

SUMMARY
To better understand the functions of non-coding enhancer RNAs (eRNAs), we annotated the estrogen-regulated eRNA transcriptome in estrogen receptor α (ERα)-positive breast cancer cells using PRO-cap and RNA sequencing. We then cloned a subset of the eRNAs identified, fused them to single guide RNAs, and targeted them to their ERα enhancers of origin using CRISPR/dCas9. Some of the eRNAs tested modulated the expression of cognate, but not heterologous, target genes after estrogen treatment by increasing ERα recruitment and stimulating p300-catalyzed H3K27 acetylation at the enhancer. We identified a ~40 nucleotide functional eRNA regulatory motif (FERM) present in many eRNAs that was necessary and sufficient to modulate gene expression, but not the specificity of activation, after estrogen treatment. The FERM interacted with BCAS2, an RNA-binding protein amplified in breast cancers. The ectopic expression of a targeted eRNA controlling the expression of an oncogene resulted in increased cell proliferation, demonstrating the regulatory potential of eRNAs in breast cancer.
Graphical Abstract
In brief
Hou and Kraus show that some eRNAs fused to sgRNAs can be targeted back to their ERα enhancers to stimulate ERα recruitment, p300-catalyzed H3K27 acetylation, and estrogen-dependent target gene expression. They discovered a conserved functional eRNA regulatory motif (FERM) in some eRNAs that mediates the effects of the functional eRNAs.
INTRODUCTION
Enhancers are genomic regulatory elements that act as nucleation sites for the binding of sequence-specific transcription factors and the formation of transcription regulatory complexes (Catarino and Stark, 2018). Enhancers are characterized by common molecular features such as (1) an open or accessible chromatin environment (Boyle et al., 2008; Buenrostro et al., 2013; Crawford et al., 2006; Giresi et al., 2007; Sheffield et al., 2013); (2) the enrichment of a common set of histone modifications, such as histone H3 lysine 4 (H3K4) monomethylation and histone H3 lysine 27 (H3K27) acetylation; (3) the binding of transcription factors (TFs), coregulators, and chromatin remodeling enzymes; and (4) looping to target gene promoters (Catarino and Stark, 2018; Gasperini et al., 2020). Enhancers are also actively transcribed, producing enhancer RNAs (eRNAs) (De Santa et al., 2010; Hah et al., 2011; Kim et al., 2010; Melgar et al., 2011; Natoli and Andrau, 2012).
The role of eRNAs in gene regulation, if any, has been debated in the literature, in part because eRNAs have short half-lives, making them difficult to study (Hou and Kraus, 2021). Three modes of potential function for enhancer transcription and eRNAs have been proposed: (1) the act of enhancer transcription promotes an open or accessible chromatin environment to allow for enhancer formation; (2) eRNAs act locally (in cis) at the enhancers from which they are transcribed to promote enhancer function; and (3) the eRNAs act distally (in trans) to control target gene transcription. Mechanistically, eRNAs are thought to function by (1) promoting the recruitment of TFs and coregulators, and regulating their activities (Bose et al., 2017; Lai et al., 2013; Rahnamoun et al., 2018; Sigova et al., 2015); (2) facilitating RNA polymerase II (RNAPII) pause-release to promote transcription elongation (Schaukowitch et al., 2014; Zhao et al., 2016); and (3) driving enhancer-promoter looping (Hah et al., 2013; Hsieh et al., 2014; Li et al., 2013; Tsai et al., 2018).
The majority of studies analyzing the function of eRNAs have relied heavily on the use of loss-of-function (subtraction) approaches, such as the small interfering RNA (siRNA)-mediated knockdown of eRNAs (Hou and Kraus, 2021; Sartorelli and Lau-berth, 2020). From an experimental standpoint, the subtraction approach raises questions, especially when examining signal-regulated enhancers and target gene transcription: How can eRNAs, which are transcribed within minutes of stimulation, be knocked down using a 24- to 48-h pre-treatment with siRNAs before the stimulus? Furthermore, how can accelerating the degradation of eRNAs, which are relatively unstable compared with mRNAs and long noncoding RNAs, impact their function? Although studies have tried to address this issue with gain-of-function (addition) approaches (Bose et al., 2017; Carullo et al., 2020; Lai et al., 2013; Lam et al., 2013; Li et al., 2013; Melo et al., 2013; Rahnamoun et al., 2018; Schaukowitch et al., 2014; Sigova et al., 2015; Tsai et al., 2018; Zhao et al., 2019), many of the eRNAs were not examined at their endogenous locus. Together, the lack of detailed eRNA annotations and an over-reliance on siRNA to deplete eRNAs have left many questions unanswered, including the sequence dependence of eRNAs and their detailed mechanisms of action. Here we use an eRNA addition approach, targeting them to their endogenous loci, to study the mechanisms and biology of estrogen-regulated eRNAs in breast cancer cells.
RESULTS
Annotating the estrogen-regulated eRNA transcriptome in breast cancer cells
To annotate the estrogen-regulated eRNA transcriptome, we generated RNA sequencing (RNA-seq) libraries using polyA-depleted and polyA-enriched fractions isolated from estrogen receptor (ERα)-positive MCF-7 human breast cancer cells cultured for 3 days in estrogen-free medium and then treated with 17β-estradiol (E2) for 45 min, which gives the maximal transcriptional response for eRNAs originating from ERα enhancers (Hah et al., 2011, 2013; Murakami et al., 2017). We assembled the universe of transcripts using StringTie and then filtered them based on their overlap with intergenic ERα binding sites (Franco et al., 2015; Hah et al., 2013) (Figure 1A), resulting in the identification of the transcribed regions (i.e., transcript bodies) of E2-regulated eRNAs (Figure S1A). Additionally, we used precision run-on of capped RNA and sequencing (PRO-cap) to identify the transcription start sites (TSSs) of eRNAs at near base pair resolution, and used them as anchoring points for annotating E2-regulated eRNAs (Figure S1A). Comparing the eRNA annotations from TSSs detected by PRO-cap with the transcripts identified from StringTie shows broad agreement in eRNA identification by these two different approaches, with most of the TSS called by PRO-cap and StringTie clustered closely with one another (Figure S1B). Owing to the lack of extensive polyadenylation on eRNAs, we were unable to use 3′ mapping strategies, such as 3P-seq (Jan et al., 2011); therefore, we relied on the transcripts assembled by StringTie to discern the 3′ ends of the eRNAs.
Figure 1. Annotation and characterization of estrogen-regulated eRNAs in MCF-7 cells.
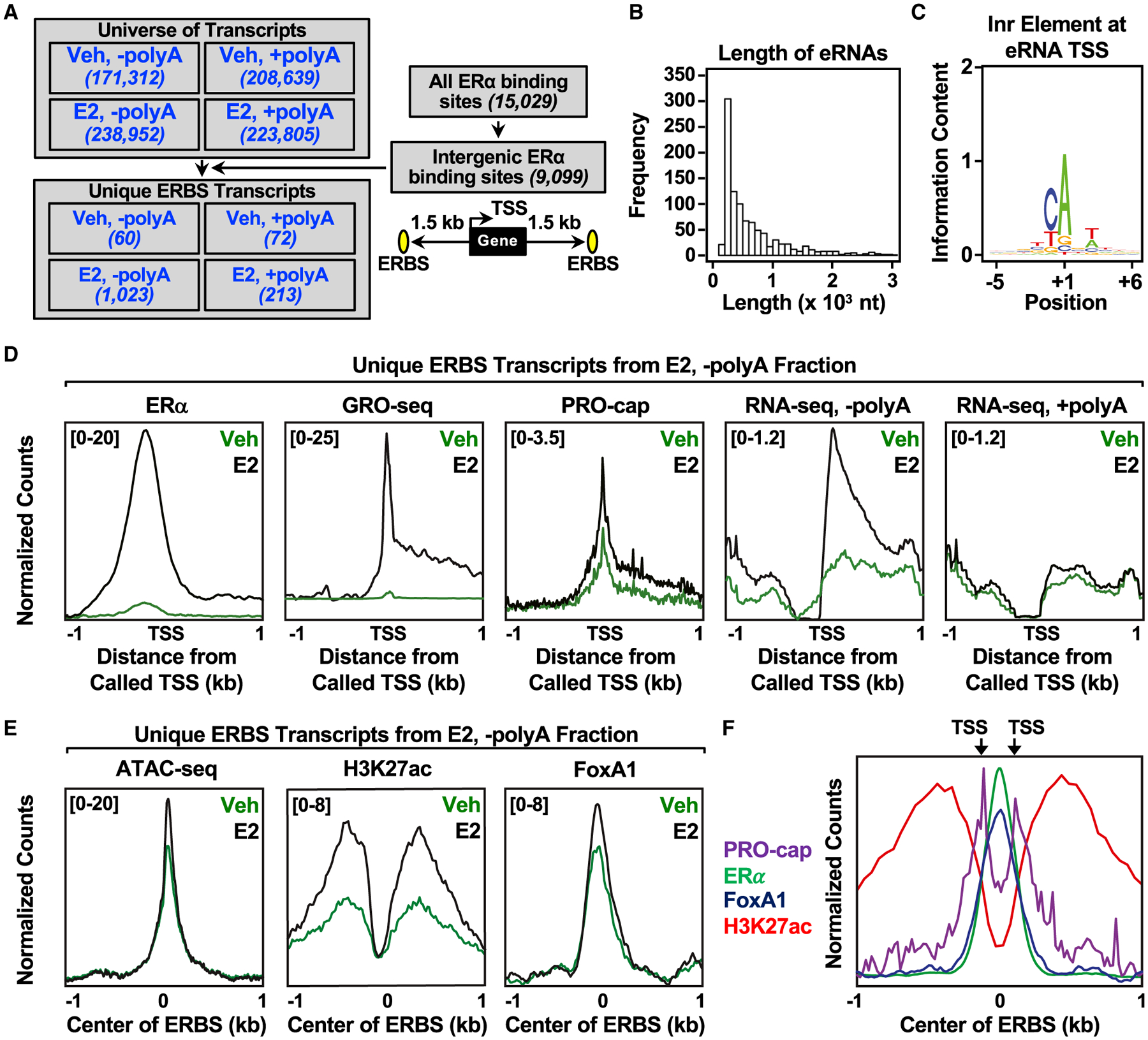
(A) Flowchart illustrating the genomic and computational analysis pipeline used to annotate estrogen-regulated eRNAs using polyA-depleted (−polyA) or polyA-enriched (+polyA) RNA-seq libraries generated from DMSO (Veh) or E2-treated MCF-7 cells.
(B) Histogram of the individual lengths of 1,023 E2-regulated eRNAs.
(C) Position weight matrix of eRNA TSSs determined using PRO-cap reveals the presence of the initiator (Inr) element.
(D) Metagene representations of normalized read counts for ERα ChIP-seq, GRO-seq, PRO-cap, RNA-seq from polyA-depleted (-polyA) fraction, and RNA-seq from polyA-enriched (+polyA) fraction around the TSSs (±1 kb) of 1,023 estrogen-regulated eRNAs from DMSO (vehicle [Veh]) or E2-treated MCF-7 cells. Antisense eRNAs were oriented the same way as the sense eRNAs.
(E) Metagene representations of average read counts for ATAC-seq, H3K27 ac, and FoxA1 ChIP-seq around the ERα binding site (ERBS) of 1,023 estrogen-regulated eRNAs from Veh or E2-treated MCF-7 cells. Antisense eRNAs were not oriented the same way as the sense eRNAs.
(F) Overlaid metagene representations of PRO-cap, ERα, FoxA1, and H3K27ac ChIP-seq from E2-treated MCF-7 cells to depict bidirectional transcription of E2-regulated eRNAs around the center of the ERBS (±1 kb). TSSs determined by PRO-cap are indicated by arrows. All genomic assays represented in this figure were performed as two biological and two technical replicates.
See also Figures S1 and S2.
With this strategy, we annotated 1,023 E2-regulated eRNAs in the polyA-depleted fraction compared with 213 in the polyA-enriched fraction, despite similar numbers in the universe of all transcripts, consistent with the notion that eRNAs are not extensively polyadenylated (Andersson et al., 2014; De Santa et al., 2010; Hah et al., 2013; Koch et al., 2011) (Figure 1A). In general, eRNAs tend to be short (median = 449 nt) (Figure 1B and S1C), similar to previous mapping by global run-on sequencing (GRO-seq) or RNA-seq (De Santa et al., 2010; Hah et al., 2011; Kim et al., 2010). An analysis of the eRNA TSSs using a position weight matrix revealed an enrichment for the initiator element (Figure 1C), a minimal core promoter for transcription initiation (Vo Ngoc et al., 2017).
Next, we integrated the genomic features of E2-stimulated enhancers, including ERα binding (ERα chromatin immunoprecipitation sequencing [ChIP-seq]), TSSs (PRO-cap), transcribed regions (GRO-seq), steady eRNAs (RNA-seq reads in both polyA-depleted and -enriched fractions) for the annotated E2-regulated eRNAs (Figure 1D). As expected, there was an increase in ERα binding, enhancer transcription, TSS signal, and eRNA reads from the polyA-depleted fraction upon E2 treatment. eRNA-producing ERα binding sites are also enriched for features associated with active enhancers upon E2 treatment, including enhanced chromatin accessibility (assay for transposase-accessible chromatin [ATAC-seq]), H3K27 acetylation (H3K27ac ChIP-seq), FoxA1 binding (FoxA1 ChIP-seq), RNAPII recruitment (RNAPII ChIP-Seq), and p300 binding (p300 ChIP-seq) (Figures 1E and S1D). We also detected TSSs (PRO-cap) on either side of ERα- and FoxA1-binding sites (Figure 1F), indicative of bidirectional transcription, a hallmark of active enhancers (Hah et al., 2013; Melgar et al., 2011).
Using our eRNA annotations with RNA-seq and GRO-seq, we have classified the ERα enhancers into three groups: (1) ERα binding without enhancer transcription or stable eRNA production (group 1); (2) ERα binding with enhancer transcription (GRO-seq), but no eRNAs (RNA-seq) (group 2); and (3) ERα binding with both enhancer transcription and eRNAs (group 3) (Figure S2A). In general, ERα binding sites with transcription and detectable eRNAs have increased ERα binding, open chromatin accessibility (ATAC-seq), H3K27 acetylation, FoxA1 binding, RNAPII recruitment, and p300 binding (Figures S2B–S2G) compared to those without them. Upon E2 treatment, all of the enhancer features examined were elevated in group 3 when compared with the other two groups. Although not all actively transcribed ERα enhancers generate stable eRNA transcripts detected by RNA-seq (1,250 of 7,495), the ERα binding sites from which the annotated E2-regulated eRNAs originate display hallmarks associated with highly active enhancers.
Directing eRNAs to their cognate enhancers modulates target gene expression
To study the functions of the annotated eRNAs, we used an addition approach based on CRISPR-Display technology (Shechner et al., 2015). We cloned and fused selected eRNAs to single guide RNAs (sgRNAs) to direct the eRNAs to their enhancers of origin in dCas9-expressing MCF-7 breast cancer cells (Figures 2A and 2B). We focused our initial characterization on eRNAs transcribed from ERα enhancers located upstream of the E2-regulated PRRX2/PTGES, UBE2E2/UBE2E1, and SEMA3C genes. These eRNAs were chosen based on the RNA-seq fragments per kilobase of transcript per million mapped reads of the annotated eRNAs and their proximity to E2-regulated genes. We used published Hi-C and ERα chromatin interaction analysis by paired-end tag sequencing data (Fullwood et al., 2009; Wang et al., 2018) to confirm that genes located near these enhancers may communicate through enhancer-promoter looping (Figures 2C and 2D). ChIP-qualitative PCR (qPCR) analyses for dCas9 demonstrated that the targeting to the enhancer was specific, since only constructs containing the targeting sgRNA sequence increased the enrichment of dCas9 at the target site (Figures 2E and 2F). Importantly, we used sgRNAs that target dCas9 to regions adjacent to, not overlapping, the ERα binding site to minimize CRISPR-mediated inhibition. We also used RNA in situ fluorescence hybridization to validate our eRNA tethering system for eRNA expression and localization under basal and E2-treated conditions (Figures 2G and 2H).
Figure 2. Characterization of the CRISPR/dCas9-based eRNA-tethering system.
