

Abstract
Kratom alkaloids have mostly been evaluated for their opioid activity but less at other targets that could contribute to their physiological effects. Here, we investigated the in vitro and in vivo activity of kratom alkaloids at serotonin receptors (5-HTRs). Paynantheine and speciogynine exhibited high affinity for 5-HT1ARs and 5-HT2BRs, unlike the principal kratom alkaloid mitragynine. Both alkaloids produced antinociceptive properties in rats via an opioid receptor-independent mechanism, and neither activated 5-HT2BRs in vitro. Paynantheine, speciogynine, and mitragynine induced lower lip retraction and antinociception in rats, effects blocked by a selective 5-HT1AR antagonist. In vitro functional assays revealed that the in vivo 5-HT1AR agonistic effects may be due to the metabolites 9-O-desmethylspeciogynine and 9-O-desmethylpaynantheine and not the parent compounds. Both metabolites did not activate 5-HT2BR, suggesting low inherent risk of causing valvulopathy. The 5-HT1AR agonism by kratom alkaloids may contribute to the mood-enhancing effects associated with kratom use.
Graphcial Abstract
INTRODUCTION
Kratom, Mitragyna speciosa (Korth.), is a widely used medicinal plant from Southeast Asia.1 Kratom’s leaves are chewed or boiled to prepare beverages that are consumed to treat diverse maladies, including fever, diarrhea, and cough.2 Kratom has been used also to enhance productivity and energy, combat anxiety, and manage opioid dependence.2 The consumption of kratom preparations appears to be well-tolerated by individuals in Southeast Asia including former and existing opioid users.3 In the United States (US), kratom use has proliferated in the past decade and is used typically in the form of capsules, powdered leaves consumed either dry or as a tea, in energy drinks, and in elixirs.2 A recent estimate of use in the US showed that around 3.3 million people consume kratom.4 In the US, kratom is used as a recreational product often to treat chronic pain, opioid use disorder symptoms, and as a remedy for anxiety and depression.5 These purported therapeutic effects of kratom products suggest potential clinical value, though no kratom-derived products have been approved for use by the US Food and Drug Administration (FDA).6−8 Currently, kratom is not federally regulated, although its possession is banned in some US jurisdictions and states. The US Drug Enforcement Administration lists kratom as a drug of concern.9
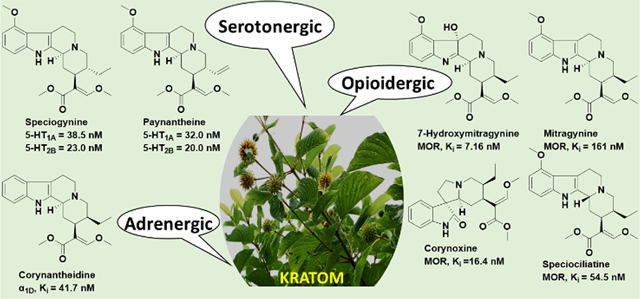
To date, about 45 different indole and oxindole alkaloids isolated from kratom leaves have been reported, including mitragynine, paynantheine, speciogynine, and speciociliatine.1,2,7 However, the major kratom alkaloids and the estimates of their relative abundance vary due to different growing conditions, origin of the kratom plant, and other factors that have not been fully elucidated.2,6−10 7-Hydroxymitragynine represents <2% of the alkaloids but has a much greater binding affinity at μ-opioid receptors (MORs) than mitragynine, which comprises 20−60% of the total alkaloid content.7,11
Functional assays at human μ-, κ-, and δ-opioid receptors (hMORs, hKORs, and hDORs, respectively) using a bioluminescence resonance energy transfer (BRET) G protein activation assay demonstrated that mitragynine is not only less potent but also a lower-efficacy hMOR agonist (EC50 = 339 nM; Emax = 34%) than 7-hydroxymitragynine (EC50 = 34 nM; Emax = 47%), whereas speciociliatine, speciogynine, and paynantheine are low-potency, competitive antagonists at hMOR; none of the compounds show appreciable activity at hKORs or hDORs.1 However, the physiological effects of kratom use likely reflect the summed actions of multiple alkaloids at a range of receptor targets, which include MOR, α-adrenergic, muscarinic, dopaminergic, and serotonergic receptors.8 The affinity of some kratom alkaloids for adrenergic α receptor subtypes has been reported previously.12 Corynantheidine has moderate nanomolar affinity for adrenergic α1D receptors (Ki = 41 nM), while mitragynine has micromolar affinity for adrenergic α1A, α1B, α1C, α2A, α2B, and α2C receptor subtypes.12 A recent report, principally using in silico methodology,13 extended those findings to predict (with some experimental corroboration) the affinities of kratom alkaloids at adrenergic (α2A, α2B, and α2C) receptors and serotonergic 5-HT1ARs and 5-HT2ARs12. The data suggested that speciogynine, mitragynine, and ajmalicine have affinity for the human 5-HT1A receptor orthosteric binding site (Ki values = 0.54, 5.8, and 0.42 μM, respectively).13 Contribution of descending serotonergic systems to the antinociceptive effects of mitragynine in mice has also been suggested.14
The current study reports experimentally obtained affinities for the major kratom alkaloids, mitragynine, speciociliatine, speciogynine, and paynantheine and minor available ones, 7-hydroxymitragynine, 9-hydroxycorynantheidine, corynantheidine, and ajmalicine (Figure 1) at serotonin receptor subtypes 5-HT1A, 5-HT1B, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, and 5-HT715. Based on their relevant affinities, we also evaluated the in vitro 5-HT1A and 5-HT2B receptor functional activity of select kratom alkaloids. Finally, we evaluated the in vivo activity of select kratom alkaloids at 5-HT1A receptors by testing their effects on lower lip retraction and on antinociception using a hotplate assay16 in rats pretreated with a vehicle or the selective 5-HT1AR antagonist WAY 100635.
Figure 1.

Chemical structures of the selected isolated kratom alkaloids and metabolites.
The independent pharmacokinetic profiles of speciogynine and paynantheine were evaluated following a single dose (i.v. administration) to rats. These pharmacokinetic parameters were compared to those of mitragynine to understand their corresponding clearance and overall contributions to the pharmacological activity of kratom products.
RESULTS AND DISCUSSION
Affinity screens of select kratom alkaloids at recombinant human 5-HTRs expressed in human embryonic kidney 293 (HEK-293) cell membranes were determined by radioligand competition binding using 100 nM and 10 μM concentrations (Table 1). Radioligand probe “displacement” values of 50% or greater at 100 nM were considered significant “hits”, and Ki values of hits were subsequently determined. Ki values of mitragynine at the 5-HT receptors were also determined, regardless of affinity screen results, as mitragynine is the most prevalent alkaloid in kratom and is often extracted from the plant and used in scientific studies independent from the other kratom alkaloids.
Table 1.
Percent Displacement of Radioligands from Human 5-HTRs by 100 nM and 10 μM Concentrations of Kratom Alkaloidsa
| binding site | ||||||
|---|---|---|---|---|---|---|
| kratom alkaloid | 5-HT1A | 5-HT1B | 5-HT2A | 5-HT2B | 5-HT2C | 5-HT3 |
| mitragynine | ||||||
| 100 nM | 9.7 | −0.2 | −0.9 | 23.0 | −8.2 | −9.1 |
| 10,000 nM | 76.7 | 8.2 | 49.9 | 90.7 | 14.7 | −12.7 |
| 7-hydroxymitragynine | ||||||
| 100 nM | 3.1 | −5.6 | −0.2 | −1.4 | −18.3 | 8.2 |
| 10,000 nM | 1.2 | −0.8 | 3.9 | 12.4 | −13.9 | 19.5 |
| speciociliatine | ||||||
| 100 nM | −4.8 | 3.8 | 1.5 | −3.4 | 0.4 | 11.1 |
| 10,000 nM | 33.3 | 16.8 | 68.1 | 47.1 | 14.3 | 0.7 |
| speciogynine | ||||||
| 100 nM | 58.9 | 2.6 | 2.8 | 70.9 | −18.6 | 11.3 |
| 10,000 nM | 98.7 | 30.4 | 82.7 | 102.2 | 77.9 | 1.5 |
| paynantheine | ||||||
| 100 nM | 69.4 | 8.9 | 16.8 | 53.8 | 12.7 | −9.8 |
| 10,000 nM | 98.7 | 19.5 | 92.8 | 101.5 | 89.3 | −0.8 |
| 9-hydroxycorynantheidine | ||||||
| 100 nM | 0.2 | 3.0 | −6.5 | 5.2 | −10.4 | −1.3 |
| 10,000 nM | 29.6 | −2.1 | 28.4 | 71.3 | 12.5 | 7.3 |
| corynantheidine | ||||||
| 100 nM | 2.5 | −4.2 | −6.8 | 14.8 | 2.5 | 6.2 |
| 10,000 nM | 77.3 | 10.7 | 28.7 | 76.3 | 3.8 | 6.8 |
| ajmalicine | ||||||
| 100 nM | 33.2 | 21.6 | 5.1 | 6.2 | −12.5 | −2.5 |
| 10,000 nM | 95.2 | 98.3 | 36.5 | 89.3 | −5.3 | 1.8 |
Bold fonts indicate ≥50% displacement of radioligands from the respective 5-HTR. Each experiment was conducted in duplicate. [3H]8-OH-DPAT, [125I]CYP (+ 30 μM isoproterenol), and [3H]imipramine were used to label 5-HT1ARs, 5-HT1BRs, and the 5-HT transporter, respectively. [125I](±)DOI was used to label 5-HT2A(h), 5-HT2B(h), and 5-HT2C(h) receptors.
At 100 nM, only speciogynine and paynantheine displaced a significant percentage (58.9 and 69.4%, respectively) of [3H]8-OH-DPAT [(±)-8-hydroxy-2-(dipropylamino)tetralin] bound to 5-HT1ARs (Table 1), while 10 μM concentrations of mitragynine, speciogynine, paynantheine, corynantheidine, and ajmalicine displaced 76.7, 98.7, 98.7, 77.3, and 95.2% (respectively) of [3H]8-OH-DPAT bound to 5-HT1ARs. As ligand affinities at 5-HT1ARs and 5-HT7Rs often correlate,17 we also determined Ki values of speciogynine and paynantheine at agonist-labeled, [3H]5-CT [3-(2-aminoethyl-1,2-t2)-1H-indole-5-carboxamide], human 5-HT7a receptors expressed in HEK-293 cells (Table 4). Alkaloid binding to recombinant 5-HT1BRs was assessed using [125I]CYP [4–2-(hydroxy-3-(tert-butylamino)propoxy)-3-iodo-1H-indole-2-carbonitrile]; no significant radioligand displacement was observed for the kratom alkaloids tested, with the exception of ajmalicine at 10 μM (Table 1). Similar to what we observed at 5-HT1ARs, 100 nM concentrations of speciogynine and paynantheine displaced, respectively, 70.9 and 53.8% of [125I]2,5-dimethoxy-4-iodoamphetamine ([125I]DOI) binding to recombinant, human 5-HT2B receptors expressed in Chinese hamster ovary K1 (CHO-K1) cells. Other alkaloids including mitragynine, 9-hydroxycorynantheidine, corynantheidine, and ajmalicine displaced ≥50% of [125I]DOI from 5-HT2BRs only at 10 μM concentrations. As ligand affinities at 5-HT2BRs, 5-HT2ARs, and 5-HT2CRs often correlate,18 we determined Ki values of speciogynine and paynantheine at agonist-labeled, [3H]LSD [(6aR,9R)-N,N-diethyl-7-(methyl-t3)-4,6,6a,7,8,9-hexahydroindolo[4,3-fg]quinoline-9-carboxamide], human 5-HT2ARs, and 5-HT2CRs expressed in HEK-293 cells (Table 2). None of the alkaloids bound to 5-HT3Rs at concentrations of 10 μM (Table 1).
Table 4.
Potencies (EC50 or IC50 ± SEM in nM) and Efficacies (Emax or Imax ± SEM) of Test Compounds at Human 5-HT1A and 5-HT2BRsa
| 5-HT1A | 5-HT2B | |||
|---|---|---|---|---|
| compounds | IC/EC50 | E/Imax | IC/EC50 | E/Imax |
| speciogynine | N.A. | N.A. | 2016 (1888) | 24.8 (6.9) |
| paynantheine | N.A. | N.A. | N.A. | N.A. |
| 9-O-desmethylspeciogynine (gambirine) | 837.6 (253.9) | 99 (0.08) | 1472 (731.6) | 45 (14.3) |
| 9-O-desmethylpaynantheine (gambireine) | 865.4 (126.4) | 100 (0.08) | 2339 (499.7) | 38 (10.3) |
| mitragynine | N.A. | N.A. | N.T. | N.T. |
| serotonin | 5.194 (2.5) | 100 | 31.31 (20.3) | 100 |
| ipsapirone | 67.10 (44.7) | 82 (0.1) | N.T. | N.T. |
| SB 206553 | N.T. | N.T. | 15.33 (4.89) | 100 |
Emax at 5-HT1ARs is the percent maximum serotonin, agonist, response; Imax at 5-HT2BRs is the percent maximum SB 206553, inverse agonist, response. N.T. = not tested. N.A. = no activation or inactivation up to 30 μM.
Table 2.
