

Abstract
Background
Blood pressure and tissue perfusion are controlled in part by the level of intrinsic (myogenic) arterial tone. However, many of the molecular determinants of this response are unknown. We previously found that mice with targeted disruption of the gene encoding the angiotensin II type 1a receptor (AT1AR) (Agtr1a), the major murine angiotensin II type 1 receptor (AT1R) isoform, showed reduced myogenic tone; however, uncontrolled genetic events (in this case, gene ablation) can lead to phenotypes that are difficult or impossible to interpret.
Methods and Results
We tested the mechanosensitive function of AT1R using tamoxifen‐inducible smooth muscle‐specific AT1aR knockout (smooth muscle‐Agtr1a −/−) mice and studied downstream signaling cascades mediated by Gq/11 and/or β‐arrestins. FR900359, Sar1Ile4Ile8‐angiotensin II (SII), TRV120027 and TRV120055 were used as selective Gq/11 inhibitor and biased agonists to activate noncanonical β‐arrestin and canonical Gq/11 signaling of the AT1R, respectively. Myogenic and Ang II‐induced constrictions were diminished in the perfused renal vasculature, mesenteric and cerebral arteries of smooth muscle‐Agtr1a −/− mice. Similar effects were observed in arteries of global mutant Agtr1a −/− but not Agtr1b −/− mice. FR900359 decreased myogenic tone and angiotensin II‐induced constrictions whereas selective biased targeting of AT1R‐β‐arrestin signaling pathways had no effects.
Conclusions
This study demonstrates that myogenic arterial constriction requires Gq/11‐dependent signaling pathways of mechanoactivated AT1R but not G protein‐independent, noncanonical pathways in smooth muscle cells.
Keywords: angiotensin II type 1a receptor, arterial smooth muscle, biased ligands, myogenic vasoconstriction
Subject Categories: ACE/Angiotension Receptors/Renin Angiotensin System, Hypertension, Vascular Biology
Nonstandard Abbreviations and Acronyms
- AT1R/AT1aR/AT1bR
angiotensin II type 1/1a/1b receptor
- GRK
G protein‐coupled receptor kinase
- MA
mesenteric arteries
- SM
smooth muscle
- SMMHC
smooth muscle myosin heavy chain
Clinical Perspective
What Is New?
Our study using novel tamoxifen‐inducible smooth muscle‐specific angiotensin II receptor type 1a knockout mice (smooth muscle‐Agtr1a −/−) demonstrates that myogenic arterial constriction requires canonical Gq/11‐dependent signaling pathways of mechanoactivated AT1R but not G protein‐independent, noncanonical pathways in smooth muscle cells.
What Are the Clinical Implications?
Our study will foster understanding and development of new diagnostic and therapeutic options for hypertension.
Our study also will provide valuable new insights for researchers working on blood pressure control‐related signaling mechanisms and for clinicians treating patients with hypertension.
Elevation of intravascular pressure causes constriction (myogenic tone) of small arteries and arterioles, and this response is a key element in regulating blood flow. This response was first described by Bayliss 1 and has been observed in various microvascular beds. 2 Many cardiovascular disorders are associated with dysfunctional arterial myogenic response and they include hypertension, chronic heart failure, ischemic stroke, and diabetes. 3 , 4 , 5 , 6 , 7 Despite the functional importance of the myogenic response, the nature of sensing intraluminal pressure that causes this response has remained elusive.
Myogenic vasoconstriction is mediated by pressure‐dependent depolarization of vascular smooth muscle (SM) cells, an event that augments Ca2+ influx through voltage‐dependent Cav1.2 channels. 8 , 9 , 10 , 11 , 12 , 13 Gq/11‐coupled receptors (GPCRs) are thought to function as the upstream sensor of membrane stretch. 14 We previously found that mice with targeted disruption of the gene encoding the angiotensin II (Ang II) type 1a receptor (Agtr1a), the major murine AT1 receptor isoform, showed reduced myogenic tone. 15 However, uncontrolled genetic events (in this case, global gene ablation) can lead to phenotypes that are difficult or impossible to interpret in terms of cardiovascular function. This is particularly relevant for mouse models of the renin‐angiotensin system. For example, mice with body‐wide elimination of angiotensin‐converting enzyme have systolic blood pressures that average 35 mm Hg below that of wild‐type mice. Surprisingly, renal arterioles and small arteries were thickened, a paradoxical finding given the very low systolic blood pressure that could have been caused by developmental processes and/or mouse strains. The renal changes in angiotensin‐converting enzyme knockout mice were completely unanticipated but have also been noted in angiotensinogen and Ang II receptor knockout mice. 16 AT1Rs are known to couple primarily to classical Gq/11 proteins to activate multiple downstream signals, including protein kinase C, extracellular signal‐regulated kinases, Raf kinases, tyrosine kinases, receptor tyrosine kinases (epidermal growth factor receptor, platelet‐derived growth factor, insulin receptor) and reactive oxygen species. 17 The AT1R activation also stimulates G protein‐independent signaling pathways such as β‐arrestin‐mediated mitogen‐activated protein kinase activation and Src‐Janus kinase‐signal transducer and activator of transcription. 17 Recently, it has been shown that the activation of intracellular signaling by mechanical stretch of the AT1R does not require the natural ligand Ang II 15 , 18 , 19 but requires the activation of the transducer ß‐arrestin. 18 Interestingly, mechanical stretch appears to allosterically stabilize specific β‐arrestin‐biased active conformations of AT1R to promote noncanonical downstream signaling mediated exclusively by the multifunctional scaffold protein, β‐arrestin. 20 Whether this noncanonical ß‐arrestin effector pathway plays a role in myogenic and ligand‐dependent vasoconstriction has yet to be ascertained.
This study explored the specific function of AT1R in the regulation of myogenic tone and whether downstream signaling pathways are dependent on canonical Gq/11 and/or noncanonical alternative signaling pathways. In this regard, we generated mice with cell specific deletion of SM AT1a receptors (SM‐Agtr1a mice) and studied the effects of biased GPCR agonists and Gq/11 protein inhibition on tone development in 3 distinct vascular beds (renal, cerebral, and mesenteric circulation). We found that the SM AT1aR coupled toward the canonical Gq/11 signaling pathway is required for the myogenic response in all 3 vascular beds. Our data argue against involvement of noncanonical G protein‐independent alternative signaling downstream of the AT1R to cause myogenic vasoconstriction.
METHODS
Data and analytic methods will be made available to other researchers upon reasonable request to the corresponding author.
Mouse Model
We used the SM myosin heavy chain (SMMHC)‐Cre‐ERT2 transgenic mouse line expressing Cre recombinase in SM cells under control of the SMMHC promoter 21 and a mouse line bearing a floxed allele of the Agtr1a gene (Agtr1a flox), encoding the major murine AT1 receptor isoform (AT1aR) 22 to generate SMMHC‐Cre+Agtr1a flox/flox (SM‐Agtr1afl/fl ) mice (Figure S1A). Genotyping was performed by polymerase chain reaction analysis of tail DNA as described previously. 21 Amplification of the SMMHC‐Cre gene was performed in a multiplex polymerase chain reaction with the primers TGA CCC CAT CTC TTC ACT CC (SMWT1), AAC TCC ACG ACC ACC TCA TC (SMWT2), and AGT CCC TCA CAT CCT CAG GTT (phCREAS1). 23 The following primers (5′–3′) were used to identify Agtr1af lox alleles: forward GCT TTC TCT GTT ATG CAG TCT and reverse ATC AGC ACA TCC AGG AAT G. Knockout (SM‐Agtr1a −/−) was induced in adult (12–16 weeks) SM‐Agtr1afl/fl male mice by intraperitoneal injection with tamoxifen (30 µg/mg body weight) on 5 consecutive days. Isolated arteries were usually obtained after 2 to 3 days after tamoxifen treatment. Tamoxifen treatment of mice did not influence myogenic tone (SM‐Agtr1a +/+ cerebral arteries without tamoxifen: 12.35±2.16%, n=6 vessels, each from individual mice; SM‐Agtr1a +/+ cerebral arteries with tamoxifen: 13.23±1.07%, n=4 vessels, each from individual mice). Figure S1B shows reduction of AT1aR expression in vascular SM cells of SM‐Agtr1a −/− arteries. The same antibody did not detect AT1R expression across the vessel wall from global Agtr1a −/− mice 15 (Figure S1C). We also studied adult (12–16 weeks) male mice with global AT1a receptor deficiency (Agtr1a −/−), 15 , 24 , 25 and with global AT1b receptor deficiency (Agtr1b −/−). 26 Age‐matched male mice were used as controls in the experiments. Animal care followed American Physiological Society guidelines, and all protocols were approved by local authority (LAGeSo, Berlin, Germany) and the animal welfare officers of the Max Delbrück Center for Molecular Medicine (approval reference number X 9011/16). All procedures conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes or the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were maintained in the Max Delbrück Center animal facility in individually ventilated cages (Tecniplast Deutschland, Hohenpeißenberg, Germany) under standardized conditions with an artificial 12‐hour dark‐light cycle, with free access to standard chow (0.25% sodium; SSNIFF Spezialitäten, Soest, Germany) and drinking water. Animals were euthanized by cervical dislocation and randomly assigned to the experimental procedures.
