

Abstract
The kidney maintains electrolyte, water, and acid-base balance, eliminates foreign and waste compounds, regulates blood pressure, and secretes hormones. There are at least 16 different highly specialized epithelial cell types in the mammalian kidney. The number of specialized endothelial cells, immune cells, and interstitial cell types might even be larger. The concerted interplay between different cell types is critical for kidney function. Traditionally, cells were defined by their function or microscopical morphological appearance. With the advent of new single-cell modalities such as transcriptomics, epigenetics, metabolomics, and proteomics we are entering into a new era of cell type definition. This new technological revolution provides new opportunities to classify cells in the kidney and understand their functions.
Keywords: kidney cell types, cell plasticity, kidney function, single-cell RNA sequencing
INTRODUCTION
The nephron, the functional unit of the mammalian kidney, consists of a filtering unit, the glomerulus, which is connected to a long tubule that is segmented into a proximal part responsible for reclaiming important nutrients; an intermediate section, important for osmolarity regulation; and the final segment, which is responsible for fine-tuning and electrolyte balance. The intimate network of epithelial cells, endothelial cells, interstitial cells, and immune cells is critical to achieve optimal function and maintain the milieu interieur (1–4). In addition to proximal-distal regional differences, there are cortical medullary spatial differences. There are large differences in interstitial sodium and urea concentrations between the cortex and medulla that are critical for water homeostasis, and the medullary region is hypoxic even at basal conditions (5). Specialized epithelial and immune cells are needed to survive such an environment. The kidney has a double capillary bed (Figure 1). Glomerular capillaries originate from the afferent arterioles, and they are able to withstand high hydrostatic pressures, which is a major driving force for filtration (6). The blood from the glomerular capillaries is collected to the efferent arterioles, which divide again to the second capillary bed, the peritubular capillaries. This second capillary system has low hydrostatic pressure and is responsible for returning the reabsorbed fluid and electrolytes to the circulation. By changing the diameter of the afferent and efferent arteries, the kidney can quickly adapt to environmental changes and uncouple glomerular filtration from blood pressure changes (6, 7).
Figure 1.
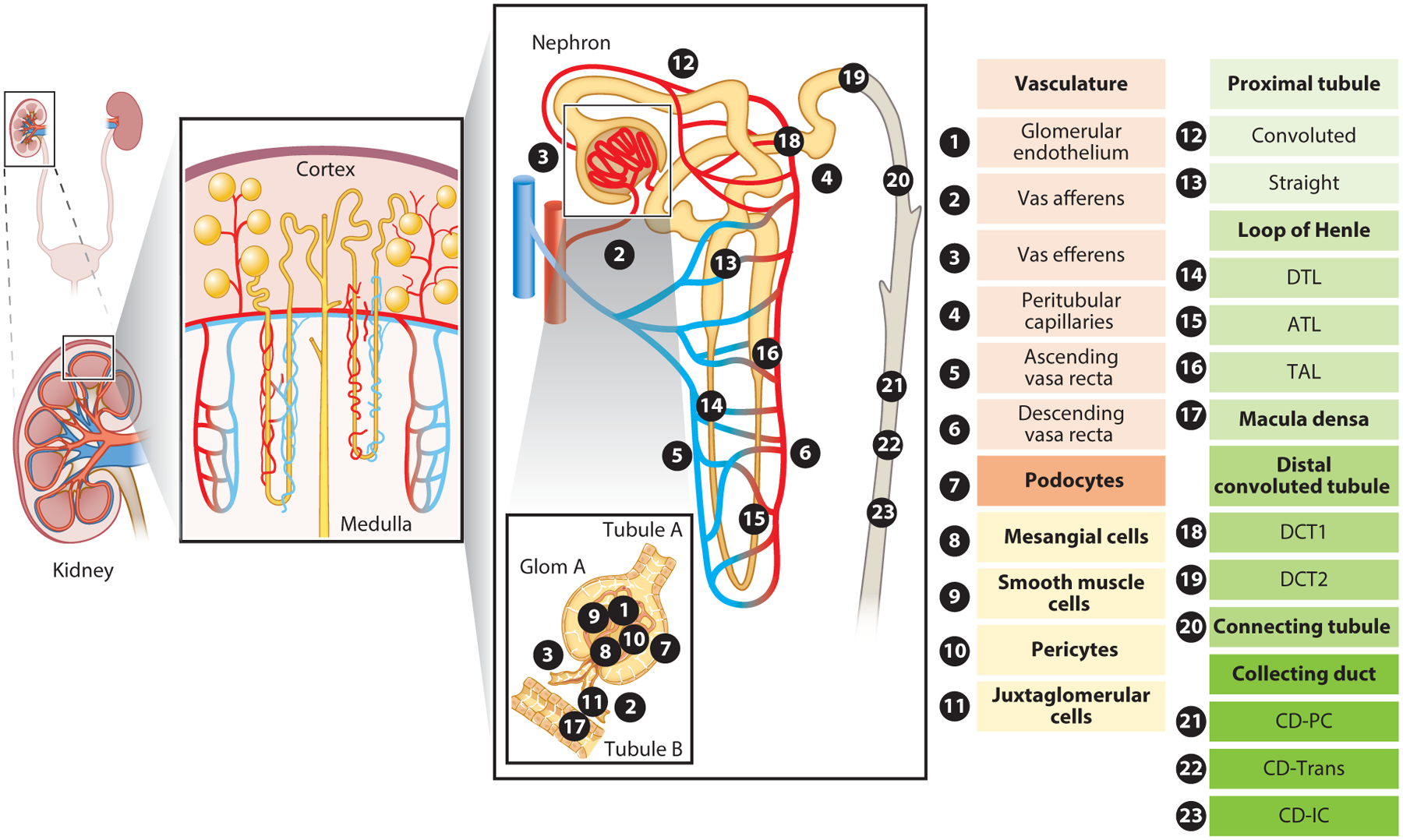
Overview of kidney epithelial and endothelial cell types. The various epithelial and endothelial cell types and subtypes of the kidney are numbered and annotated in the subpanel representing the functional unit of the kidney, the nephron. Kidney-resident immune cells are not included in the schematic. Abbreviations: ATL, ascending thin limb of loop of Henle; CD, collecting duct; CD-IC, collecting duct intercalated cell; CD-PC, collecting duct principal cell; CD-Trans, collecting duct transitional cell; DCT1/2, distal convoluted tubule 1 or 2; DTL, descending thin limb of loop of Henle; Glom, glomerulus; TAL, thick ascending limb of loop of Henle.
Recently developed single-cell methods can characterize the entire transcriptome of thousands of cells in a single experiment. Cells can then be organized into groups (clusters) based on their gene expression information (Figure 2). Single-cell clustering is mostly, but not completely, unbiased, and different resolutions of cell clustering could generate slightly different information. It is also worth noting that some cell types may be preferentially captured; therefore, some data sets might contain cells in proportions different from their natural occurrence. This review discusses this new unbiased single-cell gene expression data-based classification of kidney cell types. This new gene expression–based cell type classification matches with the traditional functional and genetic cell type definition with remarkable precision. Here, we discuss how each kidney cell type has specialized to achieve this complex function, how an individual cell’s gene expression supports this specific function, how cells change under a variety of physiological and pathological conditions, and how cells interact with one another, as predicted by recent single-cell multi-omics methods. We use two different single-cell RNA sequencing (scRNA-seq) data sets derived from mouse kidneys to illustrate our findings (Figure 3).
Figure 2.
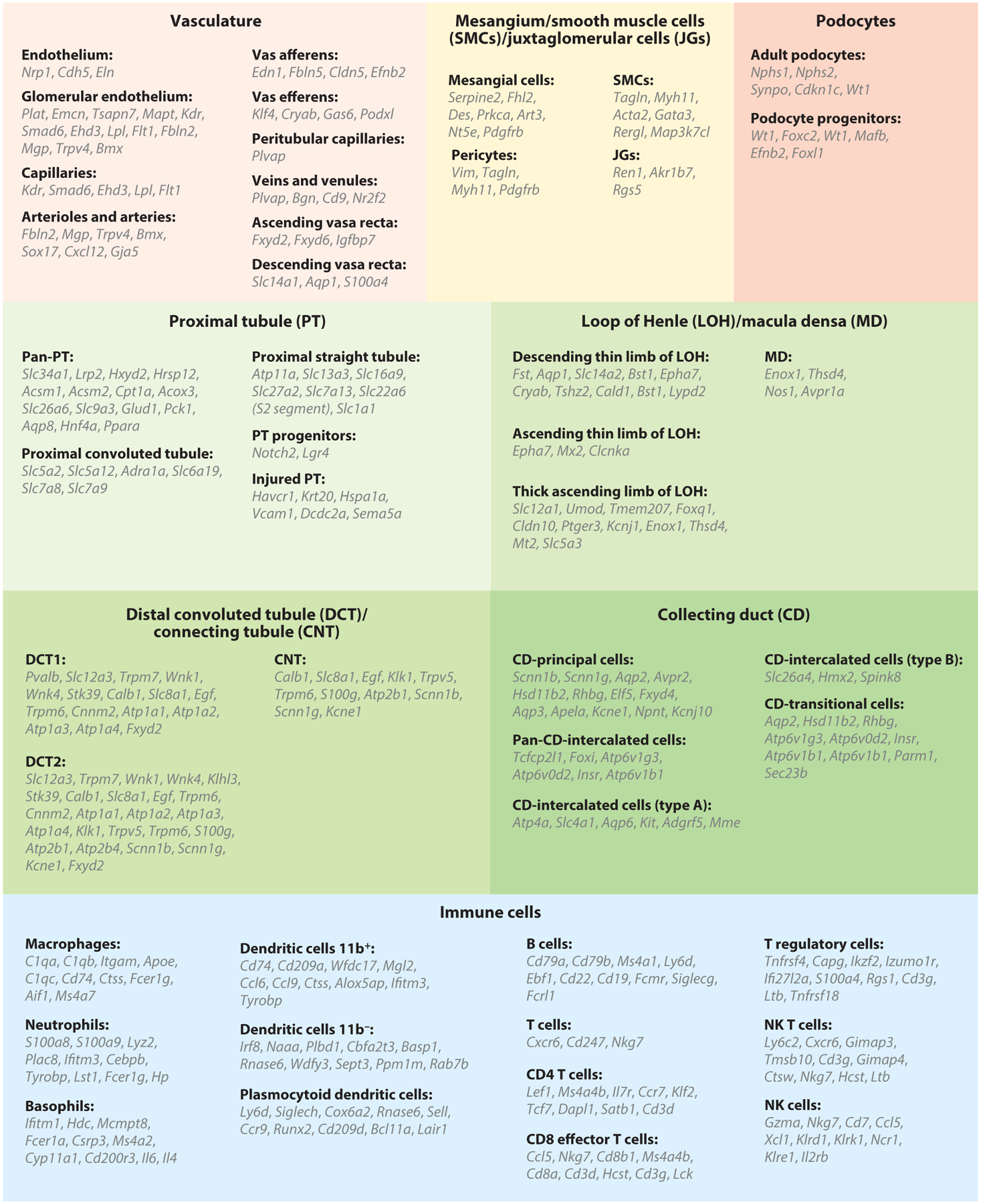
Cell type–specific marker genes of kidney epithelial, endothelial, and immune cells, as identified by single-cell RNA sequencing. Abbreviation: NK, natural killer.
Figure 3.