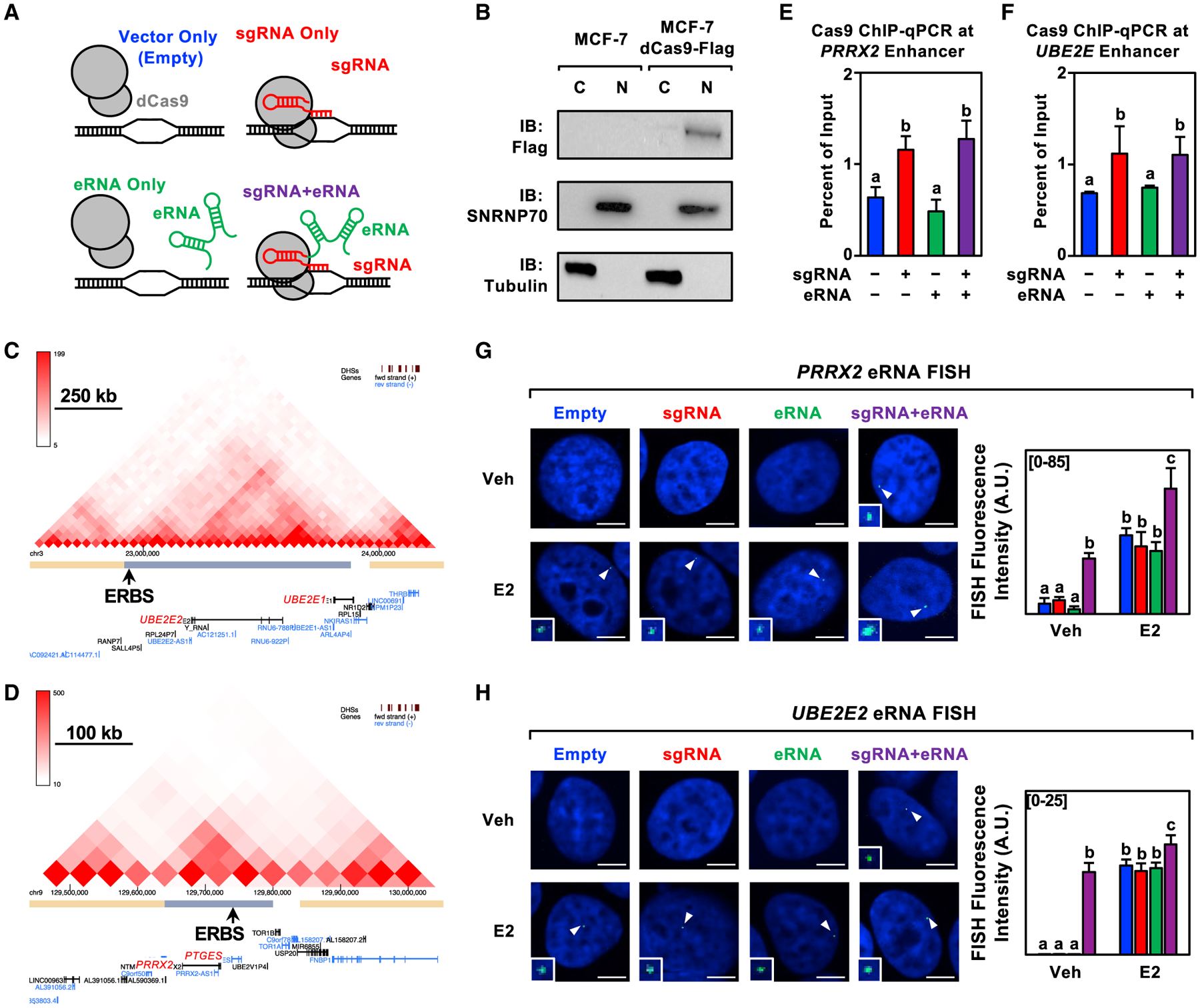
(A) Schematic representation of the four constructs (control and experimental) used in the eRNA tethering experiments.
(B) Subcellular fractionation (C = cytoplasmic, N = nuclear) followed by western blotting as indicated for MCF-7 cells expressing dCas9-Flag. This experiment was performed as two biological and two technical replicates.
(C and D) Graphical representation of Hi-C data for the (C) UBE2E2 and (D) PRRX2 loci. The main ERα binding site (ERBS) within the topologically associating domain (TAD) is designated by an arrow.
(E and F) ChIP-qPCR assays for Cas9 in dCas9-expressing MCF-7 cells expressing sgRNA, eRNA, and sgRNA fused to its cognate eRNA for the (C) PRRX2 and (D) UBE2E2 constructs. Each bar represents the mean ± standard error of the mean (n = 3 biological/technical replicates). Bars marked with different letters are significantly different (two-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05).
(G and H) Representative images of a nucleus showing a single RNA FISH spot in each condition (left) and summary of data (right) for RNA fluorescence in situ hybridization assays at E2-regulated enhancers: (E) PRRX2 constructs (n = 21–29) and (F) UBE2E2 constructs (n = 27–46) in dCas9-expressing MCF-7 cells treated with DMSO (vehicle; Veh) or E2 for 40 min. Cell nuclei were labeled with DAPI and probed with fluorescent probes targeting each eRNA. White arrows indicate RNA FISH signals (expanded in the insets). Each bar represents the mean ± standard error of the mean. Bars marked with different letters are significantly different (two-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05). Scale bar, 5 μm. This experiment was performed as two biological and two technical replicates.
Upon examination of the PRRX2/PTGES, UBE2E2/UBE2E1, and SEMA3C enhancers (Figures 3A, 3B, and S3A), we observed that eRNAs directed to their enhancer of origin using CRISPR/dCas9 augmented target gene expression in response to a time course of E2 treatment (Figures 3C, 3D, and S3A), suggesting a functional role for eRNAs in the context of E2-induced activity. In all cases, target gene expression was increased at one or more timepoints of E2 treatment, regardless of the expression construct. Recruitment of the eRNAs, however, shortened the timescale for maximal induction and/or increased the magnitude of target gene expression. Since unliganded ERα does not bind efficiently to chromatin (Caizzi et al., 2014; Franco et al., 2015; Murakami et al., 2017), we did not expect the eRNAs to exert their activating effects at these enhancers in the absence of E2 treatment. The eRNA alone construct (i.e., not fused to an sgRNA) was insufficient to enhance target gene expression (Figures 3C, 3D, S3A, and S3B), suggesting that the eRNAs examined are unable to function outside of their native genomic context in trans (Figures 3C, 3D, and S3A). Importantly, not every eRNA tested affected target gene expression (Figure S3B) and not all genes sharing a topologically associating domain with the enhancer showed enhanced expression (Figures 3C and 3D), suggesting potential diversity and specificity in the function of eRNAs.
Figure 3. Targeting an estrogen-regulated eRNA to its enhancer of origin using CRISPR-dCas9 modulates target gene expression.
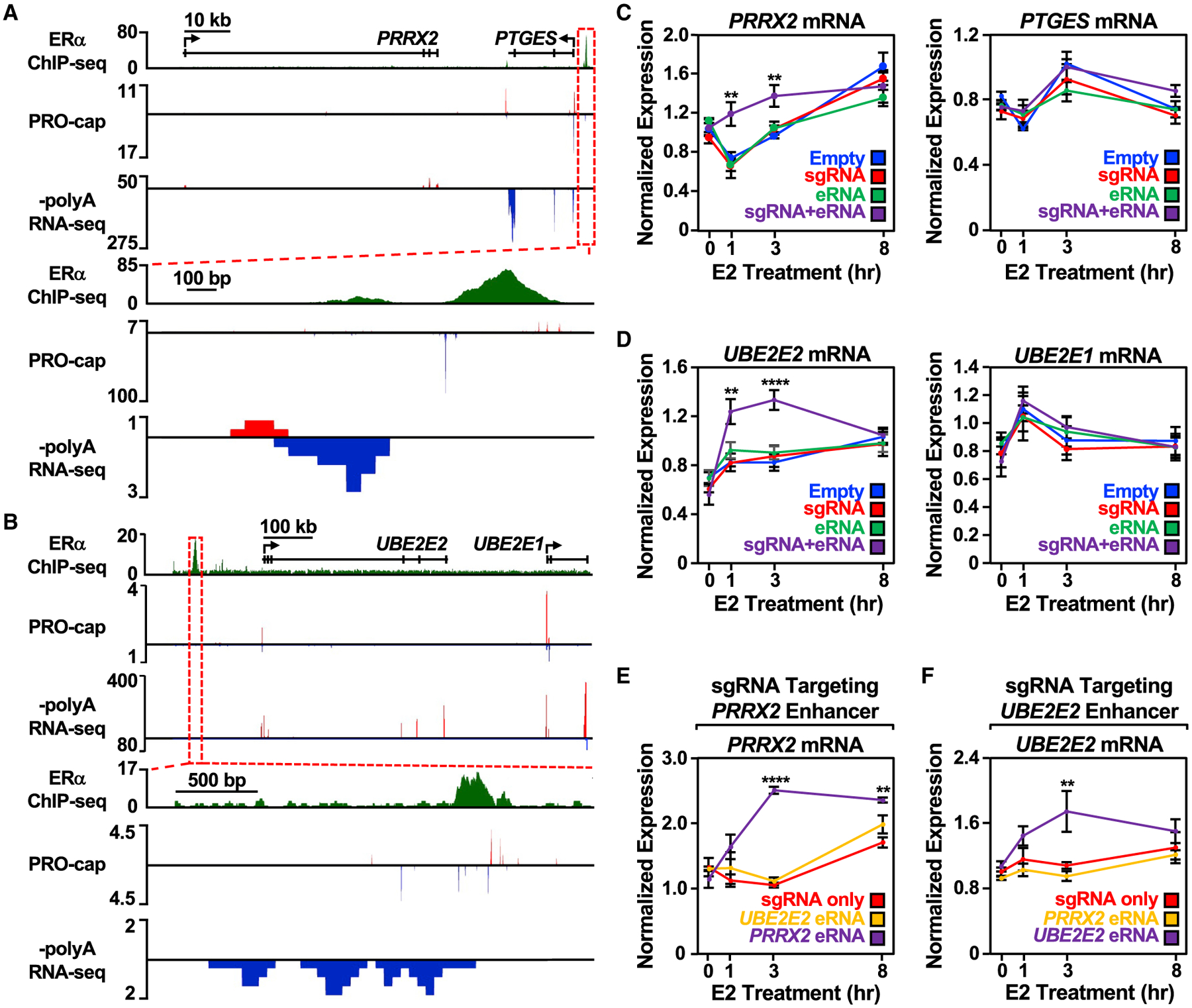
(A and B) Genome browser views of the (A) PRRX2-PTGES and (B) UBE2E2-UBE2E1 topologically associating domains (TADs). A zoomed-in view of the ERα enhancer (boxed region) is included at the bottom.
(C) Reverse transcription-quantitative PCR (RT-qPCR) analysis of the PRRX2 (left) and PTGES (right) mRNA levels normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with PRRX2 sgRNA, eRNA, and sgRNA + eRNA constructs. Cells were treated with E2 for the indicated time. Each point represents the mean ± standard error of the mean (n = 3 biological/technical replicates). Asterisks indicate significant differences from the corresponding empty control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01, ****p < 0.0001).
(D) RT-qPCR analysis of the UBE2E2 (left) and UBE2E1 (right) mRNA levels normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with UBE2E2 sgRNA, eRNA, and sgRNA + eRNA constructs. Cells were treated with E2 for the indicated time. Each point represents the mean ± standard error of the mean (n = 4 biological/technical replicates). Asterisks indicate significant differences from the corresponding empty control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01, ****p < 0.0001).
(E) RT-qPCR analysis of the PRRX2 mRNA expression level normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with PRRX2 sgRNA, sgRNA fused to UBE2E2 eRNA, or sgRNA fused to its cognate PRRX2 eRNA (PRRX2 eRNA, purple). Cells were treated with E2 for the indicated time. Each point represents the mean ± standard error of the mean (n = 3 biological/technical replicates). Asterisks indicate significant differences from the corresponding control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01, ****p < 0.0001).
(F) RT-qPCR analysis of the UBE2E2 mRNA expression level normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with UBE2E2 sgRNA, sgRNA fused to PRRX2 eRNA, or sgRNA fused to its cognate UBE2E2 eRNA. Cells were treated with E2 for the indicated time. Each point represents the mean ± standard error of the mean from four independent biological and technical replicates. The asterisks indicate significant differences from the corresponding control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01, ****p < 0.0001).
See also Figure S3.
We also determined whether eRNAs that are functional at their cognate enhancers can confer activity at a heterologous enhancer. When UBE2E2 or SEMA3C eRNA was fused to the PRRX2 sgRNA, the ability of the eRNA to enhance target gene expression was abolished (Figures 3E and S3C). This was also seen in the reciprocal experiment using the UBE2E2 sgRNA (Figures 3F and S3D), even when the heterologous eRNA was targeted to its enhancer of origin efficiently by the sgRNA (Figures S3E and S3F). Collectively, these results demonstrate that (1) some of the eRNAs tested act in cis at their enhancers of origin and modulate functional outcomes, such as shortening the timescale and/or increasing the magnitude of target gene expression upon E2 treatment and (2) the eRNAs tested are not interchangeable and may rely on locus-specific features for their activity.
eRNAs modulate enhancer formation and H3K27 acetylation
Results from previous studies have suggested that eRNAs can promote the recruitment of TFs and coregulators to enhancers (Bose et al., 2017; Lai et al., 2013; Rahnamoun et al., 2018; Sigova et al., 2015). To explore this possibility in the context of ERα enhancers, we performed ChIP-qPCR for ERα, steroid receptor coactivators (SRCs) (using a pan SRC antibody), p300, and H3K27 acetylation at the PRRX2, UBE2E2, and SEMA3C enhancers in MCF-7 cells using our targeted eRNA system (Figures 4A, 4B, and S4A). Although enhancer features were increased upon E2 treatment in all cases, directing the PRRX2 eRNA to its cognate enhancer increased the binding of ERα and SRCs 20 min after E2 treatment (Figure 4A), a time point at which a major coregulator transition occurs at ERα enhancers (Murakami et al., 2017). Interestingly, p300 recruitment was not affected by the targeted PRRX2 eRNA, but H3K27 acetylation was enriched, even in the absence of E2 treatment (Figure 4A). Directing the UBE2E2 eRNA to its cognate enhancer also enhanced ERα binding and H3K27ac enrichment, but at 45 min of E2 treatment (Figure 4B). The kinetics of enhancer activation vary from enhancer to enhancer (Hah et al., 2011, 2013; Murakami et al., 2017). Thus, it is not unexpected that different enhancers and eRNAs behave differently. These eRNA constructs had no discernable effect on enhancer assembly at the heterologous GREB1 ERα enhancer; GREB1 is an E2-regulated gene commonly used as a control to ensure proper E2 responses in breast cancer cells (Figures S4A and S4B).
Figure 4. Targeting an estrogen-regulated eRNA to its enhancer of origin modulates ERα and coregulator binding and stimulates p300 catalytic activity towards H3K27 at the enhancer.
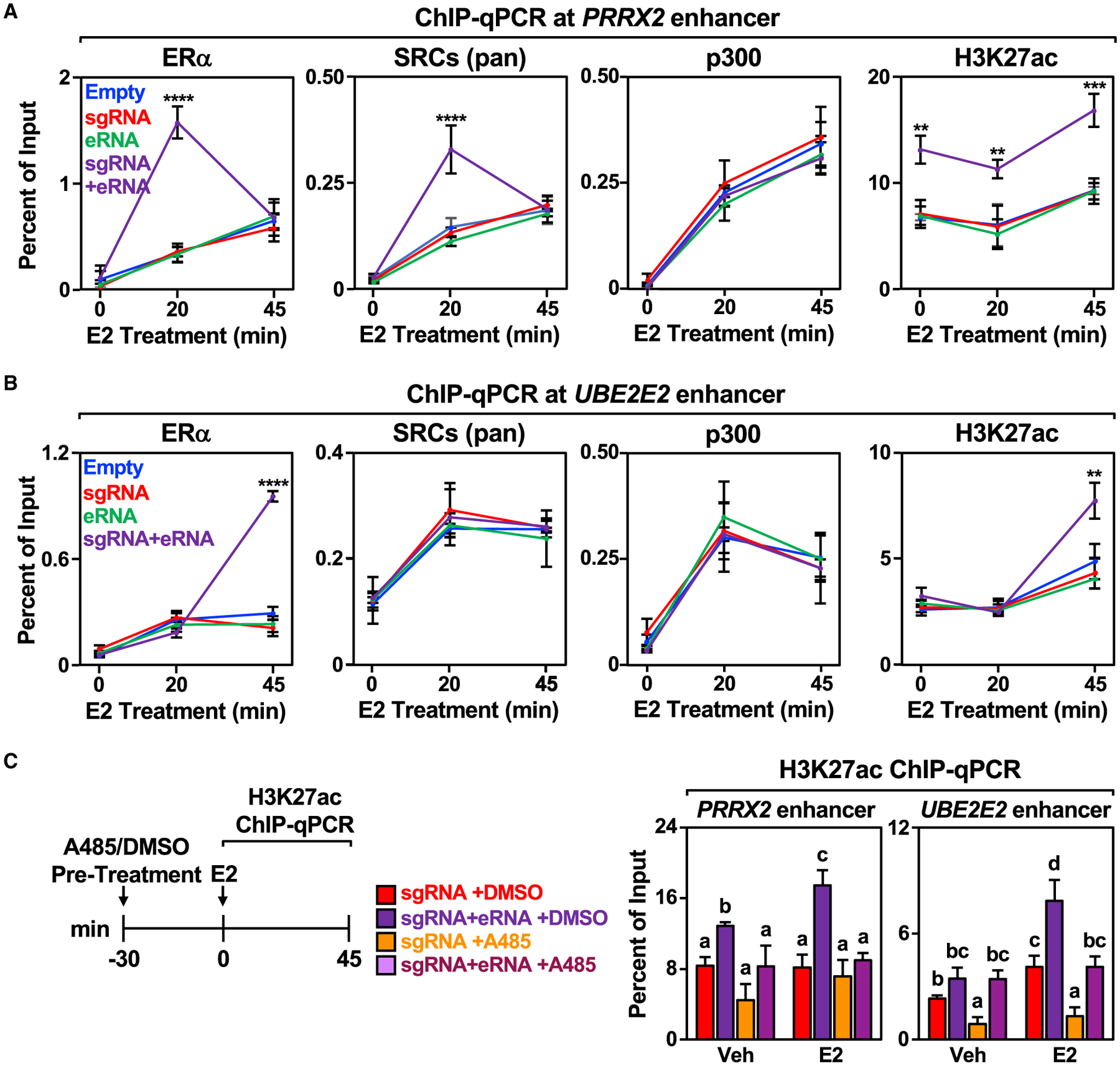
(A and B) ChIP-qPCR assays for ERα, pan SRCs), p300, and H3K27ac at the (A) PRRX2 and (B) UBE2E2 enhancers with dCas9-expressing MCF-7 cells in combination with various sgRNA/eRNA constructs. The cells were stimulated with E2 for the indicated time. Each point represents the mean ± standard error of the mean (n = 4 biological/technical replicates). The asterisks indicate significant differences from the empty control (two-way ANOVA, Fisher’s least significant difference post hoc test, **p < 0.01, ****p < 0.0001).
(C) MCF-7 cells were pre-treated with either DMSO or p300 histone acetyltransferase inhibitor A485 for 30 min before addition of DMSO (Veh) or E2 for the indicated time. Each bar represents the mean ± standard error of the mean (n = 4 biological/technical replicates). Bars marked with different letters are significantly different (two-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05).
See also Figure S4.
