

Abstract
Membrane permeabilizing peptides (MPPs) are as ubiquitous as the lipid bilayer membranes they act upon. Produced by all forms of life, most membrane permeabilizing peptides are used offensively or defensively against the membranes of other organisms. Just as nature has found many uses for them, translational scientists have worked for decades to design or optimize membrane permeabilizing peptides for applications in the laboratory and in the clinic ranging from antibacterial and antiviral therapy and prophylaxis to anti-cancer therapeutics and drug delivery. Here, we review the field of membrane permeabilizing peptides. We discuss the diversity of their sources and structures, the systems and methods used to measure their activities, and the behaviors that are observed. We discuss the fact that “mechanism” is not a discrete or a static entity for an MPP, but rather the result of a heterogeneous and dynamic ensemble of structural states that vary in response to many different experimental conditions. This has led to an almost complete lack of discrete three-dimensional active structures amongst the thousands of known peptides, and a lack of useful or predictive sequence-structure-function relationship rules. Ultimately, we discuss how it may be more useful to think of membrane permeabilizing peptides mechanisms as broad regions of a mechanistic landscape rather than discrete molecular processes.
1. Introduction
The permeability barrier of the lipid bilayer membrane is a ubiquitous and essential feature of every living organism on earth, and thus is also a critical point of vulnerability. For this reason, membrane permeabilizing proteins and peptides are a ubiquitous part of the defensive and offensive biochemical arsenals of all living things. They are also the subject of significant translational research towards development into drugs and molecular tools. Here, we review the field of membrane permeabilizing peptides.
1.1. What is a membrane permeabilizing peptide?
The hydrocarbon core of a lipid bilayer membrane, made of almost pure hydrocarbon1, is one of the most hydrophobic nano-environments that can be found in the biosphere. Although the bilayer is a two-dimensional fluid that is only two molecules thick, and the hydrocarbon core is only a portion of the total bilayer thickness, the extremely low solubility of polar compounds in the hydrocarbon core effectively prevents them from crossing the bilayer. This barrier function is essential to life and may have been an antecedent of the earliest life forms on earth. For any molecule to breach this barrier, i.e. to permeabilize the membrane, the minimum requirement is that a pathway be created that enables passage of polar molecules across the bilayer hydrocarbon core. A membrane permeabilizing peptide, by definition, is one that creates such a pathway, a process that can happen in a variety of ways. For example, a peptide can self-assemble into an explicit, membrane-spanning structure like a proteinaceous channel. In this review, we specifically refer only to explicit, discrete membrane-spanning structures as “pores” which are created by “pore-forming peptides”. Reliable evidence for such structures in membrane permeabilizing peptides is extremely rare. At a minimum, a membrane permeabilizing peptide must have “interfacial activity”2, which means it must partition into a membrane and be at least somewhat amphipathic3–6 in order to drive the rearrangement of membrane lipids3,7,8, disrupting the normally strict segregation between the bilayer hydrocarbon core, the interfacial zone of the bilayer, and the aqueous phase1,9–12. This disruption enables polar lipid and peptide moieties, as well as water molecules, to make incursions into the hydrocarbon core and reduce its hydrophobicity2. A reduction in the hydrophobicity of the core reduces the highest energy barrier to the passage of polar molecules, thus increasing the rate of the limiting step in permeation.
As we discuss in this manuscript, most of the thousands of known MPPs likely act by some form of interfacial activity, rather than by pore formation. Here, we will refer to all such peptides as “membrane permeabilizing peptides” or MPPs, a generic phrase specifically chosen as a functional description that is not meant to invoke any particular mechanism or structure. In this review, we will discuss membrane permeabilizing peptide sources and structures, and we will also discuss the ways in which one can measure their functions in model systems, in computers, and in living cells. We discuss the behaviors that have been observed in these many systems. Finally, we discuss the concept of a “mechanistic landscape” of membrane permeabilizing peptides (Figure 1), and how an MPP can sample a continuum of behaviors, functions, and structures influenced by a host of experimental conditions. This fundamental behavior must be better understood, and we must learn how to better describe it to enable the design or optimization of membrane permeabilizing peptides that might be useful tools or drugs in the laboratory or in the clinic.
Figure 1.

The mechanistic landscape of membrane permeabilizing peptides. The molecular mechanisms of membrane permeabilizing peptides have, for the most part, eluded atomic level description despite decades of intense study. As a result, their active structures and mechanisms are often drawn as cartoons like the imaginary snapshots arrayed in the cartoon bilayer above. Different colored lipids indicate changes in membrane composition, lipid tails and headgroups. Curvy tails indicate fluid phase bilayers and straight tails indicate more ordered domains. In this review we describe how it might be useful to think of membrane permeabilizing peptides on a mechanistic landscape where molecular mechanism is not a fixed entity, but instead depends on the sum of many experimental variables. The image in the center of the cartoon vesicle depicts concentric “dials”, one for each experimental variable (many are not explicitly shown). Each dial can be set to a particular “value” in the parameter space. The combinations of all the settings give rise to a point on multidimensional mechanistic landscape.
1.2. Why do we study membrane permeabilizing peptides?
Membrane-permeabilizing peptides could have utility in a wide variety of applications12–21. Foremost, there has long been promise, but also disappointment, in the possible translational applications of antimicrobial peptides (AMP) to the prevention and treatment of drug resistant bacterial infections7,21,22. Improvement in treatment outcomes may arise from antimicrobial peptides that are engineered or evolved to have improved properties, including selectivity for bacterial membrane permeabilization compared to host-cell membrane permeabilization23,24. Similarly, interfacially active peptides can have potent inhibitory activity against enveloped viruses25–30, a group that includes influenza, HIV, Ebola, Dengue, Zika virus and many others25,29. In an exciting recent paper31, Cho and colleagues showed that an amphipathic helix called AH peptide, which preferentially lyses both lipid vesicles and enveloped viruses that have high curvature, has potent, protective anti-virus activity in living animals. Systemic administration of peptide in Zika virus-infected mice protected against death, and reduced clinical symptoms, including brain damage, and reduced viral propagation. Remarkably, the peptide crossed the blood–brain barrier, without damaging it, and directly reduced viral loads in the brain.
Another potentially far-reaching application of membrane-active peptides is potent membrane permeabilization against host cells. Peptides with this property could be used to kill cancer cells19,32,33, but only if targeting and control of the peptide activity can be optimized to permeabilize only the diseased cells. Along these lines, the bee venom MPP melittin, associated with lipidic “nanocarriers” targets cancer cells in vitro and in vivo19,34.
Membrane permeabilization can also be used to deliver polar compounds35,36, including drugs, proteins, peptides, oligonucleotides and their analogs, and other molecules37, to cells. The ability to deliver polar and macromolecule drugs to cells could be very useful in the laboratory, and also in the clinic38,39 where it would significantly expand the universe of potentially druggable compounds. Although molecular mechanisms and pathways are not always well understood, cell penetrating peptides (CPPs) can sometimes deliver cargo through the plasma membrane in a process that may involve transient local permeabilization40, permeabilization of endosomal membranes following uptake, or both pathways at once41. Many CPPs utilize both pathways in a manner that depends on a variety of experimental conditions41. Examples of membrane permeabilizing peptides can be found in the relatively young field of “profection”, the delivery of whole proteins to the cell cytosol37,42. Although multiple mechanisms have been proposed, delivery of a protein through the plasma membrane and/or an endosomal membrane requires that a profection reagent must induce a transient and localized, but also locally significant permeabilization event at the membrane. An example of a peptide profection reagent has been described by Futaki and colleagues,37 who started with a classical membrane permeabilizing amphipathic α-helix from spider venom and engineered anionic charges into the sequence to make it less lytic to mammalian cell membranes at neutral pH. Ultimately, they obtained a peptide capable of passively delivering some macromolecules to the cytosol of cells in culture.
For all these important applications, and others not listed, a better understanding of the mechanisms of membrane permeabilization is essential. In fact, growth of the field has been inhibited for decades by the lack of accurate, useful, and predictive rules for membrane permeabilization, forcing the majority of researchers to utilize natural molecules without optimization, or to create novel or optimized sequences by simple trial and error.
1.3. The sting: Lessons learned from 60 years of studying melittin
Of all the known MPPs, one of the most studied, and perhaps most enigmatic, is melittin, the 26 amino acid peptide that is the main component of the venom of the European Honey Bee, Apis mellifera (Figure 2). By the 1950s, a proteinaceous, cytolytic, necrotizing component of Honey Bee venom, which was long recognized, had been named “melittin”43. The amino acid sequence of melittin was determined in 1967 by Haberman44 who also performed some of the first sequence-function relationship studies43. As early as 1967, antibacterial activity of melittin against drug resistant bacteria was reported45. By 1969, the membrane permeabilizing activity of melittin was well-established46,47 and was understood to be the consequence of its structural amphipathicity, which is two-dimensional. Melittin was immediately recognized to be amphipathic along its sequence, mostly hydrophobic from the N-terminus to residue 20, and highly polar on the cationic C-terminus, residues 21–2647. Later, melittin was also recognized to fold in membranes into an amphipathic α-helix48–50 which has polar and non-polar surfaces that are orthogonal to the helix axis51,52. Like most membrane permeabilizing peptides, melittin, by virtue of its cationic charge and amphipathicity, binds to lipid bilayer membranes and causes permeabilization by creating a pathway through the hydrocarbon core of the bilayer that polar molecules can use to cross the membrane.
Figure 2.

Melittin, the archetypal membrane permeabilizing peptide. A: European Honey Bee (apis mellifera) workers produce a defensive venom that contains many compounds, including peptide and proteins. The most abundant component by weight is melittin, a 26-residue membrane permeabilizing peptide. Photograph by William Wimley, used with permission. B: Amino acid sequences of melittin from Apis mellifera and several closely related species. Sequences are generally hydrophobic over the first 20 residues, except for lysine at position 7, and are highly polar and basic on the C-terminus. C: Helical wheel diagrams show the placement of residues on the surface of an imaginary perfect helix. D: On the surface of the ideal melittin helix, hydrophobic residues form a contiguous surface. E: In the first three-dimensional structure of melittin in solution52, and other structures50 its amphipathicity was apparent in the burial of the hydrophobic surfaces in the core of a tetrameric structure or in the membrane. F: The amphipathicity of the melittin monomers in the context of the crystal structure, in which the helices are bent and disrupted at the central Gly-X-Pro. G: Some experiments50 and biased molecular dynamics simulations429,423 demonstrate melittin forming membrane spanning equilibrium pores. However, unbiased simulations of slow insertion equilibrium requires currently unachievably long simulations528. Images courtesy of Jakob Ulmschneider. H: Under other conditions, experiments, such as electrochemical impedance spectroscopy and vesicle permeabilization, show that the permeabilization of membranes by melittin is a transient non-equilibrium process59,60,66.
But how do we describe the mechanism by which melittin permeabilizes membranes? Early electrophysiology studies by Tosteson provided data indicating that melittin formed voltage-dependent, anion-selective, membrane-spanning pores, which had a concentration dependence that indicating a tetrameric structure53. Around the same time however, DeGrado and colleagues showed that melittin releases proteins from erythrocytes54 in a transient manner indicating a larger, voltage-independent pathway through the membrane. The concept of explicit pores has also been supported by diffraction and other measurements at low hydration and high concentration showing melittin to form membrane-spanning helical structures50,55,56. However, other work, including FTIR57, oriented circular dichroism58,59, X-ray diffraction58 and EPR60 have shown that melittin has its helical axis lying mostly parallel to the membrane surface under many experimental conditions. This is not consistent with equilibrium transmembrane pore formation, unless the equilibrium pores are a minor fraction of the total population of peptide. Further, multiple studies have shown that melittin permeabilizes bilayers to different classes of molecules, sometimes including macromolecules61,62, as lipid composition and peptide concentration are varied. Therefore, melittin can form a range of different structures in the membrane, likely an ensemble of coexisting structures. At a high enough concentration, melittin solubilizes bilayers and forms peptide-lipid micelles, like a detergent63.
Perhaps most surprising, dye leakage studies64,65 and electrochemical impedance spectroscopy66 have shown that melittin often forms transient, not equilibrium, pathways through synthetic bilayers. Under many experimental conditions, these permeabilization pathways exist only for a short time after peptide first encounters the membrane surface. Within minutes after leakage begins, it slows or stops completely. As long ago as 1982, DeGrado et al. reported a transient permeabilization process in erythrocyte membranes treated with melittin. They attributed attenuation of permeabilization to peptide translocation54, an idea that is still the leading hypothesis to explain transient permeabilization. In a recent study, Weisshaar and colleagues showed that bacterial permeabilization by melittin proceeds through a complex series of steps that include stochastic membrane permeabilization after a long lag phase, followed by rapid membrane resealing67, observations that are not consistent with equilibrium pores.
Melittin is the archetypal membrane permeabilizing peptide, and also emblematic of the difficulties of the field. We have been able to speak generally of the physical chemical basis of its membrane binding and permeabilizing activity since shortly after its sequence was known. Yet, after five decades of intense study68,69,78–87,70–77, the exact molecular mechanism of melittin continues to be debated and new insights continue to appear in the literature65,67.
1.4. An introduction to the mechanistic landscapes of MPPs
Our inability to describe the action of melittin and other MPPs with single or explicit molecular models is not a failure of our techniques, but perhaps is a failure of our vocabulary. This review is inspired by the idea that the molecular mechanism of an MPP is not a fixed entity. We discuss the behaviors of membrane permeabilizing peptides, and how they might best be considered to occupy part of a continuous “mechanistic landscape” in which mechanism and behavior are both heterogeneous and plastic, dependent on the contributions of many experimental details. These include peptide sequence and structure, as well as lipid composition, peptide concentration, temperature, ionic strength, pH and many other variables. Experimental “variables” also include the assay(s) used to measure permeabilization, which are numerous, and how the observations are interpreted, which are variable. The detailed shape of the landscape is likely different for each peptide in detail, although they must broadly overlap. The mechanistic landscape is driven by the sum of a large set of physical-chemical interactions. For example, important interactions occur between the fluid phase bilayer and peptides that have partitioned into it. These interactions are driven by interfacial hydrophobicity88 and electrostatic interactions, which are non-additive78. There are also interactions between the peptides themselves in the context of the “tumultuous chemical heterogeneity”1 of the bilayer. These interactions drive peptide self-assembly89 and secondary structure formation79,90–92. There are also interactions between the highly amphipathic lipids in the dynamic milieu of the bilayer, and how these interactions are altered by interfacially active peptides partitioned into it. Finally, there are important interactions of water molecules with peptides, lipids, and the membrane itself. In fact, penetration of water into the hydrocarbon core may be the most appropriate molecular signature of membrane permeabilization93, although it is difficult to measure experimentally.
In this review we do not attempt, in most cases, to assign a particular mechanism to any membrane-permeabilizing peptide. Rather, we describe what can be observed and what has been observed in the laboratory as well as what these observations might mean. Ultimately, we hope to begin to understand the mechanistic landscapes occupied by membrane permeabilizing peptides to use this understanding to enable more rational rules for the design, engineering and optimization of their activities, and to generate a more accurate vocabulary to discuss their activity profiles.
2. Diversity of membrane permeabilizing peptides
Given the ubiquity of biomembranes in life on Earth, it is not surprising that membrane permeabilizing peptides are also abundantly found in many living things (Figure 3). There are thousands of known MPPs, many of which are host defense peptides, and there are interesting examples from many sources. Next, we will outline some of the major sources of currently known MPPs.
Figure 3.

Sources of membrane permeabilizing peptides. There are a multitude of sources for membrane-permeabilizing peptides (MPPs) as shown in the figure above. Clockwise from the top, sources include humans and other mammal host defense, bacteria and fungi, viruses, amphibian and other vertebrate host defense, insect host defense, plant host defense, bioinformatics and computational approaches, engineering and rational design, and venoms and toxins. Host defense peptides are the most ubiquitous, but there are also MPPs which can comprise part of a venom or toxin cocktail, viroporins, de novo designed peptides, synthetically evolved peptides, and other sources which are not listed. Overall, there are thousands of known MPPs and certainly many more to be discovered. Images reproduced with permission under Creative Commons CC0 license.
2.1. Host defense peptides
The innate immune systems of plants and animals are the most common source of known membrane permeabilizing peptides. They produce host defense, or antimicrobial peptides (AMPs), which were first discovered in the 1980’s in insect hemolymph94,95, mammalian neutrophil granules96 and frog skin secretions97. Now thousands of examples are known98 from an immense variety of tissues and cell types in essentially all classes of higher organisms. The number of examples continues to rapidly increase as additional organisms around the globe are examined. AMPs are wildly diverse in structure, but are nearly unified by their cationic nature98, interfacial activity2, and by their propensity to permeabilize anionic synthetic bilayers and the anionic cytoplasmic membranes of bacteria (discussed below). Many organisms produce sets of multiple MPPs that often differ only slightly from one another such as the multiple α- and β-defensins found in birds and mammals99–101, supporting the idea that different membranes have variable susceptibility to permeabilization by one peptide.
Recently, a novel function for the membrane permeabilizing activity of frog skin MPPs was proposed102 and then tested by Roelants and colleagues36. These authors noted that frog skin secretions often contain high concentrations of one or more peptide or protein toxin in addition to multiple MPPs. In Xenopus laevis for example, the membrane permeabilizing peptide caerulein precursor fragment (CPF)103 and the receptor binding toxin caerulein are both present in skin secretions at millimolar concentrations36. Roelants and colleagues pointed out that the targets of caerulein are inside the body, and they showed that it cannot spontaneously translocate into cells or permeabilize cells to gain entry. Using cells in culture and live snakes as predator models, these authors showed that CPF transiently permeabilizes eukaryotic cells at physiologically relevant concentrations and that this is both necessary and sufficient to enable entry of the toxin cearulein into the circulation of the snakes. These results suggest that these two molecules comprise a two-part chemical defense system against predators in addition to (or instead of) comprising part of the host defense system against bacteria.
Microbes also produce a vast array of defensive compounds or toxins designed to reduce competition by other microbes. Of course, these include small molecules (e.g. penicillin and many other conventional antibiotics) and specific protein toxins, such as the colicins104. However, bacteria and fungi also produce defensive MPPs, including some of the best studied MPPs such as the alamethicins105–107, the peptaibols produced by the fungus Trichoderma viride, and the gramicidins 108,109 produced by the soil bacterium Bacillus brevis. Pathogenic bacteria also produce many offensive toxins, some of which are hemolytic or cytolytic MPPs110. For example, Staphylococcus species produce δ-lysin, a 26-residue helical MPP with potent hemolytic activity110–112 that is associated with virulence110.
2.2. Toxins and venoms
Animal venoms and toxins are complex mixtures of enzymes, peptides, small molecules, and other compounds that aid in prey capture and digestion, as well as defense against other animals. Some venoms are delivered to the body by way of teeth, fangs, stingers, pincers, or spurs, while others are delivered by contact. Some venom components are membrane permeabilizing peptides which contribute to the activity through non-specific cytolysis and hemolysis, causing cell and tissue damage and exacerbating pain. As described above, the best known example is melittin, the 26-residue MPP that comprises the majority, by weight, of the venom of the European Honey Bee. Other examples are found in the venoms of bees and wasps, spiders, scorpions, ants, and more113–119.
The α-helical peptide pardaxin is an intriguing MPP that is found in the skin of the Red Sea Moses Sole (Pardachirus marmoratus) and related fish120–122. The five nearly identical pardaxin sequences are structurally and functionally similar to melittin. In the laboratory, pardaxin has potent hemolytic and antimicrobial activity123,124 and permeabilizes synthetic bilayers121,125,126. It also has activity against cancer cells127,128. However, there is evidence supporting the idea that pardaxin has shark repellent activity in its natural setting129. When introduced directly from the skin of the fish into an attacking shark’s mouth and gills, pardaxin causes apparent pain and disorientation129. Unfortunately, tests of the ability of pardaxin as a shark repellent in an open beach setting showed that it was too quickly diluted in open water to be effective130.
2.3. Viroporins
Viroporins are virally encoded proteins and peptides that permeabilize internal membranes131, viral membranes, or uninfected cell membranes132 and “customize host cells for efficient viral propagation”133. Some viroporins are MPPs. For example, the Ebola virus delta peptide, is a 40-residue peptide produced in large quantity by Ebola virus infected cells134,135. The delta peptide permeabilizes both eukaryotic cells and synthetic membranes136. The active portion is a protease resistant, disulfide crosslinked C-terminal hairpin of 15–19 residues136. The influenza M2 channel is a very different sort of viroporin that enables acidification of the viral core of a virus that has been uptaken into the endosomal pathway137. The essential part of M2 is a membrane spanning α-helix that assembles into a tetrameric channel137. M2 is one of the only MPPs known for which there is strong evidence of stoichiometric self-assembly into a specific membrane-spanning oligomer137.
The membrane permeabilizing sequences of viroporin proteins, often amphipathic α-helices, also constitute MPPs in isolated form. For example, the picornavirus 2B viroporin has several amphipathic helices that are potent MPPs138,139 in isolated form. Lentiviruses, such as HIV, contain up to three potential amphipathic helix sequences in the intraviral, C-terminal domain of their transmembrane fusion proteins140,141. Mutational analysis have shown these sequences to be critical for viral propagation,142 although their contribution to the viral lifecycle is not well understood143–146. In isolated form, these domains, called lentivirus lytic peptides (LLP), comprise highly potent membrane permeabilizing peptides. In particular, LLP2 of HIV is one of the most potent MPPs known64,143,146–148. HIV and other viruses also have membrane proximal sequences that are interfacially active, such as the membrane proximal external region, MPER, in HIV, which inserts into membranes149 and permeabilizes them150.
2.4. De novo designed MPPs
Researchers have created new MPPs which bear some relationship to natural sequences. For example, fragments of natural sequences151, hybrid combinations of known MPPs152,153 or rationally modified variants154 have been studied. Antimicrobial peptides that permeabilize anionic synthetic lipid bilayers and bacterial membranes, have also been designed de novo using first principles, based on physical chemistry, combined with trial and error or screening155–161. They have also been designed using machine learning or data-driven algorithms162–166. In almost all of these cases, the end results are classical interfacially active antimicrobial peptides that are rich in cationic and aromatic residues.
Other types of MPPs, designed de novo from first principles, are very uncommon, especially those that permeabilize zwitterionic bilayers. This is probably because the sequence-structure-function relationship rules are not understood well enough to enable intelligent design of membrane interaction, intermolecular interactions, or oligomer structure of MPPs in membranes. In fact, even if rational modulation of these individual factors were possible, it remains uncertain how they relate, mechanistically, to membrane permeabilization. The amphipathic α-helix is the simplest structure to model, and would seem to be amenable to engineering using the regular i to i+3 and i to i+4 spacings167 to design amphipathic surfaces. Along these lines, there have been a small number of successes. For example, DeGrado and colleagues created and tested a series of de novo designed peptides168,169 that were based on the concept of amphipathic α-helical bundles. One active sequence, with 3 repeats of Leu-Ser-Ser-Leu-Leu-Ser-Leu formed ion channels in lipid bilayers169 that were modelled as small bundles of transmembrane helical peptides.
The peptide GALA is perhaps one of the most well-known examples of de novo design success. It was made by Szoka and colleagues170–173 who wanted to create a pH-triggered MPP. They created an amphipathic α-helix by placing protonatable glutamate residues at positions separated by mostly i to i+4 spacings. “GALA” is named for a repeating unit of Glutamate-Alanine-Leucine-Alanine. At pH 7, GALA is inactive, has random coil structure and does not bind strongly to membranes170–173. However, at pH 5 it binds strongly171, folds into a membrane spanning α-helix,170 and permeabilizes lipid vesicles170,172,173 to small solutes at peptide to lipid ratios (P:L) as low as 1:10,000. This may be the most potent activity ever reported for a membrane permeabilizing peptide.
2.5. Synthetic molecular evolution of MPPs
In the absence of explicit sequence-structure-function rules to enable rational design of MPPs, we have turned to synthetic molecular evolution (SME), which means the iterative screening of small, rational peptide libraries based on prior knowledge and template sequences. This has proven to be especially powerful for identifying novel membrane activities and gain of function peptide sequences. For example, we evolved β-sheet MPPs with potent antimicrobial activity using two de novo libraries that were based on physical chemical first principles. These libraries were screened for peptides that permeabilized somewhat anionic, bacteria-like synthetic lipid vesicles. The “hits” did not reveal singular active sequence motifs, but rather showed that active sequences are related by overall physical chemistry5,174,175. A second generation library was designed using one 26-residue first-generation MPP as a template sequence176. This “iteration” library was screened for permeabilization of synthetic bilayers made of phosphatidylcholine (PC) lipids. Highly potent, second generation daughter sequences were identified that had previously unknown properties, β-sheet secondary structure, and highly potent, equilibrium pore forming activity176.
We have also evolved multiple families of α-helical MPPs with unique functions and properties. First, we created a second generation library based on melittin, which forms transient pores in PC bilayers59,64–66,177 and successfully screened for daughter sequences that enable potent, equilibrium permeabilization59. We discovered afterward that at least one of the these gain-of-function analogs, called MelP5, enables macromolecule release from lipid vesicles at low P:L62,178. Simultaneously, we screened the same library for loss-of-function analogs, identifying a single amino acid change, Leu 16 to Gly, in melittin which eliminated the membrane permeabilizing activity of melittin against zwitterionic synthetic bilayers and cellular membranes179. Subsequently, Hristova and colleagues used MelP5 as a template for a third generation library which was successfully screened for more potent macromolecular poration that was triggered by acidic pH180. The same library was separately screened for more potent macromolecular poration that was not triggered by pH which lead to the discovery of an uniquely potent family of MPPs called macrolittins181.
2.6. Diversity of structure of MPPs
Membrane-permeabilizing peptides (MPPs) have tremendous diversity in their secondary structures, as there is not a singular secondary structure which best enables membrane permeabilization. In this way, MPPs are unlike integral membrane proteins, which usually have architectures based on helical bundles182,183 or beta barrels184,185, and that follow established sequence-structure-function relationships. MPPs do not seem to adhere to a similarly tractable set of rules. Peptides may fall into archetypal designations of secondary structure such as α-helical, β-sheet-like, and random coil, however each of these structures are able to permeabilize lipid bilayers7,75,186–189. Explicit, or well-defined tertiary structures or oligomers are generally not found, and are not required, for the observed activity of MPPs. In Figure 4, models of secondary structure for 15 peptides have been outlined to demonstrate the secondary structure variability of MPPs. In Figure 5, spacefilling models of regional hydrophobicity for the same 15 peptides in the same dimensional orientation are outlined.
Figure 4.

Secondary structures of membrane permeabilizing peptides. Cartoon models for 15 peptides which are known membrane-permeabilizing peptides depicting the wide array of secondary structures. Some peptides are uniformly one secondary structure and other peptides appear to be a hybrid of multiple secondary structure motifs.
Figure 5.

Amphipathic structures of membrane permeabilizing peptides. Space filling models for 15 peptides which are known membrane-permeabilizing peptides depicting hydrophobicity; no peptide here is depicted as singularly hydrophilic or hydrophobic, there are elements of both when considering MPPs.
Circular dichroism (CD) spectroscopy, Fourier transform infrared (FTIR) spectroscopy and nuclear magnetic resonance (NMR) spectroscopy50,190,191 are widely-used techniques to determine secondary structure of peptides in aqueous conditions, solvents, detergents, bicelles, lipid vesicles192,193 and supported bilayers194,195. Linear peptides, for example melittin or the magainins, are often unstructured in an aqueous environment and become structured only when partitioned into lipid bilayers or membrane-like environments7,91,196–198. At the level of secondary structure, helical peptides such as melittin, alamethicin, and LL-37 adhere to the conformational patterns derived from helical hydrogen bonding, using patterns of amino acid hydrophobicity to fold into amphipathic structures58,75,199. The most common helical form is the α-helix with an i to i + 4 hydrogen bonding pattern. i to i + 3 arrangements are found in 310 helices and i to i + 5 arrangements are found in pi-helices200–202. These structural patterns can be visualized 2-dimensionally through helical wheel projections (Figure 2). Helical patterns are useful information in rational engineering172 or synthetic molecular evolution59,180,181. However, for the vast majority of helical MPPs, there is little data to suggest that they assemble into explicit membrane protein-like transmembrane helical bundles (see exceptions below). Rather, the interactions of most MPPs, helical or otherwise, are dynamic. Heterogeneous interactions with the dynamic, fluid phase interface of the lipid bilayer membrane occur where tertiary structure of the peptide is not well defined. Consequently, knowing how to engineer the amphipathic surfaces of helical peptides is not enough to knowingly engineer MPP activity.
There is also an overarching hydrogen binding pattern that defines β-sheet-like peptides 5,176,210,188,203–209. However, there are many more ways to form β-sheet-like structure compared to α-helical structure,188,211 including the possibility of both intramolecular and intermolecular hydrogen bonds. Thus, with a few exceptions, the architecture of β-sheet containing MPPs in the active state are especially difficult to describe, predict, or model computationally. For example, the defensins form rigid, globular structures that are amenable to NMR and crystallographic structure determination205,208,212,213 yet the molecular mechanism by which these globular, highly cationic structures interact with membranes to cause permeabilization remains only vaguely described.
Multiple Cys residues within an MPP can form internal disulfide linkages and change secondary structure and subsequent peptide function. Many of the β-sheet rich antimicrobial peptides, e.g. protegrin, tachyplesin, α- and β-defensins, Θ-defensins, and others have internal disulfide crosslinks that are sometimes necessary for activity. Wimley and colleagues showed that the reduction of the disulfide bonds in a human α-defensin actually greatly increased vesicle permeabilizing activity, while also abrogating biological activity214. He et al demonstrated that the viroporin activity of the Ebola virus delta peptide, which permeabilizes many cell types in vitro is entirely dependent on the presence of a disulfide cross link that forms a short C-terminal hairpin136. This indicates that the secondary structure modification elicited by the disulfide linkage is essential for membrane permeabilization by the delta peptide.
Some AMPs have secondary structures that are influenced by non-proteinogenic amino acids, such as D-amino acids, α-aminoisobutyric acid, ethylnorvaline, isovaline, phenylalinol, hydroxyproline and others to form peptaibols. Alamethicin is the classical peptaibol215. It has multiple α-aminoisobutyric acid residues which strongly promote α-helical structure. Gramicidin A, on the other hand has alternating L and D amino acids that enable formation of a unique β-helix structure216,217.
Peptide-membrane interactions are driven by physical chemistry218. Interactions between peptide and membrane are heterogeneous and such peptides often behave as molecules that have partitioned into a fluid. Explicit, discrete three-dimensional structures of MPPs are extremely rare, although many researchers have tried to determine or model them. The ability of peptides to permeabilize membranes is largely dependent on the physical chemical interactions between the membrane and peptide; the amino acid composition plays a role at least as significant as the sequence because the determinants for membrane permeabilization, under some conditions, depends on overall hydrophobicity and amphipathicity of the peptide, along with electrostatic interactions between the peptide and membrane179,219,220.
As an example of the importance of composition, Shai and colleagues made a diastereomer of melittin with four D-amino acids that thoroughly disrupted its helical secondary structure221. This melittin analog lost its ability to permeabilize red blood cells and synthetic vesicles made of PC, yet retained very high membrane permeabilizing activity against bacteria and against anionic synthetic bilayers. These results, and others,78,149,214,222,223 provide an important insight to the mechanism of actions of MPPs; The same peptide can permeabilize different membranes in very different ways. In the case of melittin, permeabilization of synthetic or biological membranes made mostly of PC and sphingomyelin is strongly dependent on the formation of amphipathic α-helical secondary structure, while the permeabilization of anionic synthetic or biological membranes can occur in the complete absence of α-helical secondary structure. Likely, the molecular mechanism of melittin simultaneously encompasses a broad ensemble of structures and mechanisms. The mechanistic landscape is dynamic and the dominant mechanisms are strongly influenced by experimental conditions.
As another example of the influence of amino acid composition on function, AMPs with scrambled sequences are sometimes as active as, or more active than, native sequences224,225. Most AMPs are amphipathic and cationic, which provides electrostatic selectivity towards anionic lipids such as phosphatidylglycerol (PG) which is abundant in bacterial membranes, or phosphatidylserine (PS) which is found in some eukaryotic internal/organelle membranes, instead of zwitterionic lipids such as PC, which are dominant in the external face of eukaryotic plasma membranes226–230. Aromatic side chains interactions with the interfacial zone of a lipid bilayer are also important in peptide interactions with membranes231,232. In keeping with the concept of a mechanistic landscape, MPP-membrane interactions, and thus function, can be dependent on other factors such as peptide concentration, salt concentration, pH, lipid composition, lipid phase state (gel/fluid), temperature, hydration and many others.
One exception to the usual lack of explicit three-dimensional structures for MPPs is gramicidin-A, an unusual peptide that is made non-ribosomally with alternating L- and D-amino acids. Gramicidin assembles into a small number of discrete membrane-spanning structures using a β-helix secondary structure architecture108,109,216,217,233,234. Protegrin, a β-sheet rich, disulfide crosslinked hairpin AMP has also been studied by NMR, and a possible three dimensional arrangement has been obtained207,235. The M2 channel peptide, is one of the only explicit structures ever determined for a helical MPP137. It forms a well-defined membrane spanning tetramer. Transmembrane helical bundles have also been detected for both alamethicin236,237 and melittin53,55,56, although it is possible that such structures are found under a limited range of experimental conditions and are unlikely to be discrete, homogeneous structures. Overall, there is not a singular secondary structure or tertiary structure that best drives peptide-membrane interaction and leads to membrane permeabilization. A multitude of MPP structures can cause membrane permeabilization.
3. Systems for studying membrane permeabilizing peptides
In this section, we discuss model systems used to dissect the physical, chemical, and biological principles of membrane permeabilizing peptides. The first model systems we will discuss are synthetic lipid bilayers, including large unilamellar vesicles (LUVs), giant unilamellar vesicles (GUVs), and planar supported bilayers. Their utility is vast as they can easily be created, controlled and manipulated in the laboratory, yet share many important properties with biological membranes238. We will then discuss molecular dynamics simulations which explore the physical chemical basis of peptide-membrane interactions through computer simulations. Finally, we will discuss biological membrane systems and compare membrane permeabilization in biosystems with permeabilization observed in synthetic model systems.
3.1. Large unilamellar vesicles
Synthetic lipid vesicles are common and useful model systems for studying membrane permeabilizing peptides. Biological membranes are complex with many components, including an immense variety of lipid species239–241 and membrane proteins242. Because of this complexity, it can be challenging to study peptide-membrane interactions in biomembranes. Synthetic lipid bilayers have only one or a few lipid species with precisely known compositions, yet can accurately recapitulate the physical chemistry and dynamics of a biological membrane, providing a way to study and interpret peptide-membrane interactions243.
Synthetic unilamellar vesicles are closed bilayer structures that have an aqueous interior space surrounded by a single lipid bilayer membrane (Figure 6). The first step toward artificial vesicle preparation was made by Bangham et al. in 1965 who showed that a suspension of lipids could encapsulate ions244. Now we have many tools and methods for making vesicles of a variety of sizes and architectures. Synthetic unilamellar (i.e. one bilayer) vesicles can be categorized by their size: SUVs (small unilamellar vesicles) are ~10–50 nm, LUVs are ~50–200 nm, and GUVs are ~> 1 μm in diameter and can be as large as 100 μM. So called oligolamellar vesicles, with a few concentric bilayers245 can be 400 nm to 1 μm. They are not widely used in MPP research.
Figure 6.

Realistic cartoons of lipid vesicles. In this cartoon, small (SUV), large (LUV) and giant (GUV) unilamellar vesicles are drawn roughly to scale in three different magnifications. Even the membrane thickness is drawn to scale in each magnifications. On the right is a simulation of a vesicle of 34 nm diameter, containing ~40,000 lipids. Image courtesy of Andrew Jewett at www.moltemplate.org.
In the 1970s, SUVs were the first type of unilamellar vesicles widely utilized. They are prepared by high powered sonication246–248 and were utilized in many studies249–251. However, their relatively high curvature created instability and artificially enhanced peptide binding. Later, LUVs were made through ethanol injection252 or detergent dilution or dialysis,253,254 but incomplete removal of those chemicals proved problematic255. In the mid 1980s, Cullis and colleagues255–257 described a method of preparing uniform large unilamellar vesicles by high pressure extrusion through Nucleopore polycarbonate filters (Figure 7). This approach had many advantages over other methods, including more stable vesicles and a lack of high curvature effects. This method is applicable, almost without limit, to bilayers of any lipid compositions. Extrusion thus quickly became the method of choice for preparation of uniform LUVs258–262 and has proven to be extraordinarily useful in studying the physicochemical interactions and functions of peptides on membranes.
