

ABSTRACT
A key step in therapeutic and endogenous humoral antibody characterization is identifying the amino acid sequence. So far, this task has been mainly tackled through sequencing of B-cell receptor (BCR) repertoires at the nucleotide level. Mass spectrometry (MS) has emerged as an alternative tool for obtaining sequence information directly at the – most relevant – protein level. Although several MS methods are now well established, analysis of recombinant and endogenous antibodies comes with a specific set of challenges, requiring approaches beyond the conventional proteomics workflows. Here, we review the challenges in MS-based sequencing of both recombinant as well as endogenous humoral antibodies and outline state-of-the-art methods attempting to overcome these obstacles. We highlight recent examples and discuss remaining challenges. We foresee a great future for these approaches making de novo antibody sequencing and discovery by MS-based techniques feasible, even for complex clinical samples from endogenous sources such as serum and other liquid biopsies.
KEYWORDS: Mass spectrometry, sequencing, endogenous antibodies
Introduction
Around the time of their initial discovery, antibodies were termed by various illustrious names, such as ‘Immunkörper’, ‘Amboceptor’, and ‘Zwischenkörper’, among many others. These terms were used more than a century ago to describe substances with antitoxin, lysin, agglutinin, and precipitin activities.12 Nowadays, the generally accepted term antibody refers to secreted immunoglobulins (Igs), whose sequence variety is several orders more diverse than the assortment of their historical names. Antibodies represent some of the most important molecules in the human immune system. Over the last century, Igs have been intensively studied because of their role in combatting infectious diseases and have taken center stage for development of therapeutics in the last decade.3–5 Beyond infectious diseases, recombinant antibodies are now also developed for cancer, rheumatoid arthritis, and various other pathological conditions.6 As key entities in the body’s defense mechanism, circulating antibodies are found in various bodily fluids, such as serum, saliva, milk, the lumen of the gut, and cerebrospinal fluid.7 New leads for biotherapeutic development of recombinant antibodies come from various sources, such as immunizing animals with specific antigens, or by discovering pathogen-neutralizing antibodies from recovered patients.8–10
The estimated diversity of Ig molecules a human body can generate extends beyond 1015 theoretical sequences,11,12 indicating that each antigen may lead to a unique antibody response. These 1015 possible antibody sequences are all unique yet highly alike, which makes their characterization and sequencing highly complex, and thus a tremendously challenging task. Ideally, one would like to sequence antibodies at the protein level instead of through B-cell receptor (BCR) sequencing,13 which is currently the standard approach, to more directly probe circulating antibody repertoires and their relative abundances in specific environments. Mass spectrometry (MS) is expected to be the method of choice to potentially achieve this feat, as MS-based protein analysis has advanced and matured considerably.14,15 However, antibodies represent a very special and rather challenging class of proteins. Consequently, while MS has been already used to characterize and sequence highly purified monoclonal antibodies (mAbs),16–18 further technical developments in sample preparation and data analysis are needed to incorporate MS fully and efficiently into an endogenous humoral antibody discovery and characterization pipeline. This review evaluates the role that MS can play in sequencing, identifying, and characterizing antibodies, focusing mainly on emerging strategies used to enable identification and characterization of endogenous neutralizing antibodies.
Nomenclature, structure, and diversity of antibodies
Humoral human antibodies are complex proteins produced by B cells.7,19 Most antibody molecules (e.g., IgGs) are made up of four protein chains: two identical light chains and two identical heavy chains, which are interconnected by disulfide bridges (Figure 1). The light- and heavy-chain form two heterodimers, which are connected via disulfide bridges in the hinge region to form the intact antibody. Functionally, the intact antibody can be divided into two antigen-binding domains (also known as Fab or fragment antigen-binding) and a constant domain (also known as Fc or fragment crystallizable)22 (Figure 1a). The Fc is the effector entity of the antibody and can bind to Fc receptors on immune cells7 and mediate immune effector responses such as phagocytosis, antibody-dependent cell-mediated cytotoxicity, respiratory burst, and cytokine release.23 In contrast to the fully conserved sequence and structure of the Fc, the Fab is responsible for the vast diversity in recognized antigens and is thus hypervariable.
Figure 1.
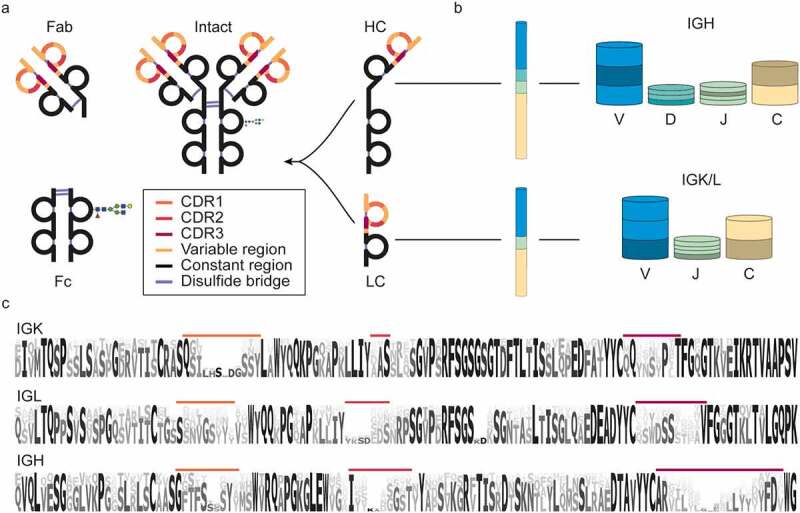
Nomenclature, structure, and diversity of IgG1 antibodies. (a) Nomenclature and protein fragments of an IgG1 molecule. The antigen-binding domain, containing light and heavy chain (LC and HC respectively) variable regions, is termed Fab (or Fab2 when dimerized). The constant part of the heavy chain carrying an N-glycosylation site is called Fc. Other IgG subclasses vary in their heavy chain constant region (Fc) and disulfide patterns. (b) The diversity in antibodies originates primarily from the V, D, J, and C-allele (each annotated with a distinct color) recombination process. In this process, each of many individual V, D, J, and C-alleles can recombine with any of the other gene segments, yielding thousands of possible combinations, in particular for the heavy chain, which incorporates the most diverse D region. (c) Sequence logo created by the alignment of in silico generated sequences of Ig kappa (IGK) and lambda (IGL) light chains and Ig heavy chain (IGH) from the international ImMunoGeneTics (IMGT) information system database.20,21 Even though the displayed sequences are part of the variable domain, large stretches of these sequences, also known as the framework regions (FRs), are relatively conserved, compared to the hypervariable complementarity determining regions (CDRs), colored in accordance with (a).
Because there is an endless and constantly evolving pool of pathogens, the antibody repertoire needs to be incredibly diverse and versatile to counteract these challenges.24,25 In humans, this enormous diversity in the potential antibody repertoire is achieved through several mechanisms. Starting at the genomic level, the light and heavy chains are encoded in four genes each: Variable (V), Diversity (D), Joining (J), and Constant (C), with the light chain lacking the D-gene. These genes are encoded in multiple alleles, which can recombine to a staggering number of combinations (Figure 1b).26 The recombination process is also error-prone, leading to insertions and deletions at the junctions between the regions, referred to as junctional diversity. By recombination alone, the number of possible variable domain sequences already reaches tens of thousands. However, the eventual antibody diversity is expanded even further by natural polymorphisms, mutations, and class switching. As the major contributor to antibody hypervariability, somatic hypermutations can occur during B-cell affinity maturation and do so at a million-fold increased rate compared to the usual mutation rates.11 These mutations are largely concentrated in the complementarity-determining regions (CDR1-3), separated by framework regions (FR1-4), which form the conserved backbone of the Fab structure (Figure 1c). Located at the tips of the Y-shaped antibody structure, CDRs are primarily responsible for antigen binding, and, therefore, elucidation of their sequences is of the utmost importance for antibody discovery.
The Fc part of Igs is used to classify antibodies into one of 5 classes: IgA, IgD, IgE, IgG, and IgM. Some of these classes are divided further into subclasses denoted by numbers, e.g., IgG1-4 or IgA1 and IgA2. Although the function of the classes and subclasses is different, their variable regions stem from the shared pool of genes. Therefore, for simplicity, in this review, we focus primarily on IgG1, the most abundant antibody subclass in serum, and the predominantly used subclass for biotherapeutic development. Still, concerning de novo sequencing by MS, different Ig classes and subclasses pose similar challenges and opportunities.
Modalities of MS-based antibody analysis
Proteomics is the large-scale study of proteins. Many different peptide- and protein-centric MS-based approaches have been developed for proteomics, whereby some of these have been adapted for de novo sequence analysis of antibodies.
Bottom-up (BU) or shotgun proteomics is by far the most widespread approach in MS-based protein analysis. In it, protein samples are digested by one or more proteases, and the resulting peptides are separated by some form of liquid chromatography (LC; usually reversed-phase high performance LC), after which their peptide masses are recorded (MS1). Highly abundant precursor ions are then selected for fragmentation, and the masses of their fragment ions (MS2) are recorded. To avoid ambiguity, we distinguish here between chemical or enzymatic cleavage, where proteins are digested into peptides before analysis by MS (referred to here as cleavage/digestion), and gas-phase fragmentation in the collision area of a mass spectrometer (referred to here as fragmentation/dissociation), where precursor ions are dissociated into fragment ions.
Because digestion and MS-based fragmentation adhere to highly specific rules, peptides and their gas-phase fragment ions can be predicted. Consequently, peptides and their parent proteins are identified by comparing recorded spectra to the spectra simulated from protein or DNA databases.27 For antibody sequencing, personalized databases are required for identification. Yet, digestion-based strategies are still widely used even without an available database. Individual spectra can be de novo sequenced, and the resulting reads can be assembled into full-length sequences.16,28,29
Additionally, intact mass analysis is a useful tool for protein analysis, providing masses that can be considered fingerprints of the species (known as proteoforms) present in the sample. Comparing different masses can lead to conclusions about relations between multiple species, for instance, if they differ by the mass of a known mutation, post-translational modification (PTM), or signal peptide.30 In the case of antibodies, such analysis can be performed with the protein in its native, and possibly complexed, state, or unfolded and separated into the comprising chains. Such approaches can provide valuable insights in the context of antibodies, e.g., by assessing the complexity of antibody repertoire or following changes in abundance of specific clones.31 When applied to de novo sequencing, the precursor mass knowledge can help determining the light and heavy-chain pairing or sequence prediction accuracy in BU sequencing.32
In addition, both denatured and native antibodies can also be fragmented to yield some sequence information, this approach is called top-down (TD) MS. Because of the much larger size and higher charge of the analyzed species, such intact-protein fragmentation spectra are more complex and harder to interpret than peptide spectra.33,34 To mitigate this, specific proteases can be used to cleave proteins into smaller subunits. This practice is called middle-down (MD) MS, and in the context of antibodies it is often performed by cleaving the hinge region of the heavy chain before MS analysis.35 Fragmentation spectra of entire chains or intact antibodies can provide valuable tools for both sequence determination and validation of sequence predictions, as fragmentation is highly specific for the precursor clone, which is often untrue in BU analysis.36
The emerging role of mass spectrometry in antibody discovery
Due to the structural complexity and immense sequence diversity of antibodies, the development of therapeutic antibodies has always been a very challenging and labor-intense task, especially when compared to small-molecule drug development. For example, the discovery of trastuzumab was achieved by using mice immunized with antigen-expressing cells. Following the generation and selection of hybridomas that showed specific activity,37 the sequence of the selected antibody was determined after cloning and expression. A humanized antibody could be produced only thereafter by adapting and modifying the sequence accordingly.38 The same approach was used in the development of other mAbs.39–42 Apart from being expensive and laborious, these early strategies required knowledge and availability of purified antigens and animal models that can produce specific antibodies in response to these antigens.43
More recently, alternative strategies for antibody discovery have been explored starting with the screening of B cells from individuals who successfully overcame an infection. In this approach, peripheral blood mononuclear cells (PBMCs) are isolated, immortalized, and screened for antigen reactivity. The reactive clones are further expanded and characterized. This method has proven effective in finding new neutralizing antibodies that can be used to combat certain infectious diseases, e.g., Ebola8,9 or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).10 These recent advances show that the discovery of antibodies from human subjects, in addition to animal models, represents a viable method for developing new avenues for therapies. However, it may be even more advantageous to discover and characterize mature antibody clones directly from clinical samples at the protein level in their functionally matured and active form.
