

Abstract
Deep annotation of a library of 4-anilinoquinolines led to the identification of 7-iodo-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine 16 as a potent inhibitor (IC50 = 14 nM) of Protein Kinase Novel 3 (PKN3) with micromolar activity in cells. Compound 16 is a potential tool compound to study the cell biology of PKN3 and its role in pancreatic and prostate cancer and T-cell acute lymphoblastic leukemia. These 4-anilinoquinolines may also be useful tools to uncover the therapeutic potential of PKN3 inhibition in a broad range of diseases.
Keywords: Protein Kinase Novel 3 (PKN3), 4-anilinoquinoline, 4-anilinoquinazoline, Homology Model
Graphical Abstract
We developed several small, focused libraries of 4-anilinoquinolines to explore structure activity relationships for the understudied kinase Protein Kinase Novel 3 (PKN3). This led to the identification of 7-iodo-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine 16 as a potent inhibitor of PKN3 with an IC50 of 1.3 μM in cells. Compound 16 is a potentially useful tool to study PKN3 biology including links to pancreatic and prostate cancer, along with T-cell acute lymphoblastic leukemia. These compounds may be useful tools to explore the therapeutic potential of PKN3 inhibition in prevention of a broad range of infectious and systemic diseases.
Protein kinase N (PKN, protein kinase novel) family genes encode for the three isozymes PKN1 (PKNα/PRK1/PAK1), PKN2 (PKNγ/PRK2/PAK2/) and PKN3 (PKNβ) that form a subfamily of AGC serine/threonine protein kinases.[1] PKNs are closely related to several members of the protein kinase C family and are therefore also named PRKs (protein kinase C-related kinases).[2,3] The catalytic domain of the mammalian PKN is homologous to protein kinase C family members at its C-terminal region and contains three repeats of an antiparallel coiled-coil (ACC) domain and a C2-like domain at its N-terminal region.[4,1] While these domains are well-conserved across the PKN isozymes, the enzymatic properties and tissue distribution of PKN1-3 show distinct differences.[1] PKN1 and PKN2 are expressed ubiquitously, but PKN3 is restricted to specific tissues including skeletal muscle, heart, liver and human cancer cell lines.[2, 4-6]
Full catalytic activity of PKN1-3 requires phosphorylation of both the activation loop by phosphoinositide-dependent kinase 1 (PDK1) and the turn motif.[2, 4] PKN1-3 differ in their responsiveness to activation by members of the small GTPase Rho, phospholipids, fatty acids and arachidonic acid.[3, 4, 7] Mukai et al. investigated the physiological role of PKN3 in normal tissue using knockout mice.[7] It was shown that PKN3 was not required for development and growth to the adult stage, but lower migratory activity of embryonic fibroblast cells was observed suggesting an involvement in the actin cytoskeleton regulation of primary fibroblastic cells. [7] Additionally, PKN3 was not required for normal vascular development but played a role in angiogenesis support. Tumor angiogenesis was not inhibited by PKN3 knock out.[1]
However, PKN3 is involved in other pathological processes. PKN3 has been shown to interact with a mediator of tumor invasion and metastasis in epithelial cancers, RhoC,[8] and with p130Cas, that are known to regulate cancer cell growth and invasiveness.[9] PKN3 acts downstream of phosphoinositide 3-kinase (PI3K). Growth factor stimulation of normal cells lead to transiently activated PI3K, which was rapidly turned off by the phosphatase and tensin homolog (PTEN).[5] PTEN is frequently inactivated in human cancer resulting in overactivation of the PI3K pathway, which in turn upregulates PKN3 and increases metastatic behavior.[9] This pathway is one of the processes that mediates malignant cell growth in prostate cancer.[5] Additionally, the malignant behavior of breast cancer cells in vitro was increased by overexpression of exogenous PKN3.[8] PKN3 inhibition has been shown to result in growth inhibition of prostate and breast cancer xenografts that were PI3K-driven.[5, 8, 10] Since PKN3 can act downstream of PI3K signaling,[5,8] therapeutics that selectively inhibit PKN3 could provide valuable insights into its the role in cancer biology and present a potential approach to target cancer types that lack tumor suppressor PTEN function or rely on chronic activation of PI3K.[5]
An siRNA formulation (Atu027) that silences PKN3 in vivo has been developed as a potential cancer therapeutic. Atu027 prevented liver and lung metastases in mouse models and inhibited prostate and pancreatic cancer growth. It has completed phase I clinical trials and has advanced to an ongoing phase I/II trial in advanced pancreatic cancer (NCT01808638).[11,12] Atu027 was well tolerated; no responses of the innate immune system, often a problem with siRNA formulations, were observed, and the treatment was not restricted to a type of cancer. It was anticipated that beneficial effects would be seen in all vascularized metastatic cancers.[12] However, dose limiting toxicity was not reached in the phase I clinical trial and the remaining drawbacks of this siRNA approach include potential enzymatic instability, off-target effects, challenges in tissue-specific access due to negative charge and size as well as rapid liver clearance and renal excretion.[12-15]
Additionally the PI3K pathway was found to be overactivated in many types of leukemia,[10,16-18] the expression level of PKN3 was increased in human T-cell acute lymphoblastic leukemia (T-ALL), and PKN3 deletion slowed T-ALL development without affecting normal hematopoiesis.[10] PKN3 was also found to be a possible regulator of neovascularization and its inhibition might be beneficial for vascular diseases like arthritis and age-related macular degeneration.[7] Another possible therapeutic use would be in bone diseases like rheumatoid arthritis and osteoporosis since, downstream of the Wnt5a-Ror2-Rho signaling pathway, PKN3 facilitates bone resorption.[19] The development of small molecule chemical probes would greatly enhance our understanding of the biology of PKN3.[20] Despite some limited reports of PKN3 inhibitors (Figure 1),[7,21] deeper structure activity relationships and kinome-wide selectivity have yet to be explored.[21-24]
Figure 1.
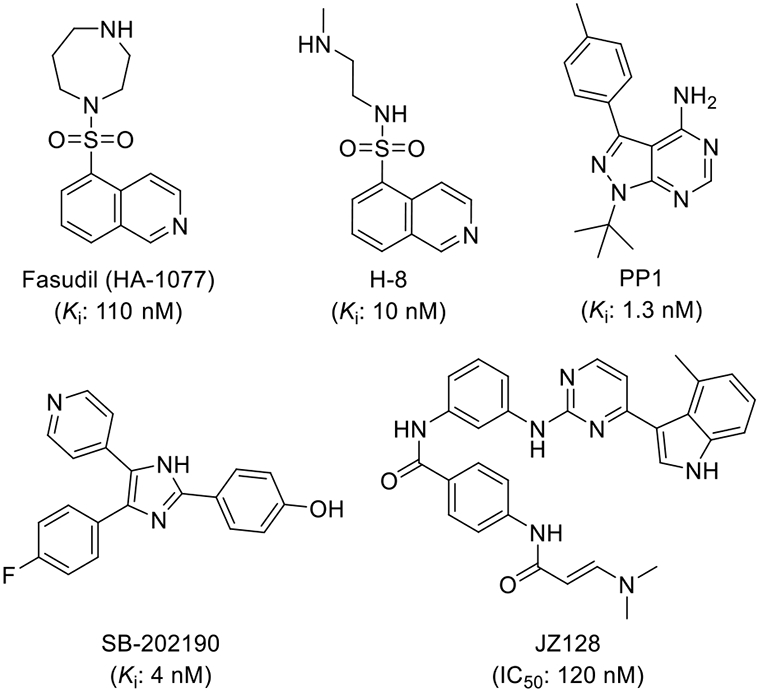
Previously reported compounds active against PKN3.
