

Abstract
Hemopexin (Hpx) is a crucial defense protein against heme liberated from degraded hemoglobin during hemolysis. High heme stress creates an imbalance in Hpx bioavailability, favoring heme accumulation and downstream pathophysiological responses leading to cardiopulmonary disease progression in sickle cell disease (SCD) patients. Here, we evaluated an model of murine SCD, which was designed to accelerate red blood cell sickling, pulmonary hypertension, right ventricular dysfunction, and exercise intolerance by exposure of the mice to moderate hypobaric hypoxia. The sequence of pathophysiology in this model tracks with circulatory heme accumulation, lipid oxidation, extensive remodeling of the pulmonary vasculature, and fibrosis. We hypothesized that Hpx replacement for an extended period would improve exercise tolerance measured by critical speed as a clinically meaningful therapeutic endpoint. Further, we sought to define the effects of Hpx on upstream cardiopulmonary function, histopathology, and tissue oxidation. Our data shows that tri-weekly administrations of Hpx for three months dose-dependently reduced heme exposure and pulmonary hypertension while improving cardiac pressure-volume relationships and exercise tolerance. Furthermore, Hpx administration dose-dependently attenuated pulmonary fibrosis and oxidative modifications in the lung and myocardium of the right ventricle. Observations in our SCD murine model are consistent with pulmonary vascular and right ventricular pathology at autopsy in SCD patients having suffered from severe pulmonary hypertension, right ventricular dysfunction, and sudden cardiac death. This study provides a translational evaluation supported by a rigorous outcome analysis demonstrating therapeutic proof-of-concept for Hpx replacement in SCD.
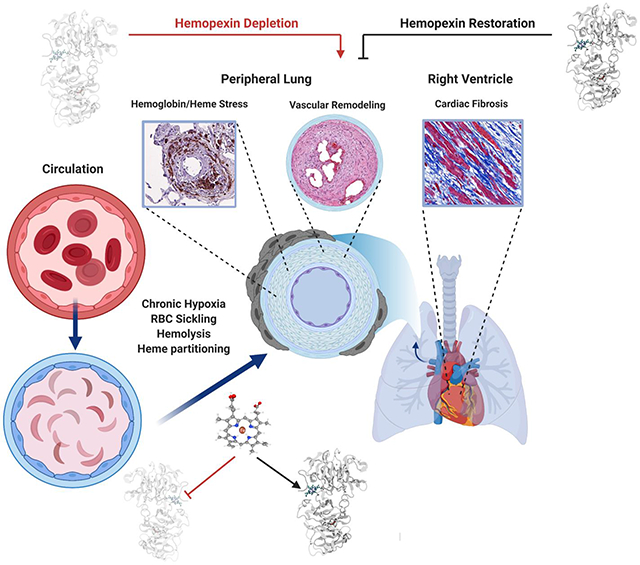
Graphical Abstract
INTRODUCTION
Sickle cell disease (SCD) is caused by a single point mutation (Glu6Val) in the hemoglobin (Hb) ß-globin gene and affects millions of people around the world. (1) This single point mutation of sickle hemoglobin (Hb-S) promotes polymerization of deoxygenated Hb within red blood cells (RBCs), causing membrane distortion and the characteristic sickled RBC morphology. (1, 2) The polymerization of Hb-S and RBC sickling is the primary cause of vascular obstruction and hemolysis, which subsequently contributes toward acute and chronic SCD sequelae, including acute chest and pain syndromes, stroke, pulmonary hypertension (PH), cardiac dysfunction, and irreversible end-organ damage. (3) Because SCD is a life-long genetic disorder, the prognosis is difficult to define, and the clinical goal is to achieve a normal life span with minimal morbidity. (4) Despite improvements in disease monitoring and therapeutics development (5-8), SCD remains a debilitating condition, and most patients are faced with repeated hospitalizations. (9)
The world health organization designates pulmonary hypertension (PH) in SCD patients into Group 5, which includes a diversity of conditions where the cause of PH is thought to be multifactorial and incompletely understood. (10) PH with pulmonary arterial pressures ≥ 25 mmHg measured by right heart catheterization occurs in 6 −10% of the SCD patient population. (10-12) and results in either pre- or post-capillary disease. (13-15) Both are linked to the pulmonary vascular pathogenesis caused by exposure to extracellular Hb, nitric oxide depletion and tissue hypoxia. (13, 16) Over nine years, the mortality rate of PH in SCD patients is 55% compared to 21% in non-PH SCD patients. (13) Despite the unacceptably high additive mortality, the disease process’s delayed onset and slow progression defines a window of opportunity for halting or reversing cardiopulmonary dysfunction and improving exercise tolerance and quality of life.
All forms of PH are associated with structural remodeling of the main pulmonary arterial branches and the peripheral pulmonary vasculature (17), which results in symptoms and outcomes primarily determined by right ventricular failure caused by increased pulmonary vascular resistance. (18) Several groups have demonstrated the importance of extracellular Hb and its degradation product heme as important multi-mechanistic causes of accelerated SCD disease progression. (19-21) Our studies suggested that Hb toxins can acutely distribute into interstitial spaces of arteries, myocardium, and lung tissues (22-24) to amplify cardiopulmonary disease in SCD. (25, 26) Further, the Hb and heme detoxification proteins, haptoglobin (Hp) and hemopexin (Hpx) are depleted in SCD patients. (24, 27) The result of Hb generated toxins and loss of Hb/heme scavenger protein homeostasis is unchecked heme transfer to circulatory proteins (28, 29), partitioning into lipoproteins (30, 31), and tissue deposition. (19, 32) Partitioning of heme enhances reactive oxygen species generation that causes protein and lipid oxidations and eventually contributes toward tissue injury and fibrosis. (33-35) Currently, there are no treatment options to specifically target the PH or right ventricular dysfunction once this process begins. Chronic replacement of Hpx in SCD may provide a new therapeutic strategy to attenuate the impact of heme-driven progression of cardiopulmonary disease complications. (24)
Using a murine model of progressive SCD, we tested the hypothesis that Hpx can dose dependently improve exercise tolerance (measured using critical speed) by attenuating the pathophysiology of PH and right ventricular dysfunction. In our experiments, young SCD mice were exposed to modest altitude (8,000 ft) for three months to accelerate murine SCD disease. This model is specifically designed to approximate PH and right ventricular dysfunction in the human SCD patient population. (36) During the 3-month time frame of altitude exposure, Hpx replacement therapy by repeated subcutaneous administration attenuated pathology in the heart and lung, normalizing physiology in the pulmonary circulation and increasing exercise tolerance.
MATERIALS AND METHODS
Human autopsy SCD PH lung tissue and citrated plasma
Deidentified human lung tissue was obtained from deceased SCD patients with identified PH from the University of Colorado Denver Anschutz Medical Center and Royal London Hospital. Use of deceased patient lung, heart and deidentified exchange transfusion citrated plasma was considered by the Colorado Multiple Institutional Review Board and deemed to be non-human subject research and given an exempt status. Apheresis samples in citrate were obtained from adult SCD patients, male (n=6-12) and female (n=6-12) undergoing regular exchange transfusion therapy at the University of Colorado Anschutz Medical Campus. Control plasma was obtained from Innovative research (Novi, MI, USA) to match the SCD patient age range (20-45 years old) and sex.
Ethical approval and animal care
Young-adult male and female C57Bl/6 WT and Berk-SS mice (8 weeks old) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Mice were housed and bred in an AAALAC accredited animal facility at the University of Colorado, Denver, Anschutz Medical campus and were maintained on a 12:12 light-dark cycle with food and water available ad libitum. Female heterozygous Berk-SS mice were bred with male homozygous Berk-SS mice to generate homozygous offspring. Specifically, Berk-SS mice with genotype Tg(Hu-miniLCR α1 Gγ Aγ δ βs) Hba0/0 Hbb0/0 and the hemizygous with genotype Tg(Hu-miniLCR α1 Gγ Aγ δ βs) Hba0/0 Hbb0 Hbb+ were littermates. Genotyping of mice used for breeding and experiments was performed by TransnetYX (Cordova, TN, USA). A total of 50 mice (WT: n=12, Berk-SS mice: n=38) were used in the present investigation and levels of discomfort and distress were monitored daily by the in-house animal care staff, with a veterinarian available as needed. Mice presented no pain or discomfort associated with hypoxia or recovery measurements (e.g., treadmill exercise and echocardiography) and were alert as well as eating, drinking, and grooming normally while housed. All experimental procedures were conducted under the guidelines recommended by The Journal of Physiology (37), the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee at the University of Colorado, Denver, Anschutz Medical Campus.
