

Abstract

Off-target side effects are major challenges hindering the clinical success of matrix metalloproteinase (MMP) inhibitors. Various targeting strategies revitalized MMP research to eliminate this drawback. Herein, we developed s-triazine-based dendrimeric architecture not only amenable to tumor targeting but also decorated with pharmacophoric entities to endow MMP-9 inhibition for halting cancer progression. The design rationale utilized hydrazide branching chains as well as carboxylic and hydroxamic acid termini as Zn-binding groups to confer substantial MMP inhibitory potential. The carboxylic acids are tetherable to tumor targeting ligands and other cargo payloads as synergistic drugs via biodegradable linkages. The synthesized series were screened for cytotoxicity against normal fibroblasts (Wi-38) and two selected cancers (MDA-MB 231 and Caco-2) via MTT assay. The most active hexacarboxylic acid dendrimer 8a was more potent and safer than Dox against MDA-MB 231 and Caco-2 cells. It intrinsically inhibited MMP-9 with selectivity over MMP-2. Docking simulations demonstrated that the extended carboxylic acid termini of 8a could possibly chelate the active site Zn of MMP-9 and form hydrogen-bonding interactions with the ligand essential backbone Tyr423. In addition, it suppressed the correlated oncogenic mediators VEGF and cyclin D, upregulated p21 expression, induced apoptosis (>75%), and inhibited the tumor cell migration (∼84%) in the treated cancer cells. Thus, up to our knowledge, it is the first triazine-based MMP-9 inhibitor dendrimer endowed with VEGF suppression potential that can be employed as a bioactive carrier.
1. Introduction
The tumor microenvironment, especially the extracellular matrix, has attracted considerable attention in cancer research. Such a matrix innately fosters various tumor progression events ranging from cancer cell proliferation to metastasis via a plethora of proteinases.1−3 Among the released proteinases, a characteristic family of zinc-dependent endopeptidases named matrix metalloproteinases (MMPs) has been reported to be obviously dysregulated in almost all human tumors.4−6 Their expression levels were found to be directly tied to the tumor stage.4 The MMP family comprises about 26 members7 classified as collagenases (MMP-1, -8, -13, -18), gelatinases (MMP-2, -9), stromelysins (MMP-3, -10), matrilysins (MMP-7, -26), membrane-type MMPs (MMP-14, -15, -16, -17, -24, -25), and other MMPs.8 Structurally, the catalytic domains of all of the family members are almost alike, comprising three α-helixes and five β-sheets. The domain is characterized by the active-site zinc ion, five calcium ions, and a shallow cleft with six binding pockets (S1, S2, S3, S1′, S2′ and S3′). The S1′ subsite slightly varies in its composition among various MMPs and is thus viewed as a selectivity pocket.7,9,10
Various MMPs promote extracellular matrix turnover, tumor growth, angiogenesis, and metastasis.11−13 Accordingly, MMPs are viewed as modulators of the tumor microenvironment and are major driving factors for carcinogenesis. No surprise, then, that MMP inhibition has been considered ideal for cancer treatment. Being validated as attractive druggable targets, numerous MMPs inhibitors have been introduced over the last decades.14−17 Early inhibitors were designed as small peptidomimetics of the endogenous MMP ligands capped with hydroxamic acid as an efficient zinc binding motif to render the enzyme inactive.17 Despite their outstanding inhibition potency and efficiency against cancer progression, hydroxamate-based peptidomimetics such as batimastat18 and marimastat failed to achieve clinical success.19 Such unexpected failure in the clinic has been mainly blamed on the pharmacokinetic challenges20,21 and the lack of selectivity-related side effects14,22 associated with the hydroxamate moiety. These doubts about the hydroxamates’ suitability as MMPs inhibitors then directed more extensive efforts to diversify the MMP inhibitors’ zinc binding groups. Hence, several non-hydroxamate inhibitors utilizing thiols, carboxylates, phosphorus-based groups, hydrazides, sulfonyl hydrazides, N-hydroxyurea, or other zinc binding moieties were introduced23 of which some potent inhibitors found their way to clinical trials with auspicious start, but eventually failed to accomplish clinical success due to their nonspecific broad spectrum activity.20,24 The growing knowledge on MMP crystal structures, particularly their catalytic domains9,10 together with computer-aided drug design,17 then revitalized MMP research in a fundamentally opposite strategy relying on avoiding zinc binding groups (ZBG) to minimize or hopefully eliminate off-target side effects.25,26 Consequently, numerous nonzinc binding MMP inhibitors were synthesized and evaluated.25−28 Although considerable success regarding selectivity has been achieved, this approach also faced a major challenge. Actually, this strategy has proven efficacy only for designing selective inhibitors for deep-pocket MMPs, particularly MMP-13, where the inhibitor relies on the intrinsic flexibility of the MMP specificity loop. The selectivity of these MMP inhibitors against other MMPs thus seems to be questionable.
The aforementioned problems associated with hydroxamic acids, non-hydroxamates, and non-zinc binding inhibitors posed a dilemma for medicinal chemists: “to bind or not to bind zinc?”.4 A pragmatic solution for optimal compromise between potency and selectivity may be proposed by targeting the tumor microenvironment itself. A recent study reported enhanced activity against MMP-2 and -9 and reduced off-target effects via targeted delivery of the hydroxamate-based MMP inhibitor, marimastat to the tumor microenvironment encapsulated in thermosensitive liposomes.29 Another fruitful targeting strategy relies on utilizing monoclonal antibodies. To date, several promising monoclonal antibodies targeting catalytic domains of specific MMPs without binding the catalytic zinc were developed.30 A prime example is DX-2400, a MMP-14 specific inhibitor with a subnanomolar inhibitory constant (Ki), by Dyax. In preclinical studies, the DX-2400 was observed to significantly decrease the tumor burden and limit metastases in liver and lung.31 Furthermore, researchers at Gilead Sciences produced andecaliximab (GS-5745), a humanized monoclonal antibody, as a highly potent and selective MMP-9 inhibitor that spares other MMPs. In mouse models, GS-5745 reduced the tumor growth and metastases incidence. Interestingly, the common side effects seen with broad-spectrum inhibitors were eliminated.32 Besides the mentioned advances in targeting strategies, the emerging role of dendrimers in the field has brought new light to MMP research.33−36 Dendrimers are repetitively branched molecules, usually adopting spherical structures with symmetry around the core.37,38 The treelike branched concentric layers of dendrimers are referred to as “generations”. Basically, dendrimers were employed as potential carriers for efficient drug delivery based on their unique properties ranging from monodispersity and small nanometer size to amenability for versatile functionalization that allow targeting to different tissues as well as enhancing bioavailability of the loaded drugs.39,40 Moreover, dendrimers could be easily decorated to make them “smart” enough to deliver its payloads to the target locus with controlled release.41 Such attractive applications encouraged synthetic chemistry studies to develop various classes of dendrimers such as poly(amidoamine) (PAMAM)42 and triazine-based ones.43 Pioneering medicinal chemistry research then introduced an “out of the box” strategy by derivatizing dendrimers with pharmacophoric entities to endow certain pharmacological activities, thus rendering them “druglike”.35,36,43−48 In this regard, some functionalized PAMAM dendrimers were designed as MMP inhibitors.35,36,43,44 On the other hand, triazine-based dendrimers and derivatives have not been popularly utilized so far in MMP research despite their wide biomedical applications.43,49−53
2. Design Rational
Motivated by recent research progress and in continuation of our previous work,54 we set out to synthesize a series of rationally designed star-shaped triazine-based dendrimers endowed with MMP inhibitory potential. Our research protocol was focused on targeting MMP-9 in two of the most widely spreading cancers, namely breast (MDA-MB 231) and colon (Caco-2) cancers,55 as it is one of the key MMP in their progression.56,57 The developed 1,3,5-triazine-based dendrimers were decorated with terminal carboxylic and hydroxamic acids as well as linker hydrazide moieties as zinc-binding groups to confer substantial MMP-inhibitory activity (Figure 1). The carboxylic acid groups will also allow feasible installation of cargo payloads such as drugs and/or tumor-targeting ligands via biodegradable linkages. Structure diversification of the designed scaffold was adopted by installing various pharmacophoric functionalities of potent selective MMP-9 as piperidine58 and morpholine59 on the terminal triazines.
Figure 1.

Design of the target dendrimers.
As a proof of concept, the synthesized dendrimers were subjected to cytotoxicity testing against normal fibroblasts (Wi-38) and two selected cancer cell lines (MDA-MB 231 and Caco-2) utilizing the MTT assay60−63 to evaluate their safety and antiproliferative profiles. Then the most promising dendrimers were evaluated for in vitro MMP-9 inhibition compared to the prototype MMP inhibitor N-isobutyl-N-(4-methoxyphenylsulfonyl)glycyl hydroxamic acid (NNGH) as a reference. Docking simulations were conducted to explore their possible binding modes within the MMP catalytic domain. Their selectivity profiles were evaluated against other members of the MMPs family namely, MMP-2, -7, -10, and -13. Mechanistic studies were extended to explore their apoptotic induction potential and antimetastatic activities as major MMP inhibition downstream events. Herein, flow cytometry analysis of the proapoptotic effect and migration inhibitory potential of the active dendrimers were investigated in the treated human cancer cell lines. The apoptotic potential of the prepared dendrimers was investigated after the treated cancer cells were stained with annexin V, which binds to phosphatidylserine. The latter is exposed on the outer leaflet of apoptotic cells to be removed by macrophages.64 The antimigration activity of the tested dendrimers was evaluated using a wound healing assay, which is a highly reproducible in vitro test used to examine the migration potential of monolayer cancer cells to reestablish cell contact following the development of a scratched wound.65 Additionally, their effects on the expression of correlated oncogenic mediators were investigated. Most importantly, vascular endothelial growth factor (VEGF) expression was evaluated in Caco-2 cells treated with the studied dendrimers given the fact that MMP-9 promotes cell migration and triggers the angiogenic switch during carcinogenesis via expression/secretion of VEGF.66,67 p21 and cyclin D expression was also quantified based on previous studies reporting that MMP-9 inhibition induces the expression of the CDK inhibitor p21.68
3. Results and Discussion
3.1. Chemistry
3.1.1. Preparation of Monochlorinated s-Triazine Derivatives 7a–c and Dendrimers 8a–c
As depicted in Scheme 1, the three Cl atoms of cyanuric chloride 1 were replaced with 4-aminobenzoic acid 2 to give the tribenzoic acid derivative 3.69 Subsequently, 3 was converted to the corresponding methyl ester derivative 4 and then to the corresponding acid hydrazide derivative 5.70 On the other hand, cyanuric chloride 1 was allowed to react with 1 equiv glycine 6 at 0–5 °C for 2 h, followed by the reaction with 1 equiv of glycine,71 morpholine,53,72 or piperidine53 at room temperature to afford the corresponding monochlorinated s-triazine derivatives 7a–c.
Scheme 1. Synthesis of Monochlorinated s-Triazine Derivatives 7a–c and Dendrimers 8a–c.
