

SUMMARY
Mutations are acquired frequently, such that each cell’s genome inscribes its history of cell divisions. Common genomic alterations involve loss of heterozygosity (LOH). LOH accumulates throughout the genome, offering large encoding capacity for inferring cell lineage. Using only single cell RNA sequencing (scRNA-seq) of mouse brain cells, we found that LOH events spanning multiple genes are revealed as tracts of monoallelically expressed, constitutionally heterozygous single nucleotide variants (SNVs). We simultaneously inferred cell lineage, marked developmental time points based on X-chromosome inactivation and the total number of LOH events, while identifying cell types from gene expression patterns. Our results are consistent with progenitor cells giving rise to multiple cortical cell types through stereotyped expansion and distinct waves of neurogenesis. This type of retrospective analysis could be incorporated into scRNA-seq pipelines and, compared to experimental approaches for determining lineage in model organisms, is applicable where genetic engineering is prohibited, such as humans.
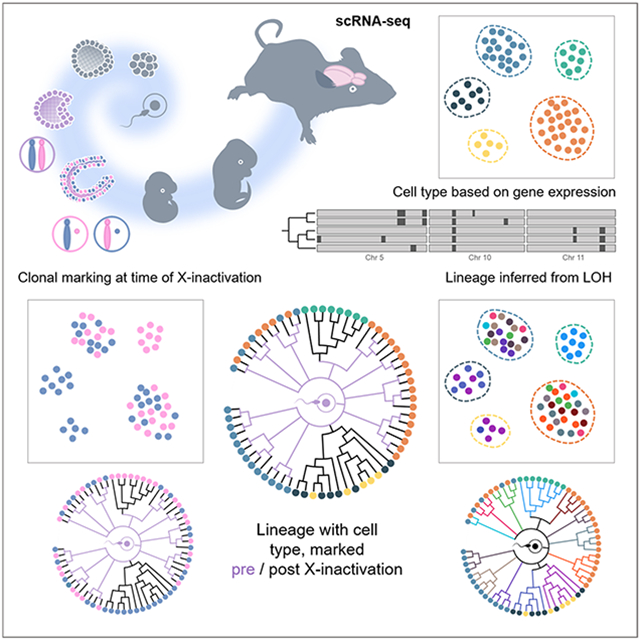
Graphical Abstract
eTOC blurb
Relying solely on scRNA-seq analysis, Anderson et al. retrospectively infer cell type based on gene expression, reconstruct lineage through detection of acquired LOH events, and temporally mark developmentally ordered cell clades from the timing of X-chromosome inactivation in females, at single cell resolution during mouse brain development.
INTRODUCTION
Developmental biology addresses how a single cell, through a repertoire limited to cell division, migration, differentiation, and death, matures into a multicellular organism. A milestone involved determination of the embryonic cell lineage of C. elegans (Sulston et al., 1983). However, mapping cell lineage in vertebrates remains daunting because there are more cells, more types of cells, longer lifespans, opaque tissues, and, unlike C. elegans lineage differs between individuals.
A cell’s descendants can be identified by labeling it with a dye or transgenic marker (Garcia-Marques et al., 2021). Still, the correspondence between clonally related cells and their lineage is ambiguous. For example, >1042 different possible lineages can describe a clone of just 32 cells (Salipante and Horwitz, 2007). A massive capacity for uniquely labeling cells is therefore required to resolve lineage.
One approach to map lineage is to retrospectively infer the sequence of mutations acquired in single cells (Carlson et al., 2011; Frumkin et al., 2008; Ju et al., 2017; Lee-Six et al., 2018; Lodato et al., 2015; Ludwig et al., 2019; Salipante and Horwitz, 2006; Spencer Chapman et al., 2021). In fact, brute force sequencing of single cell genomes enables lineage tracing (Behjati et al., 2014).
Recently, engineered recorder systems, such as GESTALT (McKenna et al., 2016), which induces mutations with CRISPR, provides for high throughput lineage reconstruction, while single cell RNA sequencing (scRNA-seq) has allowed elucidation of cell state trajectories based on gene expression patterns (Packer and Trapnell, 2018; Trapnell et al., 2014). Yet, the relationship between a cell’s developmental trajectory, as reflected in its transcriptome, and its lineage, as defined by cell division history, remains a critical question (Stadler et al., 2021; Wagner and Klein, 2020).
Engineered recorder systems can extract both cell state and lineage at single cell resolution by incorporating barcodes into a transcript read out through scRNA-seq (Raj et al., 2018). Of course, genetic engineering is ill-suited for studying development in humans, where correlation of cell state with lineage based on somatic mutations has required parallel RNA and DNA sequencing, respectively (Bizzotto et al., 2021; Huang et al., 2020).
In principle, mutation detection in RNA, compared to DNA, offers advantages. In order to perform DNA sequencing on a single cell, whole genome amplification (Frumkin et al., 2008), which is error-prone (Sabina and Leamon, 2015), or in vitro culture of clonally expanded cells (Behjati et al., 2014; Fasching et al., 2021; Salipante et al., 2008; Spencer Chapman et al., 2021), which is difficult to scale, is required. In contrast, thousands of copies of an RNA molecule may be present in a cell. However, the transcriptome comprises only a fraction of the genome. Accuracy limitations impose additional challenges: RNA polymerase has an error rate of ~10−5, which is ~5,000-fold higher than point mutation frequency (Gout et al., 2017); apparent mutations often reflect RNA edits (Ding et al., 2019); reverse transcriptase used to generate cDNA sequencing libraries has a fidelity of ~10−4 (Ji and Loeb, 1992); and next-generation sequencing misreads ~10−3 bases (Minoche et al., 2011).
Detection of loss of heterozygosity (LOH) offers a workaround. While point mutations are not uncommon, LOH, arising from recombination or other mechanisms, occurs frequently. Multiple regions of LOH of variable length are distributed throughout the genome, thereby uniquely marking cells. A vivid example involves somatic reversion of human germline keratin mutations in ichthyosis with confetti, in which innumerable revertant clones densely speckle skin (Choate et al., 2010). Measurements at marker genes indicate that LOH occurs ~10−4 - 10−5/locus/cell division (LaFave and Sekelsky, 2009; Larson et al., 2006; Moynahan and Jasin, 2010). LOH can be assayed at sites known to be germline heterozygous and are therefore predictable and more economical to survey than other mutations, arising unpredictably anywhere throughout the genome.
In the transcriptome, LOH should be recognizable as tracts of monoallelic expression stretched across adjacent genes. In fact, LOH is detectable in scRNA-seq datasets and has been used to reconstruct single cell lineages of tumors (Fan et al., 2018; Harmanci et al., 2020).
Extending such an approach to examine normal development across heterogeneous cell types imposes additional challenges because not every gene is expressed in all tissues. Here we explore a strategy for systematically reconciling gene expression with LOH determination. We employ scRNA-seq in interstrain hybrid mice to identify cells from gene expression patterns and simultaneously extract cell lineage by detection of LOH. We additionally register developmental time points based on X-chromosome inactivation, which occurs near gastrulation (Galupa and Heard, 2018), and the total number of LOH events.
In an initial application, we have studied the development of major cellular classes during mouse cerebral corticogenesis.
RESULTS
Cortical cell identification
We analyzed single cells from the cerebral cortex of eight mice (Figure 1A). In order to distinguish parental alleles, we studied F1 offspring of a cross between two different inbred strains, C57Bl/6J (B6) and CAST/EiJ (CA), whose genomes have previously been sequenced. We focused our analysis on Emx1+ cortical projection neuron and glia lineage. We utilized B6 mice transgenic for Emx1-Cre;Z/EG, such that enhanced green fluorescent protein marked cells that, at least at some point during development, had expressed the neuronal transcription factor EMX1 (Gorski et al., 2002). To control for parental sex, four mice were products of matings of female B6 and male CA parents while four mice had the reverse parentage. From each group of four, we analyzed a female and male at two developmental time points, postnatal days 0 (P0) and 42 (P42). Emx1-marked cells were isolated by flow cytometry and underwent scRNA-seq to a high depth of coverage using Smart-seq2 (Picelli et al., 2013). Approximately 50 cells from each mouse cortex (404, in total) passed quality control filtering (median of 1,735,775 unique reads per cell).
Figure 1. Isolation and transcriptome sequencing of mouse neocortical cells.

(A) B6 (blue) and CA (yellow) mice were crossed in both directions to create heterozygous F1 offspring (green). Single Emx1 expression-marked neocortical cells were isolated from two different ages and their transcriptomes sequenced.
(B) 404 cells, shown in dimensionally reduced (UMAP) space, were clustered based on gene expression and eight cell types identified. oIPC = oligodendrocytic intermediate progenitor cell.
(C) Representative allele plots of heterozygous X-chromosome SNVs in eight cells (rows) from a female P0 mouse, showing X-inactivation. Note the reciprocal allele state detected at the Xist/Tsix locus. Allele state: blue = B6, green = B6/CA, yellow = CA. N = neuron, A = astrocyte, O = oligodendrocyte.
(D) Active X-chromosome for female cells shown in dimensionally reduced space. NC = biparental/not called.
(E) Imprinting associated with the Prader-Willi/Angelman syndrome locus. Relative density histogram ridgeline plots (1 bp bins) of cells expressing maternal, paternal, or biparental variants at particular SNV locations from one mouse (P0-2, 64 cells). Chromosome coordinates, discussed genes, and Tabula Muris scRNA-seq coverage track (as in C) are shown on the x-axis. Relative density for each allele category is indicated on the y-axis.
UMAP visualizations and scRNA-seq based alleles are derived from Seurat_CountMatrix and VCFs Supplemental Data sets.
Using recognized markers and graph-based clustering (Figure S1A and Table S1), scRNA-seq allowed identification of eight cell types, depicted in dimensionally reduced (UMAP) space (Figure 1B).
Radial glial cells (RGC) are neural progenitors defined by expression of genes marking transition from neuroepithelial to mesenchymal states, including intermediate filaments Vimentin (Vim) and Nestin (Nes), extracellular matrix component Tenascin C (Tnc), neurogenic transcription factors Pax6 and Sox2, and glial markers GLAST (Slc1a3) and Blbp (Fabp7).
Three classes of neurons are apparent. Expression of microtubule-binding Doublecortin (Dcx) and transcription factor Neurod1 arises in the immature neural cluster and decreases in the more mature neuron and glutamatergic neuron clusters. Transcription of the microtubule component Beta III Tubulin (Tubb3) increases in the immature cluster and peaks in the neuron cluster. The immature neuron cluster also shows markedly increased expression of semaphorin Sema3c and Sox11 transcription factor, suggesting that these cells are radially migrating (Hoshiba et al., 2016; Wiegreffe et al., 2015), while the neuronal cluster is post-migratory. The post-mitotic neuronal marker NeuN (Rbfox3) shows low expression in immature neuron and neuron clusters, with a marked increase in the glutamatergic neuron cluster. Neuronal cytoskeleton components Nefm and Nefh also demonstrate increased transcription in glutamatergic neurons. The glutamatergic cluster is marked by the vesicular glutamate transporter vGluT1 (Slc17a7), the NMDA receptor subunit of the glutamate receptor channel GluN1 (Grin1), and glutaminase (Gls). Together these results describe the general maturation of a neuron to a functional state and are consistent with studies showing that Emx1 expression marks excitatory pyramidal projection neurons in the mouse cortex (Gorski et al., 2002).
