

Abstract
In the framework of the UK 100 000 Genomes Project, we investigated the genetic origin of a previously undescribed recessive dermatological condition, which we named LIPHAK (LTV1-associated Inflammatory Poikiloderma with Hair abnormalities and Acral Keratoses), in four affected individuals from two UK families of Pakistani and Indian origins, respectively. Our analysis showed that only one gene, LTV1, carried rare biallelic variants that were shared in all affected individuals, and specifically they bore the NM_032860.5:c.503A > G, p.(Asn168Ser) change, found homozygously in all of them. In addition, high-resolution homozygosity mapping revealed the presence of a small 652-kb stretch on chromosome 6, encompassing LTV1, that was haploidentical and common to all affected individuals. The c.503A > G variant was predicted by in silico tools to affect the correct splicing of LTV1’s exon 5. Minigene-driven splicing assays in HEK293T cells and in a skin sample from one of the patients confirmed that this variant was indeed responsible for the creation of a new donor splice site, resulting in aberrant splicing and in a premature termination codon in exon 6 of this gene. LTV1 encodes one of the ribosome biogenesis factors that promote the assembly of the small (40S) ribosomal subunit. In yeast, defects in LTV1 alter the export of nascent ribosomal subunits to the cytoplasm; however, the role of this gene in human pathology is unknown to date. Our data suggest that LIPHAK could be a previously unrecognized ribosomopathy.
Introduction
Poikiloderma is an unusual skin condition comprising pigmentation, telangiectasia and atrophy. It occurs in several rare genetic disorders with disparate pathogenesis including Kindler epidermolysis bullosa (OMIM #173650, a disorder of epithelial adhesion), Rothmund Thomson syndrome (OMIM #268400, a DNA repair disorder), dyskeratosis congenita (a group of telomeropathies) and poikiloderma with neutropenia (OMIM #604173, a disorder linked to maturation of U6 snRNA) (1). These can sometimes be differentiated clinically by the distribution of poikiloderma and associated features, but genetic analysis is diagnostic.
The ribosome is the key element of translation, an essential biological process producing proteins from messenger RNA (mRNA). In eukaryotes, it is composed of a 40S (small) and a 60S (large) subunit (2–4). The synthesis of pre-40S subunits begins with folding of ribosomal RNA (rRNA), followed by recruitment of ribosomal proteins (RPs) and assembly factors (AFs) in the nucleolus. Low temperature viability protein 1 (LTV1) is one of these AFs. It interacts with RPs, chaperoning the correct ribosomal assembly (5,6), and promotes the nuclear export of pre-40S into the cytoplasm (7,8). In yeast, Ltv1 mutations lead to reduced recruitment of some RPs to the ribosomal complex, which in turn introduces defects in translational fidelity (5,9,10). According to the mouse genome informatics database (MGI) (11), ablation of the Ltv1 gene in mouse results in preweaning lethality, but so far no LTV1-related phenotypes have been described in humans.
Using whole-genome sequencing (WGS), here we identify a mutation in LTV1 [NM_032860.5, c.503A > G, p.(Asn168Ser)] as the likely molecular cause for a previously unreported poikilodermatous recessive condition, in four patients from two different families. In vitro, this exonic DNA variant results in two different transcripts: a minor form carrying the p.(Asn168Ser) substitution (~11% of transcribed RNA, with respect to control) and a major form being aberrantly spliced and presumably degraded by nonsense-mediated mRNA decay (NMD) in vivo.
Results
Family 1: Patients 1, 2 and 3
Patients 1 (II.4), 2 (II.3) and 3 (II.2) were sisters of Pakistani origin (Fig. 1A). They also had one unaffected sister and their parents were first cousins with no other relevant family history. They were first examined aged 2, 7 and 10 years, respectively and have been followed up for 20 years.
Figure 1.

Pedigrees analyzed. (A) Family 1 originates from Pakistan and consists of parents who are first cousins, as well as four daughters, three of whom are affected. (B) Family 2, originating from India. Parents are unrelated. Their only daughter is affected. M: NM_032860.5, c.503A > G:p.(Asn168Ser); +, WT allele.
Patient 1, at 2 years, had striking livedoid erythema in a reticulate pattern particularly marked on the limbs (Fig. 2A). In some areas the erythema was scaly, atrophic and pigmented, with small, discrete patches of hypopigmentation in the spaces within the reticulate network (Fig. 2B). The upper trunk was largely spared but lower trunk, flanks and forehead were affected with mottled hyper- and hypopigmentation as were palms and soles (Fig. 2C and D). Eyebrows were absent and eyelashes were sparse, scalp hair was normal in texture but generally sparse and there were no other ectodermal or haematological or other abnormalities. Ophthalmoscopy revealed asymptomatic embryonal nuclear cataracts. Skin biopsy from the thigh at age 3 years showed a mild lichenoid lymphocytic infiltrate in the papillary dermis with melanophage accumulation and pigmentary incontinence.
