

Abstract
Reporter ion interference remains a limitation of isobaric tag-based sample multiplexing. Advances in instrumentation and data acquisition modes, such as the recently developed real-time database search (RTS), can reduce interference. However, interference persists as does the need to benchmark upstream sample preparation and data acquisition strategies. Here, we present an updated Triple yeast KnockOut (TKO) standard as well as corresponding upgrades to the TKO viewing tool (TVT2.5, http://tko.hms.harvard.edu/). Specifically, we expand the TKO standard to incorporate the TMTpro18-plex reagents (TKO18). We also construct a variant thereof which has been digested only with LysC (TKO18L). We compare proteome coverage and interference levels of TKO18 and TKO18L data that are acquired under different data acquisition modes and analyzed using TVT2.5. Our data illustrate that RTS reduces interference while improving proteome coverage and suggest that digesting with LysC alone only modestly reduces interference, albeit at the expense of proteome depth. Collectively, the two new TKO standards coupled with the updated TVT represent a convenient and versatile platform for assessing and developing methods to reduce interference in isobaric tag-based experiments.
Keywords: Eclipse, FAIMS, IFI, RTS, SPS-MS3, TMTpro18
1 |. INTRODUCTION
Isobaric labeling continues to be a widely used and well-accepted strategy for proteome-wide mass spectrometry-based quantitative proteome profiling. Sample multiplexing facilitates the simultaneous interrogation of multiple proteomes (e.g., cell lines, perturbations, time courses, and dosage responses), while improving statistical confidence, reducing missing values lost to sampling stochasticity, and increasing throughput [1, 2]. However, reporter ion “interference” – a consequence of the co-isolation, co-fragmentation, and co-analysis of multiple precursor ions – remains a major caveat of sample multiplexing [3].
We have developed an isobaric tag-based analytical standard, named the Triple yeast KnockOut (TKO) standard, that assesses interference and tracks overall instrument performance [4, 5]. The underlying concept behind this standard is simple. Specifically, a tandem mass tag (TMT) experiment is arranged using multiple replicates of three different Saccharomyces cerevisiae deletions strains – typically Δmet6, Δpfk2, and Δura2 – and often a wildtype strain. It follows that in the ideal “no interference” scenario, the TMT reporter ion signal should be zero for the protein which is absent in a given deletion strain. However, reporter ion signal that is measured in channels where the protein is absent implies a specific degree of interference, which we quantify using the interference-free index (IFI). The IFI is calculated as the difference from 1 of the ratio of the average TMT signal-to-noise of channels deficient in a specific TKO protein to that of channels in which that protein is not knocked out. Using this metric, values closer to 1 approach no interference, while values closer to 0 are indicative of a very high degree of interference.
We have described previously several variations of the TKO standard. These TKO standard variants include the original TKO9 [5] that was later expanded to incorporate two wildtype channels thereby establishing the TKO11 standard (which was commercialized as the “Pierce Protein Interference Standard”) [4, 6]. In addition, the TKO6 standard is unit-resolved and may be used to assess isobaric tag quantification in low resolution ion traps [7]. More recently, we have used the TMTpro16-plex reagents [8] to assemble the TKOpro16, as well as the unit-resolved TKOpro9 [9]. Here, we have leveraged the recent availability of the TMTpro18-plex reagents [10]. We note previously that the isobaric TMTpro16-plex molecule incorporate seven-13C and two-15N heavy isotopes and the two additional reagents (17 and 18) had eight-13C and only one-15N heavy isotopes, amounting to a 6 mDa difference that has no effect on peptide identification or quantification [10]. With these two additional reagents, we construct the TKO18 standard using the three aforementioned deletion strains, in quintuplicate, and the wildtype strain in triplicate. We prepare the TKO18 standard following the SL-TMT workflow [11] in which proteins are digested by sequential treatment with LysC and trypsin. However, we postulate that if we digest only with LysC, the reduced peptide population could potentially dampen interference. And so, we construct a variant standard (named “TKO18L”) which is assembled like TKO18 except that trypsin is omitted from the digestion workflow, that is, the sample is digested only with LysC.
Finally, we update the TKO viewing tool (TVT) [4], now TVT2.5, to facilitate the analysis of the TKO18 and TKO18L standards. The TVT is a freely accessable, web-based application designed to streamline the analysis of the TKO standard by performing an automated database search of this standard with the Comet search engine [12]. The TVT displays tables and graphics illustrating traditional figures of merit, such as protein and peptide counts, in addition to various ion statistics, and can track and compare these metrics across multiple TKO analyses. We showcase the TKO18 and TKO18L standards in two applications using TVT2.5 for data analysis. First, we determined the interference and protein depth of three common data acquisition strategies for isobaric tag-based experiments and compare the performance of both standards. Next, we use these standards to evaluate the turboTMT option, which is based on the phased spectrum deconvolution method (Φ-SDM) algorithm [13] – a computational approach for increasing resolution – for RTS-MS3 analysis. Together, the TKO18 and TKO18L standards coupled with TVT2.5 offer an integrated platform for the assessment of interference in isobaric tag experiments.
2 |. METHODS
2.1 |. Materials
TMT isobaric reagents (TMTpro) were from ThermoFisher Scientific (Waltham, MA). Trypsin was purchased from Pierce Biotechnology (Rockford, IL) and LysC from FujiFilm (Richmond, VA).
