

Abstract
HaloTag labeling technology has introduced unrivaled potential in protein chemistry and molecular and cellular biology. A wide variety of ligands have been developed to meet the specific needs of diverse applications, but only a single protein tag, DhaAHT, is routinely used for their incorporation. Following a systematic kinetic and computational analysis of different reporters, a tetramethylrhodamine- and three 4-stilbazolium-based fluorescent ligands, we showed that the mechanism of incorporating different ligands depends both on the binding step and the efficiency of the chemical reaction. By studying the different haloalkane dehalogenases DhaA, LinB, and DmmA, we found that the architecture of the access tunnels is critical for the kinetics of both steps and the ligand specificity. We showed that highly efficient labeling with specific ligands is achievable with natural dehalogenases. We propose a simple protocol for selecting the optimal protein tag for a specific ligand from the wide pool of available enzymes with diverse access tunnel architectures. The application of this protocol eliminates the need for expensive and laborious protein engineering.
Keywords: HaloTag, enzyme kinetics, molecular modeling, reaction mechanism, ligand binding, nucleophilic substitution, protein engineering, access tunnel, numerical integration
Introduction
Genetically encoded protein labeling methods are widely employed in protein chemistry and molecular and cellular biology. More recently, self-labeling protein tags designed for covalent conjugation with small-molecule ligands functionalized with biorthogonal linkers have gained widespread attention. One of the most popular self-labeling methods, HaloTag, uses engineered haloalkane dehalogenase (HLD) genetically fused to the proteins of interest, which covalently binds synthetic ligands bearing various functionalities, such as a strong light-up fluorescence response. The original concept of bifunctional linkers, developed by Janssen and co-workers1 for covalent capturing and ribosomal/phage display of HLDs, was translated to in vivo and in vitro analysis of mammalian proteins by Wood and co-workers.2−4
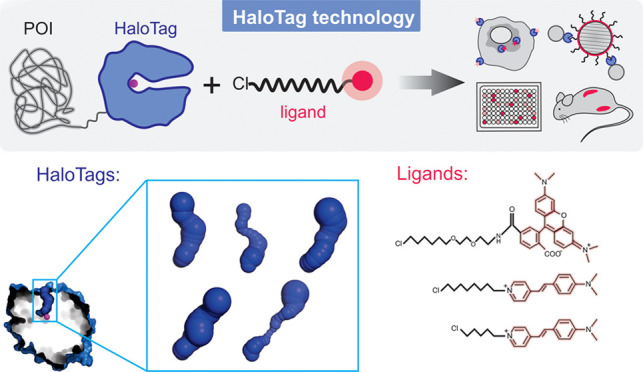
Since its development and commercialization by Promega, HaloTag has become a valuable research tool for a broad range of applications (Figure 1A) including protein purification5 and immobilization,6 enhancement of the soluble expression of recombinant proteins,7 cellular protein imaging,8,9 imaging in vivo,(10) and single-molecule studies.11−13 The technology is applicable to the analyses of protein–protein and protein–nucleic acid interactions,14,15 proteome stress,16,17 protein folding and aggregation,18,19 dynamics and hydration,20−22 or cell permeability.23 HaloTag fusions enable protein control in vivo,(24−26) including degradation27,28 or dimerization29 of proteins of interest. Further applications include high-throughput screening methods, microarrays and chip technology,30−32 intracellular detection of pH33 or biologically important ions,34,35 mechanochemistry,36,37 functionalization of nanoparticles,38 and quantum dots.39 Recently, the potential of the HaloTag technology in cell therapy was discovered, as it has been used for cell surface modification permitting angiogenesis, increased motility, and immune shielding.40
Figure 1.

HaloTag technology in chemistry, biology, biochemistry, and biophysics. (A) Widespread applications of the HaloTag technology. (B) The HaloTag ligands contain two crucial components: (i) a reactive linker that initiates the formation of a covalent bond with the HaloTag protein and (ii) a functional reporter.
A wide range of diverse HaloTag ligands have been designed and synthesized, offering a variety of properties (Figure 1B), e.g., improved photostability and brightness,12 high biocompatibility or fluorogenicity allowing “no-wash” labeling protocols41−43 or providing specific affinity handles.4 Despite the great diversity of ligands used, most of their applications always utilize the same tag protein DhaAHT, without considering the choice of another protein partner for better recognition of a specific ligand. The 10 000-fold improvement in binding efficiency of the DhaAHT tag required for the successful protein imaging was achieved using a focused directed evolution on the access tunnel residues.44 This study has already shown that the efficiency obtained by molecular evolution can differ significantly among individual ligands, despite sharing the same reactive linker. Similar effects were observed in our recent studies focused on the engineering of access tunnels in HLDs.45,46 The binding efficiency of HaloTag ligands varied across 7 orders of magnitude for HLDs with different architectures of their access tunnels. Interestingly, the change in the functional reporter strongly affected the labeling efficiency even for ligands with the same reactive linker.45,46 The results collectively suggest that the broadly used DhaAHT tag may not be the optimal tag for the incorporation of the various available ligands. Since DhaAHT was introduced, the portfolio of available dehalogenases has significantly expanded47−49 and currently offers an interesting range of variants. Some of these HLDs, which may or may not be phylogenetically close to one another,49 can display remarkably diverse tunnel properties (Figure 2 and Table 1).
Figure 2.

Tunnels and phylogenetic relationships among representative members of the HLD family. (A) Main tunnels (p1) of the tag-optimized DhaAHT and five natural HLDs. These tunnels, connecting the active sites to the surface, are shown as a full surface, and their bottleneck regions highlighted by the arrows with the corresponding radii. The respective proteins are displayed on the right-bottom side of each tunnel as a cross section of their surface, with the active sites shown by the internal pockets and the catalytic nucleophile illustrated by the stars; the rectangles show the location of the tunnels on the proteins. The proteins were aligned and are presented from the same viewpoint. The tunnels and protein images were generated with Caver Analyst 2.0.50 (B) Phylogenetic tree of the proteins in (A) (in the same colors) and other natural HLDs from Table 1 (in black). Non-HLD enzymes are marked with a * sign: the close relative Rluc (Renilla-luciferin 2-monooxygenase, from Renilla reniformis), and the outgroup sequence DehH1 (haloacetate dehalogenase from Moraxella sp. (B)). The phylogenetic tree was constructed with FireProtASR51 and represented with iTOL.52
Table 1. Structural Characteristics of the Main Tunnel in Several Natural HLDs and Their Variantsac.

The main tunnel calculated with CAVER 3.0253 in the respective structures after adding hydrogens atoms using PyMOL 2.3.2;54 the tunnel origin was defined at the carboxylic O atoms of the nucleophile Asp106 (DhaA numeration); the probe radius is 0.6 Å; shell depth is 4 Å; and shell radius is 4 Å.
The mutations affecting the p1 tunnel are marked with asterisk; NA means not applicable.
The tunnels are represented as spheres and are all shown from the same viewpoint, being the active site located at the lower end of the tunnels and the protein surface at the top.
Length is the total length of the tunnel following its central line; the bottleneck is the narrowest part of the tunnel, and its radius is important to assess the tunnel permeability to small molecules; the curvature is given by the length/distance ratio, where distance is the shortest possible distance between the starting and ending points of the tunnel and is dimensionless.53
In this study, we present a comprehensive kinetic and computational study of the mechanism of the HaloTag ligand incorporation. We have investigated the effects of two different types of functional reporters, and compared DhaAHT, which was optimized by directed evolution, with three natural dehalogenases, DhaA, LinB, and DmmA. Strikingly, the most efficient reaction was not obtained for DhaAHT and tetramethylrhodamine ligand, albeit this pair was systematically optimized by directed evolution. The wild-type enzymes LinB and DmmA showed the highest incorporation efficiency with the 4-stilbazolium probes. Our current study proposes a new concept for selecting the optimal protein tag matching specific ligands, potentially leading to an improvement of the labeling efficiency and expanding the wide variety of HaloTag applications. The selection of the optimal enzyme–ligand pairs can also significantly reduce the risk of undesirable nonspecific interactions.
