

Abstract
Femtosecond laser pulses readily produce coherent quantum beats in transient–absorption spectra. These oscillatory signals often arise from molecular vibrations and therefore may contain information about the excited-state potential energy surface near the Franck–Condon region. Here, by fitting the measured spectra of two laser dyes to microscopic models of femtosecond coherence spectra (FCS) arising from molecular vibrations, we classify coherent quantum-beat signals as fundamentals or overtones and quantify their Huang–Rhys factors and anharmonicity values. We discuss the extracted Huang–Rhys factors in the context of quantum-chemical computations. This work solidifies the use of FCS for analysis of coherent quantum beats arising from molecular vibrations, which will aid studies of molecular aggregates and photosynthetic proteins.
The topography of an excited-state potential-energy surface strongly influences the photophysical, photochemical, and photobiological properties of a molecule or molecular system,1,2 thereby impacting the development of promising device applications such as light harvesting3 or fluorescence sensing.4 Electronic spectra contain information regarding the topography of excited-state potential-energy surfaces,5,6 in particular, information about the intramolecular vibrational modes of a molecule and the coupling of those modes to the electronic transition.7,8 This information can—in principle and in practice—be extracted from measured spectra, especially near the Franck–Condon region of the potential-energy surface. Several groups, for example, have used computations and models in conjunction with steady-state electronic spectra to extract coupling parameters.9,10 In another example, Lawless and Mathies used resonance Raman spectra to find the normalized displacement, which is related to the Huang–Rhys factor,11,12 of each Franck–Condon active vibrational mode in Nile blue.13 In addition, Lee et al. extracted the reorganization energy, which is also related to the Huang–Rhys factor, of coumarin 153 from time-resolved fluorescence spectra.14 Other time-resolved spectroscopy techniques have provided similar information,15−20 and each method has benefits and limitations.
For femtosecond transient-absorption spectroscopy, researchers have measured quantum-beat signals arising from vibrational wavepackets since the 1990s,21,22 and these wavepackets are expected to contain topographical information about the excited electronic state near the Franck–Condon region. Photosynthetic pigment–protein complexes,23−25 retinal pigment–protein complexes,26−34 and laser dye molecules35−43 have been measured in detail, and other measured systems include heme proteins,44 phytochrome pigment–protein complexes,45−47 carbon nanotubes,48 charge–transfer systems,49−52 polymers,53 perovskite materials,54 and molecular aggregates and crystals.55−58 Further reports analyzed how pulse chirp and temperature affect the measurement of vibrational quantum beats.59−63 Finally, a variety of theoretical studies have aided the interpretation of the curious phase and amplitude profiles that often characterize vibrational quantum-beat signals.64−70
Despite these efforts, quantitative information is not routinely
extracted from measured vibrational quantum-beat signals in transient-absorption
spectra, other than the assessment of the presence or absence of oscillations
at a particular frequency. Therefore, in this contribution, we fit
measured quantum-beat signals to our recently developed microscopic
vibrational models of femtosecond coherence spectra (FCS),69,70 which we define as the Fourier-domain amplitude and phase profiles, A(ω) and ϕ(ω), respectively, as a function
of detection frequency, ω, for a chosen oscillation frequency,
ω0. We denote the measured spectrally resolved TA
data set as S(ω,τ2), where
τ2 is the pump–probe time interval and ω
is the detection frequency axis. After Fourier transformation of the
data set over the pump–probe time interval, the resultant complex-valued
spectrum can be denoted as M(ω, ω2) = 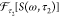 , where ω2 is the oscillation
frequency axis resulting from Fourier transformation. Some previous
works use FCS to refer to the spectrally integrated signal (1D vibronic
spectrum), A(ω2) = ∫|M(ω,ω2)|dω. Our usage of FCS
refers to the amplitude or phase profile of a selected oscillation
frequency, ω0, as a function of detection frequency, A(ω) = M(ω, ω2)|ω2 = ω0. Most FCS contain one or more sharp
amplitude nodes and abrupt phase shifts, which can be envisioned in
the 1D displaced harmonic oscillator model as arising from the features
of the vibrational eigenfunctions.69 Some
previous authors on some occasions have used FCS or the related terms
vibrational coherence spectroscopy (VCS) and broadband impulsive vibrational
spectroscopy (BB-IVS), among others, to refer to 1D vibronic spectrum
showing the amplitude of peaks as a function of oscillation frequency.31,38,44,67,71,72 Although in
principle the amplitude of each peak in a 1D vibronic spectrum is
directly related to the Huang–Rhys factor of the mode, the
amplitude of a peak is also affected by an attentuation envelope arising
from the pulse spectrum, the spectral phase of the pulse, and the
detection wavelengths over which the signal was summed.39,59−61 In this work, we normalize each mode before analyzing
its FCS—the amplitude profile as a function of detection frequency—to
characterize the anharmonicity and Huang–Rhys factor.
, where ω2 is the oscillation
frequency axis resulting from Fourier transformation. Some previous
works use FCS to refer to the spectrally integrated signal (1D vibronic
spectrum), A(ω2) = ∫|M(ω,ω2)|dω. Our usage of FCS
refers to the amplitude or phase profile of a selected oscillation
frequency, ω0, as a function of detection frequency, A(ω) = M(ω, ω2)|ω2 = ω0. Most FCS contain one or more sharp
amplitude nodes and abrupt phase shifts, which can be envisioned in
the 1D displaced harmonic oscillator model as arising from the features
of the vibrational eigenfunctions.69 Some
previous authors on some occasions have used FCS or the related terms
vibrational coherence spectroscopy (VCS) and broadband impulsive vibrational
spectroscopy (BB-IVS), among others, to refer to 1D vibronic spectrum
showing the amplitude of peaks as a function of oscillation frequency.31,38,44,67,71,72 Although in
principle the amplitude of each peak in a 1D vibronic spectrum is
directly related to the Huang–Rhys factor of the mode, the
amplitude of a peak is also affected by an attentuation envelope arising
from the pulse spectrum, the spectral phase of the pulse, and the
detection wavelengths over which the signal was summed.39,59−61 In this work, we normalize each mode before analyzing
its FCS—the amplitude profile as a function of detection frequency—to
characterize the anharmonicity and Huang–Rhys factor.
Here we analyze the measured FCS of representative Franck–Condon
active modes for two laser dyes to demonstrate the utility and applicability
of the methodology. By evaluating selected vibrational modes having
a variety of amplitudes and profiles, this fitting and analysis of
measured spectra serve as an experimental validation of our previous
theory work, and it demonstrates how quantitative parameters could
be obtained. We show that the overall shape of the FCS profile allows
us to classify the signal as arising from a fundamental, overtone,
or combination-band mode, and we show that the fits to the models
can yield reasonable parameter values. Central to the analysis is
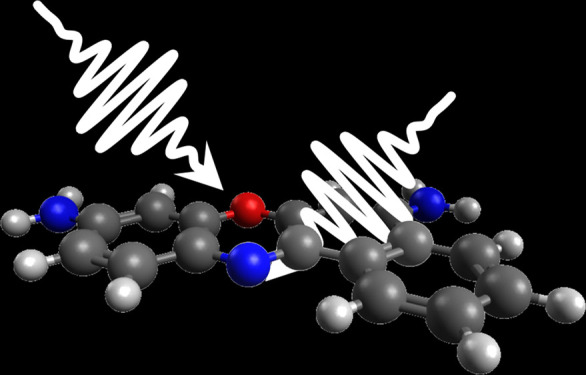
the ubiquitous displaced harmonic oscillator model shown in Figure 1a. In this model,73 both the ground (S0) and excited (S1) electronic states
are harmonic potentials of frequency ω0, offset vertically
by energy ℏωeg, and offset
horizontally by a displacement Δ, which—after normalization
relative to x0, the classical turning
point of the lowest-energy eigenfunction, Δ̃ = Δ/x0—is related to the Huang–Rhys
factor by 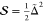 . The horizontal axis, q, is the displacement coordinate
of one specific normal mode.
. The horizontal axis, q, is the displacement coordinate
of one specific normal mode.
Figure 1.

(a) Displaced harmonic oscillator model of molecular
spectroscopy
is characterized by vibrational frequency (ω0), Huang–Rhys
factor (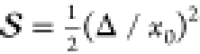 ), and electronic energy shift (ℏωeg). (b) Measured vibrational quantum beat
signals (top) can be Fourier transformed to produce femtosecond coherence
spectra (FCS) for each mode (middle), which can then be classified
as a fundamental, overtone, or combination-band mode and then fit
using the appropriate model. (bottom) The fit yields the parameters
), and electronic energy shift (ℏωeg). (b) Measured vibrational quantum beat
signals (top) can be Fourier transformed to produce femtosecond coherence
spectra (FCS) for each mode (middle), which can then be classified
as a fundamental, overtone, or combination-band mode and then fit
using the appropriate model. (bottom) The fit yields the parameters 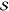 and ωeg, as well as the electronic dephasing (γ).
and ωeg, as well as the electronic dephasing (γ).
Previously we used a doorway–window method
to develop analytic
FCS expressions in terms of the normalized displacement.70 As a simplification and generalization, here
we express the fundamental (p = 1) and overtone (p = 2) FCS, respectively, directly in terms of the Huang–Rhys
factor, 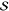 , as
, as
| 1 |
where the auxiliary functions are given by
| 2 |
In these expressions, m and n index the vibrational eigenstates of the
ground and excited
electronic states, respectively; ωa,b = (Ea – Eb)/ℏ,
which implicitly includes vertical offset ωeg ; γ is a universal phenomenological dephasing parameter
for the emitted optical coherence signals; and ω is the detection
frequency variable. When fitting measured spectra, ω arises
from the calibrated detector and ω0 is given directly
from the measured oscillation frequency of each quantum beat, and
therefore, the fit parameters are limited to only the set 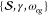 . As discussed previously for the harmonic
model,69 when p = 1 (p = 2), the amplitude profile is typically characterized
by two (three) peaks separated by one (two) node(s) in the amplitude
FCS profiles; the profiles can be interpreted as arising from the
nodes in the n = 1 or n = 2 vibrational
eigenfunctions for fundamental and overtones, respectively; in the
case of fundamental modes, the two peaks have at most a ∼20%
difference in amplitude.
