

Graphical abstract
Keywords: Gene therapy, ex vivo, retroviral vectors, stem cells, clinical approvals
Abstract
Retroviral gene therapy has emerged as a promising therapeutic modality for multiple inherited and acquired human diseases. The capability of delivering curative treatment or mediating therapeutic benefits for a long-term period following a single application fundamentally distinguishes this medical intervention from traditional medicine and various lentiviral/γ-retroviral vector-mediated gene therapy products have been approved for clinical use. Continued advances in retroviral vector engineering, genomic editing, synthetic biology and immunology will broaden the medical applications of gene therapy and improve the efficacy and safety of the treatments based on genetic correction and alteration. This review will summarize the advent and clinical translation of ex vivo gene therapy, with the focus on the milestones during the exploitation of genetically engineered hematopoietic stem cells (HSCs) tackling a variety of pathological conditions which led to marketing approval. Finally, current statue and future prospects of gene editing as an alternative therapeutic approach are also discussed.
Abbreviations
- RSV
Rous sarcoma virus
- HbS
hemoglobin S
- CMV
cytomegalovirus
- HSCs
hematopoietic stem cells
- GT
gene therapy
- NIH
National Institutes of Health
- EMA
European Medicines Agency
- FDA
Food and Drug Administration
- γ-RVs
gamma-retroviral vectors
- LVs
lentiviral vectors
- SIN-LV
self-inactivate lentiviral vector
- LTR
long terminal repeat
- CAR
chimeric antigen receptor
- ADA
adenosine deaminase
- SCID
severe combined immunodeficiency
- ALL
acute lymphoblastic leukemia
- DLBCL
diffuse large B-cell lymphoma
- FL
follicular lymphoma
- TDT
transfusion dependent β-thalassemia
- HBA
hemoglobin subunit alpha
- HBB
hemoglobin subunit beta
- HBD
hemoglobin subunit delta
- HBE
hemoglobin subunit epsilon
- HBG
hemoglobin subunit gamma
- r/r
relapsed or refractory
- MM
multiple myeloma
- MCL
mantle cell lymphoma
- BCMA
B cell maturation antigen
- GFRt
truncated growth factor receptor
- AAVs
adeno-associated viruses
- Ads
adenoviruses
- MoMLV
Moloney murine leukemia virus
- allo-BMT
allogeneic bone marrow transplantation
- allo-HSCT
allogeneic HSCs transplantation
- GvHDs
graft-versus-host diseases
- PIDs
primary immunodeficiencies
- SCID-X1
X-linked SCID
- ADA-SCID
adenosine deaminase deficiency SCID
- T-ALL
T-cell acute lymphoblastic leukemia
- AEs
adverse events
- HLA
human leukocyte antigen
- GSK
GlaxoSmithKline
- WAS
Wiskott-Aldrich syndrome
- CGD
chronic granulomatous disease
- IL2RG
interleukin 2 receptor subunit gamma
- RAG1
recombination activating gene-1
- RAG2
recombination activating gene-2
- WASP
Wiskott-Aldrich syndrome protein
- ROS
reactive oxygen species
- NADPH
nicotinamide adenine dinucleotide phosphate hydrogen
- LT-HSCs
long-term hematopoietic stem cells
- ST-HSCs
short-term hematopoietic stem cells
- MPP
multipotent progenitor
- CLP
common lymphoid progenitor
- CMP
common myeloid progenitor
- MEP
megakaryocytic erythroid progenitor
- GMP
granulomonocytic progenitor
- BBB
blood brain barrier
- RBC
red blood cell
- FANC
Fanconi anemia complementation group
- CD40LG
CD40 ligand
- DCLREIC
DNA cross-link repair 1C
- FOXP3
forkhead box protein P3
- PRF1
perforin 1
- BTK
Bruton tyrosine kinase
- F8
coagulation factor VIII
- F9
coagulation factor IX
- F10
coagulation factor X
- PKLR
pyruvate kinase L/R
- RPS19
ribosomal protein S19
- ELANE
elastase, neutrophil expressed
- CYBA
cytochrome B-245 alpha chain
- CYBB
cytochrome B-245 beta chain
- NCF1
neutrophil cytosolic factor 1
- ITGB2
integrin subunit beta 2
- ABCD1
ATP binding cassette subfamily D member 1
- ARSA
arylsulfatase A
- IDS
iduronate 2-sulfatase
- IDUA
alpha-L-iduronidase
- MPD
methyl parathion degrading
- GALC
galactosylceramidase
- SGSH
N-sulfoglucosamine sulfohydrolase
- NAGLU
N-acetyl-alpha-glucosaminidase
- HGSNAT
heparan-alpha-glucosaminide N-acetyltransferase
- GNS
glucosamine (N-acetyl)-6-sulfatase
- GALNS
galactosamine (N-acetyl)-6-sulfatase
- GLB1
beta-galactosidase-1
- ARSB
arylsulfatase B
- GUSB
glucuronidase beta
- ASAH1
N-acylsphingosine amidohydrolase 1
- NAGA
alpha-N-acetylgalactosaminidase
- GLA
alpha-galactosidase A
- GAA
alpha-glucosidase
- GBA
glucosylceramidase beta
- SCD
sickle cell disease
- LCR
locus control region
- HSs
hypersensitive sites
- Hb
blood hemoglobin
- AML
acute myeloid leukemia
- MDS
myelodysplastic syndrome
- iNMDs
inherited neurometabolic disorders
- CNS
central nervous system
- LSDs
lysosomal storage disorders
- M6P
mannose-6-phosphate
- X-ALD
X-linked adrenoleukodystrophy
- CALD
cerebral adrenoleukodystrophy
- MLD
metachromatic leukodystrophy
- MPSI
mucopolysaccharidosis Type I
- MPS IIIA
mucopolysaccharidosis Type IIIA
- NINDS
National Institute of Neurological Disorders and Stroke
- EB
Epidermolysis bullosa
- ESCs
epidermal stem cells
- MEFs
mouse embryonic fibroblasts
- EBS
EB simplex
- JEB
junctional EB
- DEB
dystrophic EB
- KEB
Kindler EB
- BMZ
epidermal basement membrane zone
- scFv
single-chain variable fragment
- MHC
major histocompatibility complex
- TCRs
T cell receptors
- CLL
chronic lymphocytic leukemia
- iPSCs
induced pluripotent stem cells
- RCV
replication competent virus
- VSV-G
vesicular stomatitis virus glycoprotein
- RRE
Rev responsive element
- cPPT/CTS
central polypurine tract/central termination sequence
- WPRE
woodchuck post-transcriptional regulatory element
- ZFNs
zinc-finger nucleases
- TALENs
transcription activator-like effector nucleases
- CRISPR/Cas
clustered regularly interspaced short palindromic repeats/CRISPR-associated protein
- PD-1
programmed cell death protein 1
- CISH
cytokine-induced SH2 protein
- B2M
β-2 microglobin
- DSBs
double-strand breaks
- NHEJ
non-homologous end joining
- HDR
homology directed repair
- NSCLC
non-small cell lung cancer
- HPFH
hereditary persistence of fetal hemoglobin
- HbF
fetal hemoglobin
- HbA
adult hemoglobin
- LAM-PCR
linear amplification-mediated PCR
1. Introduction
Gene therapy (GT) is the transfer of genetic material to target cells to treat disease [1], [2], through restoration of cell function in inherited abnormalities or rendering cells with new capability in acquired disorders. Dating back to 1953, elucidating the double helix structure of DNA by Watson&Crick [3] profoundly and naturally led to the question regarding the possibility of pinpointing and correcting faulty genes to cure genetic diseases, which underlies the early-stage conceptualization of gene therapy. With rapid growth of recombinant DNA technology during the 1960s [4], [5], the concept and approach of molecular medicine using DNA as therapeutic agent to replace defective genes continued to evolve and became increasingly concrete and attainable [6], [7], [8]. In 1972, T. Friedmann and R. Roblin comprehensively reviewed and addressed the immense potential of GT as well as various key bottlenecks for developing this revolutionary personalized medicine [8]. Translation started in 1990, when the first approved clinical study took place at the US National Institutes of Health (NIH) for a rare immunodeficiency disorder [9]. However, a number of these early clinical studies, including the first NIH trial, could not provide indisputable therapeutic results but exposed unexpected, in several cases devastating, therapy-related toxicities [7], [10]. Fortunately, these setbacks and impediments fueled more basic research and directed the focus on the biology governing the efficacy and safety of gene therapy, ultimately giving rise to successful translation in the 2000s. As a result, multiple GT products obtained approval for marketing (shown in Table 1) and more than 5,000 gene therapy trials worldwide have been registered in clinicaltrials.gov (accessed on 25th May 2022).
Table 1.
Ex vivo gene therapy products that have acquired marketing authorization.
| Product | Developer | Approval | Indication | Mechanism of action | Clinical trials | Price |
|---|---|---|---|---|---|---|
| STRIMVELIS® | GlaxoSmithKline | May 2016 (EMA) | ADA-SCID | Autologous HSCs expressing ADA; γ-RV-ADA |
NCT00598481; NCT0059978 | 594,000 $ |
| KYMRIAH® Tisagenlecleucel | Novartis | Aug 2017 (FDA) |
B-cell acute lymphoblastic leukemia (ALL), r/r diffuse large B-cell lymphoma (DLBCL) | Autologous CAR T cell targeting CD19; SIN-LV-CAR |
NCT02435849; NCT02445248; NCT02228096; NCT02445222; NCT01626495; NCT01029366; NCT01747486; NCT02030847; NCT02030834; NCT02135406 | 475,000 $ |
| YESCARTA® Axicabtagene Ciloleucel | Kite Pharma (Gilead) |
Oct 2017 (FDA) |
Non-Hodgkin lymphoma, B-cell lymphoma, high grade B-cell lymphoma and DLBCL arising from follicular lymphoma (FL) | Autologous CAR T cell targeting CD19; γ-RV-CAR |
NCT02348216 | 373,000 $ |
| ZYNTEGLOTM Betibeglogene Autotemcel | Bluebird bio | May 2019 (EMA) |
Transfusion dependent β-thalassemia (TDT) | Autologous HSCs expressing HBB; SIN-LV-HBB |
NCT01745120; NCT02151526 | ∼1.8 million $ |
| LIBMELDYTM Atidarsagene Autotemcel | Orchard Therapeutics | Dec 2020 (EMA) |
Early-onset metachromatic leukodystrophy (MLD) | Autologous HSCs expressing ARSA; SIN-LV-ARSA |
Eudract 2009-017349-77 (Study 201222) |
∼3.9 million $ |
| BREYANZI® Lisocabtagene Maraleucel |
Juno Therapeutics (Bristol-Myers Squibb) |
Feb 2021 (FDA) |
r/r large B cell lymphoma | Autologous CAR T cell targeting CD19; SIN-LV-CAR* |
NCT02631044; NCT03484702; NCT03744676; NCT03310619; NCT03483103; NCT03331198; NCT03743246; NCT03435796 | ∼ 432,055 $ |
| ABECMA® Idecabtagene Vicleucel | Celgene (Bristol-Myers Squibb) |
Mar 2021 (FDA) |
r/r multiple myeloma (MM) | Autologous CAR T cell targeting BCMA; SIN-LV-CAR |
NCT03361748; NCT03601078; NCT03651128; NCT03435796; NCT02658929; NCT02786511 | 419,500 $ |
| TECARTUS® Brexucabtagene Autoleucel | Kite Pharma (Gilead) |
Oct 2021 (FDA) |
r/r mantle cell lymphoma (MCL), r/r B-cell precursor ALL | Autologous CAR T cell targeting CD19; γ-RV-CAR# |
NCT02601313, NCT02614066, NCT02625480, NCT03624036, NCT04162756 | 373,000 $ |
| CARVYKTITM Ciltacabtagene Autoleucel | Janssen Biotech | Feb 2022 (FDA) |
r/r MM | Autologous CAR T cell targeting BCMA; SIN-LV-CAR |
NCT03548207; NCT04133636; NCT04181827 | 465,000 $ |
*The BREYANZI SIN-LV-CAR includes a nonfunctional truncated growth factor receptor (GFRt) that is co-expressed with CD19-specific CAR.
#The γ-RV-CAR applied in TECARTUS is identical to that in YESCARTA, with TECARTUS differing from YESCARTA in that T cell enrichment is performed in TECARTUS manufacture but not in YESCARTA.
γ-RV: Gamma-retroviral vector; SIN-LV: Self-inactivate lentiviral vector; CAR: Chimeric antigen receptor; ADA: Adenosine deaminase; SCID: Severe combined immunodeficiency; r/r: Relapsed or refractory; HBB: Hemoglobin subunit beta; ARSA: Arylsulfatase A; BCMA: B cell maturation antigen.
Treating diseases via gene therapy is achieved through efficient transfer and expression of the therapeutic genetic material, namely nucleic acids DNA or RNA, into the cells of interest. As the plasma membrane or endosome represents impermeable barrier for any large polyanion molecule [11], gene transfer must be conducted either by injection or electroporation (physical method), or facilitated by cationic lipids or polymers coating (chemical method), or utilize biological tools such as viral vectors [11]. Apparently, viral vectors have been chosen in the vast majority of GT-related clinical studies as well as most of the early research and development. Indeed, with the remarkable ability of introducing their genetic material into target cells during the replicative cycle, relying on just few truly simplified protein molecules encoded in the viral genome, viruses are an ideal gene delivery tool of high efficiency. Three viral vector platforms derived from retroviruses, adeno-associated viruses (AAVs) and adenoviruses (Ads) comprise around 80% of all GT applications in development (source: pharmaintelligence.informa.com).
Basically, gene therapy can be broadly divided into two classes: ex vivo and in vivo. Ex vivo gene therapy involves the harvesting of target cells from patient/third party’s body, followed by subsequent viral transduction to genetically modify the harvested cells by introducing functional copies of a desired gene or by gene editing. The transduced cells are then reinfused to the patient to correct the phenotype of the disease. Integrating retrovirus derived vectors are typically involved in the manipulation of hematopoietic or other types of stem cells in ex vivo gene therapy [12], [13], [14], [15], [16]. Conversely, AAVs and Ads are more suited for in vivo application, where the therapeutic payload is delivered into postmitotic cells and maintained in the cells as episomes [17], [18]. For the last four decades, retrovirus mediated ex vivo GT has been a major player in the field of gene therapy [19]. In early 1980s when moloney murine leukemia virus (MoMLV) was initially vectorized, yielded gamma-retroviral vectors (γ-RVs) became the first genetic tool to permit an efficient gene transfer and stable integration in mammalian cells [12], [13], [15], [20], [21]. And in 1991, γ-RV was the first vector used in the NIH clinical trial [9], and a decade later, in the first ever successful clinical study that literally achieved the cure of a genetic disease [22]. In this review, we provide historical and biological perspectives on the advent and clinical implementation of ex vivo retroviral gene therapy, focusing on the milestones during the development of HSCs-based GTs and immunotherapies that led to the clinical approvals. In the end, the rapid growth and future prospects of gene editing in the application of ex vivo gene therapy are also covered for discussion.
