

Abstract
Hydrogels are polymer networks infiltrated with water. Many biological hydrogels in animal bodies such as muscles, heart valves, cartilages, and tendons possess extreme mechanical properties including extremely tough, strong, resilient, adhesive, and fatigue-resistant. These mechanical properties are also critical for hydrogels’ diverse applications ranging from drug delivery, tissue engineering, medical implants, wound dressing, and contact lenses to sensors, actuators, electronic devices, and soft robots. Whereas numerous hydrogels have been developed over the last few decades, a set of general principles that can rationally guide the design of hydrogels using different materials and fabrication methods for various applications remain a central need in the field of soft materials. This review is aimed to synergistically report: i). general design principles for hydrogels to achieve extreme mechanical and physical properties, ii). unconventional polymer networks to implement these design principles, and iii). future directions for the orthogonal design of hydrogels to achieve multiple combined mechanical, physical, chemical, and biological properties. Since these design principles and implementation strategies are based on generic polymer networks, they are also applicable to other soft materials including elastomers and organogels. Overall, the review will not only provide a comprehensive discussion on the rational design of soft materials, but also provoke interdisciplinary discussions on a fundamental question: why does nature select soft materials with unconventional polymer networks to constitute the major parts of animal bodies?
1. Introduction
As hydrophilic polymer networks infiltrated with water1, hydrogels are the major components of animal bodies, constituting most of their cells, extracellular matrices, tissues, and organs. Not surprisingly, hydrogels have been widely used in biological and biomedical applications such as vehicles for drug delivery2–5, scaffolds for tissue engineering6–8, models for biological studies9–13, medical implants14,15, wound dressing16–18, and contact lenses15,19. More recently, intensive efforts have been devoted to exploring hydrogels’ emerging applications in devices and machines20 such as hydrogel sensors21–24, actuators25–28, electronic devices29–31, optical devices32–34, iontronic devices27,35, soft robots26,36,37, batteries38,39, super-capacitors40, adhesives41–43, and coatings44,45.
The mechanical properties of hydrogels are crucial to the survival and wellbeing of animals, and greatly affect the abovementioned applications of hydrogels. The pioneering works in the field of polymers and soft materials have laid the foundation for understanding the elasticity, swelling, poroelasticity, viscoelasticity, fracture and fatigue of hydrogels (e.g., Ref.46–59 and the references in them). However, the inverse question – how to design hydrogels that possess certain mechanical properties or certain properties in general – still poses a challenge in the field of polymers and soft materials59–62. This challenge becomes even more daunting, when one targets at hydrogels’ extreme mechanical properties, such as extremely high values of fracture toughness63, strength64,65, resilience66,67, interfacial toughness43, fatigue threshold68–70 and interfacial fatigue threshold71.
Despite the abovementioned grand challenge, the design of hydrogels with extreme mechanical properties is of both fundamental and practical importance. From the fundamental aspect, many biological hydrogels have achieved extreme mechanical properties necessary for their survival and well-being through evolution (Figure 1). For example, cartilage is a tough connective tissue that covers the surfaces of joints to provide reduced friction72. The human knee joint cartilage (i.e., articular cartilage) typically needs to sustain compressive stresses of 4–9 MPa for 1 million cycles per year, while maintaining high fracture toughness around 1,000 Jm−2 73. The high fracture toughness of articular cartilage is mainly attributed to its abundant strong collagen fibers interpenetrated with proteoglycan macromolecules, which provides both viscoelasticity and poroelasticity for mechanical dissipation74,75. The viscoelasticity of articular cartilage is mainly associated with local rearrangement of aggrecan, adhesive interactions of aggrecan, and reconfiguration of collagen75; the poroelasticity of articular cartilage is governed by the interstitial fluid movement through the porous extracellular matrix74. Tendon is a strong connective tissue that connects muscle to bone and muscle to muscle. The human patellar tendon can sustain a high tensile strength76, owing to its unique hierarchical fibrous structure that enables the simultaneous stiffening of bundles of collagen fibers before their tensile failure77,78. Heart valves generally possess both high resilience around 80% and high fracture toughness around 1,200 Jm−2 79,80, which are two seemingly contradictory properties. The elastin and crimped collagen fibers in the heart valve are elastic and non-dissipative under moderate deformation giving the heart valve the high resilience (Figure 2)81, whereas under large deformation, the stiffening and fracture of the collagen fibers dissipate substantial mechanical energy making the heart valve tough as well82. The adhesion of soft connective tissues on bones can be extremely fatigue-resistant. For example, the cartilage-bone interface in the human knee joint can sustain compressive stresses of 1 MPa along with an interfacial toughness around 800 Jm−2 over 1 million cycles of loads73,83. The fatigue-resistant adhesion of soft tissues (e.g., tendons, ligaments, and cartilages) to rigid bones is commonly achieved through nanostructured interfaces of aligned collagen nano-fibrils and ordered hydroxyapatite nanocrystals84–86. What are nature’s design principles, if any, for various biological hydrogels to achieve extreme mechanical properties? This is still a largely unanswered question, even in light of the pioneer works in the field of polymers and soft materials (e.g., Ref.46–58 and the references in them).
Figure 1. Biological hydrogels in the human body can possess extreme mechanical properties.

Aorta with tensile strength of 0.2–3.7 MPa73; heart valve with resilience above 80% and fracture toughness around 1,000 J m−2 79,80; tendon with tensile strength of 10–100 MPa96, fracture toughness of 20–30 kJ m−2 and fatigue threshold of 1,000 J m−2 97; skeletal muscle with fracture toughness around 2,490 J m−2 and fatigue threshold around 1,000 J m−2 73; articular cartilage with fracture toughness of 800–1,800 J m−2 98; and tendon/cartilage/ligament-bone interfaces with interfacial fatigue threshold around 800 J m−2 71,83.
Figure 2. Design principles and implementation strategies for various biological hydrogels to achieve extreme mechanical properties:
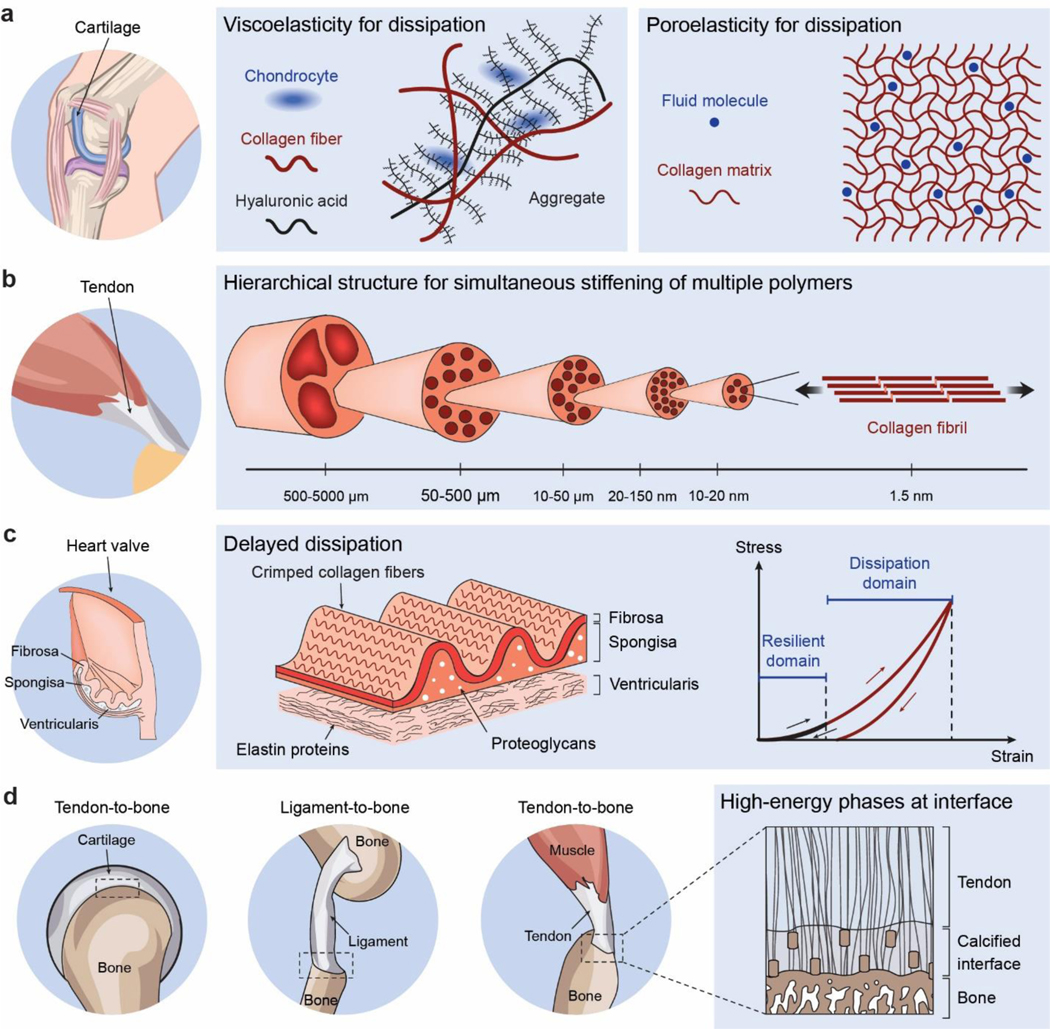
a. high toughness of cartilage due to viscoelastic and poroelastic dissipation of the polymer networks75,99,100, b. high tensile strength of tendon due to simultaneous stiffening of multiple polymers in the fibrous hierarchical structure77,96, c. high resilience and toughness of heart valve due to delayed mechanical dissipation101,102, d. high interfacial fatigue threshold of cartilage-/ligament/tendon-bone interfaces due to intrinsically high-energy phases including nano-crystals and nano-fibers strongly bonded on the interfaces71. a is adopted from Ref99. b is adopted from Ref77. c is adopted from Ref101,102. d is adopted from Ref71.
From the practical aspect, applications of hydrogels generally require the hydrogels to possess specific properties. For example, hydrogels designed with different moduli and viscoelastic properties have been used to regulate stem cell fate and activity10,12,84,87. The applications of hydrogels as artificial cartilages and spinal discs require the hydrogels to be fatigue-resistant under cyclic mechanical loading68,69,88,89. The mesh size of hydrogels’ polymer networks is critical to their applications in controlled drug delivery33,90,91. More recent applications of hydrogels as various devices require the hydrogel to possess specific properties, for instance, stimuli-sensitivity for hydrogel sensors and actuators37,92–94, strong adhesion for hydrogel coatings43, optical transparency for hydrogel optics33, electrical conductivity for hydrogel electronics30, and water absorption/release for hydrogel water harvesters95.
Over the last few decades, intensive efforts have led to the development of a plethora of hydrogels that possess extreme mechanical properties using diverse material candidates, including various natural and synthetic polymers, macro-/micro-/nano-fillers, and macro-/micro-/nano-fibers. Whereas the properties of these hydrogels are remarkable, their design often follows the Edisonian approach – trial and error with specific material candidates. As the field rapidly evolves, emerging applications of hydrogels in biomedicine pose escalating demands on the rationally-guided design of hydrogels beyond the Edisonian approach, requiring broad choices of material candidates and fabrication methods and achievements of multiple combined extreme properties. However, a set of general principles capable of rationally guiding the design of hydrogels using different materials and fabrication methods for various applications remain a central need in the field of soft materials. In this review, we aim to provide:
A set of general principles for the rational design of hydrogels to achieve extreme mechanical properties, including extremely high fracture toughness, tensile strength, resilience, interfacial toughness, fatigue threshold, and interfacial fatigue threshold; and extreme physical properties, including high electrical conductivity, patterned magnetization, high refractive index and transparency, tunable acoustic impedance, and self-healing. The design principles are generally based on fundamental mechanics and physics (beyond polymers) or inspired by biological hydrogels (e.g., muscles, cartilages, tendons, and heart valves) (Figure 3).
A set of unconventional polymer networks (UPNs) to implement the design principles discussed in i). using various material candidates and fabrication methods. The UPNs can be broadly categorized into: UPN architectures including ideal polymer networks, polymer networks with slidable crosslinks, interpenetrating and semi-interpenetrating polymer networks, polymer networks with high-functionality crosslinks, and nano-/micro-fibrous polymer networks; and UPN interactions including strong physical crosslinks, weak physical crosslinks, and dynamic covalent crosslinks (Figure 3).
A proposal of orthogonal design principles and synergistic implementation strategies for the design and fabrication of future hydrogels to achieve multiple combined mechanical, physical, chemical, and biological properties (Figure 3).
Figure 3.

This review summarizes the design principles and implementation strategies for soft materials including hydrogels, elastomers and organogels to achieve extreme properties.
Notably, since the aforementioned design principles and implementation strategies for hydrogels are based on generic polymer networks, they are also applicable to other soft materials comprised of polymer networks, including elastomers and organogels. In fact, many extreme mechanical and physical properties were first achieved in other soft materials than hydrogels. For example, high values of fracture toughness, tensile strength, resilience and interfacial toughness were realized in elastomers long before in hydrogels; ferromagnetic domains in soft materials were first programmed and 3D printed with elastomer inks as well.
The review is organized as the following. Section 2 will discuss common natural polymers, synthetic polymers, and permanent covalent crosslinks for hydrogels. Section 3 will introduce conventional polymer networks, and demonstrate that a number of mechanical properties of conventional polymer networks are coupled. Section 4 will define a set of unconventional polymer networks (UPNs), including both UPN architectures and UPN interactions, and then discuss that UPNs can provide decoupled mechanical properties. Thereafter, Sections 5 will systematically reveal the design principles for various extreme mechanical properties of hydrogels and implement strategies based on UPNs for each of the design principles; Sections 6 will briefly discuss the design principles and implementation strategies for hydrogels that possess a set of extreme physical properties. In Section 7, we will conclude the review by proposing orthogonal design principles and synergistic implementation strategies to design future hydrogels achieving multiple combined mechanical, physical, chemical and biological properties.
2. Polymers and crosslinks for hydrogels
A rich library of polymers and crosslinks have been used for the design and fabrication of various hydrogels. These polymers can be broadly categorized into natural polymers and synthetic polymers. In this section, we will briefly discuss the commonly-used natural polymers, synthetic polymers and permanent covalent crosslinks for hydrogels; we will discuss other types of crosslinks for hydrogels in Section 4.
2.1. Common natural polymers for hydrogels
Naturally derived polymers have been widely used to compose the polymer networks of hydrogels (Figure 4a). Hydrogels based on natural polymers usually possess biological properties compatible with extracellular matrices due to the similarity in their compositions. In addition, the natural polymer networks can often degrade in and be absorbed by the body through metabolism and tissue remodeling processes. Furthermore, the majority of natural polymers have reactive sites amenable to crosslinking and modification, which can endow the corresponding hydrogels with tailored biological and/or mechanical properties. In this subsection, we will briefly discuss a few natural polymers commonly-used for hydrogels. For more detailed discussions, a few classical reviews are recommended4–6,61,103.
Figure 4.

Chemical structures and schematics of typical examples of a. common natural polymers, b. common synthetic polymers, and c. permanent covalent crosslinks for hydrogels. R represents an organyl substituent or hydrogen.
Alginate.
Alginate is a polysaccharide which is usually obtained from brown-algae cell walls and two kinds of bacteria, Azotobacter and Pseudomonas104. Alginate is known to be a family of linear copolymers containing blocks of (1,4)-linked β-D-mannuronic (M) and α-L-guluronic acid (G) residues. The blocks are composed of consecutive G residues (GGGGGG), consecutive M residues (MMMMMM), and alternating M and G residues (GMGMGM)105. Alginate hydrogels can be formed with various covalent and physical crosslinks. In particular, the ionic crosslinks have been widely used for alginate hydrogels, because the G blocks106 (and GM blocks107) in alginate can be readily bound with one another by divalent cations such as Ca2+, Mg2+, Ba2+, and Sr2+ 108–110. The mechanical properties of alginate hydrogels can be easily tuned to match those of various tissues by changing different parameters, such as the molecular weight, polymer concentration, chemical modification, G/M ratio, and type or density of crosslinks106,111. Alginate hydrogels have been widely used as scaffolds in tissue engineering, such as intervertebral disk regeneration112, adipose tissue regeneration113, cardiac regeneration114, and liver regeneration115, since alginate allows the formation of hydrogels under physiological conditions and thus enables easy cell and drug encapsulation.
Hyaluronic acid.
Hyaluronic acid (also known as hyaluronan or hyaluronate) is a linear polysaccharide that consists of alternating units of a repeating disaccharide, β−1,4-D-glucuronic acid and β−1,3-N-acetyl-D-glucosamine116,117. Hyaluronic acid is present in all mammals, especially in various soft connective tissues, acting as a space filler, lubricant, and osmotic buffer118. Hyaluronic acid can be covalently crosslinked into hydrogels by various hydrazide derivatives119,120. The abundant carboxyl and hydroxyl groups on the polysaccharide structure of hyaluronic acid also offer many active sites for chemical modifications121. For example, hyaluronic acid can be modified with thiol122,123, haloacetate124, dihydrazide119,125, aldehyde126,127 and tyramine128 groups, which can react with corresponding covalent crosslinkers through addition or condensation reactions129. As another example, hyaluronic acid can also be modified by methacrylic anhydride or glycidyl methacrylate to possess reactive methacrylic groups, which can be polymerized by radical polymerization130–132. Owing to the naturally-derived, nonimmunogenic, biodegradable, and nonadhesive properties133–135, hyaluronic acid hydrogels have been widely used as scaffolds in cell therapy and tissue engineering, such as cell delivery136, molecule delivery137,138, stem cell therapy139,140, cartilage engineering137,141, cardiac repair142 and valvular engineering143.
Collagen.
Collagen is one of the major proteins in animal bodies. There are approximately 29 types of collagens discovered so far144. The structures of collagens can be defined at different levels, including primary structure (amino acid triplet), secondary structure (α-helix), tertiary structure (triple helix), and quaternary structure (fibril) 145,146. The primary structure of collagen is the tripeptide sequence of –(Gly– X–Y–)n–, where Gly is glycine, X, and Y are other amino acids than Gly. The sequence of the amino acids governs the peptide folding into a secondary structure, mainly left-handed α-helix, which is stabilized by the hydrogen bonds between amino acid residues147. Three left-handed secondary α- polypeptide chains then form a tertiary structure by the aldol condensation crosslinking, aldehyde amine condensation crosslinking, and aldol histidine crosslinking148. The triple strands can further self-assemble into a collagen fiber as the quaternary structure149.
Acid-solubilized collagens can self-assemble to form physically-crosslinked hydrogels when the collagen solutions are neutralized and heated. Since the physically-crosslinked collagen hydrogels are usually mechanically weak and thermally unstable150,151, they have been strengthened and stabilized with chemical crosslinks such as glutaraldehyde, genipin, carbodiimides or diphenylphosphoryl azide152–154. Collagens can be biodegraded by collagenases and metalloproteases; the crosslinked collagens usually have slower degradation rates than the uncrosslinked collagens155. Because collagens usually have low antigenicity, low inflammatory response, good biocompatibility and natural cell-adhesive motifs156–158, collagen hydrogels have been widely used as scaffolds for drug and protein delivery159,160 and reconstructions of liver161, skin162, blood vessel163, and small intestine164, cartilage165, vocal cord166, and spinal cord167.
Gelatin.
Gelatins are naturally derived polymers obtained through breaking the triple-helix conformation of collagens into single-strand molecules. There are two types of gelatins, type A and type B, which are obtained with acid and alkaline treatments of collagens, respectively168. Gelatins can be physically crosslinked by simply reducing the temperature of aqueous solutions of gelatins below a certain temperature169,170. The physically-crosslinked gelatins are usually unstable for long-term biomedical applications under physiological conditions. To further stabilize the physically-crosslinked gelatin hydrogels, covalent crosslinkers171 such as aldehydes (e.g., formaldehyde, glutaraldehyde, and glyceraldehyde)172,173, polyepoxides174 and isocyanates175 have been widely used to react with and bridge the free amine groups (from lysine and hydroxylysine) or free carboxylic acid (from glutamic and aspartic acid) on the gelatin molecules. Besides the introduction of covalent crosslinkers, the gelatin backbones can also be modified by methacrylates to form covalently-crosslinkable gelatin methacryloyl hydrogels176. In addition, synthetic polymers can also be coupled on gelatin chains through grafting-from177, grafting-to178, and grafting-through179 methods and enhance the mechanical properties of gelatin hydrogels. Furthermore, the gelatin molecules tend to form physical interactions with various dopants, such as carbon nanotubes180, graphene oxide181, and inorganic nanoparticles or minerals182,183. The aforementioned covalent crosslinks, modifications and/or interactions can significantly improve the mechanical properties of gelatin hydrogels172,184. The easy gelation process and the excellent biocompatibility make gelatin hydrogels attractive for biomedical applications, such as drug delivery185 and tissue engineering186,187.
Fibrin.
Fibrin is a naturally-derived polymer obtained from thrombin-treated fibrinogen188. Fibrin is involved in the natural wound healing process by forming extensive fibrous networks. Fibrin can form clots or hydrogels when mixing fibrinogen and thrombin solutions at room temperature189. The resultant fibrin hydrogels usually have weak mechanical properties due to the nature of physical crosslinks. To improve the mechanical properties of fibrin hydrogels, chemical crosslinkers such as genipin can be introduced to crosslink the amine residues on fibrin proteins and form stable covalently-crosslinked networks190. In addition, fibrin hydrogels can also be combined with synthetic polymers such as polyurethane191, polycaprolactone192, b-tricalciumphosphate193 or polyethylene glycol194 to enhance the mechanical strength of the hydrogels. Fibrin hydrogels have been widely used as sealants and adhesives to control bleeding in surgery195, and as scaffolds for cardiac tissue engineering196, neurological regeneration197, ocular therapy198, cartilage and bone reparation199,200, muscle cells engineering201, and exogenous delivery in wound healing202. In particular, fibrin hydrogels can be produced autologously from a patient’s blood, thereby reducing the risk of foreign-body reactions203.
Agarose.
Agarose is a neutral polysaccharide composed of β-D-galactopyranosyl and 3,6-anhydro-α-L-galactopyranosyl, mainly extracted from red algae (Rhodophyceae)204. As a thermoresponsive polymer, agarose can be heated to dissolve in water, and then cooled down to form a hydrogel. During this gelation process, the agarose structure changes from a random-coil configuration to bundles of associated double helices with multiple-chain aggregation in the junction zone205,206. The gelling temperature and mechanical properties of agarose hydrogels can be tuned by changing the concentration, molecular weight, and structure of the agarose in the hydrogels207,208. Agarose hydrogels have been used as scaffolds for cell encapsulation209, cartilage reparation210 and nerve regeneration211, due to its low immunoreaction in human bodies212. Notably, since the native agarose does not possess cell adhesion motifs, cell adhesion peptides have been covalently conjugated to the agarose backbone to enhance the interactions between cells and agarose hydrogels213.
Chitosan.
Chitosan is a linear polysaccharide composed of β-(1–4)-linked D-glucosamine and N-acetyl-D-glucosamine units, which is mainly prepared by partial deacetylation of chitin (obtained from crab and shrimp shells) to less than 40% of N-acetyl-D glucosamine residues214,215. The physical, chemical and biological properties of chitosan materials are highly related to the molecular weight and the degree of deacetylation216,217. Chitosan can form physically-crosslinked hydrogels by hydrophobic interaction, hydrogen bonding218,219, metal coordination (with metal ions such as Pt(II), Pd(II), Mo(VI)220,221), and electrostatic interaction (with multivalent anions such as sulfates, citrates and phosphates ions222,223; with anionic polyelectrolytes214 such as polysaccharides224,225, proteins226,227 and synthetic polymers228). These physically-crosslinked chitosan hydrogels usually have weak mechanical properties and short lifetime, which is also highly influenced by pH, temperature and environments215,229. To enhance the mechanical properties of chitosan hydrogels, covalent crosslinkers have been introduced into the hydrogels. The commonly-used covalent crosslinkers include di-aldehydes230,231, formaldehyde232, di-glycidyl ether233 and genipin234,235, which can react with the residual functional groups (such as OH, COOH, and NH2) on chitosan backbones to form the amide bonds, ester bonds, and Schiff base linkages218,236,237. In addition, chitosan can also be modified with methacrylate or aryl azide groups to form photo-crosslinkable macromers238. The gelation degree and mechanical properties of these chitosan hydrogels can be controlled by UV irradiation time and intensity239–241. Furthermore, chitosan hydrogels can be modified with biofunctional ligands such as Arg-Gly-Asp (RGD) peptides to facilitate cell adhesion and proliferation242,243. Chitosan hydrogels have been widely used in biomedical applications such as drug delivery244, cell encapsulation245, neural tissue engineering246 and bone regeneration245, owing to their excellent biocompatibility and biodegradability247.
Cellulose.
Cellulose is the most abundant natural polysaccharide, and the main constituent of plants and natural fibers such as cotton and linen248–250. Some bacteria such as acetobacter xylinum are also able to produce cellulose251. Cellulose has a chemical composite of 1,4-β-glucosidic linked glucose units, which results in high crystallinity (over 40%) and difficulty in dissolving in water and other common solvents252. Solvents such as N-methylmorpholine-N-oxide253,254, ionic liquids255,256, and alkali/urea (or thiourea) aqueous257,258 systems have been developed to dissolved native cellulose. Cellulose can also be modified through partly esterification or etherification of the hydroxyl groups on the backbones248. These cellulose derivatives, including methyl cellulose259, hydroxypropyl cellulose260, hydroxypropylmethyl cellulose261,262 and carboxymethyl cellulose263 are easier to dissolve and process compared to the native cellulose.
Cellulose and its derivatives can be chemically crosslinked to form stable three-dimensional networks. Bifunctional or multifunctional molecules, such as 1,2,3,4-butanetetracarboxylic dianhydride264, succinic anhydride265, citric acid266, epichlorohydrin267, ethylene glycol diglycidyl ether268, and divinyl sulfone269 can form covalent ester or ether bonds between cellulose chains. Cellulose chains can also be covalently crosslinked by the irradiation of electron beam and gamma rays270,271, which avoids the usage of toxic crosslinkers and allows the simultaneous sterilization of the resultant hydrogels. Cellulose and its derivatives can also be blended with natural polymers, such as chitosan272, starch273, alginates274 and hyaluronic acid275, or synthetic polymers such as polyethylene glycol276, polyvinyl alcohol277 and poly(N,N-dimethylacrylamide)278 to form interpenetrating polymer networks with excellent mechanical properties. Notably, bacterial cellulose produced from certain bacterial species such as acetobacter xylinum can directly form cellulose hydrogels with high purity and tensile strength279,280. Since cellulose-based hydrogels are proven to have superior hydrophilicity, biodegradability, biocompatibility, and transparency, they have been widely used in drug delivery281, tissue engineering282, blood purification283, strain sensor284 as well as water purification285.
2.2. Common synthetic polymers for hydrogels
In addition to the natural polymers, synthetic polymers have been widely used for the design and fabrication of hydrogels (Figure 4b). The synthetic polymer networks of hydrogels are commonly formed by copolymerization of monomers for the polymer backbones and crosslinkers, or by reactions of synthetic polymers, macromers and/or crosslinkers.
Poly(acrylic acid).
Poly(acrylic acid) (PAA) is a linear polymer prepared by radical polymerization of acrylic acid monomers. The backbone of PAA contains a large number of carboxyl groups. PAA can form hydrogels through covalent and physical crosslinking. Covalently-crosslinked PAA hydrogels are usually formed by copolymerization of di-/multi-vinyl crosslinkers together with acrylic acid monomers286. In addition, the carboxyl groups of PAA can form physical interactions with various doping agents such as clay287, graphene oxide288 and cations289, which can act as physical crosslinks for PAA hydrogels; the carboxyl groups can also form hydrogen bonds between PAA chains and introduce self-healing or self-adhesive properties to PAA hydrogels290. Furthermore, the abundant carboxyl groups on PAA can associate with water molecules to facilitate the absorption of water by PAA hydrogels291. Since the carboxyl groups are sensitive to pH and ionic strength, the equilibrium swelling ratio of PAA hydrogels is affected by the pH and ionic strength of the solutions for the hydrogels292,293. PAA hydrogels can also incorporate other linear polymers, such as biological polymers, to form various adhesives and hydrogels for biomedical applications41,294.
Poly(2-hydroxyethyl methacrylate).
Poly(2-hydroxyethyl methacrylate) (PHEMA) hydrogels can be prepared by free-radical polymerization of 2-hydroxyethyl methacrylate (HEMA) monomers with covalent crosslinkers such as trimethylene glycol dimethacrylate (TEGDMA), initiators such as sodium pyrosulfite (SMBS), and catalysts such as ammonium persulfate (APS). The HEMA monomers can also be copolymerized with acrylic or acrylamide monomers to control the swelling and mechanical properties of the resultant hydrogels295. PHEMA hydrogels are optically transparent and mechanically stable in the physiological environments. Pure PHEMA hydrogels are also resistant to cell adhesion and difficult to degrade in the physiological environments; however, various biofunctional and bioactive motifs can be coupled onto the hydrogels to improve their cell interactions and degradability296,297. PHEMA hydrogels are famous for their ophthalmic applications such as contact lens298 and artificial cornea299.
Poly(vinyl alcohol).
Poly(vinyl alcohol) (PVA) is mainly obtained from the partial hydrolysis of poly(vinyl acetate)300. PVA can form stable and elastic hydrogels through either physical or covalent crosslinking301,302. The physically-crosslinked PVA hydrogels are commonly obtained by repeated freezing and thawing of PVA solutions303, which gives elastic, tough, strong and fatigue-resistant PVA hydrogels69,71,304. PVA can also be covalently crosslinked through the use of difunctional crosslinkers such as glutaraldehyde, acetaldehyde, formaldehyde, epichlorohydrin, and other monoaldehydes305,306. Electronbeam and gamma irradiation can also crosslink PVA to avoid residual covalent crosslinkers in the hydrogels307. Pure PVA hydrogels are nonadhesive to cells, but several oligopeptide sequences can be conjugated onto the backbones of PVA hydrogels to enhance their cellular interactions308. PVA hydrogels have been extensively studied and used in biomedical applications309,310, such as articular cartilage replacement and regeneration311,312.
Poly(ethylene glycol) or poly(ethylene oxide).
Poly(ethylene glycol) (PEG) is usually obtained from the anionic or cationic polymerization of ethylene oxide. When the PEG has a molecular weight more than 10 kDa, it is also named poly(ethylene oxide) (PEO) since the end groups are negligible313. There are various methods to crosslink PEG polymers into hydrogels. The ends of PEG chains can be modified by unsaturated groups, such as acrylate or methacrylate ends, and then be used as macro-crosslinkers to form hydrogels with other unsaturated monomers by the photo-/UV-induced radical polymerization314,315. PEG can also form hydrogels by electron beam irradiation via radiation-induced free radical processes316. Furthermore, the end groups of the PEG chain can be modified with various reactive pairs, such as Nhydroxysuccinimide/NH2317, maleimide/thiol318, and acetylene/azide319. Since these functional chain-end motifs usually have high reaction efficiency and fast reaction kinetics, the obtained hydrogels by the coupling reactions of these groups can give relatively well-defined network architectures67.
PEG polymers can also form physically-crosslinked networks. Similar to the chemical crosslinking method, the ends of PEG chains can be modified with various motifs for physical crosslinking. For example, nucleobase pairs of adenines and thymines320, ureido-pyrimidinone (UPy) units321, or host-guest molecules322 can be introduced onto the chain-ends of PEG molecules to prepare physically-crosslinked PEG hydrogels. These physically-crosslinked PEG hydrogels can exhibit switchable, self-healable or stimuli-responsive properties and high mechanical strength323. Besides the modification and utilization of chain-end groups, physically-crosslinked PEG hydrogel can also be prepared by using PEG block copolymers324. PEG-b-PPG (poly(propylene glycol)) is one of the most widely used PEG-derived block copolymers to prepare thermo-responsive physical hydrogels325. These physical hydrogels are formed by the hydrophobic interaction of PPG blocks. The phase transition behavior of these hydrogels can be optimized by balancing the hydrophobic PPG block and the hydrophilic PEG block. Based on the same gelation mechanism, PEG block copolymers with poly(DL-lactic acid) (PDLLA)326, poly(dl-lactic acid-coglycolic acid) (PLGA)327,328, polylactide (PLA)329, poly(caprolactone)(PCL)330 and poly(propylene sulfide)(PPS)331 can also form physically-crosslinked hydrogels with injectable or stimuli-responsive properties. PEG, as well as its derivatives, are widely used in biomedical applications due to their non-toxic and non-immunogenic properties332. While the inert biological property of PEG hydrogels can prevent undesired interactions between native PEG hydrogels and cells333,334, PEG hydrogels can also be modified with various bioactive conjugations such as growth factors 335 and cell-adhesive peptides336 through Michael-type addition337,338or click chemistry334. PEG hydrogels with these bioactive molecules can facilitate their biomedical applications339 such as drug or cell delivery340,341 and tissue engineering342.
Poly (N-isopropylacrylamide).
Acrylamide and its derivatives have been widely used to prepare hydrogels by radical copolymerization with crosslinkers. One interesting hydrogel based on acrylamide and its derivatives is the poly(N-isopropylacrylamide) (PNIPAm) hydrogel. Uncrosslinked linear PNIPAm exhibits a coil-to-globule phase transition in aqueous solutions when the temperature is raised above a critical temperature343,344. The PNIPAm can be covalently crosslinked by crosslinkers such as bis-acrylamide derivatives through the radical polymerization process. The crosslinked PNIPAm hydrogels also possess the reversible thermo-responsive behavior with a critical temperature of around 34 °C 345, above which the hydrogel structure will collapse and exude water346,347. While the thermo-responsive behavior of PNIPAm hydrogels is usually slow, many studies have improved the phase-transition speed of PNIPAm hydrogels by incorporating porous structures during the hydrogel formation348,349. The thermo-responsive PNIPAm hydrogels can be used as actuators for soft robotics350, injectable scaffolds for tissue engineering351, and on-demand detachment of cell sheets352,353.
Silicone.
Silicone hydrogels are hydrogels that contain silicone polymers as one of its polymer components354. Silicone polymers are commonly hydrophobic355. In order to form silicone hydrogels, hydrophilic monomers and/or polymers have been introduced into the silicone matrix by blending or copolymerization to improve the hydrophilicity of silicone hydrogels356,357. For example, hydrophilic polymers such as PHEMA can be blended directly into the silicone polymer matrix, forming a hydrophilic interpenetrating polymer network358. Hydrophilic monomers such as N-vinylpyrrolidone (NVP) can be copolymerized with silicon-macromers to form hydrophilic silicone hydrogels359. Hydrophilic polymer segments such as PEG360 can also be copolymerized onto silicone segments to form block-modified361,362 or graft-modified363 hydrophilic silicone hydrogels. Since these hydrophilic silicone hydrogels usually have excellent gas permeability as well good biocompatibility, they have been used in biomedical applications such as contact lenses364,365, histological engineering materials366,367, and drug-delivery carriers368,369.
2.3. Common permanent covalent crosslinks for hydrogels
In this section, we will discuss permanent covalent crosslinks that are commonly used in hydrogels (Figure 4c); we will discuss other types of crosslinks in Section 4. The energy of permanent covalent crosslinks ranges from 220 kJ/mol to 570 kJ/mol (Figure 5)370–372.
Figure 5.

Bond energies of various types of permanent covalent crosslinks370–372, weak physical crosslinks 373–376, and dynamic covalent crosslinks372,377–381.
Carbon-carbon bonds.
The energy of the carbon-carbon bond is around 300 to 450 kJ/mol370–372. Hydrogels covalently crosslinked by carbon-carbon bonds are usually formed by radical copolymerization of monomers and di-/multi-vinyl crosslinkers. The crosslinkers can be small molecules with two double bonds such as N,N’-Methylenebisacrylamide (MBA) or macromolecules with several acrylate groups6,382. These crosslinkers are compatible with various initiation and polymerization systems4,237,383. For example, photo-radical initiators can be added into the pre-polymerization solution together with monomers and di/multi-vinyl crosslinkers302,384,385. Once the initiator is irradiated by UV light, radicals will be generated to initiate the polymerization of the double bonds on monomers as well as crosslinkers386,387. As a result, hydrogels can be formed in-situ and with patterned structures or biology functions384,388. The polymerization of vinyl monomers and crosslinkers can also be carried out with a system composed of peroxydisulfate and N,N,N′N′-tetramethylene-diamine (TEMED), where TEMED can accelerate the decomposition of peroxydisulfate to generate a large number of radicals389. This initiation and polymerization system can effectively and rapidly form various hydrogels under room temperature.
The carbon-carbon crosslinks of hydrogels can also be formed by high-energy irradiation (e.g., gamma and electron beams). Similar to UV light, high-energy radiation can be used to polymerize unsaturated compounds such as monomers and crosslinkers with vinyl groups or acrylate groups390,391. High-energy radiation can also crosslink polymers without unsaturated bonds392, because radicals can be generated from the homolytic scission of the polymer chains under high-energy radiation. The radiolysis of water molecules in the solvent can also generate hydroxyl radicals that attack polymer chains to form macroradicals393. These radicals can then undergo recombination and termination to form covalent polymer networks crosslinked by carbon-carbon bonds.
Carbon-nitrogen bonds.
The energy of the carbon-nitrogen bond is around 300 to 430 kJ/mol370,372. Hydrogels covalently crosslinked by carbon-nitrogen bonds are usually formed by highly effective chemical reactions of complementary groups. For example, the amide bonds have been widely used as the covalent crosslinks for hydrogels by the condensation reactions between amines with carboxylic acids and derivatives394. N-hydroxysuccinimide (NHS) and N,N-(3-dimethylaminopropyl)-N-ethyl carbodiimide (EDC) are widely used to facilitate the condensation reaction of amines with carboxylic acids67. The addition of NHS and EDC will also suppress possible side-reactions and give better control of the crosslink density in the hydrogels395. The carbon-nitrogen bonds can also be formed through the addition reactions of amines with electrophiles such as adipic acid dihydrazide and diisocyanates crosslinkers6,103,396. These di-functional crosslinkers have been widely used to crosslink natural macromolecules due to the high reaction efficiency. The mechanical properties of the resultant hydrogels can be controlled by tuning the concentration and ratio of the polymers and the crosslinking agents. Another category of reactions that can form carbon-nitrogen crosslinks for hydrogels is the azide-alkyne cycloaddition reaction, which is one typical click reaction to connect alkyne and azide into triazole. The click reaction has high efficiency without side reactions397. Furthermore, the azide-alkyne cycloaddition can be conducted in the absence of metal catalysis398, expanding the applicability of the azide-alkyne cycloaddition for preparing biocompatible hydrogels.
Carbon-oxygen bonds.
The energy of the carbon-oxygen bond is around 280 to 370 kJ/mol370–372. The most common carbon-oxygen bond is the ester bond formed by the reactions between hydroxyl groups and carboxylic acids or derivatives399. Such ester crosslinks can be hydrolyzed easily and make the hydrogels degradable under ambient temperature and physiological conditions. Besides the ester crosslinks, the carbon-oxygen bonds are also present in ether groups and urethane groups, which can become crosslinks due to the reaction between side groups on polymers (such as hydroxyl groups on polysaccharides or PVA) and reactive crosslinkers (such as glutaraldehyde400,401, divinyl sulfone402, dibromide403 or diisocyanate404).
Carbon-sulfide bonds.
The energy of the carbon-sulfide bond is around 220 to 310 kJ/mol372. The covalent crosslinking of hydrogels through carbon-sulfide bonds are mainly formed by the thiol-click reactions405,406. The inherent electron density of the sulfide atom makes thiols prone to react with many functional groups through a radical or catalyzed process407,408. Thiol groups can be easily converted into nucleophilic thiolates or electrophilic thiyl radicals, which then proceed with nucleophilic reactions or radical chain processes to achieve the thiol-click reactions409. Specifically, for the radical thiol-click reactions, the thiol group can be activated by heat and/or UV light to generate radicals that initiate the radical-mediated thiol-ene or thiol-yne reactions410; for the nucleophilic thiol-click reactions initiated by strong bases, the thiol groups can readily react with electron-poor ene-functional compounds through the Michael addition, with isocyanates derivatives through carbonyl addition, with halide through SN2 nucleophilic substitution, and with epoxies motifs through SN2 ring-opening reactions411–414. The thiol-click reactions commonly have high efficiency and high conversion rate without any side products, even in the presence of water, ions, and oxygen. The thiol-click reactions have been extensively used to prepare hydrogels for various biomedical applications415,416.
Silicon-oxygen bonds.
The energy of the silicon-oxygen bond is around 420 to 570 kJ/mol370–372. The silicon-oxygen bonds are mainly used in the formation of silicone-based hydrogels360,417,418, and can usually enhance the mechanical properties of the silicone-based hydrogels419. In addition, silicon-oxygen bonds have been widely used to form strong bonding between hydrogels and diverse engineering materials with modified surfaces such as salinized surfaces43.
3. Conventional polymer networks
3.1. Conventional polymer networks in the dry state
As illustrated in Figure 6, a conventional polymer network is defined as long-chain polymers crosslinked via permanent covalent bonds into a network, in which entanglements and reversible crosslinks of the polymer chains are negligible47,50,55. Conventional polymer networks have provided the basic models for the development of unentangled rubber elasticity including the affine network model and the phantom network model47,50,55. Conventional polymer networks have also been widely adopted in synthetic hydrogels, although biological hydrogels (Figures 1 and 2) usually reply on more complex polymer networks.
Figure 6.

Schematics of a conventional polymer network a. in the dry state and b. covalently bonded on a substrate, and c. in the swollen state and d. covalently bonded on a substrate.
In the dry state (Figure 6a), a conventional polymer network contains n polymer chains per unit volume, where a chain is defined as the segment of polymer between two successive covalent crosslinks. Each polymer chain contains N Kuhn monomers, and the length of each Kuhn monomer is b. The end-to-end distances of a polymer chain at the relaxed and fully stretched states are and Nb, respectively. Therefore, the stretch limit of polymer chains in the dry polymer network can be calculated as47,50,55
| (1) |
The stretch limit of the bulk polymer network scales with , and the pre-factor of the scaling relation depends on the polymer network architecture54.
Assuming that the dry polymer network follows the affine network model, the shear modulus of the network under initial deformation can be expressed as47,50,55,
| (2) |
where k is the Boltzmann constant and T is the absolute temperature.
Following the Lake-Thomas model49, the fracture toughness of the dry polymer network is its intrinsic fracture energy , which is the energy required to fracture a single layer of polymer chains per unit area,
| (3) |
where is the number of polymer chains per unit area, is the energy required to fracture a polymer chain, and is the energy required to fracture a single Kuhn monomer.
Also based on the Lake-Thomas model48,49, the fatigue threshold of the dry polymer network is the intrinsic fracture energy . If the dry polymer network is covalently bonded on a substrate (Figure 6b), both the interfacial toughness and the interfacial fatigue threshold of the adhesion are on the level of as well43,71,420.
By substituting the typical ranges of b, N, n, kT and Uf into Eqs. 1–3, we can estimate that the shear modulus G can be on the order of kilopascals to megapascals, the chain stretch limit can reach up to a few tens (without chain entanglement), and the intrinsic fracture energy can reach up to a few hundreds of joule per meter squared55.
The mechanical properties of the dry polymer network are coupled with one another. It is commonly assumed that the polymer chains occupy the major volume of the polymer network in the dry state, and therefore the volume conservation of the polymer network gives
| (4) |
where v is the volume of a Kuhn monomer.
By substituting Eq. 4 into Eqs. 1–3, we can express the chain stretch limit , shear modulus G and intrinsic fracture energy of a conventional polymer network in the dry state as functions of its polymer-chain length N,
| (5) |
From Eq. 5, it is evident that enhancing the polymer-chain length N increases the chain stretch limit and intrinsic fracture energy but decreases the shear modulus G of the conventional polymer network in the dry state. These mechanical properties of the conventional polymer network in the dry state are coupled through the following relation,
| (6) |
3.2. Conventional polymer networks in the swollen state
A dry conventional polymer network with the physical parameters discussed in Section 3.1 can imbibe water and swell into a hydrogel composed of the conventional polymer network and water (Figure 6c). The swelling of the dry polymer network stretches polymer chains in the network by a ratio of λs, named the chain stretch of swelling.
Since the swelling of the dry polymer network stretches its polymer chains by a ratio of λs, the end-to-end distance of a polymer chain in the hydrogel at the relaxed and fully stretched states are and Nb, respectively. Therefore, the stretch limit of polymer chains in the hydrogel can be calculated as,
| (7) |
The stretch limit of the bulk hydrogel scales with , and the pre-factor of the scaling relation depends on the polymer network architecture54.
The swelling of the dry polymer network reduces its shear modulus by a ratio of λs47. Therefore, the shear modulus of the hydrogel under initial deformation can be expressed as,
| (8) |
Note that n in Eq. 8 is the number of polymer chains per unit volume of the dry polymer network.
The swelling of the dry polymer network reduces the number of polymer chains per unit area by a ratio of but does not significantly change the energy required for fracturing a polymer chain in the network. Therefore, the intrinsic fracture energy of the hydrogel can be calculated as,
| (9) |
The fracture toughness and fatigue threshold of a hydrogel with the conventional polymer network are the hydrogel’s intrinsic fracture energy 48,49. If the hydrogel’s polymer network is covalently bonded on a substrate (Figure 6d), both the interfacial toughness and interfacial fatigue threshold of the adhesion are on the order of the hydrogel’s intrinsic fracture energy as well43,71,420.
By comparing Eqs. 1–3 and Eqs. 7–9, we can see that swelling the dry polymer network into the hydrogel reduces the stretch limit , shear modulus G and intrinsic fracture energy of the dry network by factors of λs, λs and , respectively47,50,55. By substituting the typical ranges of λs, b, N, n, kT and Uf into Eqs. 7–9, we estimate that the shear modulus G of the hydrogel with the conventional polymer network can be on the order of pascals to megapascals, the chain stretch limit can reach up to a few times (without chain entanglement), and the intrinsic fracture energy can reach a few tens of joule per meter square.
By substituting Eq. 4 into Eqs. 7–9, we can express the chain stretch limit , shear modulus G and intrinsic fracture energy of the hydrogel with the conventional polymer network as functions of its polymer-chain length N,
| (10) |
From Eq. 10, it is evident that enhancing the polymer-chain length N increases the chain stretch limit and the intrinsic fracture energy but decreases the shear modulus G of the hydrogel with the conventional polymer network. These mechanical properties of the hydrogel are coupled through the following relation,
| (11) |
Notably, the chain stretches due to equilibrium swelling of a conventional polymer network can be calculated. Without loss of generality, let consider a dry conventional polymer network with a cubic shape. When the polymer network reaches the equilibrium state in water, one side of the cube increases its length from the dry state by a ratio of . At the equilibrium state, the Helmholtz free energy for stretching polymer chains and for mixing polymers and water per unit volume of the dry polymer network can be expressed as47,421,
| (12a) |
| (12b) |
where χ is the Flory polymer-solvent interaction parameter and vs is the volume of a solvent molecule. Subsequently, the Helmholtz free energy per unit volume of the dry polymer network can be expressed as47,421,
| (13) |
When the polymer network reaches the equilibrium state in water, minimizes the Helmholtz free energy47,421,
| (14) |
By solving Eq. 14, one can obtain of the hydrogel at the equilibrium swollen state. The chain stretch of the hydrogel at the equilibrium swollen state scales with , and the pre-factor of the scaling relation depends on the polymer network architecture54. While Eqs. 12–14 assume that the polymer network of the hydrogel is uncharged, the effect of charges on the equilibrium swelling of hydrogels can be accounted for by introducing additional terms to the Helmholtz free energy function47,422. It should be noted that hydrogels are not necessary to reach the equilibrium swollen state in many situations, for example, when the hydrogels are insulated from water or do not have sufficient time to equilibrate with water.
4. Unconventional polymer networks
Section 3 has established that hydrogels with conventional polymer networks have intrinsically coupled mechanical properties, including shear modulus, stretch limit, fracture toughness, fatigue threshold, interfacial toughness and interfacial fatigue threshold of adhesion (Eq. 11). This section will discuss unconventional polymer networks (UPNs), which constitute most biological hydrogels (Figures 1 and 2) and have been widely used in synthetic hydrogels to achieve extreme mechanical properties.
The UPNs are defined as polymer networks that are different from the conventional polymer networks in terms of the architectures of polymer networks and/or the interactions among polymer chains in the networks63,423–431. Therefore, the UPNs can be broadly classified into two categories: the UPN architectures and the UPN interactions.
4.1. Unconventional polymer network architectures
As illustrated in Figure 7, the UPN architectures are dramatically different from the architecture of the conventional polymer networks composed of randomly-crosslinked polymer chains (Figure 4). Almost all biological tissues (Figures 1 and 2) possess UPN architectures. Over the last few decades, multiple UPN architectures have been proposed and synthesized for soft materials including elastomers and hydrogels to achieve extreme properties. Based on their topologies, the typical UPN architectures can be classified into a number of categories, including ideal polymer networks, polymer networks with slidable crosslinks, interpenetrating polymer networks, semi-interpenetrating polymer networks, polymer networks with highfunctionality crosslinks, nano-/micro-fibrous polymer networks, and bottlebrush polymer networks.
Figure 7.

