

SUMMARY
Dysregulated growth factor receptor pathways, RNA modifications, and metabolism each promote tumor heterogeneity. Here, we demonstrate that platelet-derived growth factor (PDGF) signaling induced N6-methyladenosine (m6A) accumulation in glioblastoma (GBM) stem cells (GSCs) to regulate mitophagy. PDGF ligands stimulated early growth response 1 (EGR1) transcription to induce Methyltransferase-Like 3 (METTL3) to promote GSC proliferation and self-renewal. Targeting the PDGF-METTL3 axis inhibited mitophagy by regulating m6A modification of optineurin (OPTN). Forced OPTN expression phenocopied PDGF inhibition and OPTN levels portend longer survival of GBM patients, suggesting a tumor suppressive role for OPTN. Pharmacologic targeting of METTL3 augmented anti-tumor efficacy of PDGF receptor (PDGFR) and mitophagy inhibitors in vitro and in vivo. Collectively, we define PDGF signaling as upstream of oncogenic m6A regulation driving tumor metabolism to promote cancer stem cell maintenance, highlighting PDGF-METTL3-OPTN signaling as a GBM therapeutic target.
Keywords: glioblastoma stem cell, glioblastoma, cancer stem cell, N6-Methyladenosine (m6A), mitophagy, PDGF, PDGFRβ, METTL3, OPTN, optineurin
Graphical Abstract
IN BRIEF
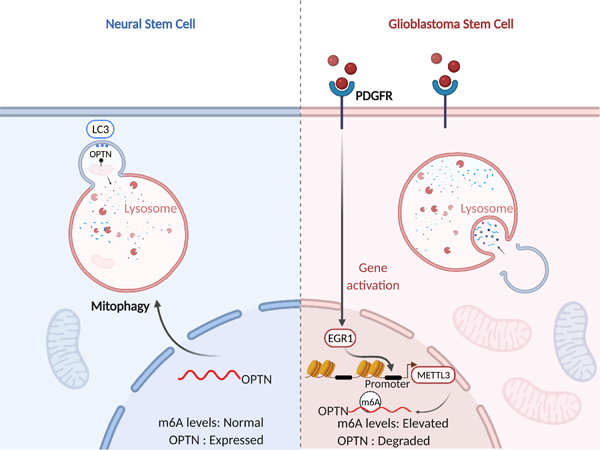
Lv et al. interrogate upstream regulation of global N6-methyladenosine (m6A) levels in glioblastoma stem cells (GSCs). PDGFR upregulates the methyltransferase, METTL3, to induce m6A modifications, including on OPTN to regulate mitophagy, revealing therapeutic combinations against GSCs.
INTRODUCTION
GBM is a highly malignant solid tumor for which conventional therapy remains palliative (Stupp et al., 2005). Although GBM ranks among the most deeply characterized cancers at the molecular level (Brennan et al., 2013; Phillips et al., 2006), precision medicine has demonstrated limited efficacy for the treatment of GBM, suggesting the need for novel molecular therapies. While genetic lesions have informed most molecular approaches, intratumoral cellular heterogeneity has presented many challenges (Nicholson and Fine, 2021). Stem-like tumor cells, GSCs, reside at the apex of a cellular hierarchy based on functionally defined capacities for self-renewal, differentiation, and tumor initiation (Chen et al., 2012). GSCs contribute to therapeutic resistance, neoangiogenesis, invasion into normal brain, and immune evasion (Gimple et al., 2019). Thus, discovering novel therapeutics targeting the molecular underpinnings of GSCs may improve GBM therapy.
N6-methyladenosine (m6A) is the most frequent transcriptional modification in eukaryotic messenger RNAs (mRNAs) (Yue et al., 2015). m6A is regulated by “writers” that are components of the methyltransferase complex [METTL3, Methyltransferase-Like 14 (METTL14), WT1 Associated Protein (WTAP), Vir-Like M6A Methyltransferase Associated (VIRMA), and RNA Binding Motif Protein 15 (RBM15)], “readers” [YTH N6-Methyladenosine RNA Binding Protein (YTHDF) and YTH Domain Containing (YTHDC) family members], and “erasers” that are demethylases [Fat mass and obesity-associated protein (FTO) and AlkB homolog 5, RNA demethylase (ALKBH5)] (Livneh et al., 2020). m6A contributes to the post-transcriptional regulation of mRNA in diverse cell types, including mRNA splicing, stability, translation, and nuclear export affecting numerous cellular process and phenotypes (Meyer and Jaffrey, 2014). In GBM, m6A and its regulators have been reported to have either oncogenic or tumor suppressive effects (Cui et al., 2017; Zhang et al., 2017). Several mRNAs modified by m6A in GBM include MYC, VEGF, FOXM1, and ADAM19, which mediate effects of m6A dysregulation (Dixit et al., 2021; Fang et al., 2021); however, the upstream regulation of m6A remains poorly understood.
A defining hallmark of GBM is activation of receptor tyrosine kinase (RTK) signaling pathways, including epidermal growth factor receptor (EGFR) or PDGFR, mediated by activating mutations, amplification, or overexpression (Furnari et al., 2015). Gliomas, especially GBM, may express all PDGF ligands and both cell surface receptors, PDGFRα and PDGFRβ (Nazarenko et al., 2012). Although inhibitors of EGFR and PDGFR have demonstrated little clinical activity against GBM (Mellinghoff et al., 2005; Raymond et al., 2008; Reardon et al., 2014), combinatorial targeting of RTKs and other molecular pathways may offer potential novel therapeutic paradigms. Considering the functional significance of m6A modification, we hypothesized that elucidating the molecular network of m6A in GBM would reveal critical dependencies in GSCs.
RESULTS
PDGF signaling increases m6A abundance in GBM
To identify upstream regulators of m6A modifications in GBM, we reasoned that leveraging an m6A regulator signature, rather than relying on the expression of a single m6A regulator, minimizes bias to detect correlations with gene expression signatures of key cellular processes. The m6A regulator signature was designed to include writers and erasers: METTL3, METTL14, WTAP, RBM15, ZC3H13, KIAA1429, METTL16, FTO, and ALKBH5. We interrogated two large glioma patient datasets, The Cancer Genome Atlas (TCGA) and the Chinese Glioma Genome Atlas (CGGA), with our m6A signature, revealing that several RTK pathways, including vascular endothelial growth factor (VEGF) receptor, EGFR, and PDGFR pathways, positively correlated with the m6A signature (Figures 1A and 1B). In contrast, enhancer-of-zeste homolog 2 (EZH2) targets negatively correlated with m6A regulators. As RTK pathways are master regulators of glioma biology, we investigated the potential of these pathways to modulate global m6A levels as a readout of m6A pathway regulation. Among the top targets, the PDGF pathway was the signature most tightly correlated with m6A regulators shared between both glioma datasets; PDGF and PDGFR signatures strongly correlated with the m6A regulator signature in each dataset (Figures S1A and S1B). The PDGFRs have differential roles in tumor heterogeneity: PDGFRα expression informs intertumoral heterogeneity with enrichment in the proneural transcriptional subgroup of gliomas (Verhaak et al., 2010); whereas PDGFRβ expression is specifically expressed in GSCs (i.e., intratumoral heterogeneity) in proneural and mesenchymal GBMs (Kim et al., 2012). Therefore, we performed correlation analysis of PDGFR signaling across the major GBM subtypes, revealing that correlations between PDGFR signaling and m6A regulation held in both proneural and mesenchymal tumors (Figure S1C). Given the relevance of PDGFRβ signaling in GSCs, we specifically considered its correlation with m6A across different subtypes, confirming a consistent association with the highest r-value in the mesenchymal subtype (Figure S1D). As mesenchymal gliomas display preferential resistance to radiation (Bhat et al., 2013), we focused on PDGFR-m6A regulation in mesenchymal GSCs. To determine if PDGF signaling induced m6A globally, we treated two patient-derived mesenchymal GSCs with multiple PDGF ligands, then measured m6A levels using m6A dot blots and immunofluorescence (IF) staining, revealing induction of m6A upon PDGF treatment, with the largest effects from the PDGF-BB ligand homodimer (Figures 1C-E). As antibody-dependent m6A detection have limitations, we developed an antibody-independent m6A detection method, leveraging Liquid Chromatography–Mass Spectrometry (LC-MS). Treatment with PDGF-BB, which is the highest-affinity ligand for PDGFRβ, increased m6A expression in GSCs (Figure 1F). Ectopic expression of either PDGFRα or PDGFRβ in GSCs promoted m6A modification with PDGFRβ inducing m6A modifications slightly more robustly than PDGFRα (Figures S1E-G). Targeting PDGFRβ expression in mesenchymal GSCs with two non-overlapping shRNAs inhibited m6A global levels (Figure 1G-J). To leverage the therapeutic potential of PDGF signaling, we treated GSCs with two PDGFR inhibitors (AG1296 and crenolanib), leading to a reduction of m6A levels (Figures S1H-J). Collectively, these data demonstrate that PDGF signaling upregulates m6A in GSCs.
Figure 1. PDGF signaling increases m6A abundance in GBM.
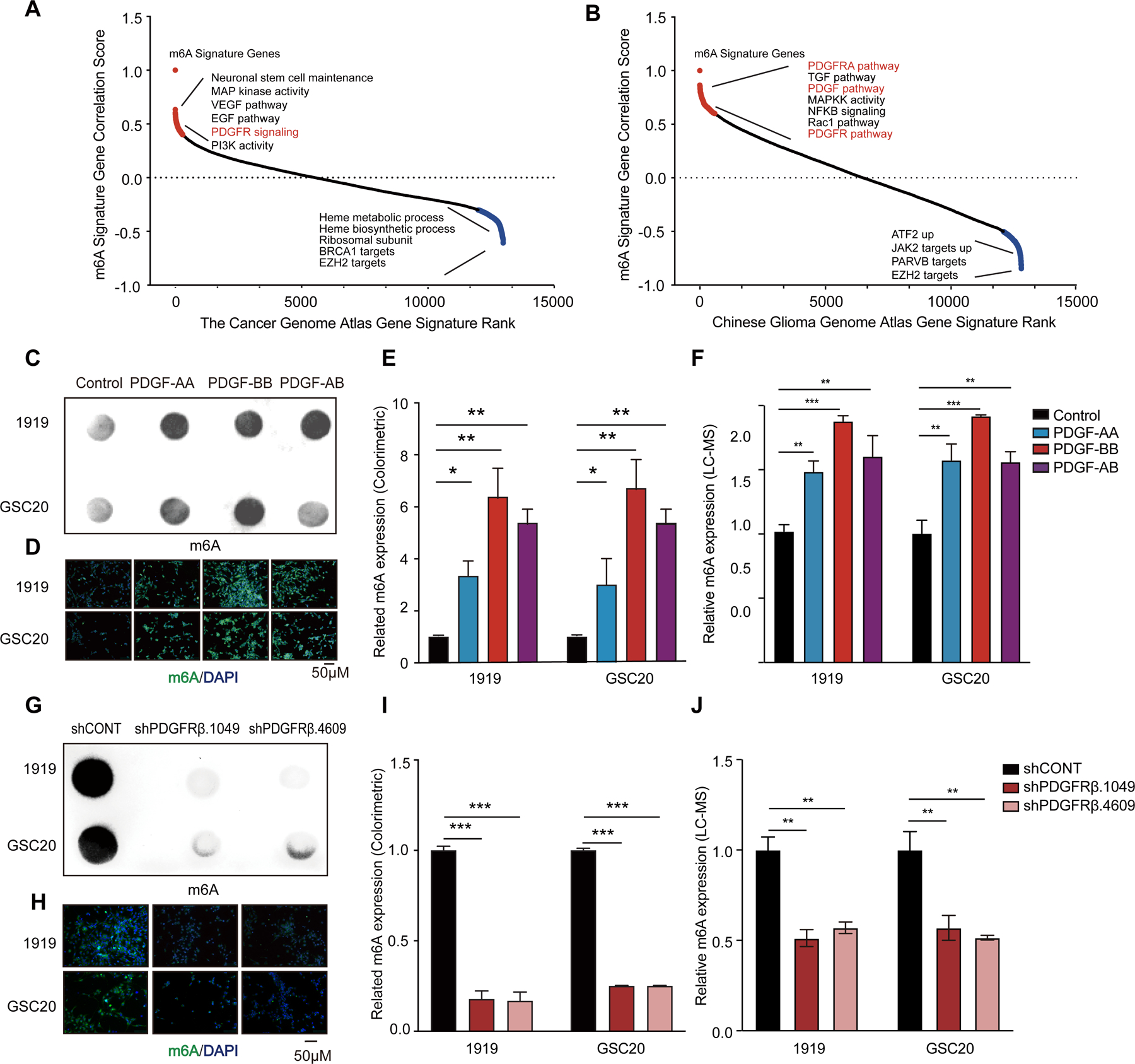
(A and B) Rank of correlation assay between m6A regulator expression with a series of gene expression signatures in TCGA (A) and CGGA (B) datasets. (C-F) PDGF treatment increases global m6A levels in GSC. Verification of the m6A abundance by dot blot (C), immunofluorescence (D), colorimetric estimation (E), and LC-MS (F). (G-J) PDGFRβ knockdown decreases global m6A levels in GSC. Verification of the m6A abundance in GSC RNA by dot blot (G), immunofluorescence (H), colorimetric estimation (I), and LC-MS (J).