Ki Values (Mean ± SEM in nM) of Mitragynine, Speciogynine, and Paynantheine at Human 5-HT1A, 5-HT7A, 5-HT2A, 5-HT2B, and 5-HT2CR Subtypesi
| binding site | |||||
|---|---|---|---|---|---|
| kratom alkaloid | 5-HT1A | 5-HT7A | 5-HT2A | 5-HT2B | 5-HT2C |
| mitragynine | 5880 ± 828a | >10,000f | 5010 ± 1150g | 1260 ± 138g | >10,000g |
| speciogynine | 38.5 ± 3.9b | 1600 ± 82f | 1320 ± 365g | 23.0 ± 5.7h | 5430 ± 922g |
| 95.5 ± 34.7a | 108 ± 39.5g | ||||
| paynantheine | 32.0 ± 3.5b | 870 ± 72f | 815 ± 192g | 20.0 ± 2.8h | 1770 ± 417g |
| 71.8 ± 13.2a | 83.1 ± 32.9g | ||||
| 9-O-desmethylspeciogynine (gambirine) | 294.1 ± 189.1a | ND | ND | 508.1 ± 96.0g | ND |
| 9-O-desmethylpaynantheine (gambireine) | 97.7 ± 44.2a | ND | ND | 335.8 ± 83.3g | ND |
| ipsapirone | 0.240c | ND | ND | ND | ND |
| 49.7 ± 10.5d | |||||
| 8-OH-DPAT | 0.75 ± 0.03b | ND | ND | ND | ND |
| DOI | ND | ND | 6.80 ± 1.00g | 2.20 ± 0.30h | ND |
| WAY 100635 | 0.90e | ND | ND | ND | ND |
| 5-HT | 4.50 ± 2.70a | 2.10 ± 0.50f | 17.2 ± 12.5g | 10.8 ± 2.60g | ND |
| SB 206553 | ND | ND | ND | ND | 2.80 ± 0.30g |
[3H]8-OH-DPAT binding to human 5-HT1ARs expressed in CHO cells in the present study.
[3H]8-OH-DPAT binding to human 5-HT1ARs expressed in HEK cells in the present study.
[3H]8-OH-DPAT binding to human 5-HT1ARs expressed in COS-7 cells.19
[3H]5-CT binding to human 5-HT1ARs expressed in CHO cells in the present study.
[3H]8-OH-DPAT binding to human 5-HT1ARs expressed in CHO cells.20
[3H]5-CT binding to human 5-HT7aRs expressed in HEK cells in the present study.
[3H]LSD binding to human 5-HT2ARs, 5-HT2BRs, or 5-HT2CRs expressed in HEK cells in the present study.
[125I]DOI binding to human 5-HT2BRs expressed in CHO cells in the present study.
Two (Eurofins) to three (Mercer University) independent experiments were conducted to calculate Ki values. In every experiment, compounds were tested at each concentration in duplicate (Eurofins) or triplicate (Mercer University). ND: not determined.
Ki values of mitragynine, speciogynine, and paynantheine at human 5-HT1A, 5-HT2A, 5-HT2B, 5-HT2C, and 5-HT7 receptors are shown in Table 2, along with Ki values of positive controls assessed in parallel. Speciogynine and paynantheine exhibited moderate (~100 nM or less) affinity at 5-HT1A and 5-HT2B receptors (Figure 2 and supplementary Figure S21 show full concentration−response curves). The next highest affinities were observed for paynantheine at 5-HT7Rs and 5-HT2ARs, though they were relatively low, i.e., 870 and 815 nM, respectively. Ki values of mitragynine, speciogynine, and paynantheine determined at each of the other 5-HTRs were >1.2 μM (Table 2). The affinity of speciogynine for 5-HT1ARs was >10-fold higher than previously reported (540 nM; the species from which the 5-HT1AR was derived was unspecified).13
Figure 2.
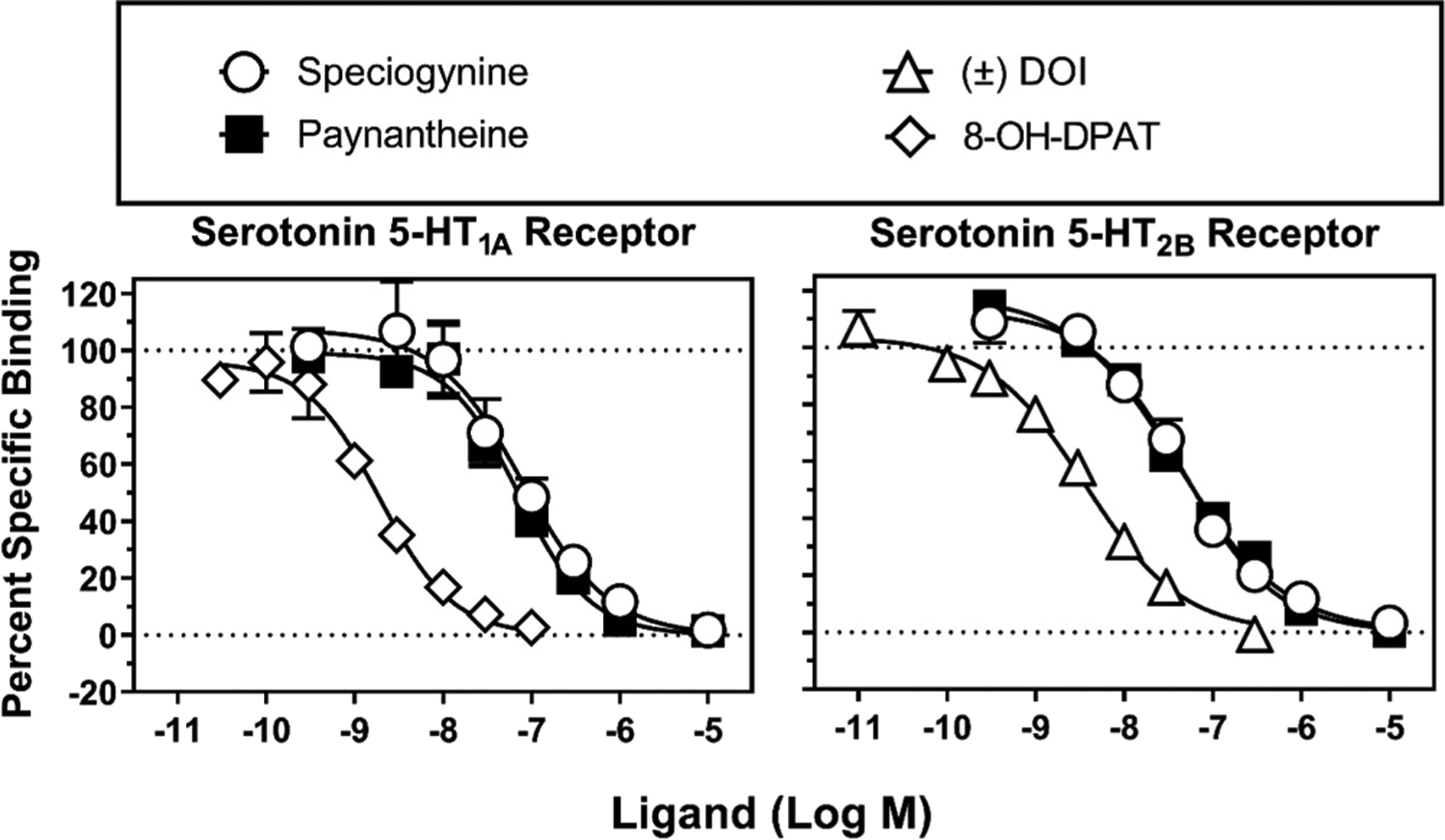
Radioligand competition binding at human 5-HT1ARs and 5-HT2BRs. Ordinates: percent specific binding of the radioligand to receptors expressed in cell membranes (described in the Experimental Section). Abscissae: concentrations of test ligands (log scale). The left panel shows percent specific binding of [3H]8-OH-DPAT at 5-HT1ARs. The right panel shows percent specific binding of [125I]DOI at 5-HT2BRs. These experiments were conducted in duplicate and repeated twice (N = 2), and the data shown were obtained by Eurofins. We conducted additional affinity tests to validate these results, and the binding curves are shown in the Supporting Information (Figure S21).
Structurally, speciogynine and mitragynine are diastereomers with different configurations at C-20, meanwhile paynantheine has the same configuration as speciogynine (speciogynine is 3S, 15S, 20R; paynantheine is 3S, 15S, 20R; mitragynine is 3S, 15S, 20S). This primary configurational difference at the C-20 position makes the β-methoxyacrylate group in speciogynine and paynantheine perpendicular to the molecule’s plane, whereas the substituent at C-20 in mitragynine is planar (see Figure 3).
Figure 3.
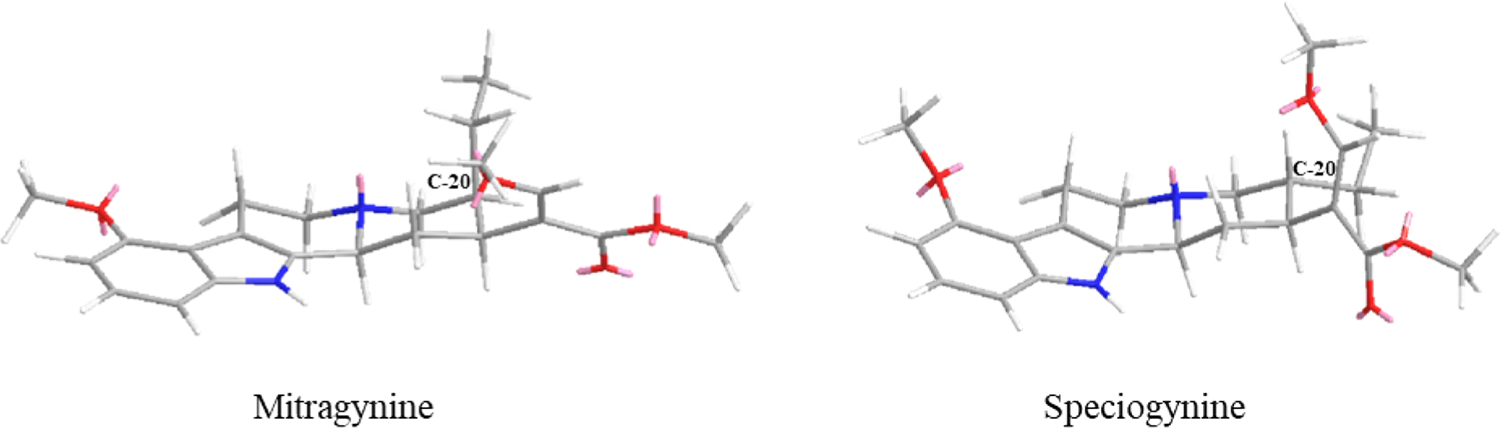
Three-dimensional structure comparison of mitragynine and speciogynine (most stable conformation, Chemdraw3D).
It is noteworthy here that a structurally related natural product, geissoschizine methyl ether, produced by the plant Uncaria rhynchopilla (yokukansan), is a Japanese traditional medicine used to treat irritability and insomnia in children and disorders such as psychosis, dementia, and anxiety. Geissoschizine methyl ether is a potent antagonist at 5-HT1A receptors19,21,22 and exhibits a similar free β-methoxyacrylate group to that of speciogynine, the motif that significantly increases the affinity of speciogynine at 5-HT1ARs relative to mitragynine (Figure 4).
Figure 4.
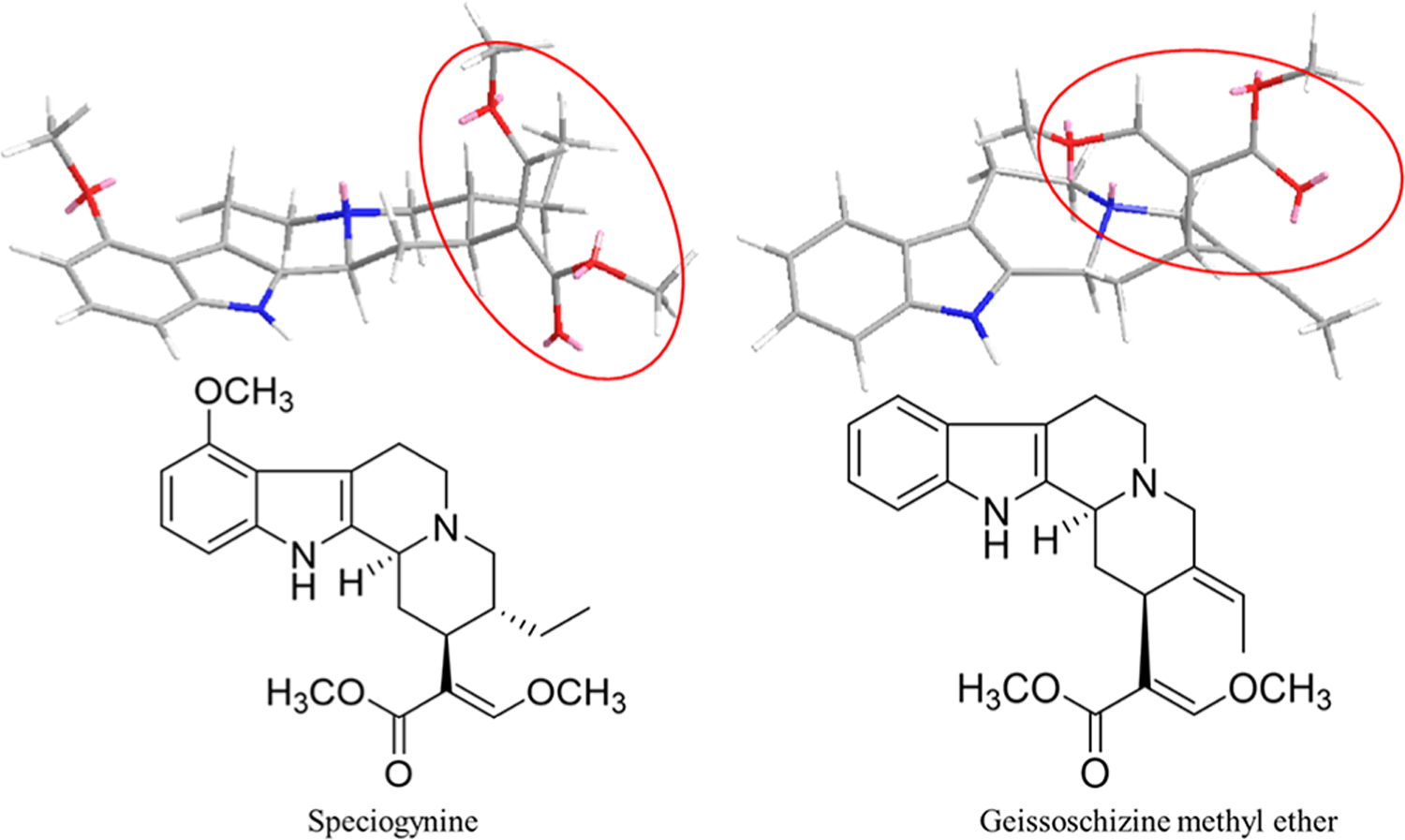
Two- and three-dimensional chemical structures for speciogynine and geissoschizine methyl ether.
