

Abstract
Pseudouridine is one of the most abundant RNA modifications, occurring when uridines are catalyzed by Pseudouridine synthase proteins. It plays an important role in many biological processes and has been reported to have application in drug development. Recently, the single-molecule sequencing techniques such as the direct RNA sequencing platform offered by Oxford Nanopore technologies have enabled direct detection of RNA modifications on the molecule being sequenced. In this study, we introduce a tool called Penguin that integrates several machine learning (ML) models to identify RNA Pseudouridine sites on Nanopore direct RNA sequencing reads. Pseudouridine sites were identified on single molecule sequencing data collected from direct RNA sequencing resulting in 723K reads in Hek293 and 500K reads in Hela cell lines. Penguin extracts a set of features from the raw signal measured by the Oxford Nanopore and the corresponding basecalled k-mer. Those features are used to train the predictors included in Penguin, which in turn, can predict whether the signal is modified by the presence of Pseudouridine sites in the testing phase. We have included various predictors in Penguin, including Support vector machines (SVM), Random Forest (RF), and Neural network (NN). The results on the two benchmark data sets for Hek293 and Hela cell lines show outstanding performance of Penguin either in random split testing or in independent validation testing. In random split testing, Penguin has been able to identify Pseudouridine sites with a high accuracy of 93.38% by applying SVM to Hek293 benchmark dataset. In independent validation testing, Penguin achieves an accuracy of 92.61% by training SVM with Hek293 benchmark dataset and testing it for identifying Pseudouridine sites on Hela benchmark dataset. Thus, Penguin outperforms the existing Pseudouridine predictors in the literature by 16 % higher accuracy than those predictors using independent validation testing. Employing penguin to predict Pseudouridine revealed a significant enrichment of “regulation of mRNA 3’-end processing” in Hek293 cell line and positive regulation of transcription from RNA polymerase II promoter involved in cellular response to chemical stimulus in Hela cell line. Penguin software and models are available on GitHub at https://github.com/Janga-Lab/Penguin and can be readily employed for predicting Ψ sites from Nanopore direct RNA-sequencing datasets.
Keywords: RNA modifications, Pseudouridine, Nanopore
1. Introduction
Pseudouridine (abbreviated by the Greek letter Ψ) is one of the most abundant RNA modifications, occurring when uridines (part of RNA chains) are catalyzed by Ψ synthase proteins. Ψ is an isomer of the nucleoside uridine in which the uracil/ pseudouracil is attached via a carbon-carbon instead of a nitrogen-carbon glycosidic bond [1]. In other words, Ψ is considered as the C5-glycoside isomer of uridine that contains a C-C bond between C1 of the ribose sugar and C5 of uracil/pseudouracil, rather than usual C1-N1 bond found in uridine. The C-C bond makes the Ψ isomer flexible to interconverts through free rotations about formally single bond [2]. Moreover, Ψ has an extra hydrogen bond donor at the N1 position [3].
Ψ plays an important role in many biological processes such as stabilizing RNA through enhancing the function of transfer RNA and ribosomal RNA [4]. The modification of uridine to Ψ has been observed in nearly all kinds of RNA, including but not limited to tRNA (transfer RNA), and mRNA (messenger RNA). Ψ has also important roles in drug development and response to various stresses [5].
Despite improvements in experimental technologies, chemical methods developed to detect Ψ sites remain both time-consuming and expensive. For example Ψ can be mapped by chemical conversion with N-cyclohexyl-N-(2-morpholinoethyl) carbodiimide metho-p-toluenesulphonate (CMC), which then blocks reverse transcription (Ψ-seq or pseudo-seq [4,6]). Ψ-seq detected 89 and 353 modified mRNA transcripts in different human cell lines. However, the number of sites identified with Ψ-seq is low compared with the spread of Ψ reported by mass spectrometry (MS) methods, and the overlap between the different studies is very modest. Another method employing a chemical pulldown enrichment step for Ψ (N3-CMC-enriched Pseudouridine sequencing, CeU-seq) recognized 1929 modified mRNAs, but whether most of these sites were missed by Ψ-seq or are false positives has not been yet determined [7]. Therefore, there is increasing evidence supports the need to address this problem using novel unbiased experimental approaches or via computational biology methodes.
In contrast to experimental methods, computational biology methods were introduced in the literature to predict Ψ sites utilizing RNA short read sequencing data. Those methods rely on developing Machine Learning (ML) or Deep Learning (DL) algorithms to identify RNA Ψ sites [8–14]. However, the existing computational biology methods in the litrature rely on genomic information for predcting RNA Ψ sites and they can’t be used to predict Ψ sites on single molecule of RNA. Moreover, they are unable to predict Ψ sites on RNA long read sequencing data. In addition, the performance results of the later group of computational methods are quite low. For example, ML models (PPUS and iRNA-PseU) have used support vector machine (SVM) as the ML classifier for predicting RNA Ψ sites in multiple species. PPUS used the nucleotides around Ψ as features for training the SVM, while iRNA-PseU model used the chemical properties of nucleotides and their occurrence frequency density distributions as features for training the SVM classifier. However, it was found that the performances of PPUS and iRNA-PseU can be improved by introducing another a computational biology method called PseUI for predicting Ψ sites based in RNA short reads sequence of multiple species [10]. This was done by generating five different kinds of features based on RNA segments including nucleotide composition, dinucleotide composition, pseudo dinucleotide composition, position-specific nucleotide propensity, and position-specific dinucleotide propensity. Then, a sequential forward feature selection strategy was used to gain the most effective subset of features. This subset of features was used to train the SVM classifier that can identify Ψ sites in the testing phase. Another research attempt introduced a deep learning model called iPseU-CNN (identifying pseudouridine by convolutional neural networks) [11] that identified Ψ sites from RNA samples by producing a real value equal to each input single RNA sequence, where the input is represented by a one-hot vector with four channels A, C, G, and U. However, there were XGBoost-based ML model, called XG-PseU that was developed in [12] to identify Ψ sites and outperformed iPseU-CNN. This model used the optimal features obtained using the forward feature selection together with increment feature selection method for training the XGBoost model. Another ML model called iPseU-NCP was introduced in [13] to predict Ψ sites on RNA sequences and outperform the performance of deep learning model presented by iPseU-CNN. It used the Random Forest (RF) algorithm combined with the nucleotide chemical properties (NCP) generated from RNA sequences. The optimal hyper-parameters of this model were determined through an exhaustive search over a specified grid of parameter values for the RF classifier using 5-fold cross-validation. Moreover, the RF model was used again in a model called RF-PseU [14] that performed also better than previous models for predicting RNA Ψ sites in multiple species. It used light gradient boosting machine algorithm and incremental feature selection strategy for selecting the optimum feature space vector for training the RF model.
Recently, the third-generation sequencing technologies such as the platform provided by Oxford Nanopore Technology (ONT) have been proposed as a new means to detect RNA modifications using long read RNA sequence data. However there were few research attemptes that used this technology to identify Ψ sites in RNA sequence data [15,16]. For example in [15], the authors developed a tool called nanoRMS that used the characteristic base-calling “error” signatures in the Nanopore data as features for training a supervised or unsupervised learning models to make them able to identify the stoichiometry of Ψ site using a threshold for base mismatch frequency in different types of RNAs in yeast. However, nanoRMS was not applied to predict Ψ sites in the RNA sequence of human cell lines which are more complex and larger than yeast. Also, the single read features used to train the predictors of nanoRMS were averaged before Ψ prediction making it not feasible to obtain the contribution of each feature in predicting Ψ sites. As an extension to the same research direction, authors in [16] developed a tool called NanoPsu to address the limitations of nanoRMS by applying their tool to predict Ψ sites in direct RNA sequence that consists of a mixture of rRNAs from human, yeast, Caenorhabditis elegans, Drosophila, and from human fecal bacteria. However, the RNA samples used in NanoPsu were not all sequenced using Nanopore technology as half of them were sequenced using the Illumina sequencing. To this end, our work aims to extend the research direction in [15,16] by introducing a tool called Penguin that integrates several developed ML models (predictors) to identify Ψ sites in Nanopore direct RNA sequencing data of human cell lines based on some features extracted from the Nanopore signals.