Figure 7.
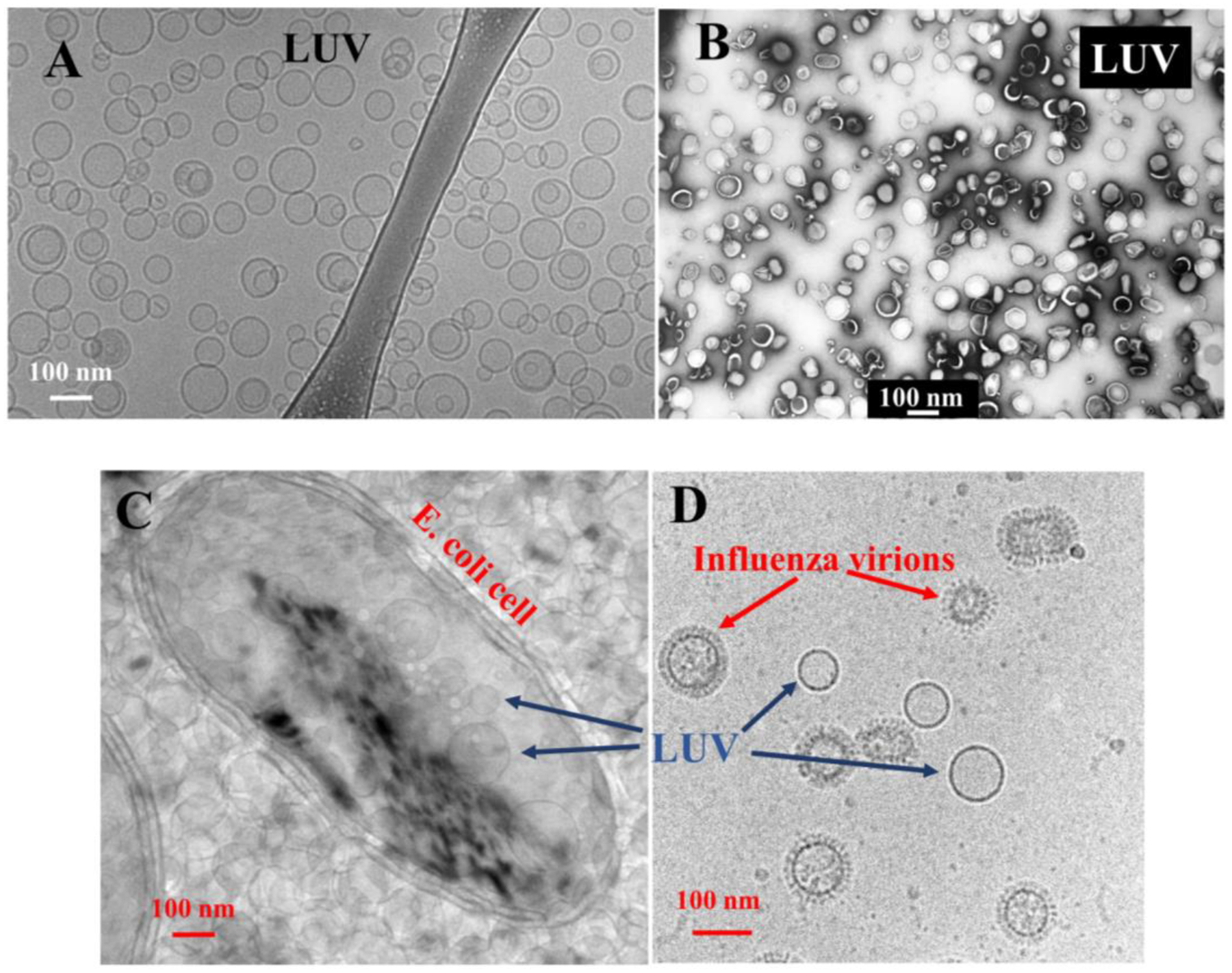
Large unilamellar vesicles. LUVs are the most commonly used synthetic models in the study of peptides in membranes. They are formed by extrusion of multilamellar lipid suspensions through Nucleopore polycarbonate filters at high pressure. A: Cryo transmission electron microscopy of a preparation of LUVs made from fluid phase PC lipids (Jibao He, Tulane University). B: Negative stain electron microscopy of LUVs showing their remarkable size and uniformity (Thomas W. Tillack, University of Virginia). A legacy image of the first extruded LUVs made by author WCW, circa 1987. C: Comparison of LUVs with the double membrane of E. coli bacteria, in the same sample, shows similar size and curvature. D: Comparison of LUVs with influenza virions in the same sample shown similar size and curvature.
By using different phospholipids, sterols, and other membrane components in a vesicle preparation, many bilayer properties can be changed. These properties include surface charge, membrane fluidity, phase behavior, and bilayer thickness. Altering these properties gives valuable insight into how peptides interact with membranes. For example, Matsuzaki et al. showed long ago that the antimicrobial peptide tachyplesin is far more active against anionic bilayers made of PG and PS than against zwitterionic bilayers made of PC263. This behavior is almost universally observed for cationic antimicrobial peptides264–268. Similarly, Ladokhin and colleagues showed that the interactions, topology, and mechanism of permeabilization of bilayers by MPPs is sensitive to lipid composition, especially headgroup charge78,222,223.
One utility in manipulating vesicle composition is to more closely mimic biological membranes. Including phosphatidylglycerol (PG) and phosphatidylethanolamine (PE) lipids in an LUV preparation, for example, increases vesicle resemblance to bacterial membranes and can be a more relevant model to use for AMP studies269. Including cholesterol and sphingomyelin increases vesicle resemblance to the outer surface of a eukaryotic cell membrane while including PE, phosphatidylserine (PS) and phosphatidylinositol (PI) increases the resemblance to the inner surface of a eukaryotic plasma membrane.
Another property of biological membranes that has been mimicked in LUVs is their transmembrane asymmetry. In an asymmetric membrane, the inner and outer leaflet compositions are different, allowing for diverse cellular processes to take place270. The outer leaflet of eukaryotic cells is mainly composed of phosphatidylcholine, sphingomyelin (SM), and cholesterol, while the inner leaflet contains mainly phosphatidylethanolamine (PE), phosphatidylinositol (PI) and phosphatidylserine (PS)271–273. Membrane asymmetry can be adjusted in synthetic vesicles, although the field is still developing. London and colleagues developed a method in which LUVs can be made with mainly sphingomyelin and phosphatidylcholine on the outer leaflet and PE and PS in the inner leaflet274. Their method utilizes (2-hydroxylpropyl)-α-cyclodextrin (HPαCD) rather than methyl-β-cyclodextrin (MβCD) to mediate lipid exchange. This allowed for the inclusion of cholesterol in the vesicles as HPαCD had much less affinity for it than for MβCD. Through this method, they were able to achieve highly asymmetric vesicles that could be a more accurate model of a biological membrane.
The purpose of using LUVs is to study membranes in a controlled, defined, and reproducible way in vitro while still mimicking the basic physical and chemical properties of biological membranes. These characteristics enable LUVs to be excellent models to study membrane permeabilizing peptides, especially because some physicochemical properties are difficult to study with biological systems or with Molecular Dynamics. Next, we discuss some techniques for studying membrane permeabilizing peptides using large unilamellar vesicles.
3.1.1. Methods of measuring LUV permeabilization
There are many ways in which permeabilization of LUVs can be measured, illustrated in Figure 8. Most share a similar fundamental concept: vesicles are prepared with reporters or reporter-related molecules entrapped within their aqueous space or in the membrane, they are then treated with peptide, and finally the rate or extent of release of the entrapped probe is measured. The readout can be through nuclear magnetic resonance, light scattering, electron spin resonance, or fluorescence spectroscopy, among other methods275, but fluorescence is used most often because it is versatile, highly sensitive, and widely available. Next, we will discuss a few commonly used types of leakage assays: small molecule and macromolecule release, flip-flop and translocation assays, along with assays that distinguish graded from all all-or-none permeabilization, and assays that distinguish transient from equilibrium permeabilization.
Figure 8.
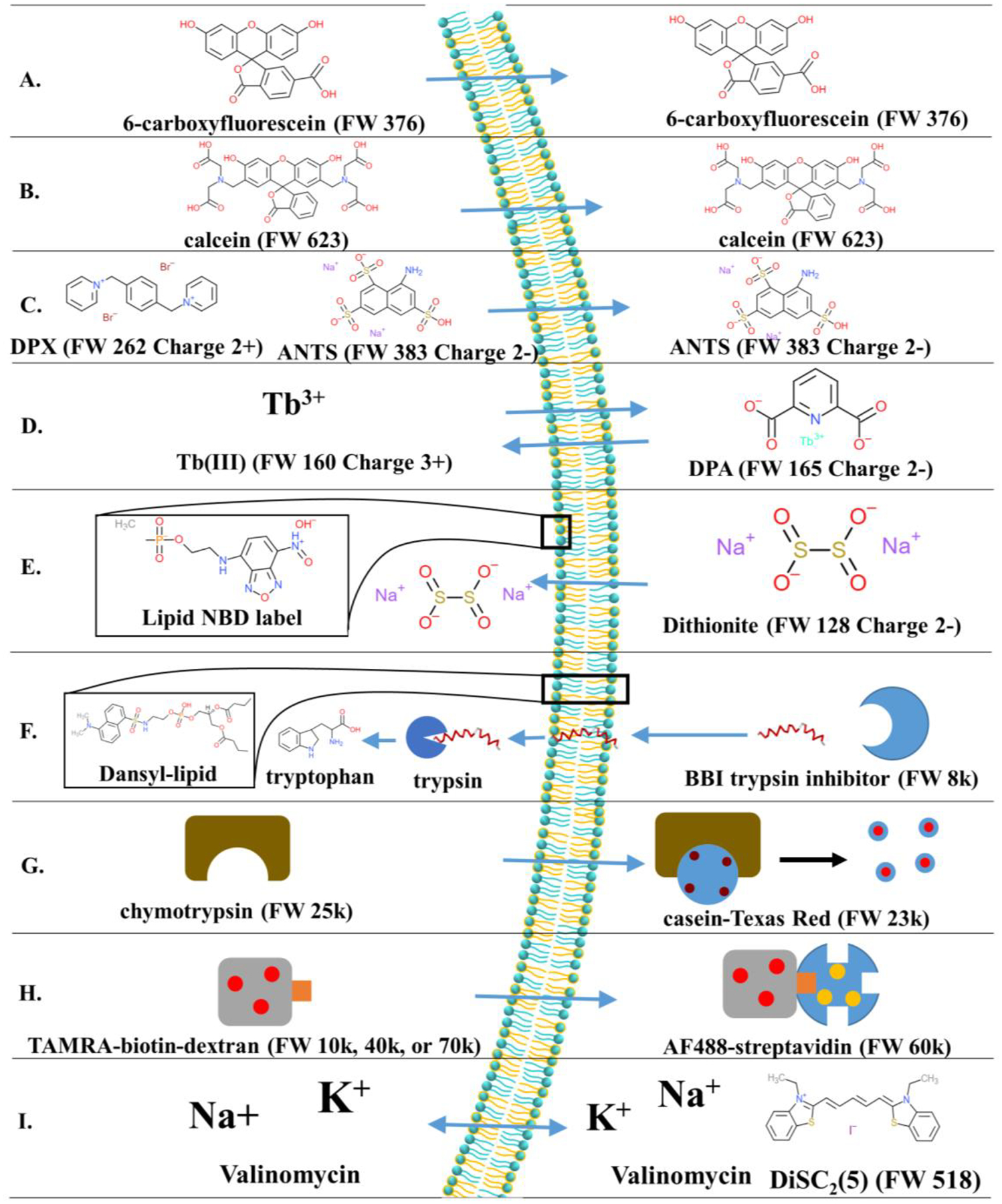
Probes used to measure permeabilization of LUVs. The chemical structures for small molecules and cartoon representations of larger molecules are shown, as well as a general theory of each assay. Only a select few assays are shown. Some assays can be relatively simple and measure leakage of a single fluorophore out of a vesicle, as in (A) and (B). Others are more elaborate, utilizing FRET [(H), (F)], macromolecules [(G), (H)], or even membrane potential [(I)]. The assays are as follows: (A) carboxyfluorescein assay, (B) calcein assay, (C) ANTS/DPX assay, (D) Tb3+/DPA assay, (E) equilibrium permeabilization assay, (F) translocation assay, (G) chymotrypsin release assay, (H) macromolecule release assay, and (I) diffusion potential assay. See text for details.
Small molecule leakage assays.
The first leakage experiments were done with entrapped carboxyfluorescein in the 1970s276–279. In this assay, liposomes are prepared in a solution with a high concentration, 50–80 mM, of the fluorophore carboxyfluorescein. Then, the vesicles are purified and extra-vesicular dye is removed through gel filtration chromatography. The high concentration of the fluorophore within the vesicles causes self-quenching and low fluorescence. Upon membrane disruption, the fluorophore leaks out of the vesicles and is diluted into the large external volume where its fluorescence is recovered. Permeabilization can be quantified through the increase in fluorescence compared to a sample completely solubilized with detergent, which amounts to 100% release. A difficulty with this assay arises from the fact that the fluorescence of carboxyfluorescein is especially pH sensitive near physiological pH. To overcome this issue, Allen and Cleland replaced the fluorophore with calcein, a pH independent fluorophore (at pH 6–8.5)280, which has since been widely used.
The binary ANTS/DPX assay uses the fluorophore 8-aminonapthalene-1,3,6 trisulfonic acid (ANTS) and its quencher p-xylene-bis-pyridinium bromide (DPX). ANTS does not self-quench and is relatively pH insensitive281 but is readily quenched by low mM concentrations of DPX. First described in 1976 by Smolarsky et. al., ANTS/DPX was used to detect complement-mediated lysis of liposomes282. It was later developed to its present form by Ellens et al.281 and others. In this assay, LUVs are made with co-encapsulated fluorophore and quencher at concentrations that causes quenching and the external ANTS and DPX is removed by gel filtration chromatography, filtration or centrifugation. Upon addition of a membrane permeabilizing peptide, ANTS and DPX escape into the external volume where the increase in ANTS fluorescence can be measured spectroscopically80. This basic form of the ANTS/DPX assay gives valuable insight into the potency of a membrane active peptide to disrupt vesicles in such a way that allows small molecules to pass through62.
The Tb3+/DPA assay is another example of a binary permeabilization assay. In this assay, the lanthanide metal Tb3+, which is weakly chelated by citrate, is encapsulated in LUVs at mM concentrations. The aromatic strong chelator dipicolinic acid (DPA) is added to the vesicle exterior at μM concentrations. Complexed with citrate, Tb3+ is very weakly luminescent, but when chelated by DPA, the complex is brightly luminescent283,284. Leakage of Tb3+ out of the vesicle, or the leakage of DPA into the vesicles, enables DPA to replace citrate, creating a very bright complex that can be measured64,284.
The equilibrium permeabilization assay is unique in that it enables the measurement of leakage, as above, but also enables the distinction between equilibrium and transient permeabilization. Krauson et al. developed this assay while studying the mechanism of action of AMPs (LL37 and dermaseptin S1) and lytic peptides (LLP1, LLP2, melittin and alamethicin)64. LUVs with entrapped Tb3+ and external DPA are prepared to measure leakage by the standard Tb3+/DPA assay, described above. However in this assay, these vesicles also contain 1 mol %, of diacyl phospholipid which are labeled on the headgroup with the dye nitrobenzoxadiazole (NBD). NBD has fluorescence that can be measured independently from Tb3+/DPA. Its fluorescence can be quenched by the membrane-impermeant reducing agent dithionite, S2O22−, which can be added at any time during the assay. NBD lipids on the vesicle inner monolayer are protected from quenching unless the membranes are permeable to dithionite at the time of its addition. In the equilibrium permeabilization assay, vesicles are treated with peptide and allowed to equilibrate for up to 8 hours; leakage usually ceases within 1 hour. Terbium fluorescence is measured to determine the total leakage. Then, dithionite is added and the degree to which the NBD fluorescence is protected is measured. When permeabilization is a transient process, Tb3+/DPA reports on net leakage after peptide addition, while dithionite only quenches external NBD-lipids, showing that the bilayers are sealed at equilibrium despite having been permeabilized initially. When permeabilization is an equilibrium process, both leakage and dithionite quenching of NBD go to completion, and dithionite can access the vesicle interior at equilibrium and quench all NBD lipids. Krauson et al. showed that the AMPs tested were transient membrane permeabilizers285 that induce only short term membrane permeabilization. On the other hand the peptaibol alamethicin and the lentivirus lytic peptides LLP1 and LLP2 showed highly potent permeabilization to Tb3+/DPA by equilibrium pores that enabled complete quenching of lipid-linked NBD by dithionite at most P:L studied, down to as low as 1:2000. Interestingly, the mechanism of melittin transitioned from transient towards equilibrium behavior over the measured concentration range of P:L of 1:2000 to P:L = 1:50.
The diffusion potential assay measures the leakage of small ions in the presence of a transmembrane potential221,286,287. Briefly, LUVs, prepared in the presence of KCl, are diluted into a K+ free buffer that contains the potential sensitive dye 3,3′-Diethylthiadicarbocyanine iodide [DiSC2(5)]. Then, valinomycin, which is a K+ ionophore, is added to the mixture, creating a negative inside transmembrane potential which causes dye quenching. After equilibrating this solution for a few minutes, the peptide treatment is then added. If there is membrane disruption, there will be a dissipation of diffusion potential as indicated by the increased fluorescence. The fluorescence recovery values can then be tracked over time or at a single time point. Kobayashi et al employed this assay to characterize the AMP buforin 2. They found that buforin 2 did not permeabilize vesicle membranes as well as magainin, with or without a membrane potential286. They went on to show that buforin 2 translocates with high efficiency across vesicle membranes, while exhibiting low vesicle permeabilization as well as low lipid translocation.
Macromolecule release assays
Macromolecule release assays provide information on the size of the permeabilization pathway based on measurements of macromolecule permeabilization61,62,78,153,203,285,288. Originally, this measurement was performed with fluorescein-labelled dextrans which self-quench when entrapped at high concentration, in the mg/ml range. Upon release, the fluorescence is recovered. Hristova et al. used this assay to study vesicle permeabilization by the rabbit defensin NP-2. They found that native NP-2 allowed for dextrans from 4 to 70 kDa to leak at the same rate which supported the hypothesis that NP-2 forms large lesions or destroys the vesicle architecture rather than forming size-specific pertubations289. Ladokhin et al. co-entrapped two dextrans of different molecular weights and used gel filtration chromatography to measure their release by melittin, finding that both sizes were released equally well61. Similar applications have been used elsewhere214,290. One potential problem with these dextran release assays is that entrapment of concentrated dextrans creates an osmotic imbalance so this should be counteracted by using external, unlabeled dextrans.
A recent adaptation of this assay uses dextrans labelled with biotin and also with the dye tetramethylrhodamine (TAMRA) which are encapsulated in LUVs at very low, μg/ml, concentration62. The low concentration used eliminates osmotic stress on the vesicles caused by mg/ml dextrans used previously. The extravesicular solution contains AlexaFluor488-labelled streptavidin. If the vesicle membranes are disrupted enough to enable TAMRA-biotin-dextran release, the dextran will complex with the external AF488-streptavidin, leading to FRET-based quenching of the AF488 by TAMRA. Using this assay, Wiedman et al. showed that MelP5, a gain of function variant of melittin59, enables macromolecule release from LUVs at low peptide concentration62. Furthermore, this assay was used in several successful screens for peptides that induce macromolecular release180,181, leading to the identification of highly potent peptides with this activity at P:L=1:1000, either in a pH-triggered or a pH-insensitive manner.
The chymotrypsin-casein assay is another macromolecular leakage assay described by Wiedman et al. during their characterization of melittin gain-of-function variants62. Here, the vesicles contain entrapped chymotrypsin. Peptide-induced release of chymotrypsin enables the proteolysis of extra-vesicular casein labelled with Texas Red (EnzChek), which can be monitored by the increase in Texas Red fluorescence.
Flip-flip and translocation assays
Lipid and peptide translocation across membranes, sometimes called “flip-flop” are a specialized kind of permeabilization wherein one measures the movement of peptide or lipid polar groups cross a lipid bilayer from one monolayer to the other. Pagano et al. developed a method in 1981 to label the inner or outer leaflet of vesicles with NBD-lipid to measure lipid asymmetry291. Matsuzaki et al. utilized this technique to detect lipid flip-flop in vesicles292. Here, NBD-lipid is first incorporated into either leaflet of the vesicles. To label the outer leaflet, pre-made LUVs are incubated in a solution with NBD-lipid. To label the inner leaflet, the NBD-lipid is added to the initial LUV lipid mixture when making the LUVs, then the outer leaflet is chemically quenched with dithionite reduction. These asymmetrically labeled vesicles can then be treated with peptide to induce translocation. Dithionite is then added to the solution at set times to quench the outer leaflet and NBD fluorescence is measured to determine the degree of protection or exposure by translocation. This assay can only be used for membrane permeabilizing peptides if they cause transient permeabilization because the bilayers must be impermeant to dithionite when it is added. A slightly modified version of this assay has been described for the study of transmembrane peptide sequences on flip-flop behavior293. Here, the transmembrane peptide is incorporated into the LUV membrane, the outer leaflet is labeled, and then flip is measured as above. It was found that less hydrophobic peptides induced less flip-flop than more hydrophobic peptides293. On the other hand, Wimley and White showed that alamethicin, even at extremely low concentrations of less than P:L = 1:2000, catalyzed very rapid lipid flip-flop294. Fuselier and Wimley used a version of this assay to show that translocating peptides also increase lipid translocation295.
Matsuzaki et al. have adapted LUVs to measure peptide translocation rate with a reporter system that involves enzymatic cleavage and FRET286,296,297. In this assay, LUVs containing fluorescent dansyl phosphatidylethanolamine are made with encapsulated trypsin. Peptides are then added to the LUVs. The tryptophan residues of bound peptides come into the vicinity of the dansyl groups, and are quenched by FRET. If the peptide translocates across the bilayer into the vesicle, it will be cleaved by the internal trypsin and FRET will be reduced (i.e. the tryptophan fluorescence increases while the dansyl fluorescence decreases). Using this technique, Matsuzaki et al. was able to determine that magainin translocates across vesicle membranes through a permeabilization mechanism that allows small molecule leakage as well297. Marks et al. developed a variant of the translocation assay to screen for spontaneous membrane-translocating peptides298. In this assay, the fluorophore aminomethylcoumarin (AMC) is added onto the C-terminal phenylalanine of the peptides. Vesicles contain entrapped chymotrypsin and Tb3+ while the extracellular space contains DPA and the α1-antitrypsin inhibitor. This assay reports independently on permeabilization by Tb3+ fluorescence, and translocation, which is reported by the chymotryopsin cleavage of the AMC group. These authors were able to identify spontaneous membrane translocating peptides that cross PC bilayers and cell membranes without any permeabilization298. Other methods of measuring peptide translocation rates have also been described38,295.
Graded vs all-or-none leakage assays
In an ensemble average measurement of leakage, there are multiple ways that leakage can be distributed across the population of vesicles examined. Understanding those mechanisms can help us understand the mechanistic landscape of membrane permeabilizing peptides. For example, leakage from vesicles can be graded, in which all vesicles release a similar portion of their contents before leakage stops. Alternately, leakage can be all-or-none, in which a fraction of the vesicles release all of their contents while the reminder of vesicles release none. In the early 1980s, a modification of the carboxyfluorescein method was used to distinguish these possibilities299. After leakage measurements were made by the standard method, vesicles in the reaction solution and any entrapped dye molecules were separated from released dye using gel filtration. In the re-purified vesicle sample, if there was graded leakage each vesicle would contain only a fraction of the initial fluorophore concentration, and there would be less quenching than in the original vesicles. On the other hand, if leakage was all-or-none, then the degree of quenching in the re-purified vesicles will be the same as the starting vesicles. Using this method, Weinstein et al. showed that HDL apolipoprotein induces an all-or-none effect at the liquid-crystalline transition temperature of the lipid bilayer299. Later, similar re-purification assays were performed using ANTS/DPX as the leakage reporter172,290,300. Both graded and all-or-none leakage mechanisms have been observed by these techniques.
A simpler method to distinguish between graded and all-or-none leakage was developed by Wimley, Ladokhin and colleagues80,214,301. This method uses “requenching” of the ANTS fluorophore in situ rather than physical separation of the vesicles from released molecules (Figure 9). Here, the ANTS/DPX assay is performed as above and fractional leakage is allowed to plateau. Different peptide concentrations can be used to give different fractional permeabilization. Then, additional DPX is titrated into the vesicle solution while fluorescence is monitored. For a control, untreated vesicles are lysed with excess detergent and fluorescence is measured as DPX is added. Through these measurements, Qin, the degree of quenching of the ANTS remaining inside the vesicles, and fout, the fraction of ANTS released are determined. If Qin remains constant with fout then leakage is all-or-none. If Qin increases with fout then leakage is graded203,302–306. Expanding on this, Ladokhin et al. showed that it is possible to calculate the relative preference for leakage of DPX over ANTS, expressed with the variable α80. Using these methods, Hristova et al. showed that for the rabbit defensin NP-5, Qin increased with fout and the α value was 1.6, indicating a graded leakage behavior with preferential DPX leakage307. Another method of distinguishing between graded and all-or-none leakage has been reported which utilizes time-resolved calcein fluorescence decay measurements as the reporter system308.
Figure 9.
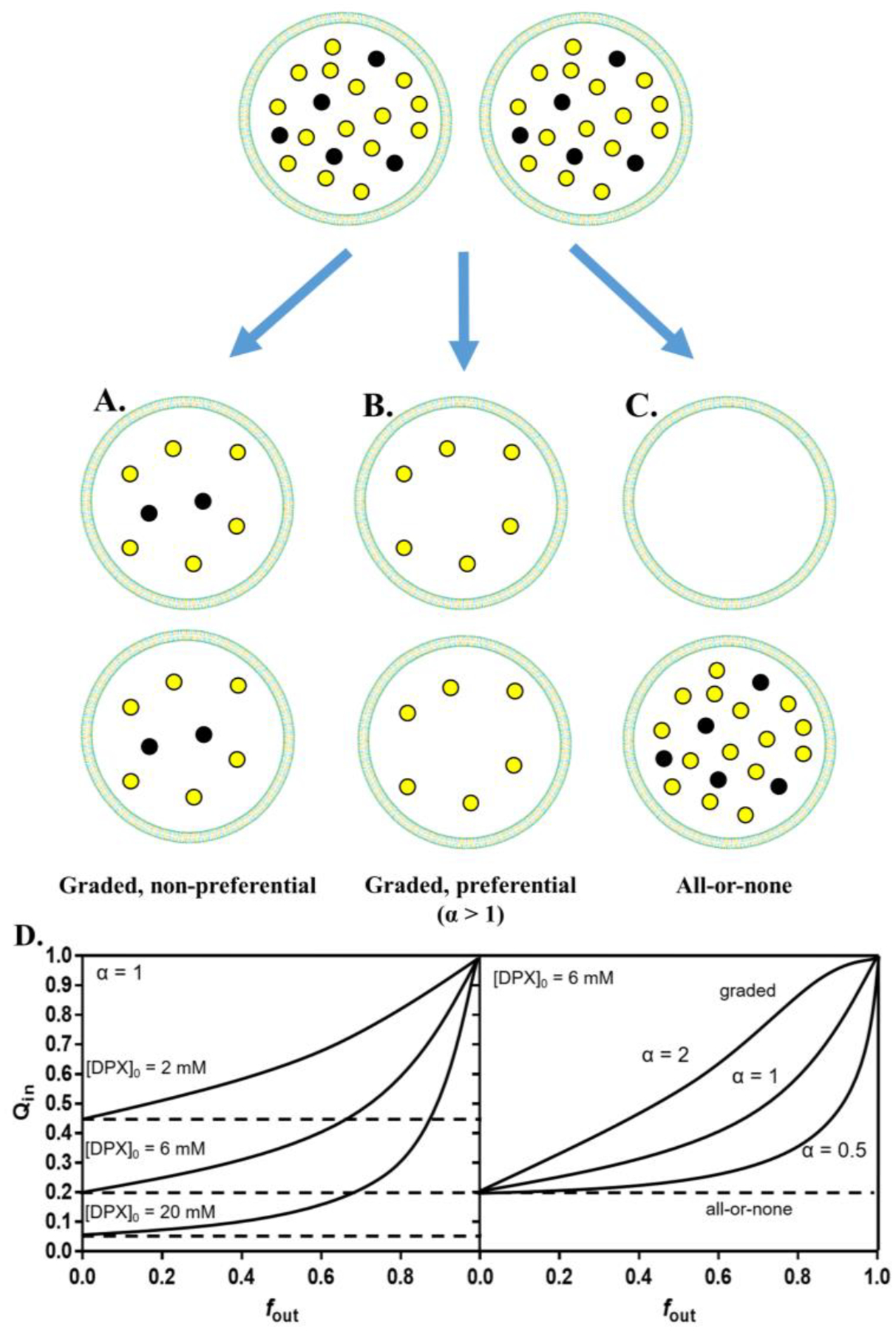
Graded and all-or-none leakage are two distinct peptide-induced leakage mechanisms on vesicles. The ANTS/DPX assay is used as an example here. (A) When all vesicles lose an equal portion of all encapsulated solutes, it is considered graded, non-preferential leakage. (B) Losing an unequal portion of encapsulated solutes is considered graded and preferential. The equations from72 can determine ANTS or DPX preferential leakage. In this example, α > 1 therefore there is preferential leakage of the cationic quencher DPX. (C) Treatment of a sample of vesicles can lead to some vesicles losing all of their contents while the remaining vesicles losing none. This is an all-or-none behavior. Yellow and black dots are ANTS and DPX molecules, respectively. (D) This simulation shows how the two mechanisms can be experimentally distinguished. When plotting Qin (internal quenching) against fout (ANTS released), a steady Qin indicates an all-or-none mechanism; an increasing Qin indicates a graded mechanism. Different encapsulated [DPX] affects the results (left) such that 4–8 mM is optimal. Here, α = kANTS/kDPX which determines the preferential nature of leakage that is occurring (right). Adapted with permission from ref 301. Copyright 1997 Elsevier, Inc.
Hoernke and colleagues309 used a statistical approach to extend the model of graded and all leakage to a more realistic continuum of states which can be described by cumulative “leakage events”. In this model, the characteristics of leakage events are a function of the membrane permeabilizing molecule and many other experimental conditions. Each event cumulatively releases a specific fraction of remaining entrapped contents so the total released contents asymptotically approaches 100%. Low release per event (≤10%), averaged over many events, gives rise to graded leakage. High leakage per event (>75%), over a low probability of event occurrence will approach all-or-none leakage. Intermediate values of leakage per event give rise to very broad distributions of entrapped probe concentration, which these authors observed in experimental systems309.
3.1.2. What behaviors have been observed with LUVs?
Many different leakage behaviors can be, and have been, observed in permeabilization assays that utilize LUVs. Leakage is not a singular process, but many possible partially overlapping processes. Understanding the intricacies and expected behaviors of each gives a better understanding of the mechanistic landscape of peptide-induced membrane permeabilization. In leakage assays, the measurement is made either continuously over time, or at a single time point. In the literature measurement of leakage is frequently, but not always, made after leakage is essentially complete. Measuring continuously provides insight into the kinetics of leakage. Since many MPPs cause a burst of leakage that slows or stops soon after it begins, single time point measurements, 30 or 60 minutes up to several hours after peptide treatment, are generally sufficient for the entire peptide-induced leakage kinetics to take place. However, the cessation of leakage should be empirically verified as some peptides cause slow leakage over many hours285.
In most assays, the fractional leakage at some time point is expressed as a function of peptide concentration, or as a function of the overall ratio of peptide to lipid, P:L in each sample. It would be much more informative for leakage to be described by the ratio of bound peptide to lipid Pbound:L as this is the relevant concentration of peptide partitioned into the bilayer. While peptide binding to bilayers can be expressed quantitatively310,311 it is rarely measured. Thus, the overall peptide to lipid ratio (P:L), which denotes the theoretical maximum of Pbound:L285 is typically used. Potency is commonly displayed as a plot of percent leakage against peptide concentration or P:L. The P:L at which there is 50% leakage, or LIC50 (for leakage inducing concentration, 50%) is a good single number representation of potency. LIC50 values for MPPs can range from ~1:1 or 1:10 for a peptide with barely measurable activity, e.g. melittin L16G179 to as high as ~1:10,000, for an extremely potent peptide, as reported for GALA172. This latter value is equivalent to about 10 peptides per LUV, and may represent the maximum possible potency of an MPP.
Transient or equilibrium permeabilization
If the peptide-induced pathway through the bilayer that enables permeabilization is an equilibrium structure, or one that continuously fluctuates in and out of existence, then leakage should be a continuous exponential approach toward 100% permeabilization312,313. As peptide concentration is changed, the rate of this approach towards 100% leakage should change accordingly314–316. While equilibrium poration is easy to describe, illustrate, or simulate, it is actually only rarely observed in the laboratory. Most vesicle leakage assays show transient leakage2, which is characterized by a burst of leakage that occurs immediately after addition of peptide, often occurring as fast as the first measurement can be made. Within minutes of the leakage burst, it slows and often stops completely as shown in Figure 2H and discussed below. In this commonly observed scenario, all peptide concentrations show similar rate behaviors, but the plateau level of leakage depends on P:L, such that increasing the peptide concentration will increase the amount of leakage that occurs during the burst. Many membrane permeabilizing peptides exhibit this behavior, including mouse and bovine PrPp314, mastoparan X from hornet venom317, melittin65 and many other families of MPPs5,65,203,223,318–320. This transience is poorly understood but is thought to be due to an initial asymmetry and imbalance (of mass, charge, or surface tension) in the membrane structure that causes a stochastic failure of the membrane, which then resolves itself of the asymmetry285 leading to a cessation of leakage. It is interesting to note that between the burst phase of rapid leakage and the equilibrium phase in which leakage has stopped, there is no change in the concentration of peptide bound to each vesicle and usually no detectible change in peptide secondary structure. Every vesicle has hundreds to thousands of peptides that remain bound to it, yet such systems very quickly transition from rapid leakage to little or no leakage.
In some experiments, melittin has been shown to cause a burst of leakage followed by a slow, almost linear rate of leakage, probably due to a change in the leakage mechanism313,321. In other experiments, melittin causes a burst of leakage with a stable plateau, the classical signature of transient leakage65. In either case, vesicle leakage by melittin can only be explained by transient leakage processes, at least until the concentration is greater than about P:L ≥ 1:200. At higher concentrations, melittin begins to behave more like an equilibrium pore former64.
Graded or all-or-none
As discussed above, graded and all-or-none leakage describes incomplete leakage on a per-vesicle basis. Graded leakage describes the case where all vesicles uniformly leak a portion of their contents and all-or-none describes the case where a fraction of the vesicles release all their contents while, the remainder release none. Both behaviors have been observed, even for transient leakage. Melittin, long thought to be an equilibrium pore-forming peptide, in fact induces graded, transient leakage in vesicles312, at least at lower P:L conditions in PC bilayers. Both graded leakage and transient leakage imply a mechanism other than equilibrium pore formation.
All-or-none leakage, often observed in conjunction with transient leakage,203 implies that a rare, stochastic, event is responsible for permeabilization. The event must be catastrophic in some way as it can lead to complete permeabilization of an individual vesicle, and sometimes leads to destruction of the vesicle architecture. But for transient leakage, the probability of the catastrophic event must decrease rapidly with time to account for the rapid leakage cessation. The AMP cecropin A exhibits all-or-none leakage304, as do defensins214 and other AMPs5,203. Rathinakumar and Wimley posited that transient, all-or-none leakage occurs because there are two ways to dissipate an initial peptide asymmetry: a stochastic, catastrophic leakage event, and a silent peptide translocation process that dissipates the asymmetry without leakage5.
Dependence on solute size and charge
The size of substances that are allowed through a permeabilized membrane is useful in discussing mechanisms and estimating the dimensions of the path that a peptide forms in a bilayer. Permeability can be selective for small molecules only, such as for protons, ions, ANTS/DPX and Tb3+, or there can be larger membrane disruptions that allow for macromolecules like dextrans of 3, 10, 40 or 70 kD to pass through. As discussed above, “permeabilization” can also be the result of catastrophic disruption of vesicle architecture, releasing all entrapped contents regardless of size. Methods for measuring macromolecule release can be very informative. For example the peptides GALA, alamethicin, and MelP5 are all highly potent MPPs in PC bilayers,64 yet GALA and alamethicin release only small molecules, while MelP5 readily releases macromolecules as large as 40 kDa62.