Materials
Antibody to α‐SM actin (α‐SMA, #ab8211) was from Abcam (Cambridge, MA). Anti‐AT1R (#PA5‐20812) and donkey antirabbit IgG (H+L) secondary antibody (A10040) were purchased from Thermo Fisher Scientific (Waltham, MA). 4′,6‐diamidino‐2‐phenylindole (DAPI, #D9542) was purchased from Sigma‐Aldrich Co. (St. Louis, MO). Ang II (#A9525), SII (#sc‐391239A), and tamoxifen (#H7904) were from Sigma‐Aldrich Co (82024 Taufkirchen, Germany). TRV120055 (#JT‐71995) and TRV120056 (#JT‐71996) were from Synpeptide Co., Ltd (Shanghai, China). TRV120027 (#HY‐P2141A) was from MedChemExpress (Monmouth Junction, NJ).
Mesenteric and Cerebral Arteries
After mice were killed, the mesenteric bed and brain were removed and placed into cold (4 °C), gassed (95% O2‐5% CO2) physiological saline solution (PSS) of the following composition (mmol/L): 119 NaCl, 4.7 KCl, 25 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, 0.03 EDTA, and 11.1 glucose. Third‐ or fourth‐order mesenteric arteries (MA) and middle cerebral arteries or posterior cerebral arteries were dissected and cleaned of adventitial connective tissue. 15 , 27 , 28 , 29
Pressure Myography
Vessel myography was performed as previously described. 14 , 15 , 21 , 28 MA or cerebral arteries were mounted on glass cannula and superfused continuously with PSS (95% O2‐5% CO2; pH, 7.4; 37 °C). The vessels were stepwise pressurized to 20, 40, 60, 80, or 100 mm Hg using a pressure servo control system (Living System Instrumentation, Burlington, VT). We measured the inner diameter of the vessels with a video microscope (Nikon Diaphot, Düsseldorf, Germany) connected to a personal computer for data acquisition and analysis (HaSoTec, Rostock, Germany). 15 , 27 , 28 , 29 , 30 Arteries were equilibrated for 45 to 60 minutes before starting experiments. A 60‐mmol/L KCl challenge was performed before any other intervention.
Isolated Perfused Kidneys
Isolated kidneys were perfused in an organ chamber using a peristaltic pump at constant flow (0.3–1.9 mL/min) of oxygenated (95% O2 and 5% CO2) PSS. 15 Drugs (Ang II or biased agonists) were added to the perfusate. Perfusion pressure was measured by a pressure transducer after an equilibration period of 60 to 90 minutes. Data were recorded and analyzed by a Powerlab acquisition system (AD Instruments, Colorado Springs, CO). Ang II‐induced pressor effects were normalized to the maximal pressor effect induced by KCl (60 mmol/L). 14 , 15 , 27
Immunofluorescence
SM‐Agtr1a +/+ and SM‐Agtr1a −/− mice MA were dissected and further fixed in 4% formaldehyde and embedded in Tissue‐Tek O.C.T. compound to be frozen in liquid nitrogen. Tissues were then sectioned and permeabilized in 1% Triton X‐100 in PBS. Sections were stained with the primary antibody overnight at 4 °C. After washing with PBS for 3×5 minutes, the secondary antibody and DAPI were applied for 2 hours at room temperature. Fluorescence images were captured by use of Olympus FV1000 confocal microscopy and images were analyzed by ImageJ analysis software.
Statistical Analysis
Data are presented as means±SEM. Statistically significant differences in mean values were determined by Student unpaired t test or 1‐way analysis of variance. P<0.05 were considered statistically significant.
RESULTS
AT1aR Is Essential for Pressure‐Induced Response in the Renal Circulation
We evaluated myogenic tone in mouse renal circulation, a highly myogenic bed regulating blood flow to the kidneys and consequently sodium excretion and systemic blood pressure. Renal vascular resistance of isolated perfused kidneys was determined by measuring perfusion pressure at fixed levels of flow. The perfusion pressure increased with flow rate in kidneys of wild‐type Agtr1a +/+ mice, reaching a value of about 160 mm Hg at a flow rate of 1.9 mL/min, which is in the physiological range (between 1.5 and 3.5 mL/min pulsatile flow) 31 (Figure 1A). Kidneys from Agtr1a −/− mice developed significantly less pressure at the same flow rate (Figure 1B and 1E). 60 mmol/L KCl‐induced increases in perfusion pressure were not altered in Agtr1a −/− kidneys (Figure 1F). At a flow rate of 1.9 mL/min, pressure in Agtr1a −/− kidneys was ≈100 mm Hg lower than in Agtr1a +/+ kidneys. Ang II (10 nmol/L) increased perfusion pressure by ≈80 mm Hg in kidneys of Agtr1a +/+ mice but had no effect in kidneys of Agtr1a −/− mice (Figure 1C); this is indicative of AT1aRs mediating Ang II‐dependent vasoconstriction. Removal of external Ca2+ nearly abolished flow‐induced myogenic constriction in perfused kidneys of Agtr1a +/+ mice but had no effect in kidneys of Agtr1a −/− mice (Figure 1D), indicating AT1aRs mediate also myogenic constriction of mouse renal arterioles. Of note, there was no difference in myogenic tone and Ang II vasoconstrictions between Agtr1b −/− versus Agtr1b +/+ kidneys (Figure 2).
Figure 1. Vasoregulation in isolated perfused kidneys of Agtr1a −/− mice.
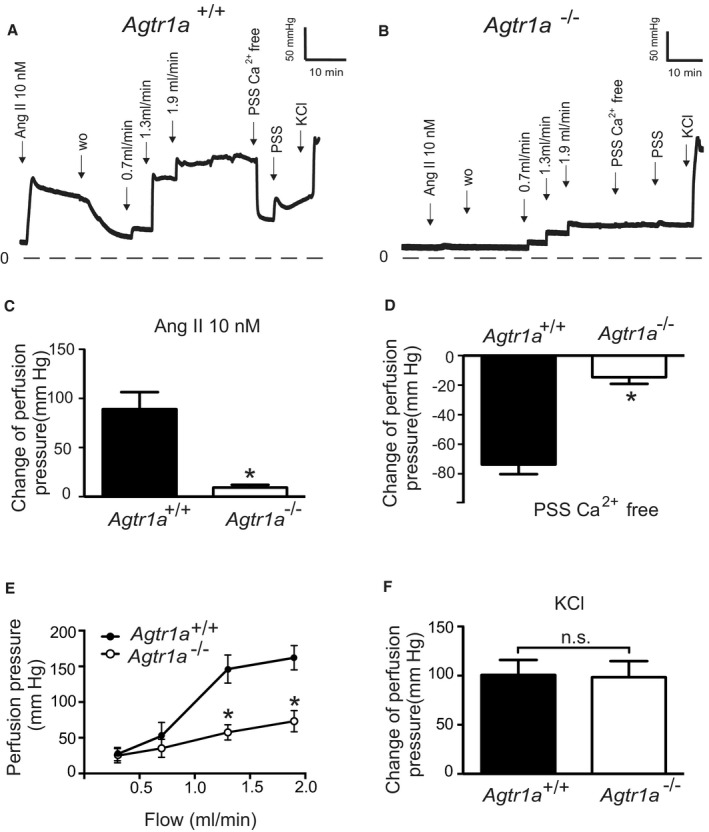
A and B, Original recordings of perfusion pressure in kidneys of Agtr1a +/+ (A) and Agtr1a −/− mice (B). C, Increase in the perfusion pressure induced by 10 nmol/L Ang II. D, Myogenic tone assessed by exposure to Ca2+ free PSS. E, Perfusion pressure at flow rates of 0.3, 0.7, 1.3, and 1.9 mL/min. F, Increase in the perfusion pressure induced by 60 mmol/L KCl. n=6 Agtr1a +/+ kidneys from 6 mice and n=7 Agtr1a −/− kidneys from 7 mice for all panels. *P<0.05. Ang II indicates angiotensin II; n.s., not significant; PSS, physiological saline solution; and wo, wash‐out.