Penguin extracts a set of features from the raw signal of Oxford Nanopore RNA Sequencing reads and the corresponding basecalled k-mers. Those features are used to train the predictors included in Penguin, which in turn, can predict whether the signal is modified by the presence of Ψ sites. The features extracted by Penguin include: the signal length, some signal statistical features including the mean and standard deviation of the signal, and one-hot encoding of reference k-mers produced from aligning Nanopore events/signals to a reference genome using the eventalign module in Nanopolish, a software for Nanopore signal analysis [17].
2. Materials and Methods
2.1. The pipeline of Penguin
The complete pipeline of Penguin is composed of various components/blocks (Figure 1). Penguin takes the fastq reads file that is generated from basecalling the fast5 files using any basecalling software (e.g., guppy, albacore, scrappy), and a reference genome as inputs. The fast5 files produced by ONT device are used to store the output of Nanopore sequencers and contains the raw electrical signal levels that come off the sequencers. Using both inputs to Penguin, the SAM file is created from the alignment stage, where the fastq reads file is aligned to reference genome to create this file. Next, using the produced SAM file and a provided BED file [18] - a file that highlights the target modified locations from the whole genome, the tool can create a coordinate file with ids of fast5 files that have the target modification. Next, the tool launches the Nanopolish software that performs signal extraction in two steps in order to produce a dataset of Nanopore signal samples. The first step takes the fast5 files folder as an input and index all files in that folder. The second step takes the fastq reads file of the indexed fasts5 files, the sorted bam file (a sorted compressed version of the SAM file that is created using samtools software [19]), and the reference genome as inputs and run Nanopolish eventalign module on these inputs to generate a dataset of Nanopore signals.
Figure 1.
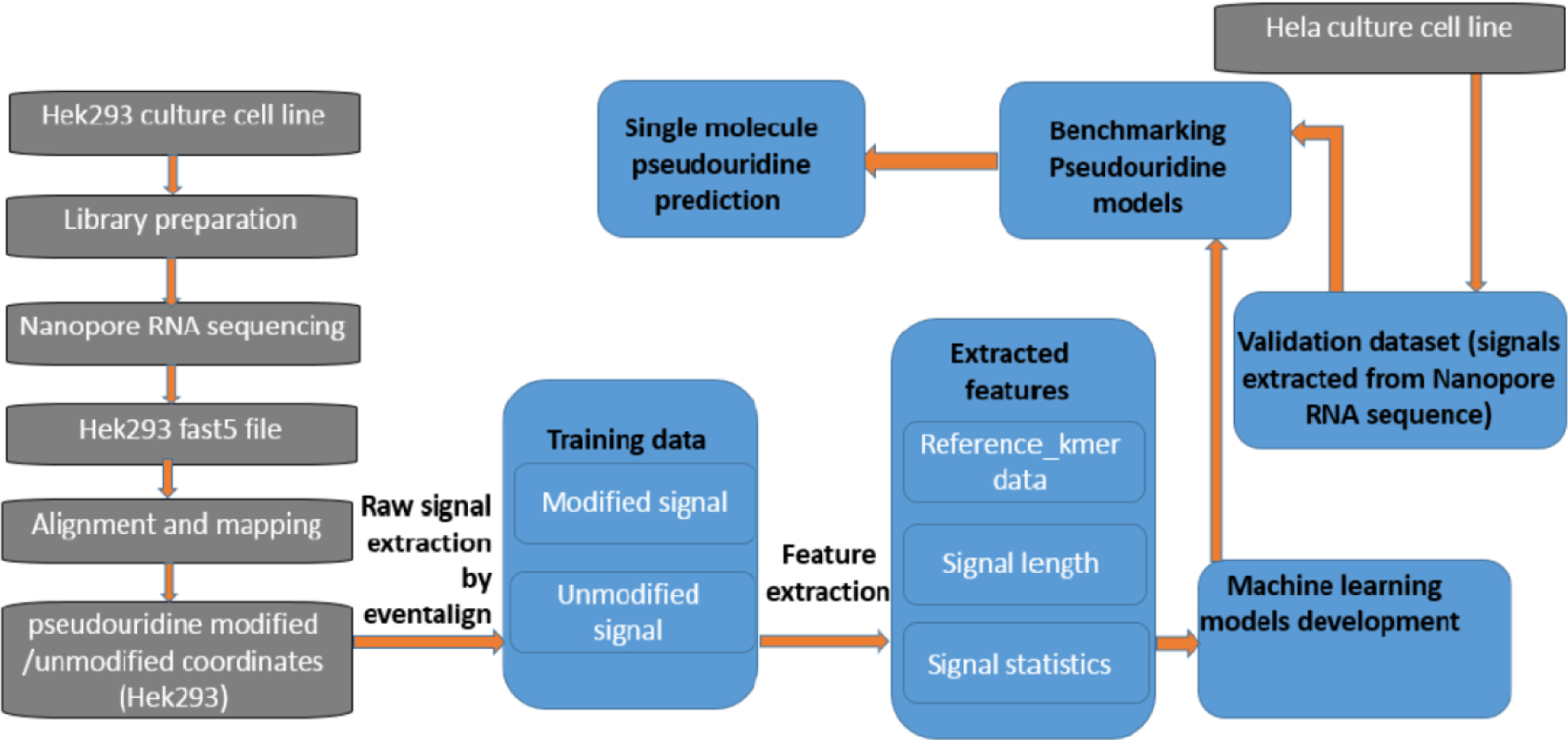
The complete pipeline of Penguin showing various steps including: RNA sequencing of a HEk293 cell line, basecalling, alignment and mapping, identifying Ψ sites and corresponding coordinates from training data, the raw signal extraction by Nanopolish eventalign module, the feature extraction from signals that address Ψ modification, machine learning model development and validation, and single molecule Ψ prediction.
Next the signal samples that are related to Ψ modification are filtered from the samples generated from signal extraction. Using the information in the coordinate file some of the filtered samples will be labeled as modified and the remaining will be labeled as unmodified, as will be described later. Next some features will be extracted from those modified and unmodified samples which are fed to one of Penguin’s machine learning classifiers in the training phase. Using those features the classifier will be able to predict the modification sites in the testing phase. The details of each Penguin component will be described in the next subsections.
2.2. Cell Culture
Culturing the cells is where we extract cells from an animal and let it grow in an artificial environment [20]. Cell culture Hek293 and Hela cell line were purchased from ATCC cell line collection, cultured in DMEM media supplemented with 10% FBS and 0.5% penicillin/streptomycin and grown at 37°C for 24–48hrs.
2.3. RNA library preparation and Nanopore RNA sequencing
The libraries that we used were prepared following Nanopore Direct RNA sequencing kit documented protocol (SQK-RNA002). Briefly, total RNA was isolated using Qiagen RNeasy Mini Kit (Cat No. /ID: 74104), followed by PolyA enrichment using Thermo Fisher Dynabeads™ mRNA DIRECT™ Micro Purification Kit (61021). 500 ng of poly (A) RNA was ligated to a poly (T) adaptor using T4 DNA ligase. Following adaptor ligation, the products were purified using Mag-Bind® TotalPure NGS Beads (M1378–00), following NGS bead purification protocol. Sequencing adaptors preloaded with motor protein were then ligated onto the overhang of the previous adaptor using T4 DNA ligase followed by NGS bead purification protocol. The RNA library was eluted from the beads in 21 μl of elution buffer and quantified using a Qubit fluorometer using the manufacturer’s RNA assay. The final RNA libraries were loaded to FLO-MIN106 flow cells andrun on MinION. Sequencing runs and base calling were performed using MinKNOW software (Oxford Nanopore Technologies Ltd.), a software that controls the MinION Nanopore sequencing device. Therefore, once we extract the RNA from the cells during library preparation, we put it through the MinION device and start generating signal data. Nanopore is a small pore in MinION device that is used to create signal on biological molecules that pass through it. The MinION is one of the Nanopore sequencing devices and it can read both DNA and RNA signals. When a strand of DNA or RNA goes through the pore it disrupts a membrane and creates a current that is measured by the device [21]. The output of the device is a fast5 file or files. These files contain the raw signals of the DNA or RNA reads.
The data output from MinKNOW for Hela cell line was 800k sequence reads using one flow cell. MinKnow generates data as pass and fail folders. FAST5 files from pass folder were again base called using Albacore 2.1.0 (Oxford Nanopore Technologies Ltd.) resulting in 723K single molecule reads for Hek293 and 500K single molecule reads for Hela cell line which corresponded to full length transcripts ranging from 50b to 8kb. The directRNA-sequencing data generated for Hek293 and Hela cell lines in this study is publicly available on SRA, under the project accession PRJNA685783 and PRJNA604314 respectively.