In recent years, MS-based proteomics has advanced tremendously in sample preparation, MS and LC instrumentation, and data analysis.14,15 Using all these advances, antibody sequencing at the protein level by MS has come within reach. Here, we review recently introduced techniques, highlighting three chiefly MS-based strategies used to determine antibody sequences (Figure 2). These three pillars are primarily classified by the source of sample material and the attainable sequencing information. The first strategy applies to highly purified recombinant antibodies that are now amenable for full sequencing with BU MS, often by combining several different proteases and advanced algorithms. Second, hybrid approaches have been introduced for analyzing endogenous antibody repertoires by combining MS-based techniques with genomics/transcriptomics, e.g., whole-genome sequencing or BCR sequencing, ideally from the same donor. The third set of techniques encompasses several MS-based de novo approaches that aim to determine complete antibody sequences of selected clones directly from clinical samples without aid from alternative omics data. While each strategy is distinct, they all share common aspects. Finally, we hypothesize which improvements are needed in the future to make comprehensive sequencing of polyclonal antibody repertoires a reality.
Figure 2.
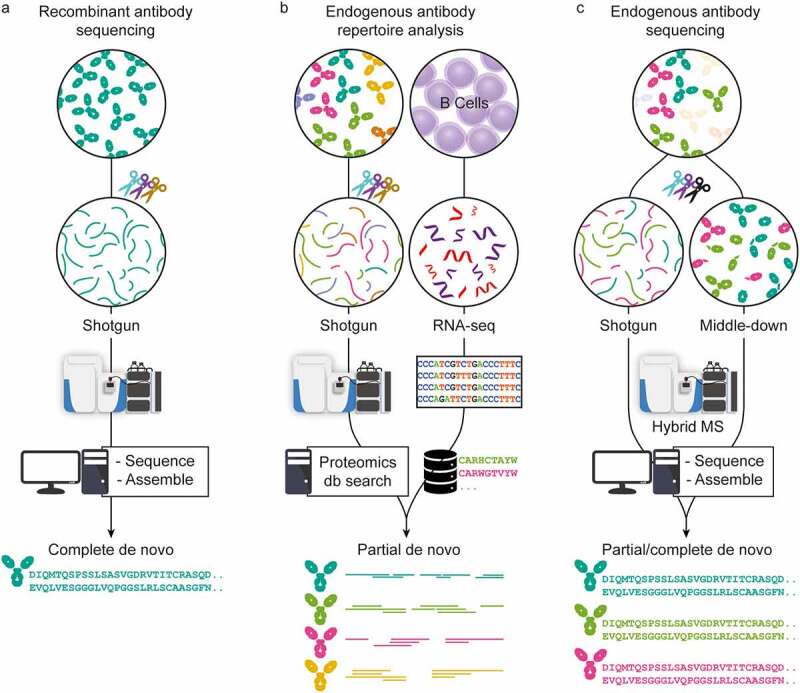
Three approaches in MS-based antibody sequencing. (a) Recombinant antibody sequencing generally starts with abundant highly purified mAbs, which can be fully sequenced through BU MS, where hundreds of peptides are generated by digesting the mAb with one or several proteases, providing multiple overlapping short sequence reads. After liquid chromatography-mass spectrometry (LC-MS) measurement, the spectra can be processed by several different de novo sequencing software solutions and assembled into full-length mAb sequences.16,17 (b) In repertoire analysis, a sequence database is generated through B-cell sequencing, and MS-data is obtained through BU MS experiments. After generation of the personalized database, a high throughput of the LC-MS is possible.44 While not strictly de novo since only hits from the sequence database are identified, it is a powerful tool for antibody repertoire analysis. (c) Endogenous antibody sequencing cannot rely on BU MS alone, as direct sequencing of endogenous humoral antibodies is hampered by inherent challenges and complexity. Emerging MD and TD MS techniques provide clone-specific sequence information highly complementary to traditional sequencing. Integrating BU MS and MD/TD MS makes it possible to achieve full-length coverage of antibody sequences.31
MS-based sequencing of recombinant monoclonal antibodies
Before delving into the topic of MS-based sequencing of endogenous antibodies from clinical samples, we first discuss the current state-of-the-art sequencing approaches developed for recombinant mAbs. Principles of mAb sequencing by MS share many technical considerations with sequencing of antibodies present in complex mixtures. Furthermore, currently available strategies for recombinant mAb sequencing provide great context for discussing limitations and bottlenecks that hamper sequencing of endogenous antibody clones.
Shotgun, bottom-up strategies used for sequencing of highly purified mAbs
Antibodies are often analyzed after digestion with one or more proteases to generate peptides (Figure 2). Such peptide-centric approaches, known as BU MS, represent the most popular type of proteomics experiments. In contrast to most shotgun proteomics experiments, de novo sequencing through BU MS necessitates a high depth of sequence coverage, i.e., each sequence position in the antibody is ideally supported by multiple overlapping unique peptides. With a typical highly specific protease such as trypsin, which cleaves C-terminally of lysine and arginine, and a low number of missed cleavages, sequence-coverage depth is often limited because only a few of the generated peptides overlap in sequence. Although this suits standard shotgun proteomics experiments, which do not require full sequence coverage of the analyzed proteins, de novo sequencing of antibodies thus requires other approaches.
Several methods to generate complete and deep sequence coverage by overlapping peptides have been introduced. For example, a shortened protease incubation time was successfully used to increase the number of peptides carrying missed cleavage sites.45 Some proteases generate a high number of overlapping peptides through nonspecific cleavage.32,46,47 Alternatively, nonspecific cleavage can be also achieved through non-enzymatic treatment, e.g., microwave-assisted hydrolysis.48 For these methods to work, digestion conditions must be tightly controlled to avoid abnormally long or short peptides and ensure reproducibility. Another elegant option is to use multiple proteases with synergistic sequence specificities. For instance, Peng et al.17 recently used a total of 9 proteases, both specific and nonspecific, to successfully de novo sequence a full-length anti-FLAG-M2 mouse mAb (Figure 3). The strength of a large panel of proteases is shown in the validation of the CDR sequences by high scoring peptides covering the entire CDR. The 6 chosen peptides are the result of digestion by 5 different proteases (trypsin, chymotrypsin, lysC, thermolysin, and elastase; Figure 3a,b).
Figure 3.
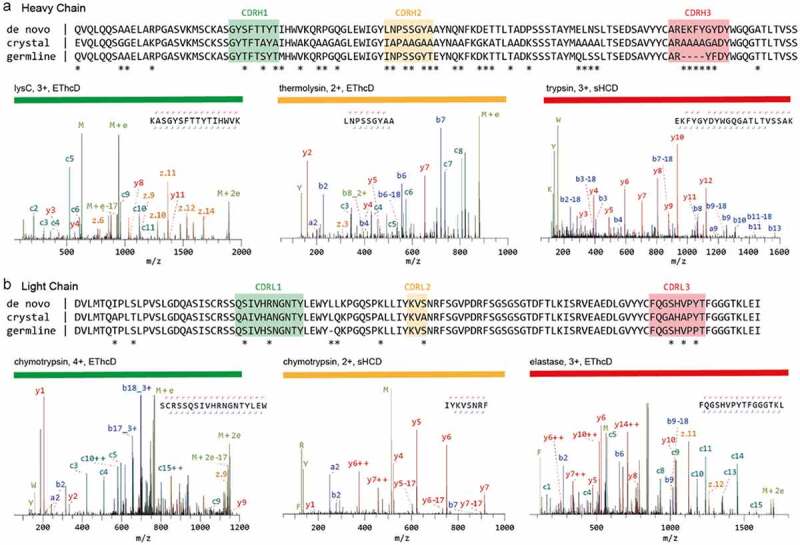
Sequencing of a monoclonal Anti-FLAG M2 antibody. The variable regions of the heavy (a) and light chains (b) are shown. The de novo sequence derived by MS is shown on top, alongside the previously published sequence used in the crystal structure of the Fab (PDB ID: 2G60) and germline sequence (IMGT-DomainGapAlign; IGHV1-04/IGHJ2; IGKV1-117/IGKJ1). Differential residues are highlighted by asterisks (*). Exemplary MS/MS spectra in support of the assigned sequences are shown below the alignments, labeled with protease, precursor charge state, and fragmentation type. The peptide sequence and fragment coverage are indicated in the top-right of each spectrum spectra, with b/c ions indicated in blue/teal and y/z ions in red/orange. The same color annotation is used for peaks in the spectra, with additional peaks such as intact/charge reduced precursors, neutral losses, and immonium ions indicated in green. Figure and caption adapted from Peng et al.17
Nowadays, most de novo sequencing solutions, such as ALPS/PeaksAB,49 GenoMS,47 SuperNovo16 and Champs50 are quite successful in obtaining full sequence coverage of highly purified antibodies. To determine the antibody sequences de novo, all these software tools require large numbers of overlapping peptides, spanning the entire sequence, which are successfully fragmented and converted into predicted peptide sequence reads (Figure 2a). This necessitates generation of BU MS data via use of multiple proteases. While complicating sample preparation and increasing the required amount, such multi-protease approaches are advantageous for de novo sequencing by alleviating the sequence assembly problem.