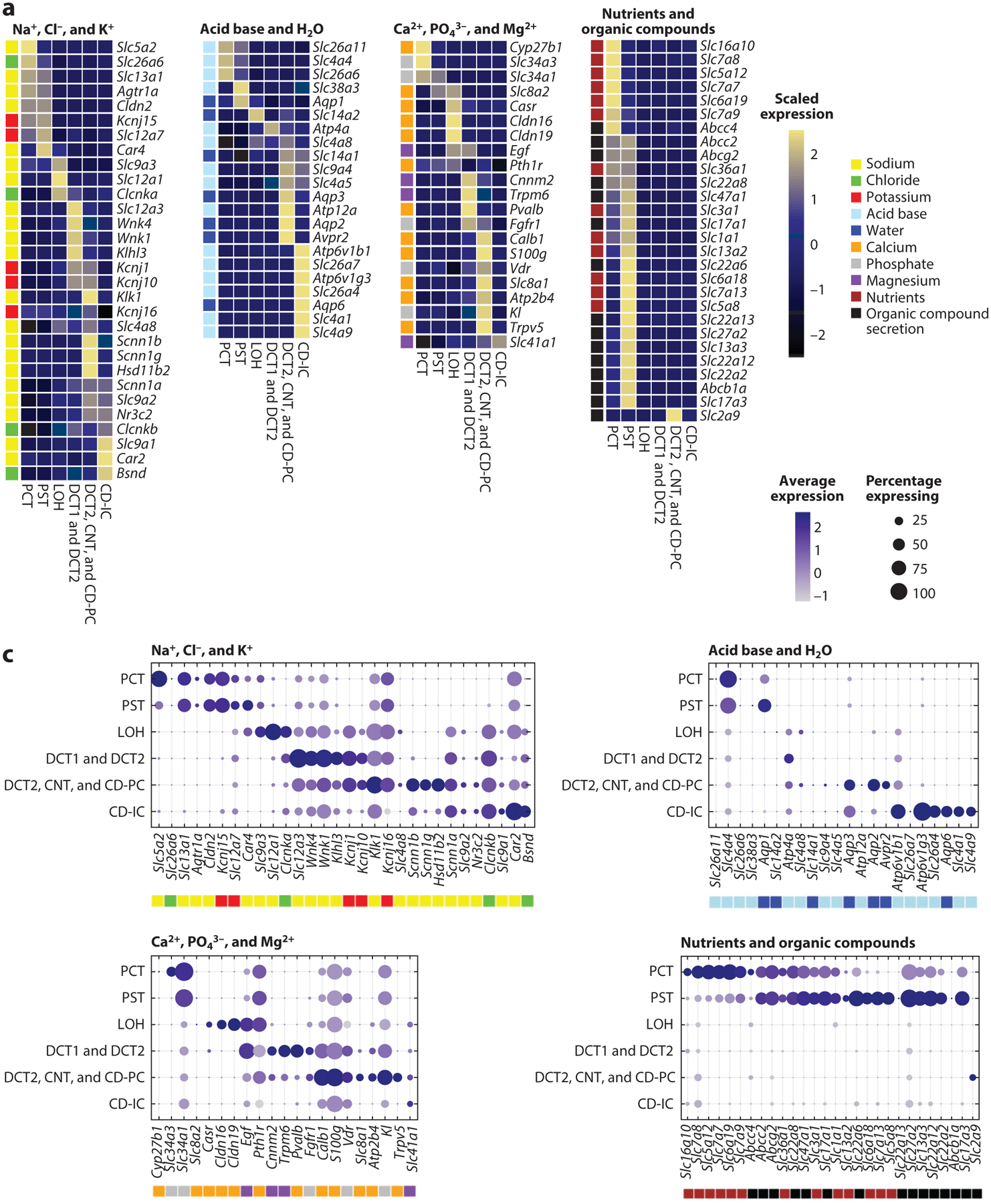
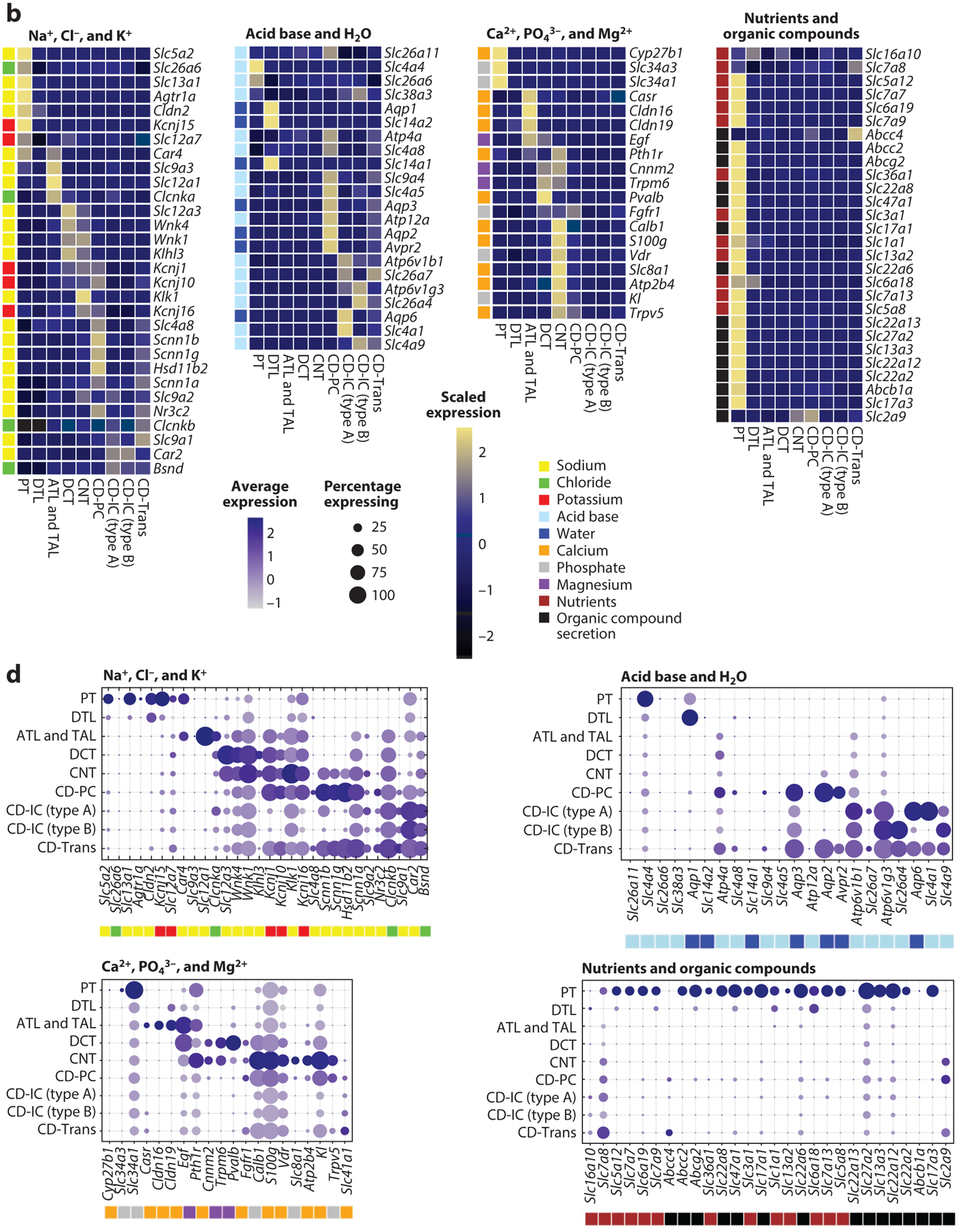
Kidney-specific single-cell expression of selected genes. Selection of representative marker genes involved in metabolite handling by the different kidney tubule segments. We present two separate single-cell RNA-sequencing data sets (panels a and c and b and d, respectively) with slightly different but complementary tubular cell clustering. Subpanels are broken down by functional handling of metabolites into genes involved in the handling of sodium (Na+), chloride (Cl−), and potassium (K+); acid base and water (H2O); calcium (Ca2+), phosphate (PO43−), and magnesium (Mg2+); and nutrients and organic compounds. Heatmaps (a,b) visualize row-scaled average expression (color scale), whereas dot plots (c,d) visualize the percentage of cells per cluster expressing the marker (dot size) and the average absolute gene expression (color scale). Color legends next to individual genes represent the metabolite handled by the corresponding transporter/protein. Abbreviations: ATL, ascending thin limb of LOH; CD-IC, collecting duct intercalated cell; CD-PC, collecting duct principal cell; CD-Trans, collecting duct transitional cell; CNT, connecting tubule; DCT, distal convoluted tubule; DTL, descending thin limb of LOH; LOH, loop of Henle; PCT, proximal convoluted tubule; PST, proximal straight tubule; PT, proximal tubule; TAL, thick ascending limb of LOH.
THE COMPLEX VASCULATURE OF THE KIDNEY
Although human kidneys only weigh approximately 100–180 g (0.5% body weight), they receive more than 20% of the cardiac output (8). The kidneys have a complex circulation, as the arterial system gives rise to two consecutive capillary beds (9). First, it branches into the afferent arterioles of the glomerulus, feeding the glomerular capillary bed, followed by the efferent arteriole, which then branches again, generating the peritubular capillary bed supplying the entire nephron. Glomerular capillaries are critical for making the primary filtrate, whereas the peritubular capillaries are critical for reabsorption via their close proximity to the tubular epithelial cells. The cortical and medullary peritubular capillary endothelial systems show important differences (10). The medullary vasa recta capillary system is right next to the loop of Henle and plays a critical role in renal concentrating ability and preserving the high osmolarity of the medulla. In addition, the parallel arrangement of the descending vasa recta (DVR) and the ascending vasa recta (AVR) in the medulla causes an oxygen shunt, creating a hypoxic renal papilla. In contrast, the cortical peritubular capillaries exhibit much higher blood flow and are critical for moving the large volume of fluid reabsorbed by the proximal and distal tubules as well as the cortical portions of the thick ascending limb of the loop of Henle back to the general circulation.
The kidney contains a large number of endothelial cells. Glomerular and pericapillary endothelial cells separate clearly even in initial whole kidney single-cell sequencing analysis (11, 12). Several studies have now performed single-cell analysis focusing exclusively on renal endothelial cells by sorting-based enrichment (13, 14). These studies highlighted some known pan-endothelial-specific markers such as Cdh5. While multiple subclusters have been identified, most of the subclusters are defined by a combination of markers rather than by just one single gene.
Glomerular endothelial cells are critical for filtration. Therefore, they are fenestrated and quite different from other endothelial beds in the body (15). Glomerular endothelial cells are the key target cells for vascular endothelial growth factor A (VEGFA, expressed by podocytes) even at basal conditions (16, 17). Single-cell studies consistently indicated high levels of Kdr [VEGF receptor 2 (VEGFR2)] expression by glomerular endothelial cells (11, 18). These observations likely explain the proteinuria development in up to 20% of patients taking VEGFA inhibitors. Several additional genes show high specificity for glomerular endothelial cells, such as Plat, Emcn, Ehd3, Tsapn7, and Lpl (11, 18, 19). One study subdivided glomerular endothelial cells into capillaries (expressing Kdr, Smad6, Ehd3, and Lpl), afferent arterioles (expressing Edn1, Fbln5, Cldn5, or Efnb2), and efferent arterioles (expressing either Klf4 and Cryab or Gas6 and Podxl) (13, 14). By profiling glomeruli, the Shaw group (20) identified capillary endothelial cells by their marker Ehd3 and afferent and efferent arteriolar endothelial cells by their expression of Fbln2, Mgp, Trpv4, and Bmx. In addition, they also identified a novel subpopulation of endothelial cells that express high levels of the Notch ligand Dlk1 and the endothelin receptor (Ednrb). A third study that also focused on endothelial cells used a different renal endothelial cell classification system (13), highlighting the expression of Ehd3, Mapt, and Sox17 in glomerular endothelial cells.
Peritubular capillary cells were found to express Igfbp3, Plvap, and Npr3. In one study, cortical peritubular capillary endothelial cells were subdivided into nine subclusters representing endothelial cells from arteries, afferent arterioles, and efferent arterioles (Sox17, Cldn5). Other clusters highly expressed capillary markers (Id3, Kdr, and Flt1) but differentiated based on high or low Apoe expression (14). One cluster showed an intermediate capillary-vein gene signature associated with postcapillary venules and another cluster that included veins [Bgn, Plvap, and Cd9 (14)].