2.2 |. Yeast strains
S. cerevisiae strains from the haploid MATalpha collection (BY4742 MATα: leu2Δ0 lys2Δ0 ura3Δ0) were purchased from Open Biosystems. Cultures were grown in standard yeast-peptone-dextrose (YPD) media to an optical density (OD) of 0.8 mL−1 and then harvested, as described previously [14]. The knockout strains selected (Δmet6, Δpfk2, Δura2) fulfilled three criteria, specifically, the deleted protein: (1) had no redundant tryptic peptides in the S. cerevisiae proteome, (2) was non-essential for yeast growth, and (3) was abundant such that tryptic peptides were observed with short (~45 min) data acquisition methods.
2.3 |. Cell lysis and protein digestion
Yeast cultures were harvested by centrifugation, washed twice with ice cold deionized water, and resuspended in lysis buffer: 50 mM EPPS pH 8.5, 8 M urea, and protease (complete mini, EDTA-free) inhibitors (Roche, Basel, Switzerland). Cells were lysed using the MiniBeadbeater (Biospec, Bartlesville, OK) in microcentrifuge tubes at a maximum speed for three cycles of 60 s each, with 1 min pauses on ice between cycles to avoid overheating of the lysates. After centrifugation, cleared lysates were transferred to new tubes. We determined the protein concentration in the lysate using the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Waltham, MA).
Proteins were subjected to disulfide reduction with 5 mM tris(2-carboxyethyl)phosphine (TCEP) at room temperature for 30 min followed by alkylation with 10 mM iodoacetamide at room temperature for 30 min in the dark. Excess iodoacetamide was quenched with 10 mM dithiotreitol at room temperature for 15 min in the dark. Methanol–chloroform precipitation was performed prior to protease digestion. In brief, four parts neat methanol was added to each sample and vortexed, then one-part chloroform was added to the sample and vortexed, and three parts water was added to the sample and vortexed. The sample was centrifuged at 12,000 rpm for 2 min at room temperature and, after removing both the aqueous and organic phases, subsequently washed once with 100% methanol. Samples were not dried, rather, all but ~10 μL of methanol was aspirated to allow for better solubilization of the precipitated proteins. Samples were resuspended in 200 mM EPPS, pH 8.5, and digested at room temperature for 16 h with LysC protease at a 100:1 protein-to-protease ratio. For the TKO18 standard (but not TKO18L standard), trypsin was then added at a 100:1 protein-to-protease ratio and the reaction was incubated 6 h at 37°C.
2.4 |. Tandem mass tag labeling
TMTpro reagents (5 mg) were dissolved in anhydrous acetonitrile (400 μL) and of this, 12 μL were added to the peptides (80 μg) along with 30 μL of acetonitrile for a final acetonitrile concentration of approximately 30% v/v. For both standards, peptides from the Δmet6 strain replicates were labeled with TMT reagents 126, 127N, 127C, 128N, and 128C; the Δpfk2 strain replicates with 129N, 129C, 130N, 130C, and 131N; the Δura2 strain replicates with 131C, 132N, 132C, 133N, and 133C; and the wildtype replicates were labeled with 134N, 134C, and 135. Following incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.3% v/v. The TMTpro-labeled samples were pooled at a 1:1 ratio across all channels. The sample was vacuum centrifuged to near dryness and subjected to C18 solid-phase extraction (SPE) (Sep-Pak, Waters).
2.5 |. Liquid chromatography and tandem mass spectrometry
Mass spectrometric data were collected on an Orbitrap Eclipse mass spectrometer with the FAIMS Pro interface (ThermoFisher Scientific, San Jose, CA) coupled to a Proxeon EASY-nLC 1200 liquid chromatograph (LC) (ThermoFisher Scientific, San Jose, CA). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ~35 cm of Accucore150 resin (2.6 μm, 150 Å, ThermoFisher Scientific, San Jose, CA). For each analysis, we loaded 0.5–1 μg of peptide onto the column and fractionated over a 45 min gradient of 7–27% acetonitrile in 0.125% formic acid at a flow rate of ~600 nL/min.
Mass spectrometric data were collected using three distinct data acquisition modes (hrMS2, SPS-MS3, and RTS-MS3), all with FAIMS. For the high-resolution MS2 (hrMS2) method, the scan sequence began with an MS1 spectrum (Orbitrap analysis; resolution, 60,000; mass range, 400−1600 Th; automatic gain control [AGC] target 100%; maximum injection time, auto). All data were acquired with FAIMS using three CVs (−40, −60, and −80 V) each with a 1 s TopSpeed method. MS2 analysis consisted of high energy collision-induced dissociation (HCD) with the following settings: resolution, 50,000; AGC target, 250%; isolation width, 0.5 Th; normalized collision energy (NCE), 37; and maximum injection time, 86 ms. For the SPS-MS3 and RTS-MS3 [15] methods, the scan sequence began with an MS1 spectrum which was collected as in the hrMS2 method. Precursors were then selected for MS2/MS3 analysis [3]. MS2 analysis consisted of collision-induced dissociation (CID) with quadrupole ion trap analysis, using the follow ing parameters: scan speed, turbo; AGC target, 100%; NCE, 35; q-value, 0.25; maximum injection time, 35 ms; and isolation window, 0.5 Th. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap with the following parameters: resolution, 50,000; NCE, 55; AGC, 250%; maximum injection time, 86 ms; maximum synchronous precursor selection (SPS) ions, 10; and isolation window, 1.2 Th. For specified methods, the turboTMTpro option was selected, the resolution was set to 30,000, and the maximum ion injection time was set to 54 or 86 ms (as indicated). The RTS node was selected from within the Thermo Method Editor software (Tune 3.4). RTS-MS3 data were collected with the “close-out” parameter set to zero (“off”).
