids for 48 h. Then they were treated with HSC (0.1, 1, 10 μM) for 1 h, and with LPS (1 μg/mL) for 4 additional hours. Cells were lyzed with IP lysis buffer. Total lysis samples (600 μg) were immunoprecipitated by using the protein A/G magnetic beads kit. Western blotting analysis and anti-FLAG magnetic beads were used to determine TLR4 dimer formation.
Immunofluorescence experiment
The protein levels of CD11b and S100A9 in colon tissues were detected by immunofluorescence staining as described previously [21]. In brief, colon was removed, fixed with 4% paraformaldehyde for 12 h, and cryoprotected in 30% sucrose for at least 24 h at 4 °C until the tissue pieces sank to the bottom. The colon with mucosal injury was sliced to 20 µm thick using a low-temperature slicer. The colon sections were infiltrated with 0.5% Triton X-100 in PBS, blocked with 3% BSA, incubated overnight at 4 °C with FITC-anti-CD11b (1:100, #CD11B01, Thermo Fisher) or FITC-anti-S100A9 (1:100, #73425S, Cell Signaling Technology) antibodies. The sections were counterstained with a secondary antibody (GB22302, Solarbio) for 1 h. The immunofluorescent cells were visualized and digital images were captured using a confocal laser scanning microscope.
Label-free quantitative proteomics analysis
Altered protein by HSC in the TNBS-induced rats was measured by label-free quantitative proteomics as described previously [21]. Briefly, on the 8th day after rats were treated with TNBS (80 mg/kg) and/or HSC, colon tissues of the rats in TNBS + saline group (n = 3) and TNBS + HSC group (2.5 mg/kg) (n = 3) were extracted and lysed by denaturing lysis buffer (8 M urea, 150 mM NaCl, 1 × protease inhibitor cocktail) on the ice for 30 min. After lysis, the supernatant was collected by centrifugation at 4 °C at 14800 × g for 10 min. The protein concentration of the samples was determined using a BCA protein quantification kit (Beyotime, Shanghai, China). The disulfide bonds of proteins were reduced with 2 mM dithiothreitol at 37 °C. After reduction, the samples were alkylated with 8 mM chloroacetamide for 1 h, and the reaction was stopped by adding 2 mM dithiothreitol. After acetone purification, the protein samples were solubilized with 8 M urea, digested with the intracellular protease Lys-C solution in a 37 °C incubator for 4–6 h, and then digested again with trypsin (TaKaRa Bio, USA) overnight at 37 °C using a trypsin/total protein ratio of 1:20 after urea was diluted to <2 M. Next, the resulting peptides were desalted with C18 Zip-tips (Merck Millipore, Massachusetts, USA) and eluted with 0.1% (v/v) trifluoroacetic acid (TFA)/50% (v/v) acetonitrile (ACN). Peptides were separated by capillary high-performance liquid chromatography and analyzed in an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Raw MS data were analyzed using MaxQuant software according to a previous procedure [22]. Proteins were qualitatively screened using protein and Peptide FDR ≦0.01. The independent sample t-test of IBM SPSS software (version 19) was used to calculate the P-values for the identified proteins. Fold changes >1.50 or <0.67 and P < 0.05 indicated a statistically significant difference.
Neutrophil purification and activation assays
Mouse bone marrow neutrophils were obtained from specific pathogen-free ICR mice and sorted using flow cytometry (BD FACSVerse™, BD Biosciences, San Jose, CA, USA). In brief, bone marrow from the femur, tibia, and sternum of mice was flushed with RPMI-1640 medium (10% FBS + Penicillin-Streptomycin Solution) with a 1 mL syringe. Then the solution was centrifuged (1800 × g, 5 min), and then resuspended in new RPMI-1640 culture medium. PE-Ly6G (#127608, BioLegend, San Diego, CA, USA) and CD11b (#101212, BioLegend) antibodies were added to label neutrophils. At last, cells were analyzed using a BD FACSVerse™ flow cytometer and Flow Jo 7.6.1 software.
The sorted neutrophils were cultured in RPMI-1640 (supplemented with 10% FBS + Penicillin-Streptomycin Solution). HSC (0.1, 1, 10 μM) is added and incubated for 1 h, and then N-formyl-methionyl-leucyl-phenyl-alanine (fMLP) (#F3506-10MG, Sigma Aldrich) (1 μM) was used to induce cell activation. The concentration of S100A9, elastase, or MMP9 (metalloproteinase-9) was detected according to the instructions of the ELISA kit (Lvye Biotechnology, Suzhou, China).
Immunohistochemistry analysis
For immunohistochemistry analysis, colon sections (5 μm) were deparaffinized and boiled for 10 min in sodium citrate buffer (10 mM, pH 6.0). Hydrogen peroxide (3.3%) in methanol was used to inhibit endogenous peroxidase activity. The sections were blocked with 2.5% normal horse serum and then were incubated overnight at 4 °C in blocking buffer with the following primary antibodies: Ly6G (1:200, #orb322983, Biorbyt, Cambridge, CB4 0WY, UK), ZO-1 (1:200, #29274, Signalway Antibody, Maryland, USA), Occludin (1:200, #27260-1-AP, Proteintech, Inc, Rosement, IL, USA) and Claudin-1 (1:200, #28674-1-AP, Proteintech) which was followed by incubation with a secondary antibody (#A0192, Beyotime). Omission of the primary antibody was used as negative control. All samples were finally visualized with a microscope and analyzed using ImageJ software. Brown or yellow particles were regarded as positive signal.
Measurement of intestinal barrier permeability
Colonic permeability was estimated in vivo by quantifying the absorbed Evans blue as previously described [23]. In brief, the rats were injected with Evans blue (1 mL/kg body weight) via the tail vein 1 h before the intestinal tissue samples were collected and mixed with dimethylformamide solution at 0.3 mL/100 mg of tissue followed by incubation for 24 h at 60 °C. The extracts were centrifuged at 837 × g for 30 min. The absorbance of the supernatant was measured by spectrophotometry at 620 nm. The results (µg/mL) were calculated based on a standard curve.
Statistical analysis
Statistical analysis was performed using Prism 5 (GraphPad). Results were expressed as mean ± standard deviation (SD). Two-way ANOVA followed by Dunnett’s multiple comparisons test was used to analyze the body weight change and DAI scores in the animal experiments. One-way ANOVA followed by Dunnett’s multiple comparison test was used for other data except the body weight change and DAI scores. A P-value <0.05 was considered statistically significant.