As kratom reportedly can produce anxiolytic effects—which can be elicited by 5-HT1AR activation—we evaluated the in vivo 5-HT1AR activity of mitragynine, speciogynine, and paynantheine using the lower-lip retraction assay in rats, a behavior elicited by 5-HT1AR activation and blocked by 5-HT1AR antagonists.23 Ipsapirone, a 5-HT1AR agonist, and the selective 5-HT1AR antagonist WAY 100635 were used to confirm in vivo 5-HT1AR-dependent effects in the assay. Intraperitoneal (i.p.) administration of ipsapirone dose-dependently increased the percent of animals showing lower lip retraction (100% at 1.0 mg/kg) (Figure 5, top left panel, circles). Details for statistical analyses are shown in Tables S2 and S3. The ED50 value (95% confidence intervals) of ipsapirone to produce lower lip retraction was 0.431 (0.261−0.585) mg/kg (Table 3), and there was no sex difference in ED50 values (data not shown). Administration of 0.01 mg/kg WAY 100635 (i.p.) completely eliminated the lower lip retraction produced by 1.0 mg/kg dose of ipsapirone (Figure 5, top left panel, squares with cross hatch; Table S3), confirming the involvement of 5-HT1ARs in this assay. There was no significant interaction of sex with ipsapirone dose × WAY 100635 (Table S3).
Figure 5.
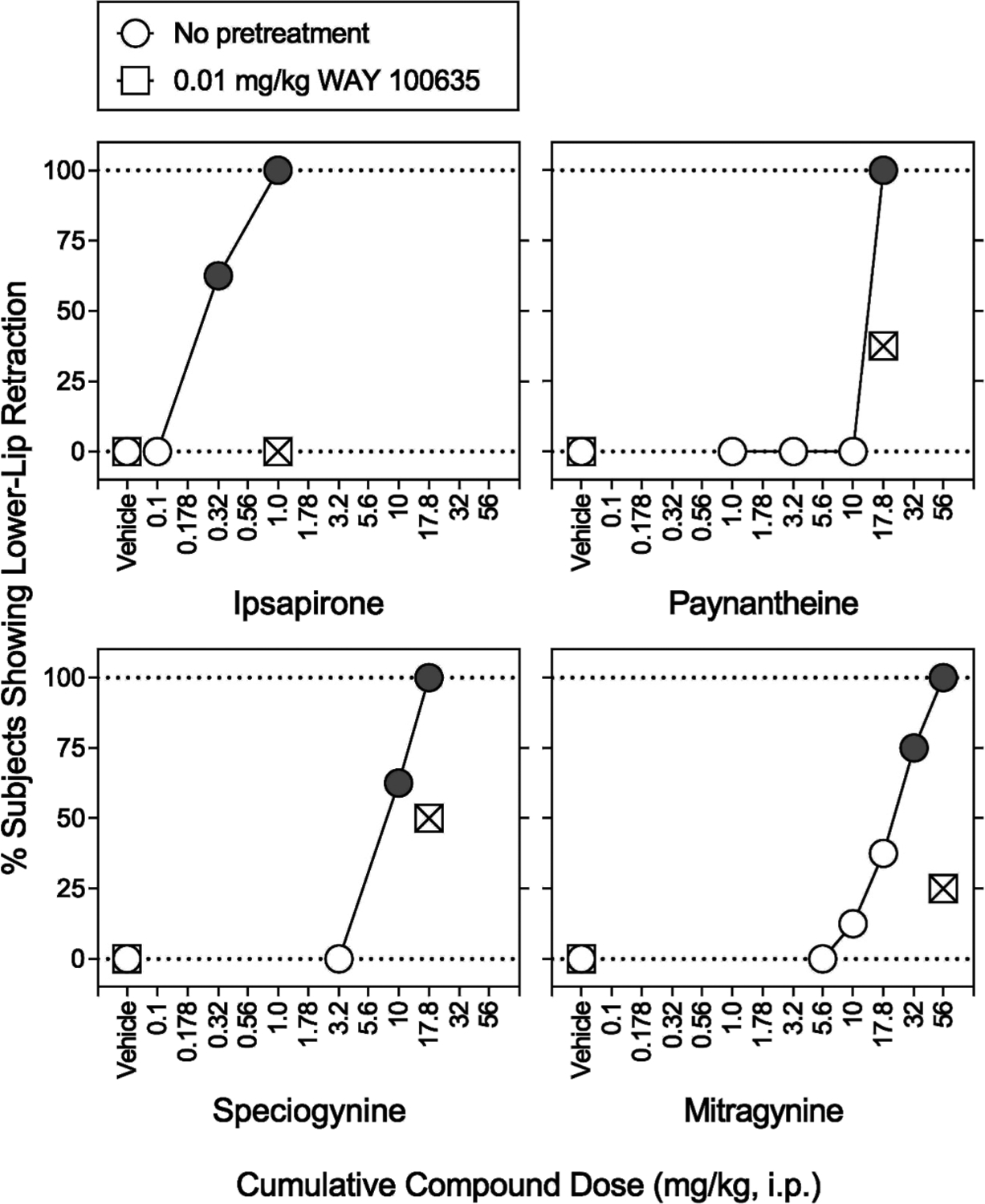
Effects of i.p. administration of various compounds alone and in combination with the 5-HT1AR antagonist WAY 100635 on expression of lower lip retraction in rats. Abscissae: vehicle and cumulative compound dose in mg/kg (i.p., log scale). Ordinates: percentage of subjects showing lower lip retraction. Each point represents the mean ± SEM (N = 8, four subjects per sex). No lower lip retraction was observed during each baseline measurement. Following the baseline measurement, a vehicle was administered i.p. 60 min prior to the next measurement (open circles). Following the second measurement, each lowest dose of test compounds was administered. Cumulative doses of each test compound were administered every 15 min prior to each measurement. Note that ipsapirone, paynantheine, speciogynine, and mitragynine produced 100% lower lip retraction. Each gray symbol indicates a significant difference from the respective vehicle. Using separate groups of rats, sensitivity of respective expression of lower lip retraction to WAY 100635 (0.01 mg/kg) was assessed. Following the baseline measurement, WAY 100635 (0.01 mg/kg) and then one of the highest doses of test compounds were administered i.p. 60 and 15 min, respectively, prior to measurement (squares). Behaviorally toxic effects were observed in a pilot observational study of higher doses. WAY 100635 significantly antagonized lower lip retraction produced by each test compound. Each square with cross hatch indicates a significant difference from the respective highest dose of test compounds. Details for statistical analyses are shown in Table 3 and Tables S2 and S3.
Table 3.
ED50 Values (95% Confidence Intervals) for the Induction of Lower Lip Retraction and Antinociceptive Effects of Test Compounds as Shown in Figures 5 and 8a
| ED50 (95% confidence intervals) in mg/kg | ||
|---|---|---|
| test compound | lower lip retraction (i.p.) | hotplate (i.v.) |
| ipsapirone | 0.431 (0.2610.585) | 3.41 (1.80–5.22) |
| ipsapirone in the presence of WAY 100635 | not assessed | 7.68 (5.72–9.59) |
| paynantheine | 12.7 (11.3–14.4) | 5.45 (4.51–6.48) |
| paynantheine in the presence of WAY 100635 | not testable | not testable |
| speciogynine | 9.10 (6.97–11.0) | 5.92 (4.13–8.35) |
| speciogynine in the presence of WAY 100635 | not testable | 11.6 (9.88–13.5) |
| mitragynine | 26.8 (21.2–32.9) | not testable |
The sample sizes are described in each figure legend. Each value represents data collected from males and females combined.
Like ipsapirone, paynantheine (17.8 mg/kg i.p.) produced lower lip retraction in 100% of animals tested (Figure 5, circles; Table S2). In the presence of 0.01 mg/kg WAY 100635, the 17.8 mg/kg dose of paynantheine resulted in 37.5% subjects showing lower lip retraction (two out of four females and one out of four males) (Figure 5, top right panel, squares with cross hatch; Table S3). These data suggest that paynantheine functions, in part, as a 5-HT1AR agonist in vivo. There was no significant interaction of sex with paynantheine dose or paynantheine dose × WAY 100635 (Tables S2 and S3).
Similar to paynantheine, speciogynine (i.p.) at 17.8 mg/kg elicited lower lip retraction in 100% of animals tested (Figure 5, lower left panel, circles; Table S2). In the presence of 0.01 mg/kg WAY 100635, the 17.8 mg/kg dose of speciogynine resulted in half of subjects (i.e., 50%) showing lower lip retraction (Figure 5, lower left panel, squares with cross hatch; Table S3). Thus, speciogynine, like paynantheine, showed effects in vivo suggestive of 5-HT1AR activation. There was no significant interaction of sex with the speciogynine dose or speciogynine dose × WAY 100635 (Tables S2 and S3).
Mitragynine (i.p.) at 56 mg/kg elicited lower lip retraction in 100% of animals tested (Figure 5, lower right panel, circles; Table S2). In the presence of 0.01 mg/kg WAY 100635, the 56 mg/kg dose of mitragynine elicited lower lip retraction in 25% of animals tested (two out of four females and zero out of four males) (Figure 5, lower right panel, squares with cross hatch; Table S3). These data suggest mitragynine likely behaves as a 5-HT1AR agonist in vivo, though with substantially lower potency than speciogynine and paynantheine, paralleling mitragynine’s much lower affinity at 5-HT1ARs. There was no significant interaction of sex with the mitragynine dose or mitragynine dose × WAY 100635 (Tables S2 and S3).
We also evaluated paynantheine’s and speciogynine’s in vitro functional activity at human 5-HT1ARs in a luminescent cAMP assay. Neither alkaloid activated or inactivated 5-HT1ARs; 5-HT and ipsapirone were tested in parallel as positive controls (Figure 6). The discrepancy observed between in vivo and in vitro results led us to hypothesize that paynantheine and speciogynine are metabolized to alkaloids that are 5-HT1AR agonists. As shown in Figure 7 and Table 4, 9-O-desmethylpaynantheine (gambireine) and 9-O-desmethylspe-ciogynine (gambirine), a metabolite of paynantheine and speciogynine, respectively, behaved as fully efficacious 5-HT1AR agonists. Also shown in Figure 7 and Tables 2 and 4, affinities and potencies of both metabolites at 5-HT1ARs were similar, though lower, than those of the parent compounds. These data provide, for the first time, evidence that metabolites of kratom alkaloids, in addition to 7-hydroxymitragynine, may have in vivo pharmacological activity.
Figure 6.