2.6 |. Data analysis
Mass spectra were processed using a Comet-based software pipeline [12, 16]. Database searching included all entries from SGD (Saccharomyces Genome Database) (March 20, 2021). This database was concatenated with one composed of all protein sequences in the reversed order. Searches were performed using a 50-ppm precursor ion tolerance and the product ion tolerance was set to 0.9 Da for SPS-MS3 and RTS-MS3 data, but this setting was 0.03 Da for hrMS2 data. Enzyme specificity was assigned as LysC and trypsin for TKO18 and to only LysC for TKO18L. We selected these wide mass tolerance windows to maximize sensitivity for the Comet searches and subsequent linear discriminant analysis [17, 18]. TMTpro labels on lysine residues and peptide N termini (+304.207) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) [19, 20]. PSM filtering was performed using a linear discriminant analysis, as described previously [18], while considering the following parameters: XCorr, peptide length, ΔCn, charge state, missed cleavages, and mass accuracy of the precursor. For TMT-based reporter ion quantitation, we extracted the signal-to-noise (S:N) ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. PSMs were identified, quantified, and collapsed to a 1% peptide FDR and then collapsed further to a final protein-level FDR of 1%. Peptide intensities were quantified by summing reporter ion counts across all matching PSMs so as to give greater weight to more intense ions [21, 22]. We required an isolation specificity ≥0.8 (i.e., peptide purity >80%) [22]. In addition, PSMs of poor quality, spectra with TMT reporter ion summed signal-to-noise measurements that were less than 200, or with no MS3 spectra (for MS3-based methods) were excluded from quantification.
2.7 |. Data access
The data have been deposited to the ProteomeXchange Consortium via the PRIDE [23] partner repository with the dataset identifier PXD029458.
3 |. RESULTS AND DISCUSSION
3.1 |. TKO18 and TKO18L interference standards were assembled using TMTpro18-plex reagents
Two TKO18 standards were designed, one of peptides resulting from a standard sequential digestion with LysC and trypsin (TKO18) and a second of peptides originating from proteins digested only with LysC (TKO18L) (Figure 1A). The deletion strains selected for these standards were Δmet6, Δpfk2, and Δura2, as was the case for several ear lier iterations of the TKO standard [5]. Samples were processed using the SL-TMT protocol [11] without fractionation. For both standards, the samples were labeled in a 5 × 5 × 5 × 3 format (i.e., five replicates each of the Δmet6, Δpfk2, and Δura2 strains, and three replicates of wild-type yeast) using TMTpro reagents to complete the 18-plex layout.
FIGURE 1.
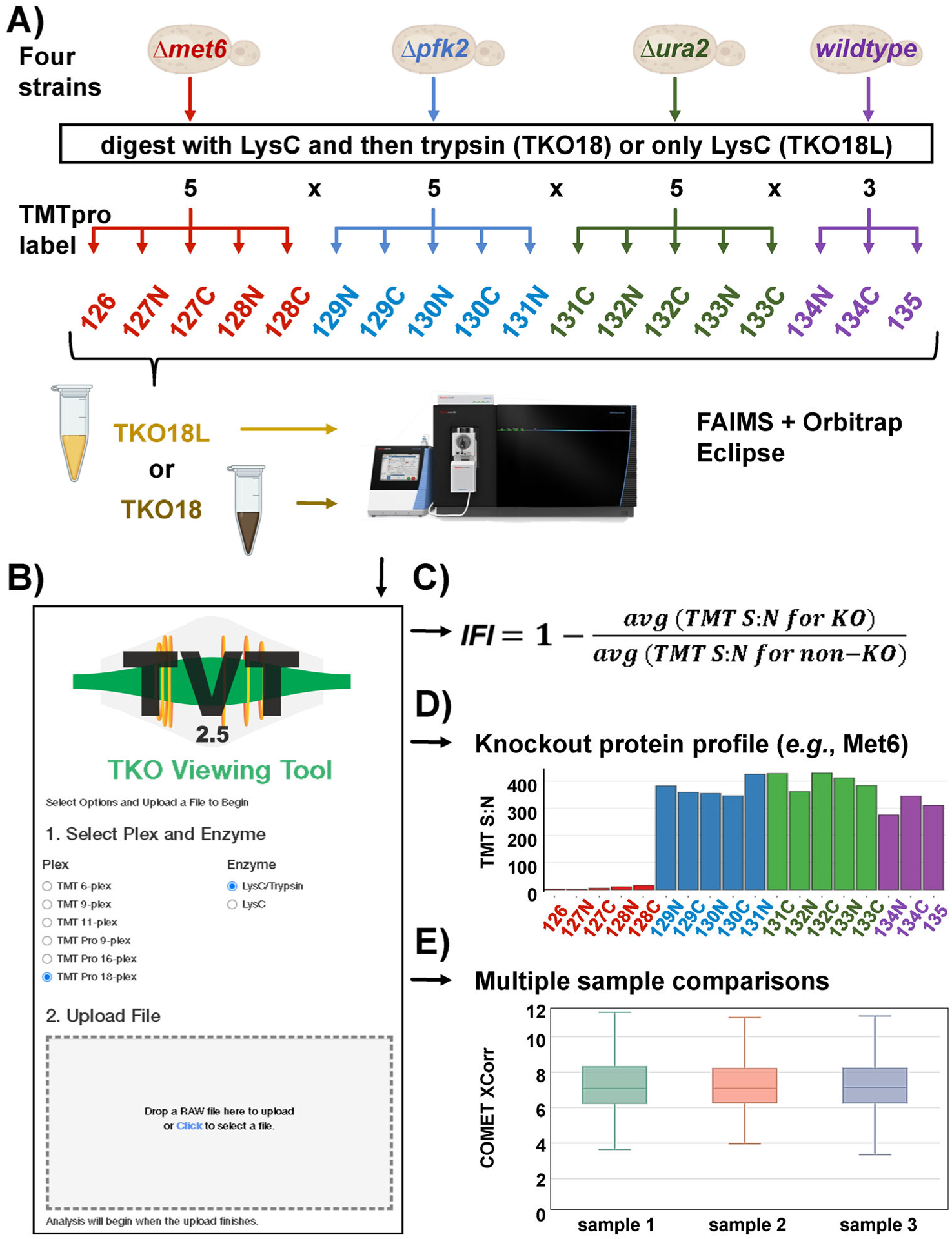
Updates to the TKO standard and the TVT companion website. (A) The TKO18 standard was constructed using three knockout (TKO) yeast strains, specifically three deletions strains were arranged in quintuplicate along with a wildtype strain in triplicate. (B) The TVT has been updated to accommodate the TKOpro18 reagents as well as LysC-only variants of any version of the TKO standard. (C) IFI calculation. (D) An example of an abundance profile for a TKO standard knockout protein (Met6). (E) Representative example of a multiple sample comparison for three different samples as viewed in TVT2.5. IFI, interference-free index; TKO, Triple yeast KnockOut; TVT, TKO viewing tool