2. Ex vivo gene therapy
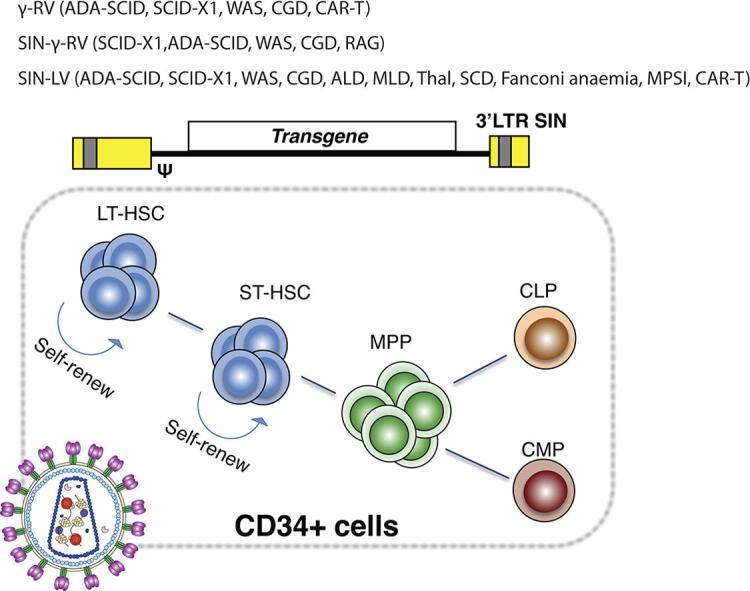
Largely due to decades of clinical management of diverse blood disorders and hematologic malignancies by applying allogeneic bone marrow transplantation (allo-BMT), the population of HSCs was chosen as the first and primary target for ex vivo gene therapy [23]. After eradication of abnormal blood/immune cells, HSCs transplantation is undertaken to reconstitute the blood/immune system and subsequently establish sustainable hematopoiesis. However, despite allogeneic HSCs transplantation (allo-HSCT) being implemented as a standard treatment [24], engineered autologous transplantation potentially exhibits multiple advantages over allogeneic approach [25], [26], such as requiring no histocompatible donor, eliminating the complications of graft-versus-host diseases (GvHDs), avoiding immunosuppressant lifelong administration, among others. Therefore, genetically modifying autologous HSCs appears to be the most promising application in the field of gene therapy tackling hematologic disorders and immune aberrations (Fig. 1).
Fig. 1.

Overview of the hematopoietic hierarchy and genetic disorders potentially curable via ex vivo gene therapy. The cell surface marker CD34 is used to enrich a mixture of HSCs for gene therapy. HSCs progressively acquire lineage specifications and differentiate into lineage-committed progenitors and eventually terminally differentiated cells. Examples of ex vivo gene therapy under investigation or with translational potential are represented along with affected cell types. LT-HSCs: long-term hematopoietic stem cells; ST-HSCs: short-term hematopoietic stem cells; MPP: multipotent progenitor; CLP: common lymphoid progenitor; CMP: common myeloid progenitor; MEP: megakaryocytic erythroid progenitor; GMP: granulomonocytic progenitor; BBB: blood brain barrier; RBC: red blood cell; FANC: Fanconi anemia complementation group; ALL: acute lymphoblastic leukemia; SCID: severe combined immunodeficiency; ADA: adenosine deaminase; CD40LG: CD40 ligand; DCLREIC: DNA cross-link repair 1C; FOXP3: forkhead box protein P3; IL2RG: interleukin 2 receptor subunit gamma; PRF1: perforin 1; RAG1: recombination activating 1; WASP: Wiskott-Aldrich syndrome protein; BTK: Bruton tyrosine kinase; F8: coagulation factor VIII; F9: coagulation factor IX; F10: coagulation factor X; HBA: hemoglobin subunit alpha; HBB: hemoglobin subunit beta; PKLR: pyruvate kinase L/R; RPS19: ribosomal protein S19; ELANE: elastase, neutrophil expressed; CYBA: cytochrome B-245 alpha chain; CYBB: cytochrome B-245 beta chain; NCF1: neutrophil cytosolic factor 1; ITGB2: integrin subunit beta 2; ABCD1: ATP binding cassette subfamily D member 1; ARSA: arylsulfatase A; IDS: iduronate 2-sulfatase; IDUA: alpha-L-iduronidase; MPD: methyl parathion hydrolase; GALC: galactosylceramidase; SGSH: N-sulfoglucosamine sulfohydrolase; NAGLU: N-acetyl-alpha-glucosaminidase; HGSNAT: heparan-alpha-glucosaminide N-acetyltransferase; GNS: glucosamine (N-acetyl)-6-sulfatase; GALNS: galactosamine (N-acetyl)-6-sulfatase; GLB1: beta-galactosidase-1; ARSB: arylsulfatase B; GUSB: glucuronidase beta; ASAH1: N-acylsphingosine amidohydrolase 1; NAGA: alpha-N-acetylgalactosaminidase; GLA: α-galactosidase A; GAA: alpha-glucosidase; GBA: glucosylceramidase beta. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.1. Severe combined immunodeficiency (SCID)
SCID is the most severe form of primary immunodeficiencies (PIDs), with an estimated incidence between 1:40,000 to 1:75,000 live births [27], [28]. Initially known as swiss-type agammaglobulinemia, SCID syndromes are characterized by the profound deficiency of T cells combined with varied abnormalities in B and NK cells. More than 20 different genes have been implicated in SCIDs [27] and the most common types are X-linked SCID (SCID-X1) and adenosine deaminase deficiency SCID (ADA-SCID), accounting for 30–40% and 15%-20% of all diagnosed [28], [29], [30], respectively. Therapeutic interventions consist of allo-HSCT [31], [32], enzyme replacement therapy and theoretically, gene therapy to correct or alleviate underlying genetic defects.
In the history of gene therapy, pioneer clinical trials were predominately driven by the goal to treat life-threatening SCIDs, counting on the vectorization of MoMLV to generate functional vehicles for gene transfer (1st generation γ-RVs). In 1990, the very first officially authorized gene therapy trial to restore ADA deficiency was conducted at the NIH, in which autologous T cells were collected from peripheral blood and transduced by γ-RV carrying human 1.5 kb ADA cDNA [9], [33], [34]. Although prolonged survival of genetically modified T lymphocytes had been observed after a decade [35], it is not possible to determine the clinical benefit from this treatment of ADA gene augmentation GT due to in parallel deployed enzyme replacement therapy.
Later on, in the late 1990s, a clinical study conducted by M. Cavazzana-Calvo and colleagues from Necker Hospital in Paris [22], [36] and Great Ormond Street Hospital in London [37] illustrated convincing therapeutic effects of gene therapy treating SCID-X1, representing the first clear GT success in human trial [23], [25], [38]. SCID-X1 is caused by loss-of-function mutations in the gene encoding the γc subunit of cytokine receptors like IL-2, -4, -7, -9, -15 and IL-21, causing the depletion of circulating T and NK cells and poorly performing B lymphocytes [29]. In these SCID-X1 trials, CD34+ cells were purified from bone marrow from 20 patients and transduced by γ-retroviruses with the expression cassette of IL2RG gene encoding γc subunit to reconstitute a range of cytokine receptors affected. Long-term immune correction and restoration of the primary immunodeficiency have been observed in most of the treated children, with substantial T-cell recovery within 2–5 months for 19/20 patients, including the capabilities of clearing of SCID-related infections, responding to standard childhood vaccinations and gain in growth [37], [39], [40], [41]. Unfortunately, T-cell acute lymphoblastic leukemia (T-ALL) developed in 6 patients 2–14 years after the treatment and one of whom did not respond well to chemotherapy and died [39], [40], [41]. In-depth analysis revealed T-ALLs were caused by insertional mutagenesis, resulted from provoking unconstrained activation of proto-oncogenes (e.g., LMO-2, CCND2, MECOM) due to integrated strong intact viral enhancer/promoter of γ-RV in proximity [42], [43], [44].
Thus, collective efforts have been accordingly devoted to improving the first generation γ-RVs to minimize the risk of adverse events (AEs), mainly through the deletion of the viral enhancer/promoter elements of the 3′-LTR to attain self-inactivate (SIN) γ-RVs and driving the expression of IL2RG transgene by other promoters, such as EF1a instead [45]. New generation of vector was tested in multiple phase I/II trials for the infant patients with confirmed mutation in IL2RG gene [45], [46], [47] and lacking HLA-identical donor or being refractory to standard therapy. The data published in a preliminary report in 2014 (NCT01410019, NCT01175239, and NCT01129544) [45] indicated comparable recoveries between the previous clinical trials applying 1st generation γ-RV and the ongoing trials adopting SIN-γ-RV system. So far, no leukemia in any of the patients has been reported, suggesting encouraging improvement in the safety as a result of the SIN-principle implementation [48], [49], [50], [51]. Meantime, during the period of γ-RVs system advancing and evolving, the SIN concept has also been introduced to improve the safety of lentiviral vectors (LVs), another retroviral vector platform that generally exhibiting safer integration pattern as well as better transduction efficiency in quiescent HSCs [52], [53], [54], [55], [56]. Very recently, multiple trials based on SIN-LVs for SCID-X1 gene therapy have been conducted and to date, no oncogenesis has been reported, albeit with relatively short follow-up [29], [39], [48], [57], [58], [59], [60].
Further on, for ADA-SCID, although the initial study failed to generate unambiguous beneficial results as described above, irrefutable success was achieved in the clinical trials performed at San Raffaele Hospital in Milan and Hadassah Hospital in Jerusalem in the early 2000s [61]. Combining the utilization of busulfan to promote the cellular engraftment and the discontinuation of enzyme replacement treatment for endowing survival advantage to genetically modified cells, autologous ex vivo gene therapy was demonstrated to be a safe, effective and durable intervention for SCID patients with ADA deficiency [62]. Notably, dissecting integration profiles found little difference between γ-RVs mediated ADA insertion and that of IL2RG, but the insertional mutagenesis that overshadowing the SCID-X1 trials were never observed in the ADA-trials, implying apart from LTR, the transgene product itself could involve in or even contribute to carcinogenesis [63]. In 2016, the treatment of ADA gene transfer mediated by γ-RV received EMA approval and became the first marketed ex vivo gene therapy in Europe [64], [65]. The product, Strimvelis® (GSK2696273), has been commercially manufactured by the GlaxoSmithKline (GSK) based in Raffaele Hospital [32], [64]. Furthermore, excellent results have been reported lately with SIN-lentiviral based approach for ADA-SCID treatment (NCT01852071, NCT02999984, and NCT01380990) [24], [66], showing robust immune reconstitution, sustained metabolic detoxification and recovered ADA activity level.
2.2. Other PIDs
PIDs are a heterogeneous group of more than 450 rare, chronic conditions associated with impaired immune development and function, globally affecting 1:10,000 live births [67], [68], [69], [70], [71]. More than 260 genes have been elucidated underlying the pathogenesis of PIDs and new genetic defects are continually identified because of improved diagnostic capability based on next-generation sequencing technologies [72]. Similar to SCIDs, other types of PIDs that can be treated by allo-HSCT are in principle eligible for autologous HSCs gene therapy [67], [68], [70], [72], such as Wiskott-Aldrich syndrome (WAS), chronic granulomatous disease (CGD), recombination activating gene severe combined immunodeficiency (RAG1, RAG2) and so on. For instance, WAS is a rare X-linked recessive PID disorder characterized by recurrent infections and elevated manifestations of autoimmune diseases and tumors. It caused by the mutations in the gene WAS encoding Wiskott-Aldrich syndrome protein (WASP) [73], [74], [75]. The first attempt to treat WAS by gene therapy was done in Germany in 2006 [75], [76]. Sustained and therapeutic WASP expression delivered by γ-RV system was achieved in transduced HSCs, myeloid and lymphoid cells, and platelets after transplantation, however, unacceptable leukaemic toxicity occurred. Recently, adopting more advanced vehicle platform showed some encouraging progresses in multiple clinical studies [73], [74], [77], [78]. Of note, SIN lentiviral vector incorporating a 1.6 kb fragment from the promoter region of WAS gene has been proved capable of mediating considerably high WASP expression, and abrogation of bleeding and sustained immunofunction were achieved with no clonal imbalance and dysregulation detected [21], [79], [80].
CGD is an another type of PID disorder, which affecting the ability of phagocytes to destruct ingested pathogens due to the incapability to generate reactive oxygen species (ROS) for germ killing [81], [82], [83], [84]. Patients with CGDs are highly susceptible to frequent and sometimes lethal opportunistic bacterial and fungal infections. For ROS production, phagocyte NADPH oxidase is first assembled by three cytosolic phox subunits, p47phox, p67phox and p40phox, and then translocated to the membrane-bound flavocytochrome, consisting of subunits gp91phox and p22phox. The most predominant form of CGDs is an X-linked condition (X-CGD) caused by the mutations in CYBB, the gene encoding the subunit gp91phox, and the rest forms of CGDs are autosomal recessive, from the mutations in CYBA, NCF1, NCF2 and NCF4, which encode p22phox, p47phox, p67phox and p40phox, respectively. Early ex vivo GT clinical trials applying γ-RVs to correct defective gp91phox and p47phox were mostly disappointing due to the lack of therapeutic efficacy [85], [86], [87] for various reasons. In the early trial studies that were initiated in the mid-1990s, bone marrow conditioning or myelosuppression was not performed prior to transplantation to make space for the genetically modified cells to engraft. Then later, other factors undermining the efficacy of GT treatment have been revealed. Persistent chronic inflammation typically associated with CGD causes the viral promoter methylation, leading to transgene expression silencing [88]. In addition, the hematopoietic proliferative stress induced by chronic inflammation reduces the fitness of harvested HSCs [89], further lowering viral transduction efficiency and engraftment. Moreover, some trials based on γ-RVs delivery system were complicated by the development of myelodysplasia and leukemia-like growth of blood cells [84], [85], [88], [90]. Notably, however, introducing SIN-LV system with some added regulatory elements resistant to epigenetic modification was recently exploited, which has shown some promising therapeutic results in several patients with severe X-linked CGDs (NCT02234934, NCT01855685) [91].
In short, the multiple clinical trials for treating SCIDs demonstrated the curative potential of gene therapy and the lesson learned from tackling SCIDs not only provided valuable guidance but also generated momentum for pursuing the translational research for a variety of PIDs [23], [68], [70], [71], [92], [93]. The properties of multipotency and readiness for in vitro manipulation and repopulation have placed HSCs at the frontline of genetically engineering ex vivo therapy (indicated in Fig. 1).
2.3. β-Hemoglobinopathies
Sickle cell anemia (also known as sickle cell disease, SCD) and β-thalassemia are two of the most common monogenic diseases [94], both caused by the mutation in the gene encoding the adult β-globin chain of hemoglobin (HBB, hemoglobin subunit beta), with high prevalence primarily found in South Asia, Middle East, Mediterranean and North Africa [95], [96], [97], [98]. SCD is characterized by a single mutation that leads to negatively charged glutamic acid replaced by hydrophobic valine [99], producing defective peptide of hemoglobin S (HbS) prone to polymerization. As a result, it causes erythrocyte distortion, hemolysis, anemia, vaso-occlusive episodes and irreversible endoorgan damage [100]. In contrast, almost 300 pathological mutations have been identified underlying β-thalassemia etiology (http://globin.cse.psu.edu), which impede the transcription, RNA processing, or translation of HBB [101], [102], [103]. The clinical presentation of β-thalassaemia varies from being mild asymptomatic to seriously severe disease, and for the latter ineffective erythropoiesis may require constant blood transfusion and iron chelation to sustain life.
