


Abstract
Dynamic regulation is an effective strategy for control of gene expression in microbial cell factories. In some pathway contexts, several metabolic modules must be controlled in a time dependent or ordered manner to maximize production, while the creation of genetic circuits with ordered regulation capacity still remains a great challenge. In this work, we develop a pathway independent and programmable system that enables multi-modular ordered control of metabolism in Bacillus subtilis. First, a series of thermosensors were created and engineered to expand their thresholds. Then we designed single-input-multi-output circuits for ordered control based on the use of thermosensors with different transition points. Meanwhile, a repression circuit was constructed by combining CRISPRi-based NOT gates. As a proof-of-concept, these genetic circuits were applied for multi-modular ordered control of 2′-fucosyllactose (2′-FL) biosynthesis, resulting in a production of 1839.7 mg/l in shake flask, which is 5.16-times that of the parental strain. In a 5-l bioreactor, the 2′-FL titer reached 28.2 g/l with down-regulation of autolysis. Taken together, this work provides programmable and versatile thermosensitive genetic toolkits for dynamic regulation in B. subtilis and a multi-modular ordered control framework that can be used to improve metabolic modules in other chassis cells and for other compounds.
INTRODUCTION
Microorganisms engineered by synthetic biology and metabolic engineering can efficiently synthesize valuable products such as fine chemicals, pharmaceuticals or functional nutraceuticals (1–3). In order to rewire metabolic fluxes and enhance production efficiency, dynamic regulation strategies have been developed to control both natural and engineered pathways (4,5). These approaches can be used to decouple cell growth and product synthesis, and help balance product synthesis without disrupting cellular homeostasis. Dynamic metabolic control requires genetic circuits that are usually designed based on different biosensors responsive to external inducers, intracellular metabolites, environmental factors or cell growth (6,7). Recently, genetic circuits have been constructed to achieve simultaneous up- and down-regulation of the metabolic modules of interest (8). However, in some pathway contexts, the efficiency of a microbial cell factory is highly dependent on the switching time or temporal control of the metabolic modules involved, which tell us that advanced genetic circuits need to be established to enable an ordered control of metabolic modules at different fermentation periods (9,10).
Layered genetic circuits have been constructed to achieve the ordered control of multiple-modules. For instance, by combing a quorum sensing system with a myo-inositol-responsive biosensor, the layered dynamic regulation could achieve the orderly down- and up-regulation of Pfk and MIOX for efficient production of glucaric acid in Escherichia coli (11). Unfortunately, not all intracellular metabolites have natural biosensors that respond to them, and in addition, the robustness of genetic circuits controlled by intercellular metabolites is limited due to the time-dependent concentration variability during the culture processes. As an alternative, pathway-independent multi-input genetic circuits have been developed, which make the switching dynamics orthogonal. For example, a genetic circuit with three inputs (anhydrotetracycline, arabinose and isopropyl beta-d-thiogalactopyranoside) was designed to achieve ordered control and divide the state of butyrate-producing bacteria into four stages. However, the use of external inducers is expensive and not economical for industrial bioprocesses (12,13).
Compared with layered or multi-inputs dynamic regulation, environmental factors such as temperature, are attractive signals for the construction of pathway-independent and ordered genetic circuits, with advantages such as easy-to-control, high robustness, good orthogonality and tunability (14,15). In previous studies, a robust thermosensitive bio-switch based on transcriptional regulator CI857 and TetR-family repressor PhlF was reported for the ordered polymerization of diblock polyhydroxyalkanoates in E. coli (16). In addition, an energy-saving cold-shock-triggered temperature control system based on the temperature-sensitive protein Gal4M9 driven by the PGal4 protomer was designed for the production of tocotrienols in Saccharomyces cerevisiae (17). Although these genetic circuits were efficient in improving product synthesis, they could not achieve ordered control of gene expression at different fermentation periods. Therefore, more efforts are required to extend the available toolbox of thermosensitive regulators to achieve multi-modular ordered control and solve complex biological problems.
In this work, we created novel programmable temperature-responsive genetic circuits for multi-modular ordered control of metabolism by combing thermosensors and Clustered Regularly Interspaced Short Palindromic Repeats interference (CRISPRi). First, we used the transcriptional regulator CI857 to construct orthogonal temperature-responsive biosensors with different transition points in Bacillus subtilis, a model microorganism widely used in the production of nutraceuticals and pharmaceuticals (18). Then we designed and created bifunctional and programmable genetic circuits by combining CRISPRi-based NOT gates as repressor system. Finally, as a proof-of-concept, we used the temperature-responsive genetic circuits for multi-modular ordered control of the production of 2′-fucosyllactose (2′-FL), which is one of the most abundant human milk oligosaccharides (HMOs) with beneficial effects on the microbiota of neonates by preventing infection from pathogenic bacteria (19,20). This work provides a novel and effective toolkit for dynamic regulation in B. subtilis and the framework here developed for the design and optimization of temperature-responsive genetic circuits may be useful for engineering the other microbes.
MATERIALS AND METHODS
Strains and cultivation conditions
The strains constructed and used in this study are listed in Table 1. The OSF medium composed of 12.5 g/l K2HPO4·3H2O, 2.5 g/l KH2PO4, 12.0 g/l yeast extract, 6.0 g/l tryptone, 10 ml/l trace metal solution, 20.0 g/l sucrose and 10.0 g/l lactose. The trace metal solution contained 4 g/l FeSO4·7H2O, 0.2 g/l ZnSO4·7H2O, 1 g/l MnSO4·5H2O, 0.4 g/l CoCl2·6H2O, 0.2 g/l NaMoO4·2H2O, 0.1 g/l AlCl3·6H2O, 0.1 g/l CuCl2·H2O, 4 g/l CaCl2 and 0.05 g/l H3BO4. Different antibiotics were added to the medium for selections: ampicillin (100 μg/ml), kanamycin (50 μg/ml), zeocin (20 μg/ml), chloromycetin (5 μg/ml) and spectinomycin (50 μg/ml).
Table 1.
Strains used in this study
| Strain | Characteristics | Ref. |
|---|---|---|
| E. coli JM109 | recA1, endA1, thi, gyrA96, supE44, hsdR17Δ (lac-proAB)/F’[traD36,proAB+, laclq, lacZΔ M15] | Lab stock |
| B. subtilis 168 | trpC2 | Lab stock |
| WY1 | B. subtilis 168, ΔamyE:: P43-CI857 | This work |
| WY2 | WY1, pHTa1-PR-GFP | This work |
| WY3 | WY1, pHTa1-2-Pveg-GFP | This work |
| WY4 | WY1, pHTa2-Pveg1-GFP | This work |
| WY5 | WY1, pHTa3-Pveg2-GFP | This work |
| WY6 | WY1, pHTa4-Pveg3-GFP | This work |
| WY7 | WY1, pHTa5-Pveg4-GFP | This work |
| WY8 | WY1, pHTa6-Pveg5-GFP | This work |
| WY9 | B. subtilis 168, pHTa6-Pveg5-GFP | This work |
| WY10 | WY1, ΔnprE:: Pveg32-CFP, ΔganA:: Pveg34-GFP-Pveg37-mCherry | This work |
| WY11 | WY1, pP43-mCherry | This work |
| WY12 | B. subtilis 168, ΔnprE:: Pveg5-dCpf1, ΔganA:: crRNAmCherry, pP43-mCherry | This work |
| WY13 | WY1, ΔnprE:: Pveg5-dCpf1, ΔganA:: crRNAmCherry, pP43-mCherry | This work |
| WY14 | WY8, ΔnprE:: Pveg5-dCpf1, ΔganA:: crRNAmCherry, pP43-mCherry | This work |
| MT0 | WY1, ΔnprE:: P43-manC-gmd-T-P43-wcaG | This work |
| MT1 | MT0, Δbpr:: P43-manB-T-P43-futC | This work |
| MT2 | MT0, Δbpr:: P43-manB-T-Pveg34-futC | This work |
| MT3 | MT0, Δbpr:: Pveg34-manB-T-Pveg34-futC | This work |
| MT4 | MT3, ΔganA | This work |
| MT5 | MT4, ΔyesZ | This work |
| MT6 | MT3, ΔganA:: Pveg-dCpf1 | This work |
| MT7 | MT6, Δepr:: Pveg5-crRNAzwf1- crRNApfkA1 | This work |
| MT8 | MT6, Δepr:: Pveg5-crRNAzwf1- crRNApfkA2 | This work |
| MT9 | MT6, Δepr:: Pveg5-crRNAzwf1- crRNApfkA3 | This work |
| MT10 | MT6, Δepr:: Pveg5-crRNAzwf2- crRNApfkA1 | This work |
| MT11 | MT6, Δepr:: Pveg5-crRNAzwf2- crRNApfkA2 | This work |
| MT12 | MT6, Δepr:: Pveg5-crRNAzwf2- crRNApfkA3 | This work |
| MT13 | MT6, Δepr:: Pveg5-crRNAzwf3- crRNApfkA1 | This work |
| MT14 | MT6, Δepr:: Pveg5-crRNAzwf3- crRNApfkA2 | This work |
| MT15 | MT6, Δepr:: Pveg5-crRNAzwf3- crRNApfkA3 | This work |
| MT16 | MT9, Δldh:: Pveg34-ndk | This work |
| MT17 | MT16, Δldh:: Pveg34-ndk-gmk | This work |
| MT18 | MT17, Δldh:: Pveg34-ndk-gmk-xpf | This work |
| MT19 | MT17, ΔlytC | This work |
| MT20 | MT17, Δapre:: Pveg5-crRNAlytC1 | This work |
| MT21 | MT17, Δapre:: Pveg5-crRNAlytC2 | This work |
| MT22 | MT17, Δapre:: Pveg5-crRNAlytC3 | This work |
To synthesize 2′-FL, the recombinant strains were pre-cultured in a shake flask with LB medium for 10 h at 30°C. Then it was inoculated into 30 ml OSF medium with 5% proportion in a 250 ml shake flask and cultured with shaking at 220 rpm for 60 h, three replicates were set for each strain. According to experimental requirements, the recombinant strains were cultured under different bioprocesses, including ordered control (0–12 h at 30°C, 12–18 h at 34°C and 18–60 h at 37°C), single-switch control (0–12 h at 30°C and 12–60 h at 37°C), and constant temperature fermentation (37°C for 60 h).