To identify a tractable chemical starting point for PKN3, we started with the second-generation GlaxoSmithKline Published Kinase Inhibitor
Set (PKIS2), a diverse and well annotated chemogenomic set of kinase inhibitors.[25-26] While PKIS2 had been screened against 80% of the human kinome, its annotation for PKN3 activity was not included due to the absence of this kinase in the DiscoverX KinomeSCAN® panel. Hence to identify PKN3 inhibitors, PKIS2 was screened against PKN3 using a split-luciferase binding assay (SLCA) at Luceome Biotechnologies.[27-29] We identified a series of 4-anilinoquinolines (1-3) with a common trimethoxyaniline motif (Figure 2) that bound to PKN3. Combining the results with the prior annotation we determined that GW577382 (2) was a promising narrow-spectrum PKN3 inhibitor. 2 had a PKN3 IC50 = 280 nM in the split-luciferase assay with Kd < 1 μM on only four kinases (GAK, RIPK2, ADCK3 and NLK) in the DiscoverX KinomeSCAN® panel. GW494610 (3) demonstrated single digit nanomolar potency on PKN3 (IC50 = 9 nM) but was more promiscuous across the human kinome (>18 kinases inhibited >90% at 1 μM). Despite its high PKN3 potency, 3 was deemed less promising as a narrow spectrum kinase inhibitor.
Figure 2.
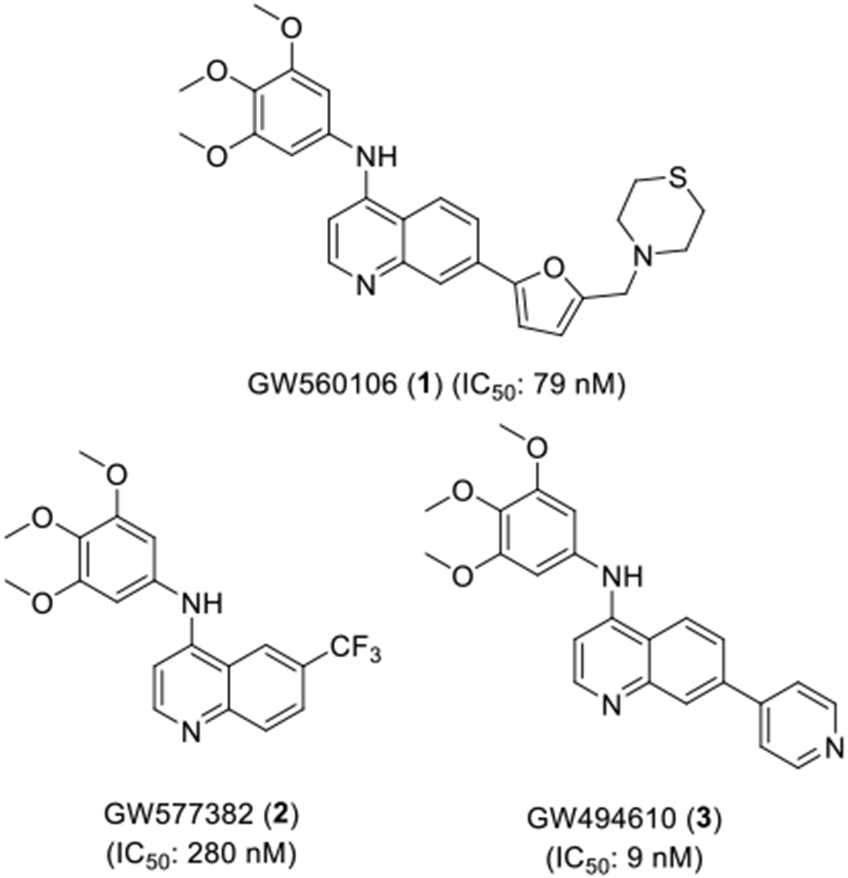
4-Anilinoquinolines identified as PKN3 inhibitors from PKIS2.
A small series of 4-anilinoquin(az)oline inhibitors described in the literature was also screened in the PKN3 binding assay (Table 1, Figure S1). While cabozantinib, sapitinib and gefitinib were weak (>10 μM) inhibitors of PKN3, lapatinib showed moderate activity. Vandetanib and tesevatinib both had IC50 <1 μM which further highlights the potential of the 4-anilinoquin(az)oline chemotype to inhibit PKN3 and the opportunity for chemical modification of 2, to improve its potency as a PKN3 inhibitor.
Table 1.
Literature 4-anilinoquin(az)oline PKN3 results.
| Compound | PKN3 IC50 (μM)a |
|---|---|
| Cabozantinib | 42 |
| Sapitinib (AZD8931) | 44 |
| Gefitinib | 23 |
| Lapatinib | 5.9 |
| Vandetanib | 0.54 |
| Tesevatinib (XL-647) | 0.24 |
To define the structure-activity for PKN3 inhibition we screened a known series of 4-anilinoquin(az)oline analogues (Scheme 1), which had been synthesized by heating the corresponding 4-chloroquinoline derivative and substituted aniline in ethanol to reflux overnight.[30] The synthesis afforded the 4-anilinoquin(az)oline analogues (2, 4-39) in good to excellent yield (24-93 %).[30-37].
Scheme 1.
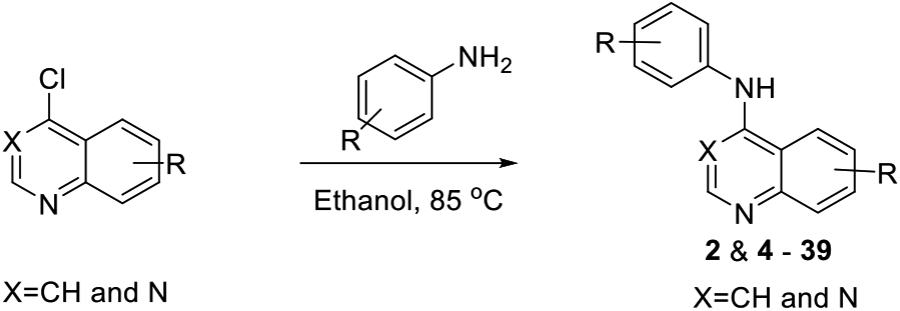
General synthetic route to analogues (2 & 4 - 39).