Figure 2.
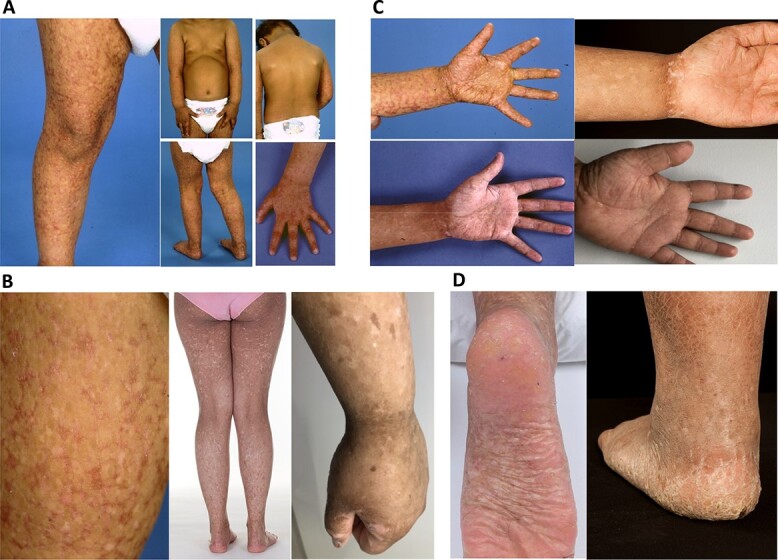
Clinical synopsis. (A) Patient 1, aged 2.5 years, showing the distribution skin changes. (B) Morphology of skin changes in (left to right) Patient 1 at 2.5 and 14 years and Patient 4 at 21 years. (C) Palms of Patient 1 (left), aged 2.5 and 7 years, and of Patient 4 (right) aged 11 and 21 years, showing reticulate pigmentation, progressive scaling and peripheral keratoses. (D) Feet of Patient 1 (left) aged 14 years and of Patient 4 (right) aged 11 years.
Patient 2, at 7 years, had mottled hyper- and hypopigmentation distributed as in her younger sister but with no erythema. She also has asymptomatic embryonal nuclear cataracts. Her palms and soles showed scaling. The other features were the same, including absent eyebrows, except that her scalp hair was normal.
Patient 3, at 10 years, had similar skin changes to patient 2; her eyebrows were absent and scalp hair was thin in patches. She had peripheral plantar keratoses. Ophthalmic examination was normal.
By 14 years all three had mottled hyper- and hypopigmentation and peripheral punctate plantar keratoses (Fig. 2D). Now aged 21, 26 and 30 years, they remain healthy and the pigmentary changes have faded considerably but remain most marked in Patient 1. Eyebrows remain absent and have been replaced by cosmetic tattoos. They avoid sun exposure which causes redness within 2 h and makes the pigmentary changes more obvious. Acral scaling and punctate keratoses persist. In all three growth, development and general health have been normal.
Family 2: Patient 4
Patient 4 (II.1) is the only child of unrelated parents whose families originate from Gujarat in India with no relevant family history (Fig. 1B). Her growth and development were normal with no haematological abnormalities. Skin changes started acrally at 6 months of age and progressed proximally. At age 6 and 9 years she had mottled hyper- and hypopigmentation affecting the face, buttocks and limbs including palmar and plantar surfaces with reticulate scaly erythema on the extremities. Her hair and eyelashes were sparse, as were eyebrows especially laterally. When last examined at age 18 years she had mottled hyper- and hypopigmentation and acral keratoderma with the same appearance and distribution as in Patients 1–3 (Fig. 2B–D). She had dry skin generally but no other ectodermal or systemic abnormalities.
A skin biopsy from the ankle region at age 4 years showed hyperkeratosis and focal parakeratosis. A mild superficial dermal chronic inflammatory infiltrate extended focally into the epidermal basal layer with some basal keratinocyte vacuolation. The papillary dermis showed scattered melanophages and focal vascular dilatation. Direct immunofluorescence showed no significant deposition of immunoglobulins, C3 or fibrin and no lupus band.
Genetic findings
Genetic testing for Rothmund Thompson syndrome and Kindler syndrome was negative in both families. Primary analysis of results from PanelApp (12) virtual gene panels applied to WGS data (Table 1), on the basis of Human Phenotype Ontology (HPO) terms entered (13), did not identify any variants considered to be causative of the phenotype.
Table 1.
