

Abstract
A novel chemoenzymatic synthon strategy has been developed to construct a comprehensive library of α2–3- and α2–6-linked sialosides containing 7-N- or 7,9-di-N-acetyl sialic acid, the stable analogue of naturally occurring 7-O-acetyl- or 7,9-di-O-acetyl-sialic acid. Diazido and triazido-mannose derivatives that were readily synthesized chemically from inexpensive galactose were shown to be effective chemoenzymatic synthons. Together with bacterial sialoside biosynthetic enzymes with remarkable substrate promiscuity, they were successfully used in one-pot multienzyme (OPME) sialylation systems for highly efficient synthesis of sialosides containing multiple azido groups. Conversion of the azido groups to N-acetyl groups generated the desired sialosides. The hydrophobic and UV-detectable benzyloxycarbonyl (Cbz) group introduced in the synthetic acceptors of sialyltransferases was used as a removable protecting group for the propylamine aglycon of the target sialosides. The resulting N-acetyl sialosides are novel stable probes for sialic acid-binding proteins such as plant lectin MAL II which bond strongly to sialyl T antigens with or without an N-acetyl at C7 or at both C7 and C9 in the sialic acid.
Keywords: biocatalysis, carbohydrate, chemoenzymatic synthesis, N-acetyl analogue, O-acetyl sialic acid
Graphical Abstract
INTRODUCTION
Sialic acids (Sias) are nine-carbon acidic monosaccharides that are called nonulosonic acids. They are commonly found as the terminal residues of glycan moieties on cell surface glycoproteins and glycolipids of all vertebrates1,2 and as components of capsular polysaccharides and lipopolysaccharides of a number of pathogenic bacteria.3,4 Sia-terminated glycans and glycoconjugates are involved in various biological and pathogenic processes which are influenced by the natural diversities of Sia structures, sialyl linkages, and internal glycans.5 More than 50 structurally distinct forms of Sias have been found in nature.1,2,5 N-Acetylneuraminic acid (Neu5Ac, 1, Figure 1) is the most common form of natural Sias. Modifications at one or more hydroxyl groups of Sias at C4, C5, C7, C8, and/or C9 are most commonly by O-acetyl group and less commonly by O-methyl, O-sulfate, or O-lactyl group.6,7 O-Acetylated sialoglycans have been identified as potential cancer markers for acute lymphoblastic leukemia and have been shown to be involved in the regulation of ganglioside-mediated apoptosis.8,9 Sialosides containing 7-O-acetyl Neu5Ac (Neu5,7Ac2, 2) have been found on human immune cells such as human lymphocytes10 and those containing 7,9-di-O-acetyl Neu5Ac (Neu5,7,9Ac3, 3) are preferentially recognized by bovine coronavirus.11–15
Figure 1.
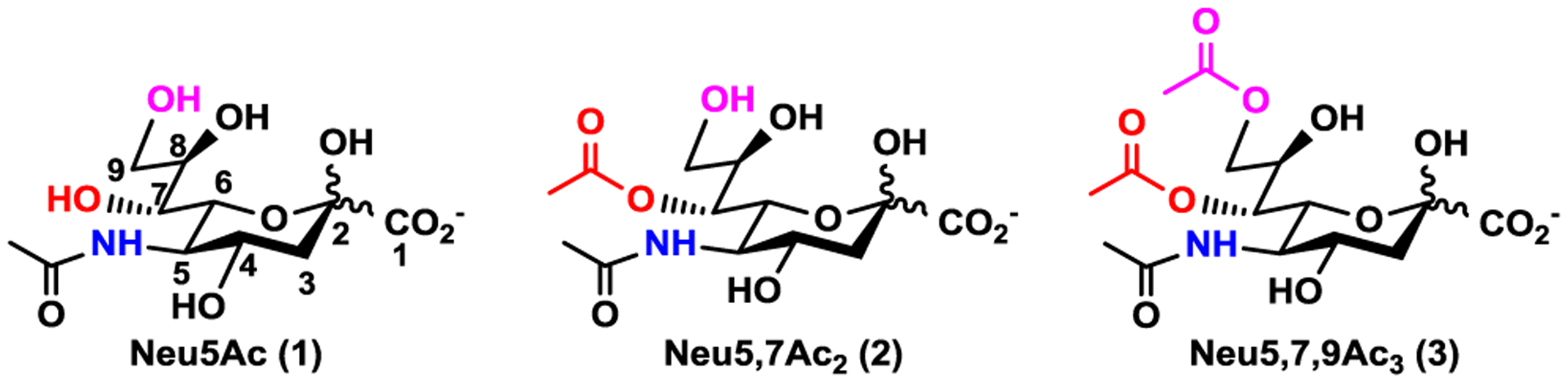
Structures of N-acetylneuraminic acid (Neu5Ac, 1), the most abundant natural sialic acid form, and its naturally occurring O-acetylated forms 7-O-acetyl-N-acetylneuraminic acid (Neu5,7Ac2, 2) and 7,9-di-O-acetyl-N-acetylneuraminic acid (Neu5,7,9Ac3, 3).
Even though O-acetylated sialoglycans were discovered in as early as 1936,16,17 the progress of elucidating their functions has been delayed due to the instability of the O-acetyl moiety.18,19 O-Acetyl group at C7-OH of Sia is notoriously challenging to study. It was shown to spontaneously migrate to C9-OH group even under physiological pH.20 A sialate O-acetyltransferase (EC 2.3.1.45) was believed to catalyze the transfer of O-acetyl group to C7-OH of the Neu5Ac in sialosides which is then migrated to C9-OH to form the sialosides containing 9-O-acetyl Neu5Ac (Neu5,9Ac2) as the most abundant O-acetylated Sia.20,21 The O-acetyl group at C7 of Neu5,7,9Ac3 (3) was also shown to migrate to C8-OH.20
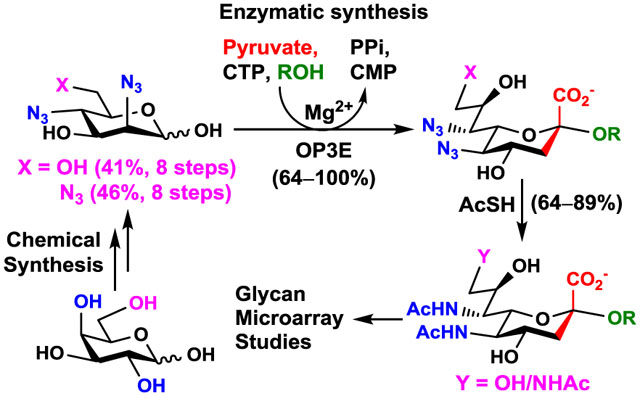
To overcome the O-acetyl instability problem, we showed previously that sialosides containing 9-N-acetyl Neu5Ac (Neu5Ac9NAc) are suitable mimics of Neu5,9Ac2-sialosides.22–24 To explore the functions and applications of more challenging sialoside targets containing Neu5,7Ac2 (2) or Neu5,7,9Ac3 (3), we report here the design and synthesis of their structurally stable mimics where the labile O-acetyl groups are replaced by N-acetyl groups. A novel chemoenzymatic synthon strategy was developed based on highly efficient one-pot three-enzyme (OP3E) sialylation systems containing sialoside biosynthetic enzymes with remarkable substrate promiscuities. The produced sialosides containing an N-acetyl sialic acid analogue were shown to be stable probes for glycan microarray-based studies for Sia-binding proteins including human Siglec 7 (hSiglec 7)25 and human Siglec 9 (hSiglec 9)26,27 as well as plant lectins Sambucus nigra lectin (SNA) and Maackia amurensis lectin II (MAL II).
RESULTS AND DISCUSSION
Synthesis Design.
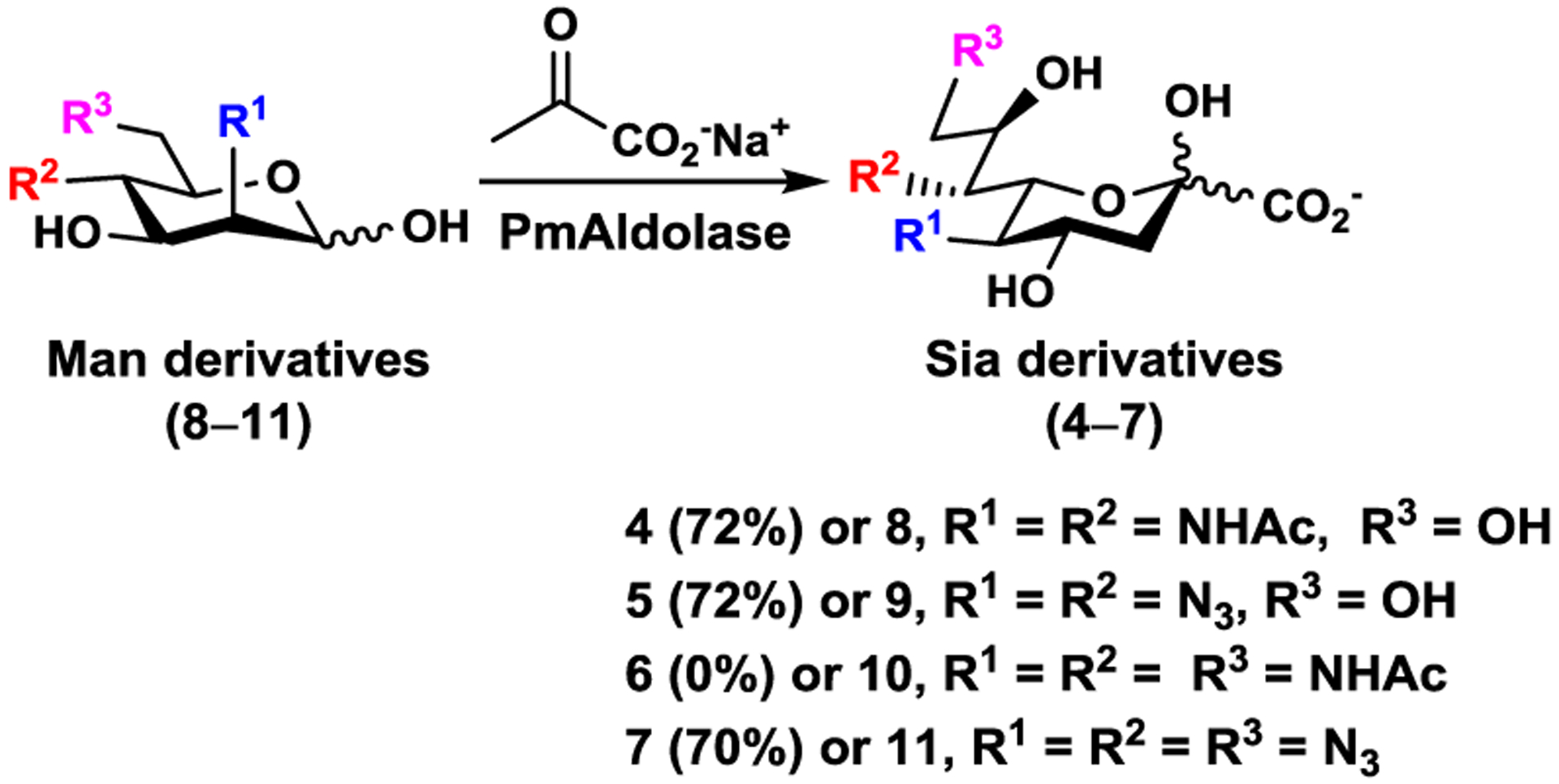
In nature, N-acetylmannosamine (ManNAc) is the six-carbon sugar precursor for Neu5Ac biosynthesis. For vertebrate cells, Neu5Ac is activated inside the nucleus to form CMP-Neu5Ac which is transported to the Golgi apparatus and used by sialyltransferases to form α-linked sialoglycoconjugates.28 Our design for synthesizing α2–3- and α2–6-linked sialosides containing modified Sia is by chemical introduction of the modifications to its six-carbon precursor and using substrate promiscuous sialoside biosynthetic enzymes to convert modified precursors to Sia derivatives which are then activated and transferred to suitable acceptors to form the target sialosides.23 Sialoside biosynthetic enzymes including a sialic acid aldolase, CMP-sialic acid synthetase, and a sialyltransferase can be used in one-pot for efficient sialylation.29,30 As shown in Figure 2, 7-N-acetyl Neu5Ac (Neu5Ac7NAc, 4) was designed as the stable mimic of Neu5,7Ac2 (2). It can be derived from the corresponding azido analogue Neu5,7diN3 (5). Similarly, 7,9-di-N-acetyl Neu5Ac (Neu5Ac7,9diNAc, 6) was proposed as the more stable mimic of Neu5,7,9Ac3 (3) and the corresponding azido analogue is Neu5,7,9triN3 (7). The corresponding six-carbon precursors for these sialic acid derivatives (4–7) of a sialic acid aldolase-catalyzed reaction are 2,4-diacetamindo-2,4-dideoxy-D-mannose (Man2,4diNAc, 8), 2,4-diazido-2,4-dideoxy-D-mannose (Man2,4diN3, 9), 2,4,6-triacetamindo-2,4,6-trideoxy-D-mannose (Man2,4,6triNAc, 10), and 2,4,6-triazido-2,4,6-trideoxy-D-mannose (Man2,4,6triN3, 11). These compounds (8–11) were chemically synthesized.
Figure 2.
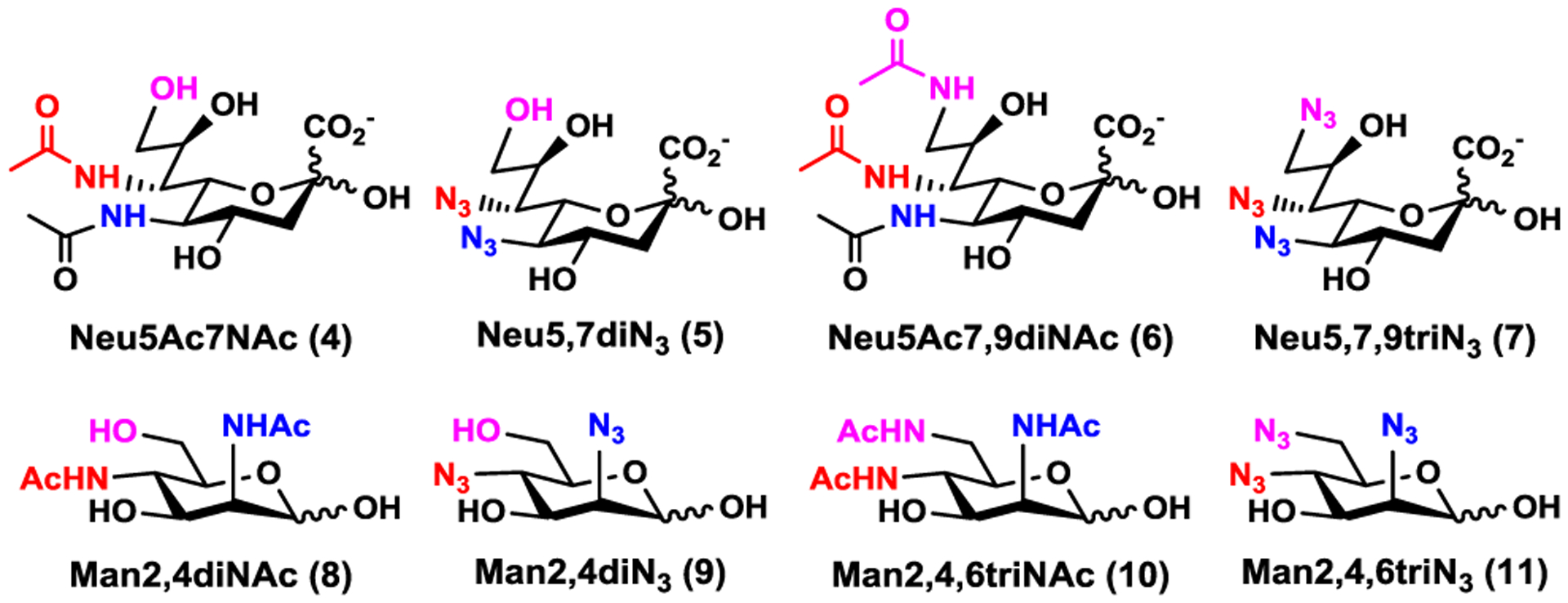
Structures of N-acetyl analogue Neu5Ac7NAc (4) and azido derivative Neu5,7diN3 (5) of Neu5,7Ac2 (2); N-acetyl analogue Neu5Ac7,9diNAc (6) and azido derivative Neu5,7,9triN3 (7) of Neu5,7,9Ac3 (3); as well as their corresponding hexose precursors Man2,4diNAc (8), Man2,4diN3 (9), Man2,4,6triNAc (10), and Man2,4,6triN3 (11), respectively.
Chemical Synthesis of Mannose Derivatives.
Numerous chemical synthetic methods have been developed to access amino sugars, rare sugars, and sugar derivatives by selective protection and epimerization.31–34 However, some of them suffer from long multi-step synthetic routes along with low to moderate yields.35 We envisioned that Man2,4diNAc (8) and Man2,4diN3 (9) could be synthesized from commercially available inexpensive D-galactose (12) by introducing two azido groups simultaneously with inversion of the stereochemistry at both C2 and C4. As shown in Scheme 1, galactoside 14 was formed in an overall 77% yield over 3 steps by per-O-acetylation of D-galactose (12) followed by nucleophilic replacement of the anomeric acetate with para-methoxyphenyl (pMP) under acidic conditions using BF3·Et2O, and de-O-acetylation using sodium methoxide. Regio-selective benzoylation of both C3 and C6-hydroxyl groups of 14 using bistributyltin oxide (Bu3Sn)2O catalyst36,37 formed a partially protected D-galactosyl-2,4-diol intermediate (15) in 79% yield. Treatment of 15 with trifluoromethanesulfonic anhydride (Tf2O) and pyridine followed by simultaneous displacement (SN2 reaction) of both triflate groups at C2 and C4 with azido groups using tetrabutylammonium azide (TBAN3)38 in dry toluene30,39 at 90 °C afforded diazido-D-mannose derivative 16 in 92% yield. The azido moieties of 16 were converted to acetamido moieties by reacting with thioacetic acid in pyridine under argon40 to produce benzoyl protected glycoside 17 in 89% yield. Debenzoylation under Zemplén reaction conditions in the presence of sodium methoxide and methanol followed by ceric ammonium nitrate (CAN)-catalyzed removal of the pMP group41 produced desired Man2,4diNAc (8). On the other hand, removal of the benzoyl groups and the anomeric pMP group from 16 formed the desired Man2,4diN3 (9). Altogether, Man2,4diNAc (8) and Man2,4diN3 (9) were synthesized from D-galactose (12) in nine and eight steps with overall yields of 32% and 41%, respectively.
Scheme 1.
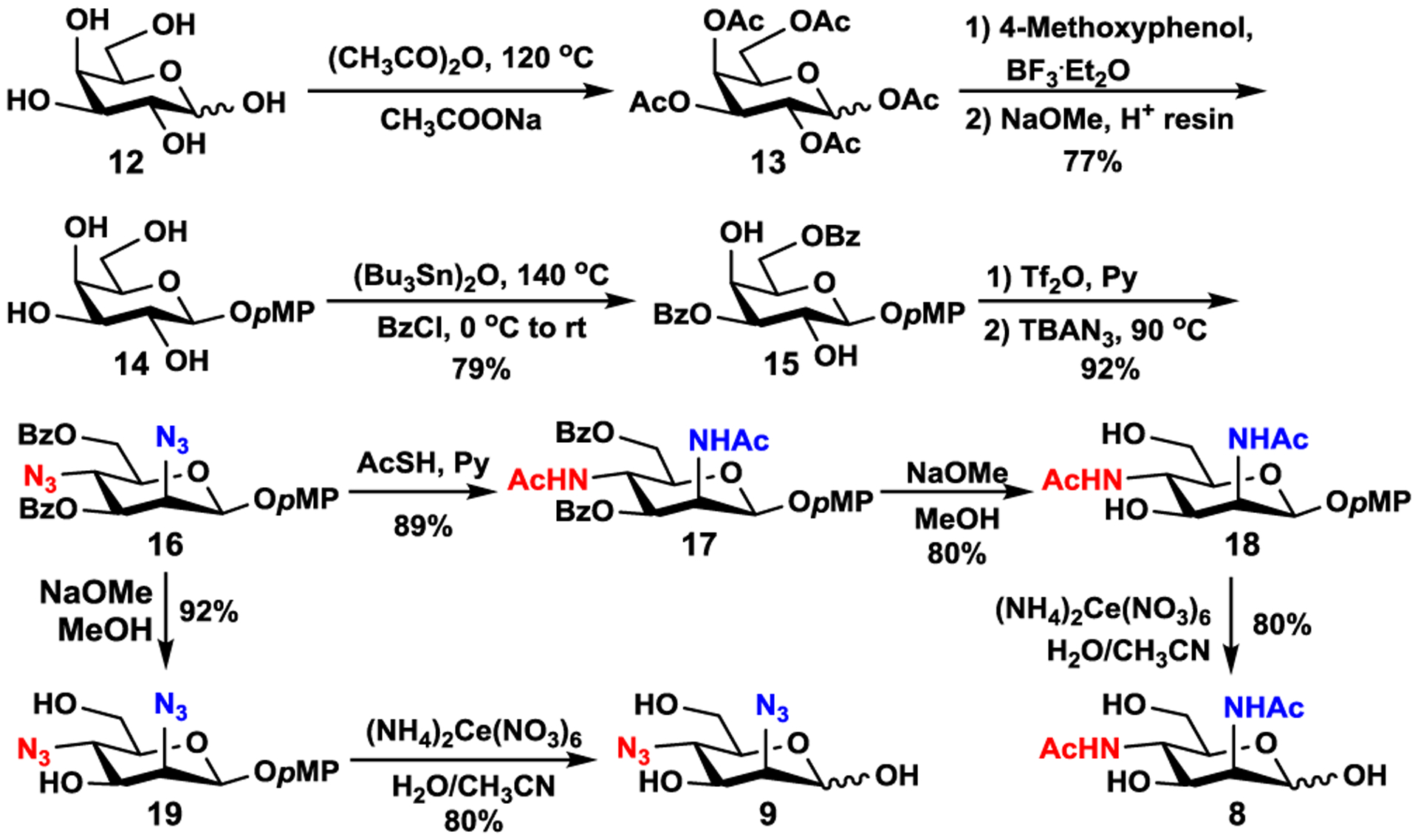
Chemical synthesis of Man2,4diNAc (8) and Man2,4diN3 (9) from galactose (12). Ac = acetyl, pMP = para-methoxyphenyl, Bz = benzoyl.