. As discussed previously for the harmonic
model,69 when p = 1 (p = 2), the amplitude profile is typically characterized
by two (three) peaks separated by one (two) node(s) in the amplitude
FCS profiles; the profiles can be interpreted as arising from the
nodes in the n = 1 or n = 2 vibrational
eigenfunctions for fundamental and overtones, respectively; in the
case of fundamental modes, the two peaks have at most a ∼20%
difference in amplitude.
We then developed a related model in
which the ground-state and
excited-state potentials are displaced and harmonic but have distinct
curvatures. Section S2 of the Supporting Information contains the details. We use an analytic, albeit complicated, expression
for the appropriate Franck–Condon coefficients73 recast in a dimensionless unit system amenable for fitting
measured spectra. The Franck–Condon coefficients can then be
inserted into the generic FCS expression. The result is that the fit
parameters for this unequal-curvature harmonic model include 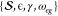 . The parameter ϵ
controls the relative
frequencies, ω0g = ϵ2ω0e, where we fix ω0e to be the measured oscillation frequency. Simulations
shown in the Supporting Information reveal
that ϵ ≠ 0 induces modest additional asymmetry to the
two main peaks.
. The parameter ϵ
controls the relative
frequencies, ω0g = ϵ2ω0e, where we fix ω0e to be the measured oscillation frequency. Simulations
shown in the Supporting Information reveal
that ϵ ≠ 0 induces modest additional asymmetry to the
two main peaks.
To quantify the anharmonicity, we also developed FCS expressions based on the displaced Morse oscillator model.70 This model introduced parameter λ, an integer representing the number of bound vibrational eigenstates in each electronic potential, which we use as a proxy for the inverse anharmonicity. This anharmonic model can produce FCS amplitude profiles in which the peaks have extremely different amplitudes. In contrast to the relatively trim FCS expression of the harmonic model, the FCS expression we derived previously for the anharmonic model was a function of both the emission frequency variable (ω) and the oscillation frequency variable (ω0). This prevented rapid fitting of measured spectra. Therefore, here we derived an FCS expression of the anharmonic model as a function of only ω by assuming that the wavepacket is composed primarily of the two lowest-energy vibrational eigenstates in the excited electronic state. In an anharmonic potential, this is the only combination of vibrational eigenstates that will produce oscillations at the fundamental oscillation frequency relevant to this FCS profile, ω2 = ω0,1. This assumption is appropriate when the normalized displacement is relatively small, Δ̃ ≲ 1, which is widely applicable to molecular systems.8,13,14 The expression for ω2 = ω0,1 is
| 3 |
where the complicated auxiliary
function, AMO(λ, Δ̃, m) is given in section S1 of the Supporting Information. Even though eq 3 arises
from assuming the excited-state wavepacket is composed of only two
vibrational eigenfunctions, all ground-state vibrational eigenfunctions m are included, and therefore, the FCS profile can still
be composed of many overlapping Lorentzian terms. Converting the normalized
displacement into an effective Huang–Rhys factor, 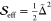 , the fit parameters for
the anharmonic
FCS model are
, the fit parameters for
the anharmonic
FCS model are 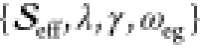 . Using this convenient
conversion requires
some caution because—in contrast to the harmonic models—in
the anharmonic model, the FCS arising from Δ̃ < 0 and
Δ̃ > 0 are distinct.
. Using this convenient
conversion requires
some caution because—in contrast to the harmonic models—in
the anharmonic model, the FCS arising from Δ̃ < 0 and
Δ̃ > 0 are distinct.
Physically, eqs 1 and 3 demonstrate that each model yields a collection of Lorentzian peaks, each peak is broadened by γ and weighted by an auxiliary function, and the peaks overlap to yield lineshapes akin to those illustrated in the middle of panel b in Figure 1. A primary aim of this work is to evaluate the utility of these one-dimensional FCS models using measured spectra. An ideal prototype molecule would
completely lack nonradiative decay mechanisms and consequently have a high fluorescence quantum yield;
have a Stokes shift large enough to separate excited-state wavepacket signals from ground-state wavepacket signals;
have absorption and fluorescence spectra in a wavelength range that is convenient for the femtosecond spectrometer;
lack Duschinsky mixing,74 which is a relative rotation between multidimensional ground-state and excited-state potential-energy surfaces, so that one-dimensional models are adequate.
One such sample seems to be rhodamine 101. Rhodamine dyes are characterized by a xanthene moiety serving as the chromophore and a benzoic-acid moiety linked at the ortho position with respect to the location of the oxygen atom in the xanthene’s central ring (see the structure in panel a of Figure 2). Rhodamine 101 has an excited-state lifetime in methanol of 4 ns75 and a reported fluorescence quantum yield of 99% in methanol over a wide temperature range.76 In methanol, rhodamine 101 has absorption and fluorescence maxima of 567 and 588 nm, respectively, which are well within the range of the ultrabroadband laser pulses produced by our femtosecond laser system. Furthermore, these values indicate a Stokes shift of 19 THz (630 cm–1), which should be adequate to distinguish between coherent wavepackets on the excited and ground electronic states. The ground-state wavepackets should be suppressed because the transition-dipole moment is likely to be coordinate-independent and the ultrabroadband laser pulses encompass the absorption spectrum.60,65,68 We found no publications reporting Duschinsky mixing in rhodamine 101. In addition to several weak peaks, the steady-state Raman spectrum displays moderate or strong peaks at 1648, 1508, 1360, and 1347 cm–1 (49.5, 45.3, and 40.5 THz) arising from aromatic C–C stretches in the xanthene moiety, and a moderate-intensity peak at 760 cm–1 (22.8 THz) arising from a C–H out-of-plane bending mode.77 Prior femtosecond transient–absorption measurements have identified multiple coherent vibrational oscillations in rhodamine 101;25,39 however, the researchers did not produce FCS profiles or compare them to a model.
Figure 2.
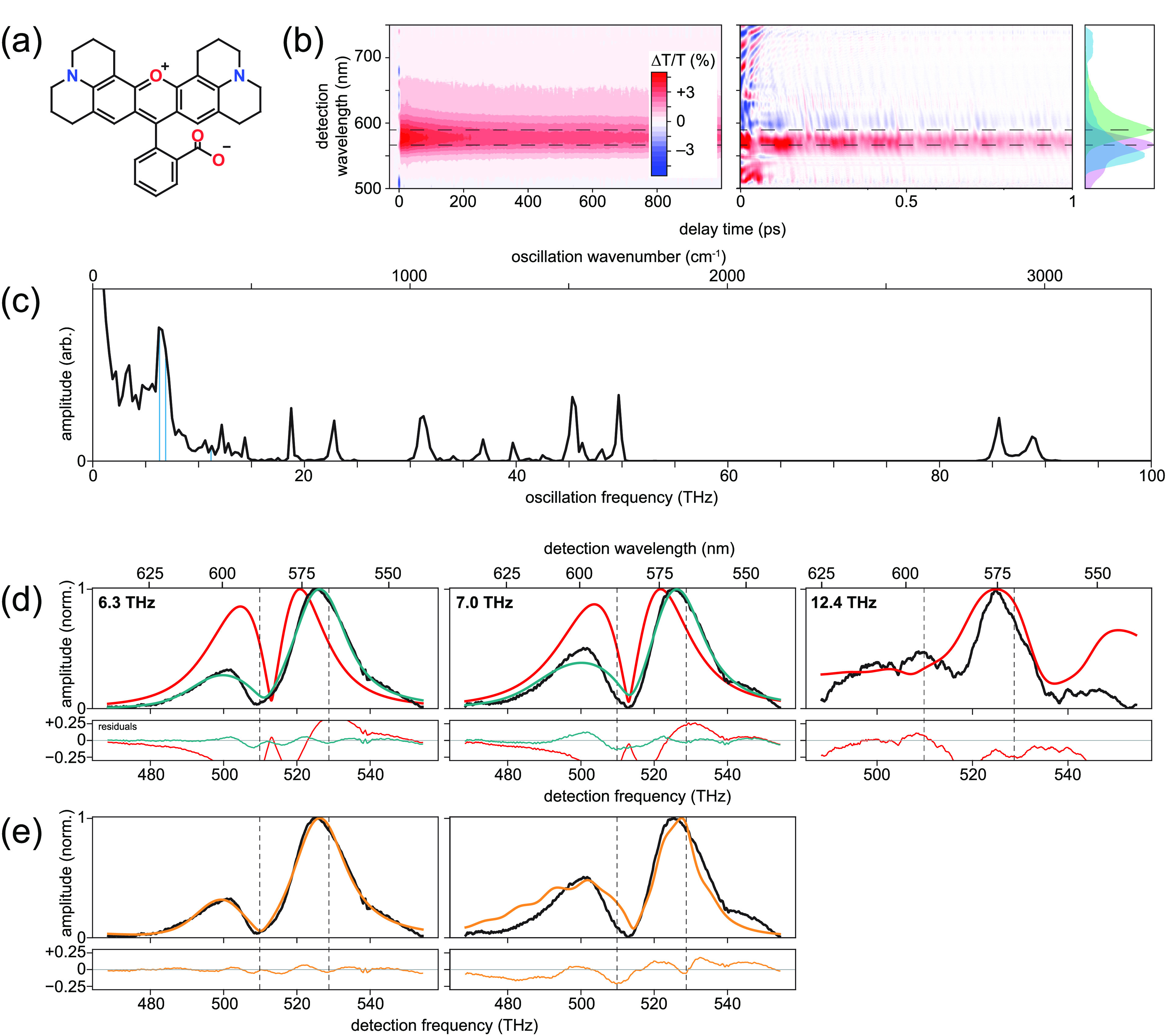
Results for rhodamine 101. (a) Molecular structure. (b) Measured transient-absorption spectra, with population decays subtracted from the 1 ps data set view. Absorption and fluorescence spectra displayed in violet and green, respectively, along with the laser pulse spectrum in cyan. (c) 1D vibronic spectrum. Vertical blue lines indicate peaks to be analyzed with FCS. (d) Measured (black) and fitted harmonic (red) and anharmonic (dark cyan) FCS amplitude profiles at indicated oscillation frequencies. (e) Measured (black) and unequal-curvature harmonic (orange) FCS amplitude profiles at indicated frequencies. Dashed lines in panels b, d, and e indicate absorption and fluorescence maxima.