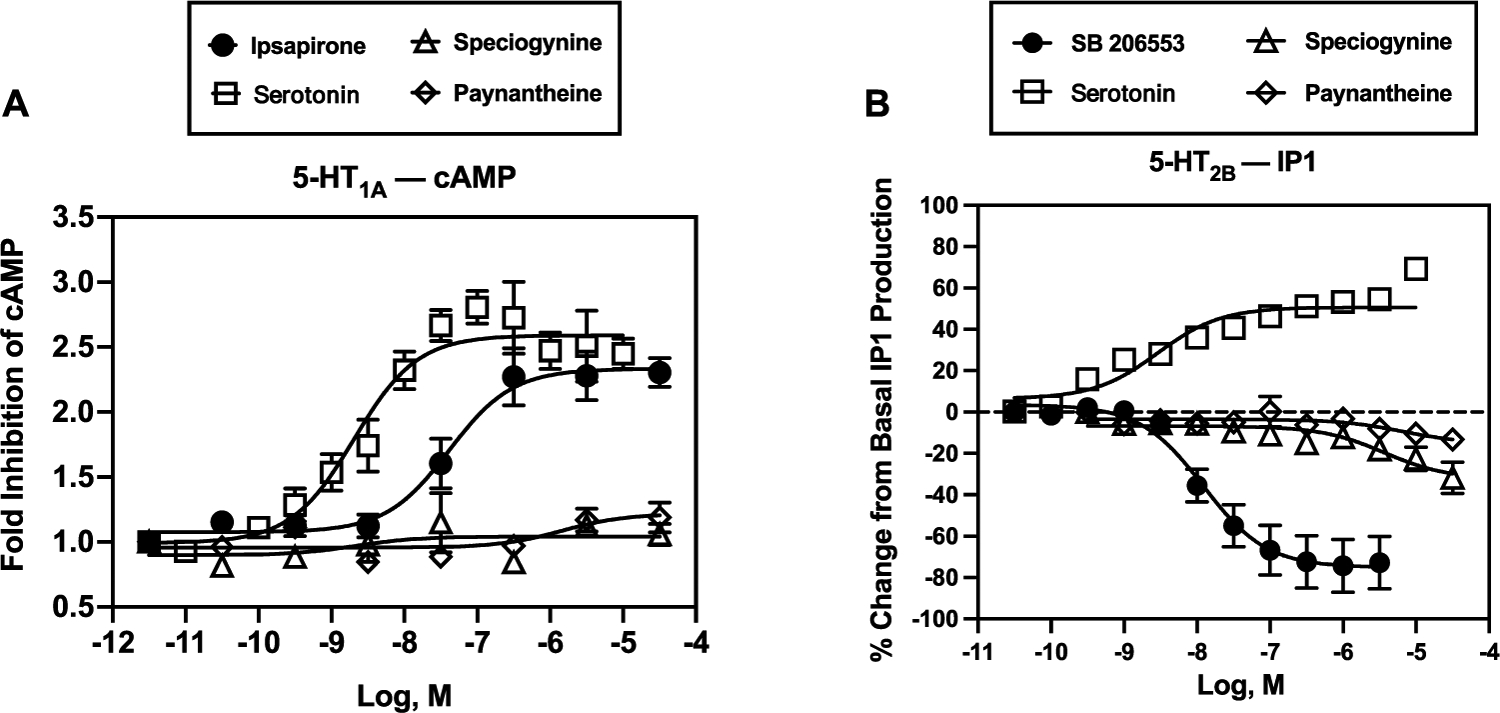
In vitro function of speciogynine and paynantheine at human 5-HT1ARs (A) and 5-HT2BRs (B). Abscissae: concentrations of test compounds (molar, log scale). Ordinates: (A) serotonin and ipsapirone activated 5-HT1ARs as measured by inhibition of forskolin-stimulated production of cAMP, whereas speciogynine and paynantheine showed no significant functional effects. (B) Serotonin activated 5-HT2BRs as measured by increased production of inositol phosphate 1 (IP1), whereas SB 206553 inactivated 5-HT2BRs; speciogynine and paynantheine showed no significant functional effects. Each symbol represents the mean ± SEM from at least three independent determinations.
Figure 7.
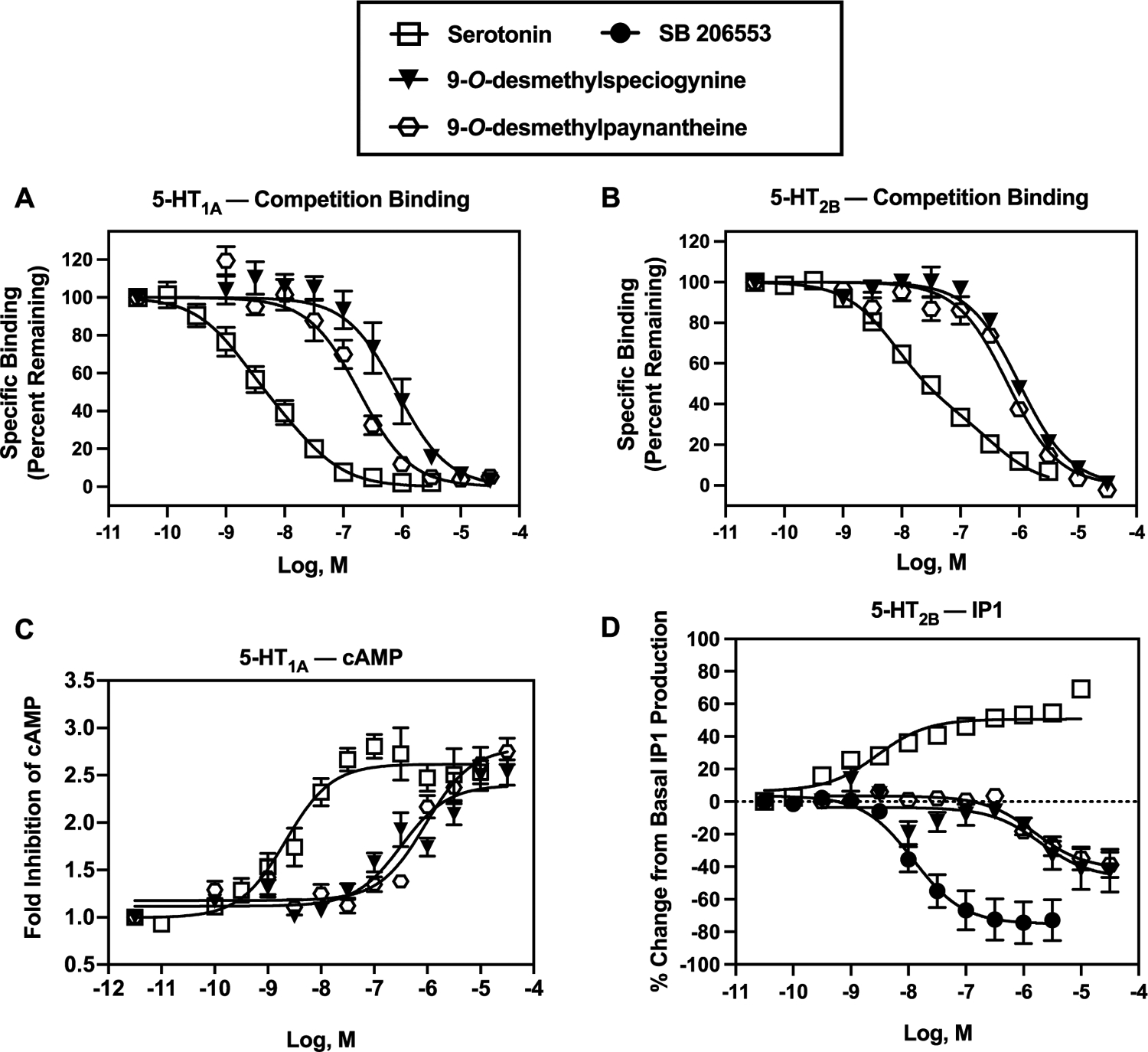
In vitro binding and function at human 5-HT1ARs (A, C) and 5-HT2BRs (B, D). Abscissae: concentrations of test compounds (molar, log scale). Ordinates: (A) 5-HT1AR competition binding with [3H]8-OH-DPAT. (B) 5-HT2BR competition binding with [3H]LSD. Note that serotonin data from both 5-HT1AR [3H]8-OH-DPAT and 5-HT2BR [3H]LSD competition binding fit best to a two-site model, relative to a one-site model (P values: <0.05). Ki values for serotonin at 5-HT1ARs were 0.41 nM (high affinity) and 6.7 nM (low affinity), and Ki values for serotonin at 5-HT2BRs were 5.0 nM (high affinity) and 230 nM (low affinity); R2 values were 0.726 and 0.924, respectively. The Ki results for serotonin at 5-HT1A and 5-HT2BRs reported in Table 2 of the manuscript are from one-site analyses, wherein R2 values were 0.715 and 0.895, respectively. (C) Serotonin, 9-O-desmethylpaynantheine, and 9-O-desmethylspeciogynine activated 5-HT1ARs as measured by inhibition of forskolin-stimulated production of cAMP. (D) Serotonin activated 5-HT2BRs as measured by increased production of IP1, whereas SB 206553, 9-O-desmethylpaynantheine, and 9-O-desmethylspeciogynine inactivated 5-HT2BRs. Table 4 shows the pIC/EC50 and E/Imax values. Each data point represents the mean (±SEM) of at least three independent experiments. In each experiment, serotonin and SB 206553 were tested in duplicate while the kratom alkaloids were tested in triplicate.
Chronic 5-HT2BR activation has been linked with valvular heart disease,24,25 and so the binding of speciogynine, paynantheine, and their 9-O-desmethyl metabolites to 5-HT2BR raised the question of a potential cardiovascular risk to chronic kratom users. To evaluate this concern, we examined the functional activity of these alkaloids using a 5-HT2BR−phosphoinositide hydrolysis assay with 5-HT and SB 206553 as the positive and inverse agonist controls (respectively, Figure 6B). Speciogynine and paynantheine showed no agonist activity at human 5-HT2BRs, while 9-O-desmethylpaynantheine and 9-O-desmethylspeciogynine metabolites clearly demonstrated an inverse partial agonist action (Figure 7D). These data suggest that speciogynine, paynantheine, and their 9-O-desmethyl metabolites are unlikely to induce the 5-HT2BR-mediated cell proliferation linked to cardiac valvulopathy.
Using the hotplate assay, cumulative mitragynine doses administered 5 min apart (up to 10 mg/kg, i.v.) did not produce antinociception, and 32 mg/kg i.v. mitragynine was lethal in our previous study.12 The lack of antinociceptive effects of mitragynine when cumulatively administered i.v. in a shorter (5 min) interval in the previous study12 may reflect less conversion of mitragynine to an active metabolite. Indeed, the CYP-450 3A metabolite 7-hydroxymitragynine was 108-fold more potent than the parent compound mitragynine in producing morphine-like discriminative-stimulus effects in rats.11,15,26,27
In contrast to i.v. mitragynine,12 a 10 mg/kg dose of i.v. ipsapirone produced 87.4% (8.78%) of the percentage of maximum possible effect in the hotplate assay; the antinociception dose-effect function of ipsapirone was shifted to the right 2.3-fold by WAY 100635 (0.01 mg/kg, i.p.) (Figure 8, squares; Table S4). Details for statistical analyses are shown in Table S4. The ED50 value of i.v. ipsapirone to produce antinociception was 3.41 (1.80−5.22) mg/kg (Table 3). Thus, the present results suggest that the antinociceptive effects of ipsapirone are 5-HT1AR mediated.
Figure 8.
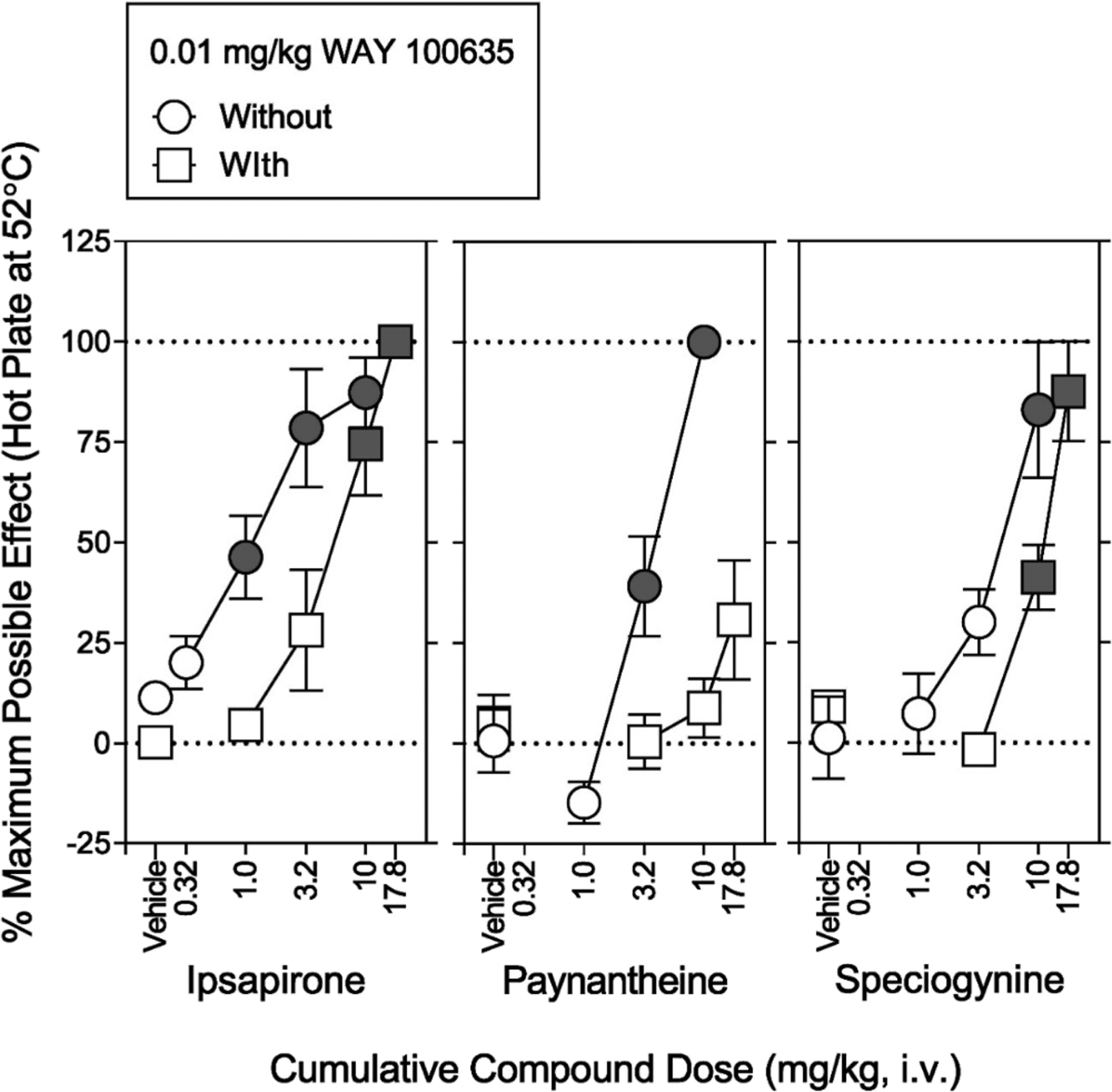
Effects of i.v. injection of ipsapirone, paynantheine, and speciogynine on hotplate latency in catheterized rats. The catheterized rats differ from rats in Figure 5. Abscissae: vehicle, cumulative compound dose in mg/kg (i.v., log scale). Ordinates: percentage of maximum possible effects. The hotplate was maintained at 52 ± 0.1 °C. Each symbol represents the mean ± SEM (N = 6, three subjects per sex). In baseline values across six groups (ipsapirone alone and in combination with WAY 100635, paynantheine alone and in combination with WAY 100635, and speciogynine alone and in combination with WAY 100635), there was no significant difference of the treatment group (F5,24 = 1.44; P = 0.245), sex (F1,24 = 3.16; P = 0.088), and interaction of the two (F5,24 = 1.54; P = 0.215). Following the baseline measurement, the vehicle was administered i.p. 60 min prior to the next measurement (open squares). Following the second measurement, cumulative doses were sequentially administered. Cumulative doses of each test compound were administered every 5 min prior to each measurement. Note that ipsapirone, paynantheine, and speciogynine significantly increased the maximum possible effect. Each gray symbol indicates a significant difference from the respective vehicle. Using separate groups of rats, sensitivity of respective antinociceptive effects to WAY 100635 (0.01 mg/kg) was assessed. Following the baseline measurement, WAY 100635 (0.01 mg/kg) was administered i.p. 60 min prior to measurement (squares). WAY 100635 shifted to the right dose-effect functions of each test compound. Paynantheine (32 mg/kg) in the presence of WAY 100635 caused lethality in all rats tested. Details for statistical analyses are shown in Table 3 and Table S4.
