

Abstract
Acetylcholinesterase (AChE) is one of the most important drug targets for Alzheimer’s disease (AD) treatment. In this work, a machine learning model was trained to rapidly and accurately screen large chemical databases for the potential inhibitors of AChE. The obtained results were then validated via in vitro enzyme assay. Moreover, atomistic simulations including molecular docking and molecular dynamics simulations were then used to understand molecular insights into the binding process of ligands to AChE. In particular, two compounds including benzyl trifluoromethyl ketone and trifluoromethylstyryl ketone were indicated as highly potent inhibitors of AChE because they established IC50 values of 0.51 and 0.33 μM, respectively. The obtained IC50 of two compounds is significantly lower than that of galantamine (2.10 μM). The predicted log(BB) suggests that the compounds may be able to traverse the blood–brain barrier. A good agreement between computational and experimental studies was observed, indicating that the hybrid approach can enhance AD therapy.
Introduction
The chronic neurodegenerative Alzheimer’s disease (AD), the most common form of dementia, causes loss of memory and cognitive abilities in several million elderly people worldwide.1−3 The disease develops slowly over 20 years or longer before showing clear symptoms. Patients’ brains are gradually damaged until their normal functions are lost, at which point extensive and intensive care are needed. Moreover, the number of patients has rapidly increased over the last few years.4 Unfortunately, it is irrefutable that AD therapy is actually unavailable2,5 despite much efforts of the scientific community.6−10
Several hypotheses have been proposed to clarify this insight into the mechanism of the disease. Among these, a hallmark called the cholinergic hypothesis, is associated with a neurotransmitter enzyme named acetylcholinesterase (AChE).11,12 The enzyme catalyzes the interruption of choline esters such as acetylcholine (ACh). The AChE enzyme has been identified as a major drug target for AD drug development.13,14 The inhibition of ACh in cholinergic neurons can hinder synaptic depression and block ACh hydrolysis. Screening inhibitors for AChE enzymes have gained some success with three commercial drugs approved by the FDA including donepezil,13 galantamine,15 and rivastigmine.16 However, these drugs have caused many side effects. Thus, the design of AChE inhibitors still attracts great interest from scientists.17−19 Many candidates for inhibiting the AChE enzyme are being investigated such as the metabolites from Zijuan tea20 and tacrine derivatives.21 Especially, some compounds are under clinical trial such as ganstigmine22 and huperzine A.23
Computer-aided drug design (CADD) approaches have been found increasingly useful in screening large databases of compounds for potential inhibitors.24,25 The accurate and efficient estimation of ligand-binding free energy is a main focus of most CADD approaches.26 To this end, many computational schemes have been developed.27 Ligand-binding affinity of a large database of ligands to a protein is often predicted by molecular docking28 or quantitative structure–activity relationship29 methods. This initial screening reduces the large ligand set to a shortlist of compounds which can be subjected to further refined calculations. More computationally expensive methods such as the molecular mechanics/Poisson–Boltzmann surface area,30−32 linear interaction energy,33,34 or fast pulling of ligand35 can be used to obtain a more accurate prediction of ligand-binding free energy for the reduced set. Moreover, recent advancements in machine learning (ML) approaches have brought many benefits to various areas of society. In particular, CADD including drug discovery and repurposing has also benefited from ML.36,37 ML has been applied in various areas because it is able to automatically find hidden patterns in large volumes of data and link them to a target variable to be predicted.38 Applied to protein–ligand binding problems, ML can learn a mapping function from molecular inputs such as structural, physical, and chemical properties to ligand-binding affinities and poses. Therefore, ML has also been widely employed in CADD.39 Popular ML algorithms used in CADD include both conventional methods such as logistic regression, random forest (RF), and support vector machine and more modern approaches such as extreme gradient boosting (XGBoost) and deep learning. They are increasingly employed in CADD to estimate strong binding ligands to enzyme targets.40−42
In this work, a combination of rigorous computational methods including conventional atomistic simulations and ML models were used to study the interaction between AChE and inhibitors. The computational outcome was then validated via an in vitro experiment. In particular, ML models were trained to predict the binding affinity of ca. 2 million compounds from the ChEMBL database43 to AChE. The ligand-binding affinity was then refined via an in vitro study. Besides, the binding pose of the top-lead substances to the enzyme was then clarified via atomistic simulations including molecular docking and molecular dynamics (MD) simulations. The important residues controlling the binding process were thus identified. Interestingly, all of the experimentally investigated compounds formed a strong binding affinity to AChE in comparison with 3′-methyl-2,2,2-trifluoroacetophenone and galantamine, which were used as positive controls. Other suggested potential inhibitors for AChE inhibition are expected to have high reliability and offer a foundation for further experimental studies. The obtained results could help enhance the development of AD therapy.
Materials and Methods
The computational and experimental strategies to estimate the binding affinity of ChEMBL compounds43 to AChE are presented in Figure 1. In particular, ML models were trained and tested to rapidly and accurately predict the ligand-binding affinity of ChEMBL compounds. The potential inhibitors for AChE, which were suggested by ML calculations, were selected for an in vitro enzyme assay. Besides, molecular docking, MD, and ADME calculations were also performed to clarify the binding mechanism of ligands to AChE and assess the ability to cross the blood–brain (BB) barrier of these compounds. Finally, the list of potential inhibitors of AChE screened from the ChEMBL database was indicated.
Figure 1.

Workflow for predicting potential inhibitors for AChE. (A) Investigation scheme was applied to estimate potential inhibitors for AChE using ML, atomistic calculations, and in vitro studies. (B) Refined investigation of the ML prediction via an in vitro enzyme assay. (C) Predicted potential inhibitors by the ML model were docked to the AChE active site via the modified AutoDock Vina.44 (D) AChE + trifluoromethylstyryl ketone complex was simulated using MD simulations to find the ligand-binding pose.
Data Set
The labeled data set was collected from the binding database.45,46 It includes 762 compounds with corresponding values of the association constant Ki. The experimental binding free energy, which was calculated as ΔG = RT ln Ki, was used as a label for training regression models. The list of labeled data with SMILES strings and the corresponding experimental binding free energy is mentioned in Table S2 of the Supporting Information. The distribution of ΔG for the labeled data set is shown in Figure 2. 600 compounds were randomly selected for the training set and the rest (162 compounds) for the test set. A best model was selected based on root-mean-square error (RMSE), Pearson’s R, and Spearman’s ρ correlation coefficients. The ChEMBL database43 was then used for screening AChE inhibitors. ChEMBL contains ca. 2 million compounds, which were downloaded in May 2020.
Figure 2.

Distribution of binding free energy from the experiment for the labeled set (left) and from prediction by the GraphConv model for the test and ChEMBL sets.
Regression Model Training
Our aim was to train a model that could predict the binding free energy of ligands to the target protein AchE with high correlation. Therefore, we framed this modeling task as a regression problem. We would then use the trained model to make predictions for the ChEMBL set and select top-lead compounds for further refined free energy calculations based on MD simulations. Four ML models were trained including linear regression (LR), RF, XGBoost,47 and a deep learning model based on the convolutional networks on graphs (GraphConv).48 Similar to RF, XGBoost is based on an ensemble of decision trees. However, unlike RF, decision trees in XGBoost are not independent. Instead, they are iteratively trained such that at each iteration, the residual prediction error of all the previous trees is used to fit the next tree.47 In a sense, each tree tries to fix the mistake made by the previous ones. Predictions from all the trees are combined using a weighted sum to output a single final prediction. GraphConv48 is a deep learning model based on convolutional networks on graphs. Input into the model is a molecule, which is represented as an undirected graph. The convolution layer will learn a fixed-length embedding vector (called a molecular fingerprint) from the graph and then input it into a densely connected layer. Both the embedding vectors and weights of densely connected layers are learned together during the training of the model. In GraphConv, manual feature extraction is not needed because it can learn features on the fly.