We screened the 4-anilinoquinazoline analogues of 2 to identify substitutions that improved potency on PKN3 and to identify opportunities to achieve selectivity over GAK, a dark kinase for which the series was originally designed.[30-37] Analogues of 2 were previously utilized in our lab to generate a selective chemical probe for GAK.[31-34] Hence, our internal GAK FRET binding data were used to guide the structure-activity for PKN3/GAK selectivity. The 4-anilinoquinazoline analogues were screened for PKN3 binding in the split-luciferase binding assay (SLCA assay),[27-29] in a 8-point dose response format to determine the IC50 values (Table 2).
Table 2.
Results of initial optimization of 2.
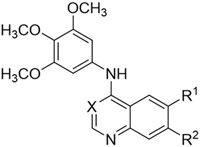
|
|||||
|---|---|---|---|---|---|
| Cmpd | X | R1 | R2 | PKN3a | GAKb |
| IC50 (μM) | Ki (μM) | ||||
| 2 | CH | CF3 | H | 0.28 | 0.0039 |
| 4 | N | CF3 | H | 4.4 | 0.037 |
| 5 | CH | H | H | 1.1 | 0.040 |
| 6 | CH | F | H | 0.70 | 0.0057 |
| 7 | N | F | H | 5.8 | 0.033 |
| 8 | CH | Cl | H | 0.070 | 0.0069 |
| 9 | CH | Br | H | 0.0093 | 0.0031 |
| 10 | CH | I | H | 0.098 | 0.0038 |
| 11 | N | I | H | 2.7 | 0.011 |
| 12 | CH | H | F | 0.39 | 0.0024 |
| 13 | N | H | F | 3.9 | 0.055 |
| 14 | CH | H | Cl | 0.027 | 0.0013 |
| 15 | CH | H | Br | 0.12 | 0.0049 |
| 16 | CH | H | I | 0.014 | 0.0044 |
| 17 | N | H | I | 0.93 | 0.021 |
| 18 | CH | tBu | H | 0.31 | 0.0081 |
| 19 | CH | CN | H | 0.21 | 0.0015 |
| 20 | CH | SO2CH3 | H | 0.60 | 0.018 |
| 21 | CH | OCH3 | H | 0.74 | 0.013 |
| 22 | CH | OCH3 | OCH3 | 0.48 | 0.00054 |
| 23 | CH | H | OCH3 | 0.92 | 0.0026 |
| 24 | CH | H | CF3 | 0.24 | 0.0062 |
| 25 | N | H | CF3 | 2.6 | 0.079 |
| 26 | CH | H | CN | 0.079 | 0.0027 |
The 6-trifluoromethyl quinoline 2 was more potent on GAK (Ki = 3.9 nM) compared to PKN3 (IC50 = 280 nM). A switch to quinazoline 4 resulted in a decrease in activity of 15-fold on PKN3 and 10-fold on GAK. Removal of the 6-trifluoromethyl moiety in unsubstituted quinoline 5 also led to a 4-fold drop on PKN3 and 10-fold drop on GAK compared. Compound 6, with a 6-fluoro substituent had a similar profile to 2. The corresponding quinazoline 7 showed a similar decrease in activity of 6-8-fold on both kinases compared to 6.
The 6-chloro quinoline analogue 8 showed a 4-fold increase in activity against PKN3 (IC50 = 70 nM) but near equipotent GAK activity compared with 2. Increasing the size of the 6-substituent with bromo derivative 9 gave single digit nanomolar activity on PKN3 (IC50 = 9.3 nM) and improved to only a 3-fold window between GAK and PKN3 activity. 9 had previously been shown to have a good selectivity profile over other human kinases.[30-33] A further increase of the halogen atom size to the 6-iodo quinoline 10 led to a decrease in activity on both PKN3 and GAK compared to the 6-bromo analogue 9. The corresponding iodo-substituted quinazoline 11 showed a further drop in both PKN3 and GAK activity. In the 7-halogen series, the fluoro quinoline analogue 12 displayed a similar profile to 2, while the 7-fluoro quinazoline analogue 13 was 10-fold less potent on both PKN3 and GAK. The 7-chloro derivative 14 showed improved potency on PKN3 (IC50 = 27 nM) without increasing GAK activity (Ki = 1.3 nM). Surprisingly, the 7-bromo derivative 15 had decreased PKN3 activity while maintaining GAK activity. The 7-iodo analogue 16 was potent on PKN3 (IC50 = 14 nM) with GAK activity decreasing 3-fold compared to 14. However, quinazoline analogue 17 showed a drop off in PKN3 and GAK activity, even though its PKN3 IC50 was still <1 μM.
The 6-tert-butyl analogue 18 had PKN3 activity equivalent to 2, but with a 2-fold decrease in GAK activity. This structure-activity was consistent with the 6-cyano analogue 19. However, 6-methyl sulfone-substituted compound 20 showed decreased GAK activity (Ki = 18 nM) and only moderate potency against PKN3 (IC50 = 600 nM). The quinoline analogues with 6-methoxy 21, 6,7-dimethoxy 22, and 7-methoxy 23 substituents showed no improvement in PKN3 activity relative to compound 2 but maintained high GAK potency. A switch of the trifluoromethyl substituent from the 6-position to the 7-position in compound 24 maintained PKN3 activity with a 2-fold reduction in GAK activity compared to 2. The quinazoline analogue 25 showed a sharp drop off in activity for both PKN3 and GAK, consistent with the structure-activity observed in the other quinazoline analogues. Finally, the 7-cyano analogue 26 was both potent on PKN3 (IC50 = 79 nM) and on GAK (Ki = 2.7 nM).
We next explored a series of halogenated analogues (27-31) (Table 3). Replacing the methoxy groups of 2 and 9 with fluoro atoms resulted in analogues 27 and 28; both quinolines showed only micromolar activity on PKN3.[30,35] Removing one of the meta-fluoro 29 and changing the substitutions 30 showed no improvement in activity.[30,35] However, switching back to the trimethoxy substitution and moving the fluoro atom to the 5- and 7-positions yielded quinoline 31 with sub-μM PKN3 potency and improved selectivity toward GAK (IC50 = 0.14 μM).[30]
Table 3.
Results of halogenated quinolines 35-39.