Experimental design
Pharmacokinetic studies were performed following Hpx SCD mice (n=38) were randomly divided into three treatment groups to receive either: (1) saline vehicle control (n=14) (VC); (2) low dose hemopexin (LD-Hpx; 100 mg/kg) (n=12); and (3) high dose hemopexin (HD-Hpx, 300 mg/kg) (n=12) by subcutaneous injection three times per week for 3 months. SCD mice were exposed continuously to a simulated moderate altitude in custom-built rodent hypobaric/hyperbaric chambers. These chambers were pressurized to mimic 8,000 ft elevation (564 mmHg, respectively) as described previously. (36) All mice were exposed to altitudes for 3 months. Investigators performing treadmill exercise, RV function and group analysis were blinded to the treatments. Additionally, two control groups were used to characterize RV function in the SCD mice exposed to moderate hypoxia. First, a cohort of 8 (n=8) wild type mice were exposed to hypoxia for comparison of RV function between a healthy and compromised SCD mouse exposed to moderate altitude. Second, a group of 6 (n=6) untreated SCD mice were exposed to sea level conditions for comparison of RV function between SCD mice exposed to sea level and moderate altitude. Unless noted these groups were not included in the primary statistical analysis for effectiveness of Hpx. To determine the Hpx dosing range used in this study, SCD mice received Hpx daily at dose levels of 50, 160 and 500 mg/kg (lateral caudal vein). After the last dose on day 5, blood was collected (lateral caudal vein) at the following time-points: 5 min, 2h, 6h, 24h. The blood was prepared for analysis of Hpx and heme-Hpx concentrations.
Hemopexin
Highly purified human plasma derived Hpx (100 mg/ml in 0.9% NaCl) was provided by CSL Behring (Kankakee, Illinois).
Total plasma heme concentration measurements
Citrated plasma was collected from the discarded blood of sickle cell anaemia patients undergoing routine exchange transfusion and approved as an exemption of human research by The University of Colorado Denver Anschutz Medical Center Institutional Review Board. Heme concentration in human and mouse plasma was determined according to manufacturer's protocols using the QuantiChrom™ Heme Assay Kit (DIHM-250, BioAssay Systems). Briefly, 50 μL of sample (diluted in water 1:2) was placed into a 96-well plate. Assay reagent (200 μL per well) was added and incubated for 5 min at room temperature. Absorbance at λ400 nm was measured using a microplate reader (Synergy H1, BioTek). Heme concentration was determined by interpolating absorbance against a hemin standard curve.
Detection of hemopexin:heme complexes in human and mouse plasma
50 microliters of plasma sample were placed into a clean Eppendorf tube followed by the addition of 150 μL Buffer A (Multiple Affinity Removal Systems, Agilent). In a first chromatography step high abundant proteins were depleted and carried out according to the manufacturer’s protocol on an Ultimate 3000SD HPLC attached to two LPG-3400SD quaternary pumps and a photodiode array detector (DAD) (ThermoFisher). Briefly, the diluted plasma sample was injected onto a multi affinity removal column depleting human albumin and IgG (Human IgG/HSA, 4.6 × 50 mm, Agilent) or mouse albumin, IgG and Transferrin (Mouse-3, 4.6 × 50 mm, Agilent) and separated with Buffer A (Multiple Affinity Removal Systems, Agilent) as the mobile phase at a flow rate of 0.25 mL/min. Depleted plasma was collected into a fresh HPLC vial and re-injected and separated on a Diol-300 (3 μm, 300 X 8.0 mm) column (YMC CO Ltd.) with PBS, pH 7.4 (Bichsel) as the mobile phase at a flow rate of 1 mL/min. For all samples two wavelengths were recorded (λ = 280 nm and λ = 414 nm). The amount of Hpx:heme complexes was determined by calculating the peak area of the complex (9 min retention time). Values from depleted plasma samples were interpolated by generating a standard curve based on peak area and plotted against the concentrations.
Determination of cell free hemoglobin concentrations
Cell free Hb was determined by SEC–high-performance liquid chromatography (SEC-HPLC) using an Agilent 1260 II HPLC attached to a quaternary pump and a photodiode array detector (DAD) (Agilent). Plasma samples and Hb standards were separated on a Diol-300 (3 μm, 300 X 8.0 mm) column (YMC CO Ltd.) with PBS, pH 7.4 (Bichsel) as the mobile phase at a flow rate of 1 mL/min. For all samples two wavelengths were recorded (λ = 280 nm and λ = 414 nm). Cell free Hb in plasma was determined by calculating the peak area (10 min retention time for cell free Hb). Values from plasma samples were interpolated by generating a standard curve based on peak area and plotted against the concentrations.
Lipoprotein peroxidation assay
Human SCD heme partitioning and oxidation was evaluated in citrated human plasma by measuring the formation of malondialdehyde (MDA), the final product of lipid peroxidation, after sample incubation with rLP with and without Hpx addition. In a 96-well plate 30 μL containing rLP (2mg/mL), Hpx (10μM) and different concentrations of Heme-albumin were incubated at 37°C for 4 hours. After incubation, the concentration of MDA was measured using a Thiobarbituric acid reactive substance (TBARS) assay.(30) 125 μL of 750 mM trichloroacetic acid in 1 M HCl was added to the samples, followed by vortexing (5 seconds) and the subsequent addition of 100 μL of 25 mM 2-thiobarbituric acid in 1 M NaOH. After an incubation period of 60 minutes at 80°C, the TBARS in the supernatant was quantified measuring the fluorescence emission at 550 nm with 510 nm used as the excitation wavelength.
Quantification of human Hpx in mouse plasma samples
Ten microliters of plasma sample were placed into a clean Eppendorf tube followed by the addition of 80 μL MeOH to precipitate the protein. The methanol was removed after centrifugation and the pellet was air-dried and afterwards re-suspended in 50 mM NH4HCO3/0.16% ProteaseMAX containing a heavy-isoptope labeled peptide, which is specific for human hemopexin and is used as internal standard. After incubation at 56°C/550 rpm for 45 min the samples were reduced by adding 0.5 M DTT (56°C/550 rpm for 20 min). The samples were then alkylated by addition of 0.5 M IAA and incubation for 20 min at RT protected from light. Tryptic digestion was carried out at 37°C/550 rpm and stopped after 3 h by addition of formic acid. After centrifugation the samples were separated immediately on a C18 column (AdvanceBio Peptide Mapping, 2.1 × 150 mm). The measurements were conducted using an Agilent 1290 Infinity II – 6550 iFunnel QTOF LC-MS system.
Data was analyzed by calculating the peak area of the analyte and the internal standard using Agilent MassHunter Quant software. A standard curve was created in Agilent MassHunter Quant by plotting the average response ratio of analyte to internal standard against concentration for each standard sample. The analyte concentration in the plasma samples was back calculated using the standard curve equation.
Blinding:
To prevent unwanted bias Investigators performing critical speed and hemodynamics were blinded to the treatment. Groups were unblinded after analysis.
Treadmill exercise and constant speed tests
Prior to the determination of CS, mice completed a treadmill familiarization phase, which consisted of four ~5 min runs on a motor-driven rodent treadmill (Exer 3/6, Columbus Instruments, Columbus, Ohio, USA). For the first several runs, the treadmill speed was maintained at 10-15 m/min (up a 5° grade, which was maintained throughout all treadmill tests). For the last several runs, the speed of the treadmill was increased progressively over the last minute to ~30-35 m/min to familiarize the mice with high speed running. Animals were encouraged to run with intermittent bursts of compressed room air aimed at the hind limbs from directly above the animal (so as not to push the mouse up the treadmill). All treadmill testing protocols were designed and conducted by experienced staff and strictly followed the guidelines set by the American Physiological Society’s resource book for the design of animal exercise protocols.(38)
The CS was determined using a modified version of the methodology used by Copp et al, 2010 for rats, as well as following the guidelines set forth by Poole et al.(38, 39) After completion of the treadmill familiarization period, each mouse performed 3-5 runs to exhaustion, in random order, at a constant speed that resulted in fatigue between 1 and 15 minutes (speeds ranging from 30-50 m/min). Each test was performed on separate days with a minimum of 24 hours between tests. For each constant-speed trial, mice were given a 2-minute warm-up period where they ran at 15-20 m/min followed by a 1-minute period of quiet resting. To start the test, the treadmill speed was increased rapidly over a 10 second period to the desired speed at which point a stopwatch was started. Testing was terminated and time to exhaustion was measured to the nearest tenth of a second whenever the mouse could no longer maintain pace with the treadmill despite obvious exertion of effort. A successful constant-speed test was determined if 1) the mouse could quickly adapt to the treadmill speed at the beginning of the test (e.g., did not waste energy), 2) a noticeable change in gait occurred preceding exhaustion (i.e., lowering of the hindlimbs and rising of the snout), and 3) the animal’s righting reflex was markedly attenuated when placed on their back in a supine position (an unexhausted quadruped will typically attempt to right themselves within ~1 second).