Preferential leakage based on solute charge has also been observed for many peptides, under some condition, including nisin (anion-selective)322, alamethicin (mildly cation-selective)323,324, PG-1 (weakly anion-selective)325, melittin (anion-selective)53, and the pentapeptide AcWLWLL (cation-selective)80. Determining the ionic preference gives insight into the structure of the peptide when it is embedded in the membrane.
Dependence on vesicle size
The size of an LUV can affect the potency of peptides as line tension and curvature of the membrane contributes to peptide-membrane interactions. Cho and colleagues showed this by using quartz crystal microbalance with dissipation monitoring (QCM-D) to demonstrate that the AH peptide, an amphipathic helix sequence found in the NS5A protein of hepatitis C virus, induced sharply different rupturing capacities for different sized extruded vesicles326. The peptide induced complete rupture for vesicles under 67 nm in diameter, incomplete rupture for those under 90 nm, and no vesicle rupture for even larger vesicles. This indicated that the AH peptide only lyses vesicles that have a maximum line tension327. They extended this work to confirm a size-dependent activity on smaller Dengue, Zika and Hepatitis C viruses compared to larger vaccinia and vesicular stomatitis virus31.
3.1.3. The problem of aggregation.
Across the field of membrane permeabilizing peptides, there is a pervasive potential artifact that is generally ignored but may influence reported vesicle leakage studies. This is the problem of vesicle aggregation and fusion caused by the addition of polycationic peptides, such as AMPs or CPPs, to liposomes containing anionic lipids. As many researchers in the field have observed, including us328, when cationic peptides are added to vesicles containing anionic lipids there is often an instantaneous and dramatic increase in the solution turbidity and a loss of opalescence of the liposomes. These observations are due to large scale vesicle aggregation, driven by the very strong, polyvalent electrostatic interactions. The resulting particles are sometimes so large, ≥ 10 μm, that they quickly precipitate from solution. Such large particles may be comprised of thousands or tens of thousands of individual vesicles. Strong adhesion between vesicles can lead to bilayer deformation, vesicle rupture, and membrane fusion328, all of which can lead to leakage of entrapped contents. This is a potential problem for the field because these effects report on a special case of peptide-induced bilayer disruption that does not effectively mimic the biological activities that are usually being studied. For example, bacterial membrane permeabilization occurs when peptides interact with isolated bacterial cytoplasmic membranes that cannot undergo aggregation and fusion, thus aggregation and fusion of PE/PG or PG LUVs may skew the observed leakage.
A simple solution to this problem is known, but is not widely utilized. The addition of 4–5 mol% PEG lipids establishes a steric barrier between bilayers329 that prevents aggregation of vesicles. We show the effect of PEG lipids on LUV aggregation in Figure 10. Currently it is not known how much aggregation and fusion influence the permeabilization reported for MPPs because the reduction in leakage by the prevention of aggregation has not been measured.
Figure 10.
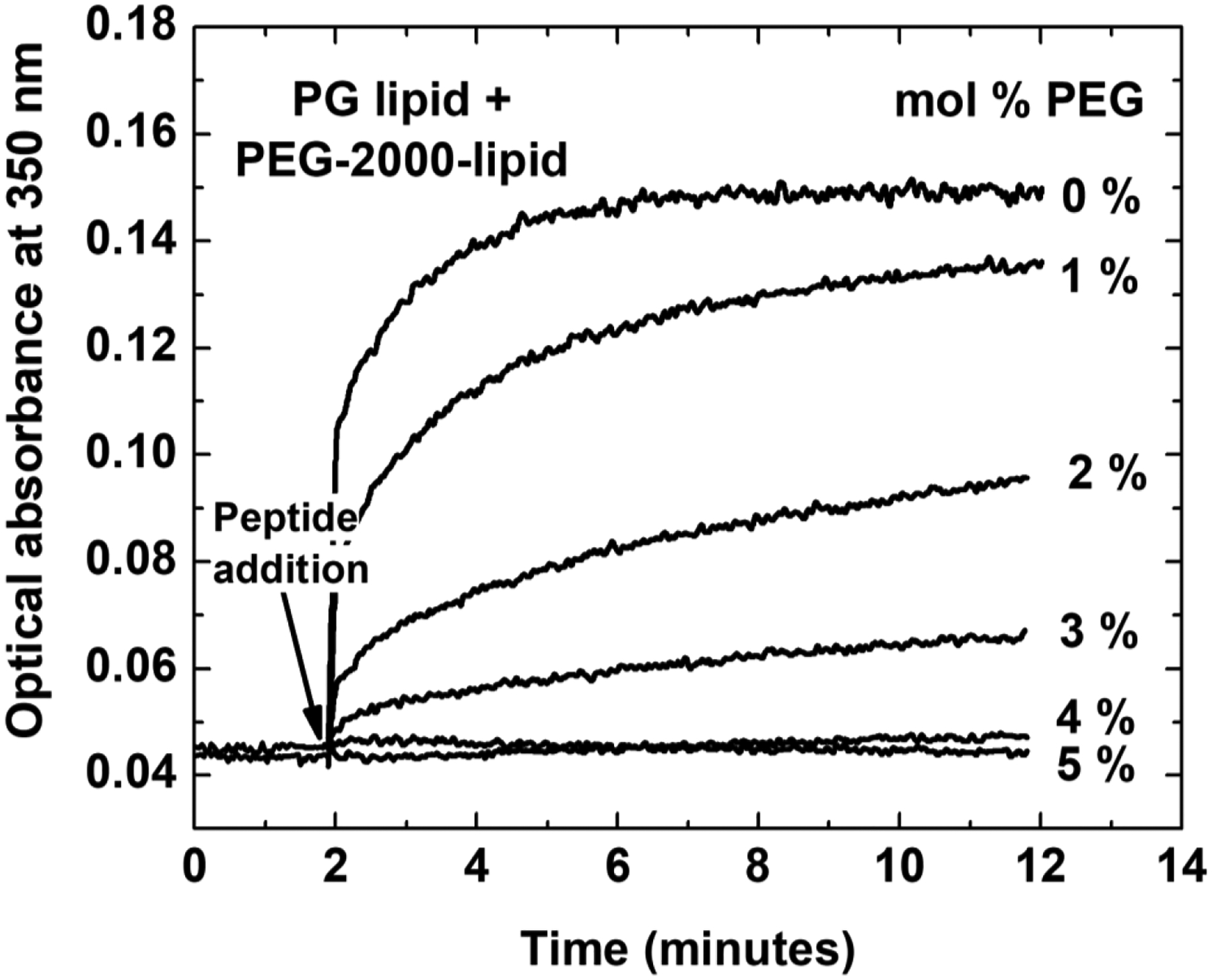
Cationic peptides rapidly aggregate anionic LUVs. Addition of the peptide *ARVA (RRGWALRLVLAY-amide) to POPG vesicles causes immediate, large scale aggregation of vesicles as shown by the increase in light scattering. Incorporation of PEG-2k-POPE lipids decreases aggregation dramatically, completely blocking it at 4–5 mol% PEG-lipids.
3.1.4. Limitations and caveats of LUVs
A limitation of leakage assays with LUVs is that the measurements are usually an ensemble average of many vesicles so that elementary processes that occur between peptide and individual vesicles cannot be accurately studied275. The structure of the LUV after permeation is usually not known, so the type of membrane disruption cannot be directly determined.275 Graded or all-or-none leakage assays partly address these questions,80,214 but only indirectly. The many behaviors that can be observed with LUVs have been very beneficial to our understanding of the mechanisms behind membrane permeabilizing peptides; even so, the use of LUVs is still developing. Over the decades that we have incorporated them into our research, new assays and interpretation of the results have continuously come to light.
3.2. Giant unilamellar vesicles
Like LUVs, Giant Unilamellar Vesicles (GUVs) are free-standing lipid bilayer models used to study membranes and membrane permeabilizing peptides. However, the diameter of a GUV is much larger than an LUV, typically 10 μm, ranging from 5 μm to 100 μm, compared to 0.1 μm for a LUV. This means that the surface area, and number of lipids, for a GUV is roughly 104 times larger than an LUV and the volume is roughly 106 larger. A GUV bilayer mimics the lipid matrix of cellular membranes and resembles the shape and size of eukaryotic cells. LUVs, on the other hand, mimic the size and shape of bacteria and viruses, as we show in Figure 7.
Giant unilamellar vesicles are commonly produced by hydration of a dried lipidic film. Initially, the gentle hydration method, originally introduced by Reeves and Dowben330, was used for preparation of GUVs, where the vesicles spontaneously formed after the addition of an aqueous solution. However, using this method, the yield of GUVs was variable and sometimes low. Even after considerable modification by Akashi et al., this method can be slow and inefficient331. Angelova and Dimitrov introduced the electroformation technique of GUV preparation which uses an AC electric field on platinum electrodes332. Later, this method was modified to work with conducting indium tin oxide (ITO) films and titanium microscope slides that further increased the unilamellarity and yield of GUVs333.
Due to their large size, single giant vesicles can be observed with optical microscopy and can be individually manipulated in real time, which is a unique capability. Smaller vesicles such as LUVs and SUVs, on the other hand, require ensemble averaging over many vesicles resulting in loss of information about individual events334–337. Hence, GUVs are currently intensively utilized in many areas of biomembrane physics and chemistry, including the study of lipid domain formation338–340 and dynamics341–343, membrane-protein interaction344–346, membrane growth, budding, and membrane fission and fusion347–349. One very useful application of GUVs is to study the function of membrane permeabilizing peptides or proteins.
3.2.1. Methods of measuring GUV permeabilization
The more traditional method of studying MPPs with GUVs is the GUV suspension method where GUVs are suspended in a solution containing a water soluble, membrane-impermeant fluorescent probe along with a peptide of interest (Figure 11). The fluorescence inside and outside the vesicle can be measured and analyzed by confocal microscopy. If the peptide permeabilizes the bilayer without rupturing the vesicle, the dye enters the GUV. Ambroggio el al. used this method to visualize and track membrane leakage induced by three antibiotic peptides: maculatin, citropin, and aurein via confocal microscopy350. Mishra et al. have also used the GUV suspension method to show how the amino acid composition of a CPP effects its membrane translocation mechanism351. And Almeida and colleagues studied the translocation of three cationic amphipathic peptides across GUV and concluded that Gibbs free energy of peptide insertion into the lipid bilayer may determine the ability of peptide to translocate352. These authors simultaneously tracked peptide translocation across bilayer and dye entry by monitoring the permeabilization of small vesicles that are often found inside larger GUVs.
Figure 11.

Peptide-induced permeabilization of GUVs. Membrane leakage of Alexa Fluor 647 hydrazide (AF647) and membrane permeabilization of carboxyfluorescein (CF)-labeled magainin 2 (CF-magainin 2) is visualized in single 40%DOPG/60%DOPC-GUVs. (A) and (B) shows confocal laser scanning microscopy (CLSM) images of (1) AF647 and (2) CF-Magainin 2 in a single GUV treated with 31 μM CF-magainin 2/magainin 2 and 20 μM CF-magainin 2/magainin 2 respectively at certain time after of CF-magainin 2/magainin 2 addition to GUV denoted in seconds under each image. (C) and (D) describe the time course of peptide-induced decrease in fluorescence intensity of AF647 in the GUV and increase in permeation of CF in the rim of GUV following the addition of CF-magainin 2/magainin 2. The solid red line corresponds to fluorescence intensity of AF647 inside the GUV while the green triangles correspond to the fluorescence intensity of CF-magainin 2 in the rim. The circles correspond to the fluorescence intensity of the outside vicinity of the GUV. FI = I(t)/I(0), where I(t) and I(0) are the fluorescence intensity of AF647 inside the GUV at time = t. (E) refers to concentration dependent average lag time between the stochastic permeabilization of magainin 2 in to the GUV and the leakage of dye from inside the GUV. These two phenomena can be studied by quantification of increase in the fluorescence intensities of the CF at rim of the GUV and decrease in the fluorescence intensity of AF647 from inside the GUV. Adapted with permission from ref 358. Copyright 2015 American Chemical Society. Images courtesy of Masahito Yamazaki.
The suspension method has also been applied by Schön et al. to determine that sphingomyelin and the coexistence of liquid-ordered and -disordered phases are necessary for permeabilization by equinatoxin II, a membrane active protein353. The GUV suspension method has also been used to demonstrate graded and all-or-none permeabilization by two different HIV-derived membrane permeabilizing peptides - CpreTM and NpreTM respectively - via confocal microscopy354.
More recently, an alternate method has been developed called the single GUV method in which the vesicles contain the water-soluble fluorescent probe, and leakage is determined by the loss of fluorescence from inside the vesicle. The structure and fluorescence intensity of the GUVs in this assay are continuously observed throughout the process by phase-contrast and confocal fluorescence microscopy. Hence, interaction of a dye-labelled peptide with a single GUV and the induced leakage of the fluorescent probe from the inside of GUV can both be monitored with time. This provides information on both membrane permeabilization and peptide translocation. Yamazaki and colleagues have used this method to describe the process of membrane permeabilization by two antimicrobial peptides, magainin 2 and lysenin355–358. They used the same method to show that translocation of a cell-penetrating peptide (CPP), transportan 10 (TP10), into a single GUV occurs without any leakage of dye from inside of the vesicle356,359,360.
3.2.2. What behaviors have been observed with GUVs?
Using the improved single GUV method with optical and fluorescence microscopy, Yamazaki and colleagues showed that magainin 2 causes leakage of calcein out of GUVs without the disruption of the vesicle architecture. This leakage occurs without interaction with any other GUVs and without change of its spherical shape indicating that the permeabilization of magainin 2 into the GUV is not due to vesicle fusion, or instability of the membrane structure275,357,361. The same group also showed that a six-residue antimicrobial peptide derived from lactoferricin B can translocate into GUVs without damaging the bilayer362. Domingues et al. studied the effect of gomesin, a potent antimicrobial peptide from a Brazilian spider species, on GUVs composed of mixtures of palmitoyloleoyl phosphatidylcholine (POPC) and either the negatively charged lipid palmitoyloleoyl phosphatidylglycerol (POPG) or cholesterol. These lipid mixtures broadly mimic bacterial and mammalian cell membranes, respectively. These authors showed that gomesin caused PG-containing GUVs to suddenly burst, lose the entrapped contents, and collapse into unresolved tubular structures363. Although not a peptide, similar bursting activity of GUVs accompanied with decrease in diameter, and vesicle transformation into indistinct compact structures was described by Yamazaki’s group, when they employed the single GUV method to study the activity of antibacterial flavonoids, e.g. tea catechins or EGCs275,364.
Many groups have been using GUVs to explore mechanistic details of the permeabilization activity of membrane-active molecules including magainin 2, melittin, LL37, and lactoferricin B365–368. In most of the peptide-induced membrane leakage studies described above, membrane permeabilization followed by the leakage of fluorescent probe from inside of a GUV starts stochastically at different times for different vesicles. Once it begins, complete leakage occurs rapidly for an individual vesicle. This suggests that the permeabilization event, and not peptide binding or contents leakage, is the rate determining step in the process356,357,364,368. For example, when magainin 2 was studied, there was a lag time of minutes between the addition of peptide and the sudden, stochastic decrease in fluorescence intensity of GUVs. Once started, the release of contents from the GUV was complete within 30 seconds356,357. In one experiment, 11 out of 16 GUVs of the same composition examined by the same technique showed sudden rapid leakage of GUV contents after lag phases, while the 5 remaining vesicles retained their contents275. This phenomenon of stochastic leakage (discussed in more detail below) is specifically detected through the use of GUVs because they can be observed individually. Similar behaviors have been observed by Cho and colleagues using surface-tethered LUVs369 and by Weisshaar and colleagues who studied the membrane permeabilization of individual bacterial cells by melittin67.
3.2.3. Limitations and caveats of GUVs
In summary, giant unilamellar vesicles constitute a unique and powerful model system that enables the process of membrane permeabilization to be studied at the level of individual cell-sized vesicles. Although one can argue that biophysical measurements of binding and secondary structure are difficult to study using GUVs due to their undefined and usually very low lipid concentration, they still have revealed a stochastic nature of membrane permeabilization by many peptides, and the catastrophic destruction of vesicle architecture by others, all of which are important clues in defining the mechanistic landscape.
3.3. Surface-supported planar bilayers
Surface-supported lipid bilayers have been successfully used for the past 30 years alongside lipid vesicles as biomimetic model systems370–373. In this technique, a supported bilayer lipid membrane (sBLM) – which is planar instead of spherical as in vesicles - is attached or adsorbed to a solid support and is generally more stable when compared to unsupported counterparts374,375. They can be designed to replicate cell surfaces of eukaryotes and microbes and are also especially useful for the study of pore and ion channel formation by peptides and proteins375,376. Planar supported lipid bilayers are assembled on substrates such as Teflon, mica, silicon, or gold, the last two of which are conductive and enable membrane electrical properties to be measured375,376. Bilayers of various lipid compositions can be assembled on surfaces by vesicle deposition377, or by Langmuir-Schaefer378, or Langmuir-Blodgett379 deposition techniques. A polymer cushion is sometimes incorporated, allowing for both sides of the membrane to be in contact with the aqueous solution and permitting the flow of ions from one side to the other376. A polymer cushion also enables integration of membrane proteins which increases cellular biomimetic fidelity375. Alternatively, tethered bilayer lipid membranes (tBLMs) are similar to sBLMs, but are covalently tethered by the lower leaflet of bifunctional phospholipids or alkyl chains, usually to a gold support platform. Tethering increases the stability of the bilayer380, but can potentially limit the degree of biomimetic fidelity as transmembrane cellular proteins cannot be incorporated as readily.
3.3.1. Applications of supported bilayers to the study of MPPs
With respect to the study of membrane permeabilizing peptides, there are numerous techniques that can be used to observe the effects of peptide-membrane interaction with high sensitivity. Atomic force microscopy (AFM) is a widely used probing technique where the surface of a sBLM can be scanned and monitored while interaction forces within the membrane can be recorded by a cantilever tip and piezoelectric scanner381 (Figure 12). When membrane-permeabilizing peptides are introduced to sBLMs, AFM images can be taken to observe the subsequent membrane rearrangement in the x, y, z planes with sub-nanometer resolution in z to show the membrane perturbations and pores181,381. This can be very useful in helping to determine the mechanism of action for membrane-permeabilizing peptides, and has been used in the study of indolicidin, gramicidin A and other MPPs178,181,382–384. AFM can be used to probe structural changes in the membrane, such as pore formation, as well as membrane thinning381,382,385. Numerous instances of membrane perturbations by peptides using AFM have been reviewed by Morandat et al381. Recently, Pittman and King published an AFM study in which they examined how fluid phase PC bilayers were affected by MelP5178, which was shown to have two effects on bilayer structure: localized bilayer thinning and formation of large pore-like voids in the membrane that form in the thinned areas of the bilayer178. The voids were dynamic, with at least some forming and dissipating within a few minutes178. The most probable diameter of the MelP5-induced voids was 1.7 nm, but they ranged as high as 4–5 nm diameter, large enough to account for the ability of MelP5 to release macromolecules from lipid vesicles178,180. Recently, King and Pittman with Hristova and colleagues showed how different pore-forming peptides exert variability in the sizes of pores formed in sBLMs by using AFM scanning and comparing the effects of MelP5 and macrolittin 70, a closely related but more potent peptide that is also capable of macromolecular poration181. Observations through AFM are not limited to these metrics; it can also measure chemical interactive forces, membrane elasticity, and can do protein receptor mapping,381 as well as map peptide membrane energy landscapes and kinetics381,386,387.
Figure 12:

Atomic Force Microscopy (AFM) demonstrating the effects of MelP5 (a melittin derivative) on POPC (Peptide:Lipid = 1:1200). A: Punctate perturbations can be visualized in this image and seem to be present across the entire bilayer plane (500 × 500 nm2). B: An image of a MelP5-treated POPC membrane at a higher magnification (290 × 290 nm2). C: A line scan through the image (dashed line in Fig 12A); numerous bilayer perturbations are scanned, and this information can be used to determine the depth of each topological depression. D: A line scan through the image (dashed line in Fig. 12B) at increased magnification. In this panel, pore-like features are highlighted in purple and thinned membrane regions are highlighted in green. Images courtesy of Gavin King. Adapted with permission from ref 178. Copyright 2018 American Chemical Society.
Neutron reflectometry and neutron scattering can also provide information on membrane structural dynamics specifically in an aqueous environment. Properties such as surface architecture, membrane thinning, composition, and hydration state are all studied with this method388–391. A useful technique that can complement AFM and neutron reflectivity data is electrochemical impedance spectroscopy (EIS) (Figure 13). Whereas AFM assesses the topological changes induced by peptide interactions with sBLMs, EIS provides measurements of bilayer electrical properties, resistance and capacitance392,393. EIS can monitor the permeability of a membrane to small ions (Na+, K+, Cl−, etc.) which can be useful in peptide ion channel studies and it can also be used to determine transiency of pores392,393. This has been demonstrated by Hristova and Searson and colleagues, who compared changes in membrane resistance and capacitance after the introduction of various membrane-permeabilizing peptides62,66,93,136,392–396. EIS is also able to provide information regarding the mode of action of peptides on membranes with respect to established mechanistic models of pore formation393. Chang et al. modelled the expected EIS signature of various models of membrane permeabilization. Several related synthetic MPPs, which have different behaviors in vesicle leakage, were then tested. When these MPPs were added to a polymer cushioned bilayer in an EIS cell, resistance decreased within 1 minute, but to a different degree for the two peptides393. The peptide that caused a much larger decrease in resistance in EIS was found to be the one that is more potent in vesicle leakage. Bilayer homogeneity, derived from capacitance, also decreased, but only for the more potent peptide, not for the other, showing that unique models can be assigned to specific peptides using EIS393.
Figure 13.
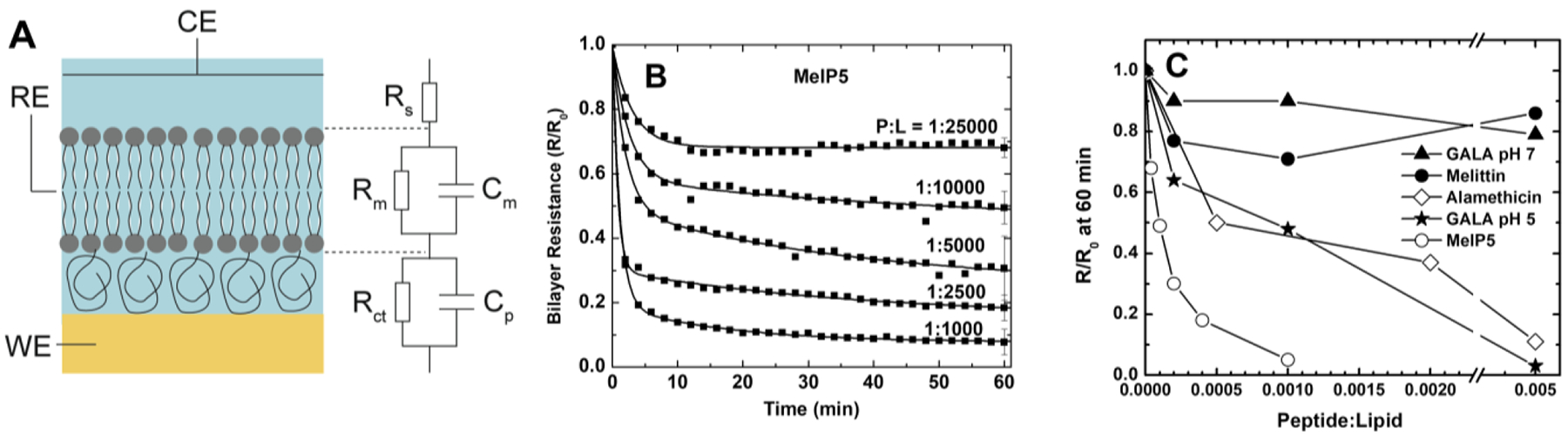
Electrochemical impedance spectroscopy. A: A polymer cushioned planar supported bilayer is adsorbed to the cleaned surface on a silicon crystal and the electrical properties are measured and modeled with an equivalent circuit. B: Resistance changes of a PC bilayer upon addition of the equilibrium pore former, MelP5. Note that the resistance does not recover as it would for a transient permeabilizing peptide66. C: Concentration dependence of the resistance drop for a variety of potent MPPs. Adapted with permission from ref 62. Copyright 2014 American Chemical Society.
Dual polarization interferometry (DPI) can also be utilized to study the effect of peptides on sBLMs. DPI uses dual optical waveguide interferometry to analyze sBLMs by collecting real-time data on membrane thickness, refractive index, mass, and anisotropy in the presence of membrane-active peptides191,397,398. Balhara et al. recently used AFM and DPI to reveal that the AMP GL13K exhibits membrane selectivity when comparing interactivity with liposomes to sBLMs398. While they found minimal evidence for binding of GL13K to DOPC liposomes, they showed that the peptide does interact with DOPC sBLMs which was confirmed through changes in membrane thickness, birefringence, and membrane mass. Their study exemplifies the ability of AFM and DPI to not only categorize, but reconcile differences in leaflet dielectric values between perturbed and unperturbed membranes. Membrane leaflet dielectric values are affected by membrane potential and architectural membrane defects, both of which may contribute to peptide binding in sBLMs, but may not be readily observable in liposomes398.
Furthermore, surface plasmon resonance (SPR) is also used often in conjunction with DPI to determine peptide binding to sBLMs by collecting data on membrane thickness, giving insight into specific peptide-membrane binding dynamics191,399,400. Papo et al. established modes-of-action for melittin and magainin by aligning SPR data to models of membrane interaction with respect to two sequential activities that occurred: 1) peptide association with the membrane and 2) insertion of the peptide into the membrane thereafter399. In this context, SPR was used to calculate binding constants in real-time after peptides were introduced to a supported lipid membrane and was able to differentiate two different peptide mechanisms of membrane interaction399. As indicated by the SPR results, in zwitterionic membranes, melittin appeared to bind preferentially and penetrate deeper than two AMPs, magainin and a melittin diastereomer. Against anionic membranes, all three peptides bound strongly, but without deep insertion, indicating a change in both membrane specificity and mechanism for melittin399.
A technique called quartz crystal microbalance with dissipation monitoring (QCM-D) is also used to probe peptide-membrane binding dynamics. QCM-D uses acoustic penetration of sBLMs at different harmonic frequencies to determine changes in membrane mass and elasticity in a leaflet-specific manner when interacting with different peptides383,401,402. Wang et al. used QCM-D signatures to profile four different AMPs (alamethicin, indolicidin, chrysophin-3, and SMAP-29) which have discrete mechanisms of action383. By using the change in dissipation (ΔD), they were able to relate the changes in membrane rigidity and viscoelasticity to the four aforementioned peptides, where a small ΔD is indicative of an unperturbed bilayer383. To map the mechanistic sequence of events, change in frequency (Δf) was used to provide metrics on peptide insertion into the membrane, the formation of pores, and the adsorption of peptides383. Using hybrid ΔD- Δf dynamic plots, each peptide was assigned a distinct mechanistic fingerprint, which outlined the step-wise interactivity of these peptides to a membrane bilayer, effectively giving insight to the modes of action under the conditions studied383. Overall, sBLMs are a robust experimental platform for numerous techniques which may elucidate mechanistic details of permeabilization of membranes by peptides and complement information obtained from lipid vesicles.
Secondary structure in supported bilayers can be determined by Fourier transform infrared spectroscopy and circular dichroism spectroscopy. More importantly, the orientation of some secondary structure elements can be assessed in surface supported bilayers by attenuated total reflectance Fourier transform infrared spectroscopy, ATR-FTIR403 and by oriented circular dichroism (OCD) spectroscopy, which is especially well suited for the determination of helix orientations in membranes, as shown in Figure 14.
Figure 14.

Oriented Circular Dichroism. In OCD, oriented multibilayers stacks containing a peptide that has mostly α-helical secondary structure are prepared on a quartz disk and hydrated through the vapor phase. Circular dichroism is measured with the bilayer plane oriented perpendicular to the beam axis. A: Bilayers with imaginary helical MPPs that have axes perpendicular or parallel to the bilayer normal, or a combination of the two. B: Theoretical spectra for perpendicular (transmembrane) and parallel (surface-oriented) α-helices are shown, along with linear combinations of the two194. These data are scaled to represent residue contributions assuming 100% helicity. C: Real experimental OCD data59 for melittin, which is parallel to the bilayer and has a helicity of ~60%, and two gain of function analogs, MelP5, which is perpendicular to the bilayer and has 90% helix, and MelP9, which is parallel to the bilayer and has 90% helix. Adapted with permission from ref 181. Copyright 2018 American Chemical Society.
3.3.2. Limitations and caveats of supported bilayers
While surface supported bilayers have been very important to the study of MPPs, there are some limitations to these techniques. A significant limitation in many sBLM experimental models is that the lipid concentration is effectively very low, and is unknown, meaning that the peptide to lipid ratio and the bound peptide to lipid ratio are not known, although they can be indirectly determined2. This same limitation also decreases the information content of GUV experiments (see above). In some cases, excess lipid vesicles and excess peptide can remain in equilibrium with the supported bilayer, to enable more precise knowledge and control of concentrations66,392,393. In other cases, such as oriented CD and some diffraction techniques, stacked oriented multibilayers are needed, and these require that samples be less than fully hydrated to remain stable.
3.4. Molecular dynamics simulations
Molecular dynamics is the simulation of interacting atoms and particles within ensembles of molecules providing predictions of the energetics and dynamics of the system. These simulations are governed by calculations based on Newton’s equations of motion404. There are three general classes of models for performing molecular dynamics on MPPs: all-atom, coarse-grained (CG), and implicit solvent models. These studies give detailed hypothetical structural and kinetic information on possible mechanisms of membrane permeabilization by peptides.
3.4.1. Methods of simulating bilayer permeabilization
The multiple classes of MD simulations arise from the degree to which each utilizes computational resources, which are usually limiting, and from the need to allow a simulated system to reach equilibrium, independent of initial conditions. All-atom models are the most comprehensive as they calculate all the bonded and non-bonded interactions between individual atoms in molecular ensembles. However, all atom simulations require very significant computer resources. Course-grained (CG) models represent molecules using a smaller number of particles, often called “beads”, that each mimic groups of connected atoms within a molecule. Thus, CG simulations must simulate fewer particles, and require far fewer computer resources to simulate longer trajectories and more extensive systems. Implicit solvent/bilayer models make the most significant deviations from reality by representing water and bilayer with implicit non-molecular field-like representations. However, implicit solvent models enable much longer trajectories and larger systems to be simulated with much less computer power. Each approach has strengths, weaknesses, and caveats that have been well discussed in the literature. We summarize the techniques below.
All-atom modeling considers all atoms as explicit particles with appropriate properties. Chemistry at Harvard Macromolecular Mechanics (CHARMM) force fields are often used here to simulate membranes, membrane proteins, and peptides. This type of modeling allows for more accurate representations of the peptide, solvent, and the lipid bilayer405. However, questions still exist about the most accurate force field406. A method sometimes employed to address the computational requirements is to increase the temperature of the system. A number of membrane interacting peptides have been found experimentally to be temperature insensitive over a wide range, making them suitable for simulations at up to 90°C or higher,93,407–410 which can decrease the time to reach equilibrium significantly411.
In the CG model, hydrogen atoms are treated implicitly and multiple non-hydrogen atoms are treated as one particle as to simplify the system and extend system size and time scales. The MARTINI force field412, which was originally described for lipid and surfactant systems413, is used most often here. It uses 4-to-1 mapping, with four non-hydrogen atoms acting as one “bead” or interaction center. There are only four main types of beads: polar, non-polar, apolar, and charged. Each of these types are further divided into subtypes414. Aggregating atoms in this way, and reducing interaction complexity can reduce computing resource needs, or increase trajectory times by as much as 3 to 4 orders of magnitude413.
Implicit solvent models further simplify the system by treating solvents, including the bilayer, as a continuum of medium rather than as individual particles. It was originally developed as a necessity when computing power was lacking405. In the implicit membrane model (IMM), the generalized Born theory of solvation is used to simulate the lipid bilayer. This allows 100x more efficient simulations as compared to explicit modeling415. In the IMM, factors such as membrane thickness416 and even poration can be calculated417,418. However, due to the simplicity of the implicit model, complex lipid bilayer composition is difficult to recapitulate.
3.4.2. What behaviors have been observed in MD simulation?
All-atom simulations have been done with many MPPs including melittin, alamethicin, and magainin and others (Figure 15). In an early monomer binding simulation of melittin, the bilayer showed thinning with water molecule penetration from both faces419. The more hydrophobic N-terminus was found to be protonated and it penetrated the bilayer. The setup had a helical melittin molecule that was set parallel in the interfacial region on a DMPC bilayer and simulated for 850 ps. Similar results have been observed in DPPC lipids420. Alamethicin simulations with POPC over 2 ns found that the peptide interacted with the polar headgroups of the lipid but not with the hydrophobic core421. A study of magainin indicated that by orienting the peptide with the hydrophobic residues close to the bilayer, it will penetrate deeper422. The same study also showed that the addition of salt to the system reduces the insertion depth of the peptide. However, these early simulations were far too short to reach equilibrium and the trajectories observed reflected mostly just the initial placement of peptide. In more recent all atom simulations, time scales in the microseconds have been observed409,423–428. Using the CHARMM22/36 protein/lipid force fields, Andersson et al. simulated melittin monomer binding to DOPC lipids for over 17 μs429. The simulation showed the peptide partitions at the depth of the glycerol groups of the lipids, but remains mostly parallel to the surface. Helicity was found to be in line with experimental observations79,430,431. Another study examined the putative transmembrane structure of melittin oligomers423. A preformed transmembrane tetramer of the peptide was inserted into DMPC lipids and simulated for 9 μs, showing formation of local disruption of the lipid bilayer with lipid head groups facing the interior of the pore. This equilibrium pore structure may mimic the structure of melittin under some experimental conditions, although as described earlier (Figure 2), melittin is not an equilibrium pore former under most experimental conditions65.
Figure 15.

Molecular dynamics simulations of MPPs show binding and structure formation on and in bilayers. (A) A simulation of melittin monomer binding over 17 μs shows that the peptide settles at a depth near the glycerol groups, which notably is consistent with X-ray diffraction results. Adapted with permission from ref 429. Copyright 2013 Elsevier, Inc. (B) Two different behaviors of multiple peptides binding to a bilayer are shown here. The interfacial S state is preferred by PGLa while a transmembrane configuration is preferred by alamethicin, a known potent pore former. Adapted with permission from ref 405. Copyright 2018 American Chemical Society. (C) Transmembrane structures of an all-or-none and graded peptide. Melittin peptides are in a U-shape that blocks water passage while magainin-2 allows water through more easily. These simulations indicate how all-or-none and graded mechanisms can be structurally different. Adapted with permission from ref 432. Copyright 2012 American Chemical Society. (D) Permeabilization is complex, as shown with the example of maculatin. The peptide does not form just one structure on a bilayer; it forms a variety of structures that assemble and disassemble over time for both DMPC and DPPC bilayers (bottom). However, it should be noted that most peptides are in the surface bound state (S). (E) Unbiased dye-conductance simulations show that P15A-E19Q, a maculatin double mutant that is thermally stable, forms lesions just large enough to allow ANTS and DPX through (bottom). Importantly, this finding was validated with experimental ANTS/DPX and dextran release assays (top). (D) and (E) are courtesy of Martin B. Ulmschneider. Adapted with permission from ref 411. Copyright 2016 Springer Nature Limited (CC) https://creativecommons.org/licenses/by/4.0/legalcode.
Coarse-grained models of melittin and magainin-2 have suggested possible permeabilization mechanisms. Melittin, which exhibits graded leakage experimentally, exhibited aggregates of 4 to 6 U-shaped peptides in the membrane, forming poorly-defined “channels” by 4.2 μs432. Magainin-2, which exhibits all-or-none leakage, formed water-filled transmembrane structures by 2.1 μs at P:L of no less than 24:512. The simulation for both peptides used the MARTINI force field, DPPC lipids, and began with interfacial placement of peptides. These two different structures show how graded and all-or-none leakage could occur in leakage assays, but they fail to explain transient leakage induced by both peptides. Another MARTINI simulation of magainin-2 showed even more dramatic activity; when a layer of peptide was placed on a DPPG/POPG bilayer, the bilayer buckles and forms a quasi-spherical vesicle by 2.4 μs, indicating yet another possible mechanism of the action of an MPP on a membrane433. A potential limitation in the study of MPPs with course grained simulations is the lack of water in channel-like structures432,434,435. Since some force fields treat four water molecules as one, this can produce water sized cavities that are empty due the artificially large “water” beads.