Man2,4,6triNAc (10) and Man2,4,6triN3 (11) were synthesized from galactoside 14 (Scheme 2) using a similar strategy except that only 3-OH group of 14 was regio-selectively protected with a benzoyl group using dimethyltin chloride (Me2SnCl2) as a catalyst38,42 to produce partially protected triol 20 in 93% yield. Simultaneous triflation of all three hydroxyl groups in 20 with Tf2O in pyridine followed by concurrent displacement of all three triflates with azido groups via an SN2-mechanism worked very well when TBAN3 in dry toluene30,39 was used at 90 °C for 2 h in the second step. The formation of D-mannose derivative 21 was achieved in an overall 88% yield. Conversion of all three azido groups in 21 to acetamido moieties using thioacetic acid and pyridine40 produced 22 in 80% yield. Debenzoylation of 22 under Zemplén reaction conditions followed by CAN-catalyzed removal of the pMP group41 produced Man2,4,6triNAc (10) in 70% yield. On the other hand, step-wise removal of the benzoyl protecting group (92% yield) and the pMP group (80% yield) from 21 produced Man2,4,6triN3 (11). Altogether, Man2,4,6triNAc (10) and Man2,4,6triN3 (11) were formed from 14 in six and five steps with overall yields of 46% and 60%, respectively.
Scheme 2.
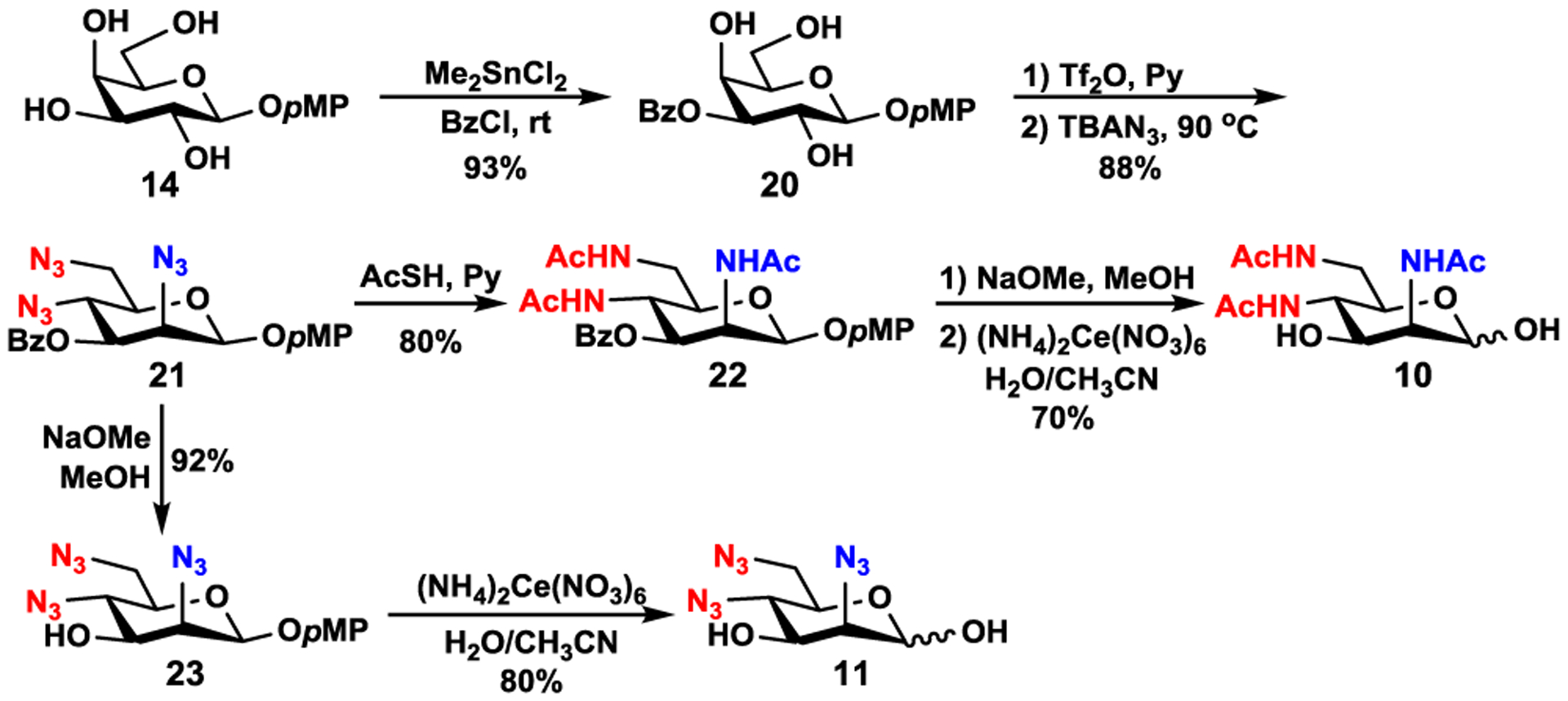
Synthesis of Man2,4,6triNAc (10) and Man2,4,6triN3 (11) from galactoside 14.
Enzymatic Synthesis of Sialic Acid Derivatives.
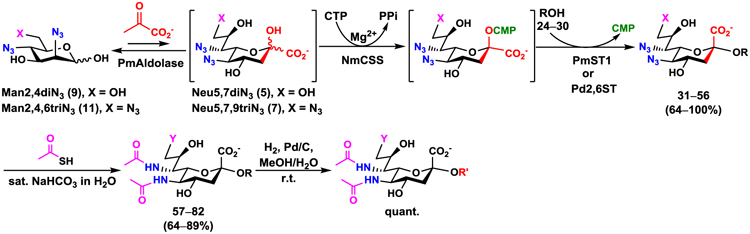
Mannose derivatives Man2,4diNAc (8), Man2,4diN3 (9), Man2,4,6triNAc (10), and Man2,4,6triN3 (11) were tested as potential substrates for Pasteurella multocida sialic acid aldolase (PmAldolase).43 Among these four compounds, Man2,4diNAc (8) and its di-azido derivative Man2,4diN3 (9), as well as the tri-azido derivative Man2,4,6triN3 (11) were suitable substrates for PmAldolase. The corresponding Sia derivatives Neu5Ac7NAc (4), Neu5,7diN3 (5), and Neu5,7,9triN3 (7) were synthesized in the presence of five equivalents of sodium pyruvate with 70–72% yields (Scheme 3). In contrast, Man2,4,6triNAc (10) was not a suitable substrate for PmAldolase, and the corresponding Neu5Ac7,9diNAc (6) could not be obtained directly from 10 by PmAldolase-catalyzed reaction.
Scheme 3.
PmAldolase-catalyzed preparative-scale synthesis of sialic acid derivatives Neu5Ac7NAc (4), Neu5,7diN3 (5), and Neu5,7,9triN3 (7) from the corresponding mannose derivatives Man2,4diNAc (8), Man2,4diN3 (9), and Man2,4,6triN3 (11), respectively. Man2,4,6triNAc (10) was not a suitable substrate for PmAldolase-catalyzed synthesis of Neu5Ac7,9diNAc (6).
One-Pot Multienzyme (OPME) Synthesis of Sialosides Containing An Azido-Modified Sialic Acid.
The Neu5Ac7NAc (4), Neu5,7diN3 (5), and Neu5,7,9triN3 (7) obtained from PmAldolase-catalyzed reaction were tested as potential substrates for Neisseria meningitidis CMP-sialic acid synthetase (NmCSS).44 While both Neu5,7diN3 (5) and Neu5,7,9triN3 (7) were suitable substrates for NmCSS, Neu5Ac7NAc (4) was not.
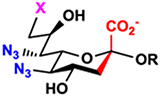
Man2,4diN3 (9) and Man2,4,6triN3 (11) were also tested as chemoenzymatic synthons30 in one-pot three-enzyme (OP3E) sialyation systems29 (Table 1) for enzymatic synthesis of α2–3- and α2–6-linked sialosides containing Neu5,7diN3 or Neu5,7,9triN3. It was remarkable that all four enzymes including PmAldolase,43 NmCSS,44 Pasteurella multocida α2–3-sialyltransferase 1 (PmST1),45 and Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST)46 were able to tolerate the diazido- and triazido-derivatives of the corresponding substrates.
Table 1.
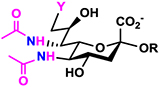
One-pot three-enzyme (OP3E) synthesis of sialosides containing Neu5,7diN3 or Neu5,7,9triN3 (31–56) from Man2,4diN3 (9) or Man2,4,6triN3 (11) and chemical conversion of the azido groups to N-acetyl moieties to form sialosides containing desired Neu5Ac7NAc or Neu5Ac7,9diNAc (57–82). The Cbz group in the resulting sialosides was removed from the NAc-sialosides in quantitative yields to produce sialosides containing a propylamine aglycone for glycan microarray studies. R’ differs from R by replacing the propyl NHCbz aglycon in R with the propylamine (ProNH2) in R’.
| ||||
|---|---|---|---|---|
|
Acceptor (ROH)
|
 31–56 (64–100%) |
 57–82 (64–89%) |
||
| α2–3-sialosidea Neu5,7diN3/ Neu5,7,9triN3α3OR |
α2–6-sialosideb Neu5,7diN3/ Neu5,7,9triN3α6OR |
α2–3-sialoside Neu5Ac7NAc/Neu5Ac7,9diNAcα3OR |
α2–6-sialoside Neu5Ac7NAc/Neu5Ac7,9diNAcα6OR |
|
|
LacβProNHCbz (24) |
X=OH, 31 (90%) X=N3, 32 (92%) |
X=OH, 33 (quant.) X=N3, 34 (quant.) |
Y=OH, 57 (68%) Y=NHAc, 58 (80%) |
Y=OH, 59 (78%) Y=NHAc, 60 (80%) |
 LacNAcβProNHCbz (25) |
X=OH, 35 (70%) X=N3, 36 (66%) |
X=OH, 37 (72%) X=N3, 38 (65%) |
Y=OH, 61 (67%) Y=NHAc, 62 (67%) |
Y=OH, 63 (77%) Y=NHAc, 64 (85%) |
|
Galβ3GalNAcβProNHCbz (26) |
X=OH, 39 (99%) X=N3, 40 (67%) |
X=OH, 41 (Quant.) X=N3, 42 (69%) |
Y=OH, 65 (64%) Y=NHAc, 66 (65%) |
Y=OH, 67 (67%) Y=NHAc, 68 (70%) |
 Galβ3GalNAcαProNHCbz (27) |
X=OH, 43 (95%) X=N3, 44 (65%) |
X=OH, 45 (68%) X=N3, 46 (79%) |
Y=OH, 69 (77%) Y=NHAc, 70 (65%) |
Y=OH, 71 (77%) Y=NHAc, 72 (72%) |
|
Galβ3GlcNAcβProNHCbz (28) |
X=OH, 47 (87%) X=N3, 48 (64%) |
X=OH, 49 (quant.) X=N3, 50 (91%) |
Y=OH, 73 (73%) Y=NHAc, 74 (66%) |
Y=OH, 75 (75%) Y=NHAc, 76 (70%) |
|
Galβ3GlcNAcαProNHCbz (29) |
X=OH, 51 (93%) X=N3, 52 (87%) |
X=OH, 53 (quant.) X=N3, 54 (96%) |
Y=OH, 77 (89%) Y=NHAc, 78 (69%) |
Y=OH, 79 (72%) Y=NHAc, 80 (68%) |
 GalNAcαProNHCbz (30) |
X=OH, 55 (65%) X=N3, 56 (65%) |
Y=OH, 81 (84%) Y=NHAc, 82 (85%) |
||
PmST1 was used as the α2–3sialyltransferase;
Pd2,6ST was used as the α2–6sialyltransferase.
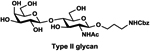
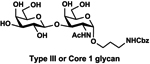
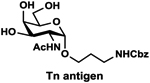
LacβProNHCbz (Type VI glycan, 24),47 LacNAcβProNHCbz (Type II glycan, 25), Galβ3GalNAcβProNHCbz (Type IV glycan, 26), Galβ3GalNAcαProNHCbz (Type III or Core 1 glycan, 27), Galβ3GlcNAcβProNHCbz (Type I glycan, 28), Galβ3GlcNAcαProNHCbz (29), and GalNAcαProNHCbz (Tn antigen, 30) were synthesized and used as sialyltransferase acceptors for the construction of a comprehensive library of sialosides (Table 1). The hydrophobic and UV-detectable benzyloxycarbonyl (Cbz) group in 24–30 was used as a removable protecting group for the propylamine aglycon of the target sialosides.
In the OP3E sialylation systems (Table 1), Man2,4diN3 (9) or Man2,4,6triN3 (11) was coupled with pyruvate by a PmAldolase-catalyzed reaction to form Neu5,7diN3 (5) or Neu5,7,9triN3 (7). The resulting azido-Sia derivative was converted by an NmCSS-catalyzed reaction in the presence of cytidine 5’-triphosphate (CTP) and magnesium cation (Mg2+) to form the corresponding CMP-activated sugar nucleotide CMP-Neu5,7diN3 or CMP-Neu5,7,9triN3, which was used by sialyltransferases such as Pasteurella multocida α2–3-sialyltransferase 1 (PmST1)45 and Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST),46 respectively, to produce α2–3- and α2–6-linked sialosides (31–56) from sialyltransferase acceptors 24–30.
Using PmST145 as the sialyltransferase, α2–3-linked Neu5,7diN3-containing (31, 35, 39, 43, 47, and 51) and Neu5,7,9triN3-containing glycosides (32, 36, 40, 44, 48, and 52) were obtained in good to excellent (64–99%) yields with galactosides 24–30 as the sialyltransferase acceptors. Similarly, using Pd2,6ST46 as the sialyltransferase, α2–6-linked Neu5,7diN3-containing (33, 37, 41, 45, 49, 53, and 55) and Neu5,7,9triN3-containing glycosides (34, 38, 42, 46, 50, 54, and 56) were obtained in good to excellent (65–100%) yields.
Chemical Conversion of The Azido Groups in Sialosides to N-Acetyl Groups and Removal of The Cbz-Protecting Group.
The azido-containing sialosides (31–56) obtained were readily converted to their N-acetyl analogues (57–82) in 64–89% yields using thioacetic acid and saturated sodium bicarbonate aqueous solution (Table 1), a condition optimized previously.30 The Cbz group in the aglycone of the sialosides 57–82 was removed by hydrogenation using Pd/C catalyst in water-methanol solution for a few hours at room temperature to form sialosides containing a propylamine aglycone in quantitative yields for downstream conjugation reactions. Compared to the previous chemoenzymatic synthon strategy30 of using sialyltransferase acceptors with a propyl chloride aglycone, the Cbz-protected propylamine aglycone has several advantages. It facilitates reaction monitoring and product purification processes due to its hydrophobic UV-detectability. It also allows selective conversion of the azido group in the enzymatic products (31–56) to the desired N-acetyl group and permits highly efficient conversion (in quantitative yields) of the aglycon in the sialosides (57–82) to a primary propylamine for glycan microarray printing.
Sialosides Containing Neu5Ac7NAc or Neu5Ac7,9diNAc Can Bind to Sia-Binding Proteins as Shown by Sialoglycan Microarray Studies.
The propylamine aglycon in the sialosides obtained made it easy to print these glycans on an array format for protein-binding studies in a high-throughput format. Glycan microarray experiments were carried out using α2–3- and α2–6-linked sialosides containing Neu5Ac7NAc or Neu5Ac7,9diNAc synthesized here as well as related sialosides containing Neu5Ac, Neu5,9Ac2, or Neu5Ac9NAc synthesized previously.22,23,45,46,48 The corresponding internal glycans (without the Sia component) were used as negative controls. As shown in Figure 3 and Table S1, the binding of hSiglec 7, hSiglec 9, and SNA to the sialosides containing Neu5Ac (grey bars) or Neu5Ac9NAc (orange bars) was blocked by substituting the C7-OH of sialic acid by a C7-NAc group (Neu5Ac7NAc red bars and Neu5Ac7,9diNAc blue bars, respectively) except for the weak binding retained for Neu5Ac7NAcα3Galβ3GalNAcβ-glycan (red bars for Js in Figure 3). Interestingly, MAL II bond to sialyl T antigens Siaα3Galβ3GalNAcα-glycans (L in Figure 3) strongly49 regardless of the Sia forms in the sialosides tested including those containing Neu5Ac (grey bar), Neu5,9Ac2 (green bar), Neu5Ac9NAc (orange bar), Neu5Ac7NAc (red bar), and Neu5Ac7,9diNAc (blue bar). On the other hand, the binding of MAL II to Neu5Acα3Galβ3GalNAcβ-glycan (grey bar in J in Figure 3), a sialoglycan differing from the sialyl T antigen only on the glycosyl linkage remotely from the Sia, was not influenced significantly by Neu5Ac 9-O-acetylation (green bar) or 9-NAc substitution (orange bar) but was blocked completely by Neu5Ac 7-NAc (red bar) or 7,9-di-NAc substitution (blue bar).
Figure 3.
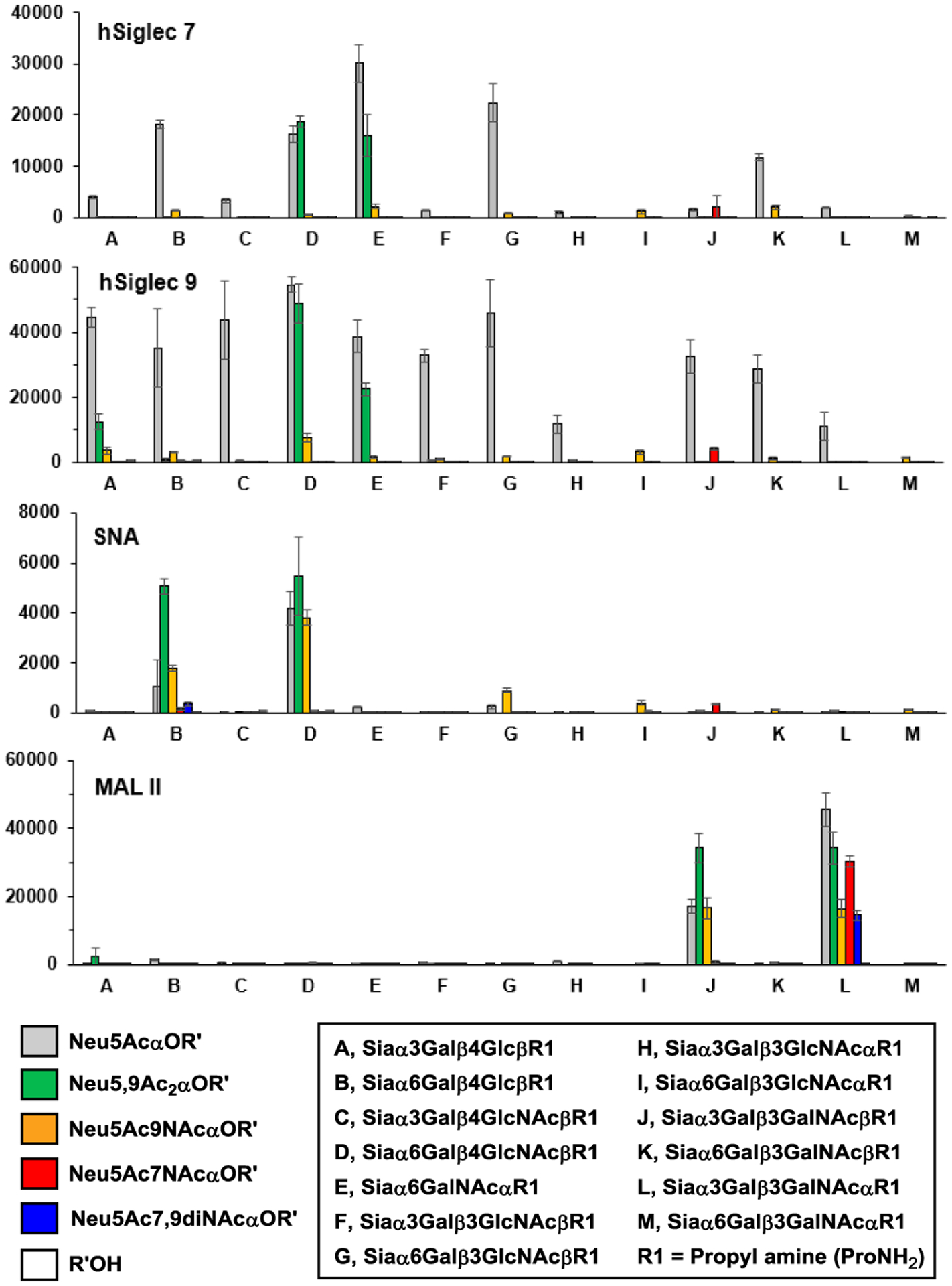
Glycan microarray study results for sialoside binding by hSiglec 7, hSiglec 9, SNA, and MAL II (Numeric data are shown in Table S1). Asialoglycans (R’OH) corresponding to the internal glycans in the sialosides are negative controls shown in white bars. R1 = ProNH2.