Astrocytes are distinguishable by the presence of GLAST (Slc1a3), the glutamate/aspartate transporter Glt-1 (Slc1a2), and glutamine synthetase (Glul). We also identified a separate cluster of astrocytes showing increased expression of calcium-binding S100b and brain-specific aldolase C (Aldoc). S100b expression marks commencement of astrocyte terminal differentiation (Raponi et al., 2007).
We identified two oligodendrocyte populations. The first, oligodendrocytic intermediate progenitor cells (oIPC), is uniquely distinguished by platelet-derived growth factor receptor Pdgfra and proteoglycan Ng2 (Cspg4). Expression of the transcription factor Sox10 increases in these cells while continuing at a lower level in oligodendrocytes. The second population, mature oligodendrocytes, exhibits increased expression of the tight junction component Claudin 11 (Cldn11) and Myelin Oligodendrocyte Glycoprotein (Mog).
In general, clusters were evenly mixed with regards to individual mouse (Figure S1B) and sex (Figure S1C). Cell types are consistent with cortical age (Figure S1D). Analysis of cell cycle related genes (Table S2) reveals that clusters identified as mature and predicted to be post-mitotic are predominantly classified as G1 phase cells (Figure S1E). Most P42 cells are classified as G1, while the oIPC of those mice exhibit populations in both S and G2/M phases. RGC, immature neurons, and neurons display more heterogeneous distributions within the cell cycle. This is expected for RGC, but not for neurons, which are post-mitotic. However, our findings are consistent with previous studies of neurons describing gradual progression to a post-mitotic state, in which cell cycle genes can be expressed without leading to productive cell division (Anda et al., 2016). We also observe a small population of neuroblasts marked by Dlx1 and Dlx2 expression (Figure S2A). These cells arise from late RGC or older equivalent pools in the subventricular zone and migrate to the olfactory bulb via the rostral migratory stream (Díaz-Guerra et al., 2013). Dlx1 and Dlx2 expressing cells appear in RGC, immature neuron, neuron, and glutamatergic neuron clusters, as expected. When compared to Figure S1D, Dlx1 and Dlx2 expressing immature neurons are similarly distributed for both P0 and P42 mice. This expected overlap of cell type in younger and older mice, along with progression of maturing cells away from the RGC progenitor population (Figure 1B), suggests our clustering methods classify cells in a functional and maturation specific manner, rather than as an artifact of age or cell cycle.
Detection of X-inactivation and imprinting
A cross between different inbred mouse strains, each homozygous throughout their diploid genome, should predictably generate heterozygosity in F1 progeny wherever parental strains differ in DNA sequence. Using published genomes of the B6 and CA parental strains, we constructed a list of 20,667,142 single nucleotide variants (SNVs) across all autosomes and the X-chromosome, to serve as a guide for determining expressed allele status in each cell.
A measure of our variant calls and filtering methods can be provided by examining regions with predictable allele-specific expression patterns. In females, one of the two X-chromosomes is randomly inactivated during embryogenesis, leading to expression predominantly from one X-chromosome or the other (Galupa and Heard, 2018). This process is regulated by the X-chromosome controlling element (Xce), for which allelic variation confers different strengths of inactivation of the opposite X-chromosome, leading to skewed ratios of active X-chromosomes in F1 mice with different Xce alleles (Calaway et al., 2013). Certain regions on the inactive chromosome escape inactivation, and, in fact, some are responsible for maintaining the inactive state. Complicating matters is that escape from X-inactivation in hybrid mice appears dependent on parental strains and can be tissue specific (Andergassen et al., 2017; Berletch et al., 2015). These exceptions offer an important test of whether biallelic expression can be identified at loci that might otherwise be interpreted as monoallelic.
Plots of the X-chromosome from eight representative cells of a B6 (female) × CA (male) F1 P0 female hybrid mouse are shown in Figure 1C. For each cell, informative SNVs (i.e., those that differ between parental strains) are plotted along the length of the chromosome. Their position and color on the vertical axis indicates which parental allele(s) are detected, with monoallelic CA (yellow) shown on top, monoallelic B6 (blue) on the bottom, and biparental expression (green) in-between. Note that SNV coverage aligns with cell type specific gene expression, as shown by transcripts mapped for relevant cell types in the Tabula Muris project (Tabula Muris Consortium et al., 2018). Four cells exhibit inactivation of the B6 X-chromosome, while the other four cells reveal inactivation of the CA X-chromosome, as indicated by the predominant expression of the opposite parental allele. A reversal of this pattern occurs predictably at the Xist/Tsix locus. Xist and Tsix are exclusively expressed, respectively, from the inactive and active X-chromosomes and contribute to X-inactivation (Galupa and Heard, 2018). Some cells show regions with inconsistent escape from X-inactivation, including genes known to exhibit leaky expression (Table S3). In general, we observe a predictable pattern of skewing of X-active chromosomes. CA mice harbor the Xcec allele, which is more resistant to inactivation than the B6 Xceb allele (Figure 1D and Figure S2B). Seventy-eight percent of cells from female mice demonstrated CA active X-chromosomes. Parental origin of the Xce allele affected this ratio, as well, increasing the frequency of CA active X-chromosomes when Xcec is maternally inherited, in agreement with prior results (Calaway et al, 2013). Interestingly, female cells in which both X-chromosomes are active are most commonly immature astrocytes, raising the possibility that this cell type escapes X-chromosome inactivation more frequently than other cell types, though we emphasize that further study is required. For the X-chromosome, our data capture the expected strain and parent of origin variation in allele-specific transcription, with most cells displaying a dominant parental variant in female mice.
A few autosomal regions exhibit imprinting, containing genes in which only one or the other parental allele is expressed, thereby offering another opportunity to assess accuracy for identifying loci with biallelic expression amidst segments of allele-specific expression. An imprinted locus is the Prader-Willi/Angelman syndrome (PWS/AS) region of mouse chromosome 7, which contains several genes and noncoding RNAs, expressing only paternal copies of Snrpn, Snurf, Ipw, and Npn, along with exclusively maternal expression of Ube3a (Bervini and Herzog, 2013). Developing neurons and glial cells, however, biallelically express Ube3a (Judson et al., 2014). scRNA-seq captures Ube3a cell type specific relaxation of imprinting (Figure 1E). As expected, expression at other loci (Snrpn, Snurf, and Npn) is predominantly paternal, though we also observe low levels of biallelic or maternal expression.
Another cluster of imprinted genes on mouse chromosome 17 includes insulin-like growth factor 2 receptor (Igf2r) and lncRNA Airn (Latos et al., 2012). Igf2r imprinting is relaxed, however, in mouse astrocytes and oligodendrocytes (Hu et al., 1998). We correspondingly detect cells expressing either maternal or paternal Igf2r alleles, along with exclusively paternal expression of Airn (Figure S2C).
These examples demonstrate that our scRNA-seq data is sufficient to produce positive and specific biallelic variant calls, even in well-studied regions of the genome where monoallelic expression otherwise predominates.
Inferring LOH in single cells
We sought to detect LOH events in single cells from four mice (one of each sex, for P0 and P42 developmental stages), resulting from a cross between inbred female B6 and male CA parents. As noted, the genome of the zygote giving rise to each of these F1 mice is necessarily heterozygous for the millions of SNVs that distinguish the two parental strains. Based on scRNA-seq, SNVs homozygous for either parental variant suggest monoallelic expression, potentially consistent with LOH, while heterozygosity would indicate that both parental alleles are present, excluding the possibility of LOH. For each mouse, we observed a mean of 200,540 (SD 67,959) informative heterozygous SNVs distributed across mapped scRNA-seq reads from all cells. Recognizing that not all cells express the same genes, as well as incomplete capture of all transcripts, the median number of transcribed SNV coordinates that passed quality filters was 10,805/cell (LQ 7,232; UQ 14,160), yielding an average median autosomal coverage density of 4 SNVs/megabase (Mb) (LQ 3; UQ 6). These results show that scRNA-seq offers allele state information at a density comparable to commercial genomic SNP microarrays clinically employed for detecting LOH in bulk DNA samples in the ~20% larger human genome (e.g., Affymetrix High Density, 750K loci; Agilent Medium Density, 30K loci; Oxford Gene Low Density, 6,186 loci).
We find that SNV detection by scRNA-seq correlates with the number of uniquely aligned reads for each cell (Figure S2D), in an exponential manner, as expected (Kishikawa et al., 2019). Note that beyond approximately 1 to 2 million unique reads per cell, there are diminishing returns in terms of the added read depth required to detect additional SNVs. This level of read depth (about 1 million) serves as a convenient benchmark because, as noted above, it yields a SNV detection frequency similar to to genomic SNV arrays (~7,000 to 25,000 SNVs), which are relied on to clinically detect LOH and other copy number variants in humans
Comparison of Tabula Muris transcriptome data for neurons and associated glia with our list of predicted heterozygous loci in B6 × CA F1 mice shows that there are approximately 3.3 million SNVs theoretically available for detection by scRNA-seq in transcripts from these cell types. In principle, the use of 3’-capture scRNA-seq methods, such as SCI-RNA-seq (Cao et al., 2017), Drop-seq (Macosko et al., 2015), or 10X Genomics Chromium (Wang et al., 2021), may suffice with lower total read requirements, as the main filtering metric is quality normalized by depth (QD), and these methods cover a smaller footprint of the transcriptome. However, there is a tradeoff between footprint and possible SNVs detected that will depend on the variation created from the particular inbred mouse cross or outbred sample and the heterogeneity of the cell types sampled, as their transcriptomic footprint will vary accordingly. As sequencing technology evolves, we anticipate that the power of our method will commensurately improve.
Compared to bulk RNA-seq, scRNA-seq may artifactually indicate monoallelic expression due to stochastic sampling biases, especially for genes with modest levels of expression, and transcriptional bursting, in which, at the time sequencing is performed, only one active allele is captured (Borel et al., 2015; Deng et al., 2014; Finn and Misteli, 2019; Larsson et al., 2021; Reinius and Sandberg, 2015; Reinius et al., 2016; Symmons et al., 2019). The latter concern is at least partly mitigated by the fact that transcriptional bursts are typically shorter than the half-lives of RNA (Finn and Misteli, 2019). Nevertheless, these phenomena complicate interpretation of allele states. The chance that an apparently monoallelic SNV reflects sampling noise due to the nature of the scRNA-seq protocol, rather than an underlying LOH event, must be considered. To take advantage of the fact that contiguous tracts of monoallelic SNVs help validate interpretation of reads at adjacent positions, we employed a hidden Markov model (HMM) to infer the most likely genotype corresponding to observed patterns of allele specific expression.