Figure 2. Vasoregulation in isolated perfused kidneys of Agtr1b −/− mice.
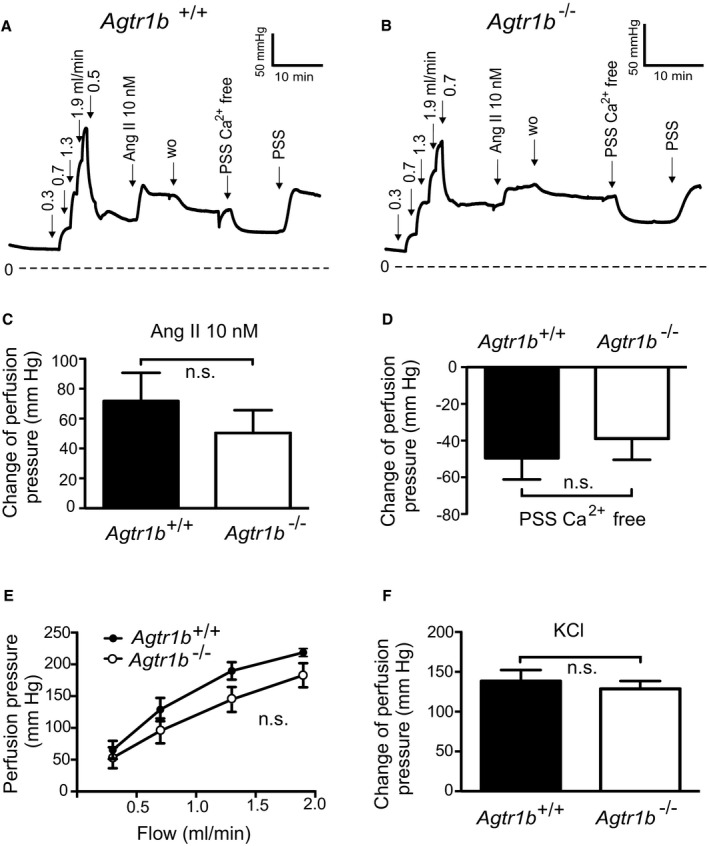
A and B, Original recordings of the perfusion pressure in kidneys of Agtr1b +/+ (A) and Agtr1b −/− mice (B). C, Increase in perfusion pressure induced by 10 nmol/L Ang II. D, Change of pressure assessed by exposure to Ca2+ free PSS. E, Perfusion pressure at flow rates of 0.3, 0.7, 1.3, and 1.9 mL/min. F, Increase in perfusion pressure induced by 60 mmol/L KCl. n=6 Agtr1b +/+ kidneys from 5 mice and n=6 Agtr1b −/− kidneys from 3 mice for all panels. Ang II indicates angiotensin II; n.s., not significant; PSS, physiological saline solution; and w.o., washout.
Next, we focused on kidneys from SM‐Agtr1a −/− mice (Figure 3). At a flow rate of 1.9 mL/min, pressure in SM‐Agtr1a −/− kidneys was ≈90 mm Hg lower than in SM‐Agtr1a +/+ kidneys (Figure 3A, 3B, and 3E). SM‐Agtr1a −/− kidneys showed largely reduced myogenic vasoconstriction as assessed by exposure of the kidneys to Ca2+ free PSS (Figure 3B and 3D), whereas wild‐type kidneys showed strong myogenic vasoconstrictions (Figure 3A and 3D). Sixty mmol/L KCl‐induced increases in perfusion pressure were normal in SM‐Agtr1a −/− kidneys (Figure 3F). Ang II (10 nmol/L) induced weaker increases in perfusion pressure in kidneys of SM‐Agtr1a −/− mice compared with controls (Figure 3C). Together, these results reveal a key role of AT1aR but not AT1bR in the flow‐induced myogenic response and Ang II‐evoked constriction of the mouse renal vasculature.
Figure 3. Vasoregulation in isolated perfused kidneys of SM‐Agtr1a −/− mice.
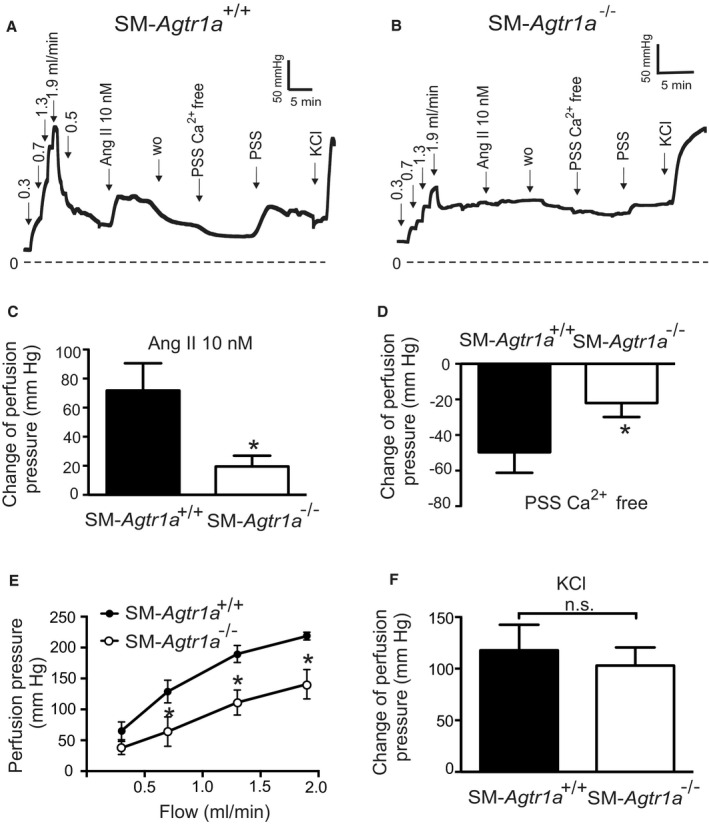
A and B, Original recordings of the perfusion pressure in kidneys of SM‐Agtr1a +/+ (A) and SM‐Agtr1a −/− mice (B). C, Increase in perfusion pressure induced by 10 nmol/L Ang II. D, Change of pressure assessed by exposure to Ca2+ free PSS. E, Perfusion pressure at flow rates of 0.3, 0.7, 1.3, and 1.9 mL/min. F, Increase in perfusion pressure induced by 60 mmol/L KCl. n=6 SM‐Agtr1a +/+ kidneys from 5 mice and n=6 SM‐Agtr1a −/− kidneys from 6 mice for all panels. *P<0.05. Ang II indicates angiotensin II; n.s., not significant; PSS, physiological saline solution; SM, smooth muscle; and w.o., washout.