Results
HSC reduced TNBS-induced colitis in rats
To determine whether intraperitoneal injection of HSC could ameliorate the intestinal damage in colitis in rats, we induced colitis by TNBS and then treated the rats with HSC or mesalazine (positive control) for 7 d. TNBS-treated rats exhibited marked colitis symptoms characterized by shaggy hair, loss body weight, diarrhea, and occult fecal blood, which were analogous to the symptoms of human IBD. Compared with the normal group, TNBS-induced rats showed a significantly higher loss of body weight from 2nd day to 9th day following a TNBS administration. However, HSC significantly decreased the loss of body weight from 8th day to 9th day (Fig. 1a). The DAI scores involving body weight loss, stool consistency, and gross bleeding, were significantly increased in colitis group following a TNBS administration for 1 day (Fig. 1b), and still gently elevated after TNBS stimulation was halted for 1 week. Accordingly, HSC (1.25, 2.5 mg/kg) treatment was implemented to the model rats at the 2nd day of post-administration of TNBS. DAI in drug-treated group was observed to be decreased sharply from day 3 to day 8.
Fig. 1. HSC ameliorated pathological phenotype of TNBS-induced colitis in rats.

Rats were enteric injected with TNBS (80 mg/kg) for once, and then intraperitoneally injected with HSC (0.625, 1.25, 2.5 mg/kg) twice per day for 7 days. Mesalazine was intragastrically administered at the dose of 200 mg/kg once per day for 7 consecutive days. Rats were sacrificed on the 8th day after colitis induction. n = 8 per group. a Body weight of rats in each group was recorded during the progression. b The disease activity index (DAI) was measured every day. DAI = weight loss score + stool characters score + hematochezia score. c Macroscopic observation of the colons in each group. d The colon length on day 8. e The colon weight on day 8. f The colon coefficient of each group of rats. Colon coefficient = colon weight/colon length (g/cm). ###P < 0.001 vs vehicle group; *P < 0.05, **P < 0.01, ***P < 0.001 vs TNBS group; $P < 0.05, $$P < 0.01, $$$P < 0.001 vs HSC (2.5 mg/kg) group. Two-way ANOVA followed by Dunnett’s multiple comparisons test was used to analyze the body weight change and DAI scores in the animal experiments.
Colon length was deemed as an important indicator of the severity of colorectal inflammation. Our experiments (Fig. 1c–e) showed that the colon length was significantly shortened and the colon weight was increased in colitis group as compared to that of the control group. By contrast, treatment with HSC ameliorated TNBS-induced colon length shortening and colon weight increase. Similarly, HSC reduced weight-to-length ratio of colon in TNBS-induced rats (Fig. 1f).
HSC reduced the productions of immune-inflammatory cytokines
TNBS-induced model of acute colitis maintains many pathological features of human colitis. During inflammation, monocytes are recruited to inflamed sites and lymphoid tissues. Therefore, we measured the effect of HSC on immune cell infiltration and inflammatory cytokine expression in TNBS-induced acute colitis in rats. The levels of inflammatory cytokines, including TNF-α, IL-6, IL-1β, CXCL-1, CXCL-2, and CXCL-5 were significantly increased in TNBS-induced colitis rats as compared to those of the control group (Fig. 2a–f). However, these elevated inflammatory cytokines were dramatically decreased by HSC in a dose-dependent manner.
Fig. 2. HSC inhibited the expression of inflammatory cytokines and chemokines in rats.

Rats were enteric injected with TNBS (80 mg/kg) for once, and then intraperitoneally injected with HSC (0.625, 1.25, 2.5 mg/kg) twice per day for 7 days. Mesalazine was intragastrically administered at the dose of 200 mg/kg once per day for 7 consecutive days. Colon of rats was collected on the 8th day after administration of TNBS. a TNF-α, b IL-6, c IL-1β were measured by ELISA kits. d CXCL-1, e CXCL-2, f CXCL-5 were assessed by quantitative real-time PCR. Data were shown as mean ± SD (n = 3). #P < 0.05, ##P < 0.01 vs vehicle group; *P < 0.05; ** P < 0.01, ***P < 0.001 vs TNBS group.
Effects of HSC on the proliferation and apoptosis of intestinal epithelial cells
To further evaluate the protective effect of HSC on TNBS-induced colon injury, we sought to examine the effects of HSC on the proliferation and apoptosis induced by TNBS. As shown in Fig. 3a, b, a decrease in the number of EdU-positive cells was observed in the TNBS-induced colitis in rats. The number of EdU-positive cells was significantly increased after HSC treatment. The results indicated that colonic epithelial cell proliferation was significantly reduced by TNBS. However, HSC partially restored the ability of regenerative proliferation.
Fig. 3. The protective effect of HSC on intestinal epithelial cells.

a Sections of colon tissue were immunostained with EdU and observed by confocal laser-scanning microscope (n = 3). b The area ratio of EdU fluorescence staining intensity to DAPI. c Sections of colon tissue were immunostained with TUNEL and observed by confocal laser-scanning microscope (n = 3). d The area ratio of TUNEL fluorescence staining intensity to DAPI. e Western blotting analysis of the levels of Bax, Bcl-2, p53, and cleaved-caspase 3. Each experiment was repeated three times. f Statistical analysis of p53, Bax, Bcl-2, and cleaved-caspase 3 protein level. #P < 0.05, ##P < 0.01 vs control group; *P < 0.05, **P < 0.01, ***P < 0.001 vs TNBS group.
In order to determine whether HSC inhibits the apoptosis of epithelial cells upon TNBS treatment, we used TUNEL kit to detect apoptotic cells. When frozen sections of colons from rats in control group were stained by TUNEL, nearly no signal was detected, indicating that apoptotic cells were fewer in normal colon. However, large numbers of TUNEL-positive cells were detected in the crypts and lamina propria of colons in TNBS-treated group (Fig. 3c, d). As predicted, HSC significantly decreased the number of the TUNEL positive spots (Fig. 3c, d), suggesting that HSC could reduce cell apoptosis in TNBS-induced colitis.
We then detected the expression of apoptosis-related proteins in colon samples. In the TNBS-treated group, the expression of Bax, p53, and cleaved caspase 3 was all significantly upregulated, but the expression of Bcl-2 was decreased. In contrast, HSC significantly reduced Bax/Bcl-2 ratio, cleaved caspase 3, and p53 protein levels in a dose-dependent manner (Fig. 3e, f). These observations clearly demonstrate that HSC can effectively alleviate acute colitis induced by TNBS in rats.