In the search for compounds with improved PKN3/GAK selectivity, we decided to screen a library of lapatinib derivatives based on the 6,7-dimethoxyquin(az)oline chemotype (Table 3, 32-39) that was related to compounds in Tables 1 and 2. Prior structure-activity studies had established that GAK activity would be removed with the addition of larger substitutions at the para-position of the aniline.[23,32,37] Gratifyingly, we found that the switch from lapatinib to the simplified quinoline derivative 32 maintained the modest activity on PKN3 (IC50 = 8.2 μM). The quinazoline derivative 33 saw a 2-fold decrease in PKN3 activity compared to lapatinib. Additional changes to increase chemical diversity (34-36) led to a further reduction in PKN3 activity. The acetamide derivative 37 showed no binding to PKN3 at the highest concentration tested, indicating that the benzyl group in 32 was at least in part important for activity. Interestingly, the removal of the 7-methoxy group in analogue 38 gave no improvement in PKN3 activity. However, removal of the 6-methoxy in analogue 39 resulted in a 4-fold increase in PKN3 activity (IC50 = 11 μM).
To further characterize the compounds with most potent PKN3 activity from the SLCA assay (8-10 & 14-16) we established a NanoBRET assay to test their in-cell target engagement (Table 5). The NanoBRET assay utilizes a fusion of Nano Luciferase (NLuc), an extremely bright and small luciferase, at the N-terminal of the PKN3 enzyme that is transiently expressed in HEK293 cells. Addition of an ATP-competitive fluorescent tracer, composed of a promiscuous kinase inhibitor tethered to Bodipy, and the NLuc enzyme substrate results in the production of a strong bioluminescence resonance energy transfer (BRET) signal. Addition of small-molecule PKN3 inhibitors that compete with the fluorescent tracer produces in a dose-dependent loss of BRET signal that allows determination of the cellular target engagement potency. It has been shown across multiple kinases that displacement of the ATP-competitive tracer correlates with potency of inhibition of the enzyme in cells.[38-39]
Table 5.
PKN3 NanoBRET results of the most potent analogues.
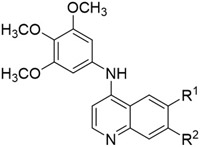
|
|||
|---|---|---|---|
| Cmpd | R1 | R2 | PKN3a |
| in-cell IC50 (μM) | |||
| 8 | Cl | H | 6.5 |
| 9 | Br | H | 3.5 |
| 10 | I | H | >10 |
| 14 | H | Cl | >10 |
| 15 | H | Br | 2.7 |
| 16 | H | I | 1.3 |
IC50 values generated in NanoBRET assay (n=2).
Interestingly, the SLCA and NanoBRET results of this small set of compounds did not have a linear relationship, which is likely due to the differences in cell penetrance and the assay formats. Compound 9 (3.5 μM), compound 15 (2.7 μM) and compound 16 (1.3 μM) all had in cell target engagement potencies under 5 μM. The 7-iodo quinoline (16) was the most potent derivative with an IC50 of 1.3 μM, representing about a 100-fold drop off activity from the cell free SLCA assay. However, compound 16 also has significant GAK activity in cells (IC50 = 0.074 μM) so further work will be required to improve selectivity over GAK.[35]
The protein crystal structure of PKN3 is not available, so to model PKN3, we first assessed the sequence similarity of PKN3 to the closely related sub-family members PKN1 and PKN2 with the idea of building a homology model. These kinases, closely related in the kinome phylogenetic tree, have high sequence homology (PKN3 vs PKN1 66% and PKN3 vs PKN2 57%).[40]
When comparing the experimental x-ray structures of the PKN1 and PKN2 kinase domains, their overall folding is almost identical (Figure S1). This observation suggests that homology modelling should result in reasonable structural quality. Experimental structures of PKN1 provide information about the apo-structure, but also about the binding mode of several co-crystallized inhibitors including tofacitinib, lestaurtinib and bisindolylmaleimide Ro-31-8220.[41]
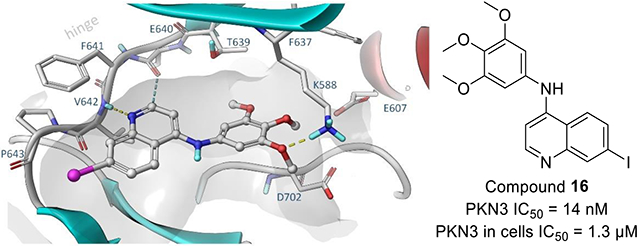
Our docking studies using the PKN3 homology model suggest that the quinoline scaffold adopts a similar binding conformation as was previously observed in the EGFR and NAK family kinases (Figure 3).[30] The quinoline scaffold binds to the hinge region via H-bond interactions between the amide of Val642 and two weaker aromatic hydrogen bonds to the carbonyl group of Glu640 and Val642. The trimethoxyaniline moiety of 16 accommodates the hydrophobic pocket and forms a hydrogen bond interaction with the catalytic lysine (Lys588) (Figure 3C).
Figure 3.

Induced fit docking of selected compounds a) 2, b) 9, c) 16, d) 39 to PKN3 made by means of comparative modelling using high homologue templates (65%, PDB: 4OTG, 4OTH).
We then assessed the kinome profile of both 2 and 16 using chemical proteomics. 2 and 16 were profiled at a concentration of 1 μM using lysates of SUM159 cells using multiplexed kinase inhibitor bead set and quantitative mass spectrometry detection of kinase peptides (MIB-MS).[30,32,42] The MIB-MS proteomics was able to detect between 350-400 kinases in the cell lysates and competitive displacement of specific kinases from the beads was used to determine the kinome profile of the PKN3 inhibitors. Compound 2 showed a narrow spectrum of kinase inhibition that was consistent with the previous DiscoverX kinome profile.
Activity was detected for 2 using MIB-MS on GAK and RIPK2, while PKN3, AKT2 and NLK were just below the threshold of significance (5-fold change) (Figure 4). Compound 16 also had a narrow spectrum kinome profile by MIB-MS. Activity was detected on PKN3, NLK, GAK, AVCR1 and RIPK2, while MINK1 and ACVR1B were just below the 5-fold threshold of significance (Figure 5). ADCK3 was not captured in any of the runs, but is likely inhibited by this series based on the DiscoverX data.[26,30-31,43] AVCR1 is not a newly detected target of this chemotype, since it was observed in the DiscoverX profiling of 2 (Kd = 220 nM),[26] and compound 9 also has reported affinity for ACVR1 (Kd = 0.98 μM).[31]
Figure 4.

MIB-MS kinome profiling of 2 (CA62) at 1 μM showed activity on only GAK and RIPK2, with PKN3, AKT2 and NLK just below the 5-fold threshold.
Figure 5.

MIB-MS kinome profiling of 16 (CA94) at 1 μM showed activity on only PKN3, NLK, GAK, AVCR1 and RIPK2, with MINK1, and ACVR1B just below the 5-fold threshold.
Discussion
Protein kinases present promising drug targets with more than 70 inhibitors targeting the ATP binding site of kinases having been approved as medicines by the FDA.[44-45] However, the high clinical efficacy of most of these drugs is based on binding to more than one kinase. The therapeutic use of these multi-kinase inhibitors has been primarily limited to oncology. Inhibitors with improved potency and selectivity profiles may be needed for the development of kinase inhibitors for the treatment of diseases outside of oncology and for more precise targeted therapy in oncology.[46] New methods to design and optimize highly selective kinase inhibitors are urgently required.