Among the four models, LR is the simplest approach, less prone to overfitting, and was used as a baseline model. Ten-fold cross-validation was applied to tune hyperparameters. The Hyperopt library49 was used to search for the optimal values of hyperparameters by minimizing mean square error. For LR, the tuned hyperparameter was L2 regularization strength (alpha). For RF, tuned hyperparameters included max_depth, min_samples_split, min_samples_leaf, and max_features. For XGBoost, tuned hyperparameters included max_depth, min_child_weight, subsample, colsample_bytree, reg_lambda, and learning_rate. For the GraphConv model, different combinations of the number of units in the graph_cov layers and dense layers, learning rate, and dropout rates were tried. We used the Python library Scikit-Learn50 to train LR and RF models and the XGBoost library for XGBoost models. We used the library DeepChem,51 which implements GraphConv48 to build and train deep learning models.
To extract features for LR, RF, and XGBoost, we used the RDKitDescriptors tool kit implemented in DeepChem,51 which computed 200 physical and chemical properties such as the numbers of hydrogen bond donors and acceptors, number of valence electrons, maximum and minimum partial charge, molecular weight, polar surface area, and so forth. Among these 200 features, there are many features that are mostly zero. Therefore, we removed those features having more than 99% zero. Furthermore, we also removed highly correlated features (an absolute value of Pearson’s R > 0.95). Eventually, we are left with 123 features. Missing values were imputed with the median of each feature. Finally, all the features were standardized to have a zero mean and a standard deviation of one. These 123 features were used to train LR, RF, and XGBoost models. The Python code for extracting features and training models is available at this GitHub URL https://github.com/nguyentrunghai/AchE_inhibitor_ML.
Molecular Docking Simulations
AutoDock Vina with modified empirical parameters44 was employed to characterize the binding pose and binding affinity of ligands to AChE.52 In particular, the structure of human AChE was downloaded from the Protein Data Bank (PDB) with an ID of 4M0E.53 AutoDockTools was employed to topologize both the receptor and ligands. The docking grid center was selected as the native ligand center of mass. The grid size was selected as 26 × 26 × 26 Å. The global exhaustiveness of modified Vina was chosen as the default value, referring to the previous assessment.44 The largest energy difference between docking modes was 7 kcal mol–1. Lowest-energy docking structures were recorded for the subsequent analysis via MD simulations.
MD Simulations
Atomistic simulations were often performed to refine the docking results.54,55 GROMACS version 201956 was thus employed to simulate the AChE + inhibitor systems. In the first step, the complexes were topologized via an all-atom force field. In particular, the Amber99SB-iLDN force field57 was used to represent AChE and neutralizing ions. The TIP3P water model58 was used for topologizing water molecules. The general Amber force field,59 also known as GAFF, was contemporaneously utilized to parameterize the ligand, in which the work was completed using a combination of AmberTools1860 and ACPYPE60 packages. During which, the geometrical and charged parameters of ligands were obtained via quantum mechanics calculation using the B3LYP/6-31G(d,p) level of theory with an implicit solvent (ε = 78.4). The atomic charges were assigned using the restrained electrostatic potential method.59 Besides, the AChE + inhibitor was put into a simulation box having a volume of 662.7 Nm3 as shown in Figure 1D. Dodecahedron periodic boundary conditions were applied. The solvated complex thus consists of ca. 65 700 atoms totally (1 AChE, 1 inhibitor, ca. 19 175 water molecules, and ca. 8 Na+ ions).
The AChE + inhibitor system was simulated via three steps including energy minimization, NVT, and NPT. A steepest descent approach was used to minimize the complex structure. The minimized system was positionally restrained over 0.1 ns of NVT simulations, in which all the atoms of AChE + inhibitors were constrained via a harmonic force with a value of ca. 240.0 kcal mol–1 nm–2 spring constant. The last snapshot of NVT simulations was used as an initial of 0.1 ns NPT simulations. The unbiased MD simulations with a length of 100 ns were then carried out to relax the AChE + inhibitor complex to the stable states.
In Vitro AChE Enzyme Assay
The selected compounds and 3′-methyl-2,2,2-trifluoroacetophenone were purchased from AK Scientific (USA) and Oakwood Product (USA), respectively. AChE, acetylthiocholine iodide (ACTI), 5,5′-dithobis-(2-nitrobenzoic acid) (DTNB), and dimethyl sulfoxide (DMSO) were obtained from Sigma (USA). Galantamine was purchased from TCI (Japan). 96-well microplates were acquired from Corning (USA). All the others chemicals were obtained at standard purity.
The AChE inhibitory activities of selected compounds were determined by the spectrophotometric method of Ellman61 with slight modification,62 using ACTI as a substrate, in 96-well microplates. All the tested compounds and the positive controls (3′-methyl-2,2,2-trifluoroacetophenone and galantamine) were dissolved in a minimum volume of DMSO (100% of concentration) and diluted to various concentrations using deionized water. The reaction mixture contained the following: 140 μL of sodium phosphate buffer (pH 8.0); 20 μL of the tested sample solution; and 20 μL of the AChE solution (0.25 IU/mL), which were mixed and incubated for 15 min at 25 °C. The reaction was initiated by adding 10 μL of DTNB 2.5 mM and 10 μL of ACTI 2.5 mM to each well and was incubated for 10 min at 25 °C. The hydrolysis of ACTI was monitored by following the formation of the yellow 5-thio-2-nitrobenzoate anion at 412 nm for 15 min, which resulted from the reaction of DTNB with thiocholine, released by the enzymatic hydrolysis of ACTI. The reaction was performed in triplicate and recorded in 96-well microplates using an Elisa microplate reader system (Biotek Instruments, Agilent, CA, USA). Each compound was evaluated at four concentrations (100, 20, 4, and 0.8 μM). The percentage of inhibitory samples was calculated as follows
The analyses were performed using Microsoft Excel (Microsoft Corp., Redmond, WA, USA) and the values are expressed as mean ± SD. The AChE inhibitory activity of each sample was expressed in terms of the IC50 value (μM required to inhibit the hydrolysis of the substrate, ACTI by 50%), as calculated using TableCurve 2Dv4 software.