The fermentation medium used for fed-batch culture consisted of 24.0 g/l yeast extract, 24.0 g/l tryptone, 60.0 g/l sucrose, 8.0 g/l urea, 30.0 g/l lactose, 12.5 g/l K2HPO4·3H2O, 2.5 g/l KH2PO4 and 10.0 ml/l trace metal solution. Seed culture was implemented in a 500 ml shake flask containing 125 ml LB medium at 30°C for 10 h. The seed culture was inoculated into a 5-l bioreactor containing 2.5 l fermentation medium and cultured by ordered control strategy (0–12 h at 30°C, 12–18 h at 34°C and 18–80 h at 37°C). The pH was maintained at 6.5 via the addition of aqueous ammonia. Dissolved oxygen was controlled ranging from 20% to 30% by adjusting the agitation speed and gas flow. In fed-batch culture, the feeding solution containing 600 g/l sucrose was pumped into the 5-l bioreactor to maintain the sucrose concentration at 10 g/l or above. In addition, the lactose concentration was maintained within 10–30 g/l by feeding with 200 g/l lactose. The feeding rates were adjusted based on the change of sucrose and lactose concentrations in the fermentation medium.
Plasmid construction and genome manipulation
Genetic design and sequence reading were carried out by SnapGene 4.2.1. Primers designed and used for genetic modification in this study are listed in Supplementary Table S1. All plasmids were constructed by Gibson Assembly toolkits (NEB, USA) through a 20 bp overlap and listed in Supplementary Table S2. The sequences of genetic parts used in this study are listed in Supplementary Table S3. E. coli JM109 was used for plasmids construction and cultivated in Bertani (LB) medium at 37°C. The cre/lox non-resistance knockout system was used to perform gene knockout or overexpression in B. subtilis genome (21). The plasmids or knockout box was transformed into B. subtilis by the method previously described (22).
Analytical methods
Cell density was measured at 600 nm wavelength after suitable dilution with deionized water. The concentration of sucrose and identification of 2′-FL were measured by LC/MS (Waters, Milford, MA, USA). The concentration of 2′-FL and lactose were assayed high-performance liquid chromatography (HPLC) equipped with a Rezex ROA Organic Acid H+ column (Phenomenex, Torrance, CA, USA) and a refractive index detector (Agilent 1260). The column was eluted with aqueous H2SO4 (5 mM) at a flow rate of 0.6 ml/min at 55°C. Specifically, fermentation samples were centrifuged at 12 000 × g for 15 min and supernatants were filtered through a 0.22-μm membrane to detect the extracellular concentration of 2′-FL. For the determination of intracellular 2′-FL, the cells were harvested by centrifuging 2-ml of the fermentation sample at 12 000 × g for 15 min and immediately stored at −80°C. Subsequently, the cells were washed thrice with 2-ml deionized water before disruption with an ultrasonic oscillation (Sonics VCX750, amplitude 25%). After removing the cellular debris by centrifugation at 12 000 × g for 10 min, the cell extracts were treated with 8% trichloroacetic acid to remove the intracellular proteins. Finally, the supernatant was measured after centrifuging at 12 000 × g for 20 min. Besides, the fluorescence characterization with cytometry was carried out on a BD FACSAria III (BD, Franklin Lakes, NJ, USA).
Fluorescence assays
The recombinant strains containing the fluorescence-reporter were cultured in a 15 ml tube with 2 ml LB medium for 10 h at 30°C and inoculated into 200 μl LB medium with 1% proportion at 96-well black plates subsequently (Corning 3603). For the static test of the activation circuits, repression circuits and bifunctional circuits, these plates were cultured at a preset temperature level (28, 30, 32, 34, 37, 39 or 42°C) with shaking at 900 rpm. Besides, these plates were cultured under different bioprocesses to explore dynamic control of the bifunctional circuits, including single-switch control (0–8 h at 32°C and 8–12 h at 39°C) and constant temperature fermentation at 32°C for 12 h. The GFP fluorescence and mCherry fluorescence were analyzed by a microplate Multi-Mode Reader (BIOTEK, Cytation 3) directly. The GFP fluorescence was measured at an excitation wavelength of 490 nm and an emission wavelength of 530 nm with a gain value 60. The mCherry fluorescence was measured at an excitation wavelength of 580 nm and an emission wavelength of 610 nm with a gain value 70. The relative fluorescence intensity is defined as the ratio of fluorescence intensity without background fluorescence to the OD without background.
RT–qPCR analysis
1-ml sample of recombinant B. subtilis was harvested during different fermentation times from the shaking flask cultures and frozen immediately in liquid nitrogen. Total RNA was eluted from the columns with RNase-free water by using the RNAprep Pure Kit (Tiangen Biotech, Beijing, China), according to the manufacturer's protocol, and measured by a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, the cDNA was obtained from the messenger RNA (mRNA) by using a the PrimeScript™ RT-PCR Kit (Takara, Dalian, China). Specifically,1 μg of RNA was mixed with 4 μl of 4× gDNA wiper Mix, and RNase-free ddH2O up to a final volume of 20 μl in a PCR tube and incubated at 42°C for 2 min then on ice for 1 min. Then, 16 μl of the above reaction mixture was mixed with 4 μl of 5× HisScript III qRT SuperMix up to a final volume of 20 μl in a PCR tube. The reactions were then incubated at 37°C for 15 min followed by 85°C for 5 s. The 96-well qPCR reaction plates were set up using SYBRH Premix ExTaq™ (TaKaRa Biotech, Beijing, China) according to the manufacturer's protocol. Each reaction was carried out in a total volume of 20 μl, containing 10 μl of 2× TB Green Premix Ex Taq II, 0.8 μl of 10 μM forward and reverse primer, respectively, 2 μl of cDNA and 6.4 μl RNase-free water. The hbsU gene was chosen as internal standard and primers utilized in the study are outlined in Supplementary Table S1. Cycling conditions were as follows: 95°C for 30 s followed by 40 cycles of 95°C for 5 s and 60°C for 20 s. All reactions were performed on a LightCycler 480 II Real-time PCR instrument (Roche Applied Science, Mannheim, Germany). Cycle threshold (Ct) values were calculated on the PCR instrument software. ΔCt was calculated by subtracting the mean Ct value for the target gene from the mean Ct value for the hbsU gene in each experiment. Next, the ΔΔCt was calculated by subtracting the ΔCt value for each sample from the ΔCt value for the respective negative control. Finally, relative gene expression was calculated by 2^(-ΔΔCt) method for each sample. Subsequent statistical analysis was performed using the statistical analysis software, Graphpad Prism 8.3.0.
