

Abstract
Inhibitors of bromodomain and extraterminal domain (BET) proteins are possible anti-severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) prophylactics as they downregulate angiotensin-converting enzyme 2 (ACE2). Here we show that BET proteins should not be inactivated therapeutically because they are critical antiviral factors at the post-entry level. Depletion of BRD3 or BRD4 in cells overexpressing ACE2 exacerbates SARS-CoV-2 infection; the same is observed when cells with endogenous ACE2 expression are treated with BET inhibitors during infection and not before. Viral replication and mortality are also enhanced in BET inhibitor-treated mice overexpressing ACE2. BET inactivation suppresses interferon production induced by SARS-CoV-2, a process phenocopied by the envelope (E) protein previously identified as a possible “histone mimetic.” E protein, in an acetylated form, directly binds the second bromodomain of BRD4. Our data support a model where SARS-CoV-2 E protein evolved to antagonize interferon responses via BET protein inhibition; this neutralization should not be further enhanced with BET inhibitor treatment.
Keywords: SARS-CoV-2, COVID-19, histone mimetic, BET proteins, BRD4, BRD3, BRD2, antiviral response, viral replication, BET inhibitors
Graphical abstract
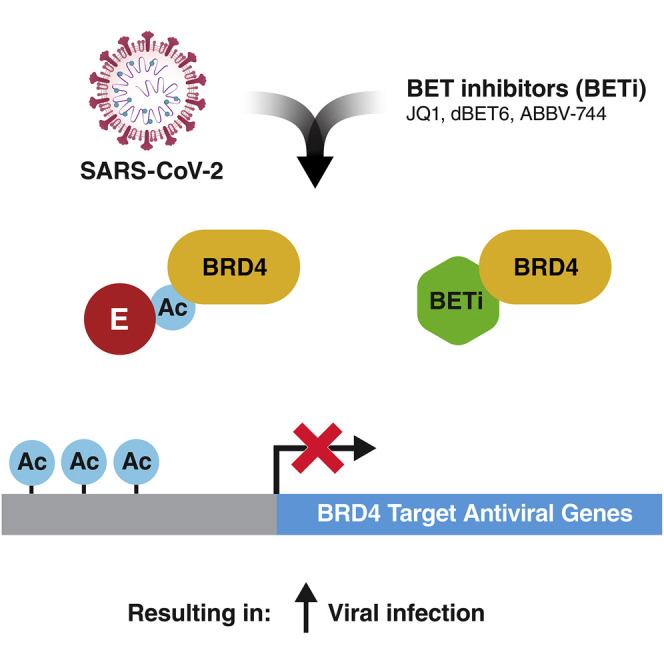
Chen et al. find that BET inactivation, by suppressing antiviral responses, enhances SARS-CoV-2 infection. This effect is phenocopied by the viral envelope (E) protein, which is acetylated in cells and has evolved to antagonize induction of the interferon response by binding and inhibiting BET proteins through their bromodomains.
Introduction
The bromodomain and extraterminal domain (BET) family of proteins consists of BRD2, BRD3, BRD4, and BRDT, with the latter only found in testes. BET proteins characteristically harbor two highly conserved N-terminal bromodomains (BDs; BD1 and BD2) and an extraterminal (ET) domain. BDs function as bone fide reader domains of acetylated lysines in histone and non-histone proteins and are the molecular targets of small-molecule BET inhibitors such as JQ1, whereas the ET domain has less defined protein binding properties (Dhalluin et al., 1999; Filippakopoulos et al., 2012; Rahman et al., 2011). Through their interaction with histones and cellular transcriptional machinery, BET proteins play an instrumental role in many cellular functions, including cell proliferation, chromatin remodeling, and gene expression (Taniguchi, 2016). BRD4 is the best-studied BET protein and exists in different splice isoforms: a so-called long isoform (amino acids [aa] 1–1362, BRD4L), a short isoform (aa 1–720, BRD4S), and an intermediate third isoform (aa 1–794, isoform B) reported only in osteosarcoma cells (Floyd et al., 2013). BRD4L has an extended C terminus that recruits the positive transcription elongation factor (PTEF-b), called the PTEF-b binding domain (PID) (Bisgrove et al., 2007). Furthermore, BRD2, BRD3, and BRD4 interact with viral proteins of herpesviruses, flaviviruses, and papillomaviruses and regulate the integration and latent viral infection of retroviruses (Conrad et al., 2017; De Rijck et al., 2013; Mourão et al., 2020; Platt et al., 1999; Sharma et al., 2013; Wu et al., 2006; You et al., 2006).
The recently emerged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic (Zhu et al., 2020). Individuals with COVID-19 are characterized by impaired type I interferon (IFN-I) responses paired with overproduction of proinflammatory cytokines (Blanco-Melo et al., 2020; Hadjadj et al., 2020; Jose and Manuel, 2020). A potent coactivator of proinflammatory and antiviral genes is BRD4. In the lungs, BRD4 coactivates IFN-stimulated genes (ISGs) during viral infection by recruiting P-TEFb and activating proinflammatory responses associated with lung fibrosis, chronic obstructive pulmonary disease, and asthma (Hargreaves et al., 2009; Stratton et al., 2017). Treatment with BET inhibitors like JQ1 attenuates transcriptional activation of the antiviral response in the context of influenza A infection (Wienerroither et al., 2014). We previously identified BRD4 and BRD2 as high-confidence interactors of the SARS-CoV-2 envelope (E) protein and described a histone-like motif similar to histone H3 within E protein that may interfere with the canonical histone:BET protein interactions (Gordon et al., 2020).
The E protein of coronaviruses is a small but critical membrane protein involved in many steps of the viral life cycle, such as virion assembly, budding, and pathogenesis. One of the reported functions of E protein is that of an ion-channeling viroporin that transports positive ions, including calcium, to trigger inflammasome activation (Schoeman and Fielding, 2019). Expression of inflammatory cytokines in animals infected with SARS-CoV (SARS) lacking the ion channel function of E protein is reduced, suggesting that E protein is important for viral pathogenicity (Nieto-Torres et al., 2014). The E proteins of SARS and SARS-CoV-2 have been reported to interact with the tight junction proteins PALS1 and ZO-1, suggesting that E protein may contribute to damage of epithelial barrier function and more severe respiratory dysfunction (Javorsky et al., 2021; Schoeman and Fielding, 2019; Shepley-McTaggart et al., 2021).
Notably, BRD2 functions as a transcriptional regulator of the viral entry receptor angiotensin-converting enzyme 2 (ACE2), where knockout of BRD2 or prophylactic application of BET inhibitors reduces ACE2 expression and de novo viral infection (Mills et al., 2021; Qiao et al., 2020). To reconcile this proviral function of BRD2 with known antiviral effects of BRD4, we tested the function of all relevant BET proteins during SARS-CoV-2 infection. Inhibition of BET proteins after viral entry or depletion of BRD3 or BRD4 in cells overexpressing ACE2 significantly enhanced viral replication, indicating a post-entry antiviral function of BET proteins on SARS-CoV-2. This enhancement of viral replication was also observed in infected, BET inhibitor-treated K18-hACE2 mice. The viral E protein in an acetylated form effectively thwarts this antiviral function as it binds to the second BD of BRD4, underscoring the relevance of BET proteins as positive regulators of antiviral gene expression and cautioning against use of BET inhibitors in an ongoing SARS-CoV-2 infection.
Results
BRD3 and BRD4 proteins antagonize SARS-CoV-2 infection
To test post-entry functions of all relevant BET proteins, we depleted them in A549 cells overexpressing ACE2 (A549-ACE2). We generated polyclonal knockout (KO) cells of BRD2 and BRD3 and knockdown (KD) cells of BRD4 via nucleofection of CRISPR-Cas9 ribonucleoprotein (RNP) complexes incorporating multiple guide RNAs against each target. As controls, cells were nucleofected with the RNP complex without guide RNAs (RNP only). Depletion of individual BET proteins was validated by western blotting before infection with SARS-CoV-2 (isolate USA/WA-1/2020) at multiplicities of infection (MOIs) of 0.01 and 0.1 (Figure 1A). Loss of BRD3 and BRD4 significantly enhanced cell-associated viral RNA titers (9-fold and 17-fold, respectively) and infectious particle production in plaque assays (1.4 log fold increase for BRD4) at MOI of 0.1, whereas only BRD4 KD significantly enhanced viral replication in cells infected at MOI of 0.01 (Figures 1B and 1C). BRD2 KO also significantly enhanced viral infection at any MOI, but not as much as BRD4 depletion, supporting a model where individual BET proteins play distinct roles in SARS-CoV-2 infection.
Figure 1.

BRD2, BRD3, and BRD4 differentially affect SARS-CoV-2 infection
(A) Representative western blots from A549-ACE2 cells with the indicated BET protein KOs and KD. Lysates were probed for the epitope indicated beside each panel. ns∗ denotes a non-specific band.
(B) qRT-PCR of SARS-CoV-2 E RNA isolated 48 h post infection (hpi) from A549-ACE2 cells with the indicated BET protein KO or KD infected with SARS-CoV-2 (MOI of 0.01 or 0.1). Data are expressed in absolute copies per microgram based on a standard curve of the E gene with known copy number. The average of three independent experiments analyzed in triplicate ± SEM is shown and compared with RNP-only samples by ANOVA: ∗p = 0.0397, ∗∗p = 0.0026, ∗∗∗∗p < 0.0001.