Analysis Tools
The protonation states of ligands in MD simulations were predicted via the chemicalize webserver, an application of ChemAxon. The statistical errors of correlation and RMSE were estimated via the bootstrapping analysis with 1000 circle resampling.63 The docking success rate p̂ was calculated via comparing the non-hydrogen atoms’ root-mean-square deviation (rmsd) between docking and experimental poses. The rmsd was calculated using the GROMACS tool “gmx rms”.56 The cluster analysis was carried out via the GROMACS tool “gmx cluster”.56 The interaction diagram between the AChE inhibitor was prepared via the free version of Maestro.64 The log(BB) is calculated using the PreADME webserver.65
Results and Discussion
Performance comparison of four ML models including LR, RF, XGBoost, and GraphConv is shown in Table 1. In addition to the two commonly used evaluation metrics, RMSE and Pearson’s R, Spearman’s ρ was also used. It measures the degree of association in rank of two variables and is useful for assessing ranking prediction. As expected, the LR model gave the poorest performance, with the highest RMSE (2.155 ± 0.160 kcal mol–1) and the lowest Pearson’s R (0.427 ± 0.070) and Spearman’s ρ (0.522 ± 0.064) among the four models (Table 1). The poor performance of LR is due to its inability to capture nonlinear relationships between features and targets. Overall, the GraphConv model shows the best predictive performance on the test set with the lowest RMSE (1.580 ± 0.137 kcal mol–1) and the highest Pearson’s R (0.721 ± 0.050) and Spearman’s ρ (0.692 ± 0.054) (Table 1). The second best model, XGBoost, is very close behind with an RMSE of 1.702 ± 0.154 kcal mol–1, Pearson’s R of 0.669 ± 0.059, and Spearman’s ρ of 0.658 ± 0.061 (Table 1). In fact, due to the sizable error bars, the difference in performance between GraphConv and XGBoost is not significant. Nevertheless, our main goal here is to obtain a reasonably accurate model for inhibitor screening but not to determine precisely which model performs best. The GraphConv model was selected to make a prediction of binding free energy for the unlabeled ChEMBL set. See Figure 2 for the distribution of predicted binding free energy. The predicted binding free energy falls in the range from −4.56 to −13.77 kcal mol–1, in which the mean value is of −8.41 kcal mol–1. The comparison of binding free energy between the experiment and prediction made by GraphConv for the test set is shown in Figure 3.
Table 1. Performance of ML Models in Predicting Binding Free Energy of 162 Test Ligands to AChEa.
| model | RMSE (kcal mol–1) | Pearson’s R | Spearman’sρ |
|---|---|---|---|
| LR | 2.155 ± 0.160 | 0.427 ± 0.070 | 0.522 ± 0.064 |
| RF | 1.648 ± 0.126 | 0.694 ± 0.055 | 0.681 ± 0.055 |
| XGBoost | 1.702 ± 0.154 | 0.669 ± 0.059 | 0.658 ± 0.061 |
| GraphConv | 1.580 ± 0.137 | 0.721 ± 0.050 | 0.692 ± 0.054 |
The error bars were estimated using bootstrapping.
Figure 3.

Comparison of binding free energy between the experiment and prediction made by the GraphConv model for 162 test compounds.
Although the training size is quite small, the trained ML model shows reasonably good predictive power. Interestingly, among the top 100 ligand-binding affinities via the ML model, 25 compounds were indicated to bind well to AChE by previous experiments (Table S3 of the Supporting Information). In particular, the association constant of 15 compounds was already reported in the respective experiments (cf. Table 2).66 It should be noted that these compounds were not included in both the training and testing sets. Over the subset of compounds, the obtained ligand-binding affinity by ML calculation is an accurate result with an RMSE of 1.36 ± 0.37 kcal mol–1. Besides, the ΔGML is underestimated in comparison with the experimental values66−70 by an amount of ca. 0.78 kcal mol–1. Moreover, the obtained Pearson’s R = 0.747 ± 0.117 is in good agreement with the corresponding value of the testing test with a value of 0.721 ± 0.050 (cf. Table 1). Overall, the outcome again confirms the realization of the ML model. A clear advantage of the ML approach is that ML models make predictions much faster than molecular docking. It took about 43 min to extract features and make predictions of binding affinity for 2 million compounds. For molecular dockings it takes about 10 min to perform one docking calculation. A limitation of the ML approach is that it requires a large data set of experimental binding affinities.
Table 2. Comparison of the ML Outcome and Available Experimentsa.
| N0 | ChEMBL ID | ΔGML | ΔGEXPb |
|---|---|---|---|
| 1 | CHEMBL89354 | –13.77 | –15.6566 |
| 2 | CHEMBL87098 | –13.75 | –15.1666 |
| 3 | CHEMBL315634 | –13.02 | –12.8866 |
| 4 | CHEMBL208599 | –12.89 | –14.4967 |
| 5 | CHEMBL140476 | –12.77 | –14.3568 |
| 6 | CHEMBL3958859 | –12.75 | –16.7469 |
| 7 | CHEMBL3785269 | –12.40 | –12.8070 |
| 8 | CHEMBL3786448 | –12.39 | –12.4370 |
| 9 | CHEMBL3787223 | –12.34 | –12.2970 |
| 10 | CHEMBL3786719 | –12.31 | –12.5270 |
| 11 | CHEMBL3786442 | –12.27 | –12.2070 |
| 12 | CHEMBL86868 | –12.16 | –11.8066 |
| 13 | CHEMBL3786873 | –12.13 | –12.1570 |
| 14 | CHEMBL3787502 | –11.85 | –12.2970 |
| 15 | CHEMBL3786516 | –11.83 | –12.5670 |
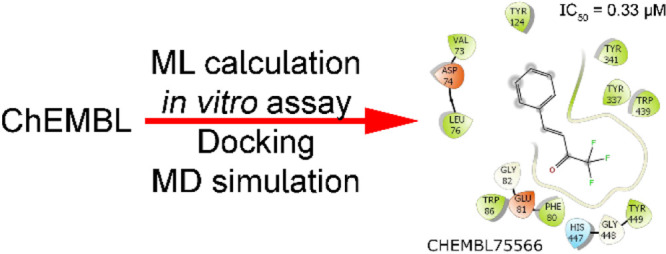
In the next step, we selected six compounds (cf. Table 3) from the top 100 lowest predicted binding free energies and did not test the AChE inhibition activities before for further refinement via the in vitro enzyme assay. The affinity of 3′-methyl-2,2,2-trifluoroacetophenone (CHEMBL86868) and galantamine (CHEMBL659) was also investigated as positive controls. It should be noted that the compound 3′-methyl-2,2,2-trifluoroacetophenone was ranked at #37 position in the list of predicted binding free energies. The compound formed ΔGEXP = −11.80 kcal mol–1 according to the previous publication in comparison with a predicted value of ΔGML = −12.16 kcal mol–1.66 Besides, although galantamine was placed at #855268 over ca. 2 million trial compounds with a predicted value of ΔGML = −8.46 kcal mol–1, the compound is a drug that was approved for AD treatment by the U.S. Food and Drug Administration.71 Moreover, the predicted-binding affinity of galantamine is in good agreement with the respective experiments because the mean of the experimental binding free energy of ΔGEXP = −8.82 kcal mol–1.72−86 Furthermore, the obtained IC50 of eight assessed compounds ranges from 0.33 ± 0.05 to 52.65 ± 4.14 μM by the in vitro enzyme assays. In particular, the largest ligand-binding affinity is the compound named trifluoromethylstyryl ketone (CHEMBL75566) with an amount of IC50 = 0.33 ± 0.05 μM. The value is slightly smaller than that of 3′-methyl-2,2,2-trifluoroacetophenone (IC50 = 0.35 ± 0.03 μM) and significantly smaller than that of galantamine (IC50 = 2.1 ± 0.17 μM). It should be noted that the obtained IC50 value of galantamine is in good agreement with the reported values from the previous works that the corresponding metric is of 2.0187 and 2.09 μM.88 Besides, the ligand-binding affinity of benzyl trifluoromethyl ketone (CHEMBL292454) and trifluoroacetophenone (CHEMBL293277) was also impressive, with an experimental value IC50 of 0.51 ± 0.09 and 4.61 ± 0.34 μM (cf. Table 3), respectively. The other ligands including 1-[4-(trifluoromethyl)phenyl]but-1-en-3-one, perflexane, and methyl nonafluorobutyl ether formed appropriate values of IC50, which measured as 26.33 ± 2.18, 50.75 ± 3.73, and 52.65 ± 4.14 μM, respectively. The obtained results indicated that six proposed ligands can inhibit the biological activity of AChE and it also reaffirmed the acceptable estimation of the ML model.