Enhanced H3K27 acetylation in the absence of enhanced p300 recruitment at the PRRX2 and UBE2E2 enhancers suggests that the PRRX2 and UBE2E2 eRNAs may stimulate p300 catalytic activity, as suggested previously (Bose et al., 2017). To explore this in greater detail, we first performed in vitro p300 histone acetyltransferase activity assays. We ran these assays under conditions chosen to eliminate potential pitfalls and artifacts described previously (Ortega et al., 2018). Incubation with PRRX2 eRNA or UBE2E2 eRNA increased p300-catalyzed H3K27 acetylation (Figure S4D). In addition, cells treated with the p300 inhibitor A485 before E2 treatment and then analyzed H3K27 acetylation by ChIP-qPCR. A485 inhibited showed inhibition of eRNA-stimulated H3K27 acetylation at both the PRRX2 and UBE2E2 enhancers (Figure 4C), as well as the GREB1 control enhancer (Figure S4E), as analyzed by ChIP-qPCR. Together, these results demonstrate that selected eRNAs, when recruited to their enhancer of origin, can increase the recruitment of ERα in response to E2 treatment and stimulate H3K27 acetylation in cells. These effects, however, occur in a locus-specific manner, suggesting additional locus-specific features for their activity.
Discovery of a FERM
Our genome-wide annotation of E2-regulated eRNAs allowed us to search for potential sequence motifs that may have a functional role. Using multiple em for motif elicitation (MEME), we found two sequences that are enriched within E2-regulated eRNAs (Figures 5A and S5A). A further analysis using find individual motif occurrences (FIMO) of the top hit, which we call the FERM element, identified 918 occurrences within 273 eRNAs (Figure 5B). The FERM element forms a predicted hairpin structure (Figures 5A and 5C). Additional analyses revealed an enrichment of the FERM element in intergenic transcript bodies in mouse cell lines and tissues (Figures S5B and S5C). An analysis of available GRO-seq datasets for human cell lines revealed an enrichment of the FERM element in intergenic enhancers, indicating that the FERM element is not exclusive to ERα enhancers (Figures S5B and S5C). Interestingly, the FERM element from ERα enhancers is found within Alu elements, as well as the related rodent B1 element, which also evolved from the 7SL RNA, but lacks the right monomer arm found in Alu elements (Dridi, 2012; Tsirigos and Rigoutsos, 2009) (Figure S5E). The lower frequency of FERM elements in mouse corresponds with the reduced fraction of B1 elements in the mouse genome (7%) compared with the fraction of Alu elements in the human genome (~10%) (Tsirigos and Rigoutsos, 2009). Thus, the FERM element could plausibly play a role in the regulation of enhancer function in non-primate species as well.
Figure 5. Identification of a FERM in E2-regulated eRNAs.
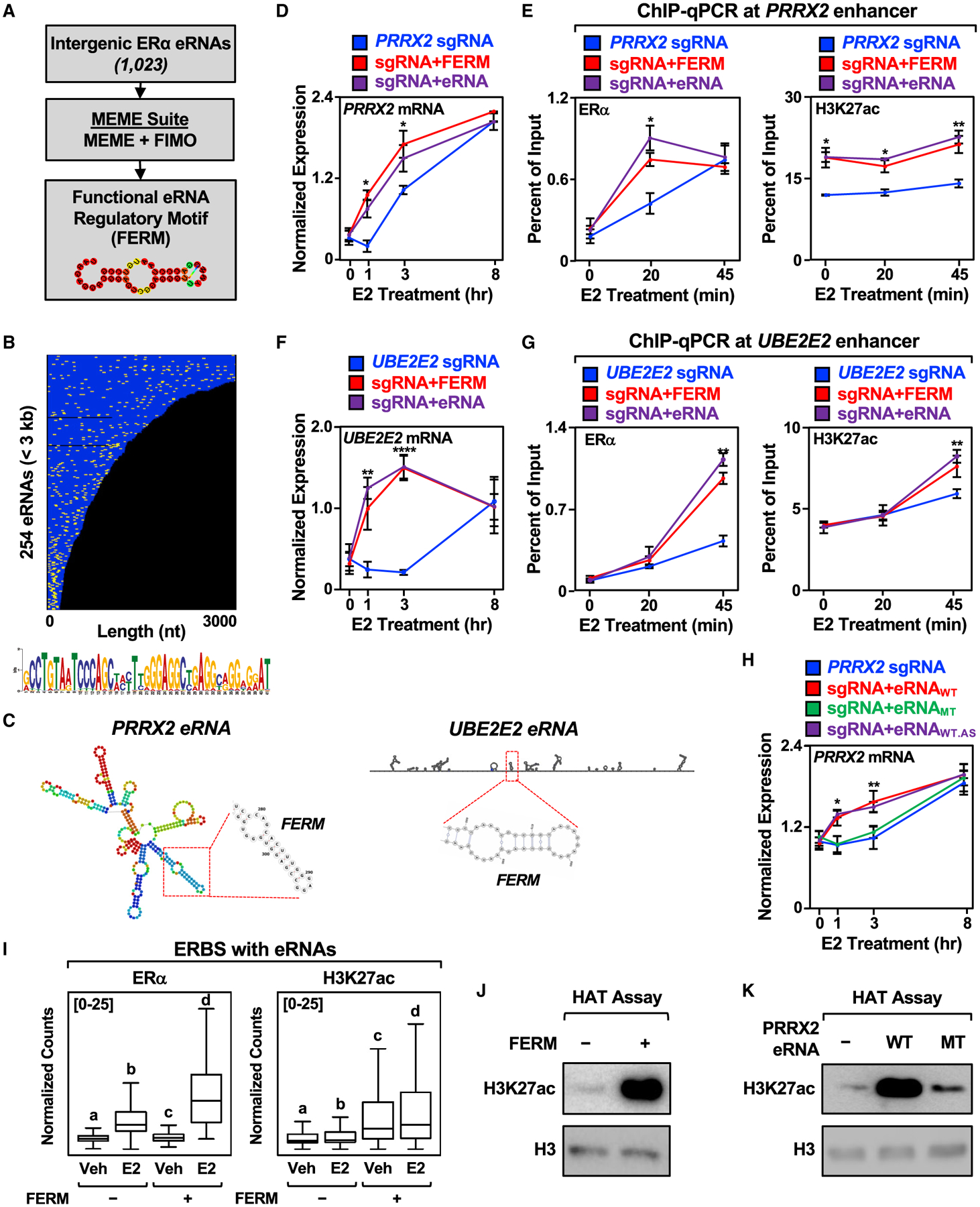
(A) Schematics of the pipeline for the discovery of FERM element within E2-regulated eRNAs using multiple em for motif elicitation (MEME) and find individual motif occurrences (FIMO) software.
(B) Graphical representation of the position of the FERM element (yellow) in the 254 E2-regulated eRNAs less than 3 kb that contain at least one FERM (273 eRNAs contain at least one FERM). Each eRNA is colored blue. The sequence for the FERM element is provided at the bottom.
(C) Predicted secondary structures of the PRRX2 eRNA (left) using RNAfold and the UBE2E2 eRNA (right) using Scanfold. The location of the FERM within the eRNA is marked with a red box.
(D) Targeting the FERM element to the PRRX2 enhancer is sufficient to modulate target gene expression. RT-qPCR analysis of PRRX2 mRNA expression levels normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with the PRRX2 sgRNA fused to the FERM element or the cognate PRRX2 eRNA. Each point represents the mean ± standard error of the mean (n = 4 biological/technical replicates). The asterisks indicate significant differences from the sgRNA control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01, ****p < 0.0001).
(E) ChIP-qPCR assays for ERα (left) and H3K27ac (right) at the PRRX2 enhancer with dCas9-expressing MCF-7 cells and various sgRNA/eRNA constructs. The cells were stimulated with E2 for the indicated time. Each point represents the mean ± standard error of the mean (n = 3 biological/technical replicates). The asterisks indicate significant differences from the corresponding control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01).
(F) Targeting the FERM element to the UBE2E2 enhancer is sufficient to modulate target gene expression. RT-qPCR analysis of the UBE2E2 mRNA expression level normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with UBE2E2 sgRNA fused to the FERM element or the cognate UBE2E2 eRNA. Each point represents the mean ± standard error of the mean (n = 4 biological/technical replicates). The asterisks indicate significant differences from the sgRNA control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01, ****p < 0.0001).
(G) ChIP-qPCR assays for ERα (left) and H3K27ac (right) at the UBE2E2 enhancer with dCas9-expressing MCF-7 cells and various sgRNA/eRNA constructs. The cells were stimulated with E2 for the indicated time. Each point represents the mean ± standard error of the mean (n = 3 biological/technical replicates). The asterisks indicate significant differences from the corresponding control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01).
(H) Mutation of the PRRX2 eRNA FERM abolishes the stimulatory effect of eRNA on target gene expression. RT-qPCR analysis of the PRRX2 mRNA expression level normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with PRRX2 sgRNA fused to wild-type, mutant, or antisense wild-type PRRX2 eRNA. The cells were stimulated with E2 for the indicated time. Each point represents the mean ± standard error of the mean (n = 3 biological/technical replicates). The asterisks indicate significant differences from the corresponding control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, **p < 0.01).
(I) Boxplot analysis of ERα (left), and H3K27ac ChIP-seq (right) of ERBS containing eRNAs with or without FERM (±4 kb). Reads were normalized to the read depth of the libraries used. The boxes represent the 25th–75th percentile (band at median), with whiskers at 1.5 × interquartile range. Bars marked with different letters are significantly different—n(without FERM) = 940, n(with FERM) = 339 (one-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05).
(J and K) In vitro histone acetyltransferase (HAT) assay using recombinant histone H3.1/H4 tetramer, purified p300, and (J) chemically synthesized FERM or (K) in vitro transcribed wild-type or mutant PRRX2 eRNA. The level of H3K27ac normalized to H3 was detected by Western blot. Each experiment was performed three independent times.
See also Figure S5.
The FERM element is found in both the PRRX2 and UBE2E2 eRNAs (Figure 5C). To determine if the FERM element is sufficient to preserve eRNA function, we fused two copies of the FERM element to the PRRX2 sgRNA, which enhanced target gene expression upon E2 treatment (Figure 5D). Similar to the full-length PRRX2 eRNA, targeting the FERM element to the enhancer stimulated ERα binding and H3K27 acetylation upon E2 treatment (Figure 5E). We observed similar results in enhancing target gene expression, ERα binding, and H3K27 acetylation when we fused two copies of the FERM element to the UBE2E2 sgRNA (Figures 5F and 5G). Interestingly, fusing the FERM elements to the SEMA3C sgRNA, which does not contain the FERM, also enhanced target gene expression (Figure S5F) by promoting ERα binding and H3K27 acetylation (Figure S5G).
To further explore the role of the FERM element in regulating enhancer function and target gene expression, we deleted the FERM from both the PRRX2 and UBE2E2 eRNAs and targeted them back to their cognate enhancers. Deletion of the FERM elements abolished the stimulatory effects of the PRRX2 and UBE2E2 eRNAs on target gene expression (Figures S5H and S5I). Next, we mutated the primary sequence of the FERM element in the PRRX2 eRNA to abolish the secondary (i.e., predicted hairpin) structure of the eRNA (PRRX2 MT, Figure S5J). Additionally, we constructed a PRRX2 eRNA containing the antisense sequence of the FERM, which is predicted to maintain the same secondary structure as wild-type eRNA (PRRX2 WT.AS, Figure S5J). In the targeted eRNA recruitment assay, disruption of the FERM element within the PRRX2 eRNA by mutation abolished the stimulatory effect of the eRNA on target gene expression (Figure 5H). In contrast, the PRRX2 eRNA with an antisense FERM element that preserves the secondary structure maintained the stimulatory effect of the PRRX2 eRNA on target gene expression (Figure 5H).
The effects of the FERM element were also observed at the PRRX2 enhancer, where mutation of the FERM element in the PRRX2 eRNA abolished its stimulatory effect on ERα binding and H3K27 acetylation, while the PRRX2 eRNA with an antisense FERM element maintained its stimulatory effects (Figure S5K). On the genomic level, ERα binding sites that produce eRNAs containing the FERM element exhibit increased ERα binding upon E2 stimulation, as well as increased H3K27 acetylation, even at the basal level (Figure 5I). These results support the prevailing observations from our locus-specific assays. The enhanced activity of FERM-containing eRNAs may be due to increased eRNA stability, as eRNAs containing the FERM element form more stable local secondary structures (Figure S5L).
The FERM element stimulated p300 histone acetyltransferase activity in an in vitro histone acetyltransferase assay (Figure 5J and S5M). Mutation of the FERM element in the PRRX2 eRNA decreased the stimulatory effect of the eRNA on p300 histone acetyltransferase activity, which was restored in a PRRX2 eRNA containing an antisense FERM element (Figure 5K and S5N and S5O). In summary, our genome-wide annotation of E2-regulated eRNAs uncovered the FERM element, which is capable of stimulating target gene expression by modulating the recruitment of ERα to its enhancer and stimulating p300-catalyzed H3K27 acetylation in locus-specific assays. While the FERM elements in tethered eRNAs may act to enhance enhancer activity, they do not contribute the enhancer specificity of eRNAs that we observed.
BCAS2 interacts with the FERM element to modulate the stimulatory effect of eRNAs
To determine the eRNA-interacting proteome, including proteins that specifically interact with the FERM element, we conducted RNA-protein pulldown coupled with mass spectrometry using the PRRX2 WT eRNA (containing the FERM), PRRX2 MT eRNA (lacking the FERM), the FERM element alone, or GFP RNA as a negative control. We identified 88 proteins enriched in the PRRX2 WT eRNA pulldown versus the MT eRNA pulldown that also overlapped with the proteins identified in the FERM element pulldown (Table S1; Figures S6A and S6B). Gene ontology (GO) analyses using the DAVID tool (Huang da et al., 2009) identified enriched terms for these 88 proteins related to splicing and gene expression (Figure S6C). One of these proteins, breast carcinoma-amplified sequence 2 (BCAS2), also known as the premRNA splicing factor SPF27, is an RNA-binding component of the activated spliceosome (Bertram et al., 2017; Zhang et al., 2017). BCAS2 has also been shown to interact with ERα and function as a coregulator in ERα-mediated transcription (Qi et al., 2005; Salmeron-Hernandez et al., 2019). We validated the interaction between BCAS2 and the FERM element by RNA-protein pull-down followed by western blotting (Figure 6A) and by electrophoretic mobility shift assay with purified BCAS2 (Figures 6B and S6D).
Figure 6. Identification of FERM-interacting protein BCAS2 and its effect on eRNA-mediated enhancer assembly and target gene expression.
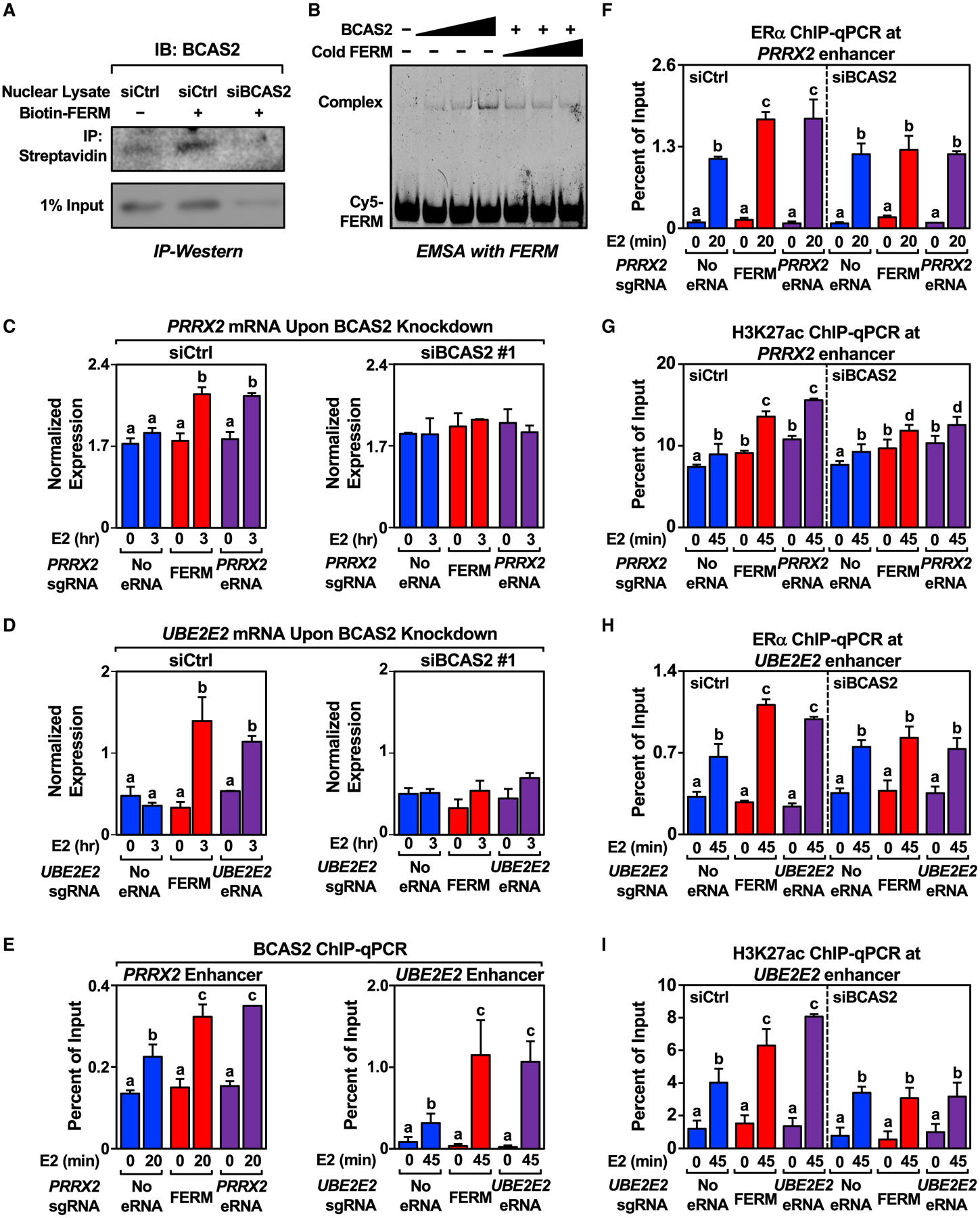
(A) In vitro pull-down of BCAS2 using biotinylated FERM incubated with nuclear lysate extracted from control or BCAS2 knockdown MCF-7 cells. This experiment was performed three independent times.