In a series of AMP studies by Lazaridis and colleagues, an implicit membrane model was modified to include cylindrical or parabolic shaped holes418,436. They showed that alamethicin, melittin, piscidin, and MG-H2 (a magainin analogue) all bind more strongly to the hole region in the membrane than to flat, intact membrane418,437. These results suggested that the peptides favor a membrane spanning pore-like geometry, a conclusion that is not consistent with all experimental measurements. They also found that alamethicin was the only one that completely embedded in the core of the membrane418, consistent with the observation that alamethicin is a very potent equilibrium pore-forming peptide that inserts across membranes under most conditions64. The authors attribute the preferential binding at hole regions to imperfect amphipathicity2.
Ulmschneider and colleagues suggested that by using experimentally guided simulations, the most correct force field could be determined, based on which produced the most accurate simulation result438. They also give an example of how setting the right time-scale is critical. They found that when simulating monomer surface binding of PGLa and melittin, the CHARMM/AA-lipids force fields gave much more helical structure compared to the OPLS/UA-lipids force fields. To determine which was more accurate, the authors looked to experimental data, which included CD and ssNMR techniques335,431,439,440.The experimental data matched the more helical conformations of the CHARMM/AA-lipid force fields. They also found that even with 2 μs simulations, maculatin 1.1 peptides that start from either coiled or partly folded conformations do not converge, indicating that simulations shorter than this time scale may not reach equilibrium and can be unreliable.
Wang et al. also utilized an experiment guided technique in a promising approach to model membrane permeabilization by the AMP maculatin411. In this study, they used circular dichroism at different temperatures to first show that a thermally stable mutant, P15A, had the same secondary structure as WT and that its structure is temperature insensitive. This enabled a simulation temperature of 90–150°C with the mutant which substantially decreased the required simulation time. They also performed ANTS/DPX and macromolecule leakage experiments to estimate lesion size; only leakage of the 400 Da ANTS/DPX, and not higher molecular weight dextrans, was observed, consistent with the simulated pore structure. Simulated initial insertion events were shown to occur through the cooperative insertion of two peptides into a transmembrane state, which allowed for more insertions events to take place. Longer simulations of 20 μs showed that maculatin forms diverse and dynamic structures in the membrane from trimers to octamers that assemble and disassemble continuously. They further note that while the leakage assays of single and double mutants had similar results, simulations showed they had different ensemble weightings of transmembrane aggregate size. Unbiased dye-conductance simulations showed that the octamers would, in fact, allow leakage of 400 Da molecules, agreeing with the experimental findings. This methodology of experimentally guided simulations bridges molecular dynamics and experimental data in a way that benefits both fields synergistically.
3.4.3. Limitations and caveats of MD simulations
Molecular dynamics simulations of membrane permeabilizing peptides in lipid bilayers remain an evolving field. It has shown tremendous growth in capabilities in recent years, a trend that can be expected to continue. The time scales that can be achieved by most researchers remain too short to ensure equilibrium in many cases and are far too short to simulate important slow processes such as peptide and lipid translocation. All atom simulations are currently limited to the nanosecond or microsecond time scale for most researchers. All atom simulations on the millisecond scale with the Anton II supercomputer437 could revolutionize the field. Longer simulations at higher P:L and with larger ensembles are needed to capture important processes that can span the stages from membrane binding to membrane translocation are desirable. Course grained simulations enable equilibration and longer processes to be studied, yet questions remain about the relevance to real molecules. At present, the most significant gap between simulations and experiment is the issue of transient permeabilization, which is currently difficult to simulate because it requires simulation of rare, slow events such as peptide translocation.
3.5. Biological systems
The study of interactions of MPPs with model membranes, and their effects on model membrane permeability, has provided many insights into their physical chemistry, structure, and mechanistic landscapes. However, the direct relevance of these model systems to biological membrane systems is uncertain. Relevance varies with each model system and depends on the information provided by the model system, and on its biases. No synthetic model system recreates all of the properties of a biological membrane. Ultimately, permeabilization of biological membranes must be measured directly and then carefully compared to other model systems to further our understanding of MPPs and to learn how to engineer them for our purposes. In this section, we will discuss MPP studies on biological membranes.
3.5.1. Eukaryotic plasma membrane permeabilization
The plasma membrane of a eukaryotic cell is the essential permeability barrier between the inside of the cell and the rest of the universe. Depending on the application, the plasma membrane can either be the target of an MPP, as in a cancer therapeutic, or an off-target membrane for which activity must be minimized, as in the case of an antimicrobial or antiviral peptide. In either case, permeabilization of the plasma membrane must be measured and we must eventually learn how to predict it. Acute permeabilization can be measured directly, or the effect of permeabilization on cell viability can be measured indirectly, because permeabilization is often cytotoxic. Perhaps the most commonly used measurement of biomembrane permeabilization is hemolysis, in which the release of protein contents from erythrocytes is measured after peptide treatment47,54. The very high concentration of hemoglobin and its strong optical absorbance provide a simple and sensitive assay measurement. The hemoglobin released into the supernatant is measured after erythrocytes are incubated with peptides and pelleted by centrifugation. The percent of hemoglobin released as a function of peptide concentration indicates membrane permeabilizing potency, which provides insights into the cytotoxicity of MPPs on erythrocytes441,442.
Permeabilization of nucleated cells in cell culture, perhaps an even more sensitive and informative assay than hemolysis, is measured by assessing the release of proteins or other polar compounds from the cell, or by assessing the entry of exogenous polar compounds into the cell (Figure 16). For example, small molecule release assays include the deoxyglucose assay that quantifies efflux of radiolabeled and intracellularly phosphorylated deoxyglucose through the plasma membrane; this allows for the evaluation of the effect of an MPP on the integrity of eukaryotic cells. ATP release has also been used to measure permeabilization. Because fluorescence-based assays are convenient and sensitive, one can preload cells with esterase-activated cytosol trapped dyes, such as calcein-acetoxymethylester, which are then released from the cytosol if a cell is permeabilized136. For protein release techniques, biochemical assays that exist for numerous enzymes can be used to quantitate release. A commonly used permeabilization assay relies on the measurement of the ubiquitous and abundant cytosolic enzyme lactate dehydrogenase that can be measured using a colorimetric detection41,441,443. Permeabilization of the plasma membrane can also be measured by assessing entry of exogenous compounds. Propidium iodide (PI) for example, is a widely used plasma membrane-impermeant nucleic acid intercalator that selectively enters compromised cells and stains their nuclei441,444. Similarly, the SYTOX family of DNA intercalating dyes only cross plasma membranes if they have been permeabilized, and then become brightly fluorescent when they interact with DNA441,445. Such assays can be performed by counting dye-positive cells, by flow cytometry, or by measuring total fluorescence in a plate reader or microscope. With the use of calcium sensitive dyes, it is also possible to assess the permeabilization of plasma membranes specifically to calcium40.
Figure 16.

Examples of permeabilization of eukaryotic plasma membranes. A: Cells in culture were treated with calcein red orange acetoxy methylester, which freely crosses the cell and is activated by cellular esterases to become entrapped and membrane impermeant (red, upper left). At the same time STYOX Green, a membrane impermeant DNA binding dye is added to the outside of the cells. Membrane permeabilization, as with MelP5 in the upper right, enables entry of SYTOX Green where it enter the nucleus and becomes fluorescent, B: In this confocal microscopy image, cell membranes are labelled green and a 3,000 Da dextran labelled with TAMRA (red) is added outside the cells. Shortly after addition of a membrane permeabilizing peptide in the corner, the first cells exposed are permeabilized to the dextran and they show osmotic swelling. The cells in the opposite corner have not yet been affected by peptide. C: Cell membranes are labelled red and cells are incubated with external SYTOX Green. A low concentration of the Ebola Virus delta peptide, a viroporin136 was added 10 min prior to taking this image. Here cells have been permeabilized to SYTOX Green, which stains the nuclei, but massive water influx and osmotic lysis are not occurring, evidenced by the lack of swelling or cell rounding at this time. D: TAMRA-labelled cell penetrating peptide (Arg9-TAMRA) and a spontaneous membrane translocating peptide TP2-TAMRA38 are incubated with cells at a low concentration of ~1 μM. The CPP gets untaken into endosomes, but cannot escape into the cytosol, in this case, because its concentration is too low to disrupt the membrane. The translocating peptide spontaneously crosses the plasma membrane and enters the cytosol. We thank Kalina Hristova for generous access to her confocal microscope.
Other live/dead assays, although indirect, are also useful indicators of acute lysis, because lysis kills cells quickly. MTT and Alamar Blue are examples of a colorimetric and a fluorescent assay respectively that detect metabolically active live cells. Both assays rely on conversion of a sensor compound by active mitochondrial reductases. For the MTT assay, reduction of MTT creates an insoluble violet-blue formazan that can be quantified by its absorbance441,446. For the Alamar Blue assay, reduction of resazurin to resorufin gives a compound that is red in color and highly fluorescent, indicating live cells441,447.
In all of these cellular leakage assays, unless care is taken to achieve osmotic balance between inside and outside54, which is inconvenient and rarely done, the assays may actually be reporting on osmotic rupture of the membrane due to the influx of water that occurs when membranes are permeabilized. The osmotic imbalance is quite large because of the very high concentration of proteins inside a cell448. In microscopy, this effect is obvious (Figure 16) as eukaryotic cells treated with MPPs swell dramatically before complete destruction of their architecture is observed.
Melittin has been widely studied using many model systems as well as many biological systems. In cells, melittin was first identified as a ‘direct lytic factor’ in bee venom as it induced hemolysis73,449. Different groups have also modified melittin and used hemolysis to better understand its binding and permeabilizing effect in erythrocytes54,73,450–453. The hemolysis and leakage assays have also been used to study potential therapeutic applications of melittin454–459. Unlike the majority of MPPs, which have some selectivity, melittin has remarkably consistent activity against various types of biological membranes. Against eukaryotic cell membranes, including erythrocytes and nucleated cells, melittin causes complete osmotic lysis at concentrations of around 5 μM46–48,54,460–462. Similarly, ~5 μM melittin has sterilizing activity against most Gram negative and Gram positive bacteria, due to membrane permeabilization24,45,174,179. These experiments, especially LDH release and hemolysis, have also been used to study another extensively investigated peptide magainin 232,33,463. The cytolytic effect of many MPPs including defensins, penetratin, and TAT have also been described using the above assays41,127,464–471.
3.5.2. Mitochondrial membrane permeabilization
Mitochondria are an important target for cellular therapeutic drug delivery. The inner membrane of mitochondria limits diffusive transport and like all membranes, it inhibits entry of therapeutics into the mitochondrial compartments. Hence, studies have been performed with membrane permeabilizing peptides and model systems mimicking mitochondrial membranes to identify peptides that are able to permeabilize or translocate into mitochondria. Further, the initiation of apoptosis can occur when the mitochondrial outer membrane is permeabilized to release cytochrome c472. This is a process that would be useful to control in some disease states, such as cancer, where apoptosis is dysregulated473–475. The permeabilization of the inner and outer membrane of mitochondria can be analyzed using various experiments476,477. The subcellular redistribution of mitochondrial proteins such as Cyt C, AIF, or adenylate kinase (ADK) to the nucleus or cytosol is used as a measurement of outer-membrane permeabilization or rupture due to matrix swelling476–479. Detection of intermembrane space metabolites or exogenous NADH oxidation, through HPLC or fluorescence respectively, can also provide insight on mitochondrial outer membrane integrity476,477,480,481. Inner mitochondrial membrane permeabilization causes dissipation of the membrane potential across the inner membrane (Δψm). Different Δψm-sensitive fluorochromes may be used to assess the peptide induced loss of Δψm476,477,482. The permeabilization of the inner mitochondrial membrane is also accompanied by flooding of water and solutes into the matrix resulting in an increase in the mitochondrial matrix volume. This osmotic swelling causes disorganization of cristae and reduction of matrix density. In case of swelling, the integrity of inner mitochondrial membrane can be monitored by measuring absorbance of the suspension in a traditional spectrophotometer476,477,483,484.
Various peptides have been shown to permeabilize mitochondrial membranes. For example, mastoporan, also known as mitoporan, has been shown to be a potent facilitator of mitochondrial permeabilization through various experiments that measure matrix swelling and inner membrane permeabilization485–487. The polycationic peptide BTM-P1 also permeabilizes mitochondria, causing matrix swelling and Δψm loss488. Amyloid-β (Aβ) is a peptide that contributes to pathogenesis of Alzheimer’s disease through apoptosis of cells. Experiments monitoring swelling of matrix and leakage of Cyt C from inner membrane disruption suggests that Aβ could induce apoptosis through mitochondrial membrane permeabilization489. Similarly, many peptides that can enter mitochondria have also been studied for their potential in cancer therapy. For example, BH3 and ABT-737 peptides have been shown to permeabilize the mitochondrial outer membrane through immunoassays for mitochondrial proteins490. In the presence of tumor cells, these peptides can permeabilize inner mitochondrial membrane causing loss of Δψm and swelling of matrix, leading to cell apoptosis490.
3.5.3. Endosomal membrane permeabilization
Many polar therapeutics, molecular tools, and other polar compounds can be endocytosed into cells when conjugated to, or complexed with, peptides and various nanomaterials, including cell penetrating peptides (CPPs) and nanoparticles. However, escape from the endosome requires membrane disruption. Endocytosed cargoes that do not escape endosomes do not reach the cytosol or nucleus where delivery would be beneficial. Failure of endosomal escape is a major bottleneck in cytosolic delivery of bioactive cargoes such as proteins.
Over the past several decades, many peptides, including CPPs and other MPPs have been used to facilitate delivery of polar compounds to cells via endosomal escape, at least partly through endosomolysis, or endosomal membrane permeabilization. The cargo delivery field is clouded by the fact that cationic CPPs that induce uptake and endosomolysis almost always simultaneously translocate and deliver cargo across the plasma membrane by a parallel process that may include transient permeabilization15,40,41. In either case, membrane permeabilization is required. Direct evidence for endosome lysis is often lacking. In biosystems, endosomal permeabilization has been inferred from cargo delivery characteristics15,41. Endosomal release has been visualized through fluorescence labeling and microscopy491. Melittin analogs have been shown to permeabilize endosomes and increase content release73,492–494. Pellois and colleagues used dfTAT, a dimerized disulfide-linked CPP, and Salerno et al. used a modified TAT peptide for better cargo delivery showing how MPPs can be modified to increase their endosomal escape efficiency495–497. Some synthetic cationic amphipathic membrane permeabilizing peptides like KALA and GALA with lysine-alanine-leucine-alanine and glutamic acid-alanine-leucine-alanine (EALA) repeats respectively have also been shown to enhance endosomal escape by membrane destabilization172,498,499. Another cationic amphipathic histidine-rich MPP, LAH4, can also efficiently cause endosomal release and permeabilize model membranes in a pH-sensitive manner500–502. Rational variation in arginine topology as well as arginine-rich MPPs have been found to induce endosomal leakage503,504. Hence, MPPs continued to be modified, evolved, or rationally designed to increase the delivery efficiency of therapeutic compounds to cells.
3.5.4. Bacterial membrane permeabilization
As discussed above, various model systems have been used to mimic the anionic cytoplasmic membranes of bacteria to study the physical chemical basis for AMP activity. These results roughly compare to what is observed in living bacteria. In bacterial systems, the majority of known AMPs kill bacteria through membrane permeabilization, following accumulation of large amounts of cationic AMP on the anionic membranes of the bacterial cells 23,24,174,505. Killing of bacteria by AMPs can be quantitated by broth dilution or agar diffusion assays, described elsewhere506. Rathinakumar and Wimley compared bacterial membrane permeabilization and sterilization by a family of evolved AMPs and found a positive correlation174.
As with eukaryotic cells, bacterial membrane permeabilization can be measured by leakage of polar compounds or proteins out of the cell or entry of polar compounds into the cell. The permeabilization of the outer and inner membrane of Gram negative bacteria can be quantified by N-phenyl-l-N-naphthylamine (NPN) uptake assay and O-nitrophenyl-3-D-galactoside (ONPG) influx method respectively507,508. For the NPN uptake assay, bacterial cells are prepared in the buffer containing NPN. The peptides are then added to the cells and NPN uptake through disrupted outer membrane is quantified using a spectrophotometer. Similarly, ONPG influx assay requires the dilution of cells in buffer containing ONPG followed by incubation with peptides, at which point the influx of ONPG into the cells can be measured and used as indicator of permeability through the inner membrane because β-galactosidase converts OPNG to a colored product509,510. Potential sensitive dyes can also be used to measure disruption of ion gradients across bacterial membranes174. Further, DNA intercalating dyes, such as propidium iodide and SYTOX family dyes also report on bacterial permeabilization, although entry of some larger dyes are restricted by the outer membrane of Gram negative bacteria and the cell wall of Gram positive bacteria67. Sani et al. used flow cytometry to show that the AMP maculatin 1.1 enabled efficient entry of a dextran of 4 kDa into bacteria while a 40 kDa dextran entered bacteria less efficiently511. These authors also showed that release of the two dextrans from bacteria-like anionic lipid vesicles recaputulated the size selectivity observed in bacteria. Eventually, bacteria that are treated with any antimicrobial peptides are physically damaged to the extent that it can be observed by electron microscopy (Figure 17).
Figure 17.

EM images of bacteria treated with AMPs. Top: SEM micrograph of E. coli ATCC 25922 (A) Control; (B-E) synthetic centrosymmetric α-helical AMP GG2, GG3, AA2 and AA3 treated; (F)MPP melittin treated. Bacterial cells were treated with 1X minimum bactericidal concentration (MBC) of peptide for 1 hour. Adapted with permission from ref 497. Copyright 2015 Nature Publishing Group. Bottom: TEM micrographs of E. coli ATCC 25922: 25922 (A) Control; (B-E) synthetic centrosymmetric α-helical AMP GG2, GG3, AA2 and AA3 treated; (F)MPP melittin treated. Bacterial cells were treated with 1X minimum bactericidal concentration (MBC) of peptide for 1 hour. Scale Bar = 500nm. Adapted with permission from ref 529 Copyright 2015 Nature Publishing Group.
Reller and colleagues performed some of these assays and described the biological activity of 44 antimicrobial agents including conventional antibiotics such as penicillin as well as modified peptides like colistin and polymyxin B512. The experiments listed above have also been used to study the antibacterial activity of natural and synthetic peptides. For example, the antibacterial activity of peptides such as LL-37, various defensins, indolicidin, and cecropins have all been studied using broth dilution, radial diffusion, hemolysis, NPN uptake, and ONPG influx assays507,513–521.
3.6. Statistics and stoichiometries in typical experimental scenarios
To appreciate what “potency” signifies in a membrane permeabilization experiment, it is useful to examine typical experimental statistics as we have done in Figure 18. An LUV of 100 nm diameter contains about 100,000 lipids, and entraps a volume of ~5×10−19 liters. The bilayer thickness is about 10% of the vesicle diameter, providing some stiffness to the vesicle. When the bound P:L is 1:50, which is typical, there are about 2000 peptides bound to each vesicle. In a typical probe release experiment, e.g. calcein, ANTS/DPX, or Tb3+/DPA, there is roughly 10 mM probe entrapped within the interior space of each vesicle. This amounts to only a few thousand probe molecules inside each vesicle. Therefore in a representative LUV permeabilization experiment there are the same number of probe molecules inside a vesicle as there are MPPs bound to it. Complete permeabilization, i.e. high potency, requires only that one probe molecule escape from a vesicle for every bound peptide over the course of the whole experiment. That is 1 or 2 probe molecules escape in 30 minutes, amounting to a flux per peptide of ~10−3 per second. Compare this to the ion flux through a classical channel protein which can be 107 per second522. Alternately, a tiny fraction of the bound peptides could be responsible for most of the leakage, if, locally, leakage flux is high but leakage events are rare. Hypothetically, the maximum possible potency of an MPP would be at P:L ~ 1:10,000 where there are only about 10 peptides bound to each vesicle, enough for one or two classical pores. The pH-sensitive peptide GALA is one of the most potent peptides known in LUV experiments with leakage reported at P:L as low as 1:10,000 or 10 peptides per vesicle171,523. Several others, including alamethicin, LLP1 and LLP2 and others are active at P:L ≤ 1:200064,147.
Figure 18.
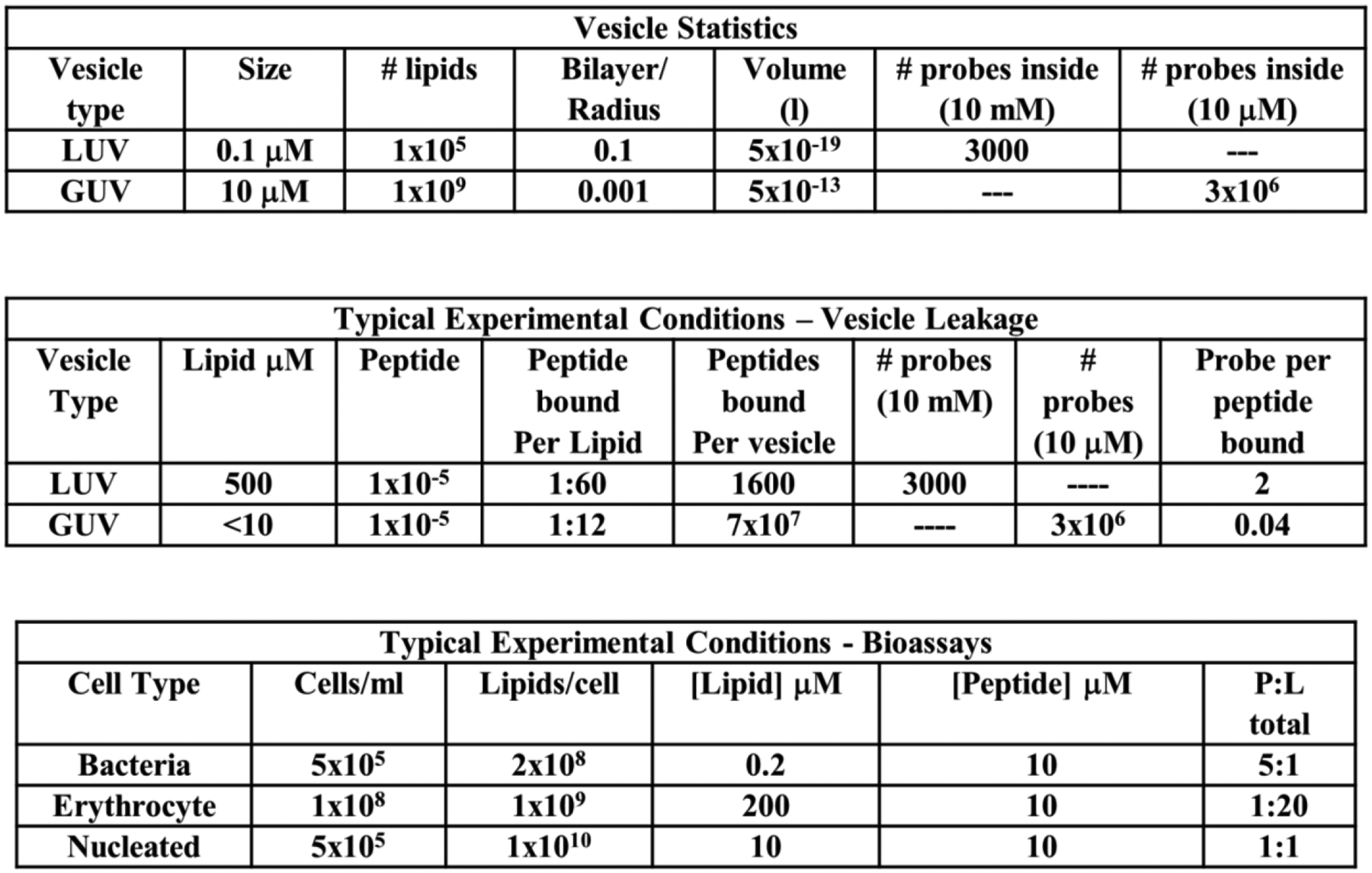
Statistics and stoichiometries in permeabilization experiments. Three tables of statistics and stoichiometries of permeabilization experiments. Top: Vesicle statistics for LUV and GUV, assuming a size of 10 μm for the GUV. Entrapped probes are assumed to be 10 mM for LUVs which use dye quenching as a probe, and 10 μM for GUVs which use dye observation in confocal microscopy as a probe. Typical experimental conditions for LUV and GUV experiments assuming 10 μM peptide concentration a mole fraction partition coefficient59 equal to that of melittin, Kx = 5×105. Lipid concentration in the GIV experiment is assumed to be very low (a few vesicles per μl. Bottom: Stoichiometries for biosystem permeabilization experiments. Lipids per cell is calculated from surface area using 70 Å2 per lipid molecule, and multiplying by 2 for each membrane present.
In a GUV the number of lipids is 10,000 times larger than in an LUV, or ~1×109 lipids, assuming a taut 10 μm vesicle. GUVs are taut because a moderate osmotic imbalance is created when they are produced, leading to a positive osmotic pressure. The thickness of the bilayer is less than 1% of the vesicle diameter. In a typical experiment, there may be ~7×107 peptides bound to a single GUV. The volume of a GUV is 106 times higher than an LUV, but the probe concentration is usually ~10 μM because microscopy is generally used. This means that there are only ~3×106 probes inside, or less than 0.1 probe per bound peptide. It is not known if the stochastic leakage events observed in GUVs are purely local. However, if it is local, then the permeabilization event has to occur only once, perhaps involving only a handful of peptides, among the 7×107 bound to the vesicle. If leakage is a global event, then it can occur when the efficiency of leakage is far less than 1 probe molecule per peptide. Explicit pores are not required to explain leakage efficiencies of ≤ 1 probe per peptide per experiment. In Figure 18, we compare these numbers to typical biomembrane experiments; with typically ~5×105 bacteria/ml (equal to 0.2 μM lipid), ~108 RBC/ml (equal to 200 μM lipid), or ~1×105 adherent nucleated cells in a 200 μl microwell plate, equal to 5×105 cells per ml or 10 μM lipid.
Taken together, we can group experiments based on effective lipid concentration. For example, bacterial sterilization, lysis of nucleated cells, and GUV permeabilization are done at very low effective lipid concentration, ≤ 10 μM. In these experiments P:L is not well defined, but the bound peptide:lipid ratio will be maximal2. LUV permeabilization and hemolysis are done at effectively much higher lipid concentrations, ≥ 200 μM, such that the system P:L will mimic the bound P:L if binding is strong.
In biosystems, as in synthetic systems, “potent” membrane permeabilizing activity does not require ion channel-like activity, but rather requires only a small number of critical metabolites to escape per peptide to be effective. For example, we showed that about 2×108 individual antimicrobial peptides are required to kill an E. coli cell. This is more than the number of ATP molecules inside an E. coli cell. So, even in the most widely studied model systems of antibacterial activity, high potency means the release of only one critical metabolite molecule per peptide over the entire course of the experiment. In hemolysis or nucleated cell lysis, even larger numbers of peptides may be bound to each cell, while biological activity requires only a moderate change in water or ion permeation across the membrane. These stoichiometries mean that it is almost never necessary to propose that an MPP forms an explicit membrane-spanning pore to explain an observed activity.
3.7. Meta-analysis of membrane permeabilizing peptides
To examine the range of behaviors and potencies observed in the permeabilization of synthetic lipid vesicles we performed a meta-analysis of published results. We collected several hundred papers describing synthetic vesicle permeabilization published between 1987 and 2018. From this collection, we selected those publications that clearly showed permeabilization data over a range of peptide concentrations that enabled the determination of the concentration that causes 50% leakage, LIC50. Papers for which lipid or peptide concentration or lipid composition could not be ascertained were rejected. We thus identified 71 high quality publications from which we extracted 524 data points. Redundant measurements of the same peptide in different papers were not removed from the database. Several hundred peptides are represented in the database, comprising many different origins. Antimicrobial peptides are common, with synthetic sequences outnumbering natural sequences. As shown in Figure 19, panel A, the database contains peptides with charges that range from −7 to greater than +9. All peptide with charge ≥ +9 are included in the bin at +9. The majority of peptides tested in these publications are basic, with a most probable value of +4. Anionic MPPs are rare, and the frequency shown in Figure 19, panel A is exaggerated because the data contain multiple measurements of a small number of peptides. The highly potent, pH triggered pore-former GALA (see above) which has a charge of −7 (at neutral pH), is the most acidic peptide in the database, and has been measured multiple times at many pH values. The assays used to measure permeabilization include most of the assays shown in Figure 8. Carboxyfluorescein and calcein entrapment are frequently used in early papers, but there is a transition towards Tb3+/DPA and ANTS/DPX in later work. Vesicles are SUV and LUV in the earliest work, transitioning to only LUV for the bulk of the papers. GUV studies cannot be included here because the lipid concentration is generally not known, and P:L cannot be calculated.
Figure 19.
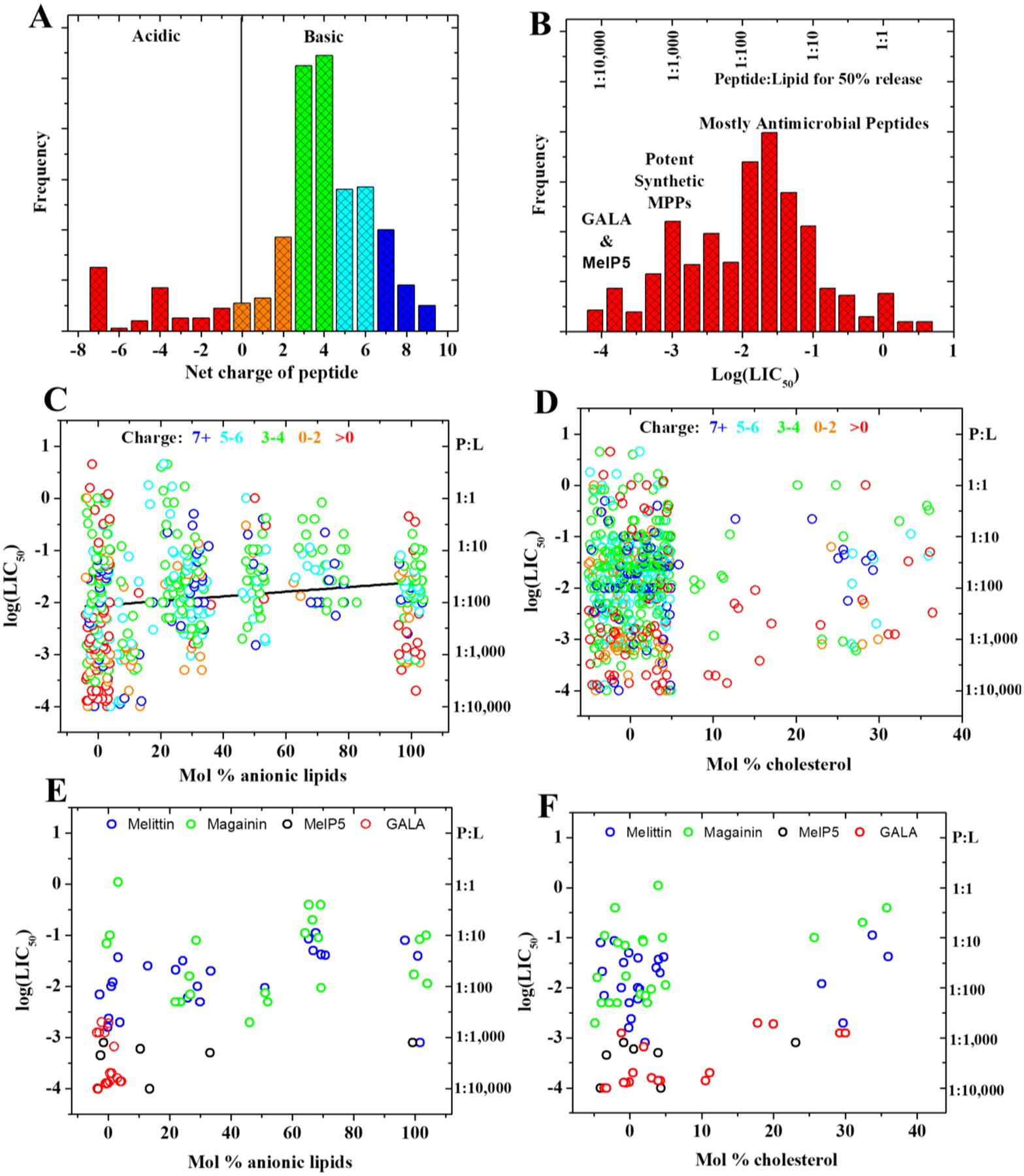
Meta-analysis of membrane permeabilizing peptides. A representative set of high quality published results was aggregated to show the range of behaviors observed for the permeabilization of synthetic vesicles by MPPs. A: Charge distribution in the dataset. The colors used here are the same colors used in panels C and D. B: Distribution of the total peptide to lipid ratio required to release 50% of vesicle-entrapped contents, or LIC50. C: LIC50 plotted as a function of acidic lipid content (PG, PS, ganglioside, cardiolipin). The peptide charge states are shown by the color of the points, as shown in panel A. Random noise is added to the X-axis values to spread out the data points. D: LIC50 plotted as a function of cholesterol content. The peptide charge states are shown by the color of the points, E: LIC50 plotted as a function of acidic lipid content (PG, PS, ganglioside, cardiolipin) for a subset of well-studied peptides. F LIC50 plotted as a function of cholesterol content for a subset of well-studied peptides.
A histogram of MPP potencies is shown in panel B. Activities range across more than 4 orders of magnitude, from LIC50 of 1:1 or greater, which is essentially inactive, down to LIC50 as low as 1:10,000. The lowest LIC50 values observed are ~1:10,000. This may be the maximum possible potency because it is equivalent to only 10 peptides per LUV vesicle. The peptide GALA at pH ≤ 5, one of the most active MPPs known, dominates the most potent portion of the plot. Also found in this region is MelP5, a gain of function analog of melittin with very high potency64. In the range of LIC50 = 1:1000, the database contains data for a number of synthetic MPPs of both α-helical and β-sheet structure, but fewer natural sequences. The bulk of the values, overall, fall between LIC50 of 1:10 and 1:100. This area contains the values for many AMPs. In panel C we show the dependence of LIC50 values on the content of acidic lipids in the vesicles. Panels C-D show the charge of the peptides by the colors of the points. It is well known that cationic AMPs are more active against anionic bilayers and are often nearly inactive against PC bilayers. However the data overall do not show this trend, because there are also peptides, such as alamethicin, melittin (and its gain of function analogs) that are highly active against both types of bilayers. In panel D we show the dependence of LIC50 values on the content of cholesterol in the bilayers. For many peptides, the increase in bilayer order caused by cholesterol decreased the permeabilizing activity significantly, but again here, as in panel C, there are peptides that are highly active against cholesterol containing bilayers. The main insight gained from the data shown in Panels C and D is that while many individual peptides are sensitive to cholesterol content or bilayer charge, there are at least some MPPs that are highly active against any lipid composition. In panels E and F of Figure 19, we show the dependence of LIC50 values on the content of acidic lipids in the vesicles or on the content of cholesterol in the bilayer for some of the most well studied peptides in the dataset, or for peptides mentioned frequently in this review, including melittin, magainin, GALA at pH ≤ 5, and MelP5.
4. The mechanistic landscape of membrane permeabilizing peptides
For six decades, researchers have studied membrane permeabilizing peptides in synthetic and biological membranes to learn about the fundamental physical chemical properties of polypeptides that interact with lipid bilayers and alter their permeability. Such information could eventually enable the rational optimization of MPPs for specific translational applications. Yet, the active molecular structures of the thousands of known MPPs in membranes, with only very few well-known exceptions, have not been described at anything resembling atomic level resolution. This is in sharp contrast to the closely related field of membrane protein structural biology, which has yielded a large and exponentially growing number of atomic resolution structures.