As with i.v. ipsapirone, a 10 mg/kg dose of i.v. paynantheine produced antinociceptive effects that were sensitive to WAY 100635 (0.01 mg/kg) (Figure 8, squares; Table S4). The ED50 value of i.v. paynantheine to produce antinociception was 5.45 (4.51−6.48) mg/kg (Table 3). Similar to i.v. ipsapirone and i.v. paynantheine, i.v. speciogynine produced antinociception in a manner sensitive to WAY 100635 (Figure 8, squares; Table S4). The ED50 value of i.v. speciogynine to produce antinociception was 5.92 (4.13−8.35) mg/kg (Table 3). Collectively, the present results suggest that the apparent antinociceptive effects of paynantheine and speciogynine may be mediated by stimulation of 5-HT1ARs.
The WAY 100635-reversible lower lip retraction illustrates involvement of 5-HT1AR in the in vivo effects of ipsapirone, paynantheine, speciogynine, and mitragynine. Though Ki values at 5-HT1AR did not significantly correlate with the ED50 values for lower-lip retraction or antinociception, the correlation approached significance (P = 0.0508, Figure 9); this may be due to the low sample size (n = 4) chosen in the experimental design. Given the apparently similar affinity of corynantheidine and ajmalicine at 5-HT1ARs to the alkaloids evaluated here (Table 1), future in vitro 5-HT1AR and in vivo lower lip retraction studies with these two alkaloids may also be warranted.
Figure 9.
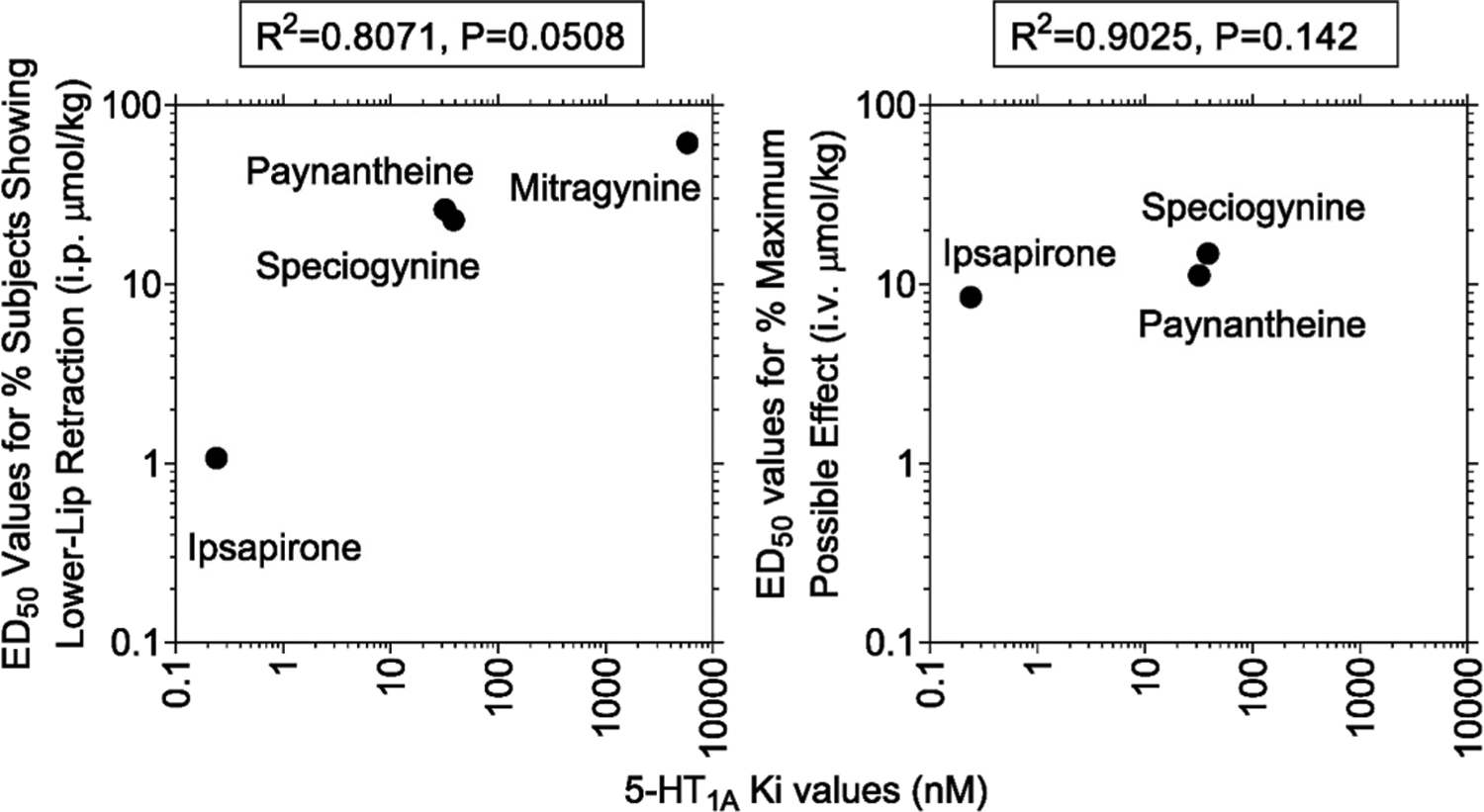
Correlations between in vitro 5-HT1AR affinity (Ki values) and potency to produce lower lip retraction and antinociception. Ordinates: (left panel) ED50 value in μmol/kg (i.p.) to produce lower lip retraction; (right panel) ED50 value in μmol/kg (i.v.) to produce antinociception. Abscissae: Ki values at 5-HT1ARs in nM. (Left panel) The R2 value was 0.807 (P = 0.0508). (Lower panels) The R2 values were 0.9025 (P = 0.142).
In vivo pharmacokinetic studies were performed to assess the systemic exposure, distribution, and elimination of i.v. paynantheine and speciogynine (Figure 10 and Table 5). Single-dose pharmacokinetic studies of paynantheine (2.5 mg/kg, n = 4) and speciogynine (1.25 mg/kg, n = 3) were performed in male Sprague Dawley rats. Blood samples (100 μL) were collected at pre-dose and 0.08, 0.25, 0.5, 0.75, 1, 2, 4, 8, 12, 18, and 24 h post-dose. The plasma concentration−time profiles for each alkaloid are shown in Figure 10, and pharmacokinetic parameters are shown in Table 5.
Figure 10.
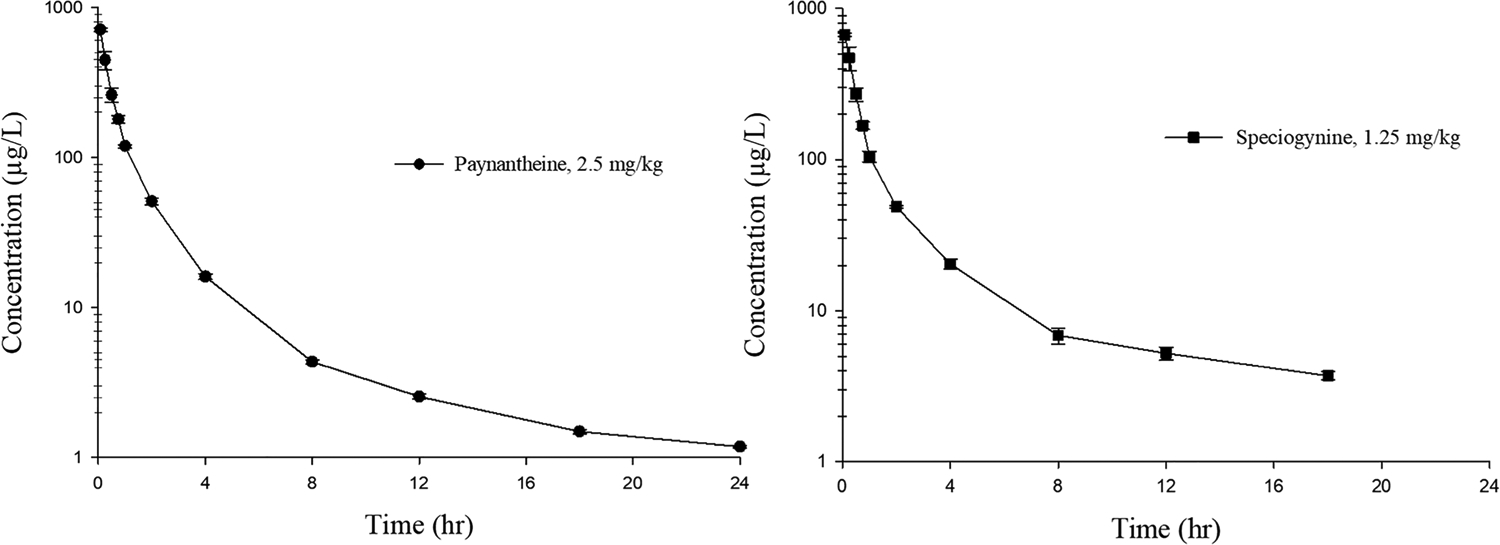
Mean plasma concentration-time profile of (left) i.v. paynantheine and (right) i.v. speciogynine in male rats. Each symbol represents mean ± SEM (n = 4 paynantheine, n = 3 speciogynine).
Table 5.
Pharmacokinetic Parameters of Speciogynine, Paynantheine, and Mitragynine Following a Single i.v. Administration in Male Ratsa
| paynantheine | speciogynine | mitragynine28 | |
|---|---|---|---|
| parameters | 2.5 mg/kg (n = 4) | 1.25 mg/kg (n = 3) | 5 mg/kg (n = 6) |
| CL (L/h/kg) | 5.3 ± 0.21 | 3.2 ± 0.01 | 1.3 ± 0.1 |
| Vz (L/kg) | 71.9 ± 6.5 | 30.1 ± 8.4 | 8.0 ± 1.5 |
| AUCinf/dose (h·μg/L/mg/kg) | 235.2 ± 8.9 | 495.1 ± 2.7 | 849.8 ± 112.8 |
| ke (1/h) | 0.08 ± 0.01 | 0.13 ± 0.05 | 0.12 ± 0.04 |
Values are mean ± SEM. Abbreviation: CL, clearance; Vz, apparent volume of distribution; AUCinf, total exposure extrapolated to infinity; ke, elimination rate constant.
The concentration−time profiles of paynantheine and speciogynine showed biexponential decay, which is typical of i.v. administered drugs. Like mitragynine, both paynantheine and speciogynine exhibited a volume of distribution (Vz) greater than the rat blood volume (0.085 L/kg),29 indicating extensive tissue distribution. Both paynantheine and speciogynine were cleared from systemic circulation more rapidly than mitragynine so that when exposure was normalized for dose (AUCinf/dose), mitragynine exposure was 3.6-fold higher than paynantheine and 1.7-fold higher than speciogynine. It is worth noting that i.p. administration was used in the in vivo studies where paynantheine and speciogynine exhibited 5-HT1AR-mediated effects, an administration route that is subject to first-pass hepatic metabolism (unlike i.v. administration) and so may well produce higher metabolite levels. Further pharmacokinetic studies examining 9-desmethylmetabolite formation following i.p. and p.o. administration of paynantheine and speciogynine are warranted given our discovery that these metabolites are more efficacious than the parent molecules as 5-HT1AR agonists.