CONCLUSIONS
In conclusion, a highly efficient chemoenzymatic synthon strategy has been successfully developed to construct a comprehensive library of sialosides containing N-acetyl analogues of 7-O- or 7,9,-di-O-acetylated Neu5Ac. Man2,4diN3 (9) and Man2,4,6triN3 (11) are effective chemoenzymatic synthons that are readily accessible via chemical synthesis from inexpensive galactose. We have shown that bacterial sialoside biosynthetic enzymes including PmAldolase, NmCSS, PmST1, and Pd2,6ST have remarkable substrate promiscuity. The OPME systems are highly efficient in using Man2,4diN3 and Man2,4,6triN3 as effective chemoenzymatic synthons to produce sialosides containing Neu5Ac7N3 or Neu5Ac7,9diN3 which are readily converted to the target NAc-containing sialosides by a facile chemical conversion process. The hydrophobic and UV-detectable Cbz group introduced in the synthetic acceptors of sialyltransferases is a convenient removable protecting group for the propylamine aglycon of the target sialosides. The application of the obtained sialosides in glycan microarray studies has demonstrated that these compounds could be valuable stable probes for Sia-binding proteins. The chemoenzymatic synthon strategy has the potential to be used for the synthesis of other glycans containing N-acetyl groups.
EXPERIMENTAL SECTION
Materials and General Methods.
Chemicals were purchased and used without further purification. Nuclear magnetic resonance (NMR) spectra were recorded in the NMR facility of the University of California, Davis on a 600 MHz and 800 MHz Bruker Avance III-NMR spectrometers and a 400 MHz Bruker Avance III HD Nanobay spectrometer. Chemical shifts are reported in parts per million (ppm) on the δ scale. High resolution electrospray ionization (ESI) mass spectra were obtained using a Thermo Electron LTQ-Orbitrap Hybrid mass spectrometer or a Thermo Scientific Q Exactive HF Orbitrap Mass Spectrometer at the mass spectrometry facility in the University of California, Davis or using a LTQ-Orbitrap Elite mass spectrometer at the Georgia State University. Column chromatography was performed using columns manually packed with silica gel 60 Å (230–400 mesh, Sorbent Technologies) or a CombiFlash® Rf 200i system with either RediSep Rf silica columns or an ODS-SM (C18) column (51 g, 50 μm, 120 Å, Yamazen). Thin layer chromatography (TLC) was performed on silica gel plates (Sorbent Technologies) using anisaldehyde sugar stain or 5% sulfuric acid in ethanol stain for detection. Gel filtration chromatography was performed with a column (100 cm × 2.5 cm) packed with Bio-Gel P-2 Fine resins (Bio-Rad). Pasteurella multocida sialic acid aldolase (PmAldolase),43 Neisseria meningitidis CMP-sialic acid synthetase (NmCSS),44 Pasteurella multocida α2–3-sialyltransferase 1 (PmST1),45 and Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST)46 were expressed and purified as described previously.
Chemical Synthesis of 2,4-Diacetamido-2,4-dideoxy-D-mannose (Man2,4diNAc, 8) and 2,4-Diazido-2,4-dideoxy-D-mannose (Man2,4diN3, 9).
p-Methoxyphenyl-β-D-galactopyranoside (14).
D-Galactopyranose (12, 7.00 g, 38.8 mmol) and sodium acetate (3.7 g, 0.045 mole) were dissolved in acetic anhydride (50 mL). The reaction mixture was heated at 120 °C for 1 h and neutralized using sodium bicarbonate. The compound was extracted by washing with dichloromethane. The dichloromethane solution was washed with brine. The organic layer was dried on sodium sulfate and the solvent was removed under a reduced pressure to produce 1,2,3,4,6-penta-O-acetyl-D-galactopyranose (13) which was used in the next step without further purification. Compound 13 (21.67 g, 0.055 mmol) and para-methoxyphenol (10.33 g, 0.083 mmol) were added to a round bottom flask (500 mL) and dissolved in dichloromethane (50 mL). The reaction mixture was cooled in an ice-water bath. Boron trifluoride diethyl etherate (11.86 g, 0.0832 mmol) was added slowly into the reaction mixture over a period of 5 minutes.30 The reaction was stirred for 6 h and quenched by adding MeOH (50 mL). The solvent was removed under a reduced pressure and the residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 3:1, by volume) to obtain p-methoxyphenyl-2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside in 81% yield. The column-purified intermediate was dissolved in dry MeOH (100 mL), and 0.2 mL of sodium methoxide solution (5.4 M) was added. After the reaction was completed, the reaction mixture was neutralized by adding H+-resin. The product was purified by silica gel column chromatography (ethyl acetate: methanol = 20:1, by volume) to produce p-methoxyphenyl-β-D-galactopyranoside (14) in 95% yield. Compound 14 (8.56 g) was obtained as a white amorphous solid in an overall 77% yield from D-galactose in 3 steps. 1H NMR (400 MHz, CD3OD) δ 7.10–7.05 (m, 2H), 6.87–6.82 (m, 2H), 4.74 (d, J = 7.7 Hz, 1H), 3.91 (dd, J = 3.4, 1.1 Hz, 1H), 3.78 (s, 3H), 3.65 (ddd, J = 6.7, 5.3, 1.1 Hz, 1H), 3.57 (dd, J = 9.7, 3.4 Hz, 1H), 3.37 (s, 1H), 3.33 (p, J = 1.7 Hz, 2H). 13C NMR (100 MHz, CD3OD) δ 156.3, 152.3, 119.3, 115.4, 108.7, 84.6, 83.5, 78.2, 72.2, 64.4, 56.0. HRMS (ESI-Orbitrap) m/z: [M + Cl]− Calcd for C13H18ClO7 321.0747; found 321.0760.
p-Methoxyphenyl-3,6-dibenzoyl-β-D-galactopyranoside (15).
Compound 14 (0.60 g, 2.10 mmol) and bis(tri-n-butyltin) oxide (1.78 g, 3.15 mmol) were dissolved in toluene (70 mL) in a round bottom flask (500 mL). The reaction mixture was heated to 140 °C in an oil bath for 15 minutes. Reaction mixture was cooled down to 0 °C in an ice-water bath, and benzoyl chloride (0.88 g, 6.30 mmol) was added. The reaction was stirred for 6 h at 0 °C and quenched by adding methanol (50 mL). The solvent was removed under a reduced pressure and the residue was purified by silica gel column chromatography (toluene:ethyl acetate = 7:1, by volume) to produce 15 as a white amorphous solid (0.82 g, 79% yield).36–38 1H NMR (400 MHz, CD3OD) δ 8.22–8.09 (m, 2H), 8.08–7.99 (m, 2H), 7.64 (dt, J = 9.7, 7.5 Hz, 2H), 7.50 (td, J = 7.6, 5.3 Hz, 4H), 7.09–6.97 (m, 2H), 6.73–6.62 (m, 2H), 5.15 (dd, J = 10.1, 3.4 Hz, 1H), 4.94 (d, J = 7.8 Hz, 1H), 4.68 (dd, J = 11.4, 8.5 Hz, 1H), 4.59 (bs, 2H), 4.50 (dd, J = 11.5, 4.0 Hz, 1H), 4.30 (d, J = 3.4 Hz, 1H), 4.20 (td, J = 7.3, 2.6 Hz, 2H), 3.69 (s, 3H). 13C{1H} NMR (100 MHz, CD3OD) δ 167.74, 167.70, 156.6, 153.0, 134.43, 134.36, 131.4, 131.3, 130.9, 130.7, 130.7, 129.6, 129.5, 119.3, 115.3, 103.7, 77.6, 74.1, 69.9, 68.0, 65.1, 56.0.
p-Methoxyphenyl-2,4-diazido-3,6-dibenzoyl-2,4-dideoxy-β-D-mannopyranoside (16).
p-Methoxyphenyl-3,6-dibenzoyl-β-D-galactopyranoside (15, 0.40 g, 0.81 mmol) was dissolved in dichloromethane (25 mL) in a round bottom flask (100 mL) at 0 °C. Pyridine (0.65 mL, 8.09 mmol) was added followed by slow addition of triflouromethanesulfonic anhydride (0.68 mL, 4.04 mmol) at 0 °C and the reaction was stirred for 30 min before it was quenched by adding sodium bicarbonate (10 mL). The organic layer was washed with hydrochloric acid (1 N) and brine. Organic layer was combined and concentrated under reduced pressure to produce a crude product, which was used for the next step without any purification. To a solution of 2,4-bistriflate in toluene (20 mL), tetrabutylammonium azide (0.76 g, 2.67 mmol) was added at room temperature. The reaction was stirred at 70 °C for 1 h and then at 90 °C for another 1 h.39 The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (toluene) to produce p-methoxyphenyl-2,4-di-azido-3,6-dibenzoyl-2,4-dideoxy-β-D-mannopyranoside (16) as a white amorphous solid (0.40 g, 92% yield). 1H NMR (400 MHz, CDCl3) δ 8.15 (ddd, J = 21.4, 8.4, 1.4 Hz, 4H), 7.71–7.60 (m, 2H), 7.52 (dt, J = 11.6, 7.8 Hz, 4H), 7.03 (d, J = 9.0 Hz, 2H), 6.75 (d, J = 9.1 Hz, 2H), 5.36–5.27 (m, 1H), 5.24 (d, J = 1.2 Hz, 1H), 4.81 (dd, J = 11.9, 2.4 Hz, 1H), 4.61 (dd, J = 11.9, 6.6 Hz, 1H), 4.57 (dd, J = 3.7, 1.1 Hz, 1H), 4.13 (t, J = 10.1 Hz, 1H), 3.76 (s, 4H). 13C{1H} NMR (100 MHz, CDCl3) δ 166.1, 165.6, 155.8, 150.4, 134.1, 133.4, 130.1, 129.9, 129.7, 129.1, 128.8, 128.5, 118.5, 114.6, 98.9, 74.1, 73.0, 63.7, 61.6, 57.4, 55.6. HRMS (ESI-Orbitrap) m/z: [M + Na]+ Calcd for C27H24N6NaO7 567.1599; found 567.1581.
p-Methoxyphenyl-2,4-diacetamido-3,6-dibenzoyl-2,4-dideoxy-β-D-mannopyranoside (17).
p-Methoxyphenyl-2,4-diazido-3,6-dibenzoyl-2,4-dideoxy-β-D-mannopyranoside (16, 0.22 g, 0.41 mmol) was dissolved in pyridine (4.0 mL) in a round bottom flask (50 mL), and thioacetic acid (1.0 mL) was added. The reaction mixture was stirred for 24 h under a reduced pressure at room temperature before the reaction was quenched by adding methanol (10 mL). The solvent was removed under a reduced pressure40 and the residue was purified by silica gel column chromatography (toluene: ethyl acetate = 2:1, by volume) to produce p-methoxyphenyl-2,4-di-acetamido-3,6-dibenzoyl-2,4-dideoxy-β-D-mannopyranoside (17) as a white amorphous solid (0.21 g, 89% yield). 1H NMR (800 MHz, CDCl3) δ 8.00 (dd, J = 8.2, 1.5 Hz, 2H), 7.95 (dd, J = 8.1, 1.5 Hz, 2H), 7.62–7.56 (m, 1H), 7.55–7.49 (m, 1H), 7.43 (t, J = 7.8 Hz, 2H), 7.36 (t, J = 7.8 Hz, 2H), 6.92–6.87 (m, 2H), 6.89–6.85 (m, 1H), 6.62–6.52 (m, 3H), 5.43 (dd, J = 10.7, 4.0 Hz, 1H), 5.19 (d, J = 1.8 Hz, 1H), 5.10 (ddd, J = 9.3, 4.0, 1.6 Hz, 1H), 4.66 (dd, J = 11.9, 2.6 Hz, 1H), 4.55 (dd, J = 11.9, 8.6 Hz, 1H), 4.51–4.44 (m, 1H), 4.01–3.95 (m, 1H), 3.65 (s, 3H), 2.06 (s, 3H), 1.89 (s, 3H). 13C{1H} NMR (200 MHz, CDCl3) δ 171.3, 171.1, 166.8, 166.3, 155.3, 150.6, 133.5, 133.3, 129.9, 129.8, 129.8, 129.3, 128.6, 128.5, 118.2, 114.4, 98.0, 74.4, 72.0, 64.4, 55.5, 50.6, 47.6, 23.5, 23.2. HRMS (ESI-Orbitrap) m/z: [M + H]+ Calcd for C31H33N2O9 577.2181; found 577.2167.
p-Methoxyphenyl-2,4-diacetamido-2,4-dideoxy-β-D-mannopyranoside (18).
p-Methoxyphenyl-2,4-diacetamido-3,6-dibenzoyl-2,4-dideoxy-β-D-mannopyranoside (17, 0.51 g, 0.88 mmol) was dissolved in dry MeOH (10 mL) in a round bottom flask (50 mL), and 0.2 mL of sodium methoxide solution (5.4 M) was added. After the reaction was completed, the reaction mixture was neutralized using H+-resin. The solvent was removed under a reduced pressure and the residue was purified by silica gel column chromatography (ethyl acetate: methanol = 9:1, by volume) to produce p-methoxyphenyl-2,4-diacetamido-2,4-dideoxy-β-D-mannopyranoside (18) as a white amorphous solid (0.26 g, 80% yield). 1H NMR (800 MHz, CD3OD) δ 7.01–6.94 (m, 2H), 6.86–6.76 (m, 2H), 5.12 (d, J = 1.6 Hz, 1H), 4.70 (dd, J = 3.8, 1.6 Hz, 1H), 3.93–3.86 (m, 2H), 3.74 (s, 3H), 3.72–3.66 (m, 2H), 3.40 (ddd, J = 9.7, 4.0, 2.4 Hz, 1H), 2.12 (s, 3H), 2.02 (s, 3H). 13C{1H} NMR (200 MHz, MeOD) δ 175.0, 174.6, 156.7, 152.4, 119.1, 115.5, 99.6, 77.5, 71.7, 62.4, 56.0, 54.5, 49.7, 22.77, 22.75. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C17H23N2O7 367.1511; found 367.1507.
2,4-Diacetamido-2,4-dideoxy-D-mannose (Man2,4diNAc, 8).
p-Methoxyphenyl-2,4-di-acetamido-2,4-dideoxy-β-D-mannopyranoside (18, 0.125 g, 0.34 mmol) was added to a round bottom flask (50 mL) and dissolved in acetonitrile (6 mL). Ammonium cerium nitrate (0.6 g) was dissolved in water (1.5 mL) and was added slowly while stirring. The reaction was carried out at room temperature for 4.5 h before the reaction mixture was concentrated. The residue was purified by silica gel column chromatography (ethyl acetate: methanol: water = 9:1:0.5, by volume) to produce Man2,4diNAc (8) as a white amorphous solid (0.071 g, 80% yield). 1H NMR (800 MHz, D2O) δ 5.14 (bs, 0.6H), 4.97 (bs, 0.4H), 4.45 (d, J = 4.8 Hz, 0.4H), 4.30 (d, J = 4.8 Hz, 0.6H), 4.10 (dd, J = 10.8, 4.0 Hz, 0.6H), 3.76–3.99 (m, 2H), 3.52–3.74 (m, 2H), 3.39–3.47 (m, 0.4H), 1.97–2.31 (m, 6H). 13C{1H} NMR (200 MHz, D2O) δ = 175.7, 174.8, 174.7, 174.6, 92.9, 92.8, 75.3, 70.8, 69.9, 66.6, 60.6, 60.5, 53.4, 52.4, 48.1, 47.8, 21.9, 21.8. HRMS (ESI-Orbitrap) m/z: [M + Na]+ Calcd for C10H18N2O6Na 285.1057; found 285.1058.
p-Methoxyphenyl-2,4-di-azido-2,4-dideoxy-β-D-mannopyranoside (19).
p-Methoxyphenyl-2,4-diazido-3,6-dibenzoyl-2,4-dideoxy-β-D-mannopyranoside (16) (0.50 g, 0.92 mmol) was dissolved in dry MeOH (5 mL) in a round bottom flask (50 mL), and 0.2 mL of sodium methoxide solution (5.4 M) was added. After the reaction was completed, the reaction mixture was neutralized using H+-resin.41 The product was purified by silica gel column chromatography (toluene: ethyl acetate = 7:1, by volume) to produce p-methoxyphenyl-2,4-diazido-2,4-dideoxy-β-D-mannopyranoside (19) as a white amorphous solid (0.28 g, 92% yield). 1H NMR (800 MHz, CDCl3) δ 6.95 (d, J = 9.0 Hz, 2H), 6.83 (d, J = 9.0 Hz, 2H), 5.13–5.09 (m, 1H), 4.21–4.15 (m, 1H), 3.93 (ddd, J = 12.4, 5.9, 2.4 Hz, 1H), 3.79 (d, J = 3.6 Hz, 1H), 3.77 (s, 3H), 3.64 (t, J = 9.9 Hz, 1H), 3.24 (ddd, J = 10.1, 4.7, 2.4 Hz, 1H), 2.61 (d, J = 9.2 Hz, 1H). 13C{1H} NMR (200 MHz, CDCl3) δ 155.6, 150.1, 117.6, 114.7, 98.8, 75.1, 72.4, 63.9, 62.0, 59.4, 55.7. HRMS (ESI-Orbitrap) m/z: [M + Cl]− Calcd for C13H16ClN6O5 371.0876; found 371.0864.
2,4-Diazido-2,4-dideoxy-D-mannose (Man2,4diN3, 9).
p-Methoxyphenyl-2,4-diazido-2,4-dideoxy-β-D-mannopyranoside (19, 0.28 g 0.84 mmol) was added to a round bottom flask (50 mL) and dissolved in acetonitrile (6 mL). Ammonium cerium nitrate (1.4 g) dissolved in water (1.5 mL) was added slowly while stirring. The reaction was carried out at room temperature41 for 2.5 h before the reaction mixture was concentrated. The residue was purified by silica gel column chromatography (toluene:ethyl acetate = 2:1, by volume) to produce Man2,4diN3 (9) as a white amorphous solid (0.15 g, 80% yield). 1H NMR (800 MHz, D2O) δ 5.29 (s, 1H), 4.99 (s, 1H), 4.21 (dd, J = 9.9, 3.8 Hz, 1H), 4.07 (d, J = 3.8 Hz, 1H), 4.03–3.97 (m, 2H), 3.87 (dd, J = 12.5, 2.2 Hz, 1H), 3.83 (dd, J = 12.4, 2.3 Hz, 1H), 3.78 (dd, J = 12.4, 4.8 Hz, 1H), 3.74 (dd, J = 12.7, 5.2 Hz, 2H), 3.66 (t, J = 10.1 Hz, 1H), 3.54 (t, J = 10.1 Hz, 1H), 3.32 (ddd, J = 10.3, 5.5, 2.2 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 93.0, 92.2, 74.7, 72.1, 70.7, 69.5, 65.6, 63.9, 61.0, 60.9, 59.1, 58.7. HRMS (ESI-Orbitrap) m/z: [M + Cl]− Calcd for C6H10ClN6O4 265.0458; found 265.0439.
Chemical Synthesis of 2,4,6-Triacetamido-2,4,6-trideoxy-D-mannose (Man2,4,6triNAc, 10) and 2,4,6-Triazido-2,4,6-trideoxy-D-mannose (Man2,4,6triN3, 11)
p-Methoxyphenyl-3-benzoyl-β-D-galactopyranoside (20).