Benefits of complementary peptide fragmentation techniques
In MS-based sequencing, extensive fragmentation of peptide ions is essential to generate arrays of adjacent fragments that reveal the amino acid sequence, often referred to as ion ladders or sequence tags (Figure 4a). The amino acid sequence of the fragmented peptide is derived by comparing the mass difference between two adjacent fragment ion peaks to the masses of amino acids and combinations thereof. The produced fragment ion series must contain very few gaps larger than a single amino acid residue because such gaps lead to exponential growth of the amino acid combinations that fit the mass difference, particularly for spectra of lower resolution.53 Since there is no universal fragmentation method that can produce uninterrupted fragment ion ladders for all possible peptides, it is highly advantageous to use various fragmentation methods with distinct mechanisms and specificities to complement each other (Figure 4b).54 While collision induced dissociation (CID) is the most used technique in shotgun proteomics experiments, multiple alternative fragmentation techniques have been introduced and have proven to be complementary. These specificities stem from the unique ion activation mechanisms used by each method. In collision-based techniques, energy is deposited to the multiply protonated peptide ions through low-energetic collisions with inert neutral atoms or gas molecules. This energy is redistributed vibrationally throughout the peptide backbone, fragmenting the most labile bonds and yielding b/y-type fragment ions, as defined by the Roepstorff-Fohlmann-Biemann ion nomenclature (Figure 4c).55 Although protonated amide bonds are usually the most susceptible to fragmentation, collisional dissociation often also leads to loss of labile PTMs, such as phosphorylation and sialyation.
Figure 4.
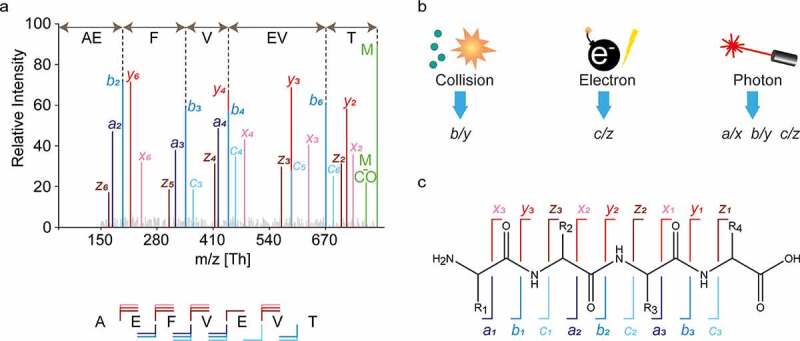
Peptide fragmentation in MS-based de novo sequencing. (a) An illustrative fragmentation spectrum. In the spectrum, fragment ion peaks are color annotated according to the type of fragment ion (a: purple, b: blue, c: light blue, x: pink, y: red, and z: brown) the unfragmented peptide (precursor ion) is shown in green as well as the precursor ion with neutral loss of CO. Adjacent fragment ions of the same type have a mass difference corresponding to a single amino acid, which is used to determine the sequence as is illustrated for b-ions above spectrum. Below the spectrum the amino acid sequence is shown together with the fragment ion annotation, n-terminal fragments (a-, b-,c-) are below the sequence and c-terminal fragments (x-, y-, z-) are shown above the sequence. (b) Three predominant gas phase fragmentation techniques with their predominantly produced fragment ion types. Collisional dissociation (CID/CAD/HCD) predominantly yield b/y ions. Electron based dissociation (ECD/ETD) yields c/z ions. Contrary the other techniques, high energy photon based dissociation (UVPD) results in all fragment ion types.51,52 (c) The Roepstorff-Fohlmann-Biemann nomenclature used for peptide fragment ions denotes different fragment ion types by italic letters a-c and x-z. The numbering indicates the position of the bond in the amino acid sequence with respect to the N- and C-termini.
In electron-based techniques, such as electron capture-induced dissociation (ECD), positively-charged peptide ions capture electrons, leading to the generation of odd-electron species that dissociate promptly without significant vibrational redistribution.56–58 In contrast to collisional dissociation, this process is not directed toward the most labile bonds, and produces distinctively c and z fragment ions through the dissociation of N-Cα bond (Figure 4c). Similarly, high-energy ultra-violet photon-based activation and dissociation techniques (UVPD) also cause bond dissociation without substantial energy redistribution. This is enabled by a number of chromophores along the peptide backbone and results in a wide array of co-occurring fragment ion types (a/x, b/y, c/z), depending on the wavelength used.51,52 Highly energetic fragmentation methods can also lead to w-type ions, which involve an amino acid side-chain dissociation.59,60 In de novo sequencing, this may be advantageous since it allows leucine and isoleucine to be distinguished, although they have identical masses.
While having multiple fragment ion types in a single spectrum can complicate ion ladder detection (Figure 4a), it can also provide insight into the direction of fragment ion series, revealing the terminus (N or C) to which peptide fragments belong. This is possible due to the characteristic mass shift patterns of consecutive a, b, c fragments and consecutive x, y, z fragments originating from the same peptide bond. Horn et al.61 pioneered this approach for de novo protein sequencing by combining CID and ECD to discern between the N- and C-terminal fragment ions, which simplified the detection of consecutive fragment ions. Subsequently, many others have used similar strategies.62–66
The report by Peng et al.17 also demonstrates the successful use of multiple fragmentation techniques. They recorded spectra using a dual fragmentation scheme of both high-energy collision dissociation (HCD) and electron-transfer high-energy collision dissociation (EThcD), resulting in a reduced number of sequencing errors when compared to using a single fragmentation method. The spectra selected to support the CDR predictions are also derived from both fragmentation techniques, showing that this versatile fragmentation strategy can benefit sequence coverage in these challenging and important regions (Figure 3).
Such multiplexing MS strategies have made de novo sequencing of mAbs feasible, at least when they are of sufficient purity. However, the procedure is quite laborious as it often involves using multiple proteases to generate overlapping peptides and multiple peptide fragmentation techniques to obtain unambiguous sequence reads, which entails longer sample preparation time, the requirement of larger sample amounts, and extensive data acquisition.
Homology-aided de novo sequencing of antibodies
To identify peptides and proteins, shotgun proteomics experiments rely on matching observed fragmentation spectra to theoretical spectra generated from sequence databases. However, complete and accurate mature sequences are not generally available for many proteins, especially for highly variable or frequently mutated proteins like antibodies. Instead, homologous sequences, primarily derived from genomic or transcriptomic experiments, can be used. For antibodies, the genes encoding each of the regions (V, D, J, and C) are available as germline sequences and can be retrieved from the international ImMunoGeneTics information system (IMGT) database.20,21 While such a database of homologous sequences can facilitate verification or guide predictions of de novo sequences, it should be noted that the exact match to the target sequence is likely not present even in the most extensive databases. Traditional database searches are thus not applicable because they require exact mass matching of fragments, and a single amino acid mutation can prevent identification. Instead, error-tolerant fragment matching algorithms, either based on sequence alignments or subsequence (i.e., sequence tag) extractions, can use homologous databases to score experimentally determined sequences.
An example of a homology-aided approach is searching BU MS data from a sample of human antibodies against a proteome database such as Swiss-Prot, whereafter the identified peptides are aligned to the IMGT database.67,68 Further reported adaptations include de novo sequencing of unidentified features from the initial search with dedicated tools, such as PEAKS, to sequence and identify hypervariable regions.69,70 Homology-aided de novo sequencing algorithms are also advantageous in identifying erroneous de novo peptide reads by comparing them against homologous sequences. In addition, they can be used as a germline template to aid in the assembly of de novo peptide reads. Alternative to scaffolds based on homologous sequences, accurate masses of the antibody clones and constituent parts, e.g., light chain or heavy chain, can create mass-based scaffolds. However, these masses need to be obtained separately by performing additional protein-centric MS experiments.
Protein-centric MS approaches
Although conventional de novo sequencing of proteins predominantly follows a peptide-centric approach, there have been various attempts to analyze recombinant mAbs intact or at the level of large domains, e.g., Fabs, bringing along a new set of challenges. First, compared to peptides, intact proteins sometimes ionize less efficiently, and LC-based separation of peptides is more established than separation of intact proteins. Furthermore, in MS analysis, mass accuracy and resolution typically diminish with increasing molecular weight, even when using the latest high-resolution mass spectrometers.30,71,72 In addition, full sequence coverage is generally unattainable for intact proteins with masses above 20 kDa. These factors have hindered implementation of protein-centric MS for de novo sequencing of antibodies. However, more recently, several advances in the field resulted in relatively high sequence coverages, reported for recombinant mAbs with available reference sequences.36,73–75 Protein-centric approaches such as TD MS33 can provide additional valuable information, including the mass of the intact antibody,30 masses of the light and heavy chains, and some predictable fragment ions, which could be used as mass constraints.18,31,76–78
Similar to peptide-centric strategies, there is the potential to combine multiple fragmentation techniques in TD MS to boost sequence coverage. In addition, intact antibody sequencing can be simplified by reducing the complexity and size of the molecules through disulfide reduction or by digestion of the antibodies using specific proteases, such as IgdE (commercially termed FabALACTICA), which cleaves above the hinge region of IgG1, specifically producing 50 kDa Fab fragments,79 or IdeS (FabRICATOR), a cysteine protease that digests antibodies at a specific site below the hinge, generating F(ab’)2 fragments of all IgG subclasses.35 Such strategies deviate from intact protein sequencing, which resulted in the introduction of the term MD MS.80 However, these MD strategies still adhere to the core principles of protein-centric MS, whereby large (50-100 kDa) domains of antibodies are analyzed.
In a large body of works, Fornelli et al.36,81–83 have shown how various factors, including sample preparation strategies, fragmentation conditions, and other improvements in instrumentation and experimental design, influence sequence coverage in the protein-centric analysis of recombinant mAbs. Recently, Shaw et al.84 demonstrated that with modern instrumentation it is possible to successfully fragment intact mAbs in their native state (Figure 5). By combining ECD and HCD in a single tandem MS experiment, 42% sequence coverage for the light chain (Figure 5a) and 20% sequence coverage for the heavy chain (Figure 5b) of trastuzumab were obtained. The resulting fragmentation spectrum contained not only the multiply charged backbone fragmentation products, but also the intact light chain, ejected from the antibody by fragmentation of the intermolecular disulfide bridge, providing information on the light- and heavy-chain pairing (Figure 5c). These and many other studies culminated in a large joint effort by the Consortium for Top-Down Proteomics, wherein they comprehensively described available approaches, techniques, and instrumentation for the analysis of recombinant mAbs.18
Figure 5.