One promising strategy to develop new kinase inhibitors with high selectivity is to utilize chemogenomic inhibitors sets composed of compounds that have already been annotated with narrow kinome-wide selectivity. This chemogenomic approach has recently become feasible due to the availability of panels of binding assays as a rapid, accurate, and robust method to assess potency and selectivity of ATP-competitive kinase inhibitors.[47-48, 22] Furthermore, these ligand binding displacement assays provide an accepted direct measurement of kinase inhibitor activity in optimization of ATP-competitive binding site inhibitors.[48-49] This is a particularly acute point in the case of dark kinases such as PKN3 and GAK, which do not have validated enzyme activity assays due to the lack of known substrates.
In addition to measuring the binding to the kinase in biochemical assays, the NanoBRET assay presents a complementary technique to measure kinase target engagement in live cells. Other reported mass spectrometry (MS)-based chemoproteomics approaches rely on the disruption of the plasma membrane and therefore suffer from dissolution of key cellular cofactors like ATP. In contrast, the NanoBRET assay is performed in intact cells requiring the compound to be cell permeable to be active. The cellular thermal shift assay (CETSA) also uses intact cells. However, in contrast to the NanoBRET assay, CETSA does not quantify the equilibrium-based inhibitor occupancy.[38-39] Hence, we opted to use a NanoBRET assay to determine the in-cell potency of potential PKN3 inhibitors.
Encouraging results from screening of PKIS2 against PKN3 demonstrated the potential of the 4-anilinoquin(az)oline chemotype and was confirmed with literature inhibitors containing the same scaffold. We explored several series of substitution patterns around the quin(az)oline core including trimethoxy and hybrid lapatinib derivatives. Structure-activity relationships were developed on PKN3 as well as the primary collateral kinase GAK. However, we were only partially successful in improving the PKN3/GAK selectivity. The trimethoxy compounds 8, 9, 15 and 16 were the most potent on PKN3 in both the in vitro SLCA and live cell NanoBRET assay. We identified 16 as a the most potent compound against PKN3 in cells (IC50 = 1.3 μM) although it was 17-fold more potent on GAK (IC50 = 0.074 μM in GAK NanoBRET). Despite its collateral activity on GAK, compound 16 represents the most selective cell active PKN3 inhibitor identified to date and may still be a useful tool compound to study PKN3 biology and can be used in complement to the recently identified covalent inhibitor.[21] However, further optimization is required to improve the selectivity over GAK to yield a high-quality chemical probe.[20] Notably, several of these quinoline-based analogues have already been shown have narrow spectrum activity across the full human kinome (e.g. 2 and 9).[30-31]
Our modelling studies with the PKN3 inhibitors suggest the traditional binding mode utilized by many quinoline and quinazoline kinase inhibitors. The modeling suggested that PKN3 may have a more flexible hydrophobic pocket area. This may give additional structural freedom for further modification of active scaffolds to improve selectivity over GAK. However, homology-based docking poses are heavily reflective of structural features derived from the template kinases and further structural biology studies with structurally related inhibitors are needed to confirm and validate current docking models.
In conclusion our results provide further evidence that PKN3 can be targeted by small molecule ATP-competitive inhibitors. Quinoline-based PKN3 inhibitors were identified with nanomolar potency in vitro and micromolar potency in cells. The 4-anilinoquinoline 16 was identified as the most potent of these PKN3 inhibitors in cells but requires additional optimization to remove the collateral activity on GAK. Importantly, however, 16 has a promising narrow spectrum by MIB-MS kinome-wide profiling, identifying it as a useful starting point for development of a high-quality chemical probe.
Experimental Section
Modelling methods
PKN3 is structurally closely related with other proteins of the serine/threonine protein kinase folding family.[50] The sequence homology to known template structures is feasible 66% - so as a consequence a homology modelling approach was conducted. The PKN3 model was constructed using structure prediction wizard of Maestro 2019.4. At the first stage a blast search was carried out in order to find suitable template structures. Sequences taken from crystal structures of PRK1 Catalytic Domain (PDB: 4OTD, 4OTG, 4OTH, 4OTI) and Human Protein Kinase N2 (PDB: 4CRS) were used for pairwise sequence alignment using clustalW methodology, which is suitable in this case, due to the structural similarity. High resolution structure, PDB:4OTH was selected as template for comparative modelling (identity 66%, positives 80%, alignment length 337 (kinase domain), resolution 1.8Å). Due to the good sequence similarity, a homology model was constructed using knowledge-based approach of the wizard employing multi template approach, where the model was mainly constructed based on PDB:4OTH, including ligand occupying the ATP binding pocket. The missing activation loop of PDB:4OTH was modeled based on other PDB structure of the same template (PDB:4OTG). Loop refinement was run to optimize the structure of C-terminal end.
Prior to docking studies, the homology model of the PKN3 structure was pre-processed by using the protein preparation wizard tool of Schrödinger Suite 2019-4 (Protein Preparation Wizard uses modules: Epik; Impact and Prime, Schrödinger, LLC, New York, NY, 2019). Structures of small molecule ligands were parametrized and minimized using Ligprep module (LigPrep, Schrödinger, LLC, New York, NY, 2019.). Molecular docking studies were computed using Induced fit workflow of Schrödinger employing SP-setting for Glide docking module and side chain 5Å were consider for conformational refinement using Prime module. Hydrogen bond constraint was added to hinge residue VAL642 to improved convergence of docking poses. Results were consistent with co-crystal binding modes in other kinases, eg GAK and the 4-anilinoquin(az)oline gefitinib.[51]
Biology
SLCA Method:
The split-luciferase kinase assays were performed by Luceome Biotechnologies. Briefly, split-firefly luciferase constructs were translated and assayed in cell-free system according to literature protocol.[27-29] Data were collected via a homogeneous competition binding assay where the displacement of an active site dependent probe by an inhibitor is measured by a change in luminescence signal. Testing was performed with either DMSO (no-inhibitor control) or a compound solution in DMSO, followed by incubation in the presence of a kinase specific probe. Luminescence was measured upon addition of a luciferin assay reagent to each assay solution. All PKIS2 library members and SAR analogues were screened at 1 μM in duplicate against PKN3.[26] IC50 values (8-point dose response format) were determined by plotting percent activity remaining against inhibitor concentration.