Panel b in Figure 2 displays measured transient–absorption spectra on 1 ns and 1 ps time scales, the latter revealing the vibrational quantum beats as oscillatory signals that appear in addition to the conventional, slowly decaying ground-state bleach and stimulated emission signals. No excited-state absorption signals appear in this spectral region. Subtraction of the slowly decaying signals, Fourier transformation, and subsequent summation over all detection frequencies lead to the 1D vibronic spectrum displayed in panel c, which reveals the oscillation frequencies of the vibrational modes. There are 16 peaks above the noise: 6.3, 7.0, 10.7, 12.4, 12.9, 14.2, 18.8, 22.9, 31.4, 34.2, 36.8, 39.8, 45.4, 49.8, 85.6, and 88.8 THz. These values are largely consistent with the peak locations that can be estimated from Figure 8 in ref (39), and the peaks above 20 THz are also largely consistent with peaks in the steady-state Raman spectrum.77 The solvent, methanol, gives rise to the peaks near 31, 85, and 88 THz.90
To evaluate the applicability and utility of the FCS models, we focus on three modes: 6.3, 7.0, and 12.4 THz (210, 233, and 413 cm–1). Specifically, we study the 7.0 THz mode because it is the highest amplitude, and we chose the 6.3 and 12.4 THz modes because the latter is at a frequency indicating it is potentially the overtone of the former, and careful inspection of Figure 8 from ref (39) reveals profiles for the 6.3 and 12.4 THz peaks that fit this anticipated classification. The measured FCS amplitude profiles (see black traces in panel d in Figure 2) reveal that the 6.3 and 7.0 THz modes have the two-peak/single-node profile of fundamental vibrational modes, while the 12.4 THz mode has the three-peak/two-node profile of an overtone. Despite the noise, the three-peak/two-node profile of the 12.4 THz profile was qualitatively distinct and was repeatable over multiple measurements. Consistent with these classifications is that the 12.4 THz mode has a frequency approximately double that of the 6.3 THz peak, given the estimated error of ±0.1 THz.
Having classified the three modes, we then fit each measured amplitude
profile to the FCS expression for the harmonic model (red traces in
panel d of Figure 2). While the fits do reproduce the main features of the measured
FCS profiles, the harmonic column of Table 1 reveals that the coefficients of determination, R2, are very poor, ranging from about 0.06 to
0.35, and the extracted Huang–Rhys values seem a bit high compared
to those of other molecules, which ranged from less than 0.01 to 0.4.8,13,14 Section S3 of the Supporting Information contains full fit results.
Because relative peak heights that are more than about 20% asymmetric
cannot be obtained with the harmonic FCS model, the quantitatively
poor fits are expected for the 6.3 THz and 7.0 THz modes. One could
hypothesize additional asymmetry even in a harmonic model from temperature
effects because the model assumes zero temperature, while we performed
the measurements at room temperature. However, Kumar and Champion
showed that even for a mode at 1.2 THz, the effects of this change
in temperature on the amplitude profiles are minor.67 In contrast to the harmonic model, simulations using the
anharmonic model have demonstrated the ability to have extremely asymmetric
peak heights.70 We therefore fit the FCS
profiles of the 6.3 and 7.0 THz modes to the anharmonic FCS model
(dark cyan traces in panel d of Figure 2). The fit errors of the 6.3 and 7.0 THz modes improved
significantly, having R2 values >0.95.
Qualitatively, the anharmonic fits reproduce the high-frequency peaks
at about 530 THz well but struggle to reproduce adequately the depth
of the nodes and the height of the lower-frequency peaks at about
500 THz. For simpler comparison between the models, we converted the
normalized displacement arising from the anharmonic model into an
effective Huang–Rhys factor, 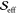 , using
, using 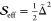 . The effective Huang–Rhys
factors
of about 1.6 and 1.2 for the 6.3 and 7.0 THz modes, respectively,
again seem too high relative to the previous reports. Unfortunately,
significant geometric changes of the benzoic acid moiety for S0 and S1 frustrated
efforts to compare the Huang–Rhys values to those arising from
quantum-chemical computations. Section S3 of the Supporting Information contains details of these attempted
quantum-chemical computations. The failure of the calculations reinforces
the need for an alternative pathway to this information. Finally,
we present the anharmonicity of the modes in Table 1 both in terms of λbest as
well as the more conventional anharmonicity value, χ, where
the two are related by χ = ω0 /(2 λbest) .
. The effective Huang–Rhys
factors
of about 1.6 and 1.2 for the 6.3 and 7.0 THz modes, respectively,
again seem too high relative to the previous reports. Unfortunately,
significant geometric changes of the benzoic acid moiety for S0 and S1 frustrated
efforts to compare the Huang–Rhys values to those arising from
quantum-chemical computations. Section S3 of the Supporting Information contains details of these attempted
quantum-chemical computations. The failure of the calculations reinforces
the need for an alternative pathway to this information. Finally,
we present the anharmonicity of the modes in Table 1 both in terms of λbest as
well as the more conventional anharmonicity value, χ, where
the two are related by χ = ω0 /(2 λbest) .
Table 1. Fit Results of Rhodamine 101 for the Three FCS Modelsa.
| harmonic |
anharmonic |
unequal
harmonic |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| ω0/(2π) | 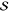 |
R2 | 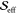 |
λbest | χ | R2 | 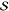 |
ϵ | R2 |
| 6.3 | 0.45 | 0.056 | 1.62 | 11 | 0.29 | 0.988 | 0.14 | 2.10 | 0.991 |
| 7.0 | 0.41 | 0.354 | 1.22 | 11 | 0.32 | 0.963 | 0.70 | 1.15 | 0.865 |
| 12.4 | 0.93 | 0.076 | |||||||
All values are dimensionless except ω0 and χ, which are in units of THz.
Using the unequal-curvature
harmonic model, we fit the measured
spectra of the two fundamental modes of interest for rhodamine 101. Table 1 contains the fit
results, and panel e in Figure 2 contains the fitted FCS. The qualitative aspects of the 6.3
THz mode fit are compelling, and the R2 value of 0.991 confirms that the fit using this model is superior
to the other two models. However, the best-fit value for ϵ is
2.10, indicating that the frequency of the ground-state potential
is more than 4× higher than the excited-state potential. Because
the value of ϵ was so large, we hypothesized this would lead
to additional peaks at lower or higher frequencies. Figure S2 in the Supporting Information contains the plot of the
fitted FCS extended across a much larger detection-frequency window.
It reveals additional peaks at lower frequencies, notably one at about
445 THz (675 nm). Although these predicted peaks are outside our detection
window, the spectrum presented in Figure 8 of ref (39) does have a peak near
the predicted detection frequency, providing tentative support of
the fit result, which at first glance seemed unreasonable. One possible
explanation is Duschinsky mixing.74 One
can envision that the projection of an  -dimensional
potential-energy model having
rotation between the ground and excited states onto a single coordinate
would indeed make it appear as if the frequencies of the two electronic
states were different.
-dimensional
potential-energy model having
rotation between the ground and excited states onto a single coordinate
would indeed make it appear as if the frequencies of the two electronic
states were different.
For the 7.0 THz mode, the fit using the unequal-curvature harmonic model is significantly better than the harmonic model, but inspection of the FCS shape reveals several discrepancies for both peaks and the node. The anharmonic model provided the best fit, although the effective Huang–Rhys factor of 1.22 remained higher than anticipated.
Although rhodamine 101 seemed to have the photophysical characteristics of an ideal test molecule, the lack of computational results prevents confirmation of the unexpectedly large parameter values obtained from the fits to the models. We therefore turn to a second laser dye, cresyl violet, that has many of the ideal photophysical characteristics and is amenable to quantum-chemical computations of its vibronic modes. The structure of cresyl violet is depicted in panel a of Figure 3. Cresyl violet is one member of the 1,4-oxazine family of dyes, which have been used in dye lasers and as fluorescent labels78−82 due in part to their favorable spectroscopic properties. Cresyl violet is a rigid, planar molecule that has four amine hydrogens. This molecule has become a standard for development of ultrafast laser spectrometers for detection of coherent quantum-beat signals.83−86 A notable feature of the members of this dye family is an extremely strongly coupled vibronic mode having a frequency of about 17.7 THz (590 cm–1).21 Measurements and analysis have generated two competing hypotheses regarding the strong vibronic mode. One hypothesis is that the mode is extremely harmonic.43,87 A second hypothesis is that the molecule is subject to nonadiabatic dynamics,42,88 and the mode is, therefore, anharmonic in the adiabatic basis.89 Here we can use the FCS models to test these hypotheses: The nonadiabatic hypothesis is supported if the anharmonic model fits the measured FCS better than both of the harmonic models. In addition, we study the 22.0 THz mode because it had the most equal-amplitude peaks representing a weak Huang–Rhys factor, and we selected the 15.6 THz because it has a similar profile as the 17.6 THz mode but without the literature attention.