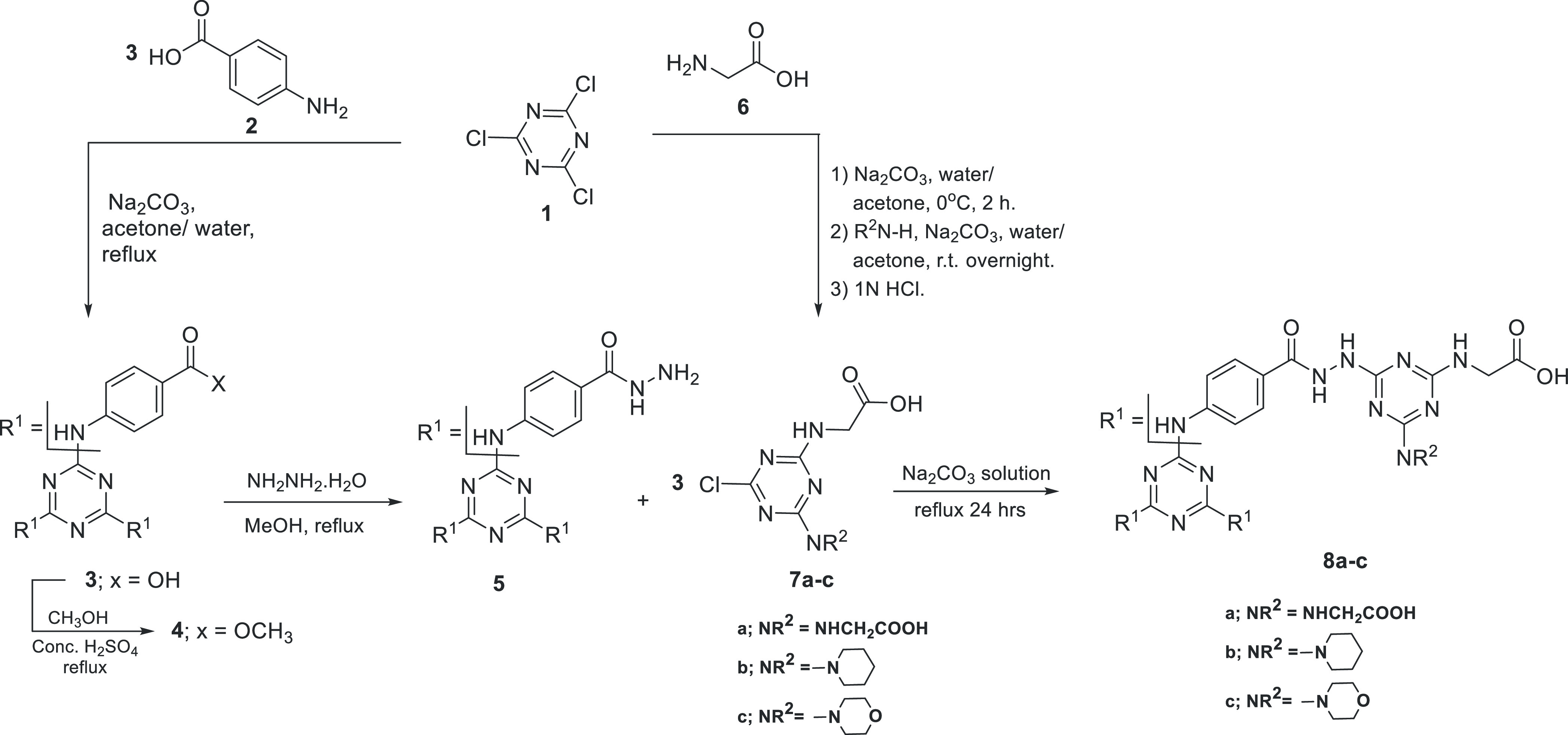
The previously prepared hydrazide derivative 5 was allowed to react with monochlorinated s-triazine derivatives 7a–c in aqueous sodium carbonate to afford the corresponding dendrimers 8a–c. IR spectra of 8a–c confirmed the presence of carboxylic OH, NH, and C=O functionalities at wavenumber ranges 3700–2400, 3412–3196, and 1753–1635 cm–1, respectively. The 1H NMR spectra of dendrimers 8a–c showed the presence of multiplet peaks at 3.75–4.01 and 7.64–8.19 ppm corresponding to glycine’s α-protons and aromatic protons, respectively. In addition, the 1H NMR spectrum of dendrimer 8b showed two multiplet peaks at chemical shifts of 1.55–1.63 and 3.79–4.01 ppm corresponding to the methylene protons of the piperidine moieties. While the 1H NMR spectrum of 8c showed two multiplet peaks at chemical shifts of 3.56–3.97 and 3.75–3.86 ppm corresponding to CH2N and CH2O of the morpholine moieties. Furthermore, 13C NMR spectra of 8a–c show the presence of signals at 41–42 and 119–130 ppm corresponding to the glycine α-carbon and aromatic carbons. Additional signals were observed at 23.27, 25.43, and 44.73 ppm for dendrimer 8b corresponding to the methylene carbons of the piperidine moieties. Moreover, the signals at 44.45 and 66.09 ppm of dendrimer 8c correspond to the CH2N and CH2O carbons, respectively, confirming the presence of morpholine moieties.
3.1.2. Preparation of Dendrimers 10–12
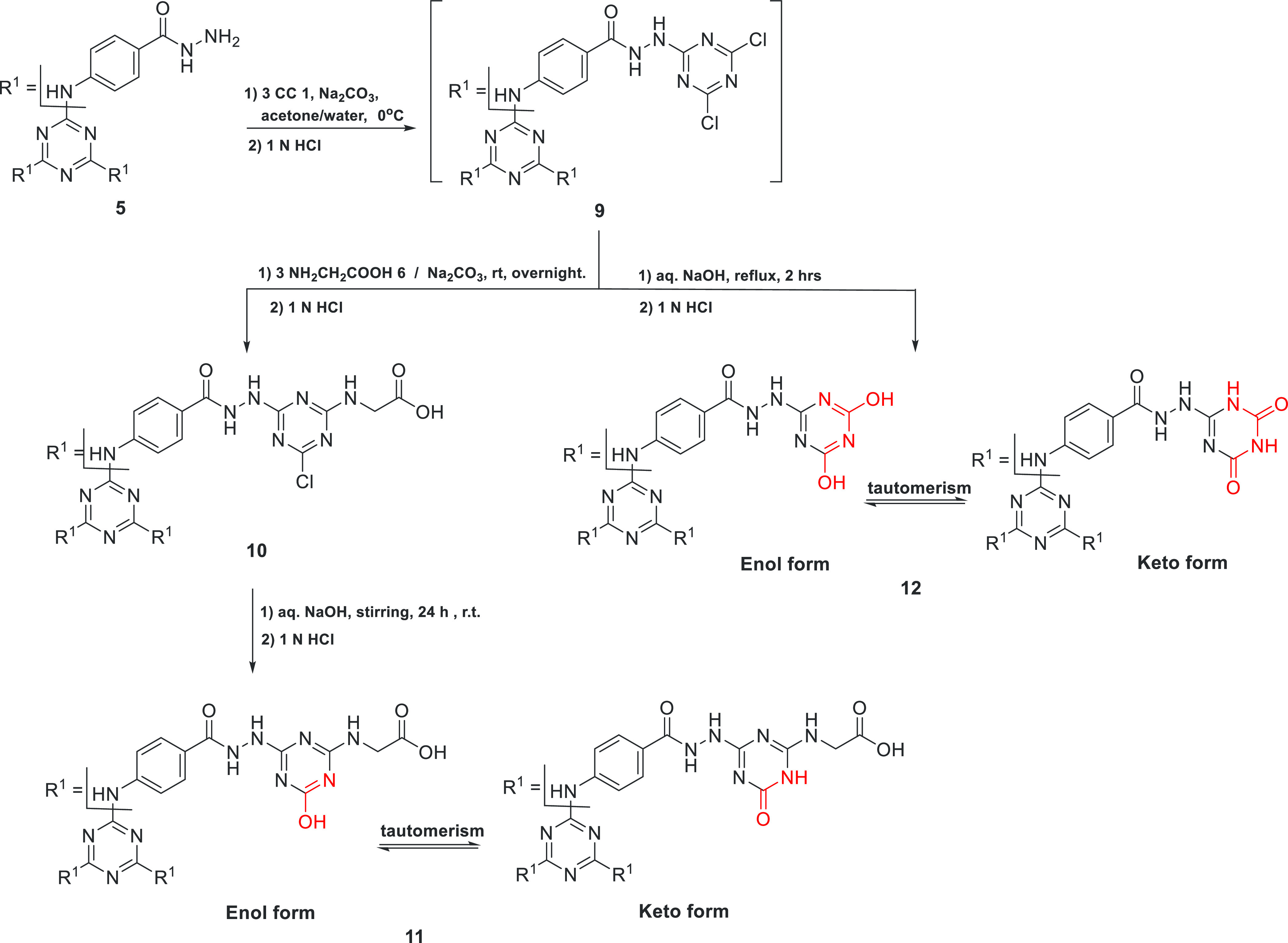
Similarly, the hydrazide derivative 5 reacted with 3 equiv of cyanuric chloride 1 to give the hexachlorinated precursor 9. Then dendrimer 9 was allowed to react with 3 equiv of glycine in the presence of sodium carbonate to give the corresponding trichlorinated s-triazine dendrimers 10. Furthermore, the trichlorinated s-triazine dendrimer 10 was converted to the corresponding trihydroxylated s-triazine dendrimer 11 by stirring with aqueous sodium hydroxide solution at room temperature for 24 h (Scheme 2). In addition, the hexachlorinated s-triazine dendrimer 9 was refluxed for 2 h in aqueous NaOH solution to afford the corresponding hexahydroxylated s-triazine dendrimer 12. The IR spectra of dendrimers 10–12 showed the presence of functionalities O-H, N-H, and C=O. The 1H NMR spectra of dendrimers 10 and 11 showed two multiplet peaks at chemical shift ranges 3.84–4.10 and 7.29–8.53 ppm corresponding to the methylene and aromatic protons, respectively. In addition, an increase in peak integration at chemical shift range 10.86–11.75 ppm by 3 protons in 1H NMR spectrum of 11, compared to that of 10, indicates the complete replacement of Cl-atoms in 10 with OH groups in 11. In addition, a carbon signal at about 42 ppm in the 13C NMR spectra of 10 and 11 confirms the presence of glycine residues. Furthermore, elemental analysis confirmed the conversion of dendrimer 10 to 11 as the Cl-atoms are absent in 11. The 1H NMR spectra of dendrimers 12 showed the presence of a multiplet peak at chemical shift range 7.37–8.04 ppm corresponding to 12 aromatic and 3 NH protons, while the remaining 6 NH and 6 OH protons appeared as two, D2O exchangeable, multiplets at chemical shift ranges 10.46–11.95 ppm. Furthermore, the overlapped, multiplet peaks at the chemical shift range 10.5–11.9 ppm of the s-triazine derivatives 11 and 12 could indicate the presence of keto and enol tautomers in a similar manner to cyanuric acid as shown in Scheme 2.73,74
Scheme 2. Synthesis of Dendrimers 10–12.