2.5. Basecalling, alignment and mapping
There are several basecallers such as scrappy, albacore, guppy and many more [22]. The basecaller uses algorithms to determine what base is being passed through the Nanopore. Some fast5 files may or may not contain sequence information, so the basecallers can provide this information for us. The median length of RNA reads of Hek293 obtained by basecalling is 574 base pairs and the Inter Quartile Range (IQR) range is 371 to 886 base pairs. Similarly, the median length of RNA reads of Hela obtained by basecalling is 548 base pairs and the IQR range is 404 to 854 base pairs. Also, the Phred quality scores of the reads of Hek293 and Hela cell lines were 8.7 and 8, respectively. Once we have the sequence information, we align the sequence to a reference genome using Minimap2 [23]. Aligning a sequence is just mapping the sequence to a certain location in the genome. It could be in any of the chromosomes and this information is saved to the SAM file. The alignment rates of the RNA reads of Hek293 and Hela cell lines were 87.31% and 95.33%, respectively.
2.6. Identifying Pseudouridine modified /unmodified coordinates
For a gold standard set of modified locations we use a BED file containing Ψ RNA modification locations that have been mentioned and verified in literature, for two cell lines: Hela and Hek239. These Ψ RNA modification locations have been obtained from publicly available databases such as Darned [24], RMBase [25], and from two primary literature sources [4,26]. These modifications are also available in an internal database site for RNA modifications called Epitomy [27] that is used to search for RNA modification locations. Penguin uses these Ψ RNA modified locations imported from Eoitomy to verify its prediction accuracy of Ψ sites. The imported Ψ BED file used by Penguin has 27839 Ψ genomic locations for Hek293 (Supplementary File 1_Hek293.bed) and 320 Ψ genomic location for Hela (Supplementary File 2_Hela.bed).
2.7. Raw signal extraction
The Nanopore direct RNA sequencing holds a great advantage in identifying RNA modifications at single base resolution by the interpretation of ONT raw signals ( i.e., events or “squiggles”) plotted over time that are corresponding to modified and unmodified base sequence contexts. To this end, the basic functionality of Penguin aims to exploit the difference in the raw signal between modified and unmodified bases in order to predict Ψ sites in RNA sequence. Thus, the signal extraction phase plays an important role in achieving Penguin functionality. For this phase, we use Nanopolish [28], the ONT analysis software. In particular, the eventalign module [29] of Nanopolish is used to extract the raw Nanopore signals and get some of its corresponding features such as the mean, standard deviation and length of the signal. In order to achieve this, eventalign first aligns events to a reference genome, so the low-level signal information can be obtained. Such information can be used to discover the differences in the current that might lead to base modifications. We refer to Nanopolish manual for more information about the Nanopolish pipeline for signal analysis [39].
2.8. Benchmark datasets generation
Two different benchmark datasets were generated for Hek293 and Hela cell lines, respectively (Supplementary Tables 1_training_hek.xlsx and 2_training_hela.xlsx). In order to generate these datasets, we considered the output of eventalign module from Nanopolish (where eventalign was run on the basecalled sequence reads of each cell line) to build associations between sequence and signal instances. In order to label the data, we filtered all samples that have a U base in the middle of the model k-mer (one column in eventalign output that refers to basecalled k-mers resulting from inferring RNA sequence reads from Nanopore signals formulated by eventalign), which is the target base for identifying Ψ modification. Next, we found the intersection between their position column on the reference genome and the position in the coordinate file (generated from Ψ BED file and SAM file for each cell line). This intersection represented the positive samples, while the remaining samples were considered as negative samples. In the end, we obtained a total of 13072 k-mer samples that have a U/ Ψ base in the middle: 6536 positive and 6536 negative samples (after sub-sampling the negative samples which were very huge in comparison with postive ones) for Hek293. Similarly, we obtained 1354 samples: 677 positive and 677 negative samples for Hela cell line. We observed that there is a total of 255 different model kmer combinations out of the possible 256 combinations, captured in the modified and unmodified signals in Hek293 training data (Supplementary Tables 3_training_model_kmer_freq_hek.xlsx) and a total of 216 different model kmer combinations captured in the modified and unmodified signal dataset in Hela training data (Supplementary Tables 4_training_model_kmer_freq_hela.xlsx).
2.9. Feature extraction
Each generated benchmark dataset has 4 columns that represent four features that were used for training the machine learning models that we developed and integrated in the Penguin platform. Those features were extracted by picking 4 columns from the evenalign output (Supplementary File 3.txt) (namely: reference_kmer, mean, stdv, and length of event) and used for training the constructed ML classifiers of Penguin. Those columns refer to the k-mers generated from aligning events to a reference genome, the mean, the standard deviation, and the length of each extracted signal, respectively.
2.10. ML Models construction
We have developed several machine learning models and integrated them into Penguin’s platform including the SVM [31], RF [32] and NN [33]. Those algorithms have been used extensively to address several problems in bioinformatics research [34], [35], [36]. The radial basis function kernel (rbf) was used in SVM training. The gamma parameter was set to ‘scale’ and the default value of C parameter was used. For RF, the seed number was set to 1234 and the number of trees was set to 30. As for NN, a two hidden layers NN was implemented. The number of neurons (nodes) was 12 in the first and 8 in the second. The ‘Adam’ optimizer with a learning rate of 0.001 was used, the number of epochs were set to 150, and the batch size was set to the length of training set.
We have used the scikit-learn toolkit [37], the free machine learning python library to implement the SVM and RF models, while Keras [38], a neural network python library with tensorflow [39,40] back end was used to implement the NN model.
2.11. Feature importance
As for deep analysis of the features that contribute mostly to the performance of machine learning model that does Ψ site identification, we have reported about the importance of each feature thorugh training and testing each developed ML model of Penguin with each of the four extracted features that were described in sec 2.9. This is acheived by buliding five versions of the ML model applied to each benchmark dataset. In the first four versions, each developed ML model was trained with one of the four extracted features, while in the fifith version each model was trained with the combination of all four features.
2.12. Random test splitting
In this approach, we randomly divided the benchmark datasets into two folds: one for training and the other for testing. The test_size was set to 0.2 meaning that 80% of the benchmark dataset was used for training the model and 20% of the dataset was kept for testing the model.
2.13. Test with independent cell line
In this approach, the two benchmark datasets were separated by cell line, one was used for training and other was used for testing.
2.14. Evaluation metric
We have used the accuracy (Acc), precision, recall, and the area under the curve (AUC) [41] as metrics to evaluate the performance of penguin predictor. Below we introduce the mathematical equation for the first three metrics:
| (1) |
| (2) |
| (3) |
where:
TP stands for true positive and refers to the number of correctly classifed Pseudouridine sites.
FP stands for false positive and refers to to the number of non Pseudouridine sites that were misclassified as Pseudouridine sites.
FN stands for false negative and refers to the number of Pseudouridine sites that were misclassified as non Pseudouridine sites
TN stands for true negative and refers to the number of correctly classifed non Pseudouridine sites.
3. Results
We have used the two validation methods: the random-test splitting and the test with independent cell line introduced in Section 2 for evaluating the performance of each predictor in Penguin platform using the metrics that we just mentioned in the previous section. In the following we present the performance results that we have got using each method.
3.1. Performance evaluation with random-test splitting
Table. 1 shows the performance of Penguin ML models that are available on its GitHub page using the extracted 4 features introduced in Section 2 from the benchmark dataset of Hek293 cell line. Clearly, SVM achieves the best performance in terms of accuracy ( 93.38%), while RF achieve best precision ( 0.98), while SVM and NN achieve the best recall (0.95).
Table 1:
The performance of Penguin’s predictors on Hek293 benchmark dataset with random-test splitting using all four extracted features.
| Classifier | accuracy (%) | precision | recall | AUC |
|---|---|---|---|---|
| SVM | 93.38 | 0.92 | 0.95 | 0.933 |
| RF | 84.59 | 0.98 | 0.72 | 0.852 |
| NN | 93.35 | 0.92 | 0.95 | 0.932 |
The learning curve of SVM, RF, and NN (see Figure 2, panels A,B, and C respectively) show the performance of SVM and NN in terms of accuracy score outperforms that of RF. Also we used receiver operating characteristic (ROC) which plots the true positive rate on the vertical coordinate versus the false positive rate on the horizontal coordinate. The ROC curve of SVM, RF and NN (see Figure 3 A,B and C) show that the percentage of true postive rate to the fasle positive rate in case of SVM and NN is more that of RF.