Schematics of unconventional polymer network architectures, including ideal polymer networks, polymer networks with slidable crosslinks, interpenetrating polymer networks, semi-interpenetrating polymer networks, polymer networks with high-functionality crosslinks, nano-/micro-fibrous polymer networks, bottlebrush polymer networks, and future new UPN architectures.
4.1.1. Ideal polymer networks.
Ideal polymer networks are polymer networks that have uniform chain length, uniform functionality, and no defect (Figure 7)317. Following the pioneer work by Sakai et al.317,432–434, the ideal polymer networks have been commonly fabricated using multi-arm macromers, where the arms of adjacent macromers are crosslinked into polymer chains. Because the lengths of the macromer arms are uniform and the reaction efficiency of the crosslinking is high, various ideal polymer networks with uniform chain length, uniform functionality, and almost no defect have been achieved67,317,435–445. The tetra-arm PEG317,446,447 is among the most frequently used macromers for the fabrication of hydrogels with ideal polymer networks. The ends of the PEG macromers are commonly modified with pairs of reaction groups such as N-hydroxysuccinimide and amine67,317,448, tetrabenzaldehyde and tetrabenzaacylhydrazide449, maleimide and thiol450, or boronic acid and diol435,439,446. Due to the almost defect-free nature, the ideal polymer networks have been made highly stretchable and resilient67. It should be noted that, although the conventional polymer networks usually have non-uniform chain lengths and topological defects, their mechanical properties are commonly calculated based on the models of ideal polymer networks as discussed in Section 3. Therefore, the ideal polymer networks by themselves still have coupled mechanical properties.
4.1.2. Polymer networks with slidable crosslinks
A slidable crosslink, commonly in the form of two covalently-crosslinked polymer rings, can interconnect two polymer chains that thread through and slide inside the rings (Figure 7)451. Polymer networks with slidable crosslinks are both mechanically stable and reconfigurable due to the permanent and slidable nature of the crosslinks, respectively. Under mechanical loads, the slidable crosslinks tend to reconfigure the polymer network in a way that the polymer chains in the network sustain the same level of forces, approximating an ideal polymer network.
The polymer networks with slidable crosslinks are mainly synthesized from cyclodextrins-based polyrotaxanes452–454. Cyclodextrins are a series of cyclic oligosaccharides with 6, 7, or 8 glucose units (named α-, β-, or γ- cyclodextrin, respectively); cyclodextrins-based polyrotaxanes are inclusion complexes composed of linear polymer chains that are threaded through the cyclodextrin molecules and then capped by bulky groups at the chain ends454–456. The formation of cyclodextrins-based polyrotaxanes mainly depends on the size matching between the interior cavities of the cyclodextrins and the cross-section of the polymer chains457. Many polymer chains have been investigated to form cyclodextrin-based polyrotaxanes including linear homopolymers, linear block copolymers as well as branched polymers457. The αcyclodextrin has the smallest cavity size and can form inclusion complexes with PEG or PCL, but not with PPO chains458,459. While the β-cyclodextrin can form complexes with PCL or PPO but not PEG458,460,461. The γ-cyclodextrin, which has the largest cavity size, can thread through a PPO chain or two chains of PEG or PCL462. The cyclodextrins can be crosslinked with each other to interconnect the threaded polymer chains and form the polymer networks with slidable crosslinks60,356. Because a polymer network with slidable crosslinks under mechanical loads approximates an ideal polymer network, the mechanical properties of the polymer network with slidable crosslinks are usually coupled with one another as discussed in Section 3451,463–465.
4.1.3. Interpenetrating and semi-interpenetrating polymer networks
Interpenetrating polymer networks are comprised of two or more interpenetrated polymer networks, which are individually crosslinked but not joint together (Figure 7); semi-interpenetrating polymer network are comprised of two or more interpenetrated polymer networks, in which at least one network is uncrosslinked and others are individually crosslinked but not joint together (Figure 7)356,383,466–473. The interpenetrating and semi-interpenetrating polymer networks are entangled or interlocked in a way that they cannot be pulled apart unless the networks are broken470–473. Hydrogels based on the interpenetrating and semi-interpenetrating polymer networks are commonly prepared following the sequential or simultaneous method. In the sequential method, one polymer network is first prepared and then immersed into a solution of monomers, initiators and/or crosslinkers for another polymer network. Thereafter, the interpenetrating or semi-interpenetrating polymer network is formed by polymerizing the second polymer network within the first network. As a remarkable example, Gong et al. have adopted the sequential method to fabricate the so-called double-network hydrogels with high fracture toughness63. In the simultaneous method, a mixture of the polymers, monomers, initiators and crosslinkers for all polymer networks form the interpenetrating or semi-interpenetrating polymer networks in one step or one pot474. This one-step or one-pot fabrication process is a merit for the simultaneous method compared to the sequential method. One remarkable example of the simultaneous method is the simple fabrication of the polyacrylamide-alginate hydrogel with high stretchability and fracture toughness475. A wide range of material candidates including both natural and synthetic polymers as discussed in Section 260,356,383,467–469,475–477 have been used to synthesize hydrogels with interpenetrating and semi-interpenetrating polymer networks via various crosslinking strategies237,383,469,478. As will be discussed in Section 4.4, the interpenetrating and semi-interpenetrating polymer networks can provide decoupled and extreme mechanical properties for hydrogels, such as extremely high stretchability and fracture toughness63,475,479–481.
4.1.4. Polymer networks with high-functionality crosslinks.
The functionality of a crosslink refers to the number of polymer chains interconnected at the crosslink. Common covalent crosslinks as discussed in Section 2.3 usually have relatively low functionality (e.g., less than 10), and there is usually a single polymer chain bridging between two adjacent covalent crosslinks. To dramatically enhance the functionality of a polymer network, various types of high-functionality crosslinks can be introduced into the polymer networks, including crystalline domains215,300,302,482, glassy nodules483,484, nano-/micro-particles356,383,477,482,485–487, and micro-phase separations488–490 (Figure 7). For example, poly(vinyl alcohol) can form crystalline domains to crosslink the polymer networks through the freeze-thaw method302,491; poly(methyl methacrylate) can form glassy spheres and crosslink poly(methyl methacrylate)-based block copolymers into networks492; exfoliated particles, such as nano-clays493, graphene oxide494 or stratified lamellar bilayers495, can crosslink polyacrylamide into mouldable or self-healable hydrogels; mixtures of styrene, butyl acrylate, and acrylic acid can form microspheres and crosslink the residual polymer chain into microsphere composite hydrogels496.
Multiple polymer chains (e.g., over 10) can be interconnected at each high-functionality crosslink (Figure 7). In addition, there can be multiple polymer chains bridging between two neighboring high-functionality crosslinks, where the lengths of the polymer chains can be highly non-uniform60,436. As will be discussed in Section 4.4, the polymer networks with high-functionality crosslinks can provide decoupled and extreme mechanical properties for hydrogels, such as extremely high fracture toughness, resilience, tensile strength and fatigue resistance.
4.1.5. Nano-/micro-fibrous polymer networks
Both synthetic and natural polymers can assemble into fibers (or fibrils referring to short fibers) with diameters ranging from nanometers to micrometers via covalent or physical bonds. The nano-/micro-fibers can further entangle, aggregate, and crosslink into percolated polymer networks6,64,497–502 (Figure 7). In biological organisms, cells can secrete proteins (e.g., collagens) and polysaccharides (e.g., celluloses), which then assemble into nano-/micro-fibrous polymer networks103,250,503–506. These naturally-derived fibers and fibrous networks have been widely harnessed for the fabrication of hydrogels with nano-/micro-fibrous polymer networks382,507–510. In addition, a wide range of natural and synthetic polymers have been fabricated into nano-/micro-fibrous polymer networks with the spinning techniques511–513, among which the electrospinning is most popular due to its simplicity, low cost, and wide applicability514. In particular, the diameter, alignment and density of the fibers can be readily controlled by tuning the parameters of the electrospinning process515–519. As will be discussed in Section 4.4, the nano-/micro-fibrous polymer networks can provide decoupled and extreme mechanical properties for hydrogels, such as extremely high fracture toughness, tensile strength, resilience and fatigue resistance520,521.
4.1.6. Other UPN architectures
Many other types of UPN architectures can provide extraordinary mechanical properties as well. For example, the bottlebrush polymer networks (Figure 7) have shown extremely low shear moduli and tissue-like stress-strain relations in the solvent-free state522,523. Although these UPN architectures have not been widely used in hydrogels, they may be exploited for the design of hydrogels in future524. Furthermore, it is also expected that new UPN architectures will be invented together with the development of polymers and hydrogels.
4.2. Unconventional polymer network interactions
As illustrated in Figure 8, the UPN interactions are defined as inter-polymer and intra-polymer interactions that are different from those in the conventional polymer networks (i.e., permanent covalent crosslinks, excluded volumes, and osmotic interactions) (Figure 8). The UPN interactions are vastly abundant in biological organisms525, and they have been intensively studied for the design of soft materials such as elastomers and hydrogels to achieve extreme mechanical properties among many other purposes237. Based on the nature of the UPN interactions, they can be broadly classified into three categories55: strong physical crosslinks, weak physical crosslinks, and dynamic covalent crosslinks.
Figure 8.

Schematics of unconventional polymer network interactions including a. strong physical crosslinks, b. weak physical crosslinks, and c. dynamic covalent crosslinks.
4.2.1. Strong physical crosslinks
In addition to the permanent covalent crosslinks discussed in Section 2.3, various types of strong physical bonds can act as effectively permanent crosslinks in polymer networks. Typical examples of strong physical crosslinks include crystalline domains, glassy nodules, and helical structures. The energy of strong physical crosslinks is similar to that of permanent covalent crosslinks.
Crystalline domain.
A specific subset of synthetic and natural polymers can form crystalline domains under appropriate conditions. A crystalline domain, with the size from nanometers to micrometers, can serve as a strong physical crosslink for multiple amorphous polymer chains connected to it (Figure 8). As an example in synthetic polymers, PVA can form crystalline domains by repeated freeze-thaw cycles or by annealing at temperatures above its glass transition temperature300,302,482. The formation of PVA crystalline domains is mainly due to the hydrogen-bonding interactions of the hydroxyl groups on PVA chains4. As an example in natural polymers, chitin and chitosan can form semi-crystalline polymer networks with crystalline domains crosslinking amorphous chains by treating the chitin and chitosan with strongly acidic or basic solutions to overcome the inter-chain electrostatic repulsions215,218. As another example in natural polymers, cellulose can also form highly crystalized nanofibers due to the strong interaction between glucose units526. These cellulose nanofibers can further aggregate and form a stable network by alkaline treatments497,527. It should be noted that heating the abovementioned semi-crystalline polymer networks above their melting temperatures can destroy the crystalline domains in the networks, although most crystalline domains are stable at room and body temperatures.
Because a crystalline domain usually interconnects multiple polymer chains, they often act as highfunctionality crosslinks in the polymer networks as discussed in Section 4.1. In addition, the energy required to pull a polymer chain out of a crystalline domain is much higher than that required to fracture the same polymer chain71; therefore, crystalline domains can also introduce intrinsically high-energy phases into the polymer networks. These attributes of crystalline domains have endowed the hydrogels containing crystalline domains with extreme mechanical properties, such as tough, strong and fatigue-resistant, which will be discussed in Section 5.
Glassy nodule.
Glassy nodules are formed by the reversible liquid-glass transition of amorphous polymers when the temperature is decreased below their glass-transition temperatures528. In order to harness glassy nodules as strong physical crosslinks, block copolymers that contain at least one segment with a high glass-transition temperature have been commonly used. As the temperature reduces to room or body temperature, the segments with the high glassy-transition temperature form glassy nodules that effectively crosslink the adjacent amorphous polymer chains (Figure 8)529. For example, the polystyrene segments in the polystyrene-b-poly(N-isopropylacrylamide)-b-polystyrene copolymers can form glassy nodules at room temperature to crosslink the block copolymers chains into a polymer network530. As another example, poly(methyl methacrylate) has a glass transition temperature around 115 °C531; therefore, the poly(methyl methacrylate) segments in the poly(methyl methacrylate)-b-poly(n-butyl acrylate) copolymers can form glassy spheres at room temperature to crosslink the polymer network492. Similar to crystalline domains, glassy nodules can also act as high-functionality crosslinks and intrinsically high-energy phases in polymer networks to give the corresponding hydrogels extreme mechanical properties, which will be discussed in Section 5.
Helical association.
Many natural polymers, due to their precisely-controlled structures, can assemble into nanometer-scale helical fibers (or fibrils), which then can aggregate or entangle to form a crosslinked network (Figure 8)103,507,509,510. For example, the well-known triple-helix structure of type I collagen is formed by the self-assembly of three peptide strands. These collagen triple helices can pack together to form collagen nanofibers and further self-assemble into an interconnected hydrogel network532,533. As another example, the linear agarose chains are disordered coils in aqueous solutions at high temperatures, and can form double-helix strings534 or simple helical chains535 when the temperature is decreased to room or body temperature. These stings or chains can associate to form agarose fibers through hydrogen bonding and further be entangled to form the interconnected hydrogel network536.
4.2.2. Weak physical crosslinks
Compared to the strong physical crosslinks, many other physical crosslinks in polymer networks are relatively weak, transient and reversible. Typical examples of weak physical crosslinks include hydrogen bond, electrostatic interaction, metal coordination, guest-host interaction, hydrophobic association, and π-π stacking. The energy of weak physical crosslinks is usually lower than that of strong physical crosslinks and permeant covalent crosslinks.
Hydrogen bond.
The energy of a single hydrogen bond ranges from 0.8 kJ mol−1 to 167 kJ mol−1 (Figure 5) 373,374. Many natural polymers can form hydrogels by the intermolecular hydrogen bonds. For example, gelatin can form polymer networks of helical structures crosslinked by hydrogen bonds170; certain types of polysaccharides, such as agarose, amylose, amylopectin and carrageenan, can also form helical structures in solutions and crosslinked into hydrogels by hydrogen bonds537. A number of synthetic polymers are also capable of forming physical hydrogels via hydrogen bonds. For example, PVA hydrogels can be obtained by forming hydrogen bonds between polymer chains through repeated freezing and thawing of PVA solutions303. PAA or polymethacrylic acid (PMA) can form complexes with PEG by hydrogen bonds between the oxygen groups of PEG and the carboxyl groups of PMA538 or PAA539.
Despite the abundance of hydrogen-bond groups (-OH, -NH, -C=O, -C-O) in natural and synthetic polymers, the hydrogen-bond interactions in hydrogels are usually screened due to the water molecules in hydrogels. To enable effective hydrogen-bond crosslinks, hydrophobic moieties with multiple self-complementary hydrogen-bond groups have been introduced478,540,541. For instance, functionalizing PEG, PHMEA and PNIPAM with amine triazine or diamino triazine groups enables the formation of triple hydrogen bonds per crosslink478,490,540,542. Similarly, the introduction of ureidopyrinimidone (UPy) groups onto PEG, PHMEA, PNIPAM, PAA and PDMAA chains gives quadruple hydrogen bonds per crosslink478,540,541. Complementary DNA base pairs (A-T, C-G) can also serve as hydrogen-bond motifs when attached to polymer chains543.
Electrostatic interaction.
The energy of electrostatic interactions ranges from 5 kJ mol−1 to 200 kJ mol−1 (Figure 5)375. Natural and synthetic polymers with fixed charges, named polyelectrolytes, can be physically crosslinked by electrostatic interactions105,475,544,545. As a typical example of the anionic polyelectrolytes, alginate has been physically crosslinked with a wide range of divalent cations such as Ca2+, Ba2+ and Mg2+. Although the energy of a single ionic bond in alginate is relatively low, multiple (e.g., over 20) adjacent ionic crosslinks on the alginate chains can form a densely-crosslinked region following the “eggbox” model6,103,105,546, giving relatively stable alginate hydrogels. As a typical example of the cationic polyelectrolytes, chitosan has been crosslinked by multivalent anions such as citrate and tripolyphosphate547–549. Electrostatic interactions of oppositely charged polyelectrolytes can also give physically-crosslinked hydrogels. For example, anionic poly(L-glutamic acid) and cationic poly(L-lysine) can form an injectable hydrogel by simply mixing them in phosphate buffered saline solutions544. As another example, poly(3-(methacryloylamino) propyl-trimethylammonium chloride) and poly(sodium p-styrenesulfonate) can form polyion complexes and give a series of tough and self-healing hydrogels by the stepwise polymerization of the oppositely charged monomers550. It should be noted that the formation of ionic crosslinks usually requires low ionic strength of the solvents for the hydrogels to avoid charge shielding.
Coordination complex.
A coordination complex consists of a central metal ion, especially transition metal ion, and a surrounding array of organic ligands551,552. The energy of coordination complexes ranges from 100 kJ mol−1 to 300 kJ mol−1 (Figure 5) 375. Coordination bonds provide structural support in many living tissues, such as human bone553, insect mandible554 as well as mussel byssal thread555. Hydrogels crosslinked by coordination complexes are primarily achieved by functionalizing polymer backbones with chelating ligands, which then form coordination complexes with metal ions. Bisphosphonate556–558, catechol559–561, histidine562–564, thiolate565,566, carboxylate567,568, pyridine569, bipyridine570, and iminodiacetate564,571 have been widely used as the chelating ligands; Cu2+, Zn2+, Fe3+, Co2+ and Ni2+ are the commonly-used metal ions. Bisphosphonate ligands can be modified onto hyaluronan572, gelatin573 or PEG557 to form coordination complex with Ca2+, Mg2+ or Ag+. Besides bisphosphonate, catechol ligands are also widely used to functionalize various polymers such as PEG574,575, gelatin576, hyaluronic acid577, chitosan578, polyacrylamide560, and PAA561. As a typical example, PEG-modified with 3,4dihydroxyphenyl-L-alanine (DOPA) residues can form coordination complexes with metal ions (Cu2+, Zn2+ and Fe3+ ions) when the pH is above 8478,575. In natural proteins, the histidine amino acid can give an imidazole ligand residue59, which is one of the most important chelators in the human body579. PEG-modified with histidine can form coordination complexes with metal ions (Cu2+, Co2+ and Ni2+ ions) to achieve the physically crosslinking of the PEG hydrogels478,580. The mechanical properties of the hydrogels crosslinked by coordination complexes can be tuned by varying the metal ions and/or the chelating ligands562,581.
Host-guest interactions.
Host-gust interactions refer to two or more molecules or ions that are held together in unique structural relationships by forces other than those of covalent bonds461,582,583. The two most common host moieties are cyclodextrins and curcubit[n]urils. Cyclodextrins (CDs) are cyclic oligosaccharides which compose of 6 to 8 D-glucose repeating units linked by α−1,4-glucosidic bonds584,585. Commonly-used CDs include α-, β-, and γ-CDs which are composed of 6, 7, and 8 D-glucose repeating units, respectively. These CDs have a truncated cone shape with the secondary and primary hydroxyl groups on the smaller cone rim exposed to the solvent583, which makes the CDs show a relatively hydrophobic inner cavity and a relatively hydrophilic outer surface. Therefore, these CDs can act as the host molecules for various hydrophobic guest molecules with appropriate molecular sizes through hydrophobic and van der Waals interactions383,457,461. For example, common guests for α-CD include azobenzene586 and ferrocene587; common guests for β-CD include adamantane588, benzimidazole589, 3-(Trimethylsilyl)propionic acid590, azobenzene586, ferrocene591, bipyridine592, phenolphthalein593 and cholesterol594; common guests for γ-CD include ferrocene587. Among various guest molecules, adamantane is widely regarded as having one of the greatest affinities due to its complementary size for β-CD and high hydrophobicity595. In addition, the complexation of azobenzene or ferrocene to CDs is responsive to light596 or redox conditions597,598, respectively.
The cucurbit[n]urils (CB[n, n=5–8]) are pumpkin-shaped macrocyclic oligomers made from the condensation reactions of formaldehyde and glycoluril583,599. The CB[n]s usually have a structure of a rigid hydrophobic cavity with two identical hydrophilic polar carbonyl groups surrounding the portals. The cavity size ranges from 4.4 to 8.8 A° (for CB[n], n = 5–8) and the portal diameter ranges from 2.4 to 6.9 A°600. The binding affinities of CBs are often greater than that of other cavitands601, mainly due to the formation of the strong charge–dipole, hydrogen bonding, and hydrophobic/hydrophilic interactions by the rigid inner cavities and the negative portals of CB[n]s602. In particular, CB[7] can form strong 1:1 complexes with positively charged amphiphilic guests including adamantane, ferrocene, p-xylylene and trimethylsilyl derivatives containing one or two amino groups, as well as viologen derivatives603,604. CB[8] also displays remarkable binding affinities towards positively charged and relatively large guests such as amantadine derivatives. Furthermore, the cavity of CB[8] is large enough to accommodate two organic guests simultaneously, thus forming highly stable ternary complexes. For example, CB[8] can form stable complexes with two doubly-charged 2,6-bis(4,5-dihydro-1Himidazol-2-yl)naphthalene molecules605, or one viologen (paraquat) and one 2,6-dihydroxynaphthalene together606.
Hydrogels crossslinked by the host-guest interactions are usually constructed with polymer networks modified by the guest molecules and/or host molecules. For example, monomers, host molecules, and guest molecules can be copolymerized into polymer networks crossslinked by the host-guest interactions607. The host/gust molecules can also be attached to the backbones or ends of polymers such as PEG, PDMAA, hyaluronic acid and PAA, and then the addition of the corresponding difunctional gust/host crosslinkers will crosslink the polymer network583. Alternatively, guest-functionalized and hostfunctionalized polymers can also be synthesized separately, and a mixture of the two types of polymers gives hydrogels crosslinked by the host-guest interactions608,609. Supramolecular hydrogels with these host-guest interactions have been extensively utilized to fabricate responsive materials610 and other dynamically assembling systems457,611.
Hydrophobic association.
The physical crosslink of hydrophobic association relies on the microphase separation and aggregation of hydrophobic domains of the polymer chains478,490. The energy of hydrophobic association ranges from 0.1 kJ mol−1 to 20 kJ mol−1 (Figure 5) 375. The hydrophobic domains can be introduced by post-polymerization modification (e.g., via the grafting-to approach) or by copolymerizing hydrophobic monomers within the polymer chains, either randomly or as blocks612. These modifications usually require the usage of non-aqueous solvents, mixed solvents, or micellar systems488,613. As a typical example of introducing the hydrophobic domains, hydrophobic stearyl acrylate monomers have been copolymerized within polyacrylamide (PAAm) chains488. Another example of introducing the hydrophobic domains is the synthesis of multiblock copolymers with hydrophobic n-alkyl acrylate end blocks and a large middle block of PEG, PAAm, PAA and PHEMA polymers614–616. Notably, because one hydrophobic association can interconnect multiple polymer chains, the hydrophobic association has also been used as high-functionality crosslinks in hydrogels290,617, although the energy of hydrophobic association is usually lower than that of crystalline domains and glassy nodules.
π-π stacking.
The π-π stacking interaction is a type of noncovalent interaction which refers specifically to the attractive interactions between π electrons in the aromatic groups618 The π-π interactions can be divided into the edge-to-face stacked (T-shaped), offset stacked, and face-to-surface stacked due to the geometry of the aromatic interactions619. The energy of π-π stacking ranges from 1 kJ to 50 kJ mol−1 (Figure 5) 376. Natural amino acids with aromatic rings, such as phenylalanine, tyrosine and tryptophan, and the other compounds with conjugate structures such as fluorenylmethyloxycarbonyl (Fmoc), 1pyrenebutyric acid, 2-naphthalene acetic acid and nitrophenyl methacrylate can be used to design and prepare polymers with aromatic moieties for gelation by the π-π stacking interaction499,620,621. For example, aromatic moiety containing short peptides and N-terminal Fmoc-amino acids can self-assemble into robust supramolecular architectures499. In addition, carbon nanotubes622,623, polythiophene30,624, and graphene-based nanomaterials625,626 (including single-layer graphene, multilayer graphene, graphene oxide, and reduced graphene oxide) are also capable to form π-π interactions, which is useful in preparing electrically conductive hydrogels627,628.
4.2.3. Dynamic covalent crosslinks
In addition to the weak physical bonds, dynamic covalent bonds can also act as reversible crosslinks that are cleavable by external stimuli. The energy of dynamic covalent bonds is usually similar to or lower than that of permanent covalent bonds629, and higher than that of weak physical bonds (Figure 5). Typical examples of dynamic covalent crosslinks in hydrogels include imine bond, boronate ester bond, disulfide bond, cyclohexenes hydrazone bond, oxime bond, and reversible Diels-Alder reaction.
Imine bond.
An imine is a compound with a carbon-nitrogen double bond, which is commonly formed by reactions between amine and aldehyde or ketone630. In particular, the imine crosslinks in hydrogels are usually formed through the Schiff base reactions, which give aliphatic Schiff bases or aromatic Schiff bases631–634. The reversible nature of the imine crosslinks endows the resultant hydrogels with properties such as mechanical dissipation, self-healing and stimuli responses635. The energy of the Schiff bases ranges from 67 kJ mol−1 to 477 kJ mol−1 (Figure 5)372,377. The aromatic Schiff bases usually have higher energy and stability than the aliphatic Schiff bases636,637.
The imide bonds are particularly useful for preparing biopolymer-based hydrogels, because most biopolymers such as proteins contain amine groups. These amines can form imide bonds with various aldehydic crosslinkers at mild conditions635,638. The obtained hydrogels with imine bonds are usually sensitive to various chemical and biological stimuli, including pH, free amine, and free aldehydes637. These hydrogels can be used as self-healing materials and injectable scaffolds in biomedical applications639–641.
Boronate ester bond.
The dynamic boronate ester bonds are formed by the reaction of diols and boronic acid642–644. The energy of boronate ester bonds ranges from 27.2 kJ mol−1 to 93.3 kJ mol−1 (Figure 5) 378, highly dependent on pH and temperature645–647. The boronic acid can be introduced into hydrogels by polymerizing boronic acid-containing monomers together with other monomers, such as acrylamide (AAm)648 and N-isopropylacrylamide (NIPAM)649. Alternatively, boronic acid functional groups can also be grafted onto pre-formed polymer chains through the carbodiimide chemistry650,651.
The boronic acid-containing polymers can react with polymers having diol functional groups. For example, polymers modified with salicyl hydroxamic acid groups or catechol groups can form the dynamic boronate ester crosslinks with boronic acid-containing polymers in acidic environment646,652 or alkaline environment574,653,654, respectively. As another example, polyhydroxy polymers such as PVA655,656, alginate657,658, and cellulose659 can also be crosslinked into dynamic hydrogels by mixing with boronic acid-containing polymers in aqueous solutions. The transient boronate ester networks usually can dynamically restructure after broken, making the resultant hydrogels injectable and self-healing660,661. In addition, the boronate ester crosslinked hydrogels are also glucose-sensitive, because glucose can compete with diol groups to form boronate–glucose complexes and therefore de-crosslink the hydrogels655. These glucosesensitivity hydrogels, based on the boronate ester bonds, have been used for self-regulated insulin release and glucose sensing383,650,655,662.
Disulfide bond.
Disulfide bonds are dynamic covalent bonds based on thiol-thiol interactions at slightly alkaline environments or at mild oxidative conditions663,664. The energy of disulfide bonds is around 425kJ mol−1 (Figure 5)372,379. Many natural polymers have disulfide bonds to stabilize their structures such as fibrinogen665 and collagen666. The disulfide bond can also be introduced into polymers by using disulphide-bond containing crosslinkers such as 3,3’-dithiobis(propanoic dihydrazide)667,668 and N, N’-cystamine-bisacrylamide669–671. The thiol-thiol reaction has relatively fast kinetics and can be used to prepare dynamic hydrogels672,673. Hydrogels crosslinked by disulfide bonds can be used to encapsulate various types of cells, due to the mild reaction conditions674,675. In addition, the disulfide bonds are sensitive to pH and can be cleaved by reducing agents such as tris(2-carboxyethyl)phosphine676, 1,4-dithiothreitol677, and glutathione672,678.
Hydrazone bond.
Hydrazone bonds are formed by the reaction of aldehyde and hydrazine groups679. Polymers with hydroxyl groups, such as PEG680, cellulose681 and polysaccharide632, can be easily modified with the aldehydes and reactive hydrazines, especially acylhydrazine or hydrazide motifs. The reversible hydrazone bonds can be formed by simply mixing the aldehyde and hydrazine containing polymers under physiological conditions682–684.
Hydrogels crosslinked by the hydrazine bonds can exhibit reversible sol-gel transition properties by changing the pH680,685. Aliphatic aldehyde-derived hydrazone has a more rapid rate of formation and hydrolysis than the aryl aldehyde-derived hydrazone at neutral pH686,687. Hydrogels crosslinked by hydrazone bonds can be used for in situ cell encapsulation due to the cytocompatibility and fast gelation kinetics of aldehyde and hydrazine coupling688,689. The mechanical properties of these hydrogels can be easily tuned, which facilitates the study of the relationships between cell behaviors and mechanics (such as stress-relaxation kinetics) of the hydrogels690. Hydrazone bonds can also be used to prepare self-healing and injectable hydrogels based on the reversibility of hydrazone bonds at the mildly acid environment (pH 4.0–6.0)682,684,686.
Oxime bond.
Oxime bonds are formed by the reaction between hydroxylamine and aldehyde or ketone with high efficiency under mild conditions691. The reactive aldehyde or ketone groups can be modified onto polymers through radical polymerization692 or oxidation693,694, while the hydroxylamine motif is mainly modified onto hydroxyl-containing polymers through a sequential N-hydroxyphthalimide induced Mitsunobu reaction and hydrazine reduction695. Then, the oxime bonds can be formed by mixing the aldehyde or ketone-containing polymers with the hydroxylamine-containing polymers at a neutral or slightly acid aqueous solution696. This reaction is biocompatible without cytotoxic side products and can be used to crosslink biopolymers into hydrogels695,697. Due to the dynamic nature, oxime bonds have been used for building self-healing and injectable hydrogels which show higher hydrolytic stability than the hydrogels crosslinked by imines and hydrazones696,698.
Reversible Diels–Alder reaction.
Diels-Alder reaction is a click reaction between diene and dienophile groups699,700. The energy of Diels-Alder bonds ranges from 37.6 kJ mol−1 to 130 kJ mol−1 (Figure 5)380,381. To harness the dynamic Diels-Alder reaction as reversible crosslinks for hydrogels, natural polymers (such as hyaluronic acid701, cellulose702 and other polysaccharides703) and synthetic polymers (such as PNIPAM704and PEG705) can be modified with diene (such as furan) functional groups and dienophile (such as maleimide) functional groups on their backbones or chain ends. The equilibrium of the Diels-Alder linkage is thermally responsive; for example, the adducted Diels-Alder linkage can reform maleimide and furan moieties when increasing the temperature706,707. The Diels-Alder reaction can be performed in aqueous media at physiologically compatible conditions without any side reactions or byproducts708–711. Therefore, the Diels-Alder reaction has been used for preparing self-healing or adaptable hydrogels for biological applications in drug delivery and tissue engineering701,711,712.
4.3. Synergy of unconventional polymer network architectures and interactions
It is not uncommon for an unconventional polymer network to simultaneously possess both UPN architectures and UPN interactions. In some cases, the UPN architectures and interactions are interdependent. The formation of certain UPN architectures requires certain UPN interactions, or certain UPN interactions naturally lead to the self-assembly of polymers into certain UPN architectures. For example, the strong physical crosslinks such as crystalline domains and glassy nodules mostly have high functionalities, giving rise to UPNs with high-functionality crosslinks. As another example, the selfassembly of polymer chains into nano-/micro-fibers usually requires the UPN interactions such as weak physical crosslinks. In other cases, specific types of UPN architectures and interactions can be intentionally designed and integrated into the same UPN. For example, weak physical crosslinks and dynamic covalent crosslinks have been introduced into various UPN architectures in order to design tough hydrogels475, because the dissociation and reformation of these reversible crosslinks can dissipate mechanical energy to toughen the hydrogels.
4.4. Decoupled mechanical properties
Decoupled mechanical properties of hydrogels with UPN architectures.
Polymer chains in the ideal polymer networks have uniform chain lengths (i.e., the same N), and the polymer networks with slidable crosslinks also tend to give relatively uniform chain lengths under mechanical loads. These polymer networks with relatively uniform chain lengths are named as unimodal polymer networks (Figure 9a)53,56,428. Because the shear moduli, stretch limits and intrinsic fracture energy of conventional polymer networks have been derived based on unimodal polymer networks, these mechanical properties are still coupled in hydrogels with the ideal polymer networks and the polymer networks with slidable crosslinks.
Figure 9. The UPN architectures decouple mechanical properties of hydrogels:

a. unimodal polymer networks such as ideal polymer networks and the polymer networks with slidable crosslinks give coupled mechanical properties; b. multimodal polymer networks such as interpenetrating polymer networks, semiinterpenetrating polymer networks and polymer networks with high-functionality crosslinks can decouple the mechanical properties.
The interpenetrating polymer networks, semi-interpenetrating polymer networks and polymer networks with high-functionality crosslinks can integrate polymer chains with dramatically varying chain lengths (i.e., different N) into the same polymer networks, which are often named as multimodal polymer networks (Figure 9b)53,56,428. Let’s classify the polymer chains in a multimodal polymer network into different types based on their chain lengths (Figure 9b). For the ith type of polymer chains, the number of the polymer chains per unit volume of the polymer network in the dry state, the number of Kuhn monomers per polymer chain, and the volume of the Kuhn monomer are denoted as ni, Ni and vi, respectively.
The multimodal polymer network (Figure 9b) can be designed such that the corresponding hydrogel can sustain its integrity up to the stretch limit of the longest polymer chains, which can be expressed as63,475,
| (15) |
where Nmax is the number of Kuhn monomers on the longest polymer chain, and accounts for the effect of swelling on the stretch limit. The stretch limit of the bulk hydrogel scales with , and the pre-factor of the scaling relation depends on the polymer network architecture54.
Based on the affine network model, the shear modulus of the hydrogel with the multimodal polymer network (Figure 9b) can be expressed as
| (16) |
where ni and account for the effects of the ith type of polymer chains and swelling on the initial shear modulus of the hydrogel.
Following the Lake-Thomas model, the intrinsic fracture energy of the hydrogel with the multimodal polymer network (Figure 9b) can be calculated as,
| (17) |
where bi and Ui are the length and fracture energy of a Kuhn monomer on the ith type of polymer chain, respectively, and accounts for the effect of swelling on the intrinsic fracture energy. It should be noted that the fracture toughness and interfacial fracture toughness of the multimodal polymer network (Figure 9b) can be much higher than the intrinsic fracture energy, which will be discussed in Section 5.
It is commonly assumed that the polymer chains occupy the major volume of the polymer network in the dry state, and therefore the volume conservation of the multimodal polymer network (Figure 9b) gives
| (18) |
Despite the relation of Eq. 18, the stretch limit, shear modulus and intrinsic fracture energy of a hydrogel with the multimodal polymer network (Figure 9b) can be decoupled and independently designed. Without loss of generality, let’s consider a hydrogel with bimodal distribution of polymer-chain lengths as an example (Figure 9b). A high density of the short polymer chains can give a high initial shear modulus of the hydrogel. While these short chains will be fractured when the hydrogel is highly stretched, the long polymer chains can still maintain the integrity and high stretch limit of the hydrogel63,475. Similarly, the long polymer chains can give high intrinsic fracture energy of the hydrogel713.
The mechanical properties of the nano-/micro-fibrous polymer networks are determined by their fibers, interactions of the fibers (e.g., crosslinks between fibers), and topologies of the fibrous polymer networks. Therefore, the stretch limit, shear modulus and intrinsic fracture energy of nano-/micro-fibrous hydrogels do not follow the coupling relations for conventional polymer networks (Eqs. 13 and 14), and they can be independently designed.
Decoupled mechanical properties of hydrogels with UPN interactions.
The crystalline domains and glassy nodules have been widely used as the high-functionality crosslinks in UPNs, whose stretch limit, shear moduli and intrinsic fracture energy are decoupled as discussed above (Figure 9b).
The weak physical crosslinks and dynamic covalent crosslinks can act as reversible crosslinks in polymer networks, leading to decoupled mechanical properties of the resultant hydrogels (Figure 10). Without loss of generality, let’s consider a conventional polymer network with long polymer chains (i.e., polymer networks sparsely crosslinked by permanent covalent bonds that give high N value), whose stretch limit, shear modulus and intrinsic fracture energy are given by Eqs. 7–9, respectively. We next introduce reversible crosslinks such as weak physical crosslinks and dynamic covalent crosslinks into the polymer network. When the polymer network undergoes initial small deformation, the reversible crosslinks act as additional crosslinks435, increasing the effective chain density of the polymer network to . Therefore, the shear modulus of the hydrogel with the reversible crosslinks under initial deformation increases to
| (19) |
Figure 10. The UPN interactions decouple the mechanical properties of hydrogels.

The reversible crosslinks give an effectively high density of short chains for the high modulus, and the sparse covalent crosslinks give long chains for high stretchability and intrinsic fracture energy.
As the hydrogel is highly stretched, the reversible crosslinks can be de-crosslinked. However, the covalently-crosslinked long polymer chains (i.e., high N ) still endow the hydrogel with high stretch limit and high intrinsic fracture energy according to Eq. 13.
5. Design of hydrogels with extreme mechanical properties
While numerous UPN architectures and interactions have been developed over the last few decades, the design of hydrogels that possess extreme mechanical properties has largely followed an Edisonian approach – trial and error with specific materials. The rational design of hydrogels with different polymers and fabrication methods for various applications remains a central need of the field. In this section, we will summarize a set of general design principles for hydrogels to achieve the corresponding extreme mechanical properties, including extremely high fracture toughness, tensile strength, resilience, interfacial toughness, fatigue-threshold, interfacial toughness, and interfacial fatigue threshold. Then, we will discuss the implementations of these design principles with the UPN architectures and/or the UPN interactions.
5.1. Tough: Build dissipation into stretchy polymer networks
Fracture toughness.
Fracture toughness has been widely used to characterize a material’s capability to resist fracture under mechanical loads. One common definition for the fracture toughness of a material is the energy required to propagate a crack in the material over a unit area measured in the undeformed state (Figure 11a) which can be quantitatively expressed as,
| (20) |
where U is the total potential energy of the system, A is the crack area measured in the undeformed state, and Gc is the critical energy release rate that drives crack propagation. According to Eq. 20, the unit for the fracture toughness is joule per meter squared (i.e., J m−2).
Figure 11. Design principle for tough hydrogels – build dissipation into stretchy polymer networks.

a. definition of fracture toughness and the pure-shear test to measure the fracture toughness. When a notched sample with height H at the undeformed state is stretched by a critical ratio of λc under pure-shear deformation, the crack begins to propagates (top). The relation of the nominal stress s and the stretch λ is measured for an un-notched sample (otherwise the same as the notched sample) under pure-shear deformation (bottom). The fracture toughness can be calculated as based on the measured λc and s vs λ relation in the pure-shear tests. b. the intrinsic fracture energy from fracturing a layer of polymer chains. c. the mechanical dissipation in the process zone around the crack tip dramatically contributes to the fracture toughness by . The mechanical dissipation manifests as a hysteresis loop on the stress-stretch curve. The total fracture toughness of the tough hydrogel is .
The fracture toughness of soft materials such as elastomers and hydrogels has been measured with many experimental methods such as the pure-shear test and the single-notch test, which have been summarized in a few recent review papers60,714,715. For example, in the pure-shear test, two identical pieces of a hydrogel are fabricated with the same thickness T, width W and height H, where W >> H >> T (Figure 11a). Both pieces of samples are clamped along their long edges (i.e., along the width direction) with rigid plates. A notch with a length of ~0.5W is introduced into the first sample, which is then gradually pulled to a stretch of λc times of its undeformed height until a crack begins to propagate from the notch (Figure 11a). The second sample without notch is uniformly stretched above the critical stretch λc to measure the nominal stress s - stretch λ relation (Figure 11a). Thereafter, the fracture toughness of the hydrogel can be calculated as , based on the measured λc and s-λ relation in the pure-shear tests.
As discussed in Section 3, the fracture toughness of a conventional polymer network is its intrinsic fracture energy , which is the energy required to fracture a layer of polymer chains over a unit area (Figure 11b). Evaluated with typical parameters of conventional polymer networks, the fracture toughness of the corresponding hydrogels is commonly limited to a few tens of joule per meter squared. In addition, the fracture toughness of hydrogels with conventional polymer networks is also coupled with their stretch limits and shear moduli (Eqs. 11). For example, in order to increase the fracture toughness of a conventional polymer network, the polymer-chain length (i.e., N) and thus the stretch limit of the polymer network need to be increased. Consequently, the polymer-chain density (i.e., n) and thus the shear modulus of the polymer network will be decreased.
Design principle.
The design principle for tough hydrogels is the same as the principle for toughening various engineering materials such as metals716, ceramics 717, composites718 and polymers57 and various biological tissues such as tendons, cartilages, muscles and blood vessels (Figure 2)719. That is to integrate both ductility and mechanical dissipation in the same material, so that a process zone with substantial mechanical dissipation develops around the crack tip prior to crack propagation (Figure 11c). The mechanical dissipation of a material manifests as the hysteresis loop on its stress-stretch curve under a loading-unloading cycle (Figure 11c); the ductility of hydrogels generally relies on the high stretchability (or the high stretch limit) of their polymer networks. Overall, the design principle for tough hydrogels is to build dissipation into stretchy polymer networks60,720. Quantitatively, the total fracture toughness of a hydrogel with the capability of mechanical dissipation can be expressed as 60,721
| (21) |
where , , and are the total fracture toughness, the intrinsic fracture energy, and the contribution of mechanical dissipation in the process zone to the total fracture toughness, respectively. While the intrinsic fracture energy for hydrogels is usually limited to a few tens of joule per meter squared, the contribution of the process-zone dissipation can be extremely high because both the dissipated energy per volume of the process zone and the size of the process zone can be large values (Figure 11c). Indeed, the fracture toughness of tough hydrogels has exceeded 10,000 Jm−2, orders of magnitude higher than that of hydrogels with conventional polymer networks60.
Implementation with UPNs.
The design principle for tough hydrogels requires: i). at least one polymer network in the hydrogel maintains a high stretch limit and thus the polymer chains in that polymer network need to have a high N value according to Eq. 7; and ii). at least one component in the hydrogel dissipates substantial mechanical energy under the deformation typically experienced in the process zone. The design principle for tough hydrogels has been widely implemented with diverse types of UPN architectures and UPN interactions. We will discuss a few examples in the following paragraphs.
The interpenetrating polymer networks and semi-interpenetrating polymer networks have been widely used for the design of tough hydrogels since the pioneer work of double-network hydrogels by Gong et al. in 2003 (Figure 12a)63. A typical double-network hydrogel interpenetrates a long-chain network (high N) and a short-chain network (low N)63. As the double-network hydrogel deforms, the short-chain network fractures and dissipates substantial mechanical energy, while the long-chain network maintains the integrity of the hydrogel even under high stretches, implementing the design principle for tough hydrogels (Figures 12f and g)63,720,722. Gong et al first demonstrated that the fracture toughness of double-network hydrogels can exceed 1,000 Jm2 63. Other interpenetrating and semi-interpenetrating polymer networks such as the triple-network architecture have also been developed for tough hydrogels724 and elastomers725, implementing the design principle. Notably, since the fracture of the short-chain network is usually irreversible, these hydrogels’ capability of mechanical dissipation may be substantially reduced after a few cycles of large deformation721.
Figure 12. Implementation of the design principle for tough hydrogels– build dissipation into stretchy polymer networks.