(C) Plaque assay titers from supernatant of infected A549-ACE2 cells with the indicated BET protein KOs and KD at MOI of 0.1. The average of three independent experiments analyzed in duplicate ± SEM is shown and compared with the RNP-only condition by Student’s t test: ∗p < 0.5.
(D) Representative western blots from Calu3 cells with the indicated BET protein KOs and KD. Lysates were probed for the epitope indicated beside each panel.
(E) qRT-PCR of SARS-CoV-2 E RNA isolated 48 hpi from Calu3 cells with the indicated BET protein KOs or KD infected with SARS-CoV-2 (MOI of 0.01 or 0.1). Data are expressed in absolute copies per microgram based on a standard curve of the E gene with known copy number. The average of three independent experiments analyzed in triplicate ± SEM is shown and compared with RNP-only samples by ANOVA: ∗∗p < 0.005, ∗∗∗∗p < 0.0001.
(F) Plaque assay titers from supernatant of infected Calu3 cells with the indicated BET protein KOs and KD at MOI of 0.1. Average of three independent experiments analyzed in duplicate ±SEM are shown and compared with the RNP-only condition by Student’s t test: ∗∗∗∗p < 0.0001.
(G) Plaque assay titers from supernatant of infected A549-ACE2 cells treated with JQ1 (500 nM) and dBET6 (500 nM) at MOI of 0.1. The average of three independent experiments analyzed in duplicate ± SEM is shown and compared with the DMSO condition by ANOVA: ∗∗∗p = 0.0005, ∗∗∗∗p < 0.0001.
(H) Plaque assay titers from supernatant of infected Calu3 cells treated with JQ1 (500 nM) and dBET6 (500 nM) at MOI of 0.1. The average of three independent experiments analyzed in duplicate ± SEM is shown and compared with the DMSO condition by ANOVA: ∗∗p = 0.0059, ∗∗∗∗p < 0.0001.
In parallel, we generated BET protein KOs and KD in Calu3 cells, airway epithelial cells with sufficient endogenous ACE2 expression to support SARS-CoV-2 infection. The KOs and KD were validated by western blotting for BET protein expression and infected with SARS-CoV-2 (Figure 1D). As expected, BRD2 KO decreased ACE2 transcript levels by ∼80%, resulting in a greater than 2 log decrease in viral RNA and greater than 1 log decrease in infectious viral titers (Figures 1E, 1F, and S1A). BRD3 KO also reduced ACE2 expression by ∼50% and viral replication about 3-fold (Figures 1E and S1A). Overall, BRD4 KD had the least effect on viral RNA expression and, in fact, increased infectious particle production despite decreasing ACE2 expression (Figures 1E, 1F, and S1A). These results show distinct effects of BET proteins on pre- and post-entry steps of SARS-CoV-2 infection and underscore BRD4’s role in regulating the antiviral state after viral entry.
Next we tested the pan-BET inhibitors JQ1 and dBET6, which disrupt BD1- and BD2-mediated interactions, in A549-ACE2 and Calu3 cells. Infectious particle production was significantly increased when JQ1 or dBET6 was added at the time of infection (Figure 1G). The same was observed with JQ1- and dBET6-treated Calu3 cells, which express ACE2 endogenously (Figure 1H). Use of the BET inhibitors at these effective concentrations was non-toxic in A549-ACE2 and Calu3 cells (Figures S1B and S1C). These results demonstrate that BRD3 and BRD4 are post-entry antagonists of SARS-CoV-2 viral replication; inactivating them genetically or chemically exacerbates infection.
Loss of BET proteins reduces IFN and proinflammatory cytokine expression
To test whether loss of BET proteins reduces IFN production during SARS-CoV-2 infection, we infected Calu3 cells, immediately treated them with JQ1 or dBET6 for 48 h, and analyzed mRNA expression of IFN-β (IFNB1), ISG15, as well as of the proinflammatory cytokine interleukin-6 (IL6). We observed robust induction of all three genes in control (DMSO-treated) cells that correlated with increasing MOI (Figure 2A). In contrast, upon JQ1 or dBET6 treatment, expression of all three genes (IFNB1, ISG15, and IL6) was suppressed.
Figure 2.

Loss of BET proteins reduces IFN and proinflammatory cytokine expression
(A) qRT-PCR of RNA isolated 48 hpi from Calu3 cells infected with SARS-CoV-2 (MOI of 0.01 or 0.1) and concurrently treated with JQ1 (500 nM) or dBET6 (500 nM). Data are expressed relative to DMSO-treated cells for each respective MOI. The average of three independent experiments analyzed in triplicate ± SEM is shown and compared with the DMSO condition by ANOVA for each MOI: ∗p < 0.05, ∗∗p < 0.005, ∗∗∗∗p < 0.0001.
(B) qRT-PCR of RNA isolated 48 hpi from Calu3 cells with the indicated BET protein KO or KD infected with SARS-CoV-2 (MOI of 0.01 or 0.1). Data are expressed relative to RNP-only cells for each respective MOI. The average of three independent experiments analyzed in triplicate ± SEM is shown and compared with RNP-only samples by ANOVA for each MOI: ∗p < 0.05, ∗∗p < 0.005,∗∗∗p < 0.001,∗∗∗∗p < 0.0001.
(C) qRT-PCR of RNA isolated from A549 cells transfected with empty vector (EV) or the SARS-CoV-2 E (E-Strep) plasmid and treated with 10 ng/mL poly(I:C) for 24 h with and without JQ1 (500 nM) or dBET6 (500 nM). Mock refers to mock transfection and DMSO treatment to mimic poly(I:C) transfection and JQ1 or dBET6 treatment. Data are expressed relative to mock-treated cells. The average of three independent experiments analyzed in triplicate ± SEM is shown with ANOVA compared with mock: ∗p = 0.0139, ∗∗p = 0.0097, ∗∗∗∗p < 0.0001.
Next we used Calu3 BET KO and KD cells to determine whether the loss of individual BET proteins was sufficient to suppress induction of these genes. After infection with SARS-CoV-2, the control (RNP only) cells showed robust induction of IFNB1, ISG15, and IL6 at all MOIs (Figure 2B). In contrast, depletion of all BET proteins significantly decreased expression of IFNB1, ISG15, and IL6 at low-MOI infection, phenocopying the effect of JQ1 and dBET6 on infected Calu3 cells (Figure 2B). Interestingly, at higher MOI, BET depletion still suppressed IFNB1 and IL6 induction, but ISG15 expression was not significantly suppressed in BRD3- and BRD4-depleted cells, implicating other ISGs as possible targets of BET action (Figure 2B). BRD2 KO cells showed the lowest IFN response, reflecting their reduced infection because of low ACE2 levels (Figure S1A). These results underscore the role of different BET proteins in activating IFN and proinflammatory responses.
SARS-CoV-2 E protein suppresses host antiviral responses and localizes to the nucleus
The recent SARS-CoV-2 protein:protein interactome identified BRD2 and BRD4 as high-confidence interactors with the viral E protein; BRD3 was also detected but just below the mass spectrometry interaction statistics (MiST) scoring threshold (Figure 3A; Gordon et al., 2020). We therefore tested whether E protein phenocopies the effect of BET inactivation. An E protein expression construct or the empty vector (EV) control was transfected into A549 cells and stimulated with poly(I:C) to induce IFN production (Figure 2C). Expression of E protein, similar to JQ1 and dBET6 treatment alone, dampened poly(I:C)-mediated induction of IFNB1, ISG15, and IL6, supporting the model where E protein evolved to suppress the antiviral response by inhibiting BET proteins.
Figure 3.

SARS-CoV-2 E protein interacts with BET proteins at the nuclear periphery
(A) Host protein interactors, including members of the BET protein family (BRD2, BRD3, and BRD4), of the SARS-CoV-2 E protein (Gordon et al., 2020). High-confidence interactors above the MiST threshold are shown as solid lines, and interactors below the threshold are shown as dashed lines.
(B) The E protein sequences from human and bat (RsHC014) coronaviruses share a histone H3-like motif. Lysine residues at position 53 and 63 are shown in red, and the histone H3-like motif is bolded in black, with the RXK motif highlighted in blue. Shown are SARS-CoV (GenBank: YP_009724392.1), SARS-CoV-2 (GenBank: NP_828854.1), bat CoV (GenBank: AGZ48809.1), and histone H3 (UniProt: P68431).
(C) Representative immunoprecipitation of overexpressed Strep-tagged SARS-CoV-2 E (E-Strep) protein with FLAG-tagged BRD4S from co-transfected HEK293T whole-cell lysates, followed by western blotting using FLAG, Strep, and GAPDH antibodies. EV, empty vector.
(D) Representative immunoprecipitation of overexpressed E-Strep protein with FLAG-tagged BRD4L from co-transfected HEK293T whole-cell lysates, followed by western blotting using FLAG, Strep, and GAPDH antibodies. EVEV, empty vector.