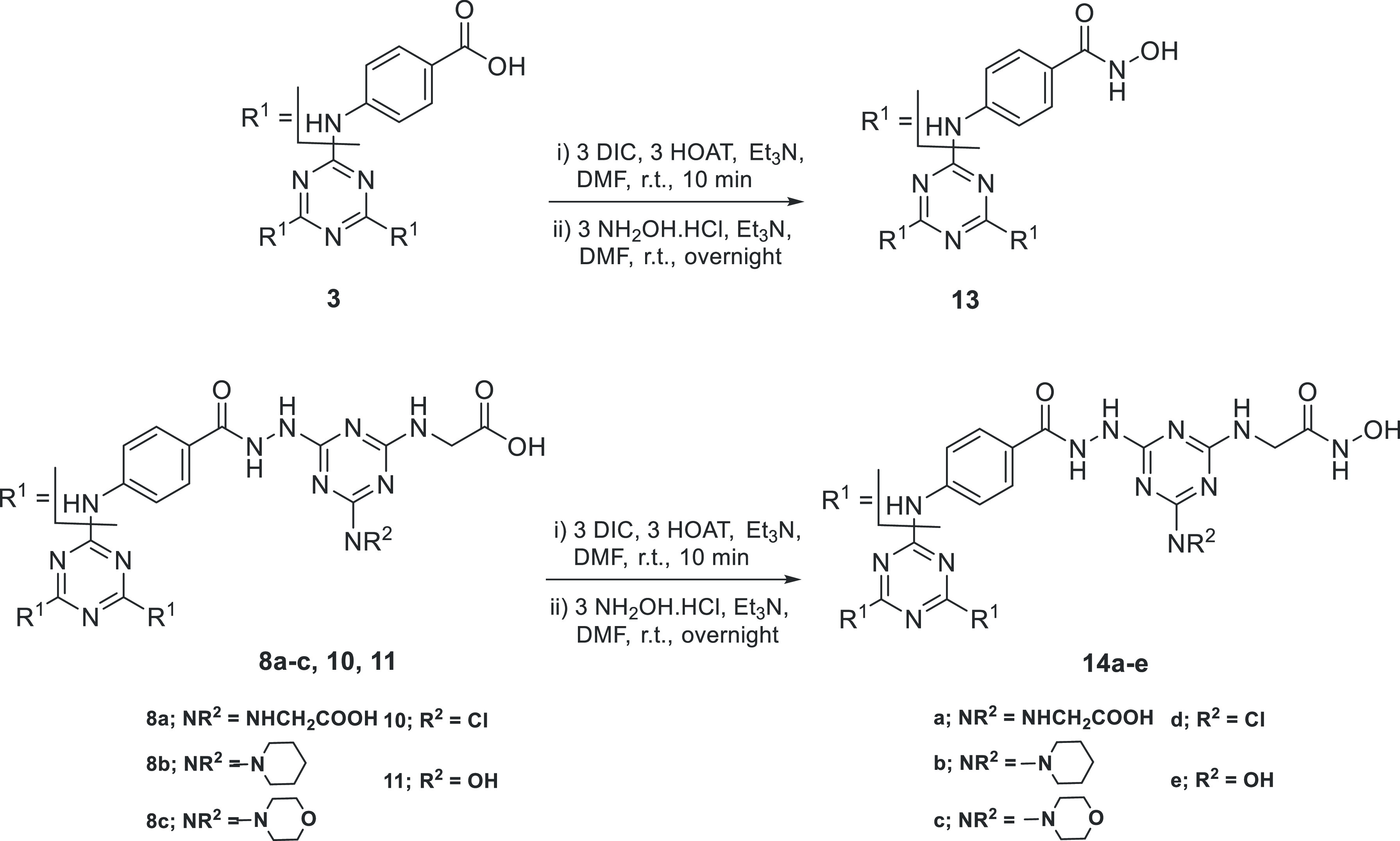
3.1.3. Preparation of Hydroxamic Acid Dendrimers 13 and 14a–e
All previously synthesized dendrimers that possess periphery carboxyl groups 3, 8a–c, 10, and 11 were allowed to react with hydroxylamine hydrochloride using DIC (N,N′-diisopropylcarbodiimide) in the presence of HOAt (1-hydroxy-7-azabenzotriazole) as coupling agent75,76 to give the corresponding hydroxamic acid dendrimers 13 and 14a–e, respectively (Scheme 3). The structure of hydroxamic acid dendrimers 13 and 14a–e was confirmed by spectroscopic methods (IR and 1H NMR and 13C NMR) as well as elemental analysis; see the Supporting Information. In the 1H NMR spectra of 13 and 14a–e, the NH and OH groups of the hydroxamic moieties, CONHOH, showed multiplet peaks at 8.20–9.13 and 10.02–12.20 ppm, respectively. In addition, elemental analysis showed an increase in N/C ratio upon conversion from dendrimers with carboxylic periphery to those with hydroxamic periphery.
Scheme 3. Synthesis of Hydroxamic Acid Dendrimers 13 and 14a–e.
3.2. Biological Evaluation
3.2.1. Cytotoxicity Screening
Cytotoxicity screening of the synthesized dendrimers, 8a–8c, 10–13, and 14a–14e on normal human fibroblasts (Wi-38) revealed that all the evaluated compounds showed higher safety profiles compared to doxorubicin except 8b, 8c, 11–13, 14c, and 14e (Table 1). Among the investigated dendrimers, the carboxyl-terminated dendrimer 10 was the safest, recording the highest EC100 value (17.121 nM), followed by the corresponding hydroxamate one 14d (15.591 nM). Promising safety profiles were also observed for the hexaacetic acid-terminated dendrimer 8a and the corresponding hydroxamate derivative 14a as well as the piperidinyl-substituted one 14b.
Table 1. Cytotoxicity Evaluation on Normal Human Fibroblasts (Wi-38).
| compd no. | EC100a (nM) |
|---|---|
| 8a | 13.8 ± 0.2 |
| 8b | 1.0 ± 0.1 |
| 8c | 1.9 ± 0.2 |
| 10 | 17.1 ± 0.1 |
| 11 | 3.0 ± 0.1 |
| 12 | 1.5 ± 0.0 |
| 13 | 5.8 ± 0.277 |
| 14a | 13.5 ± 0.2 |
| 14b | 15.0 ± 0.3 |
| 14c | 1.7 ± 0.1 |
| 14d | 15.6 ± 0.1 |
| 14e | 2.9 ± 0.2 |
| DOX | 11.5 ± 0.5 |
Values are presented as mean ± SEM.
Following assessment of the safety profiles, all of the studied compounds were evaluated for their potential antiproliferative activities against MDA-MB 231 and Caco-2 cells. As shown in Table 2, the carboxylic acid-terminated dendrimer 8a exhibited outstanding cytotoxic activities with single-digit nanomolar IC50 values against MDA-MB 231 and Caco-2 cells being more potent than doxorubicin as well as all the evaluated dendrimers in the current study. Besides this observation, its safety profile was promising. Hydoxamic acid coupling allowed slightly less potent antiproliferative activities as evidenced by the IC50 values of the respective dendrimer 14a. However, it is still superior to doxorubicin against Caco-2 cells with slightly lower potency against MDA-MB 231 cells. Obviously, Caco-2 cells were more sensitive than MDA-MB 231 to the evaluated compounds. All of the evaluated dendrimers were more active than doxorubicin against Caco-2 except 10–12. On the other hand, only 8a and 14a were superior to doxorubicin against MDA-MBA 231.
Table 2. Antiproliferative Evaluation on MDA-MB 231 and Caco-2 Cells.
| IC50a (nM) |
||
|---|---|---|
| compd no. | MDA-MB 231 | Caco-2 |
| 8a | 3.8 ± 0.7 | 3.3 ± 0.5 |
| 8b | 81.0 ± 14.3 | 42.0 ± 1.3 |
| 8c | 83.7 ± 9.0 | 26.8 ± 1.3 |
| 10 | 45.0 ± 7.6 | 54.7 ± 0.5 |
| 11 | 86.6 ± 4.1 | 64.8 ± 1.3 |
| 12 | 70.9 ± 3.7 | 52.8 ± 3.8 |
| 13 | 94.4 ± 10.6 | 38.4 ± 1.6 |
| 14a | 14.5 ± 1.6 | 10.2 ± 3.6 |
| 14b | 79.7 ± 10.2 | 39.0 ± 1.3 |
| 14c | 46.7 ± 6.9 | 39.1 ± 3.1 |
| 14d | 187.8 ± 8.1 | 21.5 ± 1.1 |
| 14e | 72.9 ± 5.3 | 34.6 ± 0.5 |
| DOX | 10.7 ± 0.3 | 48.3 ± 2.8 |
Values are presented as mean ± SEM.
3.2.2. Matrix Metalloproteinas-9 Inhibition
The selected safe and anticancer carboxylate- and hydroxamate-terminated dendrimers were in vitro evaluated for matrix metalloproteinase-9 (MMP-9) inhibition, as a possible anticancer mechanism, in comparison to NNGH as a reference MMP inhibitor (Table 3). Considering the close structural similarity of MMP-9 and MMP-2, the dendrimers were evaluated for their MMP-2 inhibitory potential as well. Interestingly, the results showed that 8a, the most active anticancer dendrimer, was a more potent MMP-9 inhibitor than the other evaluated dendrimers. Compounds 14b and 14d exhibited higher MMP-9 inhibition profiles than 14a. Although considerably active, all dendrimers were less potent than NNGH. Interestingly, 8a showed more than 2-folds selectivity to MMP-9 over MMP-2. Further investigation of the 8a selectivity profile revealed that 8a exhibited considerable potency against MMP-7, -10, and -13, being nearly equipotent against these family members. Compounds 14a and 14d were nearly 2-fold more active against MMP-9 than MMP-2, whereas 14b showed selectivity to MMP-2 over MMP-9. Notably, 14b was comparable to NNGH against MMP-2.
Table 3. MMPs Inhibitory Profiles of the Selected Dendrimers.
| IC50 (nM) |
|||||
|---|---|---|---|---|---|
| compd no. | MMP-2 | MMP-9 | MMP-7 | MMP-10 | MMP-13 |
| 8a | 630 | 156 | 200 | 145 | 124 |
| 14a | 728 | 373 | |||
| 14b | 98 | 207 | |||
| 14d | 541 | 243 | |||
| NNGH | 77 | 73 | 240 | 101 | 360 |
3.2.3. Morphological Examination of the Induced Apoptosis
The two cancer cell lines (MDA-MB 231 and Caco-2) were examined for morphological changes when treated with the most active dendrimers 8a and 14a in comparison with the untreated cancer cells and cells treated with the reference doxorubicin (Figure 2). As illustrated, all of the treated cells obviously lost their normal shapes. Additionally, their characteristic severe shrinkage indicated potent antiproliferative activities of the tested compounds, especially 8a, in comparison to doxorubicin.
Figure 2.

Morphological alterations of MDA-MB 231 and Caco-2 cells treated with the studied dendrimers 8a and 14a, at 3 nM, compared to the untreated control cells and doxorubicin (DOX)-treated cancer cells.
3.2.4. Flow Cytometric Analysis of Apoptosis
As shown in Table 4 and Figure 3; the dendrimers 8a and 14a possessed higher capability to induce apoptosis (>60%) in the tested human cancer cells than that of doxorubicin (<32%). Interestingly, the dendrimer 8a showed the highest potential among the group to induce apoptosis (>75%) in MDA-MB 231 and Caco-2. These results were consistent with the MTT assay results.