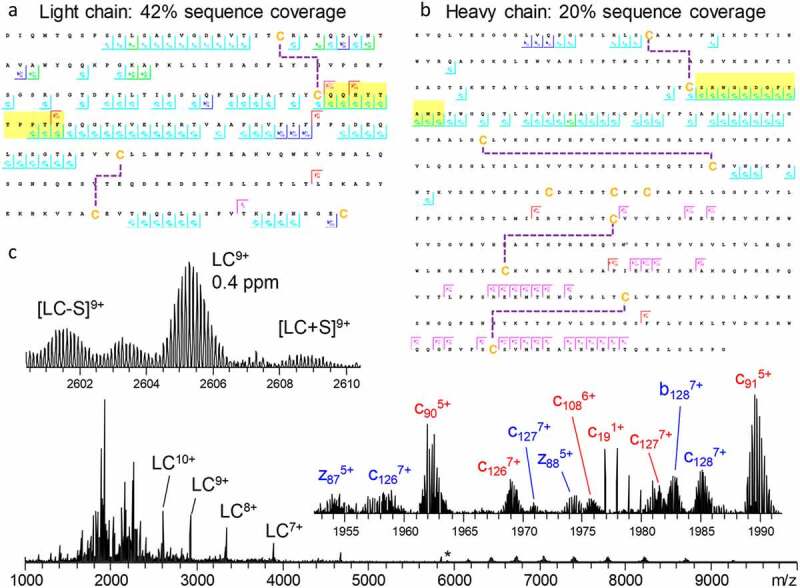
Light chain (a) and glycosylated heavy chain (b) fragmentation maps illustrate sequence coverage produced by the combination of ECD and HCD on Trastuzumab. Disulfide bonds are shown by dashed lines, CDR3 regions are highlighted in yellow. The corresponding fragmentation spectrum (c) for the 25+ charge state of intact Trastuzumab with insets displaying the zoomed in region containing the 9+ charge state of the light chain and various fragment ions. Red and blue fragment ion labels correspond to the light and heavy chain, respectively. Asterisk indicates the mass-selected precursor ion. Figure adapted from Shaw et al.84
Electron-based fragmentation of intact protein ions holds great potential for mAb sequencing. Several recent studies showed that these methods consistently yielded nearly uninterrupted c-ion ladders spanning the CDR3, which is paramount to antigen binding.31,76,77,84,85 These studies also demonstrated for various antibody isotypes (IgG1-4 and IgA1) that electron-based fragmentation methods consistently provide fragments containing the entire variable region of both the light and heavy chain. Notably, very similar fragments were formed for the intact mAb, the F(ab’)2 (produced with IdeS enzyme), and Fab molecules (produced with IgdE or Operator enzymes), showing that reducing antibody complexity through the removal of the Fc portion is not detrimental for protein-centric analysis of mAbs.
While substantial advances have been made in protein-centric sequencing of purified recombinant antibodies, studying endogenous antibodies remains much more challenging. Due to their large molecular size, and higher variability in molecular characteristics, the separation of intact proteins by LC is generally more difficult when compared to small peptides.86 This problem is exacerbated for antibody mixtures since different antibody clones are very similar and only vary in a small fraction of the overall sequence. Such minute differences are easily resolved on the peptide yet are significantly more difficult to distinguish on the level of intact antibodies with more than 1000 amino acid residues. Notwithstanding the challenges of intact protein MS, the prospects and potential benefits that protein-centric approaches bring to the de novo analytical toolbox are hard to neglect. While it is still nearly impossible to fully de novo sequence intact mAbs, protein-centric sequencing can be combined with peptide-centric methods in a hybrid MS approach, providing complementary information substantially advancing toward the goal of complete antibody sequencing by MS, as further described in the section “Combining peptide- and protein-centric MS approaches for antibody sequencing” (Figure 2b).
Dedicated software solutions for MS-based antibody sequencing
The various sample preparation methods and intricate experimental designs presented above result in extended datasets that are not feasible for manual interpretation. Thus, development of dedicated software tools for data interpretation is essential.
With regard to BU MS data, presently, two popular software suites are tailored toward de novo sequencing of antibodies, SuperNovo16 and PeaksAB.49,87 These suites can use the benefits of data generated by using multiple enzymes, multiple-fragmentation methods, and the use of a homologous antibody germline sequence database like IMGT to make a complete de novo sequence prediction based on the BU MS data. More specifically, the software iteratively screens predicted peptides against the germline gene segments of the antibody to determine the positions on the final-chain construct. Homologous germline sequence candidates represent scaffolds that are then modified to account for the highest scoring predicted peptides. This allows the prediction of both heavy- and light-chain sequences with a minimal error rate of only a few single amino acids per sequenced antibody. A disadvantage, however, is that the software currently works exclusively for sequencing single, highly purified antibodies.
Novel software solutions for de novo antibody sequencing are emerging and advancing in parallel with improvements in experimental design and instrumentation. The fast development of new de novo sequencing strategies encourages the development of new software solutions and improvement of already established tools and requires adaptable software to accommodate the frequent and considerable shifts in de novo sequencing approaches, such as the inclusion of TD or MD MS data, multiple-fragmentation methods or the analysis of polyclonal samples as opposed to mAbs.
Combining peptide- and protein-centric MS approaches for antibody sequencing
Recent advances in protein-centric MS have spawned various software tools that use these data either in a standalone manner, such as in Twister,63,88 or integrate them with BU MS data, as in TBNovo.89 Twister applies methods similar to those used for BU MS sequencing, recombining individual sequence tags (rather than peptide reads) into longer sequences using a specific implementation of de Bruijn graphs (T-Bruijn graphs) and sequence tag convolution.63,88 TBNovo uses sequence tags and precursor masses from TD MS to provide a scaffold for positioning the de novo predicted peptide reads to fill the complete sequence. Their analysis makes use of external BU de novo sequencing software, PEAKS,87 and was tested on protein mixtures. TBNovo has not achieved widespread adoption, perhaps due to the software’s complexity and because protein-centric MS was still barely practiced at the time of its first release.
Although antibody sequencing at the protein level is still not trivial, it is being applied on a steadily increasing scale in academia and industry. Efforts to extend the sequencing of antibodies to polyclonal mixtures have, however, proven extremely challenging. The first obstacle is sample availability. While recombinant mAb samples are typically available in milligram quantities, polyclonal antibody samples are often derived from clinical samples and thus only available in limited quantities. Because the median concentration of individual clones in plasma is ~1 µg/mL the available protein per individual clone is generally orders of magnitude less compared to mAbs.31 Furthermore, isolation of individual clones is extremely challenging, further complicating the sequencing process, as most software tools are exclusively designed for assembling a single antibody, and therefore fail when data represent several similar Ig sequences. Additionally, in complex endogenous polyclonal antibody mixtures, key sequence evidence on the hypervariable regions is often not detected due to a dilution effect, whereby sequence information from the conserved regions becomes amplified (as the latter is present in every clone) and thus suppresses the signal of the CDRs, which are unique for all clones. Even though the algorithms developed for mAb sequencing are not directly applicable for polyclonal antibody sequencing, they provide a great starting point for developing new tools.
Hybrid and multi-omics approaches for studying antibody repertoires
One way to further bridge the gap between sequencing of a single purified antibody and those present in bodily fluids, e.g., serum, is to use hybrid or multi-omics strategies. Using a multi-omics approach, for instance, by supplementing BU MS data with genomics or transcriptomics data derived from the same donor, allows bypassing some challenging aspects of genuine de novo sequencing, albeit at the cost of a more complex, labor- and data-intensive workflow (Figure 2c). While these methods deviate slightly from the core theme of this review, i.e., MS-based de novo sequencing, they do represent key advancements. Presently, direct de novo sequencing of antibodies from a complex mixture is still a tremendous challenge. However, integrating complementary information from multiple sources makes it possible to derive valuable data, even on endogenous antibody repertoires. Several approaches have been pioneered recently, as depicted in Figure 6 and described in more detail below.
Figure 6.
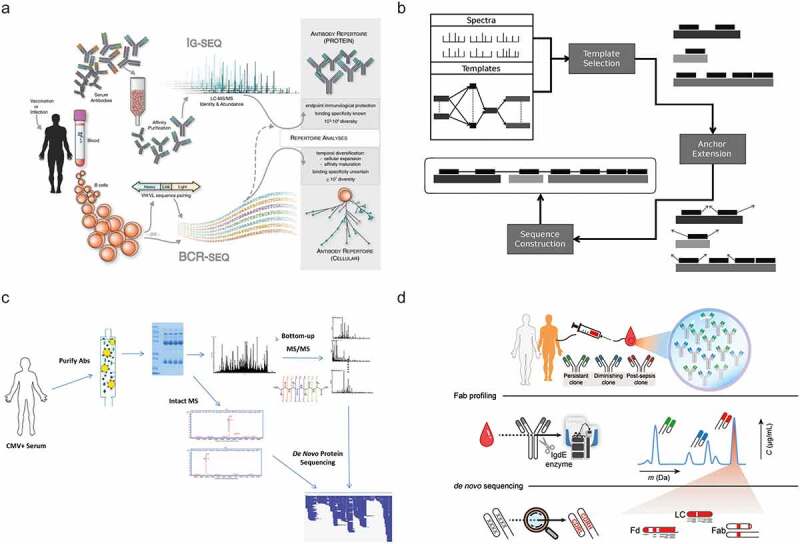
Selected recent approaches aiming toward MS-based de novo sequencing of serum antibodies. (a) In Ig-seq.90 a personalized database generated by BCR sequences is used to identify specific clones, using tryptic peptides covering the CDR3 region. Figure adapted from Lavinder et al.90 (b) GenoMS47 uses genomic data to generate template sequences. The specific construction of the templates can be defined by the user from either whole genome sequencing or BCR sequencing data. Figure adapted from Castellana et al.47 (c) PolyExtend32 helped to analyze a polyclonal mixture of antigen-specific purified antibodies measured by BU MS and intact mass measurements. Using a user-assisted algorithm, these data from different MS modalities were combined to sequence the most abundant clones. Figure adapted from Guthals et al.32 (d) Fab profiling31 measures and quantifies intact masses of Fabs to provide a view of the IgG1 clonal repertoire, enabling to quantify and monitor individual clones. Abundant serum clones are identified by using BU and MD MS data iteratively to generate full IgG de novo sequences. Figure adapted from Bondt et al.31
Ig-seq
Since the CDRs of the antibodies largely determine antigen specificity, it comes as no surprise that methods specifically targeting CDR-derived peptides have emerged. Notably, the Ig-seq method pioneered by Lavinder et al.90 in the Georgiou lab applies B-cell sequencing of a given donor to construct a database of putative CDR3 heavy-chain peptides. This database is then used to identify and quantify antibodies using CDR-specific tryptic peptides, effectively side-stepping the need for complete de novo sequencing (Figure 6a). This workflow is very effective because trypsin-targeted residues (arginine and lysine) are found to precede the CDR3 specifically and are found in the relatively conserved FR4 of the heavy chain, ensuring that tryptic peptides contain the heavy-chain CDR3 in the majority (>92%) of IgG clones.91 BU MS is highly optimized for measuring and detecting tryptic peptides, which makes this approach highly effective, as shown when this method was applied to the longitudinal monitoring of influenza antibodies over multiple years. Monitoring the effects of influenza vaccinations showed that ∼60% of the response to vaccination originated from preexisting clonotypes and highlighted the existence and relatively high abundance of broadly protective, non-neutralizing antibodies.92 Years later, follow-up studies showed that persistent antibodies account for >70% of the serum response over 5 years, further promoting the efficiency and strength of the Ig-seq method.93 It should be noted that relying solely on sequences obtained from PBMCs may provide an incomplete database,32 as it is only feasible to obtain a subset of PBMCs for analysis. Nonetheless, Ig-seq presents one of the most efficient and successful approaches to analyze and identify clones in Ig repertoires and monitor how they (dis)appear following a change in physiology, e.g., infection or vaccination.