The idea that has emerged from the long study of MPPs is not a static image of discrete pores. Peptide sequence and secondary structure are rarely enough to constrain a peptide bilayer system to a single structure or a single mechanism of action under all instances. Driven by the sum of many interactions in the context of the dynamic bilayer milieu, most MPPs form an ensemble of heterogeneous, dynamic structures. The ensemble of structures and mechanisms will shift, sometimes dramatically, with experimental conditions; peptide sequence and secondary structure, bound peptide concentration, bilayer lipid composition (i.e. bilayer charge, fluidity, thickness and more), and also with physical parameters such as temperature, ionic strength, and pH. Further, the many techniques used to study MPPs each probe a unique part of the functional space. We have referred to this phenomenon as a “mechanistic landscape”15. This leads to the concept that each MPP occupies an area across a continuum of structures and mechanisms. This may help explain why there is so little consensus on the detailed molecular mechanisms of MPPs in synthetic membranes. With only a few exceptions, our current state of knowledge provides little predictive power on the activity of MPPs in synthetic membranes, and especially in the very heterogeneous environment of the biological membrane. For this reason, researchers in the MPP field still mostly discover new peptides fortuitously, or by simple trial and error. To create an image of the properties of the landscape that need to be better understood, next we discuss the MPP behaviors that are commonly observed but are most difficult to explain.
4.1. Transient permeabilization
Many membrane permeabilizing peptides cause transient permeabilization of synthetic membranes and sometimes of biological membranes (Figure 20). The membranes become permeable shortly after peptides are added, but the leakage slows or stops before all contents have been released. This phenomenon has been noted and discussed in the literature, but its exact cause remains a mystery. It means that many MPPs are actually not membrane permeabilizing peptides at equilibrium or are much less potent at equilibrium, than the primary data would suggest. At equilibrium, such MPPs are still bound to membranes and still have the same secondary structure, but once the transient leakage event has taken place the membranes are no longer highly permeable, even in the presence of peptide.
Figure 20:

Experimental and simulated permeabilization of LUVs. We simulate vesicle leakage using simple numerical models. A: Transient leakage has a constant exponential rise toward a final value that itself depends on the P:L in the experiment. Many leakage experiments have behavior like this. The exponential rise assumes that the fraction of remaining entrapped contents released per unit time is constant. The plot on the right shows potency profile in a semi-log plot. B: Simple equilibrium leakage model assumes an exponential rise towards 100% release where the rate (the fraction of remaining entrapped contents released per unit time) is a function of peptide concentration. This behavior is rarely observed. The plots on the right shows potency profile at 30 minutes in a semi-log plot. C: Hybrid leakage, assumes that there is a major component of transient leakage, followed by a low level steady equilibrium release. Many peptides have this behavior. D: Real experimental leakage curves for various MPPs taken from the authors’ manuscripts. Real curves like all three of these simulations are seen in this set. The plots on the right shows various potent curves taken from the authors’ manuscripts. They range from high potency MPPs (e.g. alamethicin) that release ~100% of contents at P:L=1:2000, to some AMPs or analogs that release almost nothing at P:L of 1:10. Peptides, lipid compositions and other details vary in these curves.
What is the nature of the transient, permeabilization event? The leading hypothesis2,5,41,67 is that permeabilization occurs during the sudden dissipation of the initial asymmetric distribution of peptide on the membrane. In other words, upon addition, peptide accumulates on the outer monolayer of a synthetic or biological membrane, creating an imbalance of mass, charge, surface tension, lateral pressure or some combination of these. After some time, a stochastic, perhaps catastrophic, local dissipation event occurs in which the asymmetry is relieved by peptide, and possibly lipid, translocation. During the dissipation of the peptide asymmetry, the membrane is hypothesized to also become transiently permeable to polar molecules, and a burst of leakage occurs. Ensemble averaging in most vesicle experiments spreads individually rapid events across a broader time range. Although this hypothesis is consistent with what we know about peptides in membranes and can explain many observations, it has not been directly supported by many experiments or simulations.
As we show in Figure 18, the molecular efficiency of transient permeabilization can be very low and still result in efficient release. In other words, high net permeabilization can occur only when, for every bound peptide, ≤ 1 probe molecule crosses the membrane, during the entire experiment. The observation of transient all-or-none leakage5 in which some of the vesicles release all of their contents while others release none, suggests that there is also a “silent” dissipation mechanism, likely to be peptide or lipid translocation298 that can occur without causing leakage. The measured fractional leakage, in this case, denotes the fraction of vesicles that have undergone the permeabilizing dissipation event, rather than the silent one. As peptide concentration is increased, the fraction of permeabilized vesicles increases.
Transient permeabilization, like all other “mechanisms” of MPPs, is only a portion of the mechanistic landscape, although it is widely observed. MPPs that cause transient permeabilization under some conditions, such as low concentration or in the presence of anionic bilayers, may cause equilibrium permeabilization under other conditions, such as high concentration or in zwitterionic bilayers64. For example, Krauson and Wimley showed that melittin causes transient permeabilization of PC bilayers at moderate concentration, P:L < 1:200, but that the mechanism transitions towards one that is more like equilibrium permeabilization at higher P:L64. In anionic bilayers, Ladokhin and White showed that melittin caused transient, and catastrophic permeabilization across a wide range of concentrations78.
We note that a few MPPs do cause equilibrium permeabilization, rather than transient permeabilization, over a wide range of conditions. For example Krauson and Wimley showed that alamethicin and the lentivirus lytic peptides LLP1 and LLP2 cause potent equilibrium permeabilization of PC vesicles from P:L of 1:50 to P:L = 1:2000 and below. Similarly, the evolved melittin analog MelP5 and its family members also show equilibrium pore formation under a wide range of conditions59,62.
4.2. Stochastic permeabilization
Most published permeabilization experiments rely on ensemble measurements of many vesicles, often LUVs, where events at the single vesicle level are not known. However, GUV studies and experiments with surface tethered LUVs and bacteria enable individual permeabilization events to be observed. Often, when it is possible to see individual events, researchers observe that permeabilization is a sudden, catastrophic leakage event that occurs after a long lag period. For example, permeabilization of GUVs67,275,356,357, live bacteria67 and tethered LUVs369 have been shown to occur in seconds or less, following a lag phase from tens of seconds to minutes. Again, we note that stochastic permeabilization may only occur on a portion of a mechanistic landscape. A system with this behavior under one set of conditions may behave very differently under another set of conditions.
What is the nature of a stochastic permeabilization event? We note that peptide binding and folding in membranes is typically very fast, occurring in seconds111,315,524–526. Thus, the peptides are bound and structured during the entire lag phase, but there must be a significant energy barrier to the dissipation of the asymmetry of bound peptide. Presumably the hydrocarbon core still effectively blocks peptide translocation. Lipid cohesion prevents the bilayer architecture from being completely lost. Yet, in the presence of an interfacially active MPP, by definition, the bilayer order is diminished, and the hydrocarbon core is perturbed enough to lower the average energy barrier to the movement of peptides, lipids, water and polar solutes across the bilayer. We speculate that when the energy barrier is bypassed by a local fluctuation in peptide/bilayer structure and/or peptide concentration, the hydrocarbon core is transiently breached and a locally catastrophic flow of material occurs through the breach toward dissipation of the transbilayer asymmetry. Sometimes this event is accompanied by destruction of the vesicle architecture275,357,363,527. We hypothesize that the amphipathic nature of MPPs may be needed to stabilize the pathway through the membrane, at least briefly, and/or it may help catalyze the formation of an extended breach or additional breaches in the membrane. However it happens, permeabilization is a rare event, such that an individual vesicle may not undergo a permeabilization event over the course of an experimental observation, while other vesicles do. Peptide concentration and other factors determine the probability of the leakage event. Within a few seconds, or a few tens of seconds, the stochastic permeabilization event may be over. As described quantitatively by Hoernke and colleagues309, if vesicles are empty after a single event, leakage will be all-or-none. If multiple, or many individual events are required to empty a vesicle, leakage will be graded.
4.3. A challenge to computational scientists
Mathematical modelling and simulations of membrane permeabilization almost universally rely on the assumption that permeabilization is an equilibrium process. This is only occasionally true. Permeabilization is often a transient and stochastic process. In this review, we have described such behavior in detail. We challenge computational scientists and simulation scientists to explore, explain, and recreate these commonly observed behaviors to help create the next generation of testable mechanistic hypotheses.
5. Conclusions
In terms of detailed molecular mechanisms of membrane permeabilizing peptides, here, we have envisioned a mechanistic landscape that includes ensembles of overlapping structures and mechanisms of activity that depend on the sum of many experimental details. For most MPPs, it may not be possible to describe a single unifying description of mechanism. What we have done is define: the physical, chemical, and structural commonalities; the many methods used to study MPPs; and the intriguing behaviors that have been observed. While the idea of a mechanistic landscape is not new, we think it is beneficial to begin to use the concept to think and talk about the mechanisms of membrane permeabilizing peptides.
Acknowledgments
For an infinite variety of fascinating and insightful conversations, sometimes about peptides, membranes, and lipids, WCW acknowledges all of his former lab mates and all former and current members of the Wimley laboratory. The authors greatly appreciate the many people who commented on this review and significantly improved it. They include the three anonymous reviewers, and Alexey Ladokhin, Frances Separovic, Gavin King, Jose Nieva, Beatriz Apellaniz, and Charles Chen. For general discussions and insights into membrane permeabilizing peptides over many years, WCW thanks, in addition to those above, Kalina Hristova and the members of her lab, Thomas Thompson, Stephen White, Michael Selsted, Paulo Almeida, Antje Pokorny-Almeida, Anne Hinderliter, Aram Krauson, Greg Wiedman, Andrew Hoffmann, Bill Walkenhorst, Sam Landry, Pernilla Wittung-Stafshede, Bob Garry, Michael Wiener, Martin Ulmschneider, Jacob Ulmschneider, Jerome Maderdrut and many others. The Wimley lab is currently funded by NIH R01 GM111824 and NSF DMR1710053.
Biographies
Shantanu Guha was born and raised in Dallas, Texas. He obtained a B.A. in Biology in 2011 from Austin College (Sherman, Texas) and then moved to New Orleans to earn a M.P.H. with a focus on Epidemiology in 2013 from Tulane University (New Orleans, Louisiana). Currently, he is a Ph.D. student in Dr. William C. Wimley’s lab at Tulane University in the department of Biochemistry & Molecular Biology. His research is on the topic of the Ebola virus Delta peptide as a potential novel therapeutic target during pathogenesis. The Delta peptide is a viroporin produced by Ebola virus during active infections and may contribute to morbidity and mortality of infected individuals.
Jenisha Ghimire was born and raised in Kathmandu, Nepal. Upon her high school graduation, she moved to New Orleans and obtained her B.Sc. in Biology from the University of New Orleans with departmental honors. She also obtained her M.Sc. from the University of New Orleans where her work focused on understanding the role of IME4 gene in phosphate metabolism pathway of S. cerevisiae. Currently, she is a PhD student in Dr. William C. Wimley’s laboratory where her research emphasizes the discovery of novel antimicrobial peptides with high-throughput screening.
Eric Wu is currently a PhD student at Tulane University in William C. Wimley’s laboratory. He obtained a B.Sc. in microbiology in 2010 at San Jose State University and has since worked at the University of Arizona and OncoSynergy, Inc. where he studied the influence of gut commensal microorganisms on the immune system and cancer therapeutics, respectively. Currently, his research focuses on utilizing pH-triggered pore-forming peptides for macromolecule drug delivery into cells.
William C. Wimley, PhD, is from Monroe, Connecticut. He studied Biophysics at the University of Connecticut (B.S., 1985) and Biochemistry at the University of Virginia (PhD, 1990) where his dissertation work focused on the interactions between lipids in synthetic membranes. As a postdoc at the University of California, Irvine, he began a lifelong study of peptides in membranes. He is currently the George A. Adrouny Professor of Biochemistry and Molecular Biology at the Tulane University School of Medicine. The current focus of the Wimley laboratory is synthetic molecular evolution of pore-forming, cell penetrating, membrane translocating, antibacterial, and antiviral peptides.
References
- (1).Wiener MC; White SH Structure of a Fluid Dioleoylphosphatidylcholine Bilayer Determined by Joint Refinement of X-Ray and Neutron Diffraction Data. III. Complete Structure. Biophys. J 1992, 61, 434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wimley WC Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chemical Biology 2010, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bechinger B Rationalizing the Membrane Interactions of Cationic Amphipathic Antimicrobial Peptides by Their Molecular Shape. Curr. Opin. Colloid Interface Sci 2009, 14, 349–355. [Google Scholar]
- (4).Bechinger B; Lohner K Detergent-like Actions of Linear Amphipathic Cationic Antimicrobial Peptides. Biochim. Biophys. Acta - Biomembr 2006, 1758, 1529–1539. [DOI] [PubMed] [Google Scholar]
- (5).Rathinakumar R; Wimley WC Biomolecular Engineering by Combinatorial Design and High-Throughput Screening: Small, Soluble Peptides That Permeabilize Membranes. J. Am. Chem. Soc 2008, 130, 9849–9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rathinakumar R; Walkenhorst WF; Wimley WC Broad-Spectrum Antimicrobial Peptides by Rational Combinatorial Design and High-Throughput Screening: The Importance of Interfacial Activity. J. Am. Chem. Soc 2009, 131, 7609–7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jenssen H; Hamill P; Hancock REW Peptide Antimicrobial Agents. Clin. Microbiol. Rev 2006, 19, 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yount NY; Yeaman MR Multidimensional Signatures in Antimicrobial Peptides. Proc. Natl. Acad. Sci 2004, 101, 7363–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bechinger B A Dynamic View of Peptides and Proteins in Membranes. Cell. Mol. Life Sci 2008, 65, 3028–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hunter HN; Jing W; Schibli DJ; Trinh T; Park IY; Kim SC; Vogel HJ The Interactions of Antimicrobial Peptides Derived from Lysozyme with Model Membrane Systems. Biochim. Biophys. Acta - Biomembr 2005, 1668, 175–189. [DOI] [PubMed] [Google Scholar]
- (11).Jin Y; Hammer J; Pate M; Zhang Y; Zhu F; Zmuda E; Blazyk J Antimicrobial Activities and Structures of Two Linear Cationic Peptide Families with Various Amphipathic β-Sheet and α-Helical Potentials. Antimicrob. Agents Chemother 2005, 49, 4957–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shai Y; Oren Z From “Carpet” Mechanism to de-Novo Designed Diastereomeric Cell-Selective Antimicrobial Peptides. Peptides 2001, 22, 1629–1641. [DOI] [PubMed] [Google Scholar]
- (13).Govindarajan S; Sivakumar J; Garimidi P; Rangaraj N; Kumar JM; Rao NM; Gopal V Targeting Human Epidermal Growth Factor Receptor 2 by a Cell-Penetrating Peptide-Affibody Bioconjugate. Biomaterials 2012, 33, 2570–2582. [DOI] [PubMed] [Google Scholar]
- (14).Kakudo T; Chaki S; Futaki S; Nakase I; Akaji K; Kawakami T; Maruyama K; Kamiya H; Harashima H Transferrin-Modified Liposomes Equipped with a PH-Sensitive Fusogenic Peptide: An Artificial Viral-like Delivery System. Biochemistry 2004, 43, 5618–5628. [DOI] [PubMed] [Google Scholar]
- (15).Kauffman WB; Fuselier T; He J; Wimley WC Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci 2015, 40, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Komin A; Russell LM; Hristova KA; Searson PC Peptide-Based Strategies for Enhanced Cell Uptake, Transcellular Transport, and Circulation: Mechanisms and Challenges. Adv. Drug Deliv. Rev 2017, 110–111, 52–64. [DOI] [PubMed] [Google Scholar]
- (17).Kullberg M; Owens JL; Mann K Listeriolysin O Enhances Cytoplasmic Delivery by Her-2 Targeting Liposomes. J. Drug Target 2010, 18, 313–320. [DOI] [PubMed] [Google Scholar]
- (18).Oliveira S; van Rooy I; Kranenburg O; Storm G; Schiffelers RM Fusogenic Peptides Enhance Endosomal Escape Improving SiRNA-Induced Silencing of Oncogenes. Int. J. Pharm 2007, 331, 211–214. [DOI] [PubMed] [Google Scholar]
- (19).Pan H; Soman NR; Schlesinger PH; Lanza GM; Wickline SA Cytolytic Peptide Nanoparticles (‘NanoBees’) for Cancer Therapy. Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2011, 3, 318–327. [DOI] [PubMed] [Google Scholar]
- (20).Shai Y Mode of Action of Membrane Active Antimicrobial Peptides. Biopolym. - Pept. Sci. Sect 2002, 66, 236–248. [DOI] [PubMed] [Google Scholar]
- (21).Wimley WC; Hristova K Antimicrobial Peptides: Successes, Challenges and Unanswered Questions. J. Membr. Biol 2011, 239, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wolfmeier H; Pletzer D; Mansour SC; Hancock REW New Perspectives in Biofilm Eradication. ACS Infect. Dis 2018, 4, 93–106. [DOI] [PubMed] [Google Scholar]
- (23).Konai MM; Samaddar S; Bocchinfuso G; Santucci V; Stella L; Haldar J Selectively Targeting Bacteria by Tuning the Molecular Design of Membrane-Active Peptidomimetic Amphiphiles. Chem. Commun 2018, 54, 4943–4946. [DOI] [PubMed] [Google Scholar]
- (24).Starr CG; He J; Wimley WC Host Cell Interactions Are a Significant Barrier to the Clinical Utility of Peptide Antibiotics. ACS Chem. Biol 2016, 11, 3391–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Badani H; Garry RF; Wimley WC Peptide Entry Inhibitors of Enveloped Viruses: The Importance of Interfacial Hydrophobicity. Biochim. Biophys. Acta - Biomembr 2014, 1838, 2180–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hood JL; Jallouk AP; Campbell N; Ratner L; Wickline SA Cytolytic Nanoparticles Attenuate HIV-1 Infectivity. Antivir. Ther 2013, 18, 95–103. [DOI] [PubMed] [Google Scholar]
- (27).Hrobowski YM; Garry RF; Michael SF Peptide Inhibitors of Dengue Virus and West Nile Virus Infectivity. Virol. J 2005, 2, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lok SM; Costin JM; Hrobowski YM; Hoffmann AR; Rowe DK; Kukkaro P; Holdaway H; Chipman P; Fontaine KA; Holbrook MR; et al. Release of Dengue Virus Genome Induced by a Peptide Inhibitor. PLoS One 2012, 7, e50995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Spence JS; Melnik LI; Badani H; Wimley WC; Garry RF Inhibition of Arenavirus Infection by a Glycoprotein-Derived Peptide with a Novel Mechanism. J. Virol 2014, 88, 8556–8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Carravilla P; Cruz A; Martin-Ugarte I; Oar-Arteta IR; Torralba J; Apellaniz B; Pérez-Gil J; Requejo-Isidro J; Huarte N; Nieva JL Effects of HIV-1 Gp41-Derived Virucidal Peptides on Virus-like Lipid Membranes. Biophys. J 2017, 113, 1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jackman JA; Costa VV; Park S; Luiza A; Real CV; Park JH; Cardozo PL; Ferhan AR; Olmo IG; Moreira TP; Bambirra JL; Queiroz VF; Queiroz-Junior CM; Foureaux G; Souza DG; Ribeiro FM; Yoon BK; Wynendaele E; De Spiegeleer B; Teixeira MM; Cho N-J; Therapeutic Treatment of Zika Virus Infection Using a Brain-Penetrating Antiviral Peptide. 2018. Nature Materials, 971–977. [DOI] [PubMed] [Google Scholar]
- (32).Liu S; Yang H; Wan L; Cai HW; Li SF; Li YP; Cheng JQ; Lu XF Enhancement of Cytotoxicity of Antimicrobial Peptide Magainin II in Tumor Cells by Bombesin-Targeted Delivery. Acta Pharmacol. Sin 2011, 32, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lehmann J; Retz M; Sidhu SS; Suttmann H; Sell M; Paulsen F; Harder J; Unteregger G; Stöckle M Antitumor Activity of the Antimicrobial Peptide Magainin II against Bladder Cancer Cell Lines. Eur. Urol 2006, 50, 141–147. [DOI] [PubMed] [Google Scholar]
- (34).Soman NR; Baldwin SL; Hu G; Marsh JN; Lanza GM; Heuser JE; Arbeit JM; Wickline SA; Schlesinger PH Molecularly Targeted Nanocarriers Deliver the Cytolytic Peptide Melittin Specifically to Tumor Cells in Mice, Reducing Tumor Growth. J. Clin. Invest 2009, 119, 2830–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gerlach SL; Rathinakumar R; Chakravarty G; Göransson U; Wimley WC; Darwin SP; Mondal D Anticancer and Chemosensitizing Abilities of Cycloviolacin 02 from Viola Odorata and Psyle Cyclotides from Psychotria Leptothyrsa. Biopolymers 2010, 94, 617–625. [DOI] [PubMed] [Google Scholar]
- (36).Raaymakers C; Verbrugghe E; Hernot S; Hellebuyck T; Betti C; Peleman C; Claeys M; Bert W; Caveliers V; Ballet S; et al. Antimicrobial Peptides in Frog Poisons Constitute a Molecular Toxin Delivery System against Predators. Nat. Commun 2017, 8, 1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Akishiba M; Takeuchi T; Kawaguchi Y; Sakamoto K; Yu HH; Nakase I; Takatani-Nakase T; Madani F; Gräslund A; Futaki S Cytosolic Antibody Delivery by Lipid-Sensitive Endosomolytic Peptide. Nat. Chem 2017, 9, 751–761. [DOI] [PubMed] [Google Scholar]
- (38).He J; Kauffman WB; Fuselier T; Naveen SK; Voss TG; Hristova K; Wimley WC Direct Cytosolic Delivery of Polar Cargo to Cells by Spontaneous Membrane-Translocating Peptides. J. Biol. Chem 2013, 288, 29974–29986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mäe M; Langel Ü Cell-Penetrating Peptides as Vectors for Peptide, Protein. Curr. Opin. Pharmacol 2006, 6, 509–514. [DOI] [PubMed] [Google Scholar]
- (40).Melikov K; Hara A; Yamoah K; Zaitseva E; Zaitsev E; Chernomordik LV Efficient Entry of Cell-Penetrating Peptide Nona-Arginine into Adherent Cells Involves a Transient Increase in Intracellular Calcium. Biochem. J 2015, 471, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kauffman WB; Guha S; Wimley WC Synthetic Molecular Evolution of Hybrid Cell Penetrating Peptides. Nat. Commun 2018, 9, 2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yang NJ; Hinner MJ Getting across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Site-Specific Protein Labeling Methods Protoc 2015, 1266, 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Habermann E Pharmacology of Melittin. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol 1954, 222, 173–175. [PubMed] [Google Scholar]
- (44).Habermann E; Jentsch J Sequence Analysis of Melittin from Tryptic and Peptic Degradation Products. Hoppe. Seylers. Z. Physiol. Chem 1967, 348, 37–50. [PubMed] [Google Scholar]
- (45).Fennell JF; Shipman WH; Cole LJ Antibacterial Action of a Bee Venom Fraction (Melittin) against a Penicillin-Resistant Staphylococcus and Other Microorganisms. Usnrdl 1967, 1–13. [DOI] [PubMed] [Google Scholar]
- (46).Freer H Interaction of a Lytic Polypeptide, Lipid Membrane Systems. J. Biol. Chem 1969, 244, 3575–3582. [PubMed] [Google Scholar]
- (47).Weissmann G; Hirschhorn R; Krakauer K Effect of Melittin upon Cellular and Lysosomal Membranes. Biochem. Pharmacol 1969, 18, 1771–1774. [DOI] [PubMed] [Google Scholar]
- (48).Dawson CR; Drake AF; Helliwell J; Hider RC The Interaction of Bee Melittin with Lipid Bilayer Membranes. BBA - Biomembr 1978, 510, 75–86. [DOI] [PubMed] [Google Scholar]
- (49).Drake AF; Hider RC The Structure of Melittin in Lipid Bilayer Membranes. BBA - Biomembr 1979, 555, 371–373. [DOI] [PubMed] [Google Scholar]
- (50).Smith R; Separovic F; Milne T; Whittaker A; Bennett F; Cornell B; Makriyannis A Structure and Orientation of the Pore-Forming Peptide, Melittin, in Lipid Bilayers 1994, pp 456–466. [DOI] [PubMed] [Google Scholar]
- (51).Bazzo R; Tappin MJ; Pastore A; Harvey TS; Carver JA; Cambell ID The Structure of Melittin: A 1H NMR Study in Methanol. Eur. J. Biochem 1988, 173, 139–146. [DOI] [PubMed] [Google Scholar]
- (52).Terwilliger TC; Weissman L; Eisenberg D The Structure of Melittin in the Form I Crystals and Its Implication for Melittin’s Lytic and Surface Activities. Biophys. J 1982, 37, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Tosteson MT; Tosteson DC The Sting. Melittin Forms Channels in Lipid Bilayers. Biophys. J 1981, 36, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).DeGrado WF; Musso GF; Lieber M; Kaiser ET; Kézdy FJ Kinetics and Mechanism of Hemolysis Induced by Melittin and by a Synthetic Melittin Analogue. Biophys. J 1982, 37, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lee M-T; Sun T-L; Hung W-C; Huang HW Process of Inducing Pores in Membranes by Melittin. Proc. Natl. Acad. Sci 2013, 110, 14243–14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Yang L; Harroun TA; Weiss TM; Ding L; Huang HW Barrel-Stave Model or Toroidal Model? A Case Study of Melittin Pores. Biophys.~J 2001, 81, 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Frey S; Tamm LK Orientation of Melittin in Phospholipid Bilayers. A Polarized Attenuated Total Reflection Infrared Study. Biophys. J 1991, 60, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hristova K; Dempsey CE; White SH Structure, Location, and Lipid Perturbations of Melittin at the Membrane Interface. Biophys. J 2001, 80, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Krauson AJ; He J; Wimley WC Gain-of-Function Analogues of the Pore-Forming Peptide Melittin Selected by Orthogonal High-Throughput Screening. J. Am. Chem. Soc 2012, 134, 12732–12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Gordon-Grossman M; Gofman Y; Zimmermann H; Frydman V; Shai Y; Ben-Tal N; Goldfarb D Erratum: Combined Pulse EPR and Monte Carlo Simulation Study Provides Molecular Insight on Peptide-Membrane Interaction (Journal of Physical Chemistry B (2009) 113 (12687)). J. Phys. Chem. B 2009, 113, 15128. [DOI] [PubMed] [Google Scholar]
- (61).Ladokhin AS; Selsted ME; White SH Sizing Membrane Pores in Lipid Vesicles by Leakage of Co-Encapsulated Markers: Pore Formation by Melittin. Biophys. J 1997, 72, 1762–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Wiedman G; Fuselier T; He J; Searson PC; Hristova K; Wimley WC Highly Efficient Macromolecule-Sized Poration of Lipid Bilayers by a Synthetically Evolved Peptide. J. Am. Chem. Soc 2014, 136, 4724–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Dempsey CE; Sternberg B Reversible Disc-Micellization of Dimyristoylphosphatidylcholine Bilayers Induced by Melittin and [Ala-14]Melittin. BBA - Biomembr 1991, 1061, 175–184. [DOI] [PubMed] [Google Scholar]
- (64).Krauson AJ; He J; Wimley WC Determining the Mechanism of Membrane Permeabilizing Peptides: Identification of Potent, Equilibrium Pore-Formers. Biochim. Biophys. Acta - Biomembr 2012, 1818, 1625–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wimley WC How Does Melittin Permeabilize Membranes? Biophysical Journal 2018, pp 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Wiedman G; Herman K; Searson P; Wimley WC; Hristova K The Electrical Response of Bilayers to the Bee Venom Toxin Melittin: Evidence for Transient Bilayer Permeabilization. Biochim. Biophys. Acta - Biomembr 2013, 1828, 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Yang Z; Choi H; Weisshaar JC Melittin-Induced Permeabilization, Re-Sealing, and Re-Permeabilization of E. Coli Membranes. Biophys. J 2018, 114, 368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Allende D; Simon SA; McIntosh TJ Melittin-Induced Bilayer Leakage Depends on Lipid Material Properties: Evidence for Toroidal Pores. Biophys. J 2005, 88, 1828–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Benachir T; Lafleur M Study of Vesicle Leakage Induced by Melittin. BBA - Biomembr 1995, 1235, 452–460. [DOI] [PubMed] [Google Scholar]
- (70).Benachir T; Monette M; Grenier J; Lafleur M Melittin-Induced Leakage from Phosphatidylcholine Vesicles Is Modulated by Cholesterol: A Property Used for Membrane Targeting. Eur. Biophys. J 1997, 25, 201–210. [Google Scholar]
- (71).Chatterjee C; Mukhopadhyay C Melittin-GM1 Interaction: A Model for a Side-by-Side Complex. Biochem. Biophys. Res. Commun 2002, 292, 579–585. [DOI] [PubMed] [Google Scholar]
- (72).Chen X; Wang J; Boughton AP; Kristalyn CB; Chen Z Multiple Orientation of Melittin inside a Single Lipid Bilayer Determined by Combined Vibrational Spectroscopic Studies. J. Am. Chem. Soc 2007, 129, 1420–1427. [DOI] [PubMed] [Google Scholar]
- (73).Dempsey CE The Actions of Melittin on Membranes. BBA - Rev. Biomembr 1990, 1031, 143–161. [DOI] [PubMed] [Google Scholar]
- (74).Hewish D; Werkmeister J; Kirkpatrick A; Curtain C; Pantela G; Rivett DE Peptide Inhibitors of Melittin Action. J. Protein Chem 1996, 15, 395–404. [DOI] [PubMed] [Google Scholar]
- (75).Klocek G; Schulthess T; Shai Y; Seelig J Thermodynamics of Melittin Binding to Lipid Bilayers. Aggregation and Pore Formation. Biochemistry 2009, 48, 2586–2596. [DOI] [PubMed] [Google Scholar]
- (76).Kokot G; Mally M; Svetina S The Dynamics of Melittin-Induced Membrane Permeability. Eur. Biophys. J 2012, 41, 461–474. [DOI] [PubMed] [Google Scholar]
- (77).Ladokhin AS; Holloway PW Fluorescence Quenching Study of Melittin-Membrane Interactions. Ukr. Biokhim. Zh 1995, 67, 34–40. [PubMed] [Google Scholar]
- (78).Ladokhin AS; White SH “Detergent-like” Permeabilization of Anionic Lipid Vesicles by Melittin. Biochim. Biophys. Acta - Biomembr 2001, 1514, 253–260. [DOI] [PubMed] [Google Scholar]
- (79).Ladokhin AS; White SH Folding of Amphipathic α-Helices on Membranes: Energetics of Helix Formation by Melittin. J. Mol. Biol 1999, 285, 1363–1369. [DOI] [PubMed] [Google Scholar]
- (80).Ladokhin AS; Wimley WC; White SH Leakage of Membrane Vesicle Contents: Determination of Mechanism Using Fluorescence Requenching. Biophys. J 1995, 69, 1964–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Matsuzaki K; Yoneyama S; Miyajima K Pore Formation and Translocation of Melittin. Biophys. J 1997, 73, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Monette M; Lafleur M Influence of Lipid Chain Unsaturation on Melittin-Induced Micellization. Biophys. J 1996, 70, 2195–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Pott T; Paternostre M; Dufourc EJ A Comparative Study of the Action of Melittin on Sphingomyelin and Phosphatidylcholine Bilayers. Eur. Biophys. J 1998, 27, 237–245. [DOI] [PubMed] [Google Scholar]
- (84).Raghuraman H; Chattopadhyay A Melittin: A Menbrane-Active Peptide with Diverse Functions. Biosci Rep 2007, 27, 189–223. [DOI] [PubMed] [Google Scholar]