A large number of medullary endothelial cell clusters have been identified, including arterioles (Klf4, Kitl), DVR (Aqp1), and the papillary portion of DVR as a separate cluster (14). Papillary DVR cells expressed high levels of hyperosmolarity-responsive genes, consistent with the high osmolarity in the papilla, including S100a4. The AVR cells expressed Gas6, Fxyd2, and Fxyd6.
Another team used a different renal endothelial cell classification system and reported markers slightly differently (13). Arterial vessels expressed Sox17, Gja5, and Cxcl12 and low levels of Vegfr2, Kdr, Flk1, and Fbln5. Gja5 was expressed in the DVR and all preglomerular arteries, including the afferent arteriole, while absent in the efferent. The AVR was identified through the presence of Igfbp7 but a lack of Igfbp5 expression. Venous endothelial cells were classified based on Nr2f2 expression. The peritubular capillaries and venous endothelial cells expressed Plvap and Igfbp5, but not Nr2f2. Another interesting aspect of the work was that the team identified specific solute carriers for each specific endothelial bed. Pericapillary endothelial cells expressed high levels of glucose transporters, whereas DVR expressed myo-inositol, urea (Slc14a1), proton transporters, and water channels (Aqp1), indicating an interesting gene expression adaptation to function.
Upstream transcriptional regulators for endothelial cell identity were predicted by two independent groups based on the gene expression data using a method called SCENIC (single-cell regulatory network inference and clustering) (13, 14). The SCENIC algorithm predicted increased binding by Gata2 and Gata5. Barry et al. (13) also found specific enrichment for Tbx3, Gata5, Prmd1, and Pbx1 in glomerular endothelial cells. Cortical peritubular capillaries were positive for Irf1, Foxp1, Sp1, and Lef1. As expected, the regulons of arteries and veins showed an enrichment for Sox17 and Nr2f2, respectively. Direct analysis of the single-cell open chromatin region using single nucleus assay for transposase-accessible chromatin with sequencing (snATAC-Seq) indicated enrichment for Erg and Sox17 transcription factors in endothelial cells (19). Cell–cell interaction analysis highlighted important interactions of endothelial cells with a variety of epithelial cells, some of the key interaction pathways being VEGFA and its receptor KDR and the WNT and NOTCH signaling pathways (19). This again indicated the important role of epithelial cells in maintaining circulation and of endothelial cells in nurturing epithelial cells.
In summary, the kidney has a complex vascular system that includes a double capillary bed system, with the first glomerular capillary bed controlling filtration, while the second peritubular capillary system plays an important role in reabsorption. ScRNA-seq studies identified marker genes for different components of the renal vasculature. Although the glomerular and peritubular capillaries can be easily distinguished by their marker gene expression, future studies should aim at identifying a unified characterization for endothelial cells.
MESANGIAL AND SMOOTH MUSCLE CELLS
Mesangial cells provide central structural support for the glomerulus. They are defined based on their anatomical location, but their gene expression is poorly understood (21). Mesangial cells produce extracellular matrix (ECM), regulate capillary blood flow, phagocytose extracellular debris, and contribute to the homeostasis of the glomeruli by secreting growth factors. Marker genes commonly used to identify mesangial cells, such as Pdgfrb and Gata3, are not specific, as these genes are also expressed by vascular smooth muscle cells (SMCs) and stromal cells (22, 23). For example, α-smooth muscle actin (Acta2) is often used as a mesangial marker, but it is not expressed well in healthy unstimulated mesangial cells and is more specific to SMCs. Chung et al. (20) performed scRNA-seq analysis on purified glomerular cells and identified cell clusters that matched with prior mesangial markers such as Gata3 and Pdgfrb, and they separated them into two clusters. One cluster also expressed Acta2, Tagln, Myh11, and juxtaglomerular (JG) cell markers Ren1, Akr1b7, and Rgs5, indicating that this cluster likely represented JG cells. Using differential expression analysis, the team identified Serpine2, Fhl2, Des, and Pdgfrb as mesangial cell markers, as these genes were expressed at low levels in SMCs/JG cells. They reported specific mesangial markers such as Plvap, Prkca, Art3, and Nt5e and markers for SMCs/JG cells such as Acta2, Myh11, Rergl, Map3k7cl, Ren1, and Akr1b7 (20). The identified markers indicate the key contractile nature of these cells (Acta2, Myh11, and Tagln). Given the overlap between mesangial cells, vascular SMCs and interstitial cells, future validation studies or spatial transcriptomic analysis will be essential to define mesangial cell markers.
PODOCYTES
Podocytes are glomerular epithelial cells. They play a key role in establishing the filtration selectivity of the glomerulus (24) and contribute to the maintenance of glomerular basement membrane by secreting collagen (25). They also secrete VEGFA to maintain glomerular endothelial cell fenestration. Their highly specialized role in the body is reflected by their unique gene expression pattern. Podocytes are relatively easy to identify in single-cell gene expression data sets. Podocyte marker genes identified by scRNA-seq match with prior cell biological and genetic studies. For example, they highly and selectively express Nphs1, Nphs2, Synpo, Wt1, and Cdkn1c. Loss- or gain-of-function coding mutations of most podocyte-specific genes were associated with high-grade proteinuria (nephrotic syndrome) development in humans (26–28).
Combined single-cell RNA and epigenome analysis of the developing mouse kidney showed that, during development, podocyte differentiation occurs early from nephron progenitors (19). Nephron progenitor differentiation was associated with the closing of chromatin areas around nephron progenitor–specific genes such as Osr1, Gdnf, Sall1, and Pax2 and a decrease in their expression. At the same time, differentiation was associated with the opening of areas around podocyte-specific genes such as Foxc2, Wt1, Mafb, and Efnb2. At later stages, there was a strong increase in chromatin accessibility of actin filament–based processes and a significant decrease in Notch1, Notch2, and Ctnnb1 in the podocyte lineage. Although the role of several transcription factors has been established for podocyte specification, the expression of Foxl1 was newly described. Pathway analysis showed that podocytes induced programs in cytoskeletal regulation, cell adhesion, and inflammatory response, which is again highly consistent with their key function in the kidney.
PROXIMAL TUBULE
The main functions of the proximal tubule are to reabsorb all nutrients and about two-thirds of most electrolytes and water from the primary filtrate, as well as to secrete a broad range of organic anions and cations (29). Proximal tubules compose the majority of cells in the kidney (~66%) and close to half of the protein mass. Two main classification systems have been developed for the proximal tubules: One divides the proximal tubule into the S1, S2, and S3 segments, whereas the other divides it into proximal convoluted and straight segments (PCTs and PSTs, respectively). The S3 segment mostly corresponds to the straight segment of the proximal tubule. The S2 segment has been defined by its ability to secrete para-aminohippuric acid (PAH) and expresses its transporter Slc22a6 (OAT1) (30). At present, scRNA-seq and single-cell epigenome analysis are more consistent with the PCT and PST models. In mouse kidney single-cell data sets, it has been difficult to identify a segment that would clearly match with the S2 segment (11, 19), indicating that S2 might be a more anatomically defined and less functionally distinct segment.
The proximal tubule reabsorbs 60–70% of filtered water and Na+ together with either Cl− or HCO3− (31–33). Sodium reabsorption is achieved by Na+/H+ exchange (NHE3, Slc9a3) and Cl−/base exchange (CFEX, Slc26a6) with recycling of the conjugate acid, resulting in net NaCl entry. The proximal tubule exhibits a triple coupling mechanism, where Na+-sulfate cotransport (Slc13a1) runs in parallel with two anion exchangers to achieve NaCl cotransport. In addition, claudin 2 (Cldn2) is expressed at this segment to provide a cation-selective and water-permeable conductance to PCT sodium transport. Na+-coupled reabsorption of nutrients, i.e., glucose, amino acids, and mono- and dicarboxylates, with concurrent paracellular Cl− transport driven by the lumen-to-interstitial electrochemical gradient, is important for overall NaCl reabsorption (31–33). It is important to note that classic physiology studies place a strong emphasis on the role of NHE3 (Slc9a3) expression, whereas our single-cell data indicate relatively low expression of this transporter in the proximal tubule. On the other hand, Cldn2 seems to be very highly expressed by the proximal tubule. It is possible, however, that for these transporters the transcript and protein levels do not fully correlate, and the protein expression of SLC9A3 is greater than what is predicted by the transcriptomics data.
Approximately 180 g of glucose (1,000 mEq) and 50 g of amino acids (400 mEq) are filtered by the human kidney per day (34–37). The proximal tubule normally reabsorbs 99.8% of these nutrients from the luminal fluid. The PCT segment has important transporters specific to this segment, such as Slc5a2 and Slc5a12 (11, 19). Slc5a2 encodes for the sodium/glucose cotransporter 2 (SGLT2), which is one of the apical transporters responsible for reabsorption of glucose and a target for diabetes medications such as gliflozins (38, 39). This segment also expresses high levels of amino acid transporters including the SLC3, SLC7, and SLC36 families (36, 37, 40, 41). About 80% of the filtered amino acids are neutral amino acids, all of which are reabsorbed by the apical SLC6A19 (BOAT1) (42). SLC6A19 is found in the early portion of the proximal tubule and binds various neutral amino acids, including glutamine, with relatively low affinities. Mutations of SLC6A19 result in Hartnup disorder, which is characterized by a pronounced urinary loss of neutral amino acids (43, 44). The glutamate transporter (SLC1A1) reabsorbs acidic amino acids (45), whereas an antiporter (SLC7A9) mediates the uptake of basic amino acids and cysteine in exchange for a neutral amino acid (36). At the basolateral membrane, SLC7A8 is present as a heterodimeric neutral amino acid exchanger (37, 41).
The PST segment can be identified by expression of Slc27a2, Slc7a13, Slc22a6, and Slc13a3 (11, 18, 19). This segment is responsible for organic compound secretion and reabsorption of some nutrients. The segment expresses high levels of SLC22 family transporters that transport organic anions (46). Organic anion secretion is important for the elimination of foreign compounds, including drugs (47).
Metabolism of glutamine by renal proximal tubule cells plays an important role in acid-base balance (48, 49). At normal blood pH, glutamine filtered by the glomeruli is almost completely extracted from the lumen of the PCT and returned to the blood (42). The transepithelial transport utilizes apical SLC6A19 and basolateral SLC7A8 (LAT2) and SLC38A3 (50–52). During chronic acidosis, glutamine is metabolized within the early portion of the proximal tubule to generate ammonia, which serves as a urinary buffer to promote acid excretion (53, 54). Increased renal catabolism of glutamine is facilitated by increased expression of glutaminase (55, 56), glutamate dehydrogenase (GDH) (57), phospho-enolpyruvate carboxykinase (PEPCK), mitochondrial aquaporin-8 (AQP8) (58), the apical Na+/H+ exchanger (NHE3, Slc9a3) (59–61), and the basolateral glutamine transporter (Slc38a3) (50, 51, 62). Increased expression of NHE3 (Slc9a3) (59–61) contributes to the transport of ammonium ions and the acidification of the luminal fluid (63, 64).
Citrate is the only organic anion in millimolar quantities in the urine and represents the main mode of base equivalent excretion under normal circumstances (29). In addition, calcium citrate has a high association constant and solubility, making citrate the most efficient chelator of calcium, thus preventing stone formation (53, 54). The reabsorption of filtered citrate occurs in the PST via the apical membrane by Na+-dependent dicarboxylic acid cotransporters NaDC-1 (Slc13a2) and NaDC-3 (Slc13a3) (65).