Fluorescence imaging and analysis
To test the feasibility of single-input-multi-output (SIMO) genetic circuits, the recombinant strains containing the different fluorescence-reporters were cultured in a 15 ml tube with 2 ml LB medium for 10 h at 30°C and inoculated into 30 ml LB medium with 2% proportion in a 250 ml shake flask subsequently. The recombinant strains were cultured by using the ordered control (0–8 h at 30°C, 8–12 h at 32°C, 12–16 h at 34°C and 16–20 h at 37°C). The fluorescent protein CFP, GFP and mCherry were excited by 440-, 488- and 580-nm laser line of an argon-ion laser line of a He–Ne laser, respectively. Laser scanning confocal microscopy (LSCM) was carried out by Leica TCS SP8 microscope (Leica, Mannheim, Germany) fitted with a 60× oil-immersion objective. Fluorescence emissions were detected with spectral detector sets BP 465–480 for CFP, BP 515–564 for GFP, and BP 610–669 for mCherry. 1.5 ml tubes were visualized under bright field and blue illumination (440–485 nm), respectively. Image analysis was carried out on the Leica TCS SP8 software package and the ImageJ software (National Institutes of Health, Bethesda, MA).
Construction of the Hill equation
The fluorescence intensity of temperature-responsive promoters was fitted to the Hill equation as follows:
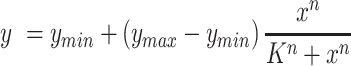 |
(1) |
where y is relative fluorescence intensity (Fl/OD) of the target promoter, the ymin and ymax are the minimum relative fluorescence intensity and maximum relative fluorescence intensity, respectively. x is the temperature of cultured state, K is the threshold and n is the Hill coefficient (23).
Statistical analysis
Three replicates were performed independently for each strain in this study and the data were expressed as mean ± SD. Statistical data analysis was carried out with t-tests in GraphPad Prism 8.3.0 and statistical significance is indicated as * P < 0.05 and ** P < 0.01.
RESULTS
Design of orthogonal temperature-responsive biosensors
Transcription factors are usually used for the construction of biosensors, while there are few reports about the temperature-responsive transcription factors in B. subtilis. The thermosensitive transcriptional regulator CI857 and its corresponding promoter PR from bacteriophage lambda is a well-known thermal regulation system (24). As shown in Figure 1A, when the temperature is low, the promoter PR is repressed by the dimmer CI857 complex bound to the operator sites (OR), which prevents the RNA polymerase from binding to the promoter. However, when the temperature is high, the DNA-binding activity of CI857 is abolished, which enables the binding of the RNA polymerase to the promoter PR turning transcription on.
Figure 1.

Construction and characterization of orthogonal temperature-responsive biosensors. (A) Reversible thermal regulation based on CI857 that is sensitive to high temperature, and the output is a reporter GFP under the temperature-responsive promoter. Dimmer CI857 complex could bind to specific sequences OR when the temperature is low and block the binding of RNA polymerase to the promoter that is necessary for transcription initiation. When the temperature is high, the DNA-binding activity of CI857 is antagonized, and RNA polymerase can bind to the promoter initiating transcription. (B) Schematic diagram of hybrid promoters, which were constructed by integrating the CI857-binding site (OR1 and OR2) into different positions of promoter Pveg. The promoter core region ‘−35’, ‘−10’ and transcription initiation site ‘+1’ are annotated. (C) The activity of each hybrid promoter in strains with CI857 was tested at the different levels of temperature. The transition point was defined as the temperature at which the relative fluorescence intensity exceeds 150. (D) The fitting of temperature and the relative fluorescence intensity of promoter Pveg5. (E) The strength of hybrid promoter Pveg5 in strains with or without CI857, respectively. (F) The activity of temperature-responsive promoters with different transition points. GFP, green fluorescent protein; RNAP, RNA polymerase; OR, operator site. Data are presented as mean ± s.d. of three replicates.
Therefore, we chose CI857 and PR for the design of novel temperature-responsive genetic circuits in B. subtilis. First, the transcriptional regulator CI857 was heterologously expressed by the strong constitutive promoter P43, generating the strain WY1. Then the expression of green fluorescent protein (GFP) was driven by the promoter PR. In previous study, the transition point was defined as the temperature at which the fluorescence intensity is reduced to 20% of the maximum (15). However, the promoters with high strength are usually accompanied with high leaky expression, which is a drawback when using this criterion for screening. To obtain promoters with low leaky expression, we chose the advanced IPTG-inducible promoter system with low leakiness developed by Tabor and colleagues for characterization purposes (25). In the absence of IPTG, this system presents a weak gene expression, 150. Therefore, in our work, the transition point was defined as the temperature at which the relative fluorescence intensity exceeds 150. We observed that the promoter PR could work at 39°C and showed a dynamic range of 2.4 at 42°C (452), indicating a poor performance that needs improvement.
In order to obtain high-performance and orthogonal temperature-responsive biosensors, we chose the strong constitutive promoter Pveg as the basal promoter for engineering due to the its simplicity, as it is only recognized by the native B. subtilis σA RNA polymerase. The specificity and sensitivity of transcription factor-based biosensor can be modulated by altering the location of the OR within the promoter region. The −10 and −35 region are the sites at which the sigma factor binds in order to mediate recruitment and assembly of the RNA polymerase complex. Besides, the zinc finger proteins-transactivation domain fusion was observed to confer transcriptional repression when bound downstream of the +1 site (26). Therefore, we placed the OR close to these core regions of the parental promoter and constructed five hybrid promoters that contained binding sites of CI857 (OR1: TACCTCTGGCGGTGATAA, OR2: TAACACCGTGCGTGTTG) in the different regions of the promoter Pveg (Figure 1B). In particular, the promoters Pveg1 and Pveg2 were constructed by inserting OR2 between −10 and −35 region and at the upstream of − 35 region, respectively. In addition, the promoter Pveg3 was designed by placing OR2 at the downstream of +1 site. These promoters were inserted in front of GFP in the trap plasmid pHTa0, which was then transformed into the strain WY1 to test their thermosensitivity. Figure 1C shows that the promoter Pveg1 had a high strength (2746) at 28°C with a dynamic range of 3.2. Compared with Pveg1, the promoter Pveg2 presented a weaker gene expression (924) at 28°C with larger dynamic range (11.5). By contrast, the promoter Pveg3 possessed constitutive expression from 28 to 42°C, which likely results from a noneffective repression effect of repressor CI857.
Based on the above results, the promoter Pveg2 was chosen for further modification. Adding OR1 into downstream of Pveg2 resulted in Pveg4, which possessed a remarkably enhanced dynamic range of 23.9 and a weak strength (300) at 28°C. Pveg5 was obtained by adding OR1 between −10 and −35 region of Pveg2. This promoter showed hardly no leaky expression below 34°C, and possessed a high dynamic range of 39.5 with temperatures ranging from 34 to 42°C. To characterize and standardize the transfer functions of Pveg5, the experimental data were fitted to the equation (1). The fit parameters to equation (1) are provided in Supplementary Table S4. As shown in Figure 1D, the relative fluorescence intensity of the Pveg5 increased with the temperature increase, suggesting that the strength of the Pveg5 positively correlated with the temperature. In addition, the strength of the promoter Pveg5 without CI857 was tested to validate whether the increase in Pveg5 promoter activity was a coincidence. Figure 1E suggests that the presence of CI857 can markedly repress the transcription of the promoter Pveg5 at low temperature. Taken together, the promoter Pveg5 with low leaky expression and high dynamic range (39.5) was chosen for further studies.