(E) Representative immunoprecipitation of overexpressed E-Strep protein constructs with GFP-tagged BRD2 from co-transfected HEK293T whole-cell lysates, followed by western blotting using BRD2, Strep, and GAPDH antibodies. EV, empty vector.
(F) Densitometry of Figure 3E showing the ratio of GFP-BRD2 to different Strep-tagged E constructs. The average of three independent experiments analyzed in triplicate ± SEM is shown and analyzed by Student’s t test: ∗p < 0.05, ∗∗p = 0.009.
(G) Representative western blotting of whole-cell lysate (WCL), cytoplasmic (CYT), nuclear (NUC), and chromatin (CHM) fractions from HEK293T cells transfected with EV or E-Strep with the indicated antibodies.
(H) Representative confocal microscopy images of HEK293T cells transfected with E-Strep or empty vector control (EV). Cells were processed for immunostaining with Strep (SARS-CoV-2 E, green), ERGIC-53 (endoplasmic reticulum-Golgi intermediate compartment; red), NPCs (nuclear pore complexes; turquoise), and Hoechst (nuclei, blue). Scale bars, 5 μm.
We previously identified two lysine residues in the C terminus of SARS-CoV-2 E protein (K53 and K63) that could be acetylated, and, in fact, K63 resides in the short motif (aa 60–64) of E protein that mimics the N terminus of histone H3 (Figure 3B; Gordon et al., 2020). This motif was conserved in the E proteins of other coronaviruses, supporting a broader model where coronavirus E proteins may neutralize the antiviral response by antagonizing BET proteins. Mechanistically, this may be achieved by inhibiting the interaction of BET proteins with chromatin. To test this model, we performed co-immunoprecipitation studies in HEK293T cells overexpressing tagged forms of E and BET proteins. We found that SARS-CoV-2 E protein (C-terminal double-streptavidin [Strep] tagged) interacted with both isoforms of BRD4 (N-terminal FLAG tagged) and BRD2 (N-terminal green fluorescent protein [GFP] tagged) (Figures 3C, 3D, and 3F). To determine whether the lysine residues within E protein interacted with BET proteins, we generated double-Strep-tagged, acetylation-null point mutants (K53R, K63R, and K53/63R) of E protein. We performed the co-immunoprecipitation studies in HEK293T cells overexpressing BRD2 and the wild type or mutant E proteins. The K53/63R mutant had decreased binding to BRD2 compared with the wild type, suggesting that the two lysine residues in E protein are involved in facilitating the interaction with BRD2 and likely other BET proteins (Figures 3E and 3F). Compared with the double mutant, the K53R and K63R single mutants had decreases in interaction with BRD2 similar to K53/63R, suggesting that the BDs are able to recognize each lysine residue individually. The retained interaction of the double mutant with BRD2 suggests possible other interactions between the E protein and BRD2, independent of BD-mediated recognition of acetylated lysine residues.
Next we performed cellular fractionation studies in HEK293T cells overexpressing Strep-tagged E protein. E protein was enriched in the nuclear and chromatin fractions (Figure 3G). These same fractions also contained the majority of BET proteins (Figure 3G). We did not observe a difference in BET protein fractionation between cells expressing E protein or the EV, excluding the possibility that E protein grossly mislocalizes BET proteins. This was confirmed by confocal microscopy, where E protein, a membrane-bound protein, showed ring-like perinuclear localization that overlapped with staining of the cellular nuclear pore complex (NPC) (Figure 3H). Previous reports of E proteins from SARS and Middle Eastern respiratory syndrome coronavirus (MERS-CoV) imaged in infected cells also showed perinuclear localization (Nal et al., 2005; Nieto-Torres et al., 2011). We also confirmed previous reports showing SARS-CoV E protein colocalizing with endoplasmic reticulum-Golgi intermediate complex (ERGIC) (Figure 3H; Schoeman and Fielding, 2019). However, reliable antibodies to detect untagged SARS-CoV-2 E protein during infection are still lacking, hampering staining of endogenous E protein during infection. Our data support a model where the E:BET protein interaction likely occurs on chromatin in the nuclear periphery.
Acetylated SARS-CoV-2 E peptide predominantly binds the second BD of BRD4
Next we tested whether E protein is acetylated in cells to act as a histone mimetic and interact with BDs. For this, we immunoprecipitated wild-type or acetylation-null (K53/63R) Strep-tagged E proteins from transfected HEK293T cells and probed for acetylation with pan-acetyl-lysine antibodies (Figure 4A). To better visualize acetylation of E protein, we treated the cells with a cocktail of cellular pan-histone deacetylase (HDAC) inhibitors and observed a signal for wild-type but not double-arginine-mutant E proteins. This indicates that E protein is reversibly acetylated in cells.
Figure 4.

Acetylated SARS-CoV-2 E peptide predominantly binds the second BD of BRD4
(A) Representative immunoprecipitation of overexpressed E-Strep constructs (E-Strep and E K53/63R-Strep) from co-transfected HEK293T WCLs with and without trichostatin A (TSA) (1 μM) and nicotinamide (NIC) (1 mM), followed by western blotting using Strep, pan-acetyl lysine, and GAPDH antibodies.
(B) 2D 1H 15N HSQC spectra measured after addition of E-K53ac (residues 48–58, acetylated), E-K63ac (residues 58–68, acetylated) or non-acetylated (residues 58–70) peptides to 15N-labeled BD1 or BD2. Arrows indicate chemical shift perturbations of peaks. The protein-to-ligand ratio is indicated.
(C) 2D 1H 15N HSQC spectra measured after addition of E-K53R (residues 48–58) and E-K63R (residues 58–68) peptides to 15N-labeled BD2. The protein-to-ligand ratio is indicated.
(D) qRT-PCR of SARS-CoV-2 E RNA isolated 48 hpi from A549-ACE2 cells infected with SARS-CoV-2 (MOI of 0.01 or 0.1) and concurrently treated with the BD2-selective inhibitor ABBV-744 (500 nM). Data are expressed in absolute copies per microgram based on a standard curve of the E gene with known copy number. The average of three independent experiments analyzed in triplicate ± SEM is shown and compared with DMSO by Student’s t test: ∗∗∗p = 0.0004.
(E) Plaque assay titers from supernatant of infected A549-ACE2 cells at MOI of 0.1 treated with ABBV-744 (500 nM). The average of three independent experiments analyzed in duplicate ± SEM is shown and compared with DMSO by Student’s t test: ∗∗∗p = 0.0001.
We used nuclear magnetic resonance (NMR) titration experiments to assess binding of acetyl lysine-containing E peptides to recombinant BD1 and BD2 domains of BRD4. The SARS-CoV-2 E protein contains two lysines, K53 and K63, and we used peptides that were acetylated at either position or were non-acetylated. In two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) spectra of BD2, we saw significant chemical shift perturbations of peaks upon addition of E-K53ac or E-K63ac peptides, indicating that BD2 is able to bind either acetyl-lysine position (Figure 4B, top panels). BD2 was found to have a higher affinity for the E-K63ac peptide (Kd value of 170 ± 60 μM) than for the E-K53ac peptide (Kd value of 610 ± 160 μM) (Figure S2A). The interaction was acetylation dependent because addition of non-acetylated E peptide did not elicit changes in chemical shifts in BD2 as observed with the acetylated peptides (Figure 4B). To determine the peptide binding site, the backbone amides most perturbed upon peptide binding were mapped onto the surface of BD2. Both peptides were found to occupy a similar binding site to where other acetylated lysine peptides have been shown to bind previously (Figures S2B and S2C). Unlike with BD2, little to no chemical shift perturbations in HSQC spectra were observed upon addition of the acetylated E peptides to BD1, indicating very weak to no interaction of acetylated E protein with BD1 (Figure 4B, bottom panels).
We further investigated whether acetylated E peptides compete with BD inhibitors for binding. We performed fluorescence polarization-based competition assays with BD2 bound to a carboxy tetramethylrhodamine (TAMRA)-conjugated JQ1 molecule. The decrease in fluorescence polarization upon increasing the peptide concentration indicates that the JQ1-TAMRA molecule is being competed off by the acetylated E peptides (Figure S2D). Next we assessed whether BD2 could bind acetylation-null peptides where the lysine residues were replaced by arginines (K53R and K63R). Similar to the observed HSQC spectra with a non-acetylated peptide, little to no chemical shift perturbations were observed upon addition of the E-K53R and E-K63R peptides to BD2, indicating very weak to no interaction (Figure 4C).
Overall, these data show that the E protein is acetylated and that the interaction of BRD4 with acetylated E peptide predominantly involves the second BD of BRD4. The relevance of the second BD to the antiviral function of BRD4 was explored in studies with ABBV-744, a BET inhibitor that specifically targets BD2-mediated interactions (Sheppard et al., 2020). In A549-ACE2 cells treated with ABBV-744 at the time of infection, SARS-CoV-2 viral RNA and infectious particle production were significantly increased (3-fold for both) compared with cells treated with vehicle alone, demonstrating that selective inhibition of BD2 alone is sufficient to suppress the antiviral function of BET proteins (Figures 4D and 4E).