Schematics of the implementation strategies with a. interpenetrating or semiinterpenetrating polymer networks, b. polymer networks with high-functionality crosslinks, c. nano-/micro-fibrous polymer networks, and d. polymer networks with reversible crosslinks. e. nominal stress s-stretch λ relations for a PAAm-alginate hydrogel under loading and unloading475. f. microscope image of the process zone around the crack in a PAMPS-PAAm hydrogel722. g. microscope image of a fibrous fibrin hydrogel723. e is adopted from Ref475. f is adopted from Ref722. g is adopted from Ref723.
The polymer networks with high-functionality crosslinks have given tough hydrogels based on various types of polymers and crosslinks. There are multiple polymer chains (e.g., over 10) bridging between two adjacent high-functionality crosslinks, and the lengths of these polymer chains are usually non-uniform (Figure 12b). As the hydrogel deforms, the relatively short polymer chains fracture or detach from the crosslinks, while the relatively long polymer chains maintain the integrity and high stretchability of the hydrogel, implementing the design principle for tough hydrogels. The bonds between the polymer chains and the high-functionality crosslinks can be permanent covalent crosslinks726, strong physical crosslinks727,728, weak physical crosslinks729,730, and dynamic covalent crosslinks731,732, or a combination of them733. Depending on the number and lengths of polymer chains between adjacent crosslinks and the types of bonds between polymer chains and crosslinks, the corresponding hydrogel can have different capabilities of mechanical dissipation and stretchability and therefore different fracture toughness.
The nano-/micro-fibrous hydrogels have also been used to implement the design principle for tough hydrogels. The nano-/micro-fibers can be made intrinsically stretchable (Figure 12c), and their reorientation and re-alignment in hydrogels under deformation further enhance the stretchability of the hydrogels (Figure 12g)723,728,731,734–736. The fracture of the nano-/micro-fibers and pull-out of the nano/micro-fibers from the hydrogel matrices can dissipate substantial mechanical energy. A combination of the high stretchability and the mechanical dissipation enabled by the nano-/micro-fibrous polymer networks implements the design principle for tough hydrogels.
In addition to the abovementioned UNP architectures, the UPN interactions have also been widely used to implement the design principle for tough hydrogels737. The strong physical crosslinks such as crystalline domains and glassy nodules naturally act as high-functionality crosslinks for the corresponding UPN architectures (Figure 12b), which lead to tough hydrogels as discussed above.
The weak physical crosslinks550,568,607,738–745 and dynamic covalent crosslinks731 have been added into polymer networks with long chains (i.e., uncrosslinked or sparsely-crosslinked polymer networks) to design tough hydrogels. The weak physical crosslinks and dynamic covalent crosslinks act as reversible crosslinks in these hydrogels (Figure 12d). As the hydrogel deforms, many of these reversible crosslinks dissociate or de-crosslink to dissipate substantial mechanical energy and the sparsely-crosslinked longchain polymer network still sustains the high stretchability of the polymer network (Figure 12d). A synergy of the mechanical dissipation and the high stretchability enabled by the hybrid reversible and covalent crosslinks implements the design principle for tough hydrogels.
The weak physical crosslinks and dynamic covalent crosslinks have also been added into UPN architectures such as the interpenetrating polymer networks (Figure 12a)475,547,746–753, polymer networks with high-functionality crosslinks (Figure 12b)727,729,730,754–757, and nano-/micro-fibrous polymer networks (Figure 12c)734,735,755,758 to further toughen the resultant hydrogels, leveraging these reversible bonds’ capability of dissipating additional mechanical energy. Furthermore, unlike irreversibly-fractured polymer chains, the dissociated weak physical crosslinks and dynamic covalent crosslinks may re-associate due to their reversible nature, potentially endowing the tough hydrogels with recoverable dissipation over cyclic loads475. For further detailed discussion on the design principle and implementation strategies for tough hydrogels, a recent review paper is recommended60.
5.2. Strong: Synchronize stiffening and fracture of multiple polymer chains
Tensile strength.
Multiple types of strengths such as tensile strength, compressive strength and shear strength have been used to characterize the strength of a material. We will focus on tensile strengths of hydrogels in this paper, because the tensile strength is easier to measure than the shear strength and less affected by boundary conditions in the measurement (such as friction) than the compressive strength. Since soft materials such as elastomers and hydrogels usually do not yield plastically, their tensile strengths are commonly defined as the stresses at which the ultimate tensile failure occurs in the uniaxial tensile test. In addition, since the hydrogel samples usually undergo large deformation before failure, the tensile strength can be defined based on either the nominal stress or the true stress (Figure 13a),
| (22) |
where is the tensile force at the failure of the sample, A and a are the cross-section areas of the sample in the reference (undeformed) and current states, respectively, and and are the nominal and true tensile strengths, respectively. The nominal tensile strengths of hydrogels with conventional polymer networks and even tough hydrogels are commonly lower than 1 MPa63,475, much lower than the tensile strengths of engineering materials such as metals and biological tissues such as tendons.
Figure 13. Design principle for strong hydrogels – synchronize stiffening and fracture of multiple polymer chains:

a. definition and measurement of the tensile strength. A and a are the cross-section areas of the sample in the undeformed and deformed states, and F is applied tensile force. b. the simultaneous stiffening and fracture of substantial polymer chains give a high tensile strength759. Ff is the tensile force at the failure of the sample. c. the nominal tensile strength sc increases with the decrease of the defect size D up to a critical value Dc, below which the tensile strength is defect-insensitive760,761.
Design principle.
A generic principle for the design of strong hydrogels is to make a substantial number of polymer chains in the polymer network to stiffen and fracture simultaneously (Figure 13b). Following this principle, the nominal and true tensile strengths of the polymer network can be evaluated as
| (23) |
where is the force required to fracture a single polymer chain, which is on the order of a few nanonewton370, and and are the numbers of simultaneously fractured polymer chains per unit area of the hydrogel at the reference and deformed states, respectively. It has been evaluated that sf and σf can reach up to 1 GPa and 10 GPa, respectively, in an ideal scenario where all polymer chains in a polymer network fracture simultaneously759.
In realistic situations, almost all materials contain defects in the forms such as notches, microcracks, cavities, impurities, and missing polymer chains or crosslinks. The presence of defects usually significantly reduces the tensile strengths of the materials760–762. Without loss of generality, let’s assume the largest defect in the tensile sample is a notch with length D in the undeformed state perpendicular to the tensile direction (Figure 13c). The tensile strength of the sample generally increases with the decrease of the defect size D up to a critical value Dc, below which the tensile strength is defect-insensitive (Figure 13c). A scaling relation for the critical defect size Dc can be expressed as760,761
| (24) |
where Wc is the work for tensile failure of a unit volume of the defect-insensitive sample, and is the fracture toughness of the sample.
In order to achieve strong hydrogels, it is highly desirable for the hydrogel samples to have defectinsensitive tensile strengths759. According to Eq. 24, a tougher material (i.e., higher ) can be insensitive to larger defects due to a larger critical defect size Dc. For example, the critical defect size is on the order of a few nanometers for glass and ceramics, a few micrometers for brittle hydrogels, and a few millimeters for tough elastomers and hydrogels760,761. Furthermore, it is a common strategy to set the characteristic size of the sample (e.g., the diameter of the sample in Figure 13c) to be similar to or smaller than the critical defect size Dc, so that the tensile strength of the sample is guaranteed to be insensitive to any possible defect in the sample759.
Implementation.
The UPNs with high-functionality crosslinks such as crystalline domains have been widely used for the design of strong hydrogels68. As the hydrogels undergo large deformation, relatively short polymer chains are gradually pulled out of the crystalline domains, so that the polymer chains bridging adjacent crystalline domains tend to have similar lengths and therefore stiffen and fracture simultaneously – implementing the design principle for strong hydrogels (Figure 14a).
Figure 14. Implementation of the design principle for strong hydrogels – synchronize stiffening and fracture of multiple polymer chains.

Schematics on the implementation with a. polymer networks with high-functionality crosslinks, b. nano-/micro-fibrous polymer networks. c. confocal (left) and SEM (right) images of a fibrous PVA hydrogel with aligned fibers69. d. nominal stress-stretch curves of the fibrous PVA hydrogels with aligned and randomly-oriented fibers69. c and d are adopted from Ref69.
The nano-/micro-fibrous polymer networks is another type of UPN architecture that implements the design principle for strong hydrogels (Figure 14b). The diameters of the nano-/micro-fibers can be readily controlled below the critical defect sizes. Bundles of polymer chains in the nano-/micro-fibers can be designed to stiffen and fracture simultaneously to endow the fibers with high tensile strengths up to the ideal strengths (Figure 14b)69,728,731,736,763. Consequently, the resultant nano-/micro-fibrous hydrogels can reach extremely high tensile strengths (Figures 14c and d). Notably, biological hydrogels such as tendons, ligaments and muscles commonly adopt nano-fibers and micro-fibers, often in hierarchical architectures, to achieve high tensile strengths (Figure 2).
In addition to the abovementioned UPN architectures, the UPN interactions facilitate the implementation of the design principles for strong hydrogels. The strong physical crosslinks such as crystalline domains allow the pull-out of polymer chains from them to achieve simultaneous stiffening and fracture of multiple polymer chains (Figure 14a)68. The weak physical crosslinks such as the hydrogen bonds can facilitate the alignment and self-assembly of polymer chains into bundles (Figure 14b), which tend to stiffen and fracture simultaneously to give high tensile strengths of the hydrogels.
On a structural level, high-strength fibers made of polymers764,765, steel766, glass767,768 and wood769 have been utilized to strengthen hydrogels, and the tensile strengths of the resultant hydrogels are primarily determined by the strengths of the fibers.
5.3. Resilient: Delay dissipation
Resilience.
Resilience of soft materials such as elastomers and hydrogels is commonly defined as the ratio of energy released in deformation recovery to the energy required to induce the deformation of the materials770. Let’s consider a cylindrical sample under the uniaxial tensile test over a loading-unloading cycle (Figure 15a). The energy released in the unloading and the dissipated energy per unit volume of the sample are denoted as and , respectively. Therefore, the resilience R and hysteresis ratio H of the material can expressed as (Figure 15a)66,770
| (25) |
Figure 15. Design principle for resilient and tough hydrogels – delay dissipation:

a. definition and measurement of resilience. The relation of nominal stress s and stretchλ of a sample is measured under uniaxial tension in a loading-unloading cycle. WR and WD are the energy released in the unloading and the dissipated energy per unit volume of the sample, respectively. The resilience can be calculated as . b. when the stretch is below a critical stretch λR, the hydrogel releases most of the stored elastic energy during deformation recovery, giving high resilience; when the stretch is above λR, the hydrogel dissipates substantial mechanical energy, giving high fracture toughness66. c. the stretch in the process zone around the crack is usually much higher than λR, dissipating substantial mechanical energy and giving high fracture toughness66. b and c are adopted from Ref66.
The resilience R and the hysteresis ratio H depend on the material properties and the loading conditions such as the applied stretch and the applied stretch rate. The resilience of soft materials has been measured with many experimental methods such as the cyclic tensile test and the dropping-ball test66,770.
Design principle.
Once a material is deformed to fracture, the elastic energy stored in the material is mostly dissipated66,67,771, giving low energy recovery and thus low resilience. Therefore, the high resilience of hydrogels can only be designed up to the failure of the hydrogels. A generic principle for the design of resilient hydrogels is to minimize the mechanical dissipation of the hydrogels within certain ranges of deformation that is commonly experienced by the hydrogels, or in short, to delay dissipation66. Without loss of generality, we define a critical stretch for polymer chains in a hydrogel λR, below which the hydrogel can release most of the stored elastic energy during deformation recovery (i.e., , Figure 15b)66. Therefore, according to Eq. 25, the hydrogel will give a high resilience under the condition,
| (26) |
where λ and are the stretch and stretch limit of polymer chains in the hydrogel.
The design principle for resilient hydrogels also reconciles a pair of seemly contradictory properties, fracture toughness and resilience in the following manner. The hydrogel is highly resilient under common deformation with (Figure 15b); however, when a crack attempts to propagate in the hydrogel, the chain stretch in the process zone around the crack tip can be higher than λR, inducing substantial mechanical dissipation to toughen the hydrogel (Figure 15c). Indeed, biological hydrogels such as heart valves delay the mechanical dissipation up to supraphysiological deformation levels to achieve both high fracture toughness (e.g., over 1,000 Jm−2) and high resilience (e.g., over 99%) (Figure 2)772,773. Synthetic elastomers774, hydrogels66 and hydrogel composites70 have also been made both tough and resilient by following the design principle of delaying dissipation.
Implementation.
The ideal polymer networks are one common UPN architecture to implement the design principle for resilient hydrogels67,771. Because the polymer chains in the ideal polymer networks have relatively uniform lengths and no entanglement, the hydrogels with the ideal polymer networks usually can be deformed without significant mechanical dissipation up to stretch limits, giving high resilience67,771. It is also expected that the polymer networks with slidable crosslinks may be able to implement the design principle for resilient hydrogels, because the energy dissipated for sliding the crosslinks during their reconfiguration may be negligibly low. Despite being resilient, the ideal polymer networks and polymer networks with slidable crosslinks are not tough, since their fracture toughness is still the intrinsic fracture energy for fracturing a layer of polymer chains as discussed in Section 4.1.
The multimodal polymer networks including the interpenetrating polymer networks, semiinterpenetrating polymer networks, and polymer networks with high-functionality crosslinks usually begin to dissipate mechanical energy at very small deformation, because of the fracture and/or de-crosslink of very short polymer chains in the polymer networks. Such “early” dissipation gives narrow ranges of deformation for the hydrogels to be resilient in practical applications66. To address the issue of “early” dissipation, Lin et al. have pre-stretched the interpenetrating polymer networks up to λR over multiple cycles to fracture and/or de-crosslink susceptible short polymer chains and thus deplete possible dissipation mechanism within the deformation range of λR (Figure 16b)66. In subsequent tests or applications, if the chain stretch in the hydrogel is below λR, the hydrogel is highly resilient, due to the lack of mechanical dissipation within this range (Figures 16b and f). However, as the polymer chains are stretched beyond λR, for example, in the process zone around the crack tip, some of the polymer chains will be further fractured and de-crosslinked to dissipate mechanical energy and toughen the hydrogel (Figures 16b and f–h). It is expected that other multimodal polymer networks such as the semi-interpenetrating polymer networks and polymer networks with high-functionality crosslinks775 can be pre-stretched in a similar way to implement the design principle for resilient hydrogels (Figure 16c). Notably, when pre-stretching the multimodal polymer networks, the fracture and de-crosslink of polymer chains should be irreversible, so that the dissipation mechanism is irrecoverable once depleted69.
Figure 16. Implementation of the design principle for resilient and tough hydrogels – delay dissipation:

a. ideal polymer networks are resilient up to fracture due to the lack of dissipation mechanism. b. pre-stretching interpenetrating polymer networks to λR can make them both resilient and tough. c. prestretching polymer networks with high-functionality crosslinks to λR can make them both resilient and tough66. d. nano-/micro-fibrous polymer networks with resilient fibers can be both resilient and tough. e. the nominal stress-stretch curve of a resilient and tough fibrous PVA hydrogels69. f. the nominal stressstretch curves of a PAAm-alginate hydrogel with λR =5 66. g. the measured strain field around a crack in the PAAm-alginate hydrogel with λR =5. h. the stretch in the process zone can be much higher than λR = 5 66. e is adopted from Ref69. f, g and h are adopted from Ref66.
The nano-/micro-fibrous polymer networks can naturally implement the design principle for resilient hydrogels by constituting them with resilient nano-/micro-fibers and eliminating dissipation mechanisms (Figures 16d and e)69. In addition, because the energy required to fracture and pull out the nano-/micro-fibers can be much higher than the energy for fracturing amorphous polymer chains, resilient nano-/micro-fibrous hydrogels can also be tough69.
Besides the abovementioned UPN architectures, some UPN interactions also facilitate the implementation of the design principles for resilient hydrogels. The strong physical crosslinks such as crystalline domains provide the high-functionality crosslinks for some UPN architectures, which can be pre-stretched to give resilient hydrogels (Figure 16c)68. On the other hand, the weak physical crosslinks and dynamic covalent crosslinks are not suitable to implement the design principle for resilient hydrogels, because of their reversible and dissipative nature66.
On a structural level, resilient elastomeric fibers have been embedded into resilient hydrogel matrices to give resilient yet tough hydrogel composites70 to implement the design principle for resilient hydrogels69.
5.4. Tough adhesion: Integrate tough dissipative hydrogels and strong interfacial linkages
Interfacial toughness.
Interfacial toughness, or so-called practical work of adhesion, has been commonly used to characterize the capability of the interface of two adhered materials to resist fracture under mechanical loads. One common definition for the interfacial toughness between two adhered materials is the energy required to propagate a crack along the interface or in either material over a unit area measured in the undeformed state of the materials (Figure 17a)776. Depending on whether the crack propagates along the interface or in either material, the failure mode is called an adhesive failure or cohesive failure, respectively (Figure 17a). Quantitatively, the interfacial toughness can be expressed as,
| (27) |
where U is the total potential energy of the system, A is the crack area measured in the undeformed state, and Gc is the critical energy release rate that drives interfacial crack propagation. According to Eq. 27, the unit for the interfacial toughness is joule per meter squared (i.e., J m−2).
Figure 17. Design principle for tough adhesion of hydrogels – integrate tough dissipative hydrogels and strong interfacial linkages.

a. definition of interfacial toughness and the 90-degree peeling test to measure the interfacial toughness. F is the peeling force, is the plateau peeling force, and W is the width of the sample. The interfacial toughness can be calculated as based on the values of and W measured in the 90-degree peeling test. b. weak interface can give the adhesive failure mode. c. brittle hydrogel matrix can give the cohesive failure mode. d. integration of tough dissipative hydrogels and strong interfacial linkages gives tough adhesion of hydrogels43. The contributions of strong interfacial linkages and mechanical dissipation in the process zone to the total interfacial toughness are and , respectively. The total interfacial toughness of the tough adhesion is d is adopted from Ref43.
The interfacial toughness of soft materials such as elastomers and hydrogels has been measured with many experimental methods such as the 90-degree peeling test, the T-peeling test and the lap-shear test714,776. For example, in the 90-degree peeling test, a layer of a hydrogel with thickness T, width W and length L (L >> W >> T) is bonded on a substrate, and a notch is introduce on the interface along the length direction (Figure 17a). The detached part of the hydrogel is further peeled off the substrate, while maintaining vertical to the substrate (Figure 17a). The measured force reaches a plateau as the peeling process enters the steady state, and the interfacial toughness is determined by dividing the plateau force by the width of the hydrogel sheet W, i.e. .
If a hydrogel with a conventional polymer network is strongly bonded on a substrate (e.g., via covalent bonds), the interfacial toughness is on the level of the hydrogel’s fracture toughness or intrinsic fracture energy . This is because the fracture toughness of the hydrogel poses an upper limit for the interfacial toughness, since the cohesive failure mode may occur (Figure 17c)43. Therefore, evaluated with typical parameters of conventional polymer networks, the interfacial toughness of the hydrogel is bounded by a few tens of joule per meter squared. If the hydrogel is adhered on the substrate via a low density of weak physical crosslinks such as hydrogen bonds and electrostatic interactions, the interfacial toughness can be even lower, since the adhesive failure mode may occur (Figure 17b)42.
Design principle.
As discussed in the previous part, if a hydrogel adheres to a substrate via a low density of weak physical crosslinks, a crack can easily propagate along the hydrogel-substrate interface, resulting in low interfacial toughness (Figure 17b). Therefore, the design of tough adhesion of hydrogels first requires strong interfacial linkages between the hydrogels and the adhered substrates, such as covalent bonds43,777,778, strong physical crosslinks42,71,779, connector polymers26,775,780,781, and mechanical interlocks782,783. In addition, because the interfacial crack can tilt into the bulk hydrogel and develop the cohesive failure mode (Figure 17c), the design of tough adhesion of hydrogels further requires high fracture toughness of the hydrogel matrices by themselves43.
Overall, the design principle for tough adhesion of hydrogels is to integrate tough dissipative hydrogel matrices and strong interfacial linkages43. When attempting to detach the tough hydrogel from the substrate, the strong interfacial linkages will hold the interfacial crack tip, allowing the bulk hydrogel to develop a process zone with substantial mechanical dissipation (Figure 17d). Quantitatively, the total interfacial toughness can be expressed as43,778
| (28) |
where , , and are the total interfacial toughness, the intrinsic interfacial toughness due to strong interfacial linkages, and the contribution of mechanical dissipation in the process zone to the total interfacial toughness, respectively.
Tough adhesion of biological hydrogels in animal bodies such as cartilages, tendons and ligaments on bones generally relies on the integration of tough hydrogels and strong interfacial linkages. However, only recently has this design principle been proposed43 and implemented26,41,43,775,777,780,781,784 for tough adhesion of synthetic hydrogels on diverse substrate materials, including metals, ceramics, glass, silicone, elastomers, hydrogels and biological tissues, partially because the role of tough dissipative hydrogel matrices has been underexplored or underestimated in adhesion43,785,786. Notably, strong interfacial linkages and/or bulk dissipation of the adherents have also been widely employed for tough bonding of engineering materials such as metals787 and rubbers788,789 on substrates.
Implementation.
Since the implementation of tough hydrogels has been discussed in Section 5.1, we will focus on how to implement the strong interfacial linkages to bond tough dissipative hydrogels on various substrates in this section. In order to achieve tough adhesion, the intrinsic interfacial toughness of the interfacial linkages should at least reach the level of the intrinsic fracture energy of tough hydrogels, i.e. over a few tens of joule per meter squared790. Given this requirement on the intrinsic interfacial toughness, the strong interfacial linkages have been commonly implemented with covalent bonds, strong physical crosslinks, connector polymers, and mechanical interlocks.
Covalent bonds have been widely adopted to strongly anchor polymer chains in tough hydrogels’ UPNs (as discussed in Section 5.1) on various substrates. The commonly-used covalent bonds for tough adhesion of hydrogels include carbon-carbon, carbon-nitrogen, carbon-sulfide, carbon-oxygen, siloxane bonds (Figure 4c)791. In order to form these covalent bonds, the hydrogels and substrates are usually designed to possess functional groups such as radical crosslinkable unsaturated bond (to form carboncarbon bond)792, amine group (to form carbon-nitrogen bond)43, thiol group (to form carbon-sulfide bond)793, hydroxyl and carboxyl group (to form carbon-oxygen bond), and silanol group (to form silicon-oxygen bond)794 (Figure 18a). According to the Lake-Thomas model, the intrinsic interfacial toughness of polymer chains covalently anchored on a substrate can be expressed as
| (29) |
where is the number of covalently-anchored polymer chains on a unit area of the substrate in the undeformed reference state, N is the number of Kuhn monomers per polymer chain, and Uf is the lower value of the energy required to fracture either the Kuhn monomer or the covalent bond on the substrate. According to Eq. 29, anchoring longer polymer chains with a higher density of covalent bonds on a substrate will give a higher value of the intrinsic interfacial toughness778,790.
Figure 18. Implementation of the design principle for tough adhesion of hydrogels – integrate tough hydrogels and strong interfacial linkages.
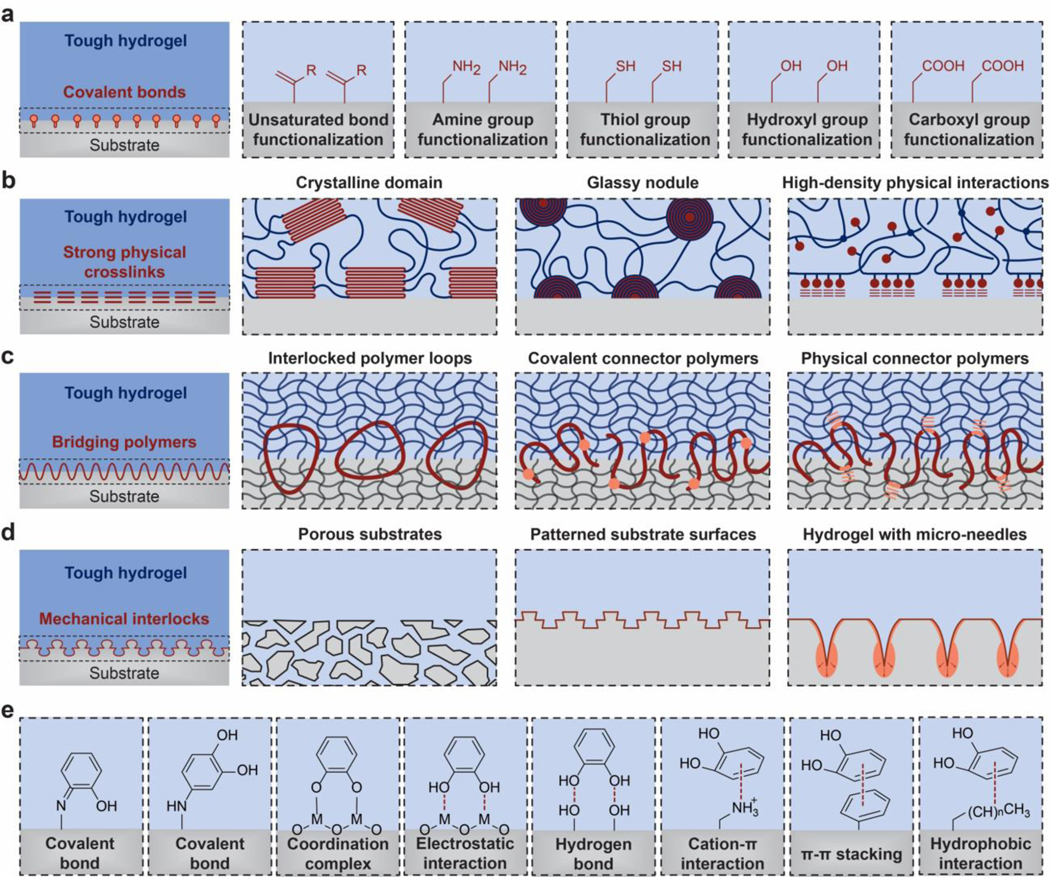
The tough UPNs are bonded on substrates via various types of strong interfacial linkages: a. covalent bonds, b. strong physical crosslinks, c. bridging polymers, and d. mechanical interlocks. e. catechol interactions can implement various types of strong interfacial linkages. e is adopted from Ref808,811.
Strong physical crosslinks including crystalline domains, glassy nodules, and high-density physical bonds such as hydrogen bonds can also strongly adhere tough hydrogels on substrates (Figure 18b)71,745,793–801. Since the crystalline domains and glassy nodules usually act as high-functionality crosslinks, each of them may anchor multiple polymer chains on the substrate, further enhancing the intrinsic interfacial toughness .
Connector polymers788,802 have been employed to strongly bond elastomers and hydrogels on substrates (Figure 18c). In this case, the substrates usually take the form of polymer networks (i.e., elastomers and hydrogels) as well. To provide strong interfacial linkages, the connector polymers can form covalent crosslinks775,803, interlocked loops780,781,793,797,804, and/or strong physical bonds781 with the polymer networks of both the hydrogels and the substrates. Specifically, strong physical interactions can be crystalline domains, glassy nodules, and/or high-density weak physical crosslinks781. The connector polymers can be polymerized from monomers across the interface of the two polymer networks780,794,803 or can be directly added on the interface to diffuse into the two polymer networks781.
Mechanical interlocks between tough hydrogels and substrates usually occur at length scales from micrometers to millimeters (Figure 18d). One commonly-used method is to impinge precursor solutions of tough hydrogels into porous substrates and then form tough hydrogels that are mechanically interlocked with the substrates782. Similarly, the surfaces of the substrates can be roughened or patterned to enhance the strength of mechanical interlocks with tough hydrogels783,805,806. As a special yet interesting case, hydrogels have been fabricated into dried microneedles, which can pierce into a soft substrate such as biological tissues and then swell to form mechanical interlocks807.
Inspired by the adhesive proteins found in mussels, catechol chemistry has been widely adopted to achieve various types of interfacial linkages between hydrogels and substrates (Figure 18e)563,779. Catechol can form both covalent and physical crosslinks with various functional groups (Figure 18e). Upon oxidation to quinone, catechol can form a covalent bond with nucleophiles (e.g, amine and thiol) via Michael addition as well as a strong coordination complex with metal oxides808. The hydroxyl groups of catechol can form electrostatic interaction with metal oxides as well as hydrogen bonds with hydrophilic substrates. The benzene ring of catechol can further form cation- π interaction with positively charged functional groups, π-π stacking with benzene functional groups, and hydrophobic interaction with hydrophobic functional groups on substrates779,808. While catechol chemistry has been widely utilized for adhesion of hydrogels to various substrates, the interfacial toughness of the adhesion achieved only by catechol-based interfacial linkages is not high809, highlighting the importance of tough dissipative hydrogel matrices on top of the interfacial linkages to achieve tough adhesion810.
5.5. Fatigue-resistant: Pin fatigue cracks by intrinsically high-energy phases
Fatigue threshold.
The word “fatigue” has been used to describe many symptoms observed in materials under prolonged loads, including materials with or without precut cracks under prolonged static or cyclic loads812,813. In this section, we will focus on the fatigue fracture of hydrogels with precut cracks under cyclic loads (Figure 19a), because this is one of the most common failure modes of hydrogels used in mechanically dynamic environments such as artificial cartilages814 and soft robots26. Fatigue threshold has been commonly used to characterize a material’s resistance to fatigue crack propagation under cyclic loads. The fatigue threshold is defined as the minimal fracture energy at which fatigue crack propagation occurs under infinite cycles of loads48,49. Quantitatively, the fatigue threshold can be expressed as,
| (30) |
where G is the energy release rate to drive crack propagation under each cycle of load, Gc is the minimal energy release rate at which crack propagation occurs under infinite cycles of loads,c is the length of the crack, N is the cycle number of the applied load, and dc /dN gives the crack extension per cycle.
Figure 19. Design principle for fatigue-resistant hydrogels – pin cracks by intrinsically high-energy phases.

a. definition of fatigue threshold and the pure-shear method to measure fatigue threshold. G is the energy release rate, c is the crack length, and N is the cycle number. The fatigue threshold is determined by intersecting the curve of dc / dN vs G with the G axis. b. dissipation mechanisms such as reversible crosslinks in tough hydrogels are depleted over cyclic loads, not contributing to the fatigue threshold. c. fatigue crack is pinned by intrinsically high-energy phases in fatigue-resistant hydrogels68. b and c are adopted from Ref68.
The fatigue threshold of soft materials such as elastomers and hydrogels have been measured with various experimental methods such as the pure-shear fatigue-fracture test and the single-notch fatigue-fracture test68. For example, in the pure-shear fatigue-fracture test, two identical pieces of a hydrogel are fabricated with the same thickness T, width W and height H, where W >> H >> T (Figure 19a). Both pieces of samples are clamped along their long edges (i.e., along the width direction) with rigid plates. The first sample is repeatedly pulled to a stretch of times of its undeformed height to measure the nominal stress s - stretch λ relation, and the corresponding energy release rate can be calculated as , which is a function of the cyclic number N (Figure 19a). Thereafter, a notch with a length of ~0.5W is introduced into the second sample, which is then repeatedly pulled to the same stretch to measure the crack length c as a function of the cyclic number N. The pure-shear fatigue-fracture tests are repeated for different values of the applied stretch (i.e., energy release rate G), and a curve of dc / dN vs. G can be obtained (Figure 19a). The fatigue threshold is then determined by intersecting the curve of dc / dN vs. G with the G axis (i.e., when dc / dN= 0). Notably, the fatigue fracture tests of hydrogels are commonly carried out in aqueous environments to avoid dehydration of the hydrogels under prolonged loads68,69. For further discussion on the theory and experiments for the fatigue of hydrogels, a comprehensive review paper on this topic is recommended813.
Design principle.
As discussed in Section 5.1, a hydrogel can be designed tough by building mechanical dissipation into stretchy polymer networks60. The mechanical dissipation in the process zone around the crack tip can dramatically enhance the fracture toughness of the hydrogel (by in Eq. 21). However, the mechanisms for irreversible dissipation such as fracturing polymer chains in the process zone are usually depleted under cyclic loads. The mechanisms for reversible dissipation such as reversible crosslinks, once depleted, usually cannot recover in time to resist fatigue crack propagation in future cycles of loads (Figure 19b)48,49,815. Consequently, the fatigue threshold of hydrogels and rubbers is their intrinsic fracture energy48,49,815,
| (31) |
Therefore, it is clear that the design of fatigue-resistant hydrogels generally cannot rely on mechanical dissipation in the bulk hydrogel matrices.
The design principle for fatigue-resistant hydrogels is to make the fatigue crack encounter and fracture objects with energies per unit area much higher than that for fracturing a single layer of polymer chains, or in short, to pin fatigue crack by intrinsically high-energy phases (Figure 19c)68. The intrinsically high-energy phases that have been exploited for the design of fatigue-resistant hydrogels include nanocrystalline domains (Figure 20a)68, nano-/micro-fibers (Figure 20b)69, microphase separations (Figure 20c)813,816, and macroscale composites (Figure 20d)70. In addition, because the design of fatigue-resistant hydrogels does not rely on mechanical dissipation in the bulk hydrogels, fatigue-resistant hydrogels usually also demonstrate low hysteresis ratio H and high resilience R (Eq. 23)68,69,817. Notably, biological hydrogels such as muscles, tendons and ligaments commonly possess intrinsically high-energy phases such as nano/micro-fibers, usually arranged in hierarchical architectures, to achieve a high fatigue threshold (Figure 2).
Figure 20. Implementation of the design principle for fatigue-resistant hydrogels – pin cracks by intrinsically high-energy phases.

Fatigue cracks can be pinned by intrinsically high-energy phases including a. nano-crystalline domains68, b. nano-/micro-fibers69, c. micro-phase separations816, d. macro-fibers70. e. Molecular dynamic simulation of the energy for pulling a polymer chain out of a PVA nano-crystalline domain and for fracturing the same polymer chain71. d is the displacement of one end of the polymer chain, and U is the energy required to achieve the displacement. f. confocal microscope image of a crack pinned by nano-fibers in a nano-fibrous PVA hydrogel, and g. measurement of the fatigue threshold of the nano-fibrous PVA hydrogel69. G is the energy release rate, c is the crack length, and N is the cycle number. a is adopted from Ref68, b is adopted from Ref69, c is adopted from Ref816, d is adopted from Ref70, e is adopted from Ref71, f and g are adopted from Ref69.
Implementation.
The design principle for fatigue-resistant hydrogels has been implemented with the UPNs that possess intrinsically high-energy phases68–70,813,816. In order to effectively pin fatigue cracks, the density of the intrinsically high-energy phases in the UPNs should be sufficiently high68.
High-functionality crosslinks such as crystalline domains can effectively play the role of intrinsically high-energy phases in the UPNs (Figure 20a). The energy required to pull out a polymer chain from a crystalline domain can be multiple times of that to fracture the same polymer chain, and the energy required to mechanically damage the crystalline domains can be multiple times of that to fracture the corresponding amorphous polymer chains (Figure 20e)71. Consequently, it has been shown that enhancing the crystallinity of a PVA hydrogel from 0.2 wt. % to 18.9 wt. % by dry-annealing can increase its fatigue threshold from 10 Jm−2 to 1,000 Jm−2, reaching the level of fatigue-resistant biological hydrogels such as cartilages for the first time68. Since the size of the crystalline domains in the PVA hydrogel has been measured to be a few nanometers, it is the nano-crystalline domains that play the role of intrinsically high-energy phases here (Figure 20a). It is expected other UPNs with sufficiently high densities of high-functionality crosslinks such as crystalline domains and glassy nodules can also implement the design principle for fatigue-resistant hydrogels. It should be further noted that hydrogels with high densities of rigid crystalline domains and glassy nodules can be much stiffer than common hydrogels68, and such high stiffness may be undesirable for many applications of hydrogels.
Nano-/micro-fibers can also act as intrinsically high-energy phases in the UPNs to implement the design principle for fatigue-resistant hydrogels (Figure 20b). The energy required to fracture a nano/micro-fiber can be much higher than that to fracture the corresponding amorphous polymer chains, because of the synergistic elongation and stiffening of bundled polymer chains in the fiber69. Based on this implementation strategy, it has been shown that introducing nano-fibers into a PVA hydrogel by freeze-thawing the hydrogel can enhance its fatigue threshold from 10 Jm−2 to 310 Jm-2. In particular, if the nanofibers are aligned perpendicular to the fatigue crack by pre-stretching the hydrogel, the measured fatigue threshold further increases to 1,250 Jm−2 (Figures 20f and g) 69. In addition, because the nano-fibers can be made compliant, stretchable and strong by using a low density of nano-crystalline domains to bundle polymer chains (Figure 14b), the resultant nano-fibrous hydrogel integrates high compliance, stretchability and strength together with high fatigue threshold – mimicking the combinational mechanical properties of biological muscles69.
Phase separations in hydrogels can also enhance the fatigue threshold of the hydrogels813,816, possibly because the energy required to fracture the separated phases is higher than that to fracture the corresponding amorphous polymer chains. The UPN interactions including reversible covalent bonds and weak physical crosslinks play critical roles in inducing the phase separations in the hydrogels813,816.
On a structural level, macroscale resilient elastomer fibers have been embedded in a resilient hydrogel to form a macroscale composite70. Since it requires much higher energy to fracture the elastomer fibers than a layer of amorphous polymer chains, a fatigue threshold over 1,000 Jm−2 has been achieved for the macroscale composite70.
5.6. Fatigue-resistant adhesion: Strongly bond intrinsically high-energy phases on interfaces
Interfacial fatigue threshold.
The interfaces of adhered materials can suffer from fatigue failure under prolonged loads, including interfaces with or without precut cracks under prolonged static or cyclic loads. In this section, we will focus on the fatigue fracture of hydrogels adhering to substrates with precut cracks on their interfaces under cyclic loads (Figure 21a). Depending on whether the fatigue crack propagates along the interface or tilts into the hydrogel under cyclic loads, the failure mode is called adhesive failure or cohesive failure, respectively (Figure 21a)71. Interfacial fatigue threshold has been commonly used to characterize the capability of adhered materials to resist interfacial fatigue crack propagation following either failure mode under cyclic loads. The interfacial fatigue threshold is defined as the minimal fracture energy at which interfacial crack propagation occurs under infinite cycles of loads71,818–820. Similar to the fatigue threshold, the interfacial fatigue threshold can be expressed as,
| (32) |
where G is the energy release rate to drive interfacial crack propagation under each cycle of load, Gc is the minimal energy release rate at which interfacial crack propagation occurs under infinite cycles of loads, c is the length of the crack, N is the cycle number of the applied load, and dc / dNgives the crack extension per cycle.
Figure 21. Design principle for fatigue-resistant adhesion of hydrogels – strongly bond intrinsically high-energy phases on interfaces:

a. definition of interfacial fatigue threshold and the 90-degree cyclic peeling test to measure the interfacial fatigue threshold. F is the applied peeling force, W is the width of the sample, G is the energy release rate, c is the crack length, and N is the cycle number. The interfacial fatigue threshold is determined by intersecting the curve of dc / dN vs G with the G axis. b. fatigue-crack propagation along the interface giving adhesive failure. c. fatigue-crack propagation in the hydrogel giving cohesive failure. d. fatigue-crack pinned by intrinsically high-energy phases on the interface and in the bulk hydrogel71. d is adopted from Ref71.
The interfacial fatigue threshold of soft materials such as elastomers and hydrogels has been measured with many experimental methods such as the cyclic 90-degree peeling test, the cyclic T-peeling test and the cyclic lap-shear test71,818–820. For example, in the cyclic 90-degree peeling test71,818, a layer of a hydrogel with thickness T, width W and length L (L >> W >> T) is bonded on a substrate, and a notch is introduce on the interface along the length direction (Figure 21a). A force F is repeatedly applied on the detached part of the hydrogel, while maintaining the detached part vertical to the substrate (Figure 21a). The applied force F gives the energy release rate , where W is the width of the hydrogel sheet. The interfacial crack length c is then measured as a function of the cyclic number N. The cyclic 90-degree peeling test are repeated for different values of the applied force F (i.e., energy release rate G), and a curve of dc / dN vs.G can be obtained (Figure 21a). The interfacial fatigue threshold is determined by intersecting the curve of dc / dN vs. G with the G axis (i.e., when dc / dN = 0). Notably, the interfacial fatigue fracture tests of hydrogels are commonly carried out in aqueous environments to avoid dehydration of the hydrogels under prolonged loads71.
Design principle.
As discussed in Section 5.4, tough adhesion of hydrogels on substrates relies on the integration of tough dissipative hydrogel matrices and strong interfacial linkages (Figure 17)43. The strong interfacial linkages can hold the interfacial crack tip, while the mechanical dissipation in the process zone around the crack tip can dramatically enhance the interfacial toughness of the adhesion. However, similar to the situation in fatigue of hydrogels48,49,815, because the mechanical dissipation in bulk hydrogel matrices is usually depleted or not timely accessible after cyclic loads, such dissipation generally cannot contribute to resisting interfacial fatigue crack propagation (Figures 21b and c)71,820. Consequently, the interfacial fatigue threshold of hydrogels and elastomers is their intrinsic interfacial toughness71,820,
| (33) |
Therefore, the design of fatigue-resistant adhesion of hydrogels generally cannot rely on mechanical dissipation in the bulk hydrogel matrices.
Because interfacial cracks can tilt into bulk hydrogels and develop the cohesive failure mode (Figure 21c), the design of fatigue-resistant adhesion of hydrogels requires fatigue-resistant hydrogel matrices that possess sufficiently high densities of intrinsically high-energy phases68. Notably, hydrogel matrices that are only tough but not fatigue-resistant are unsuitable for the design of fatigue-resistant adhesion, owing to the depletion of dissipation over cyclic loads. To further avoid the adhesive failure mode under cyclic loads (Figure 21b), fatigue cracks on the interfaces need to be pinned by intrinsically high-energy phases strongly bonded on the interfaces as well (Figure 21d).
Therefore, the design principle for fatigue-resistant adhesion of hydrogels, in short, is to strongly bond intrinsically high-energy phases on interfaces71. While the intrinsically high-energy phases that have been employed for the design of fatigue-resistant adhesion include nano-crystalline domains71 and exceptionally long polymer chains (in unfully swollen sate)819, other candidates such as nano-/micro-fibers can be explored in the future. Not surprisingly, biological hydrogels including tendons, ligaments and cartilages all rely on strongly bonding crystalline domains and nano-/micro-fibers throughout their interfaces with bones to achieve fatigue-resistant adhesion745.
Implementation.
The design of fatigue-resistant hydrogels relies on achieving sufficiently high densities of intrinsically high-energy phases in the UPNs, as discussed in Section 5.6. In this section, we will focus on how to strongly bond intrinsically high-energy phases on substrates to implement the design principle for fatigue-resistant adhesion.
High-functionality crosslinks such as crystalline domains usually can play the role of intrinsically high-energy phases in the UPNs. Nano-crystalline domains in PVA hydrogels have been strongly bonded on diverse substrates including glass, ceramics, metals and elastomers via high-density hydrogen bonds (Figure 22a)71. The molecular dynamic simulation shows that the energy required to pull a polymer chain out of the interface is even higher than that to pull the same polymer chain out of the nano-crystalline domain, implying high intrinsic interfacial toughness of the adhesion 71. As a result, the interfacial fatigue threshold of the PVA-substrate systems measured in phosphate-buffered saline reaches up to 800 Jm−2, similar to those of tendon/ligament/cartilage-bone interfaces (Figures 22d and e). In addition, the failure mode of the PVA-substrate systems observed in the interfacial fatigue fracture tests follows the cohesive failure, indicating the critical role of intrinsically high-energy phases in the bulk hydrogels (i.e., fatigue-resistant hydrogels) for the design of fatigue-resistant adhesion71.
Figure 22. Implementation of the design principle for fatigue-resistant adhesion of hydrogels – strongly bond intrinsically high-energy phases on interfaces:

The intrinsically high-energy phases can be bonded on the substrates via a. high-density physical bonds such as hydrogen bonds71, b. covalent bonds, and c. mechanical interlocks. d. measurements of the fatigue thresholds of tough adhesion and fatigueresistant adhesion of hydrogels on substrates71. e. photos of interfacial crack propagation in a cyclic peeling test for tough adhesion (top) and fatigue-resistant adhesion (bottom) of hydrogels on substrates71. a, d and e are adopted from Ref71.
Covalent bonds are also expected to be able to strongly bond the intrinsically high-energy phases such as nano-crystalline domains and nano-/micro-fibers on substrates (Figure 22b). In addition, curing precursor solutions of fatigue-resistant hydrogels on porous, roughened or patterned substrates can lead to mechanical interlocks that may strongly bond intrinsically high-energy phases on the hydrogel-substrate interfaces (Figure 22c)821.
5.7. Implementation with unconventional polymer networks
While the implementation of each design principle for the corresponding extreme property has been discussed in Sections 5.1–5.6, we will provide an overview of the design process and implementation strategy in this section.
Since the design principles discussed in Sections 5.1–5.6 are general and abstract, it is usually more intuitive to begin the design process with specific UPN architectures and/or UPN interactions than with a design principle. The commonly-used UPN architectures that can give extreme mechanical properties include interpenetrating polymer networks, semi-interpenetrating polymer networks, polymer networks with high-functionality crosslinks, and nano-/micro-fibrous polymer networks. The commonly-used UPN interactions that can give extreme mechanical properties include various types of strong physical crosslinks and weak reversible crosslinks. Let’s imagine that the selected UPN is subjected to the relevant modes of mechanical loads such as tension, compression, shear, fracture, fatigue and/or peeling. If the selected UPN under mechanical loads seems to be able to implement the design principle for the desired property, one can proceed to select polymers and crosslinks such as those discussed in Section 2 and Section 4 for the design and fabrication of the hydrogel. Furthermore, it may be difficult to initiate the design process by considering both UPN architectures and UPN interactions simultaneously; in this case, we can first test whether a UPN architecture will likely implement the design principle, and then further add UPN interactions into the UPN architecture to facilitate the implementation. For example, in order to design a fatigue-resistant hydrogel, we can begin with a polymer network with high-functionality crosslinks, because a sufficiently high density of high-functionality crosslinks can act as intrinsically high-energy phases to pin fatigue cracks. Furthermore, strong physical crosslinks such as crystalline domains and glassy nodules can be added into the polymer network as the intrinsically high-energy phases to facilitate the implementation of the design principle. Indeed, dry-annealed PVA with high densities of nano-crystalline domains has been selected to implement the design principle of fatigue-resistant hydrogels68.
Alternative design and implementation strategy are through the mimicry of the UPNs of biological hydrogels that possess the extreme mechanical properties to be designed for synthetic hydrogels (Figure 2). Because biological hydrogels have exploited various types of UPNs to implement the design principles discussed in Sections 5.1–5.6, we can simply begin the design process by replicating biological hydrogels’ UPNs (Figure 2). Notably, the biological UPNs, commonly featuring hierarchical and gradient structures (Figure 2), can be more complex than the UPNs discussed in Section 4; therefore, we should only mimic the essential characteristics of the biological UPNs that enable the desired mechanical properties. As an example, tendons, ligaments and cartilages all feature fatigue-resistant adhesion on bones, owing to nano/micro-fibers and nano-crystalline domains strongly anchored on the interfaces (Figure 2). Such fatigueresistant adhesion of PVA hydrogels on diverse solid substrates have been recently achieved by strongly anchoring nano-crystalline domains in PVA hydrogels on the substrates via densely-distributed hydrogen bonds (Figures 22d and e)71.
6. Design of hydrogels with extreme physical properties
In addition to the extreme mechanical properties discussed in Section 5, the design of hydrogels that possess extreme physical properties has attracted escalating research interests in recent years. Examples of hydrogels’ extreme physical properties under development and exploration include high electrical conductivity627, patterned magnetization822, high refractive index and transparency823,824, tunable acoustic impedance825, and self-healing826. Unlike the extreme mechanical properties discussed in Section 5, many of the extreme physical properties do not have embodiments in biological hydrogels. Nevertheless, these extreme physical properties can be of similar importance as the extreme mechanical properties to hydrogels’ various applications, especially to the nascent applications of hydrogel machines20. In this section, we will briefly discuss the design principles and implementation strategies for hydrogels to possess these extreme physical properties, while bearing in mind that many works in this field are still in the initial stage of developments.
6.1. Electrically conductive: Percolate electrically conductive phases
Electrical conductivity is critical for hydrogels’ nascent applications such as bioelectrodes for stimulation and recording of neural activities in bioelectronics627 and electrodes for supercapacitors and batteries in energy storage20,827. However, the electrical conductivity of common hydrogels is less than a few Siemens per meter, on the same level as that of water or saline water627. Compared to metals, carbon and conducting polymers, common hydrogels are usually deemed to be electrically nonconductive.
The design principle for electrically conductive hydrogels is to embed electrically conductive phases such as liquid metals, metallic nanowires, carbon nanotube, graphene and conducting polymers in hydrogel matrices and make the conductive phases form percolated networks, or in short, to percolate electrically conductive phases (Figure 23a)627,828,829. In particular, conductive hydrogels based on conducting polymers have attracted great interests recently, owing to their unique polymeric nature as well as favorable electrical and mechanical properties, stability, and biocompatibility29,30,480,624,830–833. For example, poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) has been made into pure conducting polymer hydrogels that achieve high electrical conductivity over a few thousand Siemens per meter and superior biocompatibility (Figure 23c)29,30,624. In addition to electrically conductive hydrogels, ionically conductive hydrogels have also been intensively developed as stretchable and transparent ionic conductors for various applications (Figure 23b)27. The conductive phases in ionically conductive hydrogels are usually high concentrations of salt ions. For a further detailed discussion on various types of conductive hydrogels, a recent review paper is recommended627.
Figure 23. Design of hydrogels with high electrical conductivity – percolate electrically conductive phases.

a. hydrogels with percolated electrically conductive fillers. b. hydrogels with ionically conductive salt solvents. c. hydrogels based on conducting polymers. The bottom panel of a is adopted from Ref829. The bottom panel of b is adopted from Ref27. The bottom panels of c are adopted from Ref834 (left) and Ref29 (right).
6.2. Magnetized: Embed magnetic particles and pattern ferromagnetic domains
Soft materials such as elastomers and hydrogels with ferromagnetic domains or magnetization have been intensively developed and explored for biomedical applications such as drug delivery and minimally invasive surgery28,822,835–839, owing to their mechanical compliance, potential biocompatibility, and capability of fast deformation under applied magnetic fields. Common hydrogels are usually diamagnetic and do not contain ferromagnetic domains, possessing similar magnetic properties as water. Therefore, subjected to applied magnetic fields, common hydrogels cannot be actuated to deform, exert forces, or release substances.
The design principle for hydrogels to possess patterned magnetization is to embed magnetic components such as hard-magnetic, soft-magnetic and super-paramagnetic particles in the hydrogels matrices where ferromagnetic domains may be further patterned, or in short, to embed magnetic particles and pattern ferromagnetic domains (Figure 24) 822,835–839. In particular, hard-magnetic particles such as neodymium iron boron (NdFeB) particles after magnetic saturation can retain their magnetization under actuation magnetic fields, because of the high coercivity of the hard-magnetic particles (Figure 24a). Therefore, patterned ferromagnetic domains can be programmed into elastomers and hydrogels embedded with hard-magnetic particles. Subjected to actuation magnetic fields, the elastomers and hydrogels with the patterned ferromagnetic domains can quickly transform among various shapes 835–839. Recently, 3D printing has been further exploited as an effective method to program complex 3D shapes as well as domain patterns in ferromagnetic elastomers and hydrogels (e.g., Figures 24b and c)836,839. It should be noted that magnetic particles can be corrosive in the aqueous environments of hydrogel matrices. To enhance their chemical stability in hydrogel matrices, the magnetic particles have been coated with protective layers such as silica layers (Figures 24b and c)835.
Figure 24. Design of hydrogels and elastomers with patterned magnetization – embed magnetic particles and pattern ferromagnetic domains.

a. typical relations of applied magnetic field H and magnetization M for paramagnetic, soft-magnetic, and hard-magnetic materials. Mr and Hc are the residual magnetization and coercivity of the hard-magnetic material, respectively. b. hard-magnetic particles can be embedded into an elastomer/hydrogel matrix, in which ferromagnetic domains can be patterned by 3D printing. c. Photos of the resultant magnetic soft material before and after magnetic actuation. a is adopted from Ref840. b and c are adopted from Ref841.
6.3. High reflective index and transparency: Uniformly embed high-refractive-index non-scattering nano-phases.
Various optical applications of hydrogels such as ophthalmic lenses21,364,842 and optical fibers33,843 require high refractive indices and high transparency of the hydrogels (Figure 25a). The refractive indices of common hydrogels are around 1.333, similar to that of water. One general strategy to enhance the refractive indices of hydrogels is to uniformly embed nano-phases such as nano-particles823,824 and nano-crystalline domains with high refractive indices in the hydrogel matrices. However, the refractive-index mismatch between the nano-phases and hydrogel matrices may lead to substantial undesirable light scattering, reducing the transparency of the hydrogels (Figure 25b). It has been found that decreasing the size of the nano-phases below one-tenth of the light wavelength (e.g., zinc sulfide nanoparticles with 3 nm diameter) can effectively diminish light scattering to achieve hydrogels with a high refractive index (i.e., 1.49) and high transparency (Figures 25b–d)823. Overall, the design principle for hydrogels with high refractive indices and transparency is to uniformly embed high-refractive-index non-scattering nano-phases in hydrogel matrices.
Figure 25. Design of hydrogels with high reflective indices and transparency – uniformly embed high-refractive-index non-scattering nano-phases.

a. high contrast between reflective indices of the hydrogel fiber and tissue fluid can give minimal light leakage. b. uniformly embedding nano-phases such as nano-particles with high refractive indices in the hydrogel matrices can enhance the refractive index of the hydrogel. The size of the nano-phases dNC should be much smaller than the light wavelength λ for minimal scattering and high transparency. c. hydrogels with high reflective indices and transparency can be used as optical fibers in living tissues. d. photo of a hydrogel optical fiber843. d is adopted from Ref843.
6.4. Tunable acoustic impedance: Tune densities and bulk moduli of effectively homogeneous hydrogels
Hydrogels have been widely used as the media for sound-wave transmissions such as the coupling agents for imaging and therapeutic ultrasounds. It is highly desirable to design hydrogels that possess tunable acoustic impedance to match the impedance of different materials or varying environments26,825. The acoustic impedance Z of a homogenous material can be expressed as
| (34) |
where and are the effective density and bulk modulus of the material, respectively. Because the density and bulk modulus of common hydrogels are almost the same as those of water, the acoustic impedance of common hydrogels also approximates that of water. To achieve tunable acoustic impedance, fluidic channels have been patterned into tough hydrogel matrices recently (Figure 26a)825. By infusing the air, water or liquid metal (i.e., eutectic gallium-indium) into the fluidic channels, the effective density, bulk modulus and thus acoustic impedance of the hydrogel can be dramatically varied to approximate the acoustic impedance of air, water and many solids on demand (Figure 26b) 825. In order to approximate a homogeneous material, the fluidic channels should be uniformly distributed in the hydrogel and the characteristic sizes of the fluidic channels (i.e., channel diameter and distance between adjacent channels) should be much smaller than the acoustic wavelengths. Overall, a generic design principle for hydrogels with tunable acoustic impedance is to tune densities and bulk moduli of effectively homogeneous hydrogels.
Figure 26. Design of hydrogels with tunable acoustic impedance – tune densities and bulk moduli of effectively homogeneous hydrogels.

a. by infusing air, water or liquid metal (i.e., eutectic gallium-indium) into the fluidic channels inside a hydrogel matrix, the effective density, bulk modulus and thus acoustic impedance of the hydrogel can be dramatically varied. b. the hydrogel can approximate the acoustic impedance of air, water and many solids on demand825. a and b are adopted from Ref825.
6.5. Self-healing: Form crosslinks and/or polymers at damaged regions
A salient feature of many biological hydrogels is their capability of healing after injury. The capability of self-healing can potentially bestow synthetic hydrogels with the merits such as damage mitigation and long-term robustness. However, the healing processes in biological hydrogels mostly rely on the functions of biological cells, which do not exist in synthetic hydrogels. In absence of living components, a generic strategy to achieve self-healing in engineering materials is to form new materials and/or interactions in the vicinity of damaged regions in the materials844.
Specifically for soft materials such as elastomers and hydrogels, the new materials formed in the vicinity of damaged regions are usually new crosslinks and/or polymer chains. Therefore, the design principle for self-healing hydrogels is to form new crosslinks and/or polymers at damaged regions (Figure 27a). The commonly-used crosslinks for self-healing of hydrogels include weak physical crosslinks such as hydrogen bonds738,753,755,845, ionic bonds288,481,846, metal coordination288,755,832, hydrophobic interactions847 and guest-host interactions740; and dynamic covalent bonds such as olefin metathesis848,849. Once two newly-formed surfaces in a damaged hydrogel are brought into contact with each other under certain conditions such as specific temperature and pH, new crosslinks can form on the interface, endowing the hydrogel with the self-healing capability (Figure 27c). Besides reversible crosslinks, the crack healing of hydrogels can be achieved through interdiffusion of polymer chains to form entangled chains that span the crack surfaces850,851. Furthermore, some self-healing processes in hydrogels involve both mechanisms of chain entanglement and reversible crosslinking852. When the surfaces of two intact self-healing hydrogels are in contact, new interfaces can also be formed in between to adhere the two hydrogels together. Therefore, strictly speaking, most of the existing self-healing hydrogels are self-adhesive hydrogels, because the damage of the hydrogels is not required to induce the process of self-healing. Recently, Matsuda et al. have reported a self-growing or self-reinforcing hydrogel, in which the scission of polymer chains can induce mechanoradicals that trigger the polymerization of monomers in the solvent of the hydrogel (Figure 27b)853. Consequently, the hydrogel self-grows or self-reinforces after moderate damage, analogous to mechanical training of a muscle (Figure 27d). This strategy may be adopted for the future design of truly self-healing (instead of self-adhesive) hydrogels where the healing is triggered by the damage. For a further detailed discussion on various types of self-healing hydrogels, a recent review paper is recommended826.
Figure 27. Design of self-healing hydrogels – form crosslinks and/or polymers at damaged regions.
a. reversible crosslinks form on the interfaces between two pieces of hydrogels for self-healing or selfadhesion. b. damage of a hydrogel induces new polymerization and crosslinking, giving self-reinforcement or self-growth853. c. photos of a self-healing hydrogel based on oppositely charged polyelectrolytes481,854. d. photos of a self-reinforcing or self-growing hydrogel853. b and d are adopted from Ref853. c is adopted from Ref854.
6.6. Implementation with unconventional polymer networks
While the implementation of design principles for hydrogels with extreme mechanical properties exploits various types of UPNs as discussed in Section 5, it seems hydrogels with extreme physical properties mainly reply on one implementation strategy: functional nano-/micro-/macro-fillers. The functional fillers range from percolated conductive phases for high electrical conductivity, to magnetic particles for magnetization, to high-refractive-index non-scattering nano-phases for high refractive index and transparency, to fillers with tunable densities and bulk moduli for tunable acoustic impedance, and to reversible crosslinks and polymerization triggered by damages for self-leading capability.
7. Design of hydrogels with multiple combined properties
In addition to the extreme mechanical and physical properties discussed in Sections 5 and 6, respectively, chemical and biological properties of hydrogels also play critical roles in various applications of hydrogels. In fact, many nascent applications of hydrogels such as hydrogel living devices commonly require that a set of combined mechanical, physical, chemical and biological properties simultaneously coexist in hydrogels20,24,849. In order to achieve multiple combined properties, we will propose a general strategy for the orthogonal design of hydrogels guided by the corresponding design principles, which will then be implemented with UPNs in a synergistic manner.
7.1. Orthogonal design principles
In Sections 5 and 6, we have discussed the design principles for hydrogels to achieve a variety of extreme mechanical and physical properties, which are summarized as the following.
Tough: build dissipation into stretchy polymer networks.
Strong: synchronize chain stiffening and fracture.
Resilient: delay dissipation.
Tough adhesion: integrate tough dissipative hydrogels and strong interfacial linkages.
Fatigue-resistant: pin fatigue cracks with intrinsically high-energy phases.
Fatigue-resistant adhesion: strongly bond intrinsically high-energy phases on interfaces.
Electrically conductive: percolate electrically conductive phases.
Magnetization: embed magnetic particles and pattern ferromagnetic domains.
High reflective index and transparency: uniformly embed high-refractive-index non-scattering nano-phases.
Tunable acoustic impedance: tune densities and bulk moduli of effectively homogeneous hydrogels.
Self-healing: form crosslinks and/or polymers at damaged regions.
Since the abovementioned design principles are general and material-independent, they have been widely deployed for the design of biological hydrogels, synthetic hydrogels, and other engineering materials. In addition, based on the discussions in Sections 5 and 6, these design principles do not contradict or exclude one other in general. For example, the seeming contradiction between high toughness and high resilience of hydrogels has been reconciled by the design principles of building dissipation into stretchy polymer networks and delaying dissipation, respectively. Therefore, the design of multiple combined mechanical and physical properties of hydrogels can potentially follow the corresponding design principles in an orthogonal and independent manner as illustrated in Figure 28a. For example, a tough, electrically conductive, and self-healing hydrogel can be potentially designed by following the orthogonal design principles of building dissipation into stretchy polymer networks, percolating electrically conductive phases, and forming crosslinks and/or polymers at damaged regions, respectively30 (Figure 28b). The hydrogel can further form tough adhesion on substrates by following the design principle of integrating tough hydrogels and strong interfacial linkages831.
Figure 28. Orthogonal design principles and synergistic implementation strategies for the design of hydrogels with multiple combined properties.