Alternative proteogenomics approaches
Extending beyond the Ig-seq strategy, proteogenomics approaches as taken by Castellana et al.47 incorporate personalized genomics data into the antibody sequencing workflow to identify complete antibody sequences. In their software package GenoMS47 they accept both proteomic and genomic databases as input, which are used to reconstruct antibody (sub)sequences from BU MS data. The database is used to find a homologous template sequence, whereby missing, mutated, and spliced genes are considered. The software also allows for a high degree of flexibility through user-defined constraints. In addition, users can define how the template database is used, excluding certain genes, or using multiple-gene segments (V, D, J, or C) to make up a single sequence (Figure 6b). As often occurs with hybrid approaches, the power of this proteogenomics strategy comes at a cost. While broadly applicable and very powerful, the required expertise increases because of the use and combination of multiple omics techniques. However, when successfully applied, this workflow produces exciting results, as recently shown in the analyses of antibodies from immunized rabbits94 and the characterization of neutralizing antibodies against the Ebola virus antigen95 with notable improvements in integration and visualization of the data. Unfortunately, not all these tools are currently publicly available, although several underlying protocols are open source.96–98
Protein-centric sequencing of endogenous antibodies
Some attempts have emerged aiming at novel antibody discovery by MS-based sequencing alone, directly from serum samples or other liquid biopsies, circumventing the need for genomics/transcriptomics data (multi-omics approaches). In reviewing several techniques for sequencing purified antibodies above, we pointed out that these methods are geared toward highly purified mAb samples and are therefore not directly applicable for polyclonal antibody mixtures. However, advancements in sample preparation, instrumentation, and bioinformatics make it possible to obtain partial and sometimes complete de novo sequences of endogenous antibody clones by combining different mass spectrometric techniques, as discussed further below.
Antigen-specific capture
For many pathologies, it is common to screen patient’s serum for antibodies that exhibit activity against the antigens originating from the pathogen, for example, by enzyme-linked immunosorbent assay (ELISA). Using pathogen-based antigens, it is also possible to capture specific antibody clones from serum that exhibit high affinity against the antigen. This typically reduces the complexity of the antibody mixture substantially. Nevertheless, it is still nearly impossible to reduce the complexity down to a single clone, as multiple antibodies with varying affinities for any given antigen often co-occur. An example of a capturing method whereby additional intact mass data was used to derive de novo sequences was reported by Guthals et al.32 Following affinity purification of antibodies from the serum of a cytomegalovirus-exposed individual, using the glycoprotein B antigen, both intact mass and BU MS measurements were performed. Their semi-automated software PolyExtend seeks to use the intact mass measurements to retrieve the average mass of the most abundant species in an antibody mixture, which in turn is used as a mass constraint for a sequence derived using the BU MS data (Figure 6c). PolyExtend builds further upon the meta-SPS algorithm,29 which was initially designed to extend subsequences by assembling multiple sequence predictions into longer subsequences. However, diverging extensions for the same subsequence are treated as sequencing errors with one extension selected for the output. In the case of antibodies, such divergences may indicate the presence of two similar clones. To account for this, the software displays the possible extensions as a ranked list, and the user can then select the extension. This approach aims at expanding the de novo sequencing capabilities of the previously established meta-SPS algorithm to deal with simultaneous presence of multiple clones, and Guthals et al.32 demonstrated a clear proof of concept.
Antibody profiling and sequencing in polyclonal mixtures
While it is still not possible to de novo sequence entire serum antibody repertoires, recent advances in LC-MS of intact proteins have enabled the detection and analysis of single clones from complex antibody mixtures.
For instance, developments have been made that specifically profile intact light chains from serum, even providing partial sequence information by using MD MS. Impressively these studies successfully demonstrate the analysis in serum without requiring antigen-specific capture, although they used either a spiked-in mAb as a model or worked with disease models that cause monoclonal Ig overexpression in serum (mono-gammopathy), such as multiple myeloma. Nonetheless, these studies demonstrated that detection and characterization of individual endogenous light chains is possible.99–103 Taking this one step further, Wang et al.104 developed a method to detect individual Fab fragments in serum. They were able to identify tens of heavy and light chains of serum autoantibodies. Although attempts were made to de novo sequence these antibodies at the intact protein level, the results were limited to a few sequence tags. During the SARS-CoV-2 pandemic, Melani et al.105 focused their profiling efforts on the vaccine-targeted Spike protein receptor-binding domain. The approach is named Ig-MS and features two novel metrics for capturing the intensity and complexity of the antibody response. In short, the method uses affinity purification to capture antigen-specific clones. A mAb-containing standard is spiked in for quantitation. After reduction of the disulfides, individual ion MS106 is used to measure a mass fingerprint of the sample. The ratio between the intensity of clonal peaks and the standard is used to estimate the response (“Ion Titer”), and the complexity of the response (“Degree of Clonality’) is assessed by the ratio of the most intense light-chain peak to that of the summed intensity of all light-chain peaks. Finally, these metrics are correlated to the ELISA-based antibody titer and neutralization efficiency to verify their accuracy.
In a recent study, Bondt et al.31 used an approach to generate Fab fragments exclusively from the entire IgG1 repertoire. They were able to longitudinally profile IgG1 Fabs from the serum of both healthy and sepsis-afflicted donors without the need for any enrichment of specific clones. They observed a range of 50–500 distinct detectable IgG1 Fab clones per donor and showed that most clones persist over multiple months of sampling. Contrary to widely held belief, they showed that the IgG1 repertoire is in abundance dominated by just a few hundred clones and that each donor exhibits a unique repertoire of clones. They also managed to directly de novo sequence a single highly abundant clone in one of the donors without the aid of antigen-specific capture. The de novo sequencing was achieved by a combination of protein-centric sequencing using ETD, and a BU MS approach using multiple proteases for digestion. First, closely matching light and heavy-chain germline templates were selected from the IMGT database. Subsequently, the data was used to refine these templates iteratively, yielding the final mature sequence. This provided proof of concept that de novo sequencing of clones directly from serum is feasible, although still arduous and limited to specific cases (Figure 6d). Notably, the determined sequences contained more mutations (compared to germline sequences) than expected from the reported rates from BCR sequencing studies,107 which is indicative of potential discrepancies between protein-level and gene-level sequencing. This first attempt focused exclusively on IgG1, by using an IgG1-specific protease to generate the Fab fragments. In another work, Bondt et al.108 extended their method to IgA1 by using a protease specific to the O-glycans present in IgA1 hinge region to generate Fab fragments, albeit now exclusively from IgA1. Overall, they showed that, similar to serum IgG1, just a handful of clones dominates the secretory IgA1 profile of human milk.
Using a somewhat comparable approach, Dupré et al.103 analyzed isolated light chains from the urine of a patient affected by multiple myeloma. They assembled de novo data from peptides into a full-length sequence, using the intact mass data as a scaffold. Subsequently, they used TD MS to validate their findings and further characterize the proteoforms of the light chains, including PTMs. The BU MS data further supported the resulting proteoforms, showing a similar added benefit from iteratively combining BU and TD MS data.
Additional benefits of studying antibodies at the protein level
The work described here focuses primarily on obtaining amino acid sequences of antibodies like IgG by hybrid MS-based techniques. Being biased toward MS, we do not want to undervalue the highly beneficial proteogenomics approaches that should be used when applicable. Notwithstanding, the capabilities of MS allow for antibody characterization beyond the primary amino acid sequence. Antibodies are known to harbor multiple important PTMs, including Fab- and Fc-glycosylation,109 deamidation,110 and C-terminal truncation.111 Moreover, although the disulfide bonds in IgG1 are thoroughly described, other subclasses, notably IgG2, appear to occur as structural isomers induced by different disulfide-bridge patterns. These PTMs and disulfide bridges become even more pronounced in IgA and IgM, which can form higher-order structures connected by the joining-chain in serum and other bodily fluids. All these features influence the antibody’s efficacy and stability. Such information cannot easily be obtained at the nucleotide level, requiring protein-level analysis. Altogether, these features pose additional challenges for antibody characterization that are beyond the scope of this review.
Putting the pieces together
Here, we reviewed recent progress, discussed the challenges associated with MS-based antibody de novo sequencing, and described several attempts to overcome these bottlenecks. Although it is not a routine analysis yet, MS-based de novo sequencing of antibodies is now feasible through elaborate strategies that rely on multiple proteases, multiple-fragmentation techniques, homology sequencing, or a combination thereof. Currently, these techniques have not yet matured sufficiently to become directly applicable to the analysis of endogenous antibody mixtures present in serum. Nevertheless, Ig-seq and other proteogenomics strategies have already produced reliable partial sequences for multiple clones from donor samples, although side-stepping de novo sequencing of the intact clones at the protein level.
Though the de novo assembly of sequences from an antibody mixture is still arduous, some groups have recently managed to get exciting data, either through antigen-specific capture or by targeting the most abundant clone(s). To derive a de novo sequence, a combination of intact mass measurement with BU and TD MS data seems to be highly beneficial for confident sequence predictions.
With these numerous advances in available methods, it is likely just a matter of fitting the pieces together to create an MS-based method that can be used more routinely for antibody discovery. As was seemingly already stated by Aristotle: “the whole is greater than the sum of the parts”.112 When applied to de novo sequencing it would imply that a combination of the different methods described here could have a synergistic effect resulting in a workflow that would achieve the ultimate goal. While the methods share certain common elements such as using homologous sequences, there are still a few key gaps in the pipeline that hamper routine antibody sequencing and discovery. First, nearly all de novo methods discussed here still use some form of antibody purification from the mixture present in the liquid biopsy, prior to the sequencing effort. Second, a high level of expertise is still required, both from experimental and data analytical points of view, to derive the correct de novo sequence. Especially, the bioinformatics workflows presented in these reports still require a high degree of manual curation to refine the software output. However, all these recently published proofs of concept (Figure 6) pave the way for more efficient next-generation methods.
First therapeutic antibodies discovered by MS-based de novo sequencing
The advancements made over the last decade in MS-based antibody sequencing provide an optimistic outlook for the future. We expect that a therapeutic antibody discovered by MS could be right around the corner. Looking back at the timeline of key developments in the field of antibody sequencing, we can notice several clear trends (Figure 7). Since the 1960s, rudimentary sample preparation for antibodies was available, but practical methods of obtaining sequence information appeared only in 1993, when Sanger sequencing was first applied to B cells. The first therapeutic antibody was registered in 1986, and this advent launched large-scale development of mAbs, with a hundred mAbs in advanced stage of development by 2008.4 At that point, next-generation sequencing led to high-throughput sequencing workflows and further facilitated the development of therapeutic antibodies. Wide adoption of these techniques followed shortly, and the number of deposited antibody sequences and registered antibody therapeutics has been growing exponentially ever since, with the 100th therapeutic mAb being approved by the United States Food and Drug Administration in 2021.113 Observing this trend, the popularization of MS-based proteomics has now spurred the development of platforms for de novo sequencing of antibodies heavily supported by MS. Over the last 20 years, the rapid expansion of genome-based sequencing techniques kick-started antibody discovery by allowing large-scale BCR sequencing. Similarly, we envision that the ongoing advancement of MS de novo sequencing will complement available strategies by protein-level analysis or even provide a standalone solution for future therapeutic developments.
Figure 7.