To keep up this high-activity sodium transport, proximal tubule cells have one of the highest mitochondrial content in the body and are also exceedingly sensitive to injury (66). Furthermore, recent studies using scRNA-seq indicate that proximal tubule cells are among the most plastic cells in the kidney. Recent subclustering identified many, sometimes more than 10, different proximal tubule subgroups, including progenitor cells, injured cells, failed repair cells, and profibrotic cells (18, 67). Our lab has performed scRNA-seq analysis in the folic acid nephropathy (FAN) model of kidney fibrosis (18), where proximal tubule cells showed the largest number of differentially expressed genes. Genes that showed lower expression levels in diseased proximal tubule cells were solute carriers such as Slc5a2 and Slc13a3, and these genes are highly expressed in fully differentiated proximal tubule cells. Other gene groups showing lower expression in disease samples were genes involved in fatty acid oxidation and oxidative phosphorylation (e.g., Acsm1, Acsm2, Cpt1a, and Acox3).
In the FAN disease model, we identified nine proximal tubule subclusters (18). These included clusters containing proliferating cells, immune marker (Cd74)–expressing cells, transitional cells, precursor cells expressing higher Igfbp7, and a prominent proliferating cell population. In addition, we identified a cell population expressing Notch2 and Lgr4, known markers of progenitor and transit-amplifying cells in the kidney and other organs (68, 69). We observed that PCT cells coexpressed PST markers in the disease state, suggesting that PCT cells may endure transcriptomic changes impacting their phenotypic signature (18). Cell trajectory analysis indicated that FAN samples showed a differentiation trajectory toward PCT and PST segments but followed a less organized differentiation path compared to healthy proximal tubule cells. Metabolism-associated genes such as those involved in fatty acid oxidation and oxidative phosphorylation showed strong correlation with proximal tubule cell differentiation, indicating that the metabolic need of proximal tubule cells (judged by the expression of metabolic genes) and their functional state (solute carrier expression) are linked.
Single-cell studies analyzing the ischemia reperfusion model identified eight proximal tubule subclusters, including severely injured proximal tubule marked by the expression of Krt20 and heat shock proteins (67). Another cluster was called repairing proximal tubule, which arose two days after injury and expressed cell cycle genes, including Top2a. In addition, another cluster had low expression levels of differentiated proximal tubule markers such as Slc5a12, Slc22a30, and Slc7a13 but expressed Vcam1, Dcdc2a, and Sema5a. The Humphreys group (67) has named this cluster failed repair proximal tubule and verified the VCAM1 protein expression by immunostaining. Several other research groups have performed single-cell transcriptomic studies on kidney disease models such as ischemia reperfusion (70), unilateral ureteric obstruction (71), or sepsis-induced acute kidney injury (72). Similarly, these studies have identified multiple proximal tubule subclusters. At present, however, the relationship between the subclusters described in different publications is unclear; therefore, larger integrative studies are needed to understand the consistencies and differences.
Epigenetic profiling of proximal tubule cells indicated the important role of Ppara, Esrra, Hnf1b, and Hnf4a nuclear receptor transcription factors in proximal tubule cells (19). This is consistent with the gene expression studies described above. Proximal tubule cells are highly metabolically active, as they utilize fatty acids as their energy source. The transcriptional network allows the balancing of metabolic need and gene expression changes (cellular metabolic demand). Gene expression–based transcription factor prediction by SCENIC indicated strong enrichment of Hnf1b, Maf, Hnf1a, and Hnf4a regulon activity in proximal tubule cells. During development we observed a de novo increase in chromatin accessibility of the Hnf4a and Pou3f3 motifs in the proximal tubule in parallel with cell type commitment. This coincided with expression of the cellular differentiation transcription factors Hnf4a and Hnf1a as well as their downstream target genes Ace2, Atp1a1, Dab2, Hnf1a, Hnf4a, Hsd17b2, Lrp2, Maf, Slc12a3, Slc22a12, Slc34a1, and Wnt9b (19).
LOOP OF HENLE
The key function of the loop is to improve the water concentrating ability of the kidney. Anatomically, the loop of Henle can be divided into six distinct segments: the descending thin limb, which is divided into three segments (DTL1–3), the ascending thin limb, and the thick ascending limb (TAL), which consists of medullary and cortical regions (MTAL and CTAL) (73, 74). The descending thin segment is classically defined by urea transporter Slc14a2 and Aqp1 expression (75), but additional segment markers have been identified by single-cell studies (18).
The TAL composes approximately 20% of all cells in the kidney and roughly 15% of protein mass. This segment reabsorbs 25% of the filtered Na+. A key feature of the TAL is the expression of the furosemide-sensitive Na+/K+/2Cl− cotransporter (Slc12a1). This transporter moves Na+, K+, and 2Cl− from the tubular fluid to inside the tubular cells (76, 77). Because the filtrate contains a lot more Na+ than K+, loss of K+ from the tubular fluid would become limiting without replenishing tubular K+ by the apical K+ channel ROMK (Kir1.1, Kcnj1) (78, 79). Na+ is extruded from the cell on the basolateral membrane by the Na+/K+ ATPase, and Cl− exits via the Cl− channel (Clcnkb), which requires an accessory β subunit Barttin (Bsnd). Mutations in SLC12A1, KCNJ1, CLCNKB, and BSND in humans cause Bartter’s syndrome, characterized by Na+ and K+ loss, volume depletion, and metabolic alkalosis (80).
Similar to the distal convoluted tubule (DCT), the TAL is impermeable to water, so the tubular fluid becomes diluted. A portion of the filtered Ca2+ and Mg2+ is also reabsorbed here, mainly via paracellular pathways (Cldn16, Cldn19) driven by the lumen-positive transepithelial potential difference generated by the ROMK (Kcnj) channel. Accordingly, mutations of CLDN16 and CLDN19 in patients cause renal magnesium (and calcium) wasting (81). Single-cell studies also indicated expression of the calcium-sensing receptor (Cas1) and PTH receptor (Pth1r) in this segment.
The TAL expresses uromodulin (Umod) (82), which is one of the most highly expressed genes in the kidney. Genome-wide association studies indicated that genetic variants at the UMOD locus showed the strongest association with kidney function (83–86). The complete role of UMOD in the kidney is not fully established (87–91). Uromodulin acts as a constitutive inhibitor of calcium crystallization in renal fluids (92–95). The excretion of uromodulin in urine may provide defense against urinary tract infections caused by uropathogenic bacteria (96–98). UMOD also likely plays a role in sodium balance and blood pressure regulation (99). Transgenic expression of Umod in mice causes hypertension and cardiac hypertrophy (100).
Our analysis of mouse kidney development indicated the expression of loop of Henle markers such as Cyfip2, Cytip, Esrrb, Esrrg, Irx1, Irx2, Mecom, Pla2g4a, Pou3f3, Ppargc1a, Stat3, Sytl2, Tfap2b, Thsd4, and Umod (19). The key transcription factors identified in this segment were Esrrb and Tfap2b.
A subgroup of cells in the TAL compose the macula densa, an area of closely packed specialized cells lining the wall of the distal tubule, at the point where the TAL of the loop of Henle meets the DCT (74). The macula densa is the thickening where the distal tubule touches the glomerulus. The macula densa plays a key role in regulation of estimated glomerular filtration rate by sensing distal delivery NaCl (101–104). Changes in NaCl concentration signal a state of salt deficit or excess in the body, which triggers the macula densa to reset the operating point of the tubule-glomerular feedback loop by altering the local balance of vasoconstrictors and vasodilators, particularly angiotensin II and nitric oxide (NO). Consistent with prior functional studies, the single-cell studies also indicate that the macula densa cells express high levels of Nos1 (67), the neuronal isoform of NO synthase, and are a likely source of NO acting in the juxtaglomerular apparatus. Macula densa cells also express Avpr1a.
DISTAL TUBULE
The DCT is responsible for the regulation of plasma potassium, sodium, calcium, and pH (105). The DCT is a segment defined anatomically as the portion of the nephron following the macula densa. The DCT reabsorbs roughly 5–10% of the filtered sodium load. The thiazide-sensitive NaCl cotransporter (Slc12a3) is a key marker of this segment and is responsible for sodium reabsorption (106). SLC12A3 is the key target of thiazide diuretics. The other target is the sodium-dependent chloride/bicarbonate exchanger (Slc4a8) (107). Loss-of-function mutations in SLC12A3 cause Gitelman syndrome, a disorder characterized by polyuria, Na+, K+, and Mg2+ loss and metabolic alkalosis (108). The basolateral membrane contains an inward-rectifying K1 channel Kir4.1 (Kcnj10). Patients with mutations of this channel also develop Gitelman syndrome (109) and other neurological abnormalities (110, 111). Other potassium channels are located on the basolateral membrane at KCNJ15 (Kir4.2) and KCNJ16 (Kir5.1) (112). The basal aspect of the cells expresses massive amounts of the ATP-dependent Na+/K+ pump and, owing to the high energy need of the DCT cells, these cells contain large amounts of mitochondria (113). The single-cell data show that the high mitochondrial content often limits cell type marker identification.
A basolateral Cl− transporter is also needed for transepithelial Na+ transport. Cl− exit helps to lower the intracellular Cl− concentration. Chloride depletion is a potent activator of the apical Na+-Cl− cotransporter (NCC), and maintains net Na+ reabsorption (114). Cl− exits across the DCT basolateral membrane via the CLCNKB (Clc-Kb) chloride channel. CLCNKB requires CLCNKA and an accessory subunit called Barttin (Bsnd) to be fully functional (115). Patients with CLCNKB channel mutations tend to present with a mixed Bartter–Gitelman phenotype, whereas patients lacking functional CLCNKA present with a severe neonatal salt wasting disorder that includes sensorineural deafness (115, 116). Traditionally, it was thought that both Clcnkb and Bsnd (Clc-Kb and Barttin) are expressed in the TAL. The single-cell data show some differences in their expression pattern and transcript amount, which might explain phenotypic differences observed in patients. The basolateral membrane expresses SLC12A7, another potassium chloride cotransporter (117).
Genetic studies of blood pressure extremes (low and high) highlight key regulators of the DCT. Patients with familiar hyperkalemic hypertension or Gordon syndrome carry mutations in WNK1, WNK4, CUL3, or KLHL3 (118–121). According to the current model, in the baseline inactive state, WNK4 suppresses SLC12A3 trafficking to the plasma membrane, holding the cotransporter in an intracellular storage pool (122–124). The kinase-active form of WNK1 can reverse this process (123, 125). The Kelch-like 3/Cullin-3 (Klhl3/Cul3) E3 ubiquitin ligase complex constitutively degrades the WNKs. Gordon syndrome–associated mutations in KLHL3 reduce binding to WNK1 and WNK4, increasing WNK1 and WNK4 abundance and triggering NCC activation through the WNK effector kinases SPAK and OSR1 (126, 127). Additionally, mutant WNK4 reduces its inhibitory effect on SLC12A3 traffic (125), releasing SLC12A3 from its intracellular compartment and increasing its trafficking to the cell surface (128, 129). It is important to note again that unbiased single-cell studies performed decades after the initial genetic studies not only identified these genes as being expressed in the DCT but also showed that their transcript level was high. They also showed strong enrichment for the DCT segment (11).
The DCT also plays an important role in magnesium homeostasis and expresses the magnesium channel TRPM6 (130). Loss-of-function mutations of this channel cause familial hypomagnesemia with secondary hypocalcemia (130, 131). At the luminal membrane, the K+ channel Kv1.1 (Kcna1) extrudes K+ into the tubular lumen; this process probably generates an electrical driving force for Mg2+ entry through TRPM6 located in the apical membrane. TRPM6 activity is stimulated by epidermal growth factor (EGF), which triggers an intracellular signaling cascade in the DCT after binding to basolateral EGF receptors (EGFRs), resulting in increased surface expression of TRPM6. Single-cell analysis of this segment confirmed high levels of Egf and Trpm6 (11, 18, 19). Presence of this signaling pathway in this nephron segment potentially explains the magnesiuric effect of EGF inhibitors (132, 133). Genetic studies further support the role of EGF in magnesium regulation. In a Dutch family with a recessive form of selective hypomagnesemia, affected members had a mutation in the gene encoding the precursor form of EGF (134).