Results
The labeling reaction proceeds via a two-step kinetic pathway, the binding of the ligand and the following chemical conversion, leading to a stable covalent alkyl-enzyme complex. The latter unimolecular step cannot be easily optimized by modifying the labeling protocol and it depends solely on the optimal reactive orientation of the bound ligand. In this work, we performed a comprehensive kinetic and theoretical study on the incorporation of two different HaloTag ligands, the commercial tetramethylrhodamine (TMR) and three 4-stilbazolium-based ligands (1B, 1D, and 1E) with different lengths of the reactive linker (Figure 3). The 4-stilbazolium-based dyes have shown a stronger fluorogenic response upon labeling and easier synthetic routes than any of the previous HaloTag labels, which could be highly beneficial for their applicability.42 We compared DhaAHT, optimized by directed evolution, with analogues of the three natural dehalogenases DhaA, LinB, and DmmA. DhaAH272F, LinBH272F, and DmmAH315F, correspond to the natural enzymes with a single additional mutation in the catalytic base (H272F for DhaA and LinB, H315F in DmmA). This mutation in the catalytic histidine leads to the interruption of the catalytic cycle by preventing the hydrolytic step, and to the formation of the covalent alkyl-enzyme intermediate as the final complex.
Figure 3.

Kinetic mechanism of tetramethylrhodamine (TMR) and 4-stilbazolium ligand incorporation. (A) Chemical structure of TMR ligand (left), anisotropy kinetic traces obtained upon mixing 0.001 μM TMR with 0–0.064 μM DhaAHT (middle) and the time course of the concentration of binding complex (E.L, green) and covalent alkyl-enzyme complex (E-L, red) obtained by numerical simulation (right). (B) Chemical structure of 4-stilbazolium ligands (left), anisotropy kinetic traces obtained upon mixing 0.1 μM 1E with 0–2 μM LinBH272F (middle) and the time course of the concentration of binding complex (E.L, green) and covalent alkyl-enzyme complex (E-L, red) obtained by numerical simulation (right). The anisotropy experiments were performed at 30 °C in PBS with 0.01% (w/v) CHAPS and pH 7.4. The solid lines represent the best global fit to the kinetic data. (C) Scheme of the HLD reaction with a halogenated ligand. The chemical mechanism is adopted from Verschueren et al.55 The kinetic model of HaloTag reaction: E is the enzyme, L is the ligand, E.L is enzyme–ligand binding complex, E-L is the covalent alkyl-enzyme complex, k1 and k–1 are the rates of association and dissociation of the enzyme–ligand complex, respectively, and k2 represents the rate of the chemical step (nucleophilic substitution SN2). The MALDI-TOF MS experiments with DhaAHT and LinBH272F confirmed the formation of the covalent alkyl-enzyme complex (E-L) with both tetramethylrhodamine and 4-stilbazolium-based ligands (Figure S7 and Section III). (D) Kinetic parameters obtained by numerical analysis of anisotropy data. Error bars represent the standard error of the fitted parameters. The rigorous confidence contour analysis of variance of fitted parameters is presented in the Supporting Information (Table S5). The kinetic experiments were performed in two to three independent replicates.
Protein Expression and Purification
The haloalkane dehalogenases genes linBH272F, dhaAH272F, dhaAHT, and dmmAH315F were cloned into pAQN, pET21b or pET24a vectors and transformed into Escherichia coli BL21 or BL21(DE3) (Table S1). The enzymes were overexpressed and purified by metal-affinity chromatography. The purity of proteins was analyzed by SDS-PAGE (Figure S1). A detailed description of the gene cloning, expression, and purification are provided in the Supporting Information (Section I).
Kinetic Analysis
Fluorescence intensity and anisotropy were used to systematically analyze the concentration dependence of the reactions of DhaAHT, DhaAH272F, LinBH272F, and DmmAH315F with TMR and 1B, 1D, and 1E. The conventional fitting and numerical integration methods were applied to obtain detailed information about the individual rates and equilibrium constants related to the two-step model of the HaloTag reaction (Figure 3).
Initially, the kinetic data were analyzed by conventional exponential fitting using nonlinear regression. To compare the consistency of the data with earlier published results, the apparent second-order rate constants were calculated following the procedure used originally by Los and co-workers.4 The value of the apparent rate constant obtained for the incorporation of TMR into DhaAHT, 2.3 × 106 M–1·s–1, corresponds well with the value reported by Los and co-workers of 2.7 × 106 M–1·s–1.4 Next, the concentration dependence of the kinetic data (Figure 3A) was explored to provide detailed information about the kinetic pathway and estimate the true rate and equilibrium constants. Although the single-exponential fit of DhaAHT traces obtained with TMR provided satisfying statistics (χ2/DoF = 2.29; p-value = 0.28), the use of a double-exponential function showed significantly improved goodness of fit (χ2/DoF = 1.21; p-value = 0.43) and distinguished two separate phases, well consistent with the expected two-step kinetic model for the reaction (Figure 3C).
The concentration dependence of the obtained rates was analyzed analytically by a secondary fitting to the approximate rate equations derived for the two-step model (eqs S4 and S5). The analysis provided initial estimates of the rate constant for the association (k1 = 41 ± 4 μM–1·min–1) and dissociation (k–1 = 0.08 ± 0.04 min–1) for TMR. The DhaAHT-bound complex and the rate constant for the subsequent chemical step that result in the formation of the covalent alkyl-enzyme complex (k2 = 0.06 ± 0.04 min–1). In the case of the reaction of DhaAHT with the 4-stilbazolium-based ligands (Figure 3B), the binding phase gradually disappears in the dead-time of the measurement with increasing concentration of the enzyme, and only a single-exponential fit provided reasonable estimates for the rates and amplitudes. The concentration dependence of the observed rate (eq S6) allowed us to define the initial estimates of the equilibrium dissociation constant for the enzyme–ligand bound complex (KD = k–1/k1) ranging from 0.6 to 1.3 μM and the rate of the consecutive chemical step (k2) ranging from 0.03 to 0.33 min–1 for individual 4-stilbazolium ligands.
The conventional analysis of the kinetics of DhaAHT indicated substantial differences in the reaction with the two types of ligands. The TMR probe shows a rapid binding to a tight enzyme–ligand complex followed by a relatively slow chemical conversion leading to the final covalently bound complex. The accumulation of the reversible enzyme–ligand bound complex (E.L) is well visible from the anisotropic data (Figure 3A, middle), which shows a significant concentration dependence of the equilibrium signal with amplitudes defined by the equilibrium dissociation constant of the enzyme–ligand complex. The numerical simulation of the fraction of individual reaction species (Figure 3A, right) illustrates the course of the reaction involving the rapid binding of TMR into DhaAHT associated with the accumulation of the enzyme–ligand bound complex (E.L, green), which is slowly transformed into the final covalent alkyl-enzyme complex (E-L, red) (Figure 3A, right). In contrast, the kinetics of the 4-stilbazolium ligand reaction is dominated by the chemical step leading to the dominant accumulation of the final covalent complex (E-L, red). Anisotropic traces thus reach the same level of the signal in equilibrium, which is defined by the total ligand concentration (Figure 3B).
Although the conventional approach is currently the most widely used method of kinetic data analysis, it has several limitations, such as the loss of an important relationship between velocity and amplitude, or the accumulation of errors associated with the successive calculation of a large number of temporary parameters to estimate a small number of relevant kinetic constants. To overcome these limitations, we performed a global data analysis based on numerical methods. The parameter estimates obtained by conventional analysis (Figure S2 and Table S2) were used as initial starting values for the numerical fitting. A detailed description of the conventional and numerical analysis of the kinetics data, including a rigorous statistical assessment, is provided in the Supporting Information (Section II).
The global data fitting used numerical integration of the rate equations from an input model (Figure 3C) searching a unique set of kinetic parameters (Figure S3 and Table S3) that explain the original raw data and produce a minimum χ2 value.56 The observable anisotropy signal was defined as the sum of the contributions of each species to the total signal with scaling factors for each species (Table S4). In addition to monitoring the standard errors and residuals, the global fitting of kinetic data allowed us to perform a rigorous analysis of the variance referred to as a confidence contour analysis.57 This analysis confirmed the high quality of the global fit, with all of the obtained kinetic parameters being well constrained by the experimental data (Table S5). In the same way, the complex kinetic analysis was performed systematically comparing DhaAHT with three nonoptimized natural variants DhaAH272F, LinBH272F, and DmmAH315F in the reaction with TMR, 1B, 1D, and 1E (Figure S3 and Table S3). The specific rate constants defining the velocity of individual reaction steps, i.e., the ligand binding (k1) and the chemical conversion (k2), as well as the overall labeling efficiency, defined by K1.k2, the product of the equilibrium constant for the ground-state binding K1 = 1/KD = k1/k–1 and the rate of the consecutive chemical step k2, are summarized for each enzyme variant in Figure 3D.