Compound 14 (6.14 g, 21.4 mmol) was dissolved in THF (100 mL) in a round bottom flask (500 mL). Diisopropylethylamine (DIPEA) (5.5 g, 42.9 mmol) and a catalytic amount of dimethyltin dichloride (200 mg) were added to the reaction mixture.38 Reaction mixture was cooled down to 0 °C in an ice-water bath and benzoyl chloride (3.32 g, 23.6 mmol) was added drop-wisely. The reaction mixture was stirred for 2 h at 0 °C and quenched by adding methanol (100 mL). The solvent was removed under a reduced pressure and the residue was purified by silica gel column chromatography (toluene: ethyl acetate = 3:1, by volume) to produce 20 as a white amorphous solid (7.77 g, 93% yield). 1H NMR (400 MHz, CD3OD) δ 8.20–8.13 (m, 2H), 7.69–7.60 (m, 1H), 7.51 (dd, J = 8.4, 7.1 Hz, 2H), 7.15–7.06 (m, 2H), 6.91–6.80 (m, 2H), 5.10 (dd, J = 10.1, 3.3 Hz, 1H), 4.93 (d, J = 7.7 Hz, 1H), 4.25 (d, J = 3.3 Hz, 1H), 4.19 (dd, J = 10.2, 7.7 Hz, 1H), 3.85–3.79 (m, 3H), 3.77 (s, 3H). 13C{1H} NMR (200 MHz, CD3OD) δ 167.7, 156.6, 153.1, 134.3, 130.8, 129.4, 119.3, 115.4, 104.0, 77.8, 76.6, 69.9, 67.9, 62.1, 56.0. HRMS (ESI-Orbitrap) m/z: [M + Cl]− Calcd for C20H22ClO8 425.1009; found 425.1027.
p-Methoxyphenyl-2,4,6-tri-azido-3-benzoyl-2,4,6-trideoxy-β-D-mannopyranoside (21).
p-Methoxyphenyl-3-benzoyl-β-D-galactopyranoside (20, 0.15 g, 0.38 mmol) was dissolved in dichloromethane (10 mL) at 0 °C in a round bottom flask (50 mL). Pyridine (0.31 mL, 3.84 mmol) was added followed by slow addition of trifluoromethanesulfonic anhydride (0.32 mL 1.92 mmol) at 0 °C. After stirring at the same temperature for 30 min, the reaction was quenched by adding sodium bicarbonate (10 mL). The organic layer was washed with hydrochloric acid (1 N) and brine. Organic layer was combined and concentrated under reduced pressure to produce a crude product, which was used for the next step without any purification. To a solution of 2,4,6-tristriflate in toluene (20 mL), tetrabutylammonium azide (0.6 g, 2.1 mmol) was added at room temperature to a round bottom flask (50 mL). The reaction was stirred at 70 °C for 1 h and then at 90 °C for another 1 h. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (toluene) to produce p-methoxyphenyl-2,4,6-tri-azido-3-benzoyl-2,4,6-trideoxy-β-D-mannopyranoside (21) as a colorless oil (0.15 g, 88% yield). 1H NMR (400 MHz, CD3Cl) δ 8.15 (d, J = 8 Hz, 2H), 7.67 (t, J = 7.6 Hz, 1H), 7.54 (t, J = 8 Hz, 2H), 7.05 (d, J = 8.8 Hz, 2H), 6.91–6.82 (m, 2H), 5.22, (s, 1H), 4.52 (d, J = 3.6 Hz, 1H), 4.08 (t, J = 10 Hz, 1H), 3.80 (s, 3H), 3.69–3.56 (m, 2H), 3.51–3.43 (m, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ = 165.5, 150.2, 155.9, 134.0, 130.0, 128.7, 128.4, 118.7, 114.6, 98.8, 74.1, 73.8, 61.5, 57.4, 55.6, 51.5. HRMS (ESI-Orbitrap) m/z: [M + Cl]− Calcd for C20H19ClN9O5 500.1203; found 500.1190.
p-Methoxyphenyl-2,4,6-triacetamido-3-benzoyl-2,4,6-trideoxy-β-D-mannopyranoside (22).
p-Methoxyphenyl-2,4,6-tri-azido-3-benzoyl-2,4,6-trideoxy-β-D-mannopyranoside (21, 0.18 g, 0.38 mmol) was dissolved in pyridine (4.0 mL) in a round bottom flask (50 mL) and thioacetic acid (1.0 mL) was added drop-wisely. The reaction mixture was stirred for 24 h under reduced pressure at room temperature. The reaction was quenched by adding methanol (10 mL) and the solvent was removed under a reduced pressure. The compound was purified by silica gel column chromatography (ethyl acetate: methanol = 20:1, by volume) to produce p-methoxyphenyl-2,4,6-tri-acetamido-3-benzoyl-2,4,6-trideoxy-β-D-mannopyranoside (22) as a white amorphous solid (0.16 g, 80% yield). 1H NMR (400 MHz, CD3OD) δ 7.98–7.92 (m, 2H), 7.59 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.7 Hz, 2H), 7.01–6.93 (m, 2H), 6.89–6.80 (m, 2H), 5.28 (d, J = 1.6 Hz, 1H), 5.25 (dd, J = 11.0, 4.2 Hz, 1H), 5.00 (dd, J = 4.2, 1.5 Hz, 1H), 4.34 (t, J = 10.7 Hz, 1H), 3.75 (s, 3H), 3.74–3.68 (m, 1H), 3.61–3.50 (m, 1H), 3.44 (dd, J = 14.2, 2.7 Hz, 1H), 2.06 (s, 3H), 1.96 (s, 3H), 1.88 (s, 3H). 13C{1H} NMR (200 MHz, CD3OD) δ 174.1, 173.7, 173.6, 167.1, 156.9, 152.1, 134.4, 131.1, 130.7, 129.5, 119.3, 115.5, 99.1, 75.0, 73.8, 56.1, 51.9, 48.4, 41.5, 22.7, 22.6, 22.5. HRMS (ESI-Orbitrap) m/z: [M + Na]+ Calcd for C26H31N3O8Na 536.2003; found 536.1995.
2,4,6-Triacetamido-2,4,6-trideoxy-D-mannose (Man2,4,6triNAc, 10).
p-Methoxyphenyl-2,4,6-tri-acetamido-3-benzoyl-2,4,6-trideoxy-β-D-mannopyranoside (22, 0.1 g, 0.19 mmol) was dissolved in dry MeOH (10 mL) in a round bottom flask (50 mL), and 0.3 mL of sodium methoxide solution (5.4 M) was added. After the reaction was completed, the reaction mixture was neutralized using H+-resin. The crude product without further purification was added to a round bottom flask (50 mL) and dissolved in acetonitrile (4 mL). Ammonium cerium nitrate (0.3 g) dissolved in water (1 mL) was added slowly while stirring. The reaction was run for 2.5 h at room temperature. The reaction mixture was concentrated, and the residue was purified by silica gel column chromatography (ethyl acetate: methanol: water = 5:1:0.1, by volume) to produce 2,4,6-triacetamido-2,4,6-trideoxy-D-mannopyranose (10) as a white amorphous solid (0.04 g, 70% yield). 1H NMR (800 MHz, D2O) δ 5.16 (bs, 1H), 4.97 (bs, 1H), 4.48 (dt, J = 2.8, 1.4 Hz, 1H), 4.31 (dd, J = 4.6, 1.5 Hz, 1H), 4.09 (ddd, J = 10.6, 4.6, 1.3Hz, 1H), 4.01–3.95 (m, 1H), 3.92 (t, J = 10.5 Hz, 1H), 3.88 (ddt, J = 10.7, 4.2, 0.9 Hz, 1H), 3.83 (t, J = 10.5 Hz, 1H), 3.56–3.52 (m, 1H), 3.45–3.38 (m, 3H), 3.36–3.33 (m, 1H), 2.13 (s, 3H), 2.08 (s, 3H), 2.02 (s, 6H), 2.00 (s, 6H). 13C{1H} NMR (200 MHz, D2O) δ 175.8, 174.9, 174.5, 174.2, 93.0, 73.4, 70.0, 69.0, 66.7, 53.5, 52.6, 49.3, 48.9, 40.0, 39.9, 22.0, 21.9, 21.7. HRMS (ESI-Orbitrap) m/z: [M + Na]+ Calcd for C12H21N3NaO6 326.1323; found 326.1308.
p-Methoxyphenyl-2,4,6-tri-azido-2,4,6-trideoxy-β-D-mannopyranoside (23).
p-Methoxyphenyl-2,4,6-tri-azido-3-benzoyl-2,4,6-trideoxy-β-D-mannopyranoside (21, 0.50 g, 1.1 mmol) was dissolved in dry MeOH (10 mL) in a round bottom flask (50 mL), and 0.2 mL of sodium methoxide solution (5.4 M) was added. The reaction was carried out at room temperature for 6 h. After the reaction was completed, the reaction mixture was neutralized using H+-resin. The product was purified by silica gel column chromatography (toluene: ethyl acetate = 35:1, by volume) to produce compound 23 as a white amorphous solid (0.36 g, 92% yield). 1H NMR (400 MHz, CD3Cl) δ 6.99 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 5.06 (s, 1H), 4.19 (bd, J = 3.6 Hz, 1H), 3.75 (s, 1H), 3.75 (s, 3H), 3.60–3.45 (m, 3H), 3.31–3.22 (m, 1H), 2.59 (d, J = 9.2 Hz, 1H). 13C{1H} NMR (100 MHz, CD3Cl) δ 155.9, 150.1, 118.5, 114.7, 99.4, 74.2, 72.5, 63.9, 60.5, 55.7, 51.7. HRMS (ESI-Orbitrap) m/z: [M + Cl]− Calcd for C13H15ClN9O4 396.0941; found 396.0930.
2,4,6-Triazido-2,4,6-trideoxy-D-mannose (Man2,4,6triN3, 11).
p-Methoxyphenyl-2,4,6-tri-azido-2,4,6-trideoxy-β-D-mannopyranoside (23, 0.72 g, 1.99 mmol) was added to a round bottom flask (50 mL) and dissolved in acetonitrile (12 mL). Ammonium cerium nitrate (1.1 g) dissolved in water (3 mL) was added slowly while stirring. The reaction was carried out at room temperature for 2.5 h. The reaction mixture was concentrated, and the residue was purified by silica gel column chromatography (toluene: ethyl acetate = 15:1, by volume) to produce compound 11 as a white amorphous solid (0.41 g, 80% yield). 1H NMR (400 MHz, CD3OD) δ 5.14–4.86 (m, 2H), 4.20–4.11 (m, 1H), 3.95–3.73 (m, 4H), 3.64–3.36 (m, 6H), 3.35–3.25 (m, 1H). 13C{1H} NMR (100 MHz, CD3OD) δ 93.7, 92.4, 73.6, 72.4, 69.8, 69.7, 66.2, 64.7, 60.4, 60.0, 51.7, 51.6. HRMS (ESI-Orbitrap) m/z: [M + Na]+ Calcd for C6H9N9NaO3 278.0721; found 278.0707.
Enzymatic Synthesis of Sia Derivatives 7-Acetamido-7-deoxy-N-acetylneuraminic Acid (Neu5Ac7NAc, 4), 5,7-Diazido-5,7-dideoxy-neuraminic Acid (Neu5,7diN3, 5), and 5,7,9-Triazido-5,7,9-trideoxy-neuraminic Acid (Neu5,7,9triN3, 7)
7-Acetamido-7-deoxy-N-acetylneuraminic acid (Neu5Ac7NAc, 4).
The 2,4-diacetamido-2,4-dideoxy-D-mannose (Man2,4diNAc, 8, 0.27 g, 1.03 mmol) and sodium pyruvate (0.57 g, 5.15 mmol) were dissolved in water in 50 mL centrifuge tube. After the addition of appropriate amount of PmAldolase, water was added to bring the final concentration of mannose derivative to 10 mM. The reaction was carried out by incubating the solution at 30 °C with agitation at 120 rpm in an incubator for 72 h. The product formation was observed by TLC developed with ethyl acetate: methanol: water = 7:1:0.5 (by volume) and stained with p-anisaldehyde sugar stain. The reaction was quenched by addition of methanol (15 mL) and mixture was then centrifuged. The supernatant was concentrated and passed through a BioGel P-2 gel filtration (water was used as an eluent). Then the product was further purified by silica gel chromatography (ethyl acetate: methanol: water = 7:1:0.5) to produce 5,7-di-N-acetyl-neuraminic acid (4) as a white amorphous solid (0.26 g, 72% yield). 1H NMR (400 MHz, D2O) δ 4.23 (dd, J = 10.5, 2.1 Hz, 1H), 4.00–3.90 (m, 2H), 3.81–3.68 (m, 2H), 3.63 (dd, J = 12.1, 2.7 Hz, 1H), 3.46 (dd, J = 12.1, 7.0 Hz, 1H), 2.22 (dd, J = 13.0, 4.9 Hz, 1H), 2.00 (s, 3H), 1.99 (s, 3H), 1.83 (d, J = 13.0 Hz, 1H). 13C{1H} NMR (100 MHz, D2O) δ 176.6, 174.1, 173.9, 96.5, 70.2, 69.4, 67.5, 62.5, 59.3, 54.7, 49.3, 39.6, 22.1, 21.6. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C13H21N2O9 349.1253; found 349.1275.
5,7-Diazido-5,7-dideoxy-neuraminic acid (Neu5,7diN3,5).
The 2,4-diazido-2,4-dideoxymannose (Man2,4diN3, 9, 0.05 g, 0.22 mmol) and sodium pyruvate (0.12 g, 1.1 mmol) were dissolved in water in 50 mL centrifuge tube. After the addition of appropriate amount of PmAldolase, water was added to bring the final concentration of mannose derivative to 10 mM. The reaction was carried out by incubating the solution at 30 °C with agitation at 120 rpm in an incubator for 48 h. The product formation was observed by TLC developed with ethyl acetate:methanol:water = 7:1:0.5 (by volume) and stained with p-anisaldehyde sugar stain. The reaction was quenched by addition of methanol (15 mL) and mixture was then centrifuged. The supernatant was concentrated and passed through a BioGel P-2 gel filtration (water was used as an eluent). Then the product was further purified by silica gel chromatography (ethyl acetate: methanol: water = 7:1:0.5) to produce 5,7-di-azido-5,7-dideoxy-neuraminic acid (5) as a white amorphous solid (0.05 g, 72% yield). 1H NMR (600 MHz, D2O) δ 4.11 (ddd, J = 11.6, 9.5, 5.0 Hz, 1H), 4.05 (dd, J = 10.4, 1.8 Hz, 1H), 3.98–3.91 (m, 1H), 3.82–3.74 (m, 2H), 3.70–3.65 (m, 1H), 3.60–3.56 (m, 1H), 2.21 (dd, J = 13.1, 5.0 Hz, 1H), 1.97 (d, J = 13.1 Hz, 1H). 13C{1H} NMR (150 MHz, D2O) δ 175.7, 96.4, 72.1, 69.5, 68.5, 63.1, 62.5, 61.4, 39.1. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C9H13N6O7 317.0851; found 317.0830.
5,7,9-Triazido-5,7,9-trideoxy-neuraminic acid (Neu5,7,9triN3, 7).
Man2,4,6triN3 (11, 0.04 g, 0.16 mmol) and sodium pyruvate (0.086 g, 0.78 mmol) were dissolved in water in a 50 mL centrifuge tube. After the addition of appropriate amount of PmAldolase, water was added to bring the final concentration of mannose derivative to 10 mM. The reaction was carried out by incubating the solution at 30 °C with agitation at 120 rpm in an incubator for 48 h. The product formation was observed by TLC developed with ethyl acetate:methanol:water = 7:1:0.5 (by volume) and stained with p-anisaldehyde sugar stain. The reaction was quenched by addition of methanol (15 mL) and mixture was then centrifuged. The supernatant was concentrated and passed through a Bio-Gel P-2 gel filtration (water was used as an eluent). Then the product was further purified by silica gel chromatography (ethyl acetate: methanol: water = 7:1:0.5) to produce 7 as a white amorphous solid (0.037 g, 70% yield). 1H NMR (600 MHz, D2O) δ 4.14–4.06 (m, 2H), 4.04 (dd, J = 10.4, 1.8 Hz, 1H), 3.79 (dd, J = 8.9, 1.8 Hz, 1H), 3.72 (dd, J = 13.4, 2.8 Hz, 1H), 3.60–3.53 (m, 2H), 2.20 (dd, J = 13.1, 5.0 Hz, 1H), 1.96 (dd, J = 13.2, 11.7 Hz, 1H). 13C{1H} NMR (150 MHz, D2O) δ 175.6, 96.4, 69.5, 68.5, 68.4, 64.2, 62.1, 53.9, 39.1. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C9H12N9O6 342.0916; found 342.0943.
Chemical Synthesis of NHCbz-tagged Sialyltransferase Acceptors (24–30)
LacβProNHCbz (24)
LacβProNHCbz (24) was synthesized as described previously.47 Compounds 25–30 were synthesized similarly. Briefly, in a round bottom flask, a propylazide-tagged acceptor23 (0.085 mmol) was dissolved in water, a catalytic amount of palladium (10%) Pd/C was added. The mixture was stirred under a hydrogen atmosphere for overnight. After the completion of reaction, palladium was removed by filtration. The solvent was removed in vacuo. The obtained propylamine-tagged acceptor was used directly for next reaction without any further filtration. The compound (0.085 mmol) was dissolved in 5–10 mL water in a round bottom flask, sodium carbonate (45 mg, 0.43 mmol) was added. Benzyl chloroformate (CbzCl, 30 mg, 0.17 mmol) in acetonitrile was added to the mixture in the flask immersed in ice-water bath. After the completion of reaction, the solvent was removed. The mixture was purified using a C18 column (gradient solvent of CH3CN in H2O was used for elute) to produce the Cbz-tagged acceptors (25–30).
LacNAcβProNHCbz (25).
39 mg, 80% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.43 (q, J = 6.2, 5.7 Hz, 5H), 5.11 (s, 2H), 4.47 (dd, J = 7.7, 1.9 Hz, 2H), 4.05–3.46 (m, 14H), 3.17 (d, J = 7.1 Hz, 2H), 2.00 (d, J = 11.0 Hz, 3H), 1.74 (t, J = 6.4 Hz, 2H). 13C{1H} NMR (100 MHz, D2O) δ 174.5, 158.3, 136.6, 128.8, 128.3, 127.6, 102.9, 101.0, 78.5, 75.3, 74.7, 72.5, 72.4, 71.0, 68.5, 67.6, 66.8, 61.0, 60.1, 55.1, 37.2, 28.8, 22.1. HRMS (ESI-Orbitrap) m/z: [M + FA −H]− Calcd for C26H39N2O15 619.2356; found 619.2350.
Galβ3GalNAcβProNHCbz (26).
38 mg, 78% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.48–7.38 (m, 5H), 5.12–5.08 (m, 2H), 4.45–4.40 (m, 2H), 4.16 (d, J = 3.2, 1H), 3.98 (dd, J = 10.9, 8.5 Hz, 1H), 3.90 (d, J = 3.7 Hz, 2H), 3.82 (dd, J = 10.9, 3.3 Hz, 1H), 3.81–3.70 (m, 4H), 3.68–3.63 (m, 2H), 3.62–3.58 (m, 2H), 3.52 (dd, J = 10.0, 7.8 Hz, 1H), 3.21–3.10 (m, 2H), 1.99 (s, 3H), 1.79–1.69 (m, 2H).13C{1H} NMR (200 MHz, D2O) δ 174.7, 158.3, 136.6, 128.7, 128.3, 127.6, 104.8, 101.3, 79.9, 75.0, 74.7, 72.4, 70.5, 68.5, 68.0, 67.5, 66.73, 61.0, 60.9, 51.2, 37.2, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M + FA −H]− Calcd for C26H39N2O15 619.2356; found 619.2356.
Galβ3GalNAcαProNHCbz (27).
39 mg, 81% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.52–7.34 (m, 5H), 5.12 (d, J = 2.8 Hz, 2H), 4.82 (d, J = 3.8 Hz, 1H), 4.43 (d, J = 7.6 Hz, 1H), 4.32 (dd, J = 11.0, 3.7 Hz, 1H), 4.22 (d, J = 3.1 Hz, 1H), 4.06–3.92 (m, 2H), 3.90 (d, J = 3.3 Hz, 1H), 3.81–3.66 (m, 5H), 3.65–3.57 (m, 2H), 3.55–3.40 (m, 2H), 3.25 (t, J = 6.6 Hz, 2H), 2.01 (s, 3H), 1.80 (dd, J = 9.8, 4.2 Hz, 2H). 13C{1H} NMR (100 MHz, D2O) δ 174.5, 158.4, 136.6, 128.8, 128.3, 127.5, 104.7, 97.1, 77.4, 74.9, 72.5, 70.6, 70.6, 68.8, 68.5, 66.7, 65.0, 61.2, 60.9, 48.6, 37.5, 28.5, 22.0. HRMS (ESI-Orbitrap) m/z: [M + FA −H]− Calcd for C26H39N2O15 619.2356; found 619.2351.
Galβ3GlcNAcβProNHCbz (28).
39 mg, 80% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.49–7.24 (m, 5H), 5.03 (s, 2H), 4.41 (d, J = 8.2 Hz, 1H), 4.34 (d, J = 7.7 Hz, 1H), 3.83 (tt, J = 5.8, 2.9 Hz, 3H), 3.77–3.60 (m, 6H), 3.59–3.48 (m, 2H), 3.48–3.41 (m, 2H), 3.37 (ddd, J = 9.9, 5.6, 2.1 Hz, 1H), 3.09 (hept, J = 6.9 Hz, 2H), 1.92 (s, 3H), 1.66 (p, J = 6.6 Hz, 2H). 13C{1H} NMR (100 MHz, D2O) δ 174.6, 158.3, 136.6, 128.8, 128.3, 127.6, 103.5, 100.9, 82.4, 75.3, 75.3, 72.5, 70.7, 68.7, 68.5, 67.6, 66.8, 61.0, 60.7, 54.5, 37.2, 28.8, 22.2. HRMS (ESI-Orbitrap) m/z: [M + FA −H]− Calcd for C26H39N2O15 619.2356; found 619.2353.
Galβ3GlcNAcαProNHCbz (29).