As PDGF signaling induced m6A, we investigated whether m6A regulation was a general property of RTK signaling. Although VEGF receptor 2 (VEGFR2) is traditionally regarded to be localized on endothelial cells, GSCs express VEGFRs to maintain self-renewal and tumor growth (Hamerlik et al., 2012). In contrast to PDGF, VEGF ligand treatment of GSCs did not induce global change of m6A levels (Figures S1K and S1L). Neither VEGFR inhibitors nor genetic targeting of VEGFR2 reduced global m6A levels in GSCs (Figure S1M-P). Treatment with EGF did not increase global m6A levels (data not shown), but EGF serves a role in m6A regulation by stabilizing the m6A reader YTHDF2 (Fang et al., 2021). Thus, RTK pathways display selective m6A regulation.
PDGF signaling promotes GSC stemness and proliferation through METTL3
To delineate the mechanism by which PDGF signaling regulates m6A, we analyzed the correlation of PDGF/PDGFR signatures with expression of individual m6A regulators, revealing positive correlations with several m6A regulators (Figure S2A). Next, we measured the mRNA expression levels of m6A regulators after PDGF treatment, revealing altered levels of multiple regulators, predominantly methyltransferases, with consistent induction of METTL3 mRNA levels measured by real-time quantitative polymerase chain reaction (Figures 2A and 2B). PDGF also induced METTL3 protein levels (Figure 2C). In parallel to PDGF ligand effects on global m6A levels, PDGF-BB showed the strongest effect on METTL3 expression among PDGF ligands. PDGFR overexpression increased METTL3 expression, with PDGFRβ showing greater efficacy (Figure 2D). Reciprocally, PDGFR inhibitors decreased METTL3 expression (Figure 2E) and PDGFRβ knockdown by shRNA decreased METTL3 expression (Figure 2F). Thus, among the m6A molecular regulators, the PDGF-PDGFR pathway upregulates METTL3 expression.
Figure 2. PDGF signaling promotes GSC stemness and proliferation through METTL3.

(A and B) qPCR analysis of mRNA expression of m6A regulators in 1919 (A) and GSC20 (B) after PDGF treatment with 3 technical replicates per condition. Significance was determined by Student’s t-test with Holm-Sidak’s multiple test correction. ***, p < 0.001. (C) Immunoblot of METTL3 protein levels in GSCs following treatment with PDGF. (D) Immunoblot of METTL3 protein levels in GSCs following PDGFRα and PDGFRβ overexpression. (E) Immunoblot of METTL3 protein levels in GSCs following treatment with PDGFR inhibitors. (F) After PDGFRβ knockdown with shRNAs, cell lysates from 1919 and GSC20 were used to analyze METTL3 level by immunoblot. (G) Immunoblot of METTL3 WT or D395A mutation overexpression in GSCs with, or without PDGFR knockdown. (H) Sphere formation of GSCs following transduction with METTL3 WT or D395A mutation with or without PDGFR knockdown. (I) Cell viability of METTL3 WT or D395A-transduced GSCs, in the presence or absence of PDGFRβ knockdown, up to 3 days of post-transduction. **, p < 0.01. Error bars show standard deviation. (J) In vitro limiting dilution assay in GSCs following METTL3 WT or D395A-mutant overexpression with knockdown of PDGFRβ or a non-targeting shRNA (shCONT). *, p<0.05.
Contradictory effects of METTL3 on glioma growth have been reported. Cui and co-workers suggested that METTL3 serves as a tumor suppressor, as its expression inhibits GSC growth and self-renewal (Cui et al., 2017). Other studies have shown that METTL3 promotes tumorigenesis and stemness maintenance in glioma (Lan et al., 2019; Visvanathan et al., 2018). To independently interrogate the role of METTL3 in GSCs, we targeted METTL3 expression with two independent single guide RNAs (sgRNAs). Both sgRNAs silenced METTL3 expression in GSCs (Figure S2B), inhibiting GSC proliferation (Figures S2C) and sphere formation (Figures S2D). Our results suggest that METTL3 is essential for GSC growth and stemness.
Next, we interrogated the contribution of METTL3 methyltransferase activity in PDGFR-driven GBM growth. The aspartic acid at residue 395 in the methyltransferase domain is essential for METTL3 to methylate RNA (Liu et al., 2014; Wang et al., 2016), so we mutated this residual to alanine (D395A) to disrupt METTL3 methyltransferase function. We targeted PDGFRβ expression in GSCs and attempted to rescue the loss of proliferation through overexpression of wild-type, methyltransferase active METTL3 (WT) or mutant, enzyme-dead METTL3 (D395A) (Figure 2G). Overexpression of WT, but not mutant METTL3, rescued defects in sphere formation (Figure 2H), cell proliferation (Figure 2I), and clone formation (Figure 2J) upon PDGFRβ knockdown.
To establish the directionality of signaling interactions between METTL3 and PDGFRβ, we performed loss-of-function studies with PDGFRβ knockdown (Figure S2E) or gain-of-function studies with PDGFRβ overexpression in GSCs with knockdown of METTL3 using two separate shRNAs (Figure S2F). Targeting PDGFRβ decreased METTL3 mRNA levels, which was consistent with the studies above (Figure 2D). Depletion of METTL3 in PDGFRβ-expressing cells blocked the oncogenic effects due to PDGFRβ overexpression, as measured by sphere formation (Figure S2G), cell proliferation (Figures S2H), and clone formation (Figure S2I). Thus, PDGF-PDGFRβ signaling functions upstream of METTL3 in GSCs and the methyltransferase activity of METTL3 is essential in GSC maintenance.
PDGF increases METTL3 transcription via EGR1 in GSC
To investigate the mechanisms through which PDGF affects METTL3 expression, we employed a computational approach to predict transcription factors that regulate METTL3 expression with Jaspar (Fornes et al., 2020). Early growth response 1 (EGR1) was the highest scoring transcription factor (Figure 3A). METTL3 expression positively correlated with EGR1 expression in TCGA (Figure S3A) and CGGA datasets (Figure S3B). EGR1 knockdown decreased METTL3 mRNA levels (Figure 3B) and protein levels (Figures 3C and 3D). To test whether EGR1 directly regulates METTL3 expression, we constructed a METTL3-promoter-luciferase vector. Overexpression of EGR1 increased luciferase transcription (Figure 3E), supporting EGR1 binding to the METTL3 promoter. Next, we performed chromatin immunoprecipitation followed by quantitative-PCR (ChIP-qPCR) using antibodies directed against EGR1 to measure binding to the promoter of METTL3 in GSCs. Compared with the control vector, EGR1 increased the transcription from the METTL3 promoter (Figure 3F). To confirm PDGF regulation of METTL3 expression via EGR1, we investigated the role of EGR1 in GSCs. As PDGF has been previously reported to induce EGR1 mRNA expression (Sabuda-Widemann et al., 2009; Sysol et al., 2016), we reasoned that if EGR1 were downstream of PDGF signaling, then forced EGR1 expression could rescue METTL3 expression in GSCs in which PDGFRβ was targeted (Figure 3G). Consistent with the potential role of METTL3 in PDGF-induced GSC growth, EGR1 overexpression rescued GSC proliferation impaired by PDGFRβ knockdown (Figures 3H and 3I), while transduction with an empty vector failed to rescue GSC proliferation (Figure S3C) or clone formation (Figure S3D). Supporting the functional importance of EGR1, EGR1 knockdown phenocopied PDGF loss-of-function with impaired GSC proliferation (Figure S3E) and sphere formation (Figure S3F), consistent with the oncogenic effects of EGR1 (Chen et al., 2017; Sakakini et al., 2016). These data support PDGF-PDGFRβ induction of METTL3 transcription via EGR1 in GSCs.
Figure 3. PDGF increases METTL3 transcription via EGR1 in GSC.
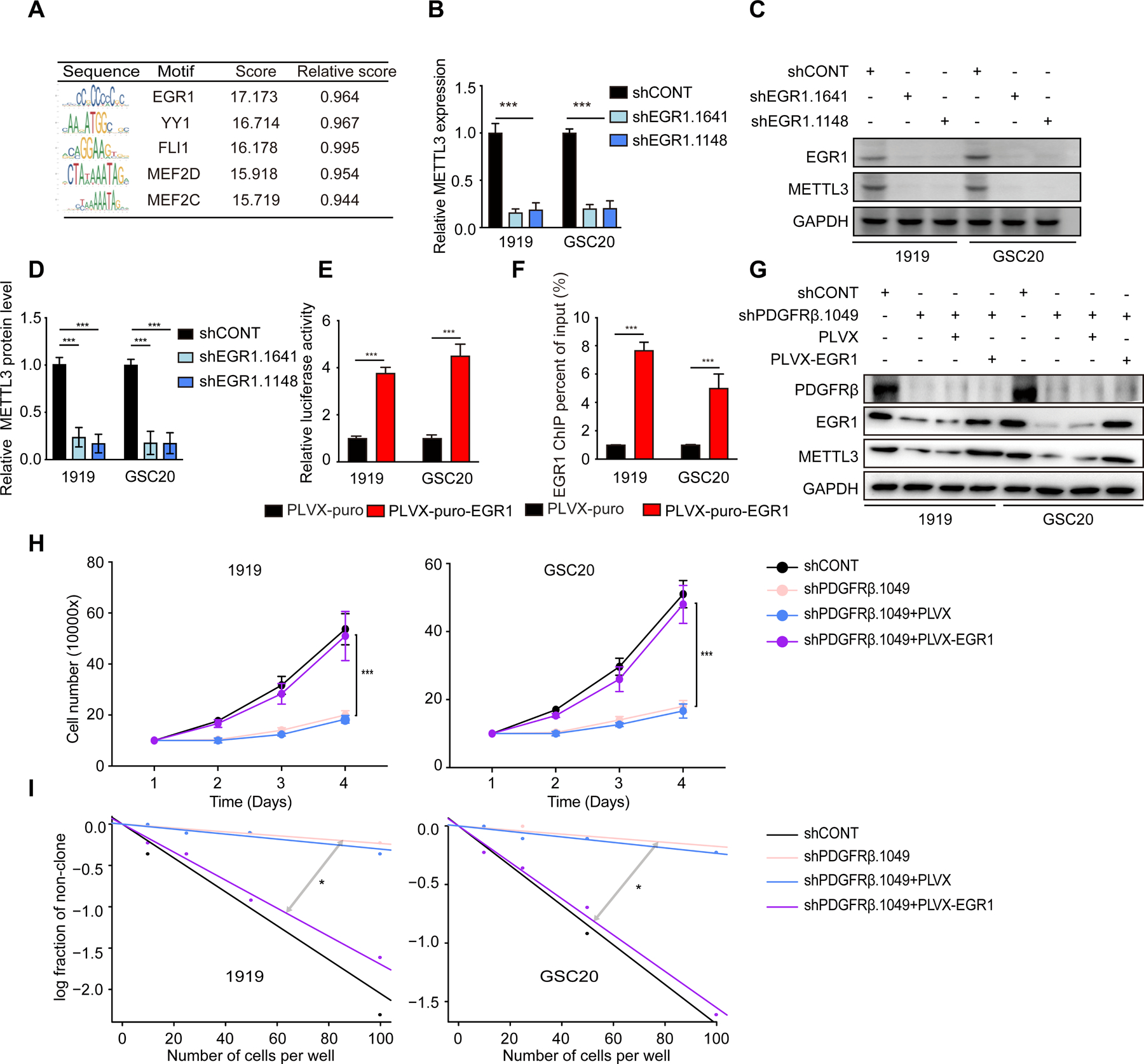
(A) Predicted binding motif of METTL3 promoter. (B) Quantitative PCR analysis of METTL3 mRNA expression following knockdown with two independent, non-overlapping shRNAs targeting EGR1 (shEGR1.1641 and shEGR1.1148) or shCONT. Three technical replicates were used for each condition. Statistical significance was assessed using a two-way ANOVA with Sidak multiple test correction, ***, p < 0.001. (C) Immunoblot of METTL3 expression following knockdown with shEGR1.1641, shEGR1.1148, or shCONT. (D) Quantification of METTL3 expression by gray value analysis. Error bars show standard deviation. ***, p < 0.001. (E) Overexpression of EGR1 activates METTL3 transcription in GSCs. GSCs transfected with METTL3 promoter-luciferase reporter with or without EGR1 overexpression. Error bars show standard deviation. Data represent two or three independent experiments with similar results. ***, p < 0.001, paired two-way Student’s t-test. (F) ChIP-qPCR of EGR1 in GSCs stable transfectant with EGR1. ***p < 0.001. (G) Immunoblot of EGR1 overexpression in GSCs with, or without PDGFR knockdown. (H) Cell viability of EGR1-transduced GSCs 1919 (left) and GSC20 (right), in the presence or absence of PDGFRΒ knockdown, up to 3 days of post-transduction. **, p < 0.01. Error bars show standard deviation. (I) In vitro limiting dilution assay in EGR1-tranduced GSCs, following PDGFRβ knockdown. *, p<0.05.