Allo-HSCT can be curative for β-hemoglobinopathies, however, less than 20% of eligible patients have HLA-matched donors [100], [102]. Being the most prevalent inherited diseases worldwide [104], both SCD and β-thalassemia have been a key focus of gene therapy to attain corrected autologous HSCs. However, the main clinical challenge to tackle β-hemoglobin aberration genetically is to express functional protein in erythroid specific lineage and in substantial amounts to reach therapeutic benefits [101]. Early studies on the ectopic expression of β-globin gene (HBB) as well as some forms of β-thalassemia led to the discovery of the locus control region (LCR) that is critical for mediating erythroid lineage specific and genomic position-independent HBB expression [105], [106], [107], [108], [109], [110]. Human β-globin locus, approximately 70 kb in length, comprises five β-like globin genes, linearly arranged in the order they are expressed during development: ε (HBE1), γG (HBG2), γA (HBG1), δ (HBD), and β (HBB) on chromosome 11 (illustrated in Fig. 2A) [105], [111], [112], [113]. The 5′ regulatory region of β-globin gene cluster spanning 34 kb, upstream to the first embryonic globin gene HBE1, includes several DNase I-hypersensitive sites (HSs; 5′HS1-HS7 in the entire region and 5′HS1-HS5 in the LCR), each 200 to 400 bp in size and separated from each other by several kb of DNA [108], [111]. As the LCR is the major element controlling expression of the downstream β-globin genes, laboratories around the world [113], [114], [115], [116] have aimed to reduce the size of LCR while maintaining most of its cis-regulatory function to confer the physiological expression of HBB to genetically modified cells [108], [112], [117], [118].
Fig. 2.

Overview of human β-globin locus and ex vivo gene therapy for the treatment of SCD and TDT. A) Schematic representation of human β-globin locus and LCR; B) Erythroid-specific expression of β-like globin transgenes using lentiviral vectors; C) Schematic representation of lentiviral gene addition therapy and HBB mutation-independent therapeutic approaches for the treatment of SCD and TDT.
In 2000, a lentiviral vector referred as TNS9 comprising mini LCR elements (840 bp for HS2, 1308 bp for HS3, and 1069 bp for HS4, in total about 3.2 kb in length, shown in Fig. 2B) was demonstrated by May and colleagues capable of effectively correcting thalassemia intermedia in mouse model [108]. In 2010, the clinical trial of LG001 study for the patients with β-thalassaemia showed one of three subjects treated by lentiviral vector HPV569 derived from TNS9 became transfusion-independent 1 year after cell infusion and the blood hemoglobin (Hb) level remained stable around 8.5 g/L for more than 8 years [119]. This vector HPV569 bears “antisickling” βT87Q amino acid substitution and various minor modifications on LCR components (646 bp for HS2, 845 bp for HS3, and 1153 bp for HS4, 2.7 kb in length) [119], [120]. Although the therapeutic benefit was found resulted from a dominant clone with lentiviral insertion occurred inside the HMGA2 gene [120], the LG001 study provided the proof of principle of tackling β-thalassaemia via engraftment of genetically modified autologous HSCs and paved a path for vector improvement and global phase I/II trials.
Further on, to increase the titre and transduction efficiency of vector HPV569, the strong and constitute CMV promoter was introduced to replace the wild-type U3 within the 5′LTR and two chromatin insulators were thereby removed, yielding SIN-LentiGlobin BB305 vector [121]. In following two phase II trials (HGB-204/NCT01745120 and HGB-205/NCT02151526), the autologous CD34+ cells from 22 patients with transfusion-dependent β-thalassemia (TDT) were transduced with BB305 vector and almost all the patients achieved sustained transfusion independence after treatment. In nine patients with the most severe form of the disease, the need for blood transfusions was dramatically reduce by 73% and completely eliminated in three patients [96], [102]. Based on the results of these two clinical trials, LentiGlobin BB305, commercially named Zynteglo (betibeglogene autotemcel), was granted conditional marketing authorisation by EMA in May 2019, for patients 12 years and above with TDT who are eligible for stem cell transplantation but do not have a matching related donor.
In comparison, the BB305 treatment for SCD subjects exhibited varied therapeutic efficacies, presumably due to the hypoxic condition, chronic inflammation, oxidative stress and dysregulated metabolism linked to SCD pathophysiology [122]. In the HGB-205 study, the treatment with BB305 vector resulted in transfusion independence, sustained clinical remission and stabilization of HbT87Q expression (contributing 47.9% and 25.8% of total Hb, respectively) in two of three patients with SCD [52], [123], [124]. However, the same treatment only produced mild improvement for the third SCD patient, with slightly reduced frequency of transfusion [52]. In the subsequent multicenter HGB-206 study (NCT02140554), the HbT87Q-globin levels in the first few SCD patients after the treatment with lovotibeglogene autotemcel (lovo-cel; formerly as BB305/LentiGlobin bb1111) were low, incapable of preventing SCD-related clinical events and delivering the discontinuation of transfusion [125]. Then, in two additional study arms (group B and C), refined transduction and transfusion protocols, optimized busulfan conditioning and plerixafor mobilization of HSCs, among others, were conducted accordingly, which resulted in complete resolution of severe vaso-occlusive episodes and sustained production of HbT87Q in the last group C [126]. Further on, in February 2021 two participants in HGB-206 trial were diagnosed with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), respectively, which raised concerns regarding lovotibeglogene autotemcel and led to the temporary suspension of HGB-206 and HGB-210 trials and additionally the marketing of betibeglogene autotemcel. The suspension was lifted by FDA several months later after further assessment showed that the trial participant diagnosed with MDS had actually developed transfusion-dependent anemia and the occurrence of AML was unlikely associated with BB305 vector [127].
Overall, the progressive understanding of the molecular, cellular and developmental processes underlying hemoglobin biology and pathology facilitates the optimization of conventional addition GT as well as lays a foundation to test various state-of-art biotechnologies, with high translational potential [96]. For instance, to improve titer and overcome the challenge of sub-optimal gene transfer efficiency to HSCs for the treatment of SCD, considerable efforts have been put on vector enhancement and redesigning, such as reducing LCR length or using “forward-oriented” lentiviral vector [128]. Very recently, Morgan et al. managed to substantially shorten LCR cis-regulatory component (∼1.2 kb) through identifying the core enhancer sequences of LCR 5’HS2-HS4 based on genomic and epigenomic analysis, generating a novel β-globin lentiviral vector with high titer and superior transduction performance [129]. In the last section of this review, HBB mutation-independent strategies based on DNA editing or RNA interference for the treatment of β-hemoglobinopathies will be discussed.
2.4. Inherited neurometabolic disorders (iNMDs)
The central nervous system (CNS) is more frequently and severely affected by the genetic abnormalities with the enzymes mediating metabolic pathways, such as lysosomal or peroxisomal enzymes [130], [131], [132], than any other organ. Build-up of toxic substrates in the CNS impairs neurodevelopment or causes neurodegeneration, which cannot be easily addressed by enzyme replacement therapy due to the obstacle of the blood–brain barrier (BBB) [133]. For over 20 years, allo-HSCT has been performed to treat such enzyme deficiency diseases as metabolically competent myeloid cells are found capable to infiltrate into the CNS and replace the resident macrophages and microglias to release enzymes (indicated in Fig. 1) [133], [134], [135], [136], [137]. In the case of lysosomal storage disorders (LSDs), once produced and released by migrated progenitor cells of transplanted allogeneic HSCs, lysosomal enzymes can be specifically recognized and bound by extracellular mannose-6-phosphate (M6P) receptors of neighboring cells to ameliorate the neuropathological severity, a mechanism known as cross-correction. Further on, it has been shown 10–15% of residual enzymatic activity is the critical threshold for the manifestations of a range of lysosomal enzyme deficiencies, indicating only a small percentage of correction is required for the physiological turnover and lysosome homeostasis [138], [139], [140]. Even so, some patients still suffer from insufficient enzymatic activity and experience neurological burden despite having successful allo-HSCT. Recent studies showed genetic engineered autologous HSCs modified by γ-RVs or LVs could substantially increase therapeutic enzyme level in the brain after engraftment and enable enhanced cross-correction effects, alleviating inflammation and oxidative stress [137], [141], [142].
For example, metachromatic leukodystrophy (MLD) is caused by the deficiency of lysosomal enzyme arylsulfatase A (ARSA), which leads to the accumulation of sphingolipid sulfatide in the oligodendrocytes, causing oligodendrocytal degeneration and progressive demyelination [140]. Previously, the efficacies of intracerebral GT with AAVs, enzyme replacement therapy and allo-HSCT have been investigated and shown could not effectively reverse the progression of MLD in clinical studies [136], [143], [144], [145], [146], [147]. In contrast, the result of a phase I/II trial (OTL-200/NCT01560182) published at the beginning of this year showed the treatment with arsa-cel (atidarsagene autotemcel, LibmeldyTM,Orchard, shown in Table 1), an ex vivo gene therapy containing autologous HSCs transduced with SIN-lentiviral vector encoding human ARSA cDNA, resulted in sustained, clinically relevant benefits in children with early-onset MLD [148].
Indeed, inherited neurometabolic disorders are individually rare, but they have a collective frequency of ∼1 in 1000 live births (∼1 in 5000 for LSDs alone) [140], [149], making them a significant public health problem worldwide [150], [151], [152]. To date, mutations in almost 700 different proteins were identified causing iNMDs (more than 70 genes for LSDs alone) [153], [154], [155], [156] and this list continues to grow. In Table 2, current ex vivo gene therapies tackling various genetic neurometabolic conditions that have entered clinical trials are summarized.
Table 2.
Ex vivo gene therapies for inherited neurometabolic disorders.
| Conditions | Clinical trial | Interventions | Sponsor/Collaborators | Phase | Enrollment | Status |
|---|---|---|---|---|---|---|
| X-ALD/CALD | NCT02559830 | Autologous HSCs transduced with lenti-ABCD1 | Shenzhen Second People's Hospital | I/II | 50 | Recruiting |
| NCT03852498 | Lenti-DTM* | Bluebird bio | III | 35 | Active, not recruiting | |
| NCT01896102 | Lenti-DTM | Bluebird bio | II/III | 32 | Completed | |
| MLD | NCT01560182 | OTL-200 | Orchard Therapeutics| | I/II | 20 | Active, not recruiting |
| NCT03725670 | Autologous HSCs transduced with lenti-ARSA | Shenzhen Geno-Immune Medical Institute | Not Applicable | 10 | Unknown status | |
| NCT02559830 | Autologous HSCs transduced with lenti-ARSA | Shenzhen Second People's Hospital | I/II | 50 | Recruiting | |
| NCT04283227 | OTL-200 | Orchard Therapeutics | III | 6 | Recruiting | |
| NCT03392987 | OTL-200 | Orchard Therapeutics | II | 10 | Active, not recruiting | |
| MPSI | NCT03488394 | OTL-203 | Orchard Therapeutics | I/II | 8 | Active, not recruiting |
| MPS IIIA | NCT04201405 | Autologous HSCs transduced lenti-SGSH | University of Manchester| Orchard Therapeutics |
I/II | 5 | Active, not recruiting |
| Fabry disease | NCT03454893 | AVR-RD-01 | AVROBIO | I/II | 11 | Terminated |
| NCT02800070 | Autologous HSCs transduced with lenti-GLA | University Health Network, Toronto|Ozmosis | I | 5 | Active, not recruiting | |
| Gaucher's disease | NCT00001234 | Autologous HSCs transduced with γ-retro-GBA | NINDS|NIH-Clinical Center | I | 120 | Completed |
| NCT04145037 | AVR-RD-02 | AVROBIO | I/II | 16 | Recruiting |
*Breakthrough Therapy Designation granted by FDA in May 2018.
X-ALD: X-linked adrenoleukodystrophy; CALD: cerebral adrenoleukodystrophy; ABCD1: ATP binding cassette subfamily D member 1; Lenti-DTM:: Autologous HSCs transduced with lenti-ABCD1; MLD: metachromatic leukodystrophy; ARSA: arylsulfatase A; OTL-200: Autologous HSCs transduced with lenti-ARSA; MPSI: mucopolysaccharidosis Type I; OTL-203: Autologous HSCs transduced with lenti-IDUA; IDUA: alpha-L-iduronidase; MPS IIIA: mucopolysaccharidosis Type IIIA; SGSH: N-sulfoglucosamine sulfohydrolase; GLA: Alpha-galactosidase A; AVR-RD-01: Autologous HSCs transduced with lenti-GLA; GBA: glucosylceramidase beta; AVR-RD-02: Autologous HSCs transduced with lenti-GBA; NINDS: National Institute of Neurological Disorders and Stroke.
2.5. Epidermolysis bullosa (EB)
Skin can be an ideal and appealing target for ex vivo gene therapy as the epidermal stem cells (ESCs) contained in the basal layer and hair follicles sustain the continuous renewal of epidermis, allowing the ESCs within epidermis to be easily harvested and subjected to ex vivo manipulation [157]. On the other hand, the advancement of in vitro cultivation of human skin cells by the co-culture with 3T3 mouse embryonic fibroblasts (MEFs) as feeder cells provided long-term expansion solution of human primary epidermal keratinocytes without immortalization, which made ESCs based ex vivo intervention to restore severe skin, mucosal and corneal defects realistic [157], [158]. Undoubtedly, applying genetically corrected ESCs to treat some rare hereditary skin diseases represented some of those unprecedented achievements in the whole field of gene therapy [159], [160], [161], [162], [163].
EB is a group of rare genetic skin fragility disorders characterized by mucocutaneous blister formation in response to minimal mechanical trauma due to reduced skin resilience [164], [165], [166]. Depending on the location of the molecular defects within the skin, EB can be divided into four major types, EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler EB (KEB). At least 16 distinct genes that encoding the structural components critically involved in maintaining the integrity and functionality of epidermis or the dermo-epidermal basement membrane zone (BMZ) have been identified underlying the causes of EBs [160], creating highly variable clinical manifestations ranging from mild localized blistering to severe and generalized erosions.
The first ex vivo gene therapy for treating EB was reported by Mavilio, de Luca and colleagues in 2006, tackling junctional subtype [159]. This landmark study demonstrated the efficacy and safety to treat an adult patient with JEB by transplantation of genetically modified epidermal graft sheet, generated from autologous keratinocytes transduced with γ-RV expressing full-length, wild-type LAMB3 cDNA. The transgenic skin has been maintained and no AEs are noted in the following 16 years follow-up [163]. And more recently, this genetic intervention has achieved the successful replacement of 80% of the body surface area for a 7-year-old patient in 2015, who had a severe form of JEB and life-threatening infection by the time of admission [158]. In particular, two years after the ex vivo gene therapy, analyzing the proviral integration sites of γ-RVs to track the clonal expansion of transduced cells verified the ESCs with holoclone-forming capability had been successfully targeted and genetically corrected, providing sustainable therapeutic benefits for the maintenance of epithelial integrity and function [158], [167].
As noted by some mechanistic studies, restoring mechano-signaling upon reintroduction of functional laminin-332 imposes survival and replicative advantages to genetically modified clones, presumably through rectifying YAP/TAZ signaling pathway [161], [168]. Therefore, although encouraging clinical outcomes obtained in the clinical trials with LAMB3-deficiency JEB, ex vivo gene addition therapy for other types of EBs remain to be greatly challenging. For instance, improved wound healing and restoration of anchoring fibrils at the BMZ were initially observed in the RDEB patients treated with regenerated epidermal sheets overexpressing COL7A1, however, type VII collagen level was found significantly decreased at the graft sites within one year [169]. Lately, an innovative strategy was deployed in GENEGRAFT project (NCT01874769; NCT04186650) [170] to transduce both autologous keratinocytes and fibroblasts using SIN-γ-RV-COL7A1, aiming to optimize anchoring fibril assembling condition. So far, one patient was treated in the early of 2021 [160].