The TVT user interface now included options for selecting the TMTpro18-based standard, as well as search parameter settings for specifying whether the peptides were generated by sequential LysC/trypsin digestion or by LysC alone. This additional “enzyme” option was needed to properly analyze the TKO18L standard. Once the specific parameters were selected, the user can upload the RAW file and enter an email address, after which a pre-defined database search was initiated [12] (Figure 1B). The user received a hyperlink to their data once the search has been completed (typically in less than 15 min). This email also served as a record of the user’s submissions, as it included the date, filename, a hyperlink to the data, and a unique search identifier (searchID). As a security feature, the link was encrypted, and linked to a specific email address. Following the provided hyperlink, the user can navigate through the menu bar to view tables and plots of traditional figures of merit (e.g., protein and peptide counts, peak width, ion times, XCorr values), as well as the TKO-specific IFI (Figure 1C). TVT2.5 also rendered plots, such as those displaying the protein abundance profiles for the deleted (“knockout”) proteins (Figure 1D). Not only can interference and instrument performance metrics be assessed for a single sample, but TVT2.5 allowed for the comparison of multiple TKO analyses. This feature can facilitate the assessment of method [24] and sample preparation optimizations [11, 25], as well as the long-term tracking of instrument performance (Figure 1E). We have included an “Easy Start Guide” to TVT2.5 in the Supplemental Material.
Moreover, we were aware that some users may choose to construct their own variants of the TKO standard with a different set of deletion strains. As such, TVT2.5 included a customization feature allowing any deletion strain to be arranged in a pre-defined format – 2 × 2 × 2 (e.g., TMT6), 3 × 3 × 3 (e.g., TKO9, TKOpro9), 3 × 3 × 3 × 2 (e.g., TKO11), 4 × 4 × 4 × 4 (e.g., TKOpro16), or 5 × 5 × 5 × 3 (e.g., TKOpro18). This customizability option was designed to facilitate a broader use of the TKO standard such that users need not purchase the commercial version of the TKO standard and can use their preferred deletion strains.
3.2 |. Data acquisition strategies may be investigated with the TKO18 and TKO18L interference standards
We showcased the TKO18 and TKO18L standards to assess the interference and protein depth (using TVT2.5) of three common data acquisition strategies for isobaric tag-based experiments. Specifically, the methods investigated were: high-resolution MS2 (hrMS2), traditional SPS-MS3 [3, 21], and real-time database search (RTS)-MS3 [26] (Figure 2A). Both standards were analyzed in triplicate for each of the three data acquisition methods using short (45-min) gradients on an FAIMS-equipped Orbitrap Eclipse mass spectrometer.
FIGURE 2.