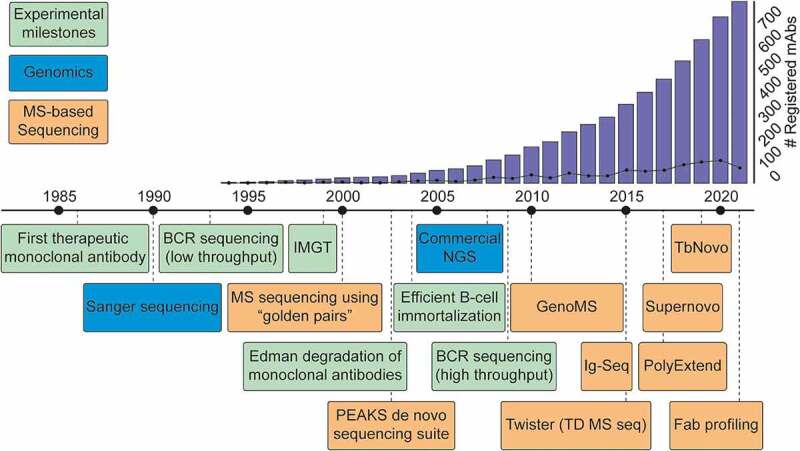
Timeline of key developments paving the way toward MS-based de novo sequencing of serum antibodies. Blue: Key developments in the field of genomic sequencing. Green: Key advances in the field of antibody research. Orange: Selected hallmark papers in the field of MS-based antibody sequencing. To visualize the impact of therapeutic antibody development, the bar graph indicates the cumulative number of registered antibody-based drugs, and the line shows the number of registrations for a given year.4
Acknowledgments
We thank the members of the Heck laboratory for their support. This research received funding through the Netherlands Organization for Scientific Research (NWO) TTW project 15575 (SCdG and AJRH), and the ENPPS.LIFT.019.001 project (AJRH).
Funding Statement
This work was supported by the nederlandse organisatie voor wetenschappelijk onderzoek [ENPPS.LIFT.019.001, 15575].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Abbreviations
| BCR | B-cell receptor |
| BU | bottom-up |
| CDR | complementarity-determining region |
| ELISA | enzyme-linked immunosorbent assay |
| FR | framework region |
| Ig | immunoglobulin |
| IMGT | the international ImMunoGeneTics information system |
| LC-MS | liquid chromatography-mass spectrometry |
| mAb | monoclonal antibody |
| MD | middle-down |
| MS | mass spectrometry |
| PBMC | peripheral blood mononuclear cell |
| PTM | post-translational modification |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| TD | top-down |
References
- 1.London ES. Der gegenwärtige Stand der Lehre von den Cytolysinen und die cytolytische Theorie der Immunität. Zentralbl f Bakteriol, Parasitenk u Infekt Abt II, Bd. 1902;32:48–17. [Google Scholar]
- 2.Lindenmann J. Origin of the terms “antibody” and “antigen”. Scand J Immunol. 1984;19(4):281–85. PMID: 6374880. doi: 10.1111/j.1365-3083.1984.tb00931.x. [DOI] [PubMed] [Google Scholar]
- 3.Marks C, Deane CM. How repertoire data are changing antibody science. J Biol Chem. 2020;295(29):9823–37. PMID: 32409582. doi: 10.1074/jbc.rev120.010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raybould MIJ, Marks C, Lewis AP, Shi J, Bujotzek A, Taddese B, Deane CM. Thera-SAbDab: the Therapeutic Structural Antibody Database. Nucleic Acids Res. 2020;48(D1):D383–D388. PMID: 31555805. doi: 10.1093/nar/gkz827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1). PMID: 33459118. doi: 10.1080/19420862.2020.1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh S, Kumar NK, Dwiwedi P, Charan J, Kaur R, Sidhu P, Chugh VK. Monoclonal antibodies: a review. Curr Clin Pharmacol. 2018;13(2):85–99. PMID: 28799485. doi: 10.2174/1574884712666170809124728. [DOI] [PubMed] [Google Scholar]
- 7.Schroeder HW, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010;125(2):S41–S52. PMID: 20176268. doi: 10.1016/j.jaci.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science. 2016;351(6277):1078–83. PMID: 26912366. doi: 10.1126/science.aad5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science. 2016;351(6279):1339–42. PMID: 26917593. doi: 10.1126/science.aad5224. [DOI] [PubMed] [Google Scholar]
- 10.Valgardsdottir R, Cattaneo I, Napolitano G, Raglio A, Spinelli O, Salmoiraghi S, Castilletti C, Lapa D, Capobianchi MR, Farina C, et al. Identification of human SARS-CoV-2 monoclonal antibodies from convalescent patients using EBV immortalization. Antibodies. 2021;10(3):26. PMID: 34287229. doi: 10.3390/antib10030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroeder HW Jr. Similarity and divergence in the development and expression of the mouse and human antibody repertoires. Dev Comp Immunol. 2006;30(1–2):119–35. PMID: 16083957. doi: 10.1016/j.dci.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Briney B, Inderbitzin A, Joyce C, Burton DR. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 2019;566(7744):393–97. PMID: 30664748. doi: 10.1038/s41586-019-0879-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hom JR, Tomar D, Tipton CM. Exploring the Diversity of the B-Cell Receptor Repertoire Through High-Throughput Sequencing. In: Rast J, Buckley K,editors. Methods in Molecular Biology: Immune Receptors. Vol. 2421. Clifton (NJ):Humana; 2022. p. 231–41.PMID: 34870823. doi: 10.1007/978-1-0716-1944-5_16 [DOI] [PubMed] [Google Scholar]
- 14.Altelaar AFM, Munoz J, Heck AJR. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet. 2013;14(1):35–48. PMID: 23207911. doi: 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- 15.Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–55. PMID: 27629641. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 16.Sen KI, Tang WH, Nayak S, Kil YJ, Bern M, Ozoglu B, Ueberheide B, Davis D, Becker C. Automated antibody de novo sequencing and its utility in biopharmaceutical discovery. J Am Soc Mass Spectrom. 2017;28(5):803–10. PMID: 28105549. doi: 10.1007/s13361-016-1580-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng W, Pronker MF, Snijder J. Mass Spectrometry-Based de novo sequencing of monoclonal antibodies using multiple proteases and a dual fragmentation scheme. J Proteome Res. 2021;20(7):3559–66. PMID: 34121409. doi: 10.1021/acs.jproteome.1c00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srzentić K, Fornelli L, Tsybin YO, Loo JA, Seckler H, Agar JN, Anderson LC, Bai DL, Beck A, Brodbelt JS, et al. Interlaboratory study for characterizing monoclonal antibodies by Top-Down and Middle-Down mass spectrometry. J Am Soc Mass Spectrom. 2020;31(9):1783–802. PMID: 32812765. doi: 10.1021/jasms.0c00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody structure and function: the basis for engineering therapeutics. Antibodies. 2019;8(4):55. PMID: 31816964. doi: 10.3390/antib8040055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lefranc MP. IMGT, the international ImMunoGeneTics database®. Nucleic Acids Res. 2003;31(1):307–10. PMID: 9847182. doi: 10.1093/nar/gkg085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lefranc M-P, Lefranc G. Immunoglobulins or Antibodies: IMGT® Bridging Genes, Structures and Functions. Biomedicines. 2020;8(9):319. PMID: 32878258. doi: 10.3390/biomedicines8090319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter RR. The hydrolysis of rabbit γ-globulin and antibodies with crystalline papain. Biochem J. 1959;73(1):119–27. PMID: 14434282. doi: 10.1042/bj0730119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herr AB, Ballister ER, Bjorkman PJ. Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. Nature. 2003;423(6940):614–20. PMID: 12768205. doi: 10.1038/nature01685. [DOI] [PubMed] [Google Scholar]
- 24.Charles A, Janeway J, Travers P, Walport M, Shlomchik MJ. The generation of diversity in immunoglobulins. In: Immunobiology: the immune system in health and disease, 5th. New York (NY): Garland Science; 2001. https://www.ncbi.nlm.nih.gov/books/NBK27140/ [Google Scholar]
- 25.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. The Generation of Antibody Diversity. In: Molecular Biology of the Cell, 4th. New York (NY): Garland Science; 2002. https://www.ncbi.nlm.nih.gov/books/NBK26860/ [Google Scholar]
- 26.Jeske DJ, Jarvis J, Milstein C, Capra JD. Junctional diversity is essential to antibody activity. J Immunol. 1984;133(3):1090–92. PMID: 6747289. [PubMed] [Google Scholar]
- 27.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. PMID: 12634793. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 28.Tran NH, Rahman MZ, He L, Xin L, Shan B, Li M. Complete De Novo Assembly of Monoclonal Antibody Sequences. Sci Rep. 2016;6(1):31730. PMID: 27562653. doi: 10.1038/srep31730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guthals A, Clauser KR, Bandeira N. Shotgun protein sequencing with Meta-contig assembly. Mol Cell Proteomics. 2012;11(10):1084–96. PMID: 22798278. doi: 10.1074/mcp.M111.015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donnelly DP, Rawlins CM, DeHart CJ, Fornelli L, Schachner LF, Lin Z, Lippens JL, Aluri KC, Sarin R, Chen B, et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat Methods. 2019;16(7):587–94. PMID: 31249407. doi: 10.1038/s41592-019-0457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bondt A, Hoek M, Tamara S, de Graaf B, Peng W, Schulte D, van Rijswijck DMH, den Boer MA, Greisch JF, Varkila MRJ, et al. Human plasma IgG1 repertoires are simple, unique, and dynamic. Cell Syst. 2021;12(12):1131–43. PMID: 34613904. doi: 10.1016/j.cels.2021.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guthals A, Gan Y, Murray L, Chen Y, Stinson J, Nakamura G, Lill JR, Sandoval W, Bandeira N. De novo MS/MS sequencing of native human antibodies. J Proteome Res. 2017;16(1):45–54. PMID: 27779884. doi: 10.1021/acs.jproteome.6b00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toby TK, Fornelli L, Kelleher NL. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu Rev Anal Chem. 2016;9(1):499–519. PMID: 27306313. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Compton PD, Zamdborg L, Thomas PM, Kelleher NL. On the scalability and requirements of whole protein mass spectrometry. Anal Chem. 2011;83(17):6868–74. PMID: 21744800. doi: 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansson BP, Shannon O, Björck L. IdeS: a bacterial proteolytic enzyme with therapeutic potential. PLoS One. 2008;3(2):e1692. PMID: 18301769. doi: 10.1371/journal.pone.0001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fornelli L, Ayoub D, Aizikov K, Beck A, Tsybin YO. Middle-Down analysis of monoclonal antibodies with electron transfer dissociation orbitrap fourier transform mass spectrometry. Anal Chem. 2014;86(6):3005–12. PMID: 24588056. doi: 10.1021/ac4036857. [DOI] [PubMed] [Google Scholar]
- 37.Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol Cell Biol. 1989;9(3):1165–72. PMID: 2566907. doi: 10.1128/mcb.9.3.1165-1172.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–89. PMID: 1350088. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsurushita N, Hinton PR, Kumar S. Design of humanized antibodies: from anti-tac to zenapax. Methods. 2005;36(1):69–83. PMID: 15848076. doi: 10.1016/j.ymeth.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 40.Khoja L, Butler MO, Kang SP, Ebbinghaus S, Joshua AM. Pembrolizumab. J Immunother Cancer. 2015;3(1):36. PMID: 26288737. doi: 10.1186/s40425-015-0078-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Busse W, Corren J, Lanier BQ, McAlary M, Fowler-Taylor A, Della Cioppa G, van As A, Gupta N. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108(2):184–90. PMID: 11496232. doi: 10.1067/mai.2001.117880. [DOI] [PubMed] [Google Scholar]