HSC inhibited activation of MAPK/NF-κB signaling pathway
The above experimental results showed that HSC had anti-inflammatory activity and inhibited the release of inflammatory factors. It is well-known that NF-κB plays an important role in inflammatory response and immune regulation. In order to further verify the anti-inflammatory activity of HSC, we studied the effect of HSC on the activation of NF-κB signaling pathway. The expression of p-p65/p65 was increased in Caco-2 cells after LPS stimulation, and the phosphorylation of proteins was activated in the upstream signaling pathways, including p-JNK, p-ERK, and p-p38, while HSC significantly reduced their phosphorylation and thus their activation (Fig. 4a, b).
Fig. 4. The inhibitory effect of HSC on LPS-induced inflammation response in Caco-2 cells.

a Caco-2 cells were pretreated with HSC (0.1, 1, 10 μM) for 1 h and then stimulated with LPS (1 µg/mL) for 2 h. The level of proteins, including p-JNK, p-ERK, p-p38, p-p65, and their nonphosphorylated forms, was determined by Western blotting analysis. Total ERK, JNK, p38, p65, and GAPDH were used as internal standards. Each experiment was repeated three times. b The relative protein levels were quantified by densitometry. c TNF-α, d IL-6, e IL-1β were measured by ELISA kits. f HEK293T cells were co-transfected with Flag-TLR4 and HA-TLR plasmids for 48 h, and were treated with HSC (1 h) and then treated with LPS (4 h). TLR4 was immunoprecipitated using anti-Flag magnetic beads. Immunocomplexes were assessed by immunoblotting with anti-HA and anti-Flag antibodies. g HEK293T cells pre-treated with HSC (1 h) were stimulated with LPS (4 h). TLR4 was immunoprecipitated using anti-Flag magnetic beads. Immunocomplexes were assessed by immunoblotting with anti-MyD88 and anti-Flag antibodies. #P < 0.05, ##P < 0.01, ###P < 0.001 vs control group; *P < 0.05, **P < 0.01 vs LPS group.
Above experiments showed that HSC inhibited MAPK/NF-κB and its downstream signaling pathway, then we used ELISA kits to detect the release of inflammatory factors. After LPS stimulation, the content of IL-6, IL-1β, and TNF-α was increased significantly, while pretreatment with HSC reduced the release of inflammatory factors (Fig. 4c–e). These results demonstrate that anti-inflammatory activity of HSC is slightly more effective than mesalazine both in vitro and in vivo under our experiment condition.
HSC inhibited TLR4 dimerization and MyD88 recruitment
TLR4 receptors are specifically present on a variety of cells and play an important role in the regulation of inflammation [24]. LPS induces TLR4 dimerization, recruits MyD88, and then activates the downstream MAPK and NF-κB signaling pathways to induce the release of inflammatory factors. To assess the effects of HSC on TLR4 dimerization, Flag-TLR4 and HA-TLR4 plasmids were co-transfected into HEK293T cells for 48 h, and Co-IP and Western blotting were performed to detect the Flag-TLR4 and HA-TLR4 complexes. HSC sharply reduced the amount of HA-TLR4 that were co-immunoprecipitated by Flag-TLR4, suggesting that HSC disrupted LPS-induced TLR4 dimerization (Fig. 4f). Additionally, HSC significantly blocked LPS-induced formation of TLR4 and MyD88 complex (Fig. 4g). These results indicated that HSC could inhibit LPS-induced TLR4 dimerization and MyD88 recruitment, thereby inhibiting the downstream pro-inflammatory cascade.
Proteomic identification of the differentially regulated proteins by HSC
The above studies showed that HSC remarkably diminished the TNBS-induced colitis and the therapeutic effect was comparable to that of mesalazine. However, the specific mechanism of HSC in alleviating IBD is still unclear. To further investigate the mechanisms of the therapeutic effect of HSC on TNBS-induced colitis, we performed a label-free quantitative proteomics to identify the differentially expressed proteins in the colon samples from TNBS-induced group and HSC-treated group.
We identified a total of 441 proteins through LC-MS/MS analysis, whose ratios were shown in the volcano plot. Among the quantified proteins, 18 protein groups with fold change <0.67 and P < 0.05 were marked with red (upregulation) dots in the HSC-treated group compared with the TNBS-induced group. Interestingly, all the differentially regulated proteins with statistical significance were down-regulated (green dots) (Fig. 5a).
Fig. 5. HSC inhibited the activation of S100A9/MAPK/NF-κB signaling pathway.

a The volcano plot for the MS-identified 441 proteins in colon tissue. Each point showed the Log2 (Fold change) in the x axis vs. their corresponding -Log10 (P-value) in the y axis. A fold change above 1.5, or a fold change below 0.67, was considered as differentially expressed, and a P-value of < 0.05 (horizontal dotted line) was considered as statistically significant. The black points indicate unaltered proteins. The green points and red points indicated the downregulated and upregulated proteins, respectively. The red circle represented S100A9. b The biological processes of the differentially regulated proteins analyzed by the Metascape database. c Western blotting analysis of S100A9 protein level in colon tissues. d Human Caco-2 cells were treated with HSC for 1 h, and then treated with recombinant S100A9 protein (1 μg/mL) for 2 h. Western blotting analysis of p-JNK, p-ERK, p-p38, p-p65 and total proteins. e Statistical analysis of p-JNK, p-ERK, p-p38, and p-p65 protein level. f TNF-α, g IL-6, h IL-1β were measured by ELISA in Caco-2 cells. i Western blotting analysis of downstream protein of S100A9, including TLR4, p-JNK, p-ERK, p-p38, and p-p65 in colon tissues. j Statistical analysis of TLR4, p-JNK, p-ERK, p-p38 and p-p65 protein level. Data were shown as mean ± SD (n = 3). Each experiment was repeated for three times. #P < 0.05, ###P < 0.001 vs control group; *P < 0.05, **P < 0.01, ***P < 0.001 vs S100A9 group or TNBS group.
Next, we analyzed the biological processes that the 18 proteins participated in through the Metascape database. The result showed that the differentially regulated proteins were mainly clustered in the neutrophil degranulation and in the regulation of blood coagulation (Fig. 5b). The S100A9 protein involved in the degranulation of neutrophils is one of the obviously reduced proteins upon HSC treatment. Therefore, Western blotting analysis was used to verify the expression of this protein. The results revealed that S100A9 was significantly increased in colon tissues treated by TNBS and the increase could be attenuated by the further treatment with HSC (Fig. 5c), consistent with the proteomic data.