(B) Electrophoresis mobility shift assay carried out with increasing amount of purified His-BCAS2 mixed with Cy5-labeled or cold FERM. This experiment was performed three independent times.
(C and D) Knockdown of BCAS2 abolishes the stimulatory effect of the FERM element and eRNA on (C) PRRX2 and (D) UBE2E2 eRNA on target gene expression levels. RT-qPCR analysis of the mRNA levels normalized to GAPDH mRNA levels in dCas9-expressing MCF-7 cells with various PRRX2 sgRNA/eRNA constructs upon control or BCAS2 knockdown. Cells were treated with E2 for the indicated time. Each bar represents the mean ± standard error of the mean (n = 3 biological/technical replicates). Bars marked with different letters are significantly different (two-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05).
(E) ChIP-qPCR assays for BCAS2 at the PRRX2 (left) and UBE2E2 (right) enhancers in dCas9-expressing MCF-7 cells expressing the sgRNA alone, sgRNA fused to the FERM element, or the sgRNA fused to the cognate eRNA. The cells were stimulated with E2 for the indicated time. Each bar represents the mean ± standard error of the mean (n = 3 biological/technical replicates). Bars marked with different letters are significantly different (two-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05).
(F and G) Knockdown of BCAS2 abolishes the stimulatory effect of FERM and PRRX2 eRNA on ERα recruitment and H3K27 acetylation. ChIP-qPCR assays for (F) ERα and (G) H3K27ac at the PRRX2 enhancer in dCas9-expressing MCF-7 cells with various PRRX2 sgRNA constructs upon control or BCAS2 knockdown. Each bar represents the mean ± standard error of the mean (n = 3 biological/technical replicates). Bars marked with different letters are significantly different (two-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05).
(H and I) Knockdown of BCAS2 abolishes the stimulatory effect of the FERM element and UBE2E2 eRNA on ERα recruitment and H3K27 acetylation. ChIP-qPCR assays for (H) ERα and (I) H3K27ac at the UBE2E2 enhancer in dCas9-expressing MCF-7 cells with various UBE2E2 sgRNA constructs upon control or BCAS2 knockdown. Each bar represents the mean ± standard error of the mean (n = 3 biological/technical replicates). Bars marked with different letters are significantly different (two-way ANOVA, Fisher’s least significant difference post hoc test, p < 0.05).
Next, we studied the functional relationship between the FERM element and BCAS2. siRNA-mediated knockdown of BCAS2 achieved depletion at both the mRNA and protein levels (Figures S6E and S6F). BCAS2 depletion inhibited PRRX2 eRNA- and FERM-mediated increases in target gene expression (Figures 6C and S6G), while having no effect on E2-regulated GREB1 gene expression (Figure S6H). Similar results were observed for the UBE2E2 eRNA (Figure 6D). BCAS2 was found to localize at both PRRX2 and UBE2E2 enhancer as determined by BCAS2 ChIP-qPCR (Figure 6E). BCAS2 depletion also inhibited PRRX2 eRNA- and FERM-mediated increases in ERα binding at the enhancer (Figure 6F) and H3K27ac enrichment (Figure 6G) upon E2 treatment. siRNA-mediated depletion of BCAS2 also significantly decreased the stimulatory effect of targeted UBE2E2 eRNA on ERα binding and H3K27ac enrichment upon E2 treatment, although the effects were modest (Figures 6H and 6I), perhaps owing to examining the effects on ERα binding at one time point when ERα enhancer kinetics are diverse (Hah et al., 2011, 2013; Murakami et al., 2017). Overall, we identified BCAS2 as an eRNA- and FERM-binding protein that plays a role in the eRNA- and E2-mediated stimulation of target gene expression, ERα binding at the enhancer, and eRNA- and FERM-mediated H3K27 acetylation.
Directing the PRRX2 eRNA to an oncogenic enhancer stimulates cell proliferation
We used our genome-wide annotation of estrogen-regulated eRNAs in breast cancer cells to determine if polyA-depleted eRNAs are present in published breast cancer databases (Zhao et al., 2014) (Figure S7A). We detected 97% of eRNAs (990 of 1023) annotated in MCF-7 cells that are also expressed in at least one breast cancer tumor sample. Since enhancer transcription is highly associated with the expression of the nearest neighboring gene, we examined eRNA/mRNA pairs in the breast cancer eRNA set and determined the Pearson correlation for the pairs. We identified 253 eRNA/mRNA pairs that had a Pearson coefficient of greater than 0.5, indicative of a strong positive correlation (Figure S7B). GO analyses revealed an enrichment of genes associated with cell growth, cell death, and signaling transduction (Figure S7C).
Using the set of breast cancer eRNA/mRNA pairs, we observed a positive correlation between PRRX2 eRNA and mRNA expression levels across nine samples (Figure 7A). Importantly, PRRX2 encodes Paired Related Homeobox 2, a TF linked to breast cancer (Lv et al., 2017). Since high expression of PRRX2 leads to poorer relapse-free survival for patients with ER+ breast cancer (Figure S7D), we asked whether directing the PRRX2 eRNA to the PRRX2 enhancer influences cell proliferation. Indeed, targeted PRRX2 eRNA increased cell proliferation (Figure 7B), while the depletion of BCAS2 abolished the effect of the targeted PRRX2 eRNA on cell proliferation (Figure 7C). This is reminiscent of how low expression of PRRX2 leads to better relapse-free survival for patients with ER+ breast cancer (Figure S7D). Interestingly, BCAS2 is not predictive of relapse-free survival in patients with ER-negative breast cancer (Figure S7E), suggesting a more specific role for BCAS2 in ER+ breast cancer biology.
Figure 7. Biological responses of eRNAs in clinical samples and cell growth.
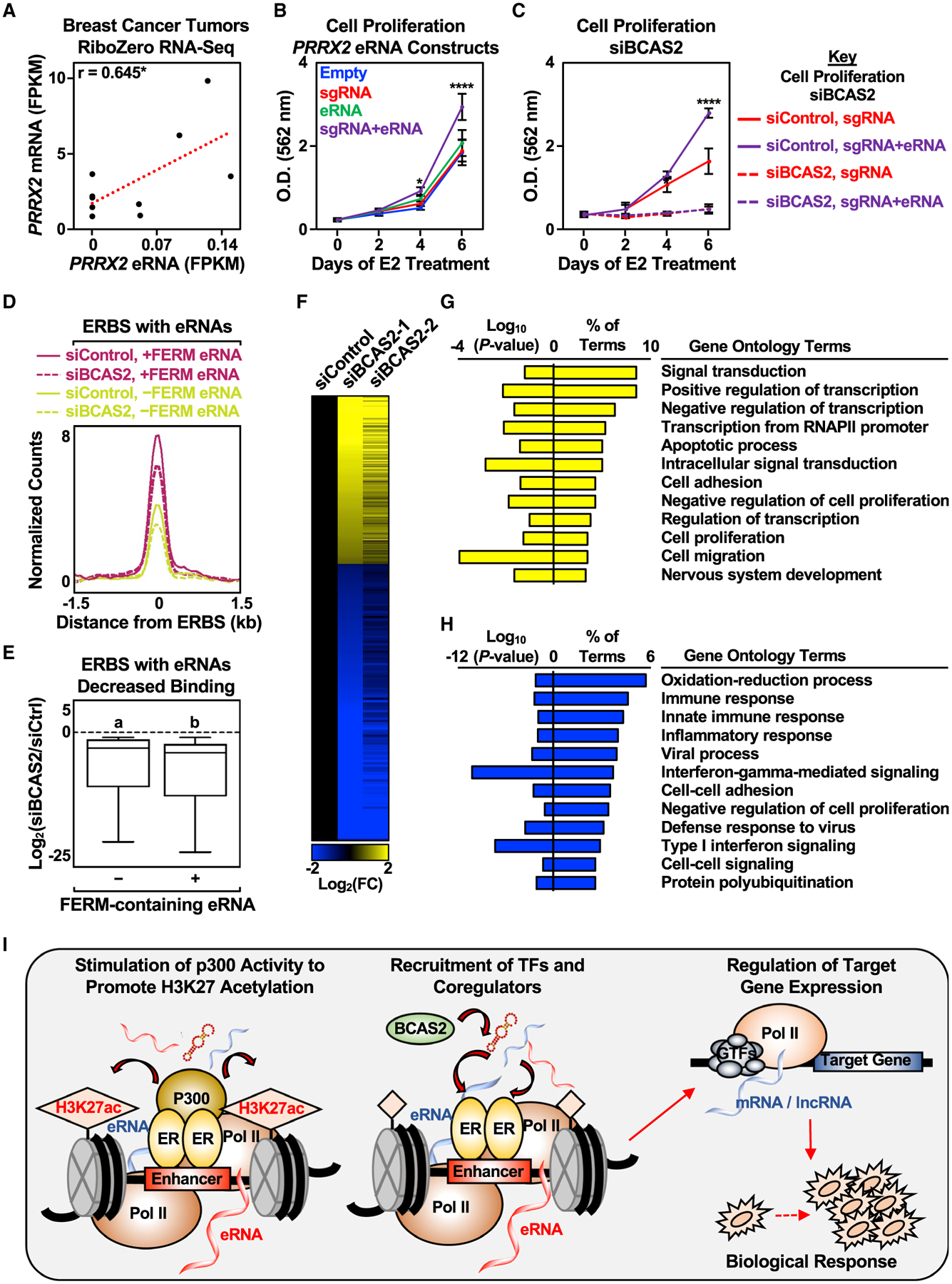
(A) Pearson’s correlation between PRRX2 mRNA and eRNA levels in nine breast cancer tumors was determined (two-tailed p value, *p < 0.05).
(B) Stimulatory effect of targeted PRRX2 eRNA on cell proliferation. Cell proliferation assay of dCas9-expressing MCF-7 cells with various PRRX2 sgRNA constructs. See (C) for a description of the statistics. Each point represents the mean ± standard error of the mean (n = 3 biological/technical replicates). The asterisks indicate significant differences from the corresponding empty control (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, ****p < 0.0001).
(C) Depletion of BCAS2 abolishes the stimulatory effect of targeted PRRX2 eRNA on cell proliferation. Cell proliferation assay of dCas9-expressing MCF-7 cells with PRRX2 sgRNA constructs upon control or BCAS2 knockdown. Each point represents the mean ± standard error of the mean (n = 3 biological/technical replicates). The asterisks indicate significant differences from the corresponding control (siControl, sgRNA at the specific time point) (two-way ANOVA, Fisher’s least significant difference post hoc test, *p < 0.05, ****p < 0.0001).
(D) Metagene representations from ERα ChIP-seq upon BCAS2 depletion between ERα binding sites (ERBS) containing eRNAs with or without FERM. The metagene plots represent the average read counts normalized to their respective input controls around the ERBS (±1.5 kb) in each group—n(without FERM) = 940, n(with FERM) = 339. The genomic assays were performed as two biological and two technical replicates.
(E) Boxplots of depleted ERα peaks upon BCAS2 depletion at ERα binding sites (ERBS) containing eRNAs with or without FERM (±1.5 kb). A cut-off of 2-fold decrease between siBCAS2/siCtrl at 45 min and at 0 min was used to define depleted ERα peaks. The boxes represent the 25th–75th percentile (band at median), with whiskers at 1.5 × interquartile range. Bars marked with different letters are significantly different—n(without FERM) = 207, n(with FERM) = 70 (two-tailed Mann-Whitney test, p = 0.0164). The genomic assays were performed as two biological and two technical replicates.
(F) Depletion of BCAS2 alters E2-regulated genes in MCF-7 cells. Heatmap of RNA-seq data representing changes in the expression of E2-regulated genes from MCF-7 cells treated with control or BCAS2 knockdowns for 48 h before E2 stimulation for 6 h. In total, 1,075 genes were differentially expressed between control and BCAS2 knockdown upon 6 h E2 treatment—n(upregulated) = 403, n(downregulated) = 672. The genomic assays were performed as two biological and two technical replicates.
(G and H) GO analysis of (G) upregulated and (H) downregulated genes upon BCAS2 knockdown in MCF-7 cells treated with E2 for 6 h.
(I) A model for estrogen-regulated eRNAs in enhancer assembly and gene expression. Details are provided in the text.
See also Figure S7.
Since knockdown of BCAS2 decreased E2-dependent cell proliferation in MCF-7 cells, we examined the effect of BCAS2 knockdown on ERα binding and subsequent gene expression. Knockdown of BCAS2 decreased ERα binding genome-wide, with modestly but significantly greater effects at binding sites that produce eRNAs containing the FERM element (Figures 7D and 7E). BCAS2 knockdown also altered the patterns of E2-regulated gene expression upon 6 h of E2 treatment in MCF-7 cells, with more genes showing decreased expression than increased expression (Figures 7F–7H). These results suggest a role for BCAS2 in regulating ERα enhancer formation and subsequent gene expression, as well as a role for eRNAs in driving biological responses, including gene expression and cell proliferation in breast cancer (Figures 7I, S7F, and S7G).
DISCUSSION
To facilitate the study of eRNAs in enhancer formation and target gene expression using a gain-of-function approach, we annotated E2-regulated eRNAs in MCF-7 breast cancer cells. Moreover, we cloned selected eRNAs and explored their molecular and cellular functions in a variety of assays. Collectively, our results describe some of the molecular mechanisms underlying the regulatory potential of eRNAs.
Annotation and gain-of-function experiments with estrogen-regulated eRNAs
We found that three of the eRNAs that we tested modulated target gene expression in a temporal and specific manner when examined using a CRISPR/dCas9-based gain-of-function enhancer targeting approach (Figure 3 and S3). Previous studies targeting eRNAs to a luciferase construct (Shechner et al., 2015) or to the natural Fos enhancer in neuronal cells (Carullo et al., 2020) have also shown that eRNAs can promote target gene expression in cis. There are, however, eRNAs that have been shown to function in trans (Stone et al., 2021; Tsai et al., 2018; Zhao et al., 2019), but these eRNAs are polyadenylated, most likely conferring greater stability and allowing enhanced function, perhaps as long non-coding RNAs. An interesting difference between these previous studies and our current results is that we observed a requirement for E2 treatment for the effects of eRNAs, except for increased H3K27ac (Figures 4A and 5I).
Our results demonstrate that eRNAs targeted to their enhancers of origin require E2 to function (Figures 3, 4, S3, and S4). While a priori one might expect targeted eRNAs to promote enhancer formation and target gene expression constitutively in the absence of E2, this is not the case. However, from an order of operations perspective, this is unsurprising. E2 stimulates ERα binding to chromatin, and liganded DNA-bound ERα recruits coregulators and RNAPII to initiate eRNA production. Thus, the context for eRNA function is not an empty enhancer, but rather a liganded ERα-bound enhancer. In this model, the initial accumulation of eRNA would be expected to reinforce ERα binding and coregulator recruitment in a virtuous cycle.
eRNAs recruit TFs and stimulate coregulator activities
We observed that some of the eRNAs tested promote the recruitment of ERα to enhancers (PRRX2 and UBE2E2 eRNAs) and stimulate p300 activity to increase H3K27 acetylation (PRRX2, UBE2E2, and SEMA3C eRNAs) (Figure 4). The ability of eRNAs to recruit TFs to enhancers is consistent with a previous study (Sigova et al., 2015), although we found a temporal aspect to the stimulatory effect of eRNAs on ERα recruitment. Recently, the histone acetyltransferase CBP/p300 has been shown to bind to eRNAs through its histone acetyltransferase domain to stimulate its catalytic activity (Bose et al., 2017), although others have failed to observe similar effects (Ortega et al., 2018). The presence of EDTA in acetyltransferase reactions was proposed to drive artefactual activation of p300 (Ortega et al., 2018); therefore, we omitted EDTA from our reactions and still observed that eRNAs stimulated p300 histone acetyltransferase activity (Figures 5J, 5K, S4D, and S5O).
We speculate that the presence of an eRNA tethered at the enhancer in the absence of E2 is sufficient to stimulate the activity of low levels of pre-bound p300 at some enhancers (Figures 4A and 5I), but is insufficient to drive the binding of ERα and subsequent recruitment of SRC (owing to the order of operations noted above). Experimentally, treatment with A485 decreased the level of H3K27ac even without E2 stimulation (Figure 4C and S4E), indicating that there is basal p300 catalytic activity at some enhancers. This is consistent with the observation of increased H3K27ac levels at ERα enhancers sites with FERM-containing eRNAs across the genome (Figure 5I). In this regard, we observed that the FERM element drives the formation of secondary structures within the eRNAs and is one feature of eRNAs that stimulates p300 histone acetyltransferase activity (Figures 5 and S5L).
Identification of the FERM element in eRNAs
Our genome-wide annotation allowed us to search for potential motifs within E2-regulated eRNAs, leading to the discovery of the FERM element that is part of the Alu and B1 elements (Figure S5E), which are sufficient to drive E2-regulated target gene expression (Figure 5). Many TF binding sites, including those for ERα, are located in Alu and related elements (Dridi, 2012; Polak and Domany, 2006; Su et al., 2014). Importantly, the nuclear receptor hormone response element (AGGTCA) is not located in the FERM element, suggesting that nature has explored the sequence space of Alu and B1 elements to develop independent regulatory mechanisms (i.e., TF binding sites and the FERM element).
The FERM element binds to BCAS2, a protein that supports the stimulatory effects of eRNAs (Figure 6). A model of eRNA function must account for enhancer specificity, as well as general molecular mechanisms that apply to many enhancers. Our studies indicate that the FERM element stimulates p300 catalytic activity and mediates interactions with BCAS2, functions that could be potentially regulated at many enhancers across the genome (Figure S7F). The specificity of the eRNAs for their cognate enhancers, however, suggests that the non-FERM portions of the eRNA act to reign in the activity of the FERM element and direct it to specific enhancers (Figure S7F). In fact, having the FERM element in the context of a full-length eRNA could change the structure of the FERM element or hold it in an inactive conformation until it binds to interacting proteins. This idea of inducible FERM activity is one that will be explored in future studies.