To construct more temperature-responsive biosensors with different transition points, a series of mutants of the promoter Pveg5 were constructed. A palindrome sequence is present in the OR2 sequence. Based on its specific sequence, we divided the OR2 sequence into three modules, including middle region sequence (CGTGC), palindrome sequence (AACAC and GTGTT) and the endmost sequence (T and G). Subsequently, three type of promoter mutation libraries at the OR2 region were built (Supplementary Figure S1). By high-throughput screening, some temperature-responsive promoter variants with different transition points were generated. When screening thermosensitive promoters, we found an enrichment in variants with high transition points. By contrast, we obtained few variants with low transition point (30°C). As shown in Figure 1F, several promoters with different dynamic ranges or transition points were selected for further characterization. In addition, the sequence features of these variants in the OR2 region were highly conserved (Supplementary Figure S2). Specifically, the middle region sequence (CGTGC) and the end region sequence (TTG) of OR2 were conserved. In summary, these biosensors with different transition points not only expanded the thermogenetic toolkits in B. subtilis, but also paved the way towards the creation of multi-modular ordered control tools.
Construction of temperature-responsive genetic circuits in B. subtilis
As shown above, the temperature-responsive promoter induced gene expression by abolishing the transcriptional inhibition of CI857 at high temperature, being therefore an activation circuit. Subsequently, we designed and constructed SIMO genetic circuits to test the feasibility of multi-modular ordered control. The genetic circuit was created by combining three activation circuits with different transition points. As shown in Figure 2A, the cyan fluorescent protein (CFP), GFP and mCherry modules were placed under the control of the temperature-responsive promoters Pveg32(with transition point at 32°C), Pveg34 (with transition point at 34°C) and Pveg37 (with transition point at 37°C), respectively. To decrease the basal leakage, the SVN degradation tag was added to the C-terminus of CFP and GFP. For comparison, we collected the recombinant strain in 1.5 ml tubes at different fermentation times, and imaged it under both bright field and blue transilluminator. Based on the different temperatures, the fermentation process was divided into four stages. As shown in Figure 2A, the cell color changed as the temperature increases, which was accompanied by a significant change in fluorescent under blue illumination.
Figure 2.
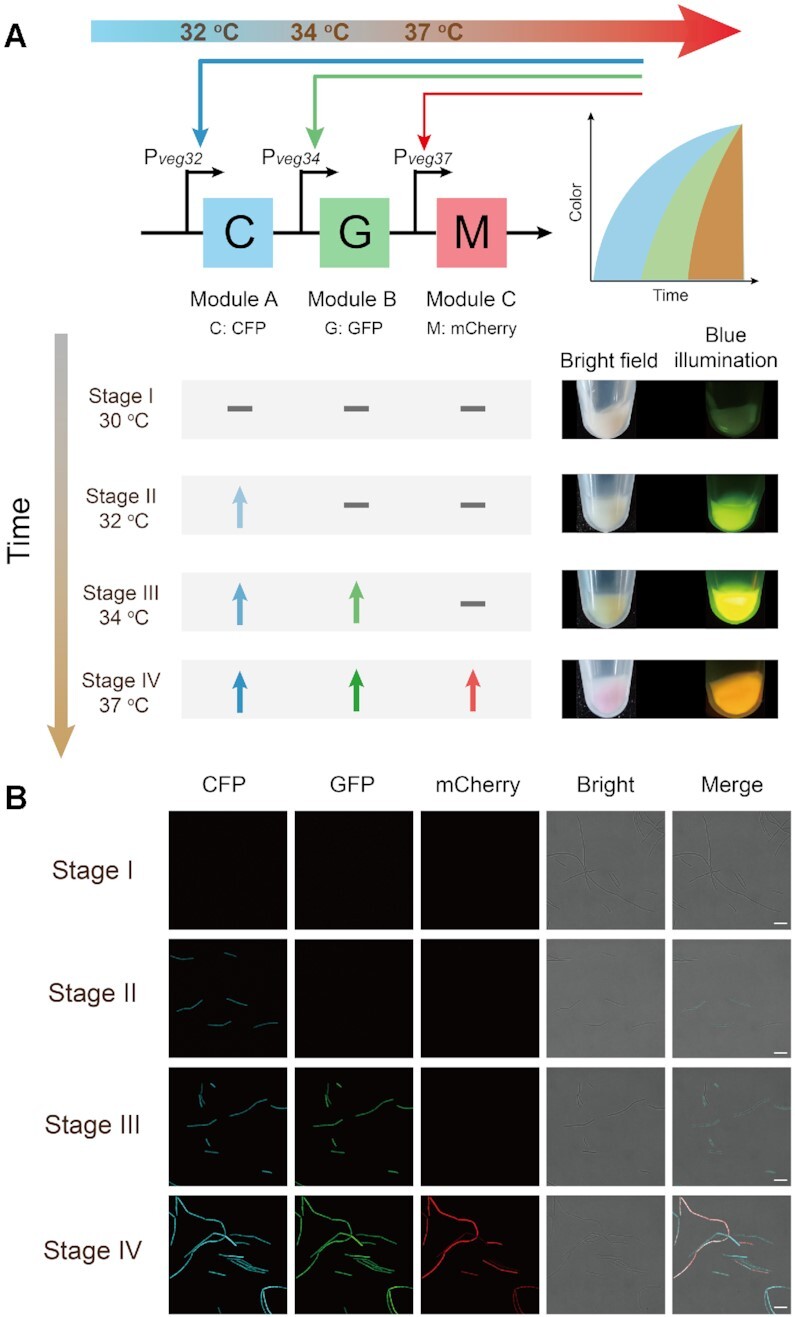
Characterization of single-input-multi-output genetic circuits. (A) Illustration of the single-input-multi-output genetic circuits. The CFP, GFP and mCherry modules were driven by the temperature-responsive promoters Pveg32 (with transition point at 32°C), Pveg34 (with transition point at 34°C) and Pveg37 (with transition point at 37°C), respectively. Depending on the temperature, the fermentation process was divided into four stages. Three modules were not expressed at 30°C during stage I, and the CFP module was up-regulated at 32°C during stage II. Both of CFP and GFP modules were expressed at 34°C during stage III, and three modules were activated at 37°C during stage IV. (B) Confocal images of the WY10 strains at different fermentation periods from the cyan channel (465–480 nm), green channel (515–564 nm) and red channel (610–669 nm). The merged images from both channels.
In addition, LSCM analysis was carried out to validate the ordered activation. As shown in Figure 2B, the CFP expression level was substantially improved by switching temperatures from 30 to 32°C, and the fluorescents GFP and mCherry were not expressed in the second stage. When the temperature was at 34°C, the GFP was clearly expressed and the CFP was persistently expressed, but the mCherry module was still silent. The three modules were all up-regulated with temperatures ranging from 34 to 37°C and cells turned pink. With the increase of temperature, the CFP, GFP and mCherry modules were expressed orderly and independently during the fermentation process. The results show that this genetic circuit is able to provide ordered control of the expression levels of multiple modules by using a single input; temperature.
For the construction of bifunctional regulation, responsive repression is also needed. We thus tried to invert the response of the activation circuit by creating a ‘NOT’ gate. In order to do so, we harnessed the CRISPRi system, in which the inactive Cpf1 (dCpf1) protein binds to and inhibits the transcription of target genes (27). To design this ‘NOT’ gate, the expression of dCpf1 was driven by the promoter Pveg5 and mCherry was controlled by the strong constitutive promoter of P43 as a reporter to test repression activity. In order to avoid the effects of temperature variation on mCherry expression, appropriate control groups were provided at different temperatures. As shown in Figure 3A, the relative fluorescence intensity gradually decreased with temperature increase in a specific temperature range. The repression activity at 34°C indirectly demonstrated the low leaky expression of the hybrid promoter Pveg5. When the temperature was 39°C, the inhibitory fold reached 2.6. However, the repression activity had no significant change from 39 to 42°C. We speculated that this might be caused by the selected crRNA and its specific repression activity.
Figure 3.

Characterization of bifunctional thermogenetic circuits. (A) The inhibitory effect of repression circuit diagram constructed by the CRISPRi based NOT gate. (B) Construction of bifunctional temperature-responsive genetic circuits by combing the CRISPRi-based NOT gates with the temperature-responsive promoter Pveg5. The GFP and dCpf1 were driven by the promoter Pveg5, and the P43 was used for the expression of mCherry. Flow cytometry scatter plots showed that the expression of GFP and mCherry are regulated by the temperature. The means of GFP and mCherry were calculated and marked in the figure. (C) Static test of bifunctional temperature-responsive genetic circuits. (D) Cytometry analysis are used to further explore dynamic control of bifunctional temperature-responsive genetic circuits. The means of GFP (green) and mCherry (red) were calculated and shown in the figure. CRISPRi, Clustered Regularly Interspaced Short Palindromic Repeats interference. The statistical analysis was based on Student's t-test. Data are presented as mean ± s.d. of three replicates. **P < 0.01.