- (85).Rex S Pore Formation Induced by the Peptide Melittin in Different Lipid Vesicle Membranes. Biophys. Chem 1996, 58, 75–85. [DOI] [PubMed] [Google Scholar]
- (86).Sengupta D; Leontiadou H; Mark AE; Marrink SJ Toroidal Pores Formed by Antimicrobial Peptides Show Significant Disorder. Biochim. Biophys. Acta - Biomembr 2008, 1778, 2308–2317. [DOI] [PubMed] [Google Scholar]
- (87).Van Den Bogaart G; Guzmán JV; Mika JT; Poolman B On the Mechanism of Pore Formation by Melittin. J. Biol. Chem 2008, 283, 33854–33857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Wimley WC; White SH Insight Experi Menta I Ly Determ i Ned Hydrophobicity Scale for Proteins at Membrane Interfaces. Nat. Struct. Biol 1996, 3, 842–848. [DOI] [PubMed] [Google Scholar]
- (89).Wimley WC; Hristova K; Ladokhin AS; Silvestro L; Axelsen PH; White SH Folding of β-Sheet Membrane Proteins: A Hydrophobic Hexapeptide Model. J. Mol. Biol 1998, 277, 1091–1110. [DOI] [PubMed] [Google Scholar]
- (90).Kaiser ET; Kézdy FJ Amphiphilic Secondary Structure: Design of Peptide Hormones. Science (80-. ). 1984, 223, 249–255. [DOI] [PubMed] [Google Scholar]
- (91).Kaiser ET; Kézdy FJ Secondary Structures of Proteins and Peptides in Amphiphilic Environments. (A Review). Proc. Natl. Acad. Sci. U. S. A 1983, 80, 1137–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).White SH; Wimley WC Membrane Protein Folding and Stability: Physical Principles. Annu. Rev. Biophys. Biomol. Struct 1999, 28, 319–365. [DOI] [PubMed] [Google Scholar]
- (93).Chen CH; Khan A; Huang JJT; Ulmschneider MB Mechanisms of Membrane Pore Formation by Amyloidogenic Peptides in Amyotrophic Lateral Sclerosis. Chem. - A Eur. J 2016, 22, 9958–9961. [DOI] [PubMed] [Google Scholar]
- (94).Okada M; Natori S Mode of Action of a Bactericidal Protein Induced in the Haemolymph of Sarcophaga Peregrina (Flesh-Fly) Larvae. Biochem. J 1984, 222, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Steiner H; Hultmark D; Engström A; Bennich H; Boman HG Sequence and Specificity of Two Antibacterial Proteins Involved in Insect Immunity. Nature 1981, 292, 246–248. [DOI] [PubMed] [Google Scholar]
- (96).Zosloff M Magainlns, a Class of Antimicrobial Peptldes from Xenopus Skin: Isolation, Characterization of Two Active Forms, and Partial CDNA Sequence of a Precursor. J. Occup. Environ. Med 1988, 30, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Patterson-Delafield J; Szklarek D; Martinez RJ; Lehrer RI Microbicidal Cationic Proteins of Rabbit Alveolar Macrophages: Amino Acid Composition and Functional Attributes. Infect. Immun 1981, 31, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Wang G; Li X; Wang Z APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res 2016, 44, D1087–D1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Arnett E; Seveau S The Multifaceted Activities of Mammalian Defensins. Curr. Pharm. Des 2011, 17, 4254–4269. [DOI] [PubMed] [Google Scholar]
- (100).Selsted ME; Ouellette AJ Mammalian Defensins in the Antimicrobial Immune Response. Nat. Immunol 2005, 6, 551–557. [DOI] [PubMed] [Google Scholar]
- (101).White SH; Wimley WC; Selsted ME Structure, Function, and Membrane Integration of Defensins. Curr. Opin. Struct. Biol 1995, 5, 521–527. [DOI] [PubMed] [Google Scholar]
- (102).König E; Bininda-Emonds ORP; Shaw C The Diversity and Evolution of Anuran Skin Peptides. Peptides 2015, 63, 96–117. [DOI] [PubMed] [Google Scholar]
- (103).Michael Conlon J; Mechkarska M Host-Defense Peptides with Therapeutic Potential from Skin Secretions of Frogs from the Family Pipidae. Pharmaceuticals 2014, 7, 58–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Stroud RM; Reiling K; Wiener M; Freymann D Ion-Channel-Forming Colicins. Cur. Opin. Struc. Biol 1998, 8, 525–533. [DOI] [PubMed] [Google Scholar]
- (105).Cafiso DS Alamethicin: A Peptide Model for Voltage Gating and Protein-Membrane Interactions. Annu. Rev. Biophys. Biomol. Struct 1994, 23, 141–165. [DOI] [PubMed] [Google Scholar]
- (106).He K; Ludtke SJ; Heller WT; Huang HW Mechanism of Alamethicin Insertion into Lipid Bilayers. Biophys. J 1996, 71, 2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Leitgeb B; Szekeres A; Manczinger L; Vágvölgyi C; Kredics L The History of Alamethicin: A Review of the Most Extensively Studied Peptaibol. Chem. Biodivers 2007, 4, 1027–1051. [DOI] [PubMed] [Google Scholar]
- (108).McKay MJ; Afrose F; Koeppe RE; Greathouse DV Helix Formation and Stability in Membranes. Biochim. Biophys. Acta - Biomembr 2018. [DOI] [PubMed] [Google Scholar]
- (109).Andersen OS; Koeppe RE; Roux B Gramicidin Channels. IEEE Trans. Nanobioscience 2005, 4, 10–19. [DOI] [PubMed] [Google Scholar]
- (110).Otto M Basis of Virulence in Community-Associated Methicillin-Resistant Staphylococcus Aureus. Annu. Rev. Microbiol 2010, 64, 143–162. [DOI] [PubMed] [Google Scholar]
- (111).Pokorny A; Almeida PFF Kinetics of Dye Efflux and Lipid Flip-Flop Induced by δ-Lysin in Phosphatidylcholine Vesicles and the Mechanism of Graded Release by Amphipathic, α-Helical Peptides. Biochemistry 2004, 43, 8846–8857. [DOI] [PubMed] [Google Scholar]
- (112).Pokorny A; Kilelee EM; Wu D; Almeida PFF The Activity of the Amphipathic Peptide δ-Lysin Correlates with Phospholipid Acyl Chain Structure and Bilayer Elastic Properties. Biophys. J 2008, 95, 4748–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Craik DJ; Fairlie DP; Liras S; Price D The Future of Peptide-Based Drugs. Chem. Biol. Drug Des 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- (114).Dubovskii PV; Vassilevski AA; Kozlov SA; Feofanov AV; Grishin EV; Efremov RG Latarcins: Versatile Spider Venom Peptides. Cell. Mol. Life Sci 2015, 72, 4501–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Dubovskii PV; Vassilevski AA; Samsonova OV; Egorova NS; Kozlov SA; Feofanov AV; Arseniev AS; Grishin EV Novel Lynx Spider Toxin Shares Common Molecular Architecture with Defense Peptides from Frog Skin. FEBS J 2011, 278, 4382–4393. [DOI] [PubMed] [Google Scholar]
- (116).Okada M; Corzo G; Romero-Perez GA; Coronas F; Matsuda H; Possani LD A Pore Forming Peptide from Spider Lachesana Sp. Venom Induced Neuronal Depolarization and Pain. Biochim. Biophys. Acta - Gen. Subj 2015, 1850, 657–666. [DOI] [PubMed] [Google Scholar]
- (117).Pluzhnikov KA; Kozlov SA; Vassilevski AA; Vorontsova OV; Feofanov AV; Grishin EV Linear Antimicrobial Peptides from Ectatomma Quadridens Ant Venom. Biochimie 2014, 107, 211–215. [DOI] [PubMed] [Google Scholar]
- (118).Ramírez-Carreto S; Jiménez-Vargas JM; Rivas-Santiago B; Corzo G; Possani LD; Becerril B; Ortiz E Peptides from the Scorpion Vaejovis Punctatus with Broad Antimicrobial Activity. Peptides 2015, 73, 51–59. [DOI] [PubMed] [Google Scholar]
- (119).Yandek LE; Pokorny A; Almeida PFF Wasp Mastoparans Follow the Same Mechanism as the Cell-Penetrating Peptide Transportan 10. Biochemistry 2009, 48, 7342–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Saberwal G; Nagaraj R A Synthetic Peptide Corresponding to the Hydrophobic Amino Terminal Region of Pardaxin Can Perturb Model Membranes of Phosphatidyl Choline and Serine. BBA - Biomembr 1989, 984, 360–364. [DOI] [PubMed] [Google Scholar]
- (121).Shai Y Pardaxin: Channel Formation by a Shark Repellant Peptide from Fish. Toxicology 1994, 87, 109–129. [DOI] [PubMed] [Google Scholar]
- (122).Shai Y; Fox J; Caratsch C; Shih YL; Edwards C; Lazarovici P Sequencing and Synthesis of Pardaxin, a Polypeptide from the Red Sea Moses Sole with Ionophore Activity. FEBS Lett 1988, 242, 161–166. [DOI] [PubMed] [Google Scholar]
- (123).Oren Z; Shai Y A Class of Highly Potent Antibacterial Peptides Derived from Pardaxin, a Pore-Forming Peptide Isolated from Moses Sole Fish Pardachirus Marmoratus. Eur. J. Biochem 1996, 237, 303–310. [DOI] [PubMed] [Google Scholar]
- (124).Thennarasu S; Nagaraj R Specific Antimicrobial and Hemolytic Activities of 18-Residue Peptides Derived from the Amino Terminal Region of the Toxin Pardaxin. “Protein Eng. Des. Sel 1996, 9, 1219–1224. [DOI] [PubMed] [Google Scholar]
- (125).Kolusheva S; Lecht S; Derazon Y; Jelinek R; Lazarovici P Pardaxin, a Fish Toxin Peptide Interaction with a Biomimetic Phospholipid/Polydiacetylene Membrane Assay. Peptides 2008, 29, 1620–1625. [DOI] [PubMed] [Google Scholar]
- (126).Vad BS; Bertelsen K; Johansen CH; Pedersen JM; Skrydstrup T; Nielsen NC; Otzen DE Pardaxin Permeabilizes Vesicles More Efficiently by Pore Formation than by Disruption. Biophys. J 2010, 98, 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Cheng H; Zhu JY; Xu XD; Qiu WX; Lei Q; Han K; Cheng YJ; Zhang XZ Activable Cell-Penetrating Peptide Conjugated Prodrug for Tumor Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 16061–16069. [DOI] [PubMed] [Google Scholar]
- (128).Wu SP; Huang TC; Lin CC; Hui CF; Lin CH; Chen JY Pardaxin, a Fish Antimicrobial Peptide, Exhibits Antitumor Activity toward Murine Fibrosarcoma in Vitro and in Vivo. Mar. Drugs 2012, 10, 1852–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Primor N Pharyngeal Cavity and the Gills Are the Target Organ for the Repellent Action of Pardaxin in Shark. Experientia 1985, 41, 693–695. [DOI] [PubMed] [Google Scholar]
- (130).Sisneros JA; Nelson DR Surfactants as Chemical Shark Repellents: Past, Present, and Future. Environ. Biol. Fishes 2001, 60, 117–129. [Google Scholar]
- (131).Hyser JM; Utama B; Crawford SE; Broughman JR; Estes MK Activation of the Endoplasmic Reticulum Calcium Sensor STIM1 and Store-Operated Calcium Entry by Rotavirus Requires NSP4 Viroporin Activity. J. Virol 2013, 87, 13579–13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Nieva JL; Madan V; Carrasco L Viroporins: Structure and Biological Functions. Nat Rev Micro 2012, 10, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Giorda KM; Hebert DN Viroporins Customize Host Cells for Efficient Viral Propagation. DNA Cell Biol 2013, 32, 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Radoshitzky SR; Warfield KL; Chi X; Dong L; Kota K; Bradfute SB; Gearhart JD; Retterer C; Kranzusch PJ; Misasi JN; Hogenbirk MA; Wahl-Jensen V; Volchkov VE; Cunningham JM; Jahrling PB; Aman MJ; Bavari S; Farzan M; Kuhn JH; Ebolavirus Δ-Peptide Immunoadhesins Inhibit Marburgvirus and Ebolavirus Cell Entry. J. Virol 2011, 85, 8502–8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Volchkova VA; Klenk HD; Volchkov VE Delta-Peptide Is the Carboxy-Terminal Cleavage Fragment of the Nonstructural Small Glycoprotein SGP of Ebola Virus. Virology 1999, 265, 164–171. [DOI] [PubMed] [Google Scholar]
- (136).He J; Melnik LI; Komin A; Wiedman G; Fuselier T; Morris CF; Starr CG; Searson PC; Gallaher WR; Hristova K; Garry RF, Wimley WC; Ebola Virus Delta Peptide Is a Viroporin. J. Virol 2017, 91, e00438–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Grigoryan G; Moore DT; DeGrado WF Transmembrane Communication: General Principles and Lessons from the Structure and Function of the M2 Proton Channel, K + Channels, and Integrin Receptors. Annu. Rev. Biochem 2011, 80, 211–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Sanchez-Martinez S; Madan V; Carrasco L; Nieva J Membrane-Active Peptides Derived from Picornavirus 2B Viroporin. Curr. Protein Pept. Sci 2012, 13, 632–643. [DOI] [PubMed] [Google Scholar]
- (139).Madan V; Sánchez-Martínez S; Vedovato N; Rispoli G; Carrasco L; Nieva JL Plasma Membrane-Porating Domain in Poliovirus 2B Protein. A Short Peptide Mimics Viroporin Activity. J. Mol. Biol 2007, 374, 951–964. [DOI] [PubMed] [Google Scholar]
- (140).Kliger Y; Shai Y A Leucine Zipper-like Sequence from the Cytoplasmic Tail of the HIV-1 Envelope Glycoprotein Binds and Perturbs Lipid Bilayers. Biochemistry 1997, 36, 5157–5169. [DOI] [PubMed] [Google Scholar]
- (141).Srinivas SK; Srinivas RV; Anantharamaiah GM; Segrest JP; Compans RW Membrane Interactions of Synthetic Peptides Corresponding to Amphipathic Helical Segments of the Human Immunodeficiency Virus Type-1 Envelope Glycoprotein. J. Biol. Chem 1992, 267, 7121–7127. [PubMed] [Google Scholar]
- (142).Tencza SB; Miller MA; Islam K; Mietzner TA; Montelaro RC Effect of Amino Acid Substitutions on Calmodulin Binding and Cytolytic Properties of the LLP-1 Peptide Segment of Human Immunodeficiency Virus Type 1 Transmembrane Protein. J. Virol 1995, 69, 5199–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Comardelle AM; Norris CH; Plymeade DR; Gatti PJ; Choi B; Fermin CD; Haislip AM; Tencza SB; Mietzner TA; Montelaro RC; Garry RF A Synthetic Peptide Corresponding to the Carboxy Terminus of Human Immunodeficiency Virus Type 1 Transmembrane Glycoprotein Induces Alterations in the Ionic Permeability of Xenopus Laevis Oocytes. AIDS Res. Hum. Retroviruses 1997, 13, 1525–1532. [DOI] [PubMed] [Google Scholar]
- (144).Moreno MR; Giudici M; Villalaín J The Membranotropic Regions of the Endo and Ecto Domains of HIV Gp41 Envelope Glycoprotein. Biochim. Biophys. Acta - Biomembr 2006, 1758, 111–123. [DOI] [PubMed] [Google Scholar]
- (145).Moreno MR; Guillén J; Pérez-Berná AJ; Amorós D; Gómez AI; Bernabeu Á; Villalaín J Characterization of the Interaction of Two Peptides from the N Terminus of the NHR Domain of HIV-1 Gp41 with Phospholipid Membranes. Biochemistry 2007, 46, 10572–10584. [DOI] [PubMed] [Google Scholar]
- (146).Tencza SB; Creighton DJ; Yuan T; Vogel HJ; Montelaro RC; Mietzner TA Lentivirus-Derived Antimicrobial Peptides: Increased Potency by Sequence Engineering and Dimerization. J. Antimicrob. Chemother 1999, 44, 33–41. [DOI] [PubMed] [Google Scholar]
- (147).Costin JM; Rausch JM; Garry RF; Wimley WC Viroporin Potential of the Lentivirus Lytic Peptide (LLP) Domains of the HIV-1 Gp41 Protein. Virol. J 2007, 4, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Phadke SM; Lazarevic V; Bahr CC; Islam K; Stolz DB; Watkins S; Tencza SB; Vogel HJ; Montelaro RC; Mietzner TA Lentivirus Lytic Peptide 1 Perturbs Both Outer and Inner Membranes of Serratia Marcescens. Antimicrob. Agents Chemother 2002, 46, 2041–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Kyrychenko A; Freites JA; He J; Tobias DJ; Wimley WC; Ladokhin AS Structural Plasticity in the Topology of the Membrane-Interacting Domain of HIV-1 Gp41. Biophys. J 2014, 106, 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Apellániz B; Ivankin A; Nir S; Gidalevitz D; Nieva JL Membrane-Proximal External HIV-1 Gp41 Motif Adapted for Destabilizing the Highly Rigid Viral Envelope. Biophys. J 2011, 101, 2426–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Thennarasu S; Tan A; Penumatchu R; Shelburne CE; Heyl DL; Ramamoorthy A Antimicrobial and Membrane Disrupting Activities of a Peptide Derived from the Human Cathelicidin Antimicrobial Peptide Ll37. Biophys. J 2010, 98, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Boman HG; Wade D; Boman IA; Wåhlin B; Merrifield RB Antibacterial and Antimalarial Properties of Peptides That Are Cecropin-Melittin Hybrids. FEBS Lett 1989, 259, 103–106. [DOI] [PubMed] [Google Scholar]
- (153).Mancheno JM; Oñaderra M; Del Pozo AM; Díaz-Achirica P; Andreu D; Rivas L; Gavilanes JG Release of Lipid Vesicle Contents by an Antibacterial Cecropin A- Melittin Hybrid Peptide. Biochemistry 1996, 35, 9892–9899. [DOI] [PubMed] [Google Scholar]
- (154).De Breij A; Riool M; Cordfunke RA; Malanovic N; De Boer L; Koning RI; Ravensbergen E; Franken M; Van Der Heijde T; Boekema BK; et al. The Antimicrobial Peptide SAAP-148 Combats Drug-Resistant Bacteria and Biofilms. Sci. Transl. Med 2018, 10. [DOI] [PubMed] [Google Scholar]
- (155).Blondelle SE; Houghten RA Design of Model Amphipathic Peptides Having Potent Antimicrobial Activities. Biochemistry 1992, 31, 12688–12694. [DOI] [PubMed] [Google Scholar]
- (156).Blondelle SE; Lohner K Combinatorial Libraries: A Tool to Design Antimicrobial and Antifungal Peptide Analogues Having Lyric Specificities for Structure-Activity Relationship Studies. Biopolymers 2000, 55, 74–87. [DOI] [PubMed] [Google Scholar]
- (157).Javadpour MM; Barkley MD Self-Assembly of Designed Antimicrobial Peptides in Solution and Micelles. Biochemistry 1997, 36, 9540–9549. [DOI] [PubMed] [Google Scholar]
- (158).Schwab U; Gilligan P; Jaynes J; Henke D; Schwab UTE In Vitro Activities of Designed Antimicrobial Peptides against Multidrug-Resistant Cystic Fibrosis Pathogens In Vitro Activities of Designed Antimicrobial Peptides against Multidrug-Resistant Cystic Fibrosis Pathogens. Antimicrob. Agents Chemother 1999, 43, 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Thennarasu S; Nagaraj R Design of 16‐residue Peptides Possessing Antimicrobial and Hemolytic Activities or Only Antimicrobial Activity from an Inactive Peptide. Int. J. Pept. Protein Res 1995, 46, 480–486. [DOI] [PubMed] [Google Scholar]
- (160).Tossi A; Tarantino C; Romeo D Design of Synthetic Antimicrobial Peptides Based on Sequence Analogy and Amphipathicity. Eur. J. Biochem 1997, 250, 549–558. [DOI] [PubMed] [Google Scholar]
- (161).Zhong L; Putnam RJ; Johnson WC Rao AG Design and Synthesis of Amphipathic Antimicrobial Peptides. Int. J. Pept. Protein Res 1995, 45, 337–347. [DOI] [PubMed] [Google Scholar]
- (162).Fjell CD; Hancock REW; Cherkasov A AMPer: A Database and an Automated Discovery Tool for Antimicrobial Peptides. Bioinformatics 2007, 23, 1148–1155. [DOI] [PubMed] [Google Scholar]
- (163).Hilpert K Sequence Requirements and a Novel Optimization Strategy for Short Antimicrobial Peptides. Chem. Biol 2006, 13, 1101–1107. [DOI] [PubMed] [Google Scholar]
- (164).Wang Z APD: The Antimicrobial Peptide Database. Nucleic Acids Res 2004, 32, 590D–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Juretić D; Vukičević D; Petrov D; Novković M; Bojović V; Lučić B; Ilić N; Tossi A Knowledge-Based Computational Methods for Identifying or Designing Novel, Non-Homologous Antimicrobial Peptides. Eur. Biophys. J 2011, 40, 371–385. [DOI] [PubMed] [Google Scholar]
- (166).Mishra B; Wang G Ab Initio Design of Potent Anti-MRSA Peptides Based on Database Filtering Technology. J. Am. Chem. Soc 2012, 134, 12426–12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Pauling L; Corey RB; Branson HR The Structure of Proteins: Two Hydrogen-Bonded Helical Configurations of the Polypeptide Chain. Proc. Natl. Acad. Sci 1951, 37, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Akerfeldt KS; Lear JD; Wasserman ZR; Chung LA; DeGrado WF Synthetic Peptides as Models for Ion Channel Proteins. Acc. Chem. Res 1993, 26, 191–197. [Google Scholar]
- (169).Lear JD; Wasserman ZR; DeGrado WF Synthetic Peptide Models for Protein Ion Channels. Science 1988, 240, 1177–1181. [DOI] [PubMed] [Google Scholar]
- (170).Goormaghtigh E; De Meutter J; Szoka F; Cabiaux V; Parente RA; Ruysschaert JM Secondary Structure and Orientation of the Amphipathic Peptide GALA in Lipid Structures: An Infrared‐spectroscopic Approach. Eur. J. Biochem 1991, 195, 421–429. [DOI] [PubMed] [Google Scholar]
- (171).Parente RA; Nadasdi L; Subbarao NK; Szoka FC Association of a PH-Sensitive Peptide with Membrane Vesicles: Role of Amino Acid Sequence. Biochemistry 1990, 29, 8713–8719. [DOI] [PubMed] [Google Scholar]
- (172).Parente RA; Nir S; Szoka FC Mechanism of Leakage of Phospholipid Vesicle Contents Induced by the Peptide GALA. Biochemistry 1990, 29, 8720–8728. [DOI] [PubMed] [Google Scholar]
- (173).Subbarao NK; Parente RA; Szoka FC; Nadasdi L; Poneracz K PH-Dependent Bilayer Destabilization by an Amphipathic Peptide. Biochemistry 1987, 26, 2964–2972. [DOI] [PubMed] [Google Scholar]
- (174).Rathinakumar R; Wimley WC High-Throughput Discovery of Broad-Spectrum Peptide Antibiotics. Faseb J 2010, 24, 3232–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Rausch JM; Marks JR; Wimley WC Rational Combinatorial Design of Pore-Forming β-Sheet Peptides. Proc. Natl. Acad. Sci 2005, 102, 10511–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Krauson AJ; He J; Wimley AW; Hoffmann AR; Wimley WC Synthetic Molecular Evolution of Pore-Forming Peptides by Iterative Combinatorial Library Screening. ACS Chem. Biol 2013, 8, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Gordon-Grossman M; Zimmermann H; Wolf SG; Shai Y; Goldfarb D Investigation of Model Membrane Disruption Mechanism by Melittin Using Pulse Electron Paramagnetic Resonance Spectroscopy and Cryogenic Transmission Electron Microscopy. J. Phys. Chem. B 2012, 116, 179–188. [DOI] [PubMed] [Google Scholar]
- (178).Pittman AE; Marsh BP; King GM Conformations and Dynamic Transitions of a Melittin Derivative That Forms Macromolecule-Sized Pores in Lipid Bilayers. Langmuir 2018, 34, 8393–8399. [DOI] [PubMed] [Google Scholar]
- (179).Krauson AJ; Hall OM; Fuselier T; Starr CG; Kauffman WB; Wimley WC Conformational Fine-Tuning of Pore-Forming Peptide Potency and Selectivity. J. Am. Chem. Soc 2015, 137, 16144–16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Wiedman G; Kim SY; Zapata-Mercado E; Wimley WC; Hristova K PH-Triggered, Macromolecule-Sized Poration of Lipid Bilayers by Synthetically Evolved Peptides. J. Am. Chem. Soc 2017, 139, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Li S; Kim SY; Pittman AE; King GM; Wimley WC; Hristova K Potent Macromolecule-Sized Poration of Lipid Bilayers by the Macrolittins, A Synthetically Evolved Family of Pore-Forming Peptides. J. Am. Chem. Soc 2018, 140, 6441–6447. [DOI] [PubMed] [Google Scholar]
- (182).Hilger D; Masureel M; Kobilka BK Structure and Dynamics of GPCR Signaling Complexes. Nat. Struct. Mol. Biol 2018, 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Hendrickson WA Atomic-Level Analysis of Membrane Protein Structure. Nat. Struct. Mol. Biol 2017, 23, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Wimley WC The Versatile β-Barrel Membrane Protein. Curr. Opin. Struct. Biol 2003, 13, 404–411. [DOI] [PubMed] [Google Scholar]
- (185).Fairman JW; Noinaj N; Buchanan SK Summary of Recent Reports 2012, 21, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Sitaram N; R. N Interaction of Antimicrobial Peptides with Biological and Model Membranes: Structural and Charge Require- Ments for Activity. Biochim. Biophys. Acta 1999, 1462, 29–54. [DOI] [PubMed] [Google Scholar]
- (187).Bechinger B The Structure, Dynamics and Orientation of Antimicrobial Peptides in Membranes by Multidimensional Solide-State NMR Spectroscopy. Biochim. Biophys. Acta 1999, 1462, 157–183. [DOI] [PubMed] [Google Scholar]
- (188).Heller WT; Waring AJ; Lehrer RI; Huang HW Multiple States of β-Sheet Peptide Protegrin in Lipid Bilayers. Biochemistry 1998, 37, 17331–17338. [DOI] [PubMed] [Google Scholar]
- (189).Heller WT; Waring AJ; Lehrer RI; Harroun TA; Weiss TM; Yang L; Huang HW Membrane Thinning Effect of the β-Sheet Antimicrobial Protegrin. Biochemistry 2000, 39, 139–145. [DOI] [PubMed] [Google Scholar]
- (190).Ketchem RR; Roux B; Cross TA High-Resolution Polypeptide Structure in a Lamellar Phase Lipid Environment from Solid State NMR Derived Orientational Constraints. Structure 1997, 5, 1655–1669. [DOI] [PubMed] [Google Scholar]
- (191).Lee TH; Sani MA; Overall S; Separovic F; Aguilar MI Effect of Phosphatidylcholine Bilayer Thickness and Molecular Order on the Binding of the Antimicrobial Peptide Maculatin 1.1. Biochim. Biophys. Acta - Biomembr 2018, 1860, 300–309. [DOI] [PubMed] [Google Scholar]
- (192).Greenfield NJ Using Circular Dichroism Spectra to Estimate Protein Secondary Structure. Nat. Protoc 2007, 1, 2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Mielke SP; Krishnan VV Characterization of Protein Secondary Structure from NMR Chemical Shifts. Prog. Nucl. Magn. Reson. Spectrosc 2009, 54, 141–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Wu Y; Huang HW; Olah GA Method of Oriented Circular Dichroism. Biophys. J 1990, 57, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Huang HW; Olah GA Uniformly Oriented Gramicidin Channels Embedded in Thick Monodomain Lecithin Multilayers. Biophys. J 1987, 51, 989–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Bürck J; Wadhwani P; Fanghänel S; Ulrich AS Oriented Circular Dichroism: A Method to Characterize Membrane-Active Peptides in Oriented Lipid Bilayers. Acc. Chem. Res 2016, 49, 184–192. [DOI] [PubMed] [Google Scholar]
- (197).Lomash S; Nagpal S; Salunke DM An Antibody as Surrogate Receptor Reveals Determinants of Activity of an Innate Immune Peptide Antibiotic. J. Biol. Chem 2010, 285, 35750–35758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Ladokhin AS; Selsted ME; White SH CD Spectra of Indolicidin Antimicrobial Peptides Suggest Turns, Not Polyproline Helix. Biochemistry 1999, 38, 12313–12319. [DOI] [PubMed] [Google Scholar]
- (199).Johansson J; Gudmundsson GH; Rottenberg ME; Berndt KD; Agerberth B Conformation-Dependent Antibacterial Activity of the Naturally Occurring Human Peptide LL-37. J. Biol. Chem 1998, 273, 3718–3724. [DOI] [PubMed] [Google Scholar]
- (200).IUPAC-IUB Commission on Biochemical Nomenclature. Abbreviations and Symbols for the Description Conformation of Polypeptide Chains. J. Biol. Chem 1970, 246, 6489–6497. [PubMed] [Google Scholar]
- (201).Kabsch W; Sander C Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen‐bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- (202).Richardson JS The Anatomy and Taxonomy of Protein Structure. Adv. Protein Chem 1981, 34, 167–339. [DOI] [PubMed] [Google Scholar]
- (203).Rausch JM; Marks JR; Rathinakumar R; Wimley WC β-Sheet Pore-Forming Peptides Selected from a Rational Combinatorial Library: Mechanism of Pore Formation in Lipid Vesicles and Activity in Biological Membranes. Biochemistry 2007, 46, 12124–12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Bach AC; Selsted ME; Pardi A Two-Dimensional NMR Studies of the Antimicrobial Peptide NP-5. Biochemistry 1987, 26, 4389–4397. [DOI] [PubMed] [Google Scholar]
- (205).Hill CP; Yee J; Selsted ME; Eisenberg D Crystal Structure of Defensin HNP-3, an Amphilphilic Dimer: Mechanism of Membrane Permeabilization. Science, 1991, 251, 1481–1485. [DOI] [PubMed] [Google Scholar]
- (206).Kawano K; Yoneya T; Miyata T; Yoshikawa K; Tokunaga F; Terada Y; Iwanaga S Antimicrobial Peptide, Tachyplesin I, Isolated from Hemocytes of the Horseshoe Crab (Tachypleus Tridentatus). NMR Determination of the β-Sheet Structure. J. Biol. Chem 1990, 265, 15365–15367. [PubMed] [Google Scholar]
- (207).Mani R; Buffy JJ; Waring AJ; Lehrer RI; Hong M Solid-State NMR Investigation of the Selective Disruption of Lipid Membranes by Protegrin-1. Biochemistry 2004, 43, 13839–13848. [DOI] [PubMed] [Google Scholar]
- (208).Pardi A; Hare DR; Selsted ME; Morrison RD; Bassolino DA; Bach AC Solution Structures of the Rabbit Neutrophil Defensin NP-5. J. Mol. Biol 1988, 201, 625–636. [DOI] [PubMed] [Google Scholar]
- (209).Roumestand C; Louis V; Aumelas A; Grassy G; Calas B; Chavanieu A Oligomerization of Protegrin-1 in the Presence of DPC Micelles. A Proton High-Resolution NMR Study. FEBS Lett 1998, 421, 263–267. [DOI] [PubMed] [Google Scholar]
- (210).Tran D; Tran PA; Tang YQ; Yuan J; Cole T; Selsted ME Homodimeric θ-Defensins from Rhesus Macaque Leukocytes. Isolation, Synthesis, Antimicrobial Activities, and Bacterial Binding Properties of the Cyclic Peptides. J. Biol. Chem 2002, 277, 3079–3084. [DOI] [PubMed] [Google Scholar]
- (211).Durkin JT; Providence LL; Koeppe RE; Andersen OS Formation of Non-Beta 6.3-Helical Gramicidin Channels between Sequence-Substituted Gramicidin Analogues. Biophys. J 1992, 62, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Westbrook EM; Lehrer RI; Selsted ME Characterization of Two Crystal Forms of Neutrophil Cationic Protein NP2, a Naturally Occurring Broad-Spectrum Antimicrobial Agent from Leukocytes. J. Mol. Biol 1984, 178, 783–785. [DOI] [PubMed] [Google Scholar]
- (213).Pardi A; Skalicky JJ; Zhang XL; Selsted ME; Yip PF NMR Studies of Defensin Antimicrobial Peptides. 2. Three-Dimensional Structures of Rabbit NP-2 and Human HNP-1. Biochemistry 1992, 31, 11357–11364. [DOI] [PubMed] [Google Scholar]
- (214).Wimley WC; Selsted ME; White SH Interactions between Human Defensins and Lipid Bilayers: Evidence for Formation of Multimeric Pores. Protein Sci 1994, 3, 1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Sansom MSP Alamethicin and Related Peptaibols - Model Ion Channels. Eur. Biophys. J 1993, 22, 105–124. [DOI] [PubMed] [Google Scholar]
- (216).Smith R; Thomas DE; Separovic F; Atkins AR; Cornell BA Determination of the Structure of a Membrane-Incorporated Ion Channel. Solid-State Nuclear Magnetic Resonance Studies of Gramicidin A. Biophys. J 1989, 56, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Ketchem RR; Hu W; Cross TA High-Resolution Conformation of Gramicidin A in a Lipid Bilayer by Solid-State NMR. Science (80-. ). 1993, 261, 1457–1460. [DOI] [PubMed] [Google Scholar]
- (218).White SH; Wimley WC Hydrophobic Interactions of Peptides with Membrane Interfaces. Biochimica et Biophysica Acta - Reviews on Biomembranes 1998, pp 339–352. [DOI] [PubMed] [Google Scholar]
- (219).Lazaridis T; He Y; Prieto L Membrane Interactions and Pore Formation by the Antimicrobial Peptide Protegrin. Biophys. J 2013, 104, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Jean-François F; Elezgaray J; Berson P; Vacher P; Dufourc EJ Pore Formation Induced by an Antimicrobial Peptide: Electrostatic Effects. Biophys. J 2008, 95, 5748–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Oren Z; Hong J; Shai Y A Repertoire of Novel Antibacterial Diastereomeric Peptides with Selective Cytolytic Activity. J. Biol. Chem 1997, 272, 14643–14649. [DOI] [PubMed] [Google Scholar]
- (222).Ladokhin AS; White SH Protein Chemistry at Membrane Interfaces: Non-Additivity of Electrostatic and Hydrophobic Interactions. J. Mol. Biol 2001, 309, 543–552. [DOI] [PubMed] [Google Scholar]
- (223).Ladokhin AS; Selsted ME; White SH Bilayer Interactions of Indolicidin, a Small Antimicrobial Peptide Rich in Tryptophan, Proline, and Basic Amino Acids. Biophys. J 1997, 72, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Hilpert K; Elliott MR; Volkmer-Engert R; Henklein P; Donini O; Zhou Q; Winkler DFH; Hancock REW Sequence Requirements and an Optimization Strategy for Short Antimicrobial Peptides. Chem. Biol 2006, 13, 1101–1107. [DOI] [PubMed] [Google Scholar]
- (225).Hilpert K; Volkmer-Engert R; Walter T; Hancock REW High-Throughput Generation of Small Antibacterial Peptides with Improved Activity. Nat. Biotechnol 2005, 23, 1008–1012. [DOI] [PubMed] [Google Scholar]
- (226).Russell AL; Kennedy AM; Spuches AM; Gibson WS; Venugopal D; Klapper D; Srouji AH; Bhonsle JB; Hicks RP Determining the Effect of the Incorporation of Unnatural Amino Acids into Antimicrobial Peptides on the Interactions with Zwitterionic and Anionic Membrane Model Systems. Chem. Phys. Lipids 2011, 164, 740–758. [DOI] [PubMed] [Google Scholar]
- (227).Lee DK; Brender JR; Sciacca MFM; Krishnamoorthy J; Yu C; Ramamoorthy A Lipid Composition-Dependent Membrane Fragmentation and Pore-Forming Mechanisms of Membrane Disruption by Pexiganan (MSI-78). Biochemistry 2013, 52, 3254–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Lam KLH; Wang H; Siaw TA; Chapman MR; Waring AJ; Kindt JT; Lee KYC Mechanism of Structural Transformations Induced by Antimicrobial Peptides in Lipid Membranes. Biochim. Biophys. Acta - Biomembr 2012, 1818, 194–204. [DOI] [PubMed] [Google Scholar]
- (229).Sychev SV; Balandin SV; Panteleev PV; Barsukov LI; Ovchinnikova TV Lipid-Dependent Pore Formation by Antimicrobial Peptides Arenicin-2 and Melittin Demonstrated by Their Proton Transfer Activity. J. Pept. Sci 2015, 21, 71–76. [DOI] [PubMed] [Google Scholar]
- (230).Nicol F; Nir S; Szoka FC Effect of Phospholipid Composition on an Amphipathic Peptide-Mediated Pore Formation in Bilayer Vesicles. Biophys. J 2000, 78, 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Yau WM; Wimley WC; Gawrisch K; White SH The Preference of Tryptophan for Membrane Interfaces. Biochemistry 1998, 37, 14713–14718. [DOI] [PubMed] [Google Scholar]
- (232).Wimley WC; White SH Experimentally Determined Hydrophobicity Scale for Proteins at Membrane Interfaces. Nature Structural Biology 1996, pp 842–848. [DOI] [PubMed] [Google Scholar]
- (233).He K; Ludtke SJ; Wu Y; Huang HW; Andersen OS; Greathouse D; Koeppe RE Closed State of Gramicidin Channel Detected by X-Ray in-Plane Scattering. Biophys. Chem 1994, 49, 83–89. [DOI] [PubMed] [Google Scholar]
- (234).Cross TA [31] Solid-State Nuclear Magnetic Resonance Characterization of Gramicidin Channel Structure. Methods Enzymol 1997, 289, 672–IN4. [DOI] [PubMed] [Google Scholar]