Figure 2.
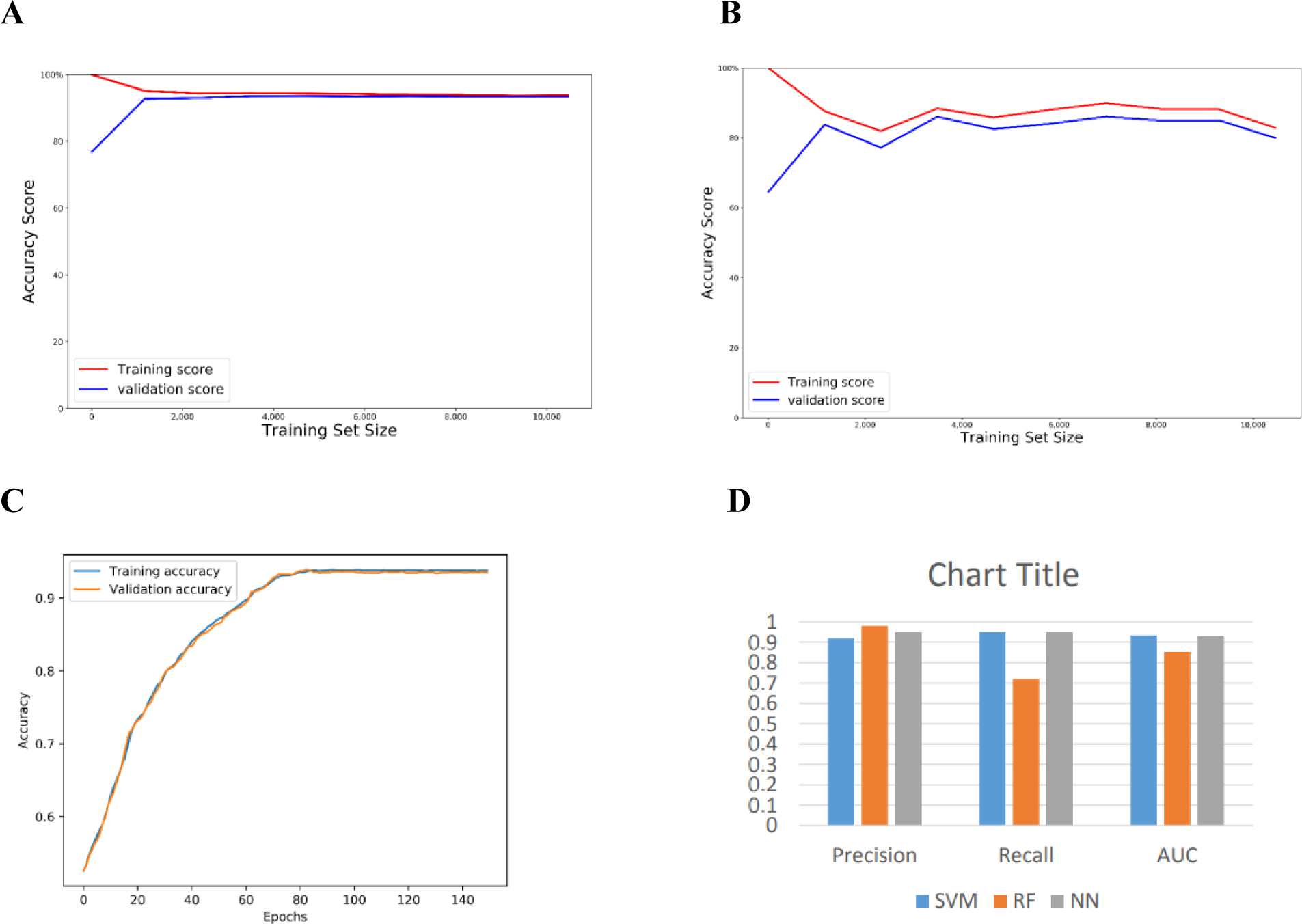
The learning curve for each of Penguin’s predictors and a bar chart of the performance evaluation using Hek293 cell line benchmark dataset with random test splitting showing the (a) learning curve of SVM (b) learning curve of RF (c) learning curve of NN (d) bar chart of the precision, recall, and AUC of Penguin’s developed ML classifiers.
Figure 3.
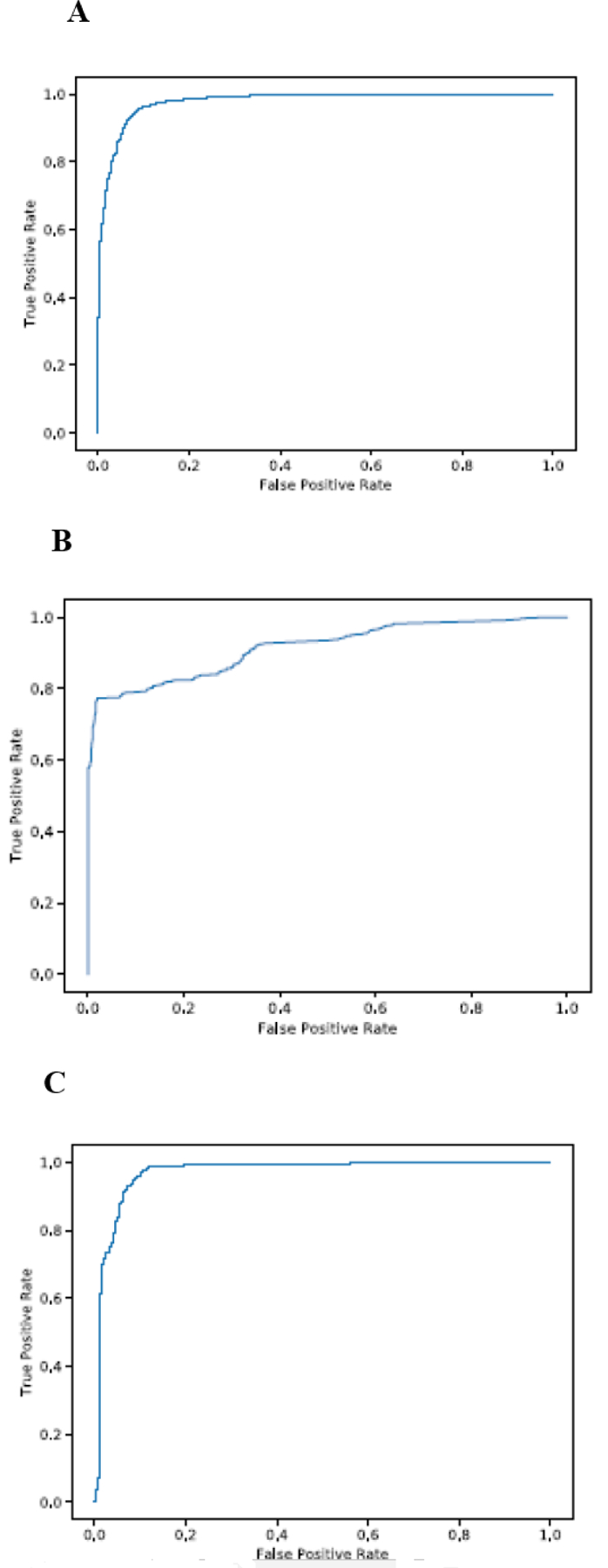
The ROC curve for each of Penguin’s machine learning models using Hek293 cell line benchmark dataset with random test splitting showing (a) The ROC curve of SVM (b) The ROC curve of RF (C) The ROC curve of NN.
3.1.1. Performance results using single type of feature
Table 2 shows the performance of ML models with random test-splitting on Hek293 benchmark dataset in terms of accuracy with single type of feature among the four extracted features introduced in Section 2.