Examples of chromosome 19 from two cells, one with evidence of LOH and one without, are shown in Figure 2A. For each cell, two plots are shown. The upper plot illustrates the observed allele state for any SNV based on scRNA-seq data, while the bottom plot shows the most likely HMM-inferred allele state. Cell 64461 demonstrates the tendency for scRNA-seq to capture SNVs within transcripts corresponding to only one allele. Most SNVs are observed as homozygous for either parental allele (colored yellow or blue by parent of origin). Nevertheless, biallelically expressed SNVs (green) and monoallelically expressed SNVs from either parent are interspersed throughout. Consequently, the inferred genotype for this chromosome is heterozygous, with no LOH events throughout its length. This interpretation is consistent with previous studies showing that in silico aggregation of what may appear to be predominately monoallelic scRNA-seq data becomes biallelic, matching bulk RNA-seq data from another sample of the same cell line (Borel et al., 2015). The second cell (64474) shows a similar stochastic monoallelic expression pattern, but two LOH events are inferred. The first is an interstitial event proximal to the centromere while the second is a more telomeric interstitial event.
Figure 2. Inferring LOH from scRNA-seq.

(A) HMM results showing two mouse cells with either no LOH event (cell 64461) or two events (cell 64474, marked with *) along chromosome 19. Detected loci are shown as in Figure 1C, with the vertical axis indicating expressed allele state (scRNA) or HMM-inferred genotype (HMM Genotype). Tabula Muris cell-type specific transcription tracks are shown at the bottom, N = neuron, A = astrocyte, O = oligodendrocyte.
(B) Length-normalized chromosome positions of all LOH events from all autosomes of one mouse.
(C) Distribution of LOH lengths (≥1 Mb) for all mice showing median (bar) and IQR (box).
(D) Violin plots of average LOH events per cell for each mouse (n = 56 (P0-1), 64 (P0-2), 56 (P42-2), 47 (P42-3)) and 10,000 randomly sampled in silico cells from the respective mouse. Mean and standard deviation are indicated for each by a black circle and vertical lines. *p-value <10−5 (two sample Z-test, mouse vs. 10K sampled cells).
(E) Autosomal barcode of 56 cells plus a virtual zygote (bottom bar) derived from one mouse (P0-2). Autosomes are shown at the top with the centromeres on the left. Black bars indicate autosomal boundaries. For all panels, blue = B6, green = B6/CA, yellow = CA.
Supplemental Data: HMM_Genotype_Tables
Applying this approach to all cells across all mice, we observe predominantly interstitial LOH events (Figure 2B). For all called LOH regions across all autosomes and cells, mean size (Mb), SD, and number of events are as follows: P0-1 - 5.30, 6.14, 795; P0-2 - 2.80, 3.65, 3447; P42-2 - 3.69, 6.44, 2469; P42-3 - 3.44, 6.93, 2381.
Allelic ratio for SNVs distinguishing parental origin in scRNA-seq datasets is noted to correlate for distances of up to ~500 kb, due in most part to SNVs residing within the same gene and hence the same sequenced RNA molecule (Borel et al., 2015). Correlation between SNVs in separate yet coordinately transcribed genes could conceivably also be observed when genes reside within the same topologically associating domain, which are ~0.2 - 1 Mb in length (Finn and Misteli, 2019). To control for such phenomena, we excluded regions of monoallelic expression <1 Mb from our analysis.
Data from high density SNP microarrays show that the predominant LOH event in healthy human tissue is interstitial (Melcher et al., 2011; Mohamedali et al., 2007; O’Keefe et al., 2010). The size distribution of LOH events we detect in mice (Figure 2C) is similar compared to what is reported for humans. For example, for the P0-1 mouse, we find a median size of 4.6 Mb, (range 1.0 - 29 Mb), compared to median estimates in humans of 1.2 Mb (range 0.3 - 6.7 Mb) (Mohamedali et al., 2007) and 8.7 Mb (range 0.3 - 65 Mb) (O’Keefe et al., 2010). In sum, our method of detecting LOH from scRNA-seq information appears to capture LOH events similar in size and chromosomal position to those observed using high density SNP microarrays based on human genomic DNA.
To determine if inferred LOH reflects noise or other sampling issues, we simulated 10,000 cells in silico for each mouse from pooled scRNA-seq data. For all four mice, we determined that the mean LOH events/cell is ~15 for actual data, compared to ~0.02 for randomly sampled data (Figure 2D), suggesting detected events are unlikely to be artifactual.
SNVs are neither uniformly distributed across a chromosome nor are the transcripts in which they are located expressed in all cells, creating ambiguity in precisely defining the boundaries of regions of LOH. We consequently assigned beginning and ending points for each region to 2 Mb bins tiled across the length of each chromosome. Using these conditions, we created an autosomal map of LOH events for each cell. This map, shown in Figure 2E, represents a cellular barcode uniquely identifying each cell. The collection of LOH events represent mitotically stable mutations that, just like other somatic mutations or genetically engineered barcodes, can be used to infer cell lineage.
We characterized the distribution of LOH events shared between cells within each mouse (Figure S3). Note that for our HMM model, ~12 consecutive monoallelic SNVs will trigger determination of an LOH event (Figure S3A-B). Most LOH regions are unique to a single cell, but as many as nine cells from a particular mouse share common events (Figure S3C). As expected, given the small proportion of sampled cells, most LOH events appear unique to a particular mouse (Figure S3D).
An LOH event should be more commonly shared by cells from the same mouse compared to cells from different mice. As a test, we devised an enrichment quotient. For each LOH event, the numerator represents the number of cells containing that allele (the particular LOH event) for one of the mice, and the denominator is the sum of all occurrences of that allele across all cells from that mouse and one other mouse. A value of one would indicate that the allele is unique to cells from a particular mouse, whereas a value of ~0.5 would indicate that the allele is just as likely to be found in cells from a different mouse. We report the mean quotient for all alleles for a particular mouse (Table S4). For each unidirectional pairwise comparison, the enrichment quotient ranges from 0.82 - 0.96. As expected, LOH events tend to recur within cells from the same mouse.
Detection of LOH by scRNA-seq in human cells
Toward the goal of using our method to map cell lineages in humans or other organisms in which genetic engineering is prohibitive, we measured our ability to detect known regions of LOH using scRNA-seq performed on human cells. We took advantage of the previously described human hepatoblastoma cell line, HepG2. HepG2 cells contain several regions of LOH that have been identified through use of haplotype-resolved whole genome sequencing and SNV microarrays (Zhou et al., 2019). scRNA-seq of HepG2 cell has been previously published (Hou et al., 2016), and we used this scRNA-seq data to examine three pairs of homologous chromosomes known to contain regions of LOH (chromosomes 6, 11, and 14). As an internal control, we compared our results to normally heterozygous diploid regions from each chromosome, as well as a few hyperdiploid segments derived from both parental homologs with 3N and 5N copy number. We called scRNA-seq based SNVs using the hg19 human reference genome, which we modified to incorporate HepG2 variant phase information. In general, we label the reference sequence as haplotype A and the alternate sequence as haplotype B.
Our approach detected the previously defined regions of LOH and did not identify LOH in chromosomal segments where it is not known to be present (Figure S4A). In heterozygous control regions we see similar ratios of monoallelic expression between haplotype A and B as well as lower levels of biallelic expression (Figure S4B). These ratios are similar to those seen in our mouse data for all cells over all autosomes (Figure S4C), where most monoallelic expression, especially when not occurring in contiguous tracts, appears to result from bursting and other sampling artifacts. When we compare the frequency of monoallelic or biallelic loci in reported LOH regions, we see a statistically significant (ANOVA with Tukey's Honest Significant Difference test, p <0.01) decrease in biallelic loci frequency (0.090 (0.031) to 0.006 (0.007), mean (SD)) as well as a shift in monoallelic frequency depending on haplotype. Haplotype A is reduced from 0.469 (0.050) to 0.085 (0.039), while haplotype B increases from 0.392 (0.062) to 0.909 (0.034) (Figure S4B). Our haplotype specific HMM model identifies LOH regions in four of the six total chromosomes examined, consistent with results reported for genomic DNA analysis (Zhou et al., 2019).
However, the regions we identified do not cover the entire reported LOH region even though we do see a decrease in heterozygosity and a shift of monoallelic SNV frequency. We attribute the small discrepancy to the presence of a few monoallelic loci within independent phase blocks with conflicting haplotypes. The phased parental haplotype blocks for this cell line, which is derived from a person from a presumably outbred population, do not span the complete chromosome. When we examine the phase set identities for the monoallelic haplotype A loci, they predominantly occur either in their own unique block or a block that is different from the next observed biallelic or monoallelic B SNV (Table S5). In other words, a challenge in extending our approach to humans involves the imprecision of linking large parental haplotype blocks together and inferring relative phase.
LOH as a marker of lineage
To deduce cellular lineage, we employed a Bayesian model of Camin-Sokal parsimony (Camin and Sokal, 1965), in which the most likely phylogeny minimizes the number of ordered LOH events from the base of the dendrogram to its tips. In order to root the lineage tree, for each mouse we added a cell representing the zygote, corresponding to the ancestral condition, with no LOH events assigned.
The consensus phylogram for a P0 (P0-2) mouse is shown in Figure 3A. The zygote occurs as an outgroup when compared to the rest of the cells (with an exception for just one other cell). Most changes occur towards the tips of the tree, and intranodal distances are short. Pairwise comparison of differences between cells exhibits a unimodal distribution (Figure S5A). Such a pattern, along with short intranodal distances, is consistent with expanding populations, as expected during embryogenesis; in contrast, populations of constant size exhibit more evenly spaced intranodal distances and monotonically decreasing pairwise-distance distributions (Rogers and Harpending, 1992). Given these results, we believe that our evolutionary model reflects the sequence of LOH events acquired during development (and is also supported by a benchmark analysis with mouse cells grown in vitro (Figure S5B) and a simulated C. elegans lineage (Figure S5C), as described further below.)
Figure 3. Phylogenetic analysis using LOH events.

(A) Lineage of 56 cortical cells plus a virtual zygote from a P0 mouse was inferred using a Camin-Sokal parsimony-inspired Bayes model. Consensus phylogram shows the relatedness and number of LOH events for each cell. Scale bar = 12.5 events. ■ RGC, ■ immature neuron, ■ neuron, ■ zygote.
(B) The same lineage in cladogram form with supporting nodal posterior probabilities ≥0.1 indicated, with mirrored “densiTree” representation of 1,500 sampled cladograms. The complete densiTree representation is shown as an inset with magnified area indicated. ● LOH event shared among all daughter cells, ▲ LOH event shared in all but one daughter cell.