CONCLUSIONS
Speciogynine and paynantheine comprise approximately 15% of the total alkaloid content of kratom.2,7 This study demonstrates that these alkaloids have a high binding affinity for 5-HT1AR and 5-HT2BRs (relative to mitragynine, the predominant alkaloid in kratom) but do not activate canonical G-protein mediated signal transduction through these receptors. The results from 5-HT1AR functional assays were surprising as we also found that speciogynine and paynantheine exert in vivo pharmacological effects consistent with 5-HT1AR activation (effects blocked in the presence of a selective 5-HT1AR antagonist). These apparently paradoxical findings are perhaps reconcilable through our discovery that paynantheine and speciogynine 9-O-desmethyl metabolites also bind to 5-HT1ARs and 5-HT2BRs and are efficacious at stimulating 5-HT1ARs and inhibiting 5-HT2BRs. As neither parent nor metabolites were agonists at 5-HT2BRs, they are unlikely to induce cardiac valvulopathy. Our results suggest that just as mitragynine’s metabolism to 7-hydroxymitragynine results in greater activity at μ-opioid receptors,11 paynantheine’s and speciogynine’s activity at 5-HT1ARs and 5-HT2BRs may be principally mediated via their metabolites, 9-O-desmethylspeciogynine and 9-O-desmethylpaynantheine. Future studies may clarify whether the serotonergic activity of these metabolites contributes to the analgesic, anxiolytic, or antidepressant effects reported by kratom users.30,31
EXPERIMENTAL SECTION
General Chemistry.
The compounds were available in our alkaloid library and were isolated and structurally elucidated through 1H and 13C NMR using a Bruker model Avance NEO 600 NMR spectrometer operating at 600 MHz in 1H and 151 MHz in 13C (Bruker Co, Billerica, MA, USA). High-resolution mass spectrometry HRMS and purity (≥98%) were determined using an Agilent 1290 Infinity series ultraperformance liquid chromatography (UPLC) system equipped with a photodiode array detector and quadrupole-time-of-flight (QTOF) Agilent 6540 mass spectrometer (Agilent Co, Santa Clara, CA, USA). The data was acquired by MassHunter data acquisition software (version B.09.00). The samples at a 10 μM concentration were injected onto a Waters Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 100 mm). The gradient elution was performed using a mobile phase of 10 mM ammonium acetate (Sigma Aldrich Co., St. Louis, MO) buffer pH 3.5 (solvent A) and acetonitrile (solvent B, Sigma Aldrich Co.) at a flow rate of 0.5 mL/min. The gradient was started at 10% B and maintained till 1 min, then linearly increased to 90% B till 3 min, maintained at 90% B till 3.5 min, then decreased to 10% B from 3.5 to 4 min, and maintained at 10% B till 5 min. The column oven and autosampler temperatures were maintained at 45 and 4 °C, respectively. The HPLC purity of samples was determined using the UV chromatogram extracted at a 254 nm wavelength. The high-resolution mass spectrometry chromatograms for each compound were extracted as base peak chromatograms at respective molecular (M + H)+ ion peaks. The isolation of mitragynine, speciociliatine, speciogynine, paynantheine, corynantheidine, and ajmalicine were conducted using a procedure described previously.32 7-Hydroxymitragynine and 9-hydroxycorynantheidine were obtained by semi-synthesis from mitragynine using published methods.12 The detailed characterization and purity determination of corynantheidine, 9-hydroxycorynantheidine, mitragynine, 7-hydroxymitragynine, and speciociliatine have been reported previously.12 The detailed characterization of ajmalicine, 9-O-desmethylpaynantheine, 9-O-desmethylspeciogynine, paynantheine, and speciogynine are shown below; the 1H and 13C NMR spectra, HR-MS spectra, and ULPC-UV chromatograms are shown in the Figures S1−S20 (Supporting information).
Methyl(4S,4aR,13bS,14aS)-4-methyl-4a,5,7,8,13,13b,14,14a-octahydro-4H-indolo[2,3-a]pyrano[3,4-g]quinolizine-1-carboxylate (Ajmalicine).
1H NMR (600 MHz, CDCl3): δ 9.20 (s, 1H), 7.54 (d, J = 1.8 Hz, 1H), 7.48−7.43 (m, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.12 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 7.09−7.03 (m, 1H), 4.43 (dd, J = 6.7, 4.0 Hz, 1H), 3.73 (s, 3H), 3.38 (dq, J = 11.3, 2.3 Hz, 1H), 3.24 (dt, J = 12.6, 3.1 Hz, 1H), 3.10 (dd, J = 11.2, 5.9 Hz, 1H), 3.05−2.98 (m, 1H), 2.95 (dd, J = 10.7, 3.0 Hz, 1H), 2.79−2.73 (m, 1H), 2.67 (td, J = 11.2, 4.4 Hz, 1H), 2.43−2.35 (m, 1H), 2.22 (t, J = 11.0 Hz, 1H), 2.16−2.09 (m, 1H), 1.28 (q, J = 11.8 Hz, 1H), 1.18 (d, J = 6.6 Hz, 3H). 13C NMR (151 MHz, CDCl3): δ 167.86, 155.07, 136.30, 134.35, 127.01, 121.14, 119.00, 117.88, 111.01, 107.18, 106.42, 73.71, 60.26, 56.65, 53.16, 51.00, 40.58, 32.37, 30.44, 21.57, 14.86. NMR data align with a previous report.33 HRMS (ESI+) calculated for C21H25N2O3+ [M + H]+: 353.1860, found: 353.1859. HPLC purity: >99%.
Methyl(E)-3-methoxy-2-((2S,3R,12bS)-8-methoxy-3-vinyl-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)acrylate (Paynantheine).
1H NMR (600 MHz, CDCl3): δ 8.00 (s, 1H), 7.35 (s, 1H), 7.01 (t, J = 7.9 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.47 (d, J = 7.7 Hz, 1H), 5.68−5.53 (m, 1H), 5.02 (ddd, J = 17.2, 2.1, 0.9 Hz, 1H), 4.97 (dd, J = 10.2, 2.0 Hz, 1H), 3.88 (s, 3H), 3.76 (s, 3H), 3.71 (s, 3H), 3.31−3.24 (m, 1H), 3.24−3.14 (m, 1H), 3.12−2.99 (m, 4H), 2.78 (td, J = 11.7, 3.8 Hz, 1H), 2.60 (td, J = 11.2, 4.4 Hz, 1H), 2.35−2.25 (m, 1H), 2.11 (d, J = 12.1 Hz, 1H), 2.02−1.93 (m, 1H). 13C NMR (151 MHz, CDCl3): δ 168.98, 159.94, 154.50, 139.43, 137.42, 133.00, 121.85, 117.53, 115.49, 111.59, 107.71, 104.35, 99.70, 61.59, 61.37, 60.09, 55.31, 53.14, 51.36, 42.79, 38.82, 33.37, 23.78. NMR data align with a previous report.34 HRMS (ESI+) calculated for C23H29N2O4+ [M + H]+: 397.2122, found: 397.2130. HPLC purity: >96%.
Methyl(E)-2-((2S,3R,12bS)-3-ethyl-8-methoxy-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (Speciogynine).
1H NMR (strong line broadening for all ring protons) (600 MHz, CDCl3): δ 7.88 (s br, 1H), 7.38 (s br, 1H), 7.00 (t, J = 7.9 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.52−6.42 (m, 1H), 3.88 (s, 5H), 3.82−3.67 (m, 5H), 3.32−2.98 (m, 5H), 2.73−2.53 (m, 2H), 2.34−2.26 (m, 1H), 2.13−1.88 (m, 3H), 1.44 (s, 1H), 0.97−0.76 (m, 3H). 13C NMR (126 MHz, CDCl3) (incomplete due to broadening of signals): δ 13C NMR (126 MHz, CDCl3): δ 160.07, 154.60, 137.46, 133.22, 122.00, 117.65, 107.83, 104.35, 99.85, 61.76, 58.54, 55.43, 51.58, 38.82, 24.51, 23.83, 11.35. For an explanation of the line broadening in the demethoxy (corynantheine) series, see ref.35 HRMS (ESI+) calculated for C23H31N2O4+ [M + H]+: 399.2278, found: 399.2286. HPLC purity: >98%.
Methyl(E)-2-((2S,3R,12bS)-8-hydroxy-3-vinyl-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (Gambireine, 9-O-desmethylpaynantheine).
AlCl3 (594 mg, 4.389 mmol) was added to a stirring solution of paynantheine (580 mg, 1.463 mmol) in anhydrous DCM (25 mL) previously cooled to 0 °C under an argon atmosphere. Ethanethiol (2.13 mL, 29.26 mmol) was slowly introduced dropwise, and the mixture was allowed to reach room temperature and then stirred at room temperature for 2 h. The mixture was quenched with sat. aq. NaHCO3 and then partitioned between water and DCM. The aqueous layer was extracted with DCM (2×), and the combined organic layers were dried with Na2SO4, filtered, and evaporated to give a yellow solid (510 mg). The crude compound was purified by flash chromatography (0−5% MeOH in DCM) to give the product as a pale yellow solid (350 mg, 71%) spectroscopically equivalent to previously reported examples.36
1H NMR (600 MHz, CDCl3): δ 7.72 (s, 1H), 7.33 (s, 1H), 6.90 (t, J = 7.8 Hz, 1H), 6.83 (dd, J = 8.2, 0.7 Hz, 1H), 6.38 (dd, J = 7.5, 0.7 Hz, 1H), 5.62−5.53 (m, 1H), 5.24 (broad s, 1H), 5.03−4.92 (m, 2H), 3.75 (s, 3H), 3.69 (s, 3H), 3.31−3.25 (m, 1H), 3.23 (ddq, J = 15.2, 8.4, 2.6 Hz, 1H), 3.11−3.00 (m, 3H), 2.98 (ddt, J = 15.5, 4.3, 1.7 Hz, 1H), 2.76 (td, J = 11.7, 3.8 Hz, 1H), 2.60 (td, J = 11.3, 4.4 Hz, 1H), 2.34−2.25 (m, 1H), 2.15−2.04 (m, 1H), 1.94 (d, J = 12.4 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 168.86, 159.95, 150.10, 139.33, 137.98, 133.27, 122.04, 116.63, 115.55, 111.43, 106.90, 104.40, 103.86, 61.59, 61.29, 59.98, 52.99, 51.36, 42.79, 38.59, 33.35, 23.48. HRMS (ESI+) calculated for C22H27N2O4+ [M + H]+: 383.1965, found: 383.1976. HPLC purity: >99%.
Methyl(E)-2-((2S,3R,12bS)-3-ethyl-8-hydroxy-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (Gambirine, 9-O-desmethylspeciogynine).
AlCl3 (510 mg, 3.765 mmol) was added to a stirring solution of speciogynine (500 mg, 1.255 mmol) in anhydrous DCM (25 mL) previously cooled to 0 °C under an argon atmosphere. Ethanethiol (1.82 mL, 25.10 mmol) was slowly introduced dropwise, and the mixture was allowed to reach room temperature and then stirred at room temperature for 2 h. The mixture was quenched with sat. aq. NaHCO3 and then partitioned between water and DCM. The aqueous layer was extracted with DCM (2×), and the combined organic layers were dried with Na2SO4, filtered, and evaporated to give a yellow solid (470 mg). The crude compound was purified by flash chromatography (0−10% MeOH in DCM) to give the product as a pale yellow solid (360 mg, 75%) spectroscopically equivalent to previously reported examples.37
1H NMR (600 MHz, MeOD): δ 7.49 (s, 1H), 6.80 (dd, J = 8.1, 7.0 Hz, 2H), 6.78 (dd, J = 8.1, 1.3 Hz, 2H), 6.32 (dd, J = 7.1, 1.3 Hz, 1H), 3.97−3.77 (m, 3H), 3.77−3.60 (m, 3H), 3.28 (broad s, 1H), 3.27−3.18 (m, 1H), 3.13 (dd, J = 11.4, 3.9 Hz, 1H), 3.08 (dd, J = 11.6, 5.8 Hz, 1H), 3.03 (ddt, J = 15.9, 4.7, 1.6 Hz, 1H), 2.70 (broad s, 1H), 2.62 (td, J = 11.6, 4.6 Hz, 1H), 2.34 (tdd, J = 11.3, 9.4, 3.6 Hz, 1H), 2.23−1.99 (m, 3H), 1.45 (broad s, 1H), 1.09 (broad s, 1H), 0.90 (t, J = 7.5 Hz, 3H). 13C NMR (151 MHz, MeOD): δ 169.73, 160.41, 151.15, 138.69, 132.16, 121.20, 116.59, 111.23, 109.68, 106.10, 102.85, 102.83, 60.82, 60.48, 53.37, 50.56, 49.86, 38.15, 38.01, 33.19, 24.08, 22.96, 9.59. HRMS (ESI+) calculated for C22H29N2O4+ [M + H]+: 385.2122, found: 385.2136. HPLC purity: >99%.
In Vitro Radioligand Competition Binding.