Table 3. Top-Lead Compounds Formed the Largest Binding Affinity to AChE by ML Calculationsa.
| N0 | ChEMBL ID | name | ΔGML | ΔGdock | ΔGEXPb | IC50 | log(BB) |
|---|---|---|---|---|---|---|---|
| 1 | CHEMBL293277 | trifluoroacetophenone | –12.18 | –9.4 | 4.61 ± 0.34 | 0.05 | |
| 2 | CHEMBL86868 | 3′-methyl-2,2,2-trifluoroacetophenone | –12.16 | –10.4 | –11.8066 | 0.35 ± 0.03 | 0.08 |
| 3 | CHEMBL292454 | benzyl trifluoromethyl ketone | –12.07 | –9.9 | 0.51 ± 0.09 | 0.05 | |
| 4 | CHEMBL1200607 | perflexane | –12.02 | –12.4 | 50.75 ± 3.73 | 1.05 | |
| 5 | CHEMBL74630 | 1-[4-(trifluoromethyl)phenyl]but-1-en-3-one | –11.95 | –11.0 | 26.33 ± 2.18 | 0.05 | |
| 6 | CHEMBL75566 | trifluoromethylstyryl ketone | –11.88 | –10.8 | 0.33 ± 0.05 | 0.07 | |
| 7 | CHEMBL500823 | methyl nonafluorobutyl ether | –11.83 | –10.5 | 52.65 ± 4.14 | 0.50 | |
| 8 | CHEMBL659 | galantamine | –8.46 | –11.7 | –8.8272−86 | 2.10 ± 0.17 | –0.24 |
In order to gain insights into the binding process of eight compounds to AChE, these ligands were docked to the enzymic binding cavity using AutoDock Vina with the modified empirical parameters.44 In order to validate our docking approach, we selected 10 compounds whose bound structures with the receptor are available in the PDB. The experimental binding affinity of the 10 compounds is also available (see Table S1 in the Supporting Information). These 10 ligands were redocked into AChE and the results are reported in Table S1. The obtained docking success rate was of ρ̂ = 90 ± 10 %, in which a docking trial is considered a success if the non-hydrogen atom rmsd between experimental and docking poses was smaller than 0.20 nm. Moreover, the Pearson’s R over these complexes was 0.863 ± 0.091 (see Figure 4). It is significantly larger than that by the default parameter, which is of R = 0.75 ± 11.89 Therefore, it may be concluded that AutoDock Vina with the modified empirical parameters is an appropriate approach to preliminarily predict the ligand binding pose and affinity of ligands to AChE. The approach was thus utilized to estimate the binding poses of eight compounds, which were indicated by ML calculation and confirmed via the in vitro enzyme assay. The docking outcome is mentioned in Table 3. Interestingly, the docking simulations provided highly accurate results compared with ML calculations, with an RMSE value of 1.94 ± 0.27 kcal mol–1.
Figure 4.
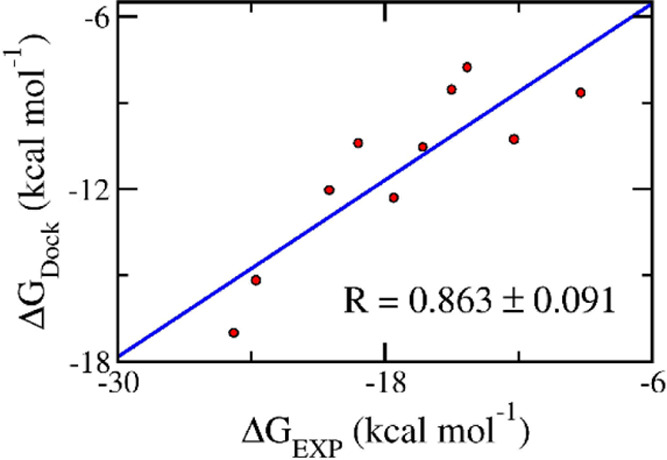
Correlation between docking and experimental data.
Although docking had a high docking success rate, the obtained pose is probably limited because the docking approach uses many approximations to enhance the computing speed.54,90 The unbiased MD simulations with a length of 100 ns were thus performed to turn the solvated complex into equilibrium states. The AChE + inhibitor complex reaches stable states after ca. 55 ns of MD simulations (Figures S1–S3 of the Supporting Information). The factors controlling the binding process of inhibitors to the AChE are then clarified by analyzing the representative structures of the AChE + inhibitor complex. The structure was obtained via clustering all of the conformational complexes within the interval of 40–100 ns with a cutoff of 0.12 nm. The interaction diagram between AChE + inhibitors was then estimated via the Maestro program.64 The diagram was mentioned in Figures 5 and S4 of the Supporting Information. In particular, we may argue that residues Trp86, Gly121, Tyr124, Ser125, Phe297, Tyr337, Phe338, and Tyr341 are important factors governing the binding process of ligands to AChE. Because the ligands frequently formed intermolecular non-bonded contacts to eight residues of AChE, which residues establish contact to inhibitors over 50% of the appraised systems. Interestingly, almost all the ligands formed non-bonded contacts with AChE and only one hydrogen bond between galantamine and Asp74 of AChE was found. Therefore, the hydrophobic interaction probably dominates over electrostatic interaction in the binding process of ligands to AChE.
Figure 5.

Interaction diagram of the AChE + inhibitor complex. The outcome was obtained via the analysis of Maestro over the representative structure of the solvated complex. The structure was obtained via clustering all of the conformational complex within the interval of 40–100 ns with a cutoff of 0.12 nm.
A potent inhibitor for preventing AChE is necessary to be able to traverse the BB barrier because the neurotransmitter is situated in the brain. The log(BB) of eight inhibitors was thus predicted by using PreADME.65 The results are also described in Table 3. Generally, a drug establishes a log(BB) in the range from −2.0 to 1.0.91 Here, eight trial inhibitors adopted log(BB) in the range from −0.24 to 1.05. The predicted log(BB) suggests that the two ligands would be able to easily traverse the BB because their log(BB) is larger than 0.3.91 The other compounds would not be very hard to encounter AChE inside the brain because their log(BB) is larger than −1.0.91 PreADME calculations also suggested that these compounds would not cause significant neurotoxicity.
Conclusions
In this work, combined computational and in vitro studies were proposed to predict the potent inhibitor for preventing AChE. In particular, the GraphConv model with a Pearson’s R on the test set of 0.721 ± 0.05 was used to screen nearly 2 million compounds in the CHEMBL database. The ligand-binding affinity would be then evaluated via the in vitro enzyme assay. The docking and MD simulations were finally performed to estimate the ligand-binding pose and serious factors governing the binding process.