a. schematics of the orthogonal design principles and synergistic implementation strategies. b. example of the design of a tough, self-healing and electrically conductive hydrogel.
In addition, although chemical and biological properties of hydrogels are beyond the scope of the current review, it is expected that the design of hydrogels’ chemical and biological properties will likely follow a set of design principles that are orthogonal with one another and with the design principles for mechanical and physical properties as well. Consequently, a set of orthogonal design principles will potentially guide the rational design of future hydrogels that possess multiple combined mechanical, physical, chemical and biological properties (Figure 28).
7.2. Synergistic implementation strategy
The orthogonal design principles for hydrogels to achieve multiple combined properties will be implemented with UPNs in a synergistic manner, meaning that one type of UPN can implement multiple design principles. As discussed in Section 4, the commonly-used UPNs include the UPN architectures:
Ideal polymer networks
Polymer networks with slidable crosslinks
Interpenetrating and semi-interpenetrating polymer networks
Polymer networks with high-functionality crosslinks
Nano-/micro-fibrous polymer networks
and the UPN interactions:
Strong physical crosslinks
Weak physical crosslinks
Dynamic covalent crosslinks.
Each UPN architecture or interaction usually can implement (or facilitate the implementation of) multiple design principles. For example, the nano-/micro-fibrous polymer networks can integrate high stretchability and mechanical dissipation (Section 5.1), delay dissipation (Section 5.3), synchronize stiffening and fracture of polymer chains (Section 5.2), and act as intrinsically high-energy phases (Section 5.5) to implement the design principles for tough, resilient, strong, and fatigue-resistant hydrogels, respectively. By strongly bonding the nano-/micro-fibers on substrates (Section 5.4 and Section 5.6), the corresponding nano-/micro-fibrous polymer networks can achieve tough and fatigue-resistant adhesion as well. Furthermore, the nano-/micro-fibers can also be made functional such as electrically conductive or high reflective index (Section 6.1) to implement the design of extreme physical properties. Not surprisingly, biological hydrogels indeed frequently employ nano-/micro-fibrous polymer networks, supplemented by other UPN architectures and interactions, to achieve multiple combined extreme mechanical and physical properties necessary for their robustness and well-being over the lifetime (Figure 2).
Last but not least, it should be emphasized that the design principles and implementation strategies for hydrogels discussed in this paper are based on generic polymer networks; therefore, they should be applicable to other soft materials comprised of polymer networks including elastomers and organogels as well. For example, the design principle and implementation strategy for tough hydrogels have been used to design tough elastomers725. We expect the current review will provide a solid foundation for rational design of various types of polymeric soft materials such as hydrogels, elastomers and organogels to achieve multiple combined extreme properties for diverse applications. Furthermore, we hope the current review will provoke interdisciplinary discussions on a fundamental question: why does nature select soft materials, especially hydrogels embodied in unconventional polymer networks (Figures 1 and 2), to constitute the major parts of animal bodies?
Table 1.
Examples of unconventional polymer network architectures.
| UPN architectures | Examples | References |
|---|---|---|
| Ideal polymer networks | Covalently-crosslinked 4-arm end-functionalized PEG; Reversibly-crosslinked 4-arm end-functionalized PEG |
67,317,356,435–450. |
| Polymer networks with slidable crosslinks | PEG with polyrotaxanes as slidable crosslinks; PCL with polyrotaxanes as slidable crosslinks; PPO with polyrotaxanes as slidable crosslinks |
60,356,451,454–458,460–465 |
| Interpenetrating polymer networks | Covalently-crosslinked PAMPS interpenetrated with covalently-crosslinked PAAm; Covalently-crosslinked polysaccharide interpenetrated with covalently-crosslinked PHEMA, PEG, PAAm, PNIPAm, and PDMA |
60,63,356,383,467–470,476 |
| Semi-interpenetrating polymer networks | Covalently-crosslinked PAAm interpenetrated with reversibly-crosslinked or uncrosslinked PVA, alginate, chitosan, and hyaluronan; Covalently-crosslinked gelatin interpenetrated with reversibly-crosslinked or uncrosslinked alginate; Covalently-crosslinked PEG-DA gelatin interpenetrated with reversibly-crosslinked or uncrosslinked alginate, chitosan, and hyaluronan; Covalently-crosslinked PAAm interpenetrated with reversibly-crosslinked or uncrosslinked PVA |
60,63,356,383,466,468,469,475,477 |
| Polymer networks with high-functionality crosslinks | PVA with crystalline domains as high-functionality crosslinks; Poly(methyl methacrylate) with glassy spheres as high-functionality crosslinks; Polyacrylamide with exfoliated particles as high-functionality crosslinks; Mixtures of polystyrene, poly(butyl acrylate), and poly(acrylic acid) with microsphere composites as high-functionality crosslinks |
60,215,300,302,356,383,436,477,478,482,485,488–496 |
| Nano-/microfibrous polymer networks | Cellulose, cellulose-derivative, collagen, gelatin, fibrin and elastin nano-/micro-fibrous networks | 6,103,382,507–521 |
| Other UPN architectures | Poly(dimethylsiloxane) bottlebrush; Poly(n-butyl acrylate) bottlebrush; Poly polylactic acid-b-polyethylene glycol-b-poly(N-isopropylacrylamide) triblock bottlebrush |
522–524 |
Table 2.
Examples of unconventional polymer network interactions.
| UPN interactions | Examples | References | |
|---|---|---|---|
| Strong physical crosslinks | Crystalline domain | PVA treated by freeze-thawing or annealing | 4,300,302,482 |
| Chitin and chitosan treated by acidic or basic solutions | 215,218 | ||
| Cellulose treated by alkalines | 497,526,527 | ||
| Glassy nodule | Polystyrene-b-poly(N-isopropylacrylamide)-bpolystyrene | 530 | |
| Poly(methyl methacrylate)-b-poly(n-butyl acrylate) | 492 | ||
| Helical association | Self-assemble of agarose or gelatin | 6,103,534,535 | |
| Self-assemble of collagen or fibrinogen | 6,103,532,533. | ||
| Self-assemble of elastin-like polypeptides | 507,510 | ||
| Weak physical crosslinks | Hydrogen bond | PAA or polymethacrylic acid (PMA) with PEG | 478,538–540 |
| PEG, PHEMA, PNIPAM with self-complementary hydrogen-bond groups (triazine moieties or ureido pyrinimidone moieties) |
478,490,540–542 | ||
| Electrostatic interaction | Alginate with Ca2+, Ba2+, Mg2+, Zn2+ | 6,103,105,546 | |
| Chitosan with tripolyphosphate, citrate ions | 547–549 | ||
| Cationic polyelectrolytes with anionic polyelectrolytes | 544,550 | ||
| Coordination complex | Bisphosphonate containing polymers with metal ions (Ca2+, Mg2+ or Ag+) | 557,572,573 | |
| Catechol containing polymers with metal ions (Cu2+, Zn2+ and Fe3+) |
478,574–577,580 560,561,578 | ||
| Histidine containing polymers with metal ions (Cu2+, Co2+ and Ni2+) |
478,579,58059 | ||
| Host-guest interaction | Polymers containing β-CD moieties with azobenzene group, adamantyl group, ferrocene group, t-butyl group, cyclohexyl(ester) group, cyclododecyl(amide) group, benzyl group, 2naphthylmethyl group, 1-pyrenylmethyl group | 383,457,478,540,586,588–594 | |
| Polymers containing α-CD moieties with n-butyl group, adamantyl group, benzyl group, trans- azobenzene group |
383,478,540,586,587; | ||
| Polymers containing curcubit[n]uril moieties with spermine, diaminohexane, viologens, naphtalenes | 383,540,605,606. | ||
| Hydrophobic association | PEG, PAAm, PNIPAM, PDMAA, PVA containing hydrophobic moieties (octylphenol-PEG acrylate, stearyl acrylate, lauryl acrylate) |
478,488,490 488,612,613 | |
| Triblock amphiphilic copolymers with PEG, PAAm, PVA, PHEMA middle blocks and n-alkyl acrylate end blocks |
614–616 | ||
| π - π stacking | Polymers modified with aromatic moiety containing short peptides and N-terminal Fmoc- amino acids |
499,620,621 | |
| Hydrogels containing carbon nanotubes, polythiophene, and graphene-based nanomaterials | 30,622–626 | ||
| Dynamic covalent crosslinks |
Imine bond | Polymers containing amine and aldehyde (or ketone) functional groups | 383,631–635,637–641 |
| Boronate ester bond | Polymers containing boronic acid and diol functional groups | 383,644,650,655,656,662 | |
| Disulfide bond | Polymers containing disulfide functional groups | 383,615,668,674,675,679 | |
| Hydrazone bond | Polymers containing hydrazide and aldehyde (or ketone) functional groups | 679,680,682–686,688–690 | |
| Oxime bond | Polymers containing hydroxylamine and aldehyde (or ketone) functional groups |
695–698,706 | |
| Reversible Diels–Alder reaction |
Polymers containing diene and dienophile functional groups | 701,711,712 | |
Acknowledgement
This work was supported by National Institutes of Health (No. 1R01HL153857-01), National Science Foundation (No. EFMA-1935291), U.S. Army Research Office through the Institute for Soldier Nanotechnologies at MIT (Grant No. W911NF-13-D-0001), and Defense Advanced Research Projects Agency through Cooperative Agreement D20AC00004.
Acronym
- APS
ammonium persulfate
- CB[n]
cucurbit[n]uril
- CD
cyclodextrin
- CNT
carbon nanotubes
- DOPA
3,4-dihydroxyphenyl-L-alanine
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- Fmoc
fluorenylmethyloxycarbonyl
- G
α-L-guluronic acid
- GO
graphene oxide
- M
β-D-mannuronic
- MBA
N,N’-Methylenebisacrylamide
- NdFeB
neodymium iron boron
- NHS
N-hydroxysuccinimide
- NVP
N-vinylpyrrolidone
- PAA
poly(acrylic acid)
- PAAm
polyacrylamide
- PCL
poly(caprolactone)
- PDLLA
poly(DL-lactic acid)
- PDMA
poly(N,N-Dimethylacrylamide)
- PEDOT
PSS: poly(3,4-ethylenedioxythiophene):poly (styrene sulfonate)
- PEG
poly(ethylene glycol)
- PEO
poly(ethylene oxide)
- PHEMA
poly(2-hydroxethyl methacrylate)
- PLA
polylactide
- PLGA
poly(DL-lactic acid-co-glycolic acid)
- PMA
polymethacrylic acid
- PNIPAm
poly(N-isopropylacrylamide)
- PPG
poly(propylene glycol)
- PPS
poly(propylene sulfide)
- PVA
poly(vinyl alcohol)
- RGD
Arg-Gly-Asp
- TEGDMA
trimethylene glycol dimethacrylate
- TEMED
N,N,N′N′-tetramethylene-diamine
- UPN
unconventional polymer network
- UPy
ureido-pyrimidinone
References
- (1).Wichterle O; LÍM D.Hydrophilic Gels for Biological Use. Nature 1960, 185 (4706), 117. [Google Scholar]
- (2).Peppas NA; Bures P; Leobandung W; Ichikawa H.Hydrogels in pharmaceutical formulations. European Journal of Pharmaceutics and Biopharmaceutics 2000, 50 (1), 27. [DOI] [PubMed] [Google Scholar]
- (3).Qiu Y; Park K.Environment-sensitive hydrogels for drug delivery. Advanced Drug Delivery Reviews 2001, 53 (3), 321. [DOI] [PubMed] [Google Scholar]
- (4).Peppas NA; Hilt JZ; Khademhosseini A; Langer R.Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Advanced Materials 2006, 18 (11), 1345. [Google Scholar]
- (5).Li J; Mooney DJ Designing hydrogels for controlled drug delivery. Nature Reviews Materials 2016, 1 (12), 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lee KY; Mooney DJ Hydrogels for Tissue Engineering. Chemical Reviews 2001, 101 (7), 1869. [DOI] [PubMed] [Google Scholar]
- (7).Nguyen KT; West JL Photopolymerizable hydrogels for tissue engineering applications. Biomaterials 2002, 23 (22), 4307. [DOI] [PubMed] [Google Scholar]
- (8).Drury JL; Mooney DJ Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials 2003, 24 (24), 4337. [DOI] [PubMed] [Google Scholar]
- (9).Discher DE; Janmey P; Wang Y.-l. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310 (5751), 1139. [DOI] [PubMed] [Google Scholar]
- (10).Engler AJ; Sen S; Sweeney HL; Discher DE Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126 (4), 677. [DOI] [PubMed] [Google Scholar]
- (11).Discher DE; Mooney DJ; Zandstra PW Growth Factors, Matrices, and Forces Combine and Control Stem Cells. Science 2009, 324 (5935), 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huebsch N; Arany PR; Mao AS; Shvartsman D; Ali OA; Bencherif SA; Rivera-Feliciano J; Mooney DJ Harnessing traction-mediated manipulation of the cell/matrix interface to control stem-cell fate. Nature Materials 2010, 9 (6), 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tibbitt MW; Anseth KS Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnology and Bioengineering 2009, 103 (4), 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Caló E; Khutoryanskiy VV Biomedical applications of hydrogels: A review of patents and commercial products. European Polymer Journal 2015, 65, 252. [Google Scholar]
- (15).Kopecek J.Hydrogels: From soft contact lenses and implants to self-assembled nanomaterials. Journal of Polymer Science Part A: Polymer Chemistry 2009, 47 (22), 5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jones A; Vaughan D.Hydrogel dressings in the management of a variety of wound types: A review. Journal of Orthopaedic Nursing 2005, 9, S1. [Google Scholar]
- (17).Boateng JS; Matthews KH; Stevens HNE; Eccleston GM Wound Healing Dressings and Drug Delivery Systems: A Review. Journal of Pharmaceutical Sciences 2008, 97 (8), 2892. [DOI] [PubMed] [Google Scholar]
- (18).Kamoun EA; Kenawy E-RS; Chen X.A review on polymeric hydrogel membranes for wound dressing applications: PVA-based hydrogel dressings. Journal of Advanced Research 2017, 8 (3), 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vanderlaan DG; Nunez IM; Hargiss M; Alton ML; Williams S; Google Patents, 1999. [Google Scholar]
- (20).Liu X; Liu J; Lin S; Zhao X.Hydrogel machines. Materials Today 2020. [Google Scholar]
- (21).Dong L; Agarwal AK; Beebe DJ; Jiang H.Adaptive liquid microlenses activated by stimuli-responsive hydrogels. Nature 2006, 442 (7102), 551. [DOI] [PubMed] [Google Scholar]
- (22).Holtz JH; Asher SA Polymerized colloidal crystal hydrogel films as intelligent chemical sensing materials. Nature 1997, 389 (6653), 829. [DOI] [PubMed] [Google Scholar]
- (23).Miyata T; Asami N; Uragami T.A reversibly antigen-responsive hydrogel. Nature 1999, 399 (6738), 766. [DOI] [PubMed] [Google Scholar]
- (24).Liu X; Tang T-C; Tham E; Yuk H; Lin S; Lu TK; Zhao X.Stretchable living materials and devices with hydrogel–elastomer hybrids hosting programmed cells. Proceedings of the National Academy of Sciences 2017, 114 (9), 2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Beebe DJ; Moore JS; Bauer JM; Yu Q; Liu RH; Devadoss C; Jo B-H Functional hydrogel structures for autonomous flow control inside microfluidic channels. Nature 2000, 404 (6778), 588. [DOI] [PubMed] [Google Scholar]
- (26).Yuk H; Lin S; Ma C; Takaffoli M; Fang NX; Zhao X.Hydraulic hydrogel actuators and robots optically and sonically camouflaged in water. Nature Communications 2017, 8, 14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Keplinger C; Sun J-Y; Foo CC; Rothemund P; Whitesides GM; Suo Z.Stretchable, Transparent, Ionic Conductors. Science 2013, 341 (6149), 984. [DOI] [PubMed] [Google Scholar]
- (28).Zhao X; Kim J; Cezar CA; Huebsch N; Lee K; Bouhadir K; Mooney DJ Active scaffolds for on-demand drug and cell delivery. Proceedings of the National Academy of Sciences 2011, 108 (1), 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu Y; Liu J; Chen S; Lei T; Kim Y; Niu S; Wang H; Wang X; Foudeh AM; Tok JBH et al. Soft and elastic hydrogel-based microelectronics for localized low-voltage neuromodulation. Nature Biomedical Engineering 2019, 3 (1), 58. [DOI] [PubMed] [Google Scholar]
- (30).Lu B; Yuk H; Lin S; Jian N; Qu K; Xu J; Zhao X.Pure PEDOT:PSS hydrogels. Nature Communications 2019, 10 (1), 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gao Y; Song J; Li S; Elowsky C; Zhou Y; Ducharme S; Chen YM; Zhou Q; Tan L.Hydrogel microphones for stealthy underwater listening. Nature Communications 2016, 7, 12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chen F; Tillberg PW; Boyden ES Expansion microscopy. Science 2015, 347 (6221), 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Guo J; Liu X; Jiang N; Yetisen AK; Yuk H; Yang C; Khademhosseini A; Zhao X; Yun SH Highly stretchable, strain sensing hydrogel optical fibers. Advanced Materials 2016, 28 (46), 10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li X-H; Liu C; Feng S-P; Fang NX Broadband Light Management with Thermochromic Hydrogel Microparticles for Smart Windows. Joule 2019, 3 (1), 290. [Google Scholar]
- (35).Kim C-C; Lee H-H; Oh KH; Sun J-Y Highly stretchable, transparent ionic touch panel. Science 2016, 353 (6300), 682. [DOI] [PubMed] [Google Scholar]
- (36).Li T; Li G; Liang Y; Cheng T; Dai J; Yang X; Liu B; Zeng Z; Huang Z; Luo Y et al. Fast-moving soft electronic fish. Science Advances 2017, 3 (4), e1602045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kim YS; Liu M; Ishida Y; Ebina Y; Osada M; Sasaki T; Hikima T; Takata M; Aida T.Thermoresponsive actuations enabled by permittivity switching in an electrostatically anisotropic hydrogel. Nature Materials 2015, 14 (10), 1002. [DOI] [PubMed] [Google Scholar]
- (38).Schroeder TBH; Guha A; Lamoureux A; VanRenterghem G; Sept D; Shtein M; Yang J; Mayer M.An electric-eel-inspired soft power source from stacked hydrogels. Nature 2017, 552, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pan L; Yu G; Zhai D; Lee HR; Zhao W; Liu N; Wang H; Tee BC-K; Shi Y; Cui Y et al. Hierarchical nanostructured conducting polymer hydrogel with high electrochemical activity. Proceedings of the National Academy of Sciences 2012, 109 (24), 9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Xiao T; Xu L; Zhou L; Sun XQ; Lin C; Wang L.Dynamic hydrogels mediated by macrocyclic host-guest interactions. J Mater Chem B 2019, 7 (10), 1526. [DOI] [PubMed] [Google Scholar]
- (41).Yuk H; Varela CE; Nabzdyk CS; Mao X; Padera RF; Roche ET; Zhao X.Dry double-sided tape for adhesion of wet tissues and devices. Nature 2019, 575 (7781), 169. [DOI] [PubMed] [Google Scholar]
- (42).Rose S; Prevoteau A; Elzière P; Hourdet D; Marcellan A; Leibler L.Nanoparticle solutions as adhesives for gels and biological tissues. Nature 2014, 505 (7483), 382. [DOI] [PubMed] [Google Scholar]
- (43).Yuk H; Zhang T; Lin S; Parada GA; Zhao X.Tough bonding of hydrogels to diverse nonporous surfaces. Nature Materials 2016, 15 (2), 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Yu Y; Yuk H; Parada GA; Wu Y; Liu X; Nabzdyk CS; Youcef‐Toumi K; Zang J; Zhao X.Multifunctional “hydrogel skins” on diverse polymers with arbitrary shapes. Advanced Materials 2019, 31 (7), 1807101. [DOI] [PubMed] [Google Scholar]
- (45).Cheng H; Yue K; Kazemzadeh-Narbat M; Liu Y; Khalilpour A; Li B; Zhang YS; Annabi N; Khademhosseini A.Mussel-inspired multifunctional hydrogel coating for prevention of infections and enhanced osteogenesis. ACS Applied Materials & Interfaces 2017, 9 (13), 11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Biot MA General theory of three‐dimensional consolidation. Journal of applied physics 1941, 12 (2), 155. [Google Scholar]
- (47).Flory PJ Principles of polymer chemistry; Cornell University Press, 1953. [Google Scholar]
- (48).Lake G; Lindley P.The mechanical fatigue limit for rubber. Journal of Applied Polymer Science 1965, 9 (4), 1233. [Google Scholar]
- (49).Lake G; Thomas A.The strength of highly elastic materials. Proceedings of the Royal Society of London. Series A. Mathematical Physical Sciences 1967, 300 (1460), 108. [Google Scholar]
- (50).Treloar LRG The physics of rubber elasticity; Oxford University Press, USA, 1975. [Google Scholar]
- (51).De Gennes P-G; Gennes P-G Scaling concepts in polymer physics; Cornell university press, 1979. [Google Scholar]
- (52).Gent A.A new constitutive relation for rubber. Rubber Chemistry Technology 1996, 69 (1), 59. [Google Scholar]
- (53).Erman B; Mark JE Structures and properties of rubberlike networks; Oxford University Press, 1997. [Google Scholar]
- (54).Boyce MC; Arruda EM Constitutive models of rubber elasticity: a review. Rubber Chemistry Technology 2000, 73 (3), 504. [Google Scholar]
- (55).Rubinstein M; Colby RH Polymer physics; Oxford university press New York, 2003. [Google Scholar]
- (56).Mark JE; Erman B.Rubberlike elasticity: a molecular primer; Cambridge University Press, 2007. [Google Scholar]
- (57).Argon AS The physics of deformation and fracture of polymers. New York.: Cambridge; 2013. [Google Scholar]
- (58).Peppas NA Hydrogels in medicine and pharmacy: fundamentals; CRC press, 2019. [Google Scholar]
- (59).Richbourg NR; Peppas NA The swollen polymer network hypothesis: Quantitative models of hydrogel swelling, stiffness, and solute transport. Progress in Polymer Science 2020, 101243. [Google Scholar]
- (60).Zhao X.Multi-scale multi-mechanism design of tough hydrogels: building dissipation into stretchy networks. Soft Matter 2014, 10 (5), 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhang YS; Khademhosseini A.Advances in engineering hydrogels. Science 2017, 356 (6337), eaaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Fan H; Gong JP Fabrication of Bioinspired Hydrogels: Challenges and Opportunities. Macromolecules 2020, 53 (8), 2769. [Google Scholar]
- (63).Gong JP; Katsuyama Y; Kurokawa T; Osada Y.Double-Network Hydrogels with Extremely High Mechanical Strength. Advanced Materials 2003, 15 (14), 1155. [Google Scholar]
- (64).Moutos FT; Freed LE; Guilak F.A biomimetic three-dimensional woven composite scaffold for functional tissue engineering of cartilage. Nature Materials 2007, 6 (2), 162. [DOI] [PubMed] [Google Scholar]
- (65).Hua J; Ng PF; Fei B.High-strength hydrogels: Microstructure design, characterization and applications. 2018, 56 (19), 1325. [Google Scholar]
- (66).Lin S; Zhou Y; Zhao X.Designing extremely resilient and tough hydrogels via delayed dissipation. Extreme Mechanics Letters 2014, 1, 70. [Google Scholar]
- (67).Kamata H; Akagi Y; Kayasuga-Kariya Y; Chung U.-i.; Sakai T.“Nonswellable” Hydrogel Without Mechanical Hysteresis. Science 2014, 343 (6173), 873. [DOI] [PubMed] [Google Scholar]
- (68).Lin S; Liu X; Liu J; Yuk H; Loh H-C; Parada GA; Settens C; Song J; Masic A; McKinley GH Anti-fatigue-fracture hydrogels. Science advances 2019, 5 (1), eaau8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Lin S; Liu J; Liu X; Zhao X.Muscle-like fatigue-resistant hydrogels by mechanical training. Proceedings of the National Academy of Sciences 2019, 116 (21), 10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Xiang C; Wang Z; Yang C; Yao X; Wang Y; Suo Z.Stretchable and fatigue-resistant materials. Materials Today 2019, DOI: 10.1016/j.mattod.2019.08.009. [DOI] [Google Scholar]
- (71).Liu J; Lin S; Liu X; Qin Z; Yang Y; Zang J; Zhao X.Fatigue-resistant adhesion of hydrogels. Nature Communications 2020, 11 (1), 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Hall BK Cartilage V1: Structure, function, and biochemistry; Academic Press, 2012. [Google Scholar]
- (73).Taylor D; O’Mara N; Ryan E; Takaza M; Simms C.The fracture toughness of soft tissues. J Mech Behav Biomed 2012, 6, 139. [DOI] [PubMed] [Google Scholar]
- (74).Mow VC; Holmes MH; Lai WM Fluid transport and mechanical properties of articular cartilage: a review. Journal of biomechanics 1984, 17 (5), 377. [DOI] [PubMed] [Google Scholar]
- (75).Han L; Frank EH; Greene JJ; Lee H-Y; Hung H-HK; Grodzinsky AJ; Ortiz C.Timedependent nanomechanics of cartilage. Biophysical journal 2011, 100 (7), 1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Tavichakorntrakool R; Prasongwattana V; Sriboonlue P; Puapairoj A; Wongkham C; Wiangsimma T; Khunkitti W; Triamjangarun S; Tanratanauijit M; Chamsuwan A.K+, Na+, Mg2+, Ca2+, and water contents in human skeletal muscle: correlations among these monovalent and divalent cations and their alterations in K+-depleted subjects. Translational Research 2007, 150 (6), 357. [DOI] [PubMed] [Google Scholar]
- (77).Connizzo BK; Yannascoli SM; Soslowsky LJ Structure–function relationships of postnatal tendon development: a parallel to healing. Matrix Biology 2013, 32 (2), 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Fratzl P.In Collagen; Springer, 2008. [Google Scholar]
- (79).Lee JM; Courtman DW; Boughner DR The glutaraldehyde‐stabilized porcine aortic valve xenograft. I. Tensile viscoelastic properties of the fresh leaflet material. Journal of Biomedical Materials Research Part A 1984, 18 (1), 61. [DOI] [PubMed] [Google Scholar]
- (80).Lee JM; Boughner DR; Courtman DW The glutaraldehyde‐stabilized porcine aortic valve xenograft. II. Effect of fixation with or without pressure on the tensile viscoelastic properties of the leaflet material. Journal of Biomedical Materials Research Part A 1984, 18 (1), 79. [DOI] [PubMed] [Google Scholar]
- (81).Vesely I.The role of elastin in aortic valve mechanics. Journal of biomechanics 1997, 31 (2), 115. [DOI] [PubMed] [Google Scholar]
- (82).Driessen NJ; Bouten CV; Baaijens FP Improved prediction of the collagen fiber architecture in the aortic heart valve. 2005. [DOI] [PubMed] [Google Scholar]
- (83).Bobyn JD; Wilson GJ; Macgregor DC; Pilliar RM; Weatherly GC Effect of Pore-Size on the Peel Strength of Attachment of Fibrous Tissue to Porous-Surfaced Implants. J Biomed Mater Res 1982, 16 (5), 571. [DOI] [PubMed] [Google Scholar]
- (84).Chaudhuri O; Gu L; Klumpers D; Darnell M; Bencherif SA; Weaver JC; Huebsch N; Lee H.-p.; Lippens E; Duda GN et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nature Materials 2016, 15 (3), 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Genin GM; Thomopoulos S.The tendon-to-bone attachment: Unification through disarray. Nature materials 2017, 16 (6), 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Rossetti L; Kuntz L; Kunold E; Schock J; Müller K; Grabmayr H; Stolberg-Stolberg J; Pfeiffer F; Sieber S; Burgkart R.The microstructure and micromechanics of the tendon–bone insertion. Nature materials 2017, 16 (6), 664. [DOI] [PubMed] [Google Scholar]
- (87).Chaudhuri O; Cooper-White J; Janmey PA; Mooney DJ; Shenoy VB Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584 (7822), 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Weightman B; Freeman M; Swanson S.Fatigue of articular cartilage. Nature 1973, 244 (5414), 303. [DOI] [PubMed] [Google Scholar]
- (89).Hansson T; Keller T; Spengler D.Mechanical behavior of the human lumbar spine. II. Fatigue strength during dynamic compressive loading. Journal of Orthopaedic Research 1987, 5 (4), 479. [DOI] [PubMed] [Google Scholar]
- (90).Peppas NA Hydrogels and drug delivery. Current Opinion in Colloid & Interface Science 1997, 2 (5), 531. [Google Scholar]
- (91).Peppas NA; Khare AR Preparation, structure and diffusional behavior of hydrogels in controlled release. Advanced drug delivery reviews 1993, 11 (1–2), 1. [Google Scholar]
- (92).Gladman AS; Matsumoto EA; Nuzzo RG; Mahadevan L; Lewis JA Biomimetic 4D printing. Nature Materials 2016, 15 (4), 413. [DOI] [PubMed] [Google Scholar]
- (93).Yang J; Li Y; Zhu L; Qin G; Chen Q.Double network hydrogels with controlled shape deformation: A mini review. 2018, 56 (19), 1351. [Google Scholar]
- (94).Peng X; Wang H.Shape changing hydrogels and their applications as soft actuators. 2018, 56 (19), 1314. [Google Scholar]
- (95).Zhao F; Zhou X; Shi Y; Qian X; Alexander M; Zhao X; Mendez S; Yang R; Qu L; Yu G.Highly efficient solar vapour generation via hierarchically nanostructured gels. Nature Nanotechnology 2018, 13 (6), 489. [DOI] [PubMed] [Google Scholar]
- (96).Shadwick RE Elastic energy storage in tendons: mechanical differences related to function and age. Journal of applied physiology 1990, 68 (3), 1033. [DOI] [PubMed] [Google Scholar]
- (97).Race A; Amis AA The mechanical properties of the two bundles of the human posterior cruciate ligament. Journal of biomechanics 1994, 27 (1), 13. [DOI] [PubMed] [Google Scholar]
- (98).Chin-Purcell MV; Lewis JL Fracture of articular cartilage. 1996. [DOI] [PubMed] [Google Scholar]
- (99).Papagiannopoulos A; Waigh T; Hardingham T; Heinrich M.Solution structure and dynamics of cartilage aggrecan. Biomacromolecules 2006, 7 (7), 2162. [DOI] [PubMed] [Google Scholar]
- (100).Hayes W; Mockros L.Viscoelastic properties of human articular cartilage. Journal of applied physiology 1971, 31 (4), 562. [DOI] [PubMed] [Google Scholar]
- (101).Rock CA; Han L; Doehring TC Complex collagen fiber and membrane morphologies of the whole porcine aortic valve. PLoS One 2014, 9 (1), e86087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Korossis S.Structure-function relationship of heart valves in health and disease. Structural Insufficiency Anomalies in Cardiac Valves 2018, 1. [Google Scholar]
- (103).Hoffman AS Hydrogels for biomedical applications. Advanced Drug Delivery Reviews 2012, 64, 18. [DOI] [PubMed] [Google Scholar]
- (104).Sabra W; Zeng AP; Deckwer WD Bacterial alginate: physiology, product quality and process aspects. Appl Microbiol Biotechnol 2001, 56 (3–4), 315. [DOI] [PubMed] [Google Scholar]
- (105).Lee KY; Mooney DJ Alginate: properties and biomedical applications. Prog Polym Sci 2012, 37 (1), 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Augst AD; Kong HJ; Mooney DJ Alginate hydrogels as biomaterials. Macromol Biosci 2006, 6 (8), 623. [DOI] [PubMed] [Google Scholar]
- (107).Donati I; Holtan S; Morch YA; Borgogna M; Dentini M; Skjak-Braek G.New hypothesis on the role of alternating sequences in calcium-alginate gels. Biomacromolecules 2005, 6 (2), 1031. [DOI] [PubMed] [Google Scholar]
- (108).Morris ER; Rees DA; Thom D.Characterization of Alginate Composition and Block-Structure by Circular-Dichroism. Carbohyd Res 1980, 81 (2), 305. [Google Scholar]
- (109).Smidsrød O; Skjåk-Br˦k G Alginate as immobilization matrix for cells. Trends in Biotechnology 1990, 8, 71. [DOI] [PubMed] [Google Scholar]
- (110).Pawar SN; Edgar KJ Alginate derivatization: a review of chemistry, properties and applications. Biomaterials 2012, 33 (11), 3279. [DOI] [PubMed] [Google Scholar]
- (111).Drury JL; Dennis RG; Mooney DJ The tensile properties of alginate hydrogels. Biomaterials 2004, 25 (16), 3187. [DOI] [PubMed] [Google Scholar]
- (112).Baer AE; Wang JY; Kraus VB; Setton LA Collagen gene expression and mechanical properties of intervertebral disc cell–alginate cultures. Journal of Orthopaedic Research 2001, 19 (1), 2. [DOI] [PubMed] [Google Scholar]
- (113).Kang SW; Cha BH; Park H; Park KS; Lee KY; Lee SH The effect of conjugating RGD into 3D alginate hydrogels on adipogenic differentiation of human adipose-derived stromal cells. Macromol Biosci 2011, 11 (5), 673. [DOI] [PubMed] [Google Scholar]
- (114).Landa N; Miller L; Feinberg MS; Holbova R; Shachar M; Freeman I; Cohen S; Leor J.Effect of injectable alginate implant on cardiac remodeling and function after recent and old infarcts in rat. Circulation 2008, 117 (11), 1388. [DOI] [PubMed] [Google Scholar]
- (115).Selden C; Khalil M; Hodgson H.Three dimensional culture upregulates extracellular matrix protein expression in human liver cell lines--a step towards mimicking the liver in vivo? Int J Artif Organs 2000, 23 (11), 774. [PubMed] [Google Scholar]
- (116).Xu X; Jha AK; Harrington DA; Farach-Carson MC; Jia X.Hyaluronic Acid-Based Hydrogels: from a Natural Polysaccharide to Complex Networks. Soft Matter 2012, 8 (12), 3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Burdick JA; Prestwich GD Hyaluronic acid hydrogels for biomedical applications. Advanced materials 2011, 23 (12), H41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Laurent TC; Laurent UB; Fraser JR The structure and function of hyaluronan: An overview. Immunol Cell Biol 1996, 74 (2), A1. [DOI] [PubMed] [Google Scholar]
- (119).Vercruysse KP; Marecak DM; Marecek JF; Prestwich GD Synthesis and in vitro degradation of new polyvalent hydrazide cross-linked hydrogels of hyaluronic acid. Bioconjug Chem 1997, 8 (5), 686. [DOI] [PubMed] [Google Scholar]
- (120).Pouyani T; Harbison GS; Prestwich GD Novel Hydrogels of Hyaluronic-Acid - Synthesis, Surface-Morphology, and Solid-State NMR. J Am Chem Soc 1994, 116 (17), 7515. [Google Scholar]
- (121).Kuo J; Prestwich G.Materials of biological origin-Materials analysis and implant uses, Comprehensive biomaterials. Ducheyne, P 2010. [Google Scholar]
- (122).Shu XZ; Liu Y; Palumbo F; Prestwich GD Disulfide-crosslinked hyaluronan-gelatin hydrogel films: a covalent mimic of the extracellular matrix for in vitro cell growth. Biomaterials 2003, 24 (21), 3825. [DOI] [PubMed] [Google Scholar]
- (123).Vanderhooft JL; Mann BK; Prestwich GD Synthesis and characterization of novel thiol-reactive poly(ethylene glycol) cross-linkers for extracellular-matrix-mimetic biomaterials. Biomacromolecules 2007, 8 (9), 2883. [DOI] [PubMed] [Google Scholar]
- (124).Serban MA; Prestwich GD Synthesis of hyaluronan haloacetates and biology of novel cross-linker-free synthetic extracellular matrix hydrogels. Biomacromolecules 2007, 8 (9), 2821. [DOI] [PubMed] [Google Scholar]
- (125).Pouyani T; Prestwich GD Functionalized derivatives of hyaluronic acid oligosaccharides: drug carriers and novel biomaterials. Bioconjug Chem 1994, 5 (4), 339. [DOI] [PubMed] [Google Scholar]
- (126).Jia XQ; Burdick JA; Kobler J; Clifton RJ; Rosowski JJ; Zeitels SM; Langer R.Synthesis and characterization of in situ cross-linkable hyaluronic acid-based hydrogels with potential application for vocal fold regeneration. Macromolecules 2004, 37 (9), 3239. [Google Scholar]
- (127).Jha AK; Hule RA; Jiao T; Teller SS; Clifton RJ; Duncan RL; Pochan DJ; Jia X.Structural Analysis and Mechanical Characterization of Hyaluronic Acid-Based Doubly Cross-Linked Networks. Macromolecules 2009, 42 (2), 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Darr A; Calabro A.Synthesis and characterization of tyramine-based hyaluronan hydrogels. J Mater Sci Mater Med 2009, 20 (1), 33. [DOI] [PubMed] [Google Scholar]
- (129).Collins MN; Birkinshaw C.Hyaluronic acid based scaffolds for tissue engineering—A review. Carbohydrate Polymers 2013, 92 (2), 1262. [DOI] [PubMed] [Google Scholar]
- (130).Burdick JA; Chung C; Jia X; Randolph MA; Langer R.Controlled degradation and mechanical behavior of photopolymerized hyaluronic acid networks. Biomacromolecules 2005, 6 (1), 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Bian L; Guvendiren M; Mauck RL; Burdick JA Hydrogels that mimic developmentally relevant matrix and N-cadherin interactions enhance MSC chondrogenesis. Proc Natl Acad Sci U S A 2013, 110 (25), 10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Bencherif SA; Srinivasan A; Horkay F; Hollinger JO; Matyjaszewski K; Washburn NR Influence of the degree of methacrylation on hyaluronic acid hydrogels properties. Biomaterials 2008, 29 (12), 1739. [DOI] [PubMed] [Google Scholar]
- (133).Chen WY; Abatangelo G.Functions of hyaluronan in wound repair. Wound Repair Regen 1999, 7 (2), 79. [DOI] [PubMed] [Google Scholar]
- (134).Kuo J.Practical aspects of hyaluronan based medical products; CRC Press, 2005. [Google Scholar]
- (135).Prestwich GD Engineering a clinically-useful matrix for cell therapy. Organogenesis 2008, 4 (1), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Toh WS; Lee EH; Guo XM; Chan JK; Yeow CH; Choo AB; Cao T.Cartilage repair using hyaluronan hydrogel-encapsulated human embryonic stem cell-derived chondrogenic cells. Biomaterials 2010, 31 (27), 6968. [DOI] [PubMed] [Google Scholar]
- (137).Anseth KS; Metters AT; Bryant SJ; Martens PJ; Elisseeff JH; Bowman CN In situ forming degradable networks and their application in tissue engineering and drug delivery. J Control Release 2002, 78 (1–3), 199. [DOI] [PubMed] [Google Scholar]
- (138).Burdick JA; Ward M; Liang E; Young MJ; Langer R.Stimulation of neurite outgrowth by neurotrophins delivered from degradable hydrogels. Biomaterials 2006, 27 (3), 452. [DOI] [PubMed] [Google Scholar]
- (139).Baksh D; Song L; Tuan RS Adult mesenchymal stem cells: characterization, differentiation, and application in cell and gene therapy. J Cell Mol Med 2004, 8 (3), 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Gerecht S; Burdick JA; Ferreira LS; Townsend SA; Langer R; Vunjak-Novakovic G.Hyaluronic acid hydrogel for controlled self-renewal and differentiation of human embryonic stem cells. Proc Natl Acad Sci U S A 2007, 104 (27), 11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Kim IL; Mauck RL; Burdick JA Hydrogel design for cartilage tissue engineering: a case study with hyaluronic acid. Biomaterials 2011, 32 (34), 8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Ifkovits JL; Tous E; Minakawa M; Morita M; Robb JD; Koomalsingh KJ; Gorman JH 3rd; Gorman RC; Burdick JA Injectable hydrogel properties influence infarct expansion and extent of postinfarction left ventricular remodeling in an ovine model. Proc Natl Acad Sci U S A 2010, 107 (25), 11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Masters KS; Shah DN; Leinwand LA; Anseth KS Crosslinked hyaluronan scaffolds as a biologically active carrier for valvular interstitial cells. Biomaterials 2005, 26 (15), 2517. [DOI] [PubMed] [Google Scholar]
- (144).Sorushanova A; Delgado LM; Wu Z; Shologu N; Kshirsagar A; Raghunath R; Mullen AM; Bayon Y; Pandit A; Raghunath M et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv Mater 2019, 31 (1), e1801651. [DOI] [PubMed] [Google Scholar]
- (145).Ferreira AM; Gentile P; Chiono V; Ciardelli G.Collagen for bone tissue regeneration. Acta Biomater 2012, 8 (9), 3191. [DOI] [PubMed] [Google Scholar]
- (146).Shoulders MD; Raines RT Collagen structure and stability. Annu Rev Biochem 2009, 78 (1), 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Banwell EF; Abelardo ES; Adams DJ; Birchall MA; Corrigan A; Donald AM; Kirkland M; Serpell LC; Butler MF; Woolfson DN Rational design and application of responsive alpha-helical peptide hydrogels. Nat Mater 2009, 8 (7), 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Que RA; Arulmoli J; Da Silva NA; Flanagan LA; Wang SW Recombinant collagen scaffolds as substrates for human neural stem/progenitor cells. J Biomed Mater Res A 2018, 106 (5), 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Liu X; Zheng C; Luo X; Wang X; Jiang H.Recent advances of collagen-based biomaterials: Multi-hierarchical structure, modification and biomedical applications. Mater Sci Eng C Mater Biol Appl 2019, 99, 1509. [DOI] [PubMed] [Google Scholar]
- (150).Schmitt FO; Hall CE; Jakus MA Electron microscope investigations of the structure of collagen. J Cell Compar Physl 1942, 20 (1), 11. [Google Scholar]
- (151).Glowacki J; Mizuno S.Collagen scaffolds for tissue engineering. Biopolymers 2008, 89 (5), 338. [DOI] [PubMed] [Google Scholar]
- (152).Chan KLS; Khankhel AH; Thompson RL; Coisman BJ; Wong KHK; Truslow JG; Tien J.Crosslinking of collagen scaffolds promotes blood and lymphatic vascular stability. J Biomed Mater Res A 2014, 102 (9), 3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Rault I; Frei V; Herbage D; Abdul-Malak N; Huc A.Evaluation of different chemical methods for cros-linking collagen gel, films and sponges. Journal of Materials Science: Materials in Medicine 1996, 7 (4), 215. [Google Scholar]
- (154).Chevallay B; Abdul-Malak N; Herbage D.Mouse fibroblasts in long-term culture within collagen three-dimensional scaffolds: Influence of crosslinking with diphenylphosphorylazide on matrix reorganization, growth, and biosynthetic and proteolytic activities. J Biomed Mater Res 2000, 49 (4), 448. [DOI] [PubMed] [Google Scholar]
- (155).Damink LHHO; Dijkstra PJ; vanLuyn MJA; vanWachem PB; Nieuwenhuis P; Feijen J.In vitro degradation of dermal sheep collagen cross-linked using a water-soluble carbodiimide. Biomaterials 1996, 17 (7), 679. [DOI] [PubMed] [Google Scholar]
- (156).Couet F; Rajan N; Mantovani D.Macromolecular biomaterials for scaffold-based vascular tissue engineering. Macromol Biosci 2007, 7 (5), 701. [DOI] [PubMed] [Google Scholar]
- (157).Marelli B; Achilli M; Alessandrino A; Freddi G; Tanzi MC; Fare S; Mantovani D.Collagenreinforced electrospun silk fibroin tubular construct as small calibre vascular graft. Macromol Biosci 2012, 12 (11), 1566. [DOI] [PubMed] [Google Scholar]
- (158).Pankajakshan D; Agrawal DK Scaffolds in tissue engineering of blood vessels. Can J Physiol Pharmacol 2010, 88 (9), 855. [DOI] [PubMed] [Google Scholar]
- (159).Thacharodi D; Rao KP Rate-controlling biopolymer membranes as transdermal delivery systems for nifedipine: Development and in vitro evaluations. Biomaterials 1996, 17 (13), 1307. [PubMed] [Google Scholar]
- (160).Lucas PA; Syftestad GT; Goldberg VM; Caplan AI Ectopic induction of cartilage and bone by water-soluble proteins from bovine bone using a collagenous delivery vehicle. J Biomed Mater Res 1989, 23 (A1 Suppl), 23. [DOI] [PubMed] [Google Scholar]
- (161).Kaufmann P.Highly porous polymer matrices as a three-dimensional culture system for hepatocytes. Cell Transplantation 1997, 6 (5), 463. [DOI] [PubMed] [Google Scholar]
- (162).Helary C; Zarka M; Giraud-Guille MM Fibroblasts within concentrated collagen hydrogels favour chronic skin wound healing. J Tissue Eng Regen Med 2012, 6 (3), 225. [DOI] [PubMed] [Google Scholar]
- (163).McGuigan AP; Sefton MV The thrombogenicity of human umbilical vein endothelial cell seeded collagen modules. Biomaterials 2008, 29 (16), 2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Voytik-Harbin SL; Brightman AO; Waisner BZ; Robinson JP; Lamar CH Small intestinal submucosa: A tissue-derived extracellular matrix that promotes tissue-specific growth and differentiation of cells in vitro. Tissue Eng 1998, 4 (2), 157. [Google Scholar]
- (165).Yuan T; Zhang L; Li KF; Fan HS; Fan YJ; Liang J; Zhang XD Collagen hydrogel as an immunomodulatory scaffold in cartilage tissue engineering. J Biomed Mater Res B 2014, 102 (2), 337. [DOI] [PubMed] [Google Scholar]
- (166).Hahn MS; Teply BA; Stevens MM; Zeitels SM; Langer R.Collagen composite hydrogels for vocal fold lamina propria restoration. Biomaterials 2006, 27 (7), 1104. [DOI] [PubMed] [Google Scholar]
- (167).Iwata A; Browne KD; Pfister BJ; Gruner JA; Smith DH Long-term survival and outgrowth of mechanically engineered nervous tissue constructs implanted into spinal cord lesions. Tissue Eng 2006, 12 (1), 101. [DOI] [PubMed] [Google Scholar]
- (168).Hudson CB In Food Hydrocolloid; Springer, 1994. [Google Scholar]
- (169).Segtnan VH; Isaksson T.Temperature, sample and time dependent structural characteristics of gelatine gels studied by near infrared spectroscopy. Food Hydrocolloid 2004, 18 (1), 1. [Google Scholar]
- (170).Duconseille A; Astruc T; Quintana N; Meersman F; Sante-Lhoutellier V.Gelatin structure and composition linked to hard capsule dissolution: A review. Food Hydrocolloid 2015, 43, 360. [Google Scholar]
- (171).Farris S; Song J; Huang Q.Alternative reaction mechanism for the cross-linking of gelatin with glutaraldehyde. J Agric Food Chem 2010, 58 (2), 998. [DOI] [PubMed] [Google Scholar]
- (172).Kuijpers AJ; Engbers GHM; Feijen J; De Smedt SC; Meyvis TKL; Demeester J; Krijgsveld J; Zaat SAJ; Dankert J.Characterization of the network structure of carbodiimide cross-linked gelatin gels. Macromolecules 1999, 32 (10), 3325. [Google Scholar]
- (173).Digenis GA; Gold TB; Shah VP Cross‐Linking of Gelatin Capsules and Its Relevance to Their in Vitro-in Vivo Performance. Journal of Pharmaceutical Sciences 1994, 83 (7), 915. [DOI] [PubMed] [Google Scholar]
- (174).Tu R; Shen SH; Lin D; Hata C; Thyagarajan K; Noishiki Y; Quijano RC Fixation of bioprosthetic tissues with monofunctional and multifunctional polyepoxy compounds. J Biomed Mater Res 1994, 28 (6), 677. [DOI] [PubMed] [Google Scholar]
- (175).Hyndman CL; Groboillot AF; Poncelet D; Champagne CP; Neufeld RJ Microencapsulation of Lactococcus lactis within cross-linked gelatin membranes. Journal of Chemical Technology & Biotechnology 1993, 56 (3), 259. [Google Scholar]
- (176).Yue K; Trujillo-de Santiago G; Alvarez MM; Tamayol A; Annabi N; Khademhosseini A.Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels. Biomaterials 2015, 73, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Morikawaand N; Matsuda T.Thermoresponsive artificial extracellular matrix: Nisopropylacrylamide-graft-copolymerized gelatin. Journal of Biomaterials Science, Polymer Edition 2002, 13 (2), 167. [DOI] [PubMed] [Google Scholar]
- (178).Rizwan M; Yao Y; Gorbet MB; Tse JW; Anderson DEJ; Hinds MT; Yim EKF One-Pot Covalent Grafting of Gelatin on Poly(Vinyl Alcohol) Hydrogel to Enhance Endothelialization and Hemocompatibility for Synthetic Vascular Graft Applications. ACS Applied Bio Materials 2020, 3 (1), 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Nichol JW; Koshy ST; Bae H; Hwang CM; Yamanlar S; Khademhosseini A.Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials 2010, 31 (21), 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Shin SR; Bae H; Cha JM; Mun JY; Chen Y-C; Tekin H; Shin H; Farshchi S; Dokmeci MR; Tang S et al. Carbon Nanotube Reinforced Hybrid Microgels as Scaffold Materials for Cell Encapsulation. ACS Nano 2012, 6 (1), 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Paul A; Hasan A; Kindi HA; Gaharwar AK; Rao VT; Nikkhah M; Shin SR; Krafft D; Dokmeci MR; Shum-Tim D et al. Injectable graphene oxide/hydrogel-based angiogenic gene delivery system for vasculogenesis and cardiac repair. ACS Nano 2014, 8 (8), 8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Bakhsheshi-Rad HR; Hadisi Z; Hamzah E; Ismail AF; Aziz M; Kashefian M.Drug delivery and cytocompatibility of ciprofloxacin loaded gelatin nanofibers-coated Mg alloy. Mater Lett 2017, 207, 179. [Google Scholar]
- (183).Nagarajan S; Belaid H; Pochat-Bohatier C; Teyssier C; Iatsunskyi I; Coy E; Balme S; Cornu D; Miele P; Kalkura NS et al. Design of Boron Nitride/Gelatin Electrospun Nanofibers for Bone Tissue Engineering. ACS Appl Mater Interfaces 2017, 9 (39), 33695. [DOI] [PubMed] [Google Scholar]