METTL3 regulates mitochondrial function via mitophagy
To investigate downstream cellular mechanisms, pathway analysis of the TCGA GBM dataset showed that METTL3 negatively correlated with a mitophagy expression signature (Figure 4A). Mitochondrial dysfunction is common in GBM and can be caused by several mechanisms, including altered mitophagy, which is important for mitochondrial quality control (Drake et al., 2017). m6A levels and autophagy are negatively correlated, with m6A modification reducing AMPK activity and subsequent autophagy inhibition (Chen et al., 2020). In cardiomyocytes, silencing METTL3 enhances autophagic flux and inhibits apoptosis (Song et al., 2019). To discover potential connections between METTL3 and mitophagy, we detected the colocalization of mitochondria to LC3 protein and lysosomes, which is a hallmark of mitophagy (Drake et al., 2017). Targeting METTL3 expression induced the mitophagy marker LC3 to the mitochondria (Figure 4B) and localization of mitochondria to lysosomes (Figures 4C–4E). To test whether METTL3 regulation of mitophagy depended on its methyltransferase activity, we rescued with shRNA-resistant METTL3 (WT) or METTL3 mutant (D395A) expression in GSCs with METTL3 knockdown. WT, but not D395A-mutant, METTL3 overexpression inhibited mitophagy in GSCs (Figures S4A and S4B). As METTL3, METTL14, and WTAP form a methyltransferase complex (Liu et al., 2014), we knocked down METTL14 to further reveal the regulation of m6A on mitophagy. METTL14 targeting increased mitochondria and lysosome co-localization in GSCs (Figures S4C-S4E). METTL3 knockdown in GSCs increased mitochondrial membrane potential (Figure 4F) and maximal respiration capacity (Figure 4G). Analysis of mitochondrial quality by staining of mitochondria and immunoblotting of mitochondria markers revealed that knocking down METTL3 decreased mitochondria quantity (Figures 4H and 4I). These data suggest that METTL3 regulates mitochondrial function through mitophagy.
Figure 4. METTL3 regulates mitochondrial function via mitophagy.
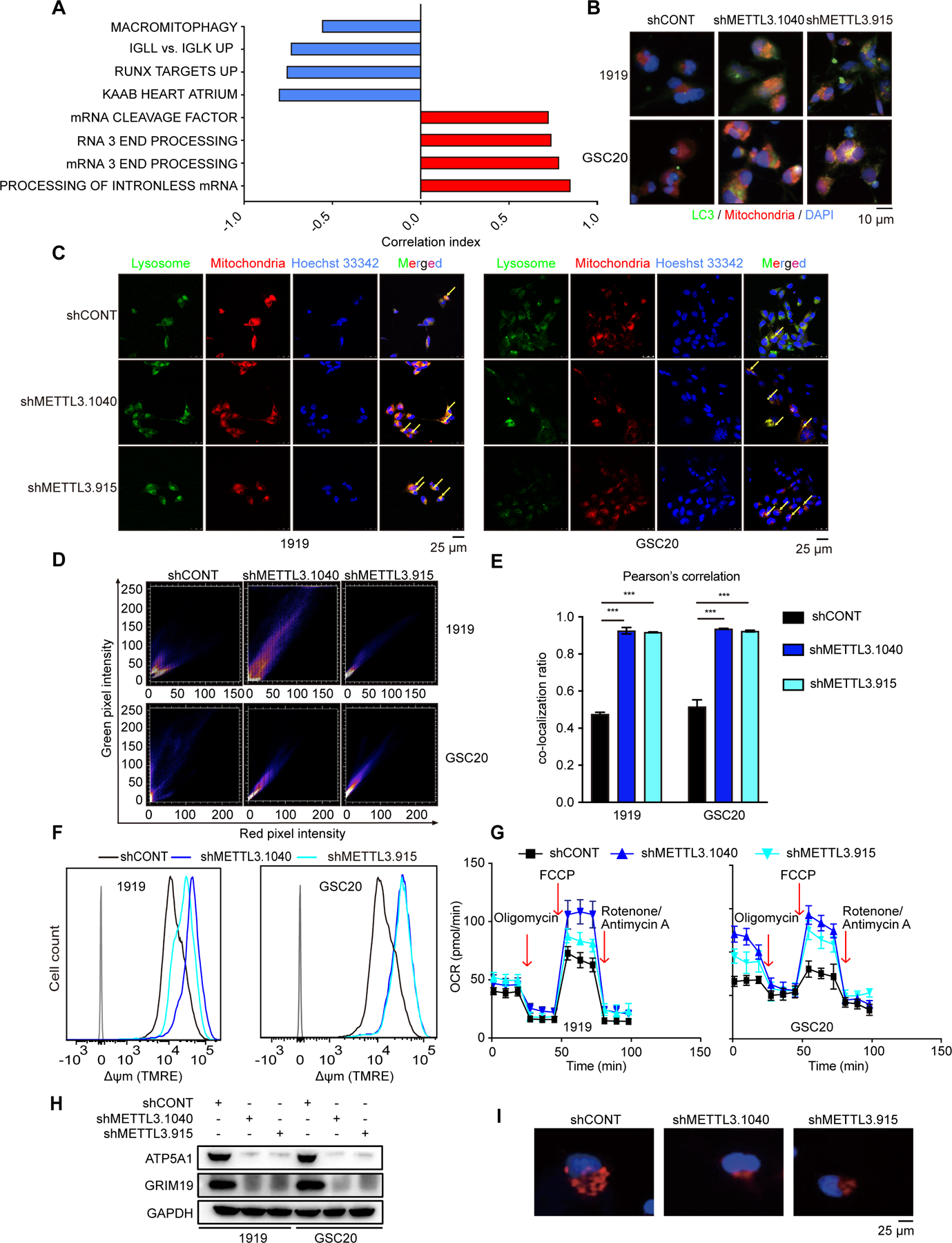
(A) Gene Ontology shows that METTL3 negatively corelates with mitophagy process with TCGA dataset. (B) Cellular colocalization and levels of LC3 and mitochondria in 1919 (above) and GSC20 (below) with METTL3 knockdown as demonstrated by immunofluorescence using LC3 antibody and mitochondrial probe. Scale bar, 10 μm. (C) Cellular colocalization of lysosome and mitochondria in 1919 (left) and GSC20 (right) after METTL3 knockdown as demonstrated by immunofluorescence using lysosomal and mitochondrial probe. Yellow arrows designate mitochondria that are being degraded. Scale bar, 25 μm. (D) Scatter plot results of mitochondria and lysosome co-localization after METTL3 knockdown. (E) Pearson’s correlation coefficient is shown in bar graph format from three independent experiments in METTL3 knockdown GSCs. Error bars show standard deviation. ***, p < 0.001. (F) METTL3 knockdown with shMETTL3.915 and shMETTL3.1040 increases 1919 (left) and GSC20 (right) cell mitochondrial membrane potential by flow cytometry analysis. (G) Representative tracings of metabolic flux. METTL3 knockdown with shMETTL3.915 and shMETTL3.1040 decreased oxygen consumption rates (OCR) in 1919 (left) and GSC20 (right). (H) Immunoblot of mitochondrial marker proteins. (I) METTL3 knockdown with shMETTL3.915 and shMETTL3.1040 decreases number of mitochondria in GSC1919.
PDGF signaling regulates autophagy in many cell types, but its role in mitophagy is not clear. Like METTL3, targeting PDGFRβ in GSCs increased mitochondria and lysosome colocalization (Figure S4F), mitochondria and LC3 colocalization (Figure S4G). Like the effects of targeting METTL3 expression, PDGFRβ knockdown increased mitochondrial membrane potential (Figure S4H) and decreased the quantity of mitochondria in GSCs as measured by IF staining and immunoblot (Figures S4I and S4J). Thus, PDGFRβ inhibits mitophagy to modulate mitochondrial function in GSCs.
METTL3 inhibits the activation of GSC mitophagy in an OPTN-dependent manner
As METTL3 regulates mitophagy, we sought the mediators of these effects. We interrogated both TCGA glioma data and our own GSC expression data (Mack et al., 2019) for correlations between METTL3 expression levels and mitophagy regulators, including PINK1, PARK2, and OPTN (Chourasia et al., 2015). METTL3 expression negatively correlated with mitophagy-related genes and positively correlated with mitochondrial biogenesis genes, such as MFN1 (Figures 5A and 5B). Targeting METTL3 expression induced expression of mitophagy-related genes (Figure S5A) (Kuppers et al., 2019). To identify the mitophagy modulator directly regulated by METTL3, we interrogated m6A-sequencing data (Cui et al., 2017). Among mitophagy-related genes, OPTN was methylated by METTL3 in GSCs (Figure S5B). Interrogating our m6A sequencing data in GSCs (Dixit et al., 2021), GSCs displayed augmented OPTN m6A modification relative to NSCs (Figure S5C). In multiple cancer types (Huang et al., 2019; Ke et al., 2017), the number of m6A peaks in the OPTN locus decreased after METTL3 knockout or knockdown (Figures S5D and S5E). To confirm that METTL3 directly methylated OPTN, we performed methylated (m6A) RNA immunoprecipitation-quantitative polymerase chain reaction (MeRIP-qPCR), demonstrating that METTL3 knockdown decreased OPTN methylation (Figure 5C). Activated OPTN is recruited to depolarized mitochondria and binds both phosphorylated ubiquitin chains and processed LC3, which is essential for mitophagy (Heo et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016). Concordantly, inhibiting METTL3 in GSCs decreased OPTN and LC3 protein levels measured by immunoblot (Figures 5D and S5F), as well as the colocalization of OPTN and LC3 as measured by IF staining (Figure 5E). To further connect METTL3 methyltransferase activity to the regulation of OPTN, we conducted complementation experiments in GSCs with METTL3 knockdown rescued with either shRNA-resistant wildtype or methyltransferase-inactivated METTL3. WT, but not D395A mutant, METTL3 rescued OPTN m6A modification (Figure 5F). Rescue with WT, but not D395A METTL3, inhibited OPTN protein expression (Figure 5G) and colocalization of LC3 with OPTN (Figure 5H). To confirm METTL3 inhibited mitophagy via OPTN, we knocked down OPTN in GSCs with METTL3 knockdown. OPTN knockdown impaired the mitophagy induced by METTL3 knockdown (Figure 5I). METTL3 promoted m6A-mediated OPTN mRNA decay (Figure S5G). Collectively, METTL3 directly inhibits OPTN expression dependent on its methyltransferase activity to inhibit mitophagy in GSCs.
Figure 5. METTL3 inhibits activation of GSC mitophagy in an OPTN-dependent manner.