- 42.Presta LG, Lahr SJ, Shields RL, Porter JP, Gorman CM, Fendly BM, Jardieu PM. Humanization of an antibody directed against IgE. J Immunol. 1993;151(5):2623–32. PMID: 8360482. [PubMed] [Google Scholar]
- 43.R-M L, Hwang Y-C, Liu I-J, Lee -C-C, Tsai H-Z, H-J L, H-C W. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1. PMID: 31894001. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32(2):158–68. PMID: 24441474. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morsa D, Baiwir D, La Rocca R, Zimmerman TA, Hanozin E, Grifnée E, Longuespée R, Meuwis M-A, Smargiasso N, De PE, et al. Multi-Enzymatic limited digestion: the Next-Generation sequencing for proteomics?. J Proteome Res. 2019;18(6):2501–13. PMID: 31046285. doi: 10.1021/acs.jproteome.9b00044. [DOI] [PubMed] [Google Scholar]
- 46.Bandeira N, Pham V, Pevzner P, Arnott D, Lill JR. Beyond edman degradation: automated de novo protein sequencing of monoclonal antibodies. Nat Biotechnol. 2008;26(12):1336–38. PMID: 19060866. doi: 10.1038/nbt1208-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castellana NE, Pham V, Arnott D, Lill JR, Bafna V. Template Proteogenomics: Sequencing Whole Proteins Using an Imperfect Database. Mol Cell Proteomics. 2010;9(6):1260–70. PMID: 20164058. doi: 10.1074/mcp.M900504-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Savidor A, Barzilay R, Elinger D, Yarden Y, Lindzen M, Gabashvili A, Adiv Tal O, Levin Y. Database-independent Protein Sequencing (DiPS) Enables Full-length de Novo Protein and Antibody Sequence Determination. Mol Cell Proteomics. 2017;16(6):1151–61. PMID: 28348172. doi: 10.1074/mcp.O116.065417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran NH, Rahman MZ, He L, Xin L, Shan B, Li M. Complete de novo assembly of monoclonal antibody sequences. Sci Rep. 2016;6(31730):1–10. PMID: 27562653. doi: 10.1038/srep31730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, Han Y, Yuen D, Ma B. Automated protein (re)sequencing with MS/MS and a homologous database yields almost full coverage and accuracy. Bioinformatics. 2009;25(17):2174–80. PMID: 19535534. doi: 10.1093/bioinformatics/btp366. [DOI] [PubMed] [Google Scholar]
- 51.Brodbelt JS. Ion activation methods for peptides and proteins. Anal Chem. 2016;88(1):30–51. PMID: 26630359. doi: 10.1021/acs.analchem.5b04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brodbelt JS, Morrison LJ, Santos I. Ultraviolet photodissociation mass spectrometry for analysis of biological molecules. Chem Rev. 2020;120(7):3328–80. PMID: 31851501. doi: 10.1021/acs.chemrev.9b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He L, Weisbrod CR, Marshall AG. Protein de novo sequencing by top-down and middle-down MS/MS: limitations imposed by mass measurement accuracy and gaps in sequence coverage. Int J Mass Spectrom. 2018;427:107–13. doi: 10.1016/j.ijms.2017.11.012. [DOI] [Google Scholar]
- 54.Macias LA, Santos IC, Brodbelt JS. Ion Activation Methods for Peptides and Proteins. Anal Chem. 2020;92(1):227–51. PMID: 31665881. doi: 10.1021/acs.analchem.9b04859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roepstorff P, Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed Mass Spectrom. 1984;11(11):601. PMID: 6525415. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 56.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci. 2004;101(26):9528–33. PMID: 15210983. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal Chem. 2000;72(3):563–73. PMID: 10695143. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 58.McLuckey SA, Stephenson JL. Ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom Rev. 1998;17(6):369–407. PMID: 10360331. doi: 10.1002/(SICI)1098-2787. [DOI] [PubMed] [Google Scholar]
- 59.Xiao Y, Vecchi MM, Wen D. Distinguishing between leucine and isoleucine by integrated LC-MS analysis using an orbitrap fusion mass spectrometer. Anal Chem. 2016;88(21):10757–66. PMID: 27704771. doi: 10.1021/acs.analchem.6b03409. [DOI] [PubMed] [Google Scholar]
- 60.Kjeldsen F, Haselmann KF, Sørensen ES, Zubarev RA. Distinguishing of Ile/Leu amino acid residues in the PP3 protein by (Hot) electron capture dissociation in fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2003;75(6):1267–74. doi: 10.1021/ac020422m. [DOI] [PubMed] [Google Scholar]
- 61.Horn DM, Zubarev RA, McLafferty FW. Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc Natl Acad Sci. 2000;97(19):10313–17. PMID: 10984529. doi: 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guthals A, Clauser KR, Frank AM, Bandeira N. Sequencing-grade de novo analysis of MS/MS triplets (CID/HCD/ETD) from overlapping peptides. J Proteome Res. 2013;12(6):2846–57. PMID: 23679345. doi: 10.1021/pr400173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vyatkina K. De Novo Sequencing of Top-Down Tandem Mass Spectra: A Next Step towards Retrieving a Complete Protein Sequence. Proteomes. 2017;5(1):6. PMID: 28248257. doi: 10.3390/proteomes5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Datta R, Bern M. Spectrum fusion: using multiple mass spectra for de novo peptide sequencing. J Comput Biol. 2009;16(8):1169–82. PMID: 19645594. doi: 10.1089/cmb.2009.0122. [DOI] [PubMed] [Google Scholar]
- 65.Horton AP, Robotham SA, Cannon JR, Holden DD, Marcotte EM, Brodbelt JS. Comprehensive de Novo Peptide Sequencing from MS/MS Pairs Generated through Complementary Collision Induced Dissociation and 351 nm Ultraviolet Photodissociation. Anal Chem. 2017;89(6):3747–53. doi: 10.1021/acs.analchem.7b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bertsch A, Leinenbach A, Pervukhin A, Lubeck M, Hartmer R, Baessmann C, Elnakady YA, Müller R, Böcker S, Huber CG, et al. De novo peptide sequencing by tandem MS using complementary CID and electron transfer dissociation. Electrophoresis. 2009;30(21):3736–47. PMID: 19862751. doi: 10.1002/elps.200900332. [DOI] [PubMed] [Google Scholar]
- 67.Schmelter C, Perumal N, Funke S, Bell K, Pfeiffer N, Grus FH. Peptides of the variable IgG domain as potential biomarker candidates in primary open-angle glaucoma (POAG). Hum Mol Genet. 2017;26(22):4451–64. PMID: 29036575. doi: 10.1093/hmg/ddx332. [DOI] [PubMed] [Google Scholar]
- 68.Singh V, Stoop MP, Stingl C, Luitwieler RL, Dekker LJ, van Duijn MM, Kreft KL, Luider TM, Hintzen RQ. Cerebrospinal-fluid-derived immunoglobulin g of different multiple sclerosis patients shares mutated sequences in complementarity determining regions. Mol Cell Proteomics. 2013;12(12):3924–34. PMID: 23970564. doi: 10.1074/mcp.M113.030346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Broodman I, de Costa D, Stingl C, Dekker LJM, VanDuijn MM, Lindemans J, van Klaveren RJ, Luider TM. Mass spectrometry analyses of κ and λ fractions result in increased number of complementarity-determining region identifications. Proteomics. 2012;12(2):183–91. PMID: 22120973. doi: 10.1002/pmic.201100244. [DOI] [PubMed] [Google Scholar]
- 70.de Costa D, Broodman I, VanDuijn MM, Stingl C, Dekker LJM, Burgers PC, Hoogsteden HC, Sillevis Smitt PAE, van Klaveren RJ, Luider TM. Sequencing and Quantifying IgG Fragments and Antigen-Binding Regions by Mass Spectrometry. J Proteome Res. 2010;9(6):2937–45. PMID: 20387908. doi: 10.1021/pr901114w. [DOI] [PubMed] [Google Scholar]
- 71.Tamara S, den Boer MA, Heck AJR. High-resolution native mass spectrometry. Chem Rev. 2021;122(8):7269–326. PMID: 34415162. doi: 10.1021/acs.chemrev.1c00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lössl P, Snijder J, Heck AJR. Boundaries of mass resolution in native mass spectrometry. J Am Soc Mass Spectrom. 2014;25(6):906–17. PMID: 24700121. doi: 10.1007/s13361-014-0874-3. [DOI] [PubMed] [Google Scholar]
- 73.Mao Y, Valeja SG, Rouse JC, Hendrickson CL, Marshall AG. Top-down structural analysis of an intact monoclonal antibody by electron capture dissociation-fourier transform ion cyclotron resonance-mass spectrometry. Anal Chem. 2013;85(9):4239–46. PMID: 23551206. doi: 10.1021/ac303525n. [DOI] [PubMed] [Google Scholar]
- 74.Tsybin YO, Fornelli L, Stoermer C, Luebeck M, Parra J, Nallet S, Wurm FM, Hartmer R. Structural Analysis of Intact Monoclonal Antibodies by Electron Transfer Dissociation Mass Spectrometry. Anal Chem. 2011;83(23):8919–27. PMID: 22017162. doi: 10.1021/ac201293m. [DOI] [PubMed] [Google Scholar]
- 75.Resemann A, Jabs W, Wiechmann A, Wagner E, Colas O, Evers W, Belau E, Vorwerg L, Evans C, Beck A, et al. Full validation of therapeutic antibody sequences by middle-up mass measurements and middle-down protein sequencing. MAbs. 2016;8(2):318–30. PMID: 26760197. doi: 10.1080/19420862.2015.1128607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Greisch J-F, den Boer MA, Beurskens F, Schuurman J, Tamara S, Bondt A, Heck AJR. Generating informative sequence tags from Antigen-Binding regions of heavily glycosylated IgA1 antibodies by native Top-Down electron capture dissociation. J Am Soc Mass Spectrom. 2021;32(6):1326–35. PMID: 33570406. doi: 10.1021/jasms.0c00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.den Boer MA, Greisch J-F, Tamara S, Bondt A, Heck AJR. Selectivity over coverage in de novo sequencing of IgGs. Chem Sci. 2020;11(43):11886–96. PMID: 33520151. doi: 10.1039/d0sc03438j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greisch J-F, den Boer MA, Lai S-H, Gallagher K, Bondt A, Commandeur J, Heck AJR. Extending native Top-Down electron capture dissociation to MDa immunoglobulin complexes provides useful sequence tags covering their critical variable Complementarity-Determining regions. Anal Chem. 2021;93(48):16068–75. PMID: 34813704. doi: 10.1021/acs.analchem.1c03740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spoerry C, Seele J, Valentin-Weigand P, Baums CG, von Pawel-Rammingen U. Identification and Characterization of IgdE, a Novel IgG-degrading Protease of Streptococcus suis with Unique Specificity for Porcine IgG. J Biol Chem. 2016;291(15):7915–25. PMID: 26861873. doi: 10.1074/jbc.M115.711440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lermyte F, Tsybin YO, O’Connor PB, Loo JA. Top or middle? Up or down? Toward a standard lexicon for protein Top-Down and allied mass spectrometry approaches. J Am Soc Mass Spectrom. 