- (235).Su Y; Waring AJ; Ruchala P; Hong M Structures of β-Hairpin Antimicrobial Protegrin Peptides in Lipopolysaccharide Membranes: Mechanism of Gram Selectivity Obtained from Solid-State Nuclear Magnetic Resonance. Biochemistry 2011, 50, 2072–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).North CL; Barranger-Mathys M; Cafiso DS Membrane Orientation of the N-Terminal Segment of Alamethicin Determined by Solid-State 15N NMR. Biophys. J 1995, 69, 2392–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Barranger-Mathys M; Cafiso DS Membrane Structure of Voltage-Gated Channel Forming Peptides by Site-Directed Spin-Labeling †. Biochemistry 1996, 35, 498–505. [DOI] [PubMed] [Google Scholar]
- (238).Islam MZ; Sharmin S; Moniruzzaman M; Yamazaki M Elementary Processes for the Entry of Cell-Penetrating Peptides into Lipid Bilayer Vesicles and Bacterial Cells. Applied Microbiology and Biotechnology 2018, pp 3879–3892. [DOI] [PubMed] [Google Scholar]
- (239).Gross RW; Han X Lipidomics at the Interface of Structure and Function in Systems Biology. Chem. Biol 2011, 18, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Shevchenko A; Simons K Lipidomics: Coming to Grips with Lipid Diversity. Nature Reviews Molecular Cell Biology 2010, pp 593–598. [DOI] [PubMed] [Google Scholar]
- (241).Van Meer G Cellular Lipidomics. EMBO J 2005, 24, 3159–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Wallin E; Von Heijne G. Genome-Wide Analysis of Integral Membrane Proteins from Eubacterial, Archaean, and Eukaryotic Organisms. Protein Sci 2008, 7, 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Fernandez-Trillo F; Grover LM; Stephenson-Brown A; Harrison P; Mendes PM Vesicles in Nature and the Laboratory: Elucidation of Their Biological Properties and Synthesis of Increasingly Complex Synthetic Vesicles. Angewandte Chemie - International Edition 2017, pp 3142–3160. [DOI] [PubMed] [Google Scholar]
- (244).Bangham AD; Standish MM; Watkins JC Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. J. Mol. Biol 1965, 13, IN26–IN27. [DOI] [PubMed] [Google Scholar]
- (245).Wimley WC; Thompson TE Exchange and Flip-Flop of Dimyristoylphosphatidylcholine in Liquid-Crystalline, Gel, and Two-Component, Two-Phase Large Unilamellar Vesicles. Biochemistry 1990, 29, 1296–1303. [DOI] [PubMed] [Google Scholar]
- (246).Huang CH Studies on Phosphatidylcholine Vesicles. Formation and Physical Characteristics. Biochemistry 1969, 8, 344–352. [DOI] [PubMed] [Google Scholar]
- (247).Huang C; Charlton JP Studies on the State of Phosphatidylcholine Molecules before and after Ultrasonic and Gel-Filtration Treatments. Biochem. Biophys. Res. Commun 1972, 46, 1660–1666. [DOI] [PubMed] [Google Scholar]
- (248).Huang C; Thompson TE Preparation of Homogeneous, Single-Walled Phosphatidylcholine Vesicles. Methods Enzymol 1974, 32, 485–489. [DOI] [PubMed] [Google Scholar]
- (249).Rothman JE; Dawidowicz EA Asymmetric Exchange of Vesicle Phospholipids Catalyzed by the Phosphatidylcholine Exchange Protein. Measurement of Inside-Outside Transitions. Biochemistry 1975, 14, 2809–2816. [DOI] [PubMed] [Google Scholar]
- (250).Newman GC; Huang C. hsien. Structural Studies on Phosphatidylcholine-Cholesterol Mixed Vesicles. Biochemistry 1975, 14, 3363–3370. [DOI] [PubMed] [Google Scholar]
- (251).Lelkes PI; Miller IR Perturbations of Membrane Structure by Optical Probes: I. Location and Structural Sensitivity of Merocyanine 540 Bound to Phospholipid Membranes. J. Membr. Biol 1980, 52, 1–15. [DOI] [PubMed] [Google Scholar]
- (252).Batzri S; Korn ED Single Bilayer Liposomes Prepared without Sonication. BBA - Biomembr 1973, 298, 1015–1019. [DOI] [PubMed] [Google Scholar]
- (253).Kagawa Y; Racker E Partial Resolution Phosphorylation of the Enzymes Catalyzing Oxidative Phosphorylation. J. Biol. Chem 1971, 246, 5477–5487. [Google Scholar]
- (254).Netzer NL; Gunawidjaja R; Hiemstra M; Zhang Q; Tsukruk VV; Jiang C Formation and Optical Properties of Compression-Induced Nanoscale Buckles on Silver Nanowires. ACS Nano 2009, 3, 1795–1802. [DOI] [PubMed] [Google Scholar]
- (255).Mayer LD; Hope MJ; Cullis PR Vesicles of Variable Sizes Produced by a Rapid Extrusion Procedure. BBA - Biomembr 1986, 858, 161–168. [DOI] [PubMed] [Google Scholar]
- (256).Nayar R; Hope MJ; Cullis PR Generation of Large Unilamellar Vesicles from Long-Chain Saturated Phosphatidylcholines by Extrusion Technique. BBA - Biomembr 1989, 986, 200–206. [Google Scholar]
- (257).Hope MJ; Bally MB; Mayer LD; Janoff AS; Cullis PR Generation of Multilamellar and Unilamellar Phospholipid Vesicles. Chem. Phys. Lipids 1986, 40, 89–107. [Google Scholar]
- (258).Walde P; Wick R; Fresta M; Mangone A; Luisi PL Autopoietic Self-Reproduction of Fatty Acid Vesicles. J. Am. Chem. Soc 1994, 116, 11649–11654. [Google Scholar]
- (259).Tzung SP; Kim KM; Basãez G; Giedt CD; Simon J; Zimmerberg J; Zhang KYJ; Hockenbery DM Antimycin A Mimics a Cell-Death-Inducing Bcl-2 Homology Domain 3. Nat. Cell Biol 2001, 3, 183–191. [DOI] [PubMed] [Google Scholar]
- (260).Kalb E; Frey S; Tamm LK Formation of Supported Planar Bilayers by Fusion of Vesicles to Supported Phospholipid Monolayers. BBA - Biomembr 1992, 1103, 307–316. [DOI] [PubMed] [Google Scholar]
- (261).Haran G; Cohen R; Bar LK; Barenholz Y Transmembrane Ammonium Sulfate Gradients in Liposomes Produce Efficient and Stable Entrapment of Amphipathic Weak Bases. BBA - Biomembr 1993, 1151, 201–215. [DOI] [PubMed] [Google Scholar]
- (262).Goldschmidt-Clermont PJ; Machesky LM; Baldassare JJ; Pollard TD The Actin-Binding Protein Profilin Binds to PIP2 and Inhibits Its Hydrolysis by Phospholipase C. Science (80-. ). 1990, 247, 1575–1578. [DOI] [PubMed] [Google Scholar]
- (263).Matsuzaki K; Fukui M; Fujii N; Miyajima K Interactions of an Antimicrobial Peptide, Tachyplesin I, with Lipid Membranes. BBA - Biomembr 1991, 1070, 259–264. [DOI] [PubMed] [Google Scholar]
- (264).Hädicke A; Blume A Binding of Cationic Model Peptides (KX)4K to Anionic Lipid Bilayers: Lipid Headgroup Size Influences Secondary Structure of Bound Peptides. Biochim. Biophys. Acta - Biomembr 2017, 1859, 415–424. [DOI] [PubMed] [Google Scholar]
- (265).Datrie M; Schumann M; Wieprecht T; Winkler A; Beyermann M; Krause E; Matsuzaki K; Murase O; Bienert M Peptide Helicity and Membrane Surface Charge Modulate the Balance of Electrostatic and Hydrophobic Interactions with Lipid Bilayers and Biological Membranes. Biochemistry 1996, 35, 12612–12622. [DOI] [PubMed] [Google Scholar]
- (266).Hancock REW; Rozek A Role of Membranes in the Activities of Antimicrobial Cationic Peptides. FEMS Microbiology Letters 2002, pp 143–149. [DOI] [PubMed] [Google Scholar]
- (267).Zhang L; Rozek A; Hancock REW Interaction of Cationic Antimicrobial Peptides with Model Membranes. J. Biol. Chem 2001, 276, 35714–35722. [DOI] [PubMed] [Google Scholar]
- (268).Manzini MC; Perez KR; Riske KA; Bozelli JC; Santos TL; Da Silva MA; Saraiva GKV; Politi MJ; Valente AP; Almeida FCL; et al. Peptide:Lipid Ratio and Membrane Surface Charge Determine the Mechanism of Action of the Antimicrobial Peptide BP100. Conformational and Functional Studies. Biochim. Biophys. Acta - Biomembr 2014, 1838, 1985–1999. [DOI] [PubMed] [Google Scholar]
- (269).Cronan JE Bacterial Membrane Lipids: Where Do We Stand? Annu. Rev. Microbiol 2003, 57, 203–224. [DOI] [PubMed] [Google Scholar]
- (270).von Heijne G; Gavel Y Topogenic Signals in Integral Membrane Proteins. Eur. J. Biochem 1988, 174, 671–678. [DOI] [PubMed] [Google Scholar]
- (271).Bretscher MS Asymmetrical Lipid Bilayer Structure for Biological Membranes. Nat. New Biol 1972, 236, 11–12. [DOI] [PubMed] [Google Scholar]
- (272).Fadeel B; Xue D The Ins and Outs of Phospholipid Asymmetry in the Plasma Membrane: Roles in Health and Disease. Critical Reviews in Biochemistry and Molecular Biology 2009, pp 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Devaux PF Static and Dynamic Lipid Asymmetry in Cell Membranes. Biochemistry 1991, 30, 1163–1173. [DOI] [PubMed] [Google Scholar]
- (274).Lin Q; London E Preparation of Artificial Plasma Membrane Mimicking Vesicles with Lipid Asymmetry. PLoS One 2014, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Yamazaki M Chapter 5 The Single Guv Method to Reveal Elementary Processes of Leakage of Internal Contents from Liposomes Induced by Antimicrobial Substances. Advances in Planar Lipid Bilayers and Liposomes 2008, pp 121–142. [Google Scholar]
- (276).Weinstein JN; Yoshikami S; Henkart P; Blumenthal R; Hagins WA Liposome-Cell Interaction: Transfer and Intracellular Release of a Trapped Fluorescent Marker. Science (80-. ). 1977, 195, 489–492. [DOI] [PubMed] [Google Scholar]
- (277).Pagano RE; Takeichi M Adhesion of Phospholipid Vesicles to Chinese Hamster Fibroblasts. Role of Cell Surface Proteins. J Cell Biol 1977, 74, 531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Ghadiri MR; Granja JR; Buehler LK Artificial Transmembrane Ion Channels from Self-Assembling Peptide Nanotubes. Nature 1994, 369, 301–304. [DOI] [PubMed] [Google Scholar]
- (279).Blumenthal R; Weinstein JN; Sharrow SO; Henkart P Liposome--Lymphocyte Interaction: Saturable Sites for Transfer and Intracellular Release of Liposome Contents. Proc. Natl. Acad. Sci 1977, 74, 5603–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Allen TM; Cleland LG Serum-Induced Leakage of Liposome Contents. BBA - Biomembr 1980, 597, 418–426. [DOI] [PubMed] [Google Scholar]
- (281).Ellens H; Bentz J; Szoka FC PH-Induced Destabilization of Phosphatidylethanolamine-Containing Liposomes: Role of Bilayer Contact. Biochemistry 1984, 23, 1532–1538. [DOI] [PubMed] [Google Scholar]
- (282).Smolarsky M; Teitelbaum D; Sela M; Gitler C A Simple Fluorescent Method to Determine Complement-Mediated Liposome Immune Lysis. J. Immunol. Methods 1977, 15, 255–265. [DOI] [PubMed] [Google Scholar]
- (283).Wilschut J; Düzgüneş N; Fraley R; Papahadjopoulos D Studies on the Mechanism of Membrane Fusion: Kinetics of Calcium Ion Induced Fusion of Phosphatidylserine Vesicles Followed by a New Assay for Mixing of Aqueous Vesicle Contents. Biochemistry 1980, 19, 6011–6021. [DOI] [PubMed] [Google Scholar]
- (284).Rausch JM; Wimley WC A High-Throughput Screen for Identifying Transmembrane Pore-Forming Peptides. Anal. Biochem 2001, 293, 258–263. [DOI] [PubMed] [Google Scholar]
- (285).Wimley WC Determining the Effects of Membrane-Interacting Peptides on Membrane Integrity. In Cell-Penetrating Peptides: Methods and Protocols; 2015; pp 89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Kobayashi S; Takeshima K; Park CB; Kim SC; Matsuzaki K Interactions of the Novel Anfimicrobial Peptide Buforin 2 with Lipid Bilayers: Proline as a Translocation Promoting Factor. Biochemistry 2000, 39, 8648–8654. [DOI] [PubMed] [Google Scholar]
- (287).Sims PJ; Waggoner AS; Wang CH; Hoffman JF Mechanism by Which Cyanine Dyes Measure Membrane Potential in Red Blood Cells and Phosphatidylcholine Vesicles. Biochemistry 1974, 13, 3315–3330. [DOI] [PubMed] [Google Scholar]
- (288).Matsuzaki K; Mitani Y; Akada KY; Murase O; Yoneyama S; Zasloff M; Miyajima K Mechanism of Synergism between Antimicrobial Peptides Magainin 2 and PGLa. Biochemistry 1998, 37, 15144–15153. [DOI] [PubMed] [Google Scholar]
- (289).Hristova K; Selsted ME; White SH Critical Role of Lipid Composition in Membrane Permeabilization by Rabbit Neutrophil Defensins. J. Biol. Chem 1997, 272, 24224–24233. [DOI] [PubMed] [Google Scholar]
- (290).Ostolaza H; Bartolomé B; de Zárate IO; de la Cruz F; Goñi FM Release of Lipid Vesicle Contents by the Bacterial Protein Toxin α-Haemolysin. BBA - Biomembr 1993, 1147, 81–88. [DOI] [PubMed] [Google Scholar]
- (291).Pagano RE; Martin OC; Schroit AJ; Struck DK Formation of Asymmetric Phospholipid Membranes via Spontaneous Transfer of Fluorescent Lipid Analogues between Vesicle Populations. Biochemistry 1981, 20, 4920–4927. [DOI] [PubMed] [Google Scholar]
- (292).Matsuzaki K; Murase O; Fujii N; Miyajima K An Antimicrobial Peptide, Magainin 2, Induced Rapid Flip-Flop of Phospholipids Coupled with Pore Formation and Peptide Translocation. Biochemistry 1996, 35, 11361–11368. [DOI] [PubMed] [Google Scholar]
- (293).Lebarron J; London E Effect of Lipid Composition and Amino Acid Sequence upon Transmembrane Peptide-Accelerated Lipid Transleaflet Diffusion (Flip-Flop). Biochim. Biophys. Acta - Biomembr 2016, 1858, 1812–1820. [DOI] [PubMed] [Google Scholar]
- (294).Wimley WC; White SH Determining the Membrane Topology of Peptides by Fluorescence Quenching. Biochemistry 2000, 39, 161–170. [DOI] [PubMed] [Google Scholar]
- (295).Fuselier T; Wimley WC Spontaneous Membrane Translocating Peptides: The Role of Leucine-Arginine Consensus Motifs. Biophys. J 2017, 113, 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Spinella SA; Nelson RB; Elmore DE Measuring Peptide Translocation into Large Unilamellar Vesicles. J. Vis. Exp 2012, No. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Matsuzaki K; Murase O; Fujii N; Miyajima K Translocation of a Channel-Forming Antimicrobial Peptide, Magainin 2, Across Lipid Bilayers by Forming a Pore. Biochemistry 1995, 34, 6521–6526. [DOI] [PubMed] [Google Scholar]
- (298).Marks JR; Placone J; Hristova K; Wimley WC Spontaneous Membrane-Translocating Peptides by Orthogonal High-Throughput Screening. J. Am. Chem. Soc 2011, 133, 8995–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Weinstein JN; Klausner RD; Innerarity T; Ralston E; Blumenthal R Phase Transition Release, a New Approach to the Interaction of Proteins with Lipid Vesicles Application to Lipoproteins. BBA - Biomembr 1981, 647, 270–284. [DOI] [PubMed] [Google Scholar]
- (300).Grant E; Beeler TJ; Taylor KMP; Gable K; Roseman MA Mechanism of Magainin 2a Induced Permeabilization of Phospholipid Vesicles. Biochemistry 1992, 31, 9912–9918. [DOI] [PubMed] [Google Scholar]
- (301).Ladokhin AS; Wimley WC, Hristova K; White SH Mechanism of Leakage of Contents of Membrane Vesicles Determined by Fluorescence Requenching. Methods Enzymol 1997, 278, 474–486. [DOI] [PubMed] [Google Scholar]
- (302).Du H; Samuel RL; Massiah MA; Gillmor SD The Structure and Behavior of the NA-CATH Antimicrobial Peptide with Liposomes. Biochim. Biophys. Acta - Biomembr 2015, 1848, 2394–2405. [DOI] [PubMed] [Google Scholar]
- (303).Fišer R; Konopásek I Different Modes of Membrane Permeabilization by Two RTX Toxins: HlyA from Escherichia Coli and CyaA from Bordetella Pertussis. Biochim. Biophys. Acta - Biomembr 2009, 1788, 1249–1254. [DOI] [PubMed] [Google Scholar]
- (304).Gregory SM; Cavenaugh A; Journigan V; Pokorny A; Almeida PFF A Quantitative Model for the All-or-None Permeabilization of Phospholipid Vesicles by the Antimicrobial Peptide Cecropin A. Biophys. J 2008, 94, 1667–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Leite NB; Aufderhorst-Roberts A; Palma MS; Connell SD; Neto JR; Beales PA PE and PS Lipids Synergistically Enhance Membrane Poration by a Peptide with Anticancer Properties. Biophys. J 2015, 109, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Wheaten SA; Lakshmanan A; Almeida PF Statistical Analysis of Peptide-Induced Graded and All-or-None Fluxes in Giant Vesicles. Biophys. J 2013, 105, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Hristova K; Selsted ME; White SH Interactions of Monomeric Rabbit Neutrophil Defensins with Bilayers: Comparison with Dimeric Human Defensin HNP-2. Biochemistry 1996, 35, 11888–11894. [DOI] [PubMed] [Google Scholar]
- (308).Patel H; Tscheka C; Heerklotz H Characterizing Vesicle Leakage by Fluorescence Lifetime Measurements. Soft Matter 2009, 5, 2849–2851. [Google Scholar]
- (309).Braun S; Pokorná Š; Šachl R; Hof M; Heerklotz H; Hoernke M Biomembrane Permeabilization: Statistics of Individual Leakage Events Harmonize the Interpretation of Vesicle Leakage. ACS Nano 2018, 12, 813–819. [DOI] [PubMed] [Google Scholar]
- (310).Wimley WC Energetics of Peptide and Protein Binding to Lipid Membranes; Adv Exp Med Biol 2010, 677:14–23. [DOI] [PubMed] [Google Scholar]
- (311).White SH; Wimley WC; Ladokhin AS; Hristova K Protein Folding in Membranes: Determining Energetics of Peptide-Bilayer Interactions. Methods Enzymol 1998, 295, 62–87. [DOI] [PubMed] [Google Scholar]
- (312).Rex S; Schwarz G Quantitative Studies on the Melittin-Induced Leakage Mechanism of Lipid Vesicles. Biochemistry 1998, 37, 2336–2345. [DOI] [PubMed] [Google Scholar]
- (313).Schwarz G; Zong R. ting; Popescu T Kinetics of Melittin Induced Pore Formation in the Membrane of Lipid Vesicles. BBA - Biomembr 1992, 1110, 97–104. [DOI] [PubMed] [Google Scholar]
- (314).Andersson A; Danielsson J; Gräslund A; Mäler L Kinetic Models for Peptide-Induced Leakage from Vesicles and Cells. Eur. Biophys. J 2007, 36, 621–635. [DOI] [PubMed] [Google Scholar]
- (315).Schwarz G; Arbuzova A Pore Kinetics Reflected in the Dequenching of a Lipid Vesicle Entrapped Fluorescent Dye. BBA - Biomembr 1995, 1239, 51–57. [DOI] [PubMed] [Google Scholar]
- (316).Schwarz G; Robert CH Kinetics of Pore-Mediated Release of Marker Molecules from Liposomes or Cells. Biophys. Chem 1992, 42, 291–296. [DOI] [PubMed] [Google Scholar]
- (317).Kristensen K; Henriksen JR; Andresen TL Quantification of Leakage from Large Unilamellar Lipid Vesicles by Fluorescence Correlation Spectroscopy. Biochim. Biophys. Acta - Biomembr 2014, 1838, 2994–3002. [DOI] [PubMed] [Google Scholar]
- (318).McLaurin J; Chakrabartty A Membrane Disruption by Alzheimer β-Amyloid Peptides Mediated through Specific Binding to Either Phospholipids or Gangliosides. Implications for Neurotoxicity. J. Biol. Chem 1996, 271, 26482–26489. [DOI] [PubMed] [Google Scholar]
- (319).McLaurin J; Chakrabartty A Characterization of the Interactions of Alzheimer β-Amyloid Peptides with Phospholipid Membranes. Eur. J. Biochem 1997, 245, 355–363. [DOI] [PubMed] [Google Scholar]
- (320).Tytler EM; Anantharamaiah GM; Walker DE; Mishra VK; Palgunachari MN; Segrest JP Molecular Basis for Prokaryotic Specificity of Magainin-Induced Lysis. Biochemistry 1995, 34, 4393–4401. [DOI] [PubMed] [Google Scholar]
- (321).Subbarao NK; MacDonald RC Lipid Unsaturation Influences Melittin-Induced Leakage of Vesicles. BBA - Biomembr 1994, 1189, 101–107. [DOI] [PubMed] [Google Scholar]
- (322).Breukink E; Van Kraaij C; Demel RA; Siezen RJ; Kuipers OP; De Kruijff B The C-Terminal Region of Nisin Is Responsible for the Initial Interaction of Nisin with the Target Membrane. Biochemistry 1997, 36, 6968–6976. [DOI] [PubMed] [Google Scholar]
- (323).Menestrina G; Voges KP; Jung G; Boheim G Voltage-Dependent Channel Formation by Rods of Helical Polypeptides. J. Membr. Biol 1986, 93, 111–132. [DOI] [PubMed] [Google Scholar]
- (324).Eisenberg M; Hall JE; Mead CA The Nature of the Voltage-Dependent Conductance Induced by Alamethicin in Black Lipid Membranes. J. Membr. Biol 1973, 14, 143–176. [DOI] [PubMed] [Google Scholar]
- (325).Mani R; Cady SD; Tang M; Waring AJ; Lehrer RI; Hong M Membrane-Dependent Oligomeric Structure and Pore Formation of a Beta-Hairpin Antimicrobial Peptide in Lipid Bilayers from Solid-State NMR. Proc. Natl. Acad. Sci 2006, 103, 16242–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Cho NJ; Dvory-Sobol H; Xiong A; Cho SJ; Frank CW; Glenn JS Mechanism of an Amphipathic α-Helical Peptide’s Antiviral Activity Involves Size-Dependent Virus Particle Lysis. ACS Chem. Biol 2009, 4, 1061–1067. [DOI] [PubMed] [Google Scholar]
- (327).Henderson JM; Waring AJ; Separovic F; Lee KYC Antimicrobial Peptides Share a Common Interaction Driven by Membrane Line Tension Reduction. Biophys. J 2016, 111, 2176–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Rausch JM, Marks JR, Rathinakumar R, and W. WC._beta-Sheet Pore-Forming Peptides Selected from a Rational Combinatorial Library- Mechanism of Pore Formation in Lipid Vesicles and Activity in Biological Membranes.Pdf. Biochemistry 2007, 46, 12124–12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Kenworthy AK; Hristova K; Needham D; McIntosh TJ Range and Magnitude of the Steric Pressure between Bilayers Containing Phospholipids with Covalently Attached Poly(Ethylene Glycol). Biophys. J 1995, 68, 1921–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Reeves JP; Dowben RM Formation and Properties of Thin‐walled Phospholipid Vesicles. J. Cell. Physiol 1969, 73, 49–60. [DOI] [PubMed] [Google Scholar]
- (331).Akashi KI; Miyata H; Itoh H; Kinosita K Preparation of Giant Liposomes in Physiological Conditions and Their Characterization under an Optical Microscope. Biophys. J 1996, 71, 3242–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Angelova MI; Dimitrov DS Liposome Electroformation. Faraday Discuss. Chem. Soc 1986, 81, 303–311. [Google Scholar]
- (333).Angelova MI; Soléau S; Méléard P; Faucon F; Bothorel P Preparation of Giant Vesicles by External AC Electric Fields. Kinetics and Applications. Trends Colloid Interface Sci. VI 1992, 131, 127–131. [Google Scholar]
- (334).Fenz SF; Merkel R; Sengupta K Diffusion and Intermembrane Distance: Case Study of Avidin and E-Cadherin Mediated Adhesion. Langmuir 2009, 25, 1074–1085. [DOI] [PubMed] [Google Scholar]
- (335).Ladokhin AS; Fernández-Vidal M; White SH CD Spectroscopy of Peptides and Proteins Bound to Large Unilamellar Vesicles. J. Membr. Biol 2010, 236, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Wieprecht T; Apostolov O; Beyermann M; Seelig J Membrane Binding and Pore Formation of the Antibacterial Peptide PGLa: Thermodynamic and Mechanistic Aspects. Biochemistry 2000, 39, 442–452. [DOI] [PubMed] [Google Scholar]
- (337).Stein H; Spindler S; Bonakdar N; Wang C; Sandoghdar V Production of Isolated Giant Unilamellar Vesicles under High Salt Concentrations. Front. Physiol 2017, 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Cicuta P; Keller SL; Veatch SL Diffusion of Liquid Domains in Lipid Bilayer Membranes. J. Phys. Chem. B 2007, 111, 3328–3331. [DOI] [PubMed] [Google Scholar]
- (339).Veatch SL; Keller SL Seeing Spots: Complex Phase Behavior in Simple Membranes. Biochim. Biophys. Acta - Mol. Cell Res 2005, 1746, 172–185. [DOI] [PubMed] [Google Scholar]
- (340).Veatch SL; Keller SL Separation of Liquid Phases in Giant Vesicles of Ternary Mixtures of Phospholipids and Cholesterol. Biophys. J 2003, 85, 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Kahya N; Scherfeld D; Bacia K; Schwille P Lipid Domain Formation and Dynamics in Giant Unilamellar Vesicles Explored by Fluorescence Correlation Spectroscopy. J. Struct. Biol 2004, 147, 77–89. [DOI] [PubMed] [Google Scholar]
- (342).Scherfeld D; Kahya N; Schwille P Lipid Dynamics and Domain Formation in Model Membranes Composed of Ternary Mixtures of Unsaturated and Saturated Phosphatidylcholines and Cholesterol. Biophys. J 2003, 85, 3758–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Kahya N; Scherfeld D; Bacia K; Poolman B; Schwille P Probing Lipid Mobility of Raft-Exhibiting Model Membranes by Fluorescence Correlation Spectroscopy. J. Biol. Chem 2003, 278, 28109–28115. [DOI] [PubMed] [Google Scholar]
- (344).Girard P; Pécréaux J; Lenoir G; Falson P; Rigaud JL; Bassereau P A New Method for the Reconstitution of Membrane Proteins into Giant Unilamellar Vesicles. Biophys. J 2004, 87, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Kahya N; Pécheur EI; De Boeij WP; Wiersma DA; Hoekstra D Reconstitution of Membrane Proteins into Giant Unilamellar Vesicles via Peptide-Induced Fusion. Biophys. J 2001, 81, 1464–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Solon J; Gareil O; Bassereau P; Gaudin Y Membrane Deformations Induced by the Matrix Protein of Vesicular Stomatitis Virus in a Minimal System. J. Gen. Virol 2005, 86, 3357–3363. [DOI] [PubMed] [Google Scholar]
- (347).Tanaka T; Yamazaki M Membrane Fusion of Giant Unilamellar Vesicles of Neutral Phospholipid Membranes Induced by La3+. Langmuir 2004, pp 5160–5164. [DOI] [PubMed] [Google Scholar]
- (348).Staneva G; Seigneuret M; Koumanov K; Trugnan G; Angelova MI Detergents Induce Raft-like Domains Budding and Fission from Giant Unilamellar Heterogeneous Vesicles: A Direct Microscopy Observation. Chem. Phys. Lipids 2005, 136, 55–66. [DOI] [PubMed] [Google Scholar]
- (349).Holopainen JM; Angelova MI; Kinnunen PKJ Vectorial Budding of Vesicles by Asymmetrical Enzymatic Formation of Ceramide in Giant Liposomes. Biophys. J 2000, 78, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Ambroggio EE; Separovic F; Bowie JH; Fidelio GD; Bagatolli LA Direct Visualization of Membrane Leakage Induced by the Antibiotic Peptides: Maculatin, Citropin, and Aurein. Biophys. J 2005, 89, 1874–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Mishra A; Lai GH; Schmidt NW; Sun VZ; Rodriguez AR; Tong R; Tang L; Cheng J; Deming TJ; Kamei DT; et al. Translocation of HIV TAT Peptide and Analogues Induced by Multiplexed Membrane and Cytoskeletal Interactions. Proc. Natl. Acad. Sci 2011, 108, 16883–16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Wheaten SA; Ablan FDO; Spaller BL; Trieu JM; Almeida PF Translocation of Cationic Amphipathic Peptides across the Membranes of Pure Phospholipid Giant Vesicles. J. Am. Chem. Soc 2013, 135, 16517–16525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Schön P; García-Sáez AJ; Malovrh P; Bacia K; Anderluh G; Schwille P Equinatoxin II Permeabilizing Activity Depends on the Presence of Sphingomyelin and Lipid Phase Coexistence. Biophys. J 2008, 95, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Apellániz B; Nieva JL; Schwille P; García-Sáez AJ All-or-None versus Graded: Single-Vesicle Analysis Reveals Lipid Composition Effects on Membrane Permeabilization. Biophysical Journal 2010, pp 3619–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Alam JM; Kobayashi T; Yamazaki M The Single-Giant Unilamellar Vesicle Method Reveals Lysenin-Induced Pore Formation in Lipid Membranes Containing Sphingomyelin. Biochemistry 2012, 51, 5160–5172. [DOI] [PubMed] [Google Scholar]
- (356).Islam MZ; Alam JM; Tamba Y; Karal MAS; Yamazaki M The Single GUV Method for Revealing the Functions of Antimicrobial, Pore-Forming Toxin, and Cell-Penetrating Peptides or Proteins. Phys. Chem. Chem. Phys 2014, 16, 15752–15767. [DOI] [PubMed] [Google Scholar]
- (357).Tamba Y; Yamazaki M Single Giant Unilamellar Vesicle Method Reveals Effect of Antimicrobial Peptide Magainin 2 on Membrane Permeability. Biochemistry 2005, 44, 15823–15833. [DOI] [PubMed] [Google Scholar]
- (358).Karal MAS; Alam JM; Takahashi T; Levadny V; Yamazaki M Stretch-Activated Pore of the Antimicrobial Peptide, Magainin 2. Langmuir 2015, 31, 3391–3401. [DOI] [PubMed] [Google Scholar]
- (359).Moghal MMR; Islam MZ; Sharmin S; Levadnyy V; Moniruzzaman M; Yamazaki M Continuous Detection of Entry of Cell-Penetrating Peptide Transportan 10 into Single Vesicles. Chemistry and Physics of Lipids 2018, pp 120–129. [DOI] [PubMed] [Google Scholar]
- (360).Islam MZ; Sharmin S; Levadnyy V; Alam Shibly SU; Yamazaki M Effects of Mechanical Properties of Lipid Bilayers on the Entry of Cell-Penetrating Peptides into Single Vesicles. Langmuir 2017, pp 2433–2443. [DOI] [PubMed] [Google Scholar]
- (361).Yamazaki M; Ito T Deformation and Instability in Membrane Structure of Phospholipid Vesicles Caused by Osmophobic Association: Mechanical Stress Model for the Mechanism of Poly(Ethylene Glycol)-Induced Membrane Fusion. Biochemistry 1990, 29, 1309–1314. [DOI] [PubMed] [Google Scholar]
- (362).Moniruzzaman M; Islam MZ; Sharmin S; Dohra H; Yamazaki M Entry of a Six-Residue Antimicrobial Peptide Derived from Lactoferricin B into Single Vesicles and Escherichia Coli Cells without Damaging Their Membranes. Biochemistry 2017, 56, 4419–4431. [DOI] [PubMed] [Google Scholar]
- (363).Domingues TM; Riske KA; Miranda A Revealing the Lytic Mechanism of the Antimicrobial Peptide Gomesin by Observing Giant Unilamellar Vesicles. Langmuir 2010, 26, 11077–11084. [DOI] [PubMed] [Google Scholar]
- (364).Tamba Y; Ohba S; Kubota M; Yoshioka H; Yoshioka H; Yamazaki M Single GUV Method Reveals Interaction of Tea Catechin (2)-Epigallocatechin Gallate with Lipid Membranes. Biophys. J 2007, 92, 3178–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Huang HW Action of Antimicrobial Peptides: Two-State Model. Biochemistry 2000, 39, 8347–8352. [DOI] [PubMed] [Google Scholar]
- (366).Lee CC; Sun Y; Qian S; Huang HW Transmembrane Pores Formed by Human Antimicrobial Peptide LL-37. Biophys. J 2011, 100, 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Lee M-T; Hung W-C; Chen F-Y; Huang HW Mechanism and Kinetics of Pore Formation in Membranes by Water-Soluble Amphipathic Peptides. Proc. Natl. Acad. Sci 2008, 105, 5087–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (368).Moniruzzaman M; Alam JM; Dohra H; Yamazaki M Antimicrobial Peptide Lactoferricin B-Induced Rapid Leakage of Internal Contents from Single Giant Unilamellar Vesicles. Biochemistry 2015, 54, 5802–5814. [DOI] [PubMed] [Google Scholar]
- (369).Jackman JA; Saravanan R; Zhang Y; Tabaei SR; Cho NJ Correlation between Membrane Partitioning and Functional Activity in a Single Lipid Vesicle Assay Establishes Design Guidelines for Antiviral Peptides. Small 2015, 11, 2372–2379. [DOI] [PubMed] [Google Scholar]
- (370).Allen LC; Kollman PA A Theory of Anomalous Water. Science (80-. ). 1970, 167, 1443–1454. [DOI] [PubMed] [Google Scholar]
- (371).Sackmann E; Tanaka M Supported Membranes on Soft Polymer Cushions: Fabrication, Characterization and Applications. Trends Biotechnol 2000, 18, 58–64. [DOI] [PubMed] [Google Scholar]
- (372).Knoll W; Naumann R; Friedrich M; Robertson JWF; Lösche M; Heinrich F; McGillivray DJ; Schuster B; Gufler PC; Pum D; et al. Solid Supported Lipid Membranes: New Concepts for the Biomimetic Functionalization of Solid Surfaces. Biointerphases 2008, 3, FA125–FA135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (373).Tamm LK; Mcconnell HM; Laboratoryfor S; Chemistry P Supported Phospholipid Bilayers. Biophys. J 1985, 47, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Purrucker O; Hillebrandt H; Adlkofer K; Tanaka M Deposition of Highly Resistive Lipid Bilayer on Silicon-Silicon Dioxide Electrode and Incorporation of Gramicidin Studied by Ac Impedance Spectroscopy. Electrochim. Acta 2001, 47, 791–798. [Google Scholar]
- (375).Lin J; Szymanski J; Searson PC; Hristova K Electrically Addressable, Biologically Relevant Surface-Supported Bilayers. Langmuir 2010, 26, 12054–12059. [DOI] [PubMed] [Google Scholar]
- (376).Phung T; Zhang Y; Dunlop J; Dalziel J Bilayer Lipid Membranes Supported on Teflon Filters: A Functional Environment for Ion Channels. Biosens. Bioelectron 2011, 26, 3127–3135. [DOI] [PubMed] [Google Scholar]
- (377).Seantier B; Kasemo B Influence of Mono- and Divalent Ions on the Formation of Supported Phospholipid Bilayers via Vesicle Adsorption. Langmuir 2009, 25, 5767–5772. [DOI] [PubMed] [Google Scholar]
- (378).Liu J; Conboy JC Structure of a Gel Phase Lipid Bilayer Prepared by the Langmuir-Blodgett/ Langmuir-Schaefer Method Characterized by Sum-Frequency Vibrational Spectroscopy. Langmuir 2005, 21, 9091–9097. [DOI] [PubMed] [Google Scholar]
- (379).Nikolov V; Lin J; Merzlyakov M; Hristova K; Searson PC Electrical Measurements of Bilayer Membranes Formed by Langmuir-Blodgett Deposition on Single-Crystal Silicon. Langmuir 2007, 23, 13040–13045. [DOI] [PubMed] [Google Scholar]
- (380).Vockenroth IK; Atanasova PP; Long JR; Jenkins ATA; Knoll W; Köper I Functional Incorporation of the Pore Forming Segment of AChR M2 into Tethered Bilayer Lipid Membranes. Biochim. Biophys. Acta - Biomembr 2007, 1768, 1114–1120. [DOI] [PubMed] [Google Scholar]
- (381).Morandat S; Azouzi S; Beauvais E; Mastouri A; El Kirat K Atomic Force Microscopy of Model Lipid Membranes. Anal. Bioanal. Chem 2013, 405, 1445–1461. [DOI] [PubMed] [Google Scholar]
- (382).Mecke A; Lee DK; Ramamoorthy A; Orr BG; Banaszak Holl MM Membrane Thinning Due to Antimicrobial Peptide Binding: An Atomic Force Microscopy Study of MSI-78 in Lipid Bilayers. Biophys. J 2005, 89, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Wang KF; Nagarajan R; Camesano TA Differentiating Antimicrobial Peptides Interacting with Lipid Bilayer: Molecular Signatures Derived from Quartz Crystal Microbalance with Dissipation Monitoring. Biophys. Chem 2015, 196, 53–57. [DOI] [PubMed] [Google Scholar]
- (384).Mou J; Czajkowsky DM; Shao Z Gramicidin A Aggregation in Supported Gel State Phosphatidylcholine Bilayers. Biochemistry 1996, 35, 3222–3226. [DOI] [PubMed] [Google Scholar]