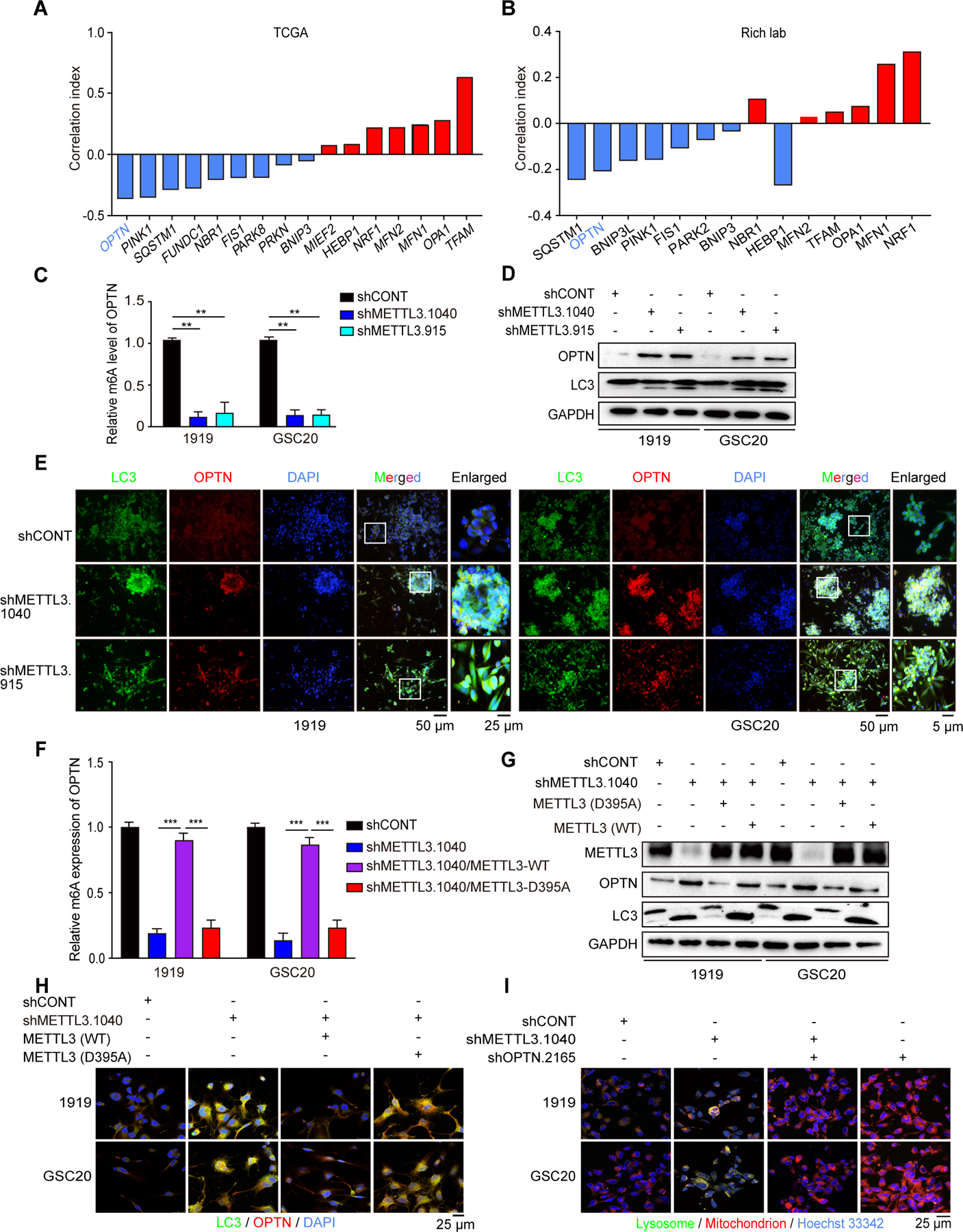
(A and B) METTL3 negatively correlates with mitophagy related genes while positively corelates with mitochondrial biogenesis related genes in TCGA data (A) and Rich lab published sequencing data (B). (C) METTL3 knockdown decreases OPTN m6A levels in GSCs. Verification of the m6A abundance in 1919 and GSC20 RNA by MeRIP-qPCR. (D) Immunoblot of OPTN and LC3 level in GSC with METTL3 knockdown. (E) Cellular colocalization and levels of LC3 and OPTN in 1919 (left) and GSC20 (right) as demonstrated by immunofluorescence using LC3 antibody and OPTN antibody. Scale bar, 50 μm. (F) Verification of the OPTN m6A abundance in shRNA-resistant METTL3 WT or D395A rescue GSCs with METTL3 knockdown. ***, p<0.001. (G) Immunoblot of LC3 and OPTN expression of shRNA-resistant METTL3 WT or D395A rescue in METTL3 knockdown GSCs. (H) Cellular colocalization and levels of LC3 and OPTN in METTL3 WT or D395A rescued GSCs as demonstrated by immunofluorescence using LC3 antibody and mitochondrial probe. Scale bar, 25 μm. (I) Cellular colocalization of lysosome and mitochondria in 1919 and GSC20 with METTL3 and OPTN knockdown as demonstrated by immunofluorescence using lysosomal and mitochondrial probe. Scale bar, 25 μm.
To determine if PDGF signaling regulates OPTN, we measured OPTN methylation after PDGF treatment, revealing increased OPTN methylation (Figures S5H and S5I). PDGF treatment also decreased OPTN expression (Figure S5J), while PDGFR inhibitors increased OPTN expression and mitophagy activation (Figure S5K). To determine if the effects of PDGF on GSC mitophagy were mediated by METTL3, we knocked down PDGFR in GSCs then attempted rescue with METTL3 expression. WT, but not D395A-mutant, METTL3 inhibited mitophagy (Figure S5L). Knockdown of OPTN impaired the mitophagy induced by PDGFRβ knockdown (Figure S5M). Thus, PDGF signaling represses mitophagy and OPTN through METTL3 methyltransferase activity.
OPTN represses tumor growth in GBM
Mitophagy-related genes can act as tumor suppressors in GBM (Agnihotri et al., 2016; Wang et al., 2017). We hypothesized that OPTN represses tumor growth in GBM, which is counteracted by the PDGF-METTL3 pathway. OPTN expression was decreased in glioma compared to normal brain (Figure 6A) and inversely correlated with tumor grade in TCGA (Figure 6B) and CGGA datasets (Figure 6C). Functionally, OPTN overexpression inhibited GSC sphere formation (Figures 6D and 6E), proliferation (Figure 6F), and clone formation (Figure 6G). In addition, knockdown of OPTN expression (Figure S6A) promoted GSC proliferation (Figure S6B) and clone formation (Figure S6C).
Figure 6. OPTN represses tumor growth in GBM.
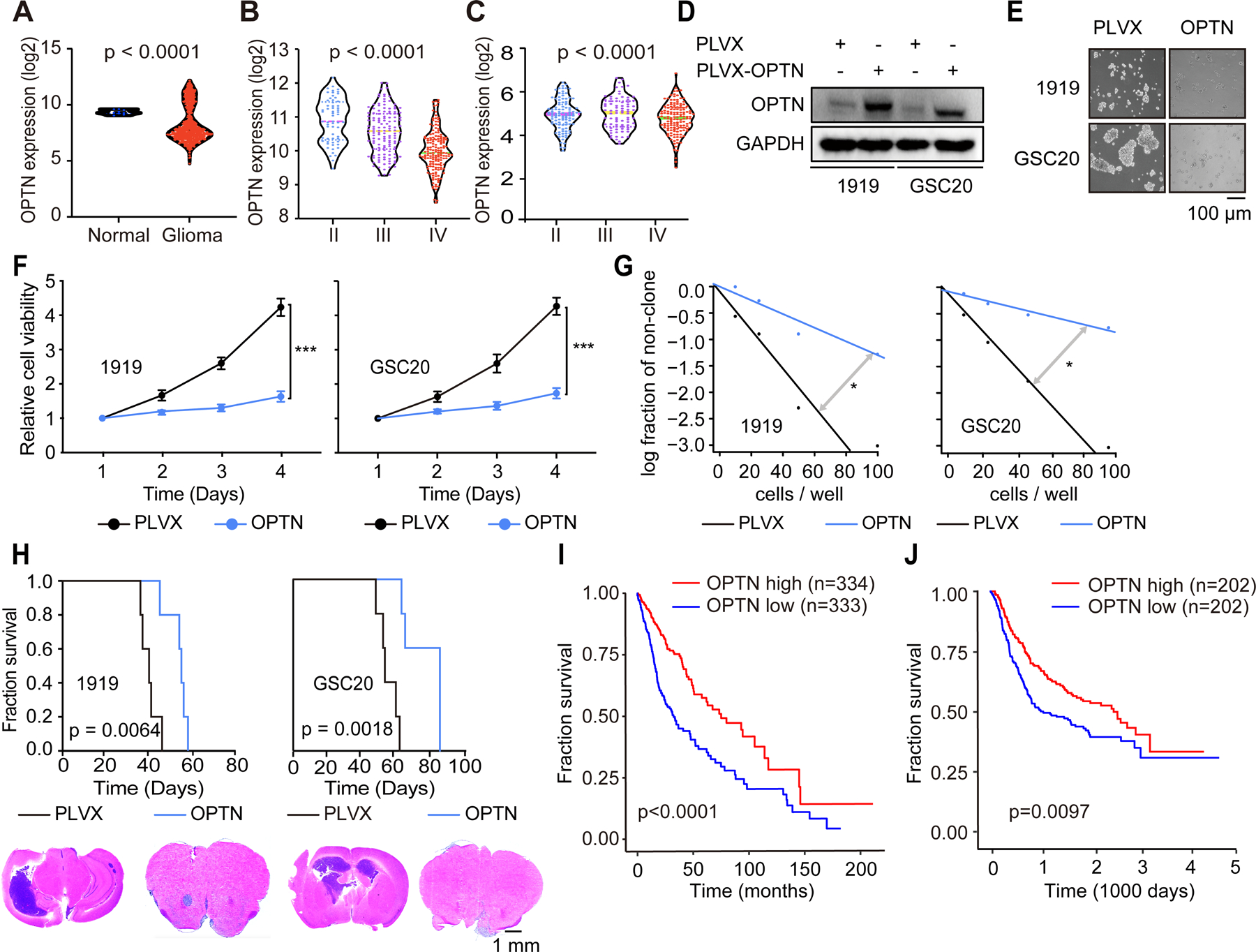
(A) Analysis of expression levels of OPTN mRNA in normal brains and GBM from TCGA dataset. P < 0.0001. (B and C) Analysis of expression levels of METTL3 mRNA across tumor grade from TCGA dataset (B) and CGGA (C). P-values were calculated by using one-way analysis of variance with Newman Keuls post-hoc test. (D) Immunoblot after OPTN overexpression in GSCs. (E) Sphere formation of GSCs with or without OPTN overexpression. Scale bar, 100 μm. (F) Cell viability of GSCs after OPTN infection, up to 3 days of post-infection. Error bars show standard deviation. ***, p < 0.001. (G) In vitro limiting dilution assay in GSCs following OPTN infection, up to 3 days of post-infection. *, p<0.05. (H) Kaplan-Meier survival curves of immunocompromised mice bearing intracranial GSC 1919 (left, above) or GSC20 (right, above) transduced with OPTN or vehicle. P = 0.0064 and p = 0.0018. Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Tumors were derived from 1919 (left, below) and GSC20 (right, below) cells transduced with OPTN or vehicle. Brains were harvested after the presentation of first neurological sign in any cohort. Scale bar, 1 mm. (I and J) Kaplan-Meier curve showing patient survival based on OPTN mRNA expression in glioma patients from the TCGA (I) and CGGA (J) dataset.
To support the potential relevance of the molecular axis mediating tumor mitophagy, we interrogated these targets in orthotopic tumor growth and association with patient prognosis. Patient-derived GSCs were transduced with OPTN or vector control then implanted into immunocompromised mice. Survival of mice bearing xenografts derived from GSCs transduced with OPTN was prolonged compared with control mice, and OPTN expression reduced in vivo tumor volume (Figure 6H).