LATER DISTAL TUBULE AND CONNECTING TUBULE
The connecting tubule (CNT) is a poorly defined kidney segment between the DCT and the collecting duct. Therefore, it is a bit difficult to match prior physiological studies with the single-cell gene expression data. Prior physiological and immunostaining studies indicated different subtypes of DCT cells such as DCT1 and DCT2. It was proposed that DCT1 and DCT2 show differential responsiveness to the mineralocorticoid aldosterone, as DCT2 is a mineralocorticoid-responsive cell type (135). Although the main site of action of aldosterone is the mineralocorticoid receptor (Nr3c2) in the principal cell of the collecting tubule, it was also expressed by a subgroup of DCT cells. In addition, DCT2 expressed an enzyme called hydroxysteroid 11-beta dehydrogenase 2 (Hsd11b2) (136, 137). HSD11B2 metabolizes cortisol to the inactive metabolite cortisone, thereby preventing circulating glucocorticoids from binding to mineralocorticoid receptors. Our single-cell data set identified a cell population that was somewhat positive for Slc12a3, expressing Wnk1 and Wnk4, but also expressing some Hsd11b2, Scnn1g (ENaC), and Nr3c2 (mineralocorticoid receptor). However, the key characteristic of this segment was not Slc12a3 or Scnn1g expression but the unique expression of calcium transporters. While these cells did not separate clearly in our first gene expression data set (Figure 3a,c), in another of our data sets the expression of Trpv5 clearly distinguished these cells in single-cell data obtained from whole kidney samples (Figure 3b,d).
Approximately 7–10% of filtered calcium is reabsorbed in the distal tubule (105). Apical calcium transport is mediated by the transient receptor potential channel vanilloid 5 (Trpv5) (138, 139). Cytosolic calcium is immediately bound by calbindin (Calb1), which shuttles calcium to the basolateral aspect of the DCT cell, where it can be transported out by the type 1 sodium/calcium exchanger (Ncx1, Slc8a1) or calcium ATPases (Atp2b4). These processes are tightly regulated by hormones such as parathyroid hormone (Pth1r) and 1,25-dihydroxyvitamin D (Vdr) (140). As predicted by physiological studies, these cells express high levels of Klotho (Kl) and Calb1, Trpv5, Pth1r, and Vdr.
A recent study reported single-cell sequencing of this specific tubule segment only (141). Unsupervised clustering revealed Slc12a3+/Pvalb+ and Slc12a3+/Pvalb− cells, identified as DCT1 and DCT2 cells, respectively. DCT1 cells appear to be heterogeneous, and one DCT subcluster was positive for cell cycle genes, which fits with the known plasticity of DCT cells. DCT2 cells expressed the ENaC subunits and transcripts associated with calcium transport (Trpv5, Calb1, S100g, and Slc8a1), almost completely matching our cluster called CNT in our single-cell gene expression studies.
We therefore propose that it is best to call CNT cells based on their unique marker expression of Trpv5, Kl, Vdr, and Calb1 in addition to the overlapping expression of both DCT (Slc12a3) and collecting duct principal cell (CD-PC) markers (ENaC). We also acknowledge their similarities to DCT2 cells and future studies should systematically compare results of prior functional studies indicating important functional heterogeneity of DCT cells and recently identified gene expression heterogeneity in this segment.
COLLECTING DUCT
There are several segments of the collecting duct system, including the CNTs, cortical collecting ducts, and medullary collecting ducts. As opposed to the proximal part of the tubule epithelial cells, the collecting duct system originates from the ureteric bud (142). The key role of the collecting duct system is to regulate free water and electrolytes, including sodium and chloride, in addition to acid-base balance. The medullary collecting ducts are divided into outer and inner segments, the latter reaching more deeply into the medulla. The variable reabsorption of water and, depending on fluid balances and hormonal influences, the reabsorption or secretion of sodium, potassium, hydrogen, and bicarbonate ions continues here. Urea passively transports out in this tubule segment and creates a 500-mOsm gradient. Papillary ducts represent the most distal portion of the collecting duct. The available single-cell data sets had limited ability to identify spatial differences between cortical medullary and papillary collecting duct cells.
A critical feature of the collecting duct is that it is known to contain two distinct cell types, principal and intercalated (143, 144). The key role of principal cells is to reabsorb water, and they express high levels of AQP2 (Aqp2) and vasopressin receptor (Avpr2). In the absence of antidiuretic hormone, water in the renal filtrate is left alone to enter the urine, promoting diuresis. When antidiuretic hormone is present, it binds to its receptor, which activates adenylate cyclase, protein kinase A, and the transcription factor CREB. Protein kinase A leads to AQP2 phosphorylation, and aquaporins translocate to the plasma membrane and allow for the reabsorption of water, thereby inhibiting diuresis. Prostaglandin E2 (PGE2) and ATP are negative regulators of the process. Mutations in AQP2 and AVPR2 in patients cause nephrogenic insipidus (145–147).
Principal cells also play a key role in regulating Na+ and K+, and this is the site where final regulation of urinary sodium excretion occurs. Aldosterone is the primary hormonal regulator of both Na+ and K+ transport (148). It acts through the mineralocorticoid receptor (Nr3c2) to increase expression of the serine-threonine kinase SGK1, ENaC (Scnn1), and the Na+/K+ ATPase (149, 150). ENaC is the primary target of aldosterone, which affects both Na+ reabsorption and K+ secretion (149). Other regulators of the principal cells are insulin (151), adenosine (152), bradykinin (153), and endothelin (154). As discussed above, single-cell gene expression analysis identified a gradient of cells expressing mineralocorticoid receptor and ENaC. Functional studies called these cells DCT2, but as we discussed above, they show some overlap with CNT based on their unique expression of Trpv5 and Trpm6.
Intercalated cells can be identified by the expression of the vacuolar proton ATPase (Atp6v1g3, Atp6v0d2, Atp6v1b1). Type A intercalated cells can secrete H+ equivalents into urine via the H1-ATPase or the H+,K+-ATPase (Atp4a) at their apical membrane (155). The latter pump exchanges one potassium ion for each extruded proton. These cells also express the Cl−/HCO3− exchanger SLC4A1 (Slc4a1) (156). The secretion of a proton into the tubular lumen, whether it is in exchange for potassium reabsorption or not, results in the generation of intracellular bicarbonate via the carbonic anhydrase II. HCO3− is transported to the interstitium in exchange for chloride by SLC4A1. In single-cell analysis, type A cells express the anion exchanger Slc4a1, whereas type B cells do not (11).
The classic type B intercalated cells express a chloride/bicarbonate exchanger, pendrin (Slc26a4), at their apical membrane and express H1-ATPase (Atp6v1g3) at their basolateral membrane (157–159). These cells are thought to be responsible for the secretion of bicarbonate equivalents. Type B intercalated cells have recently come to prominence with the discovery of SLC4A8, the Na+-driven chloride/bicarbonate exchanger (NDCBE) (107), which in scRNA-seq data is also seen in principal cells.
Unexpectedly, our single-cell profiling identified a third cell cluster in the collecting duct (11). These cells express Aqp2, Atp6v1b1, and cell type–specific markers such as Parm1 and Sec23b. Pseudotime cell trajectory analysis placed the newly identified cells between the principal cells and intercalated cells, suggesting that they are a transitional cell type. The presence of these transitional cells raised the possibility that principal cells and intercalated cells are more plastic and they could interconvert. The existence of these transitional cells and interconversion of principal cells and intercalated cells was confirmed by fluorescent linage tracing. Gene expression analysis revealed activation of the Notch signaling pathway during transition of intercalated cells to principal cells. Notch is known to regulate the cellular identity of neighboring cells by expression of its ligands or receptors. Alternating expression of ligands and receptors creates a signal-sending cell (Notch-off) and a signal-receiving cell (Notch-on). Whereas genes encoding Notch ligands (e.g., Jag1) were highly expressed by intercalated cells, expression levels were low in principal cells, which was confirmed on the protein level by immunofluorescence. In contrast, principal cells showed high expression levels of Notch2 receptor and its transcriptional target Hes1, suggesting that principal cells are Notch signal–receiving cells of the collecting duct. Using Pax8rtTA/NICD mice, which enable inducible transgenic expression of the conserved Notch intracellular domain portion of the receptor specifically in differentiated kidney tubule cells, allowed us to study intercalated cell-to-principal cell transitions occurring in adult mice only, as opposed to those occurring during embryogenesis (160). We found that Notch expression disrupted cellular patterning and that the number of cells expressing principal cell marker Aqp2 was increased, whereas cells expressing intercalated cell markers Atp6v1b1 and Adgrf5 were reduced in number. The Notch-mediated transition appeared nearly complete, given that these cells also expressed multiple principal cell markers, including Aqp3 and Hsd11b2 (11). Our recent transcription factor enrichment analysis highlighted the expression of Elf5 in principal cells and Tcfcp2l1 in intercalated cells (19).
IMMUNE CELLS
The epithelial cell lining of the nephron plays a major role in maintaining the sterility of urine, which is achieved by mucus production and urinary flow, both providing immediate defense against microbial invasion. The distal kidney segment cells are notable for the expression of Toll-like receptors, defensins (Defb1), Umod (Umod) and NGAL (Lcn2), and key molecules defending against infections.
ScRNA-seq analysis identified the diversity of the immune cells even in the healthy mouse kidney samples. One of the first publications by Park et al. (11) highlighted almost as many different immune cell types as resident epithelial cells. These included macrophages, neutrophils, B and T lymphocytes, and natural killer (NK) cells. Recently, our team has also performed single-cell analysis on a mouse chronic kidney disease (CKD) model induced by FAN (18). We found a remarkable increase in immune cell diversity upon analyzing this model. We identified 14 immune cell clusters in our FAN model. Among the newly identified cells, we observed granulocytes, macrophages, dendritic cells, and basophils. Dendritic cells were further subclustered into Cd11+ dendritic cells (Cd209a and Cd11b), Cd11− dendritic cells (Cd24a and Clec9a), and plasmacytoid dendritic cell clusters (Siglech and Cd300c). A large number of lymphoid cells was also identified, including B cells, T cells, and NK cells. T lymphocytes were subclustered into CD4+ T, T regulatory, gamma delta T, natural killer T, and CD8+ effector cells. This data set is publicly available on our interactive website (http://www.susztaklab.com/VisCello/). Unfortunately, single nuclear sequencing poorly captures immune cells, which is a major limitation of the method (161, 162).
The Clatworthy group (163) has profiled 67,000 cells from fetal and adult human kidneys. The study was based on single-cell sequencing and identified a variety of immune cells including T cells, NK cells, and granulocytes. The team identified CD14+ (Cd14) classic and CD16+ (Fcgr3) nonclassic monocytes. They were also able differentiate M1 and anti-inflammatory CD206 (Cd206)-expressing M2 macrophages. They showed the important enrichment of neutrophils in the medulla and pelvis. These cells play important antibacterial roles in this compartment and likely prevent urinary infection.
Furthermore, analyzing fetal human kidneys indicated tissue-resident immune cells with high expression of major histocompatibility complex class II genes at 87–132 days gestation (163), consistent with studies showing early colonization of the fetal kidney with macrophages (164). From postconceptional week 9 onward, other immune cell subsets, including dendritic cells and lymphocytes, increased. These findings are consistent with data from mouse studies, which suggest that tissue-resident macrophages in the kidney do not solely originate from colonization by bone marrow–derived monocytes but might be seeded early in embryonic development from erythromyeloid precursors (164).
Mass cytometry on healthy renal tissue enabled dissection of T cell and myeloid cell heterogeneity (165). The study showed an enrichment of CD4+ central memory T cells and CD4+ and CD8+ effector memory T cells in normal kidney samples; however, regulatory T cells and T cells expressing the exhaustion marker PD-1 (Pdcd1) were absent from normal samples. This study also identified classical and nonclassic monocytes in normal kidney tissue and, consistent with the single-cell transcriptional data, a population of CD206-expressing, M2-polarized tissue-resident macrophages in healthy human kidneys.