The kratom alkaloids were tested at Eurofins Cerep (Celle l’Evescault, France) and at Mercer University College of Pharmacy (Atlanta, GA) for their in vitro receptor binding affinities. 5-HT hydrochloride (Alfa Aesar, Haverhill, MA), SB 206553 hydrochloride (Tocris, Minneapolis, MN), (±)-DOI hydrochloride (Sigma-Aldrich, St. Louis, MO), M100907 hydrochloride (a gift from Dr. Kevin Murnane, synthesized by Dr. Kenner Rice), and mianserin hydrochloride (Alfa Aesar, Haverhill, MA) were used as controls and/or for nonspecific binding in tests conducted at Mercer University. Briefly, each cell membrane homogenate was incubated with a radioligand in the absence or presence of test compounds in a buffer. Nonspecific binding was determined in the presence of a specific agonist or antagonist at the target receptor. Following incubation, each sample was filtered rapidly under vacuum through glass fiber filters presoaked in a buffer and rinsed several times with an ice-cold buffer using a cell harvester. The filters were soaked with scintillation fluid, and counts per minute were detected using photodetectors. The experimental conditions are summarized in Table S1. The IC50 values (concentration causing a half-maximal inhibition of control specific binding) from the displacement data were computed using a nonlinear, least squares regression analysis (Prism; GraphPad Software, Inc., San Diego, CA) and then converted to Ki values using the Cheng−Prusoff equation.38
In Vitro 5-HT1A Receptor cAMP Signaling.
cAMP measurements were performed with a GloSensor cAMP Assay (Promega Corporation, Madison, WI) according to the manufacturer’s protocol. CHO cells were transfected with 7−11 μg of human 5-HT1AR cDNA and 1 μg of 22F Glosensor plasmid (Promega) using a TransIT-CHO reagent (Mirus Bio, Madison WI), based on established methods.39 Twenty-four hours after transfection, cells were plated in media containing 10% dialyzed FBS and DMEM in white, opaque 96-well plates (PerkinElmer, Waltham, MA) at a density of 150,000 cells/100 μL and incubated at 37 °C, 5% CO2 overnight. The next day, media were removed from plates, and cells were incubated with 90 μL of assay buffer (1× HBSS +20 mM HEPES) + 1% Glosensor substrate for 2 h in the dark at room temperature. Cells were then treated with compounds made in assay buffer for 30 min in the dark at room temperature. Then, cells were stimulated with 10 μM (final concentration) forskolin for 10 min in the dark at room temperature. Luminescence was measured with a Microbeta2 Microplate Counter (PerkinElmer). Data were analyzed as normalized means (± SEM) from at least three independent determinations, with controls and the kratom alkaloids tested at each concentration in triplicate.
5-HT2B Receptor Phosphoinositide Hydrolysis.
The activity of kratom alkaloids at human 5-HT2B receptors was measured using the IP-One homogeneous time-resolved fluorescence (HTRF) assay (Cisbio, Bedford, MA). The assay was performed per the manufacturer’s protocol with optimization. Transfection of HEK-293 cells with 7−11 μg of human 5-HT2B receptor cDNA using the TransIT 2020 reagent (Mirus) was performed as previously described.39 Forty-eight hours after transfection, cells were serum-starved for 90 min in DMEM and plated at 25,000 cells per well in 384-well plates. Cells were treated with compounds and incubated at 37 °C, 5% CO2 for 90 min. The reaction was terminated with lysis buffer containing d2-labeled IP1 and anti-IP1-Cryptate. HTRF-FRET was measured by a Mithras LB 940 Multimode Microplate Reader (Berthold Technologies, Bad Wildbad, Germany). Data were analyzed as normalized means (± SEM) from at least three independent determinations, with controls and the kratom alkaloids tested at each concentration in triplicate.
In Vivo Functional Studies.
Salt and enantiomeric forms of the compounds used in the present study were as follows: ipsapirone (Tocris), mitragynine hydrochloride, paynantheine oxalate, speciogynine free base, and WAY 100635 maleate (Sigma Aldrich Co.). Doses were expressed as the weight of the salt or free base form as listed above. Compounds were dissolved in a vehicle consisting of sterile water (HyClone Water for Injection, 1 L, Fisher Scientific, Waltham, MA) containing 5% Tween80 (polyoxyethylenesorbitanmonooleate, Fisher Scientific) and 5% propylene glycol (Sigma Aldrich Co.). Compounds and vehicle were administered i.p. or i.v. Each solution was filtered with a 0.2 μm pore size syringe filter (Millex-LG, 0.20 μm, SLLG025SS, Sigma Aldrich Co.) prior to injections. The injection volumes were 1.0−5.0 mL/kg of the subject body weight. The dose ranges and pretreatment times were chosen based on our preliminary data and published methods.12,27,40
Animals.
Adult, naïve female and male (N = 42 rats per sex) Sprague Dawley rats (Envigo, Indianapolis, IN), age 10 weeks and weighing approximately 200 and 310 g for females and males, respectively, were singly acclimated in ventilated cages for at least 3 days to a temperature- (21.9 °C ± 1.9 °C) and humidity-controlled (53% ± 14%) vivarium with a 12 h light/dark cycle (lights on at 07:00 h) during which food (2918 Teklad global 18% protein rodent diets, Envigo) and reverse osmosis water were available in the home cage at all times. Among the 82 rats, 36 rats (18 rats per sex) were pre-implanted with a catheter into a right external jugular vein prior to entering our facility. After at least 3 days of acclimation, experiments commenced. These studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Florida, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC), and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments were conducted in the light cycle (08:00−17:00 h) using drug naïve subjects.
Lower Lip Retraction Following Intraperitoneal Administration.
The sample size of each experimental group per treatment was eight (four subjects per sex). The temperature, humidity, and light/dark cycle in the experimental room were equivalent to those in the vivarium. Prior to commencement of each experiment, the body weight of each subject was measured. Each experimental session started with a baseline measurement for lower lip retraction (observation of visible lower incisors)23 in each subject (see below for more details). Vehicle and cumulative doses of a test compound were administered i.p. at intervals of 15 min to assess the effects of compounds alone. Following the baseline measurements, the vehicle or WAY 100635 (0.01 mg/kg) were administered followed by cumulative doses (every 15 min) or a single dose of a test compound i.p. at 60 and 15 min, respectively, prior to a measurement of lower lip retraction. The assessment of effects of a test compound was continued until 100% subjects showed lower lip retraction. Lower lip retraction was assessed by trained experimenters who were blind to treatment conditions. The expression of lower lip retraction was assessed by observing each subject for 5 s before or every 15 min after an injection.
Hotplate.
The whole size of the hotplate analgesia meter that included a hard-black anodized aluminum plate was 33.1 cm long, 28.8 cm wide, and 9.9 cm high and a built-in digital thermometer (1440 Analgesia Hotplate with RS-232 Port and Software, Columbus Instruments, Columbus, OH). A clear acrylic cage (26.7 cm long, 26.7 cm wide, and 31.0 cm high) was fitted over the square plate to confine an animal on the plate (total height: 40.9 cm). The size of the heated surface was 25.4 cm long, 25.4 cm wide, and 1.9 cm high; the plate surface temperature could be set between 30 and 79.9 °C with an accuracy of ±0.1 °C. To minimize biotransformation at the time of testing, tests were conducted following i.v. administration. The antinociceptive effects of ipsapirone, paynantheine, and speciogynine alone and in combination with WAY 100635 were tested. Following the baseline measurements, vehicle or WAY 100635 (0.01 mg/kg) was administered i.v. at 60 and 15 min, respectively, followed by cumulative doses (every 5 min) of a test compound. Across six treatment groups in the present study (three subjects per sex), the baseline hotplate latencies [mean (SEM) in seconds] were as follows: ipsapirone alone [11.0 (2.05)] and in combination with WAY 100635 [7.98 (1.18)], paynantheine alone [16.2 (1.69)] and in combination with WAY 100635 [10.3 (1.31)], and speciogynine alone [16.8 (2.48)] and in combination with WAY 100635 [8.72 (1.17)]. For the baseline values, two-way ANOVA indicated no significant main effect on the treatment group (F5,24 = 1.44; P = 0.245), sex (F1,24 = 3.16; P = 0.088) and interaction of the two (F5,24 = 1.54; P = 0.215). The assessment of effects of a test compound was continued until the hotplate latency reached 60 s to avoid tissue damage. Hotplate latency was assessed by trained experimenters who were blind to treatment conditions.
Data Analysis.
Lower Lip Retraction.
Percent subjects showing lower lip retraction were compared with the vehicle or WAY 100635. Across eight treatment groups, lower lip retraction was never observed at the baseline (i.e., in the absence of the drug).
Hotplate (i.p.).
The hotplate latencies were normalized to the percentage of the MPE using the following formula: % MPE = 100 × (post-injection latency − pre-injection baseline latency)/(maximum latency 60 s − pre-injection baseline latency). All data per treatment group in each figure above were presented as a mean (± an SEM). Using the SigmaPlot software (ver. 14.0; Systat Software Inc., San Jose, CA) and GraphPad Prism software (ver. 8; GraphPad Software, San Diego, CA), a two- and three-way (repeated-measures) ANOVA, with post hoc Bonferroni t tests, was used (factors were the compound dose and sex for two-way ANOVA and test compound treatment, the presence or absence of WAY 100635, and sex for three-way ANOVA). Outcomes were considered significant at P < 0.05. The ED50 values (95% confidence intervals) for hotplate latency were calculated by linear regression41 of the dose-effect functions using the GraphPad Prism software. To compare potency, potency ratios and corresponding 95% CIs were calculated.42
Correlational analysis.
Fisher’s exact test (two tails) was used to assess the association between in vivo activity (lower lip retraction and antinociceptive effects of test compounds) and receptor affinity (i.e. Ki values) for serotonin 5-HT1A receptors using GraphPad Prism software. The R2 and P values are shown.
Pharmacokinetic of Speciogynine and Paynantheine in Rats.
These procedures were approved by the University of Florida IACUC. As with the in vivo functional studies, adult, naïve male Sprague Dawley rats weighing 250 ± 25 g were housed individually in ventilated cages at the University of Florida vivarium with ad libitum access to food and water. During the pharmacokinetic tests, animals were placed inside a CulexNxT automated blood sampling system (BASi Co., West Lafayette, IN). A 1.5 mg/mL paynantheine freebase equivalent formulation was prepared by weighing an accurate amount of paynantheine oxalate and dissolving it in normal saline with 1% (v/v) Tween 80 (Sigma Aldrich Co.). A 2.5 mg/mL speciogynine formulation was prepared by weighing an appropriate amount of speciogynine freebase and dissolving it in normal saline with 2% (v/v) Tween 80 and 2% (v/v) Tween 20 (Sigma Aldrich Co.). Both solutions were filtered through a 0.2 μm syringe filter (Millex, Sigma Aldrich Co.) prior to use. Two short, sensitive, and robust bioanalytical methods were developed and partially validated according to FDA guidelines in terms of accuracy, precision, recovery, selectivity, dilution integrity, carryover, linearity, and sensitivity. The methods used a mobile phase of purified water acidified with 0.1% formic acid and either acetonitrile or acetonitrile acidified with 0.1% formic acid. All solvents were of LC−MS grade. Formic acid, acetonitrile, and water were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Blank rat plasma was purchased from VWR International (Suwanee, GA, USA). Sodium heparin-coated blood collection vials and sterile blood collection tubing were obtained from BASi Co. (West Lafayette, IN, USA). Both methods used a gradient elution, had a linearity range of 3−500 ng/mL, and a low sample volume of 20−25 μL. Verapamil hydrochloride (Sigma Aldrich Co., St. Louis, MO, USA) served as the internal standard (IS) for paynantheine, while yohimbine hydrochloride (Selleckchem, Houston, TX, USA) served as the IS for speciogynine. Chromatographic separation was achieved using a Waters Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm) with a VanGuard pre-column of the same chemistry (Waters Co, Milford, MA), and retention times for speciogynine and paynantheine were 1.0 and 2.1 min, respectively. A Waters Acquity I-class chromatograph (Waters Co, Milford, MA) was coupled to a Xevo-TQS Micro triple-quadrupole mass spectrometer (Waters Co) in positive electrospray ionization mode using multiple reaction monitoring. Following the quantification of the samples in TargetLynx (Waters Co), plasma concentration versus time functions were constructed in SigmaPlot 12.0 (Systat Software, Inc.). The data were further subjected to a noncompartmental analysis using a linear trapezoidal method in Phoenix (Certara USA, Inc., Princeton, NJ).
Supplementary Material
ACKNOWLEDGMENTS
This research was funded in part by grants UG3 DA048353 and R01 DA047855 01 from the National Institute on Drug Abuse and the University of Florida Clinical and Translational Science Institute, which is supported in part by the NIH National Center for Advancing Translational Sciences under award number UL1TR001427. C.L.-L. thanks the Program of young talent and regional impact-2018, MinCiencias-Colombia for sponsoring her abroad stage. This work was supported in part by an NIH award, S10RR031637, for magnetic resonance instrumentation. A portion of this work was performed in the McKnight Brain Institute at the National High Magnetic Field Laboratory’s AMRIS Facility, which is supported by National Science Foundation Cooperative Agreement No. DMR-1644779 and the State of Florida.
ABBREVIATIONS USED
- 8-OH-DPAT
(±)-8-Hydroxy-2-(dipropylamino)tetralin
- 9-O-desmethylpaynantheine
gambireine
- 9-O-desmethylspe-ciogynine
gambirine
- AAALAC
Association for Assessment and Accreditation of Laboratory Animal Care
- ANOVA
analysis of variance
- CHO
Chinese hamster ovary
- DOI
2,5-dimethoxy-4-iodoamphetamine
- DOR
δ-opioid receptor
- HEK
human embryonic kidney
- IACUC
Institutional Animal Care and Use Committee
- i.p.
intraperitoneally
- IS
internal standard
- i.v.
intravenous
- KOR
κ-opioid receptor
- MOR
μ-opioid receptor
- MPE
maximum possible effects
- 5-HT
serotonin
- SEM
standard of error of mean
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c00726
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00726.