HSC suppressed S100A9 downstream signaling pathway
Previous studies showed that S100A9 and S100A8 preferentially form a stable heterodimer (S100A8/A9), and through interaction with their receptor, TLR4 or RAGE, promote the activation of MAPK or NF-κB signaling pathways [25]. To further evaluate whether S100A9 is a key regulatory factor of HSC, human recombinant S100A9 protein was used to test whether it is involved in the regulation of the same pathways. S100A9 elevated the phosphorylation of JNK, ERK, p38, and p65 for their activation in Caco-2 cells. In contrast, HSC inhibited this activation by reducing their phosphorylation (Fig. 5d, e). After co-incubating HSC with cells for 1 h, they were stimulated with recombinant S100A9 protein for 2 h, and the release of inflammatory factors was detected by ELISA. The results showed that recombinant S100A9 protein stimulated the secretion of TNF-α, IL-6, and IL-1β, but HSC ameliorated the secretion of TNF-α and IL-1β. However, there was no significant difference in the IL-6 concentration (Fig. 5f–h). Hence, we could convincingly conclude that HSC achieved colonic protection by regulating S100A9 and thereby inhibiting the MAPK/NF-κB signaling pathway.
Then, we tested the activation of the S100A9/MAPK/NF-κB signaling pathway in colon tissue to verify its effect. As expected, TNBS significantly increased the downstream pathway of S100A9, such as TLR4, p-JNK, p-ERK, p-p38, and p-p65. However, the expression or phosphorylation of these proteins was significantly reduced by HSC (Fig. 5i, j).
HSC suppressed the levels of S100A9 depending on neutrophils
Neutrophil migration into the colon mucosa is a hallmark of IBD and an important source of S100A9 [26, 27]. To verify whether HSC can inhibit neutrophil migration, lymphocyte antigen 6 (Ly6G+) positive neutrophils were measured by immunohistochemistry analysis of colon samples. The results showed that Ly6G+ positive neutrophil cells in the colonic mucosa were markedly increased by the administration of TNBS at day 8. The immunohistochemical staining clearly indicated that the administration of HSC significantly decreased the numbers of Ly6G+ positive neutrophil cells in colonic mucosa (Fig. 6a).
Fig. 6. HSC attenuated the recruitment of neutrophils.

Rats were enteric injected with TNBS (80 mg/kg) once, and then intraperitoneally injected with HSC (0.625, 1.25, 2.5 mg/kg) twice per day for 7 days. Mesalazine was intragastrically administered at the dose of 200 mg/kg once a day for 7 consecutive days. Rats were sacrificed on the 8th day after colitis induction. n = 8 per group. a Neutrophils in colon tissue were analyzed by immunohistochemistry with Ly6G and the results was photographed by microscope. Black arrows indicated Ly6G. Scale bar: 100 μm. Images were from three rats in one group (n = 3 per group). b MPO in colon tissues was detected by ELISA kits. Data were shown as mean ± SD (n = 3). #P < 0.05 vs control group; *P < 0.05 vs TNBS group. c Sections of colon tissue were immunostained with FITC-S100A9 (red) and FITC-CD11b (green) and the immunofluorescence was detected by confocal laser-scanning microscope. Images were from three rats in one group (n = 3 per group).
Then, myeloperoxidase (MPO), a marker of activated neutrophils associated with the severity of inflammation in intestinal disease, which reflects the extent of neutrophil infiltration and degranulation, was greatly elevated in colonic tissue by TNBS [28]. By contrast, HSC significantly decreased the concentration of MPO in colon tissue (Fig. 6b). These data indicate that HSC significantly inhibits the recruitment of inflammatory cells, especially the chemotaxis of neutrophils.
Furthermore, we used immunofluorescence to label the inflammatory cells infiltrating the colon with CD11b (green) or S100A9 (red). Compared to normal control group, the number of CD11b or S100A9 positive cells in the intestinal wall of colitis rats was significantly increased, while HSC significantly decreased the number of positive cells, including both CD11b and S100A9 positive cells (Fig. 6c), in accordance with the above results.
To examine the co-localization of S100A9 with CD11b, colon sections were blocked and then incubated with a mouse monoclonal anti-CD11b FITC antibody and a rabbit anti-S100A9 antibody overnight at 4 °C. The results are interesting that S100A9 localization is consistent with the location of CD11b positive cells. These data indicated that neutrophils may be the major source of S100A9, and HSC reduces neutrophils recruitment and the release of S100A9.
HSC inhibited neutrophil degranulation
Accumulation of activated neutrophils in the intestine is associated with mucosal injury and debilitating IBD symptoms [29]. To further explore the effect of HSC on the activation of neutrophils, we sorted the neutrophils from mouse bone marrow by flow cytometry and used N-formyl-methionyl-leucyl-phenyl-alanine (fMLP) to induce neutrophil degranulation. ELISA assays were used to detect the activity of elastase and matrix metalloproteinase-9 (MMP-9), the biomarkers of neutrophil degranulation, and the release of S100A9. The data presented that fMLP induced mouse neutrophil degranulation, and increased the activity of elastase and MMP-9. However, HSC reduced the activity of elastase and MMP-9, suggesting that HSC inhibited the degranulation of neutrophils (Fig. 7a, b). The medium supernatant of neutrophil collected at 20 min after treatment of fMLP was added into the Caco-2 cells and cultured for 2 h. We found that the levels of p-JNK1/2, p-ERK1/2, p-p38, and p-p65 were increased by the neutrophil supernatant treated with fMLP in Caco-2 cells. While, HSC (0.1, 1, 10 µM) reduced the activation of this MAPK/NF-κB pathway (Fig. 7c, d).
Fig. 7. HSC inhibited the inflammatory response of neutrophils.

a Neutrophils were treated with HSC for 1 h and then with fMLP (1 μM) for 20 min. The activity of elastase was determined by ELISA kit. b MMP-9 was determined by ELISA kits. Data were shown as mean ± SD (n = 3). c Neutrophils were treated with HSC (0.1, 1, 10 μM) for 1 h, and then treated with fMLP (1 μM) for 20 min. The medium supernatant from neutrophils was collected and added into the Caco-2 cells for 2 h. Western blotting analysis of MAPK/NF-κB pathway protein level. d Statistical analysis of p-JNK, p-ERK, p-p38, and p-p65 protein level. #P < 0.05 vs control group; *P < 0.05 vs fMLP group.