Oncogenic eRNAs in breast cancer biology
There is an increasing interest in characterizing eRNAs in cancers with respect to their landscape of expression, associated clinical phenotypes, and therapeutic potential (Chen and Liang, 2020; Zhang et al., 2019, 2021). Our data demonstrate how an eRNA can act at an oncogenic enhancer to enhance oncogenic gene expression in breast cancer cells to increase cell proliferation. This signal can be amplified further if the eRNA regulates the expression of an oncogenic TF, such as PRRX2 (Figures 7I and S7G). Furthermore, if there are multiple eRNAs overexpressed in breast cancer, as detected in patient samples (Figure S7), this may lead to an enhanced breast cancer gene expression profile.
The cloning and study of multiple E2-regulated eRNAs revealed insights into enhancer formation (Figure 4) and target gene regulation (Figures 3 and S3). Future studies will characterize E2-regulated eRNAs on a larger scale to delineate other molecular and sequence determinants that allow eRNAs to modulate target gene expression and regulate enhancer formation and function. This will allow elucidation of the range of mechanisms by which these E2-regulated eRNAs influence target gene expression, how eRNA-interacting proteins participate in ERα enhancer formation, and, ultimately, the therapeutic potential of targeting eRNAs in breast cancer.
Limitations of the study
For these studies, we initially screened 13 different eRNAs for their ability to enhance the E2-dependent expression of their nearest-neighboring gene using 4 different sgRNAs per eRNA. From this screen, we identified three eRNAs that were effective (from the PRRX2, UBE2E2, and SEMA3C enhancers). At present, it is unclear if only a fraction of eRNAs is functional, or whether we missed some key biological component in our screen. This will require further testing with a greater number of eRNAs and target genes in the future, perhaps using a high throughput screening method with single-cell RNA-seq. We defined the eRNAs based on data from our PRO-cap and RNA-seq experiments. It will be important in the future to validate our mapping strategy using 5′ and 3′ rapid amplification of cDNA ends, although the abundance and stability of eRNAs may make this endeavor challenging.
Although our results show effects of eRNAs and the FERM element on enhancer formation and the expression of their target genes, we did not observe a single unifying effect across all ERα enhancers. Clearly, there are rules that remain to be elucidated, and the generalizability of the mechanisms observed in this study will need to be tested with additional eRNAs. We opted for an addition approach (i.e., sgRNA-fused eRNAs), which we think is more direct than the subtraction approach (i.e., knockdown of eRNAs) that has been used in the past. However, our approach is not without limitations. In future studies, we will need to find ways to effectively modify the FERM element in situ using genetic approaches. In closing, although we have not yet elucidated the complete set of distinct eRNA-dependent mechanisms operating at ERα enhancers, our results establish the functionality of eRNAs and the FERM element, as well as provide clues to the distinct mechanisms driving the different molecular features of the enhancers that are influenced by eRNA.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, W. Lee Kraus, Ph.D (Lee.Kraus@utsouthwestern.edu).
Materials availability
All cell lines and DNA constructs are available by request from W. Lee Kraus, Ph.D.
Data and code availability
Data: The PRO-cap and RNA-seq datasets generated specifically for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/). The new mass spectrometry datasets generated for this study can be accessed from the Spectrometry Interactive Virtual Environment (MassIVE) repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp). Accession numbers are listed in the Key Resources Table.
Code: This paper does not report original code.
Additional information: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ERα | Kraus and Kadonaga (1998) | N/A |
| Rabbit polyclonal anti-ERα | Millipore Sigma | Cat. No. 06-935 RRID: AB_310305 |
| Rabbit polyclonal anti-panSRC | Acevedo et al. (2004) | N/A |
| Rabbit polyclonal anti-ß-tubulin | Abcam | Cat. No. MABE1031 RRID: AB_2210370 |
| Rabbit polyclonal anti-SNRNP70 | Abcam | Cat. No.: ab83306 RRID: AB_10673827 |
| Mouse monoclonal anti-Flag | Sigma-Aldrich | Cat. No.: F3165 RRID: AB_259529 |
| Rabbit polyclonal anti-Cas9 | Diagenode | Cat. No.: C15310258 RRID: AB_2715516 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat. No.: ab4729 RRID: AB_2118291 |
| Mouse monoclonal anti-p300 | Active Motif | Cat. No.: 61401 RRID: AB_2716754 |
| Rabbit polyclonal anti-H3 | Abcam | Cat. No.: ab1791 RRID: AB_302613 |
| Rabbit polyclonal anti-BCAS2 | Bethyl | Cat. No.: A300-915A RRID:AB_661883 |
| Mouse monoclonal anti-BCAS2 | Santa Cruz | Cat. No.: sc-365346 RRID: AB_10846085 |
| Goat anti-rabbit HRP-conjugated IgG | ThermoFisher | Cat. No. 31460 RRID: AB_228341 |
| Goat anti-mouse HRP-conjugated IgG | ThermoFisher | Cat. No. 31430 RRID: AB_10960845 |
| Rabbit IgG | ThermoFisher | Cat. No. 10500C RRID: AB_2532981 |
| Chemicals, peptides, and recombinant proteins | ||
| p300 | Gupte et al. (2021) | N/A |
| 6xHis-BCAS2 | This study | N/A |
| Human H3.1/H4 tetramer | NEB | Cat. No.: M2509 |
| 17β-estradiol | Sigma | Cat. No.: E8875 |
| Doxycycline | Sigma | Cat. No.: D9891 |
| A485 | Tocaris | Cat. No.: 6387 |
| Critical commercial assays | ||
| RNA ScreenTape | Agilent Technologies | Cat. No.: 5067-5576, 5067-5577 |
| DNA ScreenTape | Agilent Technologies | Cat. No.: 5067-5584, 5067-5585 |
| Deposited data | ||
| MCF-7 ±E2, ±TAP PRO-cap | This study | GEO: GSE175469 |
| MCF-7 E2, polyA-depleted RNA-seq | This study | GEO: GSE175469 |
| MCF-7 E2, polyA-enriched RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siCtrl, ±E2 RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #1, ±E2 RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #2, ±E2 RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siCtrl, ±E2, ERα ChIP-Seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #1, ±E2, ERα ChIP-seq | This study | GEO: GSE175469 |
| MCF-7, siCtrl, ±E2, Input ChIP-Seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #1, ±E2, Input ChIP-seq | This study | GEO: GSE175469 |
| PRRX2 WT eRNA Proteomics | This study | MassIVE: MSV000087492 |
| PRRX2 MT eRNA Proteomics | This study | MassIVE: MSV000087492 |
| PRRX2 GFP RNA Proteomics | This study | MassIVE: MSV000087492 |
| FERM Proteomics | This study | MassIVE: MSV000087492 |
| Experimental models: Cell lines | ||
| 293T | ATCC | Cat. No.: CRL-3216 RRID: RRID:CVCL_0063 |
| MCF-7 | Dr. Benita Katzenellenbogen, University of Illinois, Urbana-Champaign | |
| Oligonucleotides | ||
| Primers for molecular cloning | See Table S2 | N/A |
| Recombinant DNA | ||
| pLenti-dCas9-Flag-Blast | This study | N/A |
| pINDUCER20 | Addgene | Cat. No.: 44012 |
| pIND20-sgRNA-eRNA-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-UBE2E2(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-UBE2E2(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-PRRX2(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-P2RY2(sgRNA)-bGHpA | This study | N/A |
| pIND20-P2RY2(eRNA)-bGHpA | This study | N/A |
| pIND20-P2RY2(sgRNA)-P2RY2(eRNA)-bGHpA | This study | N/A |
| pIND20-SEMA3C(sgRNA)-bGHpA | This study | N/A |
| pIND20-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-SEMA3C(sgRNA)-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-FERM-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-FERM-bGHpA | This study | N/A |
| pIND20-SEMA3C(sgRNA)-FERM-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(ΔFERM eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-UBE2E2(ΔFERM eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(MT eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(WT.AS eRNA)-bGHpA | This study | N/A |
| pcDNA3.1(+)-PRRX2(WT)eRNA | This study | N/A |
| pcDNA3.1(+)-PRRX2(MT)eRNA | This study | N/A |
| pcDNA3.1(+)-PRRX2(WT.AS)eRNA | This study | N/A |
| pcDNA3.1(+)-UBE2E2(eRNA) | This study | N/A |
| pcDNA3.1(+)-SEMA3C(eRNA) | This study | N/A |
| pcDNA3.1(+)-GFP | This study | N/A |
| pET19b-6xHis-BCAS2 | This study | N/A |
| pCMV-VSV-G | Addgene | Cat. No.: 8454 |
| psPAX2 | Addgene | Cat. No.: 12260 |
| PAdVAntage | Promega | Cat. No.: TB207 |
| Software and algorithms | ||
| FastQC | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Star | Dobin et al. (2013) |
https://github.com/alexdobin/ STAR |
| BWA | Li and Durbin (2009) | http://bio-bwa.sourceforge.net/ |
| StringTie | Pertea et al. (2016) | https://ccb.jhu.edu/software/stringtie/ |
| Cappable-seq | Ettwiller et al. (2016) | https://github.com/Ettwiller/TSS |
| BEDtools | Quinlan and Hall, (2010) | https://bedtools.readthedocs.io/en/latest/ |
| SAMtools | Li et al. (2009) | http://www.htslib.org/ |
| BEDops | Neph et al. (2012) | https://bedops.readthedocs.io/ |
| Deeptools | Ramirez et al. (2016) | https://deeptools.readthedocs.io/en/develop/ |
| SparK | Kurtenbach and Harbour (2019) | https://github.com/harbourlab/SparK |
| Subread | Liao et al., (2013) | http://subread.sourceforge.net/ |
| DESeq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MEME Suite | Bailey et al. (2009) | https://meme-suite.org/meme/index.html |
| DAVID Bioinformatics Resources | Huang da et al. (2009) | https://david.ncifcrf.gov/ |
| RNAfold | Lorenz et al., (2011) | http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi |
| Scanfold | Andrews et al. (2018) | https://github.com/moss-lab/ScanFold |
| Proteome Discoverer | ThermoFisher | Cat. No.: OPTON-30812 |
| msmsTests | Gregori et al. (2020) | https://www.bioconductor.org/packages/release/bioc/html/msmsTests.html |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
MCF-7 cells were kindly provided by Dr. Benita Katzenellenbogen (University of Illinois at Urbana-Champaign, IL) and maintained in minimal essential medium (MEM; Sigma M1018) supplemented with 5% calf serum (Sigma, C8056), 100 units/mL penicillin-streptomycin (Gibco, 15140122), and 25 μg/mL gentamicin (Gibco, 1571004). Seventy-two hours before E2 treatment, the cells were cultured in Eagle’s phenol red-free minimal essential medium (Sigma, M3024) supplemented with 5% charcoal-dextran treated calf serum (Sigma, C8056), 2 mM GlutaMAX (Gibco, 35050061), 100 units/mL penicillin-streptomycin (Gibco, 15140122), and 25 μg/mL gentamicin (Gibco, 1571004).
293T cells were obtained from the ATCC and maintained in Dulbecco’s modified Eagle medium high glucose (Sigma, D7777), supplemented with 10% FBS (Sigma, F2442) and 100 units/mL penicillin-streptomycin (Gibco, 15140122).
Fresh cell stocks were replenished within 10 passages from the original frozen stocks, verified for cell type identity using the GenePrint 24 system (Promega, B1870), and confirmed as mycoplasma-free every three months using a commercial testing kit.
Generation of cell lines
MCF-7 cell lines stably expressing dCas9 and sgRNA constructs were infected with lentiviruses generated in 293T cells. First, dCas9-Flag construct (derived from pLenti-dCas9-KRAB-Blast, Addgene, 89567), together with an expression vector for the VSV-G envelope protein (pCMV-VSV-G, Addgene plasmid no. 8454), an expression vector for GAG-Pol-Rev (psPAX2, Addgene plasmid no. 12260), and pAdVAntage (Promega, TB207) were transfected into 293T cells using GeneJuice transfection reagent (Novagen, 70967) according to the manufacturer’s protocol. The medium was changed 24 h after transfection, and the resulting viruses were used to infect MCF-7 cells in the presence of 7.5 μg/mL polybrene 48 h and 72 h after initial 293T transfection. Stably transduced dCas9-expresisng MCF-7 cells were selected with blasticidin (InvivoGen, ant-bl; 20 μg/mL). To generate the various sgRNA-, eRNA-, and sgRNA fused to eRNA-expressing cells, dCas9-expressing MCF-7 cells were infected with the constructs and selected with geneticin (Gibco, 11811031; 2 mg/mL). The viruses were titrated to ensure that the constructs for sgRNAs and sgRNAs fused to different RNAs did not suppress target gene expression. Cells were remade for each experiment and were not mixed and matched.
METHOD DETAILS
Cell treatments
For experiments, cells were treated with DMSO (Veh) or 100 nM 17β-estradiol (E2) (Sigma, E8875) for the specified amount of time. To induce the expression of sgRNA constructs, cells were treated with 1 μg/mL doxycycline (Sigma, D9891) 48 h before additional treatment. A485 (Tocaris, 6387), dissolved in DMSO, was used at a final concentration of 50 μM.
Antibodies
The custom rabbit polyclonal antiserum against ERα used for ChIP-qPCR was made in-house as described previously [first 110 amino acids of human ERα) (Kraus and Kadonaga, 1998)]. ERα ChIP-seq libraries were prepared using rabbit polyclonal anti-ERα (Millipore Sigma, 06–935). The custom rabbit polyclonal antiserum against pan-SRC was made in-house as described previously [amino acids 643–1130 of mouse SRC2 (Acevedo et al., 2004)]. The other antibodies used were as follows: rabbit polyclonal anti-ß-tubulin (Abcam, ab6046), rabbit polyclonal anti-SNRNP70 (Abcam, ab83306), mouse monoclonal anti-Flag (Sigma-Aldrich, F3165), rabbit polyclonal anti-Cas9 (Diagenode, C15310258–100), rabbit polyclonal anti-H3K27ac (Abcam, ab4729), mouse monoclonal anti-p300 (Active Motif, 61401), rabbit polyclonal anti-H3 (Abcam, ab1791), rabbit polyclonal anti-BCAS2 (Bethyl, A300–915A), mouse monoclonal anti-BCAS2 (Santa Cruz, sc-365346) for ChIP-qPCR, goat anti-rabbit HRP-conjugated IgG (Pierce, 31460), and goat anti-mouse HRP-conjugated IgG (Pierce, 31430).
siRNA-mediated knockdown
The BCAS2 siRNA oligonucleotides used were siRNA #1: SASI_Hs01_00054562, siRNA#2: SASI_Hs01_00054563 (Sigma), along with control siRNA (Sigma, SIC001). All the siRNA oligonucleotides were resuspended to 100 nM in RNase-free water and transfected at a final concentration of 10 nM using Lipofectamine RNAiMAX reagent (Invitrogen, 13778150) according to the manufacturer’s instructions. The cells were used for various assays 48 h after siRNA transfection.
Vectors for ectopic expression
The vectors described below were generated using oligonucleotide primers synthesized by Integrated DNA Technologies (IDT) and described in the next section. All constructs were verified by Sanger sequencing.
sgRNA constructs
pINDUCER20 vector (pInd20), kindly provided by Dr. Thomas Westbrook [Baylor College of Medicine, Houston, TX (Meerbrey et al., 2011)], was modified to the following configuration: NheI-sgRNA scaffold-BclI-BsiWI-NsiI-SbfI-bovine growth hormone polyA signal. Guide RNA targeting ERα enhancer was first synthesized by IDT containing the NheI overhangs, then annealed and phosphorylated according to published protocol (Ran et al., 2013). Annealed oligonucleotides were then diluted 1:200 before ligation into NheI-digested pInd20 according to the manufacturer’s protocol (Enzymatics, L6030-LC-L). The reaction was then transformed into DH5α. Clones were selected, and purified plasmids were sent for Sanger sequencing for correct directionality.
eRNA constructs
eRNA sequences, derived from hg38 assembly, were cloned from genomic DNA extracted from MCF-7 cells (Qiagen, 51304) using the four restriction sites between the sgRNA scaffold and the polyA signal. eRNAs were amplified using Phusion High-Fidelity DNA polymerase according to the manufacturer’s protocol (NEB, M0530L). The reaction was then separated on an agarose gel, and PCR product corresponding to the correct size was purified (Qiagen, 28704), then ligated into digested pInd20 (with or without sgRNA), transformed, selected, purified, and verified as above.
For PRRX2 eRNA MT and WT.AS constructs, a reverse primer encompassing the mutant or antisense sequence was used in combination with the PRRX2 eRNA forward primer. pInd20 digested with the appropriate enzymes (BclI/SbfI) was used for Gibson cloning (NEB, E2621), transformed, selected, purified, and verified as above.
For PRRX2 ΔFERM, a gBlock (IDT) containing the FERM-deletion eRNA was ordered, amplified, and cloned into pInd20. For UBE2E2 ΔFERM, two overlapping fragments without the FERM were PCR amplified and assembled using Gibson assembly. All plasmids were transformed, selected, purified, and verified as above.
FERM constructs
FERM constructs were cloned into pInd20 by annealing oligonucleotides with the appropriate cut sites similar to sgRNA constructs. In order to ensure that the FERM would not be sterically hindered by the dCas9 protein, the FERM was cloned in tandem so that the resulting construct contained an 82 nt FERM RNA. Cold FERM41, Cy5-FERM41, and biotin-triethyleneglycol(TEG)-FERM41 were all synthesized by IDT and purified by RNase-free high performance liquid chromatography, with the modification placed at the 5′ end of the RNA.
Bacterial expression construct
pET19b (Novagen, 69677–3) bacterial expression vector for 6xHis-tagged human MCF-7 was cloned out of human cDNA pools generated from total RNA extracted from MCF-7 cells (Bio Basic, BS88136). Total RNA was reverse transcribed using Superscript III Reverse Transcriptase (Invitrogen, 18080093) with Oligo(dT)21 primer (synthesized by Sigma). The resulting cDNA pools were used to amplify BCAS2 cDNA for subsequent cloning using NdeI and BamHI as cut sites for Gibson cloning (NEB, E2621).