BET inhibitors enhance SARS-CoV-2 infection in K18-hACE2 mice
To test the post-entry effects of BET inhibitors JQ1 and ABBV-744 in vivo, we used K18-hACE2 transgenic mice constitutively expressing the human ACE2 receptor in epithelial tissues from the cytokeratin 18 (K18) promoter (Jia et al., 2005). Mice were infected intranasally, and BET inhibitors were administered daily intraperitoneally or orally starting on the day of infection (Figure 5A). DMSO at the same concentration as JQ1 and ABBV-744 was used as a control. Based on the in vitro observations, we hypothesized that BET inhibitor treatment would similarly result in an increase in viral replication in mice.
Figure 5.

BET inhibitors enhance SARS-CoV-2 infection in K18-hACE2 mice
(A) Schematic of the experiment.
(B) Representative images of plaque assays at the same dilution from infected mice lungs 2 and 4 days post infection (dpi).
(C) Plaque assay titers from the lungs of infected mice. An average of 5 mice per group were analyzed, and average ± SD is shown and compared with DMSO by Student’s t test. 2 dpi: ∗∗p = 0.0041, ∗∗∗∗p < 0.0001. 4 dpi: ∗∗p = 0.0021.
(D) Representative images of H&E staining of lung tissue 7 days after infection. A box indicates the region of the inset. Scale bars, 600 μm (left panels) and 200 μm (right panels).
(E) Survival curve of mock (uninfected) and infected DMSO-treated (n = 15), JQ1-treated (n = 15), and ABBV-744-treated (n = 15) mice.
As expected, all infected mice exhibited weight loss and a change in body temperature as a sign of infection, and no significant differences between the treatment groups were observed (Figures S3A and S3B). Viral replication in all treatment groups was analyzed 2 and 4 days after infection. At these time points, we observed increased viral titers in the lungs of JQ1- and ABBV-744-treated mice (Figures 5B and 5C). Specifically, there was an 18-fold increase in infectious particle production in JQ1-treated mice and a 37-fold increase in ABBV-744-treated mice compared with DMSO 2 days after infection. Concordantly, there was increased immunohistochemical staining for SARS-CoV-2 nucleocapsid (N) protein in the lungs of JQ1-and ABBV-744-treated mice 7 days after infection compared with DMSO-treated mice (Figure S3C). Similar to the in vitro treatment, JQ1- and ABBV-744-treated mice experienced significant decreases in expression of IFNs, cytokines, and chemokines, including ISG15, IL6, RIG-I, IL1a, CCL2, CXCL9, and CXCL10 (Figure S3D). The decrease in expression of these host defense genes paired with the subsequent increase in infectious particle production was accompanied by a decrease in immune cell infiltration in the lungs 7 days after infection in JQ1- and ABBV-744-treated mice compared with DMSO-treated mice (Figure 5D). DMSO treatment showed widespread large inflammatory foci consistent with interstitial pneumonia with perivascular inflammation comprised of lymphocytes, macrophages, and neutrophils. These histological changes are attributed to SARS-CoV-2 infection because healthy, uninfected mice did not experience lung inflammation upon identical treatment with DMSO, JQ1, and ABBV-744 (Figure S3E).
Overall, 60%–80% of animals with BET inhibitor treatment reached a humane endpoint on day 6 after infection, whereas 100% of the animals in the DMSO-treated group survived (Figure 5E). These data underscore the finding that BET proteins regulate an antiviral state during viral infection that prevents exacerbation of virally induced disease and document the clinical relevance of the second BD of BET proteins, which is targeted by the viral E protein.
Discussion
We report that BET proteins (BRD4 > BRD3 > BRD2) block SARS-CoV-2 infection at the post-entry level because each of them is necessary for full induction of the type I IFN response and proinflammatory cytokines. Viral replication is exacerbated after chemical or genetic inactivation of BET proteins, underscoring the critical role of this antiviral step in controlling SARS-CoV-2 infection. We further report a so far unknown function of the SARS-CoV-2 E protein in antagonizing IFN and ISG expression. E protein is acetylated in cells and, in an acetylated form, binds to the second BD of BRD4. Finally, BET inhibitor treatment increases viral titers in the lungs of SARS-CoV-2-infected mice and results in higher morality. Because the SARS-CoV-2 E protein is functionally similar to BET antagonists in suppressing the IFN response and directly binds one of the two BDs, we propose a model where E protein evolved to neutralize the antiviral gene activation mediated by BET proteins to promote efficient SARS-CoV-2 replication, a process further enhanced with BET inhibitor treatment.
Our data highlight the unique and overlapping roles of BET proteins during SARS-CoV-2 infection, where individual BET proteins may serve proviral and antiviral roles. Previous reports have demonstrated that prophylactic treatment of cell lines with pan-BET inhibitors decreases ACE2 expression and reduces viral replication (Mills et al., 2021; Qiao et al., 2020). Here we show that BET proteins can promote or block SARS-CoV-2 infection and that this is likely dependent on the specific gene targets with which individual BET proteins interact. Comparing the effects of genetic and chemical neutralization of BET proteins in cells that endogenously express or overexpress ACE2 allows for a more nuanced parsing of the pre- and post-viral entry roles of BET proteins. With genetic neutralization of BET proteins in cells with endogenous ACE2 expression (Calu3), only KO of BRD2 reliably ablates viral replication as ACE2 expression is reduced. On the other hand, genetic neutralization of BET proteins in cells that overexpress ACE2 (A549-ACE2) does not affect constitutive ACE2 overexpression; the pro-inflammatory role of BRD4 is prominently suppressed and results in an increase in viral replication. Chemical inhibition of BET proteins at the time of infection in either cell line results in enhancement of viral replication similar to genetic neutralization of BET proteins in A549-ACE2 cells.
Among the BET proteins, BRD2 is the most proviral because it positively regulates expression of the SARS-CoV-2 entry receptor ACE2. In contrast, BRD4 has the strongest antiviral function because of its co-activator role in the induction of IFN genes. Differences between BRD2 and BRD4 lie in their domain structure; although sharing roughly 70% sequence similarity in the N terminus, including the tandem BDs, the C terminus of BRD4L contains the PID, which enhances recruitment of P-TEFb to BET target genes (Bisgrove et al., 2007; Sheppard et al., 2020). Small-molecule BET inhibitors reduce recruitment of BET proteins, along with P-TEFb, to IFN target genes upon IFN-β or Toll-like receptor ligand stimulation (Gilan et al., 2020; Malik et al., 2015; Patel et al., 2013). BRD4 plays a critical role in coordinating positive and negative regulation of paused RNA polymerase II at the transcription start sites of ISGs by recruiting negative elongation factor (NELF)/DRB sensitivity-inducing factor (DSIF) to fine-tune ISG expression (Patel et al., 2013). On the other hand, BRD2 functions as a chromatin organizer that assembles enhancer elements (Cheung et al., 2017). Depletion of BRD2 versus BRD4 results in distinct transcriptional changes, suggesting non-overlapping roles in function (Andrieu and Denis, 2018). In contrast, identification of a conserved motif B in BET proteins that facilitates heterodimerization of proteins within this family may explain the partial functional redundancy and shared chromatin occupancy of BRD2 and BRD4 (Garcia-Gutierrez et al., 2012). The fact that BRD2 and BRD4 play opposing roles in regulation of SARS-CoV-2 highlights the complexity of genes regulated by BET proteins, and future studies aimed at disentangling these regulatory networks will be of value to the field.
We also show that SARS-CoV-2 E protein predominantly targets the second BD of BRD4. Research has classically focused on histones as BD targets, but BD2 of BRD4 also binds the acetylated cyclin T1 subunit of P-TEFb (Schröder et al., 2012). Recent studies defining the unique roles of BD1 and BD2 in disease models have indicated that inhibiting BD2 preferentially blocks stimulus-mediated induction of gene expression programs without affecting pre-existing transcriptional programs (Gilan et al., 2020). BD1-specific inhibitors are effective in inhibiting cell proliferation in cancer models, whereas BD2-specific inhibitors had the greatest effect in ameliorating inflammatory and autoimmune diseases (Gilan et al., 2020). Our finding that SARS-CoV-2 E protein binds BD2 of BRD4 supports the notion that BD2 is important for rapid gene expression, especially in inflammatory disease settings such as COVID-19.
SARS-CoV-2 E protein joins a growing number of viral proteins containing histone-like motifs in their sequences. Histone tails are often mimicked by viruses because many host chromatin factors interact with histones to modulate gene expression (Schaefer et al., 2013). A prominent example of a viral histone mimetic is NSP1 of the influenza A H3N2 subtype, which contains a short histone H3-like motif capable of sequestering the transcription elongation factor PAF1 to prevent expression of antiviral genes (Qin et al., 2014). Similarly, the human immunodeficiency virus (HIV) 1 factor Tat is reversibly acetylated at multiple lysines in its basic domain to recruit key host transcriptional regulators in a coordinated manner, enhancing viral transcription (Huo et al., 2011; Kiernan et al., 1999; Ott et al., 1999; Pagans et al., 2005). A recent study identified the capsid protein of the yellow fever virus as a histone H4 mimetic with acetylated lysine residues that bind BRD4 to interfere with gene regulation (Mourão et al., 2020). Overall, BRD4 appears to be a hotspot of viral antagonism because of its critical role in positively regulating the antiviral response.