Restoration of epithelial barrier function by HSC was S100A9-dependent
Intestinal barrier dysfunction is found to result in increased intestinal permeability. To determine whether HSC reverts the increase in intestinal permeability observed upon TNBS-induced colitis, we utilized the Evans blue extravasation experiment. The concentration of Evans blue in the colon was significantly increased in the colitis rats compared to that in the control rats. While, HSC decreased Evans blue levels in the colon compared to that in the TNBS group (Fig. 8a).
Fig. 8. HSC restored intestinal barrier function in vivo and in vitro.

Rats were enteric injected with TNBS (80 mg/kg) once, and then intraperitoneally injected with HSC (0.625, 1.25, 2.5 mg/kg) twice per day for 7 days. Mesalazine was intragastrically administered at the dose of 200 mg/kg once a day for 7 consecutive days. Rats were sacrificed on the 8th day after colitis induction. n = 8 per group. a Vascular leakage of rats in each group. Data were presented as the mean ± SD (n = 8 rats per group). b Representative H&E-stained colon sections. Black arrow indicated the injury site. Scale bar: 100 μm. c Sections of colon tissue were analyzed by immunohistochemistry for ZO-1, claudin-1, occludin and the images were taken under optical microscope. The ratio of positive optical density to area. Scale bar: 100 μm. d Statistical analysis of ZO-1, claudin-1, occludin by immunohistochemistry. e The level of ZO-1, claudin-1, and occludin in colon tissue was measured by Western blotting. Each experiment was repeated three times. f Statistical analysis of ZO-1, claudin-1, occludin. g Neutrophils were treated with HSC for 1 h and again with fMLP (1 μM) for 20 min. Cell supernatant was collected to determine the content of S100A9 by an ELISA kits. h Neutrophils were treated with HSC (0.1, 1, 10 μM) for 1 h and again with fMLP (1 μM) for 20 min. The medium supernatant from neutrophils was collected and added into the Caco-2 cells for 24 h. Western blotting analysis of ZO-1, claudin-1, and occludin in Caco-2 cells. i Statistic analysis of ZO-1, claudin-1, and occludin in Caco-2 cells. j Caco-2 cells were transfected with pEnCMV-S100A9 plasmid for 24 h, and then they were treated with HSC for 1 h, compared with LPS (1 μg/mL) for another 24 h. Western blotting analysis of ZO-1, occludin in Caco-2 cells. k Statistic analysis of ZO-1 and occludin in Caco-2 cells. #P < 0.05, ###P < 0.001 vs control group; *P < 0.05, **P < 0.01 vs TNBS group or fMLP group.
To more directly assess the protective effect of HSC on the intestinal barrier, we performed morphological analyses of colon tissues. The images of H&E-staining colon tissue sections from different groups (Fig. 8b) indicated that TNBS-treated rats contained obvious damage, including mucosal epithelial necrosis, hyperemia, edema, the ulcerated mucosal layer, inflammatory cell infiltration, and the dysfunction of intestinal epithelial cells. As expected, HSC restored the ulcerated mucosal layer and dysfunction of intestinal epithelial cells, decreasing inflammatory cell infiltration induced by TNBS.
A type of intercellular junction known as tight junction is responsible for epithelial barrier functions in the gastrointestinal tract. ZO-1, claudin-1, and occludin are particularly sensitive to TNBS exposure and affect intestinal barrier functions. Therefore, the levels of ZO-1, claudin-1, and occludin were measured by Western blotting and immunohistochemistry assays. The expression of ZO-1, occludin, and claudin-1 in TNBS-treated rats was lower than that in normal rats. Compared with the model group, the HSC (1.25, 2.5 mg/kg) group exhibited significantly increased expression of ZO-1, occludin, and claudin-1 (Fig. 8c, d). Consistent with the results of immunohistochemistry assay, Western blotting showed that the level of ZO-1, claudin-1, and occludin in the colon was decreased in the TNBS group, and a recovery was observed after HSC treatment (Fig. 8e, f). These data showed that HSC could influence tight junctions.
Neutrophil chemotaxis and activation could injure epithelial barrier cells. To explore the effect of HSC on neutrophil chemotaxis and activation of epithelial cells, we also conducted a co-culture experiment to confirm whether the anti-inflammatory effect of HSC occurred by inhibiting the activation of neutrophils. For this, HSC was added for 1 h and then neutrophils were stimulated with fMLP for 20 min. The collected medium supernatant of neutrophils was added into the Caco-2 cells and the cells were cultured for 2 h or 24 h. The Caco-2 cells were collected to measure the expression of corresponding proteins. The results showed that the medium supernatant of fMLP-treated neutrophils contained higher concentration of S100A9, (Fig. 8g).
The medium supernatant of fMLP-treated neutrophils inhibited the levels of ZO-1, claudin-1, and occludin in Caco-2 cells. However, HSC could upregulate the expression of ZO-1, claudin-1, and occludin proteins (Fig. 8h). These data showed that HSC could alleviate intestinal barrier injury via junction adhesion proteins involved in action of neutrophils.
Further, the over expression of S100A9 experiment was done. In brief, Caco-2 cells were transfected with pEnCMV-S100A9 plasmid. After 24 h of transfection, cells were treated with HSC for 1 h, and then LPS (1 μg/mL) was used to stimulate the cells. Cells were lysed 24 h after administration of LPS. The results showed that HSC could increase the expression of ZO-1, occludin protein, and protect against the intestinal barrier damage caused by LPS. In exogenous over expression of S100A9 group, HSC could not increase the level of ZO-1 and occludin. These data indicated that over expression of S100A9 could reverse the protective effect of HSC on the intestinal barrier (Fig. 8j, k).
Discussion
In the present work, we conducted a series of experiments to test the protective effects of HSC on IBD through inhibiting neutrophil activity and restoring impaired intestinal barrier function in vitro and in vivo. Our experimental results showed that HSC could effectively prevent TNBS-induced colitis by inhibiting the NF-κB signaling pathway through deactivating S100A9.
Emerging experimental and clinical data have indicated that pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 play crucial roles in pathogenesis of colitis. Colitis is a chronic inflammatory disease of the gastrointestinal tract characterized by breakdown of the epithelial barrier and disruption of intestinal homeostasis. In the present study, HSC exerted its anti-inflammatory effect by inhibiting the secretion of pro-inflammatory factors such as IL-1β, IL-6, and TNF-α in colitis rats. In vitro, it also inhibited the activated NF-κB signaling pathway induced by LPS. These results are consistent with previous studies [30].