38 mg, 78% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.43 (q, J = 6.9 Hz, 5H), 5.12 (s, 2H), 4.41 (d, J = 7.7 Hz, 1H), 4.08 (dd, J = 10.6, 3.6 Hz, 1H), 3.98–3.86 (m, 2H), 3.84–3.39 (m, 12H), 3.34–3.14 (m, J = 7.0 Hz, 2H), 2.01 (s, 3H), 1.88–1.68 (m, 2H). 13C{1H} NMR (100 MHz, D2O) δ 174.4, 158.4, 136.6, 128.8, 128.3, 127.5, 103.5, 97.0, 80.6, 75.2, 72.5, 71.5, 70.6, 68.6, 68.5, 66.7, 64.9, 60.9, 60.5, 52.4, 37.4, 28.5, 21.9. HRMS (ESI-Orbitrap) m/z: [M + FA −H]− Calcd for C26H39N2O15 619.2356; found 619.2349.
GalNAcαProNHCbz (30).
42 mg, 78% yield, white amorphous solid. 1H NMR (400 MHz, CD3OD) δ 7.45–7.23 (m, 5H), 5.17–5.02 (m, 2H), 4.77 (d, J = 3.7 Hz, 1H), 4.27 (dd, J = 10.9, 3.6 Hz, 1H), 3.89 (d, J = 3.2 Hz, 1H), 3.84–3.66 (m, 5H), 3.42 (dt, J = 10.7, 6.0 Hz, 1H), 3.32–3.17 (m, 2H), 2.01 (s, 3H), 1.79 (p, J = 6.5 Hz, 2H). 13C{1H} NMR (100 MHz, CD3OD) δ 174.2, 159.0, 138.5, 129.5, 129.0, 128.8, 98.8, 72.5, 70.4, 70.0, 67.4, 65.9, 62.9, 51.6, 38.8, 30.7, 22.7. HRMS (ESI-Orbitrap) m/z: [M + FA −H]− Calcd for C20H29N2O10 457.1828; found 457.1822.
General Procedures for One-Pot Three-Enzyme (OP3E) Preparative-Scale Synthesis of Neu5,7diN3α2–3/6-Linked Sialosides (31, 33, 35, 37, 39, 41, 43, 45, 47, 49, 51, 53)
Man2,4diN3 (9, 50–70 mg, 0.22–0.30 mmol), sodium pyruvate (0.75–1.5 mmol), CTP (0.15–0.30 mmol), and an acceptor selected from LacβProNHCbz (24),47 LacNAcβProNHCbz (25), Galβ1–3GalNAcβProNHCbz (26), Galβ1–3GalNAcαProNHCbz (27), Galβ1–3GlcNAcβProNHCbz (28), Galβ1–3GlcNAcαProNHCbz (29), and GalNAcαProNHCbz (30) (0.10–0.20 mmol), were dissolved in water in a 50 mL centrifuge tube containing Tris-HCl buffer (100 mM, pH 8.5) and MgCl2 (20 mM). After adding sialic acid aldolase (0.5–3 mg), NmCSS (0.5 mg), and a sialyltransferase PmST1 (1–3 mg) or Pd2,6ST (1–4 mg), water was added to bring the final concentration of Man2,4diN3 (9) to 10 mM. The reaction mixture was incubated at 30 °C for 24–36 h. The reaction progress was monitored using TLC (ethyl acetate:methanol:water = 6:1:1, by volume) and mass spectrometry. The reaction mixture was diluted with the same volume of ethanol and incubated at 4 °C for 30 min. The mixture was then centrifuged and concentrated, which was purified using a C18 column (CH3CN in H2O gradient was used as running solvents) to produce the sialoside product.
Neu5,7diN3α2–3LacβProNHCbz (31).
107 mg, 90% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.49–7.36 (m, 5H), 5.09 (s, 2H), 4.50 (d, J = 7.9 Hz, 1H), 4.41 (d, J = 8.0 Hz, 1H), 4.06 (ddd, J = 8.8, 5.6, 2.6 Hz, 1H), 4.03 (dd, J = 9.9, 3.2 Hz, 1H), 3.96–3.89 (m, 4H), 3.81–3.73 (m, 4H), 3.71–3.52 (m, 10H), 3.28 (t, J = 8.4 Hz, 1H), 3.26–3.17 (m, 2H), 2.72 (dd, J = 12.7, 4.7 Hz, 1H), 1.90 (t, J = 12.3 Hz, 1H), 1.79 (p, J = 6.6 Hz, 2H). 13C{1H} NMR (200 MHz, D2O) δ 173.3, 158.4, 136.6, 128.8, 128.3, 127.6, 102.6, 102.1, 100.8, 78.4, 75.7, 75.1, 74.7, 74.3, 72.8, 72.4, 70.4, 69.5, 69.3, 67.9, 67.7, 66.8, 62.5, 60.9, 60.3, 60.1, 38.8, 37.3, 28.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H46N7O19 832.2854; found 832.2883.
Neu5,7diN3α2–6LacβProNHCbz (33).
140 mg, 100% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.46–7.37 (m, 5H), 5.09 (s, 2H), 4.40 (dd, J = 17.6, 7.9 Hz, 2H), 4.05 (ddd, J = 8.7, 5.5, 2.5 Hz, 1H), 3.96–3.90 (m, 5H), 3.80–3.72 (m, 5H), 3.70–3.62 (m, 4H), 3.61–3.50 (m, 5H), 3.30 (t, J = 8.7 Hz, 1H), 3.26–3.18 (m, 2H), 2.71 (dd, J = 12.6, 4.7 Hz, 1H), 1.85–1.77 (m, 3H). 13C{1H} NMR (200 MHz, D2O) δ 173.1, 158.4, 136.6, 128.8, 128.3, 127.7, 103.3, 102.0, 100.6, 80.0, 74.6, 74.5, 73.5, 72.6, 72.4, 72.1, 70.8, 70.3, 69.4, 68.3, 67.7, 66.8, 63.4, 62.5, 60.6, 60.3, 59.3, 39.8, 37.3, 28.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H46N7O19 832.2854; found 832.2879.
Neu5,7diN3α2–3LacNAcβProNHCbz (35).
26.5 mg, 70% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.46–7.40 (m, 5H), 5.11 (s, 2H), 4.54 (d, J = 7.9 Hz, 1H), 4.46 (d, J = 8.3 Hz, 1H), 4.11–4.02 (m, 2H), 4.00–3.88 (m, 4H), 3.84 (dd, J = 12.4, 5.1 Hz, 1H), 3.80–3.75 (m, 3H), 3.73–3.64 (m,7H), 3.62–3.51 (m, 4H), 3.24–3.13 (m, 2H), 2.74 (dd, J = 12.6, 4.7 Hz, 1H), 2.02 (s, 3H), 1.91 (t, J = 12.3 Hz, 1H), 1.74 (p, J = 6.5 Hz, 2H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 173.4, 158.3, 136.6, 128.8, 128.3, 127.6, 102.6, 101.1, 100.7, 78.4, 75.7, 75.1, 74.7, 72.4, 70.4, 69.5, 69.3, 67.9, 67.6, 66.7, 62.5, 61.0, 60.4, 60.1, 59.4, 55.0, 38.8, 37.2, 28.8, 22.1. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3144.
Neu5,7diN3α2–6LacNAcβProNHCbz (37).
27 mg, 72% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.51–7.39 (m, 5H), 5.20–5.09 (m, 2H), 4.53 (d, J = 8.1 Hz, 1H), 4.43 (d, J = 8.0 Hz, 1H), 4.10 (ddd, J = 9.5, 5.5, 2.6 Hz, 1H), 4.01–3.89 (m, 5H), 3.84–3.51 (m, 15H), 3.44 (t, J = 9.9 Hz, 1H), 3.25–3.12 (m, 1H), 2.69 (dd, J = 12.6, 4.8 Hz, 1H), 2.04 (s, 3H), 1.86–1.72 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.2, 173.1, 158.3, 136.6, 128.8, 128.4, 127.6, 103.5, 100.8, 100.4, 80.8, 74.5, 73.6, 72.5, 72.2, 70.8, 70.2, 69.2, 68.4, 67.6, 66.8, 63.7, 63.4, 62.5, 60.4, 60.3, 59.3, 55.0, 39.9, 37.3, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3146.
Neu5,7diN3α2–3Galβ1–3GalNAcβProNHCbz (39).
24 mg, 99% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.48–7.38 (m, 5H), 5.14–5.07 (m, 2H), 4.49 (d, J = 7.8 Hz, 1H), 4.44 (d, J = 8.5 Hz, 1H), 4.16 (d, J = 3.2 Hz, 1H), 4.09–4.04 (m, 1H), 4.02–3.96 (m, 2H), 3.94–3.88 (m, 3H), 3.83–3.70 (m, 6H), 3.70–3.60 (m, 6H), 3.58 (t, J = 9.7 Hz, 1H), 3.52 (dd, J = 9.8, 7.8 Hz, 1H), 3.23–3.12 (m, 2H), 2.73 (d, J = 12.7, 4.7 Hz, 1H), 1.99 (s, 3H), 1.88 (t, J = 12.3 Hz, 1H), 1.80–1.69 (m, 2H). 13C{1H} NMR (200 MHz, D2O) δ 174.7, 173.4, 158.3, 136.6, 128.8, 128.3, 127.6, 104.6, 101.3, 100.4, 80.3, 75.8, 74.72, 74.7, 72.2, 70.4, 69.5, 68.9, 67.8, 67.6, 67.5, 66.8, 63.2, 62.4, 60.9, 60.3, 51.1, 39.1, 37.2, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3142.
Neu5,7diN3α2–6Galβ1–3GalNAcβProNHCbz (41).
25 mg, 100% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.51–7.39 (m, 5H), 5.19–5.09 (m, 2H), 4.42 (dd, J = 8.2, 6.1 Hz, 2H), 4.19 (d, J =3.3 Hz, 1H), 4.07–4.02 (m, 1H), 4.00–3.87 (m, 5H), 3.82–3.69 (m, 8H), 3.67–3.60 (m, 4H), 3.56–3.49 (m, 2H), 3.27–3.10 (m, 2H), 2.73 (dd, J = 12.6, 4.8 Hz, 1H), 2.00 (s, 3H), 1.80–1.71 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.7, 173.0, 158.3, 136.6, 128.8, 128.3, 127.6, 104.9, 101.3, 100.8, 80.0, 74.9, 73.1, 72.5, 72.1, 70.54, 70.48, 69.3, 68.6, 67.9, 67.6, 66.8, 63.6, 63.4, 62.5, 60.9, 60.7, 51.1, 40.0, 37.2, 28.8, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3128.
Neu5,7diN3α2–3Galβ1–3GalNAcαProNHCbz (43).
47.6 mg, 95% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.50–7.39 (m, 5H), 5.14 (s, 2H), 4.85 (d, J = 3.8 Hz, 1H), 4.49 (d, J = 7.9 Hz, 1H), 4.30 (dd, J = 11.1, 3.7 Hz, 1H), 4.22 (s, 1H), 4.10–4.03 (m, 1H), 4.02–3.89 (m, 5H), 3.84–3.62 (m, 9H), 3.62–3.43 (m, 4H), 3.32–3.21 (m, 2H), 2.73 (ddd, J = 13.3, 8.6, 4.7 Hz, 1H), 2.01 (s, 3H), 1.93–1.78 (m, 3H). 13C{1H} NMR (100 MHz, D2O) δ 174.5, 173.4, 158.4, 135.7, 128.8, 128.3, 127.5, 104.5, 100.4, 97.1, 77.9, 75.8, 74.7, 72.3, 70.5, 70.4, 69.5, 69.0, 68.5, 67.7, 66.7, 65.1, 62.4, 61.2, 60.9, 60.3, 59.3, 48.5, 39.1, 37.5, 28.5, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3149.
Neu5,7diN3α2–6Galβ1–3GalNAcαProNHCbz (45).
34.1 mg, 68% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.55–7.40 (m, 5H), 5.14 (s, 2H), 4.83 (d, J = 3.7 Hz, 1H), 4.41 (d, J = 7.8 Hz, 1H), 4.31 (dd, J = 10.9, 3.7 Hz, 1H), 4.18 (s, 1H), 4.09–3.97 (m, 2H), 3.95–3.85 (m, 4H), 3.81–3.72 (m, 6H), 3.70–3.57 (m, 4H), 3.55–3.45 (m, 3H), 3.34–3.22 (m, 2H), 2.74 (dd, J = 12.6, 4.5 Hz, 1H), 2.01 (d, J = 4.2 Hz, 3H), 1.89–1.76 (m, 2H), 1.73 (t, J = 12.3 Hz, 1H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 173.0, 158.4, 136.6, 128.8, 128.3, 127.6, 104.5, 100.7, 97.1, 77.2, 73.2, 72.3, 72.1, 70.7, 70.5, 70.4, 69.3, 68.8, 68.4, 66.8, 65.0, 63.5, 62.5, 61.5, 60.6, 59.4, 48.6, 40.0, 37.6, 28.5, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3150.
Neu5,7diN3α2–3Galβ1–3GlcNAcβProNHCbz (47).
66.1 mg, 87% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.49–7.39 (m, 5H), 5.12 (s, 2H), 4.50 (dd, J = 14.0, 8.2 Hz, 2H), 4.08–4.00 (m, 2H), 3.96–3.89 (m, 4H), 3.79–3.69 (m, 7H), 3.70–3.65 (m, 3H), 3.64–3.56 (m, 2H), 3.53 (dd, J = 9.5, 7.7 Hz, 2H), 3.49–3.43 (m, 1H), 3.24–3.13 (m, 2H), 2.76 (dd, J = 12.7, 4.8 Hz, 1H), 2.02 (s, 3H), 1.89 (t, J = 12.3 Hz, 1H), 1.76 (p, J = 6.6 Hz, 2H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 173.4, 158.3, 136.6, 128.8, 128.3, 127.6, 103.5, 100.8, 100.3, 82.9, 75.8, 75.3, 75.1, 72.2, 70.4, 69.5, 69.0, 68.8, 67.7, 67.5, 66.8, 63.2, 62.4, 60.9, 60.7, 60.4, 59.3, 54.3, 39.2, 37.3, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3150.
Neu5,7diN3α2–6Galβ1–3GlcNAcβProNHCbz (49).
60.7 mg, 100% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.51–7.39 (m, 5H), 5.12 (s, 2H), 4.51 (d, J = 8.5 Hz, 1H), 4.37 (d, J = 7.8 Hz, 1H), 4.07 (ddd, J = 9.4, 5.5, 2.6 Hz, 1H), 4.00–3.89 (m, 5H), 3.82–3.76 (m, 5H), 3.74–3.66 (m, 3H), 3.65–3.48 (m, 7H), 3.24–3.12 (m, 2H), 2.72 (dd, J = 12.6, 4.8 Hz, 1H), 2.01 (s, 3H), 1.83–1.71 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.6, 173.1, 158.3, 136.6, 128.8, 128.4, 127.6, 104.0, 100.9, 100.5, 84.4, 75.3, 73.5, 72.5, 72.1, 70.6, 70.4, 69.3, 69.1, 68.4, 67.6, 66.8, 63.6, 62.5, 61.4, 61.0, 60.6, 59.3, 54.2, 39.9, 37.2, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3150.
Neu5,7diN3α2–3Galβ1–3GlcNAcαProNHCbz (51).
46.6 mg, 93% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.48–7.38 (m, 5H), 5.13 (s, 2H), 4.53–4.45 (m, 1H), 4.06 (ddt, J = 9.2, 5.6, 3.1 Hz, 2H), 4.01 (dd, J = 9.8, 3.1 Hz, 1H), 3.95–3.87 (m, 3H), 3.81–3.65 (m, 11H), 3.64–3.47 (m, 5H), 3.33–3.20 (m, 2H), 2.79–2.69 (m, 1H), 2.01 (s, 3H), 1.89 (t, J = 12.3 Hz, 1H), 1.85–1.74 (m, 2H). 13C{1H} NMR (150 MHz, D2O) δ 174.4, 173.2, 158.4, 136.7, 128.8, 128.3, 127.5, 118.9, 103.4, 100.5, 96.9, 81.1, 75.9, 75.0, 72.3, 71.5, 70.4, 69.45, 69.37, 69.1, 68.7, 67.5, 66.7, 63.2, 62.4, 60.9, 60.5, 60.4, 59.3, 52.3, 39.1, 37.4, 28.5, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3142.
Neu5,7diN3α2–6Galβ1–3GlcNAcαProNHCbz (53).
50 mg, 100% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.52–7.34 (m, 5H), 5.14 (s, 2H), 4.34 (d, J = 7.8 Hz, 1H), 4.12–4.02 (m, 2H), 3.97–3.88 (m, 4H), 3.87–3.70 (m, 9H), 3.68 (dd, J = 9.4, 2.3 Hz, 1H), 3.62–3.54 (m, 3H), 3.53–3.45 (m, 3H), 3.35–3.17 (m, 2H), 2.72 (dd, J = 12.6, 4.8 Hz, 1H), 2.01 (s, 3H), 1.86–1.74 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.4, 173.1, 158.4, 136.6, 128.8, 128.3, 127.5, 103.8, 100.6, 96.8, 82.3, 73.5, 72.5, 72.1, 71.4, 70.6, 70.3, 69.3, 68.9, 68.4, 66.7, 65.0, 63.6, 62.5, 61.4, 60.6, 59.3, 52.2, 39.9, 37.5, 28.5, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H49N8O19 873.3119; found 873.3133.
Neu5,7diN3α2–6GalNAcαProNHCbz (55).
112 mg, 65% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.46–7.38 (m, 5H), 5.17–5.06 (m, 2H), 4.12 (dd, J = 11.1, 3.6 Hz, 1H), 4.02 (ddd, J = 8.6, 5.4, 2.6 Hz, 1H), 3.99–3.87 (m, 3H), 3.87–3.80 (m, 2H), 3.79–3.58 (m, 7H), 3.50 (t, J = 9.8 Hz, 1H), 3.47–3.37 (m, 1H), 3.30–3.17 (m, 2H), 2.74 (dd, J = 12.7, 4.8 Hz, 1H), 2.02 (s, 3H), 1.82–1.71 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 172.8, 158.3, 136.6, 128.8, 128.3, 127.6, 101.0, 97.1, 72.1, 70.4, 69.4, 69.3, 68.2, 67.6, 66.7, 65.4, 63.5, 62.5, 60.6, 59.3, 49.9, 40.0, 37.5, 28.5, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C28H39N8O14 711.2591; found 711.2605.
General Procedures for One-Pot Three-Enzyme (OP3E) Preparative-Scale Synthesis of Neu5,7,9triN3α2–3/6-linked Sialosides (32, 34, 36, 38, 40, 42, 44, 46, 48, 50, 52, 54, 56)
An acceptor selected from LacβProNHCbz 24, LacNAcβProNHCbz 25, Galβ1–3GalNAcβProNHCbz 26, Galβ1–3GalNAcαProNHCbz 27, Galβ1–3GlcNAcβProNHCbz 28, Galβ1–3GlcNAcαProNHCbz 29, and GalNAcαProNHCbz 30 (0.10–0.20 mmol), Man2,4,6triN3 (11, 50–70 mg, 0.2–0.30 mmol), sodium pyruvate (1.0–1.5 mmol), CTP (0.3–0.45 mmol) were dissolved in water in a 50 mL centrifuge tube containing Tris-HCl buffer (100 mM, pH 8.5) and MgCl2 (20 mM). After adding sialic acid aldolase (0.5–3 mg), NmCSS (0.5 mg), and a sialyltransferase PmST1 (1–3 mg) or Pd2,6ST (1–4 mg) water was added to bring the final concentration of Man2,4,6triN3 (11) to 10 mM. The reaction mixture was incubated at 30 °C for 24–36 h. The reaction progress was monitored by TLC (ethyl acetate:methanol:water = 6:1:1, by volume) and mass spectrometry. The reaction mixture was diluted with the same volume of ethanol and incubated at 4 °C for 30 min. The mixture was then centrifuged and concentrated, which was purified by automated flash chromatography using a C18 column (CH3CN in H2O gradient was used as running solvents) to produce sialosides.
Neu5,7,9triN3α2–3LacβProNHCbz (32).
127 mg, 92% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.46–7.38 (m, 5H), 5.10 (s, 2H), 4.49 (d, J = 7.8 Hz, 1H), 4.42 (d, J = 8.0 Hz, 1H), 4.19 (ddd, J = 9.0, 6.2, 2.7 Hz, 1H), 4.02 (dd, J = 9.8, 3.2 Hz, 1H), 3.98–3.88 (m, 3H), 3.83–3.46 (m, 15H), 3.31–3.15 (m, 3H), 2.73 (dd, J = 12.7, 4.7 Hz, 1H), 1.89 (t, J = 12.3 Hz, 1H), 1.80 (p, J = 6.6 Hz, 2H). 13C{1H} NMR (200 MHz, D2O) δ 173.2, 158.8, 136.6, 128.8, 128.3, 127.6, 102.6, 102.6, 102.1, 78.4, 75.8, 75.1, 74.7, 74.3, 72.8, 72.2, 69.5, 69.3, 69.3, 67.8, 67.7, 66.8, 63.1, 61.3, 60.9, 60.1, 53.1, 39.0, 37.3, 28.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H45N10O18 857.2919; found 857.2948.
Neu5,7,9triN3α2–6LacβProNHCbz (34).