2.6. Chimeric antigen receptors T cells (CAR-T)
In contrast to the approach tackling monogenic diseases, which is mostly straightforward, gene therapy for the treatment of malignment conditions needs to explore and evaluate a wide range of strategies to ensure therapeutic benefits and efficacy, such as locally delivering suicidal genes like p53 or TNF-α to elicit programmed tumor cell death, applying genetically modified oncolytic viruses to enable selective replication in cancerous cells, deploying genetically engineered immune cells to elevate antigen-mediated anti-tumor cytotoxicity, and so forth. Among them, CAR-T therapy emerged as unprecedented revolutionary therapeutics for its way of engaging the immune system in the fight against cancer [171], [172], [173], [174]. Retroviral vector mediated CAR expression renders modified T lymphocytes with novel specificity, function, and metabolism. A CAR consists of a single-chain variable fragment (scFv), derived from antibody and accordingly capable of mediating non-MHC restricted surface antigen recognition, an intracellular signaling domain responsible for T cell activation (typically derived from CD3ζ), and one or more intracellular costimulatory domains to enhance T cell function and persistence (derived from either CD28 or 4‐1BB) [175]. Unlike the physiological T cell receptors (TCRs) binding to antigen, this CAR-architecture overcomes the requirement for HLA recognition and CARs can be engineered to recognize a variety of substances, substantially expanding the repertoire of T cells [176], [177].
Early design of the first generation CARs was based on the observation originated from the cloning of CD3ζ chain, which was found capable of activating T cells independently [178]. For this reason, the 1st generation CARs comprised merely the cytoplasmic CD3ζ segment of endogenous TCR as signal transmitter, without any costimulatory component. Despite some encouraging outcomes in pre‐clinical study, the 1st generation CAR-T cells exhibited limited expansion capacity as well as insufficient cytokine production, failing to induce meaningful anti-tumor effects in the following clinical studies for leukemia, lymphoma and other types of cancers [179]. Fortunately, breakthrough came after the engineering of the 2nd generation CARs by incorporating costimulatory endodomain from either CD28 or 4‐1BB to potentiate the activation, cytotoxicity and persistence of T cells. Albeit with different pharmacokinetics, the 2nd generation CARs-T therapies exhibited significantly stronger antitumor activity [180], [181], [182], [183], [184]. Specifically, CD28/CD3ζ induces prompt and intense signal, whereas 4-1BB/CD3ζ elicits more durable cytotoxic activity with improved persistence in vivo.
In 2010, National Cancer Institute reported the first clinical trail showing positive response which mediated by γ-retroviral FMC63(CD19)/CD28/CD3ζ CAR-T cells in a patient with advanced follicular lymphoma (FL) [185]. Shortly after that, another clinical trial on chronic lymphocytic leukemia (CLL) demonstrated durable and effective responses to lentiviral FMC63(CD19)/4-1BB/CD3ζ CAR-T treatment, reported by the University of Pennsylvania, Philadelphia [186]. And more early-phase studies from multiple institutions confirmed the persuasive anti-tumor efficacy in those heavily pretreated patients with B-cell malignancies. Hence, in less than 10 years, CAR-T based ex vivo gene therapy has transitioned from preclinical proof-of-concept to FDA approved therapies (demonstrated in Table 1) [187], [188], [189], [190], [191], [192], [193]. In August 2017, the FDA announced the approval of SIN-lentiviral FMC63(CD19)/4-1BB/CD3ζ CAR-T tisagenlecleucel (tisa-cel, KYMRIAH®, Novartis) for the treatment of pediatric patients and young adults with relapsed or refractory ALL. Approval was based on the results of the open-label, multicenter, single-arm Phase II ELIANA trial (CCTL019-B2202), showing 83% of patients (52 out of 63) who received treatment with tisa-cel achieved complete remission (CR). Soon after, in October 2017, γ-retroviral FMC63(CD19)/CD28/CD3ζ axicabtagene ciloleucel (axi-cel, YESCARTA®, Kite Pharma Inc.) was approved for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, based on the results from multicenter single-armed phase I/II ZUMA-1 trial [180], [181].
CAR-T cells presents an entirely new paradigm in drug development, utilizing artificial receptor to mediate therapeutic benefits for months or years following a single medical intervention. However, cytokine release syndrome, neurotoxicity, relapse due to the loss of the antigen on tumor cells and others remain to be addressed. Investigators around world currently venture into formulating next-generation CAR therapies by targeting different and/or multiple antigens, incorporating additional synergistic components, or testing alternative cell sources, such as allogeneic CAR-T cells, NK cells, or even the T/NK cells obtained from induced pluripotent stem cells (iPSCs). Furthermore, there are numerous ongoing trials exploring the potential of CAR-T therapy in other malignant disorders, including myeloma and solid tumors, which have been systemically summarized in multiple reviews [174], [175], [194], [195], [196] and are not discussed in detail here.
3. Integrating retroviral vectors
Retroviruses are enveloped integrative RNA viruses belonging to the Retroviridae family that consists of seven genera [197]. The retroviral particle contains two copies of single-stranded positive-sense molecule, 7–11 kb in length, present in a dimeric form, capable of causing disease in humans and animals. Based on splicing patterns and the presence of auxiliary proteins, retroviruses are classed as simple genera of Retroviridae (i.e., α-, β- and γ-retrovirus), and complex genera (i.e., δ- and ε-retrovirus, lenti-and spumavirus). All retroviruses harbor the minimum three essential genes gag, pol and env, and the complex retroviruses have additional accessory genes, such as nef, vif, vpu, and vpr, and regulatory genes such as tat and rev, to exert control over the virus life cycle and pathogenicity. As a result, simple γ-retrovirus gain access to host genomic DNA only after the nuclear envelope is dissolved during cell division, whereas complex lentiviruses can access host genome via active transportation through the nuclear pore.
The property of genomic integration in the infected cell has made retroviruses a valuable tool to mediate ex vivo genetic manipulation when the stable expression of transgene or the transgene inherited by the progeny of the transduced cells are preferred. The γ-RVs developed during the 1980s were the first delivery platform demonstrated with the capability of transferring exogenous gene into repopulating HSCs and thereby have been employed in almost all early clinical trials [1], [13], [20], [198], [199], [200], [201], [202], [203], [204]. However, γ-RVs system developed from simple retrovirus had an inherent limitation due to the lack of capability to infect non-dividing cells. Consequently, LVs derived from complex lentiviruse were added into the vehicles for gene therapy and have becoming the predominantly used retroviral carrier system [12], [205], [206], [207].
3.1. Vectorization of retroviruses
Early vectorization of simple retroviruse was initiated from the identification of a genome packaging signal ψ [201], [202], [203], [204]. To obtain replication incompetent virus carrying the gene of interest for transfer, the genome of MoMLV was chosen to separate into two plasmids, namely a transfer plasmid and a packaging/helper plasmid [20], [202], [203], [204]. The ψ and intact LTR that required for the transgene packaging into the virion and following host genomic integration were included into transfer plasmid, and the gag, pol, and env genes that are essential for virion assembly were contained in helper plasmid. Theoretically, due to the lack of ψ in helper construct, the transcribed viral DNA of gag, pol, and env prevented from being incorporated into the virion to produce replication competent virus (RCV). However, ψ could be relocated from transfer plasmid to helper plasmid through just a single recombination event, which bearing the significant likelihood of RCV generation. The concept of introducing more obstacles for reaching the linear alignment of LTR, ψ, gag, pol, and env led to the advent of three-plasmid γ-RV system, in which helper plasmid was further split into two: the gag and pol are remained in packaging plasmid and env transferred to third envelope plasmid. The resulting vector system showed comparable effectiveness at integration and sustaining transgene expression [12], [208], meanwhile basically eliminating RCV formation [19], [198], [209], [210], [211].
However, the performance of γ-RVs was cell-cycle dependent, incapable of transducing quiescent or terminally differentiated cells [56], [212], [213]. Lentiviral systems were developed to overcome the deficiency of γ-RVs systems. Similarly, lentiviral system has been advanced in iterative cycles toward safety improvement and reduction in the viral replicative competency (shown in the Fig. 3). The 1st generation LVs was three-plasmid system derived from HIV-1, including a ψ-containing transfer plasmid with wild-type 5′ and 3′ LTR, a packaging plasmid containing all viral genes with the exception of the env gene, and an envelope plasmid containing pseudotype env gene from vesicular stomatitis virus glycoproteins (VSV-G) to improve stability and increase tropism. In the 2nd generation, the HIV-specific accessory genes (nef, vif, vpu, and vpr) that contributing to pathogenesis but nonessential for viral assembly were removed. Further on, in the 3rd generation, the wild type 5′ LTR was modified into a chimeric promoter that is no longer tat-transactivation dependent and accordingly, tat was removed from the packaging plasmid. Apart from that, the TATA box and transcription factor-binding sites within wild type 3′ LTR were also deleted, creating a SIN packaging system. Additionally, the expression of rev is further transferred into the 4th plasmid, resulting four-plasmid packaging system. Indeed, the 3rd generation SIN-LV system utilizes only three HIV-1 genes gag, pol, and rev for production, creating the best safety profile in terms of RCV formation in all retrovirus-based delivery platforms [55], [214].
Fig. 3.

Schematic of HIV-1 and the evolution of lentiviral packaging systems. Lentiviral system derived from HIV-1 has been advanced in iterative cycles toward safety improvement and reduction in the viral replicative competency. During this process, the HIV-specific accessory genes that contributing to pathogenesis but nonessential for viral assembly are removed, then tat gene is also removed when the wild type 5′ LTR has been modified. Pseudotype env gene from vesicular stomatitis virus glycoprotein is applied to improve stability and increase tropism. RRE: Rev responsive element; cPPT/CTS: central polypurine tract/central termination sequence; WPRE: woodchuck post-transcriptional regulatory element.
4. Next-generation gene editing tools
The ex vivo gene therapy based on the addition of exogenous gene has made remarkable progress during the past four decades, demonstrating promising therapeutic results across a wide spectrum of diseases. However, multiple challenges remain to be addressed in the future, such as high variation of transgene expression, unintended effects on neighboring genes, viral packaging limits, unresolvable dominant mutation effects, and so on. Overcoming these fundamental limitations underlying augmentation strategy requires radically distinct schemes, in particular, such as directly altering nucleic acid base pairs via game-changing gene editing approach [215], [216]. Recent advances in programmable endonucleases technologies, such as ZFNs, TALENs, CRISPR/Cas and even more recently emerged base editing, allowed site-specific modifications in genome scale become possible, strategically transforming the option to harvest genetically modified therapeutic cells for ex vivo therapy [217], [218], [219], [220], [221], [222], [223].
Programmable endonucleases create site-specific double-strand breaks (DSBs) that mainly resorted into two endogenous pathways for DNA repair, the error-prone NHEJ pathway and the high-fidelity HDR pathway. NHEJ can effectively produce gene knockout, resulted from NHEJ-induced random insertion or deletion causing frameshift mutation if occurring in the coding region of a gene. For HDR, the DNA cleavage is repaired via homologous recombination with a donor DNA carrying the template of homology sequence surrounding the DSB. For this reason, HDR can be exploited to achieve precise gene modifications as designed, eliminating unexpected randomness associated nonhomologous repairs. Indeed, with the property to recognize and bind at specific genomic sites for correction and modification, gene editing approach theoretically evades the genotoxicities attributed to retroviral semi-random genomic insertion, such as proto-oncogenes ectopic activation, tumor suppressor genes knockout or perturbation of splicing [1]. Each type of programmable endonuclease consists of an enzymatic cleavage domain that is nonspecific and a customizable sequence-specific DNA binding domain. Overview of key biological features of each gene editing system have been extensively reviewed previously and briefly summarized here in Table 3 [215], [224], [225], [226].
Table 3.
Comparison of three engineered endonucleases-ZFN, TALEN, and CRISPR/Cas9.
| ZFN | TALEN | CRISPR/Cas9 | |
|---|---|---|---|
| Target sequence | 9–18 bp per ZFN monomer | 15–20 bp per TALEN monomer | 20 bp plus PAM sequence |
| Mode of recognition | Protein:DNA | Protein:DNA | RNA:DNA |
| Endonuclease | FokI | FokI | Cas |
| Delivery | Two ZFNs around the target sequence are required | Two TALENs around the target sequence are required | sgRNA with Cas9 |
| Construction | Zinc finger sequence specifically recognizing 3-nucleotide sequence linked to FokI | Protein sequence specifically recognizing 1-nucleotide sequence linked to FokI | 20-nucleotide sgRNA complementary to the target sequence fused to Cas9 |
| Reprogramming efficiency | Relatively low | Relatively low | High (easy to design, fast to synthesize) |
| Off-target effects | High | Low | Variable |
Clinically, according to the clinicaltrials.gov, the registered studies based on gene editing as treatment modality mainly concentrated on ex vivo gene therapy and overwhelmingly focused on enginerring HSCs to combat infection, malignancies and globinopathies (indicated in Table 4). Gene editing has shown a rapid increase in the recent years and there is a shift of the selected programmable endonucleases overtime. The first ever registered ex vivo genome editing trial was in 2009 (NCT00842634), which applied ZFNs. Between 2009 and 2015, only around 2 trials were registered per year. 2016 marks the beginning of TALENs and CRISPR/Cas9 clinic application when both started to be registered for trials, and since 2016 the number of registered trials has increased substantially, with CRISPR/Cas9 becoming the most selected editing system for trial studies.
Table 4.
Selected early ex vivo gene therapy trials with genome editors.
| Nuclease | Target gene and effect | Disease | Sponsor organization | Country | NCT number | Date posted |
|---|---|---|---|---|---|---|
| ZFN | CCR5 knockout | HIV-1 | University of Pennsylvania | USA | NCT00842634 | 2009 Feb |
| ZFN | CCR5 knockout | HIV-1 | Sangamo Biosciences | USA | NCT01044654 | 2010 Jan |
| ZFN | CCR5 knockout | HIV-1 | Sangamo Biosciences | USA | NCT01252641 | 2010 Dec |
| ZFN | CCR5 knockout | HIV-1 | Sangamo Biosciences | USA | NCT01543152 | 2012 Mar |
| ZFN | CCR5 knockout | HIV-1 | Sangamo Biosciences | USA | NCT02225665 | 2014 Aug |
| ZFN | CCR5 knockout | HIV-1 | University of Pennsylvania | USA | NCT02388594 | 2015 Mar |
| ZFN | CCR5 knockout | HIV-1 | City of Hope Medical Center | USA | NCT02500849 | 2015 Jul |
| ZFN | Disrupt BCL11A | β-thalassemia | Sangamo Biosciences | USA | NCT03432364 | 2018 Feb |
| ZFN | CCR5 knockout | HIV-1 | University of Pennsylvania | USA | NCT03617198 | 2018 Aug |
| ZFN | Disrupt BCL11A | SCD | Bioverativ | USA | NCT03653247 | 2018 Aug |
| ZFN | CCR5 knockout | HIV-1 | Case Western Reserve University | USA | NCT03666871 | 2018 Sep |
| TALEN |
TCRα, TCRβ, CD52 knockout; Create universal T-cells |
Advanced lymphoid malignancy | Institut de Recherches Internationales Servier |
UK, USA France |
NCT02746952 | 2016 Apr |
| TALEN |
TCRα, TCRβ, CD52 knockout; Create universal T-cells |
Refractory B-ALL | Institut de Recherches Internationales Servier |
UK, Belgium, France, USA |
NCT02808442 | 2016 Jun |
| TALEN | PD-1 and CD52 knockout | AML | Cellectis S.A. | USA | NCT03190278 | 2017 Jun |
| TALEN | PD-1 and CD52 knockout | AML | Cellectis S.A. | USA | NCT04106076 | 2019 Sep |
| TALEN | PD-1 and CD52 knockout | Multiple myeloma | Cellectis S.A. | USA | NCT04142619 | 2019 Oct |
| TALEN | PD-1 and CD52 knockout | CD22+ B cell acute lymphoblastic leukemia | Cellectis S.A. | USA | NCT04150497 | 2019 Nov |
| Cas9 | PD-1 knockout | Metastatic non-small cell lung cancer | West China Hospital, Sichuan University | China | NCT02793856 | 2016 Jun |
| Cas9 |
TCRα, TCRβ, B2M knockout; Create universal T-cells |
B-cell leukemia | Chinese PLA General Hospital |
China | NCT03166878 | 2017 May |
| Cas9 | CCR5 knockout | HIV-1 | Affiliated Hospital to Academy of Military Medical Sciences |
China | NCT03164135 | 2017 May |
| Cas9 | TCRα, TCRβ, PD-1 knockout | Various malignancies | University of Pennsylvania | USA | NCT03399448 | 2018 Jan |
| Cas9 | Disrupt BCL11A | β-thalassemia | CRISPR Therapeutics | UK, Germany | NCT03655678 | 2018 Aug |
| Cas9 | Disrupt BCL11A | SCD | Vertex Pharmaceuticals Incorporated and CRISPR Therapeutics |
USA | NCT03745287 | 2018 Nov |
| Cas9 | CISH knockout | Metastatic gastrointestinal epithelial cancer | National Cancer Institute | USA | NCT03538613 | 2018 May |
| Cas9 | CISH knockout | Metastatic gastrointestinal epithelial cancer | Masonic Cancer Center, University of Minnesota |
USA | NCT04089891 | 2019 Sep |
| TALEN |
TCRα, TCRβ, CD52 knockout; create universal T cells (15-year follow-up study) |
Advanced lymphoid malignancy | Institut de Recherches Internationales Servier |
UK, Belgium, France, USA |
NCT02735083 | 2016 Apr |
| Cas9 | Disrupt BCL11A (15-year follow-up study) |
β-thalassemia and severe SCD | Vertex Pharmaceuticals Incorporated and CRISPR Therapeutics | USA, UK, Germany |
NCT04208529 | 2019 Nov |
PD-1: programmed cell death protein 1; CISH: cytokine-induced SH2 protein; B2M: β-2 microglobin.