Upon LPS induction, TLR4 is dimerized to control the NF-κB and/or MAPK cascades via interaction of dimerized TLR4 with its downstream effector MyD88 [31]. MyD88 binds to activated TLR4, modulates NF-κB and/or MAPK cell signaling cascades to activate pro-inflammatory mediators. Thus, altering TLR4 homodimerization is considered as an alternative method for treating inflammatory disorders. In the present study, our study revealed that HSC significantly reduced TLR4/MyD88 interaction, indicating TLR4-MyD88 signaling participates in the attenuation of the inflammatory effects of HSC.
Further, moderated proteins by HSC was found by proteomic analysis. By comparing and analyzing the proteome between normal individuals and pathological individuals, we can find certain “disease or drug-specific protein molecules”, which can become molecular targets for the design of new drugs, or provide molecular markers for early diagnosis of diseases [32]. In our study, label-free quantitative proteomics identified 441 proteins in colon samples. Among these quantified proteins, the S100A9 protein, associated with the degranulation of neutrophils, is one of the proteins that was obviously reduced by HSC.
S100A9 belongs to calcium-binding proteins, also known as myeloid-related proteins. It is expressed in several types of cells, including neutrophils, macrophages, and epithelial cells, and plays a key role in the innate immune response to bacteria and other microbial pathogens via a mechanism termed as nutritional immunity [33]. S100A9 is substantially elevated in several inflammatory processes, and has been widely accepted as a biomarker of disease activity, even in COVID-19 patients. The levels of S100A9 and S100A8 were significantly higher in T, B, NK, and dendritic cells of severe COVID-19 patients than in those of moderate patients [34, 35]. S100A9 stimulates all members of the MAPK cascades, including p38, ERK, and JNK, and activates NF-κB through TLR4 or advanced glycation end products receptor (RAGE). In the present study, HSC inhibited the activation of S100A9/MAPK/NF-κB signaling pathway in vitro and in vivo. Human recombinant S100A9 protein activated MAPK/NF-κB signaling pathway, including promoting TLR4 dimerization and MyD88 recruitment and activating its downstream signaling molecules, such as increasing the phosphorylation of NF-κB/p65, and JNK, which were obviously decreased by HSC.
S100A9 is released not only by neutrophils during inflammatory reaction, but also by other cell types. Next, we want to determine which kind of cells with releasing S100A9 have the key effects during colitis induced by TNBS. Our results showed that the location of S100A9 protein almost was consistent with that of neutrophils in colon treated with TNBS, indicating neutrophils were the major source of S100A9. Besides, TNBS caused a more powerful activation of neutrophils as the TNBS-induced colitis progressed. We observed that the level of S100A9 protein, the number of neutrophils, and MPO production in the mucosa of colon was drastically decreased by HSC. These results indicated that neutrophils are the main source of S100A9 during TNBS-induced colitis, and the decrease of S100A9 protein by HSC is involved in the reduced activation of neutrophils.
Neutrophils are often the first type of immune cells recruited to the site of inflammation. Then they phagocytose pathogen, degranulate particles, produce reactive oxygen species (ROS), release neutrophil extracellular trapping nets (NETs), and eliminate extracellular pathogens [36]. Besides their antimicrobial function, deregulation of neutrophils and their hyperactivity can lead to tissue damage in severe inflammation or trauma [37]. Aggregated neutrophils will release MPO, elastase, MMP-9, and cytokines to recruit more immune cells, leading to a series of pro-inflammatory cascade reaction, thereby aggravating inflammatory diseases [38]. In our study, when fMLP was used to stimulate neutrophils, the release of S100A9 and MMP-9 was increased, accompanied by activating downstream MAPK/NF-κB signaling molecule transduction. Otherwise, HSC could inhibit the degranulation of neutrophils and reduce the release of S100A9 and MMP-9, thereby inhibiting the MAPK/NF-κB signaling pathway by fMLP in vitro. Hence, the inhibition of intestinal neutrophil infiltration may be an essential mechanism of HSC to protect colon against IBD [39, 40].
Evidence indicates that the migration of large numbers of neutrophils may destroy connexins, such as the ZO-1, β-catenin, claudin-1, E-cadherin, thereby reducing transepithelial resistance and increasing epithelium permeability. In addition, the migration of a large number of neutrophils may also form gaps between epithelial cells, thereby further weakening the epithelial barrier and promoting the development of intestinal inflammation. In this research, HSC enhanced intestinal barrier function and integrity. Further, HSC could partially improve the expression of ZO-1, claudin-1, occludin and repair the epithelial barrier. It is possible to inhibit the migration of neutrophils and the release of S100A9 to repair the damage of the epithelial barrier. Therefore, we used fMLP to activate neutrophils to test this possibility. fMLP increased the levels of S100A9 in the supernatant of degranulation of neutrophils whereas HSC alleviated the release of S100A9 protein and the activation of neutrophil. Then we used the supernatant of fMLP-activated neutrophils to co-culture the intestinal epithelial cells and found that ZO-1, claudin-1, and occludin in intestinal epithelial cells was decreased. HSC could partially recover the expression of ZO-1, claudin-1, and occludin in intestinal epithelial cells. These results suggested that HSC alleviated the damage of the epithelial barrier via decreasing the release of S100A9 from neutrophils.
In summary, Fig. 9 summarizes the underlying mechanism of HSC on inhibiting inflammation and maintaining epithelium barrier integrity in the TNBS-induced colitis model. We discovered that HSC alleviated TNBS-induced colitis through restoration of impaired intestinal barrier function and neutrophil recruitment inactivation in rats. Furthermore, our findings suggest that HSC may be used to treat colitis, which is dependent on S100A9 release from neutrophils and mediated via MAPK/NF-κB signaling pathway. Blocking the binding of S100A9/NF-κB might provide useful targets to attenuating degranulation of neutrophil‐dependent tissue damage in acute inflammation. Therefore, HSC may have the potential to prevent and treat IBD in clinic in the future. However, further studies are needed to evaluate the potential use of HSC as a therapy to relieve IBD in a clinical context and to find the direct targets of HSC on neutrophil recruitment.
Fig. 9. The schematic model for the mechanism of action of HSC in the treatment of colitis.