Table 4. Total Percentages of the Apoptotic Cell Population in the Most Active Compounds-Treated Cancer Cells Lines.
| total
% of the apoptotic cell populationa |
||
|---|---|---|
| compd no. | MDA-MBA 231 | Caco-2 |
| untreated control | 0.1 ± 0.02 | 0.1 ± 0.02 |
| 8a | 75.4 ± 2.0 | 76.8 ± 0.8 |
| 14a | 60.3 ± 2.2 | 71.3 ± 0.8 |
| DOX | 19.9 ± 1.6 | 31.7 ± 2.7 |
Values are presented as mean ± SEM.
Figure 3.

Flowcharts of annexin-PI analysis of MDA-MB 231 and Caco-2 cells treated with the studied dendrimers 8a and 14a, at 3 nM, compared to the untreated control cells and doxorubicin-treated cancer cells.
3.2.5. Tumor Cell Migration Inhibition
The antimetastatic capability of the studied dendrimers 8a and 14a was assessed utilizing the cell migration (wound healing) assay. The results (Figure 4) showed that 14a and 8a exhibited promising antimetastatic potential via inhibiting migration of Caco-2 by 83.95 ± 1.93%
Figure 4.

Antimetastatic potency of the studied dendrimers 8a and 14a via wound healing assay at their safest dose (0.1 nM) on cancer cells to avoid interfering with growth inhibitory effect. (A) Microscopic image of the scratched wounds in the untreated and 8a- and 14a-treated Caco-2 cells at 0 and 24 h and (B) migration impairment (%) in the treated cancer cells.
3.2.6. Expression of VEGF, p21, and Cyclin D
Based on the reports confirming association of MMP-9 with VEGF,65,66 p21, and cyclin D expression,67 quantitative real-time PCR analyses were performed to evaluate the regulatory potential of the studied dendrimers on these oncogenic mediators. Figure 5 demonstrates that 8a and 14a downregulated VEGF expression. Notably 14a had a unique ability to suppress VEGF expression especially in Caco-2 cells. Compound 8a upregulated p21 expression by ∼3-fold and downregulated the oncogenic expression of cyclin D by ∼4-fold in the treated MDA-MB 231 and Caco-2 cells, respectively. Compound 14a showed lower therapeutic regulatory potential against p21 and cyclin D. More importantly, the most active anticancer compounds (8a and 14a) were found to have a greater impact on the above-mentioned genes expression than DOX.
Figure 5.

Relative fold change in VEGF, p21, and cyclin D gene expression in (A) MDA-MB 231 and (B) Caco-2 cells treated with the studied dendrimers.
3.3. Molecular Docking Studies
Docking simulations were conducted to explore the possible binding mode of the most promising dendrimer 8a into the active site of MMP-9 utilizing MOE 2015.10.77 The MMP-9 catalytic domain, complexed with a reverse hydroxamate inhibitor, without fibronectin and prodomains was retrieved from the protein data bank (PDB ID: 1GKC(78)) and then subjected to structure preparation after removal of unwanted residues. The optimized domain consists of a monomer with three calcium and two zinc ions. The active dendrimer 8a was built in silico, energy minimized and docked into the cocrystallized ligand binding site. Docking simulations were conducted with various fitting protocols for validation. A rigid docking protocol was adopted applying the Triangular matcher algorithm and Alpha HB scoring function for generating the top 10 nonredundant poses of the lowest binding energy conformers of the studied dendrimer. Among the lowest binding energy conformers (ΔG = −9.72 kcal/mol), docking results (Figure 6) showed that most of the dendrimeric scaffold is likely to be extended outside the pocket, while the extended termini could reach and chelate the active site Zn via the carboxylic acid appendages as roughly expected. Interestingly, hydrogen bond interactions were also observed between the MMP-9 backbone amide groups of Tyr423 and two adjacent carboxylic acid termini posed by 8a resembling the binding mode of the cocrystallized reverse hydroxamate inhibitor.78 Additional hydrogen bonds involving the triazine-NH moiety and the backbone Met422 as well as H−π interactions between the triazine core and Tyr420 reinforced 8a fitting to the MMP-9 catalytic domain.
Figure 6.

(A) Molecular surface of MMP-9 catalytic domain (cyan) showing active site zinc ion as dark blue ball and the docked hexacarboxylic acid dendrimer 8a (yellow sticks), (B) 3D binding mode of 8a (yellow sticks), and (C) 2D binding mode of 8a in the catalytic domain of MMP-9 (PDB ID: 1GKC(78)).
3.4. Structure–Activity Relationship
In light of the aforementioned assays, the structure–activity relationship of the synthesized dendrimers can be deduced. The general cytotoxic activity pattern reflects promising antiproliferative potential displayed by the triazine-based dendrimeric scaffold (Figure 1). However, the anticancer profile of each derivative regarding activity as well as selectivity was a function of the terminal substitution. Obviously, the hexacarboxylic acid-terminated dendrimer 8a was the most potent one within the synthesized series. Diversifying the terminal triazine substitution within the carboxyl-terminated series (8a–c, 10, and 11) generally decreased the antiproliferative activity, especially against MDA-MB 231 cells. However, substituting three terminal glycine residues with morpholine moieties (8c) or with piperidine ones (8b) still confers more cytotoxic potency than doxorubicin against Caco-2 cells. The chlorinated dendrimer 10 was slightly more potent than the hydroxylated derivative 11, and both were comparable to doxorubicin. Hydoxamic acid coupling dramatically changed the cytotoxic activities of the dendrimeric architecture. As observed, complete coupling of the hexacarboxylic acid-terminated dendrimer 8a affording 14a decreased the activity by about 3-fold against MDA-MB 231 and Caco-2. However, coupling of the piperidine-substituted dendrimers yielding 14b almost conserved the intrinsic activities of its carboxylic acid precursor 8b. On the other hand, enhanced activities were detected (14c), especially against MDA-MB 231, after hydroxamic acid coupling of the morpholine-substituted precursor (8c). The chlorinated dendrimer 10 obviously lost its activity against MDA-MB 231 cells when hydroxamated (14d) while gaining considerable potency against Caco-2 cells. The transformation of dendrimer 11 also enhanced the cytotoxic potential of the hydroxylated dendrimer (14e) against Caco-2 and MDA-MB 231 cells. Obviously, the terminal chloro and carboxyl moieties in dendrimer 10, conferred the highest safety profile to the designed dendrimeric scaffold followed by its hydroxamic acid coupling product (14d), the hydroxamic acid dendrimer with terminal piperidine (14b), the hexacarboxylic acid (8a), and its hydroxamic acid derivative (14a). These notably safe dendrimers showed promising MMP-9 inhibitory activities. Fully carboxylated termini (8a) conferred the highest MMP-9 inhibition potency among the group with 2 folds selectivity over MMP-2, besides considerable potency against MMP-7, -10, and -13. Unexpectedly, hydroxamic acid coupling (14a) did not enhance the MMP-9 inhibitory potency but endowed notable potential to suppress VEGF expression. However, the hydroxamic acid dendrimer with terminal piperidine (14b) and the chlorinated derivative (14d) were comparable to 8a against MMP-9. The piperidine-derivatized hydroxamic acid dendrimer (14b) exhibited the highest detected activity against MMP-2 and was slightly less potent than the prototype MMP inhibitor NNGH.
4. Experimental Section
4.1. Material and Methods
Solvents and all reagents were purchased from Sigma-Aldrich. Unless otherwise stated, normal workup from organic solvent involved drying over Na2SO4 and rotary evaporation. TLC was performed using aluminum-backed Merck silica gel 60 F-254 plates using suitable solvent systems, with spots being visualized by a Spectroline UV Lamp (254 or 365 nm) or I2 vapor. Melting points were obtained in open capillary tubes by using a MEL-Temp II melting point apparatus and are uncorrected. Infrared (IR) spectra were recorded on a PerkinElmer 1600 series Fourier transform instrument as KBr pellets. The absorption bands (ν̅max) are given in wave numbers (cm–1). Nuclear magnetic resonance (NMR) spectra (1H NMR and 13C NMR) were recorded on JEOL 500 MHz spectrometers at ambient temperature. Chemical shifts are reported in parts per million (ppm) and are referenced relative to residual solvent (e.g., CHCl3 at δ 7.26 ppm for CDCl3, DMSO at δ 2.50 ppm for DMSO-d6). Elemental analyses were performed on a PerkinElmer 2400 elemental analyzer, and the values found were within ±0.3% of the theoretical ones.
4.2. Chemistry
4.2.1. General Procedure for Preparation of 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-(substituted-1-yl)-1,3,5-triazine-6,2-diyl))tris(azanediyl))triacetic Acid 8a–c
4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tri(benzohydrazide) 5 (0.528 g, 1 mmol) was dissolved in hot 1 N HCl (30 mL). The appropriate acid, 2-((4-substituted-6-chloro-1,3,5-triazin-2-yl)amino)acetic acid 7a–c (3 mmol), and sodium carbonate (0.636 g, 6 mmol) in water (50 mL) were added to the reaction mixture. The reaction mixture was refluxed for 24 h. During reflux, sodium carbonate (0.636 g, 6 mmol) was added portionwise to the reaction mixture after 8 h. Another amount of sodium carbonate (0.636 g, 6 mmol) was also added after 16 h of reflux. The clear solution was cooled and neutralized with 1 N HCl until complete precipitation, filtered off, washed with water, and then recrystallized from methanol to afford the pure compounds.
4.2.1.1. 2,2′,2′′,2′′′,2′′′′,2′′′′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(1,3,5-triazine-6,4,2-triyl))hexakis(azanediyl))hexaacetic acid (8a):
brown solid, 0.75 g (62.3%) yield; mp >360 °C; IR (KBr) 3600–2400 (br, O-H), 3373 (N-H), 3106 (sp2 C-H), 1677, 1635 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 3.84–4.03 (m, 12H, 6 α-CH2), 7.72–8.11 (m, 21H, 12Ar-H, 9NH), 9.89 (brs, 3H, 3NH-Ar), 11.46 (brs, 3H, 3NH-CO); 13C NMR (125 MHz, DMSO-d6) δ 41.97, 42.03, 42.63, 119.29, 122.32, 128.64, 128.69, 130.28, 156.55, 163.99, 165.56, 165.72, 165.80, 168.01, 168.24, 169.76, 171.05, 171.16, 171.21, 171.33. Anal. Calcd for C45H45N27O15: C, 44.89; H, 3.77; N, 31.41. Found: C, 45.01; H, 3.56; N, 31.59.
4.2.1.2. 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-(piperidin-1-yl)-1,3,5-triazine-6,2-diyl))tris (azanediyl))triacetic acid (8b):
brown solid, 0.69 g (56%) yield, mp >360 °C; IR (KBr) 3700–2400 (br, O-H), 3412 (N-H), 2941 (sp3 C-H), 1668 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 1.55–1.63 (m, 18H, 9 CH2-Pip), 3.79–4.01 (m, 18H, 3 α-CH2, 6 CH2N-Pip), 7.88–8.08 (m, 18H, 12Ar-H, 6NH, D2O exchangeable), 9.95–9.97 (m, 3H, 3NH-Ar, D2O exchangeable), 10.63–11.16 (m, 3H, 3NH-CO, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6) δ 23.77, 25.43, 42.21, 44.73, 119.47, 119.69, 123.55, 124.12, 124.21, 129.02, 129.09, 130.00, 130.14, 143.78, 143.89, 158.42, 163.12, 166.56, 167.14, 170.23, 170.58. Anal. Calcd for C54H63N27O9: C, 52.55; H, 5.15; N, 30.64. Found: C, 52.80; H, 5.43; N, 30.54.