To further connect OPTN to tumor suppressor activity in glioma, low OPTN expression informed poor glioma patient prognosis in both TCGA (Figure 6I) and CGGA (Figure 6J). We then analyzed OPTN expression in a pan-cancer dataset to validate its potential tumor suppressor effects. In most cancers, OPTN expression levels were decreased in tumors compared with normal tissues (Figure S6D). Supporting clinical relevance for OPTN, gliomas expressed OPTN at lower levels than normal brain (Figure 6A). The expression of OPTN in GSCs was decreased compared with astrocytes and NSCs (Figures S6E and S6F).
The PDGF-METTL3-OPTN axis drives tumorigenesis and informs prognosis of GBM patients
To interrogate the full role of PDGF-METTL3-OPTN axis in GSC mitophagy and self-renewal, we analyzed the expression of PDGFR, METTL3, and OPTN in glioma patients. PDGFRβ, but not PDGFRα, positively correlated with METTL3 and negatively correlated with OPTN (Figure S7A). METTL3 knockdown blocked PDGF or PDGFRβ-induced OPTN loss (Figures 7A and 7B). WT METTL3, but not D395A METTL3, inhibited OPTN expression (Figure 7C). Thus, PDGF signaling induces mitophagy and OPTN repression through METTL3 methyltransferase activity. Knockdown of OPTN expression impaired METTL3 knockdown-induced OPTN levels (Figure 7D) and rescued the inhibition of GSC proliferation upon METTL3 knockdown (Figure 7E). Similarly, knockdown of OPTN in GSCs blocked PDGFRβ knockdown-induced OPTN protein levels (Figure 7F) and rescued the anti-proliferative effects of PDGFRβ knockdown (Figures 7G). Together, loss of OPTN mediates the proliferative effects of PDGFRβ and METTL3 in GBM.
Figure 7. PDGF-METTL3-OPTN axis drives tumorigenesis and informs prognosis of GBM patients.

(A) Immunoblot of OPTN expression after METTL3 knockdown in 1919 and GSC20 treatment with or without PDGF. (B) Immunoblot of GSCs after transduction with shMETTL3.915, shMETTL3.1040, or shCONT with or without PDGFRΒ overexpression. (C) Immunoblot of METTL3 WT or D395A rescue in GSCs with or without PDGFRΒ knockdown. (D) Immunoblot of OPTN expression in METTL3 knockdown GSCs with or without OPTN knockdown. (E) Cell viability of 1919 (left) and GSC20 (right) cells with METTL3 and OPTN knockdown, up to 3 days of post-infection. Error bars show standard deviation. **, p < 0.01. (F) Immunoblot of OPTN expression in PDGFRΒ knockdown GSCs with or without OPTN knockdown. (G) Cell viability of 1919 (left) and GSC20 (right) cells with PDGFRΒ and OPTN knockdown, up to 3 days of post-infection. Error bars show standard deviation. **, p < 0.01. (H) Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 1919 GSC transduced with METTL3 and OPTN knockdown (above). Log-rank test was used for statistical analysis. shMETTL3 vs. shMETTL3/shOPTN (p = 0.0018). shOPTN vs. shMETTL3/shOPTN (p = 0.0018). Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains (below). Brains were harvested after the presentation of first neurological sign in any cohort. Scale bar, 1 mm. (I) Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 1919 GSC transduced with PDGFRΒ and OPTN knockdown (above). Log-rank test was used for statistical analysis. shMETTL3 vs. shMETTL3/shOPTN (p = 0.0044). shOPTN vs. shMETTL3/shOPTN (p = 0.0198). Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains (below). Tumors were derived from 1919 cells transduced with PDGFRΒ and OPTN knockdown. Brains were harvested after the presentation of first neurological sign in any cohort. Scale bar, 1 mm. (J) Kaplan-Meier curve showing patient survival based on PDGFRβ, METTL3 and OPTN mRNA expression in glioma patients from the TCGA dataset. Log-rank test was used for statistical analysis. (K and L) In vitro viability assay of 1919 (K) and MES20 (L) treated with indicated concentrations of UZH1, Crenolanib, and AICAR. (M and N) Synergy score analysis of 1919 (M) and MES20 (N) treated with UZH1 and crenolanib or AICAR. (O) Luciferase image of tumors from respective experimental groups. (P) Kaplan-Meier survival curves of tumor bearing mice from orthotopic intracranial xenograft implantation of 1919, treated with PBS, crenolanib, UZH1, AICAR, or their combined treatment. Log-rank test was used for statistical analysis. (**, p = 0.0018, UZH1 vs. vehicle, AICAR vs. vehicle, UZH1 vs. UZH1+AICRA, AICAR vs. UZH1+AICAR.)
Given the inhibitory effects of forced expression of OPTN on tumor growth in vivo, we sought to mimic the reduced levels of OPTN in patient tumors and determine the roles of OPTN in PDGFRβ- and METTL3-mediated tumor growth. Mice bearing orthotopic GSCs transduced with shOPTN displayed reduced survival until development of neurologic signs and increased tumor size than mice bearing GSCs transduced with a shCONT (Figures 7H and 7I). In contrast, mice bearing orthotopic GSCs transduced with an shRNA targeting either METTL3 or PDGFRβ displayed extended survival and reduced tumor size compared to shCONT, whereas mice bearing GSCs with targeted expression of OPTN and either METTL3 or PDGFRβ displayed nearly identical survival and tumor size as GSCs with shCONT (Figures 7H and 7I), suggesting that OPTN mediates effects of METTL3 and PDGFRβ in GBM growth.
We then applied these findings to expression analyses of GBM patients. High expression levels of PDGFRβ or METTL3 combined with low expression level of OPTN predicted a worse prognosis for patients with gliomas from the TCGA dataset (Figure 7J). Given the molecular connections between the nodes in PDGFRΒ-EGR1-METTL3-OPTN signaling, we interrogated the prognostic significance of the specific molecular targets in this axis, revealing negative prognosis associated with: high co-expression of PDGFRβ and METTL3; co-expression of EGR1 and METTL3; co-expression of PDGFRβ, EGR1, and METTL3 each correlating with shorter survival in patients with gliomas from both TCGA and CGGA datasets (Figures S7B and S7C).
To test if PDGF-METTL3-OPTN signaling was specific to GBM or universal in cancer, we performed correlation analysis in multiple cancer types within TCGA datasets. METTL3 expression positively correlated with PDGFRB and negatively correlated with OPTN in GBM, but not in other cancer types (Figure S7D). We measured METTL3 and OPTN expression in lung, breast, and hepatocellular carcinoma cell lines after PDGF-BB ligand treatment or PDGFR inhibitor treatment. Neither PDGF ligands nor PDGFR inhibitors treatment altered METTL3 or OPTN expression in these cell lines (Figures S7E and S7F), which may be due to low PDGFRB levels (Figure S7G). Given differential PDGFR function within GBM tumors, we measured METTL3 expression and OPTN m6A modification in paired GSCs and differentiated GBM cells (DGCs). METTL3 expression and m6A modifications were much lower in DGCs than in GSCs (Figures S7H and S7I). OPTN m6A levels were undetectable in DGCs (Figure S7J). These results support a pro-tumorigenic role for m6A regulation in which PDGF-PDGFRβ signaling induces METTL3 expression through EGR1, which promotes m6A-mediated control of OPTN in GSCs.
Pharmacologic targeting of METTL3 augments anti-tumor efficacy of PDGFR and mitophagy inhibitors
To evaluate the therapeutic potential of METTL3 in GBM, we tested a combination of drugs targeting METTL3 (UZH1), PDGFR (crenolanib), and mitophagy (AICAR). As expected, each agent displayed activity against GSCs in vitro (Figures 7K and 7L). Combination treatment of the METTL3 inhibitor and either crenolanib or AICAR was assayed for potential therapeutic synergy against GSCs; METTL3 inhibitor treatment increased the efficacy of both crenolanib and AICAR (Figures 7M and 7N). To test potential in vivo combinatorial benefit between UZH1 and crenolanib or AICAR, we treated orthotopic intracranial xenografts mice from GSCs with each small molecule compound as a single agent or in combination. METTL3 inhibition was the most potent of the single agents in reducing tumor growth in vivo (Figure 7O). Combinatorial treatment with UZH1 improved tumor control compared with single agents (Figure 7O), which translated into prolonged survival (Figure 7P). Taken together, METTL3 represents a potential therapeutic target with clinical utility for GBM patients, likely in combination.
DISCUSSION
Epitranscriptomics, the post-transcriptional regulation of mRNA species, has become an area of active interest in regulation of cell state and disease (Barbieri and Kouzarides, 2020). Like epigenetics, these modifications are dynamic, with complex regulation by methyltransferases, demethylases, and readers. As m6A is the most abundant post-transcriptional mRNA modification, it has been studied in many cancer types, including GBM and brain metastases, largely through consideration of the core regulatory molecules. Here, we sought to reveal upstream regulation of m6A in the highly malignant stem-like GBM population. PDGF signaling has been strongly connected to glioma biology with genetically engineered models derived from PDGF ligands and receptors (Jun et al., 2018). mRNA analysis revealed that PDGFRα is expressed by glial tumor cells, whereas PDGFβ mRNA is characteristic of GBM vasculature (Hermansson et al., 1988). However, PDGFRβ is expressed by GSCs in both proneural and mesenchymal tumors (Kim et al., 2012). We now describe a new role for PDGF in glioma biology through the regulation of the m6A methyltransferase METTL3 and subsequent effects on m6A levels. Downstream of this epitranscriptomic regulation, PDGF signaling promotes mitophagy in an m6A-dependent manner through OPTN regulation, revealing a previously unrecognized tumor suppressive mechanism.
PDGF induced expression of writers (METTL3, WTAP, VIRMA, and METTL8) but not erasers (FTO and ALKBH5) in GSCs. Although multiple PDGF ligands promoted m6A modification in GSCs, PDGF-BB showed the most robust effects on m6A expression, possibly because PDGF-BB binds both isoforms of PDGFRs. Aligned with a greater specificity to GSCs, PDGFRβ preferentially regulated m6A levels relative to PDGFRα. Other RTK pathways (e.g., VEGFR) correlate with m6A regulators but did not directly induce global m6A levels. However, growth factor pathways may regulate m6A deposition at specific sites or affect chromatin states. Indeed, EGFR/SRC/ERK signaling regulates YTHDF2 function in gliomas (Fang et al., 2021). Thus, it is likely that growth factor pathways collectively play complex roles in m6A regulation.
Metabolic alterations are central to the initiation and maintenance of tumors, including GBM. In our studies, we found a strong connection between PDGF induction of m6A and dysregulation of mitophagy. The PDGF pathway has been reported to regulate autophagy, but the role of PDGF in mitophagy has not been elucidated. Here, we found that PDGF inhibits OPTN levels and mitophagy in GSCs. PDGF promotes OPTN m6A modification via METTL3 to increase OPTN mRNA decay and reduce OPTN protein levels. The connection between signal transduction, epitranscriptomics, and metabolism demonstrates additional complexity in tumor biology that may be amenable to combinatorial therapeutic paradigms.
Although multiple RTKs, including EGFR and PDGFR, are important for glioma cellular growth, pharmacologic strategies targeting EGFR and/or PDGFR have largely proved fruitless in neuro-oncology (Mellinghoff et al., 2005; Raymond et al., 2008; Reardon et al., 2014). Treatment failure in GBM derives from myriad causes, including limitations in drug delivery and adaptive resistance mechanisms. Cancer stem cells from multiple tumor types display enhanced drug resistance due to quiescence, increased activation of DNA repair mechanisms, drug efflux, and increased defenses against reactive oxygen species (Kreso and Dick, 2014). Thus, targeting RTKs and m6A regulators may be vertical integrated for treatment in multiple cancers. Our results suggest that combinatorial targeting of GBM with RTK inhibitors with METTL3 methyltransferase blockade may improve tumor control. Targeting METTL3 may prove challenging for brain tumors, as direct enzymatic inhibitors will likely have to compete with cellular levels of S-adenosylmethionine, which is the methyl donor, so the inhibitor may be large in size and may not go across blood-brain barrier. In addition, METTL3 is essential to anti-tumor immunity. Finally, the function of m6A is dependent on downstream reader proteins. Collectively, these issues will require a nuanced consideration of METTL3 inhibitors in neuro-oncology.
In summary, we report a link between key oncogenic drivers and augmentation of m6A levels control mitophagy in stem-like tumor cells. The connections between cell signaling, epitranscriptomics and tumor cell metabolism further demonstrate the dynamic complexity between different levels of regulation connected to cell fate. Future studies will focus on the potential therapeutic benefit of targeting these critical nodes that can drive the collective dysregulation within tumors.