(C) Barcode of segregating alleles (*) from a monophyletic clade marked “Panel C” in panel above. Chromosomes are aligned with the centromere to the left. Blue and yellow regions indicate LOH events (B6 and CA, respectively). Green regions indicate heterozygosity. Node posterior probability shown on the cladogram.
Posterior probability support for a specific topology is less robust. The consensus tree, depicted as a cladogram in Figure 3B, is based on 4,502 sample trees, and the 99% credible set contains 4,457 trees. This is unsurprising, given the large polytomy at the base of the tree. Node support is indicated along with a mirrored composite visualization of 1,500 overlaid trees from the credible tree set. For nodes with a posterior probability ≥0.1, the maximum level of support was 0.60 (median 0.17; IQR 0.11 - 0.35). In general, resolved nodes correlate with the presence of at least one shared LOH event in daughter cells (●), and several clades share an allele in all but one cell (▲), shown along branches in Figure 3B. For some clades, one allele appears sufficient to distinguish a clone, as shown by large dense wedges in the composite tree, but the topology of the monophyletic group cannot be resolved due to a lack of segregating alleles or confounding effects of coincidental identity by state (homoplasy). The informative chromosomal barcodes with segregating LOH alleles are shown for a representative clade in Figure 3C. Taken together, these results indicate that there is a lineage signal driven by LOH events that is detectable using parsimony, but confounding variables such as homoplasy limit our ability to discern finer topological structure.
While we do not know the true relationship of the cells of any given mouse, pairwise analysis of any two mice can provide insight into the relationship between posterior probabilities and truly related cells, along with an estimate of homoplasy. Cells from a particular mouse should group together based on the principle of identity by descent, while grouping of cells from different mice reflects homoplasy. An analysis of pooled cells from pairwise comparisons of mice (Figure S6A) reveals that a posterior probability of 0.65 - 0.75 is sufficient for confidently distinguishing related cells from those unlikely to descend from a common progenitor in our dataset.
Benchmarking lineage reconstructions
Recently, an open competition, the “Allen Institute Cell Lineage Reconstruction DREAM Challenge,” took place to evaluate the accuracy of various phylogenetic approaches for reconstructing cell lineage from single cell mutational data (Gong et al., 2021), providing an opportunity to test how well our approach compares against algorithms devised by others. The challenge incorporates two genetic-based lineage tracing systems. The first challenge is to reconstruct “ground truth” lineage trees of mouse embryonic stem cell colonies grown in vitro while being recorded with video microscopy and labeled with the intMEMOIR integrase-based lineage reporter system (Chow et al., 2021). The second consists of reconstructing the well known C. elegans lineage based on simulated mutations induced by the GESTALT CRISPR-Cas9 lineage reporter system (McKenna et al., 2016).
For tests based on both model systems, our Camin-Sokal Bayesian model performs favorably compared to other methods of lineage reconstruction, as measured by Robinson-Foulds (RF) and triplet distance metrics of topological similarity. Using large colonies in the experimentally determined mouse cell dataset, on average, our derived phylogenies had RF and triplet distance similarity scores as good as, or better than, the top three scoring algorithms (Cassiopea, Liu Lab, and Guan Lab, respectively) (Figure S6B). For the simulated C. elegans lineage, our attempts at lineage reconstruction resulted in RF distances comparable to the winning DCLEAR method (Figure S6C). However, our algorithm yielded a poorer triplet distance score, indicating low resolution of early branching in the phylogenies, and suggesting that further optimization of “tree space” search parameters for larger datasets is needed to improve accuracy.
Although not a metric incorporated into the DREAM Challenge, the availability of benchmarked datasets independently created by other genetic-based barcoding systems provides an opportunity to explore the allele variation observed for the sort of expanding cell populations expected to take place in the embryo. We reassuringly observe similar pairwise-distance distribution patterns for both the experimentally derived in vitro mouse embryonic stem cell and simulated C. elegans datasets from the DREAM Challenge as we do for our mice (Figure S5B and C).
Stereotyped expansion and differentiation in neocortical histogenesis
Mouse neocortical development consists of a progression of progenitor cells with decreasing proliferative potential (Taverna et al., 2014). Neuroepithelial stem cells divide symmetrically to produce the initial progenitor pool in the ventricular zone of the developing neocortex. This population transitions to an asymmetrical cell division stage consisting of RGCs to create unitary clusters of neurons either directly or through transient progenitors that expand the size of the cluster. A subset of RGC (about one-sixth) transitions to a gliogenesis state where they produce astrocytes and oligodendrocytes (Gao et al., 2014). Symmetrically dividing glial progenitors are produced as well (Ge et al., 2012). This stereotyped pattern of symmetrical cell division followed by unitary asymmetrical cell division, expansion, and differentiation should produce multiple monophyletic clades of mixed cell types. We see such a pattern in the inferred phylogenies. In P0 mice, the diversity of cell types is limited, but monophyletic groups appear, consisting of neurons and RGCs. P42 mice (Figure 4A), which have increased cellular diversity, show the same topological pattern. Heterogeneous clades are predicted to be composed of neurons, immature and mature astrocytes, plus oligodendrocytes and their intermediate progenitors, in agreement with a unitary expansion model of neurogenesis (Gao et al., 2014). Interestingly, the majority of glutamatergic neurons are part of a distinct clade in both P42 mice. In contrast, RGC and neurons in P0 mice are distributed throughout different clades. Correspondingly, we note that the large polytomies in each mouse consist of a similar number of monophyletic groups (15, 14, 14, and 11, for each of the four mice, respectively), though we cannot speak to whether or not this structure is meaningful, due to sampling only a limited number of cells.
Figure 4. Stereotyped expansion in the mouse neocortex.

(A) Consensus cladograms of two P42 mice. Nodes with posterior probability of >0.05 are resolved. Posterior probabilities of >0.10 are indicated. ● LOH event shared among all daughter cells, ▲ LOH event shared in all but one daughter cell.
(B) LOH events by mouse and cell type. Total autosomal LOH events are shown for each cell on the y-axis. Violin plots show event distribution for each cell type in each mouse.
(C) The active X-chromosome is overlaid onto cell lineages of two female mice. The active X-chromosome (square) and cell type (circle) is indicated by color. NC = biparental/not called. For clades composed of either active X-chromosome, the most recent common cell (internal nodes) are inferred to occur about or before the time of gastrulation (when X-inactivation takes place) and their respective preceding branches are colored red. Nodes predicted to occur after the approximate time of gastrulation are shown in black. RGC = radial glia cell, ImmN = immature neuron, Neur = neuron, GlutN = glutamatergic neuron, ImmA = immature astrocyte, Astro = astrocyte, oIPC = oligodendrocyte intermediate progenitor cell, Oligo = oligodendrocyte.
Generation of LOH events correlates with cell division (Mehta and Haber, 2014) and therefore may reflect mitotic age. At P0, particularly for the P0-2 mouse, the distribution of the number of identified LOH events among RGC is tighter than for either group of neurons (Figure 4B). The observation of fewer LOH events in more differentiated cells is consistent with RGC serving as a stem-like population, with neurons spun off earlier during embryogenesis. Conversely, the appearance of neurons exhibiting more LOH events is consistent with their production from later RGC populations, along with subsequent additional cell divisions prior to terminal differentiation. At P42, the distribution of the number of LOH events in immature astrocytes is more tightly clustered than for mature astrocytes, and the median number of events is lower. Though we do not have concurrent RGC with which to compare, this pattern is consistent with immature astrocytes being produced from transitioning late-stage RGC, expanding, then differentiating into mature astrocytes. There appear to be at least two waves of oligodendrocyte production given that contemporary precursors are mitotically older than mature oligodendrocytes. Our findings are consistent with the concept of early vs. late populations of RGC producing neurons and glial cells, indicating that the differences we observe in P42 mice relate to sampling of both populations (Kriegstein and Alvarez-Buylla, 2009).
Timing of nodes in relation to X-chromosome inactivation
X-inactivation occurs around the time of gastrulation, depending upon tissue (Tan et al., 1993), marks clonal populations deriving from common progenitors (Linder and Gartler, 1965), and has been used to map cell fate during corticogenesis (Tan and Breen, 1993). We superimposed our previously inferred X-activation status on cell lineages of two female mice (Figure 4C). Mixed clades containing cells in which one or the other of the two X-chromosomes is active point to a shared common progenitor existing prior to when X-inactivation takes place. In the P0-1 mouse, most of these pre-X-inactivation nodes occur after the large polytomy and closer to the terminal nodes. Mouse P42-2 shows an increase in predicted post-X-inactivation nodes, overall, as well as an increase after the large polytomy. This pattern is consistent with the ages of the mice, as more cell divisions would have occurred in P42 cells relative to X-inactivation, early in embryogenesis. X-activation status also supports our observation of glutamatergic neurons being part of a distinct clade of cells, as all cell types in the relevant P42-2 clade have the same active X-chromosome. However, due to the inherent CA skew of active X-chromosomes, we cannot rule out the possibility that these cells share their status due to chance.
To investigate this pattern further, we mapped these pre-X-inactivation nodes to their corresponding phylograms (Figure S7A and B), where branch length is proportional to the number of LOH events in the cell. Assuming cell division is symmetrical and LOH generation behaves in a clock-like fashion, most branching in these two samples occurs between establishment of the inner cell mass and gastrulation, as the mixed clade nodes occur further down the tree. Combined, these results suggest that the large polytomies in all four mice correspond to the initial expansion of the epiblast during embryogenesis and are consistent with stereotyped neuronal expansion of neuroepithelial stem cells. This observation does not rule out the possibility that the glutamatergic clade is clonal with regards to X-inactivation and marks an early wave of neurogenesis; rather, it more narrowly implies that the most recent common cell of the clade occured in this developmental period.
The known timing of X-inactivation can also be used to estimate how closely LOH “barcoding” tracks with actual cell divisions. X-inactivation takes place in the mouse at about 7.5 days gestation, when experimental determination of cell number reveals there to be a mean of 14,970 cells (Snow, 1977). Since the Log2 of 14,970 is approximately equal to 14, we estimate that about 14 cell divisions have taken place by this time. The lineage phylograms for P0-1 and P42-2 female mice reveal as many as 6 to 12 LOH events, respectively. We therefore estimate that there are between 6/14 (0.4) to 12/14 (0.9) LOH events per cell division in the early embryo and that the bifurcations in the phylogram come close to marking each cell division during epiblast expansion. The total number of LOH events should therefore serve as an approximate measure of the number of cell divisions during embryogenesis.
The sparse sampling of cells (approximately 50 for each mouse) restricts our ability to identify shared LOH events occurring at later developmental time points because shared events common to more than one cell are exponentially diluted with each successive cell division. This phenomenon likely explains why fewer bifurcations are recorded on the longer branches of the phylogram occurring after X-inactivation, as shown in Figure S7.