AT1aR Contributes to Myogenic Constriction in Mesenteric and Cerebral Arteries
We first monitored myogenic constriction in resistance‐sized MA using videomicroscopy. MA were exposed to stepwise (20 mm Hg) increases in intraluminal pressure (20–100 mm Hg) in the presence and absence of external Ca2+ (1.6 mmol/L) to determine active and passive vessel diameters, respectively. Figure 4 shows representative recordings of MA from SM‐Agtr1a +/+ mice (Figure 4A) and SM‐Agtr1a −/− mice (Figure 4B) and myogenic vasoconstriction was defined as the diameter difference in the presence and absence of external Ca2+ (1.6 mmol/L) at each pressure step. 15 Increases in intraluminal pressure generated active tension that counteracted further dilation of the vessels at 60 to 80 mm Hg in MA from SM‐Agtr1a +/+ mice, reaching peak constrictions of ~50 μm at 80 to 100 mm Hg (Figure 4A). In contrast, MA from SM‐Agtr1a −/− mice produced only ≈35% of the constriction observed in wild‐type arteries (Figure 4B and 4C). Ang II strongly constricted arteries from SM‐Agtr1a+/+ mice but had no effect on arteries from SM‐Agtr1a −/− mice (Figure 4D); the latter did constrict in response to 60 mmol/L KCl (Figure 4E). This study observed a marked reduction in AT1aR expression in the media of SM‐Agtr1a −/− MA compared with wild‐type (Figure S1B), in keeping with this receptor mediating myogenic constriction in MA.
Figure 4. Myogenic tone in mesenteric arteries.
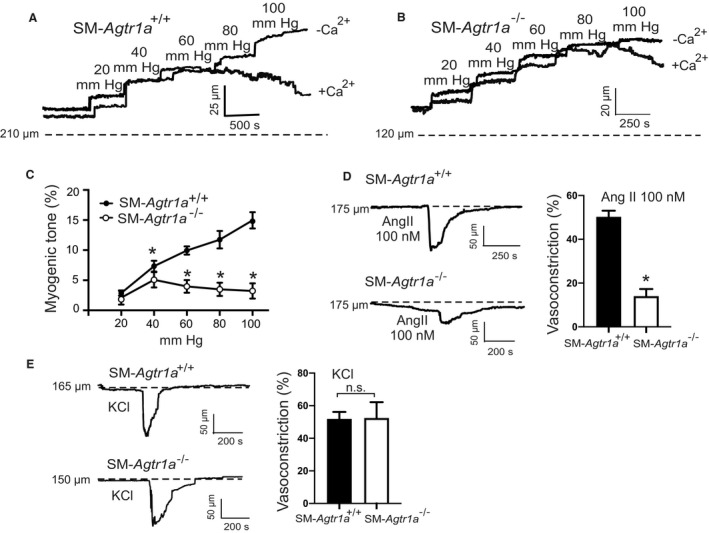
A and B, Representative recordings of MA diameter during a series of pressure steps from 20 to 100 mm Hg in 20 mm Hg increments in control conditions (+Ca2+) and in Ca2+ free solution (−Ca2+). Arteries were isolated from SM‐Agtr1a +/+ (A) and SM‐Agtr1a −/− mice (B). Note the increase in active constriction over the entire pressure range from 60 to 100 mm Hg in vessels from SM‐Agtr1a +/+, but not from SM‐Agtr1a −/− mice. Vasodilation in Ca2+‐free solution was observed in SM‐Agtr1a +/+ but not in SM‐Agtr1a −/− arteries (P<0.05). C, Average myogenic tone of mesenteric arteries in PSS expressed as dilation of vessels induced by external Ca2+ free solution (0 Ca/EGTA; SM‐Agtr1a +/+, n=9, and SM‐Agtr1a −/−, n=6 vessels, each from individual mice for both groups). D, Response to Ang II and (E) response to 60 mmol/L KCl in MA of SM‐Agtr1a +/+ and SM‐Agtr1a −/− mice. MAs were pressurized to 80 mm Hg. Responses are expressed as relative changes in vessel inner diameter. SM‐Agtr1a +/+, n=5 vessels, and SM‐Agtr1a −/−, n=4 vessels, from 5 and 4 mice, respectively. *P<0.05. Ang II indicates angiotensin II; MA, mesenteric arteries; PSS, physiological saline solution; and SM, smooth muscle.
Next, we studied the function of AT1aRs in cerebral arteries. Vessels were equilibrated at 15 mm Hg (30 minutes) and following an assessment of KCl‐induced constriction, arteries were pressurized to 80 mm Hg (Figure S2A). Ang II constrictions and myogenic constriction was significantly decreased in SM‐Agtr1a −/− arteries compared with wild‐type (Figure S2A through S2D). Both wild‐type and SM‐Agtr1a −/− arteries produced similar constrictions when exposed to 60 mmol/L KCl (Figure S2E). The results demonstrate a key role of AT1aR in the myogenic response of mouse cerebral arteries.
Gq/11 Protein Dependent Signaling Pathway Is Responsible for Myogenic Tone
To explore the role of Gq/11 and β‐arrestin signaling pathways downstream of AT1R, we used the biased agonists TRV120055, SII, and TRV120027 to activate Gq/11 and β‐arrestin signaling pathways, respectively. 32 , 33 , 34 , 35 , 36 We found that TRV120055 increased vascular tone in MA (Figure 5A and 5B), whereas SII and TRV120027 had no effect (Figure 5C through 5F). Similarly, TRV120055 and TRV120056 (another biased Gq/11 coupled AT1R agonist) enhanced dose‐dependent perfusion pressure in isolated kidneys (Figure 6A and 6C), whereas SII had no effect (Figure 6B and 6C). The removal of external Ca2+ abolished agonist‐induced vasoconstrictions in perfused kidneys (Figure 6D), indicating AT1aRs mediate vasoconstriction via canonical Gq/11 but not noncanonical β‐arrestin pathways. The biased Gq/11 coupled AT1R agonist TRV120055 and TRV120056 equally modified perfusion pressure (Figure 6E). Viability of the isolated perfused kidneys was assessed by 60 mmol/L KCl (Figure 6F). To confirm the results, we next examined the effects of FR900359, a selective Gq/11‐protein inhibitor. 37 , 38 , 39 FR900359 abolished both myogenic and Ang II‐dependent constrictions in renal arterioles (Figure 7), MA (Figure 5G and 5H) and cerebral arteries (Figure S2F). These results indicate that myogenic vasoconstriction is mediated through the mechanosensitive AT1aR and the canonical Gq/11 signaling pathway.
Figure 5. Enhancement of the vascular tone by TRV120055.
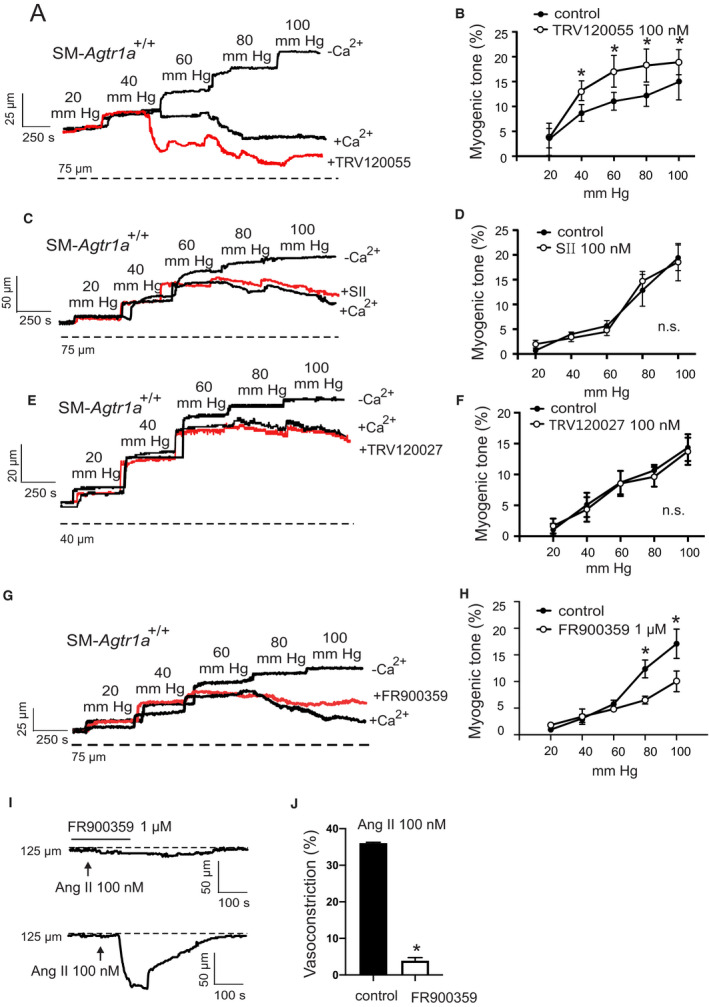
A, C, E, and G, Representative recordings of mesenteric artery diameter during a series of pressure steps from 20 to 100 mm Hg in 20 mm Hg increments in control conditions (+Ca2+), TRV120055 100 nmol/L (A), SII 100 nmol/L (C), TRV120027 100 nmol/L (E), FR900359 1 µmol/L (G) and in Ca2+‐free solution. B, D, F, and H, Average myogenic constriction of mesenteric arteries in drug‐free physiological saline solution and in PSS containing 100 nmol/L TRV120055 (B), 100 nmol/L SII (D), TRV120027 100 nmol/L (F) and 1 µmol/L FR900359 (H) (n=6, 4, 5 and 4, respectively, each from individual mice). I and J, Response to Ang II in MA in drug‐free PSS and PSS in presence of FR900359 at 80 mm Hg (n=6 each from individual mice). *P<0.05. Ang II indicates angiotensin II; n.s. indicates not significant; and SII, Sar1Ile4Ile8‐angiotensin.