Then, the temperature activation and repression circuits were assembled for the construction of the bifunctional temperature-responsive genetic circuit to either activate or repress genes of interest with temperature. GFP and mCherry were used as reporters for activation and repression, respectively (Figure 3B). First, static test was performed to verify the bifunctional temperature-responsive genetic circuit at different temperatures and the control performance of bifunctional genetic circuit is presented in Figure 3C. To further explore dynamic control, flow cytometry analysis was performed to confirm the gradual switch from high mCherry levels to high GFP levels with the temperature increase (Figure 3D and Supplementary Figure S3). The results show that the bifunctional temperature-responsive genetic circuit could achieve the up- and down-regulation, or bifunctional control, of target genes by replacing GFP and mCherry. Our results demonstrate that the bifunctional circuit was successfully constructed and it has the potential to activate and repress the expression of genes in distinct pathways in a temperature-dependent manner.
Taken these results together, advanced temperature-responsive genetic circuits could be constructed by combing bifunctional circuits and SMIO circuits to achieve the up- and down-regulation of target genes at different switching times.
Engineered 2′-fucosyllactose synthesis pathways with multi-modular ordered control
To verify whether the temperature-responsive genetic circuits could be used to control cellular metabolism, the de novo biosynthesis of 2′-FL was engineered in B. subtilis. 2′-FL is a promising infant formula ingredient and can be synthesized from mannose-6-phosphate by five enzymes: phosphomannomutase (from mannose-6-phosphate to mannose-1-phosphate), mannose-1-phosphate guanylyltransferase (from mannose-1-phosphate to GDP-D-mannose), GDP-mannose 4,6-dehydratase (from GDP-D-mannose to GDP-4-keto-6-deoxymannose), GDP-L-fucose synthase (from GDP-4-keto-6-deoxymannose to GDP-L-fucose) and α-1,2-fucosyltransferase (from GDP-L-fucose to 2′-FL). As shown in Figure 4A, we decided to divide the 2′-FL biosynthesis pathways into three functional modules: 2′-FL synthesis (module A), cofactor supply (module B) and competitive pathway (module C).
Figure 4.

Dynamic regulation of 2′-fucosyllactose biosynthesis. (A) Illustration of the modularized platform of de novo biosynthesis of 2′-fucosyllactose in B. subtilis. Module A (2′-fucosyllactose synthesis) incorporates modifications designed to divert carbon flux to 2′-fucosyllactose metabolic. Module B (GTP supply) provides cofactor GTP for producing 2′-fucosyllactose. Module C (Competitive pathway) is the endogenous pathways which competed with 2′-fucosyllactose synthesis module. FutC, α1,2-fucosyltransferase; Zwf, glucose-6-phosphate dehydrogenase; Pgi, phosphoglucose isomerase; ManA, mannose 6-phosphate isomerase; ManB, phosphomannomutase; ManC, α-d-mannose 1-phosphate guanylyltransferase; PfkA, phosphofructokinase; Gmd, GDP-mannose 6-dehydrogenase; WcaG, GDP-L-fucose synthase, Gmk, guanylate kinase; Ndk, nucleoside diphosphate kinase; PPP, pentose phosphate pathway; GTP, guanosine 5′-triphosphate; GMP, guanosine 5′-monophosphate; GDP, guanosine 5′-diphosphate. (B) The OD600 and titer of the MT1 (manB and futC were driven by P43), MT2 (manB was driven by P43 and futC was controlled by Pveg34), MT3 (manB and futC were driven by Pveg34) under the different bioprocess, including single-switch control (0–12 h at 30°C and 12–60 h at 37°C), and constant temperature fermentation at 37°C. Data are presented as mean ± s.d. of three replicates.
GDP-L-fucose is a key precursor for 2′-FL synthesis that cannot be produced from GDP-D-mannose natively in B. subtilis. In order to enable 2′ -FL synthesis in engineered B. subtilis, four genes including manB, manC, gmd and wcaG from E. coli and the gene futC from Helicobacter pylori were codon optimized and integrated into the genome of B. subtilis under the control of the constitutive promoter P43, generating the strain MT1. The biosynthesis of 2′-FL via the de novo pathway in B. subtilis was confirmed by the LC/MS analysis. Although the sucrose uptake rate of MT1 at 37°C was higher than that of the strain cultured under single-switch control (0–12 h at 30°C and 12–60 h at 37°C) before 24 h, the 2′-FL titer of MT1 was 356.5 mg/l at 37°C, which was the same as the titer when we introduced a temperature switch in the process (Figure 4B and Supplementary Figure S4). The results indicate that the control strain MT1 could stably produce similar levels of 2′-FL and that a temperature switch does not improve 2′-FL production.
Then, we decided to use the temperature-responsive promoter Pveg34 to carry out the dynamic activation of the 2′-FL synthesis module. One or both of key genes manB and futC were driven by promoter Pveg34, and other genes were controlled by the strong constitutive promoter P43. As shown in Figure 4B, by using a single-switch process (0–12 h at 30°C and 12–60 h at 37°C), the OD600 of MT2 (manB driven by P43 and futC by Pveg34) increased by 20.2% compared with MT2 cultured at 37°C from 0 to 60 h, while the 2′-FL titer decreased by 4.6%. The OD600 of MT3 (both manB and futC genes were controlled by Pveg34) was 21.32, which was 1.71-fold that of MT3 cultured at 37°C from 0 to 60 h, and it was accompanied by a sharp increase (159.5%) to 755 mg/l of 2′-FL compared with the constant-temperature fermentation at 37°C. Subsequently, a culture test was conducted by switching temperature from 30 to 37°C at 6, 9, 12, 15 and 18 h, respectively, to identify the optimal switching time for 2′-FL synthesis, and the results show that the best switch time was at 12 h (Supplementary Figure S5).
There are three natural competitive pathways for 2′-FL biosynthesis in B. subtilis: the lactose decomposition pathway (LDP), the pentose phosphate pathway (PPP) and glycolysis. First, the β-galactosidase encoded by gene ganA and yesZ were deleted directly in the strain MT3. Unexpectedly, the deletion of β-galactosidase did not further improve the 2′-FL titer (Supplementary Figure S6). We speculated that there may still be an unknown LDP in B. subtilis. PPP and glycolysis are both indispensable pathways for normal cell growth and thereby cannot be knocked out. We therefore created a repression cascade composed of dCpf1 driven by the constitutive promoter Pveg and crRNAs controlled by the temperature-responsive promoter Pveg5, targeting to zwf (glucose-6-phosphate dehydrogenase) and pfkA (phosphofructokinase) that control the endogenous competitive pathways PPP and glycolysis, respectively (Figure 5A). For fine-tuning the repressive activity, three crRNAs targeting the beginning, middle, or end of the coding region for zwf and pkfA were designed, respectively, and 9 combinations (MT6-MT15) were assembled using Golden Gate. The fermentation process of MT3 showed that the recombinant strain was still in logarithmic phase after 12 h (Supplementary Figure S7). We thus speculated that the delayed activation of the repression circuit might reduce the interference with normal cell growth and improve 2′-FL synthesis. To test the above hypothesis, the strain MT14 (crRNAzwf3 and crRNApfkA2) was randomly chosen to be cultured under different control strategies, including single-switch control, and ordered control (0–12 h at 30°C, 12–18 h at 34°C and 18–60 h 37°C). The titer of MT14 could reach 873.5 mg/l under single-switch control, and increased by 17.2% to 1023.5 mg/l under ordered control (Supplementary Figure S8).
Figure 5.