DISCUSSION
The goal of our studies is to reconcile lineage with cell state. Corticogenesis in the mouse involves a linear transition of distinct progenitors contributing to production of neurons and macroglia at different stages of development (Figure 5A). Due to the asymmetrical nature of RGC division and potential symmetrical expansion of some daughter cells, one RGC can contribute to multiple cell types and can tie cells produced at later stages of life to cells produced embryonically. Depending on when LOH occurs, several possible cell-type-specific phylogenetic topologies are possible. An event in neuroepithelial stem cells or early RGC would lead to mixed cell type clades, while asynchronous events occurring later during expansion of intermediate progenitor cells would mark more homogeneous clades. In P0 mice, we observe mixed clades consisting of RGC, immature neurons, and neurons, a pattern that is highlighted for large clades when member cells are plotted in dimensionally reduced space based on gene expression (Figure 5B). Similarly mixed clades are also seen in P42 mice, where cell types are more diverse, given their maturity.
Figure 5. Lineage across cell types and developmental time.

(A) Expansion and differentiation of Emx1+ neural stem cells (NSC) into neurons and glia, as adapted (Kriegstein and Alvarez-Buylla, 2009). Neuroendothelial (NE) cells expand (circular arrow) to form a pool of radial glial cells (RGC) that produce neurons and glia in the cortex via asymmetric cell division (red lines), expansion, and maturation (dashed lines). Time-dependent development proceeds horizontally. Observed cells in this study are indicated by vertical dotted lines along with their relative time points. oIPC = oligodendrocyte intermediate progenitor cell, SVZ = subventricular zone.
(B) Example of different clades encompassing multiple cell types. All 404 cells are shown in reduced dimensional (UMAP) space to illustrate all possible cell types, indicated by color-bound regions. Cells from four representative clades of a P0 and P42 mouse (inset cladograms) are indicated by their corresponding colors. UMAP visualizations are derived from the Seurat_CountMatrix Supplemental Data set.
Our method is based on the detection of LOH in scRNA-seq datasets. Reasons other than LOH for why transcripts corresponding to only one parental allele may be detected include inadequate depth of coverage for rare transcripts, capturing a moment in time when only one allele is actively transcribed (bursting), and imprinting or other forms of allelic exclusion, such as X-inactivation or involving antigen receptors in the immune system or olfactory receptors (Khamlichi and Feil, 2018).
We control for the first two issues, sampling and bursting, by relying on concordant uniparental expression across multiple adjacent SNVs spanning large chromosomal regions, show that regions of apparent LOH largely disappear following data permutation, and that regions interpreted as LOH are more likely to be shared between cells sampled from the same mouse compared to cells sampled from different individuals.
Regarding the latter issue, random mitotically stable monoallelic expression (Savova et al., 2016) has been proposed as a regulatory mechanism. For example, monoallelic expression of Nanog has been posited to regulate pluripotency (Miyanari and Torres-Padilla, 2012), while other reports find that its expression is biallelic and no different from other genes playing similar developmental roles (Faddah et al., 2013; Filipczyk et al., 2013).
In one study (Reinius et al., 2016), the frequency of monoallelic expression observed by scRNA-seq of primary mouse cells was 13%, but when pooled following in vitro clonal expansion was reduced to ~0.5%. The higher frequency seen in primary cells was attributed to bursting. In contrast, we observed a mean of 5.1% (range 0 - 19.1%) of the autosomal genome exhibiting monoallelic expression. One reason the value we determined may be larger is that our analysis of heterogeneous cell types requires binning of the boundaries of regions of monoallelic expression to account for differences in gene expression between individual cells, necessarily leading to overestimation. Another is that genes from regions with cell- or clone-specific chromosomal aberrations, which might underlie clonal abnormalities we interpret as LOH, were excluded from the prior study (Reinius et al., 2016). Nevertheless, for any given locus exhibiting monoallelic expression, we find the vast majority of cells exhibit a heterozygous genotype, which would not be true if random monoallelic expression were pervasive.
Several pathways can lead to LOH, whether it be copy neutral or result in loss of chromosomal material (Mehta and Haber, 2014). A mechanism of LOH consistent with our observations is gene conversion, resulting in copy neutral LOH, but we cannot exclude LOH due to deletion of the undetected allele. While the density of informative SNVs in our experiments is comparable to SNP microarrays clinically employed for detection of LOH and copy number variants in bulk human DNA samples, cell-to-cell differences in gene expression pose a challenge for interpreting copy number.
A distinct advantage to the approach described here, compared to recently introduced technologies for dynamically barcoding cells through genome editing, is that it can be performed retrospectively, permitting study of development in humans and other organisms where genetic engineering or other forms of embryonic manipulation are infeasible or where lifetimes are long. Unlike laboratory mouse strains, humans and individuals from other species with large populations are outbred, and heterozygous SNVs are abundant, making the method applicable without any special breeding strategy, provided that the underlying phased genome sequence is determinable using experimental and/or computational approaches (Choi et al., 2018; Harmanci et al., 2020).
We analyzed scRNA-seq for a human cell line, HepG2, where several regions of LOH had been previously identified using other means. While we were able to detect LOH, the boundaries did not precisely align with what was previously reported. Given that transcripts do not necessarily cover genomic breakpoints, approximation of the boundaries is not unexpected. An additional factor is the inherent imprecision of phasing the genome in an individual from an outbred population. An opportunity for refining the accuracy of genomic phasing includes sequencing parents, if available. Continuing advances in long-read genomic sequencing should also help improve methods for phasing the genome and, perhaps eventually, direct detection of LOH.
A limitation of our study is the lack of ground truth for true lineage, though X-inactivation analysis provides a spot check for clonality and permits determination of the developmental timing of early embryonic lineage branchpoints. We benchmarked our approach using Allen Institute Cell Lineage Reconstruction DREAM Challenge tests involving in vitro mouse cell colonies and the simulated C. elegans lineage. An insight gained from this exercise is that the computational resources required for our Bayesian approach to phylogenetic reconstruction may be difficult to scale for larger datasets. This becomes especially important considering the need to sample a large number of cells if one wishes to define later branches of the lineage tree. Otherwise, only mutations arising early during development and that are shared by a large proportion of sampled cells will be informative for lineage reconstruction.
A limitation common to all lineage tracing methods utilizing independently evolving loci is homoplasy, as indicated by overall modest performance current methods achieve with regards to measures of similarity to known lineage topologies. A promising future solution to this problem would be analysis of a combination of barcoding sources, much like consolidating physical character state evolution and molecular data in modern phylogenetics. In such a scenario, allele production rates that are optimal for the developmental time frames of interest could be selectively analyzed.
Another limitation of our approach is that it requires a breadth and depth of scRNA-seq sufficient to infer the allelic origin of SNVs contained within a transcript, compared to more limited sampling necessary to simply identify a transcript. As throughput increases and sequencing costs decline, this may become less problematic.
We additionally cannot entirely exclude epigenetic silencing as an explanation for monoallelic expression. Even if epigenetic phenomena were contributing to our calls of LOH, the information may still prove useful for inferring lineage. In fact, an early approach for retrospectively inferring lineage from sequencing data employed detection of cytosine methylation, involved in developmentally regulated gene silencing (Shibata and Tavaré, 2007).
In sum, we build upon earlier work analyzing LOH in tumors (Fan et al., 2018; Harmanci et al., 2020) and demonstrate the ability of scRNA-seq combined with allele-specific SNV analysis to capture the developmental trajectory of excitatory pyramidal projection neurons and glia, infer patterns of cellular expansion in mouse cortex, and correlate lineage relationships of progenitor cells with their descendants.
STAR★METHODS
RESOURCES AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marshall Horwitz (horwitz@uw.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table. All derived datasets, including the original Seurat count matrix used for cell clustering, genotype calls for all mice and human HepG2 cells, VCF files for mouse and HepG2 variants, consensus phylogenies in NEXUS format, and Allen Institute Cell Lineage Reconstruction DREAM Challenge phylogenies in Newick format are provided in a Mendeley Data archive and are available as of the date of publication. The DOI is listed in the key resources table.
All original code has been deposited in a Mendeley Data archive and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| scRNA sequencing data - mouse | Laukoter et al., 2020 | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716 |
| scRNA sequencing data - human HepG2 | Hou et al., 2016 |
GSM2039769 GSM2039770 |
| HepG2 cell line genome data | (Zhou et al., 2019) | ENCODE:ENCBS760ISV |
| GRCm38.p5 | NCBI | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.25/ |
| Ensembl | EMBL-EBI | https://www.ensembl.org/index.html?redirect=no |
| CA Mouse Genome Project sequencing data | Keane et al., 2011 | ftp://ftp-mouse.sanger.ac.uk/current_snps/strain_specific_vcfs/CAST_EiJ.mgp.v5.snps.dbSNP142.vcf.gz |
Supplemental Data
|
This study | Horwitz, Marshall (2022), “Anderson_LOH_v3”, Mendeley Data, V1, doi: 10.17632/23f7j32zmf.1 |
| Experimental models: organisms/strains | ||
| Mouse: CAST/Ei | The Jackson Laboratory | RRID:IMSR_JAX:000928 |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:00066 |
| Mouse:Emx1-Cre | The Jackson Laboratory | RRID:IMSR_JAX:005628 |
| Software and algorithms | ||
| STAR 2.5.0c | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Seurat R package | Butler et al., 2018; Stuart et al., 2019 | https://satijalab.org/seurat/ |
| GATK | Keane et al., 2011 | https://gatk.broadinstitute.org/hc/en-us |
| HMM R package | Prof. Dr. Lin M. Himmelmann, University of Applied Sciences Rapperswil, Switzerland | https://cran.r-project.org/web/packages/HMM/index.html |
| MrBayes | Ronquist et al., 2012 | http://nbisweden.github.io/MrBayes/ |
| DensiTree | Bouckaert, 2010 | https://www.cs.auckland.ac.nz/~remco/DensiTree/ |
Supplemental Code
|
This study | Horwitz, Marshall (2022), “Anderson_LOH_v3”, Mendeley Data, V1, doi: 10.17632/23f7j32zmf.1 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The dataset we used for our analysis was previously published; mouse strains, breeding strategy, cell isolation, and scRNA-sequencing approaches are as described (Laukoter et al., 2020). Briefly, B6 (Emx1-Cre;Z/EG) and CA mouse strains obtained from The Jackson Laboratory were bred and analyzed in accordance with protocols approved by Institutional Animal Care and Use Committees at IST Austria. Cells were dissociated from cerebral cortex, and Emx1+ single cells were sorted by FACS for library preparation.