NanoBRET method. Cell Transfections and BRET Measurements:
N-terminal NanoLuc/Kinase fusions were encoded in pFN31K or pFC32K expression vectors (Promega), including flexible Gly-Ser-Ser-Gly linkers between Nluc and each full-length kinase. For cellular BRET target engagement experiments, HEK-293 cells were transfected with NLuc/target fusion constructs using FuGENE HD (Promega) according to the manufacturer’s protocol. Briefly, Nluc/target fusion constructs were diluted into Opti-MEM followed by Transfection Carrier DNA (Promega) at a mass ratio of 1:10 (mass/mass), after which FuGENE HD was added at a ratio of 1:3 (mg DNA: mL FuGENE HD). 1 part (vol) of FuGENE HD complexes thus formed were combined with 20 parts (vol) of HEK-293 cells in DMEM with 10% FBS plated at a density of 2 x 105 per mL into 96-well plates (Corning), followed by incubation in a humidified, 37°C/5% CO2 incubator for 20-30 hr. BRET assays were performed in white, 96-well cell culture treated plates (Corning) at a density of 2 x 104 cells/well.
Following transfection, DMEM was exchanged for Opti-MEM. All chemical inhibitors were prepared as concentrated stock solutions in DMSO (Sigma-Aldrich) and diluted in Opti-MEM (unless otherwise noted) to prepare working stocks. Cells were equilibrated for 2 h with energy transfer probes and test compound prior to BRET measurements. Energy transfer probes were prepared at a working concentration of 20X in tracer dilution buffer (12.5 mM HEPES, 31.25% PEG-400, pH 7.5). For target engagement analysis, the energy transfer probes were added to the cells at concentrations optimized for each target. For analysis of PKN3-NL, energy transfer probe K-5 was used at a final concentration of 1000 nM. To measure BRET, NanoBRET NanoGlo Substrate and Extracellular NanoLuc Inhibitor (Promega) were added according to the manufacturer’s recommended protocol, and filtered luminescence was measured on a GloMax Discover luminometer equipped with 450 nM BP filter (donor) and 600 nM LP filter (acceptor), using 0.5 s integration time. Milli-BRET units (mBU) are calculated by multiplying the raw BRET values by 1000. Competitive displacement data were plotted with GraphPad Prism software and data were fit to Equation 1 [log(inhibitor) vs. response -- Variable slope (four parameters)] to determine the IC50 value; Y = Bottom + (Top-Bottom)/(1 + 10^((LogIC50-X)*HillSlope)). (Equation 1). If the curve was insufficiently described by the raw data, then they were normalized to the controls-BRET in the presence of only energy transfer probe, and BRET in the absence of the energy transfer probe and test compound-then plotted to fit Equation 2 [log(inhibitor) vs. normalized response -- Variable slope] to determine the IC50 value; Y=100/(1+10^((LogIC50-X)*HillSlope))). (Equation 2). For all BRET data shown, no individual data points were omitted.
MIBS Method:
Previously described.[30,32] Briefly, multiplexed inhibitor beads (MIBS) affinity chromatography/MS analysis: SUM159 cells were cultured in a 1:1 mixture of DMEM and Nutrient Mixture F-12 medium (Gibco) supplemented with 5% fetal bovine serum, 1% anti/anti, 5 mgmL−1 insulin, and 1 mgmL−1 hydrocortisone. Cells were maintained at 37°C in a humidified 5% CO2 atmosphere. SUM159 cells were grown to 80% confluency, washed twice with PBS, and harvested by scraping cells in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 1 mM EGTA, 10 mM NaF, 2.5 mM Na3VO4, complete protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail 2, and 3 (Sigma)). Lysates were sonicated then clarified by centrifugation at 14000Vg for 15 min at 4 8C. Lysate was then filtered through a 0.2 mm syringe filter and frozen at −80 °C until used. Protein concentration was quantified using a Bradford assay the day of the experiment. DMSO or the indicated concentration of 16, 17 and 26 were added to lysate containing 4 mg of total protein. Lysates were vortexed briefly then incubated for 30 min on ice. Kinases were affinity purified as previously described. [30,32] Briefly, lysates were diluted to 1.33 mgmL−1 with lysis buffer then NaCl concentration brought to 1 M. Diluted lysates were passed over a mixture of 25 mL settled beads of each of the following inhibitors conjugated to ECH Sepharose beads: Purvalanol B, PP58, VI-16832, UNC21474A, UNC8088A, and 37.5 mL settled beads conjugated to CTx-0294885 and VI-16832.[52] The kinase inhibitor–bead conjugates were previously equilibrated in high-salt buffer (50 mM HEPES pH 7.5, 1M NaCl, 0.5% Triton X-100, 1 mM EDTA, and 1 mM EGTA). MIBs columns were sequentially washed with high-salt buffer, low-salt buffer (50 mm HEPES pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, and 1 mM EGTA), and SDS buffer (50 mm HEPES pH 7.5, 150 mm NaCl, 0.5% Triton X- 100, 1 mm EDTA, 1 mm EGTA, and 0.1% SDS). Proteins were eluted by boiling samples in elution buffer (100 mM Tris·HCl pH 6.8, 0.5% SDS, and 1% b-mercaptoethanol) for 15 min twice. Dithiothreitol (DTT) was added to a final concentration of 5 mm and samples were incubated at 60°C for 25 min. Samples were then cooled to room temperature on ice and alkylated by adding iodoacetamide to a final concentration of 20 mm for 30 min in the dark at room temperature. Samples were then concentrated in 10K Amicon Ultra centrifugal concentrators (Millipore) followed by methanol and chloroform precipitation of proteins. The final protein pellets were resuspended in 50 mM HEPES pH 8.0 and incubated with trypsin at 37 °C overnight. Residual detergent was removed by three sequential ethyl acetate extractions then desalted using Pierce C18 spin columns (Thermo Scientific) according to the manufacturer’s protocol.
Chemistry
General Procedure:
4-chloroquinoline derivative (150 mg, 0.67 mmol) and aniline (0.74 mmol) was suspended in ethanol (10 mL) and heated to reflux for 16 h. The crude mixture was purified by flash chromatography using EtOAc/hexane followed by 1–5% methanol in EtOAc (or by re-crystallization). After solvent removal under reduced pressure, the product was obtained. All compounds were >98% pure by 1H/13C NMR and LCMS. Compounds 2 and 4-39 were prepared as previously described.[30-37,53]
Supplementary Material
Table 4.
Results of simplified lapatinib analogues 27-34 on PKN3.