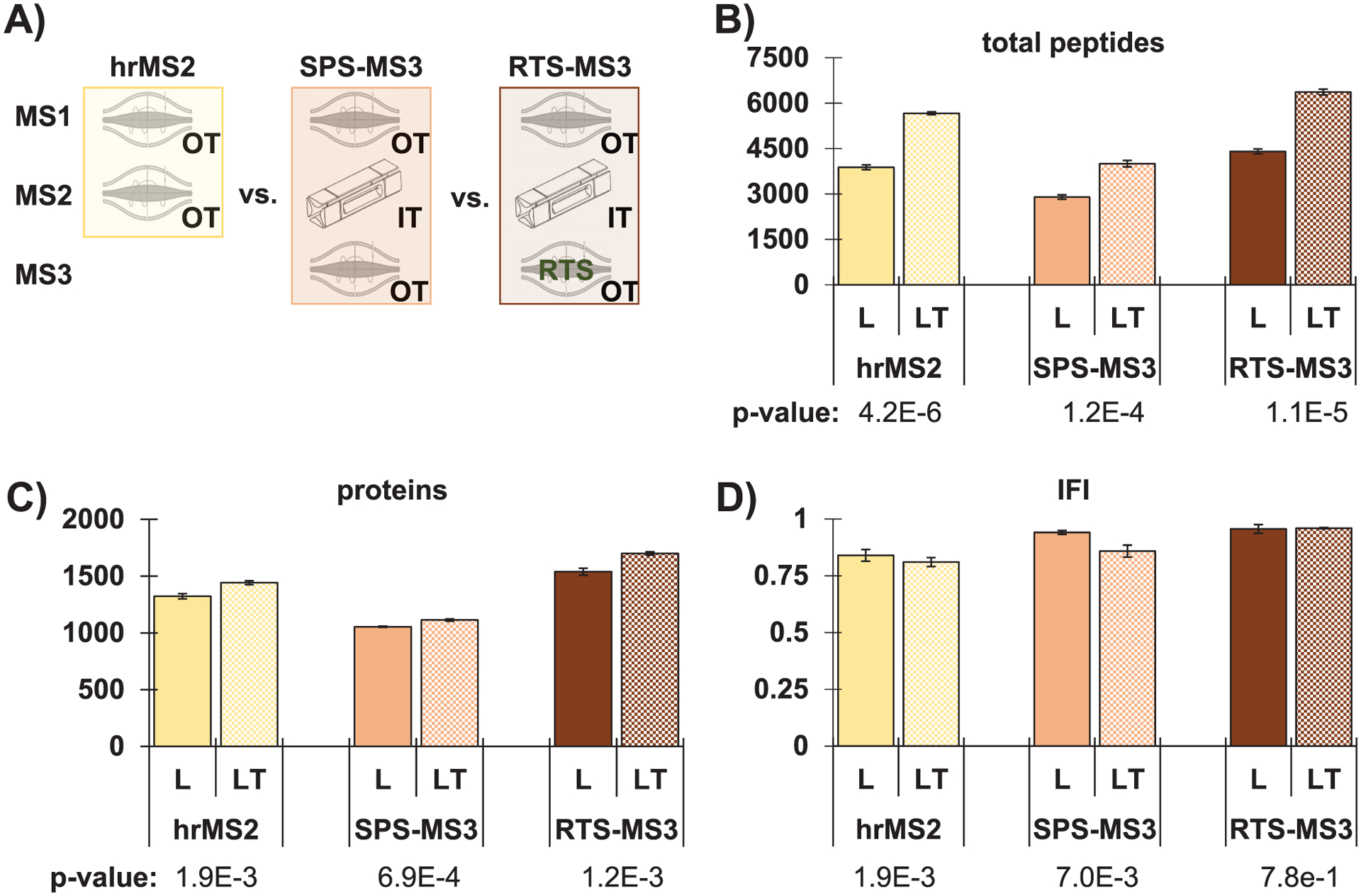
Data acquisition mode comparison between TKO18 and TKO18L. (A) Illustration of data acquisition modes investigated. Bar graphs comparing the number of (B) total peptides and (C) proteins, in addition to (D) the IFI for TKO18L (“L,” which was prepared by digestion with LysC only) and TKO18 (“LT,” which was prepared using a sequential LysC and trypsin digestion strategy) analyzed with hrMS2, SPS-MS3, and RTS-MS3 methods. Error bars represent standard deviation. p-values determined using a two-tailed Student’s t-test. hrMS2, high-resolution MS2; IFI, interference-free index; RTS, real-time database search; SPS, synchronous precursor selection; TKO, Triple yeast KnockOut
We first noted that SPS-MS3 quantified the least number of total peptides when comparing each standard individually across the three data acquisition methods. Such a result was not unexpected and was most likely due to time spent inefficiently on MS3 scans of no quantitative value (Figure 2B). Likewise, both standards showed that the RTS-MS3 method resulted in the most total peptides identified. This finding was due in part to the faster MS2 scans in the ion trap (as opposed to the Orbitrap-only hrMS2 method) and because no MS3 scans were acquired for low-quality peptides with little to no possibility of accurate quantification. This duty cycle differs from that of the SPS-MS3 method in which an MS3 scan is completed for every MS2 scan. Similar results were observed at the protein level (Figure 2C). In agreement with previous data comparing these three data acquisition methods [27, 28], here we showed that the level of interference was highest for hrMS2 and lowest for RTS-MS3 (Figure 2D). Individual data points were illustrated in Figure S1. As all three acquisition modes were performed on an Orbitrap Eclipse mass spectrometer coupled to a FAIMS Pro source, IFI values that were close to or greater than 0.75 were expected for all analyses [27, 28], as were observed. The trend with respect to data acquisition was similar for both TKO18 and TKO18L and a Student’s two-sided t-test did not show a significant difference (p-value > 0.05). These data confirmed our previous results, that with short gradients, these complex standards quantified similar numbers of proteins and peptides for RTS-MS3 and hrMS2, which were higher than those for samples analyzed by SPS-MS3 [9].