The ligand-binding affinity of eight compounds to AChE was determined via computational and experimental studies. In particular, two compounds involving 3′-methyl-2,2,2-trifluoroacetophenone and galantamine were played as positive controls. Interestingly, two compounds including benzyl trifluoromethyl ketone and trifluoromethylstyryl ketone were indicated as highly powerful inhibitors of AChE because they established IC50 values of 0.51 and 0.33 μM, respectively. The obtained IC50 of two compounds is significantly smaller than that of galantamine (2.10 μM). These compounds also formed an appropriate log(BB), which ranges from 0.05 to 0.07, letting them be able to traverse the BB barrier. Moreover, the other four compounds can also inhibit AChE in both computational and experimental studies.
In addition, besides six compounds were evaluated the ligand-binding affinity using both in silico and in vitro studies in this work and 25 compounds were indicated that they can bind well to AChE via the previous experiments, the other ligands in the top 100 lowest predicted binding free energy probably play as highly potent candidates to prevent AChE. Further experimental studies should be carried out to confirm the ligand-binding affinity of these compounds.
Acknowledgments
This research is funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under the grant number 108.05-2020.01 (P.-T.T.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00908.
Difference of docking versus experimental data, list of labeled data with SMILES strings and the corresponding experimental binding free energy, top 100 ligand-binding affinities via the ML model, rmsd of non-hydrogen atoms of the AChE + inhibitor complex, rmsd of non-hydrogen atoms of AChE, rmsd of non-hydrogen atoms of the inhibitor, and interaction diagram of the AChE + ligand complex (PDF)
Author Contributions
N.Q.A.P., D.M.H., and S.T.N. prepared the list of available compounds with the association constants; T.H.N. trained and performed the ML calculations; S.T.N. performed the docking and MD simulations; P.-T.T. and V.-H.H. carried out the in vitro assay; and all the authors drafted the manuscripts.
The authors declare no competing financial interest.
Notes
Data and software availability: all the relevant data necessary to reproduce all the results in the paper are within the main text and the Supporting Information file.
Supplementary Material
References
- Selkoe D. J. The Molecular Pathology of Alzheimer’s Disease. Neuron 1991, 6, 487–498. 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- Querfurth H. W.; LaFerla F. M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. 10.1056/nejmra0909142. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J.; Hardy J. The Amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dementia 2018, 14, 367. 10.1016/j.jalz.2018.02.001. [DOI] [Google Scholar]
- Nasica-Labouze J.; Nguyen P. H.; Sterpone F.; Berthoumieu O.; Buchete N.-V.; Coté S.; De Simone A.; Doig A. J.; Faller P.; Garcia A.; Laio A.; Li M. S.; Melchionna S.; Mousseau N.; Mu Y.; Paravastu A.; Pasquali S.; Rosenman D. J.; Strodel B.; Tarus B.; Viles J. H.; Zhang T.; Wang C.; Derreumaux P. Amyloid β Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev. 2015, 115, 3518–3563. 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo S. T.; Truong D. T.; Tam N. M.; Nguyen M. T. EGCG Inhibits the Oligomerization of Amyloid Beta (16-22) Hexamer: Theoretical Studies. J. Mol. Graphics Modell. 2017, 76, 1–10. 10.1016/j.jmgm.2017.06.018. [DOI] [PubMed] [Google Scholar]
- Dhouafli Z.; Cuanalo-Contreras K.; Hayouni E. A.; Mays C. E.; Soto C.; Moreno-Gonzalez I. Inhibition of Protein Misfolding and Aggregation by Natural Phenolic Compounds. Cell. Mol. Life Sci. 2018, 75, 3521–3538. 10.1007/s00018-018-2872-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alghazwi M.; Smid S.; Musgrave I.; Zhang W. In vitro Studies of the Neuroprotective Activities of Astaxanthin and Fucoxanthin Against Amyloid Beta (Aβ1-42) Toxicity and Aggregation. Neurochem. Int. 2019, 124, 215–224. 10.1016/j.neuint.2019.01.010. [DOI] [PubMed] [Google Scholar]
- Tran P.-T.; Hoang V.-H.; Lee J.; Hien T. T. T.; Tung N. T.; Ngo S. T. In vitro and in silico determinations of glutaminyl cyclase inhibitors. RSC Adv 2019, 9, 29619. 10.1039/C9RA05763C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armiento V.; Spanopoulou A.; Kapurniotu A. Peptide-Based Molecular Strategies To Interfere with Protein Misfolding, Aggregation, and Cell Degeneration. Angew. Chem., Int. Ed. 2020, 59, 3372–3384. 10.1002/anie.201906908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn D. M. Acetylcholinesterase: enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 1987, 87, 955–979. 10.1021/cr00081a005. [DOI] [Google Scholar]
- Drachman D. A.; Leavitt J. Human Memory and the Cholinergic System. Arch. Neurol. 1974, 30, 113–121. 10.1001/archneur.1974.00490320001001. [DOI] [PubMed] [Google Scholar]
- Kasa P.; Papp H.; Kasa P.; Torok I. Donepezil dose-dependently inhibits acetylcholinesterase activity in various areas and in the presynaptic cholinergic and the postsynaptic cholinoceptive enzyme-positive structures in the human and rat brain. Neuroscience 2000, 101, 89–100. 10.1016/s0306-4522(00)00335-3. [DOI] [PubMed] [Google Scholar]
- Sudhapriya N.; Manikandan A.; Kumar M. R.; Perumal P. T. Cu-mediated synthesis of differentially substituted diazepines as AChE inhibitors; validation through molecular docking and Lipinski’s filter to develop novel anti-neurodegenerative drugs. Bioorg. Med. Chem. Lett. 2019, 29, 1308–1312. 10.1016/j.bmcl.2019.04.002. [DOI] [PubMed] [Google Scholar]
- Sippl W.; Contreras J.-M.; Parrot I.; Rival Y. M.; Wermuth C. G. Structure-based 3D QSAR and design of novel acetylcholinesterase inhibitors. J. Comput.-Aided Mol. Des. 2001, 15, 395–410. 10.1023/a:1011150215288. [DOI] [PubMed] [Google Scholar]
- Ul-Haq Z.; Khan W.; Kalsoom S.; Ansari F. L. In silico modeling of the specific inhibitory potential of thiophene-2,3-dihydro-1,5-benzothiazepine against BChE in the formation of β-amyloid plaques associated with Alzheimer’s disease. Theor. Biol. Med. Modell. 2010, 7, 22. 10.1186/1742-4682-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y.; Kolb H. C.; Radić Z.; Sharpless K. B.; Taylor P.; Marchot P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 1449–1454. 10.1073/pnas.0308206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.-S.; Ge Y.-X.; Cheng Z.-Q.; Song J.-L.; Wang Y.-Y.; Zhu K.; Zhang H. Discovery of new multifunctional selective acetylcholinesterase inhibitors: structure-based virtual screening and biological evaluation. J. Comput.-Aided Mol. Des. 2019, 33, 521–530. 10.1007/s10822-019-00202-2. [DOI] [PubMed] [Google Scholar]