More specifically, CCR5 gene has been chosen as the very first target in human genome editing trial. A naturally occurring mutation CCR5-Δ32 that causing the loss of protein expression has been found generally harmless, but renders homozygous carriers of the null allele resistant to HIV-1 infection. This phenomenon was leveraged ingeniously by Gero Hütter 15 years ago [227] and contributed to the cure of HIV-1 infection of “Berlin Patient” [228], [229], and later on the “London Patient” [230], [231]. So far, the majority of HIV-1 therapy trials based on CCR5 knockout focused on the application of ZFNs (indicated in Table 4), and the first human clinical trial applying CRISPR for CCR5 disruption was registered in 2017 (NCT03164135) followed by two more similar clinical studies documented at the end of 2021 (NCT05144386 NCT05143307).
The first TALENs clinical application was used for generating “off-the-shelf” universal third-party CD19-CAR modified T cells by knocking out CD52, TCRα and TCRβ, and ex vivo engineered T cells have been administrated to the patients with advance lymphoid malignancy or refractory B cell acute leukemia since 2016 (NCT02746952, NCT02808442). In 2016, a lung cancer patient from China became the first person in the world to be treated with a CRISPR therapy at the West China Hospital, Sichuan University (NCT02793856). This study evaluated the safety and feasibility of PD-1-edited T cells for the treatment of refractory metastatic non-small cell lung cancer (NSCLC). Briefly, relying on electroporation of Cas9 endonuclease and gRNA plasmid to disrupt the PDCD1 gene, ex vivo engineered autologous T cells were manufactured prior to infusion into the patients with NSCLC. In April 2020, results based on 12 patients from this trial were published [232], suggesting the treatment was safe with acceptable side effects, meantime showing a tendency that the patients with higher levels of edited cells had better recovery and slower disease progression. The median editing efficiency has achieved 20.1% and the median off-target events was about 0.05%.
Another key focus of CRISPR clinical studies is the treatment of β-hemoglobinopathies. The first patient receiving CRISPR/Cas9-modified HSCs for TDT took place in Germany in February 2019 (CTX001/NCT03655678) and a few months later, the first patient with SCD was treated with the same therapy in Nashville, Tennessee in July (CTX001/NCT03745287). For β-hemoglobin disorders, CRISPR is designed mostly based on HBB mutation-independent strategies (shown in Fig. 2), with the insights into the fetal-to-adult hemoglobin switch and a rare naturally occurring condition of hereditary persistence of fetal hemoglobin (HPFH) [94], [96]. Neonates and infants with TDT or SCD are typically asymptomatic when their fetal hemoglobin (HbF/α2γ2) levels remain high, and become symptomatic during the developmental process of switching from γ- to β-globin for the adult hemoglobin (HbA/α2β2) production. On the other hand, the HPFH caused by the mutations in the regulatory region of the HBG gene has been found capable of alleviating the symptoms of SCD, due to the sustained expression of γ-globin [233], [234], [235]. Furthermore, hydroxyurea, the clinically used HbF inducer, has been applied to mitigate the severity of TDT and SCD. So that, the genome editing for the treatment of β-hemoglobinopathies has been mainly directed into two approaches: 1) perturbating the expression of BCL11A gene which encoding the fetal hemoglobin silencing factor to induce the production of HbF, or 2) introduction of HPFH or HPFH-like mutations in the HBG regulatory region or disrupting HBG(γ-globin) repressor binding sites. So far, the therapy based on BCL11A perturbation mediated by CRISPR/Case9 has entered clinical trials (NCT03655678 and NCT03745287) and the early clinical results are promising, showing normal to near-normal hemoglobin level recovery with 30%∼40% of hemoglobin as HbF, and the only immediate side effects resulted from the administration of chemotherapy [103].
Genome editing as a potential medical intervention is rapidly advancing into the clinic, however, not only it is still relatively in its translational infancy but the process has turned out to be more difficult than initially thought. Most of all, CRISPR/Cas may exhibit wider-spread off-target effects compared with ZFNs and TALENs and the improvements of CRISPR/Cas in terms of precision and specificity will be crucial for its future development.
5. Future perspectives
Since the first approved gene transfer human study, the advancement of gene therapy over the past three decades has ultimately led to multiple regulatory approvals. For ex vivo based gene therapies that have been authorized for marketing and many more to come in the future, determining the manufacture location for cell products engineering would be one of the most important issues for the long-term progression in the field. There are basically two different models under discussion regarding where it should take place, centralized model versus decentralized approach [236]. In the centralized setting, the cells from the patient/third party are harvested at local hospital, shipped to center facility for genetic manipulation, then the frozen cell product shipped back to the patient’s location, where the administering physician thaws the cell product and reinfuses it to the patient. The quality control of this manufacturing process secures every cellular product is up to a pre-defined standard, but geographical distance will certainly compromise responsiveness and personalization is usually not particularly prioritized in this model. Conversely, decentralization means cell collection and processing ex vivo genetic manipulation locally, vaguely resembling clinically conducted blood transfusion and transplantation of HSCs. Flexibility is naturally implied in decentralizing mode, potentially providing better patient-physician engaging cycles. However, standardization, cGMP manufacturing, regulatory surveillance and personnel training represent great challenges. Automation or semi-automation could be a solution to minimize variability, fluctuation and operational bias for decentralization development. Clearly, for such revolutionary medicine, upcoming innovations should balance between clinical benefit, efficacy, safety, cost-saving and therapeutic value, disease severity, availability and accessibility.
Retroviral delivery platform based on γ-RVs and LVs is instrumental in ex vivo gene therapy thanks to the capability to efficiently and permanently integrate into the host cell genome, albeit semirandomly. The pattern of integration can be broadly categorized as absolutely silent insertion, insertion that interferring but biologically irrelevant, insertion that is clinically irrelevant, and transformative harmful insertion. γ-RVs preferentially integrate near transcriptional start sites of active genes, resulting in γ-RVs have a higher chance of interfering with gene transcription contrasted to the intronic integration preference seen for LVs [55], [207], [214], [237]. Thereby, it is basically considered that LVs exhibit overall relatively safer genomic integration profile compared with γ-RVs. As semirandom integration may provide a selective clonal growth advantage, profiling insertion sites and clonal fluctuation should be routinely undertaken after infusion of transduced cells. Currently, the application of insertion-site analyses, such as linear amplification-mediated PCR (LAM-PCR), is rather limited to investigational studies or analyzing abnormal clinical findings, which should be improved in the future for the clinical practice.
The gene editing technology that capable of rewriting human genome sequences has become an entirely new treatment modality in the field of gene therapy. In principle, gene editing should be safer than retroviral vector-based gene addition as it eliminating semirandom genomic integration. However, how to predict off-target effects and how DNA DSBs triggering genome rearrangements such as inversion, translocation even deletion, remain to be answered, demanding better in-depth understanding of the molecular networks underline the genotoxicities associated with each type of programmable endonucleases in genom editing. Moreover, several next-generation editing technologies have embarked on improving the precision, accuracy, efficacy and applicability. For instance, prime editing has realized the precise alterations of DNA sequences without double-strand breaks [238], [239], thus evading DNA repairment related activities, and the development of RNA-targeted editing that conferring the temporal and reversible modification may potentially provide better safety and offer more flexible interventions [240], [241], [242]. Epigenome editing technologies may aid post-transient editor operation with the flexibility in tunability and reversibility [243]. The fast-growing versatile gene-editing technology may alter how we think about gene therapy eventually.
At last, gene therapy is considered the most complex “drugs” ever developed [1], capable of providing durable benefits for devastating, sometime incurable human diseases. The pathway was not linear, but fulfilled with iterative bench-to-bedside cycles [244]. Justified optimism, vigorous global investment, scientific consensus should facilitate their clinical translation and commercialization, increasing the applicability of genetic therapies and unfolding their true potentials.
CRediT authorship contribution statement
Xiaomo Wu: Conceptualization, Writing – original draft, Visualization, Investigation, Supervision. Xiaorong He: Investigation. Fahui Liu: Methodology, Software. Xiaochang Jiang: Methodology, Software. Ping Wang: Software, Validation. Jinyan Zhang: Software, Validation. Ju Jiang: Supervision.
Acknowledgments
Acknowledgments
This work was supported by the Nature Science Foundation of Fujian Province (Grant No. 2021J01533; Grant No. 2020GGB048) and the Science Foundation of Fuzhou (Grant No. 2021-S-188).
Author contributions
X.W wrote the draft of manuscript. All authors contributed to the reviewing and editing. All authors read and approved the final manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Contributor Information
Xiaomo Wu, Email: xiaomo.wu@gmail.com.
Ju Jiang, Email: kosan725@163.com.
References
- 1.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:6372. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 2.High K.A., Roncarolo M.G. Gene therapy. N Engl J Med. 2019;381(5):455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 3.Watson J.D., Crick F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 4.Khan S., Ullah M.W., Siddique R., Nabi G., Manan S., Yousaf M., et al. Role of recombinant DNA technology to improve life. Int J Genomics. 2016 doi: 10.1155/2016/2405954. 20162405954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen S.N., Chang A.C., Boyer H.W., Helling R.B. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci U S A. 1973;70(11):3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tatum E.L. Molecular biology, nucleic acids, and the future of medicine. Perspect Biol Med. 1966;10(1):19–32. doi: 10.1353/pbm.1966.0027. [DOI] [PubMed] [Google Scholar]
- 7.Friedmann T. A brief history of gene therapy. Nat Genet. 1992;2(2):93–98. doi: 10.1038/ng1092-93. [DOI] [PubMed] [Google Scholar]
- 8.Friedmann T., Roblin R. Gene therapy for human genetic disease? Science. 1972;175(4025):949–955. doi: 10.1126/science.175.4025.949. [DOI] [PubMed] [Google Scholar]
- 9.Culver K.W., Anderson W.F., Blaese R.M. Lymphocyte gene therapy. Hum Gene Ther. 1991;2(2):107–109. doi: 10.1089/hum.1991.2.2-107. [DOI] [PubMed] [Google Scholar]
- 10.Check E. A tragic setback. Nature. 2002;420(6912):116–118. doi: 10.1038/420116a. [DOI] [PubMed] [Google Scholar]
- 11.Giacca M., Zacchigna S. Virus-mediated gene delivery for human gene therapy. J Control Release. 2012;161(2):377–388. doi: 10.1016/j.jconrel.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Miller A.D. Retroviral vectors: from cancer viruses to therapeutic tools. Hum Gene Ther. 2014;25(12):989–994. doi: 10.1089/hum.2014.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunbar C.E. Gene transfer to hematopoietic stem cells: implications for gene therapy of human disease. Annu Rev Med. 1996:4711–4720. doi: 10.1146/annurev.med.47.1.11. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki YSY. Gene regulatable lentiviral vector system. 2011.