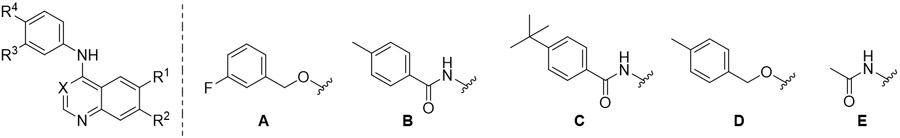
| ||||||
|---|---|---|---|---|---|---|
| Cmpd | X | R1 | R2 | R3 | R4 | PKN3a |
| IC50 (μM) | ||||||
| Lapatinib | - | - | - | - | - | 5.9 |
| 32 | CH | OCH3 | OCH3 | Cl | A | 8.2 |
| 33 | N | OCH3 | OCH3 | Cl | A | 17 |
| 34 | CH | OCH3 | OCH3 | OH | B | 38 |
| 35 | CH | OCH3 | OCH3 | OH | C | >50 |
| 36 | CH | OCH3 | OCH3 | H | D | >50 |
| 37 | CH | OCH3 | OCH3 | H | E | >50 |
| 38 | CH | OCH3 | H | OH | B | >50 |
| 39 | CH | H | OCH3 | OH | B | 11 |
Acknowledgements
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. Research reported in this publication was also supported by a grant from the National Center for Advancing Translational Sciences (NCATS) of the National Institute of Health “Tools for Accelerating R&D for Historically Understudied Protein Kinases” (1R44TR001916-01) and by the National Institute of Health “Illuminating the Druggable Genome program” (grant number 1U24DK116204-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health. In addition, we thank Biocenter Finland/DDCB for financial support and CSC – IT Center for Science Ltd. Finland for the use of their facilities, software licenses and computational resources.The authors would like to thank Prof. Timothy M. Willson (University of North Carolina at Chapel Hill) and Prof. Antti Poso (University of Eastern Finland) for supporting the work and providing constructive feedback on the manuscript. We also thank Dr. Brandie Ehrmann and Ms. Diane E. Wallace for LC-MS/HRMS support provided by in the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill. The core is supported by the National Science Foundation under Grant No. (CHE-1726291).
References
- [1].Shiga K, Takayama K, Futaki S, Hutti JE, Cantley LC, Ueki K, Ono Y, Mukai H, Biochem J. 2009, 425, 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Falk MD, Liu W, Bolaños B, Unsal-Kacmaz K, Klippel A, Grant S, Brooun A, Timofeevski S Biosci Rep. 2014, 34, 92–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hutchinson CL, Lowe PN, McLaughlin SH, Mott HR, Owen D Biochemistry 2013, 52, 7999–8011. [DOI] [PubMed] [Google Scholar]
- [4].Mukai H, J. Biochem 2003, 133, 17–27. [DOI] [PubMed] [Google Scholar]
- [5].Leenders F, Möpert K, Schmiedeknecht A, Santel A, Czauderna F, Aleku M, Penschuck S, Dames S, Sternberger M, Röhl T, Wellmann A, Arnold W, Giese K, Kaufmann J, Klippel A, EMBO J. 2004, 23, 3303–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thauerer B, Zur Nedden S, Baier-Bitterlich G, Curr. Neuropharmacol 2014, 12, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mukai H, Muramatsu A, Mashud R, Kubouchi K, Tsujimoto S, Hongu T, Kanaho Y, Tsubaki M, Nishida S, Shioi G, Danno S, Mehruba M, Satoh R, Sugiura R, Sci. Rep 2016, 6, 18979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Unsal-Kacmaz K, Ragunathan S, Rosfjord E, Dann S, Upeslacis E, Grillo M, Hernandez R, Mack F, Klippel A, Mol. Oncol 2012, 6, 284–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gemperle J, Dibus M, Koudelková L, Rosel D, Brábek J, Mol. Oncol 2019, 13, 264–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kraus M, Dolinski B, Rosahl TW, Magee JA, Leukemia 2015, 29, 255–258. [DOI] [PubMed] [Google Scholar]
- [11].Santel A, Aleku M, Röder N, Möpert K, Durieux B, Janke O, Keil O, Endruschat J, Dames S, Lange C, Eisermann M, Löffler K, Fechtner M, Fisch G, Vank C, Schaeper U, Giese K, Kaufmann J, Clin. Cancer Res 2010, 16, 5469–5480. [DOI] [PubMed] [Google Scholar]
- [12].Schultheis B, Strumberg D, Santel A, Vank C, Gebhardt F, Keil O, Lange C, Giese K, Kaufmann J, Khan M, Drevs J, J. Clin. Oncol 2014, 32, 4141–4148. [DOI] [PubMed] [Google Scholar]
- [13].Aagaard L, Rossi JJ, Adv. Drug Delivery Rev 2007, 59, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aleku M, Schulz P, Keil O, Santel A, Schaeper U, Dieckhoff B, Janke O, Endruschat J, Durieux B, Röder N, Löffler K, Lange C, Fechtner M, Möpert K, Fisch G, Dames S, Arnold W, Jochims K, Giese K, Wiedenmann B, Scholz A, Kaufmann J Cancer Res. 2008, 68, 9788–9798. [DOI] [PubMed] [Google Scholar]
- [15].Titze-de-Almeida R, David C, Titze-de-Almeida SS, Pharm. Res 2017, 34, 1339–1363. [DOI] [PubMed] [Google Scholar]
- [16].Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D, Moreau LA, Winter SS, Larson R, Zhang J, Protopopov A, Chin L, Pandolfi PP, Silverman LB, Hunger SP, Sallan SE, Look AT Blood. 2009, 114, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA, Nat. Med. (N. Y., NY, U. S.) 2010, 16, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martelli AM, Nyåkern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, Cocco L, Leukemia 2006, 20, 911–928. [DOI] [PubMed] [Google Scholar]
- [19].Uehara S, Udagawa N, Mukai H, Ishihara A, Maeda K, Yamashita T, Murakami K, Nishita M, Nakamura T, Kato S, Minami Y, Takahashi N, Kobayashi Y, Sci. Signaling 2017, 10. [DOI] [PubMed] [Google Scholar]
- [20].Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frederiksen M, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KV, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Müller S, O’Hagen RC, Overington JP, Owen DR, Rosen-berg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoicet B, Sundström M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Wilson TM, Workman P, Young RN, Zuercher WJ, Nat. Chem. Biol 2015, 11, 536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Browne CM, Jiang B, Ficarro SB, Doctor ZM, Johnson JL, Card JD, Sivakumaren SC, Alexander WM, Yaron TM, Murphy CJ, Kwiatkowski NP, Zhang T, Cantley LC, Gray NS, Marto JA J Am Chem Soc. 2019, 141, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fabian MA, Biggs III WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ, Nat Biotechnol. 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- [23].Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP, Nat Biotechnol. 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- [24].Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR Nat Biotechnol. 2011, 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Elkins JM, Fedele V, Szklarz M, Azeez KRA, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang X, Roth BL, Zen AAH, Fourches D, Muratov E, Tropsha A, Morris J, Teicher BA, Kunkel M, Polley E, Lackey KE, Atkinson FL, Overington JP, Bamborough P, Müller S, Price DJ, Willson TM, Drewry DH, Knapp S, Zuercher WJ, Nat Biotechnol. 2016, 34, 95–103. [DOI] [PubMed] [Google Scholar]