Virtual PanelApp gene panels applied to each case as part of the 100 000 Genomes Project on the basis of HPO terms entered for cases
| Case | 1, 2 and 3 | 4 |
|---|---|---|
| Panels applied | Erythropoietic protoporphyria, mild variant v1.2 Palmoplantar keratoderma and erythrokeratodermas v1.15 |
Palmoplantar keratoderma and erythrokeratodermas v1.16 Erythropoietic protoporphyria, mild variant v1.2 Hydroa vacciniforme v1.2 Hereditary haemorrhagic telangiectasia v1.51 |
We therefore utilized a gene-agnostic approach, filtering for variants in genes that segregated with disease in the affected family members and at a low population frequency (see Methods). This led to the identification of only one candidate variant, present in the LTV1 gene. The variant, NM_032860.3:c.503A > G: p.(Asn168Ser) in exon 5, was present homozygously in all four affected individuals from both families and heterozygously in parents from both pedigrees (Fig. 1). In the gnomAD database (14), this variant was reported only once in a subject of European origin, in a heterozygous state, over a total of 251 344 analyzed alleles (allele frequency = 3.98 × 10−6), of which 30 614 were from South Asian individuals. Most of the standard pathogenicity software, including SIFT (15) and PolyPhen-2 (16), predicted the missense change to be benign. However, algorithms evaluating potential impact on splicing, such as SpliceAI (17) and MaxEntScan (18), detected that the A > G transition could result in the creation of a new donor splice site. This new site could compete with the correct one, resulting in the truncation of 37 bp at the 3′ side of exon 5 by virtue of its premature fusion with exon 6, and this event would in turn lead to a frameshift and a premature termination codon in exon 6: p.(Asn168ArgfsTer47). Moreover, autozygosity mapping (19) showed that this variant was comprised within a small region of homozygosity (652 kb) that was common to all patients and was also haploidentical (Supplementary Material, Table S1, Supplementary Material, Fig. S1), implying that the mutation in the two families did not arise independently but was likely inherited from a common ancestor.
Splicing assays
To investigate the potential role on splicing of the variant detected, we designed a minigene composed of exon 5, intron 5 and exon 6 of LTV1, either including the wild-type (WT) or alternative genotype at position c.503, within exon 5. Following RNA purification, complementary DNA (cDNA) synthesis and reverse transcription polymerase chain reaction (RT-PCR), we then assessed the splicing patterns of both minigenes in transfected HEK293T cells. In addition to displaying small amounts of canonically spliced transcripts (estimated by capillary electrophoresis to represent ~ 11% of all minigene-derived RNA), minigenes carrying the c.503A > G change resulted mostly into an aberrant and shorter RNA form, in which exon 5 was prematurely fused to exon 6, exactly as predicted in silico by the software mentioned above (Fig. 3). RNA bearing this rearrangement, leading to the shift of the reading frame and the creation of a premature termination codon, would probably be subject to NMD in in vivo conditions. Of note, this aberrant form was not present in RNA from cells transfected with WT minigenes, as demonstrated by using RT-PCR primers specific for this alternative transcript (Fig. 3A).
Figure 3.

Molecular analysis. (A) Gel electrophoresis of the RT-PCR products from HEK293T cells transfected with minigene plasmids carrying exon 5 to exon 6 of LTV1 and either the WT allele or the c.503A > G mutation. The mutant form of the plasmid has two transcripts, corresponding to the correctly spliced isoform (1) and an aberrantly spliced isoform (2). This non-canonical transcript (2,3) is present only in cells transfected with minigenes bearing the mutation, as specifically shown by the use of an isoform-specific primer (CR 7326). (B) Sanger sequencing of the two transcripts, from the WT minigene (top) and the one carrying c.503A > G (bottom). (C) Schematic view of the LTV1 minigene, depicting the position of the variant identified and the splicing event resulting from its presence, as well as the position of the PCR primers used.
These results were corroborated by semi-quantitative RT-PCR analysis of LTV1 in a skin sample from Patient 4, showing reduced expression compared with control skin. cDNA sequencing across the site of the mutation also confirmed the aberrant splicing (Supplementary Material, Fig. S2).
Immunofluorescence microscopy
We then analyzed LTV1 protein expression and distribution in a skin biopsy from Patient 4 and a control. In control skin, expression of LTV1 was noted within the basal keratinocyte layer of the epidermis (Fig. 4). There was diffuse cytoplasmic staining with granular perinuclear accentuation across the basal layer. In contrast, in patient skin the intensity of the labelling was slightly reduced (Fig. 4). In addition, the pattern of staining within the basal layer was uneven with some basal keratinocytes showing near normal intensity labelling, whereas other basal keratinocytes showed markedly reduced immunostaining for LTV1.
Figure 4.