Table 2:
The performance of Penguin’s predictors on Hek293 benchmark dataaset in terms of accuracy with random test-splitting using single type of feature.
| Classifier | Mean | Stdv | Length | One-hot Encoding | Combination |
|---|---|---|---|---|---|
| SVM | 69.33 | 61.87 | 51.81 | 93.38 | 93.38 |
| RF | 69.17 | 61.49 | 50.86 | 87.92 | 84.59 |
| NN | 67.34 | 46.46 | 47.53 | 93.42 | 93.35 |
Clearly, the one-hot encoding of reference_kmer contributes more to the classifier’s accuracy than other features for all classifiers. It is by followed by mean, standard deviation, and the length for SVM and RF. In case of NN, the standard deviation and length swap places. Based on the results in Table 2, one would wonder why we do not use the one-hot encoding feature alone for prediction and avoid using features extracted from the signal as the former leads to a classification accuracy equal to that when using the combination of four features in case of SVM and outperforms the classification accuracy of combination in case of RF and NN. However, using onehot encoding feature alone is not useful when predicting new location of Ψ sites that have not seen before. Also the extracted signals are still needed even if their features are not contributing to the performance as one-hot encoding feature. This is because without signals extraction we can’t get the model_kmer that is needed to filter the samples in eventalign output that address the target modification.
3.2. Performance against independent cell line
Table 3 shows the performance of ML models against an independent cell line. (i.e., with independent test dataset), where Hek293 cell line benchmark dataset is used for training penguin’s predictors and Hela cell line benchmark dataset is used for testing them, using all the extracted features. We currently only include SVM and NN models on Penguin GitHub and execlude the RF model as it acheives very low performance against independent cell line (below 50% accuracy). Clearly as shown in Table3, NN outperforms SVM. However, SVM is more stable as it achieves reproducable results, while NN performance results changes from run to run though all NN results are still high (above 93% accuracy). See the learning curves of SVM and NN (Supplementary Figure 1 and Figure 2 respectively) and ROC curves of SVM and NN (Supplementary Figure 3 and Figure 4, respectively).
Table 3:
The performance of Penguin’s predictors against independent cell line using all extracted features.
| Classifier | accuracy (%) | precision | recall | AUC |
|---|---|---|---|---|
| SVM | 92.61 | 0.91 | 0.94 | 0.926 |
| NN | 95.35 | 0.92 | 1.00 | 0.953 |
3.2.1. Performance results using single type of feature
Table 4 shows the performance of ML models against independent cell line in terms of accuracy with single type of feature. As in the cross-validation model, the one-hot encoding of reference_kmer contributes more to the classifier accuracy than other features. It is followed by length, standard deviation, and mean.
Table 4:
The performance of Penguin’s predictors against independent cell line using single type of extracted features.
| Classifier | Mean | Stdv | Length | One-hot Encoding | Combination |
|---|---|---|---|---|---|
| SVM | 39.22 | 40.99 | 51.62 | 95.35 | 92.61 |
| NN | 32.94 | 37.59 | 51.18 | 95.35 | 95.42 |
4. Abundance of Ψ sites
In order to identify the abundance of Ψ sites in the single molecule transcriptomes of either Hek293 or Hela cell lines, we first ran Penguin’s best performing machine learning model (i.e., SVM as it achieves very high performance and reproducible results over all runs) on the complete RNA sequence reads of Hek293 and Hela cell lines. Then, we identified all U-mer samples predicted as Ψ sites in those reads. Next, we identified the number of Ψ unique genomic locations, as well as their frequencies (number of reads that support Ψ modification) in both the cell lines. We found that there are 6137606 U-mer samples predicted as Ψ U-mers from a total of 67491289 U-mers in the complete direct RNA-sequencing dataset of Hek293 cell line, with 556813 unique genomic locations corresponding to Ψ modifications (Supplementary Table 5_ps_unique_genomic_locations _hek.xlsx)1. Similarly, we found that there are 1193191 U-mers predicted as Ψ U-mers from a total of 229637931 U-mers in the complete dataset of Hela cell line, with 39384 unique genomic locations corresponding to Ψ sites (Supplementary Table 6_ps_unique_genomic_locations _Hela.xlsx). The model kmers corresponding to Ψ predictions in Hek293 and Hela cell lines can be identified as strong kmers in comparison with the model kmers corresponding to the unmodified/control U-mers, which can be considered as weak contributors to the Ψ prediction. Supplementary Tables 7_ps_model_kmer_freq_hek.xlsx and 8_ps_model_kmer_freq_Hela.xlsx). summarize the frequency of various model kmers corresponding to Ψ predictions in Hek293 and Hela cell lines, providing an overview of their abundance. Comparison of the unique genomic locations of Ψ in both cell lines for overlapping sites/genomic loci, resulted in 6482 unique genomic locations of Ψ that are common between both cell lines (Figures 4.A). We also found 7148 modified Ψ site containing genes that are common/overlapped between both cell lines. We observed 15.8% overlap between the top 1% frequently modified Ψ genes between the Hela and Hek293 cell lines (Figures 4.B).
Figure 4.
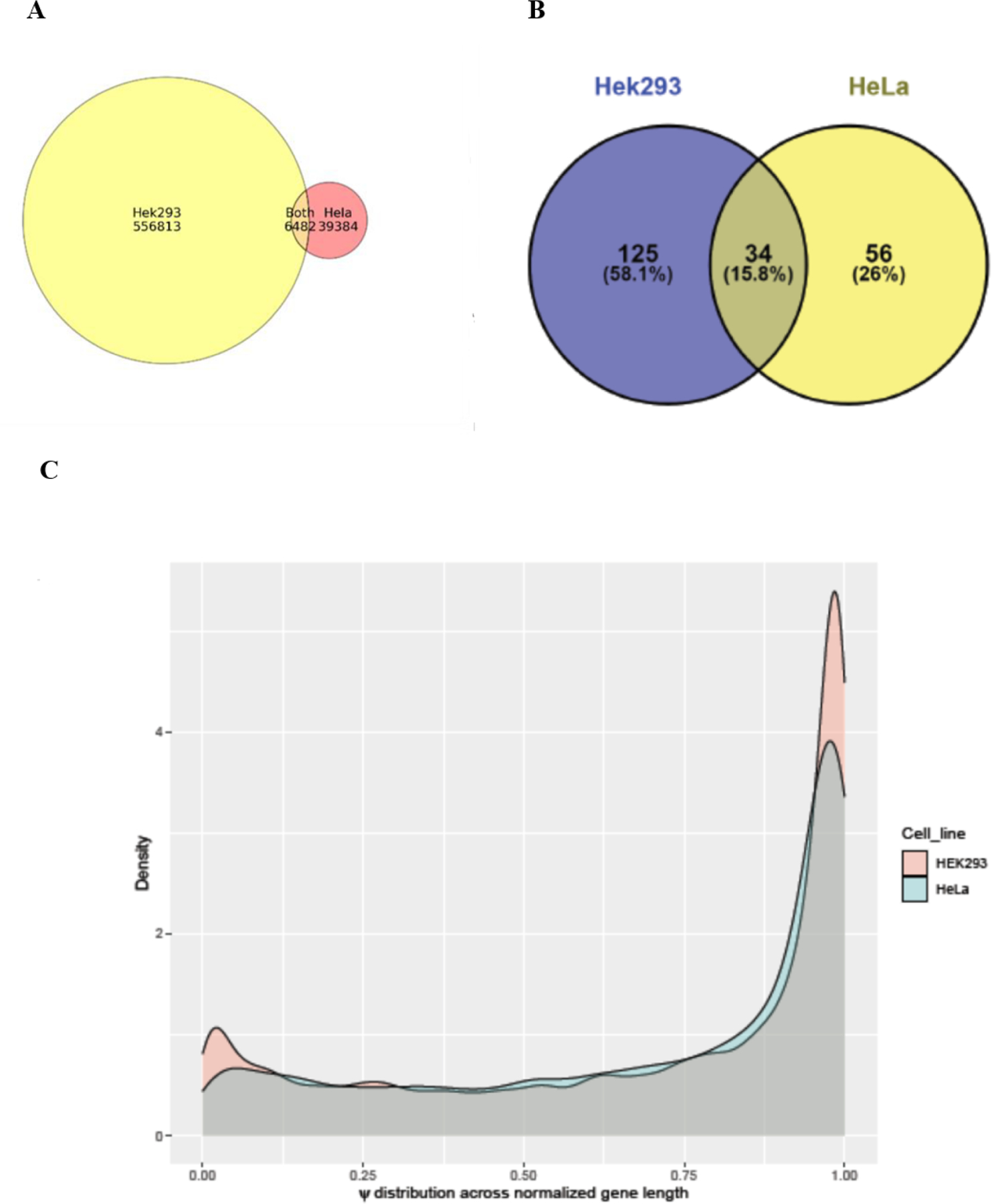
Plots showing (a) Venn diagram summarizing the overlap between unique Ψ locations predicted in complete Hek293 and Hela cell lines (b) Venn diagram showing the overlap between top frequent 1 % modified Ψ genes in Hek293 and Hela cell lines (c) Density plots representing ψ distribution across normalized gene lengths for Hek293 and Hela cell lines.