- (184).Choi YS; Hong SR; Lee YM; Song KW; Park MH; Nam YS Study on gelatin-containing artificial skin: I. Preparation and characteristics of novel gelatin-alginate sponge. Biomaterials 1999, 20 (5), 409. [DOI] [PubMed] [Google Scholar]
- (185).Yamamoto M; Tabata Y; Ikada Y.Growth factor release from gelatin hydrogel for tissue engineering. J Bioact Compat Pol 1999, 14 (6), 474. [Google Scholar]
- (186).Sajkiewicz P; Kołbuk D.Electrospinning of gelatin for tissue engineering–molecular conformation as one of the overlooked problems. Journal of Biomaterials Science, Polymer Edition 2014, 25 (18), 2009. [DOI] [PubMed] [Google Scholar]
- (187).Panzavolta S; Gioffre M; Focarete ML; Gualandi C; Foroni L; Bigi A.Electrospun gelatin nanofibers: optimization of genipin cross-linking to preserve fiber morphology after exposure to water. Acta Biomater 2011, 7 (4), 1702. [DOI] [PubMed] [Google Scholar]
- (188).Noori A; Ashrafi SJ; Vaez-Ghaemi R; Hatamian-Zaremi A; Webster TJ A review of fibrin and fibrin composites for bone tissue engineering. Int J Nanomedicine 2017, 12, 4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Perka C; Spitzer RS; Lindenhayn K; Sittinger M; Schultz O.Matrix‐mixed culture: New methodology for chondrocyte culture and preparation of cartilage transplants. Journal of Biomedical Materials Research: An Official Journal of The Society for Biomaterials and The Japanese Society for Biomaterials 2000, 49 (3), 305. [DOI] [PubMed] [Google Scholar]
- (190).Dare EV; Griffith M; Poitras P; Kaupp JA; Waldman SD; Carlsson DJ; Dervin G; Mayoux C; Hincke MT Genipin cross-linked fibrin hydrogels for in vitro human articular cartilage tissue-engineered regeneration. Cells Tissues Organs 2009, 190 (6), 313. [DOI] [PubMed] [Google Scholar]
- (191).Lee CR; Grad S; Gorna K; Gogolewski S; Goessl A; Alini M.Fibrin–Polyurethane Composites for Articular Cartilage Tissue Engineering: A Preliminary Analysis. Tissue Eng 2005, 11 (9–10), 1562. [DOI] [PubMed] [Google Scholar]
- (192).Van Lieshout M; Peters G; Rutten M; Baaijens F.A knitted, fibrin-covered polycaprolactone scaffold for tissue engineering of the aortic valve. Tissue Eng 2006, 12 (3), 481. [DOI] [PubMed] [Google Scholar]
- (193).Weinand C; Gupta R; Huang AY; Weinberg E; Madisch I; Qudsi RA; Neville CM; Pomerantseva I; Vacanti JP Comparison of hydrogels in the in vivo formation of tissue-engineered bone using mesenchymal stem cells and beta-tricalcium phosphate. Tissue Eng 2007, 13 (4), 757. [DOI] [PubMed] [Google Scholar]
- (194).Peled E; Boss J; Bejar J; Zinman C; Seliktar D.A novel poly(ethylene glycol)–fibrinogen hydrogel for tibial segmental defect repair in a rat model. Journal of Biomedical Materials Research Part A 2007, 80A (4), 874. [DOI] [PubMed] [Google Scholar]
- (195).Spotnitz WD; Prabhu R.Fibrin sealant tissue adhesive--review and update. J Long Term Eff Med Implants 2005, 15 (3), 245. [DOI] [PubMed] [Google Scholar]
- (196).Chrobak MO; Hansen KJ; Gershlak JR; Vratsanos M; Kanellias M; Gaudette GR; Pins GD Design of a Fibrin Microthread-Based Composite Layer for Use in a Cardiac Patch. Acs Biomater Sci Eng 2017, 3 (7), 1394. [DOI] [PubMed] [Google Scholar]
- (197).Schense JC; Hubbell JA Cross-linking exogenous bifunctional peptides into fibrin gels with factor XIIIa. Bioconjugate Chem 1999, 10 (1), 75. [DOI] [PubMed] [Google Scholar]
- (198).Han B; Schwab IR; Madsen TK; Isseroff RR A fibrin-based bioengineered ocular surface with human corneal epithelial stem cells. Cornea 2002, 21 (5), 505. [DOI] [PubMed] [Google Scholar]
- (199).Meinhart J; Fussenegger M; Hobling W.Stabilization of fibrin-chondrocyte constructs for cartilage reconstruction. Ann Plast Surg 1999, 42 (6), 673. [DOI] [PubMed] [Google Scholar]
- (200).Johnson TS; Xu JW; Zaporojan VV; Mesa JM; Weinand C; Randolph MA; Bonassar LJ; Winograd JM; Yaremchuk MJ Integrative repair of cartilage with articular and nonarticular chondrocytes. Tissue Eng 2004, 10 (9–10), 1308. [DOI] [PubMed] [Google Scholar]
- (201).Ye Q; Zund G; Benedikt P; Jockenhoevel S; Hoerstrup SP; Sakyama S; Hubbell JA; Turina M.Fibrin gel as a three dimensional matrix in cardiovascular tissue engineering. Eur J Cardio-Thorac 2000, 17 (5), 587. [DOI] [PubMed] [Google Scholar]
- (202).Currie LJ; Sharpe JR; Martin R.The use of fibrin glue in skin grafts and tissue-engineered skin replacements: A review. Plast Reconstr Surg 2001, 108 (6), 1713. [DOI] [PubMed] [Google Scholar]
- (203).Ahmed TA; Dare EV; Hincke M.Fibrin: a versatile scaffold for tissue engineering applications. Tissue Eng Part B Rev 2008, 14 (2), 199. [DOI] [PubMed] [Google Scholar]
- (204).Marinho-Soriano E.Agar polysaccharides from Gracilaria species (Rhodophyta, Gracilariaceae). J Biotechnol 2001, 89 (1), 81. [DOI] [PubMed] [Google Scholar]
- (205).Lahaye M; Rochas C.International workshop on gelidium, 1991; p 137. [Google Scholar]
- (206).Normand V; Lootens DL; Amici E; Plucknett KP; Aymard P.New insight into agarose gel mechanical properties. Biomacromolecules 2000, 1 (4), 730. [DOI] [PubMed] [Google Scholar]
- (207).Uludag H; De Vos P; Tresco PA Technology of mammalian cell encapsulation. Adv Drug Deliv Rev 2000, 42 (1–2), 29. [DOI] [PubMed] [Google Scholar]
- (208).Dillon GP; Yu X; Sridharan A; Ranieri JP; Bellamkonda RV The influence of physical structure and charge on neurite extension in a 3D hydrogel scaffold. J Biomater Sci Polym Ed 1998, 9 (10), 1049. [DOI] [PubMed] [Google Scholar]
- (209).Kumachev A; Greener J; Tumarkin E; Eiser E; Zandstra PW; Kumacheva E.High-throughput generation of hydrogel microbeads with varying elasticity for cell encapsulation. Biomaterials 2011, 32 (6), 1477. [DOI] [PubMed] [Google Scholar]
- (210).Clavé A; Potel JF; Servien E; Neyret P; Dubrana F; Stindel E.Third‐generation autologous chondrocyte implantation versus mosaicplasty for knee cartilage injury: 2‐year randomized trial. Journal of Orthopaedic Research 2016, 34 (4), 658. [DOI] [PubMed] [Google Scholar]
- (211).Luo Y; Shoichet MS A photolabile hydrogel for guided three-dimensional cell growth and migration. Nat Mater 2004, 3 (4), 249. [DOI] [PubMed] [Google Scholar]
- (212).Luan NM; Iwata H.Xenotransplantation of islets enclosed in agarose microcapsule carrying soluble complement receptor 1. Biomaterials 2012, 33 (32), 8075. [DOI] [PubMed] [Google Scholar]
- (213).Borkenhagen M; Clémence JF; Sigrist H; Aebischer P.Three‐dimensional extracellular matrix engineering in the nervous system. J Biomed Mater Res 1998, 40 (3), 392. [DOI] [PubMed] [Google Scholar]
- (214).Bhattarai N; Gunn J; Zhang M.Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Deliv Rev 2010, 62 (1), 83. [DOI] [PubMed] [Google Scholar]
- (215).Croisier F; Jérôme C.Chitosan-based biomaterials for tissue engineering. European Polymer Journal 2013, 49 (4), 780. [Google Scholar]
- (216).Sorlier P; Denuzière A; Viton C; Domard A.Relation between the Degree of Acetylation and the Electrostatic Properties of Chitin and Chitosan. Biomacromolecules 2001, 2 (3), 765. [DOI] [PubMed] [Google Scholar]
- (217).Foster LJR; Ho S; Hook J; Basuki M; Marcal H.Chitosan as a Biomaterial: Influence of Degree of Deacetylation on Its Physiochemical, Material and Biological Properties. Plos One 2015, 10 (8), e0135153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Berger J; Reist M; Mayer JM; Felt O; Peppas NA; Gurny R.Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur J Pharm Biopharm 2004, 57 (1), 19. [DOI] [PubMed] [Google Scholar]
- (219).Boucard N; Viton C; Domard A.New aspects of the formation of physical hydrogels of chitosan in a hydroalcoholic medium. Biomacromolecules 2005, 6 (6), 3227. [DOI] [PubMed] [Google Scholar]
- (220).Brack HP; Tirmizi SA; Risen WM A spectroscopic and viscometric study of the metal ion-induced gelation of the biopolymer chitosan. Polymer 1997, 38 (10), 2351. [Google Scholar]
- (221).Dambies L; Vincent T; Domard A; Guibal E.Preparation of chitosan gel beads by ionotropic molybdate gelation. Biomacromolecules 2001, 2 (4), 1198. [DOI] [PubMed] [Google Scholar]
- (222).Shu XZ; Zhu KJ Controlled drug release properties of ionically cross-linked chitosan beads: the influence of anion structure. Int J Pharm 2002, 233 (1–2), 217. [DOI] [PubMed] [Google Scholar]
- (223).Shen EC; Wang C; Fu E; Chiang CY; Chen TT; Nieh S.Tetracycline release from tripolyphosphate–chitosan cross-linked sponge: a preliminary in vitro study. Journal of Periodontal Research 2008, 43 (6), 642. [DOI] [PubMed] [Google Scholar]
- (224).Boddohi S; Moore N; Johnson PA; Kipper MJ Polysaccharide-based polyelectrolyte complex nanoparticles from chitosan, heparin, and hyaluronan. Biomacromolecules 2009, 10 (6), 1402. [DOI] [PubMed] [Google Scholar]
- (225).Denuziere A; Ferrier D; Damour O; Domard A.Chitosan–chondroitin sulfate and chitosan– hyaluronate polyelectrolyte complexes: biological properties. Biomaterials 1998, 19 (14), 1275. [DOI] [PubMed] [Google Scholar]
- (226).Jiang T; Zhang Z; Zhou Y; Liu Y; Wang Z; Tong H; Shen X; Wang Y.Surface functionalization of titanium with chitosan/gelatin via electrophoretic deposition: characterization and cell behavior. Biomacromolecules 2010, 11 (5), 1254. [DOI] [PubMed] [Google Scholar]
- (227).Mao JS; Cui YL; Wang XH; Sun Y; Yin YJ; Zhao HM; De Yao K.A preliminary study on chitosan and gelatin polyelectrolyte complex cytocompatibility by cell cycle and apoptosis analysis. Biomaterials 2004, 25 (18), 3973. [DOI] [PubMed] [Google Scholar]
- (228).de Oliveira HC; Fonseca JL; Pereira MR Chitosan-poly(acrylic acid) polyelectrolyte complex membranes: preparation, characterization and permeability studies. J Biomater Sci Polym Ed 2008, 19 (2), 143. [DOI] [PubMed] [Google Scholar]
- (229).Chenite A; Chaput C; Wang D; Combes C; Buschmann MD; Hoemann CD; Leroux JC; Atkinson BL; Binette F; Selmani A.Novel injectable neutral solutions of chitosan form biodegradable gels in situ. Biomaterials 2000, 21 (21), 2155. [DOI] [PubMed] [Google Scholar]
- (230).Zhang Y; Guan Y; Zhou S.Single Component Chitosan Hydrogel Microcapsule from a Layer-byLayer Approach. Biomacromolecules 2005, 6 (4), 2365. [DOI] [PubMed] [Google Scholar]
- (231).Milosavljevic NB; Kljajevic LM; Popovic IG; Filipovic JM; Krusic MTK Chitosan, itaconic acid and poly(vinyl alcohol) hybrid polymer networks of high degree of swelling and good mechanical strength. Polym Int 2010, 59 (5), 686. [Google Scholar]
- (232).Singh A; Narvi SS; Dutta PK; Pandey ND External stimuli response on a novel chitosan hydrogel crosslinked with formaldehyde. Bulletin of Materials Science 2006, 29 (3), 233. [Google Scholar]
- (233).Liu R; Xu X; Zhuang X; Cheng B.Solution blowing of chitosan/PVA hydrogel nanofiber mats. Carbohydrate Polymers 2014, 101, 1116. [DOI] [PubMed] [Google Scholar]
- (234).Mi FL; Sung HW; Shyu SS Synthesis and characterization of a novel chitosan-based network prepared using naturally occurring crosslinker. J Polym Sci Pol Chem 2000, 38 (15), 2804. [Google Scholar]
- (235).Muzzarelli RAA Genipin-crosslinked chitosan hydrogels as biomedical and pharmaceutical aids. Carbohydrate Polymers 2009, 77 (1), 1. [Google Scholar]
- (236).Hoare TR; Kohane DS Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49 (8), 1993. [Google Scholar]
- (237).Hennink WE; van Nostrum CF Novel crosslinking methods to design hydrogels. Advanced Drug Delivery Reviews 2012, 64, 223. [DOI] [PubMed] [Google Scholar]
- (238).Rickett TA; Amoozgar Z; Tuchek CA; Park J; Yeo Y; Shi R.Rapidly Photo-Cross-Linkable Chitosan Hydrogel for Peripheral Neurosurgeries. Biomacromolecules 2011, 12 (1), 57. [DOI] [PubMed] [Google Scholar]
- (239).Cho IS; Cho MO; Li Z; Nurunnabi M; Park SY; Kang S-W; Huh KM Synthesis and characterization of a new photo-crosslinkable glycol chitosan thermogel for biomedical applications. Carbohydrate Polymers 2016, 144, 59. [DOI] [PubMed] [Google Scholar]
- (240).Obara K; Ishihara M; Ishizuka T; Fujita M; Ozeki Y; Maehara T; Saito Y; Yura H; Matsui T; Hattori H et al. Photocrosslinkable chitosan hydrogel containing fibroblast growth factor-2 stimulates wound healing in healing-impaired db/db mice. Biomaterials 2003, 24 (20), 3437. [DOI] [PubMed] [Google Scholar]
- (241).Ahmadi F; Oveisi Z; Samani SM; Amoozgar Z.Chitosan based hydrogels: characteristics and pharmaceutical applications. Res Pharm Sci 2015, 10 (1), 1. [PMC free article] [PubMed] [Google Scholar]
- (242).Cui ZK; Kim S; Baljon JJ; Wu BM; Aghaloo T; Lee M.Microporous methacrylated glycol chitosan-montmorillonite nanocomposite hydrogel for bone tissue engineering. Nat Commun 2019, 10 (1), 3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Kim IY; Seo SJ; Moon HS; Yoo MK; Park IY; Kim BC; Cho CS Chitosan and its derivatives for tissue engineering applications. Biotechnol Adv 2008, 26 (1), 1. [DOI] [PubMed] [Google Scholar]
- (244).Li J; Xu Z.Physical characterization of a chitosan‐based hydrogel delivery system. Journal of pharmaceutical sciences 2002, 91 (7), 1669. [DOI] [PubMed] [Google Scholar]
- (245).Muzzarelli RA; Zucchini C; Ilari P; Pugnaloni A; Mattioli Belmonte M; Biagini G; Castaldini C.Osteoconductive properties of methylpyrrolidinone chitosan in an animal model. Biomaterials 1993, 14 (12), 925. [DOI] [PubMed] [Google Scholar]
- (246).Elcin AE; Elcin YM; Pappas GD Neural tissue engineering: Adrenal chromaffin cell attachment and viability on chitosan scaffolds. Neurol Res 1998, 20 (7), 648. [DOI] [PubMed] [Google Scholar]
- (247).Lee KY; Ha WS; Park WH Blood compatibility and biodegradability of partially N-acylated chitosan derivatives. Biomaterials 1995, 16 (16), 1211. [DOI] [PubMed] [Google Scholar]
- (248).Klemm D; Heublein B; Fink HP; Bohn A.Cellulose: fascinating biopolymer and sustainable raw material. Angew Chem Int Ed Engl 2005, 44 (22), 3358. [DOI] [PubMed] [Google Scholar]
- (249).Habibi Y; Lucia LA; Rojas OJ Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem Rev 2010, 110 (6), 3479. [DOI] [PubMed] [Google Scholar]
- (250).Moon RJ; Martini A; Nairn J; Simonsen J; Youngblood J.Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 2011, 40 (7), 3941. [DOI] [PubMed] [Google Scholar]
- (251).Ross P; Mayer R; Benziman M.Cellulose biosynthesis and function in bacteria. Microbiol Rev 1991, 55 (1), 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Czaja WK; Young DJ; Kawecki M; Brown RM Jr. The future prospects of microbial cellulose in biomedical applications. Biomacromolecules 2007, 8 (1), 1. [DOI] [PubMed] [Google Scholar]
- (253).Fink HP; Weigel P; Purz HJ; Ganster J.Structure formation of regenerated cellulose materials from NMMO-solutions. Progress in Polymer Science 2001, 26 (9), 1473. [Google Scholar]
- (254).Zhao HB; Kwak JH; Wang Y; Franz JA; White JM; Holladay JE Interactions between cellulose and N-methylmorpholine-N-oxide. Carbohydrate Polymers 2007, 67 (1), 97. [Google Scholar]
- (255).Zhang H; Wu J; Zhang J; He JS 1-Allyl-3-methylimidazolium chloride room temperature ionic liquid: A new and powerful nonderivatizing solvent for cellulose. Macromolecules 2005, 38 (20), 8272. [Google Scholar]
- (256).Zhu SD; Wu YX; Chen QM; Yu ZN; Wang CW; Jin SW; Ding YG; Wu G Dissolution of cellulose with ionic liquids and its application: a mini-review. Green Chem 2006, 8 (4), 325. [Google Scholar]
- (257).Cai J; Zhang L.Unique gelation behavior of cellulose in NaOH/urea aqueous solution. Biomacromolecules 2006, 7 (1), 183. [DOI] [PubMed] [Google Scholar]
- (258).Liang SM; Zhang LN; Li YF; Xu J.Fabrication and properties of cellulose hydrated membrane with unique structure. Macromol Chem Phys 2007, 208 (6), 594. [Google Scholar]
- (259).Li L; Thangamathesvaran PM; Yue CY; Tam KC; Hu X; Lam YC Gel network structure of methylcellulose in water. Langmuir 2001, 17 (26), 8062. [Google Scholar]
- (260).Xia XH; Tang SJ; Lu XH; Hu ZB Formation and volume phase transition of hydroxypropyl cellulose microgels in salt solution. Macromolecules 2003, 36 (10), 3695. [Google Scholar]
- (261).Weiss P; Gauthier O; Bouler JM; Grimandi G; Daculsi G.Injectable bone substitute using a hydrophilic polymer. Bone 1999, 25 (2), 67s. [DOI] [PubMed] [Google Scholar]
- (262).Silva SM; Pinto FV; Antunes FE; Miguel MG; Sousa JJ; Pais AA Aggregation and gelation in hydroxypropylmethyl cellulose aqueous solutions. J Colloid Interface Sci 2008, 327 (2), 333. [DOI] [PubMed] [Google Scholar]
- (263).Zheng WJ; Gao J; Wei Z; Zhou JX; Chen YM Facile fabrication of self-healing carboxymethyl cellulose hydrogels. European Polymer Journal 2015, 72, 514. [Google Scholar]
- (264).Kono H; Fujita S.Biodegradable superabsorbent hydrogels derived from cellulose by esterification crosslinking with 1,2,3,4-butanetetracarboxylic dianhydride. Carbohydrate Polymers 2012, 87 (4), 2582. [Google Scholar]
- (265).Yoshimura T; Matsuo K; Fujioka R.Novel biodegradable superabsorbent hydrogels derived from cotton cellulose and succinic anhydride: Synthesis and characterization. Journal of Applied Polymer Science 2006, 99 (6), 3251. [Google Scholar]
- (266).Demitri C; Del Sole R; Scalera F; Sannino A; Vasapollo G; Maffezzoli A; Ambrosio L; Nicolais L.Novel superabsorbent cellulose-based hydrogels crosslinked with citric acid. Journal of Applied Polymer Science 2008, 110 (4), 2453. [Google Scholar]
- (267).Zhou J; Chang C; Zhang R; Zhang L.Hydrogels prepared from unsubstituted cellulose in NaOH/urea aqueous solution. Macromol Biosci 2007, 7 (6), 804. [DOI] [PubMed] [Google Scholar]
- (268).Rodríguez R.a.; Alvarez-Lorenzo C; Concheiro A.Cationic cellulose hydrogels: kinetics of the cross-linking process and characterization as pH-/ion-sensitive drug delivery systems. J Control Release 2003, 86 (2–3), 253. [DOI] [PubMed] [Google Scholar]
- (269).Kabra BG; Gehrke SH; Spontak RJ Microporous, responsive hydroxypropyl cellulose gels. 1. Synthesis and microstructure. Macromolecules 1998, 31 (7), 2166. [Google Scholar]
- (270).Fei B; Wach RA; Mitomo H; Yoshii F; Kume T.Hydrogel of biodegradable cellulose derivatives. I. Radiation-induced crosslinking of CMC. Journal of Applied Polymer Science 2000, 78 (2), 278. [Google Scholar]
- (271).Liu PF; Peng J; Li JQ; Wu JL Radiation crosslinking of CMC-Na at low dose and its application as substitute for hydrogel. Radiat Phys Chem 2005, 72 (5), 635. [Google Scholar]
- (272).Long DD; vanLuyen D.Chitosan-carboxymethylcellulose hydrogels as supports for cell immobilization. J Macromol Sci Pure 1996, A33 (12), 1875. [Google Scholar]
- (273).Hebeish A; Higazy A; El-Shafei A; Sharaf S.Synthesis of carboxymethyl cellulose (CMC) and starch-based hybrids and their applications in flocculation and sizing. Carbohydrate Polymers 2010, 79 (1), 60. [Google Scholar]
- (274).Isiklan N.Controlled release of insecticide carbaryl from sodium alginate, sodium alginate/gelatin, and sodium alginate/sodium carboxymethyl cellulose blend beads crosslinked with glutaraldehyde. Journal of Applied Polymer Science 2006, 99 (4), 1310. [Google Scholar]
- (275).Sannino A; Madaghiele M; Conversano F; Mele G; Maffezzoli A; Netti PA; Ambrosio L; Nicolais L.Cellulose derivative-hyaluronic acid-based microporous hydrogels cross-linked through divinyl sulfone (DVS) to modulate equilibrium sorption capacity and network stability. Biomacromolecules 2004, 5 (1), 92. [DOI] [PubMed] [Google Scholar]
- (276).Liang S; Wu J; Tian H; Zhang L; Xu J.High-strength cellulose/poly(ethylene glycol) gels. ChemSusChem 2008, 1 (6), 558. [DOI] [PubMed] [Google Scholar]
- (277).Millon LE; Wan WK The polyvinyl alcohol-bacterial cellulose system as a new nanocomposite for biomedical applications. J Biomed Mater Res B Appl Biomater 2006, 79 (2), 245. [DOI] [PubMed] [Google Scholar]
- (278).Williamson SL; Armentrout RS; Porter RS; McCormick CL Microstructural examination of semi-interpenetrating networks of poly(N,N-dimethylacrylamide) with cellulose or chitin synthesized in lithium chloride N,N-dimethylacetamide. Macromolecules 1998, 31 (23), 8134. [Google Scholar]
- (279).Sannino A; Demitri C; Madaghiele M.Biodegradable Cellulose-based Hydrogels: Design and Applications. Materials 2009, 2 (2), 353. [Google Scholar]
- (280).Klemm D; Schumann D; Udhardt U; Marsch S.Bacterial synthesized cellulose — artificial blood vessels for microsurgery. Progress in Polymer Science 2001, 26 (9), 1561. [Google Scholar]
- (281).Chang CY; Duan B; Cai J; Zhang LN Superabsorbent hydrogels based on cellulose for smart swelling and controllable delivery. European Polymer Journal 2010, 46 (1), 92. [Google Scholar]
- (282).Vinatier C; Gauthier O; Fatimi A; Merceron C; Masson M; Moreau A; Moreau F; Fellah B; Weiss P; Guicheux J.An injectable cellulose-based hydrogel for the transfer of autologous nasal chondrocytes in articular cartilage defects. Biotechnol Bioeng 2009, 102 (4), 1259. [DOI] [PubMed] [Google Scholar]
- (283).Ye SH; Watanabe J; Iwasaki Y; Ishihara K.Antifouling blood purification membrane composed of cellulose acetate and phospholipid polymer. Biomaterials 2003, 24 (23), 4143. [DOI] [PubMed] [Google Scholar]
- (284).Hosseini H; Kokabi M; Mousavi SM Conductive bacterial cellulose/multiwall carbon nanotubes nanocomposite aerogel as a potentially flexible lightweight strain sensor. Carbohydr Polym 2018, 201, 228. [DOI] [PubMed] [Google Scholar]
- (285).Zhou D; Zhang L; Zhou J; Guo S.Cellulose/chitin beads for adsorption of heavy metals in aqueous solution. Water Res 2004, 38 (11), 2643. [DOI] [PubMed] [Google Scholar]
- (286).Elliott JE; Macdonald M; Nie J; Bowman CN Structure and swelling of poly(acrylic acid) hydrogels: effect of pH, ionic strength, and dilution on the crosslinked polymer structure. Polymer 2004, 45 (5), 1503. [Google Scholar]
- (287).Zhao L; Huang J; Zhang Y; Wang T; Sun W; Tong Z.Programmable and Bidirectional Bending of Soft Actuators Based on Janus Structure with Sticky Tough PAA-Clay Hydrogel. ACS Appl Mater Interfaces 2017, 9 (13), 11866. [DOI] [PubMed] [Google Scholar]
- (288).Zhong M; Liu Y-T; Xie X-M Self-healable, super tough graphene oxide–poly (acrylic acid) nanocomposite hydrogels facilitated by dual cross-linking effects through dynamic ionic interactions. Journal of Materials Chemistry B 2015, 3 (19), 4001. [DOI] [PubMed] [Google Scholar]
- (289).Zhao L; Huang JH; Wang T; Sun WX; Tong Z.Multiple Shape Memory, Self-Healable, and Supertough PAA-GO-Fe3+ Hydrogel. Macromol Mater Eng 2017, 302 (2), 1600359. [Google Scholar]
- (290).Gulyuz U; Okay O.Self-Healing Poly(acrylic acid) Hydrogels with Shape Memory Behavior of High Mechanical Strength. Macromolecules 2014, 47 (19), 6889. [Google Scholar]
- (291).Liu X; Steiger C; Lin S; Parada GA; Liu J; Chan HF; Yuk H; Phan NV; Collins J; Tamang S et al. Ingestible hydrogel device. Nat Commun 2019, 10 (1), 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Eichenbaum GM; Kiser PF; Dobrynin AV; Simon SA; Needham D.Investigation of the Swelling Response and Loading of Ionic Microgels with Drugs and Proteins: The Dependence on Cross-Link Density. Macromolecules 1999, 32 (15), 4867. [Google Scholar]
- (293).Scott RA; Peppas NA Compositional effects on network structure of highly cross-linked copolymers of PEG-containing multiacrylates with-acrylic acid. Macromolecules 1999, 32 (19), 6139. [Google Scholar]
- (294).Peppas NA; Wright SL Drug diffusion and binding in ionizable interpenetrating networks from poly(vinyl alcohol) and poly(acrylic acid). Eur J Pharm Biopharm 1998, 46 (1), 15. [DOI] [PubMed] [Google Scholar]
- (295).Sefton M; May M; Lahooti S; Babensee J.Making microencapsulation work: conformal coating, immobilization gels and in vivo performance. J Control Release 2000, 65 (1–2), 173. [DOI] [PubMed] [Google Scholar]
- (296).Meyvis TKL; De Smedt SC; Demeester J; Hennink WE Influence of the degradation mechanism of hydrogels on their elastic and swelling properties during degradation. Macromolecules 2000, 33 (13), 4717. [Google Scholar]
- (297).Canal T; Peppas NA Correlation between Mesh Size and Equilibrium Degree of Swelling of Polymeric Networks. J Biomed Mater Res 1989, 23 (10), 1183. [DOI] [PubMed] [Google Scholar]
- (298).Kidane A; Szabocsik JM; Park K.Accelerated study on lysozyme deposition on poly(HEMA) contact lenses. Biomaterials 1998, 19 (22), 2051. [DOI] [PubMed] [Google Scholar]
- (299).Chirila TV An overview of the development of artificial corneas with porous skirts and the use of PHEMA for such an application. Biomaterials 2001, 22 (24), 3311. [DOI] [PubMed] [Google Scholar]
- (300).Hassan CM; Peppas NA Structure and morphology of freeze/thawed PVA hydrogels. Macromolecules 2000, 33 (7), 2472. [Google Scholar]
- (301).Nuttelman CR; Mortisen DJ; Henry SM; Anseth KS Attachment of fibronectin to poly(vinyl alcohol) hydrogels promotes NIH3T3 cell adhesion, proliferation, and migration. J Biomed Mater Res 2001, 57 (2), 217. [DOI] [PubMed] [Google Scholar]
- (302).Peppas NA; Merrill EW Development of semicrystalline poly(vinyl alcohol) hydrogels for biomedical applications. J Biomed Mater Res 1977, 11 (3), 423. [DOI] [PubMed] [Google Scholar]
- (303).Stauffer SR; Peppast NA Poly(vinyl alcohol) hydrogels prepared by freezing-thawing cyclic processing. Polymer 1992, 33 (18), 3932. [Google Scholar]
- (304).Peppas NA; Stauffer SR Reinforced Uncrosslinked Poly (Vinyl Alcohol) Gels Produced by Cyclic Freezing-Thawing Processes - a Short Review. J Control Release 1991, 16 (3), 305. [Google Scholar]
- (305).Dai WS; Barbari TA Hydrogel membranes with mesh size asymmetry based on the gradient crosslinking of poly(vinyl alcohol). J Membrane Sci 1999, 156 (1), 67. [Google Scholar]
- (306).Bo J.Study on PVA hydrogel crosslinked by epichlorohydrin. Journal of applied polymer science 1992, 46 (5), 783. [Google Scholar]
- (307).Yoshii F; Zhanshan Y; Isobe K; Shinozaki K; Makuuchi K.Electron beam crosslinked PEO and PEO PVA hydrogels for wound dressing. Radiat Phys Chem 1999, 55 (2), 133. [Google Scholar]
- (308).Kobayashi H; Ikada Y.Corneal cell adhesion and proliferation on hydrogel sheets bound with cell-adhesive proteins. Curr Eye Res 1991, 10 (10), 899. [DOI] [PubMed] [Google Scholar]
- (309).Bryant SJ; Nuttelman CR; Anseth KS The effects of crosslinking density on cartilage formation in photocrosslinkable hydrogels. Biomed Sci Instrum 1999, 35, 309. [PubMed] [Google Scholar]
- (310).Shaheen SM; Yamaura K.Preparation of theophylline hydrogels of atactic poly(vinyl alcohol)/NaCl/H2O system for drug delivery system. J Control Release 2002, 81 (3), 367. [DOI] [PubMed] [Google Scholar]
- (311).Gu ZQ; Xiao JM; Zhang XH The development of artificial articular cartilage–PVA‐hydrogel. Bio-medical materials and engineering 1998, 8 (2), 75. [PubMed] [Google Scholar]
- (312).Oka M; Ushio K; Kumar P; Ikeuchi K; Hyon SH; Nakamura T; Fujita H.Development of artificial articular cartilage. Proc Inst Mech Eng H 2000, 214 (1), 59. [DOI] [PubMed] [Google Scholar]
- (313).Ostuni E; Chapman RG; Holmlin RE; Takayama S; Whitesides GM A survey of structure-property relationships of surfaces that resist the adsorption of protein. Langmuir 2001, 17 (18), 5605. [DOI] [PubMed] [Google Scholar]
- (314).Elbert DL; Hubbell JA Conjugate addition reactions combined with free-radical cross-linking for the design of materials for tissue engineering. Biomacromolecules 2001, 2 (2), 430. [DOI] [PubMed] [Google Scholar]
- (315).West JL; Hubbell JA Photopolymerized Hydrogel Materials for Drug-Delivery Applications. React Polym 1995, 25 (2–3), 139. [Google Scholar]
- (316).Lopina ST; Wu G; Merrill EW; Griffith-Cima L.Hepatocyte culture on carbohydrate-modified star polyethylene oxide hydrogels. Biomaterials 1996, 17 (6), 559. [DOI] [PubMed] [Google Scholar]
- (317).Sakai T; Matsunaga T; Yamamoto Y; Ito C; Yoshida R; Suzuki S; Sasaki N; Shibayama M; Chung U.-i. Design and Fabrication of a High-Strength Hydrogel with Ideally Homogeneous Network Structure from Tetrahedron-like Macromonomers. Macromolecules 2008, 41 (14), 5379. [Google Scholar]
- (318).Ananda K; Nacharaju P; Smith PK; Acharya SA; Manjula BN Analysis of functionalization of methoxy–PEG as maleimide–PEG. Analytical Biochemistry 2008, 374 (2), 231. [DOI] [PubMed] [Google Scholar]
- (319).Malkoch M; Vestberg R; Gupta N; Mespouille L; Dubois P; Mason AF; Hedrick JL; Liao Q; Frank CW; Kingsbury K et al. Synthesis of well-defined hydrogel networks using Click chemistry. Chemical Communications 2006, DOI: 10.1039/B603438A10.1039/B603438A(26), 2774. [DOI] [PubMed] [Google Scholar]
- (320).Tan H; Xiao C; Sun J; Xiong D; Hu X.Biological self-assembly of injectable hydrogel as cell scaffold via specific nucleobase pairing. Chemical Communications 2012, 48 (83), 10289. [DOI] [PubMed] [Google Scholar]
- (321).Bastings MM; Koudstaal S; Kieltyka RE; Nakano Y; Pape AC; Feyen DA; van Slochteren FJ; Doevendans PA; Sluijter JP; Meijer EW et al. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv Healthc Mater 2014, 3 (1), 70. [DOI] [PubMed] [Google Scholar]
- (322).Chen X; Dong C; Wei K; Yao Y; Feng Q; Zhang K; Han F; Mak AF-T; Li B; Bian L.Supramolecular hydrogels cross-linked by preassembled host–guest PEG cross-linkers resist excessive, ultrafast, and non-resting cyclic compression. NPG Asia Materials 2018, 10 (8), 788. [Google Scholar]
- (323).Dong RJ; Pang Y; Su Y; Zhu XY Supramolecular hydrogels: synthesis, properties and their biomedical applications. Biomater Sci-Uk 2015, 3 (7), 937. [DOI] [PubMed] [Google Scholar]
- (324).Xue K; Liow SS; Karim AA; Li Z; Loh XJ A Recent Perspective on Noncovalently Formed Polymeric Hydrogels. Chem Rec 2018, 18 (10), 1517. [DOI] [PubMed] [Google Scholar]
- (325).Kim HA; Lee HJ; Hong JH; Moon HJ; Ko DY; Jeong B.α,ω-Diphenylalanine-End-Capping of PEG-PPG-PEG Polymers Changes the Micelle Morphology and Enhances Stability of the Thermogel. Biomacromolecules 2017, 18 (7), 2214. [DOI] [PubMed] [Google Scholar]
- (326).Shi K; Wang YL; Qu Y; Liao JF; Chu BY; Zhang HP; Luo F; Qian ZY Synthesis, characterization, and application of reversible PDLLA-PEG-PDLLA copolymer thermogels in vitro and in vivo. Sci Rep 2016, 6, 19077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Sun J; Lei Y; Dai Z; Liu X; Huang T; Wu J; Xu ZP; Sun X.Sustained Release of Brimonidine from a New Composite Drug Delivery System for Treatment of Glaucoma. ACS Applied Materials & Interfaces 2017, 9 (9), 7990. [DOI] [PubMed] [Google Scholar]
- (328).Nagahama K; Oyama N; Ono K; Hotta A; Kawauchi K; Nishikata T.Nanocomposite injectable gels capable of self-replenishing regenerative extracellular microenvironments for in vivo tissue engineering. Biomater Sci 2018, 6 (3), 550. [DOI] [PubMed] [Google Scholar]
- (329).Cui HT; Shao J; Wang Y; Zhang PB; Chen XS; Wei Y.PLA-PEG-PLA and Its Electroactive Tetraaniline Copolymer as Multi-interactive Injectable Hydrogels for Tissue Engineering. Biomacromolecules 2013, 14 (6), 1904. [DOI] [PubMed] [Google Scholar]
- (330).Gong C; Shi S; Dong P; Kan B; Gou M; Wang X; Li X; Luo F; Zhao X; Wei Y et al. Synthesis and characterization of PEG-PCL-PEG thermosensitive hydrogel. Int J Pharm 2009, 365 (1–2), 89. [DOI] [PubMed] [Google Scholar]
- (331).Zhang J; Tokatlian T; Zhong J; Ng QK; Patterson M; Lowry WE; Carmichael ST; Segura T.Physically associated synthetic hydrogels with long-term covalent stabilization for cell culture and stem cell transplantation. Adv Mater 2011, 23 (43), 5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Zhao X; Harris JM Novel degradable poly(ethylene glycol) hydrogels for controlled release of protein. J Pharm Sci 1998, 87 (11), 1450. [DOI] [PubMed] [Google Scholar]
- (333).Burdick JA; Anseth KS Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials 2002, 23 (22), 4315. [DOI] [PubMed] [Google Scholar]
- (334).Mann BK; Tsai AT; Scott-Burden T; West JL Modification of surfaces with cell adhesion peptides alters extracellular matrix deposition. Biomaterials 1999, 20 (23–24), 2281. [DOI] [PubMed] [Google Scholar]
- (335).Kraehenbuehl TP; Zammaretti P; Van der Vlies AJ; Schoenmakers RG; Lutolf MP; Jaconi ME; Hubbell JA Three-dimensional extracellular matrix-directed cardioprogenitor differentiation: systematic modulation of a synthetic cell-responsive PEG-hydrogel. Biomaterials 2008, 29 (18), 2757. [DOI] [PubMed] [Google Scholar]
- (336).Salinas CN; Anseth KS Mixed Mode Thiol−Acrylate Photopolymerizations for the Synthesis of PEG−Peptide Hydrogels. Macromolecules 2008, 41 (16), 6019. [Google Scholar]
- (337).Metters A; Hubbell J.Network formation and degradation behavior of hydrogels formed by Michael-type addition reactions. Biomacromolecules 2005, 6 (1), 290. [DOI] [PubMed] [Google Scholar]
- (338).Hiemstra C; van der Aa LJ; Zhong ZY; Dijkstra PJ; Feijen J.Novel in situ forming, degradable dextran hydrogels by Michael addition chemistry: Synthesis, rheology, and degradation. Macromolecules 2007, 40 (4), 1165. [Google Scholar]
- (339).Lee HJ; Lee JS; Chansakul T; Yu C; Elisseeff JH; Yu SM Collagen mimetic peptide-conjugated photopolymerizable PEG hydrogel. Biomaterials 2006, 27 (30), 5268. [DOI] [PubMed] [Google Scholar]
- (340).Stevens MM; Qanadilo HF; Langer R; Prasad Shastri V.A rapid-curing alginate gel system: utility in periosteum-derived cartilage tissue engineering. Biomaterials 2004, 25 (5), 887. [DOI] [PubMed] [Google Scholar]
- (341).García AJ PEG-Maleimide Hydrogels for Protein and Cell Delivery in Regenerative Medicine. Annals of Biomedical Engineering 2014, 42 (2), 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Zhu J.Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials 2010, 31 (17), 4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Schild HG Poly(N-isopropylacrylamide): experiment, theory and application. Progress in Polymer Science 1992, 17 (2), 163. [Google Scholar]
- (344).Tanaka F; Koga T; Winnik FM Temperature-responsive polymers in mixed solvents: competitive hydrogen bonds cause cononsolvency. Phys Rev Lett 2008, 101 (2), 028302. [DOI] [PubMed] [Google Scholar]
- (345).Wu XS; Hoffman AS; Yager P.Synthesis and characterization of thermally reversible macroporous poly (N‐isopropylacrylamide) hydrogels. Journal of Polymer Science Part A: Polymer Chemistry 1992, 30 (10), 2121. [Google Scholar]
- (346).Xia LW; Xie R; Ju XJ; Wang W; Chen Q; Chu LY Nano-structured smart hydrogels with rapid response and high elasticity. Nat Commun 2013, 4 (1), 2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Kumar A; Srivastava A; Galaev IY; Mattiasson B.Smart polymers: Physical forms and bioengineering applications. Progress in Polymer Science 2007, 32 (10), 1205. [Google Scholar]
- (348).Serizawa T; Wakita K; Akashi M.Rapid deswelling of porous poly(N-isopropylacrylamide) hydrogels prepared by incorporation of silica particles. Macromolecules 2002, 35 (1), 10. [Google Scholar]
- (349).Ishida K; Uno T; Itoh T; Kubo M.Synthesis and Property of Temperature-Responsive Hydrogel with Movable Cross-Linking Points. Macromolecules 2012, 45 (15), 6136. [Google Scholar]
- (350).Breger JC; Yoon C; Xiao R; Kwag HR; Wang MO; Fisher JP; Nguyen TD; Gracias DH Self-folding thermo-magnetically responsive soft microgrippers. ACS Appl Mater Interfaces 2015, 7 (5), 3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Gutowska A; Jeong B; Jasionowski M.Injectable gels for tissue engineering. Anat Rec 2001, 263 (4), 342. [DOI] [PubMed] [Google Scholar]
- (352).Brun-Graeppi AKAS; Richard C; Bessodes M; Scherman D; Merten OW Thermoresponsive surfaces for cell culture and enzyme-free cell detachment. Progress in Polymer Science 2010, 35 (11), 1311. [Google Scholar]
- (353).Kim H; Witt H; Oswald TA; Tarantola M.Adhesion of Epithelial Cells to PNIPAm Treated Surfaces for Temperature-Controlled Cell-Sheet Harvesting. ACS Applied Materials & Interfaces 2020, 12 (30), 33516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Tighe BJ A decade of silicone hydrogel development: surface properties, mechanical properties, and ocular compatibility. Eye Contact Lens 2013, 39 (1), 4. [DOI] [PubMed] [Google Scholar]
- (355).Eddington DT; Puccinelli JP; Beebe DJ Thermal aging and reduced hydrophobic recovery of polydimethylsiloxane. Sensor Actuat B-Chem 2006, 114 (1), 170. [Google Scholar]
- (356).Peak CW; Wilker JJ; Schmidt G.A review on tough and sticky hydrogels. Colloid Polym Sci 2013, 291 (9), 2031. [Google Scholar]
- (357).van Beek M; Weeks A; Jones L; Sheardown H.Immobilized hyaluronic acid containing model silicone hydrogels reduce protein adsorption. J Biomater Sci Polym Ed 2008, 19 (11), 1425. [DOI] [PubMed] [Google Scholar]
- (358).Tang Q; Yu JR; Chen L; Zhu J; Hu ZM Poly (dimethyl siloxane)/poly (2-hydroxyethyl methacrylate) interpenetrating polymer network beads as potential capsules for biomedical use. Curr Appl Phys 2011, 11 (3), 945. [Google Scholar]
- (359).Sugimoto H; Nishino G; Tsuzuki N; Daimatsu K; Inomata K; Nakanishi E.Preparation of high oxygen permeable transparent hybrid copolymers with silicone macro-monomers. Colloid Polym Sci 2012, 290 (2), 173. [Google Scholar]
- (360).Zhang XQ; Wang L; Tao HW; Sun Y; Yang H; Lin BP The influences of poly (ethylene glycol) chain length on hydrophilicity, oxygen permeability, and mechanical properties of multicomponent silicone hydrogels. Colloid Polym Sci 2019, 297 (9), 1233. [Google Scholar]
- (361).Lin G; Zhang XJA; Kumar SR; Mark JE Modification of Polysiloxane Networks for Biocompatibility. Mol Cryst Liq Cryst 2010, 521 (1), 56. [Google Scholar]
- (362).Yao MJ; Fang J.Hydrophilic PEO-PDMS for microfluidic applications. J Micromech Microeng 2012, 22 (2), 025012. [Google Scholar]
- (363).Demming S; Lesche C; Schmolke H; Klages C-P; Büttgenbach S.Characterization of long-term stability of hydrophilized PEG-grafted PDMS within different media for biotechnological and pharmaceutical applications. physica status solidi (a) 2011, 208 (6), 1301. [Google Scholar]
- (364).Nicolson PC; Vogt J.Soft contact lens polymers: an evolution. Biomaterials 2001, 22 (24), 3273. [DOI] [PubMed] [Google Scholar]
- (365).Calo E; Khutoryanskiy VV Biomedical applications of hydrogels: A review of patents and commercial products. European Polymer Journal 2015, 65, 252. [Google Scholar]
- (366).Morales-Hurtado M; Zeng X; Gonzalez-Rodriguez P; Ten Elshof JE; van der Heide E.A new water absorbable mechanical Epidermal skin equivalent: The combination of hydrophobic PDMS and hydrophilic PVA hydrogel. J Mech Behav Biomed 2015, 46, 305. [DOI] [PubMed] [Google Scholar]
- (367).Hamid ZAA; Lim KW Evaluation of UV-crosslinked Poly(ethylene glycol) Diacrylate/Poly(dimethylsiloxane) Dimethacrylate Hydrogel: Properties for Tissue Engineering Application. Procedia Chemistry 2016, 19, 410. [Google Scholar]
- (368).Xu J; Li X; Sun F.In vitro and in vivo evaluation of ketotifen fumarate-loaded silicone hydrogel contact lenses for ocular drug delivery. Drug Deliv 2011, 18 (2), 150. [DOI] [PubMed] [Google Scholar]
- (369).Wang JJ; Liu F; Wei J.Hydrophilic silicone hydrogels with interpenetrating network structure for extended delivery of ophthalmic drugs. Polym Advan Technol 2012, 23 (9), 1258. [Google Scholar]
- (370).Grandbois M; Beyer M; Rief M; Clausen-Schaumann H; Gaub HE How Strong Is a Covalent Bond? Science 1999, 283 (5408), 1727. [DOI] [PubMed] [Google Scholar]
- (371).Beyer MK The mechanical strength of a covalent bond calculated by density functional theory. J Chem Phys 2000, 112 (17), 7307. [Google Scholar]
- (372).Luo Y-R Comprehensive handbook of chemical bond energies; CRC press, 2007. [Google Scholar]
- (373).Steiner T.The hydrogen bond in the solid state. Angew Chem Int Ed Engl 2002, 41 (1), 49. [DOI] [PubMed] [Google Scholar]
- (374).Markovitch O; Agmon N.Structure and energetics of the hydronium hydration shells. J Phys Chem A 2007, 111 (12), 2253. [DOI] [PubMed] [Google Scholar]
- (375).Varshey DB; Sander JRG; Friščić T; MacGillivray LR In Supramolecular Chemistry, 2012, DOI: 10.1002/9780470661345.smc00310.1002/9780470661345.smc003. [DOI] [Google Scholar]
- (376).Zhuang WR; Wang Y; Cui PF; Xing L; Lee J; Kim D; Jiang HL; Oh YK Applications of pi-pi stacking interactions in the design of drug-delivery systems. J Control Release 2019, 294, 311. [DOI] [PubMed] [Google Scholar]
- (377).Blanco F; Alkorta I; Elguero J.Barriers about Double Carbon-Nitrogen Bond in Imine Derivatives (Aldimines, Oximes, Hydrazones, Azines). Croat Chem Acta 2009, 82 (1), 173. [Google Scholar]
- (378).Li HF; Li HY; Dai QQ; Li H; Bredas JL Hydrolytic Stability of Boronate Ester-Linked Covalent Organic Frameworks. Adv Theor Simul 2018, 1 (2), 1700015. [Google Scholar]
- (379).Dopieralski P; Ribas-Arino J; Anjukandi P; Krupicka M; Marx D.Unexpected mechanochemical complexity in the mechanistic scenarios of disulfide bond reduction in alkaline solution. Nat Chem 2017, 9 (2), 164. [DOI] [PubMed] [Google Scholar]
- (380).Basilevskii MV; Shamov AG; Tikhomirov VA Transition state of the Diels-Alder reaction. Journal of the American Chemical Society 1977, 99 (5), 1369. [Google Scholar]
- (381).Liu S; Lei Y; Qi X; Lan Y.Reactivity for the Diels-Alder reaction of cumulenes: a distortion-interaction analysis along the reaction pathway. J Phys Chem A 2014, 118 (14), 2638. [DOI] [PubMed] [Google Scholar]
- (382).Ullah F; Othman MB; Javed F; Ahmad Z; Md Akil H.Classification, processing and application of hydrogels: A review. Mater Sci Eng C Mater Biol Appl 2015, 57, 414. [DOI] [PubMed] [Google Scholar]
- (383).Wang W; Narain R; Zeng H.Rational Design of Self-Healing Tough Hydrogels: A Mini Review. Front Chem 2018, 6, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (384).Wong RS; Ashton M; Dodou K.Effect of Crosslinking Agent Concentration on the Properties of Unmedicated Hydrogels. Pharmaceutics 2015, 7 (3), 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Malo de Molina P; Lad S; Helgeson ME Heterogeneity and its Influence on the Properties of Difunctional Poly(ethylene glycol) Hydrogels: Structure and Mechanics. Macromolecules 2015, 48 (15), 5402. [Google Scholar]
- (386).Sawhney AS; Pathak CP; Hubbell JA Bioerodible Hydrogels Based on Photopolymerized Poly(Ethylene Glycol)-Co-Poly(Alpha-Hydroxy Acid) Diacrylate Macromers. Macromolecules 1993, 26 (4), 581. [Google Scholar]
- (387).Martens P; Anseth KS Characterization of hydrogels formed from acrylate modified poly(vinyl alcohol) macromers. Polymer 2000, 41 (21), 7715. [Google Scholar]
- (388).Hubbell JA Hydrogel systems for barriers and local drug delivery in the control of wound healing. J Control Release 1996, 39 (2–3), 305. [Google Scholar]
- (389).van Dijk-Wolthuis WNE; Franssen O; Talsma H; van Steenbergen MJ; Kettenes-van den Bosch JJ; Hennink WE Synthesis, Characterization, and Polymerization of Glycidyl Methacrylate Derivatized Dextran. Macromolecules 1995, 28 (18), 6317. [Google Scholar]
- (390).Giammona G; Pitarresi G; Cavallaro G; Spadaro G.New biodegradable hydrogels based on an acryloylated polyaspartamide cross-linked by gamma irradiation. J Biomater Sci Polym Ed 1999, 10 (9), 969. [DOI] [PubMed] [Google Scholar]
- (391).Caliceti P; Salmaso S; Lante A; Yoshida M; Katakai R; Martellini F; Mei LH; Carenza M.Controlled release of biomolecules from temperature-sensitive hydrogels prepared by radiation polymerization. J Control Release 2001, 75 (1–2), 173. [DOI] [PubMed] [Google Scholar]
- (392).Charlesby A.Cross-Linking of Polythene by Pile Radiation. Proc R Soc Lon Ser-A 1952, 215 (1121), 187. [Google Scholar]
- (393).Peppas NA; Mikos AG In Hydrogels in medicine and pharmacy; CRC Press, 2019. [Google Scholar]
- (394).Reddy N; Reddy R; Jiang Q.Crosslinking biopolymers for biomedical applications. Trends in Biotechnology 2015, 33 (6), 362. [DOI] [PubMed] [Google Scholar]
- (395).Eiselt P; Lee KY; Mooney DJ Rigidity of two-component hydrogels prepared from alginate and poly(ethylene glycol)-diamines. Macromolecules 1999, 32 (17), 5561. [Google Scholar]
- (396).Moon S-Y; Jeon E; Bae J-S; Byeon M; Park J-W Polyurea networks via organic sol–gel crosslinking polymerization of tetrafunctional amines and diisocyanates and their selective adsorption and filtration of carbon dioxide. Polymer Chemistry 2014, 5 (4), 1124. [Google Scholar]
- (397).Meldal M; Tornoe CW Cu-catalyzed azide-alkyne cycloaddition. Chem Rev 2008, 108 (8), 2952. [DOI] [PubMed] [Google Scholar]
- (398).Agard NJ; Prescher JA; Bertozzi CR A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc 2004, 126 (46), 15046. [DOI] [PubMed] [Google Scholar]
- (399).Kamath KR; Park K.Biodegradable Hydrogels in Drug-Delivery. Advanced Drug Delivery Reviews 1993, 11 (1–2), 59. [Google Scholar]
- (400).Yu QA; Song YN; Shi XM; Xu CY; Bin YZ Preparation and properties of chitosan derivative/poly(vinyl alcohol) blend film crosslinked with glutaraldehyde. Carbohydrate Polymers 2011, 84 (1), 465. [Google Scholar]
- (401).Crescenzi V; Francescangeli A; Taglienti A; Capitani D; Mannina L.Synthesis and partial characterization of hydrogels obtained via glutaraldehyde crosslinking of acetylated chitosan and of hyaluronan derivatives. Biomacromolecules 2003, 4 (4), 1045. [DOI] [PubMed] [Google Scholar]
- (402).Gehrke SH; Uhden LH; McBride JF Enhanced loading and activity retention of bioactive proteins in hydrogel delivery systems. J Control Release 1998, 55 (1), 21. [DOI] [PubMed] [Google Scholar]
- (403).Coviello T; Grassi M; Rambone G; Santucci E; Carafa M; Murtas E; Riccieri FM; Alhaique F.Novel hydrogel system from scleroglucan: synthesis and characterization. J Control Release 1999, 60 (2), 367. [DOI] [PubMed] [Google Scholar]
- (404).Tronci G; Neffe AT; Pierce BF; Lendlein A.An entropy–elastic gelatin-based hydrogel system. Journal of Materials Chemistry 2010, 20 (40), 8875. [Google Scholar]
- (405).Rydholm AE; Bowman CN; Anseth KS Degradable thiol-acrylate photopolymers: polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials 2005, 26 (22), 4495. [DOI] [PubMed] [Google Scholar]
- (406).DeForest CA; Polizzotti BD; Anseth KS Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nature Materials 2009, 8 (8), 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Patai S.The chemistry of the thiol group; Wiley, 1974. [Google Scholar]
- (408).Hoyle CE; Lowe AB; Bowman CN Thiol-click chemistry: a multifaceted toolbox for small molecule and polymer synthesis. Chem Soc Rev 2010, 39 (4), 1355. [DOI] [PubMed] [Google Scholar]