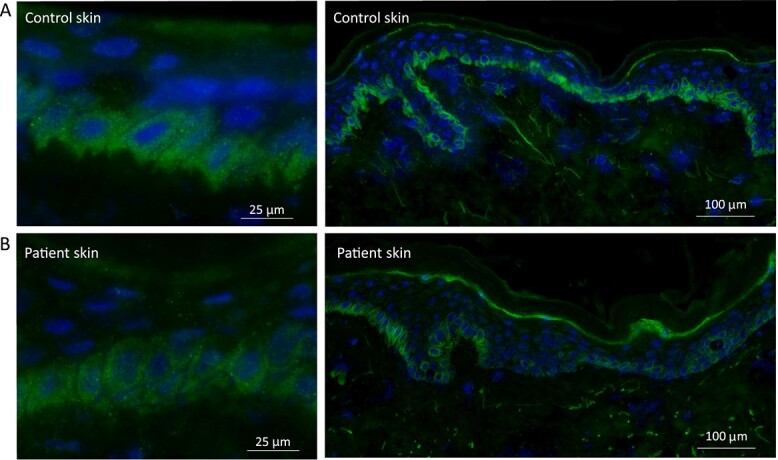
Immunostaining of LTV1 in control and patient skin. (A) In control skin there is diffuse cytoplasmic staining within the basal keratinocytes, with additional granular perinuclear labelling seen at higher magnification. The intensity of the staining is evenly distributed along the basal keratinocyte layer within the skin section. (B) In patient skin, the immunoreactivity for LTV1 is slightly reduced. Of note, staining along the basal layer is uneven with some basal keratinocytes showing barely any immunostaining.
Discussion
We report on four affected individuals from two pedigrees, all bearing a common homozygous mutation in the gene LTV1 and presenting with a homogeneous and recessive phenotype of poikiloderma associated with palmoplantar keratoses and sparse eyebrows, which we termed LIPHAK syndrome (LTV1-associated Inflammatory Poikiloderma with Hair abnormalities and Acral Keratoses). In vitro and in vivo, this variant results in a splicing defect, which in turn leads to a shift of the reading frame and to the creation of a premature termination codon. Because of its position within the primary sequence of LTV1 pre-mRNA, this stop codon should lead in vivo to the active degradation of all transcripts that bear it, by the action of the NMD machinery (20). Importantly, no additional putatively pathogenic variant that was common to both families was detected, within the ROHs shared by the affected individuals or elsewhere.
The skin condition displayed by these patients does not entirely fit the term ‘poikiloderma’. Telangiectasia and atrophy were limited and in the adults only hyper- and hypopigmentation remained. A more accurate but less concise term might be ‘reticulate poikilodermatous dyschromatosis’. The initial livedo reticularis-like pattern suggests cutaneous vasculopathy with impaired oxygenation at the periphery of arteriolar supply zones. The subsequent pigmentation may be post-inflammatory, but the final pattern of discrete hyper- and hypopigmented macules is less obviously reticulate and resembles the dyschromatosis seen in familial progressive hyper- and hypopigmentation (OMIM #145250) caused by dominant mutations in KITLG (21).
Ribosomal subunits undergo a series of processing, modifications and final maturation to be competent for protein synthesis (22). In yeast, the Ltv1 protein has been found to be central for the correct assembly and functioning of ribosomes (7,9,10,23), whereas its function in human biology has not been fully investigated. The role of Ltv1 is particularly important for biogenesis of the 40S subunit, which involves the interaction of the two 18S rRNA helices (h16 and h41) with other proteins, such as for instance Enp1 and Rps3 (22,24). These interactions induce structural rearrangement during the final maturation of small subunits and the stable incorporation of remaining RPs and AFs (5,25). In humans, Hrr25-like protein kinases, including CK1δ/ε, phosphorylate LTV1 to dissociate it from AFs of the subunit (9,26). Ltv1 mutants, in yeast and Drosophila, show reduced cell growth (6,9).
Our in vitro results indicate that c.503A > G, while leading mostly to aberrant mRNA, does not prevent completely the correct splicing of LTV1 pre-mRNA. Therefore, the patients described here, homozygous for this mutation, are likely to produce fully functional LTV1 protein, although in reduced amounts. Noteworthy, knockout mice for LTV1 die before weaning age, implying that LTV1 protein is required for survival (11). LTV1 has not previously been linked to any genetic diseases. It is expressed in >200 tissues with no particular tissue specificity, as shown in GTEx (https://www.gtexportal.org/home/gene/LTV1) (27), and its encoded protein is found in both the cytosol and the nucleoplasm. Expression of LTV1 has been noted as an unfavourable prognostic marker in renal and liver cancer (https://www.proteinatlas.org/ENSG00000135521-LTV1/pathology), but there is no known association with skin disease. Conceivably, however, this disruption of ribosome function may be more marked in tissues displaying increased oxidative stress, such as the skin. Thus, it is plausible that the poikiloderma and other features observed in our cases represent a kind of ribosomopathy, where a slight impairment of ribosome assembly can cause a mild translation defect, but only in specific cells (including certain skin cells). Another rare and syndromic poikilodermatous condition, dyskeratosis congenita (OMIM #305000), characterized by irreversible degeneration of skin tissue and bone marrow failure (28,29), while generally considered a telomeropathy, also has features of a ribosomopathy (30). It is caused by mutations in DKC1 and the encoded protein, dyskerin, is involved in the maturation of rRNAs (31). An alternative, or perhaps additional explanation is that the LTV1 mutation may impair a different cellular process altogether, for example the early endosome recycling (32). Although complex and diverse, abnormalities in endosome recycling pathways in skin cells have been implicated in certain inherited skin diseases, namely some forms of epidermolysis bullosa and Griscelli syndrome (33,34).