4.2.1.3. 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris (benzoyl))tris(hydrazine-2,1-diyl))tris(4-morpholino-1,3,5-triazine-6,2-diyl))tris(azanediyl))triacetic acid (8c):
brown solid, 0.65 g (52.5%) yield, mp >360 °C; IR (KBr) 3500–2600 (br, O-H), 3196 (N-H), 1655 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 3.56–3.79 (m, 30H, 3α-CH2, 6CH2N-Mor, 6CH2O-Mor), 7.61–8.14 (m, 18H, 12Ar-H, 6NH, D2O exchangeable), 10.00–10.02 (m, 3H, 3NH-Ar, D2O exchangeable), 10.73–11.19 (m, 3H, 3NH-CO, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6) δ 42.58, 44.42, 44.54, 66.09, 119.97, 120.02, 124.64, 128.75, 129.51, 130.53, 144.00, 144.03, 150.36, 155.47, 157.07, 160.96, 163.26, 165.91, 166.92, 167.51, 170.55, 171.10. Anal. Calcd for C51H57N27O12: C, 49.39; H, 4.63; N, 30.49. Found: C, 49.21; H, 4.76; N, 30.67.
4.2.2. Preparation of 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-chloro-1,3,5-triazine-6,2-diyl))tris(azanediyl))triacetic Acid (10)
4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tri(benzohydrazide) 5 (0.528 g, 1 mmol) was dissolved in 1 N HCl (30 mL) and then added dropwise for 30 min to a cold solution of 2,4,6-trichloro-1,3,5-triazine 1 (0.552 g, 3 mmol) in acetone (75 mL). The reaction mixture was stirred for 3 h at 0 °C. Sodium carbonate (0.636 g, 6 mmol) was added portionwise to the reaction mixture after 1 h. Subsequently, a mixture of glycine 6 (0.225 g, 3 mmol) and sodium carbonate (0.318 g, 3 mmol) dissolved in water (25 mL) was added. The mixture was stirred overnight at room temperature, cooled, and neutralized with 1 N HCl until complete precipitation, filtered off, washed with water, and then recrystallized from methanol to afford the pure compounds. The product was obtained as a brown solid: 0.91 g (83.6%) yield, mp >360 °C; IR (KBr) 3600–2500 (br, O-H, acid), 3282 (N-H), 2902 (sp3 C-H), 1659 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 3.84–3.93 (m, 6H, 3α-CH2), 7.72–8.25 (m, 18H, 12Ar-H, 6NH), 9.84–10.05 (m, 3H, 3NH-Ar), 10.33–11.11 (m, 3H, 3NH-CO); 13C NMR (125 MHz, DMSO-d6) δ 42.72, 119.71, 120.35, 124.31, 128.68, 128.85, 130.55, 130.59, 130.86, 132.71, 134.19, 141.66, 143.12, 144.55, 164.41, 166.19, 167.61, 169.43, 170.38, 171.66. Anal. Calcd for C39H33Cl3N24O9: C, 43.05; H, 3.06; N, 30.89. Found: C, 42.86; H, 3.24; N, 31.03.
4.2.3. Preparation of 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-hydroxy-1,3,5-triazine-6,2-diyl))tris(azanediyl))triacetic Acid (11)
A mixture of 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-chloro-1,3,5-triazine-6,2-diyl))tris(azanediyl))triacetic acid (10) (0.544 g, 0.5 mmol) and sodium hydroxide (0.12 g, 3 mmol) was dissolved in water (25 mL) and stirred for 24 h at room temperature. The reaction mixture was cooled and neutralized with 1 N HCl until complete precipitation, filtered off, washed with water, and then recrystallized from methanol to afford the pure compound. The product was obtained as brown solid: 0.39 g (75.5%) yield, mp >360 °C; IR (KBr) 3500–2400 (br, O-H), 3185 (N-H), 3034 (sp2 C-H), 2902 (sp3 C-H), 1720, 1624 (C=O) cm–1, 1H NMR (500 MHz, DMSO-d6) δ 3.89–4.10 (m, 6H, 3α-CH2), 7.29–8.53 (m, 18H, 12Ar-H, 6NH, D2O exchangeable), 9.90 (brs, 3H, 3NH-Ar, D2O exchangeable), 10.86–11.75 (m, 6H, 3NH-CO, 3OH, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6) δ 42.87, 119.82, 121.76, 122.24, 124.49, 125.32, 129.18, 130.91, 139.74, 143.91, 150.45, 156.38, 159.86, 163.87, 165.94, 167.15, 170.33. Anal. Calcd for C39H36N24O12: C, 45.35; H, 3.51; N, 32.55. Found: C, 45.63; H, 3.79; N, 32.27.
4.2.4. Preparation of 4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(N′-(4,6-dihydroxy-1,3,5-triazin-2-yl)benzohydrazide) (12)
4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tri(benzohydrazide) 5 (0.528 g, 1 mmol) was dissolved in 1 N HCl (30 mL) and then added dropwise for 30 min to a solution of 2,4,6-trichloro-1,3,5-triazine 1 (0.552 g, 3 mmol) in acetone (75 mL). The reaction mixture was stirred for 1 h at 0 °C. A solution of sodium hydroxide (0.24 g, 6 mmol) in water (25 mL) was then added dropwise to the reaction mixture for 2 h, keeping temperature at 0 °C. The reaction mixture was then removed from the ice-bath. Subsequently, a solution of sodium hydroxide (0.24 g, 6 mmol) in water (25 mL) was added. The mixture was refluxed for 2 h, cooled, neutralized with 1 N HCl until complete precipitation, filtered off, and washed with water and then recrystallized from acetone to afford the pure compound. The product was obtained as a brown solid: 0.77 g (89.4%) yield, mp >360 °C; IR (KBr) 3500–2400 (br, O-H), 3417 (N-H), 3027 (sp2 C-H), 1722 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 7.34–8.04 (m, 15H, 12Ar-H, 3NH), 9.98 (brs, 3H, 3 NH-Ar), 10.46–11.95 (m, 9H, 6OH, 3NH-CO); 13C NMR (125 MHz, DMSO-d6) δ 119.72, 120.04, 121.83, 122.48, 125.91, 129.32, 129.63, 130.89, 139.39, 143.71, 150.61, 156.44, 159.89, 164.10, 166.02, 166.27, 167.76. Anal. Calcd for C33H27N21O9: C, 46.00; H, 3.16; N, 34.13. Found: C, 45.86; H, 3.38; N, 34.25.
4.2.5. General Procedure for Preparation of Dendrimers 13, 14a–e
DIC (0.115 mL, 0.75 mmol) and HOAt (0.102 g, 0.75 mmol) were added to a mixture of tricarbxylic acids (3, 8a–c, 10 or 11) (0.25 mmol) and Et3N (0.105 mL, 0.75 mmol) dissolved in 3 mL of DMF with stirring for 10 min at 0 °C. Hydroxyl amine hydrochloride (0.0517 g, 0.75 mmol) and Et3N (0.105 mL, 0.75 mmol) in 2 mL of DMF were added to the reaction mixture and allowed to stir overnight at room temperature. The reaction mixture was diluted with water (70 mL), filtered off, washed with 5% citric acid (10 mL) and then with water, and then recrystallized from methanol to obtain the corresponding hydroxamic acid in good yield.
4.2.5.1. 4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(N-hydroxybenzamide) (13):
white solid, 0.078 g (58.7%) yield; mp >360 °C; IR (KBr) 3500–2400 (br, O-H), 3404, 3264, 3201 (N-H), 3071 (sp2 C-H), 1699 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 7.04–8.17 (m, 15H, 12Ar-H, 3NH, D2O exchangeable), 9.70–10.39 (m, 3H, 3NH-Ar, D2O exchangeable), 11.21 (brs, 3H, 3OH, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6) δ 119.87, 126.12, 127.92, 129.04, 130.90, 136.33, 143.66, 164.42, 164.55, 168.39, 168.46, 168.53. Anal. Calcd for C24H21N9O6: C, 54.24; H, 3.98; N, 23.72. Found: C, 54.32; H, 4.14; N, 23.52.
4.2.5.2. 2,2′,2′′,2′′′,2′′′′,2′′′′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(1,3,5-triazine-6,4,2-triyl))hexakis(azanediyl))hexakis(N-hydroxyacetamide) (14a):
brown solid, 0.050 g (61.8%) yield, mp >360 °C; IR (KBr) 3500–2500 (br, O-H), 3389, 3265 (N-H), 1652 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 3.36–3.84 (m, 12H, 6α-CH2), 7.31–8.00 (m, 25H, 12Ar-H, 6NH-gly, D2O exchangeable, 3NH, D2O exchangeable, 4NH-hydroxamic, D2O exchangeable), 8.77–8.95 (m, 2H, 2NH-hydroxamic, D2O exchangeable), 9.36–9.79 (m, 6H, 3NH-Ar, 3NH-CO, D2O exchangeable), 10.37 (brs, 4H, 4OH, D2O exchangeable), 11.12 (brs, 2H, 2OH, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6) δ 45.65, 119.23, 125.83, 127.48, 128.20, 128.59, 130.10, 142.56, 143.25, 163.86, 164.00, 164.15, 165.61. Anal. Calcd for C45H49N31O15: C, 42.76; H, 3.91; N, 34.35. Found: C, 42.64; H, 4.03; N, 34.17.
4.2.5.3. 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-(piperidin-1-yl)-1,3,5-triazine-6,2-diyl))tris(azanediyl))tris(N-hydroxyacetamide) (14b):
brown solid, 0.09 g (58.44%) yield; mp >360 °C; IR (KBr) 3500–2700 (br, O-H), 3420 (N-H), 1626 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 1.52–1.56 (m, 18H, 9 CH2-Pip), 3.42–3.71 (m, 12H, 6 CH2N Pip), 4.01–4.05 (m, 6H, 3 α-CH2), 7.92–8.00 (m, 18H, 12 Ar-H, 6 NH), 8.25–8.38 (m, 3H, 3 NH-hydroxamic), 10.70 (br s, 3H, 3 NH-Ar), 11.55 (brs, 6H, 3NH-CO, 3OH); 13C NMR (125 MHz, DMSO-d6) δ 31.05, 40.44, 47.00, 114.29, 115.23, 117.10, 117.90, 118.08, 123.72, 131.31, 158.73, 159.12, 159.51, 159.70, 160.09, 160.48, 168.00. Anal. Calcd for C54H65N29O9: C, 51.30; H, 5.18; N, 32.13. Found: C, 51.18; H, 5.30; N, 32.21.
4.2.5.4. 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-morpholino-1,3,5-triazine-6,2-diyl))tris(azanediyl))tris(N-hydroxyacetamide) (14c):
brown solid, 0.107 g (69.03%) yield; mp >360 °C; IR (KBr) 3600–2800 (br, O-H), 3434 (N-H), 1629 (C=O) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 3.60–3.75 (m, 24H, 12CH2), 4.03–4.13 (m, 6H, 3 α-CH2), 7.80–8.01 (m, 21H, 12Ar-H, 3NH Gly, 3NH-Ar, 3NH-NH-CO), 10.57 (brs, 6H, 3NH hydroxamic, 3NH-NH-CO), 11.56 (brs, 3H, 3OH); 13C NMR (125 MHz, DMSO-d6) δ 40.44, 46.80, 61.67, 114.13, 115.13, 116.99, 117.96, 121.85, 131.20, 158.96, 159.35, 159.94, 160.33, 165.04, 167.84, 168.14. Anal. Calcd for C51H59N29O12: C, 48.22; H, 4.68; N, 31.98. Found: C, 48.04; H, 4.84; N, 32.20.