- Turkan F.; Cetin A.; Taslimi P.; Karaman M.; Gulçin İ. Synthesis, biological evaluation and molecular docking of novel pyrazole derivatives as potent carbonic anhydrase and acetylcholinesterase inhibitors. Bioorg. Chem. 2019, 86, 420–427. 10.1016/j.bioorg.2019.02.013. [DOI] [PubMed] [Google Scholar]
- Wang W.; Fu X.-W.; Dai X.-L.; Hua F.; Chu G.-X.; Chu M.-J.; Hu F.-L.; Ling T.-J.; Gao L.-P.; Xie Z.-W.; Wan X.-C.; Bao G.-H. Novel acetylcholinesterase inhibitors from Zijuan tea and biosynthetic pathway of caffeoylated catechin in tea plant. Food Chem. 2017, 237, 1172–1178. 10.1016/j.foodchem.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Benchekroun M.; Romero A.; Egea J.; León R.; Michalska P.; Buendía I.; Jimeno M. L.; Jun D.; Janockova J.; Sepsova V.; Soukup O.; Bautista-Aguilera O. M.; Refouvelet B.; Ouari O.; Marco-Contelles J.; Ismaili L. The Antioxidant Additive Approach for Alzheimer’s Disease Therapy: New Ferulic (Lipoic) Acid Plus Melatonin Modified Tacrines as Cholinesterases Inhibitors, Direct Antioxidants, and Nuclear Factor (Erythroid-Derived 2)-Like 2 Activators. J. Med. Chem. 2016, 59, 9967–9973. 10.1021/acs.jmedchem.6b01178. [DOI] [PubMed] [Google Scholar]
- Bartolucci C.; Siotto M.; Ghidini E.; Amari G.; Bolzoni P. T.; Racchi M.; Villetti G.; Delcanale M.; Lamba D. Structural Determinants of Torpedo californica Acetylcholinesterase Inhibition by the Novel and Orally Active Carbamate Based Anti-Alzheimer Drug Ganstigmine (CHF-2819). J. Med. Chem. 2006, 49, 5051–5058. 10.1021/jm060293s. [DOI] [PubMed] [Google Scholar]
- Barak D.; Ordentlich A.; Kaplan D.; Kronman C.; Velan B.; Shafferman A. Lessons from functional analysis of AChE covalent and noncovalent inhibitors for design of AD therapeutic agents. Chem.-Biol. Interact. 2005, 157-158, 219–226. 10.1016/j.cbi.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Schneider G.; Fechner U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discovery 2005, 4, 649–663. 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]
- Bruno A.; Costantino G.; Sartori L.; Radi M. The In Silico Drug Discovery Toolbox: Applications in Lead Discovery and Optimization. Curr. Med. Chem. 2019, 26, 3838–3873. 10.2174/0929867324666171107101035. [DOI] [PubMed] [Google Scholar]
- Yu W.; MacKerell A. D.. Computer-Aided Drug Design Methods. In Antibiotics: Methods and Protocols; Sass P., Ed.; Springer New York: New York, NY, 2017; pp 85–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryde U.; Söderhjelm P. Ligand-Binding Affinity Estimates Supported by Quantum-Mechanical Methods. Chem. Rev. 2016, 116, 5520–5566. 10.1021/acs.chemrev.5b00630. [DOI] [PubMed] [Google Scholar]
- Gehlhaar D. K.; Verkhivker G.; Rejto P. A.; Fogel D. B.; Fogel L. J.; Freer S. T.; John R M. D.. Docking Conformationally Flexible Small Molecules into a Protein Binding Site through Evolutionary Programming. In Proceedings of the Fourth International Conference on Evolutionary Programming: 1–3 March 1995; Robert G R.; David B F., Eds.; MIT Press: San Diego, 1995.
- Yang J.; Chen J. QSAR Analysis of Purine-Type and Propafenone-Type Substrates of P-Glycoprotein Targeting β-Amyloid Clearance. Neurodegener. Dis. 2013, 10.5772/54975. [DOI] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Kollman P. A. Binding of a siverse set of ligands to avidin and streptavidin: an accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. 10.1021/jm000241h. [DOI] [PubMed] [Google Scholar]
- Wang W.; Kollman P. A. Computational study of protein specificity: the molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14937–14942. 10.1073/pnas.251265598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åqvist J.; Medina C.; Samuelsson J.-E. A New Method for Predicting Binding Affinity in Computer-Aidded Drug Design. Protein Eng. 1994, 7, 385–391. 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- Jones-Hertzog D. K.; Jorgensen W. L. Binding Affinities for Sulfonamide Inhibitors with Human Thrombin Using Monte Carlo Simulations with a Linear Response Method. J. Med. Chem. 1997, 40, 1539–1549. 10.1021/jm960684e. [DOI] [PubMed] [Google Scholar]
- Ngo S. T.; Hung H. M.; Nguyen M. T. Fast and Accurate Determination of the Relative Binding Affinities of Small Compounds to HIV-1 Protease using Non-Equilibrium Work. J. Comput. Chem. 2016, 37, 2734–2742. 10.1002/jcc.24502. [DOI] [PubMed] [Google Scholar]
- Paul D.; Sanap G.; Shenoy S.; Kalyane D.; Kalia K.; Tekade R. K. Artificial Intelligence in Drug Discovery and Development. Drug Discovery Today 2021, 26, 80–93. 10.1016/j.drudis.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh A.; Kambhampati C.; Monson J.; Drew P. Artificial intelligence in medicine. Ann. R. Coll. Surg. Engl. 2004, 86, 334–338. 10.1308/147870804290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles J. C.; Walker A. J. The Potential Application of Artificial Intelligence in Transport. Intell. Transport Syst. 2006, 153, 183–198. 10.1049/ip-its:20060014. [DOI] [Google Scholar]
- Lamberti M. J.; Wilkinson M.; Donzanti B. A.; Wohlhieter G. E.; Parikh S.; Wilkins R. G.; Getz K. A Study on the Application and Use of Artificial Intelligence to Support Drug Development. Clin. Ther. 2019, 41, 1414–1426. 10.1016/j.clinthera.2019.05.018. [DOI] [PubMed] [Google Scholar]
- Subramanian G.; Ramsundar B.; Pande V.; Denny R. A. Computational Modeling of β-Secretase 1 (BACE-1) Inhibitors Using Ligand Based Approaches. J. Chem. Inf. Model. 2016, 56, 1936–1949. 10.1021/acs.jcim.6b00290. [DOI] [PubMed] [Google Scholar]
- Chen J.-Q.; Chen H.-Y.; Dai W.-j.; Lv Q.-J.; Chen C. Y.-C. Artificial Intelligence Approach to Find Lead Compounds for Treating Tumors. J. Phys. Chem. Lett. 2019, 10, 4382–4400. 10.1021/acs.jpclett.9b01426. [DOI] [PubMed] [Google Scholar]
- Gao K.; Nguyen D. D.; Chen J.; Wang R.; Wei G.-W. Repositioning of 8565 Existing Drugs for COVID-19. J. Phys. Chem. Lett. 2020, 11, 5373–5382. 10.1021/acs.jpclett.0c01579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez D.; Gaulton A.; Bento A. P.; Chambers J.; De Veij M.; Félix E.; Magariños M. P.; Mosquera J. F.; Mutowo P.; Nowotka M.; Gordillo-Marañón M.; Hunter F.; Junco L.; Mugumbate G.; Rodriguez-Lopez M.; Atkinson F.; Bosc N.; Radoux C. J.; Segura-Cabrera A.; Hersey A.; Leach A. R. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2018, 47, D930–D940. 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T. N. H.; Nguyen T. H.; Tam N. M.; Y V. T.; Pham N. T.; Huy N. T.; Mai B. K.; Tung N. T.; Pham M. Q.; Vu V. V.; Ngo S. T. Improving Ligand-Ranking of AutoDock Vina by Changing the Empirical Parameters. J. Comput. Chem. 2022, 43, 160. 10.1002/jcc.26779. [DOI] [PubMed] [Google Scholar]
- Gilson M. K.; Liu T.; Baitaluk M.; Nicola G.; Hwang L.; Chong J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.; Guestrin C.. XGBoost: A Scalable Tree Boosting System. KDD ’16: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016; pp 785–794.