In summary, we describe a novel autosomal recessive skin disorder characterized by a reticulate poikilodermatous dyschromatosis and apparently caused by a homozygous splice variant in LTV1, categorizing it as a probable ribosomopathy. Additional research in animal models or identification of other individuals segregating LTV1 biallelic variants will be important to better understand the molecular pathology of this condition.
Materials and Methods
Patients and families
This study was performed in agreement with the tenets of the Declaration of Helsinki, following the signature of written informed consent forms (including the use of images) by the patients and their family members and the approval by the UK Health Research Authority and the Ethikkommission Nordwest- und Zentralschweiz. Families 1 and 2 were identified in routine dermatological practice and their unique but shared phenotype was noted in 2005. In 2017, in the absence of a molecular diagnosis, both families were recruited to the UK 100 000 Genomes Project (35). Consent was also obtained for WGS and further research.
WGS
WGS was performed in all affected individuals and their parents as part of the 100 000 Genomes Project. Blood samples, consent, clinical indication, and HPO terms were collected for patients at local hospitals. Blood samples were sent to regional genetics laboratories and DNA extracted, which was sent on to Illumina for WGS. Library preparation was performed using TruSeq DNA PCR-Free Library Prep, with 150 bp paired-end reads generated from the HiSeqX sequencer. Data were passed through Genomics England’s bioinformatics pipeline. Variants were filtered on the basis of quality control, allele frequency, functional impact and segregation in affected family members (36).
Autozygome and haplotype analysis
Runs of homozygosity (ROH) regions were investigated for each individual using AutoMap (19) (parameters: minimal size of ROH ≥ 0.5 Mb, minimal number of homozygous variant in window ≥15, minimal percentage of homozygous variant ≥90) and then combined in silico.
Haplotypes spanning the LTV1 region were assessed by retaining variants of good quality only (genotype quality ≥50 in the Variant Call Format (VCF) file), and their frequencies were obtained from dbSNP (37). Genotypes in polyA tails of pseudogenes were excluded from the analysis.
In silico analysis of variants
DNA variants were obtained from VCFs present in the 100 000 Genomes Project (35). For assessment of mutations, only variants with an allele frequency of 0.01 or lower, as reported by the gnomAD database (14), were retained. Missense changes were analyzed using an in silico pipeline developed internally (38).
Minigene-driven splicing assay
To assess the effect of the c.503A > G variant on pre-mRNA splicing, minigene-driven assay was performed, as aforementioned (39). Briefly, the LTV1 exon5–intron5–exon6 genomic sequences from an affected individual and a control were polymerase chain reaction (PCR)-amplified with gene-specific primers containing the recombination sites attB1 and attB2 at their 5′ ends (Forward CR7267, 5′-ggggacaagtttgtacaaaaaagcaggctgacctcgactggattttgat-3′; Reverse CR7268, 5′-ggggaccactttgtacaagaaagctgggtctatcttctcaaacctctcatcat-3′). After PCR amplification, purified PCR products were cloned into a Gateway™ pDEST™26 Vector (Thermo Fisher Scientific). The DNA sequence of both resulting WT and mutant minigenes was confirmed to be correct by direct Sanger sequencing.
Plasmids bearing minigenes were transfected into HEK293T cells using FuGENE HD (Promega) in duplicates. Cells were harvested 24 h after transfection and total RNA was extracted using the QuickPrep total RNA extraction kit (GE Healthcare). cDNA was obtained by using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems), following manufacturer’s protocols and used as a template for PCR amplification using a vector-specific attB1 forward primer (Forward CR7320, 5′-caacaagtttgtacaaaaaagcaggc-3′) in combination with either a reverse primer matching LTV1 exon 6 (Reverse CR7323, 5′-gctatctcccttctcatcatccac-3) or a reverse primer matching the aberrant exon5–exon6 junction elicited by c.503A > G and described below (Reverse CR7326, 5′-cctgttgcctctcctgttg-3). The resulting amplified fragments were displayed on a 2% agarose gel and analyzed by a QIAxcel instrument (Qiagen). Quantification of RT-PCR products separated by capillary electrophoresis was achieved by integrating the area under each peak. All products resulting from cDNA amplification were Sanger-sequenced.