2019;30(7):1149–57. PMID: 31073892. doi: 10.1007/s13361-019-02201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fornelli L, Damoc E, Thomas PM, Kelleher NL, Aizikov K, Denisov E, Makarov A, Tsybin YO. Analysis of Intact Monoclonal Antibody IgG1 by Electron Transfer Dissociation Orbitrap FTMS. Mol Cell Proteomics. 2012;11(12):1758–67. PMID: 22964222. doi: 10.1074/mcp.M112.019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fornelli L, Ayoub D, Aizikov K, Liu X, Damoc E, Pevzner PA, Makarov A, Beck A, Tsybin YO. Top-down analysis of immunoglobulin G isotypes 1 and 2 with electron transfer dissociation on a high-field orbitrap mass spectrometer. J Proteomics. 2017;159:67–76. PMID: 28242452. doi: 10.1016/j.jprot.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fornelli L, Srzentić K, Huguet R, Mullen C, Sharma S, Zabrouskov V, Fellers RT, Durbin KR, Compton PD, Kelleher NL. Accurate Sequence Analysis of a Monoclonal Antibody by Top-Down and Middle-Down Orbitrap Mass Spectrometry Applying Multiple Ion Activation Techniques. Anal Chem. 2018;90(14):8421–29. PMID: 29894161. doi: 10.1021/acs.analchem.8b00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shaw JB, Liu W, V VY, Bracken CC, Malhan N, Guthals A, Beckman JS, Voinov VG. Direct determination of antibody chain pairing by Top-down and Middle-down mass spectrometry using electron capture dissociation and ultraviolet photodissociation. Anal Chem. 2020;92(1):766–73. PMID: 31769659. doi: 10.1021/ACS.ANALCHEM.9B03129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shaw JB, Malhan N, Vasil’Ev YV, Lopez NI, Makarov A, Beckman JS, Voinov VG. Sequencing grade tandem mass spectrometry for Top-Down proteomics using hybrid electron capture dissociation methods in a benchtop orbitrap mass spectrometer. Anal Chem. 2018;90(18):10819–27. PMID: 30118589. doi: 10.1021/ACS.ANALCHEM.8B01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shen Y, Tolić N, Piehowski PD, Shukla AK, Kim S, Zhao R, Qu Y, Robinson E, Smith RD, Paša-Tolić L. High-resolution ultrahigh-pressure long column reversed-phase liquid chromatography for top-down proteomics. J Chromatogr A. 2017;1498:99–110. PMID: 28077236. doi: 10.1016/j.chroma.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 87.Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17(20):2337–42. PMID: 14558135. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 88.Vyatkina K, Wu S, Dekker LJM, VanDuijn MM, Liu X, Tolić N, Dvorkin M, Alexandrova S, Luider TM, Paša-Tolić L, et al. De Novo Sequencing of Peptides from Top-Down Tandem Mass Spectra. J Proteome Res. 2015;14(11):4450–62. PMID: 26412692. doi: 10.1021/pr501244v. [DOI] [PubMed] [Google Scholar]
- 89.Liu X, Dekker LJM, Wu S, Vanduijn MM, Luider TM, Tolić N, Kou Q, Dvorkin M, Alexandrova S, Vyatkina K, et al. De Novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra. J Proteome Res. 2014;13(7):3241–48. PMID: 24874765. doi: 10.1021/pr401300m. [DOI] [PubMed] [Google Scholar]
- 90.Lavinder JJ, Horton AP, Georgiou G, Ippolito GC. Next-generation sequencing and protein mass spectrometry for the comprehensive analysis of human cellular and serum antibody repertoires. Curr Opin Chem Biol. 2015;24:112–20. PMID: 25461729. doi: 10.1016/j.cbpa.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 91.Lavinder JJ, Wine Y, Giesecke C, Ippolito GC, Horton AP, Lungu OI, Hoi KH, DeKosky BJ, Murrin EM, Wirth MM, et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc Natl Acad Sci. 2014;111(6):2259–64. PMID: 24469811. doi: 10.1073/pnas.1317793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee J, Boutz DR, Chromikova V, Joyce MG, Vollmers C, Leung K, Horton AP, DeKosky BJ, Lee CH, Lavinder JJ, et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat Med. 2016;22(12):1456–64. PMID: 27820605. doi: 10.1038/nm.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee J, Paparoditis P, Horton AP, Frühwirth A, McDaniel JR, Jung J, Boutz DR, Hussein DA, Tanno Y, Pappas L, et al. Persistent antibody clonotypes dominate the serum response to influenza over multiple years and repeated vaccinations. Cell Host Microbe. 2019;25(3):367–376.e5. PMID: 30795981. doi: 10.1016/j.chom.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bonissone SR, Lima T, Harris K, Davison L, Avanzino B, Trinklein N, Castellana N, Patel A. Serum proteomics expands on high-affinity antibodies in immunized rabbits than deep B-cell repertoire sequencing alone. bioRxiv. 2020:833871. doi: 10.1101/833871. [DOI] [Google Scholar]
- 95.Gilchuk P, Guthals A, Bonissone SR, Shaw JB, Ilinykh PA, Huang K, Bombardi RG, Liang J, Grinyo A, Davidson E, et al. Proteo-Genomic analysis identifies two major sites of vulnerability on ebolavirus glycoprotein for neutralizing antibodies in convalescent human plasma. Front Immunol. 2021;12:706757. PMID: 34335620. doi: 10.3389/fimmu.2021.706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cha SW, Bonissone S, Na S, Pevzner PA, Bafna V. The antibody repertoire of colorectal cancer. Mol Cell Proteomics. 2017;16(12):2111–24. PMID: 29046389. doi: 10.1074/mcp.ra117.000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Safonova Y, Bonissone S, Kurpilyansky E, Starostina E, Lapidus A, Stinson J, DePalatis L, Sandoval W, Lill J, Pevzner PA. IgRepertoireConstructor: a novel algorithm for antibody repertoire construction and immunoproteogenomics analysis. Bioinformatics. 2015;31(12):i53–i61. PMID: 26072509. doi: 10.1093/bioinformatics/btv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bonissone SR, Pevzner PA. Immunoglobulin classification using the colored antibody graph. J Comput Biol. 2016;23(6):483–94. PMID: 27149636. doi: 10.1089/cmb.2016.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.He L, Anderson LC, Barnidge DR, Murray DL, Hendrickson CL, Marshall AG. Analysis of monoclonal antibodies in human serum as a model for clinical monoclonal gammopathy by use of 21 tesla FT-ICR top-down and middle-down MS/MS. J Am Soc Mass Spectrom. 2017;28(5):827–38. PMID: 28247297. doi: 10.1007/s13361-017-1602-6. [DOI] [PubMed] [Google Scholar]
- 100.He L, Anderson LC, Barnidge DR, Murray DL, Dasari S, Dispenzieri A, Hendrickson CL, Marshall AG. Classification of plasma cell disorders by 21 tesla fourier transform ion cyclotron resonance top-down and middle-down MS/MS analysis of monoclonal immunoglobulin light chains in human serum. Anal Chem. 2019;91(5):3263–69. PMID: 30801187. doi: 10.1021/acs.analchem.8b03294. [DOI] [PubMed] [Google Scholar]
- 101.Mills JR, Barnidge DR, Murray DL. Detecting monoclonal immunoglobulins in human serum using mass spectrometry. Methods. 2015;81:56–65. PMID: 25916620. doi: 10.1016/j.ymeth.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 102.Sharpley FA, Manwani R, Mahmood S, Sachchithanantham S, Lachmann HJ, Gillmore JD, Whelan CJ, Fontana M, Hawkins PN, Wechalekar AD. A novel mass spectrometry method to identify the serum monoclonal light chain component in systemic light chain amyloidosis. Blood Cancer J. 2019;9(2):16. PMID: 30718462. doi: 10.1038/s41408-019-0180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dupré M, Duchateau M, Sternke-Hoffmann R, Boquoi A, Malosse C, Fenk R, Haas R, Buell AK, Rey M, Chamot-Rooke J. De Novo Sequencing of antibody light chain proteoforms from patients with multiple myeloma. Anal Chem. 2021;93(30):10627–34. PMID: 34292722. doi: 10.1021/acs.analchem.1c01955. [DOI] [PubMed] [Google Scholar]
- 104.Wang Z, Liu X, Muther J, James JA, Smith K, Wu S. Top-down mass spectrometry analysis of human serum autoantibody antigen-binding fragments. Sci Rep. 2019;9(1):2345. PMID: 30787393. doi: 10.1038/s41598-018-38380-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Melani RD, Des Soye BJ, Kafader JO, Forte E, Hollas M, Blagojevic V, Negrão F, McGee JP, Drown B, Lloyd-Jones C, et al. Next-generation serology by mass spectrometry: readout of the SARS-CoV-2 antibody repertoire. J Proteome Res. 2022;21(1):274–88. PMID: 34268518. doi: 10.1021/acs.jproteome.1c00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kafader JO, Melani RD, Durbin KR, Ikwuagwu B, Early BP, Fellers RT, Beu SC, Zabrouskov V, Makarov AA, Maze JT, et al. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat Methods. 2020;17(4):391–94. PMID: 32123391. doi: 10.1038/s41592-020-0764-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kitaura K, Yamashita H, Ayabe H, Shini T, Matsutani T, Suzuki R. Different somatic hypermutation levels among antibody subclasses disclosed by a new next-generation sequencing-based antibody repertoire analysis. Front Immunol. 2017;8:389. PMID: 28515723. doi: 10.3389/fimmu.2017.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bondt A, Dingess KA, Hoek M, van Rijswijck DMH, Heck AJR. A direct ms-based approach to profile human milk secretory immunoglobulin A (IgA1) reveals donor-specific clonal repertoires with high longitudinal stability. Front Immunol. 2021;12:5190. PMID: 34938298. doi: 10.3389/fimmu.2021.789748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.de Haan N, Falck D, Wuhrer M. Monitoring of immunoglobulin N- and O-glycosylation in health and disease. Glycobiology. 2020;30(4):226–40. PMID: 29958069. doi: 10.1093/glycob/cwz048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yan Q, Huang M, Lewis MJ, Hu P. Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs. 2018;10(6):901–12. PMID: 29958069. doi: 10.1080/19420862.2018.1478646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Beyer B, Schuster M, Jungbauer A, Lingg N. Microheterogeneity of recombinant antibodies: analytics and functional impact. Biotechnol J. 2018;13(1):1700476. PMID: 28862393.doi: 10.1002/biot.201700476. [DOI] [PubMed] [Google Scholar]
- 112.Aristotle . Metaphysics. In: Ross WD, editor. Metaphysics. Vol. VIII. Oxford (UK): Clarendon Press; 1908. p. 1045a.8–10. [Google Scholar]
- 113.Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021;20(7):491–95. PMID: 33953368.doi: 10.1038/d41573-021-00079-7. [DOI] [PubMed] [Google Scholar]