In vitro transcription vectors
PRRX2 WT, MT, and WT.AS eRNAs were cloned from pInd20-PRRX2 eRNA plasmids as described above and ligated into pcDNA3.1(+) (Invitrogen, V79020) using EcoRI and XbaI, downstream of the T7 promoter, for Gibson cloning. UBE2E2 eRNA was cloned from pInd20-eRNA plasmids as described above and ligated into pcDNA3.1(+). GFP was cloned from an in-house GFP construct (Huang et al., 2020) using EcoRI and XbalI, downstream of the T7 promoter, for Gibson cloning.
Oligonucleotide primers used for cloning
A list of oligonucleotide primers used for cloning is provided in Table S2.
Analysis of mRNA expression by RT-qPCR
RNA expression analysis by RT-qPCR was performed as previously described (Hou et al., 2020). Total RNA was isolated from MCF-7 cells using DNAaway RNA mini-prep kit (Bio Basic, BS88136) according to manufacturer’s protocol. One μg of RNA was reverse transcribed with M-MLV reverse transcriptase (Promega, M1701) using Oligo(dT)21 primers according to the manufacturer’s protocol. cDNA, 1× SYBR Green PCR master mix, and forward and reverse primers (250 nM) were mixed and incubated at 95°C for 5 min before amplification for 45 cycles (95°C for 10 s, 60°C for 10 s, 72°C for 1 s) in a Roche LightCycler 480,384-well qPCR system. Melting curve analyses were performed to ensure that only the desired amplicon was amplified. All target gene expression was normalized to GAPDH expression.
RT-qPCR primers
BCAS2 Forward: 5′-GCCCAGTTAGAGCATCAAGCAG-3′
BCAS2 Reverse: 5′-TGAAGTTCCTTCTGTGCGTGTTC-3′
GAPDH Forward: 5′-CCACTCCTCCACCTTTGAC-3′
GAPDH Reverse: 5′-ACCCTGTTGCTGTAGCCA-3′
GREB1 Forward: 5′-CCTATTTTGGAATAAAAACTGACC-3′
GREB1 Reverse: 5′-GGGGAGAATGACACAAAAGC-3′
P2RY2 Forward: 5′-CGGTGGACTTAGCTCTGAGG-3′
P2RY2 Reverse: 5′-GCCTCCAGATGGGTCTATGA-3′
PRRX2 Forward: 5′-TTCTCGGTGAGCCACCTCCT-3′
PRRX2 Reverse: 5′-GTTGGCTGCTGTTGAACGTG-3′
PTGES Forward: 5′-AAACGGAAGCTCAGAGGATG-3′
PTGES Reverse: 5′-TGAACCAGTTTCCTCAGCTG-3′
SEMA3C Forward: 5′-ACCCACTGACTCAATGCAGAGG-3′
SEMA3C Reverse: 5′-CAGCCACTTGATAGATGCCTGC-3′
UBE2E1 Forward: 5′-TGCTTTTAGTCACCTTCTTAAGGG-3′
UBE2E1 Reverse: 5′-AATGCAGCAAGAGGCAGTTC-3′
UBE2E2 Forward: 5′-CGTGAAAGTGTTCAGCAAGAACC-3′
UBE2E2 Reverse: 5′-GGAGGGTCCAATGTGATTTCTGC-3′
Chromatin Immunoprecipitation-qPCR (ChIP-qPCR)
Chromatin immunoprecipitation was performed as previously described with some modifications (Murakami et al., 2017). Cells were grown to ~80% confluence and treated with E2 for the specified amount of time before crosslinking with 1% formaldehyde (ThermoScientific, 28,906) in PBS at 37°C for 10 min. The reaction was then quenched with glycine at a final concentration of 125 mM for 5 min at 4°C. The cells were then washed generously with ice-cold 1× PBS and collected by scraping with 1 mL ice-cold 1× PBS. The cells were pelleted by centrifugation and lysed by pipetting in Farnham Lysis Buffer [5 mM PIPES pH 8, 85 mM KCl, 0.5% NP-40, 1 mM DTT, 1× complete protease inhibitor cocktail (Roche, 11697498001)]. Nuclei were collected by brief centrifugation and resuspended in SDS Lysis Buffer (50 mM Tris-HCl pH 7.9, 1% SDS, 10 mM EDTA, 1 mM DTT, 1× complete protease inhibitor cocktail) by pipetting and incubating on ice for 10 min. Note that for H3K27ac ChIP, buffers were supplemented with 10 mM of sodium butyrate. The chromatin was sheared to ~200–500 bp DNA fragments by sonication using a Bioruptor sonicator (Diage-node) for 30 cycles of 30 s on and 30 s off. Fragment size was verified by agarose gel before quantification of protein concentrations using BCA protein assay kit (Pierce, 23225). Soluble chromatin (50 μg for H3K27ac, 500 μg for ERα, 1 mg for SRC and p300) was precleared with Protein A Dynabeads (Invitrogen, 10001D) before incubation overnight with 5 μL of polyclonal antiserum (for ERα ChIP-qPCR), 2 μg of anti-ERα (Millipore Sigma, 06–935 for ERα ChIP-seq), 1.5 μg of anti-H3K27ac, or 5 μg anti-p300, or 2 μg of anti-BCAS2 (Santa Cruz, sc-365346).
The immune complexes from the ChIP were precipitated by the addition of Protein A Dynabeads for 2 h at 4°C and washed once with each of the following in order: (1) low salt (20 mM Tris-HCl pH 7.9, 2 mM EDTA, 125 mM NaCl, 0.05% SDS, 1% Triton X-100, 1× complete protease inhibitor cocktail); (2) high salt (20 mM Tris-HCl pH 7.9, 2 mM EDTA, 500 mM NaCl, 0.05% SDS, 1% Triton X-100, 1× complete protease inhibitor cocktail); (3) LiCl (20 mM Tris-HCl pH 7.9, 1 mM EDTA, 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 1× complete protease inhibitor cocktail); (4) 1× Tris-EDTA (TE) containing 1× complete protease inhibitor cocktail. The precipitated immune complexes were transferred to a new tube in 1× TE before incubation in Elution Buffer (40 mM Tris-HCl pH = 7.9, 10 mM EDTA, 100 mM NaCl, 100 mM NaHCO3, 1% SDS) with DNase-free RNase (Roche, 11119915001) for 30 min at 37°C, followed by 50 μg proteinase K (Life Technologies, 2542) for 2 h at 55°C. The chromatin was then de-crosslinked by incubating overnight at 65°C. Genomic DNA was purified using ChIP DNA Clean & Concentrator (Zymo Research, D5201) according to the manufacturer’s protocol. The ChIPed DNA was analyzed by qPCR as described above using the primers listed below. Non-specific background signals were determined using IgG for purified antibodies. The data were expressed as the percent of input.
ChIP-qPCR primers
PRRX2 enhancer Forward: 5′-AAATCCCTGCCCTTGTTGCT-3′
PRRX2 enhancer Reverse: 5′-TCCGTCCCTGATGGTATGGT-3′
SEMA3C enhancer Forward: 5′-ACCTTTTACCTGCTACCCACTG-3′
SEMA3C enhancer Reverse: 5′-CCAACAGCGATGTTTGCCAT-3′
UBE2E2 enhancer Forward: 5′-TGCCACGATGGTATAGCACA-3′
UBE2E2 enhancer Reverse: 5′-ACAGGCTTAGCAATTTAGGGGT-3′
GREB1 enhancer Forward: 5′-TAGGCTTCAAGAGGACCACA-3′
GREB1 enhancer Reverse: 5′-AGCAGCAAAACTGCATAGGA-3′
Subcellular fractionation
Subcellular fractionation was performed as previously described (Kim et al., 2019). The cells were washed generously with ice-cold 1× PBS and collected by scraping with 1 mL ice-cold 1× PBS. Cells were pelleted by centrifugation. The packed cell volume (PCV) was then estimated and resuspended to homogeneity in 5× PCV of Isotonic Lysis Buffer (10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 0.3 M sucrose, 1 mM DTT, 1× protease inhibitor cocktail) and incubated on ice for 15 min. NP-40 was added in Isotonic Lysis Buffer to a final concentration of 0.6%. The mixture was vortexed vigorously for 10 s and centrifuged for 30 s at 11,000 RCF in a micro-centrifuge at 4°C. The supernatant was collected as the cytoplasmic fraction, and the resulting pelleted nuclei were resuspended in two-thirds PCV of ice-cold Nuclear Extraction Buffer [20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% (v/v) glycerol, 1 mM DTT, 1× protease inhibitor cocktail] and incubated with gentle mixing for 15 min for 4°C. The mixture was centrifuged at maximum speed in a microcentrifuge for 10 min at 4°C to remove the insoluble material. The supernatant was collected and an equal volume of Dilution Buffer (20 mM HEPES pH 7.6, 1.5 mM MgCl2, 0.2 mM EDTA) was added to make the soluble nuclear extract. Protein concentrations were determined using Bradford protein assay (Bio-Rad) and the nuclear extracts were used for various assays as indicated below.
Western blotting
Prior to Western blotting, lysates were mixed with water and 4× SDS Loading Buffer (250 mM Tris-HCl pH 6.8, 40% glycerol, 0.04% bromophenol blue in 10% glycerol, 4% SDS) and boiled for 10 min. Ten to 25 μg of protein (amount used depended on the target proteins) were separated on a polyacrylamide-SDS gel (percent gel used depended on the size of the target proteins), transferred onto a nitrocellulose membrane, and blocked for 1 h at room temperature in 5% nonfat dry milk dissolved in Tris-buffered saline (TBS) with 0.05% Tween (TBST). The membrane was washed with TBST before incubation with primary antibodies diluted in 5% BSA in TBST overnight at 4°C with gentle mixing (all at 1:1000 dilution). The next day, the membrane was washed with TBST before incubation with the appropriate HRP-conjugated secondary antibody in 5% nonfat dry milk for 1 h at room temperature (all at 1:5000 dilution). The membrane was washed with TBST before chemiluminescent detection using SuperSignal West Pico or Femto substrate (ThermoFisher Scientific) and ChemiDoc MP or XRS + system (Bio-Rad).
RNA fluorescence in situ hybridization (FISH)
RNA FISH was conducted with according to the Stellaris RNA FISH protocol (Biosearch Technologies, CA). Five thousand cells were seeded in phenol red-free/charcoal-dextran treated calf serum minimal essential medium (Sigma, M3024) for 72 h in poly-L-lysine coated 4-well Nunc Lab-Tek II chamber slides (ThermoFisher, 154917). Doxycycline was added 48 h prior to E2 treatment. The cells were treated with E2 for 45 min before washing with 1× PBS. They were then fixed in 3.7% (v/v) formaldehyde in 1× PBS at room temperature for 10 min, before washing twice with 1× PBS. The cells were then permeabilized in 70% (v/v) ethanol at 4°C for at least 1 h. Custom probe sets conjugated to Quasar 570® for PRRX2 and UBE2E2 eRNAs were synthesized by Biosearch Technologies and dissolved in TE buffer to make a 12.5 μM stock solution. A list of oligonucleotide sequences for the probe sets used is provided in Table S2. To hybridize the probe, 125 nM of probe solution was prepared in hybridization buffer (Biosearch Technologies, SMF-HB1–10) containing 10% deionized formamide (Millipore, S4117). Slides were washed with Wash Buffer A (Biosearch Technologies, SMFWA1–60) containing 10% deionized formamide at room temperature for 5 min. The buffer was then removed, followed by addition of the working probe solution. Slides were then placed in a humidified chamber, protected from light, and incubated at 37°C overnight. The next day, the slides were washed with Wash Buffer A at 37°C for 30 min, followed by a wash with Wash Buffer B (Biosearch Technologies, SMF-WA2–20) at room temperature for 5 min. The slides were then mounted by adding a small drop of mounting medium containing DAPI (Vector Laboratories, H-1200) and applying a coverglass (ThermoFisher, 12-548-5MP). They were then sealed with clear nail polish, allowed to dry, and imaged.
Microscopy
Images were captured using a custom W1 SoRa spinning disk confocal microscope system equipped with an Orca-Fusion sCMOS camera (Hamamatsu Photonics, Japan) on a Nikon Ti2E inverted fluorescence microscope. Images were captured using a 60× Plan Apo λ oil objective (numerical aperture = 1.4), with an λex = 405 nm and λem = 455 nm at 30% power, 1 s exposure for DAPI, and λex = 561 nm and λem = 605 nm at 50% power, 1 s exposure for Quasar 570®. All images were captured using NIS-Elements (Nikon).
Image analysis
Images were imported into Fiji (Schindelin et al., 2012) using Bio-Formats Bio-Importer (Linkert et al., 2010). Images were then adjusted with constant threshold settings for the green and blue channels for all images analyzed. Nuclei were drawn using the Analyze Particles function in Fiji. The intensity of RNA FISH spots within the nuclei were then quantified using GaussFit OnSpot with default parameters. The data were quantified for 3 to 5 fields per treatment from biological duplicate experiments.
In vitro transcription
First, 10 μg of pcDNA3.1(+)-eRNA and control pcDNA3.1(+)-GFP plasmids were linearized by overnight digestion with XbaI (NEB, R0145) and purified using DNA Clean & Concentrator kit (Zymo Research, D4033); completion of digestion was verified by agarose gel electrophoresis. For unlabeled RNAs, 1 μg of linearized plasmid was mixed with HiScribe T7 Quick High Yield RNA Synthesis kit (NEB, E2050S) overnight at 37°C. For biotinylated RNAs, 1 μg of linearized plasmid was mixed with biotin RNA labeling mix (10 mM each of ATP, CTP, GTP, 6.5 mM of UTP, and 3.5 mM of biotin-UTP; Sigma, 11685597910) and T7 RNA polymerase (Promega, P207B) and incubated overnight at 37°C. The reaction was then treated with DNase (Promega, M6101) for 15 min at 37°C, before purification with RNA Clean & Concentrator 5 (Zymo Research, R1015). The size of the transcripts was confirmed using RNA ScreenTape system (Agilent Technologies).
Histone acetyltransferase assay
10 nM FERM41 RNA, PRRX2 WT eRNA, PRRX2 MT eRNA, PRRX2 WT.AS eRNA, and UBE2E2 eRNA were folded in Folding Buffer (10 mM Tris-HCl pH 7.5, 100 mM NaCl, 50 mM KCl, 5 mM MgCl2) by heating the mixture at 65°C for 5 min, followed by gradual cooling to room temperature. The HAT reaction was assembled with the following components: 200 nM (~53 ng) human recombinant histone H3.1/H4 tetramer (NEB, M2509), 15 ng purified human full-length p300 (Martire et al., 2019), 15 μM acetyl-CoA, 0.1 nM folded RNA, 5% glycerol, 1 mM DTT, 1× complete protease inhibitor cocktail, 0.02 U/μL SUPERase inhibitor, and HAT Buffer (500 mM Tris-HCl pH 7.5, 10 mM MgCl2). Reaction without FERM41 was supplemented with the appropriate amount of Folding Buffer. The reaction was incubated for 15 min at 30°C and stopped by addition of 2× SDS Loading Buffer and boiled for 10 min. The reaction was then analyzed by Western blotting after running on a 15% SDS-PAGE gel.
Biotinylated RNA-Protein pulldowns
One hundred pmol of biotin-TEG-FERM41 (~1.3 μg), biotinylated PRRX2 WT eRNA (~10.6 μg), biotinylated PRRX2 MT eRNA (~10.6 μg), and biotinylated GFP RNA (~23 μg) were pre-folded in Folding Buffer (10 mM Tris-HCl pH 7.5, 100 mM NaCl, 50 mM KCl, 5 mM MgCl2) by heating the mixture at 65°C for 5 min, followed by gradual cooling to room temperature. A 950 μL reaction in Reaction Buffer [25 mM Tris-HCl pH 7.4, 150 mM KCl, 0.5% NP-40, 10 mM MgCl2, 2 ng/μL of poly(I:C) (Sigma, P1530), 0.5 mM DTT, 1× complete protease inhibitor cocktail, 100 U of SUPERase inhibitor (Invitrogen, AM2696)] was assembled by combining pre-folded RNA with 1.5 mg of nuclear lysate and incubated for 30 min at room temperature with rotation. In the meantime, 50 μL of M-280 streptavidin Dynabeads per reaction (ThermoFisher, 11205D) was pre-washed in Reaction Buffer. Fifty μL of washed streptavidin Dynabeads were then added to the reaction and incubated for an additional 30 min at room temperature with rotation. RNA-protein-Dynabead complexes were then washed 5 times with Reaction Buffer supplemented with 150 mM of NaCl for 10 min at 4°C per wash. To elute the proteins, Dynabeads were resuspended in 2× SDS Loading Buffer and boiled for 10 min. The reactions were then used for Western blotting or submitted for proteomics.
Liquid chromatography-tandem mass spectrometry proteomics
Samples from biotinylated RNA-protein pulldown were separated in a pre-cast 4%–12% Bis-Tris gradient gel (Invitrogen, NW04120BOX) using Bolt MES SDS running buffer (Invitrogen, B0002). The samples were run 1 cm into the gel, stained with Coomassie Blue, and cut into 1 mm cubes. Samples were then digested overnight with trypsin (Pierce) following reduction and alkylation with DTT and iodoacetamide (Sigma–Aldrich). The samples were then subjected to solid-phase extraction cleanup with an Oasis HLB plate (Waters) and the resulting samples were injected onto an Orbitrap Fusion Lumos mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were injected onto a 75 μm i.d., 75-cm long EasySpray column (Thermo) and eluted with a gradient from 0%–28% Buffer B over 90 min. Buffer A contained 2% (v/v) acetonitrile and 0.1% formic acid in water, and Buffer B contained 80% (v/v) acetonitrile, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 1.5–2.2 kV and an ion transfer tube temperature of 275°C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 25 s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v2.4 SP1 (Thermo), with peptide identification performed using Sequest HT searching against the human protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met set as a variable modification. The false-discovery rate (FDR) cutoff was 1% for all peptides.