Skin biopsy and immunofluorescence microscopy
Following written consent, an ellipse skin biopsy was obtained under local anaesthetic. The skin was snap frozen and 4 mm sections were then cut, mounted on glass slides, and allowed to dry. Sections were washed in phosphate-buffered saline (PBS; Sigma P4417, Lot SLCH5832) and then covered in 10% normal donkey serum (Sigma Aldrich G9023, Lot SLCK7210) before this was removed and the sections were exposed to the LTV1 antibody (Polyclonal rabbit anti LTV1; Abcam ab122100, Lot GR116961–8) diluted 1 in 25 in 1% bovine serum albumin (Sigma A2153, Lot SLCB5832) diluted in PBS and then incubated at 37°C for 1 h. Negative controls omitted the primary antibody. The sections were then washed twice in PBS and covered with fluorescently labelled secondary antibody (DaR AF488: Donkey anti Rabbit Alexa Fluor 488; Abcam ab150061, Lot GR3350992–3, diluted 1 in 500) and then incubated at 37°C for 1 h, before further washing in PBS. The sections were then cover slipped using Vectorshield with 4',6-diamidino-2-phenylindole (DAPI) as a mountant. The slides were viewed using an Olympus BX63 microscopy and photographed at an exposure of 127 ms (AF488) 2.49 ms (DAPI).
Semi-quantitative RT-PCR
From part of the skin biopsy, RNA was extracted using QIAGEN RNeasy Fibrous Tissue Mini kit (74704). Next, 100 ul of cDNA was converted from RNA extracted from 0.3 mm3 skin tissue, using Invitrogen SuperScript III First-Strand cDNA Synthesis System. For semi-Q-PCR, a multiplex PCR system with the target and the housekeeping gene GAPDH was designed and optimized to obtain the best-balanced efficiency between these two PCR products. Five PCR cycling points (cycle 20, 24, 28, 32 and 36) were checked to ensure that the measurements would fall within the PCR exponential phase. Each point was assessed in triplicate, and gel images were quantified using ImageJ. To assess cDNA expression, LTV1 primers were designed to amplify part of the gene within its 3′UTR: LTV1-cDNA-F, AAGGACTCACAGCAAAGCAAA; LTV1-cDNA-R, AAGGACTCACAGCAAAGCAAA. To assess the impact of the missense mutation on splicing, the following LTV1 primers were used: LTV1-c.503-C-F, GTTGCCTTCATCAGTGTTTGC; LTV1-c.503-C-R, ACATCTTCCCACTCGCTGTC.
Supplementary Material
Acknowledgements
We are grateful to Sasi Conte (King’s College London), Ed Hurt (University of Heidelberg) and Deborah Lycan (Lewis and Clark College, Portland) for helpful discussions on the potential disease relevance of the LTV1 variant. This research was made possible through access to the data and findings generated by the 100 000 Genomes Project. The 100 000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100 000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100 000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support.
Conflict of Interest statement. None declared.
Contributor Information
Ji Hoon Han, Institute of Molecular and Clinical Ophthalmology Basel (IOB), 4031 Basel, Switzerland; Department of Ophthalmology, University of Basel, 4031 Basel, Switzerland.
Gavin Ryan, West Midlands Regional Genetics Laboratory, Central and South Genomic Laboratory Hub, Birmingham B15 2TG, UK.
Alyson Guy, Viapath, St Thomas' Hospital, London SE1 7EH, UK.
Lu Liu, Viapath, St Thomas' Hospital, London SE1 7EH, UK.
Mathieu Quinodoz, Institute of Molecular and Clinical Ophthalmology Basel (IOB), 4031 Basel, Switzerland; Department of Ophthalmology, University of Basel, 4031 Basel, Switzerland; Department of Genetics and Genome Biology, University of Leicester, Leicester LE1 7RH, UK.
Ingrid Helbling, Department of Dermatology, University Hospitals of Leicester NHS Trust, Leicester LE1 5WW, UK.
Joey E Lai-Cheong, Frimley Park NHS Foundation Trust, Camberley GU16 7UJ, UK.
Genomics England Research Consortium, , https://www.genomicsengland.co.uk/wp-content/uploads/2021/05/Genomics-England-Research-Consortium-author-full-names-list-3.pdf.
Julian Barwell, Department of Genetics and Genome Biology, University of Leicester, Leicester LE1 7RH, UK; Department of Clinical Genetics, University Hospitals of Leicester NHS Trust, Leicester LE1 5WW, UK.
Marc Folcher, Institute of Molecular and Clinical Ophthalmology Basel (IOB), 4031 Basel, Switzerland; Department of Ophthalmology, University of Basel, 4031 Basel, Switzerland.
John A McGrath, NIHR Biomedical Research Centre, Guy's and St Thomas' NHS Foundation Trust and King's College London, London SE1 9RT, UK; St John's Institute of Dermatology, King's College London (Guy's campus), London SE1 9RT, UK.
Celia Moss, Department of Paediatric Dermatology, Birmingham Women’s and Children’s Hospital NHS FT, Birmingham B4 6NH, UK; College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, UK.