Figure 3.
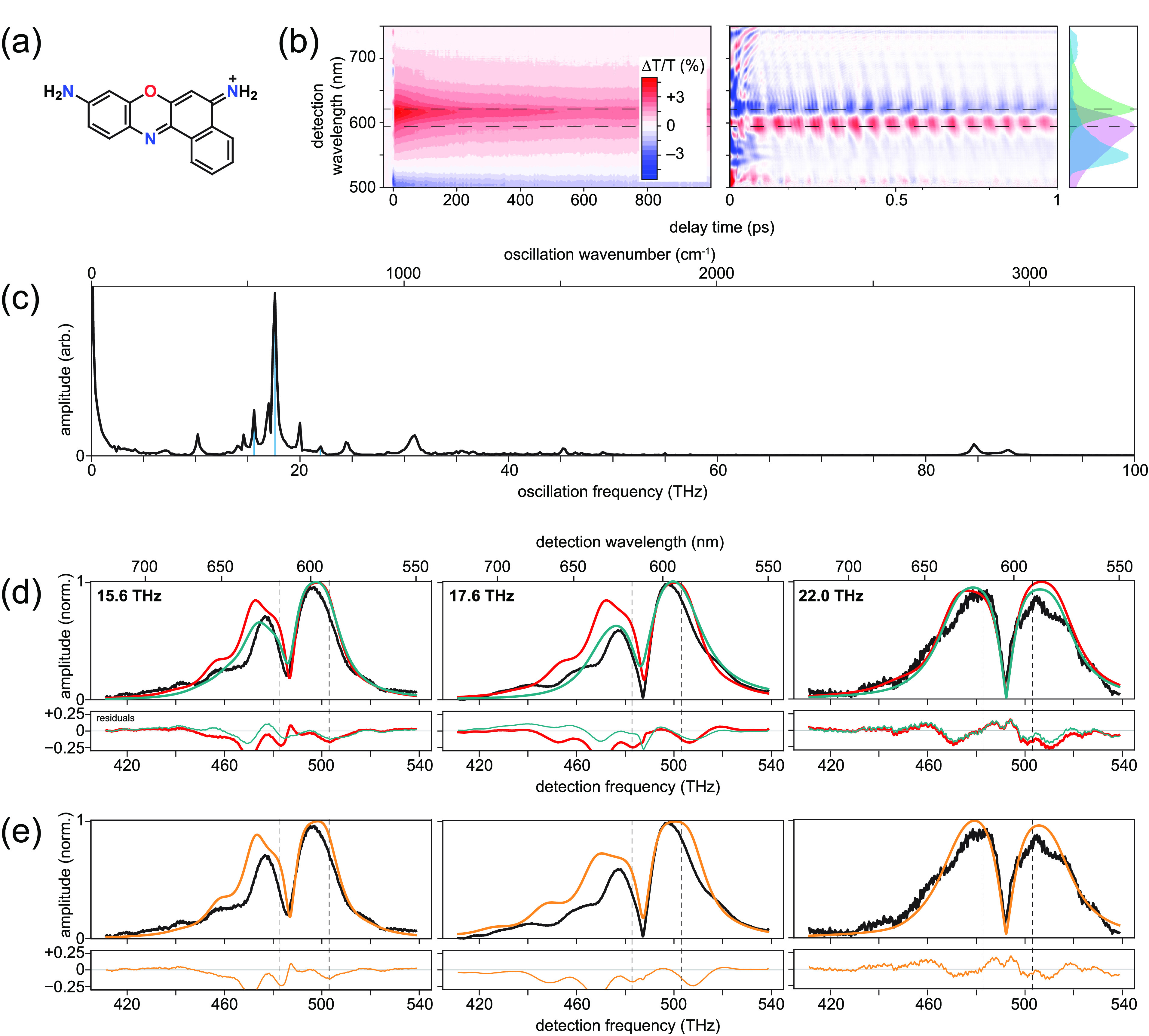
Results for cresyl violet. (a) Molecular structure. (b) Measured transient-absorption spectra, with population decays subtracted from the 1 ps data set view. Absorption and fluorescence spectra displayed in violet and green, respectively, along with the laser pulse spectrum in cyan. (c) 1D vibronic spectrum. Vertical blue lines indicate peaks to be analyzed with FCS. (d) Measured (black) and fitted harmonic (red) and anharmonic (dark cyan) FCS amplitude profiles at indicated oscillation frequencies. (e) Measured (black) and unequal-curvature harmonic (dashed orange) FCS amplitude profile at indicated frequencies. Dashed lines in panels b, d, and e indicate absorption and fluorescence maxima.
Panel b in Figure 3 displays measured transient–absorption spectra on 1 ns and 1 ps time scales, the latter revealing the vibrational quantum beats as oscillatory signals that appear in addition to the conventional, slowly decaying ground-state bleach and stimulated emission signals. An excited-state absorption band appears at detection wavelengths below about 510 nm (588 THz). Subtraction of the slowly decaying signals, Fourier transformation, and subsequent summation over all detection frequencies lead to the 1D vibronic spectrum displayed in panel c, which reveals the oscillation frequencies of the vibrational modes. There are 14 peaks above the noise: 7.2, 10.2, 14.6, 15.6, 17.0, 17.6, 20.0, 22.0, 24.4, 31.0, 45.2, 49.0, 84.6, and 87.6 THz. These values are consistent with prior spectroscopic measurements of cresyl violet,83−85 and again the peaks near 31, 85, and 88 THz arise from methanol.
Parallel to the rhodamine 101 analysis, we focus on three representative modes. The measured FCS displayed in black traces in panel d in Figure 3 reveal that all three modes have the two-peak/single-node profile of fundamental vibrational modes. We therefore first apply fits using the harmonic model (red traces in panel d of Figure 3). The fits detailed in Table 2 have R2 ranging from 0.7 to over 0.9 and yielded extracted Huang–Rhys factors of 0.25, 0.24, and 0.01 for the 15.6, 17.6, and 22.0 THz modes, respectively. These values seem reasonable when compared to values of similar molecules;13,14 however, the fits of the 15.6 and 17.6 THz modes struggled to reproduce the low-frequency peaks at about 470 THz. The results of the unequal-curvature harmonic model were almost identical to the harmonic model—the ϵ values were very nearly unity; hence, we do not evaluate this model further. Finally, we apply fits using the anharmonic model. This model improved the results for the 15.6 and 17.6 THz modes and produced negligible changes for the 22.0 THz mode. The R2 values for all modes rise to above 0.9, and the effective Huang–Rhys parameters extracted were, respectively, 0.32, 0.15, and 0.20. These values also seem plausible.
Table 2. Fit Results of Cresyl Violet for the Three FCS Modelsa.
| harmonic |
anharmonic |
unequal
harmonic |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| ω0/(2π) | 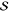 |
R2 | 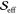 |
λbest | χ | R2 | 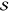 |
ϵ | R2 |
| 15.6 | 0.25 | 0.855 | 0.32 | 5 | 1.56 | 0.947 | 0.27 | 0.98 | 0.856 |
| 17.6 | 0.24 | 0.710 | 0.15 | 2 | 4.4 | 0.919 | 0.22 | 1.09 | 0.718 |
| 22.0 | 0.01 | 0.946 | 0.20 | 28 | 0.4 | 0.951 | 0.00 | 0.95 | 0.955 |
All values are dimensionless
except ω0 and χ, which are in units of THz.
The value of 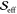 for the 22.0 THz mode arises from a negative
for the 22.0 THz mode arises from a negative 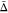 value.
value.
Because of the planarity of the cresyl violet molecule and its modest S0–S1 geometry changes, quantum-chemical calculations of the vibronic modes were more successful than those for rhodamine 101. Section S4 of Supporting Information contains further details. Briefly, of the 11 nonsolvent peaks observed in the 1D vibronic spectrum, 10 appear in the computations and their frequencies agree to within <10% of the measured frequencies. The very weak 22.0 THz mode did not appear in the computations, and we analyze it further below. The salient results are that the computed Huang–Rhys factors were 0.07 and 0.28 for the 15.6 and 17.6 THz modes, respectively. While quantitatively there is some disagreement between values extracted from the measurements and the computational results, the general trend is the same.
Comparing the fit results between the harmonic and anharmonic models suggests that the 22.0 THz mode is harmonic and 15.6 and 17.6 THz have some anharmonicity. To bolster this confirmation, we can inspect R2(λ), which is the coefficient of determination at each value of λ for the anharmonic model. Figure 4 displays the R2(λ) results for both molecules. In the case of a very harmonic potential, there should be negligible variation in the coefficient of determination for values of λ above a certain threshold. In contrast, an anharmonic potential should yield fits that are worse both above and below a preferred λ value. In Figure 4, the R2(λ) curves for both modes of rhodamine have maxima near λ ≈ 10, supporting the earlier assessment that these modes have significant anharmonicity. In sharp contrast to these maxima, the R2(λ) curve for the 22.0 THz mode of cresyl violet is essentially flat, having no visible maximum. The R2(λ) data sets for the 15.6 THz mode of cresyl violet displays a weak maximum at λ = 5. Finally, the R2(λ) data set for the 17.6 THz mode of cresyl violet shows a maximum at λ = 2. These data for cresyl violet support the conclusion that the 22.0 THz mode is harmonic and that the 15.6 and 17.6 THz modes have anharmonicity.
Figure 4.