Unlike TMR, which provides only the possibility of instrumentally more complex anisotropy/polarization measurements, the 4-stilbazolium-based ligands provide the additional advantage of tracking the fluorescence intensity signal (Figure S5), commonly available in most laboratories. The increase in fluorescence intensity observed upon the incorporation of ligands into the enzymes was 5-, 2-, and 10-fold for 1B, 1D, and 1E ligands, respectively. Such increases provide sufficient signal for in vitro enzymology studies. Additionally, the strong signal change observed especially for the 1E ligand provides a promising alternative to TMR in no-wash applications for cell labeling experiments.
We further analyzed nonspecific ligand binding by comparing the interaction of the ligands with the active free enzyme and with the enzyme with the blocked active site after reaction with a typical nonfluorescent HLDs substrate, 1-chlorohexane. We tested the effects of both types of ligands, TMR and the 4-stilbazolium ligand 1E, in the reaction with DhaAH272F (Figure S6). For the weaker nonoptimal interactions of DhaA272F with 1E, when the reaction needs to be performed at a high ligand concentration, the nonspecific binding was more pronounced in comparison to the strong interactions found with TMR. In the case that nonspecific binding might cause adverse effects on the target applications of the HaloTag technology, it is desirable to examine the extent of nonspecific interactions via this simple procedure. The results of such analysis should be considered for the selection of the optimal enzyme–ligand pair.
Computational Studies
DhaAHT, DhaAH272F, LinBH272F, and DmmAH315F were modeled from the crystal structures by in silico mutagenesis with Rosetta,58 and the respective access tunnels from the active site to the surface were calculated using CAVER 3.02.53 The tunnels found in DhaAHT, DhaAH272F, LinBH272F, and DmmAH315F showed considerably different geometric properties, especially the main tunnel, p1 (Figures 4A and S8). P1 is considerably wider in DhaAHT (bottleneck radius of 1.68 Å) than in DhaAH272F (1.29 Å) or LinBH272F (1.35 Å), and slightly wider than in DmmAH315F (1.62 Å). This fact suggests a higher accessibility of the DhaAHT active site compared to DhaAH272F and LinBH272F, which provides a first explanation for the generally higher binding rates of the probes to DhaAHT and DmmAH315F than to the other two proteins. Moreover, the orientation of p1 is very different in LinBH272F and DmmAH315F compared to that of the DhaA variants. This suggests that LinBH272F and DmmAH315F may have different chemical and geometric preferences for the ligands that they can bind, compared to DhaAHT and DhaAH272F.
Figure 4.

Molecular modeling of DhaAHT and LinBH272F and their binding to TMR and 1E. (A) The main access tunnel p1 (blue) and the slot tunnel p2 (green). (B) Structures of the complexes in the bound state obtained from Markov state analysis of the molecular dynamics simulations with a superimposition of the respective probes (magenta). (C) Potential energy difference (ΔEp) of the complex during the SN2 reaction between DhaAHT and TMR, obtained from a QM/MM adiabatic mapping of the distance between the reacting atoms of the protein (D106-COO–) and the TMR probe (CH2Cl), with respect to its ground state. ΔG‡ is the activation barrier of the reaction, where the ground state (GS), transition state (TS), and ligand–enzyme covalent complex (CC) are depicted. TMR is shown as magenta sticks, the chloride ion is shown as the green ball, and the nucleophile D106 and the halide-stabilizing residues N41 and W107 are shown as gray sticks.
To understand the large differences found in the experimental kinetic measurements, we selected two representative probes (TMR and 1E) and two proteins (DhaAHT and LinBH272F) to study their molecular binding in more detail. These systems were chosen because of the high binding specificity found among two of the corresponding pairs, i.e., DhaAHT with TMR and LinBH272F with 1E. The TMR and 1E ligands were modeled and then refined with the Density Function Theory, which provided the energy-minimized structures and the partial atomic charges (see Supporting Information, Section IV, for details). The binding of both probes to DhaAHT and LinBH272F was studied by molecular dynamics (MD), using the adaptive sampling approach.59 The simulations started with the probes located in the bulk solvent, and consecutive rounds (epochs) of multiple MD simulations were performed. According to the adaptive sampling method, the starting points for the new MDs in each epoch were chosen from the previously sampled states based on the distance between the reacting groups in the probe and the enzyme. Each system was simulated for a total time of 20 μs. Markov state model (MSM) analysis was performed to obtain the relevant kinetic ensembles describing the binding of the molecular probes to the active sites of the proteins. Four Markov states could describe well the binding process, consisting of one fully bound state, two intermediates, and a fully unbound state, in which the reacting carbon atom of the probe was well inserted in the active site (Figures 4B, S12, and S13). The kinetic parameters were calculated for the transitions between the most unbound state and the fully bound state. The results showed that the estimated binding rates (DhaAHT: k1 = 2.99 ± 0.45 × 108 M–1·s–1 for TMR, 1.09 ± 0.23 × 108 M–1·s–1 for 1E; LinBH272F: k1 = 7.6 ± 2.5 × 107 M–1·s–1 for TMR, 1.22 ± 0.21 × 108 M–1·s–1 for 1E; see Table S7) followed the exact same order as the experimental ones, and k1 was highest for DhaAHT with TMR, and lowest for LinBH272F with TMR. The computational and experimental rates, however, differed by several orders of magnitude, being higher for the theoretical values. Such discrepancy is not unprecedented59,60 and will be discussed below. Regarding the unbinding rates (k–1; Table S7), they were all slower than the binding, which is consistent with the majority of the experimental results obtained here, although the order was not strictly observed. The slowest unbinding was predicted for TMR with DhaAHT, while the experiments showed the lowest unbinding rate for TMR with LinBH272F. The predicted binding affinity, given by K1 = 1/KD, also partially followed the experimental trends, where DhaAHT and TMR were correctly predicted with the highest affinity. However, the predicted order among the other pairs was incorrect, where LinBH272F and 1E showed experimentally the second strongest affinity. Interestingly, the highest probability of the bound state (Pbound) was obtained for TMR with DhaAHT, with 0.260 ± 0.043, and the lowest was found for 1E with LinBH272F, with 0.047 ± 0.008 (Table S7).
Inspecting visually the bound states, we found that, in every system, the probes used exclusively the p1 tunnel (Figures 4B and S13). We also observed that the 4-stilbazolium aromatic system of 1E was partially inserted in the tunnel, both in DhaAHT and LinBH272F. Conversely, due to its longer linker, TMR presented its aromatic moiety completely outside of the protein. Moreover, the bound conformations of TMR followed the natural orientation of the p1 tunnel in DhaAHT, while 1E followed the tilted orientation of the p1 tunnel in LinBH272F (Figure 4). This interesting finding is in line with the fact that 1E is a better binder with LinBH272F and DmmAH315F than TMR. It also suggests a higher complementarity of the 4-stilbazolium-based probes with the LinB and DmmA variants in comparison with TMR. Analyzing in more detail the interactions found in the LinBH272F-1E complex, we found that in the bound state the aromatic system formed close hydrophobic contacts with L150, V173, L177, and L248, located in the tunnel, and with L179, P245, A247, and A271, located on the extension of the tunnel mouth. Interestingly, the negatively charged D147 residue also formed electrostatic interactions with the 4-stilbazolium system due to its delocalized positive charge. We expect that the many interactions and constraints of the aromatic system of 1B, 1D, and 1E within the access tunnel contribute to strong fluorescence effects upon binding, as Clark and co-workers42 have previously suggested.
Next, we predicted the reactivity of the TMR and 1E probes toward DhaAHT and LinBH272F and compared them with the experimental results. We started by analyzing the pre-reactive complexes found during the respective MDs, hereafter termed as near-attack conformation (NAC). We estimated the constant of formation of this pre-reactive complex, KNAC, based on the total number of NACs found and the probability of the bound state, Pbound. Surprisingly, the highest KNAC was obtained for 1E with LinBH272F, and the lowest for TMR with DhaAHT (Table S7), with a difference of nearly 2 orders of magnitude. This suggests that, despite the binding of TMR with DhaAHT being extremely fast, the probability of the system adopting a potentially reactive conformation is very low. In contrast, once 1E and LinBH272F reach the bound state, the reactive conformation is achieved much faster than for any of the other systems. This perfectly follows the trends of the experimental kinetic rates obtained for the chemical step (k2 in Figure 3 and Table S7). We then applied a hybrid QM/MM adiabatic mapping (the QM region was described with the semiempirical PM6 level of theory61) to estimate the energy barriers of the SN2 reaction, ΔGSN2‡ (Figure 4C). The predicted ΔGSN2‡ values (Table S7) showed the lowest activation barrier for LinBH272F-TMR (12.1 ± 1.9 kcal·mol–1), followed by LinBH272F-1E (13.8 ± 1.8 kcal·mol–1), and the highest barrier for DhaAHT-TMR (15.5 ± 1.3 kcal·mol–1). This indicates that once the NAC has been achieved, LinBH272F provides a better environment for performing the SN2 step with both of the probes than DhaAHT.