Data modeling for the determination of CS
Following successful completion of the constant speed treadmill tests, the CS and finite distance capacity (D’) were calculated for each mouse using the linear 1/time model (Speed = D′×1/time+CS) as described previously.(54-56) In this model, the treadmill speed used for the constant speed test is plotted as a function of the inverse of time to exhaustion and the y-intercept of the regression line yields the CS and the slope is the D’.
Open chest solid state catheterization for right ventricular function analysis
After 3 months, mice underwent terminal open chest right ventricular (RV) function measurements with a 1.2F, FTE-1212B-4018 pressure volume catheter (Transonic Systems Inc., Ithaca, NY) inserted by direct cardiac puncture. Mice were induced inhaled isoflurane (4-5%), and tracheal incision (~ 1 cm) was performed. Next, a tracheal tube was inserted and connected to an Anesthesia Workstation or Hallowell EMC Microvent and an anesthestic plain was maintained at 1.0-2.5% isoflurane in 100% oxygen. After which, a thoracotomy was performed exposing the heart, the pericardium was resected and a small hole made at the base of the RV with a 30g needle for insertion of the pressure-volume catheter. Steady state hemodynamics are collected with short pauses in ventilation (up to 10 seconds) or high frequency oscillatory ventilation to eliminate ventilator artifact from the pressure-volume recordings. Occlusions of the inferior vena cava were performed by applying pressure to the inferior vena cava (up to 10 seconds) through the abdominal opening. After pressure volume and hemodynamic measurements completed mice were humanely euthanized by exsanguination and cervical dislocation. Data was recorded continuously using LabScribe2 and analyzed offline.
Blood and organ collection
At the end of the experimental protocol, 0.8 mL of blood was collected and placed in an EDTA-K+ vacutainer and a hematocrit (Hct) tube for analysis of plasma and hematocrit, respectively. The plasma was collected, snap frozen in liquid nitrogen, and immediately stored at −80 °C until analysis. Tissues were collected after PBS perfusion as reported previously.(36) The hearts were removed, and the right ventricle (RV) and left ventricle with septum (LV+S) were weighed for the assessment of the Fulton Index (RV/LV+S).
Histology and morphology
Tissue sections were dewaxed and rehydrated and processed as previously described.(36) Five-micron lung sections were stained with hematoxylin and eosin (H&E) by standard procedures to assess the accumulation of perivascular cells as well as vessel wall thickness as previously described.(40)
Perls iron staining:
As previously described (37), tissue sections were incubated with Perls iron reagent containing 5% potassium ferrocyanide and 2% hydrochloric acid for 45 min at room temperature and rinsed in deionized water. Sections were then incubated with 0.3% hydrogen peroxide and 0.01 M sodium azide in methanol for 30 min at room temperature. All sections were then rinsed in 0.1 M phosphate buffer, pH 7.4, incubated with DAB (SigmaFast DAB, Sigma) for 3 min, washed in deionized water, and lightly counterstained with Gill’s II hematoxylin.
Elastin staining:
The Verhoeff Van Gieson Elastin stain kit by astral diagnostics from polysciences, Inc (item# 25089-1, lot# 0126, exp 2022/04/30) was used for elastin visualization. Briefly, slides were deparaffinized, rehydrated, and stained according to the recommended procedure provided by the manufacturer. Verhoeff’s solution was used for 1 hour, each stain was followed by a tap water rinse, subsequent differentiation was achieved using 2% ferric chloride solution, 5% sodium thiosulphate, and finally Van Gieson counterstain I, for 4 mins. The slides were quickly dehydrated, cleared with xylenes, and coverslipped using ctyoseal 60 (Thermo Scientific, catalog # 8310-16).
4HNE staining:
Slides bearing tissues were deparaffinized, rehydrated, and underwent antigen retrieval using 0.01M citrate buffer. The tissues were blocked using 10% horse serum in PBST. The primary antibody (Genox, USA. Catalog # MHN-100P, lot 011 MHN-100P) concentration was 20ug/mL and was left on the tissue overnight at 4°C. After rinsing, the secondary antibody (Hourse anti-mouse, Vector laboratories, catalog # PI-2000, lot ZGO211) was applied at 1:300 dilution for 1 hour at room temperature. Signal amplification was produced using Vector Laboratories DAB peroxidase substrate kit (Catalog # SK-4100). Slides were counterstained with Harris hematoxylin, followed by dehydration, and coverslip application.
Masson’s Trichrome staining:
Tissue was re-fixed in Bouin’s solution, washed in deionized water, stained in Weigert’s iron hematoxylin, Biebrich scarlet-acid fuchsin solution and finally phosphomolybdic-phosphotungstic acid differentiation.
Hemoglobin and Hemopexin cy5 conjugation, delivery and imaging
Plasma derived hemoglobin and hemopexin, at a concentration of 100mg/mL, were labeled with 1.04mg of Sulfo-cyanine5 NHS ester (Lumiprobe, Catalog #43320, batch 4FW7C 008, 25mg) per manufacturer’s instructions and incubated at room temperature for 4 hours. The labelling solution was filtered and concentrated using Amicon® Ultra 0.5mL, ultracel® 10K centrifugal filter (Millipore, Ref # UFC501096, lot R8KA10692), washed with normal saline, and spun at 14,000g for 25 mins to retrieve final volume.
SCD mice were anesthetized with inhaled isoflurane (4-5%), and tracheal incision (~ 1 cm) was performed. Next, a tracheal tube was inserted and connected to an Anesthesia Workstation or Hallowell EMC Microvent and an anesthestic plain was maintained at 1.0-2.5% isoflurane in 100% oxygen. After which, a thoracotomy was performed exposing the heart. Next, 100 ul of either Hb or Hpx conjugated to cy5 or saline as control was injected into the left ventricle and allowed to circulate for 30 minutes before mice were humanely euthanized. The heart was perfused free of blood and fixed in Formalin for 18 h and switched to 70% ethanol before being paraffin embedded. Five-micron sections of the RV were visualized in the University of Colorado Light Microscopy Imaging Core using an Oympus FV1000 FC with laser excitation at 635 nm and emission at at 640-680 nm.
Morphology
Vasculature in the lungs was quantified for the H&E stained lungs. Scanned images were equally divided into 100 equal segments. Images that did not have sufficient tissue in the segment were excluded and from the remaining images, 10 randomly selected segments were used for analysis. Using the program STEPanizer (Tschanz & Weibel, 2011), a grid was overlaid on each image and was used to quantify the vasculature within each image. The percentage of vascular tissue to parenchymal tissue in the lung was calculated.
Statistical analysis
Data are presented as a mean ± standard error of the mean (SEM). Statistical comparisons for data measurements were completed with the analysis of variance (ANOVA). Main effects between hemopexin and vehicle control were determined by pooling all SCD Hpx dosed mice into a single cohort. Post-hoc analyses were completed with the Tukey-Kramer multiple comparison tests. Strain effects between wild type and SCD were completed using a students t test between SCD vehicle control and wild type cohorts. Statistical analysis was completed using the statistical software package GraphPad (version 6.0). Statistical significance was defined as P ≤ 0.05.
RESULTS
Pulmonary and right ventricle tissue remodeling in SCD patients with PH shows pathology consistent with oxidation and fibrosis.
First, we aimed to explore whether advanced remodeling processes in the pulmonary vasculature of SCD patients with PH coincide with oxidative tissue modifications, which would be consistent with a heme-driven pathway of disease progression.