Single-cell analysis has also been performed in human kidney biopsy samples obtained from kidney allografts with acute antibody-mediated rejection (166). In the allograft sample, the team identified distinct clusters of activated endothelial cells consistent with the diagnosis of antibody-mediated rejection. Monocytes, B cells, plasma cells, and T cells were evident in the immune compartment. The team identified plasma cells in the kidney, which was compatible with the local generation of donor-specific antibodies. Lymphoid aggregates or tertiary lymphoid follicles have been previously described in transplant biopsy samples in the context of rejection (167). A limitation of the study was that immune cells were not identified in the healthy control kidney sample, which might be related to the platforms (nuclear sequencing) used to generate these data and the relatively modest number of analyzed cells (166).
RENAL INTERSTITIUM
One of the most interesting and understudied compartments in the kidney is the interstitium. The interstitium contains fibroblasts, myofibroblasts, perivascular cells, erythropoietin- and renin-producing fibroblasts, renin-producing juxtaglomerular cells, and many other cell types, but their full entity remains elusive (168). A recent article by the Kramann group (169) has addressed interstitial cell heterogeneity using single-cell methods. Given that the cell types remain somewhat elusive, the team has developed an ECM score, which included collagens, glycoproteins, and proteoglycans, and validated this gene expression score in a published CKD data set. Although ACTA2 has been used as a myofibroblast marker previously, the team defined myofibroblasts as cells with high ECM scores. This is important because the ECM score will need external validation. Myofibroblasts were identified as cells expressing periostin (POSTN). Trajectory analysis of high ECM-expressing mesenchymal cells suggested that myofibroblasts arise from both pericytes and fibroblasts, and even from epithelial cells. Injured proximal tubule cells showed the highest expression of ECM genes in mice. The vast majority of ECM in human kidney fibrosis originated from mesenchymal cells, with a minor contribution from dedifferentiated proximal tubule cells. Unsupervised clustering of sorted human PDGFRβ+ cells identified mesenchymal populations. ECM gene expression again dominated the pericyte, fibroblast, and myofibroblast clusters. Some macrophage, monocyte, endothelial, and injured epithelial populations also expressed COL1A1 and PDGFRB, albeit at much lower levels. Pseudotime trajectory analysis of ECM-expressing cells indicated three main sources of myofibroblasts in human kidneys: (a) NOTCH3+RGS5+PDGFRA− pericytes, (b) MEG3+PDGFRA+ fibroblasts, and (c) COLEC11+CXCL12+ fibroblasts. The team also mapped the differentiation trajectory from low-ECM, healthy pericytes and fibroblasts to high-ECM diseased myofibroblasts.
A mouse ischemia reperfusion single-cell data set showed consistent results such as the multiple stromal clusters, including pericytes and vascular SMCs (67). Both of these clusters were positive for Notch ligand Jag1 and receptor Notch3. The team identified Dapk2 as a marker of cortical fibroblasts. In addition, the team also highlighted myofibroblasts expressing Acta2 and Col1a1. The number of cells in this cluster strongly increased following ischemia reperfusion. In the data set there were cells that only transiently expressed Acta2 and Col1a1, indicating the plasticity of fibroblasts and maybe even fibrosis.
Our single-cell analysis of the developing mouse kidney highlighted two different stromal clusters expressing high levels of Twist1 and Nr2f2 (19), although further subclustering of stromal cells did not yield robust results. Future studies should compare results from the different data sets and identify consistent cell types, changes, and key drivers.
CONCLUSION
Recent unbiased scRNA-seq studies have begun to dissect the molecular determinants of the various cell types in the kidney. The data show remarkable correlations with established molecular mechanisms in kidney physiology, validating both earlier findings and this new method. ScRNA-seq uncovers the surprising diversity of immune cells in normal kidneys, and this technique also yields novel knowledge about the plasticity of renal cell types and their transition into each other. The characterization of the changes in renal pathology has also begun, and it is expected to provide unparalleled insights into the molecular mechanisms of kidney disease. Future studies that integrate spatial transcriptomics information will be critical to define cell types and changes during development and disease. Single-cell RNA sequencing may also uncover novel mechanisms in kidney physiology, as well as identify novel biomarkers for disease and targets for therapeutic intervention.
ACKNOWLEDGMENTS
Work in the Susztak lab is supported by the National Institutes of Health (NIH) grants DK076077, DK087635, and DK105821. M.S.B. is supported by German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) grant BA6205/2-1.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Bernard C 1878. Leçons sur les pheńomeǹes de la vie communs aux animaux et aux veǵet́aux. Paris: J.B. Baillier̀e [Google Scholar]
- 2.Smith HW. 1953. From Fish to Philosopher. Boston: Little, Brown [Google Scholar]
- 3.Smith HW. 1956. Principles of Renal Physiology. New York: Oxford Univ. Press [Google Scholar]
- 4.Smith HW. 1959. The fate of sodium and water in the renal tubules. Bull. N. Y. Acad. Med 35:293–316 [PMC free article] [PubMed] [Google Scholar]
- 5.Hoenig MP, Zeidel ML. 2014. Homeostasis, the milieu intérieur, and the wisdom of the nephron. Clin. J. Am. Soc. Nephrol 9:1272–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong R 2008. The renal circulations and glomerular ultrafiltration. In Brenner and Rector’s The Kidney, pp. 91–129. Philadelphia: WB Saunders. 8th ed. [Google Scholar]
- 7.Stein JH, Boonjarern S, Wilson CB, Ferris TF. 1973. Alterations in intrarenal blood flow distribution. Methods of measurement and relationship to sodium balance. Circ. Res 32(Suppl. 1):61–72 [PubMed] [Google Scholar]
- 8.Pagel H, Jelkmann W, Weiss C. 1989. O2-supply to the kidneys and the production of erythropoietin. Respir. Physiol 77:111–17 [DOI] [PubMed] [Google Scholar]
- 9.Tisher C, Madsen K. 2008. Anatomy of the kidney. In Brenner and Rector’s The Kidney, pp. 25–90. Philadelphia: WB Saunders. 8th ed. [Google Scholar]
- 10.Stolz DB, Sims-Lucas S. 2015. Unwrapping the origins and roles of the renal endothelium. Pediatr. Nephrol 30:865–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park J, Shrestha R, Qiu C, Kondo A, Huang S, et al. 2018. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360:758–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molema G, Aird WC. 2012. Vascular heterogeneity in the kidney. Semin. Nephrol 32:145–55 [DOI] [PubMed] [Google Scholar]
- 13.Barry DM, McMillan EA, Kunar B, Lis R, Zhang T, et al. 2019. Molecular determinants of nephron vascular specialization in the kidney. Nat. Commun 10:5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dumas SJ, Meta E, Borri M, Goveia J, Rohlenova K, et al. 2020. Single-cell RNA sequencing reveals renal endothelium heterogeneity and metabolic adaptation to water deprivation. J. Am. Soc. Nephrol 31:118–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanabe K, Wada J, Sato Y. 2020. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat. Rev. Nephrol 16:289–303 [DOI] [PubMed] [Google Scholar]
- 16.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, et al. 2008. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med 358:1129–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, et al. 2003. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig 111:707–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhillon P, Park J, Hurtado Del Pozo C, Li L, Doke T, et al. 2021. The nuclear receptor ESRRA protects from kidney disease by coupling metabolism and differentiation. Cell Metab. 33:379–94.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao Z, Balzer MS, Ma Z, Liu H, Wu J, et al. 2021. Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat. Commun 12:2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung JJ, Goldstein L, Chen YJ, Lee J, Webster JD, et al. 2020. Single-cell transcriptome profiling of the kidney glomerulus identifies key cell types and reactions to injury. J. Am. Soc. Nephrol 31:2341–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollak MR, Quaggin SE, Hoenig MP, Dworkin LD. 2014. The glomerulus: the sphere of influence. Clin. J. Am. Soc. Nephrol 9:1461–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grigorieva IV, Oszwald A, Grigorieva EF, Schachner H, Neudert B, et al. 2019. A novel role for GATA3 in mesangial cells in glomerular development and injury. J. Am. Soc. Nephrol 30:1641–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marciano DK. 2019. Mesangial cells: the tuft guys of glomerular development. J. Am. Soc. Nephrol 30:1551–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber TB, Benzing T. 2005. The slit diaphragm: a signaling platform to regulate podocyte function. Curr. Opin. Nephrol. Hypertens 14:211–16 [DOI] [PubMed] [Google Scholar]
- 25.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. 2007. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 17:428–37 [DOI] [PubMed] [Google Scholar]
- 26.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, et al. 2000. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet 24:349–54 [DOI] [PubMed] [Google Scholar]
- 27.Tryggvason K, Patrakka J, Wartiovaara J. 2006. Hereditary proteinuria syndromes and mechanisms of proteinuria. N. Engl. J. Med 354:1387–401 [DOI] [PubMed] [Google Scholar]
- 28.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, et al. 1998. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol. Cell 1:575–82 [DOI] [PubMed] [Google Scholar]
- 29.Curthoys NP, Moe OW. 2014. Proximal tubule function and response to acidosis. Clin. J. Am. Soc. Nephrol 9:1627–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Nieto CE, You G, Bush KT, Barros EJ, Beier DR, Nigam SK. 1997. Molecular cloning and characterization of NKT, a gene product related to the organic cation transporter family that is almost exclusively expressed in the kidney. J. Biol. Chem 272:6471–78 [DOI] [PubMed] [Google Scholar]
- 31.Preisig PA, Rector FC Jr. 1988. Role of Na+-H+ antiport in rat proximal tubule NaCl absorption. Am. J. Physiol 255:F461–65 [DOI] [PubMed] [Google Scholar]
- 32.Moe OW, Preisig PA, Alpern RJ. 1990. Cellular model of proximal tubule NaCl and NaHCO3 absorption. Kidney Int. 38:605–11 [DOI] [PubMed] [Google Scholar]
- 33.Aronson PS. 2002. Ion exchangers mediating NaCl transport in the renal proximal tubule. Cell Biochem. Biophys 36:147–53 [DOI] [PubMed] [Google Scholar]
- 34.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, et al. 2011. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol 22:104–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM. 2011. Glucose transport by human renal Na+/d-glucose cotransporters SGLT1 and SGLT2. Am. J. Physiol. Cell Physiol 300:C14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verrey F, Singer D, Ramadan T, Vuille-dit-Bille RN, Mariotta L, Camargo SM. 2009. Kidney amino acid transport. Pflügers Arch. 458:53–60 [DOI] [PubMed] [Google Scholar]
- 37.McGivan JD, Bungard CI. 2007. The transport of glutamine into mammalian cells. Front. Biosci 12:874–82 [DOI] [PubMed] [Google Scholar]
- 38.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, et al. 2013. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Ren. Physiol 304:F156–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bakris GL, Fonseca VA, Sharma K, Wright EM. 2009. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney Int. 75:1272–77 [DOI] [PubMed] [Google Scholar]
- 40.Torrents D, Estévez R, Pineda M, Fernández E, Lloberas J, et al. 1998. Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J. Biol. Chem 273:32437–45 [DOI] [PubMed] [Google Scholar]
- 41.Rossier G, Meier C, Bauch C, Summa V, Sordat B, et al. 1999. LAT2, a new basolateral 4F2hc/CD98-associated amino acid transporter of kidney and intestine. J. Biol. Chem 274:34948–54 [DOI] [PubMed] [Google Scholar]