Coefficient of determination dependence as a function of anharmonicity, R2(λ), for two modes of rhodamine 101 (green) and three modes of cresyl violet (black/gray). The 22.0 THz mode of cresyl violet is essentially flat, indicating no perferred value of λ.
These results contribute to the debate surrounding the strongly coupled vibronic mode (17.6 THz) of cresyl violet, using the 22.0 THz mode as an internal control. The asymmetric peak heights in the FCS profile of the 17.6 THz mode suggest that the harmonic models will fail, and indeed, these models struggle both qualitatively and quantitatively to fit the measured spectrum. In contrast, the relative symmetry of the two peaks in the FCS profile of the 22.0 THz mode suggest that the harmonic models will fit well, and this is borne out both qualitatively and quantitatively. The anharmonic model yields essentially no improvement to the fit of the 22.0 THz mode, whereas this model yields a substantial improvement for the 17.6 THz mode. Further, the R2(λ) function has a maximum at a low value of λ = 2. Taken together, these observations are inconsistent with the hypothesis that the 17.6 THz mode is strongly harmonic and support the hypothesis that this mode has significant anharmonicity. More broadly, the data presented here indicate that anharmonicity plays a key role in most of the analyzed vibrational modes. Hence, while the displaced harmonic oscillator model is very useful for pedagogical and qualitative understanding of vibrational spectroscopy signals, it seems inadequate for quantitative analysis of vibrational quantum beats in femtosecond transient–absorption spectra, which may require an anharmonic analysis such as the Morse oscillator model derived and used in this work.
In conclusion,
we have refined the harmonic and anharmonic (Morse
oscillator) models of FCS profiles arising from vibrational quantum
beats, and we have developed a new harmonic model for the case of
the ground and excited states having distinct curvatures. The harmonic
models of fundamentals and overtones include explicit dependence on
the Huang–Rhys factor, 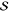 , and
conventional anharmonicity, χ.
These models are useful for studying excited-state vibrational modes
and, in particular, low-frequency excited-state vibrational modes
that are challenging to study with frequency-domain vibrational spectroscopy
methods such as Raman spectroscopy. The data and analyses of two laser
dye samples demonstrate that combined use of the harmonic and anharmonic
FCS models can provide qualitative insights into the vibrational quantum
beats and the topography of the excited-state potential energy surface
near the Franck–Condon region. The extracted Huang–Rhys
values for three modes of cresyl violet were somewhat consistent with
values produced by quantum-chemical calculations, which were restricted
to the harmonic approximation. The key observable related to the extracted
Huang–Rhys factors and anharmonicity is the asymmetry of the
two peaks on either side of the node in the amplitude profile of the
FCS. The ratio of peak heights strongly affects the values obtained
for these two microscopic vibrational parameters. We conducted a preliminary
analysis of three additional factors—the pump-pulse spectrum,
adding a second mode, and nonzero temperature—that could potentially
affect the ratio; thus far, no adjustment to the model has produced
a strong effect. Therefore, future work is needed to re-evaluate the
assumptions of the models to assess the accuracy of the extracted
values. For example, one could consider limitations of the Condon
approximation or the evolution of the system during the pump pulse.
We found that an effective assessment of anharmonicity should include
evaluating the coefficient of determination R2(λ), which is the dependency of the anharmonic fit on
the number of bound vibrational eigenstates. Despite these promising
advances, the models do not yet meet quantitative expectations. Therefore,
future efforts will be needed to reassess the assumptions underlying
the models and to understand additional challenges that complicated
this analysis. A salient issue is to resolve any effects of using
1D models to fit data arising from measurements of a molecule having
a ∼3N-dimensional potential-energy surface.
We previously showed that including a second mode affects the peak
widths,69 but a future detailed study may
reveal changes to the extracted Huang–Rhys factors. Furthermore,
including at least one more mode would allow the introduction of a
Duschinsky rotation matrix and thereby more reliably modeling spectra
from molecules such as rhodamine 101. Despite the nuances, the results
indicate the utility of the FCS models for studies of vibrational
quantum beats in femtosecond transient-absorption spectroscopy measurements
of molecules and molecular aggregates.
, and
conventional anharmonicity, χ.
These models are useful for studying excited-state vibrational modes
and, in particular, low-frequency excited-state vibrational modes
that are challenging to study with frequency-domain vibrational spectroscopy
methods such as Raman spectroscopy. The data and analyses of two laser
dye samples demonstrate that combined use of the harmonic and anharmonic
FCS models can provide qualitative insights into the vibrational quantum
beats and the topography of the excited-state potential energy surface
near the Franck–Condon region. The extracted Huang–Rhys
values for three modes of cresyl violet were somewhat consistent with
values produced by quantum-chemical calculations, which were restricted
to the harmonic approximation. The key observable related to the extracted
Huang–Rhys factors and anharmonicity is the asymmetry of the
two peaks on either side of the node in the amplitude profile of the
FCS. The ratio of peak heights strongly affects the values obtained
for these two microscopic vibrational parameters. We conducted a preliminary
analysis of three additional factors—the pump-pulse spectrum,
adding a second mode, and nonzero temperature—that could potentially
affect the ratio; thus far, no adjustment to the model has produced
a strong effect. Therefore, future work is needed to re-evaluate the
assumptions of the models to assess the accuracy of the extracted
values. For example, one could consider limitations of the Condon
approximation or the evolution of the system during the pump pulse.
We found that an effective assessment of anharmonicity should include
evaluating the coefficient of determination R2(λ), which is the dependency of the anharmonic fit on
the number of bound vibrational eigenstates. Despite these promising
advances, the models do not yet meet quantitative expectations. Therefore,
future efforts will be needed to reassess the assumptions underlying
the models and to understand additional challenges that complicated
this analysis. A salient issue is to resolve any effects of using
1D models to fit data arising from measurements of a molecule having
a ∼3N-dimensional potential-energy surface.
We previously showed that including a second mode affects the peak
widths,69 but a future detailed study may
reveal changes to the extracted Huang–Rhys factors. Furthermore,
including at least one more mode would allow the introduction of a
Duschinsky rotation matrix and thereby more reliably modeling spectra
from molecules such as rhodamine 101. Despite the nuances, the results
indicate the utility of the FCS models for studies of vibrational
quantum beats in femtosecond transient-absorption spectroscopy measurements
of molecules and molecular aggregates.
Methods
Laboratory Methods. We acquired rhodamine 101 (C32H31N2O3·ClO4; Rhodamine 640 Perchlorate) and cresyl violet (C16H11N3O2·HClO4; Cresyl Violet 670 Perchlorate) from Luxottica Exciton and dissolved each solid dye sample in anhydrous spectroscopic-grade methanol (Sigma-Aldrich). To reduce scatter, we filtered each solution using a 0.45 μm poly(ether sulfone) syringe filter (VWR) prior to optical measurements. The peak optical density (OD) was about 0.25. We used Cary 5000 and Horiba FL3-21 instruments to collect the absorption and fluorescence spectra, respectively.
The output of a commercial 1 kHz amplified Ti:sapphire laser producing ∼150 fs pulses centered at 806 nm pumped a home-built noncollinear optical parametric amplifier identical to one used in prior works.4,91 The ultrabroadband laser pulses spanned from approximately 510 to 750 nm, and the shot-to-shot stability was ∼1% relative standard deviation. Two sets of dispersion-compensating mirror pairs adjusted the temporal dispersion of the pulse, which had a pulse duration of about 8.8 fs based on second-harmonic generation frequency-resolved optical gating measurements (see Figure S5), conducted using a 10 μm thick beta barium borate crystal (Newlight Photonics).
The pump–probe spectrometer included a computer-controlled delay stage (Newport XMS50-S) to adjust the timing of the pump pulse relative to the probe pulse, and we performed the scans with 1 fs steps to minimize the presence of scatter in the FCS profiles. The only transmissive optic was a 1 mm thick ultraviolet fused silica 10% reflective beamsplitter that was antireflection coated for 440–1020 nm on the back side (Layertec), which separated the pump and probe beams. An identical optic placed in the probe arm equalized the dispersion and created a reference beam used for balanced detection. The electronics package included a phase-locked rotary optical chopper (New Focus 3502) in the pump arm, a data-acquisition board (NI PCI-6281), and an amplified photoreceiver (New Focus 2001-FS) to implement the optimal balanced detection algorithm.92 The spectrally resolved detector consisted of an Andor Kymera 193i and Zyla 5.5 sCMOS camera, calibrated to an estimated ±1 nm using a linear fit to multiple peaks from an atomic lamp. We report transient-absorption spectra as percent transient transmittance, ΔT/T.
We quantify the error between the fitted, F(ω), and measured, M(ω), FCS using the coefficient of determination
| 4 |
where M̅ is the mean of the measured spectrum. We used the curve fit function of Python and SciPy to perform the nonlinear least-squares fits of the models to the measured FCS spectra.
Acknowledgments
The Department of the Navy, Office of Naval Research (ONR) via ONR Award No. N00014-19-1-2615 supported the research conducted at Boise State University, including the collection, analysis, and interpretation of the data, as well as manuscript preparation. Additional funding sources supported specific equipment usage. The National Science Foundation NSF MRI Award No. 0923541 supported the femtosecond laser amplifier. A U.S. Department of Energy (DOE), Idaho National Laboratory, Laboratory Directed Research and Development project through blanket master contract No. 154754 Release 15, between Battelle Energy Alliance and Boise State University, supported the construction of the noncollinear optical parametric amplifier and compressor. The DOE, Office of Basic Energy Sciences, Division of Materials Science and Engineering through the Established Program to Stimulate Competitive Research (EPSCoR), via Award No. DE-SC0020089, provided support to upgrade the pump–probe spectrometer and enable femtosecond coherence spectroscopy measurements. Finally, we acknowledge use of the Borah (DOI: 10.18122/oit/3/boisestate)93 compute cluster provided by Boise State University’s Research Computing Department.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.1c04162.
(PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Michl J.; Bonačić-Koutecký V.. Electronic Aspects of Organic Photochemistry; Wiley, 1990. [Google Scholar]
- Yarkony D. R. Nonadiabatic Quantum Chemistry–Past, Present, and Future. Chem. Rev. 2012, 112, 481–498. 10.1021/cr2001299. [DOI] [PubMed] [Google Scholar]
- Tiwari V.; Peters W. K.; Jonas D. M. Electronic Resonance with Anticorrelated Pigment Vibrations Drives Photosynthetic Energy Transfer Outside the Adiabatic Framework. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1203–1208. 10.1073/pnas.1211157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Pacheco B.; Carbery W. P.; Khan S.; Turner D. B.; Buccella D. Fluorescence Quenching Effects of Tetrazines and Their Diels-Alder Products: Mechanistic Insight Toward Fluorogenic Efficiency. Angew. Chem., Int. Ed. 2020, 59, 22140–22149. 10.1002/anie.202008757. [DOI] [PubMed] [Google Scholar]
- Azumi T.; Matsuzaki K. What Does the Term “Vibronic Coupling” Mean?. Photochem. Photobiol. 1977, 25, 315–326. 10.1111/j.1751-1097.1977.tb06918.x. [DOI] [Google Scholar]
- Heller E. J. The Semiclassical Way to Molecular Spectroscopy. Acc. Chem. Res. 1981, 14, 368–375. 10.1021/ar00072a002. [DOI] [Google Scholar]
- Stock G. Classical Description of Nonadiabatic Photoisomerization Processes and Their Real-Time Detection via Femtosecond Spectroscopy. J. Chem. Phys. 1995, 103, 10015–10029. 10.1063/1.469905. [DOI] [Google Scholar]
- Ohta K.; Larsen D. S.; Yang M.; Fleming G. R. Influence of Intramolecular Vibrations in Third-Order, Time-Domain Resonant Spectroscopies. II. Numerical Calculations. J. Chem. Phys. 2001, 114, 8020–8039. 10.1063/1.1359241. [DOI] [Google Scholar]
- Reimers J. R.; Cai Z.-L.; Kobayashi R.; Ratsep M.; Freiberg A.; Krausz E. Assignment of the Q-Bands of the Chlorophylls: Coherence Loss via Qx - Qy Mixing. Sci. Rep. 2013, 3, 2761. 10.1038/srep02761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean J. C.; Navotnaya P.; Parobek A. P.; Clayton R. M.; Zwier T. S. Ultraviolet Spectroscopy of Fundamental Lignin Subunits: Guaiacol, 4-Methylguaiacol, Syringol, and 4-Methylsyringol. J. Chem. Phys. 2013, 139, 144313. 10.1063/1.4824019. [DOI] [PubMed] [Google Scholar]
- Huang K.; Rhys A. Theory of Light Absorption and Non-Radiative Transitions in F-Centres. Proc. R. Soc. London A 1950, 204, 406–423. 10.1098/rspa.1950.0184. [DOI] [Google Scholar]
- de Jong M.; Seijo L.; Meijerink A.; Rabouw F. T. Resolving the Ambiguity in the Relation Between Stokes Shift and Huang-Rhys Parameter. Phys. Chem. Chem. Phys. 2015, 17, 16959. 10.1039/C5CP02093J. [DOI] [PubMed] [Google Scholar]
- Lawless M. K.; Mathies R. A. Excited-State Structure and Electronic Dephasing Time of Nile Blue from Absolute Resonance Raman Intensities. J. Chem. Phys. 1992, 96, 8037–8045. 10.1063/1.462355. [DOI] [Google Scholar]
- Lee G.; Kim J.; Kim S. Y.; Kim D. E.; Joo T. Vibrational Spectrum of an Excited State and Huang-Rhys Factors by Coherent Wave Packets in Time-Resolved Fluorescence Spectroscopy. ChemPhysChem 2017, 18, 670–767. 10.1002/cphc.201601295. [DOI] [PubMed] [Google Scholar]
- Walmsley I. A.; Wise F. W.; Tang C. L. On the Difference Between Quantum Beats in Impulsive Stimulated Raman Scattering and Resonance Raman Scattering. Chem. Phys. Lett. 1989, 154, 315–320. 10.1016/0009-2614(89)85362-X. [DOI] [Google Scholar]
- Hamaguchi H.-o.; Gustafson T. L. Ultrafast Time-Resolved Spontaneous and Coherent Raman Spectroscopy: The Structure and Dynamics of Photogenerated Transient Species. Annu. Rev. Phys. Chem. 1994, 45, 593–622. 10.1146/annurev.pc.45.100194.003113. [DOI] [Google Scholar]
- Kukura P.; McCamant D. W.; Mathies R. A. Femtosecond Stimulated Raman Spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. 10.1146/annurev.physchem.58.032806.104456. [DOI] [PubMed] [Google Scholar]
- Zhu R.; Zou J.; Wang Z.; Chen H.; Weng Y. Electronic State-Resolved Multimode-Coupled Vibrational Wavepackets in Oxazine 720 by Two-Dimensional Electronic Spectroscopy. J. Phys. Chem. A 2020, 124, 9333–9342. 10.1021/acs.jpca.0c06559. [DOI] [PubMed] [Google Scholar]
- Gaynor J. D.; Weakly R. B.; Khalil M. Multimode Two-Dimensional Vibronic Spectroscopy. II. Simulating and Extracting Vibronic Coupling Parameters from Polarization-Selective Spectra. J. Chem. Phys. 2021, 154, 184202. 10.1063/5.0047727. [DOI] [PubMed] [Google Scholar]
- Begusic T.; Vanicek J. Finite-Temperature, Anharmonicity, and Duschinsky Effects on the Two-Dimensional Electronic Spectra from Ab Initio Thermo-Field Gaussian Wavepacket Dynamics. J. Phys. Chem. Lett. 2021, 12, 2997–3005. 10.1021/acs.jpclett.1c00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard W. T.; Mathies R. A. Analysis of Femtosecond Dynamic Absorption Spectra of Nonstationary States. Annu. Rev. Phys. Chem. 1992, 43, 497–523. 10.1146/annurev.pc.43.100192.002433. [DOI] [PubMed] [Google Scholar]
- Mukamel S.Principles of Nonlinear Optical Spectroscopy; Oxford University Press: New York, 1995. [Google Scholar]
- McClure S. D.; Turner D. B.; Arpin P. C.; Mirkovic T.; Scholes G. D. Coherent Oscillations in the PC577 Cryptophyte Antenna Occur in the Excited Electronic State. J. Phys. Chem. B 2014, 118, 1296–1308. 10.1021/jp411924c. [DOI] [PubMed] [Google Scholar]
- Arpin P. C.; Turner D. B.; McClure S. D.; Jumper C. C.; Mirkovic T.; Challa J. R.; Lee J.; Teng C. Y.; Green B. R.; Wilk K. E.; et al. Spectroscopic Studies of Cryptophyte Light Harvesting Proteins: Vibrations and Coherent Oscillations. J. Phys. Chem. B 2015, 119, 10025–10034. 10.1021/acs.jpcb.5b04704. [DOI] [PubMed] [Google Scholar]
- Jumper C. C.; Arpin P. C.; Turner D. B.; McClure S. D.; Rafiq S.; Dean J. C.; Cina J. A.; Kovac P. A.; Mirkovic T.; Scholes G. D. Broadband Pump-Probe Spectroscopy Quantifies Ultrafast Solvation Dynamics of Proteins and Molecules. J. Phys. Chem. Lett. 2016, 7, 4722–4731. 10.1021/acs.jpclett.6b02237. [DOI] [PubMed] [Google Scholar]
- Mathies R. A.; Cruz C. H. B.; Pollard W. T.; Shank C. V. Direct Observation of the Femtosecond Excited-State cis-trans Isomerization in Bacteriorhodopsin. Science 1988, 240, 777–779. 10.1126/science.3363359. [DOI] [PubMed] [Google Scholar]
- Pollard W. T.; Dexheimer S. L.; Wang Q.; Peteanu L. A.; Shank C. V.; Mathies R. A. Theory of Dynamic Absorption Spectroscopy of Nonstationary States. 4. Application to 12-fs Resonant Impulsive Raman Spectroscopy of Bacteriorhodopsin. J. Phys. Chem. 1992, 96, 6147–6158. 10.1021/j100194a013. [DOI] [Google Scholar]
- Vos M. H.; Rappaport F.; Lambry J.-C.; Breton J.; Martin J.-L. Visualization of Coherent Nuclear Motion in a Membrane Protein by Femtosecond Spectroscopy. Nature 1993, 363, 320–325. 10.1038/363320a0. [DOI] [Google Scholar]
- Wang Q.; Schoenlein R. W.; Peteanu L. A.; Mathies R. A.; Shank C. V. Vibrationally Coherent Photochemistry in the Femtosecond Primary Event of Vision. Science 1994, 266, 422–424. 10.1126/science.7939680. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Saito T.; Ohtani H. Real-Time Spectroscopy of Transition States in Bacteriorhodopsin during Retinal Isomerization. Nature 2001, 414, 531–534. 10.1038/35107042. [DOI] [PubMed] [Google Scholar]
- Kahan A.; Nahmias O.; Friedman N.; Sheves M.; Ruhman S. Following Photoinduced Dynamics in Bacteriorhodopsin with 7-fs Impulsive Vibrational Spectroscopy. J. Am. Chem. Soc. 2007, 129, 537–546. 10.1021/ja064910d. [DOI] [PubMed] [Google Scholar]