Representative sections from tissues obtained at autopsy are shown for control (no SCD and no PH), SCD without PH (SCD PH–), and SCD with PH (SCD PH+) (Figure 1). Main pulmonary artery sections are shown in Figure 1A. Intimal hyperplasia, thickening, collagen accumulation and elastin fragmentation can be visualized in SCD PH+, indicating severe pulmonary arterial remodeling compared to control and SCD PH – tissues. Peripheral lung vasculature tissue sections demonstrate pronounced distal lung remodeling, seen as plexiform lesions with recanalized vessels, a hallmark of severe PH (Figure 1B). Further, iron-loaded macrophages, oxidation (4-HNE immunoreactivity) and vessel fibrosis are prominent features of distal lung pulmonary arterial pathology in SCD PH+ tissues. Unique to peripheral lung tissue is the observation of iron within vascular adventitial macrophages, which is a typical finding in SCD patients with progressive PH. (22) We have also observed some indication of lipid peroxidation (4-HNE immunoreactivity) in the peripheral lung vessels of patients with SCD and no PH, but to a lesser extent than in SCD PH+ tissue.
Figure 1: Pulmonary hypertension in SCD results in lung vascular and right ventricle tissue remodeling.

(A) Human main pulmonary artery (MPA) tissue sections from SCD patients with PH (SCD PH (+)) demonstrate distinctive pathology when compared to SCD patients without PH (SCD PH (−)) and controls. H&E staining (a-c), Masson’s trichrome (d-f) and Verhoeff Van Gieson elastic stain, EVG (g-i ). SCD PH + patient H&E stained tissues sections show intimal hyperplasia (black arrows), medial thickening and loss of vascular smooth muscle cell integrity (c, inset). Collagen deposition (f) and elastin fragmentation (i) are observed in the MPA media. (B) Human peripheral lung tissue sections are shown from top to bottom stained for H&E (a-c), Perls iron (non-heme iron) (d-f), 4-hydroxynonenal (4-HNE) (g-i) and Masson’s trichrome (j-l). Staining in SCD PH + patient peripheral lung tissue shows arterial recanalization, vascular plexiform lesions, and intimal thickening (c). Iron positive cellular staining is observed in lung vascular adventitia (f) and immune reactivity to 4-HNE within outer medial and adventitia accumulated cells consistent with iron localization (i). Peripheral arterial lung fibrosis is a prominent pathologic feature of SCD PH and visually observed across all structures of the vessel (l). Limited indicators of pathological processes are visualized in SCD PH - patient tissue sections, except for oxidative stress visually observed as 4-HNE immune reactivity in adventitia accumulated cells (h). No indications of peripheral lung vascular pathology are observed in control tissue sections. (C) Right ventricular pathology is observed in SCD PH (+) as loss of cardiac muscle striations (black arrows) and eosinophilia (back box) (c). Cardiac iron was not observed in any of the tissue sections (e-f). 4-HNE immune reactivity was most intense visually in SCD PH + patient tissue sections (i) and to a lesser extent in SCD PH – tissue sections (h). Interstitial collagen accumulation is visualized in SCD PH + patient tissue sections (l), to a lesser extent in SCD PH – tissue sections (k) and not at all in control in tissue sections. Magnification main PA - 10x; distal PA- 10x; right ventricle- 20x
Right ventricular tissue sections are shown in Figure 1C. Hematoxylin and eosin-staining suggests a loss of cardiac muscle striations (black arrows) and eosinophilia (back box) throughout the right ventricle. As expected, non-heme iron did not accumulate in the right ventricle, consistent with SCD pathophysiology. However, intense staining for the lipid oxidation marker 4-HNE is present in SCD PH+ tissue sections, indicating that heme partitioning into the RV could function as a relevant pro-oxidant source. Like observations in the distal lung, limited 4-HNE immunoreactivity is also found in SCD PH– right ventricle, but not in the control. Further, the right ventricle of SCD PH+ tissue sections are fibrotic as seen following Masson’s Trichrome histochemistry, which reveals intense blue staining of interstitial spaces between cardiomyocytes. This intensity of collagen staining was not observed in SCD PH– or control tissues.
Collectively, our findings are consistent with a pathway of hemolysis-induced oxidative tissue injury progressing from early or mild disease without concomitant cardiopulmonary changes to severe fibrosis and structural remodeling found in SCD patients with advanced PH and right heart failure.
Heme partitioning in SCD is defined by a persistent hemopexin deficit.
We next evaluated heme partitioning in plasma of patients with SCD to determine the availability of free heme and concomitant lipid oxidation activity to rationalize the conceptual framework of a Hpx replacement therapy.
Figure 2A shows three main components of total plasma-heme as cell free Hb, heme-Hpx and heme bound to other macromolecules in citrated plasma from SCD patients. The amount of heme-Hpx is shown prior to (top) and after the addition of Hpx (bottom). Based on HPLC and UV-visible spectrophotometry, the amount of heme-Hpx complexes is more than 5-fold increased after the addition of plasma derived Hpx (bottom). These data suggest a significant imbalance in the heme:Hpx ratio in SCD patient plasma favoring excess free heme available for oxidative reactions. Figure 2B Shows the differences in healthy control versus SCD sample Hpx levels. This data is from an existing sample set (24) , also shown here to illustrate the extent of Hpx depletion in SCD. Figure 2C shows the amount of thiobarbituric acid reactive substances (TBARS) as lipid oxidation byproducts generated in a mixture of SCD patient plasma with a reconstituted lipoprotein (rLP) containing a high ratio of unsaturated fatty-acids. The addition of Hpx significantly attenuates TBARS in this mixture, suggesting that free heme is a critical driver of oxidative stress in the plasma of SCD patients. In the absence of heme, rLP is not oxidized and generates the same level of TBARS as SCD plasma + rLP +Hpx. Collectively, tissue pathology, plasma Hpx depletion and excess transferable heme suggest that heme-driven oxidation could be critical driver of tissue injury and functional impairment in SCD, rationalizing Hpx replacement as a therapeutic strategy.
Figure 2: Heme transfer to Hpx and lipid oxidation in heme rich biological samples.

(A) Shows heme binding in citrated plasma obtained at the end of exchange transfusions, before and after the addition of Hpx (n=6 samples). Light grey - no Hb or holo-Hpx, red - holo-Hpx and dark grey cell-free Hb. Prior to exogenous Hpx addition, holo-Hpx concentration was 6.20 ± 2.5 mmol/L (Mean ± SD). After Hpx addition, holo-Hpx increased to 34.0 ± 9.6 mmol/L (Mean ± SD) (p <0.001), suggesting a 5-fold increase in heme transfer from non-Hpx proteins and lipids. (B) Shows differences in healthy control donor plasma (390 μg/ml ± 88.7, n=10) and SCD (77.3 ± 55.0, n=10) (p <0.001), data from the same sample cohort shown for quick reference (PMID: 30047285). (C) Shows citrated plasma in the presence of reconstituted lipoprotein (rLP) as a source for heme partitioning and oxidation. Box plots with highest to lowest individual values (n=12-24 per group) show the accumulation of lipid peroxidation products (thiobarbituric acid reactive substances, TBARS). SCD samples + saline + rLP show the greatest generation of TBARS (left), which is significantly reduced (p=0.004) with the addition of Hpx. Alone, rLP does not increase TBARS generation (right). (D) Dose finding for the primary murine study is based on the efficiency of Hpx dosing (50, 160 and 500 mg/kg/day) to bind heme in SCD mice (n=4) after five daily doses followed by blood collections at 0, 2, 6, 24 hours after the final dose. Red circles represent total Hpx, blue circles represent heme bound Hpx, solid blue and red lines indicate mean values and bars indicate SD. Data indicates that doses equal to 50 and 160 mg/kg/day x 5 days consume Hpx within a heme-Hpx complex, while dosing at 500 mg/kg/day exceeds transferable heme availability and suggests a steady state transferable heme concentration of approximately 40 - 80 uM.
Pilot study to optimize main study dosing in SCD mice
To rationalize a dosing strategy for our main study, we conducted a pilot study that dosed SCD mice (n=4/group) at 50, 160 and 500 mg/kg, 3-times weekly over a duration of 0-14 days. Dosing of SCD mice in the abbreviated duration pilot allowed for us to assess the extent of Hpx heme binding and evaluate apoHpx and Hpx-heme plasma concentrations relative to dose ( not to characterize the full pharmacokinetic profile apoHpx and Hpx-heme). After administration of the last dose, plasma samples were obtained and analyzed at 0, 2, 6 and 24 hours by size exclusion chromatography (SEC) to quantify Hpx-heme complexes and by LC MS/MS to quantify human Hpx protein concentrations (Figure 2D). After dosing with 500 mg/kg human Hpx, the transfer of heme to Hpx was maintained at approximately 40 μM heme-Hpx, suggesting that Hpx concentrations exceed free-heme availability. These data suggest that a low (100 mg/kg) and a high (300 mg/kg) dose of human Hpx administered 3-times weekly would demonstrate a dose dependent effect on heme binding and subsequently an attenuation of heme-induced cardiopulmonary disease progression in SCD mice.