- 15.Vu H.N., Ramsey J.D., Pack D.W. Engineering of a stable retroviral gene delivery vector by directed evolution. Mol Ther. 2008;16(2):308–314. doi: 10.1038/sj.mt.6300350. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y., Stepto H., Schneider C.K. Development of the First World Health Organization lentiviral vector standard: toward the production control and standardization of lentivirus-based gene therapy products. Hum Gene Ther Methods. 2017;28(4):205–214. doi: 10.1089/hgtb.2017.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naso M.F., Tomkowicz B., Perry W.L., 3rd, Strohl W.R. Adeno-Associated Virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31(4):317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wold W.S., Toth K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr Gene Ther. 2013;13(6):421–433. doi: 10.2174/1566523213666131125095046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barquinero J., Eixarch H., Perez-Melgosa M. Retroviral vectors: new applications for an old tool. Gene Ther. 2004;11 doi: 10.1038/sj.gt.3302363. Suppl 1S3-9. [DOI] [PubMed] [Google Scholar]
- 20.Cone R.D., Mulligan R.C. High-efficiency gene transfer into mammalian cells: generation of helper-free recombinant retrovirus with broad mammalian host range. Proc Natl Acad Sci U S A. 1984;81(20):6349–6353. doi: 10.1073/pnas.81.20.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan M.A., Galla M., Grez M., Fehse B., Schambach A. Retroviral gene therapy in Germany with a view on previous experience and future perspectives. Gene Ther. 2021 doi: 10.1038/s41434-021-00237-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cavazzana-Calvo M., Hacein-Bey S., de Saint Basile G., Gross F., Yvon E., Nusbaum P., et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288(5466):669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 23.Ferrari G., Thrasher A.J., Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet. 2021;22(4):216–234. doi: 10.1038/s41576-020-00298-5. [DOI] [PubMed] [Google Scholar]
- 24.Snowden J.A., Sanchez-Ortega I., Corbacioglu S., Basak G.W., Chabannon C., de la Camara R., et al. Indications for haematopoietic cell transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2022. Bone Marrow Transplant. 2022 doi: 10.1038/s41409-022-01691-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohn L.A., Kohn D.B. Gene therapies for primary immune deficiencies. Front Immunol. 2021 doi: 10.3389/fimmu.2021.648951. 12648951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alhakamy N.A., Curiel D.T., Berkland C.J. The era of gene therapy: From preclinical development to clinical application. Drug Discov Today. 2021;26(7):1602–1619. doi: 10.1016/j.drudis.2021.03.021. [DOI] [PubMed] [Google Scholar]
- 27.Kumrah R., Vignesh P., Patra P., Singh A., Anjani G., Saini P., et al. Genetics of severe combined immunodeficiency. Genes Dis. 2020;7(1):52–61. doi: 10.1016/j.gendis.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tasher D., Dalal I. The genetic basis of severe combined immunodeficiency and its variants. Appl Clin Genet. 2012:567–580. doi: 10.2147/TACG.S18693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pai S.Y., Thrasher A.J. Gene therapy for X-linked severe combined immunodeficiency: historical outcomes and current status. J Allergy Clin Immunol. 2020;146(2):258–261. doi: 10.1016/j.jaci.2020.05.055. [DOI] [PubMed] [Google Scholar]
- 30.Bradford K.L., Moretti F.A., Carbonaro-Sarracino D.A., Gaspar H.B., Kohn D.B. Adenosine Deaminase (ADA)-Deficient Severe Combined Immune Deficiency (SCID): molecular pathogenesis and clinical manifestations. J Clin Immunol. 2017;37(7):626–637. doi: 10.1007/s10875-017-0433-3. [DOI] [PubMed] [Google Scholar]
- 31.Parkman R. The application of bone marrow transplantation to the treatment of genetic diseases. Science. 1986;232(4756):1373–1378. doi: 10.1126/science.3520819. [DOI] [PubMed] [Google Scholar]
- 32.Ferrua F., Aiuti A. Twenty-five years of gene therapy for ADA-SCID: from bubble babies to an approved drug. Hum Gene Ther. 2017;28(11):972–981. doi: 10.1089/hum.2017.175. [DOI] [PubMed] [Google Scholar]
- 33.Blaese R.M., Culver K.W., Miller A.D., Carter C.S., Fleisher T., Clerici M., et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270(5235):475–480. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 34.Bordignon C., Notarangelo L.D., Nobili N., Ferrari G., Casorati G., Panina P., et al. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 1995;270(5235):470–475. doi: 10.1126/science.270.5235.470. [DOI] [PubMed] [Google Scholar]
- 35.Muul L.M., Tuschong L.M., Soenen S.L., Jagadeesh G.J., Ramsey W.J., Long Z., et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood. 2003;101(7):2563–2569. doi: 10.1182/blood-2002-09-2800. [DOI] [PubMed] [Google Scholar]
- 36.Hacein-Bey-Abina S., Le Deist F., Carlier F., Bouneaud C., Hue C., De Villartay J.P., et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346(16):1185–1193. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 37.Gaspar H.B., Parsley K.L., Howe S., King D., Gilmour K.C., Sinclair J., et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364(9452):2181–2187. doi: 10.1016/S0140-6736(04)17590-9. [DOI] [PubMed] [Google Scholar]
- 38.Sagoo P., Gaspar H.B. The transformative potential of HSC gene therapy as a genetic medicine. Gene Ther. 2021 doi: 10.1038/s41434-021-00261-x. [DOI] [PubMed] [Google Scholar]
- 39.Cavazzana M., Six E., Lagresle-Peyrou C., Andre-Schmutz I., Hacein-Bey-Abina S. Gene therapy for X-linked severe combined immunodeficiency: where do we stand? Hum Gene Ther. 2016;27(2):108–116. doi: 10.1089/hum.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rans TS, England R. The evolution of gene therapy in X-linked severe combined immunodeficiency. Ann Allergy Asthma Immunol. 2009;102(5):357-362; quiz 363-355, 402. [DOI] [PubMed]
- 41.Hacein-Bey-Abina S., Hauer J., Lim A., Picard C., Wang G.P., Berry C.C., et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363(4):355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 43.Howe S.J., Mansour M.R., Schwarzwaelder K., Bartholomae C., Hubank M., Kempski H., et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hacein-Bey-Abina S., Garrigue A., Wang G.P., Soulier J., Lim A., Morillon E., et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hacein-Bey-Abina S., Pai S.Y., Gaspar H.B., Armant M., Berry C.C., Blanche S., et al. A modified gamma-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371(15):1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thornhill S.I., Schambach A., Howe S.J., Ulaganathan M., Grassman E., Williams D., et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol Ther. 2008;16(3):590–598. doi: 10.1038/sj.mt.6300393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clarke E.L., Connell A.J., Six E., Kadry N.A., Abbas A.A., Hwang Y., et al. T cell dynamics and response of the microbiota after gene therapy to treat X-linked severe combined immunodeficiency. Genome Med. 2018;10(1):70. doi: 10.1186/s13073-018-0580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanco E., Izotova N., Booth C., Thrasher A.J. Immune reconstitution after gene therapy approaches in patients with X-linked severe combined immunodeficiency disease. Front Immunol. 2020 doi: 10.3389/fimmu.2020.608653. 11608653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Perez L., Ordas A., Cante-Barrett K., Meij P., Pike-Overzet K., Lankester A., et al. Preclinical development of autologous hematopoietic stem cell-based gene therapy for immune deficiencies: a journey from mouse cage to bed side. Pharmaceutics. 2020;12(6) doi: 10.3390/pharmaceutics12060549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fischer A., Hacein-Bey-Abina S. Gene therapy for severe combined immunodeficiencies and beyond. J Exp Med. 2020;217(2) doi: 10.1084/jem.20190607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tucci F., Galimberti S., Naldini L., Valsecchi M.G., Aiuti A. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat Commun. 2022;13(1):1315. doi: 10.1038/s41467-022-28762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magrin E., Semeraro M., Hebert N., Joseph L., Magnani A., Chalumeau A., et al. Long-term outcomes of lentiviral gene therapy for the beta-hemoglobinopathies: the HGB-205 trial. Nat Med. 2022;28(1):81–88. doi: 10.1038/s41591-021-01650-w. [DOI] [PubMed] [Google Scholar]
- 53.Ferreira M.V., Cabral E.T., Coroadinha A.S. Progress and perspectives in the development of lentiviral vector producer cells. Biotechnol J. 2021;16(1):e2000017. doi: 10.1002/biot.202000017. [DOI] [PubMed] [Google Scholar]
- 54.Buchschacher G.L., Jr., Wong-Staal F. Development of lentiviral vectors for gene therapy for human diseases. Blood. 2000;95(8):2499–2504. [PubMed] [Google Scholar]
- 55.Milone M.C., O'Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32(7):1529–1541. doi: 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bulcha J.T., Wang Y., Ma H., Tai P.W.L., Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021;6(1):53. doi: 10.1038/s41392-021-00487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mamcarz E., Zhou S., Lockey T., Abdelsamed H., Cross S.J., Kang G., et al. Lentiviral gene therapy combined with low-dose Busulfan in infants with SCID-X1. N Engl J Med. 2019;380(16):1525–1534. doi: 10.1056/NEJMoa1815408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Ravin S.S., Wu X., Moir S., Anaya-O'Brien S., Kwatemaa N., Littel P., et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2016;8(335) doi: 10.1126/scitranslmed.aad8856. 335ra357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greene M.R., Lockey T., Mehta P.K., Kim Y.S., Eldridge P.W., Gray J.T., et al. Transduction of human CD34+ repopulating cells with a self-inactivating lentiviral vector for SCID-X1 produced at clinical scale by a stable cell line. Hum Gene Ther Methods. 2012;23(5):297–308. doi: 10.1089/hgtb.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou S., Mody D., DeRavin S.S., Hauer J., Lu T., Ma Z., et al. A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells. Blood. 2010;116(6):900–908. doi: 10.1182/blood-2009-10-250209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aiuti A., Slavin S., Aker M., Ficara F., Deola S., Mortellaro A., et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 62.Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360(5):447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 63.Carriglio N., Klapwijk J., Hernandez R.J., Vezzoli M., Chanut F., Lowe R., et al. Good laboratory practice preclinical safety studies for GSK2696273 (MLV vector-based ex vivo gene therapy for adenosine deaminase deficiency severe combined immunodeficiency) in NSG mice. Hum Gene Ther Clin Dev. 2017;28(1):17–27. doi: 10.1089/humc.2016.191. [DOI] [PubMed] [Google Scholar]
- 64.Aiuti A., Roncarolo M.G., Naldini L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol Med. 2017;9(6):737–740. doi: 10.15252/emmm.201707573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cicalese M.P., Ferrua F., Castagnaro L., Pajno R., Barzaghi F., Giannelli S., et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128(1):45–54. doi: 10.1182/blood-2016-01-688226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kohn D.B., Booth C., Shaw K.L., Xu-Bayford J., Garabedian E., Trevisan V., et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med. 2021;384(21):2002–2013. doi: 10.1056/NEJMoa2027675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Notarangelo L.D. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S182-194. [DOI] [PubMed]
- 68.Fischer A., Hacein-Bey-Abina S., Cavazzana-Calvo M. Gene therapy for primary immunodeficiencies. Immunol Allergy Clin North Am. 2010;30(2):237–248. doi: 10.1016/j.iac.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 69.Rhim J.W. Importance of neonatal screening for primary immunodeficiencies. Clin Exp Pediatr. 2021;64(10):519–520. doi: 10.3345/cep.2021.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mardy A.H., Norton M.E. In utero gene therapy for primary immunodeficiencies. Clin Obstet Gynecol. 2021;64(4):886–897. doi: 10.1097/GRF.0000000000000652. [DOI] [PubMed] [Google Scholar]
- 71.Demirdag Y., Fuleihan R., Orange J.S., Yu J.E. New primary immunodeficiencies 2021 context and future. Curr Opin Pediatr. 2021;33(6):657–675. doi: 10.1097/MOP.0000000000001075. [DOI] [PubMed] [Google Scholar]
- 72.Tani K. Current status of ex vivo gene therapy for hematological disorders: a review of clinical trials in Japan around the world. Int J Hematol. 2016;104(1):42–72. doi: 10.1007/s12185-016-2030-2. [DOI] [PubMed] [Google Scholar]
- 73.Coleman A., Gern J.E. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-Aldrich syndrome. Pediatrics. 2014;134 doi: 10.1542/peds.2014-1817HHHH. Suppl 3S182-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341(6148):1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boztug K., Schmidt M., Schwarzer A., Banerjee P.P., Diez I.A., Dewey R.A., et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363(20):1918–1927. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boztug K., Dewey R.A., Klein C. Development of hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Curr Opin Mol Ther. 2006;8(5):390–395. [PubMed] [Google Scholar]
- 77.Consiglieri G., Ferrua F., San Raffaele Hospital C., Aiuti A., Cicalese M.P. A Case of two adult brothers with wiskott-aldrich syndrome, one treated with gene therapy and one with HLA-identical hematopoietic stem cell transplantation. J Clin Immunol. 2021 doi: 10.1007/s10875-021-01157-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Avedillo D.I., Zychlinski D., Coci E.G., Galla M., Modlich U., Dewey R.A., et al. Development of novel efficient SIN vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy. Mol Pharm. 2011;8(5):1525–1537. doi: 10.1021/mp200132u. [DOI] [PubMed] [Google Scholar]
- 79.Hacein-Bey A.S., Gaspar H.B., Blondeau J., Caccavelli L., Charrier S., Buckland K., et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313(15):1550–1563. doi: 10.1001/jama.2015.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Charrier S., Dupre L., Scaramuzza S., Jeanson-Leh L., Blundell M.P., Danos O., et al. Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients. Gene Ther. 2007;14(5):415–428. doi: 10.1038/sj.gt.3302863. [DOI] [PubMed] [Google Scholar]
- 81.Jofra H.R., Calabria A., Sanvito F., De Mattia F., Farinelli G., Scala S., et al. Hematopoietic tumors in a mouse model of X-linked chronic granulomatous disease after lentiviral vector-mediated gene therapy. Mol Ther. 2021;29(1):86–102. doi: 10.1016/j.ymthe.2020.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bradshaw B., Jaffal H., Wysocki C.A., Grover L.A., Mitchell R.B., Ulualp S., et al. Chronic granulomatous disease of the upper airway. Ear Nose Throat J. 2021 doi: 10.1177/01455613211054635. 1455613211054635. [DOI] [PubMed] [Google Scholar]
- 83.De Ravin S.S., Brault J., Meis R.J., Li L., Theobald N., Bonifacino A.C., et al. NADPH oxidase correction by mRNA transfection of apheresis granulocytes in chronic granulomatous disease. Blood Adv. 2020;4(23):5976–5987. doi: 10.1182/bloodadvances.2020003224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaufmann K.B., Chiriaco M., Siler U., Finocchi A., Reichenbach J., Stein S., et al. Gene therapy for chronic granulomatous disease: current status and future perspectives. Curr Gene Ther. 2014;14(6):447–460. doi: 10.2174/1566523214666140918113201. [DOI] [PubMed] [Google Scholar]
- 85.Ott M.G., Schmidt M., Schwarzwaelder K., Stein S., Siler U., Koehl U., et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 86.Grez M., Reichenbach J., Schwable J., Seger R., Dinauer M.C., Thrasher A.J. Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther. 2011;19(1):28–35. doi: 10.1038/mt.2010.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stein S., Siler U., Ott M.G., Seger R., Grez M. Gene therapy for chronic granulomatous disease. Curr Opin Mol Ther. 2006;8(5):415–422. [PubMed] [Google Scholar]
- 88.Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16(2):198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 89.Weisser M., Demel U.M., Stein S., Chen-Wichmann L., Touzot F., Santilli G., et al. Hyperinflammation in patients with chronic granulomatous disease leads to impairment of hematopoietic stem cell functions. J Allergy Clin Immunol. 2016;138(1):219–228 e219. doi: 10.1016/j.jaci.2015.11.028. [DOI] [PubMed] [Google Scholar]
- 90.Jafarian A., Shokri G., Shokrollahi B.M., Moin M., Pourpak Z., Soleimani M. Recent advances in gene therapy and modeling of chronic granulomatous disease. Iran J Allergy Asthma Immunol. 2019;18(2):131–142. [PubMed] [Google Scholar]