Limitations of the Study
Epitranscriptional regulation of stem-like tumor cells is certainly regulated by multiple pathways, and growth factor receptor pathways, including PDGFR, activate multiple intracellular signal transduction events. Our initial discovery efforts focused on transcriptional correlations with m6A readers and writers collectively, but important regulation occurs on single genes and on other m6A regulators, including readers. Finally, translation of preclinical therapeutic paradigms faces substantial challenges due to drug delivery that may not be fully replicated in mouse models.
STAR★METHODS
LEAD CONTACT
Further information and requests for resources and reagents should be directed to Jeremy Rich (drjeremyrich@gmail.com).
MATERIALS AVAILABILITY
Plasmids in this study are available upon a completed Material Transfer Agreement.
DATA AVAILABILITY
All data accessed from external sources and prior publications have been referenced in the text and corresponding figure legends. Additional data will be made available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
GSC derivation
GBM tissues were obtained from excess surgical resection samples from patients after review from a neuropathologist and used in accordance with an approved protocol by the Institutional Review Board at Cleveland Clinic. As previously described (Bao et al., 2006), GBM cells were derived immediately after dissociation of primary patient tumor. The 1919 GSC model was derived from a GBM from a 53-year-old male patient. The GSC20 was provided as a generous gift by Dr. Erik Sulman (Bhat et al., 2013). To minimize the incidence of cell culture-based artifacts, patient derived xenografts were produced and propagated as a renewable source of tumor cells for this study. Short Tandem Repeat (STR) analyses were performed on the tumor model for authentication on a yearly basis. Cells were frozen and stored at −196 °C (liquid nitrogen) when not being actively cultured.
Cell culture
Human HEK293T cells were purchased from American Type Culture Collection and cultured in DMEM with 10 % fetal bovine serum. The astrocyte cells were purchased from ScienCell and cultured in astrocyte medium. The nonmalignant NSC models ENSA, and NSC11 were used in this study. NSCs and GSCs were cultured in Neurobasal media supplemented with serum-free B-27 supplement, 20 ng/mL EGF and 20 ng/mL FGF, sodium pyruvate, and GlutaMAX supplement. All cells were cultured in CO2 incubator at 37 °C with 20 % oxygen and 5 % carbon dioxide. Short tandem repeat analyses were performed to authenticate the identity of each tumor model used at least annually. Mycoplasma testing with qPCR was performed on supernatants from cell culture at least annually. Cells were stored in liquid nitrogen when not being actively cultured.
Plasmid Isolation and Site-Directed Mutagenesis
Plasmids were amplified by transformation into DH5α competent cells (ThermoFisher) by following manufacturer’s instructions. Bacteria was then grown overnight in 10 mL cultures in lysogeny broth medium with respective selectable marker antibiotic in incubator shaker (ThermoFisher). Plasmids were then isolated using GeneJET Plasmid Miniprep Kit (ThermoFisher) by following manufacturer’s instructions. Plasmid concentrations were determined via absorbance at 260 nm using a Nanodrop one device (Thermo Scientific). Site directed mutagenesis was performed on pcDNA3-METTL3 by using the Quick-change Site Directed mutagenesis kit (Agilent Technologies) according to manufacturer’s instructions. Mutations were verified via commercial sanger sequencing (Eton). The primer for METTL3 (D395A) is 5’-TGTGATGGCTGCCCCACCCTGGGATATTCACAT-3’ and 5’-CCAGGGTGGGGCAGCCATCACAACTGCAAACTT-3’, the primer for shRNA-resistant METTL3 is 5’-TGTCGCAAGCTCCATTTTAGACGAATTATCAATA-3’ and 5’-TTCGTCTAAAATGGAGCTTGCGACAGG GGTCGATC-3’.
In vivo tumorigenesis
To test the roles of OPTN in vivo, intracranial xenografts were generated by implanting 50,000 human-derived GSCs into the right cerebral cortex of NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; The Jackson Laboratory) at a depth of 3.5 mm under a University of California, San Diego, Institutional Animal Care and Use Committee–approved protocol. Healthy, wild-type male or female mice of NSG background, 4 to 6 weeks old, with no prior treatment were randomly selected and used in this study for intracranial injection. Mice were maintained in 14-hour light/10-hour dark cycle by animal husbandry staff with no more than 5 mice per cage. Housing conditions and animal status were supervised by a veterinarian. Animals were monitored until neurologic symptoms which included hunched posture, gait changes, or lethargy were observed, at which point they were sacrificed. Brains were harvested and fixed in 4 % formaldehyde for 48 hours, stored in 70 % ethanol, and sectioned. Hematoxylin and eosin staining was performed on sections for histologic analysis.
Patient database and bioinformatics
For survival analyses, TCGA data was downloaded using the “TCGA2STAT” R package. CGGA data was downloaded directly from website (http://cgga.org.cn/). The Kaplan-Meier survival analysis with the log-rank test was used to assess prognostic significance of every gene in the TCGA GBM HG-U133A microarray and GBM or GBMLGG RNA-seq datasets. The processed UCSC TOIL analysis of TCGA and GTEx RNA-seq data were used to determine genes that were differentially expressed between GBM specimens and normal brain specimens (Vivian et al., 2017). The Cox Proportional Hazards model and log-rank analysis were used to assess prognostic significance of each selected gene in the TCGA GBM HG-U133A microarray dataset.
METHODS DETAILS
Retroviral packaging and infection
To obtain cell lines with stable knockdown (shPDGFRβ, shEGR1, shMETTL3, shOPTN, and shVEGFR2) or overexpression [PDGFRα/β, METTL3 (WT), METTL3 (D395A), EGR1, and OPTN] of selected molecular targets, or relevant controls (shCONT, EV), we packaged the recombinant lentivirus. Constructs were co-transfected with pxpax2 and plp/vsvg into HEK293T cells with transfection reagent LipoD293 (Signagen Laboratories). Medium were collected in 48 and 72 hours after transfection. Lentivirus is extracted from the medium with lenti-X concentrator according to the manufacturer’s video protocol.Lentivirus and 10 μg/mL polybrene were added to cell suspension for co-culture and changed the medium in 24 hours. Infected cells were selected by puromycin for 3 days. The efficiency of infection was detected by western blotting.
Cell viability
GSC proliferation and viability assays were performed after gene transfection or drug treatment. The cell proliferation assay was performed using hemocytometer. The cells were incubated with basal medium for 12 hours then seeded 1×105 cells in each well of 12-well plate. The cells were counted by trypan blue staining. The cell viability assay was performed using CellTiterGlo according to the manufacturer’s instructions. Briefly, 4 × 104 cells were seeded in one well of a 96-well plate. After treatment, equal amounts of medium of the mixture of regents A and B were added to the 96-well plates and incubated for 15 minutes with gentle shaking. The luminescence was analyzed in a plate reader according to the manufacturer’s instructions.
In vitro clone formation assays
Sphere formation by in vitro limiting dilution was performed as previously described (Gimple et al., 2019b). Briefly, decreasing numbers of cells per well (100, 50, 25, and 10) were plated into 96-well plates. The presence and number of neurospheres in each well were recorded seven days after plating. The experiments had 10 technical replicates for each group and 3 biologic replicates .Extreme limiting dilution analysis was performed using software available at http://bioinf.wehi.edu.au/software/elda, as previously described (Hu and Smyth, 2009).
Real-time quantitative polymerase chain reaction
Real-time quantitative polymerase chain reaction was performed using the TaqMan technology (Applied Biosystems) as previously described (Gimple et al., 2019b). Briefly, total RNA was extracted with direct-zol RNA kits and digested with DNase-I. Reverse transcription was performed using PrimeScript™ RT Master Mix. Real-time qPCR was performed using the standard protocol for Taqman assay with SYBR™ Green Master Mix on a C1000 Touch thermal cycler (BioRad). All data were normalized to ACTB. The primer used in this study are listed as follows: ALKBH5 5’-CGGCGAAGGCTACACTTACG-3’ and 5’-CCACCAGCTTTTGGATCACCA-3’; FTO 5’-ACTTGGCTCCCTTATCTGACC-3’ and 5’-TGTGCAGTGTGAGAAAGGCTT-3’; METTL3 5’-TTGTCTCCAACCTTCCGTAGT-3’ and 5’-CCAGATCAGAGAGGTGGTGTAG-3’; METTL14 5’-AGTGCCGACAGCATTGGTG-3’ and 5’-GGAGCAGAGGTATCATAGGAAGC-3’; METTL16 5’-CTCTGACGTGTACTCTCCTAAGG-3’ and 5’-CTCTGACGTGTACTCTCCTAAGG-3’; WTAP 5’-CTTCCCAAGAAGGTTCGATTGA-3’ and 5’-TCAGACTCTCTTAGGCCAGTTAC-3’; ZC3H13 5’-TCTGATAGCACATCCCGAAGA-3’ and 5’-TCTGATAGCACATCCCGAAGA-3’; RBM15 5’-TACACGGAGGCTACCAGTACA-3’ and 5’-TACACGGAGGCTACCAGTACA-3’; VIRAMA 5’-AAGTGCCCCTGTTTTCGATAG-3’ and 5’-ACCAGACCATCAGTATTCACCT-3’; EGR1 5’-GGTCAGTGGCCTAGTGAGC-3’ and 5’-GTGCCGCTGAGTAAATGGGA-3’; OPTN 5’-CCAAACCTGGACACGTTTACC-3’ and 5’-CCTCAAATCTCCCTTTCATGGC-3’; ACTB 5’-GTCTGCCTTGGTAGTGGATAATG-3’ and 5’-TCGAGGACGCCCTATCATGG-3’.
Western blot
Briefly, cells were lysed with RIPA lysate buffer(50 mM Tris (pH 7.4),150 mM NaCl,1 % Triton X-100,1 % sodium deoxycholate,0.1 % SDS,1 mM sodium orthovanadate,10 mM sodium fluoride,and 0.5 M EDTA (pH 8.0))on ice after being treated under varying conditions as indicated. Lysates were separated by SDS-PAGE and transferred to PVDF membrane (IPVH00010, MilliporeSigma). The PVDF membrane was blocked with 5 % non-fat dry milk dissolved in TBST, then incubated with the primary antibody of METTL3 (1:1000), METTL14 (1:1000), WTAP (1:1000), METTL16 (1:1000), RBM15 (1:1000), ZC3H13 (1:1000), VIRMA (1:1000), ALKBH5 (1:1000), FTO (1:1000), PDGFRα (1:1000), PDGFRβ (1:1000), EGR1 (1:1000), OPTN (1:1000), LC3 (1:3000), ATP5A1 (1:1000), GRIM19 (1:1000), GAPDH (1:10000) for 2 hours at room temperature or 4 °C overnight. PVDF membranes were washed with TBST for 1 hour and incubated with anti-rabbit lgG HRP-linked antibody (1:1000) or anti-mouse lgG HRP-linked antibody (1:1000) for another 2 hours. Images were captured by a BIO-RAD workstation and gray values were analyzed using image-analysis software AlphaImager 2200 (ProteinSimple, San Jose, CA, USA).