Finally, the KNAC and ΔGSN2‡ were combined (Supporting Information, Section IV) to estimate the overall activation energy of the second kinetic step, ΔG2‡. This step 2 (Figure 3C) is a direct measure of reactivity, and the estimated and experimental values of ΔG2‡ can be directly compared. As a result, the highest calculated ΔG2‡ value was obtained for DhaAHT with TMR, 2.5 kcal·mol–1 above the second highest energy barrier of DhaAHT-1E and 4.4 kcal·mol–1 above the ΔG2‡ value for LinBH272F-1E (Table S7). This is in reasonably good agreement with the experimental values, where DhaAHT with TMR also showed the highest ΔG2‡ value, 2.7 kcal·mol–1 above that of LinBH272F with 1E.
Discussion
We conducted a detailed kinetic and computational study of the reaction of a tetramethylrhodamine-based (TMR) and three 4-stilbazolium-based ligands (1B, 1D, and 1E) with reacting linkers of different lengths, and the haloalkane dehalogenase optimized by directed evolution (DhaAHT) and the analogues of three natural dehalogenases (DhaAH272F, LinBH272F, and DmmAH315F). The kinetic study showed substantial differences in the reaction kinetics between the individual enzymes as well as among the different ligands. The TMR probe showed a very high rate of binding toward the engineered DhaAHT (k1 = 39.7 ± 0.6 μM–1·min–1), which is in high contrast with the other studied enzymes, which showed rates of binding ranging between 0.001 and 0.4 μM–1·min–1. In comparison to the nonoptimized DhaAH272F, the engineering of the access tunnel of DhaA44 led to a significant improvement of the ligand binding, while it did not compromise the catalytic step. The engineered DhaAHT showed even a slightly decreased activation barrier of the chemical step (ΔΔG2‡ = −0.6 kcal·mol–1), although the ground state energy (ΔΔG0) of the enzyme–ligand complex was significantly lower (−3.3 kcal·mol–1) in comparison to DhaAH272F (Table S6 and Figure S4). Interestingly, the nonoptimized LinBH272F showed a slow binding toward TMR, but it displayed the highest rate of the consecutive chemical conversion leading to the formation of the covalent enzyme-TMR complex.
The importance of the chemical step for the efficiency of the HaloTag labeling reactions is more pronounced with the 4-stilbazolium-based ligands. Even though the binding of 1E into LinBH272F is orders of magnitude slower and weaker, the elevated velocity of the following chemical step ensures the fully comparable labeling efficiency of 1E with LinBH272F (K1·k2 = 3.0 μM–1·min–1) to that observed for the reaction of TMR with the engineered DhaAHT (K1·k2 = 3.1 μM–1·min–1). The weaker binding of 1E to LinBH272F (ΔΔG0 = 2.8 kcal·mol–1) is compensated by a lower activation energy, resulting in a fast consecutive chemical conversion (ΔΔG‡ = −2.8 kcal·mol–1). The reaction of 1E with LinBH272F illustrates that the desired labeling efficiency can be achieved not only by an improved ligand binding, but also by an acceleration of the chemical reaction. The reaction mechanism observed for LinBH272F can be explained by the specific architecture of its access tunnel. The narrower tunnel bottleneck compromises to some extent the ligand transport, but at the same time reduces the active site solvation and makes the productive binding more probable. Moreover, the tunnel lining residues lower the initial entropy and promote the contact of the reacting atoms, possibly through specific interactions with the ligand. All of these effects may have a positive effect on increasing the rate of the carbon-halogen bond cleavage (SN2), and have been described in previous studies focused on the engineering of access tunnels, for both DhaA and LinB.62−65 However, the narrow architecture of the access tunnel in LinB makes this enzyme more sensitive to the length of the reactive linker. This is important to allow the formation of favorable interactions between the aromatic system of the probes and the residues lining the tunnel. The eight-carbon linker of 1E was the only one providing optimal length for LinB labeling since shorter linkers have not been able to achieve even remotely the efficiency of 1E.
Wider and more accessible access tunnels seem to be more universal, as evidenced by the reaction of the nonoptimized DmmAH315F with 4-stilbazolium ligands. DmmA has the most open main tunnel ensuring easy access of the substrates to its active site. The reaction of DmmAH315F with all 4-stilbazolium-based ligands showed rapid binding, but also rapid chemical steps. The resulting labeling efficiency thus surpasses the commercial reaction of TMR with DhaAHT. The reaction of LinBH272F with 1E and DmmAH315F with all of the 4-stilbazolium ligands showed that natural variants can provide high efficiency useful for HaloTag applications without time-demanding protein optimization by directed evolution. It is also interesting that, just as DhaAHT is highly specific for the reaction with TMR, DmmAH315F showed high efficiency only in reaction with the 4-stilbazolium-based ligands, but not with TMR. Clearly, it is important to select an appropriate protein for the binding of a specific ligand. Selection of the appropriate protein can lead to a significant improvement in labeling efficiency without the need for costly enzyme or ligand optimization. An example is the laborious and time-consuming chemical development of the dimerization-inducing HaXS ligand29 for improved reactivity with DhaAHT. This might have been avoided by exploring the diverse pool of natural HLDs.
Using several computational methods, we simulated and predicted the kinetics and thermodynamics of the two-step binding process of four representative fluorescent probe/protein systems, namely, for the TMR and 1E probes with DhaAHT and LinBH272F. We found disparities in the absolute values of the calculated kinetic rates. Such differences have been reported previously59,60 and can be attributed to the bias intrinsic to the simulation method (adaptive sampling) or the conditions used in our MD simulations, namely, the force field and the solvent model. The ligand transport in proteins is highly influenced by the solvent and its respective bulk properties, such as diffusivity. In spite of being one of the most widely used water models in molecular simulations, the TIP3P model has a higher diffusivity than pure water. It is also known to overestimate the diffusion properties of amino acids,66 and we can presume that that same holds for many other solvated molecules. Importantly, our results showed significant correlations with some of the experimental parameters and revealed important clues for different aspects of the molecular binding on focus here. We could qualitatively replicate the order in the k1 binding rates, with DhaAHT-TMR showing the highest k1 value, followed by LinBH272F-1E, and partially the order of affinities. The unbinding rates, however, were less consistent with the experimental results. This may be due to a possible undersampling of the unbinding events. While the binding of the probes was always relatively fast (τ1 ≈ 102–103 ns), sometimes the unbinding took place in time scales near that of the total simulation time. This was also reflected in relatively high standard deviations associated with some constants (e.g., k–1 and KD). Nonetheless, since our main goal was to investigate the binding of the probes, we did not extend the MDs further. The simulation of the binding process also revealed how the probes interacted differently with the proteins. Both used solely p1 tunnel to bind the proteins, but the preferred orientation of TMR was more compatible with the geometry of the p1 tunnel in the DhaA variants, while 1E adopted an orientation more similar to the p1 tunnel found in the native LinBH272F and DmmAH315F. This reveals a complementarity intrinsic to those two pairs that seem to explain the labeling efficiencies described above. The large number of interactions formed between 1E and the residues lining the tunnel of LinBH272F supports this hypothesis.
The overall chemical step was dissected into the pre-organization of the bound state to form the pre-reactive complex (NAC) and the SN2 reaction. We estimated these two partial steps from our computational approach and calculated the total activation energy of the second kinetic step (ΔG2‡), which can be compared with the parameter determined experimentally. We found that DhaAHT-TMR displayed the lowest overall reactivity (with the highest ΔG2‡ value), which is in good agreement with the experimental data. It showed not only the worst efficiency in achieving a productive binding mode (lowest KNAC) but also presented the highest activation barrier to the SN2 reaction. Conversely, LinBH272F-1E was the most efficient system in adopting the pre-reactive conformation after the binding (highest KNAC). A low ΔGSN2‡ also resulted in LinBH272F-1E having a rather low activation barrier to the overall chemical step, with an estimated ΔG2‡ value below that of DhaAHT-TMR by 4.4 kcal·mol–1. Some of the discrepancies between the theoretical and experimental values were likely due to a poor sampling of the fully bound states, which may have not been sufficient to provide an accurate ensemble distribution of the pre-reactive state. However, our results provided sufficient clues to explain why, although DhaAHT-TMR presented the highest binding rate, its reactivity is very far from ideal. In contrast, although the LinBH272F-1E system had poorer binding rates, it is much more efficient on the chemical step. Overall, the binding of the 1E probe to the nonoptimized LinBH272F protein revealed a reasonable binding/reactivity trade-off, which resulted in a labeling efficiency very close to that of DhaAHT-TMR.