Carlo Rivolta, Institute of Molecular and Clinical Ophthalmology Basel (IOB), 4031 Basel, Switzerland; Department of Ophthalmology, University of Basel, 4031 Basel, Switzerland; Department of Genetics and Genome Biology, University of Leicester, Leicester LE1 7RH, UK.
Data Availability
The data to support the findings of this study are available from the corresponding author upon request.
Funding
Swiss National Science Foundation (grant no. 204285 to C.R.).
References
- 1. Rayinda, T., vanSteensel, M. and Danarti, R. (2021) Inherited skin disorders presenting with poikiloderma. Int. J. Dermatol., 60, 1343–1353. [DOI] [PubMed] [Google Scholar]
- 2. Ben-Shem, A., Garreau de Loubresse, N., Melnikov, S., Jenner, L., Yusupova, G. and Yusupov, M. (2011) The structure of the eukaryotic ribosome at 3.0 a resolution. Science, 334, 1524–1529. [DOI] [PubMed] [Google Scholar]
- 3. Melnikov, S., Ben-Shem, A., Garreau de Loubresse, N., Jenner, L., Yusupova, G. and Yusupov, M. (2012) One core, two shells: bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol., 19, 560–567. [DOI] [PubMed] [Google Scholar]
- 4. Khatter, H., Myasnikov, A.G., Natchiar, S.K. and Klaholz, B.P. (2015) Structure of the human 80S ribosome. Nature, 520, 640–645. [DOI] [PubMed] [Google Scholar]
- 5. Collins, J.C., Ghalei, H., Doherty, J.R., Huang, H., Culver, R.N. and Karbstein, K. (2018) Ribosome biogenesis factor Ltv1 chaperones the assembly of the small subunit head. J. Cell Biol., 217, 4141–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim, W., Kim, H.D., Jung, Y., Kim, J. and Chung, J. (2015) Drosophila low temperature viability protein 1 (LTV1) is required for ribosome biogenesis and cell growth downstream of drosophila Myc (dMyc). J. Biol. Chem., 290, 13591–13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seiser, R.M., Sundberg, A.E., Wollam, B.J., Zobel-Thropp, P., Baldwin, K., Spector, M.D. and Lycan, D.E. (2006) Ltv1 is required for efficient nuclear export of the ribosomal small subunit in Saccharomyces cerevisiae. Genetics, 174, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merwin, J.R., Bogar, L.B., Poggi, S.B., Fitch, R.M., Johnson, A.W. and Lycan, D.E. (2014) Genetic analysis of the ribosome biogenesis factor Ltv1 of Saccharomyces cerevisiae. Genetics, 198, 1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghalei, H., Schaub, F.X., Doherty, J.R., Noguchi, Y., Roush, W.R., Cleveland, J.L., Stroupe, M.E. and Karbstein, K. (2015) Hrr25/CK1delta-directed release of Ltv1 from pre-40S ribosomes is necessary for ribosome assembly and cell growth. J. Cell Biol., 208, 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fassio, C.A., Schofield, B.J., Seiser, R.M., Johnson, A.W. and Lycan, D.E. (2010) Dominant mutations in the late 40S biogenesis factor Ltv1 affect cytoplasmic maturation of the small ribosomal subunit in Saccharomyces cerevisiae. Genetics, 185, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bult, C.J., Blake, J.A., Smith, C.L., Kadin, J.A., Richardson, J.E., Database, M.G. and G. (2019) Mouse genome database (MGD) 2019. Nucleic Acids Res., 47, D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martin, A.R., Williams, E., Foulger, R.E., Leigh, S., Daugherty, L.C., Niblock, O., Leong, I.U.S., Smith, K.R., Gerasimenko, O., Haraldsdottir, E.et al. (2019) PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet., 51, 1560–1565. [DOI] [PubMed] [Google Scholar]
- 13. Kohler, S., Doelken, S.C., Mungall, C.J., Bauer, S., Firth, H.V., Bailleul-Forestier, I., Black, G.C., Brown, D.L., Brudno, M., Campbell, J.et al. (2014) The human phenotype ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res., 42, D966–D974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alfoldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ng, P.C. and Henikoff, S. (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adzhubei, I., Jordan, D.M. and Sunyaev, S.R. (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7, Unit7, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., Darbandi, S.F., Knowles, D., Li, Y.I., Kosmicki, J.A., Arbelaez, J., Cui, W., Schwartz, G.B.et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 535–548 e524. [DOI] [PubMed] [Google Scholar]
- 18. Yeo, G. and Burge, C.B. (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol., 11, 377–394. [DOI] [PubMed] [Google Scholar]
- 19. Quinodoz, M., Peter, V.G., Bedoni, N., Royer Bertrand, B., Cisarova, K., Salmaninejad, A., Sepahi, N., Rodrigues, R., Piran, M., Mojarrad, M.et al. (2021) AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun., 12, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hentze, M.W. and Kulozik, A.E. (1999) A perfect message: RNA surveillance and nonsense-mediated decay. Cell, 96, 307–310. [DOI] [PubMed] [Google Scholar]