- Polli D.; Altoe P.; Weingart O.; Spillane K. M.; Manzoni C.; Brida D.; Tomasello G.; Orlandi G.; Kukura P.; Mathies R. A.; et al. Conical Intersection Dynamics of the Primary Photoisomerization Event in Vision. Nature 2010, 467, 440–443. 10.1038/nature09346. [DOI] [PubMed] [Google Scholar]
- Johnson P. J. M.; Halpin A.; Morizumi T.; Brown L. S.; Prokhorenko V. I.; Ernst O. P.; Miller R. The Photocycle and Ultrafast Vibrational Dynamics of Bacteriorhodopsin in Lipid Nanodiscs. Phys. Chem. Chem. Phys. 2014, 16, 21310–21320. 10.1039/C4CP01826E. [DOI] [PubMed] [Google Scholar]
- Liebel M.; Schnedermann C.; Bassolino G.; Taylor G.; Watts A.; Kukura P. Direct Observation of the Coherent Nuclear Response after the Absorption of a Photon. Phys. Rev. Lett. 2014, 112, 238301. 10.1103/PhysRevLett.112.238301. [DOI] [PubMed] [Google Scholar]
- Fragnito H. L.; Bigot B.-Y.; Becker P. C.; Shank C. V. Evolution of the Vibronic Absorption Spectrum in a Molecule Following Impulsive Excitation with a 6 fs Optical Pulse. Chem. Phys. Lett. 1989, 160, 101–104. 10.1016/0009-2614(89)87564-5. [DOI] [Google Scholar]
- Scherer N.; Jonas D. M.; Fleming G. R. Femtosecond Wave Packet and Chemical Reaction Dynamics of Iodine in Solution: Tunable Probe Study of Motion Along the Reaction Coordinate. J. Chem. Phys. 1993, 99, 153–168. 10.1063/1.465795. [DOI] [Google Scholar]
- Yang T.-S.; Chang M.-S.; Chang R.; Hayashi M.; Lin S. H.; Vöinger P.; Dietz W.; Scherer N. Femtosecond Pump-Probe Study of Molecular Vibronic Structures and Dynamics of a Cyanine Dye in Solution. J. Chem. Phys. 1999, 110, 12070–12081. 10.1063/1.479142. [DOI] [Google Scholar]
- Liebel M.; Kukura P. Broad-Band Impulsive Vibrational Spectroscopy of Excited Electronic States in the Time Domain. J. Phys. Chem. Lett. 2013, 4, 1358–1364. 10.1021/jz4004203. [DOI] [PubMed] [Google Scholar]
- Liebel M.; Schnedermann C.; Wende T.; Kukura P. Principles and Applications of Broadband Impulsive Vibrational Spectroscopy. J. Phys. Chem. A 2015, 119, 9506–9517. 10.1021/acs.jpca.5b05948. [DOI] [PubMed] [Google Scholar]
- Bishop M. M.; Roscioli J. D.; Ghosh S.; Mueller J. J.; Shepherd N. C.; Beck W. F. Vibrationally Coherent Preparation of the Transition State for Photoisomerization of the Cyanine Dye Cy5 in Water. J. Phys. Chem. B 2015, 119, 6905–6915. 10.1021/acs.jpcb.5b02391. [DOI] [PubMed] [Google Scholar]
- Dean J. C.; Rafiq S.; Oblinsky D. G.; Cassette E.; Jumper C. C.; Scholes G. D. Broadband Transient Absorption and Two-Dimensional Electronic Spectroscopy of Methylene Blue. J. Phys. Chem. A 2015, 119, 9098–9108. 10.1021/acs.jpca.5b06126. [DOI] [PubMed] [Google Scholar]
- Brazard J.; Bizimana L. A.; Gellen T.; Carbery W. P.; Turner D. B. Experimental Detection of Branching at a Conical Intersection in a Highly Fluorescent Molecule. J. Phys. Chem. Lett. 2016, 7, 14–19. 10.1021/acs.jpclett.5b02476. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick C.; Odhner J. H.; Levis R. J. Spectral Signatures of Ground- and Excited-State Wavepacket Interference after Impulsive Excitation. J. Phys. Chem. A 2020, 124, 6856–6866. 10.1021/acs.jpca.0c03912. [DOI] [PubMed] [Google Scholar]
- Rosca F.; Kumar A. T. N.; Ionascu D.; Sjodin T.; Demidov A. A.; Champion P. M. Wavelength Selective Modulation in Femtosecond Pump-Probe Spectroscopy and its Application to Heme Proteins. J. Chem. Phys. 2001, 114, 10884. 10.1063/1.1363673. [DOI] [Google Scholar]
- Müller M. G.; Lindner I.; Martin I.; Gärtner W.; Holzwarth A. R. Femtosecond Kinetics of Photoconversion of the Higher Plant Photoreceptor Phytochrome Carrying Native and Modified Chromophores. Biophys. J. 2008, 94, 4370–4382. 10.1529/biophysj.106.091652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizimana L. A.; Epstein J.; Brazard J.; Turner D. B. Conformational Homogeneity in the Pr Isomer of Phytochrome Cph1. J. Phys. Chem. B 2017, 121, 2622–2630. 10.1021/acs.jpcb.7b02180. [DOI] [PubMed] [Google Scholar]
- Bizimana L. A.; Epstein J.; Turner D. B. Inertial Water Response Dominates Protein Solvation Dynamics. Chem. Phys. Lett. 2019, 728, 1–5. 10.1016/j.cplett.2019.04.069. [DOI] [Google Scholar]
- Lüer L.; Gadermaier C.; Crochet J.; Hertel T.; Brida D.; Lanzani G. Coherent Phonon Dynamics in Semiconducting Carbon Nanotubes: A Quantitative Study of Electron-Phonon Coupling. Phys. Rev. Lett. 2009, 102, 127401. 10.1103/PhysRevLett.102.127401. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Wang Z.; Otsubo T. Classification of Dynamic Vibronic Couplings in Vibrational Real-Time Spectra of a Thiophene Derivative by Few-Cycle Pulses. J. Phys. Chem. A 2007, 111, 12985–12994. 10.1021/jp076750d. [DOI] [PubMed] [Google Scholar]
- Rafiq S.; Dean J. C.; Scholes G. D. Observing Vibrational Wavepackets during an Ultrafast Electron Transfer Reaction. J. Phys. Chem. A 2015, 119, 11837–11846. 10.1021/acs.jpca.5b09390. [DOI] [PubMed] [Google Scholar]
- Rury A. S.; Sorenson S. A.; Dawlaty J. M. Evidence of Ultrafast Charge Transfer Driven by Coherent Lattice Vibrations. J. Phys. Chem. Lett. 2017, 8, 181–187. 10.1021/acs.jpclett.6b02523. [DOI] [PubMed] [Google Scholar]
- Rafiq S.; Fu B.; Kudisch B.; Scholes G. D. Interplay of Vibrational Wavepackets During an Ultrafast Electron Transfer Reaction. Nat. Chem. 2021, 13, 70–76. 10.1038/s41557-020-00607-9. [DOI] [PubMed] [Google Scholar]
- Du J.; Wang Z.; Feng W.; Yoshino K.; Kobayashi T. Simultaneous Measurement of Electronic and Vibrational Dynamics to Clarify a Geometrical Relaxation Process in a Conjugated Polymer. Phys. Rev. B 2008, 77, 195205. 10.1103/PhysRevB.77.195205. [DOI] [Google Scholar]
- Thouin F.; Valverde-Chavez D. A.; Quarti C.; Cortecchia D.; Bargigia I.; Beljonne D.; Petrozza A.; Silva C.; Kandada A. R. S. Phonon Coherences Reveal the Polaronic Character of Excitons in Two-Dimensional Lead Halide Perovskites. Nat. Mater. 2019, 18, 349–356. 10.1038/s41563-018-0262-7. [DOI] [PubMed] [Google Scholar]
- Kano H.; Saito T.; Kobayashi T. Observation of Herzberg-Teller-Type Wave Packet Motion in Porphyrin J-Aggregates Studied by sub-5-fs Spectroscopy. J. Phys. Chem. A 2002, 106, 3445–3453. 10.1021/jp012493f. [DOI] [Google Scholar]
- Musser A. J.; Liebel M.; Schnedermann C.; Wende T.; Kehoe T. B.; Rao A.; Kukura P. Evidence for Conical Intersection Dynamics Mediating Ultrafast Singlet Exciton Fission. Nat. Phys. 2015, 11, 352–357. 10.1038/nphys3241. [DOI] [Google Scholar]
- Rury A. S.; Sorenson S.; Driscoll E.; Dawlaty J. M. Electronic State-Resolved Electron-Phonon Coupling in an Organic Charge Transfer Material from Broadband Quantum Beat Spectroscopy. J. Phys. Chem. Lett. 2015, 6, 3560–3564. 10.1021/acs.jpclett.5b01706. [DOI] [PubMed] [Google Scholar]
- Rury A. S.; Sorenson S. A.; Dawlaty J. M. Coherent Vibrational Probes of Hydrogen Bond Structure Following Ultrafast Electron Transfer. J. Phys. Chem. C 2016, 120, 21740–21750. 10.1021/acs.jpcc.6b05239. [DOI] [Google Scholar]
- Bardeen C. J.; Wang Q.; Shank C. V. Selective Excitation of Vibrational Wave Packet Motion Using Chirped Pulses. Phys. Rev. Lett. 1995, 75, 3410–3413. 10.1103/PhysRevLett.75.3410. [DOI] [PubMed] [Google Scholar]
- Jonas D. M.; Fleming G. R. In Ultrafast Processes in Ultrafast Processes in and Photobiology; El-Sayed M. A., Tanaka I., Molin Y., Eds.; Chemistry for the 21st Century; Blackwell Scientific Publications: Oxford, 1995; p 225. [Google Scholar]