Immunofluorescence analysis
Cells were plated on 13-mm round glass coverslips in a 24-well plate with Matrigel in a humidified chamber at 37 °C and 5 % CO2 for 12 hours. For lysosome studies, cells were cultured with mitochondrial dye, lysoView dye and Hoechst 33342 for 30 minutes. For immunofluorescence studies, cells were washed with chilled PBS (pH 7.2) twice and fixed in 4% paraformaldehyde for 20 minutes on ice and then permeabilized with 0.2% Triton X-100 for 10 minutes. After PBS washes, cells were blocked with goat serum for 30 minutes, then incubated overnight with selected antibodies: LC3 (1:100), OPTN (1:50), m6A (1:100) or fluorogenic dyes MitoTracker. Cells were incubated with Alexa Fluor 488 goat anti-rabbit IgG or 568 goat anti-mouse lgG for 2 hours. After washing, the cells were treated with a 1:1000 solution of DAPI in PBS for 5 minutes, washed and blocked with PVF medium. Images were acquired with fluorescence microscope or Zeiss LSM710 confocal microscope (40x oil objective).
mRNA Purification
75 μg of the total RNA was heated to 65 °C for 2 minutes to disrupt secondary structures then the sample was placed on ice. Total RNA was added to the prepared Dynabeads™/binding buffer suspension. The combination was mixed thoroughly and rotated on a roller for 5 minutes at room temperature to allow the mRNA to anneal to the oligo (dT)25 on the beads. The tube was placed on a magnet then the supernatant was removed. The mRNA-bead complex was washed twice with 200 μL washing buffer. The magnet was used to remove all traces of supernatant between each washing. 10–20 μL of 10 mM Tris-HCl, pH 7.5 was added to elute the mRNA. The sample was heated at 65 °C for 2 minutes and the tube was placed immediately on the magnet. Eluted mRNA was transferred to a new RNase-free tube and the concentration detected by nanodrop one.
m6A dot blot
RNA from selected cells (100 ng for gain-of-function studies and 500 ng for loss-of-function studies) was denatured at 95 °C for 3 minutes then chilled on ice immediately for 2 minutes. The mRNA was then dropped directly onto Hybond-N+ membranes. Spotted mRNA was crosslinked to membranes in a UV crosslinker twice using the following mode (1,200 microjoules [x100]; 25–50 sec). Membranes were washed in 10 mL of PBS for 5 minutes at room temperature with gentle shaking to wash off the unbound mRNA. The membranes were then incubated in 5 % 10 mL of goat serum for 1 hour at room temperature with gentle shaking. After blocking, the membranes were incubated with anti-m6A antibody (1:500) overnight at 4 °C with gentle shaking. Membranes were washed three times for 5 minutes each in 10 mL of PBS with gentle shaking. Membranes were then incubated with goat anti-rabbit IgG-HRP (1:1000) for 1 hour at room temperature with gentle shaking. Membranes were imaged after being washed four times for 10 minutes each in 10 mL of PBS.
Quantitative analysis of m6A
The levels of m6A were quantified with EpiQuik™ m6A RNA Methylation Quantification Kit (Epigentek) according to the manufacturer’s protocol. Briefly, 80 μl of binding solution was added to strip each well before use. 2 μL of negative control (NC), 2 μl of diluted positive control (PC), or 200 ng of sample RNA was then plated into the wells and the solution mixed by gently tilting from side-to-side. The strip plate was sealed with plate sealing and incubated at 37 °C for 90 minutes. Each well was washed with 150 μL of diluted wash buffer by pipetting for a total of three washes. 50 μL of diluted capture antibody was added to each well, then incubated at room temperature for 60 minutes. The capture antibody solution was removed from each well and each well was washed with 150 μL of diluted wash buffer three times. 50 μL of diluted detection antibody was added to each well, then incubated at room temperature for 30 minutes. Each well was washed with 150 μL of wash buffer four times; 50 μL of enhancer solution was added to each well, then incubated at room temperature for 30 minuets. Each well was washed with 150 μL of wash buffer five times. 100 μL of developer solution was added to each well and incubated at room temperature for 10 minutes away from light. 100 μL of stop solution was added to each well to stop enzyme reaction and absorbance was measured on a microplate reader at 450 nm. m6A quantity was calculated according to the following formula:
Where S is the amount of input sample RNA in ng. P is the amount of input positive control (PC) in ng.
Quantitative analysis of m6A RNA modification by LC-MS
Liquid Chromatography–Mass Spectrometry (LC-MS) assay was developed based on a previous protocol (Mathur et al., 2021). Briefly, total RNA was extracted with direct-zol RNA kits and purified with Invitrogen™ Dynabeads™ mRNA Purification Kit. The purified mRNA was digested with nucleoside digestion mix (M0649S, NEB). The N6-Methyladenosine and adenosine was detected by Q Exactive Mass Spectrometers (Thermo Fisher).
Promoter reporter and dual luciferase assays
The METTL3 promoter including −2000bp to +100bp of the METTL3 transcription start site was amplified by Q5 DNA polymerase using PCR with primers: 5′-TAGCCCGGGCTCGAGGATCAAATTACATGGAATTCTTTCAAAAC-3′ and 5′-CGGAATGCCAAGCTTAGCGGAGACCGTTCCTCGCACC-3′ and genomic DNA template. The promoter was then inserted into pGL3-Basic vector (E1751, Promega). For normalization of transfection efficiency, the pRL-TK-Renilla luciferase (E2241, Promega) reporter plasmid was added to each transfection. The activities of firefly luciferase and Renilla luciferase were quantified using a dual-specific luciferase assay kit according to manufacturer’s instruction (E1910, Promega).
ChIP-qPCR
ChIP was performed using the Chromatin Immunoprecipitation Kit (Millipore-Upstate) according to the manufacturer’s instructions. For cross linking and cell lysis, cells were fixed in 1 % formaldehyde by adding 550 μL of 37 % formaldehyde to 20 mL of growth media and gently swirled to mix. Cells were incubated at room temperature for 10 minutes. 2 mL of 10 x glycine was added to each dish to quench the unreacted formaldehyde and the mixture was incubated at room temperature for 5 minutes. Culture media was aspirated, removing as much medium as possible without disturbing the cells. Cells were washed with PBS containing protease inhibitors and collected using a sterile cell scraper. Cells were pelleted at 800 x g at 4 °C for 5 minutes, supernatant was removed, and cells were lysed in cell lysis buffer. Cell lysates were incubated on ice for 15 minutes, vortexed briefly, and centrifuged at 800 x g at 4 °C for 5 minutes. The supernatant was carefully removed, and lysates were resuspended in 0.5 mL of nuclear lysis buffer. Chromatin was sonicated to shear DNA and quality was assessed using agarose gel analysis. 1 % of each sample were stored as an “input” fraction. Immunoprecipitation of cross inked chromatin was performed by incubating with the selected antibody and protein A/G magnetic beads according to the manufacturer’s instructions. Immunoprecipitated chromatin was isolated using magnetic separation and washed. Chromatin was de-crosslinked in elution buffer and incubated at 62 °C for 2 hours with shaking followed by a 10-minute incubation at 95 °C. Beads were removed using magnetic separation and DNA was purified using spin columns. After elution, the pure DNA was used for qRT-PCR. Primers of METTL3: 5′-AAGCCCTCTTAGTGTCGGAAC-3′ and 5′-GGTGGTCCCCAATCTGAGGT −3′.
MeRIP-qPCR
The MeRIP was performed with the Magna MeRIP™ m6A Kit (17–1094, Millipore-Sigma) according to the manufacturer’s instructions. Briefly, RNA was fragmented by repeated heating at 94 °C for 5 minutes and vortexing. RNA was precipitated in ethanol with incubation at −80 °C overnight, allowed to air dry, and resuspended in RNAase-free water. Magna ChIP Protein A/G Magnetic Beads (Part # CS203152) were used for immunoprecipitation with 10 μg of the anti m6A antibody (Part # MABE1006) and 300 μg of total RNA for each reaction. 30 μg of total RNA was preserved as “RNA input”. Immunoprecipitation tubes were incubated for 2 hours at 4 °C. Beads were washed and RNA was eluted and purified using a RNeasy mini kit. The RNA levels were analyzed by quantitative RT-PCR. Primers of OPTN: 5′-GCTGAGTCCGCACATAGAAG-3′ and 5′-TCCTGTGGAAAAGTCACTCCA-3′.
Metabolic profiling
Mitochondrial function was detected Seahorse XF Cell Mito Stress Test Kit (Agilent) according to the manufacturer’s protocol. Assembled sensor cartridge and utility plate were placed in a non-CO2 37 °C incubator overnight with 200 μL water in each well. Water was discarded and each well of the utility plate was filled with 200 μL of pre-warmed Seahorse XF Calibrant. The assembled sensor cartridge with utility plate was placed in a non-CO2 37 °C incubator for 45 – 60 minutes prior to loading drug ports of the sensor cartridge. 3×104 GSCs were plated on a Seahorse plate coated with Matrigel (Corning) in XF DMEM medium. Oxygen consumption was recorded by a Seahorse XFe96 Analyzer (Agilent) using the mito stress test protocol, in which cells were sequentially perturbed by 2 μM oligomycin, 0.5 μM CCCP and 0.5 μM Rot/AA.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses are described in the figure legends. For TCGA GBM vs. normal brain RNAseq calculations, four-way ANOVA controlling for sex, age, and ethnicity with the Benjamini and Hochberg false discovery rate (FDR) method was used for statistical analysis. For survival analyses, Cox proportional hazards and log-rank analyses were used. For qPCR analyses, Student’s t-test was used to assess statistical significance, when appropriate. Two-way repeated measures ANOVA was used for statistical analysis with Dunnett’s multiple hypothesis test correction. For proliferation assays and limiting dilution assay, two-way repeated measures ANOVA was used for statistical analysis with Dunnett’s multiple hypothesis test correction.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
Antibodies
| ||
| METTL3 | Proteintech | Cat#15073–1-AP; RRID:AB_2142033 |
| ALKBH5 | Proteintech | Cat# 16837–1-AP; RRID:AB_2242665 |
| METTL14 | Proteintech | Cat#26158–1-AP; RRID:AB_2800447 |
| METTL16 | Proteintech | Cat#19924–1-AP; RRID:AB_10639364 |
| FTO | Proteintech | Cat#27226–1-AP; RRID:AB_2880809 |
| ZC3H13 | Thermo Fisher Scientific | Cat# PA5–36515; RRID:AB_2553564 |
| WTAP | Proteintech | Cat# 60188–1-Ig; RRID:AB_10859484 |
| RBM15 | Abcam | Cat# ab70549; RRID:AB_2253428 |
| VIRMA | Proteintech | Cat# 25712–1-AP; RRID:AB_2880204 |
| PDGFRα | Proteintech | Cat#13449–1-AP; RRID:AB_2162644 |
| PDGFRβ | Proteintech | Cat# 60045–1-Ig; RRID:AB_2162343 |
| EGR1 | Proteintech | Cat#22008–1-AP; RRID:AB_11182923 |
| OPTN | Proteintech | Cat#60293–1-Ig; RRID:AB_2881408 |
| LC3 | Abcam | Cat#ab229327 |
| ATP5A1 | Proteintech | Cat#66037–1-Ig; RRID:AB_11044196 |
| GRIM19 | Proteintech | Cat#10986–1-AP; RRID:AB_2150609 |
| GAPDH | Proteintech | Cat#HRP-60004; RRID:AB_2737588 |
| m6A Antibody | Sigma | Cat#ABE572-I-100UG; RRID:AB_2892214 |
| HRP-linked anti-Rabbit lgG | Cell Signaling Technology | Cat#7074S; RRID:AB_2099233 |
| HRP-linked anti-Mouse lgG | Cell Signaling Technology | Cat#7076S; RRID:AB_209923 |
| Goat anti-Rabbit IgG, Fluor 488 | ThermoFisher | Cat#A32731TR; RRID:AB_2866491 |
| Goat anti-Mouse lgG, Fluor 568 | ThermoFisher | Cat#A-11031; RRID: AB_144696 |
|
Chemicals, Peptides and Recombinant Proteins | ||
| Recombinant Human EGF | R&D Systems | Cat#236-EG-01M |
| Recombinant Human FGF | R&D Systems | Cat#4114-TC-01M |
| Recombinant Human PDGF-AA | R&D Systems | Cat#221-AA |
| Recombinant Human PDGF-BB | R&D Systems | Cat#220-BB |
| Recombinant Human PDGF-AB | R&D Systems | Cat#222-AB |
| B27 supplement | ThermoFisher | Cat#17504044 |
| Neurobasal media | ThermoFisher | Cat#12348017 |
| Sodium pyruvate | ThermoFisher | Cat#11360070 |
| GlutaMAX | ThermoFisher | Cat#35050061 |
| Astrocyte medium | ScienCell | Cat#1801 |
| DMEM | ThermoFisher | Cat#11995073 |
| Fetal bovine serum | ThermoFisher | Cat#26140079 |
| Matrigel | Corning | Cat#354277 |
| AG1296 | Selleck | Cat#S8024 |
| Crenolanib | Selleck | Cat#S2730 |
| UZH1 | MCE | Cat#HY-134673 |
| AICAR | Cayman | Cat#10010241 |
| Foretinib | Selleck | Cat#S1111 |
| Fruquintinib | Selleck | Cat#S5667 |
| Lenti-X concentrator | Takara | Cat#631232 |
| Polybrene | Sigma | Cat#TR-1003 |
| MitoTracker | ThermoFisher | Cat#M7512 |
| Mitochondrial dye | Biotium | Cat#70075 |
| LysoView dye | Biotium | Cat#70067 |
| DAPI | Sigma | Cat#10236276001 |
| Hoechest 33342 | ThermoFisher | Cat#H3570 |
| TMRE | Cayman Chem | Cat#21426 |
| LipoD293 | SignaGen Laboratories | Cat#SL100668 |