The E proteins of coronaviruses are similar in domain structure: a short, hydrophilic N terminus followed by a hydrophobic transmembrane domain and a subsequent C terminus that comprises the majority of the protein and contains a PDZ-binding motif (PBM). Among the E proteins of human pathogenetic coronaviruses, those from SARS and SARS-CoV-2 share ∼95% sequence similarity along with the histone H3-like motif, whereas the MERS-CoV E protein has only ∼35% similarity with SARS and SARS-CoV-2 E proteins and does not have the histone-like motif (Schoeman and Fielding, 2020). The histone-like motif of interest is also present in the common coronavirus NL63 and bat coronavirus SHC014, suggesting that the function of E:BET proteins could also be conserved in these viruses. Future work should address whether E proteins from these coronaviruses are similarly capable of derailing antiviral responses through BRD4 antagonism.
Since the beginning of the pandemic, there has been growing interest in prophylactic application of BET inhibitors to prevent SARS-CoV-2 infection because they reduce ACE2 expression. All previous studies relied on multi-day pretreatment of cell lines, organoids, and primary cells with BET inhibitors to inhibit infection (Gilham et al., 2021; Mills et al., 2021; Qiao et al., 2020). In contrast, our study focuses on concurrent administration of the BET inhibitors JQ1 and ABBV-744 at the time of infection. Similar to our observations, Mills et al. found that therapeutic application in K18-hACE2 mice of the BET inhibitor INCB054329 resulted in severe lung pathology and significant viral RNA in the lungs (Mills et al., 2021). We demonstrate that therapeutic application of BET inhibitors worsens viral pathogenesis, as evidenced by enhanced immune infiltration, a marked increase in infectious particle production, and lower probability of survival, underscoring the clinical significance of post-entry restriction of SARS-CoV-2 by BET proteins.
Our study sheds light on the antiviral function of BET proteins during SARS-CoV-2 infection, highlighting their contrasting functions at different stages of the viral life cycle. Whether SARS-CoV-2 evolved the anti-BET protein function of the E protein because of the unique influence of BRD2 on ACE2 receptor levels or the necessity for a more surgical inactivation of BET proteins as transcriptional coactivators necessary for antagonizing the virus remains to be determined. Because most BET inhibitors do not distinguish between the different BET proteins and inhibit all of them indiscriminately, our study urges caution with clinical use, prophylactically or therapeutically, of pan-BET inhibitors in people at risk for or afflicted with COVID-19.
Limitations of the study
This study explores the nuanced role of BET proteins in pre- and post-entry steps of SARS-CoV-2 infection. Although we characterize the cellular interaction between SARS-CoV-2 E protein with BRD2 and BRD4 proteins and their effect on the viral life cycle, additional work is necessary to translate this into clinical application. First, although we focus on K53 and K63 residues in SARS-CoV-2 E protein, a comprehensive analysis of E protein acetylation, ideally in infected cells, is missing and would give better insight into the functional consequences of this modification. Second, the nature of the cellular lysine acetyltransferases and deacetylases that modify the E protein are not yet known; this would help to potentially harness available inhibitors for these enzymes for COVID-19 treatment. Finally, continued development of more targeted small molecules for each BET protein will allow specific tuning of cellular signaling pathways to achieve a more desirable therapeutic outcome. For example, a molecule that specifically inhibits BRD2 but not BRD4 could be useful for therapeutic applications for COVID-19 in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BRD2 | Abcam | Cat#ab139690; RRID:AB_2737409 |
| BRD3 | Abcam | Cat#ab50818; RRID:AB_868478 |
| BRD4 | Abcam | Cat#ab128874; RRID:AB_11145462 |
| Nuclear Pore Complex Proteins AlexaFluor 647 | BioLegend | Cat#682204; RRID:AB_2728508 |
| PARP | Cell Signaling | Cat#9532S |
| ERGIC-53 AlexaFluor 594 | Santa Cruz Biotechnology | Cat#sc-365158 AF594 |
| Hoescht 33342 | Invitrogen | Cat#H3570 |
| GAPDH | Cell Signaling | Cat#5174S |
| Strep | Qiagen | Cat#1023944 |
| FLAG | Sigma | Cat#F3165; RRID:AB_259529 |
| Rabbit IgG-HRP | Bethyl Laboratories | Cat#A120-201P; RRID:AB_67265 |
| Mouse IgG-HRP | Bethyl Laboratories | Cat#A90-516P; RRID:AB_10631212 |
| Pan-acetyl | Cell Signaling | Cat#9441S |
| Pan-acetyl | Abcam | Cat#ab80178; RRID:AB_1640674 |
| Histone H4 | Cell Signaling | Cat#13919S |
| Mouse IgG-AlexaFluor549 | Invitrogen | Cat#A11005; RRID:AB_2534073 |
| Rabbit IgG-AlexaFluor488 | Invitrogen | Cat#A11008; RRID:AB_143165 |
| Bacterial and virus strains | ||
| Stbl3 cells | Invitrogen | Cat#C737303 |
| LOBSTR E. coli | Kerafast | Cat#EC1002 |
| SARS-CoV-2 (Isolate USA-WA1/2020) | BEI | Cat#NR-52281 |
| Chemicals, peptides, and recombinant proteins | ||
| JQ1 | Selleckchem | Cat#S7110 |
| dBET6 | Selleckchem | Cat#S8762 |
| ABBV-744 | Selleckchem | Cat#S8723 |
| RNA-STAT 60 | AMSBIO | Cat#CS-110 |
| iSCRIPT | Bio-Rad | Cat#1708841 |
| Micrococcal Nuclease | NEB | Cat#M0247S |
| Halt Protease & Phosphatase Inhibitor Cocktail | Thermo Scientific | Cat#1861282 |
| Avicel | DuPont | Cat#RC-591 |
| Crystal Violet | Sigma | Cat#C0775 |
| NP-40 | Sigma | Cat#9002-93-1 |
| Trichostatin A (TSA) | Selleckchem | Cat#S1045 |
| Nicotinamide (NIC) | Sigma | Cat#N3376 |
| X-tremeGENE 9 DNA Transfection Reagent | Roche | Cat#XTG9-RO |
| DNA QuickExtract | Lucigen | Cat#QE09050 |
| AmpliTaq Gold 360 polymerase | Applied Biosystems | Cat#4398881 |
| Streptococcus Pyogenes NLS-Sp.Cas9-NLS (SpCas9) nuclease | Aldevron | Cat#9212 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat#4309155 |
| (2-Hydroxypropyl)-β-cyclodextrin | Sigma | Cat#C0926 |
| Polyethylene Glycol 400 | Sigma | Cat#8074850050 |
| Phosal 50 PG | MedChemExpress | Cat#HY-Y1903 |
| SARS-CoV-2 E Peptides | See Table S4 for peptide sequences. | N/A |
| His-SUMO-BD1 (42-168) | This Paper. | N/A |
| His-SUMO-BD2 (349-460) | This Paper. | N/A |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat#23225 |
| Direct-zol RNA Miniprep Kit | Zymo Research | Cat#R2052 |
| Plasmid DNA Minipreps Kit | BioBasic | Cat#BS614 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat#78835 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7571 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| Vero E6 | ATCC | Cat#CRL-1586; RRID:CVCL_0574 |
| A549-ACE2 | Gordon et al. (2020) | N/A |
| A549-ACE2 BRD2 KO | This Paper. | N/A |
| A549-ACE2 BRD3 KO | This Paper. | N/A |
| A549-ACE2 BRD4 KD | This Paper. | N/A |
| Calu3 | ATCC | Cat#HTB-55; RRID:CVCL_0609 |
| Calu3 BRD2 KO | This Paper. | N/A |
| Calu3 BRD3 KO | This Paper. | N/A |
| Calu3 BRD4 KD | This Paper. | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: K18-hACE2 C57BL/6J: B6.Cg-Tg(K18-ACE2)2Primn/J | The Jackson Laboratory | Cat#034860; RRID:IMSR_JAX:034860 |
| Oligonucleotides | ||
| sgRNAs | See Table S1 for sgRNA sequences. | N/A |
| Genotyping primers | See Table S2 for genotyping primers. | N/A |
| qPCR primers | See Table S3 for qPCR primers. | N/A |
| Recombinant DNA | ||
| GFP-BRD2 | Gong et al., 2015 | Addgene plasmid # 65376; RRID:Addgene_65376 |
| FLAG-BRD4L | This Paper. | N/A |
| FLAG-BRD4S | This Paper. | N/A |
| Strep-SARS-CoV-2 E | Gordon et al. (2020) | N/A |
| Strep-SARS-CoV-2 E K53R | This Paper. | N/A |
| Strep-SARS-CoV-2 E K63R | This Paper. | N/A |
| Strep-SARS-CoV-2 E K53/63R | This Paper. | N/A |
| Strep-EV | Gordon et al. (2020) | N/A |
| Software and algorithms | ||
| Prism8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Imaris | Oxford Instruments | https://imaris.oxinst.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Inference of CRISPR Edits (ICE) | Synthego Corporation | http://ice.synthego.com |
| Origin | OriginLab Corporation | https://www.originlab.com/ |
| CCPNmr Analysis | CCPN | https://ccpn.ac.uk/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Melanie Ott (melanie.ott@gladstone.ucsf.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mammalian cell lines and culture conditions
HEK293T and Vero-E6 were obtained from ATCC were cultured in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS) (GeminiBio), 1% glutamine (Corning), and 1% penicillin-streptomycin (Corning) at 37°C, 5% CO2.