Some of the effects discussed above can be extrapolated to the DmmAH315F-1E system, which presented the best binding efficiency among all of the tested pairs. Hence, we hypothesize that the binding of 1E to DmmAH315F is fast due to the combination of a sufficiently wide access tunnel and a good complementarity of its architecture with the 1E probe, which lead to a high number of favorable interactions. Second, strong probe-enzyme interactions can contribute to a stable and highly reactive DmmAH315F-1E complex, thus leading to a fast chemical step. The combination of a fast binding and a fast chemical step resulted in a system with the highest labeling efficiency.
Conclusions
Here we have demonstrated that not only the ligand accessibility is important for the binding of diverse probes to HaloTag proteins, but also the subsequent chemical step can significantly affect the ligand specificity and labeling efficiency. We have identified substantial differences in the kinetics of binding and chemical reaction between individual enzymes with different ligands. The TMR probe showed a rapid binding to DhaAHT, which was followed by a slow chemical conversion to the alkyl-enzyme complex. In contrast, the binding of the 4-stilbazolium-based ligands to DhaAHT and other tag proteins was much slower than with TMR, but the chemical step was greatly improved in most cases. Interestingly, we found that the best efficiencies for the incorporation of several 4-stilbazolium-based probes (namely, 1D and 1E) were achieved with the analogues of natural nonoptimized dehalogenases, LinBH272F and DmmAH315F, which provided high kinetic rates for both binding and chemistry. This demonstrates that different natural proteins can be effective for the incorporation of specific probes without the need for demanding protein engineering procedures. Moreover, the 4-stilbazolium-based ligands, due to a better light-up response upon binding, may provide better detection limits and thus could be preferable to the traditional probes, e.g., for simple fluorescence assays, analysis of binding interactions, or microscopy imaging.
We propose that, before conducting laborious optimization rounds by directed evolution, a rapid screening of the available natural dehalogenases could lead to the identification of potential candidates for optimal tag proteins. Thus, one could benefit from the very diverse pool of tunnel architectures already available among the known haloalkane dehalogenases. Calculation of the respective access tunnels with CAVER and molecular docking could provide good first filters for this selection. The subsequent utilization of more robust computational methods, like molecular dynamics and quantum mechanics, can help identify the ideal enzyme–probe pairs. The HaloTag optimization strategy described here should lead to a significant improvement of the labeling efficiency in a wide range of HaloTag applications. Moreover, the selection of the optimal enzyme–ligand pair can also significantly reduce the risk of undesirable nonspecific interactions.
Materials and Methods
The materials and methods are described in detail in the Supporting Information.
Protein Expression and Purification
All of the studied dehalogenases were expressed in E. coli BL21 or BL21(DE3) cells and purified from cell-free extracts by metal-affinity chromatography using Ni-NTA Superflow column (Qiagen, Germany). Dialysis or gel permeation chromatography was used for buffer exchange to phosphate-buffered saline (pH 7.4).
Fluorescence Intensity and Anisotropy Measurements
The fluorescence was monitored using an Infinite F500 plate reader (Tecan, Switzerland) equipped with polarization filters with excitation/emission wavelengths 544/620 nm or 544/580 nm at 30 °C. The reaction mixture contained 0.001–0.1 μM of the fluorescent ligand and 0.001–8 μM of the enzyme in PBS (pH 7.4) with 0.01% of CHAPS. The signal from the enzyme-free sample was measured as a negative control. To rule out the effects of nonspecific interactions on the kinetic data, we analyzed the signal from DhaAH272F and LinBH272 with TMR and 1E ligand after blocking their active sites with 1-chlorohexane.
Kinetic Data Analysis and Statistics
The conventional analysis was performed by fitting the kinetic data with a nonlinear regression using exponential functions in the KinTek Explorer software56 (KinTek). The dependence of the observed rates on the enzyme concentration was analyzed using the Origin 6.0 software (OriginLab), to derive the kinetic constants.
The global analysis of the kinetic data was performed using the Kintek Explorer software56 (KinTek). Numerical integration of the rate equations from an input reaction model was used to search for the set of parameters which produced the minimum χ2 values (calculated based on the Levenberg–Marquardt method). The correctness of the obtained kinetic constants was verified using the FitSpace Explorer57 (KinTek).
MALDI-TOF Mass Spectrometry
MALDI-TOF MS experiments were performed with the proteins DhaAHT, LinBH272F, and DhaA31H272F, and the probes 1D and TMR, using an Ultraflextreme instrument (Bruker Daltonics, Billerica, Germany) operated in linear mode for detecting positive ions.
Computational Analysis
The structures of DhaAHT, DhaAH272F, LinBH272F, and DmmAH315F were modeled using the ddg_monomer module of Rosetta.58 For that, we used the crystal structures of the respective wild-types, obtained from the RCSB Protein Data Bank67 (PDB entry 4E46 for DhaA, 1MJ5 for LinB, and 3U1T for DmmA), and the talaris2014(68,69) force field. The access tunnels were calculated on the static structures of the different proteins using CAVER 3.02,53 defining the origin as the carboxylic O atoms of the catalytic aspartate.
The ligands were constructed and minimized with Avogadro 270 using the UFF force field.71TMR or 1E were further minimized with Gaussian 09,72 with the B3LYP hybrid functional, the 6-311+G(d,p) basis set, and implicit solvent (the Polarizable Continuum Model). The protonation state of DhaAHT and LinBH272F at pH 7.5 was predicted by the H++ server.73 The systems were prepared using scripts from the High Throughput Molecular Dynamics (HTMD)74 package. The TMR or 1E ligands were randomly placed at least 5 Å from the protein, and we added a cubic water box of TIP3P75 molecules with the edges at least 10 Å away from the protein, and Cl– and Na+ ions to neutralize the system and achieve 0.1 M concentration of salt. The proteins were described by the ff14SB76 AMBER force field, and the ligands by the General Amber force field (GAFF).
Molecular dynamics (MD) simulations were performed with HTMD.74 An equilibration cycle included several steps of minimization and dynamics (5 ns in total), with the Berendsen barostat, Langevin thermostat at 300 K, periodic boundary conditions, and 4 fs time steps. Adaptive sampling MDs were then performed using as the adaptive metric the distance between the reacting groups in the ligands and the proteins. A total of 50 epochs of 10 individual MDs of 40 ns each were performed, corresponding to a cumulative simulation time of 20 μs. The binding process was studied by analyzing the simulations with Markov state models (MSM), projecting the same metric used in the adaptive MDs. This allowed the estimation of kinetic rates and equilibrium populations of bound and unbound states, as previously described.59 The pre-reactive complexes present in the MDs were identified using geometric criteria according to Hur et al.77 To calculate the energy barriers of the SN2 reaction (ΔG‡) between the ligands and the enzymes, the pre-reactive complexes were submitted to adiabatic mapping along the reaction coordinate (decreasing the C–O distance), using hybrid QM/MM calculations78 with AMBER 16.79 The QM part of the system was described by the semiempirical PM661 Hamiltonian and the MM part by the ff14SB76 force field.
Acknowledgments
This work was supported by the Czech Ministry of Education (Grants INBIO CZ.02.1.01/0.0/0.0/16_026/0008451, ESFRI RECETOX LM2018121, and ESFRI ELIXIR LM2018131) and the European Union (SinFonia 814418 and TEAMING 857560). The computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures. M.M. acknowledges support from the internal Grant Agency of the Masaryk University (MUNI/H/1561/2018) and the Czech Science Foundation (22-09853S). E.T.K. acknowledges support from the U.S. National Cancer Institute (CA217809).
Glossary
Abbreviations
- HLD
haloalkane dehalogenase
- TMR
tetramethylrhodamine
- PBS
phosphate-buffered saline
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- MSM
Markov state model
- NAC
near-attack conformation
- QM/MM
quantum mechanics/molecular mechanics
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00002.