- (409).Gress A; Volkel A; Schlaad H.Thio-click modification of poly [2-(3-butenyl)-2-oxazoline]. Macromolecules 2007, 40 (22), 7928. [Google Scholar]
- (410).Dondoni A.The Emergence of Thiol–Ene Coupling as a Click Process for Materials and Bioorganic Chemistry. Angewandte Chemie International Edition 2008, 47 (47), 8995. [DOI] [PubMed] [Google Scholar]
- (411).Grazu V; Abian O; Mateo C; Batista-Viera F; Fernandez-Lafuente R; Guisan JM Novel bifunctional epoxy/thiol-reactive support to immobilize thiol containing proteins by the epoxy chemistry. Biomacromolecules 2003, 4 (6), 1495. [DOI] [PubMed] [Google Scholar]
- (412).Dyer E; Glenn JF The Kinetics of the Reactions of Phenyl Isocyanate with Certain Thiols1. Journal of the American Chemical Society 1957, 79 (2), 366. [Google Scholar]
- (413).Becer CR; Babiuch K; Pilz D; Hornig S; Heinze T; Gottschaldt M; Schubert US Clicking Pentafluorostyrene Copolymers: Synthesis, Nanoprecipitation, and Glycosylation. Macromolecules 2009, 42 (7), 2387. [Google Scholar]
- (414).Hurd CD; Gershbein LL Reactions of Mercaptans with Acrylic and Methacrylic Derivatives. Journal of the American Chemical Society 1947, 69 (10), 2328. [Google Scholar]
- (415).Chen X; Li R; Wong SHD; Wei K; Cui M; Chen H; Jiang Y; Yang B; Zhao P; Xu J et al. Conformational manipulation of scale-up prepared single-chain polymeric nanogels for multiscale regulation of cells. Nat Commun 2019, 10 (1), 2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (416).Nair DP; Podgorski M; Chatani S; Gong T; Xi WX; Fenoli CR; Bowman CN The ThiolMichael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem Mater 2014, 26 (1), 724. [Google Scholar]
- (417).Tao H; Zhang X; Sun Y; Yang H; Lin B.The influence of molecular weight of siloxane macromere on phase separation morphology, oxygen permeability, and mechanical properties in multicomponent silicone hydrogels. Colloid Polym Sci 2017, 295 (1), 205. [Google Scholar]
- (418).Weinhold F; West R.The Nature of the Silicon–Oxygen Bond. Organometallics 2011, 30 (21), 5815. [Google Scholar]
- (419).Si L; Zheng X; Nie J; Yin R; Hua Y; Zhu X.Silicone-based tough hydrogels with high resilience, fast self-recovery, and self-healing properties. Chemical Communications 2016, 52 (54), 8365. [DOI] [PubMed] [Google Scholar]
- (420).Ahagon A; Gent A.Effect of interfacial bonding on the strength of adhesion. Journal of Polymer Science: Polymer Physics Edition 1975, 13 (7), 1285. [Google Scholar]
- (421).Hong W; Zhao X; Zhou J; Suo Z.A theory of coupled diffusion and large deformation in polymeric gels. Journal of the Mechanics and Physics of Solids 2008, 56 (5), 1779. [Google Scholar]
- (422).Hong W; Zhao X; Suo Z.Large deformation and electrochemistry of polyelectrolyte gels. Journal of the Mechanics and Physics of Solids 2010, 58 (4), 558. [Google Scholar]
- (423).Roland C.Unconventional rubber networks: Circumventing the compromise between stiffness and strength. Rubber Chemistry Technology 2013, 86 (3), 351. [Google Scholar]
- (424).Richtering W; Saunders BR Gel architectures and their complexity. Soft Matter 2014, 10 (21), 3695. [DOI] [PubMed] [Google Scholar]
- (425).Wang H; Heilshorn SC Adaptable hydrogel networks with reversible linkages for tissue engineering. Advanced Materials 2015, 27 (25), 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Petka WA; Harden JL; McGrath KP; Wirtz D; Tirrell DA Reversible hydrogels from self-assembling artificial proteins. Science 1998, 281 (5375), 389. [DOI] [PubMed] [Google Scholar]
- (427).Rosales AM; Anseth KS The design of reversible hydrogels to capture extracellular matrix dynamics. Nature Reviews Materials 2016, 1 (2), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Mark JE Elastomeric networks with bimodal chain-length distributions. Accounts of chemical research 1994, 27 (9), 271. [Google Scholar]
- (429).Kloxin AM; Kasko AM; Salinas CN; Anseth KS Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science 2009, 324 (5923), 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Creton C.50th anniversary perspective: Networks and gels: Soft but dynamic and tough. Macromolecules 2017, 50 (21), 8297. [Google Scholar]
- (431).Liu Y; He W; Zhang Z; Lee BP Recent developments in tough hydrogels for biomedical applications. Gels 2018, 4 (2), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Sakai T.In Physics of Polymer Gels, 2020, DOI: 10.1002/9783527346547.ch3. [DOI]
- (433).Sakai T.In Physics of Polymer Gels, 2020, DOI: 10.1002/9783527346547.ch4. [DOI]
- (434).Sakai T.In Physics of Polymer Gels, 2020, DOI: 10.1002/9783527346547.ch5. [DOI]
- (435).Parada GA; Zhao X.Ideal reversible polymer networks. Soft Matter 2018, 14 (25), 5186. [DOI] [PubMed] [Google Scholar]
- (436).Akagi Y; Matsunaga T; Shibayama M; Chung U.-i.; Sakai T.Evaluation of Topological Defects in Tetra-PEG Gels. Macromolecules 2010, 43 (1), 488. [Google Scholar]
- (437).Yigit S; Sanyal R; Sanyal A.Fabrication and functionalization of hydrogels through “click” chemistry. Chemistry–An Asian Journal 2011, 6 (10), 2648. [DOI] [PubMed] [Google Scholar]
- (438).Huang X; Nakagawa S; Li X; Shibayama M; Yoshie N.A Simple and Versatile Method for the Construction of Nearly Ideal Polymer Networks. Angew Chem Int Edit 2020, 59 (24), 9646. [DOI] [PubMed] [Google Scholar]
- (439).Marco-Dufort B; Iten R; Tibbitt MW Linking Molecular Behavior to Macroscopic Properties in Ideal Dynamic Covalent Networks. J Am Chem Soc 2020, 142 (36), 15371. [DOI] [PubMed] [Google Scholar]
- (440).Matsunaga T; Sakai T; Akagi Y; Chung UI; Shibayama M.SANS and SLS Studies on Tetra-Arm PEG Gels in As-Prepared and Swollen States. Macromolecules 2009, 42 (16), 6245. [Google Scholar]
- (441).Hild G.Model networks based on èndlinking’ processes: synthesis, structure and properties. Progress in Polymer Science 1998, 23 (6), 1019. [Google Scholar]
- (442).Zhong M; Wang R; Kawamoto K; Olsen BD; Johnson JA Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353 (6305), 1264. [DOI] [PubMed] [Google Scholar]
- (443).Okaya Y; Jochi Y; Seki T; Satoh K; Kamigaito M; Hoshino T; Nakatani T; Fujinami S; Takata M; Takeoka Y.Precise Synthesis of a Homogeneous Thermoresponsive Polymer Network Composed of Four-Branched Star Polymers with a Narrow Molecular Weight Distribution. Macromolecules 2020, 53 (1), 374. [Google Scholar]
- (444).Apostolides DE; Patrickios CS; Sakai T; Guerre M; Lopez G; Ameduri B; Ladmiral V; Simon M; Gradzielski M; Clemens D et al. Near-Model Amphiphilic Polymer Conetworks Based on Four-Arm Stars of Poly(vinylidene fluoride) and Poly(ethylene glycol): Synthesis and Characterization. Macromolecules 2018, 51 (7), 2476. [Google Scholar]
- (445).Oshima K; Fujimoto T; Minami E; Mitsukami Y.Model Polyelectrolyte Gels Synthesized by End-Linking of Tetra-Arm Polymers with Click Chemistry: Synthesis and Mechanical Properties. Macromolecules 2014, 47 (21), 7573. [Google Scholar]
- (446).Yesilyurt V; Webber MJ; Appel EA; Godwin C; Langer R; Anderson DG Injectable Self-Healing Glucose-Responsive Hydrogels with pH-Regulated Mechanical Properties. Adv Mater 2016, 28 (1), 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Shibayama M; Li X; Sakai T.Precision polymer network science with tetra-PEG gels—a decade history and future. Colloid Polym Sci 2018, 297 (1), 1. [Google Scholar]
- (448).Hashimoto K; Fujii K; Nishi K; Sakai T; Shibayama M.Nearly Ideal Polymer Network Ion Gel Prepared in pH-Buffering Ionic Liquid. Macromolecules 2016, 49 (1), 344. [Google Scholar]
- (449).Apostolides DE; Sakai T; Patrickios CS Dynamic Covalent Star Poly(ethylene glycol) Model Hydrogels: A New Platform for Mechanically Robust, Multifunctional Materials. Macromolecules 2017, 50 (5), 2155. [Google Scholar]
- (450).Hayashi K; Okamoto F; Hoshi S; Katashima T; Zujur DC; Li X; Shibayama M; Gilbert EP; Chung U; Ohba S et al. Fast-forming hydrogel with ultralow polymeric content as an artificial vitreous body. Nature Biomedical Engineering 2017, 1 (3), 0044. [Google Scholar]
- (451).Okumura Y; Ito K.The polyrotaxane gel: A topological gel by figure‐of‐eight cross‐links. Advanced materials 2001, 13 (7), 485. [Google Scholar]
- (452).Wenz G; Han BH; Muller A.Cyclodextrin rotaxanes and polyrotaxanes. Chem Rev 2006, 106 (3), 782. [DOI] [PubMed] [Google Scholar]
- (453).Bin Imran A; Esaki K; Gotoh H; Seki T; Ito K; Sakai Y; Takeoka Y.Extremely stretchable thermosensitive hydrogels by introducing slide-ring polyrotaxane cross-linkers and ionic groups into the polymer network. Nature Communications 2014, 5 (1), 5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Loethen S; Kim J-M; Thompson DH Biomedical Applications of Cyclodextrin Based Polyrotaxanes. Polymer Reviews 2007, 47 (3), 383. [Google Scholar]
- (455).Mayumi K; Ito K.Structure and dynamics of polyrotaxane and slide-ring materials. Polymer 2010, 51 (4), 959. [Google Scholar]
- (456).Huang FH; Gibson HW Polypseudorotaxanes and polyrotaxanes. Progress in Polymer Science 2005, 30 (10), 982. [Google Scholar]
- (457).Liu G; Yuan Q; Hollett G; Zhao W; Kang Y; Wu J.Cyclodextrin-based host–guest supramolecular hydrogel and its application in biomedical fields. Polymer Chemistry 2018, 9 (25), 3436. [Google Scholar]
- (458).Harada A; Kamachi M.Complex formation between poly (ethylene glycol) and α-cyclodextrin. Macromolecules 1990, 23 (10), 2821. [Google Scholar]
- (459).Harada A; Li J; Kamachi M.The molecular necklace: a rotaxane containing many threaded α-cyclodextrins. Nature 1992, 356 (6367), 325. [Google Scholar]
- (460).Xie D; Yang K; Sun W.Formation and characterization of polylactide and β-cyclodextrin inclusion complex. Curr Appl Phys 2007, 7, e15. [Google Scholar]
- (461).Harada A; Takashima Y; Yamaguchi H.Cyclodextrin-based supramolecular polymers. Chem Soc Rev 2009, 38 (4), 875. [DOI] [PubMed] [Google Scholar]
- (462).Gao P; Wang J; Ye L; Zhang A. y.; Feng Z. g. Stable and Unconventional Conformation of Single PEG Bent γ‐CD‐Based Polypseudorotaxanes. Macromol Chem Phys 2011, 212 (21), 2319. [Google Scholar]
- (463).Granick S; Rubinstein M.Polymers: a multitude of macromolecules. Nat Mater 2004, 3 (9), 586. [DOI] [PubMed] [Google Scholar]
- (464).Oku T; Furusho Y; Takata T.A Concept for Recyclable Cross‐Linked Polymers: Topologically Networked Polyrotaxane Capable of Undergoing Reversible Assembly and Disassembly. Angewandte Chemie International Edition 2004, 43 (8), 966. [DOI] [PubMed] [Google Scholar]
- (465).Ooya T; Eguchi M; Yui N.Enhanced accessibility of peptide substrate toward membranebound metalloexopeptidase by supramolecular structure of polyrotaxane. Biomacromolecules 2001, 2 (1), 200. [DOI] [PubMed] [Google Scholar]
- (466).Li J; Suo Z; Vlassak JJ Stiff, strong, and tough hydrogels with good chemical stability. J Mater Chem B 2014, 2 (39), 6708. [DOI] [PubMed] [Google Scholar]
- (467).Nonoyama T; Gong JP Double-network hydrogel and its potential biomedical application: A review. Proc Inst Mech Eng H 2015, 229 (12), 853. [DOI] [PubMed] [Google Scholar]
- (468).Costa AMS; Mano JF Extremely strong and tough hydrogels as prospective candidates for tissue repair – A review. European Polymer Journal 2015, 72, 344. [Google Scholar]
- (469).Dragan ES Design and applications of interpenetrating polymer network hydrogels. A review. Chem Eng J 2014, 243, 572. [Google Scholar]
- (470).Sperling LH Interpenetrating polymer networks and related materials; Springer Science & Business Media, 2012. [Google Scholar]
- (471).Myung D; Waters D; Wiseman M; Duhamel PE; Noolandi J; Ta CN; Frank CW Progress in the development of interpenetrating polymer network hydrogels. Polym Adv Technol 2008, 19 (6), 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Sperling LH In Interpenetrating Polymer Networks; American Chemical Society, 1994; Vol. 239. [Google Scholar]
- (473).Visscher KB; Manners I; Allcock HR Synthesis and Properties of Polyphosphazene Interpenetrating Polymer Networks. Macromolecules 1990, 23 (22), 4885. [Google Scholar]
- (474).Wang JJ; Liu F.Enhanced adsorption of heavy metal ions onto simultaneous interpenetrating polymer network hydrogels synthesized by UV irradiation. Polym Bull 2013, 70 (4), 1415. [Google Scholar]
- (475).Sun JY; Zhao X; Illeperuma WR; Chaudhuri O; Oh KH; Mooney DJ; Vlassak JJ; Suo Z.Highly stretchable and tough hydrogels. Nature 2012, 489 (7414), 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Haque MA; Kurokawa T; Gong JP Super tough double network hydrogels and their application as biomaterials. Polymer 2012, 53 (9), 1805. [Google Scholar]
- (477).Hong S; Sycks D; Chan HF; Lin S; Lopez GP; Guilak F; Leong KW; Zhao X.3D Printing of Highly Stretchable and Tough Hydrogels into Complex, Cellularized Structures. Adv Mater 2015, 27 (27), 4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Seiffert S.Suppramolecular Polymer Networks and Gels; Springer: Berlin, 2015. [Google Scholar]
- (479).Tong XM; Yang F.Engineering interpenetrating network hydrogels as biomimetic cell niche with independently tunable biochemical and mechanical properties. Biomaterials 2014, 35 (6), 1807. [DOI] [PubMed] [Google Scholar]
- (480).Feig VR; Tran H; Lee M; Bao Z.Mechanically tunable conductive interpenetrating network hydrogels that mimic the elastic moduli of biological tissue. Nature communications 2018, 9 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Darnell MC; Sun JY; Mehta M; Johnson C; Arany PR; Suo Z; Mooney DJ Performance and biocompatibility of extremely tough alginate/polyacrylamide hydrogels. Biomaterials 2013, 34 (33), 8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Kumar A; Han SS PVA-based hydrogels for tissue engineering: A review. International Journal of Polymeric Materials and Polymeric Biomaterials 2016, 66 (4), 159. [Google Scholar]
- (483).Henderson KJ; Zhou TC; Otim KJ; Shull KR Ionically Cross-Linked Triblock Copolymer Hydrogels with High Strength. Macromolecules 2010, 43 (14), 6193. [Google Scholar]
- (484).Vancaeyzeele C; Fichet O; Boileau S; Teyssié D.Polyisobutene–poly(methylmethacrylate) interpenetrating polymer networks: synthesis and characterization. Polymer 2005, 46 (18), 6888. [Google Scholar]
- (485).Schexnailder P; Schmidt G.Nanocomposite polymer hydrogels. Colloid Polym Sci 2008, 287 (1), 1. [Google Scholar]
- (486).Fu J.Strong and tough hydrogels crosslinked by multi-functional polymer colloids. 2018, 56 (19), 1336. [Google Scholar]
- (487).Liu Z; Faraj Y; Ju X-J; Wang W; Xie R; Chu L-Y Nanocomposite smart hydrogels with improved responsiveness and mechanical properties: A mini review. 2018, 56 (19), 1306. [Google Scholar]
- (488).Tuncaboylu DC; Sari M; Oppermann W; Okay O.Tough and Self-Healing Hydrogels Formed via Hydrophobic Interactions. Macromolecules 2011, 44 (12), 4997. [Google Scholar]
- (489).Okay O.Self-Healing Hydrogels Formed via Hydrophobic Interactions. Adv Polym Sci 2015, 268, 101. [Google Scholar]
- (490).Jiang HC; Duan LJ; Ren XY; Gao GH Hydrophobic association hydrogels with excellent mechanical and self-healing properties. European Polymer Journal 2019, 112, 660. [Google Scholar]
- (491).Peppas NA; Merrill EW Poly(vinyl alcohol) hydrogels: Reinforcement of radiationcrosslinked networks by crystallization. Journal of Polymer Science: Polymer Chemistry Edition 1976, 14 (2), 441. [Google Scholar]
- (492).Seitz ME; Martina D; Baumberger T; Krishnan VR; Hui CY; Shull KR Fracture and large strain behavior of self-assembled triblock copolymer gels. Soft Matter 2009, 5 (2), 447. [Google Scholar]
- (493).Okay O; Oppermann W.Polyacrylamide−Clay Nanocomposite Hydrogels: Rheological and Light Scattering Characterization. Macromolecules 2007, 40 (9), 3378. [Google Scholar]
- (494).Liu RQ; Liang SM; Tang XZ; Yan D; Li XF; Yu ZZ Tough and highly stretchable graphene oxide/polyacrylamide nanocomposite hydrogels. Journal of Materials Chemistry 2012, 22 (28), 14160. [Google Scholar]
- (495).Haque MA; Kurokawa T; Kamita G; Gong JP Lamellar Bilayers as Reversible Sacrificial Bonds To Toughen Hydrogel: Hysteresis, Self-Recovery, Fatigue Resistance, and Crack Blunting. Macromolecules 2011, 44 (22), 8916. [Google Scholar]
- (496).Huang T; Xu HG; Jiao KX; Zhu LP; Brown HR; Wang HL A novel hydrogel with high mechanical strength: A macromolecular microsphere composite hydrogel. Advanced Materials 2007, 19 (12), 1622. [Google Scholar]
- (497).Abe K; Yano H.Formation of hydrogels from cellulose nanofibers. Carbohydrate Polymers 2011, 85 (4), 733. [Google Scholar]
- (498).Gao Y; Kuang Y; Guo ZF; Guo Z; Krauss IJ; Xu B.Enzyme-instructed molecular selfassembly confers nanofibers and a supramolecular hydrogel of taxol derivative. J Am Chem Soc 2009, 131 (38), 13576. [DOI] [PubMed] [Google Scholar]
- (499).Ma M; Kuang Y; Gao Y; Zhang Y; Gao P; Xu B.Aromatic-aromatic interactions induce the self-assembly of pentapeptidic derivatives in water to form nanofibers and supramolecular hydrogels. J Am Chem Soc 2010, 132 (8), 2719. [DOI] [PubMed] [Google Scholar]
- (500).Kohler K; Forster G; Hauser A; Dobner B; Heiser UF; Ziethe F; Richter W; Steiniger F; Drechsler M; Stettin H et al. Self-assembly in a bipolar phosphocholine-water system: the formation of nanofibers and hydrogels. Angew Chem Int Ed Engl 2004, 43 (2), 245. [DOI] [PubMed] [Google Scholar]
- (501).Yang G; Lin H; Rothrauff BB; Yu S; Tuan RS Multilayered polycaprolactone/gelatin fiberhydrogel composite for tendon tissue engineering. Acta Biomater 2016, 35, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (502).Ma L; Yang G; Wang N; Zhang P; Guo F; Meng J; Zhang F; Hu Z; Wang S; Zhao Y.Trap Effect of Three-Dimensional Fibers Network for High Efficient Cancer-Cell Capture. Adv Healthc Mater 2015, 4 (6), 838. [DOI] [PubMed] [Google Scholar]
- (503).D’Amore A; Stella JA; Wagner WR; Sacks MS Characterization of the complete fiber network topology of planar fibrous tissues and scaffolds. Biomaterials 2010, 31 (20), 5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Toshima M; Ohtani Y; Ohtani O.Three-dimensional architecture of elastin and collagen fiber networks in the human and rat lung. Arch Histol Cytol 2004, 67 (1), 31. [DOI] [PubMed] [Google Scholar]
- (505).Weber KT Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 1989, 13 (7), 1637. [DOI] [PubMed] [Google Scholar]
- (506).Nakagaito AN; Iwamoto S; Yano H.Bacterial cellulose: the ultimate nano-scalar cellulose morphology for the production of high-strength composites. Appl Phys a-Mater 2005, 80 (1), 93. [Google Scholar]
- (507).Yan C; Pochan DJ Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem Soc Rev 2010, 39 (9), 3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Woolfson DN Building fibrous biomaterials from alpha-helical and collagen-like coiled-coil peptides. Biopolymers 2010, 94 (1), 118. [DOI] [PubMed] [Google Scholar]
- (509).Viebke C; Piculell L; Nilsson S.On the Mechanism of Gelation of Helix-Forming Biopolymers. Macromolecules 1994, 27 (15), 4160. [Google Scholar]
- (510).Prince E; Kumacheva E.Design and applications of man-made biomimetic fibrillar hydrogels. Nature Reviews Materials 2019, 4 (2), 99. [Google Scholar]
- (511).Tamayol A; Akbari M; Annabi N; Paul A; Khademhosseini A; Juncker D.Fiber-based tissue engineering: Progress, challenges, and opportunities. Biotechnol Adv 2013, 31 (5), 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Iwamoto S; Isogai A; Iwata T.Structure and mechanical properties of wet-spun fibers made from natural cellulose nanofibers. Biomacromolecules 2011, 12 (3), 831. [DOI] [PubMed] [Google Scholar]
- (513).Ren L; Pandit V; Elkin J; Denman T; Cooper JA; Kotha SP Large-scale and highly efficient synthesis of micro- and nano-fibers with controlled fiber morphology by centrifugal jet spinning for tissue regeneration. Nanoscale 2013, 5 (6), 2337. [DOI] [PubMed] [Google Scholar]
- (514).Agarwal S; Greiner A; Wendorff JH Functional materials by electrospinning of polymers. Progress in Polymer Science 2013, 38 (6), 963. [Google Scholar]
- (515).Rogina A.Electrospinning process: Versatile preparation method for biodegradable and natural polymers and biocomposite systems applied in tissue engineering and drug delivery. Appl Surf Sci 2014, 296, 221. [Google Scholar]
- (516).Hong Y; Huber A; Takanari K; Amoroso NJ; Hashizume R; Badylak SF; Wagner WR Mechanical properties and in vivo behavior of a biodegradable synthetic polymer microfiberextracellular matrix hydrogel biohybrid scaffold. Biomaterials 2011, 32 (13), 3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (517).Ekaputra AK; Prestwich GD; Cool SM; Hutmacher DW Combining electrospun scaffolds with electrosprayed hydrogels leads to three-dimensional cellularization of hybrid constructs. Biomacromolecules 2008, 9 (8), 2097. [DOI] [PubMed] [Google Scholar]
- (518).Thorvaldsson A; Silva-Correia J; Oliveira JM; Reis RL; Gatenholm P; Walkenstrom P.Development of Nanofiber-Reinforced Hydrogel Scaffolds for Nucleus Pulposus Regeneration by a Combination of Electrospinning and Spraying Technique. Journal of Applied Polymer Science 2013, 128 (2), 1158. [Google Scholar]
- (519).Ramakrishna S.An introduction to electrospinning and nanofibers; World Scientific, 2005. [Google Scholar]
- (520).Yoo HS; Kim TG; Park TG Surface-functionalized electrospun nanofibers for tissue engineering and drug delivery. Adv Drug Deliv Rev 2009, 61 (12), 1033. [DOI] [PubMed] [Google Scholar]
- (521).Boudriot U; Dersch R; Greiner A; Wendorff JH Electrospinning approaches toward scaffold engineering--a brief overview. Artif Organs 2006, 30 (10), 785. [DOI] [PubMed] [Google Scholar]
- (522).Vatankhah-Varnosfaderani M; Daniel WF; Everhart MH; Pandya AA; Liang H; Matyjaszewski K; Dobrynin AV; Sheiko SS Mimicking biological stress–strain behaviour with synthetic elastomers. Nature 2017, 549 (7673), 497. [DOI] [PubMed] [Google Scholar]
- (523).Daniel WF; Burdyńska J; Vatankhah-Varnoosfaderani M; Matyjaszewski K; Paturej J; Rubinstein M; Dobrynin AV; Sheiko SS Solvent-free, supersoft and superelastic bottlebrush melts and networks. Nature materials 2016, 15 (2), 183. [DOI] [PubMed] [Google Scholar]
- (524).Vohidov F; Milling LE; Chen QX; Zhang WX; Bhagchandani S; Nguyen HVT; Irvine DJ; Johnson JA ABC triblock bottlebrush copolymer-based injectable hydrogels: design, synthesis, and application to expanding the therapeutic index of cancer immunochemotherapy. Chem Sci 2020, 11 (23), 5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Bell GI Models for the specific adhesion of cells to cells. Science 1978, 200 (4342), 618. [DOI] [PubMed] [Google Scholar]
- (526).Delmer DP; Amor Y.Cellulose biosynthesis. Plant Cell 1995, 7 (7), 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Abe K; Yano H.Cellulose nanofiber-based hydrogels with high mechanical strength. Cellulose 2012, 19 (6), 1907. [Google Scholar]
- (528).Debenedetti PG; Stillinger FH Supercooled liquids and the glass transition. Nature 2001, 410 (6825), 259. [DOI] [PubMed] [Google Scholar]
- (529).Burns AB; Register RA Mechanical Properties of Star Block Polymer Thermoplastic Elastomers with Glassy and Crystalline End Blocks. Macromolecules 2016, 49 (24), 9521. [Google Scholar]
- (530).Nykanen A; Nuopponen M; Laukkanen A; Hirvonen SP; Rytela M; Turunen O; Tenhu H; Mezzenga R; Ikkala O; Ruokolainen J.Phase behavior and temperature-responsive molecular filters based on self-assembly of Polystyrene-block-poly(N-isopropylacrylamide)-blockpolystyrene. Macromolecules 2007, 40 (16), 5827. [Google Scholar]
- (531).Roth CB; Pound A; Kamp SW; Murray CA; Dutcher JR Molecular-weight dependence of the glass transition temperature of freely-standing poly(methyl methacrylate) films. Eur Phys J E Soft Matter 2006, 20 (4), 441. [DOI] [PubMed] [Google Scholar]
- (532).Ottani V; Martini D; Franchi M; Ruggeri A; Raspanti M.Hierarchical structures in fibrillar collagens. Micron 2002, 33 (7–8), 587. [DOI] [PubMed] [Google Scholar]
- (533).O’Leary LE; Fallas JA; Bakota EL; Kang MK; Hartgerink JD Multi-hierarchical selfassembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel. Nat Chem 2011, 3 (10), 821. [DOI] [PubMed] [Google Scholar]
- (534).Arnott S; Fulmer A; Scott WE; Dea IC; Moorhouse R; Rees DA The agarose double helix and its function in agarose gel structure. J Mol Biol 1974, 90 (2), 269. [DOI] [PubMed] [Google Scholar]
- (535).Foord SA; Atkins EDT New X-Ray-Diffraction Results from Agarose - Extended Single Helix Structures and Implications for Gelation Mechanism. Biopolymers 1989, 28 (8), 1345. [Google Scholar]
- (536).Armisen R.Agar and Agarose Biotechnological Applications. Hydrobiologia 1991, 221 (1), 157. [Google Scholar]
- (537).Van Vlierberghe S; Dubruel P; Schacht E.Biopolymer-based hydrogels as scaffolds for tissue engineering applications: a review. Biomacromolecules 2011, 12 (5), 1387. [DOI] [PubMed] [Google Scholar]
- (538).Eagland D; Crowther NJ; Butler CJ Complexation between polyoxyethylene and polymethacrylic acid—the importance of the molar mass of polyoxyethylene. European Polymer Journal 1994, 30 (7), 767. [Google Scholar]
- (539).Kim IS; Kim SH; Cho CS Drug release from pH-sensitive interpenetrating polymer networks hydrogel based on poly (ethylene glycol) macromer and poly (acrylic acid) prepared by UV cured method. Arch Pharm Res 1996, 19 (1), 18. [Google Scholar]
- (540).Voorhaar L; Hoogenboom R.Supramolecular polymer networks: hydrogels and bulk materials. Chem Soc Rev 2016, 45 (14), 4013. [DOI] [PubMed] [Google Scholar]
- (541).Zhang GZ; Chen YH; Deng YH; Ngai T; Wang CY Dynamic Supramolecular Hydrogels: Regulating Hydrogel Properties through Self-Complementary Quadruple Hydrogen Bonds and Thermo-Switch. Acs Macro Lett 2017, 6 (7), 641. [DOI] [PubMed] [Google Scholar]
- (542).Cui J; del Campo A.Multivalent H-bonds for self-healing hydrogels. Chem Commun (Camb) 2012, 48 (74), 9302. [DOI] [PubMed] [Google Scholar]
- (543).Li F; Tang JP; Geng JH; Luo D; Yang DY Polymeric DNA hydrogel: Design, synthesis and applications. Progress in Polymer Science 2019, 98, 101163. [Google Scholar]
- (544).Cui H; Zhuang X; He C; Wei Y; Chen X.High performance and reversible ionic polypeptide hydrogel based on charge-driven assembly for biomedical applications. Acta Biomater 2015, 11, 183. [DOI] [PubMed] [Google Scholar]
- (545).Ji DY; Kuo TF; Wu HD; Yang JC; Lee SY A novel injectable chitosan/polyglutamate polyelectrolyte complex hydrogel with hydroxyapatite for soft-tissue augmentation. Carbohydr Polym 2012, 89 (4), 1123. [DOI] [PubMed] [Google Scholar]
- (546).Lee KY; Rowley JA; Eiselt P; Moy EM; Bouhadir KH; Mooney DJ Controlling mechanical and swelling properties of alginate hydrogels independently by cross-linker type and cross-linking density. Macromolecules 2000, 33 (11), 4291. [Google Scholar]
- (547).Yang Y; Wang X; Yang F; Wang L; Wu D.Highly elastic and ultratough hybrid ionic–covalent hydrogels with tunable structures and mechanics. Advanced Materials 2018, 30 (18), 1707071. [DOI] [PubMed] [Google Scholar]
- (548).Mohammadzadeh Pakdel P; Peighambardoust SJ Review on recent progress in chitosan-based hydrogels for wastewater treatment application. Carbohydrate Polymers 2018, 201, 264. [DOI] [PubMed] [Google Scholar]
- (549).Mi F-L; Sung H-W; Shyu S-S; Su C-C; Peng C-K Synthesis and characterization of biodegradable TPP/genipin co-crosslinked chitosan gel beads. Polymer 2003, 44 (21), 6521. [Google Scholar]
- (550).Luo F; Sun TL; Nakajima T; Kurokawa T; Zhao Y; Sato K; Ihsan AB; Li X; Guo H; Gong JP Oppositely charged polyelectrolytes form tough, self‐healing, and rebuildable hydrogels. Advanced materials 2015, 27 (17), 2722. [DOI] [PubMed] [Google Scholar]
- (551).Mozhdehi D; Ayala S; Cromwell OR; Guan Z.Self-healing multiphase polymers via dynamic metal-ligand interactions. J Am Chem Soc 2014, 136 (46), 16128. [DOI] [PubMed] [Google Scholar]
- (552).Zhang JY; Su CY Metal-organic gels: From discrete metallogelators to coordination polymers. Coordin Chem Rev 2013, 257 (7–8), 1373. [Google Scholar]
- (553).Fantner GE; Hassenkam T; Kindt JH; Weaver JC; Birkedal H; Pechenik L; Cutroni JA; Cidade GA; Stucky GD; Morse DE et al. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat Mater 2005, 4 (8), 612. [DOI] [PubMed] [Google Scholar]
- (554).Hillerton JE; Vincent JFV The Specific Location of Zinc in Insect Mandibles. J Exp Biol 1982, 101 (Dec), 333. [Google Scholar]
- (555).Harrington MJ; Masic A; Holten-Andersen N; Waite JH; Fratzl P.Iron-clad fibers: a metalbased biological strategy for hard flexible coatings. Science 2010, 328 (5975), 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (556).Nejadnik MR; Yang X; Bongio M; Alghamdi HS; van den Beucken JJ; Huysmans MC; Jansen JA; Hilborn J; Ossipov D; Leeuwenburgh SC Self-healing hybrid nanocomposites consisting of bisphosphonated hyaluronan and calcium phosphate nanoparticles. Biomaterials 2014, 35 (25), 6918. [DOI] [PubMed] [Google Scholar]
- (557).Lopez-Perez PM; da Silva RMP; Strehin I; Kouwer PHJ; Leeuwenburgh SCG; Messersmith PB Self-healing hydrogels formed by complexation between calcium ions and bisphosphonate-functionalized star-shaped polymers. Macromolecules 2017, 50 (21), 8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Shi LY; Carstensen H; Holzl K; Lunzer M; Li H; Hilborn J; Ovsianikov A; Ossipov DA Dynamic Coordination Chemistry Enables Free Directional Printing of Biopolymer Hydrogel. Chem Mater 2017, 29 (14), 5816. [Google Scholar]
- (559).Li Q; Barrett DG; Messersmith PB; Holten-Andersen N.Controlling Hydrogel Mechanics via Bio-Inspired Polymer-Nanoparticle Bond Dynamics. ACS Nano 2016, 10 (1), 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Hou S; Ma PX Stimuli-responsive supramolecular hydrogels with high extensibility and fast self-healing via precoordinated mussel-inspired chemistry. Chem Mater 2015, 27 (22), 7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (561).Wang WN; Xu YS; Li A; Li T; Liu MM; von Klitzing R; Ober CK; Kayitmazer AB; Li L; Guo XH Zinc induced polyelectrolyte coacervate bioadhesive and its transition to a Self-Healing hydrogel. Rsc Adv 2015, 5 (82), 66871. [Google Scholar]
- (562).Grindy SC; Learsch R; Mozhdehi D; Cheng J; Barrett DG; Guan Z; Messersmith PB; Holten-Andersen N.Control of hierarchical polymer mechanics with bioinspired metalcoordination dynamics. Nat Mater 2015, 14 (12), 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Fullenkamp DE; He L; Barrett DG; Burghardt WR; Messersmith PB Mussel-inspired histidine-based transient network metal coordination hydrogels. Macromolecules 2013, 46 (3), 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (564).Wang C; Stewart RJ; Kopecek J.Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains. Nature 1999, 397 (6718), 417. [DOI] [PubMed] [Google Scholar]
- (565).Qin HL; Zhang T; Li HN; Cong HP; Antonietti M; Yu SH Dynamic Au-Thiolate Interaction Induced Rapid Self-Healing Nanocomposite Hydrogels with Remarkable Mechanical Behaviors. Chem-Us 2017, 3 (4), 691. [Google Scholar]
- (566).Casuso P; Odriozola I; Perez-San Vicente A; Loinaz I; Cabanero G; Grande HJ; Dupin D.Injectable and Self-Healing Dynamic Hydrogels Based on Metal(I)-Thiolate/Disulfide Exchange as Biomaterials with Tunable Mechanical Properties. Biomacromolecules 2015, 16 (11), 3552. [DOI] [PubMed] [Google Scholar]
- (567).Peng F; Li G; Liu X; Wu S; Tong Z.Redox-responsive gel-sol/sol-gel transition in poly(acrylic acid) aqueous solution containing Fe(III) ions switched by light. J Am Chem Soc 2008, 130 (48), 16166. [DOI] [PubMed] [Google Scholar]
- (568).Zheng SY; Ding H; Qian J; Yin J; Wu ZL; Song Y; Zheng Q.Metal-coordination complexes mediated physical hydrogels with high toughness, stick–slip tearing behavior, and good processability. Macromolecules 2016, 49 (24), 9637. [Google Scholar]
- (569).Buwalda SJ; Dijkstra PJ; Feijen J.Poly(ethylene glycol)-poly(L-lactide) star block copolymer hydrogels crosslinked by metal-ligand coordination. Journal of Polymer Science Part A: Polymer Chemistry 2012, 50 (9), 1783. [Google Scholar]
- (570).Chujo Y; Sada K; Saegusa T.Iron(Ii) Bipyridyl-Branched Polyoxazoline Complex as a Thermally Reversible Hydrogel. Macromolecules 1993, 26 (24), 6315. [Google Scholar]
- (571).Weng G; Thanneeru S; He J.Dynamic Coordination of Eu-Iminodiacetate to Control Fluorochromic Response of Polymer Hydrogels to Multistimuli. Adv Mater 2018, 30 (11), 1706526. [DOI] [PubMed] [Google Scholar]
- (572).Zhang KY; Feng Q; Xu JB; Xu XY; Tian F; Yeung KWK; Bian LM Self-Assembled Injectable Nanocomposite Hydrogels Stabilized by Bisphosphonate-Magnesium (Mg2+) Coordination Regulates the Differentiation of Encapsulated Stem Cells via Dual Crosslinking. Adv Funct Mater 2017, 27 (34), 1701642. [Google Scholar]
- (573).Diba M; Camargo WA; Brindisi M; Farbod K; Klymov A; Schmidt S; Harrington MJ; Draghi L; Boccaccini AR; Jansen JA et al. Composite Colloidal Gels Made of BisphosphonateFunctionalized Gelatin and Bioactive Glass Particles for Regeneration of Osteoporotic Bone Defects. Adv Funct Mater 2017, 27 (45), 1703438. [Google Scholar]
- (574).He L; Fullenkamp DE; Rivera JG; Messersmith PB pH responsive self-healing hydrogels formed by boronate–catechol complexation. Chemical Communications 2011, 47 (26), 7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (575).Holten-Andersen N; Harrington MJ; Birkedal H; Lee BP; Messersmith PB; Lee KY; Waite JH pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. Proc Natl Acad Sci U S A 2011, 108 (7), 2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (576).Chan Choi Y; Choi JS; Jung YJ; Cho YW Human gelatin tissue-adhesive hydrogels prepared by enzyme-mediated biosynthesis of DOPA and Fe(3+) ion crosslinking. J Mater Chem B 2014, 2 (2), 201. [DOI] [PubMed] [Google Scholar]
- (577).Lee J; Chang K; Kim S; Gite V; Chung H; Sohn D.Phase Controllable Hyaluronic Acid Hydrogel with Iron(III) Ion–Catechol Induced Dual Cross-Linking by Utilizing the Gap of Gelation Kinetics. Macromolecules 2016, 49 (19), 7450. [Google Scholar]
- (578).Yavvari PS; Srivastava A.Robust, self-healing hydrogels synthesised from catechol rich polymers. J Mater Chem B 2015, 3 (5), 899. [DOI] [PubMed] [Google Scholar]
- (579).Sundberg RJ; Martin RB Interactions of Histidine and Other Imidazole Derivatives with Transition-Metal Ions in Chemical and Biological-Systems. Chemical Reviews 1974, 74 (4), 471. [Google Scholar]
- (580).Shi L; Ding P; Wang Y; Zhang Y; Ossipov D; Hilborn J.Self-Healing Polymeric Hydrogel Formed by Metal-Ligand Coordination Assembly: Design, Fabrication, and Biomedical Applications. Macromol Rapid Commun 2019, 40 (7), e1800837. [DOI] [PubMed] [Google Scholar]
- (581).Cook TR; Zheng YR; Stang PJ Metal-organic frameworks and self-assembled supramolecular coordination complexes: comparing and contrasting the design, synthesis, and functionality of metal-organic materials. Chem Rev 2013, 113 (1), 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (582).Rodell CB; Mealy JE; Burdick JA Supramolecular Guest-Host Interactions for the Preparation of Biomedical Materials. Bioconjug Chem 2015, 26 (12), 2279. [DOI] [PubMed] [Google Scholar]
- (583).Appel EA; del Barrio J; Loh XJ; Scherman OA Supramolecular polymeric hydrogels. Chem Soc Rev 2012, 41 (18), 6195. [DOI] [PubMed] [Google Scholar]
- (584).Szejtli J.Introduction and General Overview of Cyclodextrin Chemistry. Chem Rev 1998, 98 (5), 1743. [DOI] [PubMed] [Google Scholar]
- (585).Harada A; Takashima Y; Nakahata M.Supramolecular polymeric materials via cyclodextringuest interactions. Acc Chem Res 2014, 47 (7), 2128. [DOI] [PubMed] [Google Scholar]
- (586).Yamaguchi H; Kobayashi Y; Kobayashi R; Takashima Y; Hashidzume A; Harada A.Photoswitchable gel assembly based on molecular recognition. Nat Commun 2012, 3 (1), 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Wu JS; Toda K; Tanaka A; Sanemasa I.Association constants of ferrocene with cyclodextrins in aqueous medium determined by solubility measurements of ferrocene. B Chem Soc Jpn 1998, 71 (7), 1615. [Google Scholar]
- (588).Voskuhl J; Waller M; Bandaru S; Tkachenko BA; Fregonese C; Wibbeling B; Schreiner PR; Ravoo BJ Nanodiamonds in sugar rings: an experimental and theoretical investigation of cyclodextrin-nanodiamond inclusion complexes. Org Biomol Chem 2012, 10 (23), 4524. [DOI] [PubMed] [Google Scholar]
- (589).Lezcano M; Al-Soufi W; Novo M; Rodriguez-Nunez E; Tato JV Complexation of several benzimidazole-type fungicides with alpha- and beta-cyclodextrins. J Agric Food Chem 2002, 50 (1), 108. [DOI] [PubMed] [Google Scholar]
- (590).Huh KM; Tomita H; Lee WK; Ooya T; Yui N.Synthesis of α-Cyclodextrin-Conjugated Poly(ɛ-lysine)s and Their Inclusion Complexation Behavior. Macromolecular Rapid Communications 2002, 23 (3), 179. [Google Scholar]
- (591).Tomatsu I; Hashidzume A; Harada A.Redox-Responsive Hydrogel System Using the Molecular Recognition ofβ-Cyclodextrin. Macromolecular Rapid Communications 2006, 27 (4), 238. [Google Scholar]
- (592).Nelissen HF; Feiters MC; Nolte RJ Synthesis and self-inclusion of bipyridine-spaced cyclodextrin dimers. J Org Chem 2002, 67 (17), 5901. [DOI] [PubMed] [Google Scholar]
- (593).Hetzer M; Fleischmann C; Schmidt BVKJ; Barner-Kowollik C; Ritter H.Visual recognition of supramolecular graft polymer formation via phenolphthalein–cyclodextrin association. Polymer 2013, 54 (19), 5141. [Google Scholar]
- (594).Ravichandran R; Divakar S.Inclusion of Ring A of Cholesterol Inside the β-Cyclodextrin Cavity: Evidence from Oxidation Reactions and Structural Studies. Journal of Inclusion Phenomena and Molecular Recognition in Chemistry 1998, 30 (3), 253. [Google Scholar]
- (595).Rekharsky MV; Inoue Y.Complexation Thermodynamics of Cyclodextrins. Chem Rev 1998, 98 (5), 1875. [DOI] [PubMed] [Google Scholar]
- (596).Bortolus P; Monti S.cis. dblharw. trans Photoisomerization of azobenzene-cyclodextrin inclusion complexes. The Journal of Physical Chemistry 1987, 91 (19), 5046. [Google Scholar]
- (597).Matsue T; Evans DH; Osa T; Kobayashi N.Electron-Transfer Reactions Associated with Host Guest Complexation - Oxidation of Ferrocenecarboxylic Acid in the Presence of BetaCyclodextrin. Journal of the American Chemical Society 1985, 107 (12), 3411. [Google Scholar]
- (598).Kaifer AE Interplay between molecular recognition and redox chemistry. Accounts of Chemical Research 1999, 32 (1), 62. [Google Scholar]
- (599).Freeman WA; Mock WL; Shih NY Cucurbituril. Journal of the American Chemical Society 1981, 103 (24), 7367. [Google Scholar]
- (600).Lagona J; Mukhopadhyay P; Chakrabarti S; Isaacs L.The cucurbit[n]uril family. Angew Chem Int Ed Engl 2005, 44 (31), 4844. [DOI] [PubMed] [Google Scholar]
- (601).Liu S; Ruspic C; Mukhopadhyay P; Chakrabarti S; Zavalij PY; Isaacs L.The cucurbit[n]uril family: prime components for self-sorting systems. J Am Chem Soc 2005, 127 (45), 15959. [DOI] [PubMed] [Google Scholar]
- (602).Mock WL; Shih NY Structure and Selectivity in Host Guest Complexes of Cucurbituril. Journal of Organic Chemistry 1986, 51 (23), 4440. [Google Scholar]
- (603).Jeon WS; Moon K; Park SH; Chun H; Ko YH; Lee JY; Lee ES; Samal S; Selvapalam N; Rekharsky MV et al. Complexation of ferrocene derivatives by the cucurbit[7]uril host: a comparative study of the cucurbituril and cyclodextrin host families. J Am Chem Soc 2005, 127 (37), 12984. [DOI] [PubMed] [Google Scholar]
- (604).Moghaddam S; Yang C; Rekharsky M; Ko YH; Kim K; Inoue Y; Gilson MK New ultrahigh affinity host-guest complexes of cucurbit[7]uril with bicyclo[2.2.2]octane and adamantane guests: thermodynamic analysis and evaluation of M2 affinity calculations. J Am Chem Soc 2011, 133 (10), 3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (605).Kim J; Jung I-S; Kim S-Y; Lee E; Kang J-K; Sakamoto S; Yamaguchi K; Kim K.New Cucurbituril Homologues: Syntheses, Isolation, Characterization, and X-ray Crystal Structures of Cucurbit[n]uril (n= 5, 7, and 8). Journal of the American Chemical Society 2000, 122 (3), 540. [Google Scholar]
- (606).Lee JW; Kim K; Choi S; Ko YH; Sakamoto S; Yamaguchi K; Kim K.Unprecedented hostinduced intramolecular charge-transfer complex formation. Chem Commun (Camb) 2002, DOI: 10.1039/b208280b10.1039/b208280b(22), 2692. [DOI] [PubMed] [Google Scholar]
- (607).Takashima Y; Sawa Y; Iwaso K; Nakahata M; Yamaguchi H; Harada A.Supramolecular Materials Cross-Linked by Host–Guest Inclusion Complexes: The Effect of Side Chain Molecules on Mechanical Properties. Macromolecules 2017, 50 (8), 3254. [Google Scholar]
- (608).Jin J; Cai L; Jia YG; Liu S; Chen Y; Ren L.Progress in self-healing hydrogels assembled by host-guest interactions: preparation and biomedical applications. J Mater Chem B 2019, 7 (10), 1637. [DOI] [PubMed] [Google Scholar]
- (609).Yang X; Yu H; Wang L; Tong R; Akram M; Chen Y; Zhai X.Self-healing polymer materials constructed by macrocycle-based host-guest interactions. Soft Matter 2015, 11 (7), 1242. [DOI] [PubMed] [Google Scholar]
- (610).Takashima Y; Hatanaka S; Otsubo M; Nakahata M; Kakuta T; Hashidzume A; Yamaguchi H; Harada A.Expansion-contraction of photoresponsive artificial muscle regulated by host-guest interactions. Nat Commun 2012, 3 (1), 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (611).Zhou LP; Li JX; Luo Q; Zhu JY; Zou HX; Gao YZ; Wang L; Xu JY; Dong ZY; Liu JQ Dual stimuli-responsive supramolecular pseudo-polyrotaxane hydrogels. Soft Matter 2013, 9 (18), 4635. [Google Scholar]
- (612).Jeong B; Kim SW; Bae YH Thermosensitive sol–gel reversible hydrogels. Advanced Drug Delivery Reviews 2012, 64, 154. [DOI] [PubMed] [Google Scholar]
- (613).Mihajlovic M; Staropoli M; Appavou MS; Wyss HM; Pyckhout-Hintzen W; Sijbesma RP Tough Supramolecular Hydrogel Based on Strong Hydrophobic Interactions in a Multiblock Segmented Copolymer. Macromolecules 2017, 50 (8), 3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (614).Patrickios CS; Georgiou TK Covalent amphiphilic polymer networks. Current Opinion in Colloid & Interface Science 2003, 8 (1), 76. [Google Scholar]
- (615).Klaikherd A; Nagamani C; Thayumanavan S.Multi-stimuli sensitive amphiphilic block copolymer assemblies. J Am Chem Soc 2009, 131 (13), 4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (616).Zhang HJ; Sun TL; Zhang AK; Ikura Y; Nakajima T; Nonoyama T; Kurokawa T; Ito O; Ishitobi H; Gong JP Tough Physical Double-Network Hydrogels Based on Amphiphilic Triblock Copolymers. Adv Mater 2016, 28 (24), 4884. [DOI] [PubMed] [Google Scholar]
- (617).Tuncaboylu DC; Argun A; Sahin M; Sari M; Okay O.Structure optimization of self-healing hydrogels formed via hydrophobic interactions. Polymer 2012, 53 (24), 5513. [Google Scholar]
- (618).Wheeler SE Understanding substituent effects in noncovalent interactions involving aromatic rings. Acc Chem Res 2013, 46 (4), 1029. [DOI] [PubMed] [Google Scholar]
- (619).Butterfield SM; Patel PR; Waters ML Contribution of aromatic interactions to alpha-helix stability. J Am Chem Soc 2002, 124 (33), 9751. [DOI] [PubMed] [Google Scholar]
- (620).Singh V; Snigdha K; Singh C; Sinha N; Thakur AK Understanding the self-assembly of Fmoc-phenylalanine to hydrogel formation. Soft Matter 2015, 11 (26), 5353. [DOI] [PubMed] [Google Scholar]
- (621).Yan X; Zhu P; Li J.Self-assembly and application of diphenylalanine-based nanostructures. Chem Soc Rev 2010, 39 (6), 1877. [DOI] [PubMed] [Google Scholar]
- (622).Mehra NK; Palakurthi S.Interactions between carbon nanotubes and bioactives: a drug delivery perspective. Drug Discov Today 2016, 21 (4), 585. [DOI] [PubMed] [Google Scholar]
- (623).Kovtyukhova NI; Mallouk TE; Pan L; Dickey EC Individual single-walled nanotubes and hydrogels made by oxidative exfoliation of carbon nanotube ropes. J Am Chem Soc 2003, 125 (32), 9761. [DOI] [PubMed] [Google Scholar]
- (624).Yuk H; Lu B; Lin S; Qu K; Xu J; Luo J; Zhao X.3D printing of conducting polymers. Nat Commun 2020, 11 (1), 1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (625).Ghawanmeh AA; Ali GAM; Algarni H; Sarkar SM; Chong KF Graphene oxide-based hydrogels as a nanocarrier for anticancer drug delivery. Nano Res 2019, 12 (5), 973. [Google Scholar]
- (626).Loh KP; Bao QL; Ang PK; Yang JX The chemistry of graphene. Journal of Materials Chemistry 2010, 20 (12), 2277. [Google Scholar]
- (627).Yuk H; Lu B; Zhao X.Hydrogel bioelectronics. Chem Soc Rev 2019, 48 (6), 1642. [DOI] [PubMed] [Google Scholar]
- (628).Dhanjai; Sinha A; Kalambate PK; Mugo SM; Kamau P; Chen JP; Jain R. Polymer hydrogel interfaces in electrochemical sensing strategies: A review. Trac-Trend Anal Chem 2019, 118, 488. [Google Scholar]
- (629).Krishnakumar B; Sanka RVSP; Binder WH; Parthasarthy V; Rana S; Karak N.Vitrimers: Associative dynamic covalent adaptive networks in thermoset polymers. Chem Eng J 2020, 385, 123820. [Google Scholar]
- (630).Cordes EH; Jencks WP The Mechanism of Hydrolysis of Schiff Bases Derived from Aliphatic Amines. Journal of the American Chemical Society 1963, 85 (18), 2843. [Google Scholar]
- (631).Lu S; Gao C; Xu X; Bai X; Duan H; Gao N; Feng C; Xiong Y; Liu M.Injectable and Self-Healing Carbohydrate-Based Hydrogel for Cell Encapsulation. ACS Appl Mater Interfaces 2015, 7 (23), 13029. [DOI] [PubMed] [Google Scholar]
- (632).Wei Z; Yang JH; Liu ZQ; Xu F; Zhou JX; Zrinyi M; Osada Y; Chen YM Novel Biocompatible Polysaccharide-Based Self-Healing Hydrogel. Adv Funct Mater 2015, 25 (9), 1352. [Google Scholar]
- (633).Karimi AR; Khodadadi A.Mechanically Robust 3D Nanostructure Chitosan-Based Hydrogels with Autonomic Self-Healing Properties. ACS Appl Mater Interfaces 2016, 8 (40), 27254. [DOI] [PubMed] [Google Scholar]
- (634).Wu X; He C; Wu Y; Chen X.Synergistic therapeutic effects of Schiff’s base cross-linked injectable hydrogels for local co-delivery of metformin and 5-fluorouracil in a mouse colon carcinoma model. Biomaterials 2016, 75, 148. [DOI] [PubMed] [Google Scholar]
- (635).Ding F; Wu S; Wang S; Xiong Y; Li Y; Li B; Deng H; Du Y; Xiao L; Shi X.A dynamic and self-crosslinked polysaccharide hydrogel with autonomous self-healing ability. Soft Matter 2015, 11 (20), 3971. [DOI] [PubMed] [Google Scholar]