- [26].Drewry DH, Wells CI, Andrews DM, Angell R, Al-Ali H, Axtman AD, Capuzzi SJ, Elkins JM, Ettmayer P, Frederiksen M, Gileadi O, Gray N, Hooper A, Knapp S, Laufer S, Luecking U, Michaelides M, Müller S, Muratov E, Denny RA, Saikatendu KS, Treiber DK, Zuercher WJ, Willson TM, PLoS One. 2017, 12, e0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jester BW, Cox KJ, Gaj A, Shomin CD, Porter JR, Ghosh I, Am J. Chem Soc. 2010, 132, 11727–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jester BW, Gaj A, Shomin CD, Cox KJ, Ghosh I, J. Med. Chem 2012, 55, 1526–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Deng Q, Wang D, Xiang X, Gao X, Hardwidge PR, Kaushik RS, Wolff T, Chakravarty S, Li F J Virol Methods. 2011, 176, 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Asquith CRM, Laitinen T, Bennett JM, Godoi PH, East MP, Tizzard GJ, Graves LM, Johnson GL, Dornsife RE, Wells CI, Elkins JM, Willson TM, Zuercher WJ, ChemMedChem 2018, 13, 48–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Asquith CRM, Berger BT, Wan J, Bennett JM, Capuzzi SJ, Crona DJ, Drewry DH, East MP, Elkins JM, Fedorov O, Godoi PH, Hunter DM, Knapp S, Muüller S, Torrice CD, Wells CI, Earp HS, Willson TM, Zuercher WJ, J. Med. Chem 2019, 62, 2830–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Asquith CRM, Naegeli KM, East MP, Laitinen T, Havener TM, Wells CI, Johnson GL, Drewry DH, Zuercher WJ, Morris DC, J. Med. Chem 2019, 62, 4772–4778. [DOI] [PubMed] [Google Scholar]
- [33].Asquith CRM, Treiber DK, Zuercher WJ, Bioorg. Med. Chem. Lett 2019, 29, 1727–1731. [DOI] [PubMed] [Google Scholar]
- [34].Asquith CRM, Bennett JM, Su L, Laitinen T, Elkins JM, Pickett JE, Wells CI, Li Z, Willson TM, Zuercher WJ, Molecules. 2019, 24, pii: E4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Asquith CRM, Laitinen T, Bennett JM, Wells CI, Elkins JM, Zuercher WJ, Tizzard GJ, Poso A, ChemMedChem. 2020, 15, 26–49. [DOI] [PubMed] [Google Scholar]
- [36].Asquith CRM, Tizzard GJ, Bennett JM, Wells CI, Elkins JM, Willson TM, Poso A, Laitinen T ChemMedChem. 2020, 15, 1200–1215. [DOI] [PubMed] [Google Scholar]
- [37].Asquith CRM, Maffuid KA, Laitinen T, Torrice CD, Tizzard GJ, Crona DJ, Zuercher WJ, ChemMedChem. 2019, 14, 1693–1700. [DOI] [PubMed] [Google Scholar]
- [38].Machleidt T, Woodroofe CC, Schwinn MK, Méndez J, Robers MB, Zimmerman K, Otto P, Daniels DL, Kirkland TA, Wood KV, ACS Chem. Biol 2015, 10, 1797–1804. [DOI] [PubMed] [Google Scholar]
- [39].Vasta JD, Corona CR, Wilkinson J, Zimprich CA, Hartnett JR, Ingold MR, Zimmerman K, Machleidt T, Kirkland TA, Huwiler KG, Ohana RF, Slater M, Otto P, Cong M, Wells CI, Berger B-T, Hanke T, Glas C, Ding K, Drewry DH, Huber KVM, Willson TM, Knapp S, Müller S, Meisenheimer PL, Fan F, Wood KV, Robers MB, Cell Chem. Biol 2018, 25, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mukai H, J Biochem. 2003, 133, 17–27. [DOI] [PubMed] [Google Scholar]
- [41].Chamberlain P, Delker S, Pagarigan B, Mahmoudi A, Jackson P, Abbasian M, Muir J, Raheja N, Cathers B, PLoS One 2014, 9, e103638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, Usary J, Kuan P, Smalley DM, Major B, He X, Hoadley KA, Zhou B, Sharpless NE, Perou CM, Kim WY, Gomez SM, Chen X, Jin J, Frye SV, Earp HS, Graves LM, Johnson GL, Cell. 2012, 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Asquith CRM, Murray NH, Pagliarini DJ Nat Rev Drug Discov. 2019. 18, 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Roskoski R Jr Pharmacol Res. 2022, 175, 106037. [DOI] [PubMed] [Google Scholar]
- [45].Attwood MM, Fabbro D, Sokolov AV, Knapp S, Schiöth HB Nat Rev Drug Discov. 2021, 20, 839–861. [DOI] [PubMed] [Google Scholar]
- [46].Cohen P, Alessi DR, ACS Chem. Biol 2013, 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP, Nat. Biotechnol 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- [48].Ma H, Deacon S, Horiuchi K, Expert Opin. Drug Discovery 2008, 3, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rudolf AF, Skovgaard T, Knapp S, Jensen LJ, Berthelsen J, PLoS One 2014, 9, e98800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Endicott JA, Noble ME, Johnson LN Annu Rev Biochem. 2012, 81, 587–613. [DOI] [PubMed] [Google Scholar]
- [51].a) Ohbayashi N, Murayama K, Kato-Murayama M, Kukimoto-Niino M, Uejima T, Matsuda T, Ohsawa N, Yokoyoma S, Nojima H, Shirouzu M, ChemistryOpen. 2018, 7, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schrödinger Maestro software package (Small-Molecule Drug Discovery Suite 2018-4, Schrödinger, LLC, New York, NY, 2018). [Google Scholar]
- [52].Collins KAL, Stuhlmiller TJ, Zawistowski JS, East MP, Pham TT, Hall CR, Goulet DR, Bevill SM, Angus SP, Velarde SH, Sciaky N, Oprea TI, Graves LM, Johnson GL, Gomez SM. Oncotarget. 2018, 9, 15480–15497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].a) Asquith CRM, Fleck N, Torrice CD, Crona DJ, Grundner C, Zuercher WJ, Bioorg. Med. Chem. Lett 2019, 18, 2695–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carabajal MA, Asquith CRM, Laitinen T, Tizzard GJ, Yim L, Rial A, Chabalgoity JA, Zuercher WJ, Véscovi EG. Antimicrob Agents Chemother. 2019, 64, e01744–19. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Saul S, Pu SY, Zuercher WJ, Einav SS, Asquith CRM. Bioorg Med Chem Lett. 2020, 30, 127284. [DOI] [PubMed] [Google Scholar]; d) Saul S, Huang P, Einav S, Asquith CRM. Bioorg Med Chem Lett. 2021, 52, 128407. [DOI] [PubMed] [Google Scholar]; e) Huang P, Saul S, Einav S, Asquith CRM. Molecules. 2021, 26, 7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.