Hemopexin therapy improves exercise tolerance in progressive murine SCD.
In SCD patients, exercise tolerance and quality of life are assessed via a 6-minute walk test. The rigor of critical speed analysis allows for a meaningful proof-of-concept outcome that defines a potential therapeutic effect of chronic Hpx replacement. Further, dyspnea/fatigue syndrome is known to limit exercise capacity in RV failure (41), and is commonly observed in SCD PH. Thus, we first sought to understand if Hpx replacement therapy would improve the outcome of a rigorously applied exercise tolerance measurement conducive to quality of life. As such, studies were designed to compare the running/speed/duration relationship, known as the critical speed (38, 41-43), in SCD mice exposed to moderate hypoxia and administered tri weekly subcutaneous injections of either (saline: vehicle control; VC), low dose Hpx (100 mg/kg: LD-Hpx) or high dose Hpx (300 mg/kg: HD-Hpx).
Critical speed is a fundamental cross-species exercise parameter that defines the tolerable duration of exercise intensity and yields a functional framework to support changes in RV and pulmonary vascular function. (43) Figure 3A and B illustrates the hyperbolic modeling of the critical speed obtained from each treatment group and the corresponding relationship with the anaerobic work capacity, representing the changes brought abo ut by Hpx treatment. The bar graphs in Figure3C and D below the schematics show the determinant critical speed and anaerobic work capacity values for animals in each group. Specifically, data in Figure 3C demonstrate that relative to the VC cohort, there is a significant improvement of (~ 12%) in the HD but not LD Hpx group (VC- 22.4±0.89 meters•min−1 vs. HD Hpx- 25.5±0.56 meters•min−1; p=0.042) demonstrating a dose response effect. Combining all Hpx dosed animals into a single group shows a stronger simple main effect between VC and Hpx treated mice (VC, 22.4±0.89 meters•min−1 vs. Hpx dosed 25.1±0.56 meters•min−1; p=0.01). Similarly, Figure 3D shows HD-Hpx moderately increased the glycolytic state of SCD mice referred to as the anaerobic work capacity (AWC; p=0.087 t-test). A significantly increased main effect became apparent when all Hpx treated animals were pooled into a single group (VC, 1487±115 VC vs. Hpx treated, 1081±128; p=0.04).
Figure 3: Hemopexin improves exercise tolerance in SCD mice.

(A) Hyperbolic curve fit for critical speed in Hpx treated and untreated SCD mice. (B) Anerobic work capacity demonstrates the liner relationship between exercise intensity vs. . (C) Dose and main effect of Hpx treatment on exercise tolerance as determined from the the quantitative value for critical speed plotted as bar graphs representing animals in each cohort. (D) Dose and main effects of Hpx treatment on the glycolytic state during exercise as quantitatified by the value or slope of the anerobic work capacity. The anerobic work capacity is plotted as a bar graphs representing animals for each cohort. (E) Packed red blood cell volume as measured by the hemotocrit and plotted as bar graphs representing each cohort. (F) High spleen weights associate with red blood hemolysis. Absence any difference in packed red cell volume and spleen weights suggests Hpx therapy is not altering red blood cell oxygen delivery. * p<0.05
The Hemopexin dosing effect on exercise tolerance is not driven by changes in anemia.
Improvement in exercise tolerance is a result of either an increase in oxygen delivery or a more efficient exchange of oxygen at the tissue/muscular level. We next evaluated the physiological parameters that contributed to the improved exercise tolerance. To assess the mechanism(s) by which Hpx improves critical speed we first evaluated the packed red cell volume and spleen weights of each animal to determine if Hpx improved anemia. The lack of any changes in either the hematocrit or spleen weights between treatment groups shown in Figure 3E, F demonstrates improvement in exercise tolerance was not a consequence of increased erythropoiesis, p > 0.05 between groups based on a multiple comparisons ANOVA. (36)
Moderate hypoxia accelerates pulmonary and right-ventricular disease progression in SCD mice.
After confirming that Hpx does not alter circulation RBC mass, we sought to determine its effects on pulmonary vascular and RV functions in our augmented disease model (SCD-HX), which takes advantage of the oxygenation-dependent sickling characteristics of HbS. To this end, we designed studies to verify that RV and pulmonary functional properties in SCD mice exposed to 3 months of moderate hypoxia (8,000 ft) are consistent with accelerated cardiopulmonary disease. The RV function was analyzed by solid state catheter measurements of pressure-volume loops with occlusion. This method is advantageous compared to simple closed chest techniques because it provides understanding of right ventricular (RV) contractility and stiffness. We compared: (1) sickle cell disease mice exposed to 3 months moderate hypoxia (SCD-HX), (2) sickle cell disease mice maintained 3 months at sea level (SCD-SL) conditions, and (3) wild type mice maintained at moderate hypoxia (WT-HX). Data shown in Figure 4 show pressure-volume (PV) loop analysis. Figure 4A are representations of PV tracings and how they differ in each cohort, and Figure 4B provides a schematic visualization of the changes in functional end points obtained from a PV loop. Quantitative assessment of right ventricular systolic pressures (RVSP), stiffness and elastance, which describes contractility is shown Figure 4C,D and E. Compared to SCD-SL and WT-HX, SCD-HX mice have greater RVSPs, ventricular stiffness, and contractility. However, from the PV loops we also observe in Figure 4F,G,and H that SCD-HX mice exhibit unique functional changes consistent with lower stroke volumes (SV), cardiac outputs (CO) and heart rates (HR) that accompany a stiff RV and compromised pre-load volumes. Further, SCD-HX mice have greater pulmonary vascular resistance which is described by the afterload (Figure 4I) and poor ventricular to vascular coupling ratio of < 1 (Figure 4J). The high afterload is a result of the reduced SV coupled to elevated (compared to sea level SCD mice) pulmonary arterial pressures as measured by RVSP (Afterload= Afterload=RVSP/SV) . The ventricular to pulmonary vascular coupling ratio is determined by contractility divided by the afterload. While not a clinical marker it is useful in animal models for describing the efficiency of energy transfer between ventricular contractility and arterial afterload. The coupling ratio is preserved across species with a ratio between 1 and 2 in healthy humans. (41) Uncoupling is a hallmark of cardiopulmonary vascular disease that is defined by right ventricular to pulmonary vascular ratio < 1. (41) Finally, these changes in RV and pulmonary function in SCD-HX mice are reflected in greater distal pulmonary arterial thickening, RV hypertrophy (as determined by the Fulton index (RV/LV+S), and reduced exercise performance (Figure 4K,L,M and N).
Figure 4: SCD mice housed for 3 months at moderate hypoxia (8,000 ft) demonstrate progressive pulmonary vascular disease.

(A) Representative tracings of pressure volume (PV) loops during an occlusion for wild type and SCD mice housed at either sea level or moderate hypoxia (8,000 ft) (B) shows the corresponding schematics of the pressure volume relationships for wild type and SCD mice for each cohort. (C-J) Right ventricular functional analysis in wild type and SCD mice showing right ventricular systolic pressure, stiffness, contractility, stroke volume, cardiac output, heart rate, afterload and right ventricle to pulmonary vascular coupling ratio. (K) Pulmonary vascular thickening (L) right ventricular hypertrophy. (M) Critical speed. (N) Anerobic work capacity. Data is represented as means ± standard error of measurement. RVSP- right ventricular pressures; Ea-Afterload; EDPVR-end diastolic pressure volume relationship; ESPVR- end systolic pressure volume relationship. *p<0.05 vs. SCD mice at 8,000 ft; † p < 0.05 vs SCD mice t-test
Collectively, these data are indicative of an accelerated progression of pulmonary arterial as well as RV dysfunction in SCD mice exposed to moderate hypoxia. The data suggests that this model may demonstrate optimized sensitivity and specificity for proof-of-concept therapeutic studies (Figure 4).
Hemopexin replacement improves right ventricular mechanical and functional properties.
We next sought to estimate the efficacy of chronic Hpx replacement therapy toward improving the RV and pulmonary vascular function in SCD-HX mice. In these studies, SCD-HX mice were administered tri weekly subcutaneous injections of either (saline- vehicle control; VC), low dose Hpx (100 mg/kg, LD-Hpx) or high dose Hpx (300 mg/kg, HD-Hpx). The data were analyzed per dose of and per main treatment effect with all Hpx treated animals merged in a single group.