- 21. Wang, J., Li, W., Zhou, N., Liu, J., Zhang, S., Li, X., Li, Z., Yang, Z., Sun, M. and Li, M. (2021) Identification of a novel mutation in the KITLG gene in a Chinese family with familial progressive hyper- and hypopigmentation. BMC Med. Genet., 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mitterer, V., Murat, G., Rety, S., Blaud, M., Delbos, L., Stanborough, T., Bergler, H., Leulliot, N., Kressler, D. and Pertschy, B. (2016) Sequential domain assembly of ribosomal protein S3 drives 40S subunit maturation. Nat. Commun., 7, 10336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ameismeier, M., Cheng, J., Berninghausen, O. and Beckmann, R. (2018) Visualizing late states of human 40S ribosomal subunit maturation. Nature, 558, 249–253. [DOI] [PubMed] [Google Scholar]
- 24. Mitterer, V., Shayan, R., Ferreira-Cerca, S., Murat, G., Enne, T., Rinaldi, D., Weigl, S., Omanic, H., Gleizes, P.E., Kressler, D.et al. (2019) Conformational proofreading of distant 40S ribosomal subunit maturation events by a long-range communication mechanism. Nat. Commun., 10, 2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Panse, V.G. and Johnson, A.W. (2010) Maturation of eukaryotic ribosomes: acquisition of functionality. Trends Biochem. Sci., 35, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schafer, T., Maco, B., Petfalski, E., Tollervey, D., Bottcher, B., Aebi, U. and Hurt, E. (2006) Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature, 441, 651–655. [DOI] [PubMed] [Google Scholar]
- 27. Consortium, G.T. (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knight, S.W., Heiss, N.S., Vulliamy, T.J., Greschner, S., Stavrides, G., Pai, G.S., Lestringant, G., Varma, N., Mason, P.J., Dokal, I.et al. (1999) X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am. J. Hum. Genet., 65, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heiss, N.S., Knight, S.W., Vulliamy, T.J., Klauck, S.M., Wiemann, S., Mason, P.J., Poustka, A. and Dokal, I. (1998) X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet., 19, 32–38. [DOI] [PubMed] [Google Scholar]
- 30. Nakhoul, H., Ke, J., Zhou, X., Liao, W., Zeng, S.X. and Lu, H. (2014) Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord, 7, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bellodi, C., McMahon, M., Contreras, A., Juliano, D., Kopmar, N., Nakamura, T., Maltby, D., Burlingame, A., Savage, S.A., Shimamura, A.et al. (2013) H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep., 3, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. MacDonald, C. and Piper, R.C. (2017) Genetic dissection of early endosomal recycling highlights a TORC1-independent role for rag GTPases. J. Cell Biol., 216, 3275–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bare, Y., Chan, G.K., Hayday, T., McGrath, J.A. and Parsons, M. (2021) Slac2-b coordinates extracellular vesicle secretion to regulate keratinocyte adhesion and migration. J. Invest. Dermatol., 141, 523–532.e2. [DOI] [PubMed] [Google Scholar]
- 34. Hume, A.N., Collinson, L.M., Rapak, A., Gomes, A.Q., Hopkins, C.R. and Seabra, M.C. (2001) Rab27a regulates the peripheral distribution of melanosomes in melanocytes. J. Cell Biol., 152, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Caulfield, M., Davies, J., Dennys, M., Elbahy, L., Fowler, T., Hill, S., Hubbard, T., Jostins, L., Maltby, N. and Mahon-Pearson, J. (2020) The National Genomic Research Library v5.1. 10.6084/m9.figshare.4530893.v6. [DOI] [Google Scholar]
- 36. Turnbull, C., Scott, R.H., Thomas, E., Jones, L., Murugaesu, N., Pretty, F.B., Halai, D., Baple, E., Craig, C., Hamblin, A.et al. (2018) The 100 000 genomes project: bringing whole genome sequencing to the NHS. BMJ, 361, k1687. [DOI] [PubMed] [Google Scholar]
- 37. Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M. and Sirotkin, K. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peter, V.G., Nikopoulos, K., Quinodoz, M., Granse, L., Farinelli, P., Superti-Furga, A., Andreasson, S. and Rivolta, C. (2019) A novel missense variant in IDH3A causes autosomal recessive retinitis pigmentosa. Ophthalmic Genet., 40, 177–181. [DOI] [PubMed] [Google Scholar]
- 39. Peter, V.G., Quinodoz, M., Pinto-Basto, J., Sousa, S.B., Di Gioia, S.A., Soares, G., Ferraz Leal, G., Silva, E.D., Pescini Gobert, R., Miyake, N.et al. (2019) The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in thePISD gene. Genet. Med., 21, 2734–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data to support the findings of this study are available from the corresponding author upon request.