Improving 2′-fucosyllactose production by ordered control of competitive pathways and enhancing GTP supply. (A) A bifunctional thermosensitive genetic circuit was constructed using the temperature-responsive biosensors and CRISPRi based NOT gate. The 2′-fucosyllactose synthesis and GTP supply were upregulated by the temperature-responsive biosensor, and the competitive pathways were downregulated by the temperature -responsive repression cascade. G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; M6P, mannose-6- phosphate; M1P, mannose-1-phosphate. (B) Effects of weakening PPP and glycolysis on 2′-fucosyllactose production. (C) Trends of cell growth, lactose, sucrose, extracellular and intracellular 2′-fucosyllactose concentration of the strain MT9 under ordered control from 30 to 34 to 37°C (changing the temperature to 34 and 37°C orderly after 12- and 18-h growth). (D) The metabolic pathway of GTP synthesis in B. subtilis. (E) The cell growth and titer of GTP recycling-modified strains. In addition, trends of cell growth, sucrose, extracellular and intracellular 2′-fucosyllactose concentration of the strain MT17 are shown. Data are presented as mean ± s.d. of three replicates.
Based on these results, all recombinant strains were cultured using ordered control. As shown in Figure 5B, the 2′-FL titers of the recombinant strains were improved when compared with the strain without downregulation of PPP and glycolysis, with the exception of strain MT8. The OD600 of MT8 (crRNAzwf1 and crRNApfkA2) decreased by 21.8% when compared with MT3, which was accompanied by a decrease in 2′-FL titer (13.6%). We speculated that the excessive inhibition on PPP and glycolysis could interfere with the normal cell growth, and therefore with total 2′-FL production. The OD600 of strain MT9 (crRNAzwf1and crRNApfkA3) was 26.5 at 60 h, and this strain could produce the highest 2′-FL titer (1399.5 mg/l), with an increase of 85.4% compared with the control strain MT3. This result suggests that weakening the fluxes through the so called competitive module can increase 2′-FL synthesis. In addition, the sucrose concentration was 4.6 g/l at 24 h, which means that the sucrose uptake rate for MT9 was lower than for the recombinant strains without the down-regulation of competitive pathways (Figure 5C and Supplementary Figure S4). Interestingly, in the highest producing strain MT9, 151.5 and 1248 mg/l 2′-FL were produced in the intracellular and extracellular fractions, respectively, indicating that almost 90% of the synthesized 2′-FL was transported to the medium (Figure 5C).
In addition, since GTP is involved in GDP-L-fucose biosynthesis, we decided to increase GTP availability by enhancing the GTP supply module. In order to do so, the expression of three genes, guanylate kinase (gmk), nucleoside diphosphate kinase (ndk) and xanthine phosphoribosyl transferase (xpt), were modulated (Figure 5D). In particular, the expression of these genes were controlled by the promoter Pveg34 to activate the GTP and 2′-FL synthesis modules simultaneously. As shown in Figure 5E, the strain MT17 overexpressing both ndk and gmk exhibited a total 2′-FL production of 1839.7 mg/l, increasing by 31.5% compared with the the 2′-FL titer of MT9. Therefore, an in accordance to previous studies, the overexpression of ndk and gmk can optimize GTP supply and improve the production of 2′-FL (28). In addition, we have tried to extend the fermentation period at 37°C for the overproduction of 2′-FL. However, there were no obvious increases in the 2′-FL production of strain MT17 at 72 h or 84 h, which suggested that the optimal fermentation time was 60 h (Supplementary Figure S9).
We noticed that the recombinant strains suffered from the cell autolysis in our experiments (Supplementary Figure S10A), which limits biomass accumulation in fed-batch fermentations (29). To address this challenge, the peptidoglycan hydrolases encoded by the gene lytC was knocked out in the strain MT17, generating the strain MT19. The cell growth of MT19 was slightly lower than that of the strain MT17 before post-log-phase, which suggested that the complete deletion of lytC may affect the normal cell growth (Supplementary Figure S10A). As an alternative, three crRNAs targeting to different positions on lytC, driven by the promoter Pveg5, were integrated into the genome of MT17 to generate the strains MT20, MT21 and MT22. Compared to the strain MT17, the cell growth of the strains MT20, MT21, and MT22 was undisturbed and the final OD600 was higher. In addition, no sharp decrease in OD600 at 60 h was observed after knocking down lytC. By contrast, the 2′-FL productions of the strains MT20, MT21 and MT22 decreased and we speculate that the limited carbon source was preferentially used to make higher biomass (Supplementary Figure S10B).
2′-Fucosyllactose production in a 5-l bioreactor
Based on the shake-flask results, the recombinant strains MT17 with the highest 2′-FL production and MT22 (crRNAlytC3) with the lowest cell autolysis (OD600 was 35.06 at 36 h and 32.74 at 60 h) were cultured in a 5-l bioreactor in order to test the stability and robustness of the temperature-responsive genetic circuits at larger scale. For scale-up production of 2′-FL in a 5-l bioreactor, both the sucrose and lactose concentrations were maintained above 10 g/l by pluse fed-batch fermentation.
The relative transcription levels of the genes controlled in MT17 and MT22 in different fermentation periods were measured using qPCR. In strain MT17, the mRNA levels of manB, futC, ndk and gmk showed a rapid increase by switching temperatures from 30 to 34°C after 12-h growth, and continued to increase by switching the temperature from 34 to 37°C. Meanwhile, the mRNA levels of zwf and pfkA at 37°C were reduced by 75.5% and 54.5%, respectively, when compared with those of MT17 at 30°C (Supplementary Figure S11). The changes of mRNA levels of target genes in strain MT22 were almost similar to the strain MT17 expect for lytC. In strain MT22, the mRNA level of lytC at 37 oC reduced by 87.6% compared with that at 30°C (Figure 6).
Figure 6.

Relative normalized expression of genes manB, futC, ndk, gmk, lytC, zwf and pfkA in strain MT22 by using multi-modular ordered control (0–12 h at 30°C, 12–18 h at 34°C and 18–80 h at 37°C). Data are presented as mean ± s.d. of three replicates.
As shown in Figure 7A, in MT17, there was no production of 2′-FL before 12 h. Then, at 16 h, the titer reached 1.6 g/l of 2′-FL, which coincided with the phase where the mRNA levels were controlled. The maximal OD600 reached 114.5 at 44 h and decreased by 25.2% to 85.6 at the end of the fermentation. The strain MT17 could produce 20.6 g/l 2′-FL in 5-l bioreactor, which was 11.2-times that in shake flask. The OD600 of strain MT22 reached a maximum value of 138.6 and maintained a high level until the end of fermentation (128.7 at 80 h). In addition, the 2′-FL titer reached 28.2 g/l (36.9% higher) (Figure 7B). Given the above results, we can conclude that temperature-responsive genetic circuits perform well in bioreactor conditions.
Figure 7.
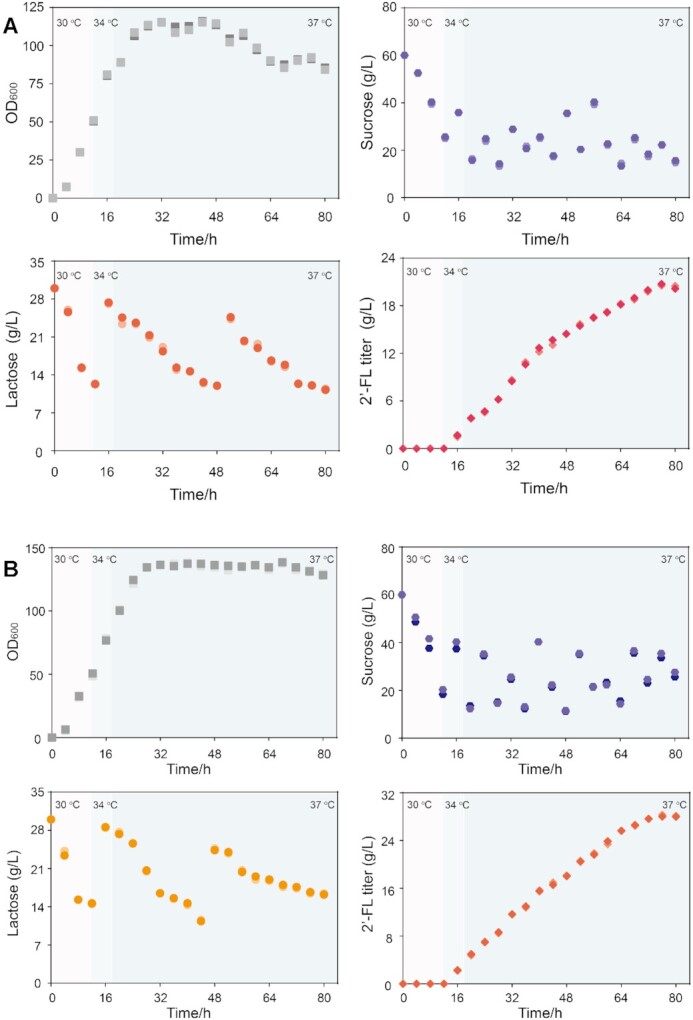
Production of 2′-fucosyllactose in a 5-l bioreactor under ordered control from 30 to 34 to 37°C (changing the temperature to 34 and 37°C orderly after 12- and 18-h growth). (A) Production of 2′-fucosyllactose by MT17 in a 5-l bioreactor under ordered control from 30 to 34 to 37°C. Trends of cell growth, sucrose, lactose, and 2′-fucosyllactose titer during pulse fed-batch fermentation in a 5-l bioreactor (n = 2 biologically independent samples). (B) Production of the 2′-fucosyllactose by MT22 in a 5-l bioreactor by using ordered control. Trends of cell growth, sucrose, lactose, and 2′-fucosyllactose titer during pulse fed-batch fermentation in a 5-l bioreactor (n = 2 biologically independent samples).