The right ventricular PV relationships of each group are shown in Figure 5A-B. The schematics in Figure 5B below each PV loop tracing represent the changes that occur in RV stiffness, contractility, and afterload that lead to improved stroke work determined at the end of Hpx dosing. Figure 5C, D shows the quantified comparisons between VC, LD-Hpx and HD-Hpx SCD mice and Hpx dosing dependent effects on both improvement in RV stiffness and contractility. The insets in Figure 5C-D show the main treatment effects of Hpx (independent of dose) on stiffness (0.036±0.004 Hpx vs. 0.06±0.007 VC; p=0.008) and contractility (1.21±0.12 Hpx vs. 2.1±0.51; p=0.02).
Figure 5: Right ventricular functional analysis in hemopexin treated and untreated SCD mice experiencing progressive cardiopulmonary vascular disease.

(A) Representative tracings of pressure volume (PV) loops during an occlusion (B) shows the corresponding schematics of the pressure volume relationships for hemopexin treated and untreated SCD mice. (C-J) Right ventricular stiffness, contractility, stroke volume, cardiac output, heart rate, preload and ejection fraction. Data is represented as means ± standard error of measurement. Inset graphs show low and high dose Hpx treated mice combined into one group. RVSP- right ventricular pressures; Ea-Afterload; EDPVR-end diastolic pressure volume relationship; ESPVR- end systolic pressure volume relationship. *p<0.05, **p<0.005
Right ventricular functional parameters demonstrate Hpx dose dependent improvements in RV stiffness and contractility that translate to improved stroke volume (SV) and cardiac output with no change in heart rate (HR). We compared these parameters across cohorts (VC, LD-Hpx and HD-Hpx). Group comparisons show a 50% improvement in both SV (p=0.0047) and cardiac output (p=0.027) specific to the HD-Hpx cohort (Figure 5E,F) in the absence of either dose dependent or main effects on HR as shown in Figure 5G. These data demonstrate that while LD-Hpx administration was sufficient to suppress RV stiffness, this dose was not effective at improving either pre-load volume or ejection fraction to enhance cardiac output. Conversely, these data demonstrate the effectiveness of HD-Hpx at attenuating diastolic stiffness, while improving pre-load volume (26%; p=0.06), ejection fraction (~30%, p=0.026) and ultimately cardiac output.
Interestingly analysis of Hpx dosing on pulmonary vasculature responses (Figure 6A) shows that neither LD-Hpx nor HD-Hpx changed RVSP, but in contrast to this observation HD-Hpx reduced RV afterload ~ 25% (HD-Hpx- 1.1±0.08 mm Hg•ul−1 vs. VC 1.52±0.10 mmHg•ul−1; p=0.02, Figure 6B). Thus, we observed a reduction in afterload without a corresponding decrease in PA pressures. This data suggest that Hpx dosing improves the reactive afterload (a reflection of the improved SV) that is driven by pulmonary vascular and RV stiffness, but not resistive afterload that is defined by increase pulmonary pressures and reflected in RVSP.
Figure 6: Pulmonary vascular analysis in hemopexin treated and untreated SCD mice experiencing progressive cardiopulmonary vascular disease.

(A) Right ventricular systolic pressures (B) Right ventricular afterload (C) Ventricular to vascular coupling ration (VVCR); (D) Pulmonary vascular thickening (E) Right ventricular weight (F)Fulton index a determinant of RV hypertrophy*p<0.05, †p<0.05 by t test.
A reduction in both right ventricular contractility and afterload are observed following repeated HD-Hpx administrations. The Hpx dose dependent increase in RV to PV coupling is shown in Figure 6C, indicating therapeutic improvements toward a normalized ratio (0.92±0.12 HD-Hpx vs.0.58 ±0.06 VC p=0.02;).
Collectively, we established RV mechanical parameters in response to a three-month Hpx treatment of SCD mice by solid state catheter measurements. These parameters defined a reduction in stiffness and contractility as the primary therapeutic effects. The RV functional properties that changed in response to Hpx dosing are subsequently defined by an increased stroke volume and cardiac output, reduced afterload and an increased RV to PV ratio. These changes are consistent with a therapeutic activity of Hpx improving critical speed as a hard proof of concept endpoint.
Hemopexin effects on pulmonary arterial wall thickening and right ventricular hypertrophy.
Pulmonary arterial wall thickening reflects an advanced disease state that is more pronounced in SCD-HX mice than in sea level housed SCD cohorts. (36) The significance of this pathophysiology is impaired blood flow and increased myocardial workload in hypoxia exposed SCD mice. Therefore, we evaluated the ability of Hpx dosing to attenuate vascular wall thickness in lung tissue sections of SCD-HX mice. The lung tissue from six animals per cohort were randomly selected and stained with hematoxylin and eosin to visually and quantitatively assess pulmonary vascular remodeling in response to Hpx dosing (determined by the ratio between vascular and parenchyma tissue). This analysis demonstrate a Hpx dose dependent attenuation of pulmonary vascular remodeling, shown in Figure 6D. HD-Hpx, but not LD-Hpx reduced pulmonary vascular wall thickening by 30% (p=0.037 t test; 0.10 ANOVA) when compared to vehicle control (VC).
Increased right ventricular stiffness, afterload, and uncoupling from the pulmonary artery occurs in ventricles with pathological hypertrophy. Although Hpx therapy improved RV function, reduced afterload, and a normalized the coupling ratio, these improvements did not translate into a reduction in RV hypertrophy. Our data shows neither a Hpx dose nor main effect in either RV weights or the Fulton index in SCD mice administered Hpx when compared with VC cohorts as shown in Figure 6E, F. This is an important finding suggesting that ventricular function improved without full regression of cellular hypertrophy.
Hemopexin replacement attenuates heme-driven oxidative protein modification in the lungs and right ventricle.
Having established that Hpx dosing 3 times per week is effective at improving exercise tolerance, RV and pulmonary vascular function in the SCD-HX model, we sought to determine the mechanistic underpinnings of the adverse effect of heme related to oxidative stress and fibrosis as observed in autopsy tissue of SCD patients with PH. To this end, we stained lung tissue and RV sections from vehicle control and Hpx treated SCD-HX mice for markers of iron accumulation oxidative stress (4-HNE) and collagen (Masson's Trichrome) to identify pulmonary and RV vascular fibrosis. Perls-DAB iron staining in the lungs is shown in Figure 7A and shows that SCD-HX mice have iron loaded macrophages distributed throughout the adventitia and perivascular regions of the lung, demonstrating similarities with human pathology. Similar to the RV, lung histology shows robust staining for 4-HNE and collagen in the SCD-HX mice administered saline, which was reduced or nearly absent in the Hpx dosed mice (Figure 7A). Congruent with human autopsy tissue, we did not observe iron in the RV tissue of SCD-HX mice (Figure 7B). In contrast, the RV of SCD-HX mice treated with saline show 4-HNE-generation, indicating oxidative stress (Figure 7B, panel 2). Visualization of Masons Trichrome in RV tissue shows diffuse collagen deposition surrounding coronary arteries and interspersed between myocardial cells in SCD-HX, but was reduced with both doses of Hpx treatment (Figure 7B, panel 3)
Figure 7: Microscopic examination of lung and right ventricle tissues in hemopexin treated and untreated SCD mice.

(A) Histopathology of lung tissue . Top row- Perls iron staining; Middle rows show 4HNE staining for lipid peroxidation; Bottom row- Trichrome staining as a marker for fibrosis; (B) Histopathology of right ventricle tissue Top row- Perls iron staining; Middle rows show 4HNE staining for lipid peroxidation (C) Confocal microscopy images of showing the distribution of hemoglobin and hemopexin extravasation into cardiac tissue 30 min after administration.
Overall, these data suggest that Hpx treatment protects against heme-driven oxidative tissue injury and fibrosis in the peripheral lung vasculature and fibrosis and right ventricle stiffening.
Hemopexin and cell-free hemoglobin extravasation in the heart.
Up to this point, the functional and molecular data provides strong evidence that Hpx attenuates cardiopulmonary disease by preventing oxidative stress and fibrosis in the peripheral lung vasculature and oxidation in the right ventricle. We have previously demonstrated that extravasation of cell-free Hb is a prerequisite for its NO and redox reactions that contribute toward tissue injury. (23) Therefore, we hypothesized that Hpx also extravasates into the same regions with Hb within the cardiac interstitium to neutralize heme and its down-stream effects. Supporting our hypothesis, we observed strong and congruent fluorescent signals in the interstitial spaces around cardiomyocytes and in muscular coronary arteries after i.v. infusion of either cyanine-5 labeled Hb or Hpx in SCD mice, shown in Figure 7C. Taken together with our functional and molecular data this provides evidence that Hpx can directly target adverse oxidative effects of cell-free Hb in exposed tissues.