- Duvenaud D. K.; Maclaurin D.; Iparraguirre J.; Bombarell R.; Hirzel T.; Aspuru-Guzik A.; Adams R. P.. Convolutional Networks on Graphs for Learning Molecular Fingerprints. In Advances in Neural Information Processing Systems; Cortes C., Lawrence N., Lee D., Sugiyama M., Garnett R., Eds.; Curran Associates, Inc., 2015; Vol. 28. [Google Scholar]
- Bergstra J.; Yamins D.; Cox D.. Making a Science of Model Search: Hyperparameter Optimization in Hundreds of Dimensions for Vision Architectures. Proceedings of the 30th International Conference on Machine Learning, 2013;Vol. 28; 115–123.
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay É. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Ramsundar B.; Eastman P.; Walters P.; Pande V.; Leswing K.; Wu Z.. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More; O’Reilly Media, 2019. [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J.; Gary E. N.; Shiomi K.; Rosenberry T. L. Structures of Human Acetylcholinesterase Bound to Dihydrotanshinone I and Territrem B Show Peripheral Site Flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. 10.1021/ml400304w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo S. T.; Tam N. M.; Pham M. Q.; Nguyen T. H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 2302–2312. 10.1021/acs.jcim.1c00159. [DOI] [PubMed] [Google Scholar]
- Zhang C.-H.; Stone E. A.; Deshmukh M.; Ippolito J. A.; Ghahremanpour M. M.; Tirado-Rives J.; Spasov K. A.; Zhang S.; Takeo Y.; Kudalkar S. N.; Liang Z.; Isaacs F.; Lindenbach B.; Miller S. J.; Anderson K. S.; Jorgensen W. L. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1-2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Aliev A. E.; Kulke M.; Khaneja H. S.; Chudasama V.; Sheppard T. D.; Lanigan R. M. Motional Timescale Predictions by Molecular Dynamics Simulations: Case Study using Proline and Hydroxyproline Sidechain Dynamics. Proteins: Struct., Funct., Bioinf. 2014, 82, 195–215. 10.1002/prot.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. C. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Huang Y.; Izadi S.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; SalomonFerrer R.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 18; University of California: San Francisco, 2018.
- Ellman G. L.; Courtney K. D.; Andres V. Jr; Featherstone R. M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Pohanka M.; Hrabinova M.; Kuca K.; Simonato J.-P. Assessment of acetylcholinesterase activity using indoxylacetate and comparison with the standard Ellman’s method. Int. J. Mol. Sci. 2011, 12, 2631–2640. 10.3390/ijms12042631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B. Bootstrap Methods: Another Kook at the Jackknife. Ann. Stat. 1979, 7, 1–26. 10.1214/aos/1176344552. [DOI] [Google Scholar]
- Schrödinger LLC . Schrödinger Release 2020-4. Maestro; August, 2020.
- Lee S. K.; Lee I. H.; Kim H. J.; Chang G. S.; Chung J. E.; No K. T.. The PreADME approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties, EuroQSAR 2002 Designing Drugs and Crop Protectants: Processes, Problems and Solutions; Blackwell Publishing: Maldenh, MA, 2003, pp 418–420. [Google Scholar]
- Nair H. K.; Quinn D. M. m-Alkyl, α,α,α-trifluoroacetophenones: A new class of potent transition state analog inhibitors of acetylcholinesterase. Bioorg. Med. Chem. Lett. 1993, 3, 2619–2622. 10.1016/s0960-894x(01)80727-7. [DOI] [Google Scholar]
- Savini L.; Gaeta A.; Fattorusso C.; Catalanotti B.; Campiani G.; Chiasserini L.; Pellerano C.; Novellino E.; McKissic D.; Saxena A. Specific Targeting of Acetylcholinesterase and Butyrylcholinesterase Recognition Sites. Rational Design of Novel, Selective, and Highly Potent Cholinesterase Inhibitors. J. Med. Chem. 2003, 46, 1–4. 10.1021/jm0255668. [DOI] [PubMed] [Google Scholar]
- Camps P.; Gómez E.; Muñoz-Torrero D.; Badia A.; Clos M. V.; Curutchet C.; Muñoz-Muriedas J.; Luque F. J. Binding of 13-Amidohuprines to Acetylcholinesterase: Exploring the Ligand-Induced Conformational Change of the Gly117-Gly118 Peptide Bond in the Oxyanion Hole. J. Med. Chem. 2006, 49, 6833–6840. 10.1021/jm060945c. [DOI] [PubMed] [Google Scholar]
- Tommonaro G.; García-Font N.; Vitale R. M.; Pejin B.; Iodice C.; Cañadas S.; Marco-Contelles J.; Oset-Gasque M. J. Avarol Derivatives as Competitive AChE inhibitors, Non Hepatotoxic and Neuroprotective Agents for Alzheimer’s Disease. Eur. J. Med. Chem. 2016, 122, 326–338. 10.1016/j.ejmech.2016.06.036. [DOI] [PubMed] [Google Scholar]
- Özgeriş B.; Göksu S.; Polat Köse L.; Gülçin İ.; Salmas R. E.; Durdagi S.; Tümer F.; Supuran C. T. Acetylcholinesterase and Carbonic Anhydrase Inhibitory Properties of Novel Urea and Sulfamide Derivatives Incorporating Dopaminergic 2-Aminotetralin Scaffolds. Bioorg. Med. Chem. 2016, 24, 2318–2329. 10.1016/j.bmc.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Thompson C. A. FDA Approves Galantamine for Alzheimer’s Disease. Am. J. Health-Syst. Pharm. 2001, 58, 649. 10.1093/ajhp/58.8.649a. [DOI] [PubMed] [Google Scholar]
- McNulty J.; Nair J. J.; Little J. R. L.; Brennan J. D.; Bastida J. Structure–Activity Studies on Acetylcholinesterase Inhibition in the Lycorine Series of Amaryllidaceae Alkaloids. Bioorg. Med. Chem. Lett. 2010, 20, 5290–5294. 10.1016/j.bmcl.2010.06.130. [DOI] [PubMed] [Google Scholar]
- Heller L.; Kahnt M.; Loesche A.; Grabandt P.; Schwarz S.; Brandt W.; Csuk R. Amino Derivatives of Platanic Acid Act as Selective and Potent Inhibitors of Butyrylcholinesterase. Eur. J. Med. Chem. 2017, 126, 652–668. 10.1016/j.ejmech.2016.11.056. [DOI] [PubMed] [Google Scholar]