4.2.5.5. 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-chloro-1,3,5-triazine-6,2-diyl))tris(azanediyl))tris(N-hydroxyacetamide) (14d):
brown solid, 0.19 g (67.0%) yield; mp >360 °C; IR (KBr) 3600–2500 (br, O-H), 3282 (N-H), 2968 (sp3 CH), 1652 (C=O, amide) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 3.83–4.19 (m, 6H, 3 α-CH2), 7.57–8.11 (m, 18H, 12Ar-H, 6NH), 8.42–8.72 (m, 3H, 3NH), 9.71–9.80 (m, 3H, 3 NH-Ar), 10.27–10.41 (m, 3H, 3NH-CO), 11.10 (brs, 3H, 3OH); 13C NMR (125 MHz, DMSO-d6) δ 41.16, 44.25, 44.26, 119.64, 120.02, 123.53, 127.76, 128.64, 129.09, 130.55, 137.25, 150.43, 154.83, 162.89, 164.25, 164.41, 166.48, 168.16. Anal. Calcd for C39H35Cl3N26O9: C, 41.89; H, 3.15; N, 32.57. Found: C, 41.75; H, 3.40; N, 32.71.
4.2.5.6. 2,2′,2′′-((6,6′,6′′-(2,2′,2′′-(4,4′,4′′-((1,3,5-Triazine-2,4,6-triyl)tris(azanediyl))tris(benzoyl))tris(hydrazine-2,1-diyl))tris(4-hydroxy-1,3,5-triazine-6,2-diyl))tris(azanediyl))tris(N-hydroxyacetamide) (14e):
brown solid, 0.24 g (87.6%) yield; mp >360 °C; IR (KBr) 3600–2500 (br, O-H), 3411 (N-H), 2904 (sp3 CH), 1677 (C=O, amide) cm–1; 1H NMR (500 MHz, DMSO-d6) δ 3.60–3.96 (m, 6H, 3α-CH2), 7.35–7.95 (m, 18H, 12Ar-H, 3NH-Gly, 3NH-Ar), 8.49–8.83 (m, 3H, 3NH-NH-CO), 9.51–9.75 (m, 6H, 3NH 3NH-NH-CO, 3NH-hydroxamic), 10.33–10.66 (m, 6H, 6OH); 13C NMR (125 MHz, DMSO-d6) δ 42.96, 119.86, 121.85, 122.55, 125.80, 128.37, 129.51, 130.78, 140.15, 144.01, 146.88, 147.96, 148.89, 150.45, 156.49, 159.78, 164.06, 165.94, 167.15, 167.62, 170.27. Anal. Calcd for C39H38N26O12: C, 44.07; H, 3.60; N, 34.26. Found: C, 43.93; H, 3.72; N, 34.44.
4.3. Biological Evaluation
4.3.1. Cytotoxicity Screening
Cytotoxicity was performed utilizing MTT assay60−63 as detailed in the Supporting Information
4.3.2. In Vitro MMPs Inhibition Assay
The assay was performed utilizing MMP-9 Colorimetric Assay Kit for Drug Discovery - AK-410a A BIOMOL QuantiZyme, MMP-2 Inhibitor Screening Assay Kit (Fluorometric) 09/19 (K2017-100), MMP-7 Inhibitor Screening Assay Kit (ab139445), MMP-10 Assay Kit (ab139457), and MMP-13 Inhibitor Screening Assay Kit (ab139451), respectively, following the manufacturers’ instructions.
4.3.3. Apoptosis Studies
Morphological examination and flow cytometric analysis of apoptosis79 are detailed in the Supporting Information.
4.3.4. Tumor Cell Migration inhibition
In vitro wound healing assay80 was performed for evaluating the potential of the studied dendrimers to inhibit tumor cell migration. The procedure is detailed in the Supporting Information.
4.3.5. Quantitative Real-Time PCR Analyses of VEGF, Cyclin D, and p21 Genes
Total RNAs of untreated and MDA-MB-231 and Caco-2 cells treated with the studied dendrimers were extracted using Gene JET RNA Purification Kit (Thermo Scientific, USA). The cDNA was synthesized from mRNA using cDNA Synthesis Kit (Thermo Scientific, USA). Real time PCR analyses of VEGF81 cyclin D82 and p2183 genes were performed as detailed in the Supporting Information.
4.3.6. Docking
MOE 2015.1077 was employed for performing docking studies. The structural coordinates of MMP-9 catalytic domain complexed with a reverse hydroxamate inhibitor were downloaded from the protein data bank (PDB ID: 1GKC(78)). Unwanted residues were removed. The protein structure was prepared and refined utilizing the default settings of the MOE “structure preparation” module. The active dendrimer 8a was built in silico and energy minimized employing Amber10:EHT force field with reaction-field electrostatics (an interior dielectric constant of 1 and an exterior dielectric of 80) using an 8–10 Å cutoff distance. Then docking was conducted into the cocrystallized ligand binding site with various fitting protocols for validation. Rigid docking protocol was adopted. The ligand placement method was set to apply the Triangular matcher algorithm and Alpha HB scoring function as the default scoring function generating the top 10 nonredundant poses of the lowest binding energy conformers of the studied dendrimer for investigation.
4.3.7. Statistical Analysis
The statistical analysis throughout the study was performed as detailed in the Supporting Information.
5. Conclusion
The current study portrays the design, synthesis, and evaluation of 1,3,5-triazine-based dendrimers as the first-in-class MMP-9 inhibitorsto the best of our knowledge. The design rationale relied on decorating 1,3,5-triazine dendrimers with zinc-binding entities to endow MMP-9 inhibition potential. MTT assay generally revealed the promising antiproliferative potential of the dendrimeric scaffold. Diversification of the dendrimers’ terminal substitutions enriched the deduced structure–activity relationship within the synthesized series. Obviously, the hexacarboxylic acid-terminated dendrimer 8a was the most potent derivative with single-digit nanomolar IC50 and significant apoptotic induction (>75%) in MDA-MB 231 and Caco-2 cells, in addition to promising safety profile. MMP-9 inhibition assay results were consistent with the MTT assay and flow cytometric analysis data, where 8a exhibited the most potent MMP-9 inhibitory activity among the evaluated dendrimers with MMP-9 over MMP-2 selectivity. Docking simulations demonstrated the possible Zn-binding mode of the studied dendrimer 8a via its carboxylic acid termini. Compound 8a also showed considerable potency against MMP-7, -10, and -13. Further mechanistic studies revealed the potential of 8a to suppress VEGF, upregulate p21, and downregulate cyclin D expression in the treated tumor cells. Therefore, the antiproliferative potency of 8a is better judged collectively as the resultant outcome of all these modulatory activities. With these results, it could be concluded that the hexacarboxylic acid-terminated dendrimer 8a may be considered as a lead triazine-based dendrimer in its class. Furthermore, it could be employed in further studies as smart bioactive carrier via installation of tumor targeting ligands as well as other cargo payloads such as drugs on the amenable carboxylic acid termini via biodegradable linkages.
Acknowledgments
The authors thank the Science, Technology & Innovation Funding Authority (STDF), Cairo, Egypt, for funding this work through the Young Researcher Grant (Proposal ID 43024). The authors also thank the graphic designer Fatma Moustafa Mohamed for her technical support in improving the figures.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01949.
NMR (1H, 13C) and IR spectra of the synthesized dendrimers and precursors as well as biological evaluation experimental procedures (PDF)
Author Contributions
Nesreen S. Haiba, Ahmed Bergas, Marwa M. Abu-Serie: Methodology, Validation. Sherine N. Khattab: Conceptualization. Sherine N. Khattab, Mohamed Teleb, Hosam H. Khalil: Formal analysis, Supervision, Resources. Nesreen S. Haiba, Marwa M. Abu-Serie: Visualization, Investigation. Nesreen S. Haiba, Mohamed Teleb: Writing main manuscript text. Sherine N. Khattab: Writing - Review & Editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Stetler-Stevenson W. G.; Aznavoorian S.; Liotta L. A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 1993, 9, 541–573. 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- Walker C.; Mojares E.; Del Río Hern andez A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028–3058. 10.3390/ijms19103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K.; Plaks V.; Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010, 141 (1), 52–67. 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathcart J.; Pulkoski-Gross A.; Cao J. Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Gnes Dis. 2015, 2 (1), 26–34. 10.1016/j.gendis.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran S.; Dundas S. R.; Buxton J.; Leeman M. F.; Ramsay R.; Murray G. I. Matrix metalloproteinase/tissue inhibitors of matrix metalloproteinase phenotype identifies poor prognosis colorectal cancers. Clin. Cancer Res. 2004, 10 (24), 8229–8234. 10.1158/1078-0432.CCR-04-0424. [DOI] [PubMed] [Google Scholar]
- Forget M. A.; Desrosiers R. R.; Béliveau R. Physiological roles of matrix metalloproteinases: implications for tumor growth and metastasis. Can. J. Physiol. Pharmacol. 1999, 77 (7), 465–480. 10.1139/y99-055. [DOI] [PubMed] [Google Scholar]
- Adhikari N.; Mukherjee A.; Saha A.; Jha T. Arylsulfonamides and selectivity of matrix metalloproteinase-2: An overview. Eur. J. Med. Chem. 2017, 129, 72–109. 10.1016/j.ejmech.2017.02.014. [DOI] [PubMed] [Google Scholar]
- Visse R.; Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 2003, 92 (8), 827–839. 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- Maskos K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie 2005, 87 (3–4), 249–263. 10.1016/j.biochi.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Schechter T. On the size of the acitive site in proteases, I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- Bhowmick N. A.; Neilson E. G.; Moses H. L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432 (7015), 332–337. 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers A. F.; Matrisian L. M. Changing views of the role of matrix metalloproteinases in metastasis. J. Natl. Cancer Inst. 1997, 89, 1260–1270. 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- Kalluri R.; Zeisberg M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Brown S.; Meroueh S. O.; Fridman R.; Mobashery S. Quest for selectivity in inhibition of matrix metalloproteinases. Curr. Top. Med. Chem. 2004, 4, 1227–1238. 10.2174/1568026043387854. [DOI] [PubMed] [Google Scholar]
- Nuti E.; Tuccinardi T. Matrix metalloproteinase inhibitors: new challenges in the era of post broad-spectrum inhibitors. Curr. Pharmaceut. Des. 2007, 13, 2087–2100. 10.2174/138161207781039706. [DOI] [PubMed] [Google Scholar]
- Georgiadis D.; Yiotakis A. Specific targeting of metzincin family members with small-molecule inhibitors: progress toward a multifarious challenge. Bioorg. Med. Chem. 2008, 16, 8781–8794. 10.1016/j.bmc.2008.08.058. [DOI] [PubMed] [Google Scholar]
- Rao B. G. Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr. Pharmaceut. Des. 2005, 11, 295–322. 10.2174/1381612053382115. [DOI] [PubMed] [Google Scholar]
- Gialeli C.; Theocharis A. D.; Karamanos N. K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. Eur. J. Biochem. 2011, 278, 16e27. [DOI] [PubMed] [Google Scholar]