We noticed that the extent of Ψ modification (the number of U-mers samples predicted as Ψ samples to the total number of U-mer samples in the complete RNA sequence of the cell line) in RNA sequences of Hek293 cell line is much greater than its counterpart for Hela cell line (9% for Hek293 versus 0. 5 % for Hela cell line). This is due to the existence of more modified Ψ genes with extensive Ψ locations in the complete RNA sequence reads of the Hek293 cell line compared to the number of modified Ψ genes with fewer unique Ψ locations found in the complete RNA sequence reads of the Hela cell line. Therefore, the ψ distribution across normalized gene length for Hek293 cell line is larger than its equivalent in Hela cell line (Figures 4.C).
5. Functional enrichment analysis
To investigate the potential functional role of Ψ modification in RNA, we performed functional enrichment analysis for the most frequently modified Ψ genes (top 1%) across Hek293 and Hela cell lines. A total of 159 genes from Hek293 and 90 genes from Hela cell lines were identified to have the most abundance of Ψ RNA modification. The short-listed genes from both cell lines were plugged into Cytsoscape ClueGo [42] application to obtain enriched ontologies and pathways at high confidence (p<0.05). Enrichment observations from this analysis are visualized in Figure 5.
Figure 5.
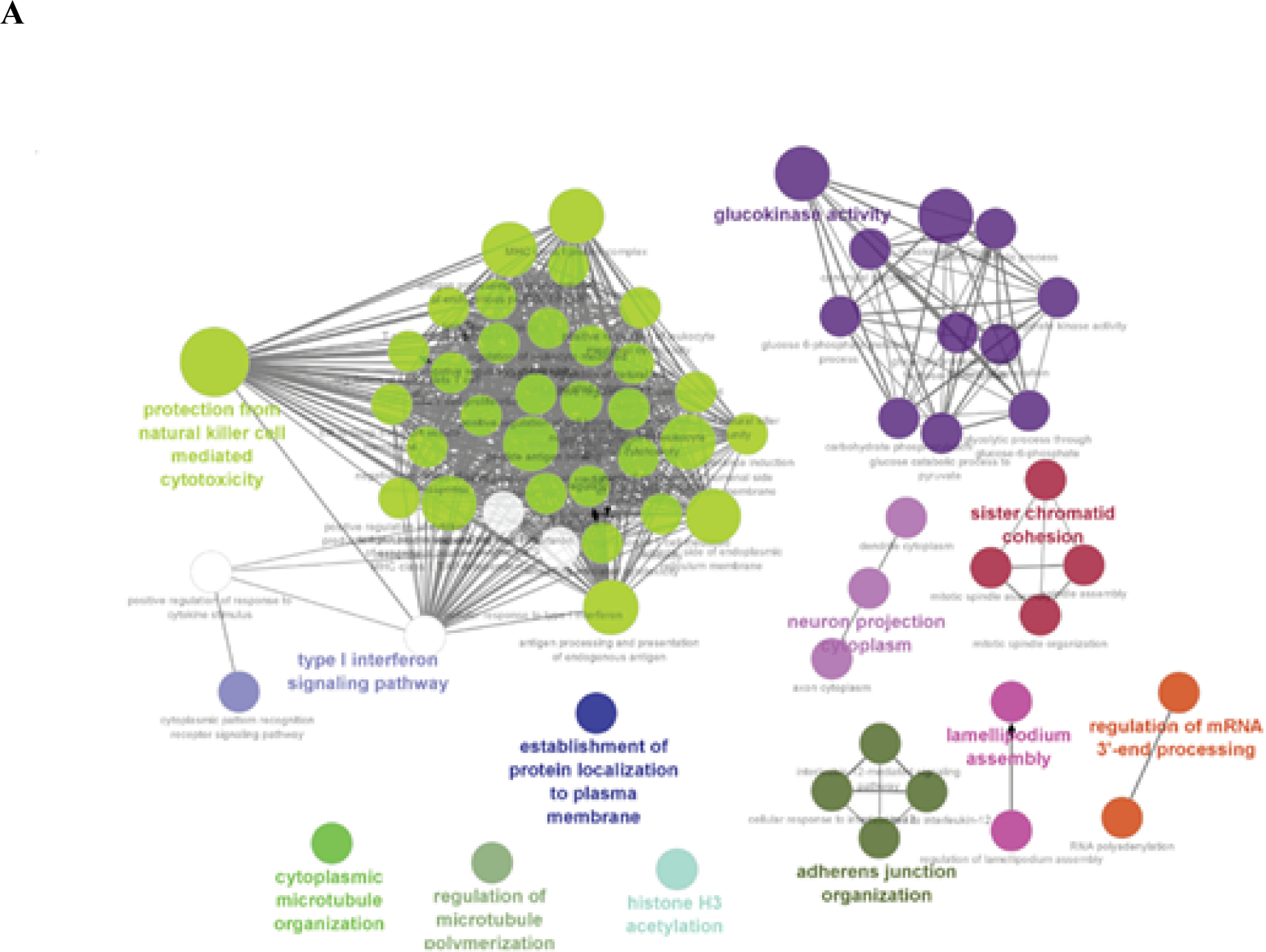
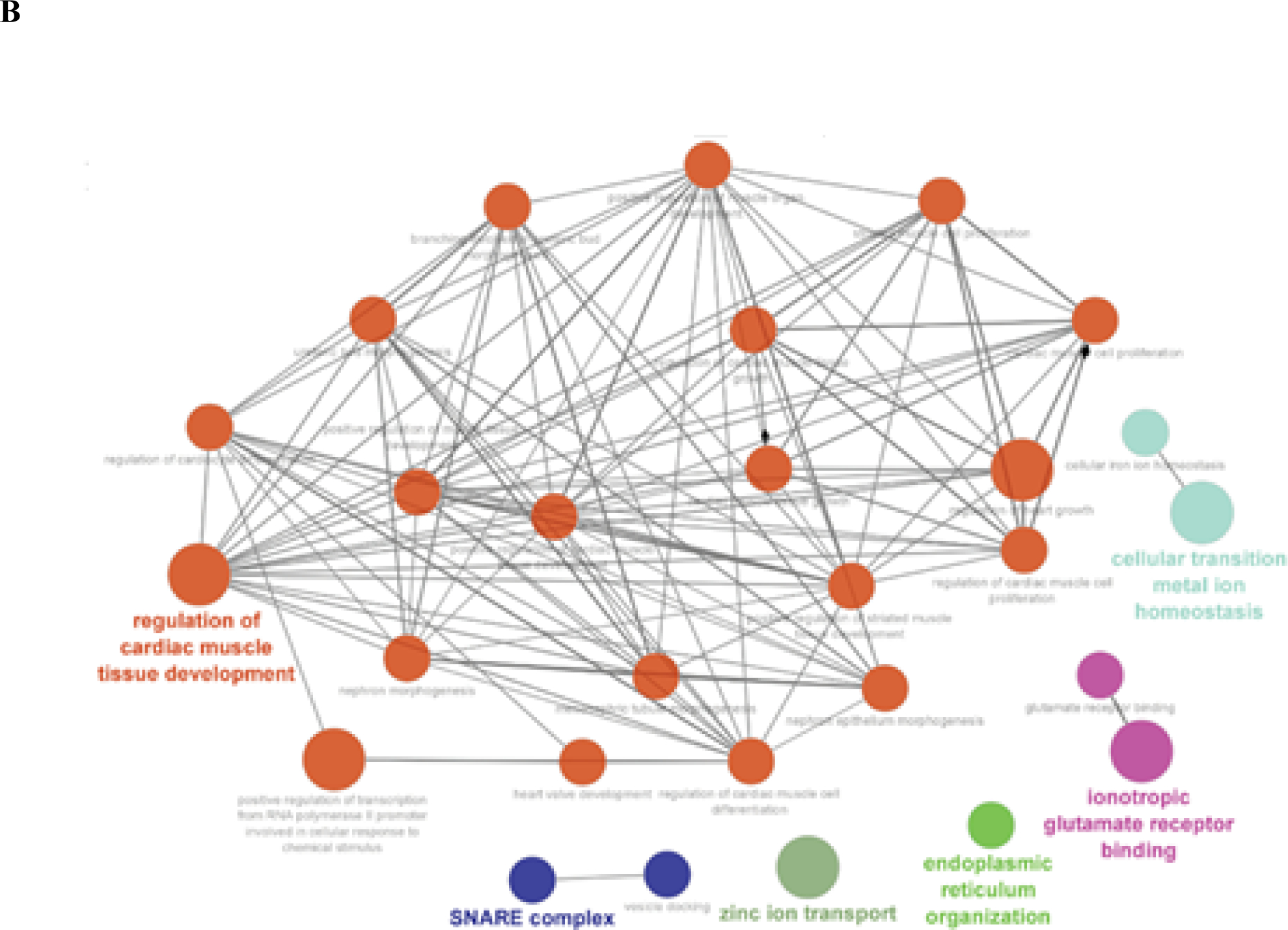
Functional enrichment analysis of most frequenlty Ψ modified genes across (a) Hek293 cell line and (b) Hela cell line, visualizing high confident (p-val<0.05) set of ontologies and pathways potentially associated with Ψ RNA modification. Cliques clustered by functional grouping of the GO-terms based on GO hierarchy using Cytoscape ClueGO application. Size of the nodes are representative of the significance of the association with respect to genes per GO-term.