- 91.Kohn D.B., Booth C., Kang E.M., Pai S.Y., Shaw K.L., Santilli G., et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med. 2020;26(2):200–206. doi: 10.1038/s41591-019-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuo C.Y., Kohn D.B. Overview of the current status of gene therapy for primary immune deficiencies (PIDs) J Allergy Clin Immunol. 2020;146(2):229–233. doi: 10.1016/j.jaci.2020.05.024. [DOI] [PubMed] [Google Scholar]
- 93.Tucci F., Galimberti S., Naldini L., Valsecchi M.G., Aiuti A. Gene therapy with hematopoietic stem and progenitor cell for monogenic disorders: a systematic review and meta-analysis. 2021. [DOI] [PMC free article] [PubMed]
- 94.Tisdale J.F., Thein S.L., Eaton W.A. Treating sickle cell anemia. Science. 2020;367(6483):1198–1199. doi: 10.1126/science.aba3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharma A., Jagannath V.A., Puri L. Hematopoietic stem cell transplantation for people with beta-thalassaemia. Cochrane Database Syst Rev. 2021;4CD008708. [DOI] [PMC free article] [PubMed]
- 96.Zittersteijn H.A., Harteveld C.L., Klaver-Flores S., Lankester A.C., Hoeben R.C., Staal F.J.T., et al. A small key for a heavy door: genetic therapies for the treatment of hemoglobinopathies. Front Genome Editing. 2021;2 doi: 10.3389/fgeed.2020.617780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mansilla-Soto J., Riviere I., Boulad F., Sadelain M. Cell and gene therapy for the beta-thalassemias: advances and prospects. Hum Gene Ther. 2016;27(4):295–304. doi: 10.1089/hum.2016.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang J., Yan J., Zeng F. Recent progress on genetic diagnosis and therapy for beta-thalassemia in china and around the world. Hum Gene Ther. 2018;29(2):197–203. doi: 10.1089/hum.2017.228. [DOI] [PubMed] [Google Scholar]
- 99.Pauling L., Itano H.A., et al. Sickle cell anemia, a molecular disease. Science. 1949;109(2835):443. [PubMed] [Google Scholar]
- 100.Ware R.E., de Montalembert M., Tshilolo L., Abboud M.R. Sickle cell disease. The Lancet. 2017;390(10091):311–323. doi: 10.1016/S0140-6736(17)30193-9. [DOI] [PubMed] [Google Scholar]
- 101.Persons D.A. Gene therapy: targeting beta-thalassaemia. Nature. 2010;467(7313):277–278. doi: 10.1038/467277a. [DOI] [PubMed] [Google Scholar]
- 102.Thompson A.A., Walters M.C., Kwiatkowski J., Rasko J.E.J., Ribeil J.A., Hongeng S., et al. Gene therapy in patients with transfusion-dependent beta-thalassemia. N Engl J Med. 2018;378(16):1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 103.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N Engl J Med. 2021;384(3):252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 104.Piel F.B. The present and future global burden of the inherited disorders of hemoglobin. Hematol Oncol Clin North Am. 2016;30(2):327–341. doi: 10.1016/j.hoc.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 105.Liang S., Moghimi B., Yang T.P., Strouboulis J., Bungert J. Locus control region mediated regulation of adult beta-globin gene expression. J Cell Biochem. 2008;105(1):9–16. doi: 10.1002/jcb.21820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Q., Peterson K.R., Fang X., Stamatoyannopoulos G. Locus control regions. Blood. 2002;100(9):3077–3086. doi: 10.1182/blood-2002-04-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pawliuk R., Westerman K.A., Fabry M.E., Payen E., Tighe R., Bouhassira E.E., et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294(5550):2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- 108.May C., Rivella S., Callegari J., Heller G., Gaensler K.M., Luzzatto L., et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406(6791):82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 109.Porcu S., Kitamura M., Witkowska E., Zhang Z., Mutero A., Lin C., et al. The human beta globin locus introduced by YAC transfer exhibits a specific and reproducible pattern of developmental regulation in transgenic mice. Blood. 1997;90(11):4602–4609. [PubMed] [Google Scholar]
- 110.Bouhassira E.E., Westerman K., Leboulch P. Transcriptional behavior of LCR enhancer elements integrated at the same chromosomal locus by recombinase-mediated cassette exchange. Blood. 1997;90(9):3332–3344. [PubMed] [Google Scholar]
- 111.Talbot D., Collis P., Antoniou M., Vidal M., Grosveld F., Greaves D.R. A dominant control region from the human beta-globin locus conferring integration site-independent gene expression. Nature. 1989;338(6213):352–355. doi: 10.1038/338352a0. [DOI] [PubMed] [Google Scholar]
- 112.Dzierzak E.A., Papayannopoulou T., Mulligan R.C. Lineage-specific expression of a human beta-globin gene in murine bone marrow transplant recipients reconstituted with retrovirus-transduced stem cells. Nature. 1988;331(6151):35–41. doi: 10.1038/331035a0. [DOI] [PubMed] [Google Scholar]
- 113.Drakopoulou E., Papanikolaou E., Georgomanoli M., Anagnou N.P. Towards more successful gene therapy clinical trials for beta-thalassemia. Curr Mol Med. 2013;13(8):1314–1330. doi: 10.2174/15665240113139990064. [DOI] [PubMed] [Google Scholar]
- 114.Davis R., Gurumurthy A., Hossain M.A., Gunn E.M., Bungert J. Engineering globin gene expression. Mol Ther Methods Clin Dev. 2019:12102–21210. doi: 10.1016/j.omtm.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ferrari G., Cavazzana M., Mavilio F. Gene therapy approaches to hemoglobinopathies. Hematol Oncol Clin North Am. 2017;31(5):835–852. doi: 10.1016/j.hoc.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 116.Goodman M.A., Malik P. The potential of gene therapy approaches for the treatment of hemoglobinopathies: achievements and challenges. Ther Adv Hematol. 2016;7(5):302–315. doi: 10.1177/2040620716653729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Karlsson S., Papayannopoulou T., Schweiger S.G., Stamatoyannopoulos G., Nienhuis A.W. Retroviral-mediated transfer of genomic globin genes leads to regulated production of RNA and protein. Proc Natl Acad Sci U S A. 1987;84(8):2411–2415. doi: 10.1073/pnas.84.8.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grosveld F., van Assendelft G.B., Greaves D.R., Kollias G. Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987;51(6):975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 119.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Negre O., Eggimann A.V., Beuzard Y., Ribeil J.A., Bourget P., Borwornpinyo S., et al. Gene Therapy of the beta-hemoglobinopathies by lentiviral transfer of the beta(A(T87Q))-globin gene. Hum Gene Ther. 2016;27(2):148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Negre O., Bartholomae C., Beuzard Y., Cavazzana M., Christiansen L., Courne C., et al. Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of beta-thalassemia and sickle cell disease. Curr Gene Ther. 2015;15(1):64–81. doi: 10.2174/1566523214666141127095336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jagadeeswaran R., Rivers A. Evolving treatment paradigms in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2017;2017(1):440–446. doi: 10.1182/asheducation-2017.1.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ribeil J.A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 124.Elisa Magrin P., Michaela Semeraro, MD PhD, Alessandra Magnani, MD PhD, Hervé Puy, MD PhD, Annarita Miccio, PhD, Nicolas Hebert, Jean-Sebastien Diana, MD, Francois Lefrere, MD, Felipe Suarez, MD PhD, Olivier Hermine, MD PhD, Valentine Brousse, MD, Catherine Poirot, MD PhD, Philippe Bourget, PharmD, PhD, Wassim El Nemer, PhD, Isabelle Guichard, MD, Despina Moshous, MD PhD, Benedicte Neven, MD, Fabrice Monpoux, MD, Marilyne Poirée, MD, Pablo Bartolucci, MD PhD, Jean-François Meritet, MD, David Grévent, MD, Thibaud Lefebvre, Pharm D.,PhD, Mohammed Asmal, MD PhD, Erin Whitney, MBA, Marisa Gayron, MS, Wenmei Huang, PhD MS, Isabelle Funck-Brentano, PsyD, Mariane de Montalembert, MD PhD, Laure Joseph, MD, Jean-Antoine Ribeil, MD PhD, Marina Cavazzana. Results from the Completed Hgb-205 Trial of Lentiglobin for β-Thalassemia and Lentiglobin for Sickle Cell Disease Gene Therapy. Blood. 2019;134 (Supplement_1): 3358. https://doi.org/10.1182/blood-2019-127393.
- 125.Walters M.C., Tisdale J.F., Kwiatkowski J.L., Krishnamurti L., Mapara M.Y., Schmidt M., et al. Exploring the drivers of potential clinical benefit in initial patients treated in the Hgb-206 study of lentiglobin for Sickle Cell Disease (SCD) gene therapy. Blood. 2019;134(Supplement_1):2061. [Google Scholar]
- 126.Kanter J., Walters M.C., Krishnamurti L., Mapara M.Y., Kwiatkowski J.L., Rifkin-Zenenberg S., et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N Engl J Med. 2022;386(7):617–628. doi: 10.1056/NEJMoa2117175. [DOI] [PubMed] [Google Scholar]
- 127.Goyal S., Tisdale J., Schmidt M., Kanter J., Jaroscak J., Whitney D., et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med. 2022;386(2):138–147. doi: 10.1056/NEJMoa2109167. [DOI] [PubMed] [Google Scholar]
- 128.Uchida N., Hsieh M.M., Raines L., Haro-Mora J.J., Demirci S., Bonifacino A.C., et al. Development of a forward-oriented therapeutic lentiviral vector for hemoglobin disorders. Nat Commun. 2019;10(1):4479. doi: 10.1038/s41467-019-12456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Morgan R.A., Unti M.J., Aleshe B., Brown D., Osborne K.S., Koziol C., et al. Improved titer and gene transfer by lentiviral vectors using novel, small beta-globin locus control region elements. Mol Ther. 2020;28(1):328–340. doi: 10.1016/j.ymthe.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Honsho M., Okumoto K., Tamura S., Fujiki Y. Peroxisome biogenesis disorders. Adv Exp Med Biol. 2020:129945–129954. doi: 10.1007/978-3-030-60204-8_4. [DOI] [PubMed] [Google Scholar]
- 131.Waterham H.R., Ferdinandusse S., Wanders R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta. 2016;1863(5):922–933. doi: 10.1016/j.bbamcr.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 132.Cotrim A.P., Baum B.J. Gene therapy: some history, applications, problems, and prospects. Toxicol Pathol. 2008;36(1):97–103. doi: 10.1177/0192623307309925. [DOI] [PubMed] [Google Scholar]
- 133.Piguet F., Alves S., Cartier N. Clinical gene therapy for neurodegenerative diseases: past, present, and future. Hum Gene Ther. 2017;28(11):988–1003. doi: 10.1089/hum.2017.160. [DOI] [PubMed] [Google Scholar]
- 134.Neumann H. Microglia: a cellular vehicle for CNS gene therapy. J Clin Invest. 2006;116(11):2857–2860. doi: 10.1172/JCI30230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Issa S.S., Shaimardanova A.A., Valiullin V.V., Rizvanov A.A., Solovyeva V.V. Mesenchymal stem cell-based therapy for lysosomal storage diseases and other neurodegenerative disorders. Front Pharmacol. 2022 doi: 10.3389/fphar.2022.859516. 13859516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cabanillas Stanchi K.M., Bohringer J., Strolin M., Groeschel S., Lenglinger K., Treuner C., et al. Hematopoietic stem cell transplantation with mesenchymal stromal cells in children with metachromatic leukodystrophy. Stem Cells Dev. 2022;31(7–8):163–175. doi: 10.1089/scd.2021.0352. [DOI] [PubMed] [Google Scholar]
- 137.Privolizzi R., Chu W.S., Tijani M., Ng J. Viral gene therapy for paediatric neurological diseases: progress to clinical reality. Dev Med Child Neurol. 2021;63(9):1019–1029. doi: 10.1111/dmcn.14885. [DOI] [PubMed] [Google Scholar]
- 138.Edelmann M.J., Maegawa G.H.B. CNS-targeting therapies for lysosomal storage diseases. Curr Adv Challenges Front Mol Biosci. 2020 doi: 10.3389/fmolb.2020.559804. 7559804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Conzelmann E., Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6(1):58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- 140.Platt F.M., d'Azzo A., Davidson B.L., Neufeld E.F., Tifft C.J. Lysosomal storage diseases. Nat Rev Dis Primers. 2018;4(1):27. doi: 10.1038/s41572-018-0025-4. [DOI] [PubMed] [Google Scholar]
- 141.Bonam S.R., Wang F., Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discov. 2019;18(12):923–948. doi: 10.1038/s41573-019-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fernandez-Pereira C., San M.-T., Gallardo-Gomez M., Perez-Marquez T., Alves-Villar M., Melcon-Crespo C., et al. Therapeutic approaches in lysosomal storage diseases. Biomolecules. 2021;11(12) doi: 10.3390/biom11121775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dali C.I., Sevin C., Krageloh-Mann I., Giugliani R., Sakai N., Wu J., et al. Safety of intrathecal delivery of recombinant human arylsulfatase A in children with metachromatic leukodystrophy: results from a phase 1/2 clinical trial. Mol Genet Metab. 2020;131(1–2):235–244. doi: 10.1016/j.ymgme.2020.07.002. [DOI] [PubMed] [Google Scholar]
- 144.Beschle J., Doring M., Kehrer C., Raabe C., Bayha U., Strolin M., et al. Early clinical course after hematopoietic stem cell transplantation in children with juvenile metachromatic leukodystrophy. Mol Cell Pediatr. 2020;7(1):12. doi: 10.1186/s40348-020-00103-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.van den Broek B.T.A., Page K., Paviglianiti A., Hol J., Allewelt H., Volt F., et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv. 2018;2(1):49–60. doi: 10.1182/bloodadvances.2017010645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Martin H.R., Poe M.D., Provenzale J.M., Kurtzberg J., Mendizabal A., Escolar M.L. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol Blood Marrow Transplant. 2013;19(4):616–624. doi: 10.1016/j.bbmt.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 147.Smith N.J., Marcus R.E., Sahakian B.J., Kapur N., Cox T.M. Haematopoietic stem cell transplantation does not retard disease progression in the psycho-cognitive variant of late-onset metachromatic leukodystrophy. J Inherit Metab Dis. 2010;33 Suppl:3S471-475. doi: 10.1007/s10545-010-9240-1. [DOI] [PubMed] [Google Scholar]
- 148.Kurtzberg J. Gene therapy offers new hope for children with metachromatic leukodystrophy. Lancet. 2022;399(10322):338–339. doi: 10.1016/S0140-6736(22)00057-5. [DOI] [PubMed] [Google Scholar]
- 149.Karimzadeh P., Ghofrani M., Nasiri S. Approach to patients with neurometabolic diseases who show characteristic signs and symptoms. Iran J Child Neurol. 2020;14(3):19–32. [PMC free article] [PubMed] [Google Scholar]
- 150.Koto Y., Sakai N., Lee Y., Kakee N., Matsuda J., Tsuboi K., et al. Prevalence of patients with lysosomal storage disorders and peroxisomal disorders: a nationwide survey in Japan. Mol Genet Metab. 2021;133(3):277–288. doi: 10.1016/j.ymgme.2021.05.004. [DOI] [PubMed] [Google Scholar]
- 151.Poletti V., Biffi A. Gene-based approaches to inherited neurometabolic diseases. Hum Gene Ther. 2019;30(10):1222–1235. doi: 10.1089/hum.2019.190. [DOI] [PubMed] [Google Scholar]
- 152.Pampols T. Inherited metabolic rare disease. Adv Exp Med Biol. 2010:686397–686431. doi: 10.1007/978-90-481-9485-8_23. [DOI] [PubMed] [Google Scholar]
- 153.Ballabio A., Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793(4):684–696. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 154.Bosch M.E., Kielian T. Neuroinflammatory paradigms in lysosomal storage diseases. Front Neurosci. 2015;9417 doi: 10.3389/fnins.2015.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cassis L., Cortes-Saladelafont E., Molero-Luis M., Yubero D., Gonzalez M.J., Ormazabal A., et al. Review and evaluation of the methodological quality of the existing guidelines and recommendations for inherited neurometabolic disorders. Orphanet J Rare Dis. 2015;10164 doi: 10.1186/s13023-015-0376-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tabarki B, Ortigoza-Escobar JD, Lee WT, AlFadhel M. Editorial: Pediatric Neurometabolic Disorders. Front Neurol. 2021;12737398. [DOI] [PMC free article] [PubMed]
- 157.Hynds R.E., Bonfanti P., Janes S.M. Regenerating human epithelia with cultured stem cells: feeder cells, organoids and beyond. EMBO Mol Med. 2018;10(2):139–150. doi: 10.15252/emmm.201708213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hirsch T., Rothoeft T., Teig N., Bauer J.W., Pellegrini G., De Rosa L., et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680):327–332. doi: 10.1038/nature24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mavilio F., Pellegrini G., Ferrari S., Di Nunzio F., Di Iorio E., Recchia A., et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12(12):1397–1402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- 160.Welponer T., Prodinger C., Pinon-Hofbauer J., Hintersteininger A., Breitenbach-Koller H., Bauer J.W., et al. Clinical perspectives of gene-targeted therapies for epidermolysis bullosa. Dermatol Ther (Heidelb) 2021;11(4):1175–1197. doi: 10.1007/s13555-021-00561-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Koller U., Bauer J.W. Gene replacement therapies for genodermatoses: a status quo. Front Genet. 2021;12658295. [DOI] [PMC free article] [PubMed]