METHOD DETAILS
scRNA-seq
RNAseq cDNA libraries were prepared from single cells using Smart-seq2 (Picelli et al., 2013). Single-end 50 base pair (bp) reads were mapped to GRCm38.p5 (mm10) and expression determined using Ensmbl [91, Dec 2017] with STAR 2.5.0c (Dobin et al., 2013), as previously described (Laukoter et al., 2020). A million or greater total reads and a range of 10,000 - 30,000 total mRNAs were used to filter high quality single cell samples. 1,735,775 unique reads were mapped to the median cell (Lower Quantile (LQ) 990,582, Upper Quantile (UQ) 2,858,522). RNA sequencing data can be accessed using the GEO Series accession number GSE152716 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716). HepG2 RNAseq cDNA libraries (GSM2039769 (HepG2_1) and GSM2039770 (HepG2_2)) were aligned to hg19 as above, resulting in 3.6 and 3.8 million uniquely mapped reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cell Identification
Cell identity was determined through the Seurat R package (Butler et al., 2018; Stuart et al., 2019). Gene level expression data from all 404 cells from 8 mice were combined and filtered. Genes expressed in a minimum of three cells were analyzed. Cells with a minimum of 200 genes detected were kept for further inspection. All 404 cells passed the criteria above resulting in a 404 × 23,373 feature count matrix (Supplemental Data). The count matrix was normalized with the NormalizeData function, setting normalization.method to “LogNormalize” and scale.factor to 10,000. Each cell was assigned a cell cycle state using the CellCycleScoring function. The list of S and G2/M genes is provided in Table S2. For clustering of cells we reduced the dimensionality of the expression data via principal component analysis (PCA) of the top 2,000 variably expressed genes (selection.method = “vst”). This expression matrix was scaled (mean expression = 0, variance = 1) so that highly-expressed genes do not dominate downstream analysis. We also removed variation due to mitochondrial contamination, library preparation (individual mouse), mouse age, and cell cycle stage (S or G2/M) using the vars.to.regress option in the ScaleData function (Figure S1C). Cells were clustered, based on the first 15 components, using Seurat’s graph-based clustering tools. A K-nearest neighbor graph, based on Euclidean distance in PCA space, was constructed for all cells using the FindNeighbors function. The Louvain algorithm (FindClusters(resolution = 0.8)) was then used to iteratively group cells together. Clustered cells were then annotated based on the initially normalized marker gene expression patterns (Table S1).
scRNA-seq Variant Calling
Heterozygous sites were identified using the CA Mouse Genome Project sequencing data (CAST_EiJ.mgp.v5.dbSNP142.vcf) (Keane et al., 2011). Briefly, homozygous SNV loci for CA, when compared to mm10 (B6), passing GATK hard filtering metrics for SNV detection based on DNA sequencing were identified and used as a guide (ROD file) for loci to be analyzed in GATK. 20,667,142 SNV loci were identified. On average, this provides 8,392 loci per Mb coverage of autosomes and X chromosomes. RNA-based SNV calls were made at predicted sites of heterozygosity in B6:CA F1 mice using the GATK (Auwera et al., 2013) variant calling tool (-T GenotypeGVCFs -stand_call_conf 20 -stand_emit_conf 10) and hard filtered using recommended settings for scRNA-seq variants (-T VariantFiltration -window 35 -cluster 3 -filterName FS -filter “FS > 30.0” -filterName QD -filter “QD < 2.0”). SNV loci containing variants due to alignment errors near splice sites that passed these filters (1,144 total) were removed.
For HepG2 samples, variants were called as above using a phase specific version of hg19 for chromosomes 6, 11, and 14 constructed from previously reported phased DNAseq data (ENCODE:ENCBS760ISV; ENCFF853HDD). Variants at heterozygous loci that were found in phase with the hg19 reference sequence (1∣0) were incorporated into a HepG2 version of hg19 using the GATK tool FastaAlternateReferenceMaker. A ROD file was then constructed using heterozygous SNV loci across the chromosomes as well as homozygous variant loci in the reported LOH regions.
Genotype Determination
Studies focused on a subset of four mice (P0-1, 56 cells; P0-2, 64 cells; P42-2, 56 cells; and P42-3, 47 cells). A cell's genotype at a particular locus was inferred by modeling the scRNA-seq variant data as a HMM process with the underlying genotype at a particular locus corresponding to the hidden state and the scRNA-seq-based variant status as the emission or observation state.
We posit three hidden states relating to a cell’s genotype: heterozygous, homozygous B6, or homozygous CA. A transition from the heterozygous state to either homozygous state is 10,000-fold less likely than remaining in the heterozygous state, based on observed frequencies of interhomolog chromosomal exchanges generating LOH (Larson et al., 2006). Within a stretch of homozygosity created by recombination, transitioning back to the heterozygous state would require a second recombination event, which observations suggest is 10-fold less likely than a single event (i.e., overall 100,000-fold less likely than remaining in the particular state) (Larson et al., 2006). Transitions from one homozygous genotype to the other cannot be generated through recombination. The ultimate effect of these transition rates is that, given an observed homozygous run of either allele, there is a small bias for continuation of calling a homozygous state, instead of transitioning back and forth between heterozygous and homozygous states. The transition matrix between states is defined as:
| Homozygous B6 | Heterozygous | Homozygous CA | |
|---|---|---|---|
| Homozygous B6 | 0.99999 | 0.00001 | 0 |
| Heterozygous | 0.00010 | 0.99980 | 0.00010 |
| Homozygous CA | 0 | 0.00001 | 0.99999 |
The probability of observing a particular SNV in the heterozygous state or either homozygous state is described by the emission state matrix. Due to both bursting and incomplete sampling, we estimated that biallelic SNVs will be observed correctly as heterozygous in the scRNA-seq data with a probability of 0.10 but will be more frequently incorrectly observed as monoallelic for the maternal allele with probability 0.45 or the paternal allele also with a probability of 0.45, based on comparisons of bulk to single cell sequencing (Borel et al., 2015) and our own results, where we observed for all 404 cells a mean (SD) proportion of SNVs as 0.438 (0.007) monoallelic B6, 0.105 (0.012) biallelic, and 0.456 (0.012) monoallelic CA. A truly homozygous locus should only be observed as monoallelic. An observation of either heterozygous SNVs or homozygosity for a SNV from the opposite allele would switch the hidden state back to heterozygosity. The observed scRNA-seq-based genotype (monoallelic B6, biallelic B6:CA, or monoallelic CA) is based on the emission state probability of the underlying hidden state given here:
| Hidden\Emission state | Monoallelic B6 | Biallelic | Monoallelic CA |
|---|---|---|---|
| Homozygous B6 | 1 | 0 | 0 |
| Heterozygous | 0.45 | 0.10 | 0.45 |
| Homozygous CA | 0 | 0 | 1 |
The Markov-chain's starting state probability is given as 0.05 for each of the homozygous states and as 0.90 for the heterozygous state. In our model, the heterozygous to homozygous transition probability only modestly influences determination of tracts of LOH. A ten-fold decrease in probability (i.e., from 10−4 to 10−5) results in a requirement for just three more continuously observed monoallelic SNVs necessary to assign a region of LOH (Figure S3A). We therefore selected an intermediate transition probability to account for genome-wide variance in observed frequencies of LOH (Larson et al. 2006). In fact, most LOH events would still be called using this model if the transition probability were set to 10−8 (Figure S3B). For HepG2 samples the same model was applied. In this analysis we substituted the HepG2-specific version of hg19 (haplotype A) for B6 while the hg19 variant sequence (haplotype B) replaced CA.
For any given chromosome of any given cell the underlying genotypes of scRNA-seq detected loci were determined by the most probable order of hidden states (Viterbi path) based on the scRNA-seq variant data using the Viterbi algorithm contained in the HMM R package (https://cran.r-project.org/web/packages/HMM/index.html). The Viterbi path describes the amalgamation of both homologous chromosomes and is assumed to have a copy number of two. Hemizygosity will appear as LOH, and duplications will appear as heterozygous (Supplemental Data).
Identification of LOH Events
A single transition from the hidden heterozygous state to either homozygous state could indicate recombination between two homologous chromosomes that extends to the telomere. A subsequent transition from the homozygous state back to the heterozygous state would describe an interstitial event such as a double crossover or non-crossover strand invasion. Partial or micro-chromosomal deletions would also trigger these transitions. Transcriptional bursting kinetics or epigenetic silencing on one allele could account for short runs of either B6 or CA alleles, as well, confounding our results. To control for such events we removed homozygous runs that spanned a region <1 Mb.
Due to non-uniform gene expression and scRNA-seq coverage across cells we grouped LOH events in 2 Mb bins, starting at the beginning of each chromosome. Events were characterized by their beginning and ending bins along with their parental allele identity.
Permutation Analysis
For each mouse, a subset of 10,000 randomly sampled “cells” was created in silico based on the observed scRNA-seq variants. For each locus, an scRNA-seq allele state (no coverage, biallelic, monoallelic B6, or monoallelic CA) was randomly sampled with replacement from the pool of allele states at that locus across all cells from the mouse in question. This process was repeated for all analyzed loci along the length of each autosome. These virtual cells were then genotyped as described above, and LOH events were identified for each generated cell (Supplemental Data).
Encoding LOH Events for Phylogenetic Analysis
Tracts of LOH ≥1 Mb are described by four elements: their chromosome location, the 2 Mb bin in which they start, the strain identity of the tract (B6 or CA), and their ending 2 Mb bin. For example, a homozygous run on chromosome 19, starting at position 5,000,000 and ending at position 9,000,000, and consisting exclusively of CA variants would be described as chr19_3CA5. A chromosome with no LOH event is coded as HT, designating the chromosome as apparently heterozygous (e.g., for chromosome 19 the designation would be chr19_HT). A chromosome for any one cell can contain multiple LOH events.
Each chromosome is considered its own independently evolving region within a mouse, with the cell specific combination of LOH events defining its state. An LOH event occurring over a specific region of the chromosome does not preclude another event happening nor does it affect the probability of that event happening. Once a heterozygous stretch of chromosome is converted to a homozygous run it cannot revert back to the homozygous state. For any chromosome, each LOH event is treated as independent and irreversible. The multiple character states for any chromosome in any mouse can be factored into binary characters representing the presence (1) or absence (0) of any chromosome specific LOH event. For example, consider the state of chromosome 19 in three fictitious cells:
Cell01: chr19_HT
Cell02: chr19_2B65, chr19_20CA30
Cell03: chr19_2B65, chr19_15B626
There are three unique LOH events with one cell lacking any event. These cells would be factored and encoded as:
| Cell\State | chr19_2B65 | chr19_15B626 | chr19_20CA30 |
|---|---|---|---|
| Cell01 | 0 | 0 | 0 |
| Cell02 | 1 | 0 | 1 |
| Cell03 | 1 | 1 | 0 |
Allen Institute Cell Lineage Reconstruction DREAM Challenge benchmark data (Gong et al., 2021) were encoded as follows:
intMEMOIR alleles: Sub-challenge 1 alleles recorded from videomicroscopy of in vitro grown mouse embryonic stem cells based on the intMEMOIR recording cassette were flattened into a similar binary matrix with either allele (inversion or deletion) coded as present or absent (not edited) for each of the ten sites. Each site was treated as independently evolving, edits as irreversible, and the unedited state as ancestral.