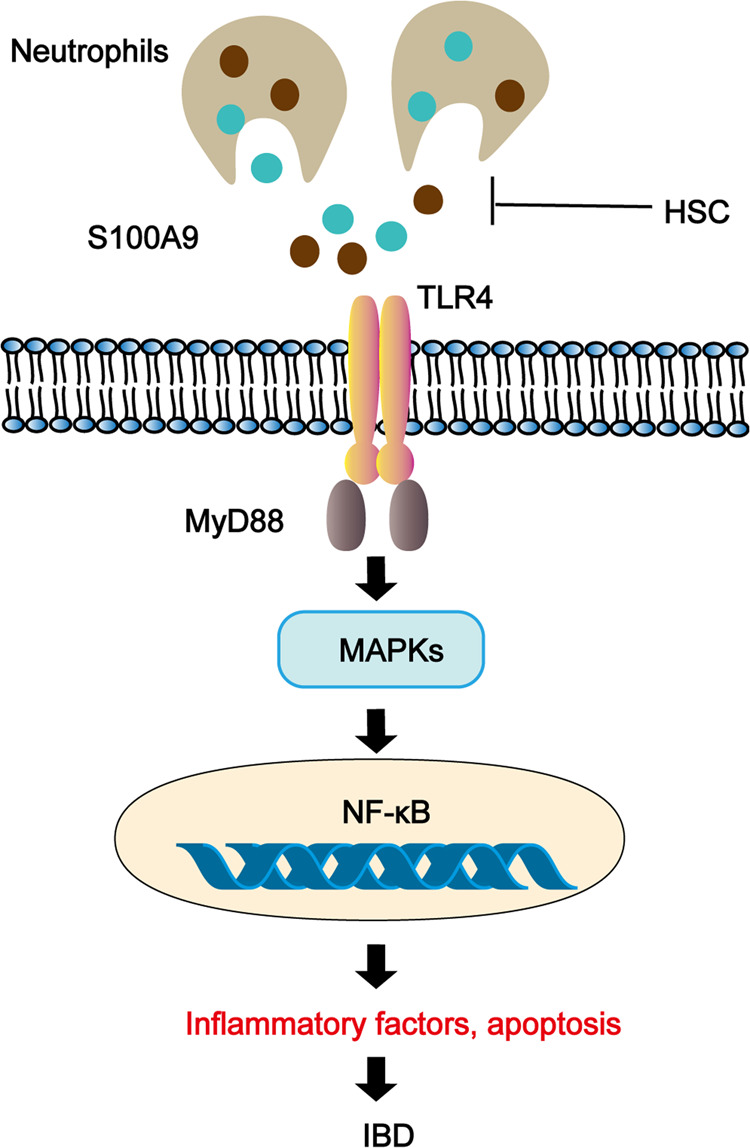
HSC could reduce the recruitment and activation of neutrophils, down-regulated S100A9, inhibit the signal molecular conduction of MAPK/NF-κB, and downregulate the secretion of inflammatory factors, inhibit epithelial cell apoptosis, and repair the epithelial barrier to alleviate colitis.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82073912), Suzhou Science and Technology Plan Project (SYS2019032), and a project funded by Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Author contributions
YLL and QMX designed the research. ZXZ, YL, KXW, and DL conducted the experiments. ZXZ, YL, YLZ, and YLL wrote the manuscript. GQX, QMX, YL, YLZ, and YLL revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Zheng-xia Zha, Yu Lin
References
- 1.Windsor JW, Kaplan GG. Evolving epidemiology of IBD. Curr Gastroenterol Rep. 2019;21:40. doi: 10.1007/s11894-019-0705-6. [DOI] [PubMed] [Google Scholar]
- 2.Flynn S, Eisenstein S. Inflammatory bowel disease presentation and diagnosis. Surg Clin North Am. 2019;99:1051–62. doi: 10.1016/j.suc.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Metzger JC, Kurz E, Von Spee-Mayer C, Kolck G, Bogumil A, Galle PR, et al. Chronic granulomatous disease as a rare differential diagnosis of inflammatory bowel disease. Z Gastroenterol. 2018;56:1507–12. doi: 10.1055/a-0732-5865. [DOI] [PubMed] [Google Scholar]
- 4.Zhou M, He J, Shen Y, Zhang C, Wang J, Chen Y. New frontiers in genetics, gut microbiota, and immunity: A rosetta stone for the pathogenesis of inflammatory bowel disease. Biomed Res Int. 2017;2017:8201672. doi: 10.1155/2017/8201672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebbo M, Crinier A, Vely F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol. 2017;17:665–78. doi: 10.1038/nri.2017.86. [DOI] [PubMed] [Google Scholar]
- 6.Pickard JM, Zeng MY, Caruso R, Nunez G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279:70–89. doi: 10.1111/imr.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94:155–65. doi: 10.1016/j.mayocp.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–53. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 9.Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124:3–20. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. 2016;17:564–80. doi: 10.1038/nrm.2016.80. [DOI] [PubMed] [Google Scholar]
- 11.Blander JM. Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J. 2016;283:2720–30. doi: 10.1111/febs.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coskun M. Intestinal epithelium in inflammatory bowel disease. Front Med. 2014;1:24. doi: 10.3389/fmed.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonovas S, Fiorino G, Allocca M, Lytras T, Nikolopoulos GK, Peyrin-Biroulet L, et al. Biologic therapies and risk of infection and malignancy in patients with inflammatory bowel disease: A systematic review and network meta-analysis. Clin Gastroenterol Hepatol. 2016;14:1385–97.e10. doi: 10.1016/j.cgh.2016.04.039. [DOI] [PubMed] [Google Scholar]
- 14.Xu HC, Wu B, Ma YM, Xu H, Shen ZH, Chen S. Hederacoside-C protects against AGEs-induced ECM degradation in mice chondrocytes. Int Immunopharmacol. 2020;84:106579. doi: 10.1016/j.intimp.2020.106579. [DOI] [PubMed] [Google Scholar]
- 15.Akhtar M, Shaukat A, Zahoor A, Chen Y, Wang Y, Yang M, et al. Hederacoside-C inhibition of staphylococcus aureus-induced mastitis via TLR2 & TLR4 and their downstream signaling NF-kappaB and MAPKs pathways in vivo and in vitro. Inflammation. 2020;43:579–94. doi: 10.1007/s10753-019-01139-2. [DOI] [PubMed] [Google Scholar]
- 16.Kang NX, Zhu YJ, Zhao JP, Zhu WF, Liu YL, Xu QM, et al. Antischistosomal activity of hederacochiside C against schistosoma japonicum harbored in experimentally infected animals. J Asian Nat Prod Res. 2017;19:402–15.. doi: 10.1080/10286020.2016.1208181. [DOI] [PubMed] [Google Scholar]
- 17.Joh EH, Jeong JJ, Kim DH. Kalopanaxsaponin B inhibits LPS-induced inflammation by inhibiting IRAK1 Kinase. Cell Immunol. 2012;279:103–8. doi: 10.1016/j.cellimm.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Requena P, Daddaoua A, Martinez-Plata E, Gonzalez M, Zarzuelo A, Suarez MD, et al. Bovine glycomacropeptide ameliorates experimental rat ileitis by mechanisms involving downregulation of interleukin 17. Br J Pharmacol. 2008;154:825–32. doi: 10.1038/bjp.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H, Yang J, Zhang W, Wang Q, Du Y, Sun Q, et al. Characterisation of hederacoside C metabolites using ultrahigh-performance liquid chromatography quadrupole orbitrap mass spectrometry based on automatic fragment ion search. Phytochem Anal. 2020;31:395–407. doi: 10.1002/pca.2908. [DOI] [PubMed] [Google Scholar]