Figure 6. Function of biased AT1R agonists to vasoregulation in isolated perfused kidneys from SM‐Agtr1a +/+ mice.
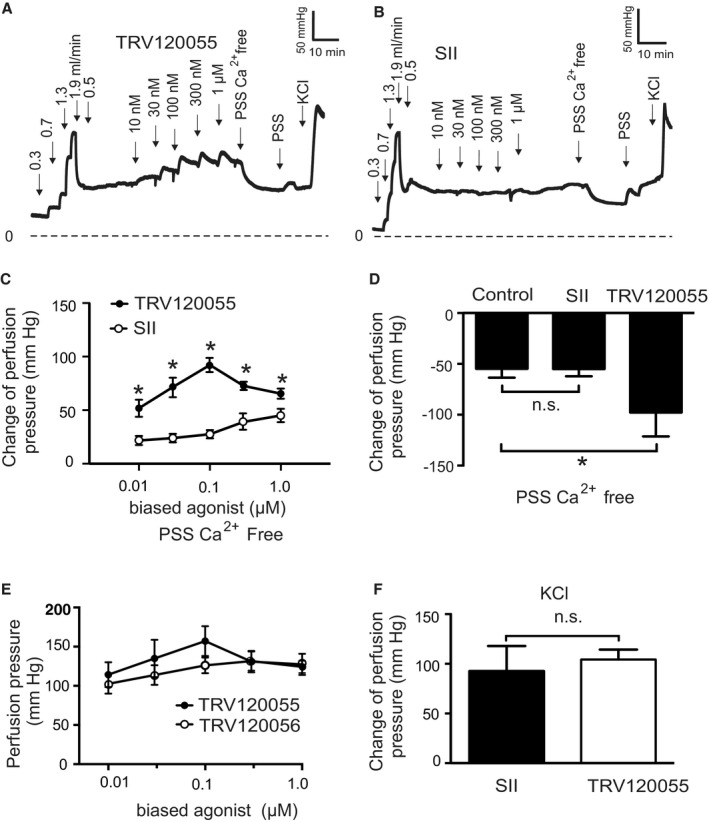
A and B, Original recordings of perfusion pressure in response to various flow rates (in mL/min), TRV120055 (A) or Sar1Ile4Ile8‐SII (SII) (B), Ca2+ free perfusion solution (PSS Ca2+ free) and reexposure of the kidneys to PSS. C, Increase in perfusion pressure induced by TRV120055 and SII in various concentrations (10 nmol/L to 1 µmol/L). D, Change of perfusion pressure assessed by exposure of the kidneys to Ca2+ free PSS at the presence of TRV120055 or Sar‐Ile II at the concentration of 100 nmol/L. E, Dose‐response relationships for TRV120055 and TRV120056. F, Increase in perfusion pressure induced by 60 mmol/L KCl. TRV120055, TRV120056, SII. n=6 kidneys from 5 mice in each group; n=6 kidneys from 5 mice in the control group. *P<0.05. AT1R, angiotensin II type 1 receptor; Control, SM‐Agtr1a +/+ without biased ligand; n.s., not significant; PSS, physiological saline solution; and SM, smooth muscle.
Figure 7. Vasoregulation in isolated perfused kidneys of SM‐Agtr1a +/+ mice pretreated with 300 nmol/L Gq/11 blocker FR900359.
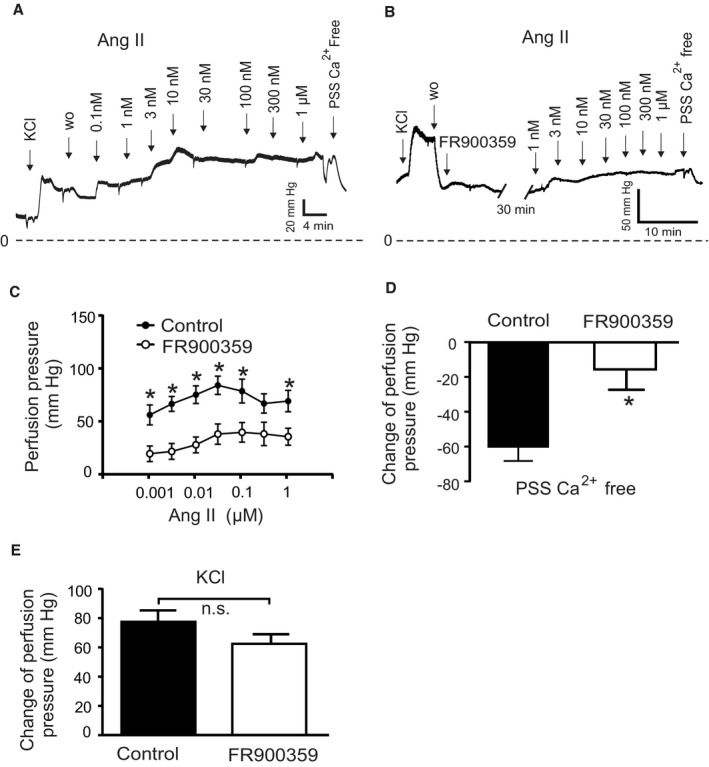
A, Original recordings of perfusion pressure in kidneys of Agtr +/+ mice in response to various concentrations of Ang II (B) same as (A) but pretreated with 300 nmol/L FR900359 for 30 minutes. C, Increases in perfusion pressure induced by Ang II (1 nmol/L to 1 µmol/L). D, Myogenic tone assessed by exposure of the kidneys to Ca2+‐free PSS. E, Increase in perfusion pressure induced by 60 mmol/L KCl. n=15 SM‐Agtr1a +/+ kidneys from 12 mice and n=6 SM‐Agtr1a +/+ kidneys from 4 mice pretreated with FR900359 for all panels. *P<0.05. Ang II indicates angiotensin II; n.s., not significant; PSS, physiological saline solution; SM, smooth muscle; and w.o., wash‐out.