- Coussens L. M.; Fingleton B.; Matrisian L. M. Matrix metalloproteinase inhibitors and cancer-trials and tribulations. Science 2002, 295, 2387–2392. 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Breuer E.; Frant J.; Reich R. Recent non-hydroxamate matrix metalloproteinase inhibitors. Expert Opin. Ther. Pat. 2005, 15, 253–269. 10.1517/13543776.15.3.253. [DOI] [Google Scholar]
- Whittaker M.; Floyd C. D.; Brown P.; Gearing A. J. H. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999, 99, 2735–2776. 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- Overall C. M.; Kleifeld O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- Jacobsen J. A.; Major J. L.; Jourden M. T.; Miller S. M. Cohen, To bind zinc or not to bind zinc: an examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. 10.1016/j.bbamcr.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Folgueras A. R.; Pendas A. M.; Sanchez L. M.; Lopez-Otin C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int. J. Dev. Biol. 2004, 48, 411–424. 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- Dublanchet A. C.; Ducrot P.; Andrianjara C.; O’Gara M.; Morales R.; Compere D.; Denis A.; Blais S.; Cluzeau Ph.; Courté K.; Hamon J.; Moreau F.; Prunet M. L.; Tertre A. Structure-based design and synthesis of novel non-zinc chelating MMP-12 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3787–3790. 10.1016/j.bmcl.2005.05.079. [DOI] [PubMed] [Google Scholar]
- Johnson A. R.; Pavlovsky A. G.; Ortwine D. F.; Prior F.; Man C.-F.; Bornemeier D. A.; Banotai C. A.; Mueller W. T.; McConnell P.; Yan Ch.; Baragi V.; Lesch Ch.; Roark W. H.; Wilson M.; Datta K.; Guzman R.; Han Hyo-K.; Dyer R. D. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 2007, 282, 27781–27791. 10.1074/jbc.M703286200. [DOI] [PubMed] [Google Scholar]
- Li J. J.; Nahra J.; Johnson A. R.; Bunker A.; O’Brien P.; Yue W.-S.; Ortwine D. F.; Man C.-F.; Baragi V.; Kilgore K.; Dyer R. D.; Han H.-K. Quinazolinones and pyrido [3, 4-d] pyrimidin-4-ones as orally active and specific matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. J. Med. Chem. 2008, 51 (4), 835–841. 10.1021/jm701274v. [DOI] [PubMed] [Google Scholar]
- Morales R.; Perrier S.; Florent J.-M.; Beltra J.; Dufour S.; De Mendez I.; Manceau P.; Tertre A.; Moreau F.; Compere D.; Dublanchet A. C.; O’.Gara M. Crystal structures of novel non-peptidic, non-zinc chelating inhibitors bound to MMP-12. J. Mol. Biol. 2004, 341, 1063–1076. 10.1016/j.jmb.2004.06.039. [DOI] [PubMed] [Google Scholar]
- Lyu Y.; Xiao Q.; Yin L.; Yang L.; He W. Potent delivery of an MMP inhibitor to the tumor microenvironment with thermosensitive liposomes for the suppression of metastasis and angiogenesis. Signal Transduct. Target Ther. 2019, 2019 (4), 1–9. 10.1038/s41392-019-0054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer T.; Riedl R. Inhibitory Antibodies Designed for Matrix Metalloproteinase Modulation. Molecules 2019, 24, 2265. 10.3390/molecules24122265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devy L.; Huang L.; Naa L.; Yanamandra N.; Pieters H.; Frans N.; Chang E.; Tao Q.; Vanhove M.; Lejeune A.; Gool R. V.; Sexton D. J.; Kuang G.; Rank D.; Hogan Sh.; Pazmany C.; Ma Y. L.; Schoonbroodt S.; Nixon A. E.; Ladner R. C.; Hoet R.; Henderikx P.; Tenhoor Ch.; Rabbani Sh. A.; Valentino M. L.; Wood C. R.; Dransfield D. T. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009, 69, 1517–1526. 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- Marshall D. C.; Lyman S. K.; McCauley S.; Kovalenko M.; Spangler R.; Liu C.; Lee M.; O’Sullivan C.; Barry-Hamilton V.; Ghermazien H.; Mikels-Vigdal A.; Garcia C. A.; Jorgensen B.; Velayo A. C.; Wang R.; Adamkewicz J. I.; Smith V. Selective Allosteric Inhibition of MMP9 Is Efficacious in Preclinical Models of Ulcerative Colitis and Colorectal Cancer. PLoS One 2015, 10, 127063. 10.1371/journal.pone.0127063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar S. G.; Yarema K. J. Targeting cancer cells with dendrimers. Chem. Biol. 2005, 12, 5–6. 10.1016/j.chembiol.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Cerofolini L.; Baldoneschi V.; Dragoni E.; Storai A.; Mamusa M.; Berti D.; Fragai M.; Richichi B.; Nativi C. Synthesis and binding monitoring of a new nanomolar PAMAM-based matrix metalloproteinases inhibitor (MMPIs). Bioorg. Med. Chem. 2017, 25, 523–527. 10.1016/j.bmc.2016.11.028. [DOI] [PubMed] [Google Scholar]
- Yao H.; Veine D. M.; Fay K. S.; Staszewski E. D.; Zeng Z.; Livant D. L. The PHSCN dendrimer as a more potent inhibitor of human breast cancer cell invasion, extravasation, and lung colony formation. Breast Cancer Res. Treat. 2011, 125, 363–375. 10.1007/s10549-010-0826-y. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Shan T.; Zhao M.; Mai S.; Gu L. The inhibitory effect of carboxyl-terminated polyamidoamine dendrimers on dentine host-derived matrix metalloproteinases in vitro in an etch-and-rinse adhesive system. R. Soc. Open Sci. 2019, 6 (10), 182104. 10.1098/rsos.182104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomalia D. A.; Naylor A. M.; Goddard W. A. Starburst Dendrimers: Molecular-Level Control of Size, Shape, Surface Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter. Angew. Chem., Int. Ed. Engl. 1990, 29, 138–175. 10.1002/anie.199001381. [DOI] [Google Scholar]
- Newkome G. R.; Moorefield C. N.; Vögtle F.. Dendritic molecules: Concepts, syntheses, perspectives; Wiley-VCH Verlag GmbH, 2008. [Google Scholar]
- Svenson S.; Tomalia D. A. Dendrimers in biomedical applications-reflections on the field. Adv. Drug Delivery Rev. 2005, 57, 2106–2129. 10.1016/j.addr.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Kannan R. M.; Nance E.; Kannan S.; Tomalia D. A. Emerging concepts in dendrimer-based nanomedicine: from design principles to clinical applications. J. Int. Med. 2014, 276, 579–617. 10.1111/joim.12280. [DOI] [PubMed] [Google Scholar]
- Saluja V.; Mankoo A.; Saraogi G. K.; Tambuwala M. M.; Mishra V. Smart dendrimers: Synergizing the targeting of anticancer bioactives. J. Drug Deliv Sci. Technol. 2019, 52, 15–26. 10.1016/j.jddst.2019.04.014. [DOI] [Google Scholar]
- Lyu Z.; Ding L.; Huang A. T.; Kao C. L.; Peng L. Poly (amidoamine) dendrimers: covalent and supramolecular synthesis. Mater. Today Chem. 2019, 13, 34–48. 10.1016/j.mtchem.2019.04.004. [DOI] [Google Scholar]
- Lim J.; Simanek E. E. Triazine dendrimers as drug delivery systems: From synthesis to therapy. Adv. Drug Delivery Rev. 2012, 64, 826–835. 10.1016/j.addr.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Richichi B.; Baldoneschi V.; Burgalassi S.; Fragai M.; Vullo D.; Akdemir A.; Dragoni E.; Louka A.; Mamusa M.; Monti D.; Berti D.; Novellino E.; De Rosa G.; Supuran C. T.; Nativi C. A Divalent PAMAM-Based Matrix Metalloproteinase/Carbonic Anhydrase Inhibitor for the Treatment of Dry Eye Syndrome. Chem.—Eur. J. 2016, 22 (5), 1714–1721. 10.1002/chem.201504355. [DOI] [PubMed] [Google Scholar]
- Akbarzadeh A.; Khalilov R.; Mostafavi E.; Annabi N.; Abasi E.; Kafshdooz T.; Herizchi R.; Kavetskyy T.; Saghfi S.; Nasibova A.; Davaran S. Role of dendrimers in advanced drug delivery and biomedical applications: a review. Exp. Oncol. 2018, 40 (3), 178–183. 10.31768/2312-8852.2018.40(3):178-183. [DOI] [PubMed] [Google Scholar]
- Carta F.; Osman S. M.; Vullo D.; Gullotto A.; Winum J. Y.; AlOthman Z.; Masini E.; Supuran C. T. Poly (amidoamine) dendrimers with carbonic anhydrase inhibitory activity and antiglaucoma action. J. Med. Chem. 2015, 58 (9), 4039–4045. 10.1021/acs.jmedchem.5b00383. [DOI] [PubMed] [Google Scholar]
- Carta F.; Osman S. M.; Vullo D.; AlOthman Z.; Del Prete S.; Capasso C.; Supuran C. T. Poly (amidoamine) dendrimers show carbonic anhydrase inhibitory activity against α-, β-, γ-and η-class enzymes. Bioorg. Med. Chem. 2015, 23, 6794–6798. 10.1016/j.bmc.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Carta F.; Osman S. M.; Vullo D.; AlOthman Z.; Supuran C. T. Dendrimers incorporating benzenesulfonamide moieties strongly inhibit carbonic anhydrase isoforms I-XIV. Org. Biomol. Chem. 2015, 13 (23), 6453–6457. 10.1039/C5OB00715A. [DOI] [PubMed] [Google Scholar]
- Lo S.-T.; Stern S.; Clogston J. D.; Zheng J.; Adiseshaiah P. P.; Dobrovolskaia M.; Lim J.; Patri A. K.; Sun X.; Simanek E. E. Biological assessment of triazine dendrimers as candidate platforms for nanomedicine: Toxicological profiles, solution behavior, biodistribution, and drug release and efficacy in a PEGylated, paclitaxel construct. Mol. Pharmaceutics 2010, 7 (4), 993. 10.1021/mp100104x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab Sh.N.; Abdel Naim S. E.; El-Sayed M.; El Bardan A. A.; Elzoghby A. O.; Bekhit A. A.; El-Faham A. Design and synthesis of new s-triazine polymers and their application as nanoparticulate drug delivery systems. New J. Chem. 2016, 40 (11), 9565–9578. 10.1039/C6NJ02539K. [DOI] [Google Scholar]
- EL Massry A. M.; Asal A. M.; Khattab Sh.N.; Haiba N. S.; Awney H. A.; Helmy M.; Langer V.; Amer A. Synthesis and structure elucidation of novel fused 1,2,4-triazine derivatives as potent inhibitors targeting CYP1A1 activity. Bioorg. Med. Chem. 2012, 20, 2624–2637. 10.1016/j.bmc.2012.02.041. [DOI] [PubMed] [Google Scholar]
- Khattab Sh.N.; Khalil H. H.; Bekhit A. A.; Abd El-Rahman M. M.; de la Torre B. G.; El-Faham A.; Albericio F. 1,3,5-Triazino-Peptide Derivatives: Synthesis, Characterization and Preliminary Antileishmanial Activity. Chem. Med. Chem. 2018, 13, 725–735. 10.1002/cmdc.201700770. [DOI] [PubMed] [Google Scholar]
- Khattab Sh.N.; Khalil H. H.; Bekhit A. A.; Abd El-Rahman M. M.; El-Faham A.; Albericio F. Synthesis and Preliminary Biological Evaluation of 1,3,5-Triazine Amino Acid Derivatives to Study Their MAO Inhibitors. Molecules. 2015, 20, 15976–15988. 10.3390/molecules200915976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil H. H.; Osman H. A.; Teleb M.; Darwish A. I.; Abu-Serie M. M.; Khattab Sh. N.; Haiba N. S. Engineered s-Triazine-Based Dendrimer-Honokiol Conjugates as Targeted MMP-2/9 Inhibitors for Halting Hepatocellular Carcinoma. Chem. Med. Chem. 2021, 16, 1–20. 10.1002/cmdc.202100465. [DOI] [PubMed] [Google Scholar]
- WHO Report on Cancer: setting Priorities, Investing Wisely and Providing Care for All, WHO, Geneva; WHO: Geneva, 2020.