Mass spectrometry data analysis
In order to filter out non-specific RNA-protein interactions, samples from PRRX2 WT or MT eRNA pulldown with spectral counts less than GFP RNA pulldown were converted to 0. Next, the spectral counts were analyzed using the LC-MS/MS Differential Expression Tests available from Bioconductor (Gregori et al., 2020). The data were analyzed using the Poisson-based GLM regression. Enriched proteins in PRRX2 WT eRNA pulldown were plotted in a volcano plot as log 2 of the fold change over PRRX2 MT eRNA pulldown against the negative log of the adjusted p values using the Benjamini-Hochberg method.
Expression and purification of recombinant BCAS2
pET19b-6xHis-BCAS2 was expressed in Rosetta (DE3) E. coli competent cells (Sigma-Aldrich, 70954) by induction with 1 mM of isopropyl β-D-1-thiogalactopyranoside during log phase growth for 3 h at 37°C. The bacteria were pelleted and resuspended in Lysis Buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.1% NP-40, 10% glycerol, 1 mM PMSF, 2 μL/mL aprotinin, 2 μL/mL leupeptin, 7 μL/100 mL β-mercaptoethanol) and sonicated with a Branson digital sonifier at 65% amplitude 10 s on/30 s off for 10 min. The sample was then centrifuged at 20,000 RCF for 30 min at 4°C, and the supernatant was then combined with 50% Ni-NTA glutathione Sepharose resin pre-washed with Lysis Buffer (Qiagen, 30210). The resin was incubated with the extract for 3 h at 4°C, before washing 3 times with Wash Buffer (10 mM Tris-HCl pH 7.5, 300 mM NaCl, 0.2% NP-40, 10% glycerol, 15 mM imidazole, 1 mM PMSF, 7 μL/100 mL β-mercaptoethanol). The 6xHis-BCAS2 was then eluted from the resin by incubating with Elution Buffer (10 mM Tris-HCl pH 7.5, 200 mM NaCl, 0.1% NP-40, 10% glycerol, 500 mM imidazole, 1 mM PMSF, 7 μL/100 mL β-mercaptoethanol) twice. The two eluates were then dialyzed overnight at 4°C in Dialysis Buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% glycerol, 1 mM PMSF, 7 μL/100 mL β-mercaptoethanol). The next morning, samples were transferred to a fresh Dialysis Buffer and dialyzed once more for 3 h at 4°C. The supernatants were then collected and measured by Bradford protein assay. Purity was checked by running the eluates on a 12% SDS-PAGE gel, followed by 0.1% Coomassie blue R250 staining [(Sigma, 27816); prepared in 10% acetic acid, 50% methanol, 40% water]. Destaining was done in 10% acetic acid, 50% methanol, 40% water solution.
Electrophoresis mobility shift assays
Buffer exchange for 6xHis-BCAS2 was done by using Amicon Ultra-0.5 Centrifugal Filter device (Millipore, UFC501024) with 50% glycerol twice to eliminate excess salt. Concentration was measured once again with Bradford protein assay. Before binding, 100 nM Cy5-FERM41 was folded in Folding Buffer (25 mM HEPES pH 7.6, 12.5 mM KCl, 0.5 mM MgCl2) by heating the mixture at 65°C for 5 min, followed by gradual cooling to room temperature, protected from light. The binding reaction was then assembled with the following components: purified 6xHis-BCAS2 (0, 25 ng, 250 ng, 500 ng), 10 nM pre-folded Cy5-FERM41, 2 ng/μL of poly(I:C) (Sigma, P1530), 1 mg/mL bovine serum albumin (Invitrogen, AM2616), 1 mM DTT, 1× complete protease inhibitor cocktail, 0.02 U/μL SUPERase inhibitor (Invitrogen, AM2696). For cold FERM41 reactions, the FERM41 was folded as described above, and added into the reaction at 10 nM, 100 nM, and 1 μM. The reactions were then incubated for 15 min at 30°C before separation with 6% native PAGE gel supplemented with 1% glycerol and resolved with 1× TBE supplemented with 1% glycerol for 45 min at 4°C. Note that no loading dye was added to the reactions to avoid changing the salt concentration. The gel was then imaged directly with ChemiDoc MP system (Bio-Rad).
Cell proliferation assays
dCas9-expressing MCF-7 cells were seeded at 1 × 104 cells per well in a 24-well plate (USA Scientific, CC7682–7524) and cultured in Eagle’s minimal essential medium (Sigma, M3024) supplemented with 5% charcoal-dextran treated calf serum (Sigma, C8056), 2 mM GlutaMAX (Gibco, 35050061), 100 units/mL penicillin-streptomycin (Gibco, 15140122), and 25 μg/mL gentamicin (Gibco, 1571004). The cells were allowed to rest overnight before they were transfected with siCtrl (Sigma, SIC001) or siBCAS2 using Lipofectamine RNAiMax (Invitrogen, 13778150) at 10 nM final concentration according to the manufacturer’s protocol. The cells were also treated with 1 μg/mL doxycycline (Sigma, D9891) for 48 h to induce the expression of the various constructs. Seventy-two hours after initial seeding (Day 0), cells were treated with 100 nM E2. The medium was changed daily with either supplementation of E2 (Days 0, 2, 4) or doxycycline (Days 1, 3, 5). The cells were fixed in 10% formaldehyde for 10 min and stored at 4°C at the specified day. Once all the samples were collected, they were stained with 0.5% crystal violet (Sigma, C0775) in ddH2O for 30 min at room temperature. Plates were then washed with distilled water thoroughly to remove unincorporated stain and allowed to dry at room temperature. Crystal violet was then extracted using 10% glacial acetic acid and the absorbance was read at 595 nm after 1:10 dilution in ddH2O. All growth assays were performed at least three times using different passages of cells.
Preparation of PRO-Cap libraries
PRO-cap libraries were generated as previously described (Mahat et al., 2016) with the following modifications. Nuclear run-on was performed with the 1-biotin (biotin-CTP). In order to increase the efficiency of the 3′ adaptor ligation due to the biotin-CTP near the end of the transcripts, RNA sample was resuspended in 10 μL mixed with 1× poly(A) polymerase buffer, 1 mM ATP, 1 μL poly(A) polymerase (NEB, M0276) in 20 μL reaction volume for 8 min at 37°C. The reaction was stopped by addition of Trizol (Invitrogen, 15596026) before proceeding to RNA extraction, precipitation, and 3′ adaptor ligation. Analyses using 32P autoradiography and FastQC suggested ~4 A’s added to the 3′ end. This resulted in an improvement of at least 2 PCR cycles used for the final library amplification.
Analysis of PRO-cap data
The following steps were used to analyze the PRO-cap data: (1) Raw data were analyzed using FastQC tool for quality control; (2) Reads were subjected to trimming using Cutadapt to remove the polyA tails and adapter sequences to maximize mappability; (3) Trimmed PRO-cap reads were aligned to the human reference genome (GRCh38/hg38) using the BWA aligner (Li and Durbin, 2009); (4) Output files were then converted into BED files using SAMtools (Li et al., 2009) and BEDtools (Quinlan and Hall, 2010); and (5) bigWig files for visualization were also generated using BEDtools. For figure presentation, we used SparK to generate the browser tracks (Kurtenbach and Harbour, 2019).
Transcription start site calling
The TSS workflow for cappable-seq was applied to PRO-cap data (Ettwiller et al., 2016). For bam2firstbasegtf.pl, the option –absolute 1 was used. The data were further processed using cluster_tss.pl with default –cutoff. The resulting called TSSs were used as an anchoring point for the annotation of eRNA transcripts.
Position weight matrix
The TSS closest to the 1,023 eRNAs assembled by StringTie (see below) was extended 50 bp up- and downstream using –bedtools slop -b 50. The sequences were then extracted using –bedtools getfasta -s. Sequences were then imported into Biostrings using R, a consensus matrix was computed, and the sequence enrichment was analyzed using seqLogo.
Metagene analyses
Metagenes were used to illustrate the distribution of PRO-cap reads in 1 kb windows around the called TSSs of the eRNAs. The average of the normalized counts was determined using computeMatrix and plotProfile functions in the deeptools package (Ramirez et al., 2016).
Preparation of RNA-Sequencing libraries
RNA-seq libraries were generated as previously described (Hou et al., 2020) with the following modifications. Total RNA from MCF-7 cells treated with DMSO (Veh) or 100 nM E2 for 45 min was isolated using DNAaway RNA mini-prep kit (Bio Basic, BS88136) according to the manufacturer’s protocol. Five μg of total RNA was then used for ribosomal RNA depletion using Ribo-Zero Plus rRNA depletion kit (Illumina, MRZG126). The total RNA was then enriched for polyA + RNA using Dynabeads Oligo(dT)25 (Invitrogen, 61002). All the supernatants, including the washes, were collected and precipitated by supplementation with 300 mM NaCl, 1 μL glycoblue (Invitrogen, AM9515), and 3× volume of 100% ethanol. This was the polyA-depleted fraction and was used in parallel with the polyA-enriched fraction for library construction. Further fragmentation, first-strand cDNA synthesis, and subsequent procedures have been described by Zhong et al. (2011) (Zhong et al., 2011).
The libraries were subjected to quality control analyses, such as optimizing the number of PCR cycles required to amplify each set of libraries, quantifying the final library yield by Qubit fluorometer (Invitrogen, Q32851), and checking the size distribution of the final library by DNA ScreenTape system (Agilent Technologies, G2964AA, 5067–5584, and 5067–5603). The libraries were barcoded with indices as described for the Illumina TruSeq RNA Prep kit, multiplexed, and sequenced in Illumina HiSeq (2000) by 150 bp paired-end sequencing. RNA-seq libraries from vehicle-treated MCF-7 cells were published previously and can be accessed from GEO using accession numbers GSM3188708, GSM3188709, GSM3188710, and GSM3188711 (Kim et al., 2019). QC steps for the siBCAS2 libraries were performed as described above prior to sequencing (75 bp single-end).
Analysis of RNA-seq data for eRNA annotations – Paired-end sequencing
The following steps were used to analyze the RNA-seq data: (1) Raw data was analyzed using FastQC tool for quality control; (2) Paired-end total RNA-seq reads were aligned to the human reference genome (GRCh38/hg38) using Star with default parameters (Dobin et al., 2013); (3) Output files were then converted into BED files using SAMtools and BEDtools; and (4) bigWig files for visualization were generated using BEDtools.
Assembly of eRNA transcripts
StringTie was used to assemble eRNA transcripts (Pertea et al., 2016) by optimizing the following three parameters: (1) -m, minimum length allowed for the predicted transcripts; (2) -c, minimum read coverage allowed for the predicted transcripts; (3) -g, minimum locus gap separation value. The option -t, which disables trimming at the ends of the assembled transcripts, was used. The output files were then converted to BED files using BEDOPS (Neph et al., 2012) gtf2bed and inspected with corresponding bigWig tracks. Using the called TSS from PRO-cap, the assembled transcripts were intersected with the TSS to ensure that the called TSS from both methods was normally distributed around 0, as shown in Figure S1B.
Metagene analyses
Similar to PRO-cap, metagenes were used to illustrate the distribution of RNA-seq reads in 1 kb windows around the called TSSs of the eRNAs. The average of the normalized counts was determined using computeMatrix and plotProfile functions in the deeptools package.
Discovery of the functional enhancer RNA motif
1,023 eRNA sequences were analyzed with MEME for de novo motif discovery (Bailey et al., 2009). The position specific frequency matrix from MEME for the top motif was then imported into FIMO in order to look for the FERM element in the large dataset that may have been missed during the de novo motif discovery. The location of the FERM element was then plotted along the eRNAs using computeMatrix.
Analysis of secondary structures using scanfold
eRNAs were first separated based on the presence of the FERM element. For the analysis, sequences less than 1 kb were analyzed using Scanfold (Andrews et al., 2018) with default parameters. Z-scores were calculated based on the average of the scanning window for each eRNA.
Analysis of RNA-seq data for differential gene expression – Single-end sequencing
RNA-seq data were prepared and analyzed as described in paired-end sequencing above, except for alignment in single-end sequencing mode. Differential gene analysis for siControl, siBCAS2 #1, siBCAS2 #2 was performed by first counting reads using –featureCounts in the subread package using default parameters. The count matrix was then imported into DESeq2 (Love et al., 2014) and differential gene analysis was done based on the interaction between siRNA and Treatment. Heatmap was drawn using Morpheus (https://software.broadinstitute.org/morpheus). Differential up- and downregulated genes were then used for downstream gene ontology analysis (see below).
Preparation of ChIP-seq libraries
ChIP-seq libraries from MCF-7 cells treated with vehicle (DMSO) or 100 nM E2 for 45 min were prepared as described previously with some modifications (Franco et al., 2015; Murakami et al., 2017). ChIPed DNA samples were purified using Agencourt AMPure XP beads (Beckman Coulter, A63881). Five ng of purified DNA was used to prepare libraries. After adaptor ligation, the libraries were purified with 0.8× of AMPure XP beads two times in order to eliminate unligated adaptors. DNA was then PCR-amplified for 11 to 13 cycles and purified using 2 rounds of 1× of AMPure XP beads. QC was performed in the same manner as RNA-seq libraries.
Analysis of ChIP-seq data
ChIP-seq data were processed as previously described (Franco et al., 2015; Murakami et al., 2017): (1) Raw data were analyzed using FastQC tool for quality control; (2) reads were aligned to the human reference genome (GRCh38/hg38) using the BWA aligner (Li and Durbin, 2009); (3) Output files were then converted into BED files using SAMtools (Li et al., 2009) and BEDtools (Quinlan and Hall, 2010); and (4) bigWig files for visualization were also generated using BEDtools. Metagene analysis was done using computeMatrix and plotProfile functions in the deeptools package, with estrogen receptor binding sites generating eRNAs with or without the FERM element. Boxplots were generated for quantitative assessment of read distribution around the peak center for each group by using HOMER software (Gupte et al., 2021). Briefly, tag directories were made for siControl and siBCAS2 treated with either Vehicle or 45 min E2 using makeTagDirectory. Counts were then determined using annotatePeaks.pl hg38 -size 200 -hist 25 -ghist with regions corresponding to ERα binding sites (ERBSs) generating eRNAs with or without the FERM element. Reads were then subtracted by their corresponding Input control, and fold change was calculated using the following formula: Fc = log[(E2-treated siBCAS2/siControl)/(Vehicle-treated siBCAS2/siControl)]. We considered differences greater than 2-fold for analysis and plotting using the boxplot function in R.
Analysis of publicly available datasets
The following published GRO-seq, ChIP-seq, and ATAC-seq datasets from MCF-7 cells (±E2 treatment) were used for the analyses. All datasets are available from the NCBI’s Gene Expression Omnibus repository (GEO) (http://ncbi.nlm.nih.gov/geo/) using the accession numbers listed below:
ERα (−E2, +E2) GSM1534720, GSM1534721, GSM1534722, GSM1534723.
ATAC-Seq (−E2, +E2) GSM3315603, GSM3315604, GSM3315605, GSM3315606, GSM3315607, GSM3315608.
H3K27ac (−E2, +E2) GSM1115993, GSM1115992.
FOXA1 (−E2, +E2) GSM1534736, GSM1534737, GSM1534738, GSM1534739.
GRO-seq datasets were processed using previously an established data analysis pipeline (Hou et al., 2020), while ChIP-seq and ATAC-seq datasets were processed similarly to RNA-seq data described above. Metagene analyses were used to illustrate the distribution of reads in 4 kb windows around the center of the ERα binding sites (ERBSs). In order to minimize the bias that can be caused by outliers in the metagene analyses and for efficient comparison across different groups, the read distributions in 4 kb windows around the ERBS were calculated and plotted using the boxplot function in R.
Analysis of the FERM element in MCF-7 cells
We downloaded the broadPeak bed files for following ChIP-seq MCF-7 transcription factor binding sites from ENCODE/HAIB. The data sets and the NCBI GEO accession numbers are listed below:
CEBFB GSM1010889.
CTCF GSM1010734.
FOSL2 GSM1010764.
GABP GSM1010864.
GATA3 GSM1010764.
HDAC2 GSM1020936.
JUND GSM1010892.
MAX GSM1010863.
NRSF GSM1010891.
PML GSM1010838.
SRF GSM1010839.
RAD21 GSM1010791.
TCF12 GSM1010861.
TEAD4 GSM1010860.
Genomic coordinates were first converted from hg19 to hg38 using liftOver. Peaks were then divided into three regions: (1) inter-genic; (2) intronic; and (3) exonic using Gencode v38 annotation. We extracted sequences by taking 1.5 kb from the center of the binding site in 5’→3′ direction, mimicking the transcription of the enhancer. FERM element was then searched using FIMO as described above. Results were calculated as percent of sequences analyzed.
Analysis of FERM element in human versus mouse
We used the following GRO-seq datasets to look for transcription units. The data sets and the NCBI GEO accession numbers are listed below∷
Human breast cancer cell lines GSE96859.
Mouse liver GSM1437737, GSM1437742.
Mouse macrophages SRX651735.
Mouse uterus (−E2, +E2) Manuscript in preparation
GRO-seq datasets were processed using a previously established data analysis pipeline (Hou et al., 2020). Intergenic transcription units were defined using Gencode v38 annotation for human, and Gencode v10 for mouse. The FERM element was then searched using FIMO as described above. The results were calculated as percent of sequences analyzed.
Gene ontology (GO) analyses
Gene ontology analyses were performed using DAVID (Database for Annotation, Visualization, and Integrated Discovery) (Huang da et al., 2009). Inputs for GO analyses included the proteomic studies and the mRNA/eRNA correlation analysis. DAVID returns clusters of related ontological terms that are ranked according to an enrichment score. The top GO biological process terms from these clusters based on the enrichment score were listed.
Breast cancer tumor samples
FPKM for mRNAs and eRNAs were calculated using –featureCounts in the subread package followed by normalization based on the number of fragments per kilobase mapped. The eRNAs were then used as anchoring points to locate the nearest expressed gene expressed in all samples. The FPKMs were then paired and the Pearson’s coefficients for individual mRNA/eRNA pairings for the 10 samples were then determined. The mRNA/eRNA pairings that had a Pearson’s coefficient of greater than 0.5 were then used for gene ontology analysis.
Kaplan-Meier analyses
Kaplan-Meier analysis was performed using Kaplan-Meier Plotter (Nagy et al., 2021). Plots were generated for PRRX2 and BCAS2 for ER+ and ER− breast cancer patients that were identified using array. Patients were split by the median expression, while all other options were set to default.