DISCUSSION
AT1Rs Are Primary Mechanosensors in Intact Arteries
Multiple GPCRs have been proposed to act as mechanosensors to regulate myogenic tone in resistance arteries. Although stretch induces activation of purinergic P2Y6 UDP receptors, thromboxane A2 receptors and sphingosine‐1‐phosphate receptors in certain vascular beds, 40 , 41 , 42 the AT1R remains one of the best characterized mechanosensor in the vasculature. 19 , 43 Humans express a single type of AT1R, whereas 2 isoforms (AT1aR and AT1bR) are present in rodents. 44 , 45 Using Agtr1a −/− mice and inverse AT1R agonist, our previous data suggested that ligand‐independent AT1aR activation is required for myogenic response in resistance MA and renal arterioles. 15 Two recent studies reported also a possible role of AT1bRs in myogenic constriction in certain vessels. 46 , 47 We found that Ang II and myogenic constrictions were normal in Agtr1b −/− perfused kidneys, which is consistent with AT1A receptor being the major murine AT1 receptor isoform in the renal circulation. 48 All data were, however, obtained in global mutant mice, which often display compensatory mechanisms for the lack of AT1Rs and can lead to phenotypes that are difficult or impossible to interpret in terms of cardiovascular function. For example, AT1aR and AT1bR are expressed at similar levels in cerebral parenchymal arterioles and genetic knockout of AT1aR (but not AT1bR) blunted the ability of these vessels to generate myogenic tone. 49 The latter effect is opposite to cerebral arteries where genetic knockout of AT1bR blunted the ability to develop myogenic tone. 46 To overcome these potential limitations, we generated tamoxifen‐inducible SM‐Agtr1a (SMMHC‐Cre+Agtr1aflox /flox) mice for careful phenotypic investigation. We found that myogenic constriction was impaired in cerebral, mesenteric, and renal arteries isolated from SM AT1aR‐deficient mice. Our data provide firm evidence that AT1Rs play a key role as mechanosensors mediating myogenic constriction in the murine vasculature.
AT1Rs Downstream Signaling to Cause Vasoconstriction
We next explored downstream signaling pathways mediated by Gq/11 and/or β‐arrestin of AT1R in the vascular response. In cell culture, osmotic cell stretch has been found to increase the binding affinity and potency of the β‐arrestin‐biased agonist TRV120023 with no effect on the balanced agonist Ang II through AT1R to induce a conformation change of β‐arrestin 2, similar to that induced by β‐arrestin‐biased agonists. 20 Similarly, hypo‐osmotic stretch induced β‐arrestin‐biased signaling of AT1Rs in the absence of G protein activation. 18 We failed to observe β‐arrestin‐mediated enhancement of myogenic constriction with the β‐arrestin biased agonists SII and TRV120027 in intact arteries (mesenteric and renal arteries: Figure 8). The discrepancy might be caused by differences between hypo‐osmotic cell swelling and tensile stretch on the SM cell layer in intact arteries to cause mechanoactivation of AT1Rs in situ. AT1R is one of the best characterized GPCR enabling biased receptor signaling. It can be activated in either a canonical G protein‐dependent signaling mode 14 , 50 or noncanonical β‐arrestin‐mediated signaling mode. 18 , 20 In line, we found that the natural biased agonist Ang II was able to increase G protein signaling of mechanoactivated AT1R receptors to enhance the vasoconstrictor response. A recent study reported that GPCRs, including AT1R, can promote a direct interaction between Gαi protein subtype family members and β‐arrestins, regardless of their canonical G protein subtype coupling, that might be prerequisite for certain β‐arrestin signaling pathways. 51
Figure 8. Schematic illustration of angiotensin II type 1a receptor (AT1aR) biased signaling cascade regulating myogenic arterial tone.
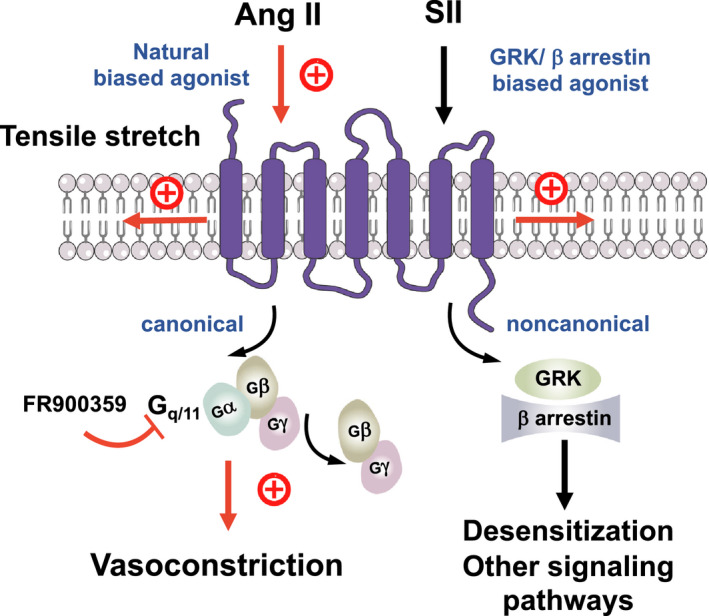
Canonical Gq/11 signaling pathway of the AT1R (purple blue) causes myogenic vasoconstriction whereas noncanonical β‐arrestin‐biased signaling is not involved in this process. Gq/11 proteins are heterotrimeric G proteins, which are made up of alpha (α), beta (β), and gamma (γ) subunits. The alpha subunit is attached to either a guanosine triphosphate (GTP) or guanosine diphosphate (GDP), which serves as an on‐off switch for the activation of the G‐protein. Upon activation of the AT1aR by either ligand‐independent mechanical stretch or the natural‐biased ligand Ang II, the Gβγ complex is released from the Gα subunit after its GDP‐GTP exchange for canonical G protein signaling to cause myogenic and/or humoral (Ang II‐mediated) vasoconstriction. This pathway is inhibited by the Gq/11 inhibitor FR900359. Although, GRKs and arrestins play a role in multiple noncanonical signaling pathways in cells, this pathway is unlikely engaged by mechanoactivated AT1Rs in response to tensile stretch or their natural ligand angiotensin II to cause vasoconstriction. Ang II indicates angiotensin II; GRK, G protein‐coupled receptor kinase; and SII, Sar1Ile4Ile8‐angiotensin.
We hypothesized that Gq/11 signaling contributes to myogenic tone in MA and renal arteries and consistent with this idea, we found that the vasoconstrictor responses were strongly increased by the Gq/11 AT1R biased agonists TRV120055 and TRV20056 (Figure 8). Moreover, we found that the Gq/11 blocker FR900359 inhibited both myogenic tone and Ang II‐induced constrictions in MA and renal arterioles (Figure 8). The data imply that myogenic vasoconstriction requires canonical Gq/11 signaling of the AT1R. Consistently, myogenic tone is increased in the absence of regulator of G‐protein signaling 2, which is an endogenous terminator of Galphaq/11 (Gαq/11) signaling. 14 , 30 The data align with findings indicating that mechanically activated AT1Rs generate diacylglycerol, which in turn activates protein kinase C to induce the actin cytoskeleton reorganization necessary for pressure‐induced vasoconstriction. 52 Finally, our conclusions are supported by findings indicating that another Gq/11‐protein inhibitor YM 254890 profoundly reduced myogenic tone in MA 43 (but see Ref. 50). Our study provides firm evidence that AT1Rs coupled to Gq/11 signaling is an essential component of dynamic mechanochemical signaling in arterial vascular SM cells causing myogenic tone (Figure 8).
Signaling of most GPCRs via G proteins is terminated (desensitization) by the phosphorylation of active receptor by specific kinases (GPCR kinases) and subsequent binding of ß‐arrestins that selectively recognize active phosphorylated receptors. Although, GPCR kinases and ß‐arrestins also play a role in multiple noncanonical signaling pathways in the cell, both GPCR initiated and receptor independent, 53 , 54 our study demonstrates that this pathway is not involved in the myogenic response (Figure 8). Thus, blood pressure‐lowering effects of ß‐arrestin biased AT1R agonists, for example, Trevena 120027, 55 are unlikely caused by direct effects of this GPCR in arterial SM cells.
CONCLUSIONS
In summary, we provide new and firm evidence for a mechanosensitive function of AT1R in myogenic constriction in mesenteric, renal, and cerebral arteries, that is, in 3 different highly myogenic vascular beds. Our study clearly shows that mechanical stress activates AT1R in arterial SM cells, which subsequently triggers canonical Gq/11 signaling, irrespective of GPCR kinases/β‐arrestin signaling, to cause myogenic vasoconstriction. Our results argue against the idea of multiple mechanosensors coupled to noncanonical β‐arrestin pathways generating myogenic arterial tone. These findings lay ground for future studies to characterize the molecular mechanisms of mechanoactivated AT1R coupled to Gq/11 signaling in intact arteries, which may reveal new molecular targets for drug development to alleviate increased or dysregulated arterial tone in hypertension and other cardiovascular diseases.