DISCUSSION
SCD patients suffering from progressive malignant cardiopulmonary dysfunction demonstrate a unique pathology signature that can be characterized at autopsy. (22) Human SCD-PH patients’ lung and heart tissue examined as part of this study demonstrate PA neointimal proliferation, elastin fragmentation and fibrosis as well as peripheral pulmonary arterial remodeling, iron positive adventitial macrophage accumulation, medial and intimal oxidative stress, and fibrosis as notable characteristics. Further, apo-Hpx added to plasma samples collected from SCD patients after exchange transfusion rapidly converted to holo-Hpx. Lipid peroxidation was also increased in these samples suggesting a state of heme stress exacerbated by depleted Hpx. Based on these observations we hypothesized that dosing of plasma derived Hpx (3 doses/week x 12 weeks) to SCD mice with progressive cardiopulmonary dysfunction could attenuate pathophysiology and improve exercise tolerance as a rigorous measure of pre-clinical proof-of-concept.
As a therapeutic endpoint, treadmill speed-duration relationships allow for a unique evaluation of progressive cardiopulmonary dysfunction and the effect of interventions in SCD mice. (36, 42) Our study suggests a 40-50% improvement in stroke volume and cardiac output with Hpx administration and a 12% increase in critical speed, demonstrating that changes in stroke volume / cardiac output and exercise tolerance are not linear. Ultimately, exercise tolerance in health and in SCD is regulated by oxygen delivery to skeletal muscle, which can be adapted centrally (at the heart), peripherally (local control of blood flow), or by a combination of the two. (44) The improvements in our critical speed endpoint is supported by reductions in right ventricular stiffness, afterload, pulmonary vascular wall thickening, and normalized right ventricle to pulmonary artery coupling. The right ventricular mechanical and functional parameters were associated with a Hpx dose dependent attenuation of pulmonary vascular remodeling highlighting a mechanistic sequence underpinning the cardiopulmonary pathophysiology of heme excess. Moreover, the right ventricular phenotype described herein shares strong similarities to a unique left ventricular cardiomyopathy previously described in human SCD patients and mice, highlighting the translational potential of the present findings. (33, 45) These similarities include a restrictive physiology superimposed on a hyperdynamic physiology that is characterized by diastolic dysfunction and preserved systolic function. In both cases, the diastolic ventricular dysfunction is associated with oxidative stress and hypertrophy. This is supported by the overall effectiveness of Hpx to normalize stiffness end diastolic pulmonary vascular resistance values in the right ventricle, while simultaneously reducing the histopathological markers for oxidative stress. In our murine model we cannot suggest that right ventricle hypertrophy is associated with a fibrotic response, which is occasionally observed in patients with PH. (46)
In progressive SCD PH there is a dramatic pulmonary vascular remodeling and the right ventricle is faced with a high pressure load. (47) In early phases, right ventricle contractility increases in proportion to the afterload.(41) This response maintains an efficient energy transfer between the ventricle and pulmonary vasculature, which can be measured by the coupling ratio defined as contractility divided by afterload. Right ventricle to pulmonary vascular coupling ratio between 1 and 2 is considered normal and is conserved across species. (41) In progressive pulmonary hypertension, preserved contractility reaches an upper limit, while afterload continues to rise, reducing the coupling ratio to <1. (41) While there is no clinical standard for coupling ratio to be assigned to severity of pulmonary hypertension, a ratio below 0.8 is considered the cutoff for defining uncoupling between the right ventricle and pulmonary artery, and a sign of an inefficient heart. (41) Finally, in the last stage of pulmonary hypertension high contractility can no longer be preserved as the right ventricle fails, resulting in a drastic decline in the coupling ratio.(41) Our data demonstrate that despite having a preserved right ventricle systolic function over the study time frame, SCD mice administered vehicle control remained in a state of progressive pulmonary hypertension compared to Hpx dosed cohorts. This is evidence from the elevated contractility and low coupling ratio of <0.8. In contrast, HD-Hpx reduced right ventricle contractility normalizing the coupling ratio to ~ 1. Interestingly, while correcting right ventricle diastolic dysfunction LD-Hpx administration was not sufficient to decrease the afterload and improve the energy transfer between the right ventricle and pulmonary artery, as a result the coupling ratio remained below optimal.
Hpx dosing was effective at attenuating right ventricle and pulmonary oxidation (determined by 4-HNE accumulation) and pulmonary arterial fibrogenesis (determined by Masson’s Trichrome staining). It is well recognized that pulmonary vascular fibrosis contributes to the development of PH. (48, 49) Previous studies have demonstrated that mild hypoxia in SCD mice increases markers of hemolysis and heme partitioning, enhancing tissue oxidative stress. (24, 36) This is congruent with a mechanistic sequence of hemolysis-accelerated cardiopulmonary disease, which includes release of cell-free Hb from RBCs, dissociation of Hb into small Hb-dimers, delocalization of dimers across vascular barriers into interstitial tissue spaces, where heme partitioning into lipid-rich compartments initiates oxidative injury. This process can be blocked when Hb is sequestered in a large Hb-haptoglobin complex. (23) In contrast to haptoglobin, Hpx does not form macromolecular complexes with cell-free Hb and can, therefore, not reduce tissue Hb exposures. However, our labeling studies suggested that Hpx, which is considerably smaller than haptoglobin or Hb-haptoglobin complexes, can also extravasate and distribute into the same tissue compartments as cell-free Hb. There, Hpx is perfectly positioned to locally suppress heme-facilitated lipid peroxidation. Accordingly, we found in our study consistent patterns of lipid peroxidation in the RV and lung of control SCD mice, which were suppressed by Hpx replacement therapy.
EXPERIMENTAL CONSIDERATIONS
This is the first preclinical study that evaluates chronic Hpx replacement as a treatment to attenuate cardiopulmonary complications of SCD. Our data provide compelling proof-of-concept that the restoration of Hpx plasma levels protects pulmonary and right ventricular tissue from heme-mediated oxidant stress and pulmonary fibrogenesis. Our study shows that Hpx replacement attenuates the progressive decline of specific cardiopulmonary functions resulting in improved exercise performance assessed by a rigorous exercise tolerance endpoint. A limitation of this study is that absolute dose dependent effects are not possible to discern across all parameters. For example, despite evidence of a positive Hpx effect on cardiac parameters, there are a few Hpx dose dependent discrepancies that warrant greater discussion. For example Hpx at 100 mg/kg suppressed pulmonary and RV 4HNE and collagen formation, improved RV stiffness and contractility, and improved exercise tolerance. However, improvement in pulmonary vascular remodeling was not observed. This data suggests that dosing with 100 mg/kg Hpx would further improve the overall pathophysiology and endpoint parameters with dosing beyond three months. Nonetheless, the effect of replenishment of depleted Hpx in SCD PH demonstrates a strong dosing effect in this animal model. This concept warrants further dose finding and efficacy studies in the SCD PH population. Moreover, SCD is typically not associated with the cardiac iron overload that would be present in thalassemia, particularly in patients requiring transfusion. Our human autopsy samples did not show iron accumulation in RV tissue but did show distinct lipid peroxidation. This data was consistent in SCD mice subjected to 3 months of hypobaric hypoxia and Hpx was able to attenuate RV lipid oxidation. We suggest that this is an off target effect of Hpx that may be tied to circulating lipid oxidation and distribution to RV tissue. We specifically observed this effect in human SCD PH patients (24). This data suggests a unique therapeutic potential of Hpx-based therapeutics for improving quality of life in newly diagnosed PH in SCD patients. Like many other proof-of-principle cardiopulmonary studies testing therapeutic effects before clinical evaluation, our experimental design started treatment at the time of stress instead of determining if the therapeutic would reverse the disease process after it began. Therefore, optimal timing of Hpx replacement therapy is warranted in human studies.
CONCLUSIONS
In summary, we show that Hpx replacement therapy in a murine model of hypoxia accelerated SCD improved exercise tolerance, right ventricular function, and pulmonary vascular remodeling that were caused by heme mediated oxidant stress. These data support our hypothesis that depletion of the heme scavenger protein Hpx allows for excess heme to be a primary driver of cardiopulmonary dysfunction in SCD and provides compelling evidence that Hpx therapeutics warrant further evaluation.