132 mg, 100% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.41 (dd, J = 25.3, 7.7 Hz, 5H), 5.08 (s, 2H), 4.40 (t, J = 8.9 Hz, 2H), 4.17 (ddd, J = 8.7, 5.8, 2.6 Hz, 1H), 3.96–3.90 (m, 4H), 3.80–3.71 (m, 6H), 3.71–3.63 (m, 4H), 3.61–3.51 (m, 5H), 3.31 (t, J = 8.7 Hz, 1H), 3.23 (dt, J = 12.5, 6.7 Hz, 2H), 2.71 (dd, J = 12.6, 4.8 Hz, 1H), 1.85–1.74 (m, 3H). 13C{1H} NMR (200 MHz, D2O) δ 173.1, 158.3, 136.5, 128.8, 128.3, 127.7, 103.3, 102.0, 100.6, 80.0, 74.6, 74.5, 73.5, 72.6, 72.5, 72.0, 70.8, 69.5, 69.1, 68.3, 67.7, 66.8, 63.4, 63.3, 61.6, 60.4, 53.2, 39.9, 37.4, 28.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H45N10O18 857.2919; found 857.2939.
Neu5,7,9triN3α2–3LacNAcβProNHCbz (36).
12.9 mg, 66% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.52–7.37 (m, 5H), 5.11 (s, 2H), 4.53 (d, J = 7.8 Hz, 1H), 4.47 (d, J = 8.3 Hz, 1H), 4.21 (ddd, J = 9.1, 6.3, 2.7 Hz, 1H), 4.04 (dd, J = 9.9, 3.2 Hz, 1H), 3.99 (dd, J = 12.4, 2.4 Hz, 1H), 3.95–3.87 (m, 2H), 3.84 (dd, J = 12.4, 5.1 Hz, 1H), 3.81–3.75 (m, 3H), 3.73–3.65 (m, 7H), 3.65–3.52 (m, 5H), 3.23–3.08 (m, 2H), 2.74 (dd, J = 12.6, 4.7 Hz, 1H), 2.02 (s, 3H), 1.90 (t, J= 12.3 Hz, 1H), 1.75 (t, J = 6.5 Hz, 2H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 173.3, 158.3, 136.6, 128.8, 128.4, 127.6, 102.6, 101.1, 100.7, 78.5, 75.8, 75.1, 74.7, 72.4, 72.2, 71.0, 69.5, 69.4, 69.3, 67.8, 66.8, 61.3, 60.9, 60.1, 59.3, 55.0, 53.2, 39.0, 37.3, 28.8, 22.1. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3200.
Neu5,7,9triN3α2–6LacNAcβProNHCbz (38).
25.4 mg, 65% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.50–7.39 (m, 5H), 5.17–5.08 (m, 2H), 4.52 (d, J = 8.2 Hz, 1H), 4.42 (d, J = 7.9 Hz, 1H), 4.23 (ddd, J = 8.8, 5.9, 2.7 Hz, 1H), 4.01–3.88 (m, 3H), 3.86–3.76 (m, 4H), 3.73–3.51 (m, 12H), 3.44 (t, J = 12 Hz, 1H), 3.25–3.12 (m, 2H), 2.69 (dd, J = 12.6, 4.7 Hz, 1H), 2.06 (s, 3H), 1.84–1.71 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.2, 173.0, 158.3, 136.6, 128.8, 128.4, 127.6, 103.5, 102.9, 100.8, 80.8, 74.5, 73.6, 72.51, 72.47, 72.1, 70.8, 69.2, 68.9, 68.3, 67.6, 66.8, 63.6, 61.2, 60.4, 59.3, 55.0, 53.2, 39.9, 37.3, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3202.
Neu5,7,9triN3α2–3Galβ1–3GalNAcβProNHCbz (40).
7.3 mg, 67% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.47–7.38 (m, 5H), 5.15–5.09 (m, 2H), 4.48 (d, J = 7.8 Hz, 1H), 4.44 (d, J = 8.6 Hz, 1H), 4.19–4.15 (m, 2H), 4.01–3.96 (m, 2H), 3.94–3.90 (m, 1H), 3.89 (d, J = 3.2 Hz, 1H), 3.82–3.73 (m, 5H), 3.72–3.55 (m, 9H), 3.52 (dd, J = 9.8, 7.8 Hz, 1H), 3.23–3.11 (m, 2H), 2.73 (dd, J = 12.7, 4.7 Hz, 1H), 2.00 (s, 3H), 1.87 (t, J = 12.3 Hz, 1H), 1.77–1.72 (m, 2H). 13C{1H} NMR (200 MHz, D2O) δ 174.7, 173.3, 158.3, 136.6, 128.8, 128.3, 127.6, 104.5, 101.3, 100.3, 80.3, 75.8, 74.7, 72.1, 69.5, 69.3, 68.9, 67.8, 67.5, 66.7, 61.2, 60.92, 60.9, 59.3, 53.1, 51.1, 39.2, 37.2, 29.6, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3206.
Neu5,7,9triN3α2–6Galβ1–3GalNAcβProNHCbz (42).
17.2 mg, 69% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.55–7.36 (m, 5H), 5.25–5.05 (m, 2H), 4.43 (ddd, J = 15.5, 11.6, 8.0 Hz, 2H), 4.24–4.12 (m, 2H), 3.93–3.69 (m, 10H), 3.66–3.50 (m, 9H), 3.43–3.29 (m, 1H), 2.90 (s, 1H), 2.77–2.70 (m, 1H), 2.01 (s, 3H), 1.84–1.71 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 173.02, 172.97, 171.0, 158.2, 136.5, 128.8, 128.4, 127.6, 104.9, 104.4, 100.8, 75.0, 72.5, 72.31, 72.28, 72.1, 70.5, 70.4, 69.3, 69.2, 69.1, 68.6, 63.4, 63.2, 61.3, 60.9, 53.2, 51.2, 45.7, 40.0, 28.8, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3212.
Neu5,7,9triN3α2–3Galβ1–3GalNAcαProNHCbz (44).
29.2 mg, 65% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.44 (q, J = 7.3 Hz, 5H), 5.14 (s, 2H), 4.85 (d, J = 3.7 Hz, 1H), 4.48 (d, J = 7.8 Hz, 1H), 4.29 (dd, J = 11.1, 3.7 Hz, 1H), 4.24–4.13 (m, 2H), 4.04–3.86 (m, 4H), 3.81–3.46 (m, 14H), 3.35–3.19 (m, 2H), 2.74 (dd, J = 12.7, 4.7 Hz, 1H), 2.01 (s, 3H), 1.92–1.76 (m, 3H). 13C{1H} NMR (100 MHz, D2O) δ 174.5, 173.4, 158.4, 136.5, 128.8, 128.3, 127.4, 104.5, 100.3, 97.1, 77.9, 77.3, 75.9, 74.7, 72.1, 70.6, 69.5, 69.2, 69.0, 68.5, 67.5, 66.7, 65.1, 63.1, 61.2, 60.9, 53.1, 48.6, 39.3, 37.4, 28.4, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3201.
Neu5,7,9triN3α2–6Galβ1–3GalNAcαProNHCbz (46).
40.7 mg, 79% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.52–7.31 (m, 5H), 5.13 (s, 2H), 4.82–4.81 (m, 1H), 4.39 (d, J = 7.8 Hz, 1H), 4.29 (dd, J = 11.1, 3.7 Hz, 1H), 4.20–4.07 (m, 2H), 3.98 (dd, J = 11.0, 3.1 Hz, 1H), 3.94–3.82 (m, 3H), 3.79 (dd, J = 10.3, 2.3 Hz, 1H), 3.78–3.65 (m, 6H), 3.63 (t, J = 6.2 Hz, 1H), 3.60–3.52 (m, 3H), 3.52–3.43 (m, 3H), 3.26 (dq, J = 13.5, 6.9 Hz, 2H), 2.71 (dd, J = 12.6, 4.8 Hz, 1H), 2.00 (s, 3H), 1.80 (qt, J = 13.0, 7.6 Hz, 2H), 1.71 (t, J = 12.3 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 174.5, 173.0, 158.4, 136.6, 128.8, 128.3, 127.6, 104.5, 100.7, 97.1, 77.2, 73.1, 72.3, 72.0, 70.7, 70.5, 69.3, 69.2, 68.8, 68.3, 66.7, 65.0, 63.4, 61.5, 61.5, 59.3, 53.1, 48.6, 40.0, 37.6, 28.4, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3216.
Neu5,7,9triN3α2–3Galβ1–3GlcNAcβProNHCbz (48).
50 mg, 64% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.71–7.20 (m, 5H), 5.29–5.04 (m, 2H), 4.49 (d, J = 8.5 Hz, 1H), 4.46 (d, J = 7.8 Hz, 1H), 4.15 (ddd, J = 9.0, 6.4, 2.8 Hz, 1H), 4.00 (dd, J = 9.8, 3.2 Hz, 1H), 3.96–3.87 (m, 3H), 3.84–3.77 (m, 1H), 3.75–3.55 (m, 12H), 3.54–3.48 (m, 2H), 3.48–3.40 (m, 1H), 3.24–3.08 (m, 2H), 2.74 (dd, J = 12.6, 4.7 Hz, 1H), 2.01 (s, 3H), 1.86 (t, J = 12.3 Hz, 1H), 1.74 (p, J = 6.5 Hz, 2H). 13C{1H} NMR (200 MHz, D2O) δ 174.5, 173.3, 158.3, 136.6, 128.8, 128.3, 127.6, 103.4, 100.8, 100.3, 82.9, 75.9, 75.3, 75.0, 72.1, 69.5, 69.3, 69.0, 68.8, 67.6, 67.4, 66.7, 63.1, 61.3, 60.9, 60.7, 54.3, 53.1, 39.3, 37.3, 28.7, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3201.
Neu5,7,9triN3α2–6Galβ1–3GlcNAcβProNHCbz (50).
57.1 mg, 91% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.50–7.41 (m, 5H), 5.16–5.08 (m, 2H), 4.50 (d, J = 8.5 Hz, 1H), 4.37 (d, J = 7.8 Hz, 1H), 4.20 (ddd, J = 9.0, 6.0, 2.7 Hz, 1H), 4.01–3.88 (m, 4H), 3.84–3.74 (m, 6H), 3.71–3.66 (m, 2H), 3.65–3.56 (m, 4H), 3.55–3.45 (m, 4H), 3.26–3.05 (m, 2H), 2.71 (dd, J = 12.6, 4.8 Hz, 1H), 2.01 (s, 3H), 1.82–1.70 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.6, 173.0, 158.3, 136.6, 128.8, 128.4, 127.6, 103.9, 100.9, 100.6, 84.4, 75.3, 73.5, 72.5, 72.0, 70.6, 69.3, 69.1, 69.1, 68.4, 67.6, 66.8, 63.5, 63.5, 61.5, 61.0, 59.3, 54.2, 53.2, 39.9, 37.2, 28.8, 22.2. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3213.
Neu5,7,9triN3α2–3Galβ1–3GlcNAcαProNHCbz (52).
44.8 mg, 87% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.53–7.37 (m, 5H), 5.19–5.08 (m, 2H), 4.52–4.39 (m, 1H), 4.21–4.13 (m, 1H), 4.12–4.02 (m, 1H), 3.99 (dd, J = 9.8, 3.2 Hz, 1H), 3.96–3.85 (m, 2H), 3.87–3.81 (m, 1H), 3.81–3.71 (m, 5H), 3.73–3.63 (m, 4H), 3.64–3.43 (m, 7H), 3.35–3.16 (m, 2H), 2.78–2.69 (m, 1H), 2.02 (s, 3H), 1.88 (t, J = 12.3 Hz, 1H), 1.86–1.73 (m, 2H). 13C{1H} NMR (150 MHz, D2O) δ 174.4, 173.3, 158.3, 136.7, 128.8, 128.3, 127.5, 103.5, 100.3, 96.9, 81.3, 75.9, 75.0, 72.5, 72.1, 71.5, 70.7, 69.5, 69.3, 69.1, 68.8, 67.4, 66.7, 63.1, 61.3, 60.9, 60.5, 53.1, 52.3, 39.3, 28.5, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3209.
Neu5,7,9triN3α2–6Galβ1–3GlcNAcαProNHCbz (54).
49.5 mg, 96% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.51–7.38 (m, 5H), 5.14 (s, 2H), 4.34 (d, J = 7.9 Hz, 1H), 4.23–4.16 (m, 1H), 4.06 (dd, J = 10.6, 3.6 Hz, 1H), 3.95–3.87 (m, 3H), 3.87–3.78 (m, 3H), 3.78–3.66 (m, 7H), 3.64–3.53 (m, 4H), 3.53–3.43 (m, 3H), 3.39–3.16 (m, 2H), 2.71 (dd, J = 12.6, 4.8 Hz, 1H), 2.01 (s, 3H), 1.90–1.70 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.4, 173.0, 158.4, 136.6, 128.8, 128.3, 127.5, 103.8, 100.6, 96.8, 82.2, 73.4, 72.5, 72.0, 71.4, 70.6, 69.3, 69.1, 68.9, 68.3, 66.7, 65.0, 63.5, 63.3, 61.5, 60.7, 59.3, 53.2, 52.2, 40.0, 37.5, 28.5, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H48N11O18 898.3184; found 898.3209.
Neu5,7,9triN3α2–6GalNAcαProNHCbz (56).
108 mg, 65% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.72–7.09 (m, 5H), 5.32–5.02 (m, 2H), 4.22–4.05 (m, 2H), 3.95 (s, 2H), 3.89–3.46 (m, 11H), 3.43 (dt, J = 11.3, 6.1 Hz, 1H), 3.29–3.06 (m, 2H), 2.79–2.63 (m, 1H), 2.00 (s, 3H), 1.82–1.65 (m, 3H). 13C{1H} NMR (200 MHz, D2O) δ 174.5, 158.2, 136.6, 128.7, 128.3, 127.6, 101.3, 97.1, 72.0, 69.4, 69.2, 68.1, 67.6, 66.7, 65.4, 63.4, 63.3, 63.2, 61.5, 53.2, 53.1, 49.9, 40.0, 37.5, 28.5, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C28H38N11O13 736.2656; found 736.2672.
General Procedures for Converting Azido-containing Glycosides (31–56) to N-Acetyl-containing Glycosides (57–82).
To a sodium bicarbonate saturated solution in water in a round bottom flask (100 mL), an azido-containing glycoside (30–50 mg) was added followed by drop-wise addition of 12–24 equivalents of thioacetic acid under argon at room temperature and stir at 70 °C for 20 h. After the completion of the reaction, the solvent was removed under vacuum. The mixture was passed through a Bio-Gel P-2 gel filtration (water was used as an eluent). The product-containing fractions were concentrated and further purified by silica gel chromatography using a mixed solvent (ethyl acetate: methanol: water = 10:1:0.1, by volume) as an eluent, followed by C18 purification (CH3CN in H2O gradient was used as running solvents) to obtain the pure product.
Neu5Ac7NAcα2–3LacβProNHCbz (57).
38 mg, 68% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.55–7.37 (m, 5H), 5.10 (s, 2H), 4.51 (d, J = 7.9 Hz, 1H), 4.43 (d, J = 8.0 Hz, 1H), 4.13 (ddd, J = 10.0, 3.1, 1.0 Hz, 1H), 4.00–3.87 (m, 5H), 3.82–3.67 (m, 7H), 3.66–3.61 (m, 3H), 3.61–3.54 (m, 3H), 3.53–3.49 (m, 1H), 3.29 (t, J = 8.4 Hz, 1H), 3.27–3.18 (m, 2H), 2.79 (dd, J = 12.5, 4.5 Hz, 1H), 1.98 (s, 3H), 1.93 (s, 3H), 1.80 (p, J = 6.6 Hz, 2H), 1.76 (t, J = 12.2 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 174.0, 173.8, 173.8, 158.4, 136.6, 128.8, 128.3, 127.6, 102.6, 102.1, 99.6, 78.3, 75.4, 75.1, 74.7, 74.3, 72.8, 71.8, 71.6, 69.4, 68.6, 67.7, 66.9, 66.8, 62.4, 61.0, 60.1, 51.8, 49.2, 40.1, 37.3, 28.8, 22.1, 21.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C36H54N3O21 864.3255; found 864.3282.
Neu5Ac7,9diNAcα2–3LacβProNHCbz (58).
42 mg, 80% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.47–7.41 (m, 5H), 5.12 (s, 2H), 4.51 (d, J = 7.8 Hz, 1H), 4.45 (d, J = 8.1 Hz, 1H), 4.12 (dd, J = 9.9, 3.2 Hz, 1H), 3.99–3.95 (m, 4H), 3.87–3.53 (m, 13H), 3.48 (dd, J = 14.1, 2.4 Hz, 1H), 3.34–3.23 (m, 3H), 3.09 (dd, J = 14.1, 7.5 Hz, 1H), 2.80 (dd, J = 12.4, 4.6 Hz, 1H), 2.01 (s, 3H), 2.01 (s, 3H), 1.96 (s, 3H), 1.85–1.74 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.2, 174.0, 173.83, 173.79, 158.4, 136.6, 128.8, 128.3, 127.6, 102.6, 102.1, 99.6, 78.3, 75.5, 75.1, 74.7, 74.3, 72.8, 71.8, 69.7, 69.4, 68.7, 67.7, 67.0, 66.8, 61.0, 60.1, 51.9, 50.6, 41.8, 40.1, 37.3, 28.8, 22.1, 21.90, 21.88. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3554.
Neu5Ac7NAcα2–6LacβProNHCbz (59).
42 mg, 78% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.51–7.34 (m, 5H), 5.10 (s, 2H), 4.43 (dd, J = 19.3, 8.0 Hz, 2H), 4.04–3.87 (m, 7H), 3.84–3.72 (m, 3H), 3.71–3.52 (m, 9H), 3.49 (dd, J = 12.2, 6.7 Hz, 1H), 3.34 (t, J = 8.6 Hz, 1H), 3.27–3.20 (m, 2H), 2.73 (dd, J = 12.4, 4.6 Hz, 1H), 2.00 (s, 3H), 1.93 (s, 3H), 1.81 (p, J = 6.7 Hz, 2H), 1.75 (t, J = 12.2 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 173.93, 173.86, 173.3, 158.4, 136.6, 128.8, 128.3, 127.6, 103.2, 101.9, 100.4, 79.5, 74.6, 73.6, 72.8, 72.4, 71.6, 71.5, 70.7, 68.7, 68.4, 67.7, 66.8, 63.4, 62.4, 60.2, 51.9, 49.2, 40.1, 37.3, 28.8, 22.1, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C36H54N3O21 864.3255; found 864.3280.
Neu5Ac7,9diNAcα2–6LacβProNHCbz (60).
36 mg, 80% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.49–7.37 (m, 5H), 5.12 (s, 2H), 4.45 (dd, J = 15.6, 8.0 Hz, 2H), 4.04–3.90 (m, 8H), 3.86–3.54 (m, 12H), 3.45 (dd, J = 14.1, 2.5 Hz, 1H), 3.36 (t, J = 8.6 Hz, 1H), 3.09 (dd, J = 14.1, 7.6 Hz, 1H), 2.75 (dd, J = 12.4, 4.6 Hz, 1H), 2.02 (s, 3H), 2.01 (s, 3H), 1.96 (s, 3H), 1.83 (p, J = 6.5 Hz, 2H), 1.76 (t, J = 12.2 Hz, 1H). 13C{1H} NMR (150 MHz, D2O) δ 174.2, 174.0, 173.8, 173.3, 158.4, 136.6, 128.8, 128.3, 127.6, 103.2, 102.0, 100.4, 79.5, 74.64, 74.62, 73.6, 72.9, 72.4, 71.6, 70.7, 69.5, 68.7, 68.5, 67.7, 66.8, 63.4, 60.2, 51.9, 50.6, 41.8, 40.1, 37.3, 28.9, 22.14, 22.09, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3555.
Neu5Ac7NAcα2–3LacNAcβProNHCbz (61).
10 mg, 67% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.44 (dq, J = 14.1, 7.3 Hz, 5H), 5.12 (s, 2H), 4.59–4.53 (m, 1H), 4.48 (d, J = 8.0 Hz, 1H), 4.19–4.11 (m, 1H), 4.03–3.96 (m, 2H), 3.91 (ddd, J = 9.5, 6.7, 2.3 Hz, 2H), 3.87–3.80 (m, 2H), 3.78–3.64 (m, 9H), 3.63–3.50 (m, 5H), 3.26–3.11 (m, 2H), 2.80 (dd, J = 12.4, 4.6 Hz, 1H), 2.03–1.93 (m, 9H), 1.80–1.73 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 174.0, 173.9, 173.8, 158.4, 136.6, 128.8, 128.3, 127.6, 102.5, 101.1, 99.5, 78.4, 75.4, 75.1, 74.7, 72.4, 71.9, 71.6, 69.4, 68.6, 67.6, 67.0, 66.8, 62.4, 61.0, 60.1, 55.0, 51.9, 49.2, 40.1, 37.2, 28.7, 22.1, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3546.
Neu5Ac7,9diNAcα2–3LacNAcβProNHCbz (62).
8.5 mg, 67% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.56–7.34 (m, 5H), 5.14 (d, J = 20.6 Hz, 2H), 4.54 (dd, J = 7.9, 2.3 Hz, 1H), 4.50–4.45 (m, 1H), 4.12 (dd, J = 9.9, 3.1 Hz, 1H), 4.03–3.88 (m, 5H), 3.87–3.80 (m, 2H), 3.80–3.65 (m, 7H), 3.65–3.54 (m, 4H), 3.49 (dd, J = 14.0, 2.3 Hz, 1H), 3.24–3.13 (m, 2H), 3.09 (dd, J = 14.1, 7.5 Hz, 1H), 2.80 (dd, J = 12.4, 4.6 Hz, 1H), 2.05–2.02 (m, 6H), 2.01 (s, 3H), 1.96 (s, 3H), 1.87–1.72 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.2, 174.0, 173.8, 158.4, 136.8, 128.8, 128.3, 127.6, 102.5, 101.1, 100.0, 78.3, 75.5, 75.1, 74.7, 72.4, 71.8, 69.7, 69.4, 68.6, 67.6, 67.0, 66.8, 61.0, 60.1, 55.0, 51.9, 50.6, 41.8, 40.1, 37.2, 28.6, 22.12, 22.10, 21.90, 21.88. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3804.