Calu3 cells were obtained from ATCC and cultured in AdvancedMEM (Gibco) supplemented with 2.5% FBS, 1% GlutaMax, and 1% penicillin-streptomycin at 37°C, 5% CO2.
A549 cells stably expressing ACE2 (A549-ACE2) were a gift from O. Schwartz. A549-ACE2 cells were cultured in DMEM supplemented with 10% FBS, blasticidin (20 μg/mL) (Sigma) and maintained at 37°C with 5% CO2. Short Terminal Repeat (STR) analysis by the Berkeley Cell Culture Facility on 17 July 2020 authenticates these as A549 cells with 100% probability.
Generation of CRISPR A549-ACE2 and Calu3 KO cell lines
sgRNAs were designed according to Synthego’s multi-guide gene knockout. Briefly, three sgRNAs are bioinformatically designed to work in a cooperative manner to generate small fragment deletions in early exons causing knock out. These fragment deletions are larger than standard indels generated from single guides. The genomic repair patterns from a multi-guide approach are highly predictable based on the spacing of and on design constraints that limit off-targets, resulting in a higher probability of protein knockout phenotype.
To induce gene knockout, 10 pmol Streptococcus Pyogenes NLS-Sp.Cas9-NLS (SpCas9) nuclease (Aldevron, 9212) was combined with 30 pmol total synthetic sgRNA (10 pmol each sgRNA) (Synthego) to form ribonucleoproteins (RNPs) in 20uL total volume with SE Buffer for Calu-3 and A549-ACE2 cells. The RNP assembly reaction was mixed by pipetting up and down and incubated at room temperature for 10 min. Cells were resuspended in transfection buffer according to cell type and added to the preformed RNP solution and gently mixed. Nucleofections were performed on a Lonza nucleofector system (Lonza, AAU-1001) using program CM-130 and CM-120 for Calu3 and A549-ACE2 cells, respectively. Two days post-nucleofection, DNA was extracted with DNA QuickExtract (Lucigen, QE09050). Amplicons for indel analysis were generated by PCR using AmpliTaq Gold 360 polymerase (Thermo Fisher Scientific, 4398813). PCR products were cleaned-up and analyzed by Sanger sequencing. Sanger data files and sgRNA target sequences were input into Inference of CRISPR Edits (ICE) analysis (ice.synthego.com) to determine editing efficiency and to quantify generated indels (Conant et al., 2022). A list of sgRNA sequences and genotyping primers can be found in Tables S1 and S2.
Mice
All protocols concerning animal use were approved (AN169239-01B) by the Institutional Animal Care and Use committees at the University of California, San Francisco and Gladstone Institutes and conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animal (National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011). Studies were conducted with 6–8 week old male and female K18-hACE2 C57BL/6J mice (strain B6.Cg-Tg(K18-ACE2)2Primn/J) (The Jackson Laboratory, 034860). Mice were housed in a temperature- and humidity-controlled pathogen-free facility with 12-hour light/dark cycle and ad libitum access to water and standard laboratory rodent chow.
SARS-CoV-2 culture
SARS-CoV-2 Isolate USA-WA1/2020 (BEI NR-52281) was used for all infection studies. All live virus experiments were performed in a Biosafety Level 3 laboratory. SARS-CoV-2 stocks were propagated in Vero-E6 cells and their sequence verified by next-generation sequencing. Viral stock titer was calculated using plaque forming assays.
Plasmids
The plasmids expressing the envelope protein of SARS-CoV-2 were generous gifts from Dr. Nevan Krogan (UCSF, The Gladstone Institutes). The majority of BRD4 plasmids were generous gifts from Dr. Eric Verdin (The Buck Institute for Research on Aging, Novato, CA). GFP-BRD2 was a gift from Kyle Miller (Addgene plasmid # 65376 ; http://n2t.net/addgene:65376 ; RRID:Addgene_65376). All plasmids and corresponding sequence information are available upon request.
Method details
Cell fractionation
Cell fractionation was performed with the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, 78835) with additional modifications to extract the chromatin fraction. Following the extraction of the nuclear fraction per manufacturer’s instructions, the resulting pellet was resuspended in NER with MNase (NEB, M0247S), Halt protease inhibitor cocktail (Thermo Fisher Scientific, 1861282), and CaCl2 (Sigma, C4901). Samples were vortexed on the highest setting and sheared on the Bioruptor (Diagenode) with 10 cycles of 10 seconds on and 10 seconds off on medium setting. The samples were pelleted by centrifugation and the resulting supernatant was the chromatin fraction.
Western blot analysis
Cells were lysed in RIPA lysis buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, supplemented with Halt protease inhibitor cocktail) to obtain whole-cell lysates or lysed using the NE-PER nuclear and cytoplasmic extraction kit to obtain cytoplasmic, nuclear, and chromatin fractions. Protein concentrations of cell fractions were determined using a BCA Assay (Thermo Fisher Scientific, 23225), and normalized among samples per experiment before analysis via western blotting using standard techniques. Proteins were visualized by chemiluminescent detection with ECL on ChemiDoc MP (Bio-Rad). Antibodies: BRD2 (Abcam, ab139690), BRD3 (Abcam, ab50818), BRD4 (Abcam, ab128874), LaminB1 (Abcam, ab16048), PARP (Cell Signaling, 9532S), GAPDH (Cell Signaling, 5174S), Strep (Qiagen, 1023944), FLAG (Sigma, F3165), Histone H4 (Cell Signaling, 13919S), pan acetyl lysine (Cell Signaling, 9441S; Abcam, ab80178), Rabbit IgG-HRP (Bethyl, A120–201P), and Mouse IgG-HRP (Bethyl, A90–516P).
Quantitative polymerase chain reaction
RNA was extracted from cells or supernatants using RNA STAT-60 (AMSBIO, CS-110) and the Direct-Zol RNA Miniprep Kit (Zymo Research, R2052). RNA from cells or supernatant was reverse-transcribed to cDNA with iScript cDNA Synthesis Kit (Bio-Rad, 1708890). qPCR reaction was performed with cDNA and SYBR Green Master Mix (Thermo Scientific) using the CFX384 Touch Real-Time PCR Detection System (Bio-Rad). See Table S3 for primers sequences. E gene standards were used to generate a standard curve for copy number quantification. E gene standard was generated by PCR using extracted genomic SARS-CoV-2 RNA as template. A single product was confirmed by gel electrophoresis and DNA was quantified by Nanodrop.
Immunoprecipitation
Transfected HEK293T cells were lysed in IP buffer (50mM Tris-HCl, 150mM NaCl, 1mM EDTA, 1% Triton X-, supplemented with Halt protease inhibitor cocktail) and 1mg of lysate was incubated with Strep-Tactin Sepharose resin (iba Life Science, 2-1201-002) overnight rotating at 4°C. Bound protein was washed five times with IP buffer and eluted with Strep-Tactin elution buffer (iba Life Sciences, 2-1000-025). Eluted samples were analyzed by western blot.
Immunofluorescence microscopy
Transfected HEK293T cells were plated onto rat tail collagen (EMS, 72295) coated, 22- by 22-mm no. 1.5 coverslips. Cells were fixed in 4% paraformaldehyde, permeabilized with methanol on ice for 10 min, and blocked in 3% bovine serum albumin. Cells were then immunostained with the indicated antibodies: Strep (Qiagen, 1023944), ERGIC-53 (Santa Cruz Biotechnology, sc-365158), Nuclear Pore Complex (Biolegend, 682204), Hoescht 33342 (Invitrogen, H3570), Mouse IgG-AlexaFluor549 (Invitrogen, A11005), and Rabbit IgG-AlexaFluor488 (Invitrogen, A11008). Coverslips were mounted onto glass slides using ProLong Gold Antifade Mountant (Invitrogen, P36934) and analyzed by confocal microscopy (Olympus FV3000RS) using a Olympus UPLAN S-APO 60× OIL OBJ,NA1.35,WD0.15MM objective. The resulting Z-stack was reconstructed and rendered in 3D using Imaris software (Oxford Instruments).
Expression and purification of Brd4 bromodomains
His-SUMO-BD1 (42–168) and BD2 (349–460) constructs were expressed in LOBSTR E. coli cells (Kerafast, EC1002). Expression and purification of both constructs followed the same protocol. For NMR studies the His-SUMO-BD constructs were expressed in M9 minimal media containing 15N ammonium chloride. Cells were induced with 0.1 mM IPTG and grown at 16°C overnight before the pellet was collected. The cells were resuspended in lysis buffer (50 mM Tris, 200 mM NaCl, pH 7.5), lysed by sonication and centrifuged. The supernatant was purified using Ni resin equilibrated in binding buffer (50 mM Tris, 200 mM NaCl, 20 mM imidazole, pH 7.5), washed first with lysis buffer and 30 mM imidazole and then a final wash with lysis buffer and 50 mM imidazole. The protein was eluted from the Ni resin after incubation with SUMO protease Ulp1 in order to remove the His-SUMO tag. The cleaved proteins were then further purified by size-exclusion chromatography using a Hiload 26/60 Superdex 75 gel filtration column (GE, GE28-9893-34) in a buffer of 50 mM sodium phosphate, 100 mM NaCl pH 7.4 buffer before being concentrated and flash–frozen.