DISCUSSION
The development of high-performance circuits that enable precise and dynamic control of multiple modules is essential for the construction of efficient microbial cell factories for bioproduction. Although several genetic circuits have recently been designed for dynamic regulation, it is still challenging to finely modulate gene expression at different fermentation periods (30,31). Temperature-responsive genetic circuits offer a promising strategy to address the above-mentioned challenge. These circuits have the ability to effectively and rapidly control metabolic enzyme expression even in a post-log-phase of cell growth. In this work, we demonstrated that the target genes driven by temperature-responsive promoter can be upregulated after 18-h (Fig. 6), which is crucial for scale-up fermentations. Another advantage of these genetic circuits is their ability to achieve multi-modular ordered control of target genes by using thermosensors with different transition points. Ordered control is a desired characteristic of programmed genetic circuits that enable the control of multiple modules over time (32). In this study, a SIMO genetic circuit was created, and three modules could be up-regulated orderly during the fermentation process. The module previously expressed would not interfere with the other inactivated modules, and it maintained the on-state for the rest of fermentation, indicating the orthogonality and programmability of temperature-responsive genetic circuits. Therefore, temperature-responsive SIMO genetic circuits can be an easier and more convenient approach to achieve ordered control than using multi-layered dynamic regulation.
One of the challenges when constructing temperature-responsive genetic circuits is the availability of thermosensor (33). In E. coli, the CI857 and its corresponding promoter PR enable unrivaled temporal control of gene expression. However, in B. subtilis, the CI857 cannot be expressed by the PM promoter (data no shown) and PR worked poorly. For improving the availability of thermosensor in B. subtilis, the promoter P43 was chosen to control CI857, and then we designed and created a series of orthogonal temperature-responsive promoter based on Pveg that is a typical constitutive promoter in B. subtilis. The engineered thermosensors were able to sense the temperature and control the timing and levels of expression of enzyme with low leakiness (non-expression below 34°C) and high dynamic range (39.5) with temperatures ranging from 34 to 42°C. In addition, different applications in biotechnology require unique temperature functional ranges for activation, thus a series of thermosensors with different transition points (30, 32, 34 and 37°C) were identified.
To verify the performance of temperature-responsive genetic circuits in metabolic engineering, the de novo biosynthesis of 2′-FL was chosen as a case of study, dividing the pathway engineering strategy in 3 modules. We upregulated the 2′-FL synthesis module at 12 h, and delayed the repression of PPP and glycolysis until 18 h. The significant increase of 2′-FL titer from 356.5 to 1399.5 mg/l in shake flask culture confirmed the benefit of multi-modular ordered control. In addition, the simultaneous activation of the 2′-FL synthesis module and the GTP supply module showed a noticeably increased titer (1839.7 mg/l) compared with the constitutive overexpression of GTP supply module, which provide a potential strategy of cofactor engineering (Supplementary Figure S12). Although almost 90% of the synthesized 2′-FL was transported to the medium, the transporters capable of exporting 2′-FL in B. subtilis are still unknown. To maximize the extracellular concentration of 2′-FL, overexpressing heterologous transporters like the one identified in yeast or changing the culture media may be promising strategies (34,35). Besides, the low cell autolysis of the recombinant strains after the down-regulation of lytC proved the potential use of temperature-responsive genetic circuits to control autolysis. In addition, the successful performance of the system in a fed-batch fermentation (5-l bioreactor) demonstrated the stability and robustness of temperature-responsive genetic circuits in larger scale fermentations.
Despite their favorable features, temperature-responsive genetic circuits still present some limitations. One of the critical challenges is to modify the dynamic range, and to address this, the effects of the CI857 expression on the biosensor response should be explored for in future works (36). Additionally, the performances of genetic circuits are highly dependent on the physiological conditions of the cells. Therefore, temperature compensation should be applied to maintain host's metabolism when encountering external temperature fluctuations for counteracting the temperature effects on genetic circuit performance (37).
In conclusion, our results suggest that the microbial cell factory equipped with a robust temperature-responsive genetic circuit with multi-modular ordered control can achieve high-level production. Strategies like this one will be useful for the fine-tuning of metabolic modules aimed to produce other products and in other microbial species.
DATA AVAILABILITY
Datasets from flow cytometry experiments were submitted to the FlowRepository. Repository ID: FR-FCM-Z564.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Yang Gu (Nanjing Normal University) for the gift of plasmids, Dr Xianhao Xu and Dr Shixiu Cui for helpful discussion. We are also grateful for the State Key Laboratory of Food Science and Technology, Jiangnan University for the LC/MS analysis.
Contributor Information
Wenwen Yu, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Ke Jin, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Yaokang Wu, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Quanwei Zhang, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Yanfeng Liu, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Jianghua Li, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Guocheng Du, Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Jian Chen, Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Xueqin Lv, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
Rodrigo Ledesma-Amaro, Department of Bioengineering and Centre for Synthetic Biology, Imperial College London, London SW7 2AZ, UK.