Additional and detailed materials and methods; additional results and discussions; and the respective schemes, figures, and tables (PDF)
Author Contributions
⊥ S.M.M. and M.S. contributed equally to this study.
The authors declare no competing financial interest.
Supplementary Material
References
- Pieters R. J.; Fennema M.; Kellogg R. M.; Janssen D. B. Design and Synthesis of Reagents for Phage Display Screening of Dehalogenases. Bioorg. Med. Chem. Lett. 1999, 9, 161–166. 10.1016/s0960-894x(98)00697-0. [DOI] [PubMed] [Google Scholar]
- Los G. V.; Darzins A.; Karassina N.; Zimprich C.; Learish R.; McDougall M. G.; Encell L. P.; Friedman-Ohana R.; Wood M.; Vidugiris G.; Zimmerman K.; Otto P.; Klaubert D. H.; Wood K. V. HaloTag Interchangeable Labeling Technology for Cell Imaging and Protein Capture. Cell Notes 2005, 11, 2–6. [Google Scholar]
- Los G. V.; Wood K.. The HaloTagTM: A Novel Technology for Cell Imaging and Protein Analysis. In High Content Screening: A Powerful Approach to Systems Cell Biology and Drug Discovery; Taylor D. L.; Haskins J. R.; Giuliano K. A., Eds.; Humana Press: Totowa, NJ, 2006; pp 195–208. [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Mishra V. Affinity Tags for Protein Purification. Curr. Protein Pept. Sci. 2020, 21, 821–830. 10.2174/1389203721666200606220109. [DOI] [PubMed] [Google Scholar]
- Döbber J.; Pohl M. HaloTag: Evaluation of a Covalent One-Step Immobilization for Biocatalysis. J. Biotechnol. 2017, 241, 170–174. 10.1016/j.jbiotec.2016.12.004. [DOI] [PubMed] [Google Scholar]
- Sun C.; Li Y.; Taylor S. E.; Mao X.; Wilkinson M. C.; Fernig D. G. HaloTag Is an Effective Expression and Solubilisation Fusion Partner for a Range of Fibroblast Growth Factors. PeerJ 2015, 3, e1060 10.7717/peerj.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Wang S.; Kool E. T. Genetically Encoded Multispectral Labeling of Proteins with Polyfluorophores on a DNA Backbone. J. Am. Chem. Soc. 2013, 135, 6184–6191. 10.1021/ja4004393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelzel C. A.; Zhang X. Visualizing and Manipulating Biological Processes by Using HaloTag and SNAP-Tag Technologies. ChemBioChem 2020, 21, 1935–1946. 10.1002/cbic.202000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N.; Ogawa M.; Choyke P. L.; Karassina N.; Corona C.; McDougall M.; Lynch D. T.; Hoyt C. C.; Levenson R. M.; Los G. V.; Kobayashi H. In Vivo Stable Tumor-Specific Painting in Various Colors Using Dehalogenase-Based Protein-Tag Fluorescent Ligands. Bioconjugate Chem. 2009, 20, 1367–1374. 10.1021/bc9001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Zhang Z.; Li L.; Chen B.-C.; Revyakin A.; Hajj B.; Legant W.; Dahan M.; Lionnet T.; Betzig E.; Tjian R.; Liu Z. Single-Molecule Dynamics of Enhanceosome Assembly in Embryonic Stem Cells. Cell 2014, 156, 1274–1285. 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; English B. P.; Chen J.; Slaughter J. P.; Zhang Z.; Revyakin A.; Patel R.; Macklin J. J.; Normanno D.; Singer R. H.; Lionnet T.; Lavis L. D. A General Method to Improve Fluorophores for Live-Cell and Single-Molecule Microscopy. Nat. Methods 2015, 12, 244–250. 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas-Pardo J. A.; Li Y.; Mártonfalvi Z.; Tapia-Rojo R.; Unger A.; Fernández-Trasancos Á.; Herrero-Galán E.; Velázquez-Carreras D.; Fernández J. M.; Linke W. A.; Alegre-Cebollada J. A HaloTag-TEV Genetic Cassette for Mechanical Phenotyping of Proteins from Tissues. Nat. Commun. 2020, 11, 2060 10.1038/s41467-020-15465-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urh M.; Hartzell D.; Mendez J.; Klaubert D. H.; Wood K. Methods for Detection of Protein-Protein and Protein-DNA Interactions Using HaloTag. Methods Mol. Biol. 2008, 3, 191–209. 10.1007/978-1-59745-582-4_13. [DOI] [PubMed] [Google Scholar]
- Minner-Meinen R.; Weber J.-N.; Albrecht A.; Matis R.; Behnecke M.; Tietge C.; Frank S.; Schulze J.; Buschmann H.; Walla P. J.; Mendel R.-R.; Hänsch R.; Kaufholdt D. Split-HaloTag Imaging Assay for Sophisticated Microscopy of Protein-Protein Interactions in Planta. Plant Commun. 2021, 2, 100212 10.1016/j.xplc.2021.100212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Fares M.; Dunham N. P.; Gao Z.; Miao K.; Jiang X.; Bollinger S. S.; Boal A. K.; Zhang X. AgHalo: A Facile Fluorogenic Sensor to Detect Drug-Induced Proteome Stress. Angew. Chem., Int. Ed. 2017, 56, 8672–8676. 10.1002/anie.201702417. [DOI] [PubMed] [Google Scholar]
- Fares M.; Li Y.; Liu Y.; Miao K.; Gao Z.; Zhai Y.; Zhang X. A Molecular Rotor-Based Halo-Tag Ligand Enables a Fluorogenic Proteome Stress Sensor to Detect Protein Misfolding in Mildly Stressed Proteome. Bioconjugate Chem. 2018, 29, 215–224. 10.1021/acs.bioconjchem.7b00763. [DOI] [PubMed] [Google Scholar]
- Wolstenholme C. H.; Hu H.; Ye S.; Funk B. E.; Jain D.; Hsiung C.-H.; Ning G.; Liu Y.; Li X.; Zhang X. AggFluor: Fluorogenic Toolbox Enables Direct Visualization of the Multi-Step Protein Aggregation Process in Live Cells. J. Am. Chem. Soc. 2020, 142, 17515–17523. 10.1021/jacs.0c07245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K. H.; Kim S. F.; Liu Y.; Zhang X. A Fluorogenic AggTag Method Based on Halo- and SNAP-Tags to Simultaneously Detect Aggregation of Two Proteins in Live Cells. ChemBioChem 2019, 20, 1078–1087. 10.1002/cbic.201800782. [DOI] [PubMed] [Google Scholar]
- Sykora J.; Brezovsky J.; Koudelakova T.; Lahoda M.; Fortova A.; Chernovets T.; Chaloupkova R.; Stepankova V.; Prokop Z.; Smatanova I. K.; Hof M.; Damborsky J. Dynamics and Hydration Explain Failed Functional Transformation in Dehalogenase Design. Nat. Chem. Biol. 2014, 10, 428–430. 10.1038/nchembio.1502. [DOI] [PubMed] [Google Scholar]
- Amaro M.; Brezovský J.; Kováčová S.; Sýkora J.; Bednář D.; Němec V.; Lišková V.; Kurumbang N. P.; Beerens K.; Chaloupková R.; Paruch K.; Hof M.; Damborský J. Site-Specific Analysis of Protein Hydration Based on Unnatural Amino Acid Fluorescence. J. Am. Chem. Soc. 2015, 137, 4988–4992. 10.1021/jacs.5b01681. [DOI] [PubMed] [Google Scholar]
- Popa I.; Rivas-Pardo J. A.; Eckels E. C.; Echelman D. J.; Badilla C. L.; Valle-Orero J.; Fernández J. M. A HaloTag Anchored Ruler for Week-Long Studies of Protein Dynamics. J. Am. Chem. Soc. 2016, 138, 10546–10553. 10.1021/jacs.6b05429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peier A.; Ge L.; Boyer N.; Frost J.; Duggal R.; Biswas K.; Edmondson S.; Hermes J. D.; Yan L.; Zimprich C.; Sadruddin A.; Kristal Kaan H. Y.; Chandramohan A.; Brown C. J.; Thean D.; Lee X. E.; Yuen T. Y.; Ferrer-Gago F. J.; Johannes C. W.; Lane D. P.; Sherborne B.; Corona C.; Robers M. B.; Sawyer T. K.; Partridge A. W. NanoClick: A High Throughput, Target-Agnostic Peptide Cell Permeability Assay. ACS Chem. Biol. 2021, 16, 293–309. 10.1021/acschembio.0c00804. [DOI] [PubMed] [Google Scholar]