We next compared the two standards to determine if the reduced peptide population that resulted from digestion with only LysC also reduced interference. As expected, LysC-only digestion produced fewer identified peptides for TKO18L, as the enzyme cuts only after lysine residues rather than after both lysine and arginine residues as did trypsin (Figure 2B). This difference was not as substantial at the protein level, yet TKO18L also quantified fewer proteins (Figure 2C). Finally, we compared the IFI values between the two standards (Figure 2D). Interestingly, for all three data acquisition methods, the IFI values of TKO18L trended higher than those of TKO18, thereby indicating less interference, but the difference was not significant (p-values > 0.05). That said, the improvement in IFI may be considered negligible as the loss in the number of proteins quantified remained high. The decrease in proteome depth for TKO18L was expected as we have shown previously that samples prepared by LysC followed by trypsin digestion resulted in greater proteome coverage than digestion with LysC alone [29]. As such, data acquired with RTS-MS3 on samples digested with LysC followed by trypsin achieved an acceptable compromise of high protein/peptide quantification and low interference.
TurboTMT showed a slight advantage in the number of proteins quantified when using RTS-MS3 data acquisition and did not increase interference. As a second application, we tested a method implementing the turboTMT option with 30,000 resolution (t30K) for the MS3 scans when using RTS-MS3 and compared it to standard 50,000 resolution (50K) for RTS-MS3 scans. We again used both the TKO18 and TKO18L and analyzed the data using TVT2.5. The data acquired with t30K in place of the typical 50K resolution setting showed a slight, but not statistically significant, increase in both the number of MS/MS scans acquired (Figure 3A) and the number of quantified peptides (Figure 3B). We observed no difference in the average MS2 scan time (Figure 3C) or the XCorr (Figure 3D) values (p-values > 0.05), indicating that the outcomes at the MS2 level were unaffected by differences in the MS3 scan settings, as expected. However, we did observe differences at the MS3 stage. For most scans, the ion times reached the default maximum of 86 ms for the 50K settings and 54 ms for the t30K setting, as anticipated with the relatively high AGC setting of 250% (Figure 3E). In concordance with the ion time differences, the average TMT reporter ion S:N value was higher for the 50K resolution setting than for the t30K setting (Figure 3F) (p-value << 0.05). It follows that fewer peptides were able to pass the TMT reporter ion S:N threshold of 200 and, thus, potential gains resulting from the faster MS3 scans were negated. The end result was merely a slight increase in quantified protein numbers when using the t30K setting compared to the 50K setting (Figure 3G) with negligible difference in IFI (Figure 3H) (p-values > 0.05). We showed individual points for these data in Figure S2. Additionally, we observed that the trends in the data were nearly identical for the TKO18 and TKO18L standards when comparing the 50K and t30K resolution settings (Figure 3A–H).
FIGURE 3.
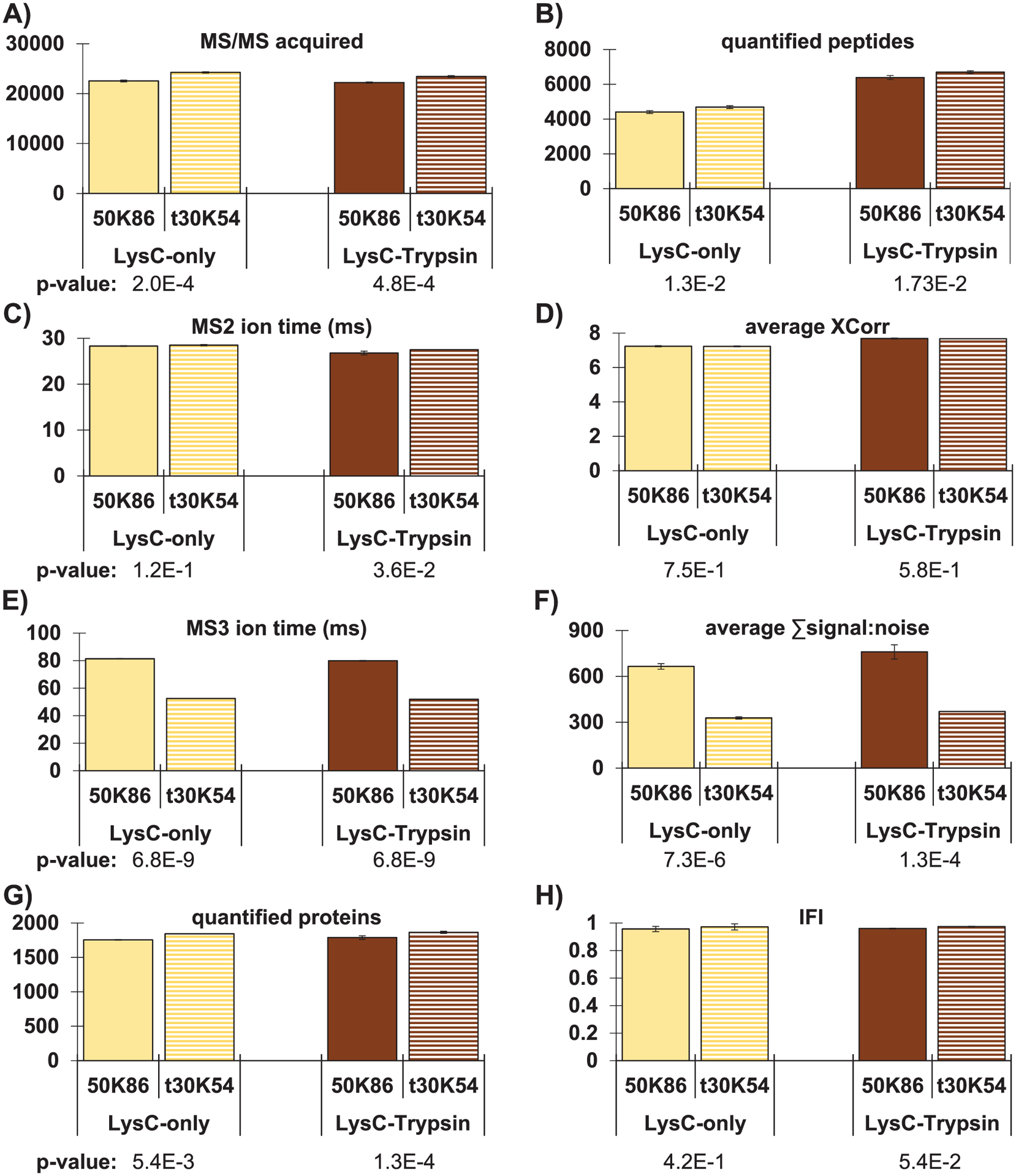
Evaluation of turboTMT using TKO18 and TKO18L. Bar graphs comparing standard RTS-MS3 analysis with resolution of 50,000 (50K86) to the application of turboTMT at resolution 30,000 (t30K54) for both TKO18 and TKO18L. Comparisons include (A) total MS/MS acquired, (B) quantified proteins, (C) MS2 ion injection time, (D) average XCorr, (E) MS3 ion injection time, (F) average TMT summed signal-to-noise, (G) number of quantified proteins, and (H) the IFI. Error bars represent standard deviation. p-values determined using a two-tailed Student’s t-test. IFI, interference-free index; RTS, real-time database search; TKO, Triple yeast KnockOut; TMT, tandem mass tag