- 42.Romeo E, Dave MH, Bacic D, Ristic Z, Camargo SM, et al. 2006. Luminal kidney and intestine SLC6 amino acid transporters of B0AT-cluster and their tissue distribution in Mus musculus. Am. J. Physiol. Ren. Physiol 290:F376–83 [DOI] [PubMed] [Google Scholar]
- 43.Seow HF, Bröer S, Bröer A, Bailey CG, Potter SJ, et al. 2004. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat. Genet 36:1003–7 [DOI] [PubMed] [Google Scholar]
- 44.Kleta R, Romeo E, Ristic Z, Ohura T, Stuart C, et al. 2004. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat. Genet 36:999–1002 [DOI] [PubMed] [Google Scholar]
- 45.Shayakul C, Kanai Y, Lee WS, Brown D, Rothstein JD, Hediger MA. 1997. Localization of the high-affinity glutamate transporter EAAC1 in rat kidney. Am. J. Physiol. Ren. Physiol 273:F1023–29 [DOI] [PubMed] [Google Scholar]
- 46.Nigam SK. 2018. The SLC22 transporter family: a paradigm for the impact of drug transporters on metabolic pathways, signaling, and disease. Annu. Rev. Pharmacol. Toxicol 58:663–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nigam SK. 2015. What do drug transporters really do? Nat. Rev. Drug Discov 14:29–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Curthoys NP, Gstraunthaler G. 2001. Mechanism of increased renal gene expression during metabolic acidosis. Am. J. Physiol. Ren. Physiol 281:F381–90 [DOI] [PubMed] [Google Scholar]
- 49.Boron WF. 2006. Acid-base transport by the renal proximal tubule. J. Am. Soc. Nephrol 17:2368–82 [DOI] [PubMed] [Google Scholar]
- 50.Moret C, Dave MH, Schulz N, Jiang JX, Verrey F, Wagner CA. 2007. Regulation of renal amino acid transporters during metabolic acidosis. Am. J. Physiol. Ren. Physiol 292:F555–66 [DOI] [PubMed] [Google Scholar]
- 51.Christensen HN. 1990. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev 70:43–77 [DOI] [PubMed] [Google Scholar]
- 52.Chaudhry FA, Reimer RJ, Edwards RH. 2002. The glutamine commute: take the N line and transfer to the A. J. Cell Biol 157:349–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moe OW. 2006. Kidney stones: pathophysiology and medical management. Lancet 367:333–44 [DOI] [PubMed] [Google Scholar]
- 54.Moe OW, Preisig PA. 2006. Dual role of citrate in mammalian urine. Curr. Opin. Nephrol. Hypertens 15:419–24 [DOI] [PubMed] [Google Scholar]
- 55.Curthoys NP, Lowry OH. 1973. The distribution of glutaminase isoenzymes in the various structures of the nephron in normal, acidotic, and alkalotic rat kidney. J. Biol. Chem 248:162–68 [PubMed] [Google Scholar]
- 56.Wright PA, Knepper MA. 1990. Phosphate-dependent glutaminase activity in rat renal cortical and medullary tubule segments. Am. J. Physiol. Ren. Physiol 259:F961–70 [DOI] [PubMed] [Google Scholar]
- 57.Wright PA, Knepper MA. 1990. Glutamate dehydrogenase activities in microdissected rat nephron segments: effects of acid-base loading. Am. J. Physiol. Ren. Physiol 259:F53–59 [DOI] [PubMed] [Google Scholar]
- 58.Molinas SM, Trumper L, Marinelli RA. 2012. Mitochondrial aquaporin-8 in renal proximal tubule cells: evidence for a role in the response to metabolic acidosis. Am. J. Physiol. Ren. Physiol 303:F458–66 [DOI] [PubMed] [Google Scholar]
- 59.Preisig PA, Alpern RJ. 1988. Chronic metabolic acidosis causes an adaptation in the apical membrane Na/H antiporter and basolateral membrane Na(HCO3)3 symporter in the rat proximal convoluted tubule. J. Clin. Investig 82:1445–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ambühl PM, Amemiya M, Danczkay M, Lötscher M, Kaissling B, et al. 1996. Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney. Am. J. Physiol. Ren. Physiol 271:F917–25 [DOI] [PubMed] [Google Scholar]
- 61.Wu MS, Biemesderfer D, Giebisch G, Aronson PS. 1996. Role of NHE3 in mediating renal brush border Na+-H+ exchange. Adaptation to metabolic acidosis. J. Biol. Chem 271:32749–52 [DOI] [PubMed] [Google Scholar]
- 62.Solbu TT, Boulland JL, Zahid W, Lyamouri Bredahl MK, Amiry-Moghaddam M, et al. 2005. Induction and targeting of the glutamine transporter SN1 to the basolateral membranes of cortical kidney tubule cells during chronic metabolic acidosis suggest a role in pH regulation. J. Am. Soc. Nephrol 16:869–77 [DOI] [PubMed] [Google Scholar]
- 63.Horie S, Moe O, Tejedor A, Alpern RJ. 1990. Preincubation in acid medium increases Na/H antiporter activity in cultured renal proximal tubule cells. PNAS 87:4742–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tannen RL, Ross BD. 1979. Ammoniagenesis by the isolated perfused rat kidney: the critical role of urinary acidification. Clin. Sci 56:353–64 [DOI] [PubMed] [Google Scholar]
- 65.Pajor AM. 1995. Sequence and functional characterization of a renal sodium/dicarboxylate cotransporter. J. Biol. Chem 270:5779–85 [DOI] [PubMed] [Google Scholar]
- 66.Qiu C, Huang S, Park J, Park Y, Ko YA, et al. 2018. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med 24:1721–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kirita Y, Wu H, Uchimura K, Wilson PC, Humphreys BD. 2020. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. PNAS 117:15874–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Lau W, Barker N, Low TY, Koo BK, Li VS, et al. 2011. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476:293–97 [DOI] [PubMed] [Google Scholar]
- 69.Walker JV, Zhuang H, Singer D, Illsley CS, Kok WL, et al. 2019. Transit amplifying cells coordinate mouse incisor mesenchymal stem cell activation. Nat. Commun 10:3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rudman-Melnick V, Adam M, Potter A, Chokshi SM, Ma Q, et al. 2020. Single-cell profiling of AKI in a murine model reveals novel transcriptional signatures, profibrotic phenotype, and epithelial-to-stromal crosstalk. J. Am. Soc. Nephrol 31:2793–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Conway BR, O’Sullivan ED, Cairns C, O’Sullivan J, Simpson DJ, et al. 2020. Kidney single-cell atlas reveals myeloid heterogeneity in progression and regression of kidney disease. J. Am. Soc. Nephrol 31:2833–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Janosevic D, Myslinski J, McCarthy TW, Zollman A, Syed F, et al. 2021. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife 10:e62270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen L, Clark JZ, Nelson JW, Kaissling B, Ellison DH, Knepper MA. 2019. Renal-tubule epithelial cell nomenclature for single-cell RNA-sequencing studies. J. Am. Soc. Nephrol 30:1358–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mount DB. 2014. Thick ascending limb of the loop of Henle. Clin. J. Am. Soc. Nephrol 9:1974–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nielsen S, Pallone T, Smith BL, Christensen EI, Agre P, Maunsbach AB. 1995. Aquaporin-1 water channels in short and long loop descending thin limbs and in descending vasa recta in rat kidney. Am. J. Physiol. Ren. Physiol 268:F1023–37 [DOI] [PubMed] [Google Scholar]
- 76.Hebert SC, Mount DB, Gamba G. 2004. Molecular physiology of cation-coupled Cl− cotransport: the SLC12 family. Pflügers Arch. 447:580–93 [DOI] [PubMed] [Google Scholar]
- 77.Burg M, Stoner L, Cardinal J, Green N. 1973. Furosemide effect on isolated perfused tubules. Am. J. Physiol 225:119–24 [DOI] [PubMed] [Google Scholar]
- 78.Xu JZ, Hall AE, Peterson LN, Bienkowski MJ, Eessalu TE, Hebert SC. 1997. Localization of the ROMK protein on apical membranes of rat kidney nephron segments. Am. J. Physiol. Ren. Physiol 273:F739–48 [DOI] [PubMed] [Google Scholar]
- 79.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, et al. 1993. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362:31–38 [DOI] [PubMed] [Google Scholar]
- 80.Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, et al. 1996. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet 14:152–56 [DOI] [PubMed] [Google Scholar]
- 81.Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, et al. 2006. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am. J. Hum. Genet 79:949–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee JW, Chou CL, Knepper MA. 2015. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J. Am. Soc. Nephrol 26:2669–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gorski M, Tin A, Garnaas M, McMahon GM, Chu AY, et al. 2015. Genome-wide association study of kidney function decline in individuals of European descent. Kidney Int. 87:1017–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Böger CA, Gorski M, Li M, Hoffmann MM, Huang C, et al. 2011. Association of eGFR-related loci identified by GWAS with incident CKD and ESRD. PLOS Genet. 7:e1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Köttgen A, Glazer NL, Dehghan A, Hwang SJ, Katz R, et al. 2009. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet 41:712–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Olden M, Corre T, Hayward C, Toniolo D, Ulivi S, et al. 2014. Common variants in UMOD associate with urinary uromodulin levels: a meta-analysis. J. Am. Soc. Nephrol 25:1869–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cavallone D, Malagolini N, Serafini-Cessi F. 2001. Mechanism of release of urinary Tamm-Horsfall glycoprotein from the kidney GPI-anchored counterpart. Biochem. Biophys. Res. Commun 280:110–14 [DOI] [PubMed] [Google Scholar]
- 88.Padmanabhan S, Graham L, Ferreri NR, Graham D, McBride M, Dominiczak AF. 2014. Uromodulin, an emerging novel pathway for blood pressure regulation and hypertension. Hypertension 64:918–23 [DOI] [PubMed] [Google Scholar]
- 89.Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O. 2011. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int. 80:338–47 [DOI] [PubMed] [Google Scholar]
- 90.Scolari F, Izzi C, Ghiggeri GM. 2015. Uromodulin: from monogenic to multifactorial diseases. Nephrol. Dial. Transplant 30:1250–56 [DOI] [PubMed] [Google Scholar]
- 91.Vyletal P, Bleyer AJ, Kmoch S. 2010. Uromodulin biology and pathophysiology—an update. Kidney Blood Pressure Res. 33:456–75 [DOI] [PubMed] [Google Scholar]
- 92.Liu Y, Mo L, Goldfarb DS, Evan AP, Liang F, et al. 2010. Progressive renal papillary calcification and ureteral stone formation in mice deficient for Tamm-Horsfall protein. Am. J. Physiol. Ren. Physiol 299:F469–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mo L, Liaw L, Evan AP, Sommer AJ, Lieske JC, Wu XR. 2007. Renal calcinosis and stone formation in mice lacking osteopontin, Tamm-Horsfall protein, or both. Am. J. Physiol. Ren. Physiol 293:F1935–43 [DOI] [PubMed] [Google Scholar]
- 94.Mo L, Huang HY, Zhu XH, Shapiro E, Hasty DL, Wu XR. 2004. Tamm-Horsfall protein is a critical renal defense factor protecting against calcium oxalate crystal formation. Kidney Int. 66:1159–66 [DOI] [PubMed] [Google Scholar]
- 95.Grant AM, Baker LR, Neuberger A. 1973. Urinary Tamm-Horsfall glycoprotein in certain kidney diseases and its content in renal and bladder calculi. Clin. Sci 44:377–84 [DOI] [PubMed] [Google Scholar]
- 96.Bates JM, Raffi HM, Prasadan K, Mascarenhas R, Laszik Z, et al. 2004. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection: rapid communication. Kidney Int. 65:791–97 [DOI] [PubMed] [Google Scholar]
- 97.Mo L, Zhu XH, Huang HY, Shapiro E, Hasty DL, Wu XR. 2004. Ablation of the Tamm-Horsfall protein gene increases susceptibility of mice to bladder colonization by type 1-fimbriated Escherichia coli. Am. J. Physiol. Ren. Physiol 286:F795–802 [DOI] [PubMed] [Google Scholar]
- 98.Orskov I, Ferencz A, Orskov F. 1980. Tamm-Horsfall protein or uromucoid is the normal urinary slime that traps type 1 fimbriated Escherichia coli. Lancet 1:887. [DOI] [PubMed] [Google Scholar]
- 99.Graham LA, Padmanabhan S, Fraser NJ, Kumar S, Bates JM, et al. 2014. Validation of uromodulin as a candidate gene for human essential hypertension. Hypertension 63:551–58 [DOI] [PubMed] [Google Scholar]
- 100.Trudu M, Janas S, Lanzani C, Debaix H, Schaeffer C, et al. 2013. Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat. Med 19:1655–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nielsen S, Maunsbach AB, Ecelbarger CA, Knepper MA. 1998. Ultrastructural localization of Na-K-2Cl cotransporter in thick ascending limb and macula densa of rat kidney. Am. J. Physiol. Ren. Physiol 275:F885–93 [DOI] [PubMed] [Google Scholar]