|
Critical commercial assays | ||
| CellTiterGlo | Promega | Cat#G7571 |
| EpiQuik™ m6A RNA Methylation Quantification Kit | Epigentek | Cat#P-9005-96 |
| Chromatin Immunoprecipitation Kit | Millipore sigma | Cat#17-295 |
| Cell Mito Stress Test Kit | Agilent | Cat#103708-100 |
| Direct-zol RNA kits | ZYMO research | Cat#R2061 |
| Dual-Luciferase® Reporter Assay | Promega | Cat#E1910 |
| Magna MeRIP™ m6A Kit | Millipore-sigma | Cat#17-1094 |
| SYBR™ Green Master Mix | ThermoFisher | Cat#A25742 |
| PrimeScript™ RT Master Mix | Takara | Cat#RR036A |
| GeneJET Plasmid Miniprep Kit | ThermoFisher | Cat#K0502 |
| Site-Directed Mutagenesis Kit | Agilent | Cat#200523 |
| mRNA Purification Kit | ThermoFisher | Cat#61006 |
| Nucleoside Digestion Mix | NEB | Cat#M0649S |
| SDS-PAGE | Invitrogen | Cat#NP0323BOX |
|
Experimental models: Cell lines | ||
| Cell line | 1919 | Patient derived |
| Cell line | GSC20 | Patient derived |
| Cell line | Human Astrocyte | ScienCell, Cat#1850 |
| Cell line | NSC11 | Alstem, Cat#hNSC11 |
| Cell line | ENSA | Millipore Sigma, Cat# SCC003 |
| Cell line | HEK293T | ATCC, CRL-11268 |
| competent cell | DH5α | ThermoFishere, Cat#18265017 |
|
Experimental models: organisms/strains | ||
| NSG mice | The Jackson Laboratory | NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ |
|
Recombinant DNA | ||
| pGL3-Basic | Promega | Cat#E1751 |
| pRL-TK-Renilla luciferase | Promega | Cat#E2241 |
| pcDNA3/Flag-METTL3 | Addgene | Cat#53739 |
| PLVX-puro-METTL3 (WT) | Clontech | METTL3, WT |
| PLVX-puro-METTL3 (D395A) | Clontech | METTL3, D395A |
| pHAGE-PDGFRA | Addgene | Cat#116769 |
| pHAGE-PDGFRB | Addgene | Cat#116770 |
| pMXs-hs-EGR1 | Addgene | Cat#52724 |
| PLVX-puro-EGR1 | Clontech | EGR1 |
| PLVX-puro-OPTN | Clontech | OPTN |
| shPDGFRβ.1049 | Sigma | TRCN0000002000 |
| shPDGFRβ.4533 | Sigma | TRCN0000001999 |
| shMETTL3.1040 | Sigma | TRCN0000034718 |
| shMETTL3.915 | Sigma | TRCN0000034717 |
| shEGR1.1148 | Sigma | TRCN0000013837 |
| shEGR1.1641 | Sigma | TRCN0000013835 |
| ShVEGFR2.852 | Sigma | TRCN0000001688 |
| ShVEGFR2.1536 | Sigma | TRCN0000001689 |
| shOPTN.2165 | Sigma | TRCN0000083743 |
| shOPTN.1115 | Sigma | TRCN0000083744 |
|
Software and algorithms | ||
| Endnote X9 | https://endnote.com | Clarivate |
| Graphpad Prism v6.01 | https://www.graphpad.com | Graphpad Prism |
| Adobe illustrator | https://www.adobe.com | Adobe |
| Image J | https://imagej.nih.gov/ij | National Institutes of Health |
| FLOWJO | https://www.flowjo.com | BD |
| CGGA | http://cgga.org.cn | CGGA |
| JASPAR | http://jaspar.genereg.net | JASPAR |
| GEPIA | http://gepia.cancer-pku.cn | GEPIA |
HIGHLIGHTS.
PDGF signaling upregulates m6A levels in GBM through transcriptional control of METTL3
PDGF-METTL3 represses OPTN by m6A modification to inhibit GSC mitophagy
OPTN inhibits GBM tumor growth in vitro and in vivo
Pharmacologic METTL3 inhibition synergizes with PDGFR and mitophagy inhibitors
ACKNOWLEDGMENTS
We appreciate the UCSD Histology Core, and University of Pittsburgh Mass Spectrometry Lab for their work on histologic experiments, and Liquid Chromatography–Mass Spectrometry assays, respectively. We thank the scientists from Agilent for providing technical support. We thank Dr. Lu He and Dr. Guoliang Chai from Howard Hughes Medical Institute, University of California for assistance in promoter-luciferase system construction. We thank Matt Halbert from UPMC John G. Rangos Sr. Research Center for helping edit the manuscript. This work was supported by NIH grants (CA197718, CA238662, and NS103434 to J.N. Rich; NS115831 to S. Agnihotri; CA217066 to B.C. Prager; CA217065 to R.C. Gimple). K. Yang was supported by the Computational Genomic Epidemiology of Cancer (CoGEC) Program at Case Comprehensive Cancer Center (T32CA094186), Young Investigator Award in Glioblastoma from ASCO Conquer Cancer Foundation, and RSNA Research Resident Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, Chalil A, Smith CA, Krumholtz SL, Mackenzie D, et al. (2016). PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res 76, 4708–4719. 10.1158/0008-5472.CAN-15-3079. [DOI] [PubMed] [Google Scholar]
- Barbieri I, and Kouzarides T (2020). Role of RNA modifications in cancer. Nat Rev Cancer 20, 303–322. 10.1038/s41568-020-0253-2. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DG, Zhu B, Lv SQ, Zhu H, Tang J, Huang C, Li Q, Zhou P, Wang DL, and Li GH (2017). Inhibition of EGR1 inhibits glioma proliferation by targeting CCND1 promoter. J Exp Clin Cancer Res 36, 186. 10.1186/s13046-017-0656-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, McKay RM, and Parada LF (2012). Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 149, 36–47. 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang J, Xu D, Xiang Z, Ding J, Yang X, Li D, and Han X (2020). m(6)A mRNA methylation regulates testosterone synthesis through modulating autophagy in Leydig cells. Autophagy, 1–19. 10.1080/15548627.2020.1720431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Boland ML, and Macleod KF (2015). Mitophagy and cancer. Cancer Metab 3, 4. 10.1186/s40170-015-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG, et al. (2017). m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep 18, 2622–2634. 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, Qiu Z, Kidwell RL, Kim LJY, Xie Q, et al. (2021). The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer Discov 11, 480–499. 10.1158/2159-8290.CD-20-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake LE, Springer MZ, Poole LP, Kim CJ, and Macleod KF (2017). Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol 47, 110–124. 10.1016/j.semcancer.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Chen X, Zhang S, Shi H, Ye Y, Zou Z, Li P, Guo Q, Ma L, He C, and Huang S (2021). EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat Commun 12, 177. 10.1038/s41467-020-20379-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92. 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Cloughesy TF, Cavenee WK, and Mischel PS (2015). Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer 15, 302–310. 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimple RC, Bhargava S, Dixit D, and Rich JN (2019). Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev 33, 591–609. 10.1101/gad.324301.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M, Moudry P, Bartek J Jr., Fischer W, Lukas J, et al. (2012). Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J. Exp. Med 209, 507–520. 10.1084/jem.20111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, and Harper JW (2015). The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell 60, 7–20. 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermansson M, Nister M, Betsholtz C, Heldin CH, Westermark B, and Funa K (1988). Endothelial cell hyperplasia in human glioblastoma: coexpression of mRNA for platelet-derived growth factor (PDGF) B chain and PDGF receptor suggests autocrine growth stimulation. Proc Natl Acad Sci U S A 85, 7748–7752. 10.1073/pnas.85.20.7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F, et al. (2019). Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567, 414–419. 10.1038/s41586-019-1016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HJ, Appleman VA, Wu HJ, Rose CM, Pineda JJ, Yeo AT, Delcuze B, Lee C, Gyuris A, Zhu H, et al. (2018). A PDGFRα-driven mouse model of glioblastoma reveals a stathmin1-mediated mechanism of sensitivity to vinblastine. Nat Commun 9, 3116. 10.1038/s41467-018-05036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, Hanna JH, Black DL, Darnell JE Jr., and Darnell RB (2017). m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31, 990–1006. 10.1101/gad.301036.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, Serwanski D, Sloan AE, Weil RJ, Lee J, et al. (2012). Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev 26, 1247–1262. 10.1101/gad.193565.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, and Dick JE (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291. 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Kuppers DA, Arora S, Lim Y, Lim AR, Carter LM, Corrin PD, Plaisier CL, Basom R, Delrow JJ, Wang S, et al. (2019). N(6)-methyladenosine mRNA marking promotes selective translation of regulons required for human erythropoiesis. Nat Commun 10, 4596. 10.1038/s41467-019-12518-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, and Liu T (2019). The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res 79, 1285–1292. 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10, 93–95. 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, and Dominissini D (2020). The m(6)A epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci 21, 36–51. 10.1038/s41583-019-0244-z. [DOI] [PubMed] [Google Scholar]
- Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, Bernatchez JA, Zhu Z, Gimple RC, Kim LJY, et al. (2019). Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J. Exp. Med 216, 1071–1090. 10.1084/jem.20190196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, et al. (2005). Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353, 2012–2024. 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- Meyer KD, and Jaffrey SR (2014). The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15, 313–326. 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, and Holzbaur EL (2016). Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A 113, E3349–3358. 10.1073/pnas.1523810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarenko I, Hede SM, He X, Hedren A, Thompson J, Lindstrom MS, and Nister M (2012). PDGF and PDGF receptors in glioma. Ups J Med Sci 117, 99–112. 10.3109/03009734.2012.665097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JG, and Fine HA (2021). Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov 11, 575–590. 10.1158/2159-8290.CD-20-1474. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. (2006). Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173. 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PM, Frenay M, Rampling R, Stupp R, Kros JM, et al. (2008). Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J Clin Oncol 26, 4659–4665. 10.1200/JCO.2008.16.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DA, Wen PY, and Mellinghoff IK (2014). Targeted molecular therapies against epidermal growth factor receptor: past experiences and challenges. Neuro Oncol 16 Suppl 8, viii7–13. 10.1093/neuonc/nou232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 113, 4039–4044. 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabuda-Widemann D, Grabensee B, Schwandt C, and Blume C (2009). Mycophenolic acid inhibits the autocrine PDGF-B synthesis and PDGF-BB-induced mRNA expression of Egr-1 in rat mesangial cells. Nephrol Dial Transplant 24, 52–61. 10.1093/ndt/gfn462. [DOI] [PubMed] [Google Scholar]
- Sakakini N, Turchi L, Bergon A, Holota H, Rekima S, Lopez F, Paquis P, Almairac F, Fontaine D, Baeza-Kallee N, et al. (2016). A Positive Feed-forward Loop Associating EGR1 and PDGFA Promotes Proliferation and Self-renewal in Glioblastoma Stem Cells. J Biol Chem 291, 10684–10699. 10.1074/jbc.M116.720698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, Jin J, Ding X, Wu S, Huang H, et al. (2019). METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15, 1419–1437. 10.1080/15548627.2019.1586246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352, 987–996. 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Sysol JR, Natarajan V, and Machado RF (2016). PDGF induces SphK1 expression via Egr-1 to promote pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 310, C983–992. 10.1152/ajpcell.00059.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, and Somasundaram K (2018). Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 37, 522–533. 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- Wang H, Jiang Z, Na M, Ge H, Tang C, Shen H, and Lin Z (2017). PARK2 negatively regulates the metastasis and epithelial-mesenchymal transition of glioblastoma cells via ZEB1. Oncol Lett 14, 2933–2939. 10.3892/ol.2017.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, and Nam Y (2016). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell 63, 306–317. 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev 29, 1343–1355. 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bogler O, et al. (2017). m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 31, 591–606 e596. 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data accessed from external sources and prior publications have been referenced in the text and corresponding figure legends. Additional data will be made available upon request.