We also performed a separate experiment to investigate the effects of increasing the maximum ion injection time for the t30K setting from the default of 54 ms to the default for the 50K setting, namely 86 ms. Compared to the data acquired using t30K resolution with 54 ms, the results for t30K with 86 ms were as expected. Signal-to-noise increased (Figure S3B) due to the additional IT (Figure S3E), but the number of quantified peptides (Figure S3F) decreased slightly possibly due to fewer MS2 acquired (Figure S3A). Overall, the number of quantified proteins remained similar, for TKO18, while a slight decrease was observed for the TKO18L standard. Further optimizations may be warranted by balancing the maximum ion time and measured signal-to-noise for the t30K method to maximize the gain in the number of peptides (and essentially proteins) passing the signal-to-noise threshold. Moreover, the findings for these experiments may differ if highly fractionated datasets were used in in lieu of the unfractionated standards analyzed here.
4 |. CONCLUSION
Here, we introduced the TKO18 and TKO18L standards along with an updated TVT website that facilitated the automated analysis of these quality control standards. We showcased the TKO18 and TKO18L standards by comparing three data acquisition methods and evaluating turboTMT for RTS-MS3 analysis. Our data illustrated the advantage of RTS in reducing interference while improving proteome coverage. We also showed that despite the reduced peptide population, LysC alone only modestly reduced interference. However, any advantage of LysC was diminished due to the lower protein and peptide coverage obtained when compared to the common sequential digestion strategy of using LysC followed by trypsin. Of course, instances exist when certain peptides cannot be identified if trypsin was used, for example, due to exceptionally high density of arginine residues in a given region of a protein. Such a scenario may result in peptides that were too short, hydrophobic, or hydrophilic, or have lower ionization efficiency properties for standard tandem mass spectrometry methods and such associated protein regions may be more readily detected when proteins were digested only with LysC. Therefore, we cannot dismiss the utility of LysC digestion for particular experiments or for targeting specific peptides. Applications of the TKO standards extend greatly beyond what was presented here. As such, we stress that the flexibility of the TVT can accommodate custom versions of the TKO standard, many which may be geared toward a specific procedural optimization, such as the use of lower abundance, difficult-to-detect proteins to assay the lower limit of quantification for targeted assays. Moreover, TVT2.5 offers a user-friendly and nearly effortless database searching and visualization platform for all currently available TKO standards. Collectively, the TKO standards and TVT2.5 comprise an innovative and scalable platform for quality control assessment and standardization, while serving as a conduit for the development of methodology to limit interference in isobaric tag-based quantitative proteomics experiments.
Supplementary Material
Significance Statement.
Isobaric labeling is a well-accepted strategy for proteome-wide quantitative proteome profiling by mass spectrometry. Reporter ion interference resulting from the co-isolation, co-fragmentation and co-analysis of multiple precursor ions is a major caveat of sample multiplexing. Here, we introduce the TKO18 interference standard along with an updated companion website that facilitates the automated analysis of this and other TKO-based quality control standards. This platform may facilitate the development of methodology designed to mitigate interference.
ACKNOWLEDGEMENTS
We would like to thank the members of the Gygi Lab at Harvard Medical School. This work was funded in part by NIH/NIGMS grant R01 GM132129 (J.A.P.) and GM67945 (S.P.G.).
Funding information
National Institute of General Medical Sciences, Grant/Award Numbers: GM132129, GM67945
Glossary
- FAIMS
high-field asymmetric waveform ion mobility spectrometry
- hrMS2
high-resolution MS2
- IFI
interference-free index
- RTS
real-time database search
- SPS
synchronous precursor selection
- TKO
Triple yeast KnockOut
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online https://doi.org/10.1002/pmic.202100317 in the Supporting Information section at the end of the article.
DATA AVAILABILITY STATEMENT
RAW files will be made available upon request, in addition, the data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD029458.