Sources of Funding
The Deutsche Forschungsgemeinschaft (DFG, GO766/12‐3, GO766/15‐2, GO766/18‐2, SFB1365) (Gollasch, Schleifenbaum, Alenina, and Bader), Germany/Hong Kong Joint Research Scheme (G‐CUHK408/18 and Deutscher Akademischer Austauschdienst [DAAD]) (Gollasch, Huang), and Hong Kong Research Grants Council (SRFS2021‐4S0) (Huang) supported our study.
Disclosures
None.
Supporting information
Figures S1–S2
Acknowledgments
We thank Thomas Coffman for providing Agtr1a −/−, Agtr1b −/− and Agtr1a flox mice. We thank Gabriele M. König and Evi Kostenis for FR900359.
Author contributions: Conceptualization: Gollasch, Huang; Pressure myography: Cui; Perfused kidneys: Cui, Nickel, Immunohistochemistry: Zhang; Formal analysis: Cui, Nickel, Zhang, Kassmann, Gollasch; Funding acquisition: Gollasch; Investigation: Cui, Nickel, Zhang; Methodology: Cui, Zhang, Alenina; Visualization: Cui, Zhang; Writing—original draft: Cui, Gollasch; Writing—review and editing: Cui, Kassmann, Schleifenbaum, Bader, Welsh, Huang.
Preprint posted on BioRxiv September 11, 2020. https://doi.org/10.1101/2020.09.09.289280.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.022070
For Sources of Funding and Disclosures, see page 14.
See Editorial by Chen and Sonkusare
References
- 1. Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davis MJ. Perspective: physiological role(s) of the vascular myogenic response. Microcirculation. 2012;19:99–114. doi: 10.1111/j.1549-8719.2011.00131.x [DOI] [PubMed] [Google Scholar]
- 3. Cipolla MJ, Curry AB. Middle cerebral artery function after stroke: the threshold duration of reperfusion for myogenic activity. Stroke. 2002;33:2094–2099. doi: 10.1161/01.STR.0000020712.84444.8D [DOI] [PubMed] [Google Scholar]
- 4. Gschwend S, Henning RH, Pinto YM, de Zeeuw D, van Gilst WH, Buikema H. Myogenic constriction is increased in mesenteric resistance arteries from rats with chronic heart failure: instantaneous counteraction by acute AT1 receptor blockade. Br J Pharmacol. 2003;139:1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ledoux J, Gee DM, Leblanc N. Increased peripheral resistance in heart failure: new evidence suggests an alteration in vascular smooth muscle function. Br J Pharmacol. 2003;139:1245–1248. doi: 10.1038/sj.bjp.0705366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pires PW, Jackson WF, Dorrance AM. Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am J Physiol Heart Circ Physiol. 2015;309:H127–H136. doi: 10.1152/ajpheart.00168.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sauvé M, Hui SK, Dinh DD, Foltz WD, Momen A, Nedospasov SA, Offermanns S, Husain M, Kroetsch JT, Lidington D, et al. Tumor necrosis factor/sphingosine‐1‐phosphate signaling augments resistance artery myogenic tone in diabetes. Diabetes. 2016;65:1916–1928. doi: 10.2337/db15-1450 [DOI] [PubMed] [Google Scholar]
- 8. Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3 [DOI] [PubMed] [Google Scholar]
- 9. Coats P, Johnston F, MacDonald J, McMurray JJ, Hillier C. Signalling mechanisms underlying the myogenic response in human subcutaneous resistance arteries. Cardiovasc Res. 2001;49:828–837. doi: 10.1016/S0008-6363(00)00314-X [DOI] [PubMed] [Google Scholar]
- 10. Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387 [DOI] [PubMed] [Google Scholar]
- 11. Hansen PB, Jensen BL, Andreasen D, Skøtt O. Differential expression of T‐ and L‐type voltage‐dependent calcium channels in renal resistance vessels. Circ Res. 2001;89:630–638. doi: 10.1161/hh1901.097126 [DOI] [PubMed] [Google Scholar]
- 12. Harder DR. Pressure‐dependent membrane depolarization in cat middle cerebral artery. Circ Res. 1984;55:197–202. doi: 10.1161/01.RES.55.2.197 [DOI] [PubMed] [Google Scholar]
- 13. Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F, Klugbauer N. Dominant role of smooth muscle L‐type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003;22:6027–6034. doi: 10.1093/emboj/cdg583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mederos y Schnitzler MM, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq‐coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008;27:3092–3103. doi: 10.1038/emboj.2008.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schleifenbaum J, Kassmann M, Szijarto IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N, et al. Stretch‐activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res. 2014;115:263–272. doi: 10.1161/CIRCRESAHA.115.302882 [DOI] [PubMed] [Google Scholar]
- 16. Xiao HD, Fuchs S, Frenzel K, Cole JM, Bernstein KE. Newer approaches to genetic modeling in mice: tissue‐specific protein expression as studied using angiotensin‐converting enzyme (ACE). Am J Pathol. 2003;163:807–817. doi: 10.1016/S0002-9440(10)63441-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Balakumar P, Jagadeesh G. A century old renin‐angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell Signal. 2014;26:2147–2160. doi: 10.1016/j.cellsig.2014.06.011 [DOI] [PubMed] [Google Scholar]
- 18. Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. Beta‐arrestin‐biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3:ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137 [DOI] [PubMed] [Google Scholar]
- 20. Tang W, Strachan RT, Lefkowitz RJ, Rockman HA. Allosteric modulation of β‐arrestin‐biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem. 2014;289:28271–28283. doi: 10.1074/jbc.M114.585067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaßmann M, Szijártó IA, García‐Prieto CF, Fan G, Schleifenbaum J, Anistan Y‐M, Tabeling C, Shi Y, le Noble F, Witzenrath M, et al. Role of ryanodine type 2 receptors in elementary Ca signaling in arteries and vascular adaptive responses. J Am Heart Assoc. 2019;8:e010090. doi: 10.1161/JAHA.118.010090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sparks MA, Parsons KK, Stegbauer J, Gurley SB, Vivekanandan‐Giri A, Fortner CN, Snouwaert J, Raasch EW, Griffiths RC, Haystead TAJ, et al. Angiotensin II type 1A receptors in vascular smooth muscle cells do not influence aortic remodeling in hypertension. Hypertension. 2011;57:577–585. doi: 10.1161/HYPERTENSIONAHA.110.165274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Groneberg D, Konig P, Wirth A, Offermanns S, Koesling D, Friebe A. Smooth muscle‐specific deletion of nitric oxide‐sensitive guanylyl cyclase is sufficient to induce hypertension in mice. Circulation. 2010;121:401–409. doi: 10.1161/CIRCULATIONAHA.109.890962 [DOI] [PubMed] [Google Scholar]
- 24. Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci USA. 1995;92:3521–3525. doi: 10.1073/pnas.92.8.3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jarve A, Todiras M, Lian X, Filippelli‐Silva R, Qadri F, Martin RP, Gollasch M, Bader M. Distinct roles of angiotensin receptors in autonomic dysreflexia following high‐level spinal cord injury in mice. Exp Neurol. 2019;311:173–181. doi: 10.1016/j.expneurol.2018.10.003 [DOI] [PubMed] [Google Scholar]
- 26. Oliverio MI, Kim H‐S, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, et al. Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor‐mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci USA. 1998;95:15496–15501. doi: 10.1073/pnas.95.26.15496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijártó IA, Schleifenbaum J, Vitzthum H, Gollasch M, Ehmke H, et al. Disruption of vascular Ca2+‐activated chloride currents lowers blood pressure. J Clin Invest. 2014;124:675–686. doi: 10.1172/JCI70025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ercu M, Markó L, Schächterle C, Tsvetkov D, Cui Y, Maghsodi S, Bartolomaeus TUP, Maass PG, Zühlke K, Gregersen N, et al. Phosphodiesterase 3A and arterial hypertension. Circulation. 2020;142:133–149. doi: 10.1161/CIRCULATIONAHA.119.043061 [DOI] [PubMed] [Google Scholar]