- Samadi A.; Marco-Contelles J.; Soriano E.; Álvarez-Pérez M.; Chioua M.; Romero A.; González-Lafuente L.; Gandía L.; Roda J. M.; López M. G.; Villarroya M.; García A. G.; Ríos C. d. l. Multipotent Drugs with Cholinergic and Neuroprotective Properties for the Treatment of Alzheimer and Neuronal Vascular Diseases. I. Synthesis, Biological Assessment, and Molecular Modeling of Simple and Readily Available 2-Aminopyridine-, and 2-Chloropyridine-3,5-Dicarbonitriles. Bioorg. Med. Chem. 2010, 18, 5861–5872. 10.1016/j.bmc.2010.06.095. [DOI] [PubMed] [Google Scholar]
- Ahmed E.; Nawaz S. A.; Malik A.; Choudhary M. I. Isolation and Cholinesterase-Inhibition Studies of Sterols from Haloxylon Recurvum. Bioorg. Med. Chem. Lett. 2006, 16, 573–580. 10.1016/j.bmcl.2005.10.042. [DOI] [PubMed] [Google Scholar]
- Wen H.; Lin C.; Que L.; Ge H.; Ma L.; Cao R.; Wan Y.; Peng W.; Wang Z.; Song H. Synthesis and Biological Evaluation of Helicid Analogues as Novel Acetylcholinesterase Inhibitors. Eur. J. Med. Chem. 2008, 43, 166–173. 10.1016/j.ejmech.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Schwarz S.; Lucas S. D.; Sommerwerk S.; Csuk R. Amino Derivatives of Glycyrrhetinic Acid as Potential Inhibitors of Cholinesterases. Bioorg. Med. Chem. 2014, 22, 3370–3378. 10.1016/j.bmc.2014.04.046. [DOI] [PubMed] [Google Scholar]
- Heller L.; Schwarz S.; Obernauer A.; Csuk R. Allobetulin Derived Seco-Oleananedicarboxylates Act as Inhibitors of Acetylcholinesterase. Bioorg. Med. Chem. Lett. 2015, 25, 2654–2656. 10.1016/j.bmcl.2015.04.086. [DOI] [PubMed] [Google Scholar]
- Loesche A.; Wiese J.; Sommerwerk S.; Simon V.; Brandt W.; Csuk R. Repurposing N,N’-bis-(arylamidino)-1,4-piperazinedicarboxamidines: An unexpected class of potent inhibitors of cholinesterases. Eur. J. Med. Chem. 2017, 125, 430–434. 10.1016/j.ejmech.2016.09.051. [DOI] [PubMed] [Google Scholar]
- Schwarz S.; Loesche A.; Lucas S. D.; Sommerwerk S.; Serbian I.; Siewert B.; Pianowski E.; Csuk R. Converting Maslinic Acid into an Effective Inhibitor of Acylcholinesterases. Eur. J. Med. Chem. 2015, 103, 438–445. 10.1016/j.ejmech.2015.09.007. [DOI] [PubMed] [Google Scholar]
- Pietsch M.; Gütschow M. Synthesis of Tricyclic 1,3-Oxazin-4-ones and Kinetic Analysis of Cholesterol Esterase and Acetylcholinesterase Inhibition. J. Med. Chem. 2005, 48, 8270–8288. 10.1021/jm0508639. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Peng D.-Y.; Yang S.-G.; Zhu X.-L.; Yang W.-C.; Yang G.-F. Syntheses of Coumarin–Tacrine Hybrids as Dual-Site Acetylcholinesterase Inhibitors and Their Activity Against Butylcholinesterase, Aβ Aggregation, and β-Secretase. Bioorg. Med. Chem. 2014, 22, 4784–4791. 10.1016/j.bmc.2014.06.057. [DOI] [PubMed] [Google Scholar]
- Peng D.-Y.; Sun Q.; Zhu X.-L.; Lin H.-Y.; Chen Q.; Yu N.-X.; Yang W.-C.; Yang G.-F. Design, Synthesis, and Bioevaluation of Benzamides: Novel Acetylcholinesterase Inhibitors with Multi-Functions on Butylcholinesterase, Aβ Aggregation, and β-Secretase. Bioorg. Med. Chem. 2012, 20, 6739–6750. 10.1016/j.bmc.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Rahman A.-u.; Khalid A.; Sultana N.; Nabeel Ghayur M.; Ahmed Mesaik M.; Riaz Khan M.; Gilani A. H.; Iqbal Choudhary M. New Natural Cholinesterase Inhibiting and Calcium Channel Blocking Quinoline Alkaloids. J. Enzyme Inhib. Med. Chem. 2006, 21, 703–710. 10.1080/14756360600889708. [DOI] [PubMed] [Google Scholar]
- Akıncıoğlu A.; Akıncıoğlu H.; Gülçin İ.; Durdagi S.; Supuran C. T.; Göksu S. Discovery of Potent Carbonic Anhydrase and Acetylcholine Esterase Inhibitors: Novel Sulfamoylcarbamates and Sulfamides Derived from Acetophenones. Bioorg. Med. Chem. 2015, 23, 3592–3602. 10.1016/j.bmc.2015.04.019. [DOI] [PubMed] [Google Scholar]
- Vanzolini K. L.; Vieira L. C. C.; Corrêa A. G.; Cardoso C. L.; Cass Q. B. Acetylcholinesterase Immobilized Capillary Reactors–Tandem Mass Spectrometry: An On-Flow Tool for Ligand Screening. J. Med. Chem. 2013, 56, 2038–2044. 10.1021/jm301732a. [DOI] [PubMed] [Google Scholar]
- Bolognesi M. L.; Banzi R.; Bartolini M.; Cavalli A.; Tarozzi A.; Andrisano V.; Minarini A.; Rosini M.; Tumiatti V.; Bergamini C.; Fato R.; Lenaz G.; Hrelia P.; Cattaneo A.; Recanatini M.; Melchiorre C. Novel Class of Quinone-Bearing Polyamines as Multi-Target-Directed Ligands To Combat Alzheimer’s Disease. J. Med. Chem. 2007, 50, 4882–4897. 10.1021/jm070559a. [DOI] [PubMed] [Google Scholar]
- Arumugam N.; Almansour A. I.; Kumar R. S.; Kotresha D.; Saiswaroop R.; Venketesh S. Dispiropyrrolidinyl-Piperidone Embedded Indeno[1,2-b]quinoxaline Heterocyclic Hybrids: Synthesis, Cholinesterase Inhibitory Activity and Their Molecular Docking Simulation. Bioorg. Med. Chem. 2019, 27, 2621–2628. 10.1016/j.bmc.2019.03.058. [DOI] [PubMed] [Google Scholar]
- Thai Q. M.; Pham T. N. H.; Hiep D. M.; Pham M. Q.; Tran P.-T.; Nguyen T. H.; Ngo S. T. Searching for AChE inhibitors from natural compounds by using machine learning and atomistic simulations. J. Mol. Graphics Modell. 2022, 115, 108230. 10.1016/j.jmgm.2022.108230. [DOI] [PubMed] [Google Scholar]
- Tam N. M.; Pham M. Q.; Ha N. X.; Nam P. C.; Phung H. T. T. Computational Estimation of Potential Inhibitors from Known Drugs Against the Main Protease of SARS-CoV-2. RSC Adv. 2021, 11, 17478–17486. 10.1039/d1ra02529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood–brain barrier penetration. J. Pharm. Sci. 1999, 88, 815–821. 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.