- 162.Jayarajan V., Kounatidou E., Qasim W., Di W.L. Ex vivo gene modification therapy for genetic skin diseases-recent advances in gene modification technologies and delivery. Exp Dermatol. 2021;30(7):887–896. doi: 10.1111/exd.14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Keith A.R., Twaroski K., Ebens C.L., Tolar J. Leading edge: emerging drug, cell, and gene therapies for junctional epidermolysis bullosa. Expert Opin Biol Ther. 2020;20(8):911–923. doi: 10.1080/14712598.2020.1740678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Has C., Bauer J.W., Bodemer C., Bolling M.C., Bruckner-Tuderman L., Diem A., et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–627. doi: 10.1111/bjd.18921. [DOI] [PubMed] [Google Scholar]
- 165.Guttmann-Gruber C., Bauer J.W., Piñón H.J. Hereditary bullous diseases: current and innovative models to study the skin blistering disease epidermolysis bullosa. Drug Discovery Today: Disease Models. 2020:3217–3225. [Google Scholar]
- 166.Bardhan A., Bruckner-Tuderman L., Chapple I.L.C., Fine J.D., Harper N., Has C., et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6(1):78. doi: 10.1038/s41572-020-0210-0. [DOI] [PubMed] [Google Scholar]
- 167.Kueckelhaus M., Rothoeft T., De Rosa L., Yeni B., Ohmann T., Maier C., et al. Transgenic epidermal cultures for junctional epidermolysis bullosa - 5-year outcomes. N Engl J Med. 2021;385(24):2264–2270. doi: 10.1056/NEJMoa2108544. [DOI] [PubMed] [Google Scholar]
- 168.Uitto J. Toward treatment and cure of epidermolysis bullosa. Proc Natl Acad Sci U S A. 2019 doi: 10.1073/pnas.1919347117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Siprashvili Z., Nguyen N.T., Gorell E.S., Loutit K., Khuu P., Furukawa L.K., et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA. 2016;316(17):1808–1817. doi: 10.1001/jama.2016.15588. [DOI] [PubMed] [Google Scholar]
- 170.Gaucher S., Lwin S.M., Titeux M., Abdul-Wahab A., Pironon N., Izmiryan A., et al. EBGene trial: patient preselection outcomes for the European GENEGRAFT ex vivo phase I/II gene therapy trial for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2020;182(3):794–797. doi: 10.1111/bjd.18559. [DOI] [PubMed] [Google Scholar]
- 171.June C.H., Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.June C.H., O'Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 173.Sadelain M. CD19 CAR T cells. Cell. 2017;171(7):1471. doi: 10.1016/j.cell.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 174.Weber E.W., Maus M.V., Mackall C.L. The emerging landscape of immune cell therapies. Cell. 2020;181(1):46–62. doi: 10.1016/j.cell.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Waldman A.D., Fritz J.M., Lenardo M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Huang R., Li X., He Y., Zhu W., Gao L., Liu Y., et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86. doi: 10.1186/s13045-020-00910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sterner R.C., Sterner R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Irving B.A., Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64(5):891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- 179.Cross D., Burmester J.K. Gene therapy for cancer treatment: past, present and future. Clin Med Res. 2006;4(3):218–227. doi: 10.3121/cmr.4.3.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., et al. Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Locke F.L., Neelapu S.S., Bartlett N.L., Siddiqi T., Chavez J.C., Hosing C.M., et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Park J.H., Geyer M.B., Brentjens R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312–3320. doi: 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zheng P.P., Kros J.M., Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today. 2018;23(6):1175–1182. doi: 10.1016/j.drudis.2018.02.012. [DOI] [PubMed] [Google Scholar]
- 184.Sadelain M. From the guest editor: the rise of CAR therapy: the CD19 paradigm, and beyond. Introduction Cancer J. 2014;20(2):105–106. doi: 10.1097/PPO.0000000000000042. [DOI] [PubMed] [Google Scholar]
- 185.Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Weisel K., Martin T., Krishnan A., Jagannath S., Londhe A., Nair S., et al. Comparative efficacy of ciltacabtagene autoleucel in CARTITUDE-1 vs Physician's choice of therapy in the long-term follow-up of POLLUX, CASTOR, and EQUULEUS clinical trials for the treatment of patients with relapsed or refractory multiple myeloma. Clin Drug Investig. 2022;42(1):29–41. doi: 10.1007/s40261-021-01100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Iacoboni G., Rejeski K., Villacampa G., van Doesum J.A., Chiappella A., Bonifazi F., et al. Real-world evidence of brexucabtagene autoleucel for the treatment of relapsed or refractory mantle cell lymphoma. Blood Adv. 2022 doi: 10.1182/bloodadvances.2021006922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Frey N.V. Approval of brexucabtagene autoleucel for adults with relapsed and refractory acute lymphocytic leukemia. Blood. 2022 doi: 10.1182/blood.2021014892. [DOI] [PubMed] [Google Scholar]
- 190.Cohen A.D., Parekh S., Santomasso B.D., Gallego P.-L., van de Donk N., Arnulf B., et al. Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J. 2022;12(2):32. doi: 10.1038/s41408-022-00629-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Anderson M.K., Torosyan A., Halford Z. Brexucabtagene autoleucel: a novel chimeric antigen receptor T-cell therapy for the treatment of mantle cell lymphoma. Ann Pharmacother. 2022;56(5):609–619. doi: 10.1177/10600280211026338. [DOI] [PubMed] [Google Scholar]
- 192.Khurana A., Dalland J.C., Young J.R., Inwards D.J., Paludo J. Brexucabtagene autoleucel therapy induces complete remission in a primary refractory blastoid mantle cell lymphoma with neurolymphomatosis. Am J Hematol. 2021;96(8):E298–E301. doi: 10.1002/ajh.26233. [DOI] [PubMed] [Google Scholar]
- 193.Berdeja J.G., Madduri D., Usmani S.Z., Jakubowiak A., Agha M., Cohen A.D., et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–324. doi: 10.1016/S0140-6736(21)00933-8. [DOI] [PubMed] [Google Scholar]
- 194.Cerrano M., Ruella M., Perales M.A., Vitale C., Faraci D.G., Giaccone L., et al. The advent of CAR T-cell therapy for lymphoproliferative neoplasms: integrating research into clinical practice. Front Immunol. 2020:11888. doi: 10.3389/fimmu.2020.00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sermer D., Brentjens R. CAR T-cell therapy: full speed ahead. Hematol Oncol. 2019;37 doi: 10.1002/hon.2591. Suppl 195-100. [DOI] [PubMed] [Google Scholar]
- 196.Mohanty R., Chowdhury C.R., Arega S., Sen P., Ganguly P., Ganguly N. CAR T cell therapy: a new era for cancer treatment (Review) Oncol Rep. 2019;42(6):2183–2195. doi: 10.3892/or.2019.7335. [DOI] [PubMed] [Google Scholar]
- 197.Gifford R.J., Blomberg J., Coffin J.M., Fan H., Heidmann T., Mayer J., et al. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology. 2018;15(1):59. doi: 10.1186/s12977-018-0442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sheridan P.L., Bodner M., Lynn A., Phuong T.K., DePolo N.J., de la Vega D.J., et al. Generation of retroviral packaging and producer cell lines for large-scale vector production and clinical application: improved safety and high titer. Mol Ther. 2000;2(3):262–275. doi: 10.1006/mthe.2000.0123. [DOI] [PubMed] [Google Scholar]
- 199.Clay T.M., Custer M.C., Spiess P.J., Nishimura M.I. Potential use of T cell receptor genes to modify hematopoietic stem cells for the gene therapy of cancer. Pathol Oncol Res. 1999;5(1):3–15. doi: 10.1053/paor.1999.0003. [DOI] [PubMed] [Google Scholar]
- 200.Onodera M., Nelson D.M., Yachie A., Jagadeesh G.J., Bunnell B.A., Morgan R.A., et al. Development of improved adenosine deaminase retroviral vectors. J Virol. 1998;72(3):1769–1774. doi: 10.1128/jvi.72.3.1769-1774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Alford R.L., Honda S., Lawrence C.B., Belmont J.W. RNA secondary structure analysis of the packaging signal for Moloney murine leukemia virus. Virology. 1991;183(2):611–619. doi: 10.1016/0042-6822(91)90990-s. [DOI] [PubMed] [Google Scholar]
- 202.Adam M.A., Miller A.D. Identification of a signal in a murine retrovirus that is sufficient for packaging of nonretroviral RNA into virions. J Virol. 1988;62(10):3802–3806. doi: 10.1128/jvi.62.10.3802-3806.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bender M.A., Palmer T.D., Gelinas R.E., Miller A.D. Evidence that the packaging signal of Moloney murine leukemia virus extends into the gag region. J Virol. 1987;61(5):1639–1646. doi: 10.1128/jvi.61.5.1639-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mann R., Mulligan R.C., Baltimore D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell. 1983;33(1):153–159. doi: 10.1016/0092-8674(83)90344-6. [DOI] [PubMed] [Google Scholar]
- 205.Gandara C., Affleck V., Stoll E.A. Manufacture of third-generation lentivirus for preclinical use, with process development considerations for translation to good manufacturing practice. Hum Gene Ther Methods. 2018;29(1):1–15. doi: 10.1089/hgtb.2017.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Vargas J.E., Chicaybam L., Stein R.T., Tanuri A., Delgado-Canedo A., Bonamino M.H. Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med. 2016;14(1):288. doi: 10.1186/s12967-016-1047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Marini B., Kertesz-Farkas A., Ali H., Lucic B., Lisek K., Manganaro L., et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015;521(7551):227–231. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 208.Yi Y., Noh M.J., Lee K.H. Current advances in retroviral gene therapy. Curr Gene Ther. 2011;11(3):218–228. doi: 10.2174/156652311795684740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Evans M.J., Bacharach E., Goff S.P. RNA sequences in the Moloney murine leukemia virus genome bound by the Gag precursor protein in the yeast three-hybrid system. J Virol. 2004;78(14):7677–7684. doi: 10.1128/JVI.78.14.7677-7684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dubensky T.W., Jr., Sauter S.L. Generation of retroviral packaging and producer cell lines for large-scale vector production with improved safety and titer. Methods Mol Med. 2003:76309–76330. doi: 10.1385/1-59259-304-6:309. [DOI] [PubMed] [Google Scholar]
- 211.Li Z., Dullmann J., Schiedlmeier B., Schmidt M., von Kalle C., Meyer J., et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296(5567):497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- 212.Soldi M., Sergi S.L., Unali G., Kerzel T., Cuccovillo I., Capasso P., et al. Laboratory-scale lentiviral vector production and purification for enhanced ex vivo and in vivo genetic engineering. Mol Ther Methods Clin Dev. 2020:19411–21925. doi: 10.1016/j.omtm.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bauler M., Roberts J.K., Wu C.C., Fan B., Ferrara F., Yip B.H., et al. Production of lentiviral vectors using suspension cells grown in serum-free media. Mol Ther Methods Clin Dev. 2020:1758–1768. doi: 10.1016/j.omtm.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Poletti V., Mavilio F. Designing lentiviral vectors for gene therapy of genetic diseases. Viruses. 2021;13(8) doi: 10.3390/v13081526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.O'Leary K. Gene editing advances on all fronts. Nat Med. 2021;27(12):2056. doi: 10.1038/s41591-021-01607-z. [DOI] [PubMed] [Google Scholar]
- 216.Elisseeff J., Badylak S.F., Boeke J.D. Immune and genome engineering as the future of transplantable tissue. N Engl J Med. 2021;385(26):2451–2462. doi: 10.1056/NEJMra1913421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhang Y., Zhang F., Li X., Baller J.A., Qi Y., Starker C.G., et al. Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol. 2013;161(1):20–27. doi: 10.1104/pp.112.205179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Beurdeley M., Bietz F., Li J., Thomas S., Stoddard T., Juillerat A., et al. Compact designer TALENs for efficient genome engineering. Nat Commun. 2013;41762 doi: 10.1038/ncomms2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sanjana N.E., Cong L., Zhou Y., Cunniff M.M., Feng G., Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7(1):171–192. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 225.Wang H., La Russa M., Qi L.S. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem. 2016:85227–85264. doi: 10.1146/annurev-biochem-060815-014607. [DOI] [PubMed] [Google Scholar]
- 226.Bortesi L., Fischer R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv. 2015;33(1):41–52. doi: 10.1016/j.biotechadv.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 227.Hutter G., Nowak D., Mossner M., Ganepola S., Mussig A., Allers K., et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 228.Lederman M.M., Pike E. Ten years HIV free: an interview with “The Berlin Patient,” Timothy Ray Brown. Pathog Immun. 2017;2(3):422–430. doi: 10.20411/pai.v2i3.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Brown T.R. I am the Berlin patient: a personal reflection. AIDS Res Hum Retroviruses. 2015;31(1):2–3. doi: 10.1089/aid.2014.0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Brown T.R. The London patient. AIDS Res Hum Retroviruses. 2020;36(4):251–252. doi: 10.1089/AID.2020.0058. [DOI] [PubMed] [Google Scholar]
- 231.Gupta R.K., Peppa D., Hill A.L., Galvez C., Salgado M., Pace M., et al. Evidence for HIV-1 cure after CCR5Delta32/Delta32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV. 2020;7(5):e340–e347. doi: 10.1016/S2352-3018(20)30069-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lu Y., Xue J., Deng T., Zhou X., Yu K., Deng L., et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med. 2020;26(5):732–740. doi: 10.1038/s41591-020-0840-5. [DOI] [PubMed] [Google Scholar]
- 233.Ravi N.S., Wienert B., Wyman S.K., Bell H.W., George A., Mahalingam G., et al. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. Elife. 2022;11 doi: 10.7554/eLife.65421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Eridani S., Mosca A. Fetal hemoglobin reactivation and cell engineering in the treatment of sickle cell anemia. J Blood Med. 2011:223–230. doi: 10.2147/JBM.S14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Akinsheye I., Alsultan A., Solovieff N., Ngo D., Baldwin C.T., Sebastiani P., et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.A. Shahryari, M. Saghaeian Jazi, S. Mohammadi, H. Razavi Nikoo, Z. Nazari, E. S. Hosseini, et al., Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front Genet 2019 Vol. 10 Pages 868Accession Number: 31608113 PMCID: PMC6773888 DOI: 10.3389/fgene.2019.00868. [DOI] [PMC free article] [PubMed]
- 237.Hayakawa J., Washington K., Uchida N., Phang O., Kang E.M., Hsieh M.M., et al. Long-term vector integration site analysis following retroviral mediated gene transfer to hematopoietic stem cells for the treatment of HIV infection. PLoS ONE. 2009;4(1):e4211. doi: 10.1371/journal.pone.0004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Matsoukas I.G. Prime editing: genome editing for rare genetic diseases without double-strand breaks or donor DNA. Front Genet. 2020:11528. doi: 10.3389/fgene.2020.00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Xu W., Biswas J., Singer R.H., Rosbash M. Targeted RNA editing: novel tools to study post-transcriptional regulation. Mol Cell. 2021 doi: 10.1016/j.molcel.2021.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Katrekar D., Chen G., Meluzzi D., Ganesh A., Worlikar A., Shih Y.R., et al. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat Methods. 2019;16(3):239–242. doi: 10.1038/s41592-019-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Abudayyeh O.O., Gootenberg J.S., Franklin B., Koob J., Kellner M.J., Ladha A., et al. A cytosine deaminase for programmable single-base RNA editing. Science. 2019;365(6451):382–386. doi: 10.1126/science.aax7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bloomer H., Khirallah J., Li Y., Xu Q. CRISPR/Cas9 ribonucleoprotein-mediated genome and epigenome editing in mammalian cells. Adv Drug Deliv Rev. 2021;114087 doi: 10.1016/j.addr.2021.114087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Morgan R.A., Gray D., Lomova A., Kohn D.B. Hematopoietic stem cell gene therapy: progress and lessons learned. Cell Stem Cell. 2017;21(5):574–590. doi: 10.1016/j.stem.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]