GESTALT alleles: Sub-challenge 2 alleles synthesized in silico from the C. elegans lineage based on the GESTALT recording cassette were flattened into a similar binary matrix with all observed edited alleles (A-Z, a-e, and “-”) for 200 sites coded as present or absent. Each site was treated as independently evolving, edits as irreversible, and the unedited state as ancestral.
LOH Evolution Model
We implement a Camin-Sokal (Camin and Sokal, 1965) parsimony-inspired Bayesian model of LOH evolution to infer lineage relationships, using the restriction site model in the MrBayes software package (Ronquist et al., 2012) with default mcmc tool settings and ending analysis when the average standard deviation of split frequencies approached 0.01. LOH events for any one chromosome can be described as a discrete and irreversible event. We coded autosomal LOH events occurring on the same chromosome as a series of two-state characters, with heterozygosity being the ancestral character. It is assumed that the zygote for any F1 mouse is heterozygous across the assayed SNVs for any particular autosome (a state of 0 in the coding scheme described above), and that this represents the ancestral condition. An LOH event creates a new character state (1), and the transition from 0 to 1 is 100-fold more likely than the reverse (prset statefreqr=fixed(0.01,0.99)). The zygote branch length is expected to be close to zero and placed as the outgroup in the resultant dendrogram. We excluded the sex chromosomes. Though LOH events can occur between two X-chromosomes, sex chromosomes are not considered here because two mice are male, and X-inactivation in female mice changes assumptions of the HMM used to infer genotypes based on RNA. Visualization of the Bayesian posterior distribution of cladograms employs the DensiTree algorithm (Bouckaert, 2010). For Allen Institute Cell Lineage Reconstruction DREAM Challenge data, the phylogenies were determined similarly with the following changes. No representative zygote was added to the analysis. The temperature setting for the GESTALT based lineages was set to 0.1 for the “Horwitz-1” phylogeny and 0.05 for the “Horwitz-2” phylogeny.
Benchmark Analysis Utilizing Allen Institute Cell Lineage Reconstruction DREAM Challenge Data
Lineage recording data for in vitro cell colonies and simulated recording data for C. elegans development was obtained from the Allen Institute Cell Lineage Reconstruction DREAM Challenge repository (https://www.synapse.org/Portal.html#!Synapse:syn20692755/wiki/597064). intMEMOIR in vitro lineage barcode data (sub-challenge 1) for the five largest test cell colonies (test_11, test_14, test_25, test_28, and test_29) were retrieved, along with simulated GESTALT lineage barcode data (sub-challenge 2), and analyzed using our Camin-Sokal Bayes model. Inferred rooted phylogenies were compared to provided ground-truth cladograms for topological similarity using both Robinson-Foulds and Triplet distances. Scoring was accomplished using both the treeMan R package along with the provided custom DREAM Challenge scoring method.
Supplementary Material
Table S5. HepG2 cells phase blocks, Related to STAR Methods Section, scRNA-seq Variant Calling, xlsx file.
Table S4. LOH Allele Enrichment, Related to Figure 3, xlsx file.
Table S3. Genes Escaping X-Inactivation, Related to Figure 1, xlsx file.
Table S2. Cell Cycle Related Genes, Related to Figure 1, xlsx file.
Table S1. Genes Used to Identify Cell Types, Related to Figure 1, xlsx file.
Highlights.
Temporally resolved single cell lineage reveals developmental patterning of mouse brain.
Acquired LOH allows for inference of cell lineage and is identifiable through scRNA-seq.
Determining the active X-chromosome in females registers timing of lineage branchpoints.
ACKNOWLEDGEMENTS
DJA thanks Wayne K. Potts, Alan R. Rogers, Kristen Hawkes, Ryk Ward, and Jon Seger for inspiring a young undergraduate to apply evolutionary theory to intra-organism development. Supported by the Paul G. Allen Frontiers Group (University of Washington); NIH R00HG010152 (Dartmouth); and NÖ Forschung und Bildung n[f+b] life science call grant (C13-002) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program 725780 LinPro to SH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anda F.C. de, Madabhushi R, Rei D, Meng J, Gräff J, Durak O, Meletis K, Richter M, Schwanke B, Mungenast A, et al. (2016). Cortical neurons gradually attain a post-mitotic state. Cell Res. 26, 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D, Dotter CP, Wenzel D, Sigl V, Bammer PC, Muckenhuber M, Mayer D, Kulinski TM, Theussl H-C, Penninger JM, et al. (2017). Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. Elife 6, e25125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols in Bioinformatics 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, et al. (2014). Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513, 422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berletch JB, Ma W, Yang F, Shendure J, Noble WS, Disteche CM, and Deng X (2015). Escape from X inactivation varies in mouse tissues. PLoS Genet. 11, e1005079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bervini S, and Herzog H (2013). Mouse models of Prader-Willi Syndrome: a systematic review. Front. Neuroendocrinol 34, 107–119. [DOI] [PubMed] [Google Scholar]
- Bizzotto S, Dou Y, Ganz J, Doan RN, Kwon M, Bohrson CL, Kim SN, Bae T, Abyzov A, NIMH Brain Somatic Mosaicism Network, et al. (2021). Landmarks of human embryonic development inscribed in somatic mutations. Science 371, 1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel C, Ferreira PG, Santoni F, Delaneau O, Fort A, Popadin KY, Garieri M, Falconnet E, Ribaux P, Guipponi M, et al. (2015). Biased allelic expression in human primary fibroblast single cells. Am. J. Hum. Genet 96, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert RR (2010). DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26, 1372–1373. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calaway JD, Lenarcic AB, Didion JP, Wang JR, Searle JB, McMillan L, Valdar W, and Pardo-Manuel de Villena F (2013). Genetic architecture of skewed X inactivation in the laboratory mouse. PLoS Genet. 9, e1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camin JH, and Sokal RR (1965). A Method for Deducing Branching Sequences in Phylogeny. Evolution 19, 311–326. [Google Scholar]
- Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CA, Kas A, Kirkwood R, Hays LE, Preston BD, Salipante SJ, and Horwitz MS (2011). Decoding cell lineage from acquired mutations using arbitrary deep sequencing. Nat. Methods 9, 78–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choate KA, Lu Y, Zhou J, Choi M, Elias PM, Farhi A, Nelson-Williams C, Crumrine D, Williams ML, Nopper AJ, et al. (2010). Mitotic Recombination in Patients with Ichthyosis Causes Reversion of Dominant Mutations in KRT10. Science 330, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chan AP, Kirkness E, Telenti A, and Schork NJ (2018). Comparison of phasing strategies for whole human genomes. PLoS Genet. 14, e1007308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow K-HK, Budde MW, Granados AA, Cabrera M, Yoon S, Cho S, Huang T-H, Koulena N, Frieda KL, Cai L, et al. (2021). Imaging cell lineage with a synthetic digital recording system. Science 372. [DOI] [PubMed] [Google Scholar]
- Deng Q, Ramsköld D, Reinius B, and Sandberg R (2014). Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196. [DOI] [PubMed] [Google Scholar]
- Díaz-Guerra E, Pignatelli J, Nieto-Estévez V, and Vicario-Abejón C (2013). Transcriptional regulation of olfactory bulb neurogenesis. Anat. Rec 296, 1364–1382. [DOI] [PubMed] [Google Scholar]
- Ding J, Lin C, and Bar-Joseph Z (2019). Cell lineage inference from SNP and scRNA-Seq data. Nucleic Acids Res. 47, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faddah DA, Wang H, Cheng AW, Katz Y, Buganim Y, and Jaenisch R (2013). Single-cell analysis reveals that expression of nanog is biallelic and equally variable as that of other pluripotency factors in mouse ESCs. Cell Stem Cell 13, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Lee H-O, Lee S, Ryu D-E, Lee S, Xue C, Kim SJ, Kim K, Barkas N, Park PJ, et al. (2018). Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 28, 1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasching L, Jang Y, Tomasi S, Schreiner J, Tomasini L, Brady MV, Bae T, Sarangi V, Vasmatzis N, Wang Y, et al. (2021). Early developmental asymmetries in cell lineage trees in living individuals. Science 371, 1245–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipczyk A, Gkatzis K, Fu J, Hoppe PS, Lickert H, Anastassiadis K, and Schroeder T (2013). Biallelic expression of nanog protein in mouse embryonic stem cells. Cell Stem Cell 13, 12–13. [DOI] [PubMed] [Google Scholar]
- Finn EH, and Misteli T (2019). Molecular basis and biological function of variability in spatial genome organization. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumkin D, Wasserstrom A, Itzkovitz S, Harmelin A, Rechavi G, and Shapiro E (2008). Amplification of multiple genomic loci from single cells isolated by laser micro-dissection of tissues. BMC Biotechnol. 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galupa R, and Heard E (2018). X-Chromosome Inactivation: A Crossroads Between Chromosome Architecture and Gene Regulation. Annu. Rev. Genet 52, 535–566. [DOI] [PubMed] [Google Scholar]
- Gao P, Postiglione MP, Krieger TG, Hernandez L, Wang C, Han Z, Streicher C, Papusheva E, Insolera R, Chugh K, et al. (2014). Deterministic progenitor behavior and unitary production of neurons in the neocortex. Cell 159, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marques J, Espinosa-Medina I, and Lee T (2021). The art of lineage tracing: From worm to human. Prog. Neurobiol 199, 101966. [DOI] [PubMed] [Google Scholar]
- Ge W-P, Miyawaki A, Gage FH, Jan YN, and Jan LY (2012). Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Granados AA, Hu J, Jones MG, Raz O, Salvador-Martínez I, Zhang H, Chow K-HK, Kwak I-Y, Retkute R, et al. (2021). Benchmarked approaches for reconstruction of in vitro cell lineages and in silico models of C. elegans and M. musculus developmental trees. Cell Syst 12, 810–826.e4. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, and Jones KR (2002). Cortical Excitatory Neurons and Glia, But Not GABAergic Neurons, Are Produced in the Emx1-Expressing Lineage. The Journal of Neuroscience 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout J-F, Li W, Fritsch C, Li A, Haroon S, Singh L, Hua D, Fazelinia H, Smith Z, Seeholzer S, et al. (2017). The landscape of transcription errors in eukaryotic cells. Sci Adv 3, e1701484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci AS, Harmanci AO, and Zhou X (2020). CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nature Communications 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiba Y, Toda T, Ebisu H, Wakimoto M, Yanagi S, and Kawasaki H (2016). Sox11 Balances Dendritic Morphogenesis with Neuronal Migration in the Developing Cerebral Cortex. J. Neurosci 36, 5775–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, et al. (2016). Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J-F, Oruganti H, Vu TH, and Hoffman AR (1998). Tissue-Specific Imprinting of the Mouse Insulin-Like Growth Factor II Receptor Gene Correlates with Differential Allele-Specific DNA Methylation. Molecular Endocrinology 12, 220–232. [DOI] [PubMed] [Google Scholar]