- 102.He XR, Greenberg SG, Briggs JP, Schnermann J. 1995. Effects of furosemide and verapamil on the NaCl dependency of macula densa-mediated renin secretion. Hypertension 26:137–42 [DOI] [PubMed] [Google Scholar]
- 103.Ito S, Carretero OA. 1990. An in vitro approach to the study of macula densa-mediated glomerular hemodynamics. Kidney Int. 38:1206–10 [DOI] [PubMed] [Google Scholar]
- 104.Lapointe JY, Laamarti A, Bell PD. 1998. Ionic transport in macula densa cells. Kidney Int. 54(Suppl. 67):S58–64 [DOI] [PubMed] [Google Scholar]
- 105.Subramanya AR, Ellison DH. 2014. Distal convoluted tubule. Clin. J. Am. Soc. Nephrol 9:2147–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hierholzer K, Wiederholt M. 1976. Some aspects of distal tubular solute and water transport. Kidney Int. 9:198–213 [DOI] [PubMed] [Google Scholar]
- 107.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, et al. 2010. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J. Clin. Investig 120:1627–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, et al. 1996. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet 12:24–30 [DOI] [PubMed] [Google Scholar]
- 109.Bandulik S, Schmidt K, Bockenhauer D, Zdebik AA, Humberg E, et al. 2011. The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflügers Arch. 461:423–35 [DOI] [PubMed] [Google Scholar]
- 110.Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, et al. 2009. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. PNAS 106:5842–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, et al. 2009. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med 360:1960–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hamilton KL, Devor DC. 2012. Basolateral membrane K+ channels in renal epithelial cells. Am. J. Physiol. Ren. Physiol 302:F1069–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dørup J 1985. Ultrastructure of distal nephron cells in rat renal cortex. J. Ultrastruct. Res 92:101–18 [DOI] [PubMed] [Google Scholar]
- 114.Pacheco-Alvarez D, Cristóbal PS, Meade P, Moreno E, Vazquez N, et al. 2006. The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem 281:28755–63 [DOI] [PubMed] [Google Scholar]
- 115.Estévez R, Boettger T, Stein V, Birkenhäger R, Otto E, et al. 2001. Barttin is a Cl− channel β-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 414:558–61 [DOI] [PubMed] [Google Scholar]
- 116.Thakker RV. 1999. Chloride channels in renal disease. Adv. Nephrol. Necker Hosp 29:289–98 [PubMed] [Google Scholar]
- 117.Velázquez H, Silva T. 2003. Cloning and localization of KCC4 in rabbit kidney: expression in distal convoluted tubule. Am. J. Physiol. Ren. Physiol 285:F49–58 [DOI] [PubMed] [Google Scholar]
- 118.Hadchouel J, Delaloy C, Fauré S, Achard JM, Jeunemaitre X. 2006. Familial hyperkalemic hypertension. J. Am. Soc. Nephrol 17:208–17 [DOI] [PubMed] [Google Scholar]
- 119.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, et al. 2001. Human hypertension caused by mutations in WNK kinases. Science 293:1107–12 [DOI] [PubMed] [Google Scholar]
- 120.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, et al. 2012. Mutations in Kelch-like 3 and Cullin 3 cause hypertension and electrolyte abnormalities. Nature 482:98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, et al. 2012. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet 44:456–60 [DOI] [PubMed] [Google Scholar]
- 122.Subramanya AR, Liu J, Ellison DH, Wade JB, Welling PA. 2009. WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J. Biol. Chem 284:18471–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Golbang AP, Cope G, Hamad A, Murthy M, Liu CH, et al. 2006. Regulation of the expression of the Na/Cl cotransporter by WNK4 and WNK1: evidence that accelerated dynamin-dependent endocytosis is not involved. Am. J. Physiol. Ren. Physiol 291:F1369–76 [DOI] [PubMed] [Google Scholar]
- 124.Cai H, Cebotaru V, Wang YH, Zhang XM, Cebotaru L, et al. 2006. WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int. 69:2162–70 [DOI] [PubMed] [Google Scholar]
- 125.Yang CL, Angell J, Mitchell R, Ellison DH. 2003. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J. Clin. Investig 111:1039–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vitari AC, Deak M, Morrice NA, Alessi DR. 2005. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J 391:17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, et al. 2013. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. PNAS 110:14366–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. 2013. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. PNAS 110:7838–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, et al. 2013. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 3:858–68 [DOI] [PubMed] [Google Scholar]
- 130.Schlingmann KP, Weber S, Peters M, Nejsum LN, Vitzthum H, et al. 2002. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat. Genet 31:166–70 [DOI] [PubMed] [Google Scholar]
- 131.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, et al. 2002. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat. Genet 31:171–74 [DOI] [PubMed] [Google Scholar]
- 132.Pietropaolo G, Pugliese D, Armuzzi A, Guidi L, Gasbarrini A, et al. 2020. Magnesium absorption in intestinal cells: evidence of cross-talk between EGF and TRPM6 and novel implications for cetuximab therapy. Nutrients 12:3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kimura M, Usami E, Yoshimura T. 2020. Effects of type of antibody to EGFR and hypomagnesemia on overall survival in first-line treatment of patients with unresectable advanced/recurrent colorectal cancer. Anticancer Res. 40:7135–40 [DOI] [PubMed] [Google Scholar]
- 134.Groenestege WM, Thébault S, van der Wijst J, van den Berg D, Janssen R, et al. 2007. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J. Clin. Investig 117:2260–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, et al. 1987. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237:268–75 [DOI] [PubMed] [Google Scholar]
- 136.Meneton P, Loffing J, Warnock DG. 2004. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am. J. Physiol. Ren. Physiol 287:F593–601 [DOI] [PubMed] [Google Scholar]
- 137.Hunter RW, Ivy JR, Flatman PW, Kenyon CJ, Craigie E, et al. 2015. Hypertrophy in the distal convoluted tubule of an 11β-hydroxysteroid dehydrogenase type 2 knockout model. J. Am. Soc. Nephrol 26:1537–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, et al. 2003. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J. Clin. Investig 112:1906–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, et al. 1999. Molecular identification of the apical Ca2+ channel in 1,25-dihydroxyvitamin D3-responsive epithelia. J. Biol. Chem 274:8375–78 [DOI] [PubMed] [Google Scholar]
- 140.Hoenderop JG, Nilius B, Bindels RJ. 2005. Calcium absorption across epithelia. Physiol. Rev 85:373–422 [DOI] [PubMed] [Google Scholar]
- 141.Chen L, Chou C-L, Knepper MA. 2021. Targeted single-cell RNA-seq identifies minority cell types of kidney distal nephron. J. Am. Soc. Nephrol 32:886–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Davies JA, Davey MG. 1999. Collecting duct morphogenesis. Pediatr. Nephrol 13:535–41 [DOI] [PubMed] [Google Scholar]
- 143.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. 2015. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol 10:135–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Roy A, Al-bataineh MM, Pastor-Soler NM. 2015. Collecting duct intercalated cell function and regulation. Clin. J. Am. Soc. Nephrol 10:305–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.van den Ouweland AM, Dreesen JC, Verdijk M, Knoers NV, Monnens LA, et al. 1992. Mutations in the vasopressin type 2 receptor gene (AVPR2) associated with nephrogenic diabetes insipidus. Nat. Genet 2:99–102 [DOI] [PubMed] [Google Scholar]
- 146.Gao C, Higgins PJ, Zhang W. 2020. AQP2: mutations associated with congenital nephrogenic diabetes insipidus and regulation by post-translational modifications and protein-protein interactions. Cells 9:2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.García Castaño A, Pérez de Nanclares G, Madariaga L, Aguirre M, Chocron S, et al. 2015. Novel mutations associated with nephrogenic diabetes insipidus: a clinical-genetic study. Eur. J. Pediatr 174:1373–85 [DOI] [PubMed] [Google Scholar]
- 148.Pearce D, Bhalla V, Funder J, Stokes J. 2012. Aldosterone regulation of ion transport. In Brenner and Rector’s The Kidney, pp. 202–25. Philadelphia: WB Saunders/Elsevier. 9th ed. [Google Scholar]
- 149.Soundararajan R, Wang J, Melters D, Pearce D. 2010. Glucocorticoid-induced Leucine zipper 1 stimulates the epithelial sodium channel by regulating serum- and glucocorticoid-induced kinase 1 stability and subcellular localization. J. Biol. Chem 285:39905–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu M, Wang J, Jones KT, Ives HE, Feldman ME, et al. 2010. mTOR complex-2 activates ENaC by phosphorylating SGK1. J. Am. Soc. Nephrol 21:811–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rocchini AP. 2000. Obesity hypertension, salt sensitivity and insulin resistance. Nutr. Metab. Cardiovasc. Dis 10:287–94 [PubMed] [Google Scholar]
- 152.Wei Y, Sun P, Wang Z, Yang B, Carroll MA, Wang WH. 2006. Adenosine inhibits ENaC via cytochrome P-450 epoxygenase-dependent metabolites of arachidonic acid. Am. J. Physiol. Ren. Physiol 290:F1163–68 [DOI] [PubMed] [Google Scholar]
- 153.Zaika O, Mamenko M, O’Neil RG, Pochynyuk O. 2011. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. Am. J. Physiol. Ren. Physiol 300:F1105–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Flores D, Liu Y, Liu W, Satlin LM, Rohatgi R. 2012. Flow-induced prostaglandin E2 release regulates Na and K transport in the collecting duct. Am. J. Physiol. Ren. Physiol 303:F632–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, et al. 2013. Renal intercalated cells are rather energized by a proton than a sodium pump. PNAS 110:7928–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Alper SL, Natale J, Gluck S, Lodish HF, Brown D. 1989. Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. PNAS 86:5429–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Brown D, Hirsch S, Gluck S. 1988. An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature 331:622–24 [DOI] [PubMed] [Google Scholar]
- 158.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, et al. 2001. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. PNAS 98:4221–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, et al. 2003. Localization of pendrin in mouse kidney. Am. J. Physiol. Ren. Physiol 284:F229–41 [DOI] [PubMed] [Google Scholar]
- 160.Werth M, Schmidt-Ott KM, Leete T, Qiu A, Hinze C, et al. 2017. Transcription factor TFCP2L1 patterns cells in the mouse kidney collecting ducts. eLife 6:e24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, et al. 2019. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat. Commun 10:2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wu H, Kirita Y, Donnelly EL, Humphreys BD. 2019. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol 30:23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, et al. 2019. Spatiotemporal immune zonation of the human kidney. Science 365:1461–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, et al. 2012. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90 [DOI] [PubMed] [Google Scholar]
- 165.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, et al. 2017. An immune atlas of clear cell renal cell carcinoma. Cell 169:736–49.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wu H, Malone AF, Donnelly EL, Kirita Y, Uchimura K, et al. 2018. Single-cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J. Am. Soc. Nephrol 29:2069–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.de Leur K, Clahsen-van Groningen MC, van den Bosch TPP, de Graav GN, Hesselink DA, et al. 2018. Characterization of ectopic lymphoid structures in different types of acute renal allograft rejection. Clin. Exp. Immunol 192:224–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zeisberg M, Kalluri R. 2015. Physiology of the renal interstitium. Clin. J. Am. Soc. Nephrol 10:1831–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, et al. 2021. Decoding myofibroblast origins in human kidney fibrosis. Nature 589:281–86 [DOI] [PMC free article] [PubMed] [Google Scholar]