- Parvez S.; Fu Y.; Li J.; Long M. J. C.; Lin H.-Y.; Lee D. K.; Hu G. S.; Aye Y. Substoichiometric Hydroxynonenylation of a Single Protein Recapitulates Whole-Cell-Stimulated Antioxidant Response. J. Am. Chem. Soc. 2015, 137, 10–13. 10.1021/ja5084249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S.; Long M. J. C.; Lin H.-Y.; Zhao Y.; Haegele J. A.; Pham V. N.; Lee D. K.; Aye Y. T-REX on-Demand Redox Targeting in Live Cells. Nat. Protoc. 2016, 11, 2328–2356. 10.1038/nprot.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X.; Fu Y.; Long M. J. C.; Haegele J. A.; Ge E. J.; Parvez S.; Aye Y. Temporally Controlled Targeting of 4-Hydroxynonenal to Specific Proteins in Living Cells. J. Am. Chem. Soc. 2013, 135, 14496–14499. 10.1021/ja405400k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K.; Tae H. S.; Schneekloth A. R.; Stulberg M. J.; Corson T. W.; Sundberg T. B.; Raina K.; Holley S. A.; Crews C. M. Small-Molecule Hydrophobic Tagging-Induced Degradation of HaloTag Fusion Proteins. Nat. Chem. Biol. 2011, 7, 538–543. 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Raina K.; Darricarrere N.; Hines J.; Gustafson J. L.; Smith I. E.; Miah A. H.; Harling J. D.; Crews C. M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhart D.; Zimmermann M.; Jacques O.; Wittwer M. B.; Ernst B.; Constable E.; Zvelebil M.; Beaufils F.; Wymann M. P. Chemical Development of Intracellular Protein Heterodimerizers. Chem. Biol. 2013, 20, 549–557. 10.1016/j.chembiol.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Gu L.; Li C.; Aach J.; Hill D. E.; Vidal M.; Church G. M. Multiplex Single-Molecule Interaction Profiling of DNA-Barcoded Proteins. Nature 2014, 515, 554–557. 10.1038/nature13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noblin D. J.; Page C. M.; Tae H. S.; Gareiss P. C.; Schneekloth J. S.; Crews C. M. A HaloTag-Based Small Molecule Microarray Screening Methodology with Increased Sensitivity and Multiplex Capabilities. ACS Chem. Biol. 2012, 7, 2055–2063. 10.1021/cb300453k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki J.; Galli M.; Kim A. Y.; Nito K.; Aleman F.; Chang K. N.; Carvunis A.-R.; Quan R.; Nguyen H.; Song L.; Alvarez J. M.; Huang S.-S. C.; Chen H.; Ramachandran N.; Altmann S.; Gutiérrez R. A.; Hill D. E.; Schroeder J. I.; Chory J.; LaBaer J.; Vidal M.; Braun P.; Ecker J. R. Mapping Transcription Factor Interactome Networks Using HaloTag Protein Arrays. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E4238–E4247. 10.1073/pnas.1603229113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benink H.; McDougall M.; Klaubert D.; Los G. Direct PH Measurements by Using Subcellular Targeting of 5(and 6-) Carboxyseminaphthorhodafluor in Mammalian Cells. BioTechniques 2009, 47, 769–774. 10.2144/000113220. [DOI] [PubMed] [Google Scholar]
- Taguchi R.; Terai T.; Ueno T.; Komatsu T.; Hanaoka K.; Urano Y. A Protein-Coupled Fluorescent Probe for Organelle-Specific Imaging of Na+. Sens. Actuators, B 2018, 265, 575–581. 10.1016/j.snb.2018.03.090. [DOI] [Google Scholar]
- Li D.; Liu L.; Li W.-H. Genetic Targeting of a Small Fluorescent Zinc Indicator to Cell Surface for Monitoring Zinc Secretion. ACS Chem. Biol. 2015, 10, 1054–1063. 10.1021/cb5007536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa I.; Berkovich R.; Alegre-Cebollada J.; Badilla C. L.; Rivas-Pardo J. A.; Taniguchi Y.; Kawakami M.; Fernandez J. M. Nanomechanics of HaloTag Tethers. J. Am. Chem. Soc. 2013, 135, 12762–12771. 10.1021/ja4056382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Caballero A.; Echelman D. J.; Tapia-Rojo R.; Haldar S.; Eckels E. C.; Fernandez J. M. Protein Folding Modulates the Chemical Reactivity of a Gram-Positive Adhesin. Nat. Chem. 2021, 13, 172–181. 10.1038/s41557-020-00586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbujo S.; Galbiati E.; Salvioni L.; Mazzucchelli M.; Frascotti G.; Sun X.; Megahed S.; Feliu N.; Prosperi D.; Parak W. J.; Colombo M. Functionalization of Colloidal Nanoparticles with a Discrete Number of Ligands Based on a “HALO-Bioclick” Reaction. Chem. Commun. 2020, 56, 11398–11401. 10.1039/d0cc04355a. [DOI] [PubMed] [Google Scholar]
- Liu D. S.; Phipps W. S.; Loh K. H.; Howarth M.; Ting A. Y. Quantum Dot Targeting with Lipoic Acid Ligase and HaloTag for Single-Molecule Imaging on Living Cells. ACS Nano 2012, 6, 11080–11087. 10.1021/nn304793z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakariya H.; Takashima I.; Takemoto M.; Noda N.; Sato S.-I.; Uesugi M. Non-Genetic Cell-Surface Modification with a Self-Assembling Molecular Glue. Chem. Commun. 2021, 57, 1470–1473. 10.1039/d0cc07171d. [DOI] [PubMed] [Google Scholar]
- Lukinavičius G.; Umezawa K.; Olivier N.; Honigmann A.; Yang G.; Plass T.; Mueller V.; Reymond L.; Corrêa I. R.; Luo Z.-G.; Schultz C.; Lemke E. A.; Heppenstall P.; Eggeling C.; Manley S.; Johnsson K. A Near-Infrared Fluorophore for Live-Cell Super-Resolution Microscopy of Cellular Proteins. Nat. Chem. 2013, 5, 132–139. 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- Clark S. A.; Singh V.; Vega Mendoza D.; Margolin W.; Kool E. T. Light-Up “Channel Dyes” for Haloalkane-Based Protein Labeling in Vitro and in Bacterial Cells. Bioconjugate Chem. 2016, 27, 2839–2843. 10.1021/acs.bioconjchem.6b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockalova V.; Sanchez-Carnerero E. M.; Dunajova Z.; Palao E.; Slanska M.; Buryska T.; Damborsky J.; Klán P.; Prokop Z. Fluorescent Substrates for Haloalkane Dehalogenases: Novel Probes for Mechanistic Studies and Protein Labeling. Comput. Struct. Biotechnol. J. 2020, 18, 922–932. 10.1016/j.csbj.2020.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encell L. P.; Friedman Ohana R.; Zimmerman K.; Otto P.; Vidugiris G.; Wood M. G.; Los G. V.; McDougall M. G.; Zimprich C.; Karassina N.; Learish R. D.; Hurst R.; Hartnett J.; Wheeler S.; Stecha P.; English J.; Zhao K.; Mendez J.; Benink H. A.; Murphy N.; Daniels D. L.; Slater M. R.; Urh M.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. Development of a Dehalogenase-Based Protein Fusion Tag Capable of Rapid, Selective and Covalent Attachment to Customizable Ligands. Curr. Chem. Genomics 2013, 6, 55–71. 10.2174/1875397301206010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S.; Prokop Z.; Damborsky J.; Chaloupkova R. Kinetics of Binding of Fluorescent Ligands to Enzymes with Engineered Access Tunnels. FEBS J. 2017, 284, 134–148. 10.1111/febs.13957. [DOI] [PubMed] [Google Scholar]
- Kaushik S.; Marques S. M.; Khirsariya P.; Paruch K.; Libichova L.; Brezovsky J.; Prokop Z.; Chaloupkova R.; Damborsky J. Impact of the Access Tunnel Engineering on Catalysis Is Strictly Ligand-Specific. FEBS J. 2018, 285, 1456–1476. 10.1111/febs.14418. [DOI] [PubMed] [Google Scholar]
- Vanacek P.; Sebestova E.; Babkova P.; Bidmanova S.; Daniel L.; Dvorak P.; Stepankova V.; Chaloupkova R.; Brezovsky J.; Prokop Z.; Damborsky J. Exploration of Enzyme Diversity by Integrating Bioinformatics with Expression Analysis and Biochemical Characterization. ACS Catal. 2018, 8, 2402–2412. 10.1021/acscatal.7b03523. [DOI] [Google Scholar]
- Vasina M.; Vanacek P.; Hon J.; Kovar D.; Faldynova H.; Kunka A.; Badenhorst C.; Buryska T.; Mazurenko S.; Bednar D.; Stavros S.; Bornscheuer U.; deMello A.; Damborsky J.; Prokop Z.. Functional Annotation of an Enzyme Family by Integrated Strategy Combining Bioinformatics with Microanalytical and Microfluidic Technologies, 2021, ChemRxiv 10.26434/chemrxiv.13621517.v1. [DOI] [Google Scholar]
- Nagata Y.; Ohtsubo Y.; Tsuda M. Properties and Biotechnological Applications of Natural and Engineered Haloalkane Dehalogenases. Appl. Microbiol. Biotechnol. 2015, 99, 9865–9881. 10.1007/s00253-015-6954-x. [DOI] [PubMed] [Google Scholar]
- Jurcik A.; Bednar D.; Byska J.; Marques S. M.; Furmanova K.; Daniel L.; Kokkonen P.; Brezovsky J.; Strnad O.; Stourac J.; Pavelka A.; Manak M.; Damborsky J.; Kozlikova B. CAVER Analyst 2.0: Analysis and Visualization of Channels and Tunnels in Protein Structures and Molecular Dynamics Trajectories. Bioinformatics 2018, 34, 3586–3588. 10.1093/bioinformatics/bty386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil M.; Khan R. T.; Beier A.; Stourac J.; Konegger H.; Damborsky J.; Bednar D. FireProtASR: A Web Server for Fully Automated Ancestral Sequence Reconstruction. Brief. Bioinform. 2020, 22, bbaa337 10.1093/bib/bbaa337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I.; Bork P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chovancová E.; Pavelka A.; Benes P.; Strnad O.; Brezovsky J.; Kozlikova B.; Gora A.; Sustr V.; Klvana M.; Medek P.; Biedermannova L.; Sochor J.; Damborsky J. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8, e1002708 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.3.2; Schrödinger, LLC, 2019.