Neu5Ac7NAcα2–6LacNAcβProNHCbz (63).
11.3 mg, 77% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.68–7.29 (m, 5H), 5.14 (d, J = 12.8 Hz, 2H), 4.60–4.32 (m, 2H), 4.18–3.39 (m, 21H), 3.36–2.84 (m, 2H), 2.72 (dd, J = 12.5, 4.6 Hz, 1H), 2.05 (s, 3H), 2.00 (s, 3H), 1.95 (s, 3H), 1.87–1.56 (m, 3H). 13C{1H} NMR (100 MHz, D2O) δ 174.5, 173.8, 173.7, 173.4, 158.6, 138.3, 128.8, 128.3, 127.6, 103.3, 100.9, 100.3, 80.0, 74.6, 73.6, 72.4, 71.7, 71.5, 70.7, 68.7, 68.3, 67.6, 66.8, 63.2, 62.5, 60.3, 55.1, 51.9, 49.2, 40.1, 36.9, 28.6, 22.2, 22.1, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3529.
Neu5Ac7,9diNAcα2–6LacNAcβProNHCbz (64).
19.3mg, 85% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.44 (td, J = 7.4, 4.1 Hz, 5H), 5.14 (d, J = 12.9 Hz, 2H), 4.56–4.43 (m, 2H), 4.24–3.31 (m, 20H), 3.27–2.87 (m, 3H), 2.71 (dd, J = 12.6, 4.7 Hz, 1H), 2.19–1.91 (m, 12H), 1.77 (h, J = 11.7 Hz, 3H). 13C{1H} NMR (100 MHz, D2O) δ 175.2, 174.2, 173.8, 173.7, 173.4, 166.2, 136.1, 128.8, 128.3, 127.6, 103.3, 101.0, 100.4, 79.9, 74.6, 73.5, 72.4, 71.9, 71.6, 70.7, 69.5, 68.7, 68.4, 67.6, 66.8, 63.3, 55.1, 51.9, 50.6, 41.8, 40.1, 36.0, 27.6, 22.1, 22.0, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3805.
Neu5Ac7NAcα2–3Galβ1–3GalNAcβProNHCbz (65).
5.7 mg, 64% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.59–7.34 (m, 5H), 5.15 (d, J = 22.3 Hz, 2H), 4.50 (d, J = 7.8 Hz, 1H), 4.43 (dd, J = 15.4, 8.6 Hz, 1H), 4.16 (d, J = 3.5 Hz, 1H), 4.10 (dd, J = 9.9, 3.2 Hz, 1H), 4.05–3.96 (m, 2H), 3.95–3.86 (m, 3H), 3.85–3.71 (m, 6H), 3.69–3.55 (m, 6H), 3.51 (dd, J = 12.0, 6.8 Hz, 1H), 3.37–3.17 (m, 1H), 2.93 (d, J = 32.4 Hz, 2H), 2.81 (dd, J = 12.3, 4.6 Hz, 1H), 2.01–1.94 (m, 9H), 1.87–1.69 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.1, 173.6, 173.52, 173.45, 158.0, 136.3, 128.4, 127.9, 127.1, 104.1, 99.0, 96.7, 76.9, 75.2, 74.4, 71.4, 71.3, 70.2, 68.6, 68.3, 68.2, 66.5, 66.3, 64.6, 62.0, 60.9, 60.6, 51.4, 48.9, 48.3, 39.9, 37.1, 28.1, 21.72, 21.66, 21.5. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3546.
Neu5Ac7,9diNAcα2–3Galβ1–3GalNAcβProNHCbz (66).
4.4 mg, 65% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.44 (dtt, J = 18.5, 6.5, 3.0 Hz, 5H), 5.15 (d, J = 22.8 Hz, 2H), 4.49 (d, J = 8.0 Hz, 1H), 4.43 (dd, J = 14.7, 8.6 Hz, 1H), 4.17 (d, J = 3.1 Hz, 1H), 4.09–3.91 (m, 6H), 3.84–3.72 (m, 7H), 3.67–3.57 (m, 5H), 3.51–3.45 (m, 1H), 3.36–3.13 (m, 2H), 3.09–2.98 (m, 1H), 2.80 (dd, J = 12.4, 4.5 Hz, 1H), 2.02–1.99 (m, 6H), 1.95 (s, 6H), 1.84–1.80 (m, 1H), 1.78–1.72 (m, 2H). 13C{1H} NMR (150 MHz, D2O) δ 174.1, 173.80, 173.76, 173.61, 173.58, 158.1, 136.4, 128.5, 128.1, 127.3, 103.2, 100.6, 99.2, 82.0, 75.4, 75.2, 74.8, 71.5, 69.5, 68.8, 68.42, 68.36, 67.5, 66.6, 66.5, 60.8, 60.5, 54.3, 51.6, 50.2, 41.4, 40.0, 37.1, 28.5, 21.91, 21.88, 21.7, 21.6. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3803.
Neu5Ac7NAcα2–6Galβ1–3GalNAcβProNHCbz (67).
10.4 mg, 67% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.61–7.33 (m, 5H), 5.15 (d, J = 20.8 Hz, 2H), 4.50–4.36 (m, 2H), 4.20 (dd, J = 19.0, 3.2 Hz, 1H), 4.02–3.83 (m, 8H), 3.78–3.59 (m, 9H), 3.56–3.48 (m, 2H), 3.43–3.10 (m, 1H), 3.00–2.87 (m, 2H), 2.84–2.70 (m, 1H), 2.01 (s, 3H), 1.98 (s, 3H), 1.96 (s, 3H), 1.89–1.63 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.0, 173.8, 173.7, 173.3, 157.9, 136.7, 128.8, 128.4, 127.6, 104.9, 104.4, 100.4, 75.0, 74.7, 73.2, 72.5, 72.4, 71.70, 71.67, 71.5, 71.4, 70.6, 70.4, 68.6, 68.5, 67.4, 66.8, 62.5, 61.2, 61.0, 52.0, 51.2, 49.23, 49.16, 45.7, 40.3, 22.2, 22.1, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3530.
Neu5Ac7,9diNAcα2–6Galβ1–3GalNAcβProNHCbz (68).
10.4 mg, 70% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.55–7.34 (m, 5H), 5.23–5.09 (m, 2H), 4.43 (ddd, J = 14.7, 9.2, 5.6 Hz, 2H), 4.23–4.11 (m, 1H), 4.01–3.89 (m, 6H), 3.84–3.53 (m, 11H), 3.47–3.33 (m, 2H), 3.29–3.02 (m, 2H), 2.98–2.88 (m, 1H), 2.80–2.70 (m, 1H), 2.04–1.98 (m, 9H), 1.96 (s, 3H), 1.84–1.62 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.2, 173.99, 173.97, 173.7, 173.3, 158.3, 136.6, 128.8, 128.3, 127.6, 104.9, 100.5, 100.4, 75.0, 73.21, 73.16, 72.5, 71.61, 71.58, 70.6, 70.4, 69.6, 69.4, 68.6, 67.7, 67.4, 66.8, 61.0, 52.0, 51.2, 50.6, 45.7, 41.9, 40.3, 34.3, 28.9, 22.2, 22.1, 21.90, 21.87. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3805.
Neu5Ac7NAcα2–3Galβ1–3GalNAcαProNHCbz (69).
20 mg, 77% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.50–7.35 (m, 5H), 5.17–5.09 (m, 2H), 4.84 (d, J = 3.8 Hz, 1H), 4.56–4.48 (m, 1H), 4.37–4.30 (m, 1H), 4.25–4.19 (m, 1H), 4.11–4.06 (m, 1H), 4.00–3.92 (m, 3H), 3.88 (ddd, J = 9.5, 6.8, 2.4 Hz, 1H), 3.82 (dd, J = 10.5, 3.1 Hz, 1H), 3.80–3.67 (m, 7H), 3.63–3.54 (m, 4H), 3.53–3.46 (m, 2H), 3.32–3.20 (m, 2H), 2.81 (dd, J = 12.4, 4.6 Hz, 1H), 1.99 (s, 6H), 1.95 (s, 3H), 1.85–1.79 (m, 2H), 1.76 (t, J = 12.1 Hz, 1H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 174.0, 173.9, 173.8, 158.4, 136.7, 128.8, 128.3, 127.5, 104.5, 99.4, 97.1, 77.2, 75.6, 74.8, 71.73, 71.70, 70.6, 69.0, 68.7, 68.6, 66.9, 66.7, 65.0, 62.3, 61.2, 60.9, 51.8, 49.2, 48.7, 40.2, 37.5, 28.4, 22.1, 22.0, 21.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3536.
Neu5Ac7,9diNAcα2–3Galβ1–3GalNAcαProNHCbz (70).
10.2 mg, 65% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.53–7.31 (m, 5H), 5.17–5.09 (m, 2H), 4.85 (d, J = 3.8 Hz, 1H), 4.52–4.42 (m, 1H), 4.32 (dd, J = 11.1, 3.8 Hz, 1H), 4.26–4.20 (m, 1H), 4.10–4.00 (m, 2H), 3.99–3.89 (m, 4H), 3.83 (dd, J = 10.4, 3.1 Hz, 1H), 3.78–3.68 (m, 6H), 3.63–3.53 (m, 3H), 3.52–3.44 (m, 2H), 3.34–3.21 (m, 2H), 3.04 (dd, J = 14.0, 7.9 Hz, 1H), 2.80 (dd, J = 12.4, 4.6 Hz, 1H), 2.02–1.94 (m, 12H), 1.87–1.71 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.4, 174.04, 173.99, 173.8, 158.4, 136.7, 128.8, 128.3, 127.4, 104.4, 99.5, 97.0, 77.4, 75.6, 74.7, 71.7, 70.6, 69.7, 69.1, 68.6, 66.8, 66.7, 65.0, 61.2, 60.9, 51.8, 50.5, 48.6, 41.7, 40.2, 37.5, 28.4, 22.1, 22.0, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3804.
Neu5Ac7NAcα2–6Galβ1–3GalNAcαProNHCbz (71).
13.5 mg, 77% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.61–7.29 (m, 5H), 5.28–5.04 (m, 2H), 4.84 (d, J = 3.7 Hz, 1H), 4.43 (d, J = 7.8 Hz, 1H), 4.32 (dd, J = 11.0, 3.8 Hz, 1H), 4.24 (s, 1H), 4.11–3.40 (m, 19H), 3.38–3.19 (m, 2H), 2.78 (dd, J = 12.5, 4.6 Hz, 1H), 2.07–1.91 (m, 9H), 1.89–1.75 (m, 2H), 1.65 (t, J = 12.1 Hz, 1H). 13C{1H} NMR (100 MHz, D2O) δ 174.3, 173.9, 173.8, 173.3, 158.4, 136.6, 128.8, 128.3, 127.5, 103.9, 100.4, 97.0, 77.7, 73.2, 72.4, 71.7, 71.4, 70.4, 68.6, 68.5, 68.3, 66.7, 65.1, 63.7, 62.4, 61.6, 52.0, 49.1, 48.7, 40.4, 37.6, 28.5, 22.1, 22.0. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3533.
Neu5Ac7,9diNAcα2–6Galβ1–3GalNAcαProNHCbz (72).
14.6 mg, 72% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.35 (q, J = 7.5 Hz, 5H), 5.05 (s, 2H), 4.76 (d, J = 3.7 Hz, 1H), 4.34 (d, J = 7.7 Hz, 1H), 4.23 (dd, J = 11.1, 3.8 Hz, 1H), 4.15 (s, 1H), 3.98–3.29 (m, 18H), 3.26–3.11 (m, 2H), 3.00 (dd, J = 14.1, 7.3 Hz, 1H), 2.69 (dd, J = 12.4, 4.6 Hz, 1H), 1.98–1.81 (m, 12H), 1.79–1.65 (m, 2H), 1.56 (t, J = 12.1 Hz, 1H). 13C{1H} NMR (100 MHz, D2O) δ 174.5, 174.2, 173.9, 173.7, 173.3, 158.4, 136.6, 128.8, 128.3, 127.5, 103.9, 100.4, 97.2, 77.7, 73.2, 72.4, 71.6, 70.4, 69.4, 68.6, 68.5, 68.3, 66.7, 64.7, 63.7, 61.6, 52.0, 50.5, 48.7, 41.8, 40.4, 22.1, 22.00, 21.96, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3793.
Neu5Ac7NAcα2–3Galβ1–3GlcNAcβProNHCbz (73).
24.2 mg, 73% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.43 (h, J = 7.3 Hz, 5H), 5.12 (s, 2H), 4.53–4.45 (m, 2H), 4.15–4.07 (m, 1H), 3.98–3.44 (m, 20H), 3.25–3.11 (m, 2H), 2.81 (dd, J = 12.3, 4.5 Hz, 1H), 2.04–1.92 (m, 9H), 1.80–1.72 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 174.0, 173.9, 173.8, 158.3, 136.6, 128.8, 128.3, 127.5, 103.6, 100.8, 99.3, 82.4, 75.6, 75.4, 75.1, 71.7, 71.7, 68.9, 68.62, 68.57, 67.7, 66.8, 66.7, 62.3, 61.1, 60.7, 54.5, 51.7, 49.2, 40.2, 37.3, 28.7, 22.2, 22.1, 21.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3534.
Neu5Ac7,9diNAcα2–3Galβ1–3GlcNAcβProNHCbz (74).
11.6 mg, 66% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.55–7.35 (m, 5H), 5.12 (s, 2H), 4.48 (dd, J = 11.2, 8.1 Hz, 2H), 4.26–3.34 (m, 20H), 3.28–3.10 (m, 2H), 3.02 (dd, J = 14.2, 7.7 Hz, 1H), 2.80 (dd, J = 12.5, 4.6 Hz, 1H), 2.07–1.88 (m, 12H), 1.76 (d, J = 10.2 Hz, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.3, 174.03, 173.99, 173.84, 173.81, 158.3, 136.6, 128.8, 128.3, 127.5, 103.4, 100.8, 99.5, 82.2, 75.6, 75.4, 75.1, 71.7, 69.7, 69.0, 68.7, 68.6, 67.7, 66.8, 66.7, 61.0, 60.7, 54.5, 51.8, 50.5, 41.7, 40.2, 37.3, 28.7, 22.14, 22.11, 21.88, 21.86. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3807.
Neu5Ac7NAcα2–6Galβ1–3GlcNAcβProNHCbz (75).
25.4 mg, 75% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.84–7.09 (m, 5H), 5.13 (d, J = 10.4 Hz, 2H), 4.52 (d, J = 8.5 Hz, 1H), 4.39 (d, J = 7.8 Hz, 1H), 4.22–3.35 (m, 21H), 3.28–3.09 (m, 2H), 2.74 (dd, J = 12.4, 4.7 Hz, 1H), 2.10–1.88 (m, 9H), 1.76 (tq, J = 6.7, 3.1 Hz, 3H). 13C{1H} NMR (100 MHz, D2O) δ 174.7, 173.9, 173.7, 173.5, 158.3, 136.6, 128.8, 128.4, 127.6, 104.0, 100.9, 100.1, 84.4, 75.6, 73.5, 72.5, 71.6, 71.5, 70.5, 69.0, 68.6, 68.3, 67.6, 66.8, 63.2, 62.4, 60.9, 54.2, 51.9, 49.2, 40.1, 37.2, 28.7, 22.15, 22.13, 22.10. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21 905.3521; found 905.3531.
Neu5Ac7,9diNAcα2–6Galβ1–3GlcNAcβProNHCbz (76).
23.7 mg, 70% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.50–7.39 (m, 5H), 5.13 (d, J = 10.8 Hz, 2H), 4.51 (d, J = 8.5 Hz, 1H), 4.39 (d, J = 7.7 Hz, 1H), 4.23–3.34 (m, 20H), 3.29–2.99 (m, 3H), 2.73 (dd, J = 12.4, 4.6 Hz, 1H), 2.17–1.91 (m, 12H), 1.86–1.64 (m, 3H). 13C{1H} NMR (100 MHz, D2O) δ 174.6, 174.2, 173.9, 173.6, 173.5, 158.3, 136.6, 128.8, 128.4, 127.6, 104.0, 100.9, 100.1, 84.3, 75.6, 73.5, 72.5, 71.6, 70.5, 69.5, 69.0, 68.7, 68.3, 67.6, 66.8, 63.1, 60.9, 54.2, 51.9, 50.6, 41.8, 40.1, 37.2, 28.7, 22.1, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3799.
Neu5Ac7NAcα2–3Galβ1–3GlcNAcaProNHCbz (77).
18.4 mg, 89% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.48–7.38 (m, 5H), 5.19–5.09 (m, 2H), 4.48 (d, J = 7.9 Hz, 1H), 4.13–4.06 (m, 2H), 3.98 (dd, J = 9.8, 3.0 Hz, 1H), 3.95–3.68 (m, 12H), 3.66–3.43 (m, 7H), 3.34–3.17 (m, 2H), 2.81 (dd, J = 12.4, 4.6 Hz, 1H), 2.02–1.91 (m, 9H), 1.86–1.70 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.3, 174.0, 173.9, 173.8, 158.4, 136.7, 128.8, 128.3, 127.5, 103.4, 99.3, 97.0, 80.2, 75.6, 75.0, 71.75, 71.68, 71.6, 69.0, 68.57, 68.55, 66.8, 66.7, 64.9, 62.3, 61.0, 60.5, 52.5, 51.7, 49.2, 40.2, 37.4, 28.4, 22.1, 22.0, 21.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C38H57N4O21− 905.3521; found 905.3540.
Neu5Ac7,9diNAcα2–3Galβ1–3GlcNAcαProNHCbz (78).
14.5 mg, 69% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.47–7.37 (m, 5H), 5.17–5.09 (m, 2H), 4.52–4.40 (m, 1H), 4.08 (td, J = 9.8, 3.4 Hz, 2H), 4.00–3.87 (m, 4H), 3.87–3.68 (m, 8H), 3.62–3.45 (m, 6H), 3.40 (s, 1H), 3.27 (ddt, J = 26.8, 13.3, 6.6 Hz, 2H), 3.03 (dd, J = 14.2, 7.9 Hz, 1H), 2.80 (dd, J = 12.4, 4.6 Hz, 1H), 2.07–1.91 (m, 12H), 1.86–1.78 (m, 2H), 1.75 (t, J = 12.1 Hz, 1H). 13C{1H} NMR (150 MHz, D2O) δ 174.2, 174.0, 173.84, 173.80, 158.4, 136.7, 128.8, 128.3, 127.4, 103.3, 99.5, 96.9, 80.4, 78.5, 75.6, 74.9, 71.7, 71.6, 69.7, 69.1, 68.62, 68.59, 66.8, 66.7, 64.9, 61.0, 60.5, 55.3, 52.4, 51.8, 50.5, 41.7, 40.3, 37.4, 28.4, 22.1, 21.94, 21.88, 21.86. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3808.
Neu5Ac7NAcα2–6Galβ1–3GlcNAcαProNHCbz (79).
19.3 mg, 72% yield, white amorphous solid. 1H NMR (400 MHz, D2O) δ 7.50–7.36 (m, 5H), 5.13 (s, 2H), 4.36 (d, J = 7.8 Hz, 1H), 4.20–3.40 (m, 22H), 3.34–3.17 (m, 2H), 2.74 (dd, J = 12.4, 4.7 Hz, 1H), 2.00 (d, J = 6.3 Hz, 6H), 1.98–1.91 (m, 3H), 1.86–1.71 (m, 3H). 13C{1H} NMR (100 MHz, D2O) δ 174.5, 173.9, 173.7, 173.5, 158.4, 136.6, 128.8, 128.3, 127.5, 103.9, 100.1, 96.8, 82.5, 73.4, 72.5, 71.7, 71.6, 71.5, 70.5, 68.8, 68.7, 68.3, 66.7, 65.0, 63.1, 62.4, 60.6, 52.1, 51.9, 49.2, 40.1, 37.5, 28.5, 22.13, 22.09, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calced for C38H57N4O21 905.3521; found 905.3531.
Neu5Ac7,9diNAcα2–6Galβ1–3GlcNAcαProNHCbz (80).
13.8 mg, 68% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.51–7.37 (m, 5H), 5.14 (t, J = 9.1 Hz, 2H), 4.38 (dd, J = 16.6, 7.9 Hz, 1H), 4.10 (dd, J = 10.5, 3.6 Hz, 1H), 4.05–3.82 (m, 8H), 3.81–3.70 (m, 4H), 3.68–3.38 (m, 8H), 3.37–3.19 (m, 2H), 3.08 (dd, J = 14.0, 7.3 Hz, 1H), 2.73 (dd, J = 12.4, 4.7 Hz, 1H), 2.07–1.99 (m, 9H), 1.96 (s, 3H), 1.90–1.70 (m, 3H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 174.2, 173.9, 173.6, 173.5, 158.4, 136.6, 128.8, 128.3, 127.5, 103.9, 100.1, 96.9, 82.4, 73.4, 72.5, 71.7, 71.5, 70.5, 69.5, 68.8, 68.7, 68.3, 66.7, 65.0, 63.0, 60.6, 52.1, 51.9, 50.6, 41.8, 40.1, 37.5, 28.5, 22.2, 22.1, 22.0, 21.9. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C40H60N5O21 946.3786; found 946.3794.