- Li H.; Qiu Z.; Li F.; Wang Ch. The relationship between MMP-2 and MMP-9 expression levels with breast cancer incidence and prognosis. Oncol. Lett. 2017, 14, 5865–5870. 10.3892/ol.2017.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapral M.; Wawszczyk J.; Jurzak M.; Dymitruk D.; Weglarz L. Evaluation of the expression of metalloproteinases 2 and 9 and their tissue inhibitors in colon cancer cells treated with phytic acid. Acta Polym. Pharm. 2010, 67 (6), 625–629. [PubMed] [Google Scholar]
- Aranapakam V.; Davis J. M.; Grosu G. T.; Baker J.; Ellingboe J.; Zask A.; Levin J. I.; Sandanayaka V. P.; Du M.; Skotnicki J. S.; DiJoseph J. F.; Sung A.; Sharr M. A.; Killar L. M.; Walter T.; Jin G.; Cowling R.; Tillett J.; Zhao W.; McDevitt J.; Xu Z. B. Synthesis and structure-activity relationship of N-substituted 4-arylsulfonylpiperidine-4-hydroxamic acids as novel, orally active matrix metalloproteinase inhibitors for the treatment of osteoarthritis. J. Med. Chem. 2003, 46, 2376–2396. 10.1021/jm0205550. [DOI] [PubMed] [Google Scholar]
- Levin J. I.; Chen M.; Du M. T.; Nelson F. C.; Wehr T.; DiJoseph J. F.; Killar L. M.; Skala S.; Sung A.; Sharr M. A.; Roth C. E.; Jin G.; Cowling R.; Di L.; Sherman M.; Xu Z. B.; March C. J.; Mohler K. M.; Black R. A.; Skotnicki J. S. The discovery of anthranilic acid-based MMP inhibitors. Part 3: incorporation of basic amines. Bioorg. Med. Chem. Lett. 2001, 11, 2975–2978. 10.1016/S0960-894X(01)00601-1. [DOI] [PubMed] [Google Scholar]
- Rizk O. H.; Teleb M.; Abu-Serie M. M.; Shaaban O. G. Dual VEGFR-2/PIM-1 kinase inhibition towards surmounting the resistance to antiangiogenic agents via hybrid pyridine and thienopyridine-based scaffolds: Design, synthesis and biological evaluation,. Bioorg. Chem. 2019, 92, 103189. 10.1016/j.bioorg.2019.103189. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Abdelmoneem M. A.; Abd Elwakil M. M.; Khattab Sh.N.; Helmy M. W.; Bekhit A. A.; Abdulkader M. A.; Zaky A.; Teleb M.; Elkhodairy K. A.; Albericio F.; Elzoghby A. O. Lactoferrin-dual drug nanoconjugate: Synergistic anti-tumor efficacy of docetaxel and the NF-κB inhibitor celastrol. Materials Science and Engineering: C 2021, 118, 111422. 10.1016/j.msec.2020.111422. [DOI] [PubMed] [Google Scholar]
- Metawea O. R.; Abdelmoneem M. A.; Haiba N. S.; Khalil H. H.; Teleb M.; Elzoghby A. O.; Khafaga A. F.; Noreldin A. E.; Albericio F.; Khattab Sh.N. A novel ‘smart’ PNIPAM-based copolymer for breast cancer targeted therapy: Synthesis, and characterization of dual pH/temperature-responsive lactoferrin-targeted PNIPAM-co-AA. Colloids Surf., B 2021, 202, 111694. 10.1016/j.colsurfb.2021.111694. [DOI] [PubMed] [Google Scholar]
- Koopman G.; Reutelingsperger C. P. M.; Kuijten G. A. M.; Keehnen R. M. J.; Pals S. T. M. H. van Oers. Annexin V for Flow Cytometric Detection of Phosphatidylserine Expression on B Cells Undergoing Apoptosis. Blood 1994, 84 (5), 1415–1420. 10.1182/blood.V84.5.1415.1415. [DOI] [PubMed] [Google Scholar]
- Freitas J. T.; Jozic I.; Bedogni B.. Wound healing assay for melanoma cell migration. In Melanoma; Humana: New York, NY, 2021; pp 65–71. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Zhou C.; Chellaiah M. Osteopontin and MMP9: associations with VEGF expression/secretion and angiogenesis in PC3 prostate cancer cells. Cancers. 2013, 5, 617–38. 10.3390/cancers5020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero-Fabián S.; Arreola R.; Becerril-Villanueva E.; Torres-Romero J. C.; Arana-Argáez V.; Lara-Riegos J.; Ramírez-Camacho M. A.; Alvarez-Sánchez M. E. Role of matrix metalloproteinases in angiogenesis and cancer. Front. Oncol 2019, 9, 1370. 10.3389/fonc.2019.01370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao J. S.; Bhoopathi P.; Chetty C.; Gujrati M.; Lakka S. S. MMP-9 short interfering RNA induced senescence resulting in inhibition of medulloblastoma growth via p16INK4a and mitogen-activated protein kinase pathway. Cancer res. 2007, 67 (10), 4956–4964. 10.1158/0008-5472.CAN-07-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc Z. E. Iron(III) and Chromium(III) Salen and Salophen Schiff Bases with Bridging 1,3,5-Triazine Derived Multidirectional Ligands. J. Heterocyclic Chem. 2011, 48, 769–775. 10.1002/jhet.577. [DOI] [Google Scholar]
- Zhang J.; Yu H.; Zhang C.; He C.; Duan C. Cerium-based M4L4 tetrahedrons containing hydrogen bond groups as functional molecular flasks for selective reaction prompting. New J. Chem. 2014, 38, 3137–3145. 10.1039/C4NJ00062E. [DOI] [Google Scholar]
- Ramadan D. R.; Elbardan A. A.; Bekhit A. A.; El-Faham A.; Khattab Sh.N. Synthesis and characterization of novel dimeric s-triazine derivatives as potential anti-bacterial agents against MDR clinical isolates. New. J. Chem. 2018, 42, 10676–10688. 10.1039/C8NJ01483C. [DOI] [Google Scholar]
- Dugar S.; Hollinger F. P.; Mahajan D.; Sen S.; Kuila B.; Arora R.; Pawar Y.; Shinde V.; Rahinj M.; Kapoor K. K.; Bhumkar R. Discovery of Novel and Orally Bioavailable Inhibitors of PI3 Kinase Based on Indazole Substituted Morpholino-Triazines. ACS Med. Chem. Lett. 2015, 6 (12), 1190–1194. 10.1021/acsmedchemlett.5b00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Pu X.; Zhou H.; Wong N.; Tian A. Keto-enol tautomerization of cyanuric acid in the gas phase and in water and methanol. J. Mol. Struct: THEOCHEM 2007, 816, 125–136. 10.1016/j.theochem.2007.04.010. [DOI] [Google Scholar]
- Pérez-Manríquez L.; Cabrera A.; Sansores L. E.; Salcedo R. Aromaticity in cyanuric acid. J. Mol. Model. 2011, 17 (6), 1311–1315. 10.1007/s00894-010-0825-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab Sh.N. Sulfonate Esters of 1-Hydroxypyridin-2(1H)-one and ethyl 2-cyano-2-(hydroxyimino)acetate (Oxyma) as Effective Peptide Coupling Reagents for Replacing HOBt and HOAt. Chem. Pharm. Bull. 2010, 58 (4), 501–506. 10.1248/cpb.58.501. [DOI] [PubMed] [Google Scholar]
- Cherkupally P.; Acosta G. A.; Nieto-Rodriguez L.; Rodriguez H.; Khattab Sh.N.; El-Faham A.; Spengler J.; Shamis M.; Luxembourg Y.; Prohens R.; Subiros-Funosas R.; Albericio F. K-Oxyma: a strong acylation-promoting, 2-CTC resin-friendly coupling additive. Eur. J. Org. Chem. 2013, 2013, 6372–6378. 10.1002/ejoc.201300777. [DOI] [Google Scholar]
- Molecular Operating Environment (MOE); 2015.10 Chemical Computing Group ULC: Montreal, QC, Canada, 2015. [Google Scholar]
- Rowsell S.; Hawtin P.; Minshull C. A.; Jepson H.; Brockbank S. M.; Barratt D. G.; Slater A. M.; McPheat W. L.; Waterson D.; Henney A. M.; Pauptit R. A. Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J. Mol. Biol. 2002, 319 (1), 173–181. 10.1016/S0022-2836(02)00262-0. [DOI] [PubMed] [Google Scholar]
- Crowley L. C.; Marfell B. J.; Scott A. P.; Waterhouse N. J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb Protoc. 2016, 10.1101/pdb.prot087288. [DOI] [PubMed] [Google Scholar]
- Wang X.; Decker C. C.; Zechner L.; Krstin S.; Wink M. In vitro wound healing oftumor cells: inhibition of cell migration by selected cytotoxic alkaloids. BMC Pharmacol Toxicol 2019, 20, 1–12. 10.1186/s40360-018-0284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Huang Y.; Wang X.; Liang Q.; Li Y.; Li F.; Fu X.; Huang C.; Liu H. Dianhydrogalactitol, a potential multitarget agent, inhibits glioblastoma migration, invasion, and angiogenesis. Biomed Pharmacother 2017, 91, 1065–1074. 10.1016/j.biopha.2017.05.025. [DOI] [PubMed] [Google Scholar]
- Albelwi F. F.; Teleb M.; Abu-Serie M. M.; Moaty M. N. A. A.; Alsubaie M. S.; Zakaria M. A.; El Kilany Y.; Aouad M. R.; Hagar M.; Rezki N. Halting Tumor Progression via Novel Non-Hydroxamate Triazole-Based Mannich Bases MMP-2/9 Inhibitors; Design, Microwave-Assisted Synthesis, and Biological Evaluation. Int. J. Mol. Sci. 2021, 22 (19), 10324. 10.3390/ijms221910324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Shi J. J.; Thakur K.; Hu F.; Zhang J. G.; Wei Z. J. Anti-cancerous potential of polysaccharide fractions extracted from peony seed dreg on various human cancer cell lines via cell cycle arrest and apoptosis. Front. Pharmacol 2017, 3 (8), 102. 10.3389/fphar.2017.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.