- (636).Engel AK; Yoden T; Sanui K; Ogata N.Synthesis of Aromatic Schiff-Base Oligomers at the Air-Water-Interface. Journal of the American Chemical Society 1985, 107 (26), 8308. [Google Scholar]
- (637).Zhang Y; Tao L; Li S; Wei Y.Synthesis of multiresponsive and dynamic chitosan-based hydrogels for controlled release of bioactive molecules. Biomacromolecules 2011, 12 (8), 2894. [DOI] [PubMed] [Google Scholar]
- (638).Huang W; Wang Y; Huang Z; Wang X; Chen L; Zhang Y; Zhang L.On-Demand Dissolvable Self-Healing Hydrogel Based on Carboxymethyl Chitosan and Cellulose Nanocrystal for Deep Partial Thickness Burn Wound Healing. ACS Appl Mater Interfaces 2018, 10 (48), 41076. [DOI] [PubMed] [Google Scholar]
- (639).Boehnke N; Cam C; Bat E; Segura T; Maynard HD Imine Hydrogels with Tunable Degradability for Tissue Engineering. Biomacromolecules 2015, 16 (7), 2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (640).Xu Y; Li Y; Chen Q; Fu L; Tao L; Wei Y.Injectable and Self-Healing Chitosan Hydrogel Based on Imine Bonds: Design and Therapeutic Applications. Int J Mol Sci 2018, 19 (8), 2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (641).Wang H; Heilshorn SC Adaptable hydrogel networks with reversible linkages for tissue engineering. Adv Mater 2015, 27 (25), 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (642).Bull SD; Davidson MG; van den Elsen JM; Fossey JS; Jenkins AT; Jiang YB; Kubo Y; Marken F; Sakurai K; Zhao J et al. Exploiting the reversible covalent bonding of boronic acids: recognition, sensing, and assembly. Acc Chem Res 2013, 46 (2), 312. [DOI] [PubMed] [Google Scholar]
- (643).Guan Y; Zhang Y.In Boron; The Royal Society of Chemistry, 2015, DOI: 10.1039/9781782622123-0026810.1039/9781782622123-00268. [DOI] [Google Scholar]
- (644).Nishiyabu R; Kubo Y; James TD; Fossey JS Boronic acid building blocks: tools for sensing and separation. Chem Commun (Camb) 2011, 47 (4), 1106. [DOI] [PubMed] [Google Scholar]
- (645).Pizer R; Babcock L.Mechanism of the complexation of boron acids with catechol and substituted catechols. Inorganic Chemistry 1977, 16 (7), 1677. [Google Scholar]
- (646).Jay JI; Shukair S; Langheinrich K; Hanson MC; Cianci GC; Johnson TJ; Clark MR; Hope TJ; Kiser PF Modulation of Viscoelasticity and HIV Transport as a Function of pH in a Reversibly Crosslinked Hydrogel. Adv Funct Mater 2009, 19 (18), 2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (647).Mahalingam A; Jay JI; Langheinrich K; Shukair S; McRaven MD; Rohan LC; Herold BC; Hope TJ; Kiser PF Inhibition of the transport of HIV in vitro using a pH-responsive synthetic mucin-like polymer system. Biomaterials 2011, 32 (33), 8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (648).Sartain FK; Yang X; Lowe CR Holographic lactate sensor. Anal Chem 2006, 78 (16), 5664. [DOI] [PubMed] [Google Scholar]
- (649).Kataoka K; Miyazaki H; Bunya M; Okano T; Sakurai Y.Totally Synthetic Polymer Gels Responding to External Glucose Concentration: Their Preparation and Application to On−Off Regulation of Insulin Release. Journal of the American Chemical Society 1998, 120 (48), 12694. [Google Scholar]
- (650).Asher SA; Alexeev VL; Goponenko AV; Sharma AC; Lednev IK; Wilcox CS; Finegold DN Photonic crystal carbohydrate sensors: low ionic strength sugar sensing. J Am Chem Soc 2003, 125 (11), 3322. [DOI] [PubMed] [Google Scholar]
- (651).Zhang Y; Guan Y; Zhou S.Synthesis and volume phase transitions of glucose-sensitive microgels. Biomacromolecules 2006, 7 (11), 3196. [DOI] [PubMed] [Google Scholar]
- (652).Roberts MC; Hanson MC; Massey AP; Karren EA; Kiser PF Dynamically restructuring hydrogel networks formed with reversible covalent crosslinks. Advanced Materials 2007, 19 (18), 2503. [Google Scholar]
- (653).Nakahata M; Mori S; Takashima Y; Hashidzume A; Yamaguchi H; Harada A.pH-and sugarresponsive gel assemblies based on boronate–catechol interactions. Acs Macro Lett 2014, 3 (4), 337. [DOI] [PubMed] [Google Scholar]
- (654).Shan M; Gong C; Li B; Wu G.A pH, glucose, and dopamine triple-responsive, self-healable adhesive hydrogel formed by phenylborate–catechol complexation. Polymer Chemistry 2017, 8 (19), 2997. [Google Scholar]
- (655).Kitano S; Hisamitsu I; Koyama Y; Kataoka K; Okano T; Sakurai Y.Effect of the incorporation of amino groups in a glucose‐responsive polymer complex having phenylboronic acid moieties. Polym Advan Technol 1991, 2 (5), 261. [Google Scholar]
- (656).Ivanov A; Larsson H; Galaev IY; Mattiasson B.Synthesis of boronate-containing copolymers of N, N-dimethylacrylamide, their interaction with poly (vinyl alcohol) and rheological behaviour of the gels. Polymer 2004, 45 (8), 2495. [Google Scholar]
- (657).Hong SH; Kim S; Park JP; Shin M; Kim K; Ryu JH; Lee H.Dynamic Bonds between Boronic Acid and Alginate: Hydrogels with Stretchable, Self-Healing, Stimuli-Responsive, Remoldable, and Adhesive Properties. Biomacromolecules 2018, 19 (6), 2053. [DOI] [PubMed] [Google Scholar]
- (658).Pettignano A; Grijalvo S; Haring M; Eritja R; Tanchoux N; Quignard F; Diaz Diaz D.Boronic acid-modified alginate enables direct formation of injectable, self-healing and multistimuli-responsive hydrogels. Chem Commun (Camb) 2017, 53 (23), 3350. [DOI] [PubMed] [Google Scholar]
- (659).An H; Bo YY; Chen DY; Wang Y; Wang HJ; He YN; Qin JL Cellulose-based Self-Healing hydrogel through boronic ester bonds with excellent biocompatibility and conductivity. Rsc Adv 2020, 10 (19), 11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (660).Jay JI; Langheinrich K; Hanson MC; Mahalingam A; Kiser PF Unequal stoichiometry between crosslinking moieties affects the properties of transient networks formed by dynamic covalent crosslinks. Soft Matter 2011, 7 (12), 5826. [Google Scholar]
- (661).Phadke A; Zhang C; Arman B; Hsu CC; Mashelkar RA; Lele AK; Tauber MJ; Arya G; Varghese S.Rapid self-healing hydrogels. Proc Natl Acad Sci U S A 2012, 109 (12), 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (662).Deng CC; Brooks WLA; Abboud KA; Sumerlin BS Boronic Acid-Based Hydrogels Undergo Self-Healing at Neutral and Acidic pH. Acs Macro Lett 2015, 4 (2), 220. [DOI] [PubMed] [Google Scholar]
- (663).Patenaude M; Smeets NM; Hoare T.Designing Injectable, Covalently Cross‐Linked Hydrogels for Biomedical Applications. Macromolecular rapid communications 2014, 35 (6), 598. [DOI] [PubMed] [Google Scholar]
- (664).Wilson JM; Bayer RJ; Hupe D.Structure-reactivity correlations for the thiol-disulfide interchange reaction. Journal of the American Chemical Society 1977, 99 (24), 7922. [Google Scholar]
- (665).Madrazo J; Brown JH; Litvinovich S; Dominguez R; Yakovlev S; Medved L; Cohen C.Crystal structure of the central region of bovine fibrinogen (E5 fragment) at 1.4-A resolution. Proc Natl Acad Sci U S A 2001, 98 (21), 11967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (666).Cheung DT; DiCesare P; Benya PD; Libaw E; Nimni ME The presence of intermolecular disulfide cross-links in type III collagen. J Biol Chem 1983, 258 (12), 7774. [PubMed] [Google Scholar]
- (667).Zhang Y; Heher P; Hilborn J; Redl H; Ossipov DA Hyaluronic acid-fibrin interpenetrating double network hydrogel prepared in situ by orthogonal disulfide cross-linking reaction for biomedical applications. Acta Biomater 2016, 38, 23. [DOI] [PubMed] [Google Scholar]
- (668).Shu XZ; Liu Y; Luo Y; Roberts MC; Prestwich GD Disulfide cross-linked hyaluronan hydrogels. Biomacromolecules 2002, 3 (6), 1304. [DOI] [PubMed] [Google Scholar]
- (669).Lee H; Park TG Reduction/oxidation induced cleavable/crosslinkable temperature-sensitive hydrogel network containing disulfide linkages. Polym J 1998, 30 (12), 976. [Google Scholar]
- (670).Meng F; Hennink WE; Zhong Z.Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials 2009, 30 (12), 2180. [DOI] [PubMed] [Google Scholar]
- (671).Chen X; Lai NC; Wei K; Li R; Cui M; Yang B; Wong SHD; Deng Y; Li J; Shuai X et al. Biomimetic Presentation of Cryptic Ligands via Single-Chain Nanogels for Synergistic Regulation of Stem Cells. ACS Nano 2020, 14 (4), 4027. [DOI] [PubMed] [Google Scholar]
- (672).Lei ZQ; Xiang HP; Yuan YJ; Rong MZ; Zhang MQ Room-Temperature Self-Healable and Remoldable Cross-linked Polymer Based on the Dynamic Exchange of Disulfide Bonds. Chem Mater 2014, 26 (6), 2038. [Google Scholar]
- (673).Ryu JH; Chacko RT; Jiwpanich S; Bickerton S; Babu RP; Thayumanavan S.Self-crosslinked polymer nanogels: a versatile nanoscopic drug delivery platform. J Am Chem Soc 2010, 132 (48), 17227. [DOI] [PubMed] [Google Scholar]
- (674).Choh SY; Cross D; Wang C.Facile synthesis and characterization of disulfide-cross-linked hyaluronic acid hydrogels for protein delivery and cell encapsulation. Biomacromolecules 2011, 12 (4), 1126. [DOI] [PubMed] [Google Scholar]
- (675).Wu D-C; Loh XJ; Wu Y-L; Lay CL; Liu Y.‘Living’controlled in situ gelling systems: thiol− disulfide exchange method toward tailor-made biodegradable hydrogels. Journal of the American Chemical Society 2010, 132 (43), 15140. [DOI] [PubMed] [Google Scholar]
- (676).Burns JA; Butler JC; Moran J; Whitesides GM Selective Reduction of Disulfides by Tris(2Carboxyethyl)Phosphine. Journal of Organic Chemistry 1991, 56 (8), 2648. [Google Scholar]
- (677).Konigsberg W.In Methods in Enzymology; Academic Press, 1972; Vol. 25. [Google Scholar]
- (678).Pleasants JC; Guo W; Rabenstein DL A Comparative-Study of the Kinetics of Selenol Diselenide and Thiol Disulfide Exchange-Reactions. Journal of the American Chemical Society 1989, 111 (17), 6553. [Google Scholar]
- (679).Deng G; Li F; Yu H; Liu F; Liu C; Sun W; Jiang H; Chen Y.Dynamic Hydrogels with an Environmental Adaptive Self-Healing Ability and Dual Responsive Sol–Gel Transitions. Acs Macro Lett 2012, 1 (2), 275. [DOI] [PubMed] [Google Scholar]
- (680).Deng G; Tang C; Li F; Jiang H; Chen Y.Covalent Cross-Linked Polymer Gels with Reversible Sol−Gel Transition and Self-Healing Properties. Macromolecules 2010, 43 (3), 1191. [Google Scholar]
- (681).Yang XF; Liu GQ; Peng L; Guo JH; Tao L; Yuan JY; Chang CY; Wei Y; Zhang LN Highly Efficient Self-Healable and Dual Responsive Cellulose-Based Hydrogels for Controlled Release and 3D Cell Culture. Adv Funct Mater 2017, 27 (40), 1703174. [Google Scholar]
- (682).Dirksen A; Dirksen S; Hackeng TM; Dawson PE Nucleophilic catalysis of hydrazone formation and transimination: implications for dynamic covalent chemistry. J Am Chem Soc 2006, 128 (49), 15602. [DOI] [PubMed] [Google Scholar]
- (683).Dirksen A; Yegneswaran S; Dawson PE Bisaryl Hydrazones as Exchangeable Biocompatible Linkers. Angew Chem Int Edit 2010, 49 (11), 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (684).Skene WG; Lehn JM Dynamers: polyacylhydrazone reversible covalent polymers, component exchange, and constitutional diversity. Proc Natl Acad Sci U S A 2004, 101 (22), 8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (685).Liu FY; Li FY; Deng GH; Chen YM; Zhang BQ; Zhang J; Liu CY Rheological Images of Dynamic Covalent Polymer Networks and Mechanisms behind Mechanical and Self-Healing Properties. Macromolecules 2012, 45 (3), 1636. [Google Scholar]
- (686).Kool ET; Park DH; Crisalli P.Fast Hydrazone Reactants: Electronic and Acid/Base Effects Strongly Influence Rate at Biological pH. Journal of the American Chemical Society 2013, 135 (47), 17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (687).Nguyen R; Huc I.Optimizing the reversibility of hydrazone formation for dynamic combinatorial chemistry. Chem Commun (Camb) 2003, DOI: 10.1039/b211645f10.1039/b211645f(8), 942. [DOI] [PubMed] [Google Scholar]
- (688).Dahlmann J; Krause A; Moller L; Kensah G; Mowes M; Diekmann A; Martin U; Kirschning A; Gruh I; Drager G.Fully defined in situ cross-linkable alginate and hyaluronic acid hydrogels for myocardial tissue engineering. Biomaterials 2013, 34 (4), 940. [DOI] [PubMed] [Google Scholar]
- (689).Patenaude M; Hoare T.Injectable, mixed natural-synthetic polymer hydrogels with modular properties. Biomacromolecules 2012, 13 (2), 369. [DOI] [PubMed] [Google Scholar]
- (690).McKinnon DD; Domaille DW; Brown TE; Kyburz KA; Kiyotake E; Cha JN; Anseth KS Measuring cellular forces using bis-aliphatic hydrazone crosslinked stress-relaxing hydrogels. Soft Matter 2014, 10 (46), 9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (691).Jung H; Park JS; Yeom J; Selvapalam N; Park KM; Oh K; Yang JA; Park KH; Hahn SK; Kim K.3D tissue engineered supramolecular hydrogels for controlled chondrogenesis of human mesenchymal stem cells. Biomacromolecules 2014, 15 (3), 707. [DOI] [PubMed] [Google Scholar]
- (692).Mukherjee S; Hill MR; Sumerlin BS Self-healing hydrogels containing reversible oxime crosslinks. Soft Matter 2015, 11 (30), 6152. [DOI] [PubMed] [Google Scholar]
- (693).Sanchez-Moran H; Ahmadi A; Vogler B; Roh KH Oxime Cross-Linked Alginate Hydrogels with Tunable Stress Relaxation. Biomacromolecules 2019, 20 (12), 4419. [DOI] [PubMed] [Google Scholar]
- (694).Buffa R; Sedova P; Basarabova I; Moravcova M; Wolfova L; Bobula T; Velebny V.alpha,beta-Unsaturated aldehyde of hyaluronan--Synthesis, analysis and applications. Carbohydr Polym 2015, 134, 293. [DOI] [PubMed] [Google Scholar]
- (695).Grover GN; Lam J; Nguyen TH; Segura T; Maynard HD Biocompatible hydrogels by oxime Click chemistry. Biomacromolecules 2012, 13 (10), 3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (696).Grover GN; Braden RL; Christman KL Oxime Cross‐Linked Injectable Hydrogels for Catheter Delivery. Advanced Materials 2013, 25 (21), 2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (697).Kalia J; Raines RT Hydrolytic stability of hydrazones and oximes. Angew Chem Int Ed Engl 2008, 47 (39), 7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (698).Lin F; Yu J; Tang W; Zheng J; Defante A; Guo K; Wesdemiotis C; Becker ML Peptidefunctionalized oxime hydrogels with tunable mechanical properties and gelation behavior. Biomacromolecules 2013, 14 (10), 3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (699).Kloetzel MC The D iels‐A lder Reaction with Maleic Anhydride. Organic Reactions 2004, 4, 1. [Google Scholar]
- (700).Holmes H.The D iels‐A lder Reaction Ethylenic and Acetylenic Dienophiles. Organic Reactions 2004, 4, 60. [Google Scholar]
- (701).Nimmo CM; Owen SC; Shoichet MS Diels-Alder Click cross-linked hyaluronic acid hydrogels for tissue engineering. Biomacromolecules 2011, 12 (3), 824. [DOI] [PubMed] [Google Scholar]
- (702).Shao C; Wang M; Chang H; Xu F; Yang J.A Self-Healing Cellulose Nanocrystal-Poly(ethylene glycol) Nanocomposite Hydrogel via Diels–Alder Click Reaction. ACS Sustainable Chemistry & Engineering 2017, 5 (7), 6167. [Google Scholar]
- (703).Tan H; Rubin JP; Marra KG Direct synthesis of biodegradable polysaccharide derivative hydrogels through aqueous Diels-Alder chemistry. Macromol Rapid Commun 2011, 32 (12), 905. [DOI] [PubMed] [Google Scholar]
- (704).Wei H-L; Yang Z; Chu H-J; Zhu J; Li Z-C; Cui J-S Facile preparation of poly(Nisopropylacrylamide)-based hydrogels via aqueous Diels–Alder click reaction. Polymer 2010, 51 (8), 1694. [Google Scholar]
- (705).Kirchhof S; Brandl FP; Hammer N; Goepferich AM Investigation of the Diels-Alder reaction as a cross-linking mechanism for degradable poly(ethylene glycol) based hydrogels. J Mater Chem B 2013, 1 (37), 4855. [DOI] [PubMed] [Google Scholar]
- (706).Adzima BJ; Kloxin CJ; Bowman CN Externally triggered healing of a thermoreversible covalent network via self‐limited hysteresis heating. Advanced Materials 2010, 22 (25), 2784. [DOI] [PubMed] [Google Scholar]
- (707).Chen X; Dam MA; Ono K; Mal A; Shen H; Nutt SR; Sheran K; Wudl F.A thermally remendable cross-linked polymeric material. Science 2002, 295 (5560), 1698. [DOI] [PubMed] [Google Scholar]
- (708).Gacal B; Durmaz H; Tasdelen M; Hizal G; Tunca U; Yagci Y; Demirel A.Anthracene− maleimide-based Diels− Alder “click chemistry” as a novel route to graft copolymers. Macromolecules 2006, 39 (16), 5330. [Google Scholar]
- (709).Moses JE; Moorhouse AD The growing applications of click chemistry. Chem Soc Rev 2007, 36 (8), 1249. [DOI] [PubMed] [Google Scholar]
- (710).Nandivada H; Jiang XW; Lahann J.Click chemistry: Versatility and control in the hands of materials scientists. Advanced Materials 2007, 19 (17), 2197. [Google Scholar]
- (711).Wei Z; Yang JH; Du XJ; Xu F; Zrinyi M; Osada Y; Li F; Chen YM Dextran‐Based Self-Healing Hydrogels Formed by Reversible Diels–Alder Reaction under Physiological Conditions. Macromolecular rapid communications 2013, 34 (18), 1464. [DOI] [PubMed] [Google Scholar]
- (712).Koehler KC; Anseth KS; Bowman CN Diels–alder mediated controlled release from a poly (ethylene glycol) based hydrogel. Biomacromolecules 2013, 14 (2), 538. [DOI] [PubMed] [Google Scholar]
- (713).Zhou Y; Zhang W; Hu J; Tang J; Jin C; Suo Z; Lu T.The Stiffness-Threshold Conflict in Polymer Networks and a Resolution. Journal of Applied Mechanics 2020, 87 (3). [Google Scholar]
- (714).Creton C; Ciccotti M.Fracture and adhesion of soft materials: a review. Reports on Progress in Physics 2016, 79 (4), 046601. [DOI] [PubMed] [Google Scholar]
- (715).Long R; Hui C-Y Fracture toughness of hydrogels: measurement and interpretation. Soft Matter 2016, 12 (39), 8069. [DOI] [PubMed] [Google Scholar]
- (716).Rice JR; Thomson R.Ductile versus brittle behaviour of crystals. The Philosophical Magazine: A Journal of Theoretical Experimental and Applied Physics 1974, 29 (1), 73. [Google Scholar]
- (717).McMeeking R; Evans A.Mechanics of transformation‐toughening in brittle materials. Journal of the American Ceramic Society 1982, 65 (5), 242. [Google Scholar]
- (718).Mallick PK Fiber-reinforced composites: materials, manufacturing, and design; CRC press, 2007. [Google Scholar]
- (719).Fung Y.-c. Biomechanics: mechanical properties of living tissues; Springer Science & Business Media, 2013. [Google Scholar]
- (720).Gong JP Why are double network hydrogels so tough? Soft Matter 2010, 6 (12), 2583. [Google Scholar]
- (721).Zhang T; Lin S; Yuk H; Zhao X.Predicting fracture energies and crack-tip fields of soft tough materials. Extreme Mechanics Letters 2015, 4, 1. [Google Scholar]
- (722).Yu QM; Tanaka Y; Furukawa H; Kurokawa T; Gong JP Direct observation of damage zone around crack tips in double-network gels. Macromolecules 2009, 42 (12), 3852. [Google Scholar]
- (723).Brown AE; Litvinov RI; Discher DE; Purohit PK; Weisel JW Multiscale mechanics of fibrin polymer: gel stretching with protein unfolding and loss of water. Science 2009, 325 (5941), 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (724).Yang F; Tadepalli V; Wiley BJ 3D printing of a double network hydrogel with a compression strength and elastic modulus greater than those of cartilage. Acs Biomater Sci Eng 2017, 3 (5), 863. [DOI] [PubMed] [Google Scholar]
- (725).Ducrot E; Chen Y; Bulters M; Sijbesma RP; Creton C.Toughening elastomers with sacrificial bonds and watching them break. Science 2014, 344 (6180), 186. [DOI] [PubMed] [Google Scholar]
- (726).Du J; Xu S; Feng S; Yu L; Wang J; Liu Y.Tough dual nanocomposite hydrogels with inorganic hybrid crosslinking. Soft Matter 2016, 12 (6), 1649. [DOI] [PubMed] [Google Scholar]
- (727).Chen Y; Shull KR High-toughness polycation cross-linked triblock copolymer hydrogels. Macromolecules 2017, 50 (9), 3637. [Google Scholar]
- (728).He Q; Huang Y; Wang S.Hofmeister Effect‐Assisted One Step Fabrication of Ductile and Strong Gelatin Hydrogels. Adv Funct Mater 2018, 28 (5), 1705069. [Google Scholar]
- (729).Sun Y.-n.; Gao G.-r.; Du G.-l.; Cheng Y.-j.; Fu J.Super tough, ultrastretchable, and thermoresponsive hydrogels with functionalized triblock copolymer micelles as macro-crosslinkers. Acs Macro Lett 2014, 3 (5), 496. [DOI] [PubMed] [Google Scholar]
- (730).Zhong M; Liu X-Y; Shi F-K; Zhang L-Q; Wang X-P; Cheetham AG; Cui H; Xie X-M Self-healable, tough and highly stretchable ionic nanocomposite physical hydrogels. Soft Matter 2015, 11 (21), 4235. [DOI] [PubMed] [Google Scholar]
- (731).Foster EM; Lensmeyer EE; Zhang B; Chakma P; Flum JA; Via JJ; Sparks JL; Konkolewicz D.Effect of polymer network architecture, enhancing soft materials using orthogonal dynamic bonds in an interpenetrating network. Acs Macro Lett 2017, 6 (5), 495. [DOI] [PubMed] [Google Scholar]
- (732).Cao L; Fan J; Huang J; Chen Y.A robust and stretchable cross-linked rubber network with recyclable and self-healable capabilities based on dynamic covalent bonds. Journal of Materials Chemistry A 2019, 7 (9), 4922. [Google Scholar]
- (733).Fu J.Strong and tough hydrogels crosslinked by multi‐functional polymer colloids. Journal of Polymer Science Part B: Polymer Physics 2018, 56 (19), 1336. [Google Scholar]
- (734).Nakayama A; Kakugo A; Gong JP; Osada Y; Takai M; Erata T; Kawano S.High mechanical strength double‐network hydrogel with bacterial cellulose. Adv Funct Mater 2004, 14 (11), 1124. [Google Scholar]
- (735).Toivonen MS; Kurki-Suonio S; Schacher FH; Hietala S; Rojas OJ; Ikkala O.Waterresistant, transparent hybrid nanopaper by physical cross-linking with chitosan. Biomacromolecules 2015, 16 (3), 1062. [DOI] [PubMed] [Google Scholar]
- (736).Ye D; Cheng Q; Zhang Q; Wang Y; Chang C; Li L; Peng H; Zhang L.Deformation drives alignment of nanofibers in framework for inducing anisotropic cellulose hydrogels with high toughness. ACS applied materials & interfaces 2017, 9 (49), 43154. [DOI] [PubMed] [Google Scholar]
- (737).Wang W; Zhang Y; Liu W.Bioinspired fabrication of high strength hydrogels from noncovalent interactions. Progress in Polymer Science 2017, 71, 1. [Google Scholar]
- (738).Dai X; Zhang Y; Gao L; Bai T; Wang W; Cui Y; Liu W.A Mechanically Strong, Highly Stable, Thermoplastic, and self‐healable supramolecular polymer hydrogel. Advanced Materials 2015, 27 (23), 3566. [DOI] [PubMed] [Google Scholar]
- (739).Zhang Y; Li Y; Liu W.Dipole–dipole and h‐bonding interactions significantly enhance the multifaceted mechanical properties of thermoresponsive shape memory hydrogels. Adv Funct Mater 2015, 25 (3), 471. [Google Scholar]
- (740).Nakahata M; Takashima Y; Harada A.Highly flexible, tough, and self‐healing supramolecular polymeric materials using host–guest interaction. Macromolecular rapid communications 2016, 37 (1), 86. [DOI] [PubMed] [Google Scholar]
- (741).Gonzalez MA; Simon JR; Ghoorchian A; Scholl Z; Lin S; Rubinstein M; Marszalek P; Chilkoti A; López GP; Zhao X.Strong, tough, stretchable, and self‐adhesive hydrogels from intrinsically unstructured proteins. Advanced Materials 2017, 29 (10), 1604743. [DOI] [PubMed] [Google Scholar]
- (742).Mayumi K; Guo J; Narita T; Hui CY; Creton C.Fracture of dual crosslink gels with permanent and transient crosslinks. Extreme Mechanics Letters 2016, 6, 52. [Google Scholar]
- (743).Zhang XN; Wang YJ; Sun S; Hou L; Wu P; Wu ZL; Zheng Q.A tough and stiff hydrogel with tunable water content and mechanical properties based on the synergistic effect of hydrogen bonding and hydrophobic interaction. Macromolecules 2018, 51 (20), 8136. [Google Scholar]
- (744).Chang X; Geng Y; Cao H; Zhou J; Tian Y; Shan G; Bao Y; Wu ZL; Pan P.Dual‐Crosslink Physical Hydrogels with High Toughness Based on Synergistic Hydrogen Bonding and Hydrophobic Interactions. Macromolecular rapid communications 2018, 39 (14), 1700806. [DOI] [PubMed] [Google Scholar]
- (745).Zhang Y; Yong Y; An D; Song W; Liu Q; Wang L; Pardo Y; Kern VR; Steen PH; Hong W.A drip-crosslinked tough hydrogel. Polymer 2018, 135, 327. [Google Scholar]
- (746).Bakarich SE; Gorkin III R; Panhuis MIH; Spinks GM 4D printing with mechanically robust, thermally actuating hydrogels. Macromolecular rapid communications 2015, 36 (12), 1211. [DOI] [PubMed] [Google Scholar]
- (747).Li J; Illeperuma WR; Suo Z; Vlassak JJ Hybrid hydrogels with extremely high stiffness and toughness. Acs Macro Lett 2014, 3 (6), 520. [DOI] [PubMed] [Google Scholar]
- (748).Hu X; Vatankhah‐Varnoosfaderani M; Zhou J; Li Q; Sheiko SS Weak hydrogen bonding enables hard, strong, tough, and elastic hydrogels. Advanced materials 2015, 27 (43), 6899. [DOI] [PubMed] [Google Scholar]
- (749).Zheng WJ; An N; Yang JH; Zhou J; Chen YM Tough Al-alginate/poly (Nisopropylacrylamide) hydrogel with tunable LCST for soft robotics. ACS applied materials & interfaces 2015, 7 (3), 1758. [DOI] [PubMed] [Google Scholar]
- (750).Li C; Rowland MJ; Shao Y; Cao T; Chen C; Jia H; Zhou X; Yang Z; Scherman OA; Liu D.Responsive double network hydrogels of interpenetrating DNA and CB [8] host–guest supramolecular systems. Advanced materials 2015, 27 (21), 3298. [DOI] [PubMed] [Google Scholar]
- (751).Chen Q; Yan X; Zhu L; Chen H; Jiang B; Wei D; Huang L; Yang J; Liu B; Zheng J.Improvement of mechanical strength and fatigue resistance of double network hydrogels by ionic coordination interactions. Chem Mater 2016, 28 (16), 5710. [Google Scholar]
- (752).Chen Q; Chen H; Zhu L; Zheng J.Engineering of tough double network hydrogels. Macromol Chem Phys 2016, 217 (9), 1022. [Google Scholar]
- (753).Jia H; Huang Z; Fei Z; Dyson PJ; Zheng Z; Wang X.Unconventional tough double-network hydrogels with rapid mechanical recovery, self-healing, and self-gluing properties. ACS applied materials & interfaces 2016, 8 (45), 31339. [DOI] [PubMed] [Google Scholar]
- (754).Guo M; Pitet LM; Wyss HM; Vos M; Dankers PY; Meijer E.Tough stimuli-responsive supramolecular hydrogels with hydrogen-bonding network junctions. Journal of the American Chemical Society 2014, 136 (19), 6969. [DOI] [PubMed] [Google Scholar]
- (755).Shao C; Chang H; Wang M; Xu F; Yang J.High-strength, tough, and self-healing nanocomposite physical hydrogels based on the synergistic effects of dynamic hydrogen bond and dual coordination bonds. ACS Applied Materials & Interfaces 2017, 9 (34), 28305. [DOI] [PubMed] [Google Scholar]
- (756).Gao G; Du G; Sun Y; Fu J.Self-healable, tough, and ultrastretchable nanocomposite hydrogels based on reversible polyacrylamide/montmorillonite adsorption. ACS Applied Materials & Interfaces 2015, 7 (8), 5029. [DOI] [PubMed] [Google Scholar]
- (757).Yang J; Han C-R; Zhang X-M; Xu F; Sun R-C Cellulose nanocrystals mechanical reinforcement in composite hydrogels with multiple cross-links: correlations between dissipation properties and deformation mechanisms. Macromolecules 2014, 47 (12), 4077. [Google Scholar]
- (758).Cui K; Sun TL; Liang X; Nakajima K; Ye YN; Chen L; Kurokawa T; Gong JP Multiscale energy dissipation mechanism in tough and self-healing hydrogels. Physical review letters 2018, 121 (18), 185501. [DOI] [PubMed] [Google Scholar]
- (759).Zhao X.Designing toughness and strength for soft materials. Proceedings of the National Academy of Sciences 2017, 114 (31), 8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (760).Gao H; Ji B; Jäger IL; Arzt E; Fratzl P.Materials become insensitive to flaws at nanoscale: lessons from nature. Proceedings of the national Academy of Sciences 2003, 100 (10), 5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (761).Chen C; Wang Z; Suo Z.Flaw sensitivity of highly stretchable materials. Extreme Mechanics Letters 2017, 10, 50. [Google Scholar]
- (762).Griffith AA VI. The phenomena of rupture and flow in solids. Philosophical transactions of the royal society of london. Series A, containing papers of a mathematical or physical character 1921, 221 (582–593), 163. [Google Scholar]
- (763).Keten S; Xu Z; Ihle B; Buehler MJ Nanoconfinement controls stiffness, strength and mechanical toughness of β-sheet crystals in silk. Nature materials 2010, 9 (4), 359. [DOI] [PubMed] [Google Scholar]
- (764).Liao IC; Moutos FT; Estes BT; Zhao X; Guilak F.Composite three‐dimensional woven scaffolds with interpenetrating network hydrogels to create functional synthetic articular cartilage. Adv Funct Mater 2013, 23 (47), 5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (765).Lin S; Cao C; Wang Q; Gonzalez M; Dolbow JE; Zhao X.Design of stiff, tough and stretchy hydrogel composites via nanoscale hybrid crosslinking and macroscale fiber reinforcement. Soft matter 2014, 10 (38), 7519. [DOI] [PubMed] [Google Scholar]
- (766).Illeperuma WR; Sun J-Y; Suo Z; Vlassak JJ Fiber-reinforced tough hydrogels. Extreme Mechanics Letters 2014, 1, 90. [Google Scholar]
- (767).Agrawal A; Rahbar N; Calvert PD Strong fiber-reinforced hydrogel. Acta biomaterialia 2013, 9 (2), 5313. [DOI] [PubMed] [Google Scholar]
- (768).King DR; Sun TL; Huang Y; Kurokawa T; Nonoyama T; Crosby AJ; Gong JP Extremely tough composites from fabric reinforced polyampholyte hydrogels. Materials horizons 2015, 2 (6), 584. [Google Scholar]
- (769).Kong W; Wang C; Jia C; Kuang Y; Pastel G; Chen C; Chen G; He S; Huang H; Zhang J.Muscle‐Inspired Highly Anisotropic, Strong, Ion‐Conductive Hydrogels. Advanced Materials 2018, 30 (39), 1801934. [DOI] [PubMed] [Google Scholar]
- (770).Gent A.Mechanical properties of rubber. The Pneumatic Tire; 2006, 28. [Google Scholar]
- (771).Cui J; Lackey MA; Madkour AE; Saffer EM; Griffin DM; Bhatia SR; Crosby AJ; Tew GN Synthetically simple, highly resilient hydrogels. Biomacromolecules 2012, 13 (3), 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (772).Sacks MS; Yoganathan AP Heart valve function: a biomechanical perspective. Philosophical Transactions of the Royal Society B: Biological Sciences 2007, 362 (1484), 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (773).Sacks MS; Merryman WD; Schmidt DE On the biomechanics of heart valve function. Journal of biomechanics 2009, 42 (12), 1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (774).Le Cam J-B Energy storage due to strain-induced crystallization in natural rubber: the physical origin of the mechanical hysteresis. Polymer 2017, 127, 166. [Google Scholar]
- (775).Li J; Celiz A; Yang J; Yang Q; Wamala I; Whyte W; Seo B; Vasilyev N; Vlassak J; Suo Z.Tough adhesives for diverse wet surfaces. Science 2017, 357 (6349), 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (776).Volinsky A; Moody N; Gerberich WW Interfacial toughness measurements for thin films on substrates. Acta materialia 2002, 50 (3), 441. [Google Scholar]
- (777).Yuk H; Zhang T; Parada GA; Liu X; Zhao X.Skin-inspired hydrogel–elastomer hybrids with robust interfaces and functional microstructures. Nature communications 2016, 7 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (778).Zhang T; Yuk H; Lin S; Parada GA; Zhao X.Tough and tunable adhesion of hydrogels: experiments and models. Acta Mechanica Sinica 2017, 33 (3), 543. [Google Scholar]
- (779).Lee H; Dellatore SM; Miller WM; Messersmith PB Mussel-inspired surface chemistry for multifunctional coatings. science 2007, 318 (5849), 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (780).Wirthl D; Pichler R; Drack M; Kettlguber G; Moser R; Gerstmayr R; Hartmann F; Bradt E; Kaltseis R; Siket CM Instant tough bonding of hydrogels for soft machines and electronics. Science advances 2017, 3 (6), e1700053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (781).Yang J; Bai R; Suo Z.Topological adhesion of wet materials. Advanced Materials 2018, 30 (25), 1800671. [DOI] [PubMed] [Google Scholar]
- (782).Kurokawa T; Furukawa H; Wang W; Tanaka Y; Gong JP Formation of a strong hydrogel– porous solid interface via the double-network principle. Acta biomaterialia 2010, 6 (4), 1353. [DOI] [PubMed] [Google Scholar]
- (783).Rao P; Sun TL; Chen L; Takahashi R; Shinohara G; Guo H; King DR; Kurokawa T; Gong JP Tough hydrogels with fast, strong, and reversible underwater adhesion based on a multiscale design. Advanced Materials 2018, 30 (32), 1801884. [DOI] [PubMed] [Google Scholar]
- (784).Yang J; Bai R; Chen B; Suo Z.Hydrogel adhesion: A supramolecular synergy of chemistry, topology, and mechanics. Adv Funct Mater 2020, 30 (2), 1901693. [Google Scholar]
- (785).Ghobril C; Grinstaff M.The chemistry and engineering of polymeric hydrogel adhesives for wound closure: a tutorial. Chemical Society Reviews 2015, 44 (7), 1820. [DOI] [PubMed] [Google Scholar]
- (786).Pizzi A; Mittal KL Handbook of adhesive technology, revised and expanded; CRC press, 2003. [Google Scholar]
- (787).Evans AG; Hutchinson JW; Wei Y.Interface adhesion: effects of plasticity and segregation. Acta Materialia 1999, 47 (15), 4093. [Google Scholar]
- (788).Raphael E; De Gennes P.Rubber-rubber adhesion with connector molecules. The Journal of Physical Chemistry 1992, 96 (10), 4002. [Google Scholar]
- (789).Gent A; Petrich R.Adhesion of viscoelastic materials to rigid substrates. Proceedings of the Royal Society of London. A. Mathematical and Physical Sciences 1969, 310 (1502), 433. [Google Scholar]
- (790).Yuk H; Zhang T; Lin S; Parada GA; Zhao X.Tough bonding of hydrogels to diverse nonporous surfaces. Nature Materials 2016, 15 (2), 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (791).Walia R; Akhavan B; Kosobrodova E; Kondyurin A; Oveissi F; Naficy S; Yeo GC; Hawker M; Kaplan DL; Dehghani F.Hydrogel− Solid Hybrid Materials for Biomedical Applications Enabled by Surface‐Embedded Radicals. Adv Funct Mater 2020, 2004599. [Google Scholar]
- (792).Mao S; Zhang D; Zhang Y; Yang J; Zheng J.A Universal Coating Strategy for Controllable Functionalized Polymer Surfaces. Adv Funct Mater 2020, 2004633. [Google Scholar]
- (793).Wei K; Chen X; Zhao P; Feng Q; Yang B; Li R; Zhang Z-Y; Bian L.Stretchable and Bioadhesive Supramolecular Hydrogels Activated by a One-Stone–Two-Bird Postgelation Functionalization Method. ACS applied materials & interfaces 2019, 11 (18), 16328. [DOI] [PubMed] [Google Scholar]
- (794).Liu Q; Nian G; Yang C; Qu S; Suo Z.Bonding dissimilar polymer networks in various manufacturing processes. Nature communications 2018, 9 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (795).Chen H; Liu Y; Ren B; Zhang Y; Ma J; Xu L; Chen Q; Zheng J.Super bulk and interfacial toughness of physically crosslinked double‐network hydrogels. Adv Funct Mater 2017, 27 (44), 1703086. [Google Scholar]
- (796).Liu J; Tan CSY; Scherman OA Dynamic interfacial adhesion through cucurbit [n] uril molecular recognition. Angewandte Chemie 2018, 130 (29), 8992. [DOI] [PubMed] [Google Scholar]
- (797).Gao Y; Wu K; Suo Z.Photodetachable adhesion. Advanced Materials 2019, 31 (6), 1806948. [DOI] [PubMed] [Google Scholar]
- (798).Liu X; Zhang Q; Duan L; Gao G.Tough Adhesion of Nucleobase‐Tackifed Gels in Diverse Solvents. Adv Funct Mater 2019, 29 (17), 1900450. [Google Scholar]
- (799).Fan H; Wang J; Tao Z; Huang J; Rao P; Kurokawa T; Gong JP Adjacent cationic–aromatic sequences yield strong electrostatic adhesion of hydrogels in seawater. Nature communications 2019, 10 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (800).Gao Y; Chen J; Han X; Pan Y; Wang P; Wang T; Lu T.A Universal Strategy for Tough Adhesion of Wet Soft Material. Adv Funct Mater 2020, 30 (36), 2003207. [Google Scholar]
- (801).Tian K; Bae J; Suo Z; Vlassak JJ Adhesion between hydrophobic elastomer and hydrogel through hydrophilic modification and interfacial segregation. ACS applied materials & interfaces 2018, 10 (49), 43252. [DOI] [PubMed] [Google Scholar]
- (802).Ji H; De Gennes P.Adhesion via connector molecules: the many-stitch problem. Macromolecules 1993, 26 (3), 520. [Google Scholar]
- (803).Yuk H; Lin S; Ma C; Takaffoli M; Fang NX; Zhao X.Hydraulic hydrogel actuators and robots optically and sonically camouflaged in water. Nature communications 2017, 8 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (804).Tamesue S; Endo T; Ueno Y; Tsurumaki F.Sewing hydrogels: adhesion of hydrogels utilizing in situ polymerization of linear polymers inside gel networks. Macromolecules 2019, 52 (15), 5690. [Google Scholar]
- (805).Takahashi R; Shimano K; Okazaki H; Kurokawa T; Nakajima T; Nonoyama T; King DR; Gong JP Tough particle‐based double network hydrogels for functional solid surface coatings. Advanced Materials Interfaces 2018, 5 (23), 1801018. [Google Scholar]
- (806).Cho H; Wu G; Jolly JC; Fortoul N; He Z; Gao Y; Jagota A; Yang S.Intrinsically reversible superglues via shape adaptation inspired by snail epiphragm. Proceedings of the National Academy of Sciences 2019, 116 (28), 13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (807).Yang SY; O’Cearbhaill ED; Sisk GC; Park KM; Cho WK; Villiger M; Bouma BE; Pomahac B; Karp JM A bio-inspired swellable microneedle adhesive for mechanical interlocking with tissue. Nature communications 2013, 4 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (808).Saiz‐Poseu J; Mancebo‐Aracil J; Nador F; Busqué F; Ruiz‐Molina D.The chemistry behind catechol‐based adhesion. Angewandte Chemie International Edition 2019, 58 (3), 696. [DOI] [PubMed] [Google Scholar]
- (809).Ahn BK; Das S; Linstadt R; Kaufman Y; Martinez-Rodriguez NR; Mirshafian R; Kesselman E; Talmon Y; Lipshutz BH; Israelachvili JN High-performance mussel-inspired adhesives of reduced complexity. Nature communications 2015, 6 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (810).Zhang W; Wang R; Sun Z; Zhu X; Zhao Q; Zhang T; Cholewinski A; Yang FK; Zhao B; Pinnaratip R.Catechol-functionalized hydrogels: biomimetic design, adhesion mechanism, and biomedical applications. Chemical Society Reviews 2020, 49 (2), 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (811).Kord Forooshani P; Lee BP Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. Journal of Polymer Science Part A: Polymer Chemistry 2017, 55 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (812).Suresh S.Fatigue of materials; Cambridge university press, 1998. [Google Scholar]
- (813).Bai R; Yang J; Morelle XP; Suo Z.Flaw‐Insensitive Hydrogels under Static and Cyclic Loads. Macromolecular rapid communications 2019, 40 (8), 1800883. [DOI] [PubMed] [Google Scholar]
- (814).Baker MI; Walsh SP; Schwartz Z; Boyan BD A review of polyvinyl alcohol and its uses in cartilage and orthopedic applications. Journal of Biomedical Materials Research Part B: Applied Biomaterials 2012, 100 (5), 1451. [DOI] [PubMed] [Google Scholar]
- (815).Bai R; Yang Q; Tang J; Morelle XP; Vlassak J; Suo Z.Fatigue fracture of tough hydrogels. Extreme Mechanics Letters 2017, 15, 91. [Google Scholar]
- (816).Li X; Cui K; Sun TL; Meng L; Yu C; Li L; Creton C; Kurokawa T; Gong JP Mesoscale bicontinuous networks in self-healing hydrogels delay fatigue fracture. Proceedings of the National Academy of Sciences 2020, 117 (14), 7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (817).Wang Z; Xiang C; Yao X; Le Floch P; Mendez J; Suo Z.Stretchable materials of high toughness and low hysteresis. Proceedings of the National Academy of Sciences 2019, 116 (13), 5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (818).Baumard T; Thomas A; Busfield J.Fatigue peeling at rubber interfaces. Plastics, rubber and composites 2012, 41 (7), 296. [Google Scholar]
- (819).Zhang W; Gao Y; Yang H; Suo Z; Lu T.Fatigue-resistant adhesion I. Long-chain polymers as elastic dissipaters. Extreme Mechanics Letters 2020, 100813. [Google Scholar]
- (820).Ni X; Chen C; Li J.Interfacial fatigue fracture of tissue adhesive hydrogels. Extreme Mechanics Letters 2020, 34, 100601. [Google Scholar]
- (821).Ushio K; Oka M; Hyon SH; Hayami T; Yura S; Matsumura K; Toguchida J; Nakamura T.Attachment of artificial cartilage to underlying bone. Journal of Biomedical Materials Research Part B: Applied Biomaterials: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 2004, 68 (1), 59. [DOI] [PubMed] [Google Scholar]
- (822).Zrinyi M; Barsi L; Büki A.Ferrogel: a new magneto-controlled elastic medium. Polymer Gels and Networks 1997, 5 (5), 415. [Google Scholar]
- (823).Zhang Q; Fang Z; Cao Y; Du H; Wu H; Beuerman R; Chan-Park MB; Duan H; Xu R.High refractive index inorganic–organic interpenetrating polymer network (IPN) hydrogel nanocomposite toward artificial cornea implants. Acs Macro Lett 2012, 1 (7), 876. [DOI] [PubMed] [Google Scholar]
- (824).Lü C; Yang B.High refractive index organic–inorganic nanocomposites: design, synthesis and application. Journal of Materials Chemistry 2009, 19 (19), 2884. [Google Scholar]
- (825).Zhang K; Ma C; He Q; Lin S; Chen Y; Zhang Y; Fang NX; Zhao X.Metagel with Broadband Tunable Acoustic Properties Over Air–Water–Solid Ranges. Adv Funct Mater 2019, 29 (38), 1903699. [Google Scholar]
- (826).Taylor DL; in het Panhuis M.Self‐healing hydrogels. Advanced Materials 2016, 28 (41), 9060. [DOI] [PubMed] [Google Scholar]
- (827).Guo Y; Bae J; Fang Z; Li P; Zhao F; Yu G.Hydrogels and Hydrogel-Derived Materials for Energy and Water Sustainability. Chemical Reviews 2020. [DOI] [PubMed] [Google Scholar]
- (828).Takahashi R; Sun TL; Saruwatari Y; Kurokawa T; King DR; Gong JP Creating stiff, tough, and functional hydrogel composites with low‐melting‐point alloys. Advanced Materials 2018, 30 (16), 1706885. [DOI] [PubMed] [Google Scholar]
- (829).Shin SR; Jung SM; Zalabany M; Kim K; Zorlutuna P; Kim S. b.; Nikkhah M; Khabiry M; Azize M; Kong J.Carbon-nanotube-embedded hydrogel sheets for engineering cardiac constructs and bioactuators. ACS Nano 2013, 7 (3), 2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (830).Zhang S; Chen Y; Liu H; Wang Z; Ling H; Wang C; Ni J; Çelebi‐Saltik B; Wang X; Meng X.Room‐Temperature‐Formed PEDOT: PSS Hydrogels Enable Injectable, Soft, and Healable Organic Bioelectronics. Advanced Materials 2020, 32 (1), 1904752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (831).Inoue A; Yuk H; Lu B; Zhao X.Strong adhesion of wet conducting polymers on diverse substrates. Science advances 2020, 6 (12), eaay5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (832).Shi Y; Wang M; Ma C; Wang Y; Li X; Yu G.A conductive self-healing hybrid gel enabled by metal–ligand supramolecule and nanostructured conductive polymer. Nano letters 2015, 15 (9), 6276. [DOI] [PubMed] [Google Scholar]
- (833).Shi Y; Ma C; Peng L; Yu G.Conductive “smart” hybrid hydrogels with PNIPAM and nanostructured conductive polymers. Adv Funct Mater 2015, 25 (8), 1219. [Google Scholar]
- (834).Yuk H; Lu B; Lin S; Qu K; Xu J; Luo J; Zhao X.3D printing of conducting polymers. Nature Communications 2020, 11 (1), 1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (835).Kim Y; Parada GA; Liu S; Zhao X.Ferromagnetic soft continuum robots. Science Robotics 2019, 4 (33), eaax7329. [DOI] [PubMed] [Google Scholar]
- (836).Jeon S; Hoshiar AK; Kim K; Lee S; Kim E; Lee S; Kim J.-y.; Nelson BJ; Cha H-J; Yi BJ et al. A Magnetically Controlled Soft Microrobot Steering a Guidewire in a Three-Dimensional Phantom Vascular Network. Soft Robotics 2018, 6 (1), 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (837).Edelmann J; Petruska AJ; Nelson BJ Magnetic control of continuum devices. The International Journal of Robotics Research 2017, 36 (1), 68. [Google Scholar]
- (838).Hu W; Lum GZ; Mastrangeli M; Sitti M.Small-scale soft-bodied robot with multimodal locomotion. Nature 2018, 554, 81. [DOI] [PubMed] [Google Scholar]
- (839).Xu T; Zhang J; Salehizadeh M; Onaizah O; Diller E.Millimeter-scale flexible robots with programmable three-dimensional magnetization and motions. Science Robotics 2019, 4 (29), eaav4494. [DOI] [PubMed] [Google Scholar]
- (840).Wang L; Kim Y; Guo CF; Zhao X.Hard-magnetic elastica. Journal of the Mechanics and Physics of Solids 2020, 142, 104045. [Google Scholar]
- (841).Kim Y; Yuk H; Zhao R; Chester SA; Zhao X.Printing ferromagnetic domains for untethered fast-transforming soft materials. Nature 2018, 558 (7709), 274. [DOI] [PubMed] [Google Scholar]
- (842).de Groot JH; van Beijma FJ; Haitjema HJ; Dillingham KA; Hodd KA; Koopmans SA; Norrby S.Injectable Intraocular Lens Materials Based upon Hydrogels. Biomacromolecules 2001, 2 (3), 628. [DOI] [PubMed] [Google Scholar]
- (843).Choi M; Humar M; Kim S; Yun SH Step‐index optical fiber made of biocompatible hydrogels. Advanced Materials 2015, 27 (27), 4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (844).Blaiszik BJ; Kramer SL; Olugebefola SC; Moore JS; Sottos NR; White SR Self-healing polymers and composites. Annual review of materials research 2010, 40, 179. [Google Scholar]
- (845).Han L; Yan L; Wang K; Fang L; Zhang H; Tang Y; Ding Y; Weng L-T; Xu J; Weng J.Tough, self-healable and tissue-adhesive hydrogel with tunable multifunctionality. NPG Asia Materials 2017, 9 (4), e372. [Google Scholar]
- (846).Zhang H; Wang C; Zhu G; Zacharia NS Self-Healing of Bulk Polyelectrolyte Complex Material as a Function of pH and Salt. ACS Applied Materials & Interfaces 2016, 8 (39), 26258. [DOI] [PubMed] [Google Scholar]
- (847).Kurt B; Gulyuz U; Demir DD; Okay O.High-strength semi-crystalline hydrogels with Self-Healing and shape memory functions. European Polymer Journal 2016, 81, 12. [Google Scholar]
- (848).Neal JA; Mozhdehi D; Guan Z.Enhancing mechanical performance of a covalent self-healing material by sacrificial noncovalent bonds. Journal of the American Chemical Society 2015, 137 (14), 4846. [DOI] [PubMed] [Google Scholar]
- (849).Liu X; Yuk H; Lin S; Parada GA; Tang TC; Tham E; de la Fuente‐Nunez C; Lu TK; Zhao X.3D printing of living responsive materials and devices. Advanced Materials 2018, 30 (4), 1704821. [DOI] [PubMed] [Google Scholar]
- (850).Yamaguchi M; Ono S; Okamoto K.Interdiffusion of dangling chains in weak gel and its application to self-repairing material. Materials Science and Engineering: B 2009, 162 (3), 189. [Google Scholar]
- (851).Sirajuddin N; Jamil M.Self-healing of poly (2-hydroxyethyl methacrylate) hydrogel through molecular diffusion. Sains Malaysiana 2015, 44 (6), 811. [Google Scholar]
- (852).Zhang H; Xia H; Zhao Y.Poly(vinyl alcohol) Hydrogel Can Autonomously Self-Heal. Acs Macro Lett 2012, 1 (11), 1233. [DOI] [PubMed] [Google Scholar]
- (853).Matsuda T; Kawakami R; Namba R; Nakajima T; Gong JP Mechanoresponsive self-growing hydrogels inspired by muscle training. Science 2019, 363 (6426), 504. [DOI] [PubMed] [Google Scholar]
- (854).Sun TL; Kurokawa T; Kuroda S; Ihsan AB; Akasaki T; Sato K; Haque MA; Nakajima T; Gong JP Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nature Materials 2013, 12 (10), 932. [DOI] [PubMed] [Google Scholar]