- Jonas D. M.; Bradforth S. E.; Passino S. A.; Fleming G. R. Femtosecond Wavepacket Spectroscopy: Influence of Temperature, Wavelength, and Pulse Duration. J. Phys. Chem. 1995, 99, 2594–2608. 10.1021/j100009a018. [DOI] [Google Scholar]
- Bardeen C.; Wang Q.; Shank C. Femtosecond Chirped Pulse Excitation of Vibrational Wave Packets in LD690 and Bacteriorhodopsin. J. Phys. Chem. A 1998, 102, 2759–2766. 10.1021/jp980346k. [DOI] [Google Scholar]
- Wand A.; Kallush S.; Shoshanim O.; Bismuth O.; Kosloff R.; Ruhman S. Chirp Effects on Impulsive Vibrational Spectroscopy: A Multimode Perspective. Phys. Chem. Chem. Phys. 2010, 12, 2149–2163. 10.1039/b920356g. [DOI] [PubMed] [Google Scholar]
- Yan Y. J.; Fried L. E.; Mukamel S. Ultrafast Pump-Probe Spectroscopy: Femtosecond Dynamics in Liouville Space. J. Phys. Chem. 1989, 93, 8149–8162. 10.1021/j100362a006. [DOI] [Google Scholar]
- Yan Y. J.; Mukamel S. Femtosecond Pump-Probe Spectroscopy of Polyatomic Molecules in Condensed Phases. Phys. Rev. A 1990, 41, 6485–6404. 10.1103/PhysRevA.41.6485. [DOI] [PubMed] [Google Scholar]
- Pollard W. T.; Fragnito H. L.; Bigot J.-Y.; Shank C. V.; Mathies R. A. Quantum-Mechanical Theory for 6 fs Dynamic Absorption Spectroscopy and its Application to Nile Blue. Chem. Phys. Lett. 1990, 168, 239–245. 10.1016/0009-2614(90)85603-A. [DOI] [Google Scholar]
- Kumar A. T. N.; Rosca F.; Widom A.; Champion P. M. Investigations of Amplitude and Phase Excitation Profiles in Femtosecond Coherence Spectroscopy. J. Chem. Phys. 2001, 114, 701–724. 10.1063/1.1329640. [DOI] [Google Scholar]
- Cina J. A.; Kovac P. A.; Jumper C. C.; Dean J. C.; Scholes G. D. Ultrafast Transient Absorption Revisited: Phase-Flips, Spectral Fingers, and Other Dynamical Features. J. Chem. Phys. 2016, 144, 175102. 10.1063/1.4947568. [DOI] [PubMed] [Google Scholar]
- Turner D. B.; Arpin P. C. Basis Set Truncation Further Clarifies Vibrational Coherence Spectra. Chem. Phys. 2020, 539, 110948. 10.1016/j.chemphys.2020.110948. [DOI] [Google Scholar]
- Arpin P. C.; Turner D. B. Signatures of Vibrational and Electronic Quantum Beats in Femtosecond Coherence Spectra. J. Phys. Chem. A 2021, 125, 2425–2435. 10.1021/acs.jpca.0c10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo M.; Gruia F.; Benabbas A.; Barabanschikov A.; Montfort W. R.; Maes E. M.; Champion P. M. Low-Frequency Mode Activity of Heme: Femtosecond Coherence Spectroscopy of Iron Porphine Halides and Nitrophorin. J. Am. Chem. Soc. 2008, 130, 9800–9811. 10.1021/ja800916d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran V.; Benabbas A.; Youn H.; Champion P. M. Vibrational Coherence Spectroscopy of the Heme Domain in the CO-Sensing Transcriptional Activator CooA. J. Am. Chem. Soc. 2011, 133, 18816–18827. 10.1021/ja206152m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iachello F.; Ibrahim M. Analytic and Algebraic Evaluation of Franck-Condon Overlap Integrals. J. Phys. Chem. A 1998, 102, 9427–9432. 10.1021/jp981911m. [DOI] [Google Scholar]
- Duschinsky F. Zur Deutung der Elektronenspektren mehratomiger Moleküle. Acta Physiochimica U.R.S.S. 1937, VII, 551–566. [Google Scholar]
- Frank A. J.; Otvos J. W.; Calvin M. Quenching of Rhodamine 101 Emission in Methanol and in Colloidal Suspensions of Latex Particles. J. Phys. Chem. 1979, 83, 716–722. 10.1021/j100469a016. [DOI] [Google Scholar]
- Karstens T.; Kobs K. Rhodamine B and Rhodamine 101 as Reference Substances for Fluorescence Quantum Yield Measurements. J. Phys. Chem. 1980, 84, 1871–1872. 10.1021/j100451a030. [DOI] [Google Scholar]
- Vosgröne T.; Meixner A. J. Surface- and Resonance-Enhanced Micro-Raman Spectroscopy of Xanthene Dyes: From the Ensemble to Single Molecules. ChemPhysChem 2005, 6, 154–163. 10.1002/cphc.200400395. [DOI] [PubMed] [Google Scholar]
- Runge P. K. A Continuous Mode-Locked Dye Laser Pumped in the Red. Opt. Commun. 1971, 4, 195–198. 10.1016/0030-4018(71)90070-8. [DOI] [Google Scholar]
- Isak S. J.; Eyring E. M. Fluorescence Quantum Yield of Cresyl Violet in Methanol and Water as a Function of Concentration. J. Phys. Chem. 1992, 96, 1738–1742. 10.1021/j100183a045. [DOI] [Google Scholar]
- Knemeyer J.-P.; Marme N.; Sauer M. Probes for Detection of Specific DNA Sequences at the Single-Molecule Level. Anal. Chem. 2000, 72, 3717–3724. 10.1021/ac000024o. [DOI] [PubMed] [Google Scholar]
- Klar T. A.; Jakobs S.; Dyba M.; Egner A.; Hell S. W. Fluorescence Microscopy with Diffraction Resolution Barrier Broken by Stimulated Emission. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 8206–8210. 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q.; Song Y.; Li Z.; Gao X.; Ma H. In vivo Monitoring of Hydrogen Sulfide Using a Cresyl Violet-Based Ratiometric Fluorescence Probe. Chem. Commun. 2013, 49, 502–504. 10.1039/C2CC37725J. [DOI] [PubMed] [Google Scholar]
- Heisler I. A.; Moca R.; Camargo F. V. A.; Meech S. R. Two-Dimensional Electronic Spectroscopy Based on Conventional Optics and Fast Dual Chopper Data Acquisition. Rev. Sci. Instrum. 2014, 85, 063103. 10.1063/1.4879822. [DOI] [PubMed] [Google Scholar]
- Spokoyny B.; Koh C. J.; Harel E. Stable and High-Power Few Cycle Supercontinuum for 2D Ultrabroadband Electronic Spectroscopy. Opt. Lett. 2015, 40, 1014–1017. 10.1364/OL.40.001014. [DOI] [PubMed] [Google Scholar]
- Bizimana L. A.; Brazard J.; Carbery W. P.; Gellen T.; Turner D. B. Resolving Molecular Vibronic Structure using High-Sensitivity Two-Dimensional Electronic Spectroscopy. J. Chem. Phys. 2015, 143, 164203. 10.1063/1.4934717. [DOI] [PubMed] [Google Scholar]
- Turner D. B. Standardized Specifications of 2D Optical Spectrometers. Results in Chemistry 2019, 1, 100001. 10.1016/j.rechem.2019.100001. [DOI] [Google Scholar]
- Rafiq S.; Scholes G. D. Slow Intramolecular Vibrational Relaxation Leads to Long-Lived Excited-State Wavepackets. J. Phys. Chem. A 2016, 120, 6792–6799. 10.1021/acs.jpca.6b07796. [DOI] [PubMed] [Google Scholar]
- Carbery W. P.; Pinto-Pacheco B.; Buccella D.; Turner D. B. Resolving the Fluorescence Quenching Mechanism of an Oxazine Dye Using Ultrabroadband Two-Dimensional Electronic Spectroscopy. J. Phys. Chem. A 2019, 123, 5072–5080. 10.1021/acs.jpca.9b03632. [DOI] [PubMed] [Google Scholar]
- Farfan C. A.; Turner D. B. A Systematic Model Study Quantifying how Conical Intersection Topography Modulates Photochemical Reactions. Phys. Chem. Chem. Phys. 2020, 22, 20265–20283. 10.1039/D0CP03464A. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Wang Y.; Lin K.; Hu N.; Zhou X.; Liu S. Complete Raman Spectral Assignment of Methanol in the C-H Stretching Region. J. Phys. Chem. A 2013, 117, 4377–4384. 10.1021/jp400886y. [DOI] [PubMed] [Google Scholar]
- Petkov B. K.; Gellen T. A.; Farfan C. A.; Carbery W. P.; Hetzler B. E.; Trauner D.; Li X.; Glover W. J.; Ulness D. J.; Turner D. B. Two-Dimensional Electronic Spectroscopy Reveals the Spectral Dynamics of Förster Resonance Energy Transfer. Chem. 2019, 5, 2111. 10.1016/j.chempr.2019.05.005. [DOI] [Google Scholar]
- Brazard J.; Bizimana L. A.; Turner D. B. Accurate Convergence of Transient-Absorption Spectra using Pulsed Lasers. Rev. Sci. Instrum. 2015, 86, 053106. 10.1063/1.4921479. [DOI] [PubMed] [Google Scholar]
- Boise State’s Research Computing Department . 2020. Borah: Dell HPC (High Performance Computing Cluster). Boise State University, Boise, ID. 10.18122/oit/3/boisestate. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.