REFERENCES
- 1.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, & Pappin DJ (2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Molecular and Cellular Proteomics, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- 2.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, & Hamon C (2003). Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- 3.Ting L, Rad R, Gygi SP, & Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature Methods, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gygi JP, Yu Q, Navarrete-Perea J, Rad R, Gygi SP, & Paulo JA (2019). Web-based search tool for visualizing instrument performance using the triple knockout (TKO) proteome standard. Journal of Proteome Research, 18, 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulo JA, O’connell JD, & Gygi SP (2016). A triple knockout (TKO) proteomics standard for diagnosing ion interference in isobaric labeling experiments. Journal of the American Society for Mass Spectrometry, 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myers SA, Klaeger S, Satpathy S, Viner R, Choi J, Rogers J, Clauser K, Udeshi ND, & Carr SA (2018). Evaluation of advanced precursor determination for tandem mass tag (TMT)-based quantitative proteomics across instrument platforms. Journal of Proteome Research, 18, 542–547. [DOI] [PubMed] [Google Scholar]
- 7.Paulo JA, Navarrete-Perea J, Guha Thakurta S, & Gygi SP (2018). TKO6: A peptide standard to assess interference for unit-resolved isobaric labeling platforms Journal of Proteome Research, 18, 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Van Vranken JG, Pontano Vaites L, Schweppe DK, Huttlin EL, Etienne C, Nandhikonda P, Viner R, Robitaille AM, Thompson AH, Kuhn K, Pike I, Bomgarden RD, Rogers JC, Gygi SP, & Paulo JA (2020). TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples Nature Methods, 17, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gygi JP, Rad R, Navarrete-Perea J, Younesi S, Gygi SP, & Paulo JA (2020). A triple knockout isobaric-labeling quality control platform with an integrated online database search. Journal of the American Society for Mass Spectrometry, 31, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Cai Z, Bomgarden RD, Pike I, Kuhn K, Rogers JC, Roberts TM, Gygi SP, & Paulo JA (2021). TMTpro-18plex: The expanded and complete set of TMTpro reagents for sample multiplexing. Journal of Proteome Research, 20, 2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navarrete-Perea J, Yu Q, Gygi SP, & Paulo JA (2018). Streamlined tandem mass tag (SL-TMT) protocol: An efficient strategy for quantitative (Phospho)proteome profiling using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research, 17, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eng JK, Jahan TA, & Hoopmann MR (2013). Comet: An open-source MS/MS sequence database search tool. Proteomics, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- 13.Grinfeld D, Aizikov K, Kreutzmann A, Damoc E, & Makarov A (2017). Phase-constrained spectrum deconvolution for Fourier transform mass spectrometry. Analytical Chemistry, 89, 1202–1211. [DOI] [PubMed] [Google Scholar]
- 14.Paulo JA, O’connell JD, Everley RA, O’brien J, Gygi MA, & Gygi SP (2016). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. Journal of Proteomics, 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schweppe DK, Eng JK, Yu Q, Bailey D, Rad Ramin, Navarrete-Perea J, Huttlin EL, Erickson BK, Paulo JA, & Gygi SP (2020). Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics Journal of Proteome Research, 19, 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eng JK, Hoopmann MR, Jahan TA, Egertson JD, Noble WS, & MacCoss MJ (2015). A deeper look into comet—implementation and features Journal of the American Society for Mass Spectrometry, 26, 1865–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beausoleil SA, Villén J, Gerber SA, Rush J, & Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nature Biotechnology, 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- 18.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, & Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell, 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elias JE, & Gygi SP (2010). Target-decoy search strategy for mass spectrometry-based proteomics. Methods in Molecular Biology, 604, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elias JE, & Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- 21.Mcalister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, & Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Analytical Chemistry, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mcalister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, & Gygi SP (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Analytical Chemistry, 84, 7469–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yılmaz Ş, Tiwary S, Cox J, Audain E, Walzer M, … Vizcaíno JA (2019). The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Research, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schweppe DK, Prasad S, Belford MW, Navarrete-Perea J, Bailey DJ, Huguet R, Jedrychowski MP, Rad R, Mcalister G, Abbatiello SE, Woulters ER, Zabrouskov V, Dunyach J-J, Paulo JA, & Gygi SP (2019). Characterization and optimization of multiplexed quantitative analyses using high-field asymmetric-waveform ion mobility mass spectrometry. Analytical Chemistry, 91, 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paulo JA, Navarrete-Perea J, & Gygi SP (2020). Multiplexed proteome profiling of carbon source perturbations in two yeast species with SL-SP3-TMT. Journal of Proteomics, 210, 103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erickson BK, Mintseris J, Schweppe DK, Navarrete-Perea J, Erickson AR, Nusinow DP, Paulo JA, & Gygi SP (2019). Active instrument engagement combined with a real-time database search for improved performance of sample multiplexing workflows. Journal of Proteome Research, 18, 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navarrete-Perea J, Gygi SP, & Paulo JA (2021). HYpro16: A twoproteome mixture to assess interference in isobaric tag-based sample multiplexing experiments. Journal of the American Society for Mass Spectrometry, 32, 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang T, Keele GR, Churchill GA, Gygi SP, & Paulo JA (2021). Strain-specific peptide (SSP) interference reference sample: A genetically encoded quality control for isobaric tagging strategies. Analytical Chemistry, 93, 5241–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navarrete-Perea J, Gygi SP, & Paulo JA (2021). Growth media selection alters the proteome profiles of three model microorganisms. Journal of Proteomics, 231, 104006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RAW files will be made available upon request, in addition, the data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD029458.