From the functional enrichment analysis of the gene set form Hek293 cell line, we observed a wide range of functional processes like (as seen in Figure 5A): “protection from natural killer cell mediated cytotoxicity”, “T cell mediated cytotoxicity”, “glucokinase activity”, “histone H3 acetylation”, “type I interferon signaling pathway”, and “regulation of mRNA 3’-end processing” being significantly enriched (adjusted p-val < 0.0013). Essentially highlighting the diverse regulatory role of Ψ modification, from its involvement in cell immune signaling to mRNA 3’-end processing.
In Hela cell line, we observed several high confidence (adjusted p-val < 0.0014) enriched ontologies that were more representative of Ψ modification role in cellular development and homeostasis via post-transcriptional regulation (as seen in Figure 5B): “regulation of cardiac muscle tissue development”, “cellular transition metal ion homeostasis”, “positive regulation of transcription from RNA polymerase II promoter involved in cellular response to chemical stimulus”, “SNARE complex”, “Zinc ion transport”, and “endoplasmic reticulum organization”.
6. Usage of Penguin
Penguin tool is implemented in python 3.x and the tool has to be run on Linux environment by running the following command from Penguin main directory on the user’s local machine after cloning the code from Penguin GitHub repository:
python main.py -r ref.fa -f reads.fastq
Where the penguin tool needs the following two inputs files when running it:
The absolute path to the reference Genome file (ref.fa)
The absolute path to fastq reads file (reads.fastq)
Once the user runs the penguin tool main file, then the tool pipeline (Figure 1) that accepts the aforementioned inputs will start execution and the user will be asked to enter the bed file name with the absolute path and extension that is needed to generate the coordinate file that is needed for labeling the U-mers Nanopore signals samples as modified and unmodified ones. Next, the tool will extract the raw Nanopore signals from the input fast5 file(s) as well as extracting some of its corresponding features that are used later to train the three different machine learning models (SVM, RF, and NN) integrated in Penguin platform for predicting Ψ sites in direct Nanopore RNA sequence. More extensive usage guide with all the required software that should be installed before running penguin is available on Penguin GitHub README file.
7. Discussions and Conclusion
In this study, we have proposed a new tool called Penguin that represents a complete pipeline for predicting Ψ sites from direct Nanopore RNA sequencing datasets. We note that our proposed tool outperforms the existing non-Nanopore tools for Ψ sites prediction in the literature [8–14] in terms of accuracy. However, we found the comparison between penguin and those tools does not make sense in terms of implementation, but it makes sense in terms of accuracy. This is because those tools were only applied to predict Ψ sites in short reads of RNA sequences, while ours can predict Ψ sites on long reads of RNA sequencs. As for comparison of the performance of Penguin with Nanopore based tools for Ψ sites prediction such as nanoRMS [15] and NanoPsu tools [16] that are developed for predicting Ψ sites from direct RNA sequencing data, we found that nanoRMS was only tested on predicting Ψ in direct RNA sequences of yeast and it was not tested on predicting the Ψ sites on direct RNA sequencing data of human cell lines which are more complex than lower eukaryotes like yeast. In addition, the single read features used to train the predictors of nanoRMS were averaged before Ψ prediction, making it not feasible to obtain the contribution of each feature in predicting Ψ sites. Hence, it is not possible to directly compare the accuracy of the tool on human cell line data. Since only the accuracy values of predicting the stoichiometry of Ψ for each read and no other performance statistics such as precision and recall are reported in nanoRMS study, we can only compare the average of the accuracy values using KNN (the best supervised classifier employed in nanoRMS) to the accuracy of predictors distributed in Penguin. Based on this comparison, we conclude that the accuracies of different ML models employed in Penguin significantly outperform the average accuracy of KNN (66.8%), the best supervised predictor employed in nanoRMS. As for NanoPsu predictor, it was reported about its performance in identifying Ψ sites in terms of AUC (Area Under The Curve) and ROC curve, but the accuracy of prediction, precision and recall were not explicilty mentioned in the study. So currently it is not possible to directly compare the performance of the Nanopore based Ψ prediction tools against a controlled and common dataset of eukaryotic transcriptomes.
It was also observed that generating direct RNA sequence data for Hela and Hek293 cell lines in our study allows us for generating two Ψ benchmark datasets for the respective cell lines. This in turn allowed Penguin to predict Ψ sites on RNAsequence of independent cell line which is superior to the standard single dataset benchmarking in which training, and testing occurs on the same benchmark dataset. This also helped in obtaining biological results from performing functional enrichment analysis for the most frequently modified Ψ genes (top 1%) across both cell lines. These biological results can be observed for a wide range of functional processes in Hek293 and Hela cell lines. In Hek293, it essentially highlights the diverse regulatory role of Ψ modification, from its involvement in cell immune signaling to mRNA 3’-end processing, while in Hela it was observed that Ψ modified genes were enriched for several high confident (adjusted p-val < 0.0014) ontologies associated with cellular development and homeostasis.
We expect that Penguin will become a useful tool for accurate identification of Ψ sites in direct RNA sequencing datasets of human genome and other species. Penguin platform can be also adopted for predicting other types of RNA modifications.
With the improvement of Nanopore sequencing protocols and Nanopore basecalling algorithms, we anticipate improved accuracies in read mapping and Nanopore signal association in the upcoming years. Such an improvement in addition to the parallel development of deep learning models for Nanopore basecalling could contribute to refine the basecalling models which would result in more precise accuracies of RNA modification prediction tools such as Penguin.
Supplementary Material
HIGHLIGHTS.
Penguin integrates several Machine learning models (i.e., predictors) to identify RNA Ψ sites in Nanopore direct RNA sequencing reads.
The pipeline of penguin automates the data preprocessing including Nanopore direct RNA read alignment using Minimap2, and Nanopore signal extraction using Nanopolish, feature extraction from raw Nanopore signal for training ML predictors integrated in its platform, and the prediction of RNA Ψ sites with those predictors.
Penguin outperforms the state-of-the-art Ψ prediction methods in accuracy.
Penguin framework can be adopted to be used for predicting other types of RNA modifications.
Only a small fraction of 0.01% (6482 unique genomic locations) of Ψ sites were detected to be common (overlapped) between both Hek293 and Hela cell lines.
The extent of Ψ modifications (the number of U-mers predicted as Ψ sites to the total number of U-mers in the complete set of RNA sequences of the cell line) in Hek293 cell line is much higher than its counterpart for Hela cell line (9% for Hek293 versus 0.5 % for Hela cell line).