- Huang AY, Li P, Rodin RE, Kim SN, Dou Y, Kenny CJ, Akula SK, Hodge RD, Bakken TE, Miller JA, et al. (2020). Parallel RNA and DNA analysis after deep sequencing (PRDD-seq) reveals cell type-specific lineage patterns in human brain. Proc. Natl. Acad. Sci. U. S. A 117, 13886–13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JP, and Loeb LA (1992). Fidelity of HIV-1 reverse transcriptase copying RNA in vitro. Biochemistry 31, 954–958. [DOI] [PubMed] [Google Scholar]
- Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, Wedge DC, Davies HR, Ramakrishna M, Fullam A, et al. (2017). Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543, 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, and Philpot BD (2014). Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol 522, 1874–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamlichi AA, and Feil R (2018). Parallels between Mammalian Mechanisms of Monoallelic Gene Expression. Trends Genet. 34, 954–971. [DOI] [PubMed] [Google Scholar]
- Kishikawa T, Momozawa Y, Ozeki T, Mushiroda T, Inohara H, Kamatani Y, Kubo M, and Okada Y (2019). Empirical evaluation of variant calling accuracy using ultra-deep whole-genome sequencing data. Sci. Rep 9, 1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, and Alvarez-Buylla A (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci 32, 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave MC, and Sekelsky J (2009). Mitotic recombination: why? when? how? where? PLoS Genet. 5, e1000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson JS, Yin M, Fischer JM, Stringer SL, and Stringer JR (2006). Expression and loss of alleles in cultured mouse embryonic fibroblasts and stem cells carrying allelic fluorescent protein genes. BMC Mol. Biol 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson AJM, Ziegenhain C, Hagemann-Jensen M, Reinius B, Jacob T, Dalessandri T, Hendriks G-J, Kasper M, and Sandberg R (2021). Transcriptional bursts explain autosomal random monoallelic expression and affect allelic imbalance. PLoS Comput. Biol 17, e1008772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latos PA, Pauler FM, Koerner MV, Şenergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, et al. (2012). Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338, 1469–1472. [DOI] [PubMed] [Google Scholar]
- Laukoter S, Pauler FM, Beattie R, Amberg N, Hansen AH, Streicher C, Penz T, Bock C, and Hippenmeyer S (2020). Cell-Type Specificity of Genomic Imprinting in Cerebral Cortex. Neuron 107, 1160–1179.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H, Øbro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, Osborne RJ, Huntly BJP, Martincorena I, Anderson E, et al. (2018). Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder D, and Gartler SM (1965). Glucose-6-Phosphate Dehydrogenase Mosaicism: Utilization as a Cell Marker in the Study of Leiomyomas. Science 150, 67–69. [DOI] [PubMed] [Google Scholar]
- Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D’Gama AM, Cai X, et al. (2015). Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, Pelka K, Ge W, Oren Y, Brack A, et al. (2019). Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, and Shendure J (2016). Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, and Haber JE (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol 6, a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher R, Hartmann E, Zopf W, Herterich S, Wilke P, Müller L, Rosler E, Kudlich T, Al-Taie O, Rosenwald A, et al. (2011). LOH and copy neutral LOH (cnLOH) act as alternative mechanism in sporadic colorectal cancers with chromosomal and microsatellite instability. Carcinogenesis 32, 636–642. [DOI] [PubMed] [Google Scholar]
- Minoche AE, Dohm JC, and Himmelbauer H (2011). Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 12, R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y, and Torres-Padilla M-E (2012). Control of ground-state pluripotency by allelic regulation of Nanog. Nature 483, 470–473. [DOI] [PubMed] [Google Scholar]
- Mohamedali A, Gäken J, Twine NA, Ingram W, Westwood N, Lea NC, Hayden J, Donaldson N, Aul C, Gattermann N, et al. (2007). Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood 110, 3365–3373. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, and Jasin M (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol 11, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe C, McDevitt MA, and Maciejewski JP (2010). Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood 115, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer J, and Trapnell C (2018). Single-Cell Multi-omics: An Engine for New Quantitative Models of Gene Regulation. Trends Genet. 34, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Raj B, Wagner DE, McKenna A, Pandey S, Klein AM, Shendure J, Gagnon JA, and Schier AF (2018). Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol 36, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raponi E, Agenes F, Delphin C, Assard N, Baudier J, Legraverend C, and Deloulme J-C (2007). S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia 55, 165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B, and Sandberg R (2015). Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat. Rev. Genet 16, 653–664. [DOI] [PubMed] [Google Scholar]
- Reinius B, Mold JE, Ramsköld D, Deng Q, Johnsson P, Michaëlsson J, Frisén J, and Sandberg R (2016). Analysis of allelic expression patterns in clonal somatic cells by single-cell RNA–seq. Nature Genetics 48, 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AR, and Harpending H (1992). Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution 9, 552–569. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, and Huelsenbeck JP (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol 61, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabina J, and Leamon JH (2015). Bias in Whole Genome Amplification: Causes and Considerations. Methods Mol. Biol 1347, 15–41. [DOI] [PubMed] [Google Scholar]
- Salipante SJ, and Horwitz MS (2006). Phylogenetic fate mapping. Proc. Natl. Acad. Sci. U. S. A 103, 5448–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salipante SJ, and Horwitz MS (2007). A phylogenetic approach to mapping cell fate. Curr. Top. Dev. Biol 79, 157–184. [DOI] [PubMed] [Google Scholar]
- Salipante SJ, Thompson JM, and Horwitz MS (2008). Phylogenetic Fate Mapping: Theoretical and Experimental Studies Applied to the Development of Mouse Fibroblasts. Genetics 178, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savova V, Patsenker J, Vigneau S, and Gimelbrant AA (2016). dbMAE: the database of autosomal monoallelic expression. Nucleic Acids Res. 44, D753–D756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow MHL (1977). Gastrulation in the mouse: Growth and regionalization of the epiblast. Development 42, 293–303. [Google Scholar]
- Spencer Chapman M, Ranzoni AM, Myers B, Williams N, Coorens THH, Mitchell E, Butler T, Dawson KJ, Hooks Y, Moore L, et al. (2021). Lineage tracing of human development through somatic mutations. Nature. [DOI] [PubMed] [Google Scholar]
- Stadler T, Pybus OG, and Stumpf MPH (2021). Phylodynamics for cell biologists. Science 371. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, and Thomson JN (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology 100, 64–119. [DOI] [PubMed] [Google Scholar]
- Symmons O, Chang M, Mellis IA, Kalish JM, Park J, Suszták K, Bartolomei MS, and Raj A (2019). Allele-specific RNA imaging shows that allelic imbalances can arise in tissues through transcriptional bursting. PLoS Genet. 15, e1007874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, Cell type annotation, Writing group, Supplemental text writing group, and Principal investigators (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SS, and Breen S (1993). Radial mosaicism and tangential cell dispersion both contribute to mouse neocortical development. Nature 362, 638–640. [DOI] [PubMed] [Google Scholar]
- Tan SS, Williams EA, and Tam PP (1993). X-chromosome inactivation occurs at different times in different tissues of the post-implantation mouse embryo. Nat. Genet 3, 170–174. [DOI] [PubMed] [Google Scholar]
- Taverna E, Götz M, and Huttner WB (2014). The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell Dev. Biol 30, 465–502. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DE, and Klein AM (2020). Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet 21, 410–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, He Y, Zhang Q, Ren X, and Zhang Z (2021). Direct Comparative Analyses of 10X Genomics Chromium and Smart-seq2. Genomics Proteomics Bioinformatics 19, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegreffe C, Simon R, Peschkes K, Kling C, Strehle M, Cheng J, Srivatsa S, Liu P, Jenkins NA, Copeland NG, et al. (2015). Bcl11a (Ctip1) Controls Migration of Cortical Projection Neurons through Regulation of Sema3c. Neuron 87, 311–325. [DOI] [PubMed] [Google Scholar]
- Zhou B, Ho SS, Greer SU, Spies N, Bell JM, Zhang X, Zhu X, Arthur JG, Byeon S, Pattni R, et al. (2019). Haplotype-resolved and integrated genome analysis of the cancer cell line HepG2. Nucleic Acids Res. 47, 3846–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S5. HepG2 cells phase blocks, Related to STAR Methods Section, scRNA-seq Variant Calling, xlsx file.
Table S4. LOH Allele Enrichment, Related to Figure 3, xlsx file.
Table S3. Genes Escaping X-Inactivation, Related to Figure 1, xlsx file.
Table S2. Cell Cycle Related Genes, Related to Figure 1, xlsx file.
Table S1. Genes Used to Identify Cell Types, Related to Figure 1, xlsx file.
Data Availability Statement
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table. All derived datasets, including the original Seurat count matrix used for cell clustering, genotype calls for all mice and human HepG2 cells, VCF files for mouse and HepG2 variants, consensus phylogenies in NEXUS format, and Allen Institute Cell Lineage Reconstruction DREAM Challenge phylogenies in Newick format are provided in a Mendeley Data archive and are available as of the date of publication. The DOI is listed in the key resources table.
All original code has been deposited in a Mendeley Data archive and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| scRNA sequencing data - mouse | Laukoter et al., 2020 | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716 |
| scRNA sequencing data - human HepG2 | Hou et al., 2016 |
GSM2039769 GSM2039770 |
| HepG2 cell line genome data | (Zhou et al., 2019) | ENCODE:ENCBS760ISV |
| GRCm38.p5 | NCBI | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.25/ |
| Ensembl | EMBL-EBI | https://www.ensembl.org/index.html?redirect=no |
| CA Mouse Genome Project sequencing data | Keane et al., 2011 | ftp://ftp-mouse.sanger.ac.uk/current_snps/strain_specific_vcfs/CAST_EiJ.mgp.v5.snps.dbSNP142.vcf.gz |
Supplemental Data
|
This study | Horwitz, Marshall (2022), “Anderson_LOH_v3”, Mendeley Data, V1, doi: 10.17632/23f7j32zmf.1 |
| Experimental models: organisms/strains | ||
| Mouse: CAST/Ei | The Jackson Laboratory | RRID:IMSR_JAX:000928 |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:00066 |
| Mouse:Emx1-Cre | The Jackson Laboratory | RRID:IMSR_JAX:005628 |
| Software and algorithms | ||
| STAR 2.5.0c | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Seurat R package | Butler et al., 2018; Stuart et al., 2019 | https://satijalab.org/seurat/ |
| GATK | Keane et al., 2011 | https://gatk.broadinstitute.org/hc/en-us |
| HMM R package | Prof. Dr. Lin M. Himmelmann, University of Applied Sciences Rapperswil, Switzerland | https://cran.r-project.org/web/packages/HMM/index.html |
| MrBayes | Ronquist et al., 2012 | http://nbisweden.github.io/MrBayes/ |
| DensiTree | Bouckaert, 2010 | https://www.cs.auckland.ac.nz/~remco/DensiTree/ |
Supplemental Code
|
This study | Horwitz, Marshall (2022), “Anderson_LOH_v3”, Mendeley Data, V1, doi: 10.17632/23f7j32zmf.1 |