Experimental procedures, statistical analysis of in vivo data, additional binding data at 5HT1A and 5HT2B receptors, and spectroscopic data for the kratom alkaloids and metabolites (PDF)
Molecular formula strings (CSV)
The authors declare no competing financial interest.
The views and opinions expressed in this manuscript are those of the authors only and do not necessarily represent the views, official policy, or position of the U.S. Department of Health and Human Services or any of its affiliated institutions or agencies.
Contributor Information
Francisco León, Department of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States; Department of Drug Discovery and Biomedical Sciences, College of Pharmacy, University of South Carolina, Columbia, South Carolina 29208, United States.
Samuel Obeng, Department of Medicinal Chemistry, College of Pharmacy and Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Marco Mottinelli, Department of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Yiming Chen, Department of Pharmaceutical Sciences, College of Pharmacy, Mercer University, Atlanta, Georgia 30341, United States.
Tamara I. King, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
Erin C. Berthold, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States; Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
Shyam H. Kamble, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States; Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
Luis F. Restrepo, Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States
Avi Patel, Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Lea R. Gamez-Jimenez, Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States
Carolina Lopera-Londoño, Department of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Takato Hiranita, Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Abhisheak Sharma, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States; Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
Aidan J. Hampson, Division of Therapeutics and Medical Consequences, National Institute on Drug Abuse, National Institutes of Health, Bethesda, Maryland 20892, United States
Clinton E. Canal, Department of Pharmaceutical Sciences, College of Pharmacy, Mercer University, Atlanta, Georgia 30341, United States.
Lance R. McMahon, Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Christopher R. McCurdy, Department of Medicinal Chemistry, College of Pharmacy and Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
REFERENCES
- (1).Kruegel AC; Gassaway MM; Kapoor A; Váradi A; Majumdar S; Filizola M; Javitch JA; Sames D Synthetic and receptor signaling explorations of the mitragyna alkaloids: mitragynine as an atypical molecular framework for opioid receptor modulators. J. Am. Chem. Soc 2016, 138, 6754–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Brown PN; Lund JA; Murch SJ A botanical, phytochemical and ethnomedicinal review of the genus Mitragyna korth: implications for products sold as kratom. J. Ethnopharmacol 2017, 202, 302–325. [DOI] [PubMed] [Google Scholar]
- (3).Singh D; Chear NJY; Narayanan S; Leon F; Sharma A; McCurdy CR; Avery BA; Balasingam V Patterns and reasons for kratom (Mitragyna speciosa) use among current and former opioid poly-drug users. J. Ethnopharmacol 2020, 249, 112462. [DOI] [PubMed] [Google Scholar]
- (4).Schimmel J; Amioka E; Rockhill K; Haynes CM; Black JC; Dart RC; Iwanicki JL Prevalence and description of kratom (Mitragyna speciosa) use in the United States: a cross-sectional study. Addiction 2020, 116, 176–181. [DOI] [PubMed] [Google Scholar]
- (5).Han C; Schmitt J; Gilliland KM DARK classics in chemical neuroscience: kratom. ACS Chem. Neurosci 2019, 11, 3870–3880. [DOI] [PubMed] [Google Scholar]
- (6).Chear NJ-Y; León F; Sharma A; Kanumuri SRR; Zwolinski G; Abboud KA; Singh D; Restrepo LF; Patel A; Hiranita T; Ramanathan S; Hampson AJ; McMahon LR; McCurdy CR Exploring the chemistry of alkaloids from Malaysian Mitragyna speciosa (Kratom) and the role of oxindoles on human opioid receptors. J. Nat. Prod 2021, 84, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gogineni V; Leon F; Avery BA; McCurdy C; Cutler SJ Phytochemistry of Mitragyna speciosa. In Kratom and Other Mitragynines; CRC Press: 2014; pp. 77–93. [Google Scholar]
- (8).Kruegel AC; Grundmann O The medicinal chemistry and neuropharmacology of kratom: a preliminary discussion of a promising medicinal plant and analysis of its potential for abuse. Neuropharmacology 2018, 134, 108–120. [DOI] [PubMed] [Google Scholar]
- (9).Eastlack SC; Cornett EM; Kaye AD Kratom—pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther. 2020, 9, 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang M; Sharma A; León F; Avery B; Kjelgren R; McCurdy CR; Pearson BJ Effects of nutrient fertility on growth and alkaloidal content in Mitragyna speciosa (kratom). Front. Plant Sci 2020, 11, 2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kruegel AC; Uprety R; Grinnell SG; Langreck C; Pekarskaya EA; Le Rouzic V; Ansonoff M; Gassaway MM; Pintar JE; Pasternak GW 7-Hydroxymitragynine is an active metabolite of mitragynine and a key mediator of its analgesic effects. ACS Cent. Sci 2019, 5, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Obeng S; Kamble SH; Reeves ME; Restrepo LF; Patel A; Behnke M; Chear NJ-Y; Ramanathan S; Sharma A; Leon F; Hiranita T; Avery BA; McMahon LR; McCurdy CR Investigation of the adrenergic and opioid binding affinities, metabolic stability, plasma protein binding properties, and functional effects of selected indole-based kratom alkaloids. J. Med. Chem 2020, 63, 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ellis CR; Racz R; Kruhlak NL; Kim MT; Zakharov AV; Southall N; Hawkins EG; Burkhart K; Strauss DG; Stavitskaya L Evaluating kratom alkaloids using PHASE. PLoS One 2020, 15, No. e0229646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Matsumoto K; Mizowaki M; Suchitra T; Murakami Y; Takayama H; Sakai S.-i.; Aimi N; Watanabe H Central antinociceptive effects of mitragynine in mice: contribution of descending noradrenergic and serotonergic systems. Eur. J. Pharmacol 1996, 317, 75–81. [DOI] [PubMed] [Google Scholar]
- (15).Kamble SH; Sharma A; King TI; León F; McCurdy CR; Avery BA Metabolite profiling and identification of enzymes responsible for the metabolism of mitragynine, the major alkaloid of Mitragyna speciosa (kratom). Xenobiotica 2019, 49, 1279–1288. [DOI] [PubMed] [Google Scholar]
- (16).Koek W; Assié M-B; Zernig G; France CP In vivo estimates of efficacy at 5-HT 1A receptors: effects of EEDQ on the ability of agonists to produce lower-lip retraction in rats. Psychopharmacology 2000, 149, 377–387. [DOI] [PubMed] [Google Scholar]
- (17).Perry CK; Casey AB; Felsing DE; Vemula R; Zaka M; Herrington NB; Cui M; Kellogg GE; Canal CE; Booth RG Synthesis of novel 5-substituted-2-aminotetralin analogs: 5-HT1A and 5-HT7 G protein-coupled receptor affinity, 3D-QSAR and molecular modeling. Bioorg. Med. Chem 2020, 28, 115262. [DOI] [PubMed] [Google Scholar]
- (18).Canal CE Serotonergic psychedelics: experimental approaches for assessing mechanisms of action. Handb. Exp. Pharmacol 2018, 252, 227–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Boess FG; Martin IL Molecular biology of 5-HT receptors. Neuropharmacology 1994, 33, 275–317. [DOI] [PubMed] [Google Scholar]
- (20).Zhou D; Stack GP; Lo J; Failli AA; Evrard DA; Harrison BL; Hatzenbuhler NT; Tran M; Croce S; Yi S; Golembieski J; Hornby GA; Lai M; Lin Q; Schechter LE; Smith DL; Shilling AD; Huselton C; Mitchell P; Beyer CE; Andree TH Synthesis, potency, and in vivo evaluation of 2-piperazin-1-ylquinoline analogues as dual serotonin reuptake inhibitors and serotonin 5-HT1A receptor antagonists. J. Med. Chem 2009, 52, 4955–4959. [DOI] [PubMed] [Google Scholar]
- (21).Ikarashi Y; Sekiguchi K; Mizoguchi K Serotonin receptor binding characteristics of geissoschizine methyl ether, an indole alkaloid in Uncaria hook. Curr. Med. Chem 2018, 25, 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ueda T; Ugawa S; Ishida Y; Shimada S Geissoschizine methyl ether has third-generation antipsychotic-like actions at the dopamine and serotonin receptors. Eur. J. Pharmacol 2011, 671, 79–86. [DOI] [PubMed] [Google Scholar]
- (23).Berendsen HH; Broekkamp CL; Van Delft AM Antagonism of 8-OH-DPAT-induced behaviour in rats. Eur. J. Pharmacol 1990, 187, 97–103. [DOI] [PubMed] [Google Scholar]
- (24).Hutcheson JD; Setola V; Roth BL; Merryman WD Serotonin receptors and heart valve disease–it was meant 2B. Pharmacol. Ther 2011, 132, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Roth BL Drugs and valvular heart disease. N. Engl. J. Med 2007, 356, 6–9. [DOI] [PubMed] [Google Scholar]
- (26).Kamble SH; León F; King TI; Berthold EC; Lopera-Londoño C; Siva Rama Raju K; Hampson AJ; Sharma A; Avery BA; McMahon LR; McCurdy CR Metabolism of a kratom alkaloid metabolite in human plasma increases its opioid potency and efficacy. ACS Pharmacol. Transl. Sci 2020, 3, 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Obeng S; Wilkerson JL; León F; Reeves ME; Restrepo LF; Gamez-Jimenez LR; Patel A; Pennington AE; Taylor VA; Ho NP; Braun T; Fortner JD; Crowley ML; Williamson MR; Pallares VL; Mottinelli M; Lopera-Londoño C; McCurdy CR; McMahon LR; Hiranita T Pharmacological comparison of mitragynine and 7-hydroxymitragynine: in vitro affinity and efficacy for mu-opioid receptor and opioid-like behavioral effects in rats. J. Pharmacol. Exp. Ther 2021, 376, 410–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Avery BA; Boddu SP; Sharma A; Furr EB; Leon F; Cutler SJ; McCurdy CR Comparative pharmacokinetics of mitragynine after oral administration of Mitragyna speciosa (kratom) leaf extracts in rats. Planta Med. 2019, 85, 340–346. [DOI] [PubMed] [Google Scholar]
- (29).Davies B; Morris T Physiological parameters in laboratory animals and humans. Pharm. Res 1993, 10, 1093–1095. [DOI] [PubMed] [Google Scholar]
- (30).Johnson LE; Balyan L; Magdalany A; Saeed F; Salinas R; Wallace S; Veltri CA; Swogger MT; Walsh Z; Grundmann O The potential for kratom as an antidepressant and antipsychotic. Yale J. Biol. Med 2020, 93, 283–289. [PMC free article] [PubMed] [Google Scholar]
- (31).Fluyau DF; Revadigar N Biochemical benefits, diagnosis, and clinical risks evaluation of kratom. Front. Psychiatry 2017, 8, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sharma A; Kamble SH; León F; Chear NJY; King TI; Berthold EC; Ramanathan S; McCurdy CR; Avery BA Simultaneous quantification of ten key Kratom alkaloids in Mitragyna speciosa leaf extracts and commercial products by ultra-performance liquid chromatography−tandem mass spectrometry. Drug Test. Anal 2019, 11, 1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wenkert E; Chang C-J; Chawla HPS; Cochran DW; Hagaman EW; King JC; Orito K General methods of synthesis of indole alkaloids. 14. Short routes of construction of yohimboid and ajmalicinoid alkaloid systems and their carbon-13 nuclear magnetic resonance spectral analysis. J. Am. Chem. Soc 1976, 98, 3645–3655. [Google Scholar]
- (34).Kitajima M; Misawa K; Kogure N; Said IM; Horie S; Hatori Y; Murayama T; Takayama H A new indole alkaloid, 7-hydroxyspeciociliatine, from the fruits of Malaysian Mitragyna speciosa and its opioid agonistic activity. J. Nat. Med 2006, 60, 28–35. [Google Scholar]
- (35).Stærk D; Norrby P-O; Jaroszewski JW Conformational analysis of indole alkaloids corynantheine and dihydrocorynantheine by dynamic 1H NMR spectroscopy and computational methods: steric effects of ethyl vs vinyl group. J. Org. Chem 2001, 66, 2217–2221. [DOI] [PubMed] [Google Scholar]
- (36).Kam T-S; Lee K-H; Goh S-H Alkaloid distribution in Malaysian Uncaria. Phytochemistry 1992, 31, 2031–2034. [Google Scholar]
- (37).Goh SH; Asiah S; Junan A Alkaloids of Uncaria callophylla. Phytochemistry 1985, 24, 880–881. [Google Scholar]
- (38).Yung-Chi C; Prusoff WH Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (39).Chen Y; Canal CE Structure-activity relationship study of psychostimulant synthetic cathinones reveals nanomolar antagonist potency of alpha-pyrrolidinohexiophenone at human muscarinic M2 receptors. ACS Chem. Neurosci 2020, 11, 960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hiranita T; Leon F; Felix JS; Restrepo LF; Reeves ME; Pennington AE; Obeng S; Avery BA; McCurdy CR; McMahon LR; Wilkerson JL The effects of mitragynine and morphine on schedule-controlled responding and antinociception in rats. Psychopharmacology 2019, 236, 2725–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Snedecor GW; Cochran WG Statistical Methods. Ames, Iowa. In Iowa State University Press: 1967. [Google Scholar]
- (42).Tallarida RJ The interaction index: a measure of drug synergism. Pain 2002, 98, 163–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.