NMR binding experiments
For 2D 1H, 15N HSQC peptide titration experiments, 50 μM 15N-labeled BD1 or BD2 in 50 mM sodium phosphate, 100 mM NaCl, 5% D2O, pH 7.4 buffer was used, and spectra were measured after each addition of E protein peptide. The 2D 15N-FHSQC spectra for the E protein peptide titration series were recorded on a Bruker 500 MHz AVANCE DRX spectrometer equipped with an actively shielded Z-gradient QCI cryoprobe (15N/13C/31P, 1H) using programs from the pulse program library (TopSpin 1.3pl10) at 298 K. Chemical shift perturbations of HSQC peaks, upon addition of E peptide, were calculated with the following equation:
Dissociation constants (K d) were determined after the change in chemical shift was plotted against peptide concentration. Data were then fit to the following equation:
where [P] is the concentration of BD2, [L] is the peptide concentration, K d is the dissociation constant and CSPmax is the maximum chemical shift perturbation. The majority of residues were assigned after data from BMRB entry 50146 were transferred to our spectra. See Table S4 for peptide sequences.
Fluorescence polarization assays
E protein peptide binding to purified BRD4 bromodomains was determined by competition–based fluorescence polarization (FP) using a JQ1-TAMRA molecule. Synthesis of JQ1-TAMRA followed a protocol previously described (Olp et al., 2017). For competition–based FP assays, His-SUMO-BD1 and His-SUMO-BD2, at a concentration equal to the K d value for JQ1-TAMRA, were incubated with 10 nM of JQ1-TAMRA and different concentrations of unlabeled peptides were used as competitors. See Table S4 for peptide sequences.
Compound treatments
Compounds, JQ1 (Selleckchem, S7110), dBET6 (Selleckchem, S8762), and ABBV-744 (Selleckchem, S8732), were dissolved in DMSO as per manufacturer’s instructions. Cells were treated at the time of infection for 48 hours with media changes with fresh compound-containing media every 24 hours.
Compound cytotoxicity measurements
A549-ACE2 and Calu3 cells were seeded into 96-well plates and treated with identical compound concentrations used in the infection assays for 48 hours, where fresh compound-containing media was added every 24 hours. At the end of 48 hours, cell viability was assayed following the manufacturer’s protocol of CellTiter-Glo (Promega, G7571). Luminescence was recorded with an Infinite M Plex plate reader (Tecan) using an integration time of 1 second.
Viral infection studies
A549-ACE2 and Calu3 cells were seeded into 12-well plates and rested for at least 24 hours prior to infection. At the time of infection, media containing compound and/or viral inoculum (MOI 0.01 or 0.1) was added on the cells for 24 hours. At 24 hours post infection, fresh compound-containing media or media only was added on the cells. The supernatant and cells were harvested by adding STAT-60 for downstream quantification of genes.
Plaque-forming assays
Viral inoculations were harvested from experiments and serially diluted in DMEM (Corning). At the time of infection, the media on Vero-E6 cells were replaced with viral inoculation for one hour. After the one-hour absorption period, 2.5% Avicel (Dupont, RC-591) was layered on the cells and incubated for 72 hours. Avicel was then removed and cells were fixed in 10% formalin for one hour, stained with crystal violet for 10 minutes, and washed multiple times with water. Plaques were counted and averaged from two technical replicates.
Preparation of compounds for animal study
A stock solution of JQ1 (50mg/mL in DMSO) and ABBV-744 (50mg/mL in DMSO) was prepared. JQ1 was then diluted to a working concentration of 5mg/mL in an aqueous carrier ((2-Hydroxypropyl)-β-cyclodextrin [Sigma C0926]) using vigorous vortexing. Mice were injected at a dose of 50mg/kg given intraperitoneally once daily. ABBV-744 was diluted to a working concentration of 5mg/mL in an carrier ((V/V): 2% DMSO, 30% PEG-400 [Sigma, 8074850050], 68% Phosal-50PG [MedchemExpress, HY-Y1903]) using vigorous vortexing. Mice were treated at a dose of 20mg/kg given orally once daily.
SARS-CoV-2 K18-hACE2 mouse infection model
A total of 50 animals were used in the study. Forty five animals were anesthetized and intranasally infected with 5 × 104 PFU/mL (50μL) of SARS-CoV-2 WA1 strain, five animals were mock infected and used as a control. The infected animals were divided in three groups each of 15 animals. Each group were treated with either JQ1 (50mg/kg) or ABBV-744 (20mg/kg), while DMSO at a vehicle concentration was used as a control. The animals were dosed from day 0 to day 7 post infection. All animals were monitored for their temperature and weight loss on daily basis. At 2 and 4 days post infection five animals from each group were euthanized by cervical dislocation and their lung tissue were homogenized for downstream analysis. Rest of the animals were monitored for their survival after infection. Lungs tissues from virus and mock infected animals were further processed for histological observations.
Histology
Mouse lung tissues were fixed in 4% paraformaldehyde (Sigma, 47608) for 24 hours, washed three times with phosphate buffer saline, and stored in 70% ethanol. Briefly, tissues were processed and embedded in paraffin, and tissue sections were stained for SARS-CoV-2 Nucleocapsid (Genetex, GTX135357). The sections were then imaged using Leica Aperio ImageScope.
Quantification and statistical analysis
The number of experiments and replicates are indicated in individual figure legends. Data were processed and visualized using GraphPad Prism. All quantified data are represented as mean ± SEM or SD, as indicated, and quantification details are available in figure legends. Western blot band intensities were quantified using ImageJ.
Acknowledgments
We thank all members of the Ott and Fujimori laboratories for sharing reagents, expertise, and feedback in the preparation of this manuscript. We thank Lauren Weiser and Veronica Fonseca for administrative support, Dr. Francoise Chanut for editorial support, and John Carroll and Sarah Gardner for graphics support. This research is funded by grants from the National Institutes of Health: NCI R01CA250459 and NIAID R01AI137270 (to D.G.F.) and NIAID R37AI083139 (to M.O.). I.P.C. was supported by NIH/NIAID (F31 AI164671-01). S.M. was supported by TRDRP (T30DT1006) and the UCSF Discovery Fellowship. M.O. acknowledges support from the Roddenberry Foundation, Pam and Ed Taft, and the Gladstone Institutes. We are grateful for philanthropic support from QCRG philanthropic donors and the James B. Pendleton Charitable Trust. Histology, imaging, and image processing were performed at the Gladstone Institutes’ Histology and Light Microscopy Core.
Author contributions
I.P.C., J.E.L., S.M., D.G.F., and M.O. conceived and designed the study. I.P.C., J.E.L., R.K.S., and M.M.K. performed experiments and data analysis. I.P.C. and R.K.S. designed and conducted the animal study. J.C.-S. and J.O. generated the knockout cell lines. M.Y.Z. contributed to binding studies. S.M., J.M.H., T.Y.T., T.T., and F.W.S. provided experimental support. M.G., M.Y.Z., V.L.L., Y.L., Z.Y., E.W.T., and A.D. provided protein expression support. D.G.F. and M.O. supervised the study design and provided technical guidance. K.H., N.K., D.G.F., and M.O. secured funding. I.P.C., J.E.L., D.G.F., and M.O. wrote the manuscript.
Declaration of interests
J.C.-S., J.O., and K.H. are employees and shareholders of Synthego Corporation.
Published: June 27, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111088.