Neu5Ac7NAcα2–6GalNAcαProNHCbz (81).
32 mg, 84% yield, white amorphous solid. 1H NMR (800 MHz, D2O) δ 7.47–7.36 (m, 5H), 5.19–5.07 (m, 2H), 4.82 (d, J = 3.7 Hz, 1H), 4.12 (dd, J = 11.1, 3.7 Hz, 1H), 4.03–3.93 (m, 3H), 3.91–3.84 (m, 4H), 3.75 (dt, J = 11.5, 5.8 Hz, 1H), 3.71–3.66 (m, 1H), 3.64 (dd, J = 12.1, 2.3 Hz, 1H), 3.60–3.51 (m, 2H), 3.50–3.44 (m, 2H), 3.31–3.19 (m, 2H), 2.75 (dd, J = 12.5, 4.6 Hz, 1H), 2.00 (s, 3H), 1.96 (s, 3H), 1.94 (s, 3H), 1.81 (p, J = 6.3 Hz, 2H), 1.65 (t, J = 12.2 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 174.5, 174.0, 173.7, 173.3, 158.4, 136.6, 128.8, 128.3, 127.6, 100.2, 97.0, 71.6, 71.5, 69.5, 68.6, 68.5, 67.6, 66.7, 65.4, 64.3, 62.4, 52.0, 49.9, 49.2, 40.4, 37.6, 28.4, 22.1, 21.9, 21.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H47N4O16 743.2993; found 743.3022.
Neu5Ac7,9diNAcα2–6GalNAcαProNHCbz (82).
33 mg, 85% yield, white amorphous solid. 1H NMR (600 MHz, D2O) δ 7.48–7.41 (m, 5H), 5.14 (q, J = 12.7 Hz, 2H), 4.83 (d, J = 3.9 Hz, 1H), 4.17–4.10 (m, 1H), 3.98–3.86 (m, 6H), 3.80–3.66 (m, 3H), 3.62–3.53 (m, 2H), 3.52–3.43 (m, 2H), 3.33–3.21 (m, 2H), 3.08 (dd, J = 14.1, 7.3 Hz, 1H), 2.76 (dd, J = 12.5, 4.6 Hz, 1H), 2.03 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H), 1.96 (s, 3H), 1.83 (p, J = 6.3 Hz, 2H), 1.67 (t, J = 12.2 Hz, 1H). 13C{1H} NMR (150 MHz, D2O) δ 174.5, 174.2, 174.0, 173.7, 173.3, 158.4, 136.6, 128.8, 128.3, 127.6, 100.3, 97.0, 71.4, 69.7, 69.5, 68.63, 68.56, 67.6, 66.7, 65.5, 64.3, 52.0, 50.7, 49.9, 41.9, 40.4, 37.6, 28.4, 22.1, 21.91, 21.87, 21.8. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C34H50N5O16 784.3258; found 784.3285.
General Procedures for Synthesis of Neu5Ac7NAc- and Neu5Ac7,9diNAc-terminated Propylamino-glycosides by Reduction.
To a stirred solution of a glycoside (selected from 57–82, 1.0–6.0 mg) in water-methanol solution (2 mL, 1:1 by volume), a catalytic amount of 10% palladium on charcoal was added to a round botom flask (50 mL). The mixture was stirred under a hydrogen environment for 2–5 h. The solution was passed through filter to remove the catalyst. The solvent was removed under vacuum and lyophilzed and used directly for microaray stuides.
Neu5Ac7NAcα2–3LacβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C28H48N3O19 730.2887; found 730.2896.
Neu5Ac7,9diNAcα2–3LacβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 (M-H) 771.3153; found 771.3170.
Neu5Ac7NAcα2–6LacβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C28H48N3O19 730.2887; found 730.2898.
Neu5Ac7,9diNAcα2–6LacβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3172.
Neu5Ac7NAcα2–3LacNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3157.
Neu5Ac7,9diNAcα2–3LacNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3427.
Neu5Ac7NAcα2–6LacNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3151
Neu5Ac7,9diNAcα2–6LacNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3416.
Neu5Ac7NAcα2–3Galβ1–3GalNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3172.
Neu5Ac7,9diNAcα2–3Galβ1–3GalNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3427.
Neu5Ac7NAcα2–6Galβ1–3GalNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3154.
Neu5Ac7,9diNAcα2–6Galβ1–3GalNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3421.
Neu5Ac7NAcα2–3Galβ1–3GalNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3150.
Neu5Ac7,9diNAcα2–3Galβ1–3GalNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3424.
Neu5Ac7NAcα2–6Galβ1–3GalNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3147.
Neu5Ac7,9diNAcα2–6Galβ1–3GalNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3422.
Neu5Ac7NAcα2–3Galβ1–3GlcNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3158.
Neu5Ac7,9diNAcα2–3Galβ1–3GlcNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3421.
Neu5Ac7NAcα2–6Galβ1–3GlcNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3159.
Neu5Ac7,9diNAcα2–6Galβ1–3GlcNAcβProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3421.
Neu5Ac7NAcα2–3Galβ1–3GlcNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3161.
Neu5Ac7,9diNAcα2–3Galβ1–3GlcNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3421.
Neu5Ac7NAcα2–6Galβ1–3GlcNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C30H51N4O19 771.3153; found 771.3155.
Neu5Ac7,9diNAcα2–6Galβ1–3GlcNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C32H54N5O19 812.3418; found 812.3421.
Neu5Ac7NAcα2–6GaNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C24H41N4O14 609.2625; found 609.2631.
Neu5Ac7,9diNAcα2–6GalNAcαProNH2 white amorphous solid. HRMS (ESI-Orbitrap) m/z: [M − H]− Calcd for C26H44N5O14 650.2890; found 650.2901.
Sialoglycan Microarray Studies.
Glycans were printed on PolyAn Glass Slide, 3D-NHS (Automate Scientific) at 100 μM in 4 replicates each in phosphate buffer (300 mM, pH 8.4). Printed glycan microarray slides were blocked with ethanolamine (0.05 M) in Tris-HCl (0.1 M, pH 9.0), washed and dried. Slides were fitted in a multiwell microarray hybridization cassette (AHC4X8S, ArrayIt, Sunnyvale, CA, USA) to divide into 8 subarrays. The subarrays were blocked with 1% ovalbumin in PBS, pH 7.4 at room temperature for 1 h with gentle shaking (all incubations were carried out in a humid chamber). The blocking solution was removed followed by incubation at room temperature for 2 h with gentle shaking with 30 μg/mL of Fc-fused human Siglec 7 (hSiglec 7) or Fc-fused human Siglec 9 (hSiglec 9), or 20 μg/mL of Sambucus nigra lectin (SNA) or Maackia amurensis lectin II (MAL II) (Vector Laboratories, Burlingame, CA) in blocking buffer. The slides were washed and incubated at room temperature for 1 h with goat anti-human IgG-Cy3 (1.5 μg/mL in PBS) (Jackson ImmunoResearch Laboratories, 109-165-088) for Siglecs or streptavidin-Cy3 (1.5 μg/mL in PBS) for lectins (Jackson ImmunoResearch, 109-160-084). The slides were subsequently washed and dried. The microarray slides were then scanned by Genepix 4000B microarray scanner (Molecular Devices Corp., Union City, CA, USA) and data analysis was performed using Genepix Pro 7.0 analysis software (Molecular Devices Corp., Union City, CA).
Supplementary Material
ACKNOWLEDGMENTS
Research reported in theis publication was supported by the National Institute of Allergy and Infectious Dieseases of the United States National Institutes of Health (NIH) under Award Number R01AI130684. The Bruker Avance-800 NMR spectrometer was purchased with a United States National Science Foundation (NSF) Shared Instrumentation Grant (grant no. DBI-0722538). The Thermo Scientific Q Exactive HF Orbitrap Mass Spectrometer was purchased with a United States NIH Shared Instrumentation Grant (grant no. S10OD025271). The authors would like to thank Dr. Lei Li and Dr. Ding Liu at Georgia State University for their help in obtaining high-resolution mass spectrometry (HRMS) data for some of the products.
Footnotes
Supporting Information.
Microarray data, detailed synthetic procedures, nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS) data, and NMR spectra of the products. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Schauer R Sialic Acids as Regulators of Molecular and Cellular Interactions. Curr. Opin. Struct. Biol 2009, 19, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chen X; Varki A Advances in The Biology and Chemistry of Sialic Acids. ACS Chem. Biol 2010, 5, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vimr ER; Kalivoda KA; Deszo EL; Steenbergen SM Diversity of Microbial Sialic Acid Metabolism. Microbiol. Mol. Biol. Rev 2004, 68, 132–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Severi E; Hood DW; Thomas GH Sialic Acid Utilization by Bacterial Pathogens. Microbiol. 2007, 153, 2817–2822. [DOI] [PubMed] [Google Scholar]
- (5).Angata T; Varki A Chemical Diversity in The Sialic Acids and Related alpha-Keto Acids: An Evolutionary Perspective. Chem. Rev 2002, 102, 439–469. [DOI] [PubMed] [Google Scholar]
- (6).Varki A Diversity in The Sialic Acids. Glycobiology 1992, 2, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schauer R Achievements and Challenges of Sialic Acid Research. Glycoconj. J 2000, 17, 485–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mandal C; Schwartz-Albiez R; Vlasak R Functions and Biosynthesis of O-Acetylated Sialic Acids. Top. Curr. Chem 2015, 366, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Visser EA; Moons SJ; Timmermans S; de Jong H; Boltje TJ; Bull C Sialic Acid O-Acetylation: From Biosynthesis to Roles in Health and Disease. J. Biol. Chem 2021, 297, 100906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Erdmann M; Wipfler D; Merling A; Cao Y; Claus C; Kniep B; Sadick H; Bergler W; Vlasak R; Schwartz-Albiez R Differential Surface Expression and Possible Function of 9-O- and 7-O-Acetylated GD3 (CD60 b and c) During Activation and Apoptosis of Human Tonsillar B and T Lymphocytes. Glycoconj. J 2006, 23, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Vlasak R; Luytjes W; Spaan W; Palese P Human And Bovine Coronaviruses Recognize Sialic Acid-Containing Receptors Similar to Those of Influenza C Viruses. Proc. Natl. Acad. Sci. U. S. A 1988, 85, 4526–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Smits SL; Gerwig GJ; van Vliet AL; Lissenberg A; Briza P; Kamerling JP; Vlasak R; de Groot RJ Nidovirus Sialate-O-Acetylesterases: Evolution and Substrate Specificity of Coronaviral and Toroviral Receptor-Destroying Enzymes. J. Biol. Chem 2005, 280, 6933–6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schwegmann-Wessels C; Herrler G Sialic Acids as Receptor Determinants for Coronaviruses. Glycoconj. J 2006, 23, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wasik BR; Barnard KN; Ossiboff RJ; Khedri Z; Feng KH; Yu H; Chen X; Perez DR; Varki A; Parrish CR Distribution of O-Acetylated Sialic Acids among Target Host Tissues for Influenza Virus. mSphere 2017, 2, e00379–16, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li Z; Lang Y; Liu L; Bunyatov MI; Sarmiento AI; de Groot RJ; Boons GJ Synthetic O-Acetylated Sialosides Facilitate Functional Receptor Identification for Human Respiratory Viruses. Nat. Chem 2021, 13, 496–503. [DOI] [PubMed] [Google Scholar]
- (16).Lundblad A Gunnar Blix and His Discovery of Sialic Acids. Fascinating Molecules in Glycobiology. Ups J. Med. Sci 2015, 120, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gottschalk A The Influenza Virus Neuraminidase. Nature 1958, 181, 377–378. [DOI] [PubMed] [Google Scholar]
- (18).Klein A; Roussel P O-Acetylation of Sialic Acids. Biochimie 1998, 80, 49–57. [DOI] [PubMed] [Google Scholar]
- (19).Ji Y; Sasmal A; Li W; Oh L; Srivastava S; Hargett AA; Wasik BR; Yu H; Diaz S; Choudhury B; Parrish CR; Freedberg DI; Wang LP; Varki A; Chen X, Reversible O-Acetyl Migration within the Sialic Acid Side Chain and Its Influence on Protein Recognition. ACS Chem. Biol 2021, In press. doi: 10.1021/acschembio.0c00998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kamerling JP; Schauer R; Shukla AK; Stoll S; Van Halbeek H; Vliegenthart JF Migration of O-Acetyl Groups in N,O-Acetylneuraminic Acids. Eur. J. Biochem 1987, 162, 601–607. [DOI] [PubMed] [Google Scholar]
- (21).Vandamme-Feldhaus V; Schauer R Characterization of The Enzymatic 7-O-Acetylation of Sialic Acids and Evidence for Enzymatic O-Acetyl Migration from C-7 to C-9 in Bovine Submandibular Gland. J. Biochem 1998, 124, 111–121. [DOI] [PubMed] [Google Scholar]
- (22).Khedri Z; Xiao A; Yu H; Landig CS; Li W; Diaz S; Wasik BR; Parrish CR; Wang LP; Varki A; Chen X A Chemical Biology Solution to Problems with Studying Biologically Important But Unstable 9-O-Acetyl Sialic Acids. ACS Chem. Biol 2017, 12, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Li W; Xiao A; Li Y; Yu H; Chen X Chemoenzymatic Synthesis of Neu5Ac9NAc-Containing alpha2–3- and alpha2–6-Linked Sialosides and Their Use for Sialidase Substrate Specificity Studies. Carbohydr. Res 2017, 451, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li W; Battistel MD; Reeves H; Oh L; Yu H; Chen X; Wang LP; Freedberg DI A Combined NMR, MD and DFT Conformational Analysis of 9-O-Acetyl Sialic Acid-Containing GM3 Ganglioside Glycan and its 9-N-Acetyl Mimic. Glycobiology 2020, 30, 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Angata T; Varki A Siglec-7: A Sialic Acid-Binding Lectin of The Immunoglobulin Superfamily. Glycobiology 2000, 10, 431–438. [DOI] [PubMed] [Google Scholar]
- (26).Angata T; Varki A Cloning, Characterization, and Phylogenetic Analysis of Siglec-9, A New Member of The CD33-Related Group of Siglecs. Evidence for Co-Evolution with Sialic Acid Synthesis Pathways. J. Biol. Chem 2000, 275, 22127–22135. [DOI] [PubMed] [Google Scholar]
- (27).Zhang JQ; Nicoll G; Jones C; Crocker PR Siglec-9, A Novel Sialic Acid Binding Member of Tthe Immunoglobulin Superfamily Expressed Broadly on Human Blood Leukocytes. J. Biol. Chem 2000, 275, 22121–22126. [DOI] [PubMed] [Google Scholar]
- (28).Li Y; Chen X Sialic Acid Metabolism and Sialyltransferases: Natural Functions and Applications. Appl. Microbiol. Biotechnol 2012, 94, 887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yu H; Chokhawala HA; Huang S; Chen X One-Pot Three-Enzyme Chemoenzymatic Approach to The Synthesis of Sialosides Containing Natural and Non-Natural Functionalities. Nat. Protoc 2006, 1, 2485–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Santra A; Xiao A; Yu H; Li W; Li Y; Ngo L; McArthur JB; Chen X A Diazido Mannose Analogue as a Chemoenzymatic Synthon for Synthesizing Di-N-Acetyllegionaminic Acid-Containing Glycosides. Angew. Chem. Int. Ed. Engl 2018, 57, 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dong H; Pei Z; Angelin M; Bystrom S; Ramstrom O Efficient Synthesis of beta-D-Mannosides and beta-D-Talosides by Double Parallel or Double Serial Inversion. J. Org. Chem 2007, 72, 3694–3701. [DOI] [PubMed] [Google Scholar]
- (32).Dong H; Pei Z; Ramstrom O Stereospecific Ester Activation in Nitrite-Mediated Carbohydrate Epimerization. J. Org. Chem 2006, 71, 3306–3309. [DOI] [PubMed] [Google Scholar]
- (33).Hale KJ; Hough L; Manaviazar S; Calabrese A An Update of The Rules for Pyranoside Sulfonate Displacement. Org. Lett 2014, 16, 4838–4841. [DOI] [PubMed] [Google Scholar]
- (34).Cai Y; Ling CC; Bundle DR Concise and Efficient Synthesis of 2-Acetamido-2-deoxy-beta-D-hexopyranosides of Diverse Aminosugars from 2-Acetamido-2-deoxy-beta-D-glucose. J. Org. Chem 2009, 74, 580–589. [DOI] [PubMed] [Google Scholar]
- (35).Kulkarni SS; Wang CC; Sabbavarapu NM; Podilapu AR; Liao PH; Hung SC “One-Pot” Protection, Glycosylation, and Protection-Glycosylation Strategies of Carbohydrates. Chem. Rev 2018, 118, 8025–8104. [DOI] [PubMed] [Google Scholar]
- (36).Ogawa T; Matsui M A New Approach to Regioselective Acylation of Polyhydroxy Compounds. Carbohydr. Res 1977, 56, c1–c6. [Google Scholar]
- (37).Ogawa T; Matsui M Regioselective Stannylation: Acylation of Carbohydrates: Coordination Control. Tetrahedron 1981, 37, 2363–2369. [Google Scholar]
- (38).Sanapala SR; Kulkarni SS Expedient Route To Access Rare Deoxy Amino L-Sugar Building Blocks for the Assembly of Bacterial Glycoconjugates. J. Am. Chem. Soc 2016, 138, 4938–4947. [DOI] [PubMed] [Google Scholar]
- (39).Tsvetkov YE; Shashkov AS; Knirel YA; Zahringer U Synthesis and NMR Spectroscopy of Nine Stereoisomeric 5,7-Diacetamido-3,5,7,9-Tetradeoxynon-2-ulosonic Acids. Carbohydr. Res 2001, 335, 221–243. [DOI] [PubMed] [Google Scholar]
- (40).Shangguan N; Katukojvala S; Greenberg R; Williams LJ The Reaction of Thio Acids with Azides: A New Mechanism and New Synthetic Applications. J. Am. Chem. Soc 2003, 125, 7754–7755. [DOI] [PubMed] [Google Scholar]
- (41).Ghosh S; Nishat S; Andreana PR Synthesis of an Aminooxy Derivative of the Tetrasaccharide Repeating Unit of Streptococcus dysgalactiae 2023 Polysaccharide for a PS A1 Conjugate Vaccine. J. Org. Chem 2016, 81, 4475–4484. [DOI] [PubMed] [Google Scholar]
- (42).Demizu Y; Kubo Y; Miyoshi H; Maki T; Matsumura Y; Moriyama N; Onomura O Regioselective Protection of Sugars Catalyzed by Dimethyltin Dichloride. Org. Lett 2008, 10, 5075–5077. [DOI] [PubMed] [Google Scholar]
- (43).Li Y; Yu H; Cao H; Lau K; Muthana S; Tiwari VK; Son B; Chen X Pasteurella multocida Sialic Acid Aldolase: A Promising Biocatalyst. Appl. Microbiol. Biotechnol 2008, 79, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Yu H; Yu H; Karpel R; Chen X Chemoenzymatic Synthesis of CMP-Sialic Acid Derivatives by A One-Pot Two-Enzyme System: Comparison of Substrate Flexibility of Three Microbial CMP-Sialic Acid Synthetases. Bioorg. Med. Chem 2004, 12, 6427–6435. [DOI] [PubMed] [Google Scholar]
- (45).Yu H; Chokhawala H; Karpel R; Yu H; Wu B; Zhang J; Zhang Y; Jia Q; Chen X A Multifunctional Pasteurella multocida Sialyltransferase: A Powerful Tool for The Synthesis of Sialoside Libraries. J. Am. Chem. Soc 2005, 127, 17618–17619. [DOI] [PubMed] [Google Scholar]
- (46).Yu H; Huang S; Chokhawala H; Sun M; Zheng H; Chen X Highly Efficient Chemoenzymatic Synthesis of Naturally Occurring and Non-Natural alpha-2,6-Linked Sialosides: A P. damsela alpha-2,6-Sialyltransferase with Extremely Flexible Donor-Substrate Specificity. Angew. Chem. Int. Ed. Engl 2006, 45, 3938–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Li W; Ghosh T; Bai Y; Santra A; Xiao A; Chen X A Substrate Tagging and Two-Step Enzymatic Reaction Strategy for Large-Scale Synthesis of 2,7-Anhydro-Sialic Acid. Carbohydr. Res 2019, 479, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McArthur JB; Santra A; Li W; Kooner AS; Liu Z; Yu H; Chen X L. pneumophila CMP-5,7-di-N-Acetyllegionaminic Acid Synthetase (LpCLS)-Involved Chemoenzymatic Synthesis of Sialosides and Analogues. Org. Biomol. Chem 2020, 18, 738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Padler-Karavani V; Song X; Yu H; Hurtado-Ziola N; Huang S; Muthana S; Chokhawala HA; Cheng J; Verhagen A; Langereis MA; et al. Cross-comparison of protein recognition of sialic acid diversity on two novel sialoglycan microarrays. J. Biol. Chem 2012, 287, 22593–22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.