Long Liu, Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China; Science Center for Future Foods, Jiangnan University, Wuxi 214122, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2021YFD2100700]; National Natural Science Foundation of China [31930085, 32021005]; Fundamental Research Funds for the Central Universities [JUSRP52019A, JUSRP121010, JUSRP221013]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Luo X., Reiter M.A., Espaux L., Wong J., Denby C.M., Lechner A., Zhang Y., Grzybowski A.T., Harth S., Lin W.et al.. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature. 2019; 567:123–126. [DOI] [PubMed] [Google Scholar]
- 2. Qin J.F., Krivoruchko A., Ji B.Y., Chen Y., Kristensen M., Ozdemir E., Keasling J.D., Jensen M.K., Nielsen J.. Engineering yeast metabolism for the discovery and production of polyamines and polyamine analogues. Nat. Cat. 2021; 4:498–509. [Google Scholar]
- 3. Fujiwara R., Noda S., Tanaka T., Kondo A.. Metabolic engineering of escherichiacoli for shikimate pathway derivative production from glucose-xylose co-substrate. Nat. Commun. 2020; 11:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Santos-Moreno J., Tasiudi E., Stelling J., Schaerli Y.. Multistable and dynamic CRISPRi-based synthetic circuits. Nat. Commun. 2020; 11:2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fontanarrosa P., Doosthosseini H., Borujeni A.E., Dorfan Y., Voigt C.A., Myers C.. Genetic circuit dynamics: hazard and glitch analysis. ACS Synth. Biol. 2020; 9:2324–2338. [DOI] [PubMed] [Google Scholar]
- 6. Chen Y., Zhang S.Y., Young E.M., Jones T.S., Densmore D., Voigt C.A.. Genetic circuit design automation for yeast. Nat. Microbiol. 2020; 5:1349–1360. [DOI] [PubMed] [Google Scholar]
- 7. Fang Y., Wang J., Ma W., Yang J., Zhang H., Zhao L., Chen S., Zhang S., Hu X., Li Y.et al.. Rebalancing microbial carbon distribution for L-threonine maximization using a thermal switch system. Metab. Eng. 2020; 61:33–46. [DOI] [PubMed] [Google Scholar]
- 8. Wu Y., Chen T., Liu Y., Tian R., Lv X., Li J., Du G., Chen J., Ledesma-Amaro R., Liu L.. Design of a programmable biosensor-CRISPRi genetic circuits for dynamic and autonomous dual-control of metabolic flux in bacillussubtilis. Nucleic Acids Res. 2020; 48:996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou S., Yuan S.F., Nair P.H., Alper H.S., Deng Y., Zhou J.. Development of a growth coupled and multi-layered dynamic regulation network balancing malonyl-CoA node to enhance (2S)-naringenin biosynthesis in escherichiacoli. Metab. Eng. 2021; 67:41–52. [DOI] [PubMed] [Google Scholar]
- 10. Dinh C.V., Prather K.L.J.. Development of an autonomous and bifunctional quorum-sensing circuit for metabolic flux control in engineered escherichiacoli. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:25562–25568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doong S.J., Gupta A., Prather K.L.J.. Layered dynamic regulation for improving metabolic pathway productivity in escherichiacoli. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:2964–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mannan A.A., Bates D.G.. Designing an irreversible metabolic switch for scalable induction of microbial chemical production. Nat. Commun. 2021; 12:3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo L., Diao W., Gao C., Hu G., Ding Q., Ye C., Chen X., Liu J., Liu L.. Engineering escherichiacoli lifespan for enhancing chemical production. Nat. Cat. 2020; 3:307–318. [Google Scholar]
- 14. Chee W.K.D., Yeoh J.W., Dao V.L., Poh C.L.. Thermogenetics: applications come of age. Biotechnol. Adv. 2022; 55:107907. [DOI] [PubMed] [Google Scholar]
- 15. Zheng Y., Meng F., Zhu Z., Wei W., Sun Z., Chen J., Yu B., Lou C., Chen G.Q.. A tight cold-inducible switch built by coupling thermosensitive transcriptional and proteolytic regulatory parts. Nucleic Acids Res. 2019; 47:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X., Han J.N., Zhang X., Ma Y.Y., Lin Y.N., Wang H., Li D.J., Zheng T.R., Wu F.Q., Ye J.W.et al.. Reversible thermal regulation for bifunctional dynamic control of gene expression in escherichiacoli. Nat. Commun. 2021; 12:1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen B., Zhou P.P., Jiao X., Yao Z., Ye L.D., Yu H.W.. Fermentative production of vitamin e tocotrienols in Saccharomycescerevisiae under cold-shock-triggered temperature control. Nat. Commun. 2020; 11:5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y., Liu L., Li J., Du G., Chen J.. Synthetic biology toolbox and chassis development in bacillussubtilis. Trends Biotechnol. 2019; 37:548–562. [DOI] [PubMed] [Google Scholar]
- 19. Mortaz E., Nomani M., Adcock I., Folkerts G., Garssen J.. Galactooligosaccharides and 2′-fucosyllactose can directly suppress growth of specific pathogenic microbes and affect phagocytosis of neutrophils. Nutrition. 2022; 96:111601. [DOI] [PubMed] [Google Scholar]
- 20. Liu J.J., Kwak S., Pathanibul P., Lee J.W., Yu S., Yun E.J., Lim H., Kim K.H., Jin Y.S.. Biosynthesis of a functional human milk oligosaccharide, 2′-Fucosyllactose, and l-Fucose using engineered saccharomycescerevisiae. ACS Synth. Biol. 2018; 7:2529–2536. [DOI] [PubMed] [Google Scholar]
- 21. Yan X., Yu H.J., Hong Q., Li S.P.. Cre/lox system and PCR-based genome engineering in bacillussubtilis. Appl. Environ. Microb. 2008; 74:5556–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gu Y., Lv X., Liu Y., Li J., Du G., Chen J., Rodrigo L.A., Liu L.. Synthetic redesign of central carbon and redox metabolism for high yield production of N-acetylglucosamine in bacillussubtilis. Metab. Eng. 2019; 51:59–69. [DOI] [PubMed] [Google Scholar]
- 23. Meyer A.J., Segall-Shapiro T.H., Glassey E., Zhang J., Voigt C.A.. Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 2019; 15:196–204. [DOI] [PubMed] [Google Scholar]
- 24. Johnson A.D., Poteete A.R., Lauer G., Sauer R.T., Ackers G.K., Ptashne M.. lambda repressor and cro–components of an efficient molecular switch. Nature. 1981; 294:217–223. [DOI] [PubMed] [Google Scholar]
- 25. Castillo-Hair S.M., Fujita M., Igoshin O.A., Tabor J.J.. An engineered B. subtilis inducible promoter system with over 10000-Fold dynamic range. ACS Synth. Biol. 2019; 8:1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Younger A.K., Dalvie N.C., Rottinghaus A.G., Leonard J.N.. Engineering modular biosensors to confer metabolite-responsive regulation of transcription. ACS Synth. Biol. 2017; 6:311–325. [DOI] [PubMed] [Google Scholar]
- 27. Kim D., Lim K., Kim D.E., Kim J.S.. Genome-wide specificity of dCpf1 cytidine base editors. Nat. Commun. 2020; 11:4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deng J., Gu L., Chen T., Huang H., Yin X., Lv X., Liu Y., Li N., Liu Z., Li J.et al.. Engineering the substrate transport and cofactor regeneration systems for enhancing 2′-Fucosyllactose synthesis in bacillussubtilis. ACS Synth. Biol. 2019; 8:2418–2427. [DOI] [PubMed] [Google Scholar]
- 29. Abe K., Toyofuku M., Nomura N., Obana N.. Autolysis-mediated membrane vesicle formation in Bacillussubtilis. Environ. Microbiol. 2021; 23:2632–2647. [DOI] [PubMed] [Google Scholar]
- 30. Xu X., Li X., Liu Y., Zhu Y., Li J., Du G., Chen J., Ledesma-Amaro R., Liu L.. Pyruvate-responsive genetic circuits for dynamic control of central metabolism. Nat. Chem. Biol. 2020; 16:1261–1268. [DOI] [PubMed] [Google Scholar]
- 31. Dong X., Li N., Liu Z., Lv X., Shen Y., Li J., Du G., Wang M., Liu L.. CRISPRi-Guided multiplexed fine-tuning of metabolic flux for enhanced Lacto-N-neotetraose production in bacillussubtilis. J. Agric. Food Chem. 2020; 68:2477–2484. [DOI] [PubMed] [Google Scholar]
- 32. Dinh C.V., Prather K.L.. Layered and multi-input autonomous dynamic control strategies for metabolic engineering. Curr. Opin. Biotechnol. 2020; 65:156–162. [DOI] [PubMed] [Google Scholar]
- 33. Vennettilli M., Saha S., Roy U., Mugler A.. Precision of protein thermometry. Phys. Rev. Lett. 2021; 127:098102. [DOI] [PubMed] [Google Scholar]
- 34. Hollands K., Baron C.M., Gibson K.J., Kelly K.J., Krasley E.A., Laffend L.A., Lauchli R.M., Maggio-Hall L.A., Nelson M.J., Prasad J.C.et al.. Engineering two species of yeast as cell factories for 2′-fucosyllactose. Metab. Eng. 2019; 52:232–242. [DOI] [PubMed] [Google Scholar]
- 35. Lee J.W., Kwak S., Liu J.J., Yu S., Yun E.J., Kim D.H., Liu C., Kim K.H., Jin Y.S.. Enhanced 2′-fucosyllactose production by engineered Saccharomycescerevisiae using xylose as a co-substrate. Metab. Eng. 2020; 62:322–329. [DOI] [PubMed] [Google Scholar]
- 36. Chee W.K.D., Yeoh J.W., Dao V.L., Poh C.L.. Highly reversible tunable thermal-repressible split-t7 RNA polymerases (thermal-T7RNAPs) for dynamic gene regulation. ACS Synth. Biol. 2022; 11:921–937. [DOI] [PubMed] [Google Scholar]
- 37. Hussain F., Gupta C., Hirning A.J., Ott W., Matthews K.S., Josic K., Bennett M.R.. Engineered temperature compensation in a synthetic genetic clock. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:972–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets from flow cytometry experiments were submitted to the FlowRepository. Repository ID: FR-FCM-Z564.