Supplemental information
Data and code availability
-
•
All data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Andrieu G.P., Denis G.V. BET proteins exhibit transcriptional and functional opposition in the epithelial-to-mesenchymal transition. Mol. Cancer Res. 2018;16:580–586. doi: 10.1158/1541-7786.mcr-17-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove D.A., Mahmoudi T., Henklein P., Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA. 2007;104:13690–13695. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K.L., Zhang F., Jaganathan A., Sharma R., Zhang Q., Konuma T., Shen T., Lee J.-Y., Ren C., Chen C.-H., et al. Distinct roles of Brd2 and Brd4 in potentiating the transcriptional program for Th17 cell differentiation. Mol. Cell. 2017;65:1068–1080.e5. doi: 10.1016/j.molcel.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant D., Hsiau T., Rossi N., Oki J., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., et al. Inference of CRISPR Edits from sanger trace data. CRISPR J. 2022;5:123–130. doi: 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
- Conrad R.J., Fozouni P., Thomas S., Sy H., Zhang Q., Zhou M.-M., Ott M. The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol. Cell. 2017;67:1001–1012.e6. doi: 10.1016/j.molcel.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rijck J., de Kogel C., Demeulemeester J., Vets S., El Ashkar S., Malani N., Bushman F.D., Landuyt B., Husson S.J., Busschots K., et al. The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell Rep. 2013;5:886–894. doi: 10.1016/j.celrep.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C., Carlson J.E., Zeng L., He C., Aggarwal A.K., Zhou M.M., Zhou M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.-P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T., et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd S.R., Pacold M.E., Huang Q., Clarke S.M., Lam F.C., Cannell I.G., Bryson B.D., Rameseder J., Lee M.J., Blake E.J., et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498:246–250. doi: 10.1038/nature12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gutierrez P., Mundi M., Garcia-Dominguez M. Association of bromodomain BET proteins with chromatin requires dimerization through the conserved motif B. J. Cell Sci. 2012;125:3671–3680. doi: 10.1242/jcs.105841. [DOI] [PubMed] [Google Scholar]
- Gilan O., Rioja I., Knezevic K., Bell M.J., Yeung M.M., Harker N.R., Lam E.Y.N., Chung C.-W., Bamborough P., Petretich M., et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. 2020;368:387–394. doi: 10.1126/science.aaz8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilham D., Smith A.L., Fu L., Moore D.Y., Muralidharan A., Reid S.P.M., Stotz S.C., Johansson J.O., Sweeney M., Wong N.C.W., et al. Bromodomain and extraterminal protein inhibitor, apabetalone (RVX-208), reduces ACE2 expression and attenuates SARS-cov-2 infection in vitro. Biomedicines. 2021;9:437. doi: 10.3390/biomedicines9040437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves D.C., Horng T., Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L., Li D., Sun X., Shi X., Karna P., Yang W., Liu M., Qiao W., Aneja R., Zhou J. Regulation of Tat acetylation and transactivation activity by the microtubule-associated deacetylase HDAC6. J. Biol. Chem. 2011;286:9280–9286. doi: 10.1074/jbc.m110.208884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javorsky A., Humbert P.O., Kvansakul M. Structural basis of coronavirus E protein interactions with human PALS1 PDZ domain. Commun. Biol. 2021;4:724. doi: 10.1038/s42003-021-02250-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H.P., Look D.C., Shi L., Hickey M., Pewe L., Netland J., Farzan M., Wohlford-Lenane C., Perlman S., McCray P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005;79:14614–14621. doi: 10.1128/jvi.79.23.14614-14621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose R.J., Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir. Med. 2020;8:e46–e47. doi: 10.1016/s2213-2600(20)30216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan R.E., Vanhulle C., Schiltz L., Adam E., Xiao H., Maudoux F., Calomme C., Burny A., Nakatani Y., Jeang K.-T., et al. HIV-1 Tat transcriptional activity is regulated by acetylation. EMBO J. 1999;18:6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N., Vollmer S., Nanda S.K., Lopez-Pelaez M., Prescott A., Gray N., Cohen P. Suppression of interferon β gene transcription by inhibitors of bromodomain and extra-terminal (BET) family members. Biochem. J. 2015;468:363–372. doi: 10.1042/bj20141523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R.J., Humphrey S.J., Fortuna P.R.J., Lor M., Foster S.R., Quaife-Ryan G.A., Johnston R.L., Dumenil T., Bishop C., Rudraraju R., et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell. 2021;184:2167–2182.e22. doi: 10.1016/j.cell.2021.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourão D., Chen S., Schaefer U., Bozzacco L., Carneiro L.A., Gerber A., Adura C., Dill B.D., Molina H., Carroll T., et al. A histone-like motif in yellow fever virus contributes to viral replication. bioRxiv. 2020 doi: 10.1101/2020.05.05.078782. Preprint at. [DOI] [Google Scholar]
- Nal B., Chan C., Kien F., Siu L., Tse J., Chu K., Kam J., Staropoli I., Crescenzo-Chaigne B., Escriou N., et al. Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E. J. Gen. Virol. 2005;86:1423–1434. doi: 10.1099/vir.0.80671-0. [DOI] [PubMed] [Google Scholar]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals . National Academies Press (US); 2011. Guide for the Care and Use of Laboratory Animals. [PubMed] [Google Scholar]
- Nieto-Torres J.L., Dediego M.L., Alvarez E., Jiménez-Guardeño J.M., Jiménez-Guardeño J.M., Regla-Nava J.A., Llorente M., Kremer L., Shuo S., Enjuanes L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology. 2011;415:69–82. doi: 10.1016/j.virol.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Torres J.L., DeDiego M.L., Verdiá-Báguena C., Jimenez-Guardeño J.M., Regla-Nava J.A., Fernandez-Delgado R., Castaño-Rodriguez C., Alcaraz A., Torres J., Aguilella V.M., Enjuanes L. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014;10:e1004077. doi: 10.1371/journal.ppat.1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olp M.D., Zhu N., Smith B.C. Metabolically derived lysine acylations and neighboring modifications tune the binding of the BET bromodomains to histone H4. Biochemistry. 2017;56:5485–5495. doi: 10.1021/acs.biochem.7b00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M., Schnölzer M., Garnica J., Fischle W., Emiliani S., Rackwitz H.R., Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999;9:1489–1493. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- Pagans S., Pedal A., North B.J., Kaehlcke K., Marshall B.L., Dorr A., Hetzer-Egger C., Henklein P., Frye R., McBurney M.W., et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M.C., Debrosse M., Smith M., Dey A., Huynh W., Sarai N., Heightman T.D., Tamura T., Ozato K. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell Biol. 2013;33:2497–2507. doi: 10.1128/mcb.01180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt G.M., Simpson G.R., Mittnacht S., Schulz T.F. Latent nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 1999;73:9789–9795. doi: 10.1128/jvi.73.12.9789-9795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y., Wang X.-M., Mannan R., Pitchiaya S., Zhang Y., Wotring J.W., Xiao L., Robinson D.R., Wu Y.-M., Tien J.C.-Y., et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA. 2020;118 doi: 10.1073/pnas.2021450118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S., Liu Y., Tempel W., Eram M.S., Bian C., Liu K., Senisterra G., Crombet L., Vedadi M., Min J. Structural basis for histone mimicry and hijacking of host proteins by influenza virus protein NS1. Nat. Commun. 2014;5:3952. doi: 10.1038/ncomms4952. [DOI] [PubMed] [Google Scholar]
- Rahman S., Sowa M.E., Ottinger M., Smith J.A., Shi Y., Harper J.W., Howley P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 2011;31:2641–2652. doi: 10.1128/mcb.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer U., Ho J.S.Y., Prinjha R.K., Tarakhovsky A. The “histone mimicry” by pathogens. Cold Spring Harb. Symp. Quant. Biol. 2013;78:81–90. doi: 10.1101/sqb.2013.78.020339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeman D., Fielding B.C. Coronavirus envelope protein: current knowledge. Virol. J. 2019;16:69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeman D., Fielding B.C. Is there a link between the pathogenic human coronavirus envelope protein and immunopathology? A review of the literature. Front. Microbiol. 2020;11:2086. doi: 10.3389/fmicb.2020.02086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder S., Cho S., Zeng L., Zhang Q., Kaehlcke K., Mak L., Lau J., Bisgrove D., Schnölzer M., Verdin E., et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012;287:1090–1099. doi: 10.1074/jbc.m111.282855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A., Larue R.C., Plumb M.R., Malani N., Male F., Slaughter A., Kessl J.J., Shkriabai N., Coward E., Aiyer S.S., et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA. 2013;110:12036–12041. doi: 10.1073/pnas.1307157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepley-McTaggart A., Sagum C.A., Oliva I., Rybakovsky E., DiGuilio K., Liang J., Bedford M.T., Cassel J., Sudol M., Mullin J.M., Harty R.N. SARS-CoV-2 Envelope (E) protein interacts with PDZ-domain-2 of host tight junction protein ZO1. PLoS One. 2021;16:e0251955. doi: 10.1371/journal.pone.0251955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard G.S., Wang L., Fidanze S.D., Hasvold L.A., Liu D., Pratt J.K., Park C.H., Longenecker K., Qiu W., Torrent M., et al. Discovery of N-Ethyl-4-[2-(4-fluoro-2, 6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2, 3-c]pyridine-2-carboxamide (ABBV-744), a BET bromodomain inhibitor with selectivity for the second bromodomain. J. Med. Chem. 2020;63:5585–5623. doi: 10.1021/acs.jmedchem.0c00628. [DOI] [PubMed] [Google Scholar]
- Stratton M.S., Haldar S.M., McKinsey T.A. BRD4 inhibition for the treatment of pathological organ fibrosis. F1000Res. 2017;6 doi: 10.12688/f1000research.11339.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y. The bromodomain and extra-terminal domain (BET) family: functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016;17:1849. doi: 10.3390/ijms17111849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienerroither S., Rauch I., Rosebrock F., Jamieson A.M., Bradner J., Muhar M., Zuber J., Müller M., Decker T. Regulation of NO synthesis, local inflammation, and innate immunity to pathogens by BET family proteins. Mol. Cell Biol. 2014;34:415–427. doi: 10.1128/mcb.01353-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-Y., Lee A.-Y., Hou S.Y., Kemper J.K., Erdjument-Bromage H., Tempst P., Chiang C.-M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J., Srinivasan V., Denis G.V., Harrington W.J., Jr., Ballestas M.E., Kaye K.M., Howley P.M. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 2006;80:8909–8919. doi: 10.1128/jvi.00502-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/nejmoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.