- Verschueren K. H.; Seljée F.; Rozeboom H. J.; Kalk K. H.; Dijkstra B. W. Crystallographic Analysis of the Catalytic Mechanism of Haloalkane Dehalogenase. Nature 1993, 363, 693–698. 10.1038/363693a0. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. Global Kinetic Explorer: A New Computer Program for Dynamic Simulation and Fitting of Kinetic Data. Anal. Biochem. 2009, 387, 20–29. 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. FitSpace Explorer: An Algorithm to Evaluate Multidimensional Parameter Space in Fitting Kinetic Data. Anal. Biochem. 2009, 387, 30–41. 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Kellogg E. H.; Leaver-Fay A.; Baker D. Role of Conformational Sampling in Computing Mutation-Induced Changes in Protein Structure and Stability. Proteins 2011, 79, 830–838. 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S. M.; Bednar D.; Damborsky J. Computational Study of Protein-Ligand Unbinding for Enzyme Engineering. Front. Chem. 2019, 6, 650 10.3389/fchem.2018.00650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce N. J.; Ganotra G. K.; Kokh D. B.; Sadiq S. K.; Wade R. C. New Approaches for Computing Ligand-Receptor Binding Kinetics. Curr. Opin. Struct. Biol. 2018, 49, 1–10. 10.1016/j.sbi.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P. Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements. J. Mol. Model. 2007, 13, 1173–1213. 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klvana M.; Pavlova M.; Koudelakova T.; Chaloupkova R.; Dvorak P.; Prokop Z.; Stsiapanava A.; Kuty M.; Kuta-Smatanova I.; Dohnalek J.; Kulhanek P.; Wade R. C.; Damborsky J. Pathways and Mechanisms for Product Release in the Engineered Haloalkane Dehalogenases Explored Using Classical and Random Acceleration Molecular Dynamics Simulations. J. Mol. Biol. 2009, 392, 1339–1356. 10.1016/j.jmb.2009.06.076. [DOI] [PubMed] [Google Scholar]
- Brezovsky J.; Babkova P.; Degtjarik O.; Fortova A.; Gora A.; Iermak I.; Rezacova P.; Dvorak P.; Smatanova I. K.; Prokop Z.; Chaloupkova R.; Damborsky J. Engineering a de Novo Transport Tunnel. ACS Catal. 2016, 6, 7597–7610. 10.1021/acscatal.6b02081. [DOI] [Google Scholar]
- Kokkonen P.; Sykora J.; Prokop Z.; Ghose A.; Bednar D.; Amaro M.; Beerens K.; Bidmanova S.; Slanska M.; Brezovsky J.; Damborsky J.; Hof M. Molecular Gating of an Engineered Enzyme Captured in Real Time. J. Am. Chem. Soc. 2018, 140, 17999–18008. 10.1021/jacs.8b09848. [DOI] [PubMed] [Google Scholar]
- Marques S. M.; Dunajova Z.; Prokop Z.; Chaloupkova R.; Brezovsky J.; Damborsky J. Catalytic Cycle of Haloalkane Dehalogenases Toward Unnatural Substrates Explored by Computational Modeling. J. Chem. Inf. Model. 2017, 57, 1970–1989. 10.1021/acs.jcim.7b00070. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Yin C.; Jiang Y.; van der Spoel D. Force Field Benchmark of Amino Acids: I. Hydration and Diffusion in Different Water Models. J. Chem. Inf. Model. 2018, 58, 1037–1052. 10.1021/acs.jcim.8b00026. [DOI] [PubMed] [Google Scholar]
- Rose P. W.; Bi C.; Bluhm W. F.; Christie C. H.; Dimitropoulos D.; Dutta S.; Green R. K.; Goodsell D. S.; Prlić A.; Quesada M.; Quinn G. B.; Ramos A. G.; Westbrook J. D.; Young J.; Zardecki C.; Berman H. M.; Bourne P. E. The RCSB Protein Data Bank: New Resources for Research and Education. Nucleic Acids Res. 2012, 41, D475–D482. 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Tyka M.; Leaver-Fay A.; Thompson J.; Baker D. Structure-Guided Forcefield Optimization. Proteins 2011, 79, 1898–1909. 10.1002/prot.23013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara M. J.; Leaver-Fay A.; Tyka M. D.; Stein A.; Houlihan K.; DiMaio F.; Bradley P.; Kortemme T.; Baker D.; Snoeyink J.; Kuhlman B. Combined Covalent-Electrostatic Model of Hydrogen Bonding Improves Structure Prediction with Rosetta. J. Chem. Theory Comput. 2015, 11, 609–622. 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe A. K.; Casewit C. J.; Colwell K. S.; Goddard W. A.; Skiff W. M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. 10.1021/ja00051a040. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision E.01; Gaussian, Inc.: Wallingford, CT, 2009.
- Gordon J. C.; Myers J. B.; Folta T.; Shoja V.; Heath L. S.; Onufriev A. H++: A Server for Estimating PKas and Adding Missing Hydrogens to Macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr S.; Harvey M. J.; Noé F.; De Fabritiis G. HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 2016, 12, 1845–1852. 10.1021/acs.jctc.6b00049. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur S.; Kahn K.; Bruice T. C. Comparison of Formation of Reactive Conformers for the SN2 Displacements by CH3CO– in Water and by Asp124-CO– in a Haloalkane Dehalogenase. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 2215–2219. 10.1073/pnas.242721799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R. C.; Crowley M. F.; Case D. A. The Implementation of a Fast and Accurate QM/MM Potential Method in Amber. J. Comput. Chem. 2008, 29, 1019–1031. 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; Izadi S.; Janowski P.; Kaus J.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Luchko T.; Luo R.; Madej B.; Mermelstein D.; Merz K. M.; Monard G.; Nguyen H.; Nguyen H. T.; Omelyan I.; Onufriev A.; Roe D. R.; Roitberg A.; Sagui C.; Simmerling C. L.; Botello-Smith W. M.; Swails J.; Walker R. C.; Wang J.; Wolf R. M.; Wu X.; Xiao L.; Kollman P. A.. AMBER 16; University of California: San Francisco, 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.