- 29. Fan G, Kassmann M, Cui Y, Matthaeus C, Kunz S, Zhong C, Zhu S, Xie Y, Tsvetkov D, Daumke O, et al. Age attenuates the T‐type Cav 3.2‐RyR axis in vascular smooth muscle. Aging Cell. 2020;19:e13134. doi: 10.1111/acel.13134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hercule HC, Tank J, Plehm R, Wellner M, da Costa Goncalves AC, Gollasch M, Diedrich A, Jordan J, Luft FC, Gross V. Regulator of G protein signalling 2 ameliorates angiotensin II‐induced hypertension in mice. Exp Physiol. 2007;92:1014–1022. doi: 10.1113/expphysiol.2007.038240 [DOI] [PubMed] [Google Scholar]
- 31. Janssen BJA, Smits JFM. Autonomic control of blood pressure in mice: basic physiology and effects of genetic modification. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1545–R1564. doi: 10.1152/ajpregu.00714.2001 [DOI] [PubMed] [Google Scholar]
- 32. Kendall RT, Strungs EG, Rachidi SM, Lee M‐H, El‐Shewy HM, Luttrell DK, Janech MG, Luttrell LM. The beta‐arrestin pathway‐selective type 1A angiotensin receptor (AT1A) agonist [Sar1, Ile4, Ile8] angiotensin II regulates a robust G protein‐independent signaling network. J Biol Chem. 2011;286:19880–19891. doi: 10.1074/jbc.M111.233080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li W, Xu J, Kou X, Zhao R, Zhou W, Fang X. Single‐molecule force spectroscopy study of interactions between angiotensin II type 1 receptor and different biased ligands in living cells. Anal Bioanal Chem. 2018;410:3275–3284. doi: 10.1007/s00216-018-0956-3 [DOI] [PubMed] [Google Scholar]
- 34. Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta‐arrestin 2 and G protein‐mediated pathways for angiotensin II activation of extracellular signal‐regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu C‐H, Gong Z, Liang Z‐L, Liu Z‐X, Yang F, Sun Y‐J, Ma M‐L, Wang Y‐J, Ji C‐R, Wang Y‐H, et al. Arrestin‐biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nat Commun. 2017;8:14335. doi: 10.1038/ncomms14335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Santos GA, Duarte DA, Parreiras‐E‐Silva LT, Teixeira FR, Silva‐Rocha R, Oliveira EB, Bouvier M, Costa‐Neto CM. Comparative analyses of downstream signal transduction targets modulated after activation of the AT1 receptor by two β‐arrestin‐biased agonists. Front Pharmacol. 2015;6:131. doi: 10.3389/fphar.2015.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inamdar V, Patel A, Manne BK, Dangelmaier C, Kunapuli SP. Characterization of UBO‐QIC as a Galphaq inhibitor in platelets. Platelets. 2015;26:771–778. doi: 10.3109/09537104.2014.998993 [DOI] [PubMed] [Google Scholar]
- 38. Schrage R, Schmitz A‐L, Gaffal E, Annala S, Kehraus S, Wenzel D, Büllesbach KM, Bald T, Inoue A, Shinjo Y, et al. The experimental power of FR900359 to study Gq‐regulated biological processes. Nat Commun. 2015;6:10156. doi: 10.1038/ncomms10156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lian X, Beer‐Hammer S, Konig GM, Kostenis E, Nurnberg B, Gollasch M. RXFP1 receptor activation by relaxin‐2 induces vascular relaxation in mice via a Galphai2‐protein/PI3Kß/gamma/nitric oxide‐coupled pathway. Front Physiol. 2018;9:1234. doi: 10.3389/fphys.2018.01234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kauffenstein G, Laher I, Matrougui K, Guérineau NC, Henrion D. Emerging role of G protein‐coupled receptors in microvascular myogenic tone. Cardiovasc Res. 2012;95:223–232. doi: 10.1093/cvr/cvs152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kauffenstein G, Tamareille S, Prunier F, Roy C, Ayer A, Toutain B, Billaud M, Isakson BE, Grimaud L, Loufrani L, et al. Central role of P2y6 UDP receptor in arteriolar myogenic tone. Arterioscler Thromb Vasc Biol. 2016;36:1598–1606. doi: 10.1161/ATVBAHA.116.307739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kroetsch JT, Bolz S‐S. The TNF‐α/sphingosine‐1‐phosphate signaling axis drives myogenic responsiveness in heart failure. J Vasc Res. 2013;50:177–185. doi: 10.1159/000350528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Storch U, Blodow S, Gudermann T, Mederos Y, Schnitzler M. Cysteinyl leukotriene 1 receptors as novel mechanosensors mediating myogenic tone together with angiotensin II type 1 receptors‐brief report. Arterioscler Thromb Vasc Biol. 2015;35:121–126. doi: 10.1161/ATVBAHA.114.304844 [DOI] [PubMed] [Google Scholar]
- 44. Madhun ZT, Ernsberger P, Ke FC, Zhou J, Hopfer U, Douglas JG. Signal transduction mediated by angiotensin ii receptor subtypes expressed in rat renal mesangial cells. Regul Pept. 1993;44:149–157. doi: 10.1016/0167-0115(93)90238-4 [DOI] [PubMed] [Google Scholar]
- 45. Zhou J, Ernsberger P, Douglas JG. A novel angiotensin receptor subtype in rat mesangium. Coupling to adenylyl cyclase. Hypertension. 1993;21:1035–1038. [DOI] [PubMed] [Google Scholar]
- 46. Pires PW, Ko EA, Pritchard HAT, Rudokas M, Yamasaki E, Earley S. The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells. J Physiol. 2017;595:4735–4753. doi: 10.1113/JP274310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blodow S, Schneider H, Storch U, Wizemann R, Forst AL, Gudermann T, Mederos y Schnitzler M. Novel role of mechanosensitive AT1B receptors in myogenic vasoconstriction. Pflugers Arch. 2014;466:1343–1353. doi: 10.1007/s00424-013-1372-3 [DOI] [PubMed] [Google Scholar]
- 48. Le TH, Fogo AB, Salzler HR, Vinogradova T, Oliverio MI, Marchuk DA, Coffman TM. Modifier locus on mouse chromosome 3 for renal vascular pathology in AT1A receptor‐deficiency. Hypertension. 2004;43:445–451. doi: 10.1161/01.HYP.0000112423.28987.00 [DOI] [PubMed] [Google Scholar]
- 49. Yamasaki E, Thakore P, Krishnan V, Earley S. Differential expression of angiotensin II type 1 receptor subtypes within the cerebral microvasculature. Am J Physiol Heart Circ Physiol. 2020;318:H461–H469. doi: 10.1152/ajpheart.00582.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chennupati R, Wirth A, Favre J, Li R, Bonnavion R, Jin Y‐J, Wietelmann A, Schweda F, Wettschureck N, Henrion D, et al. Myogenic vasoconstriction requires G/G and LARG to maintain local and systemic vascular resistance. Elife. 2019;8:e49374. doi: 10.7554/eLife.49374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith JS, Pack TF, Inoue A, Lee C, Zheng K, Choi I, Eiger DS, Warman A, Xiong X, Ma Z, et al. Noncanonical scaffolding of G and β‐arrestin by G protein‐coupled receptors. Science. 2021;371:eaay1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hong K, Zhao G, Hong Z, Sun Z, Yang Y, Clifford PS, Davis MJ, Meininger GA, Hill MA. Mechanical activation of angiotensin II type 1 receptors causes actin remodelling and myogenic responsiveness in skeletal muscle arterioles. J Physiol. 2016;594:7027–7047. doi: 10.1113/JP272834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lefkowitz RJ. A brief history of G‐protein coupled receptors (Nobel Lecture). Angew Chem Int Ed Engl. 2013;52:6366–6378. doi: 10.1002/anie.201301924 [DOI] [PubMed] [Google Scholar]
- 54. Gurevich VV, Gurevich EV. GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol. 2019;10:125. doi: 10.3389/fphar.2019.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC Jr. Cardiorenal actions of TRV120027, a novel ß‐arrestin‐biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail. 2011;4:770–778. doi: 10.1161/CIRCHEARTFAILURE.111.962571 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S2