- 20.Peeters L, Beirnaert C, Van der Auwera A, Bijttebier S, De Bruyne T, Laukens K, et al. Revelation of the metabolic pathway of hederacoside C using an innovative data analysis strategy for dynamic multiclass biotransformation experiments. J Chromatogr A. 2019;1595:240–7. doi: 10.1016/j.chroma.2019.02.055. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Zha Z, Shen W, Li D, Kang N, Chen Z, et al. Anemoside B4 ameliorates TNBS-induced colitis through S100A9/MAPK/NF-kappaB signaling pathway. Chin Med. 2021;16:11. doi: 10.1186/s13020-020-00410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan Q, Li D, Xiong L, Chang Z, Xu G. SILAC Quantitative proteomics and biochemical analyses reveal a novel molecular mechanism by which ADAM12S promotes the proliferation, migration, and invasion of small cell lung cancer cells through upregulating hexokinase 1. J Proteome Res. 2019;18:2903–14. doi: 10.1021/acs.jproteome.9b00208. [DOI] [PubMed] [Google Scholar]
- 23.Liu P, Bian Y, Liu T, Zhong J, Zhong Y, Zhuang S, et al. Huai hua san alleviates dextran sulphate sodium-induced colitis and modulates colonic microbiota. J Ethnopharmacol. 2020;259:112944. doi: 10.1016/j.jep.2020.112944. [DOI] [PubMed] [Google Scholar]
- 24.Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Wei L, Wang J, Qin Z, Wang J, Lu Y, et al. Suppression colitis and colitis-associated colon cancer by anti-S100a9 antibody in mice. Front Immunol. 2017;8:1774. doi: 10.3389/fimmu.2017.01774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bressenot A, Salleron J, Bastien C, Danese S, Boulagnon-Rombi C, Peyrin-Biroulet L. Comparing histological activity indexes in UC. Gut. 2015;64:1412–8. doi: 10.1136/gutjnl-2014-307477. [DOI] [PubMed] [Google Scholar]
- 27.Wera O, Lancellotti P, Oury C. The dual role of neutrophils in inflammatory bowel diseases. J Clin Med. 2016;5:118. doi: 10.3390/jcm5120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J, Gao W, Cai X, Xu J, Zou D, Li Z, et al. Nanozyme-mediated catalytic nanotherapy for inflammatory bowel disease. Theranostics. 2019;9:2843–55. doi: 10.7150/thno.33727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou G, Yu L, Fang L, Yang W, Yu T, Miao Y, et al. CD177+ neutrophils as functionally activated neutrophils negatively regulate IBD. Gut. 2018;67:1052–63.. doi: 10.1136/gutjnl-2016-313535. [DOI] [PubMed] [Google Scholar]
- 30.Akhtar M, Shaukat A, Zahoor A, Chen Y, Wang Y, Yang M, et al. Anti-inflammatory effects of Hederacoside-C on staphylococcus aureus induced inflammation via TLRs and their downstream signal pathway in vivo and in vitro. Micro Pathog. 2019;137:103767. doi: 10.1016/j.micpath.2019.103767. [DOI] [PubMed] [Google Scholar]
- 31.Tsukamoto H, Fukudome K, Takao S, Tsuneyoshi N, Kimoto M. Lipopolysaccharide-binding protein-mediated Toll-like receptor 4 dimerization enables rapid signal transduction against lipopolysaccharide stimulation on membrane-associated CD14-expressing cells. Int Immunol. 2010;22:271–80. doi: 10.1093/intimm/dxq005. [DOI] [PubMed] [Google Scholar]
- 32.Sands BE. Biomarkers of inflammation in inflammatory bowel disease. Gastroenterology. 2015;149:1275–85.e2. doi: 10.1053/j.gastro.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Xu X, Chen H, Zhu X, Ma Y, Liu Q, Xue Y, et al. S100A9 promotes human lung fibroblast cells activation through receptor for advanced glycation end-product-mediated extracellular-regulated kinase 1/2, mitogen-activated protein-kinase and nuclear factor-kappaB-dependent pathways. Clin Exp Immunol. 2013;173:523–35. doi: 10.1111/cei.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. 2018;9:1298. doi: 10.3389/fimmu.2018.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2020;182:1401–18.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nemeth T, Sperandio M, Mocsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. 2020;19:253–75. doi: 10.1038/s41573-019-0054-z. [DOI] [PubMed] [Google Scholar]
- 37.Mortaz E, Alipoor SD, Adcock IM, Mumby S, Koenderman L. Update on neutrophil function in severe inflammation. Front Immunol. 2018;9:2171. doi: 10.3389/fimmu.2018.02171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buechler C, Pohl R, Aslanidis C. Pro-resolving molecules-new approaches to treat sepsis? Int J Mol Sci. 2017;18:476. doi: 10.3390/ijms18030476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang LC, Wang Y, Tong LC, Sun S, Liu WY, Zhang S, et al. Berberine alleviates dextran sodium sulfate-induced colitis by improving intestinal barrier function and reducing inflammation and oxidative stress. Exp Ther Med. 2017;13:3374–82.. doi: 10.3892/etm.2017.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bian X, Yang L, Wu W, Lv L, Jiang X, Wang Q, et al. Pediococcus pentosaceus LI05 alleviates DSS-induced colitis by modulating immunological profiles, the gut microbiota, and short-chain fatty acid levels in a mouse model. Micro Biotechnol. 2020;13:1228–44. doi: 10.1111/1751-7915.13583. [DOI] [PMC free article] [PubMed] [Google Scholar]