- (385).Askou HJ; Jakobsen RN; Fojan P An Atomic Force Microscopy Study of the Interactions Between Indolicidin and Supported Planar Bilayers. J. Nanosci. Nanotechnol 2008, 8, 4360–4369. [DOI] [PubMed] [Google Scholar]
- (386).Matin TR; Sigdel KP; Utjesanovic M; Marsh BP; Gallazzi F; Smith VF; Kosztin I; King GM Single-Molecule Peptide-Lipid Affinity Assay Reveals Interplay between Solution Structure and Partitioning. Langmuir 2017, 33, 4057–4065. [DOI] [PubMed] [Google Scholar]
- (387).Sigdel KP; Pittman AE; Matin TR; King GM High-Resolution AFM-Based Force Spectroscopy. Methods Mol. Biol 2018, 1814, 49–62. [DOI] [PubMed] [Google Scholar]
- (388).Andersson J; Köper I Tethered and Polymer Supported Bilayer Lipid Membranes: Structure and Function. Membranes (Basel) 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Dante S; Hauß T; Steitz R; Canale C; Dencher NA Nanoscale Structural and Mechanical Effects of Beta-Amyloid (1–42) on Polymer Cushioned Membranes: A Combined Study by Neutron Reflectometry and AFM Force Spectroscopy. Biochim. Biophys. Acta - Biomembr 2011, 1808, 2646–2655. [DOI] [PubMed] [Google Scholar]
- (390).Junghans A; Champagne C; Cayot P; Loupiac C; Köper I Probing Protein-Membrane Interactions Using Solid Supported Membranes. Langmuir 2011, 27, 2709–2716. [DOI] [PubMed] [Google Scholar]
- (391).Fernandez DI; Le Brun AP; Lee TH; Bansal P; Aguilar MI; James M; Separovic F Structural Effects of the Antimicrobial Peptide Maculatin 1.1 on Supported Lipid Bilayers. Eur. Biophys. J 2013, 42, 47–59. [DOI] [PubMed] [Google Scholar]
- (392).Lin J; Motylinski J; Krauson AJ; Wimley WC; Searson PC; Hristova K Interactions of Membrane Active Peptides with Planar Supported Bilayers: An Impedance Spectroscopy Study. Langmuir 2012, 28, 6088–6096. [DOI] [PubMed] [Google Scholar]
- (393).Chang WK; Wimley WC; Searson PC; Hristova K; Merzlyakov M Characterization of Antimicrobial Peptide Activity by Electrochemical Impedance Spectroscopy. Biochim. Biophys. Acta - Biomembr 2008, 1778, 2430–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Cruz J; Mihailescu M; Wiedman G; Herman K; Searson PC; Wimley WC; Hristova K A Membrane-Translocating Peptide Penetrates into Bilayers without Significant Bilayer Perturbations. Biophys. J 2013, 104, 2419–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Lin J; Szymanski J; Chsearson P; Hristova K Effect of a Polymer Cushion on the Electrical Properties and Stability of Surface-Supported Lipid Bilayers. Langmuir 2010, 26, 3544–3548. [DOI] [PubMed] [Google Scholar]
- (396).Lin J; Merzlyakov M; Hristova K; Searson PC Impedance Spectroscopy of Bilayer Membranes on Single Crystal Silicon. Biointerphases 2008, 3, FA33–FA40. [DOI] [PubMed] [Google Scholar]
- (397).Mashaghi A; Swann M; Popplewell J; Textor M; Reimhult E Optical Anisotropy of Supported Lipid Structures Probed by Waveguide Spectroscopy and Its Application to Study of Supported Lipid Bilayer Formation Kinetics. Anal. Chem 2008, 80, 3666–3676. [DOI] [PubMed] [Google Scholar]
- (398).Balhara V; Schmidt R; Gorr SU; Dewolf C Membrane Selectivity and Biophysical Studies of the Antimicrobial Peptide GL13K. Biochim. Biophys. Acta - Biomembr 2013, 1828, 2193–2203. [DOI] [PubMed] [Google Scholar]
- (399).Papo N; Shai Y Exploring Peptide Membrane Interaction Using Surface Plasmon Resonance: Differentiation between Pore Formation versus Membrane Disruption by Lytic Peptides. Biochemistry 2003, 42, 458–466. [DOI] [PubMed] [Google Scholar]
- (400).Lee TH; Hall KN; Swann MJ; Popplewell JF; Unabia S; Park Y; Hahm KS; Aguilar MI The Membrane Insertion of Helical Antimicrobial Peptides from the N-Terminus of Helicobacter Pylori Ribosomal Protein L1. Biochim. Biophys. Acta - Biomembr 2010, 1798, 544–557. [DOI] [PubMed] [Google Scholar]
- (401).Wang KF; Nagarajan R; Mello CM; Camesano TA Characterization of Supported Lipid Bilayer Disruption by Chrysophsin-3 Using QCM-D. J. Phys. Chem. B 2011, 115, 15228–15235. [DOI] [PubMed] [Google Scholar]
- (402).Mechler A; Praporski S; Atmuri K; Boland M; Separovic F; Martin LL Specific and Selective Peptide-Membrane Interactions Revealed Using Quartz Crystal Microbalance. Biophys. J 2007, 93, 3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Shai Y ATR-FTIR Studies in Pore Forming and Membrane Induced Fusion Peptides. Biochim. Biophys. Acta 2013, 1828, 2306–2313. [DOI] [PubMed] [Google Scholar]
- (404).Newton I; Philosophiæ Naturalis Principia Mathematica 1667, Royal Society, London, UK. [Google Scholar]
- (405).Ulmschneider JP; Ulmschneider MB Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res 2018, 51, 1106–1116. [DOI] [PubMed] [Google Scholar]
- (406).Lyubartsev AP; Rabinovich AL Force Field Development for Lipid Membrane Simulations. Biochim. Biophys. Acta - Biomembr 2016, 1858, 2483–2497. [DOI] [PubMed] [Google Scholar]
- (407).Ulmschneider MB; Doux JPF; Killian JA; Smith JC; Ulmschneider JP Mechanism and Kinetics of Peptide Partitioning into Membranes from All-Atom Simulations of Thermostable Peptides. J. Am. Chem. Soc 2010, 132, 3452–3460. [DOI] [PubMed] [Google Scholar]
- (408).Ulmschneider JP; Smith JC; White SH; Ulmschneider MB In Silico Partitioning and Transmembrane Insertion of Hydrophobic Peptides under Equilibrium Conditions. J. Am. Chem. Soc 2011, 133, 15487–15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Chen CH; Wiedman G; Khan A; Ulmschneider MB Absorption and Folding of Melittin onto Lipid Bilayer Membranes via Unbiased Atomic Detail Microsecond Molecular Dynamics Simulation. Biochim. Biophys. Acta - Biomembr 2014, 1838, 2243–2249. [DOI] [PubMed] [Google Scholar]
- (410).Ulmschneider MB; Ulmschneider JP; Schiller N; Wallace BA; Von Heijne G; White SH Spontaneous Transmembrane Helix Insertion Thermodynamically Mimics Translocon-Guided Insertion. Nat. Commun 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Wang Y; Chen CH; Hu D; Ulmschneider MB; Ulmschneider JP Spontaneous Formation of Structurally Diverse Membrane Channel Architectures from a Single Antimicrobial Peptide. Nat. Commun 2016, 7, 13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Marrink SJ; Risselada HJ; Yefimov S; Tieleman DP; De Vries AH The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- (413).Marrink SJ; de Vries AH; Mark AE Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar]
- (414).Marrink SJ; Tieleman DP Perspective on the Martini Model. Chem. Soc. Rev 2013, 42, 6801–6822. [DOI] [PubMed] [Google Scholar]
- (415).Ulmschneider JP; Ulmschneider MB Sampling Efficiency in Explicit and Implicit Membrane Environments Studied by Peptide Folding Simulations. Proteins Struct. Funct. Bioinforma 2009, 75, 586–597. [DOI] [PubMed] [Google Scholar]
- (416).Panahi A; Feig M Dynamic Heterogeneous Dielectric Generalized Born (DHDGB): An Implicit Membrane Model with a Dynamically Varying Bilayer Thickness. J. Chem. Theory Comput 2013, 9, 1709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Lazaridis T Structural Determinants of Transmembrane β-Barrels. J. Chem. Theory Comput 2005, 1, 716–722. [DOI] [PubMed] [Google Scholar]
- (418).Mihajlovic M; Lazaridis T Antimicrobial Peptides Bind More Strongly to Membrane Pores. Biochim. Biophys. Acta - Biomembr 2010, 1798, 1494–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Bernèche S; Nina M; Roux B Molecular Dynamics Simulation of Melittin in a Dimyristoylphosphatidylcholine Bilayer Membrane. Biophys. J 1998, 75, 1603–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Bachar M; Becker OM Protein-Induced Membrane Disorder: A Molecular Dynamics Study of Melittin in a Dipalmitoylphosphatidylcholine Bilayer. Biophys. J 2000, 78, 1359–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Tieleman DP; Berendsen HJC; Sanson MSP Surface Binding of Alamethicin Stabilizes Its Helical Structure: Molecular Dynamics Simulations. Biophys. J 1999, 76, 3186–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Kandasamy SK; Larson RG Effect of Salt on the Interactions of Antimicrobial Peptides with Zwitterionic Lipid Bilayers. Biochim. Biophys. Acta - Biomembr 2006, 1758, 1274–1284. [DOI] [PubMed] [Google Scholar]
- (423).Leveritt JM; Pino-Angeles A; Lazaridis T The Structure of a Melittin-Stabilized Pore. Biophys. J 2015, 108, 2424–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Bennett WFD; Hong CK; Wang Y; Tieleman DP Antimicrobial Peptide Simulations and the Influence of Force Field on the Free Energy for Pore Formation in Lipid Bilayers. J. Chem. Theory Comput 2016, 12, 4524–4533. [DOI] [PubMed] [Google Scholar]
- (425).Sahoo BR; Fujiwara T Membrane Mediated Antimicrobial and Antitumor Activity of Cathelicidin 6: Structural Insights from Molecular Dynamics Simulation on Multi-Microsecond Scale. PLoS One 2016, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Ulmschneider JP; Smith JC; Ulmschneider MB; Ulrich AS; Strandberg E Reorientation and Dimerization of the Membrane-Bound Antimicrobial Peptide Pgla from Microsecond All-Atom MD Simulations. Biophys. J 2012, 103, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Perrin BS; Fu R; Cotten ML; Pastor RW Simulations of Membrane-Disrupting Peptides II: AMP Piscidin 1 Favors Surface Defects over Pores. Biophys. J 2016, 111, 1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Perrin BS; Pastor RW Simulations of Membrane-Disrupting Peptides I: Alamethicin Pore Stability and Spontaneous Insertion. Biophys. J 2016, 111, 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Andersson M; Ulmschneider JP; Ulmschneider MB; White SH Conformational States of Melittin at a Bilayer Interface. Biophys. J 2013, 104, 12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Vogel H Incorporation of Melittin into Phosphatidylcholine Bilayers. Study of Binding and Conformational Changes. FEBS Lett 1981, 134, 37–42. [DOI] [PubMed] [Google Scholar]
- (431).Vogel H; Jähnig F The Structure of Melittin in Membranes. Biophys. J 1986, 50, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Santo KP; Berkowitz ML Difference between Magainin-2 and Melittin Assemblies in Phosphatidylcholine Bilayers: Results from Coarse-Grained Simulations. J. Phys. Chem. B 2012, 116, 3021–3030. [DOI] [PubMed] [Google Scholar]
- (433).Woo HJ; Wallqvist A Spontaneous Buckling of Lipid Bilayer and Vesicle Budding Induced by Antimicrobial Peptide Magainin 2: A Coarse-Grained Simulation Study. J. Phys. Chem. B 2011, 115, 8122–8129. [DOI] [PubMed] [Google Scholar]
- (434).Thøgersen L; Schiøtt B; Vosegaard T; Nielsen NC; Tajkhorshid E Peptide Aggregation and Pore Formation in a Lipid Bilayer: A Combined Coarse-Grained and All Atom Molecular Dynamics Study. Biophys. J 2008, 95, 4337–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Bond PJ; Chze LW; Sansom MSP Coarse-Grained Molecular Dynamics Simulations of the Energetics of Helix Insertion into a Lipid Bilayer. Biochemistry 2008, 47, 11321–11331. [DOI] [PubMed] [Google Scholar]
- (436).He Y; Prieto L; Lazaridis T Modeling Peptide Binding to Anionic Membrane Pores. J. Comput. Chem 2013, 34, 1463–1475. [DOI] [PubMed] [Google Scholar]
- (437).Lipkin R; Lazaridis T Computational Studies of Peptide-Induced Membrane Pore Formation. Philosophical Transactions of the Royal Society B: Biological Sciences 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Wang Y; Zhao T; Wei D; Strandberg E; Ulrich AS; Ulmschneider JP How Reliable Are Molecular Dynamics Simulations of Membrane Active Antimicrobial Peptides? Biochim. Biophys. Acta - Biomembr 2014, 1838, 2280–2288. [DOI] [PubMed] [Google Scholar]
- (439).Almeida PF; Pokorny A Mechanisms of Antimicrobial, Cytolytic, and Cell-Penetrating Peptides: From Kinetics to Thermodynamics. Biochemistry 2009, pp 8083–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440).Dempsey CE; Butler GS Helical Structure and Orientation of Melittin in Dispersed Phospholipid Membranes from Amide Exchange Analysis in Situ. Biochemistry 1992, 31, 11973–11977. [DOI] [PubMed] [Google Scholar]
- (441).Lundin P; EL Andaloussi S; Langel Ü Toxicity Methods for CPPs. Cell-Penetrating Pept 2011, 195–205. [DOI] [PubMed] [Google Scholar]
- (442).Javadpour MM; Juban MM; Lo WCJ; Bishop SM; Alberty JB; Cowell SM; Becker CL; McLaughlin ML De Novo Antimicrobial Peptides with Low Mammalian Cell Toxicity. J. Med. Chem 1996, 39, 3107–3113. [DOI] [PubMed] [Google Scholar]
- (443).Korzeniewski C; Callewaert DM An Enzyme-Release Assay for Natural Cytotoxicity. J. Immunol. Methods 1983, 64, 313–320. [DOI] [PubMed] [Google Scholar]
- (444).Walev I; Martin E; Jonas D; Mohamadzadeh M; Muller-Klieser W; Kunz L; Bhakdi S Staphylococcal Alpha-Toxin Kills Human Keratinocytes by Permeabilizing the Plasma Membrane for Monovalent Ions. Infect. Immun 1993, 61, 4972–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Nekhotiaeva N; Elmquist A; Rajarao GK; Hallbrink M; Langel U; Good L Cell Entry and Antimicrobial Properties of Eukaryotic Cell-Penetrating Peptides. FASEB J 2004, 18, 394–396. [DOI] [PubMed] [Google Scholar]
- (446).Mosmann T Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
- (447).O’Brien J; Wilson I; Orton T; Pognan F Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem 2000, 267, 5421–5426. [DOI] [PubMed] [Google Scholar]
- (448).Musiani F; Giorgetti A Protein Aggregation and Molecular Crowding: Perspectives From Multiscale Simulations. Int. Rev. Cell Mol. Biol 2017, 329, 49–77. [DOI] [PubMed] [Google Scholar]
- (449).Neumann W; Habermann E; Hansen H Differentiation of Two Hemolytic Factors in the Bee’s Venom. Naunyn. Schmiedebergs. Arch. Exp. Pathol. Pharmakol 1953, 217, 130–143. [PubMed] [Google Scholar]
- (450).Rudenko SV; Nipot EE [Modulation of Melittin-Induced Hemolysis of Erythrocytes]. Biokhimiia 1996, 61, 2116—2124. [PubMed] [Google Scholar]
- (451).Raghuraman H; Chattopadhyay A Cholesterol Inhibits the Lytic Activity of Melittin in Erythrocytes. Chem. Phys. Lipids 2005, 134, 183–189. [DOI] [PubMed] [Google Scholar]
- (452).Maher S; Devocelle M; Ryan S; McClean S; Brayden DJ Impact of Amino Acid Replacements on in Vitro Permeation Enhancement and Cytotoxicity of the Intestinal Absorption Promoter, Melittin. Int. J. Pharm 2010, 387, 154–160. [DOI] [PubMed] [Google Scholar]
- (453).Oren Z; Shai Y Selective Lysis of Bacteria but Not Mammalian Cells by Diastereomers of Melittin: Structure-Function Study. Biochemistry 1997, 36, 1826–1835. [DOI] [PubMed] [Google Scholar]
- (454).Kim YM; Kim NH; Lee JW; Jang JS; Park YH; Park SC; Jang MK Novel Chimeric Peptide with Enhanced Cell Specificity and Anti-Inflammatory Activity. Biochem. Biophys. Res. Commun 2015, 463, 322–328. [DOI] [PubMed] [Google Scholar]
- (455).Maher S; McClean S Investigation of the Cytotoxicity of Eukaryotic and Prokaryotic Antimicrobial Peptides in Intestinal Epithelial Cells in Vitro. Biochem. Pharmacol 2006, 71, 1289–1298. [DOI] [PubMed] [Google Scholar]
- (456).Bacalum M; Radu M Cationic Antimicrobial Peptides Cytotoxicity on Mammalian Cells: An Analysis Using Therapeutic Index Integrative Concept. Int. J. Pept. Res. Ther 2015, 21, 47–55. [Google Scholar]
- (457).Liu S; Yu M; He Y; Xiao L; Wang F; Song C; Sun S; Ling C; Xu Z Melittin Prevents Liver Cancer Cell Metastasis through Inhibition of the Rac1-Dependent Pathway. Hepatology 2008, 47, 1964–1973. [DOI] [PubMed] [Google Scholar]
- (458).Russell PJ; Hewish D; Carter T; Sterling-Levis K; Ow K; Hattarki M; Doughty L; Guthrie R; Shapira D; Molloy PL; et al. Cytotoxic Properties of Immunoconjugates Containing Melittin-like Peptide 101 against Prostate Cancer: In Vitro and in Vivo Studies. Cancer Immunol. Immunother 2004, 53, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Lee G; Bae H Anti-Inflammatory Applications of Melittin, a Major Component of Bee Venom: Detailed Mechanism of Action and Adverse Effects. Molecules 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Tosteson MT; Holmes SJ; Razin M; Tosteson DC Melittin Lysis of Red Cells. J. Membr. Biol 1985, 87, 35–44. [DOI] [PubMed] [Google Scholar]
- (461).HG PJ and The L Use of Bee Venom Melittin to Assess the Topography of Membrane Vesicles Derived from Paracoccus Denitrificans. Can.J.Biochem 1980, No. 58, 996–1003. [DOI] [PubMed] [Google Scholar]
- (462).Williams JC; Bell RM Membrane Matrix Disruption by Melittin. BBA - Biomembr 1972, 288, 255–262. [DOI] [PubMed] [Google Scholar]
- (463).Imura Y; Choda N; Matsuzaki K Magainin 2 in Action: Distinct Modes of Membrane Permeabilization in Living Bacterial and Mammalian Cells. Biophysical Journal 2008, pp 5757–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Sioud M; Mobergslien A Selective Killing of Cancer Cells by Peptide-Targeted Delivery of an Anti-Microbial Peptide. Biochem. Pharmacol 2012, 84, 1123–1132. [DOI] [PubMed] [Google Scholar]
- (465).Chu DSH; Bocek MJ; Shi J; Ta A; Ngambenjawong C; Rostomily RC; Pun SH Multivalent Display of Pendant Pro-Apoptotic Peptides Increases Cytotoxic Activity. J. Control. Release 2015, 205, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Yang H; Liu S; Cai H; Wan L; Li S; Li Y; Cheng J; Lu X Chondroitin Sulfate as a Molecular Portal That Preferentially Mediates the Apoptotic Killing of Tumor Cells by Penetratin-Directed Mitochondria-Disrupting Peptides. J. Biol. Chem 2010, 285, 25666–25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Song J; Zhang Y; Zhang W; Chen J; Yang X; Ma P; Zhang B; Liu B; Ni J; Wang R Cell Penetrating Peptide TAT Can Kill Cancer Cells via Membrane Disruption after Attachment of Camptothecin. Peptides 2015, 63, 143–149. [DOI] [PubMed] [Google Scholar]
- (468).Oguiura N; Boni-Mitake M; Affonso R; Zhang G In Vitro Antibacterial and Hemolytic Activities of Crotamine, a Small Basic Myotoxin from Rattlesnake Crotalus Durissus. J. Antibiot. (Tokyo) 2011, 64, 327–331. [DOI] [PubMed] [Google Scholar]
- (469).Yang N; Stensen W; Svendsen JS; Rekdal Ø Enhanced Antitumor Activity and Selectivity of Lactoferrin-Derived Peptides. J. Pept. Res 2002, 60, 187–197. [DOI] [PubMed] [Google Scholar]
- (470).Jung S; Mysliwy J; Spudy B; Lorenzen I; Reiss K; Gelhaus C; Podschun R; Leippe M; Grötzinger J Human β-Defensin 2 and β-Defensin 3 Chimeric Peptides Reveal the Structural Basis of the Pathogen Specificity of Their Parent Molecules. Antimicrob. Agents Chemother 2011, 55, 954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Klüver E; Schulz-Maronde S; Scheid S; Meyer B; Forssmann WG; Adermann K Structure-Activity Relation of Human β-Defensin 3: Influence of Disulfide Bonds and Cysteine Substitution on Antimicrobial Activity and Cytotoxicity. Biochemistry 2005, 44, 9804–9816. [DOI] [PubMed] [Google Scholar]
- (472).Burke PJ Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends in Cancer 2017, 3, 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Li K; Lv XX; Hua F; Lin H; Sun W; Bin Cao W; Fu XM; Xie J; Yu JJ; Li Z; et al. Targeting Acute Myeloid Leukemia with a Proapoptotic Peptide Conjugated to a Toll-like Receptor 2-Mediated Cell-Penetrating Peptide. Int. J. Cancer 2013, 134, 692–702. [DOI] [PubMed] [Google Scholar]
- (474).Lin YC; Lim YF; Russo E; Schneider P; Bolliger L; Edenharter A; Altmann KH; Halin C; Hiss JA; Schneider G Multidimensional Design of Anticancer Peptides. Angew. Chemie - Int. Ed 2015, 54, 10370–10374. [DOI] [PubMed] [Google Scholar]
- (475).Valero JG; Sancey L; Kucharczak J; Guillemin Y; Gimenez D; Prudent J; Gillet G; Salgado J; Coll J-L; Aouacheria A Bax-Derived Membrane-Active Peptides Act as Potent and Direct Inducers of Apoptosis in Cancer Cells. J. Cell Sci 2011, 124, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Galluzzi L; Zamzami N; De La Motte Rouge T; Lemaire C; Brenner C; Kroemer G Methods for the Assessment of Mitochondrial Membrane Permeabilization in Apoptosis. Apoptosis 2007, 12, 803–813. [DOI] [PubMed] [Google Scholar]
- (477).Kroemer G; Galluzzi L; Brenner C Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Reveiw 2007, 99–163. [DOI] [PubMed] [Google Scholar]
- (478).Susin S. a; Lorenzo HK; Zamzami N; Marzo I; Brenner C; Larochette N; Prévost MC; Alzari PM; Kroemer G Mitochondrial Release of Caspase-2 and −9 during the Apoptotic Process. J. Exp. Med 1999, 189, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Köhler C; Gahm A; Noma T; Nakazawa A; Orrenius S; Zhivotovsky B Release of Adenylate Kinase 2 from the Mitochondrial Intermembrane Space during Apoptosis. FEBS Lett 1999, 447, 10–12. [DOI] [PubMed] [Google Scholar]
- (480).Vander Heiden MG; Chandel NS; Li XX; Schumacker PT; Colombini M; Thompson CB Outer Mitochondrial Membrane Permeability Can Regulate Coupled Respiration and Cell Survival. Proc. Natl. Acad. Sci 2000, 97, 4666–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Rostovtseva TK; Tan W; Colombini M On the Role of VDAC in Apoptosis: Fact and Fiction. J. Bioenerg. Biomembr 2005, 37, 129–142. [DOI] [PubMed] [Google Scholar]
- (482).Castedo M; Ferri K; Roumier T; Métivier D; Zamzami N; Kroemer G Quantitation of Mitochondrial Alterations Associated with Apoptosis. J. Immunol. Methods 2002, 265, 39–47. [DOI] [PubMed] [Google Scholar]
- (483).Lecoeur H; Langonné A; Baux L; Rebouillat D; Rustin P; Prévost MC; Brenner C; Edelman L; Jacotot E Real-Time Flow Cytometry Analysis of Permeability Transition in Isolated Mitochondria. Exp. Cell Res 2004, 294, 106–117. [DOI] [PubMed] [Google Scholar]
- (484).Nishihara M; Miura T; Miki T; Tanno M; Yano T; Naitoh K; Ohori K; Hotta H; Terashima Y; Shimamoto K Modulation of the Mitochondrial Permeability Transition Pore Complex in GSK-3β-Mediated Myocardial Protection. J. Mol. Cell. Cardiol 2007, 43, 564–570. [DOI] [PubMed] [Google Scholar]
- (485).García AJ; Takagi J; Boettiger D Two-Stage Activation for Integrin Binding to Surface-Adsorbed Fibronectin. Journal of Biological Chemistry 1998, pp 34710–34715. [DOI] [PubMed] [Google Scholar]
- (486).Yamamoto T; Ito M; Kageyama K; Kuwahara K; Yamashita K; Takiguchi Y; Kitamura S; Terada H; Shinohara Y Mastoparan Peptide Causes Mitochondrial Permeability Transition Not by Interacting with Specific Membrane Proteins but by Interacting with the Phospholipid Phase. FEBS J 2014, 281, 3933–3944. [DOI] [PubMed] [Google Scholar]
- (487).Jones S; Martel C; Belzacq-Casagrande AS; Brenner C; Howl J Mitoparan and Target-Selective Chimeric Analogues: Membrane Translocation and Intracellular Redistribution Induces Mitochondrial Apoptosis. Biochim. Biophys. Acta - Mol. Cell Res 2008, 1783, 849–863. [DOI] [PubMed] [Google Scholar]
- (488).Lemeshko VV; Arias M; Orduz S Mitochondria Permeabilization by a Novel Polycation Peptide BTM-P1. J. Biol. Chem 2005, 280, 15579–15586. [DOI] [PubMed] [Google Scholar]
- (489).Rodrigues CM; Solá S; Silva R; Brites D Bilirubin and Amyloid-Beta Peptide Induce Cytochrome c Release through Mitochondrial Membrane Permeabilization. Mol. Med 2000, 6, 936–946. [PMC free article] [PubMed] [Google Scholar]
- (490).Buron N; Porceddu M; Brabant M; Desgué D; Racoeur C; Lassalle M; Péchoux C; Rustin P; Jacotot E; Borgne-Sanchez A Use of Human Cancer Cell Lines Mitochondria to Explore the Mechanisms of BH3 Peptides and ABT-737-Induced Mitochondrial Membrane Permeabilization. PLoS One 2010, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Gillmeister MP; Betenbaugh MJ; Fishman PS Cellular Trafficking and Photochemical Internalization of Cell Penetrating Peptide Linked Cargo Proteins: A Dual Fluorescent Labeling Study. Bioconjug. Chem 2011, 22, 556–566. [DOI] [PubMed] [Google Scholar]
- (492).Meyer M; Philipp A; Oskuee R; Schmidt C; Wagner E Breathing Life into Polycations: Functionalization with PH-Responsive Endosomolytic Peptides and Polyethylene Glycol Enables SiRNA Delivery. J. Am. Chem. Soc 2008, 130, 3272–3273. [DOI] [PubMed] [Google Scholar]
- (493).Boeckle S; Fahrmeir J; Roedl W; Ogris M; Wagner E Melittin Analogs with High Lytic Activity at Endosomal PH Enhance Transfection with Purified Targeted PEI Polyplexes. J. Control. Release 2006, 112, 240–248. [DOI] [PubMed] [Google Scholar]
- (494).Ogris M; Carlisle RC; Bettinger T; Seymour LW Melittin Enables Efficient Vesicular Escape and Enhanced Nuclear Access of Nonviral Gene Delivery Vectors. J. Biol. Chem 2001, 276, 47550–47555. [DOI] [PubMed] [Google Scholar]
- (495).Najjar K; Erazo-Oliveras A; Pellois J-P Delivery of Proteins, Peptides or Cell-Impermeable Small Molecules into Live Cells by Incubation with the Endosomolytic Reagent DfTAT. J. Vis. Exp 2015, No. 103, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (496).Erazo-Oliveras A; Najjar K; Dayani L; Wang TY; Johnson GA; Pellois JP Protein Delivery into Live Cells by Incubation with an Endosomolytic Agent. Nat. Methods 2014, 11, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Salerno JC; Ngwa VM; Nowak SJ; Chrestensen CA; Healey AN; McMurry JL Novel Cell-Penetrating Peptide-Adaptors Effect Intracellular Delivery and Endosomal Escape of Protein Cargos. Journal of Cell Science 2016, pp 2473–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Li W; Nicol F; Szoka FC GALA: A Designed Synthetic PH-Responsive Amphipathic Peptide with Applications in Drug and Gene Delivery. Adv. Drug Deliv. Rev 2004, 56, 967–985. [DOI] [PubMed] [Google Scholar]
- (499).Wyman TB; Nicol F; Zelphati O; Scaria PV; Plank C; Szoka FC Design, Synthesis, and Characterization of a Cationic Peptide That Binds to Nucleic Acids and Permeabilizes Bilayers. Biochemistry 1997, 36, 3008–3017. [DOI] [PubMed] [Google Scholar]
- (500).Kichler A; Leborgne C; Marz J; Danos O; Bechinger B Histidine-Rich Amphipathic Peptide Antibiotics Promote Efficient Delivery of DNA into Mammalian Cells. Proc. Natl. Acad. Sci 2003, 100, 1564–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Langlet-Bertin B; Leborgne C; Scherman D; Bechinger B; Mason AJ; Kichler A Design and Evaluation of Histidine-Rich Amphipathic Peptides for SiRNA Delivery. Pharm. Res 2010, 27, 1426–1436. [DOI] [PubMed] [Google Scholar]
- (502).Kichler A; Mason AJ; Bechinger B Cationic Amphipathic Histidine-Rich Peptides for Gene Delivery. Biochim. Biophys. Acta - Biomembr 2006, 1758, 301–307. [DOI] [PubMed] [Google Scholar]
- (503).Appelbaum JS; Larochelle JR; Smith BA; Balkin DM; Holub JM; Schepartz A Arginine Topology Controls Escape of Minimally Cationic Proteins from Early Endosomes to the Cytoplasm. Chem. Biol 2012, 19, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).El-Sayed A; Futaki S; Harashima H Delivery of Macromolecules Using Arginine-Rich Cell-Penetrating Peptides: Ways to Overcome Endosomal Entrapment. AAPS J 2009, 11, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (505).Savini F; Bobone S; Roversi D; Mangoni ML; Stella L From Liposomes to Cells: Filling the Gap between Physicochemical and Microbiological Studies of the Activity and Selectivity of Host-Defense Peptides. Pept. Sci 2018, e24041. [Google Scholar]
- (506).Wiegand I; Hilpert K; Hancock REW Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (MIC) of Antimicrobial Substances. Nat. Protoc 2008, 3, 163–175. [DOI] [PubMed] [Google Scholar]
- (507).Lehrer RI; Barton A; Daher KA; Harwig SSL; Ganz T; Selsted ME Interaction of Human Defensins with Escherichia Coli. Mechanism of Bactericidal Activity. J. Clin. Invest 1989, 84, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Loh B; Grant C; Hancock RE Use of the Fluorescent Probe 1-N-Phenylnaphthylamine to Study the Interactions of Aminoglycoside Antibiotics with the Outer Membrane of Pseudomonas Aeruginosa. Antimicrob. Agents Chemother 1984, 26, 546–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (509).Subbalakshmi C; Krishnakumari V; Nagaraj R; Sitaram N Requirements for Antibacterial and Hemolytic Activities in the Bovine Neutrophil Derived 13-Residue Peptide Indolicidin. FEBS Lett 1996, 395, 48–52. [DOI] [PubMed] [Google Scholar]
- (510).Skerlavaj B; Romeo D; Gennaro R Rapid Membrane Permeabilization and Inhibition of Vital Functions of Gram-Negative Bacteria by Bactenecins. Infect. Immun 1990, 58, 3724–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Sani MA; Whitwell TC; Gehman JD; Robins-Browne RM; Pantarat N; Attard TJ; Reynolds EC; O’Brien-Simpson NM; Separovic F Maculatin 1.1 Disrupts Staphylococcus Aureus Lipid Membranes via a Pore Mechanism. Antimicrob. Agents Chemother 2013, 57, 3593–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Reimer LG; Stratton CW; Reller LB Minimum Inhibitory and Bactericidal Concentrations of 44 Antimicrobial Agents against Three Standard Control Strains in Broth with and without Human Serum. Antimicrob. Agents Chemother 1981, 19, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (513).Turner J; Cho Y; Dinh N; Alan J; Lehrer RI; Waring AJ Activities of LL-37, a Cathelin-Associated Antimicrobial Peptide of Human Neutrophils Antimicrob. Agents Chemother 1998, 42, 2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).García JRC; Jaumann F; Schulz S; Krause A; Rodríguez-Jiménez J; Forssmann U; Adermann K; Klüver E; Vogelmeier C; Becker D; et al. Identification of a Novel, Multifunctional β-Defensin (Human β-Defensin 3) with Specific Antimicrobial Activity: Its Interaction with Plasma Membranes of Xenopus Oocytes and the Induction of Macrophage Chemoattraction. Cell Tissue Res 2001, 306, 257–264. [DOI] [PubMed] [Google Scholar]
- (515).Falla TJ; Nedra Karunaratne D; Hancock REW Mode of Action of the Antimicrobial Peptide Indolicidin. J. Biol. Chem 1996, 271, 19298–19303. [DOI] [PubMed] [Google Scholar]
- (516).Van Abel RJ; Tang Y-Q; Rao VSV; Dobbs CH; Tran D; Barany G; Selsted ME Synthesis and Characterization of Indocilidin, a Tryptophan-Rich Antimicrobial Peptide from Bovine Neutrophils. Int. J. Pept. Protein Res 1995, 45, 401–409. [DOI] [PubMed] [Google Scholar]
- (517).Shin YP; Park HJ; Shin SH; Lee YS; Park S; Jo S; Lee YH; Lee IH Antimicrobial Activity of a Halocidin-Derived Peptide Resistant to Attacks by Proteases. Antimicrob. Agents Chemother 2010, 54, 2855–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (518).Schroeder BO; Ehmann D; Precht JC; Castillo PA; Küchler R; Berger J; Schaller M; Stange EF; Wehkamp J Paneth Cell α-Defensin 6 (HD-6) Is an Antimicrobial Peptide. Mucosal Immunol 2015, 8, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Moore AJ; Beazley WD; Bibby MC; Devine DA Antimicrobial Activity of Cecropins. J. Antimicrob. Chemother 1996, 37, 1077–1089. [DOI] [PubMed] [Google Scholar]
- (520).Silvestro L; Weiser JN; Axelsen PH Antibacterial and Antimembrane Activities of Cecropin A in Escherichia Coli. Antimicrob. Agents Chemother 2000, 44, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Takemura H; Kaku M; Kohno S; Hirakata Y; Tanaka H; Yoshida R; Tomono K; Koga H; Wada A; Hirayama T; et al. Evaluation of Susceptibility of Gram-Positive and -Negative Bacteria to Human Defensins by Using Radial Diffusion Assay. Antimicrob. Agents Chemother 1996, 40, 2280–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Hille B Ionic Channels in Nerve Membranes. Prog. Biophys. Mol. Biol 1970, 21, 1–32. [DOI] [PubMed] [Google Scholar]
- (523).Nicol F; Nir S; Szoka FC Effect of Cholesterol and Charge on Pore Formation in Bilayer Vesicles by a PH-Sensitive Peptide. Biophys. J 1996, 71, 3288–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Almeida PF; Pokorny A Binding and Permeabilization of Model Membranes by Amphipathic Peptides. Methods Mol. Biol 2010, 618, 155–169. [DOI] [PubMed] [Google Scholar]
- (525).Gregory SM; Pokorny A; Almeida PFF Magainin 2 Revisited: A Test of the Quantitative Model for the All-or-None Permeabilization of Phospholipid Vesicles. Biophys. J 2009, 96, 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (526).Schwarz G; Gerke H; Rizzo V; Stankowski S Incorporation Kinetics in a Membrane, Studied with the Pore-Forming Peptide Alamethicin. Biophys. J 1987, 52, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Dos Santos Cabrera MP; Alvares DS; Leite NB; Monson De Souza B; Palma MS; Riske KA; Ruggiero Neto J New Insight into the Mechanism of Action of Wasp Mastoparan Peptides: Lytic Activity and Clustering Observed with Giant Vesicles. Langmuir 2011, 27, 10805–10813. [DOI] [PubMed] [Google Scholar]
- (528).Upadhyay SK; Wang Y; Zhao T; Ulmschneider JP Insights from Micro-second Atomistic Simulations of Melittin in Thin Lipid Bilayers. J Membr Biol 2015, 248, 497–503. [DOI] [PubMed] [Google Scholar]
- (529).Wang J; Chou S; Xu L; Zhu X; Dong N; Shan A; Chen Z High Specific Selectivity and Membrane-Active Mechanism of the Synthetic Centrosymmetric α-Helical Peptides with Gly-Gly Pairs. Sci. Rep 2015, 5,15963. [DOI] [PMC free article] [PubMed] [Google Scholar]