ACKNOWLEDGMENTS:
The present study was supported by the National Heart, Lung and Blood Institute Grants 1R01HL125642-01 (Irwin DC), Colorado Sickle Cell Treatment and Research Center (Hassell K and Nuss R).
REFERENCES
- 1.McCavit TL. Sickle cell disease. Pediatr Rev. 2012;33(5):195–204; quiz 5-6. [DOI] [PubMed] [Google Scholar]
- 2.Simpson S Sickle cell disease: a new era. Lancet Haematol. 2019;6(8):e393–e4. [DOI] [PubMed] [Google Scholar]
- 3.Quinn CT. Sickle cell disease in childhood: from newborn screening through transition to adult medical care. Pediatr Clin North Am. 2013;60(6):1363–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.E MJ, and T TA. Sickle Cell Anemia. Medscape. 2020. [Google Scholar]
- 5.Fushimi N, Sawada H, Udaka F, and Kameyama M. [Five-year follow-up studies of laboratory data in the elderly group]. Nihon Ronen Igakkai Zasshi. 1989;26(2):179–80. [DOI] [PubMed] [Google Scholar]
- 6.Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med. 2018;379(3):226–35. [DOI] [PubMed] [Google Scholar]
- 7.Salinas Cisneros G, and Thein SL. Recent Advances in the Treatment of Sickle Cell Disease. Front Physiol. 2020;11:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med. 2019;381(6):509–19. [DOI] [PubMed] [Google Scholar]
- 9.Liem RI, Reddy M, Pelligra SA, Savant AP, Fernhall B, Rodeghier M, et al. Reduced fitness and abnormal cardiopulmonary responses to maximal exercise testing in children and young adults with sickle cell anemia. Physiol Rep. 2015;3(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gladwin MT, and Kato GJ. Cardiopulmonary Complications of Sickle Cell Disease: Role of Nitric Oxide and Hemolytic Anemia. Hematology. 2005;2005(1):51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary Hypertension as a Risk Factor for Death in Patients with Sickle Cell Disease. New England Journal of Medicine. 2004;350(9):886–95. [DOI] [PubMed] [Google Scholar]
- 13.Gordeuk VR, Castro OL, and Machado RF. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood. 2016;127(7):820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaer DJ, Vinchi F, Ingoglia G, Tolosano E, and Buehler PW. Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front Physiol. 2014;5:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Telen MJ, Malik P, and Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discov. 2019;18(2):139–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rother RP, Bell L, Hillmen P, and Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. Jama. 2005;293(13):1653–62. [DOI] [PubMed] [Google Scholar]
- 17.Tuder RM, Stacher E, Robinson J, Kumar R, and Graham BB. Pathology of pulmonary hypertension. Clin Chest Med. 2013;34(4):639–50. [DOI] [PubMed] [Google Scholar]
- 18.Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res. 2012;110(5):739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, and Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014;123(24):3818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gladwin MT, and Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol. 2012;59(13):1123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redinus K, Baek JH, Yalamanoglu A, Shin HKH, Moldova R, Harral JW, et al. An Hb-mediated circulating macrophage contributing to pulmonary vascular remodeling in sickle cell disease. JCI Insight. 2019;4(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaer CA, Deuel JW, Schildknecht D, Mahmoudi L, Garcia-Rubio I, Owczarek C, et al. Haptoglobin Preserves Vascular Nitric Oxide Signaling during Hemolysis. Am J Respir Crit Care Med. 2016;193(10):1111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yalamanoglu A, Deuel JW, Hunt RC, Baek JH, Hassell K, Redinius K, et al. Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease. Am J Physiol Lung Cell Mol Physiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. 2013;123(11):4809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klings ES, Machado RF, Barst RJ, Morris CR, Mubarak KK, Gordeuk VR, et al. An official American Thoracic Society clinical practice guideline: diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med. 2014;189(6):727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller-Eberhard U, Javid J, Liem HH, Hanstein A, and Hanna M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood. 1968;32(5):811–5. [PubMed] [Google Scholar]
- 28.Ascenzi P, di Masi A, Fanali G, and Fasano M. Heme-based catalytic properties of human serum albumin. Cell Death Discov. 2015;1:15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunn HF, and Jandl JH. Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem. 1968;243(3):465–75. [PubMed] [Google Scholar]
- 30.Deuel JW, Vallelian F, Schaer CA, Puglia M, Buehler PW, and Schaer DJ. Different target specificities of haptoglobin and hemopexin define a sequential protection system against vascular hemoglobin toxicity. Free Radic Biol Med. 2015;89:931–43. [DOI] [PubMed] [Google Scholar]
- 31.Miller YI, and Shaklai N. Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim Biophys Acta. 1999;1454(2):153–64. [DOI] [PubMed] [Google Scholar]
- 32.Pimstone NR. Renal degradation of hemoglobin. Semin Hematol. 1972;9(1):31–42. [PubMed] [Google Scholar]
- 33.Bakeer N, James J, Roy S, Wansapura J, Shanmukhappa SK, Lorenz JN, et al. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci U S A. 2016;113(35):E5182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buehler PW, Baek JH, Lisk C, Connor I, Sullivan S, Kominsky D, et al. Free Hemolgobin Induction of Pulmonary Vascular Disease: Evidence for and inflammatory Mechanism. Am J Physiol Lung Cell Mol Physiol. 2012;303(4):312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irwin DC, Hyen Baek J, Hassell K, Nuss R, Eigenberger P, Lisk C, et al. Hemoglobin-induced lung vascular oxidation, inflammation, and remodeling contribute to the progression of hypoxic pulmonary hypertension and is attenuated in rats with repeated-dose haptoglobin administration. Free Radic Biol Med. 2015;82:50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferguson SK, Redinius K, Yalamanoglu A, Harral JW, Hyen Baek J, Pak D, et al. Effects of living at moderate altitude on pulmonary vascular function and exercise capacity in mice with sickle cell anaemia. J Physiol. 2019;597(4):1073–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Principles Grundy D. and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol. 2015;593(12):2547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poole DC, Copp SW, Colburn TD, Craig JC, Allen DL, Sturek M, et al. Guidelines for animal exercise and training protocols for cardiovascular studies. Am J Physiol Heart Circ Physiol. 2020;318(5):H1100–H38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Copp SW, Hirai DM, Musch TI, and Poole DC. Critical speed in the rat: implications for hindlimb muscle blood flow distribution and fibre recruitment. J Physiol. 2010;588(Pt 24):5077–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(3):261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tello K, Dalmer A, Axmann J, Vanderpool R, Ghofrani HA, Naeije R, et al. Reserve of Right Ventricular-Arterial Coupling in the Setting of Chronic Overload. Circ Heart Fail. 2019;12(1):e005512. [DOI] [PubMed] [Google Scholar]
- 42.Ferguson SK, Redinius KM, Harral JW, Pak DI, Swindle DC, Hirai DM, et al. The effect of dietary nitrate supplementation on the speed-duration relationship in mice with sickle cell disease. J Appl Physiol (1985). 2020;129(3):474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poole DC, Burnley M, Vanhatalo A, Rossiter HB, and Jones AM. Critical Power: An Important Fatigue Threshold in Exercise Physiology. Med Sci Sports Exerc. 2016;48(11):2320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirai DM, Copp SW, Ferguson SK, Holdsworth CT, McCullough DJ, Behnke BJ, et al. Exercise training and muscle microvascular oxygenation: functional role of nitric oxide. J Appl Physiol (1985). 2012;113(4):557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niss O, Quinn CT, Lane A, Daily J, Khoury PR, Bakeer N, et al. Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease. JACC Cardiovasc Imaging. 2016;9(3):243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersen S, Nielsen-Kudsk JE, Vonk Noordegraaf A, and de Man FS. Right Ventricular Fibrosis. Circulation. 2019;139(2):269–85. [DOI] [PubMed] [Google Scholar]
- 47.Thenappan T, Ormiston ML, Ryan JJ, and Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egemnazarov B, Crnkovic S, Nagy BM, Olschewski H, and Kwapiszewska G. Right ventricular fibrosis and dysfunction: Actual concepts and common misconceptions. Matrix Biol. 2018;68-69:507–21. [DOI] [PubMed] [Google Scholar]
- 49.Samokhin AO, Stephens T, Wertheim BM, Wang RS, Vargas SO, Yung LM, et al. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci Transl Med. 2018;10(445). [DOI] [PMC free article] [PubMed] [Google Scholar]