Acknowledgement
We thank Rajashekar Varma Kadumuri form whom we borrow the code that we slightly updated to generate the coordinate file for Hela and Hek293. We also thank Alexander Krohannon, Ratanond Koonchanok and Padma Poojitha Alla at IUPUI for giving valuable comments on this work and valuable discussions. This work is supported by the National Science Foundation (NSF) grant # 1940422 and #1908992 as well as the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM123314 (SCJ).
Footnotes
Conflict of interest
The authors report no financial or other conflict of interest relevant to the subject of this article.
Author statement
All authors acknowledge that the material presented in this manuscript has not been previously published, except in abstract form, nor is it simultaneously under consideration by any other journal.
U-mers samples are rows (with U in the middle of their reference kmers column) in the eventalign output that corresponds to the result of aligning the events (signals or squiggles) of RNA sequence of a specific cell line to a reference genome.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hamma Tomoko; Ferré-D’Amaré Adrian R. (November 2006). “Pseudouridine Synthases”. Chemistry & Biology. 13 (11): 1125–1135. doi: 10.1016/j.chembiol.2006.09.009. ISSN 1074–5521. [DOI] [PubMed] [Google Scholar]
- [2].Gray Michael Charette, Michael W (2000-May-01). “Pseudouridine in RNA: What, Where, How, and Why”. IUBMB Life. 49 (5): 341–351. doi: 10.1080/152165400410182. ISSN 1521–6543. [DOI] [PubMed] [Google Scholar]
- [3]. https://en.wikipedia.org/wiki/Pseudouridine .
- [4].Carlile T, Rojas-Duran M, Zinshteyn B et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146 (2014). 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhao BS and He C (2015). Pseudouridine in a new era of RNA modifications. CellRes.25,153–154.doi: 10.1038/cr.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schwartz Schraga, Bernstein Douglas A., Mumbach Maxwell R., Jovanovic Marko, Herbst Rebecca H., León-Ricardo Brian X., Engreitz Jesse M., Guttman Mitchell, Satija Rahul, Lander Eric S., Fink Gerald, and Regev Aviv, Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA, Cell, Volume 159, Issue 1, 2014, Pages 148–162, ISSN 0092–8674, 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Anreiter I, Mir Q, Simpson JT, Janga SC, Soller M. New Twists in Detecting mRNA Modification Dynamics. Trends Biotechnol. 2020. Jul 1;S0167–7799(20)30166–9.doi: 10.1016/j.tibtech.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li YH, Zhang G, Cui Q. PPUS: a web server to predict PUS-specific pseudouridine sites. Bioinformatics. 2015; 31(20):3362–3364. doi: 10.1093/bioinformatics/btv366 [DOI] [PubMed] [Google Scholar]
- [9].Chen Wei, Tang Hua, Ye Jing, Lin Hao and Chou Kuo-Chen. iRNA-PseU: Identifying RNA pseudouridine sites. Molecular Therapy-Nucleic Acids (2016), 5, Official journal of the American Society of Gene & Cell Therapy, July 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].He J, Fang T, Zhang Z et al. PseUI: Pseudouridine sites identification based on RNA sequence information. BMC Bioinformatics 19, 306 (2018). 10.1186/s12859-018-2321-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tahir M, Tayara H, and Chong KT. iPseU-CNN: identifying RNA pseudouridine sites using convolutional neural networks. Molecular Therapy—Nucleic Acids, 16, 463–470. doi: 10.1016/j.omtn.2019.03.010, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu Kewei, Chen Wei, and Lin Hao. XG-PseU: an eXtreme Gradient Boosting based method for identifying pseudouridine sites. Molecular Genetics and Genomics,295, 13–21 (2020). 10.1007/s00438-019-01600-9. [DOI] [PubMed] [Google Scholar]
- [13].Nguyen-Vo Thanh-Hoang et al. iPseU-NCP: Identifying RNA pseudouridine sites using random forest and NCP-encoded features. BMC Genomics, 20 (Suppl 10):971, 2019. 10.1186/s12864-019-6357-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lv Zhibin, Zhang Jun, Ding Hui and Zou Quan. RF-PseU: A Random Forest Predictor for RNA Pseudouridine Sites. Frontiers in Bioengineering and Biotechnology, Volume 8, Article 134, February 2020. doi: 10.3389/fbioe.2020.00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Begik O, Lucas MC, Pryszcz LP, Ramirez JM, Medina R, Milenkovic I, Cruciani S, Liu H, Vieira HGS, Sas-Chen A, Mattick JS, Schwartz S, Novoa EM. Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat Biotechnol. 2021. Oct;39(10):1278–1291. doi: 10.1038/s41587-021-00915-6. [DOI] [PubMed] [Google Scholar]
- [16].Huang S, Zhang W, Katanski CD, Dersh D, Dai Q, Lolans K, Yewdell J, Eren AM, Pan T. Interferon inducible pseudouridine modification in human mRNA by quantitative nanopore profiling. Genome Biol. 2021. Dec 6;22(1):330. doi: 10.1186/s13059-021-02557-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. https://github.com/jts/nanopolish .
- [18]. http://genome.ucsc.edu/FAQ/FAQformat#format1 .
- [19]. http://www.htslib.org/
- [20].Lynn Dwight E.. Cell Culture. In Encyclopedia of Insects (Second Edition), 2009. [Google Scholar]
- [21].How Does nanopore DNA/RNA sequencing work. Oxford Nanopore Technologies, 2020. [Google Scholar]
- [22]. https://github.com/rrwick/Basecalling-comparison/
- [23].Li Heng, Minimap2: pairwise alignment for nucleotide sequences, Bioinformatics, Volume 34, Issue 18, 15 September 2018, Pages 3094–3100, 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kiran Anmol, Baranov Pavel V., DARNED: a DAtabase of RNa EDiting in humans, Bioinformatics, Volume 26, Issue 14, 15 July 2010, Pages 1772–1776, 10.1093/bioinformatics/btq285 [DOI] [PubMed] [Google Scholar]
- [25].Xuan Jia-Jia, Sun Wen-Ju, Lin Peng-Hui, Zhou Ke-Ren, Liu Shun, Zheng Ling-Ling, Qu Liang-Hu, Yang Jian-Hua, RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data, Nucleic Acids Research, Volume 46, Issue D1, 4 January 2018, Pages D327–D334, 10.1093/nar/gkx934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li X, Ma S, Yi C. Pseudouridine: the fifth RNA nucleotide with renewed interests. Curr Opin Chem Biol. 2016;33:108–16. [DOI] [PubMed] [Google Scholar]
- [27]. https://epitomy.soic.iupui.edu/
- [28]. https://github.com/jts/nanopolish .
- [29].Quickstart - how to align events to a reference genome. Available at https://nanopolish.readthedocs.io/en/latest/quickstart_eventalign.html
- [30]. https://nanopolish.readthedocs.io/en/latest/manual.html .
- [31].Cortes C, and Vapnik V Support-vector networks. Mach. Learn. 20, 273–297, 1995. [Google Scholar]
- [32].Breiman L Random forests. Machine learning. 45:5–32, 2001. [Google Scholar]
- [33].Gurney K An introduction to neural network. UCL Press (Taylor & Francis group), 1997. [Google Scholar]
- [34].Chicco Davide. Support Vector Machines in Bioinformatics: a Survey. TECHNICAL REPORT, [TP-2012/01], published online: 12th October, 2012. [Google Scholar]
- [35].Qi Y (2012). Random Forest for Bioinformatics. In Ensemble Machine Learning, pp. 307–323, Springer, 2012. [Google Scholar]
- [36].Rozenberg G et al. Neural Networks in Bioinformatics Handbook of Natural Computing, Springer-Verlag; Berlin Heidelberg, 2012. [Google Scholar]
- [37]. https://scikit-learn.org/
- [38].Keras: Deep learning library for theano and tensorflow. Available at: https://github.com/keras-team/keras
- [39]. https://github.com/tensorflow/tensorflow .
- [40].Abadi Martin et al. TensorFlow: A system for large-scale machine learning. In Proceedings of 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), pp. 265–283, 2016. [Google Scholar]
- [41].Bradley Andrew E. The Use of the Area under the Roc Curve in the Evaluation of Machine Learning Algorithms. Pattern Recognition, Vol. 30, No. 7, pp. 1145–1159, 1997. [Google Scholar]
- [42].Bindea Gabriela et al. “ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks.” Bioinformatics (Oxford, England) vol. 25,8 (2009): 1091–3. doi: 10.1093/bioinformatics/btp101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.