QUANTIFICATION AND STATISTICAL ANALYSIS
All sequencing-based genomic experiments were performed with two independent biological samples. Statistical analyses for the genomic experiments were performed using standard genomic statistical tests as described above. Proteomic experiments were performed with two independent biological samples on two different days. qPCR-based experiments were performed as indicated in the corresponding figure legends. Western blotting experiments with quantification were performed a minimum of three times with independent biological samples and analyzed by Image Lab 6.1 (Bio-Rad). EMSA experiment was performed a minimum of three times on three different days. Cell proliferation assays were performed a minimum of three times with independent biological samples. Statistical analyses were performed using GraphPad Prism 9. All tests and p values are provided in the corresponding figures or figure legends.
Supplementary Material
Highlights.
eRNAs fused to sgRNAs can be targeted to enhancers of origin using CRISPR/dCas9
Some eRNAs targeted to ERα enhancers can enhance target gene expression with estrogen
Functional eRNAs stimulate ERα recruitment and p300-catalyzed H3K27 acetylation
A FERM in some eRNAs drives target gene expression
ACKNOWLEDGMENTS
The authors thank the members of the Kraus lab for helpful feedback on this work, especially Shino Murakami, Rebecca Gupte, Aarin Jones, Andrea Edwards, Cristel Camacho, and David Owen; the members of the Green Center for Reproductive Biology Sciences Computational Core Facility for assistance with data analysis; Dr. Gary Hon and Dr. Shiqi Xie (University of Texas Southwestern Medical Center, Dallas) for helpful discussions and the dCas9-Flag construct; Rebecca Gupte for the purified p300; and Shu-Ping Chiu for assistance with the 6xHIs-BCAS2 purification. The also acknowledge the following core facilities at University of Texas Southwestern Medical Center: the McDermott Center Next Generation Sequencing Core (Dr. Ralf Kittler and Vanessa Schmid) and the UT Southwestern Proteomics Core (Dr. Andrew Lemoff). The breast cancer datasets used for the analyses described in this manuscript were obtained from dbGaP at www.ncbi.nlm.nih.gov/gap through dbGaP accession number phs000676. This work was supported by a grant from the NIH/NIDDK (R01 DK058110), grants from the Cancer Prevention and Research Institute of Texas (CPRIT; RP160318 and RP190235), and funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment to W.L.K.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110944.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acevedo ML, Lee KC, Stender JD, Katzenellenbogen BS, and Kraus WL (2004). Selective recognition of distinct classes of coactivators by a ligand-inducible activation domain. Mol. Cell 13, 725–738. 10.1016/s1097-2765(04)00121-2. [DOI] [PubMed] [Google Scholar]
- Andersson R, Refsing Andersen P, Valen E, Core LJ, Bornholdt J, Boyd M, Heick Jensen T, and Sandelin A (2014). Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat. Commun 5, 5336. 10.1038/ncomms6336. [DOI] [PubMed] [Google Scholar]
- Andrews RJ, Roche J, and Moss WN (2018). ScanFold: an approach for genome-wide discovery of local RNA structural elements-applications to Zika virus and HIV. PeerJ 6, e6136. 10.7717/peerj.6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). Meme SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–W208. 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, and Luhrmann R (2017). Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542, 318–323. 10.1038/nature21079. [DOI] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, and Berger SL (2017). RNA binding to CBP stimulates histone acetylation and transcription. Cell 168, 135–149.e22. 10.1016/j.cell.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, and Crawford GE (2008). High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322. 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caizzi L, Ferrero G, Cutrupi S, Cordero F, Ballare C, Miano V, Reineri S, Ricci L, Friard O, Testori A, et al. (2014). Genome-wide activity of unliganded estrogen receptor-alpha in breast cancer cells. Proc. Natl. Acad. Sci. U S A 111, 4892–4897. 10.1073/pnas.1315445111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carullo NVN, Phillips III RA, Simon RC, Soto SAR, Hinds JE, Salisbury AJ, Revanna JS, Bunner KD, Ianov L, Sultan FA, et al. (2020). Enhancer RNAs predict enhancer-gene regulatory links and are critical for enhancer function in neuronal systems. Nucleic Acids Res 48, 9550–9570. 10.1093/nar/gkaa671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino RR, and Stark A (2018). Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev 32, 202–223. 10.1101/gad.310367.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, and Liang H (2020). A high-resolution map of human enhancer RNA loci characterizes super-enhancer activities in cancer. Cancer Cell 38, 701–715.e5. 10.1016/j.ccell.2020.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, et al. (2006). Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16, 123–131. 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, and Natoli G (2010). A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 8, e1000384. 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dridi S (2012). Alu mobile elements: from junk DNA to genomic gems. Scientifica (Cairo) 2012, 545328. 10.6064/2012/545328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettwiller L, Buswell J, Yigit E, and Schildkraut I (2016). A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome. BMC Genom 17, 199. 10.1186/s12864-016-2539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Nagari A, and Kraus WL (2015). TNFα signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol. Cell 58, 21–34. 10.1016/j.molcel.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. (2009). An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462, 58–64. 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini M, Tome JM, and Shendure J (2020). Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet 21, 292–310. 10.1038/s41576-019-0209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, and Lieb JD (2007). FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17, 877–885. 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori J, Sanchez A, and Villaneuva J (2020). msmsTests: LC-MS/MS Differential Expression Tests. R Package Version 1.28.0 [Google Scholar]
- Gupte R, Nandu T, and Kraus WL (2021). Nuclear ADP-ribosylation drives IFNγ-dependent STAT1α enhancer formation in macrophages. Nat. Commun 12, 3931. 10.1038/s41467-021-24225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, and Kraus WL (2011). A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145, 622–634. 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, and Kraus WL (2013). Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23, 1210–1223. 10.1101/gr.152306.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou TY, and Kraus WL (2021). Spirits in the material world: enhancer RNAs in transcriptional regulation. Trends Biochem. Sci 46, 138–153. 10.1016/j.tibs.2020.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou TY, Nandu T, Li R, Chae M, Murakami S, and Kraus WL (2020). Characterization of basal and estrogen-regulated antisense transcription in breast cancer cells: role in regulating sense transcription. Mol. Cell. Endocrinol 506, 110746. 10.1016/j.mce.2020.110746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Fei T, Chen Y, Li T, Gao Y, Wang X, Sun T, Sweeney CJ, Lee GS, Chen S, et al. (2014). Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. U S A 111, 7319–7324. 10.1073/pnas.1324151111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang D, Camacho CV, Setlem R, Ryu KW, Parameswaran B, Gupta RK, and Kraus WL (2020). Functional interplay between histone H2B ADP-ribosylation and phosphorylation controls adipogenesis. Mol. Cell 79, 934–949.e14. 10.1016/j.molcel.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, and Bartel DP (2011). Formation, regulation and evolution of Caenorhabditis elegans 3’UTRs. Nature 469, 97–101. 10.1038/nature09616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, and Kraus WL (2019). Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol. Cell 75, 1270–1285.e14. 10.1016/j.molcel.2019.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch F, Fenouil R, Gut M, Cauchy P, Albert TK, Zacarias-Cabeza J, Spicuglia S, de la Chapelle AL, Heidemann M, Hintermair C, et al. (2011). Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat. Struct. Mol. Biol 18, 956–963. 10.1038/nsmb.2085. [DOI] [PubMed] [Google Scholar]
- Kraus WL, and Kadonaga JT (1998). p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev 12, 331–342. 10.1101/gad.12.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtenbach S, and Harbour WJ (2019). SparK: a publication-quality NGS visualization tool. Preprint at bioRxiv 10.1101/845529. [DOI] [Google Scholar]
- Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, and Shiekhattar R (2013). Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494, 497–501. 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. (2013). Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498, 511–515. 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. (2013). Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520. 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108. 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Lor-anger B, Moore J, Neves C, Macdonald D, et al. (2010). Metadata matters: access to image data in the real world. J. Cell Biol 189, 777–782. 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and ViennaRNA Package 2.0. Algorithms; and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms. Mol Biol 6, 26. 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv ZD, Wang HB, Liu XP, Jin LY, Shen RW, Wang XG, Kong B, Qu HL, Li FN, and Yang QF (2017). Silencing of Prrx2 inhibits the invasion and metastasis of breast cancer both in vitro and in vivo by reversing epithelialmesenchymal transition. Cell. Physiol. Biochem 42, 1847–1856. 10.1159/000479542. [DOI] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc 11, 1455–1476. 10.1038/nprot.2016.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng YC, Tastemel M, and Banaszynski LA (2019). Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat. Genet 51, 941–946. 10.1038/s41588-019-0428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U S A 108, 3665–3670. 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melgar MF, Collins FS, and Sethupathy P (2011). Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol 12, R113. 10.1186/gb-2011-12-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrie-link JA, Elkon R, Melo SA, Leveille N, Kalluri R, et al. (2013). eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 49, 524–535. 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nagari A, and Kraus WL (2017). Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes Dev 31, 1535–1548. 10.1101/gad.302182.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Munkacsy G, and Gyorffy B (2021). Pancancer survival analysis of cancer hallmark genes. Sci. Rep 11, 6047. 10.1038/s41598-021-84787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G, and Andrau JC (2012). Noncoding transcription at enhancers: general principles and functional models. Annu. Rev. Genet 46, 1–19. 10.1146/annurev-genet-110711-155459. [DOI] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. (2012). BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920. 10.1093/bioinformatics/bts277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega E, Rengachari S, Ibrahim Z, Hoghoughi N, Gaucher J, Hole-house AS, Khochbin S, and Panne D (2018). Transcription factor dimerization activates the p300 acetyltransferase. Nature 562, 538–544. 10.1038/s41586-018-0621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, and Salzberg SL (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc 11, 1650–1667. 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, and Domany E (2006). Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genom 7, 133. 10.1186/1471-2164-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, Zhu YT, Chang J, Yeldandi AV, Sambasiva Rao M, and Zhu YJ (2005). Potentiation of estrogen receptor transcriptional activity by breast cancer amplified sequence 2. Biochem. Biophys. Res. Commun 328, 393–398. 10.1016/j.bbrc.2004.12.187. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnamoun H, Lee J, Sun Z, Lu H, Ramsey KM, Komives EA, and Lauberth SM (2018). RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat. Struct. Mol. Biol 25, 687–697. 10.1038/s41594-018-0102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeron-Hernandez A, Noriega-Reyes MY, Jordan A, Baranda-Avila N, and Langley E (2019). BCAS2 enhances carcinogenic effects of estrogen receptor alpha in breast cancer cells. Int. J. Mol. Sci 20, 966. 10.3390/ijms20040966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, and Lauberth SM (2020). Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol 27, 521–528. 10.1038/s41594-020-0446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, and Kim TK (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. 10.1016/j.molcel.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechner DM, Hacisuleyman E, Younger ST, and Rinn JL (2015). Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods 12, 664–670. 10.1038/nmeth.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, Lenhard B, Crawford GE, and Furey TS (2013). Patterns of regulatory ac tivity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res 23, 777–788. 10.1101/gr.152140.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, Jangi M, Giallourakis CC, Sharp PA, and Young RA (2015). Transcription factor trapping by RNA in gene regulatory elements. Science 350, 978–981. 10.1126/science.aad3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JK, Vukadin L, and Ahn EE (2021). eNEMAL, an enhancer RNA transcribed from a distal MALAT1 enhancer, promotes NEAT1 long isoform expression. PLoS One 16, e0251515. 10.1371/journal.pone.0251515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Han D, Boyd-Kirkup J, Yu X, and Han JD (2014). Evolution of Alu elements toward enhancers. Cell Rep 7, 376–385. 10.1016/j.celrep.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Tsai PF, Dell’Orso S, Rodriguez J, Vivanco KO, Ko KD, Jiang K, Juan AH, Sarshad AA, Vian L, Tran M, et al. (2018). A muscle-specific enhancer RNA mediates cohesin recruitment and regulates transcription in trans. Mol. Cell 71, 129–141.e8. 10.1016/j.molcel.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigos A, and Rigoutsos I (2009). Alu and B1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLoS Comput. Biol 5, e1000610. 10.1371/journal.pcbi.1000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo Ngoc L, Cassidy CJ, Huang CY, Duttke SH, and Kadonaga JT (2017). The human initiator is a distinct and abundant element that is precisely positioned in focused core promoters. Genes Dev 31, 6–11. 10.1101/gad.293837.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, Li D, Choudhary MNK, Li Y, Hu M, et al. (2018). The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 19, 151. 10.1186/s13059-018-1519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yan C, Hang J, Finci LI, Lei J, and Shi Y (2017). An atomic structure of the human spliceosome. Cell 169, 918–929.e14. 10.1016/j.cell.2017.04.033. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hong W, Ruan H, Jing Y, Li S, Liu Y, Wang J, Li W, Diao L, and Han L (2021). HeRA: an atlas of enhancer RNAs across human tissues. Nucleic Acids Res 49, D932–D938. 10.1093/nar/gkaa940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lee JH, Ruan H, Ye Y, Krakowiak J, Hu Q, Xiang Y, Gong J, Zhou B, Wang L, et al. (2019). Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun 10, 4562. 10.1038/s41467-019-12543-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, and Perou CM (2014). Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom 15, 419. 10.1186/1471-2164-15-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang L, Ren S, Wang L, Blackburn PR, McNulty MS, Gao X, Qiao M, Vessella RL, Kohli M, et al. (2016). Activation of P-TEFb by androgen receptor-regulated enhancer RNAs in castration-resistant prostate cancer. Cell Rep 15, 599–610. 10.1016/j.celrep.2016.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhou J, He L, Li Y, Yuan J, Sun K, Chen X, Bao X, Esteban MA, Sun H, and Wang H (2019). MyoD induced enhancer RNA interacts with hnRNPL to activate target gene transcription during myogenic differentiation. Nat. Commun 10, 5787. 10.1038/s41467-019-13598-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, Xiang JZ, Fei Z, and Giovannoni JJ (2011). High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc 2011, 940–949. 10.1101/pdb.prot5652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data: The PRO-cap and RNA-seq datasets generated specifically for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/). The new mass spectrometry datasets generated for this study can be accessed from the Spectrometry Interactive Virtual Environment (MassIVE) repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp). Accession numbers are listed in the Key Resources Table.
Code: This paper does not report original code.
Additional information: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ERα | Kraus and Kadonaga (1998) | N/A |
| Rabbit polyclonal anti-ERα | Millipore Sigma | Cat. No. 06-935 RRID: AB_310305 |
| Rabbit polyclonal anti-panSRC | Acevedo et al. (2004) | N/A |
| Rabbit polyclonal anti-ß-tubulin | Abcam | Cat. No. MABE1031 RRID: AB_2210370 |
| Rabbit polyclonal anti-SNRNP70 | Abcam | Cat. No.: ab83306 RRID: AB_10673827 |
| Mouse monoclonal anti-Flag | Sigma-Aldrich | Cat. No.: F3165 RRID: AB_259529 |
| Rabbit polyclonal anti-Cas9 | Diagenode | Cat. No.: C15310258 RRID: AB_2715516 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat. No.: ab4729 RRID: AB_2118291 |
| Mouse monoclonal anti-p300 | Active Motif | Cat. No.: 61401 RRID: AB_2716754 |
| Rabbit polyclonal anti-H3 | Abcam | Cat. No.: ab1791 RRID: AB_302613 |
| Rabbit polyclonal anti-BCAS2 | Bethyl | Cat. No.: A300-915A RRID:AB_661883 |
| Mouse monoclonal anti-BCAS2 | Santa Cruz | Cat. No.: sc-365346 RRID: AB_10846085 |
| Goat anti-rabbit HRP-conjugated IgG | ThermoFisher | Cat. No. 31460 RRID: AB_228341 |
| Goat anti-mouse HRP-conjugated IgG | ThermoFisher | Cat. No. 31430 RRID: AB_10960845 |
| Rabbit IgG | ThermoFisher | Cat. No. 10500C RRID: AB_2532981 |
| Chemicals, peptides, and recombinant proteins | ||
| p300 | Gupte et al. (2021) | N/A |
| 6xHis-BCAS2 | This study | N/A |
| Human H3.1/H4 tetramer | NEB | Cat. No.: M2509 |
| 17β-estradiol | Sigma | Cat. No.: E8875 |
| Doxycycline | Sigma | Cat. No.: D9891 |
| A485 | Tocaris | Cat. No.: 6387 |
| Critical commercial assays | ||
| RNA ScreenTape | Agilent Technologies | Cat. No.: 5067-5576, 5067-5577 |
| DNA ScreenTape | Agilent Technologies | Cat. No.: 5067-5584, 5067-5585 |
| Deposited data | ||
| MCF-7 ±E2, ±TAP PRO-cap | This study | GEO: GSE175469 |
| MCF-7 E2, polyA-depleted RNA-seq | This study | GEO: GSE175469 |
| MCF-7 E2, polyA-enriched RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siCtrl, ±E2 RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #1, ±E2 RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #2, ±E2 RNA-seq | This study | GEO: GSE175469 |
| MCF-7, siCtrl, ±E2, ERα ChIP-Seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #1, ±E2, ERα ChIP-seq | This study | GEO: GSE175469 |
| MCF-7, siCtrl, ±E2, Input ChIP-Seq | This study | GEO: GSE175469 |
| MCF-7, siBCAS2 #1, ±E2, Input ChIP-seq | This study | GEO: GSE175469 |
| PRRX2 WT eRNA Proteomics | This study | MassIVE: MSV000087492 |
| PRRX2 MT eRNA Proteomics | This study | MassIVE: MSV000087492 |
| PRRX2 GFP RNA Proteomics | This study | MassIVE: MSV000087492 |
| FERM Proteomics | This study | MassIVE: MSV000087492 |
| Experimental models: Cell lines | ||
| 293T | ATCC | Cat. No.: CRL-3216 RRID: RRID:CVCL_0063 |
| MCF-7 | Dr. Benita Katzenellenbogen, University of Illinois, Urbana-Champaign | |
| Oligonucleotides | ||
| Primers for molecular cloning | See Table S2 | N/A |
| Recombinant DNA | ||
| pLenti-dCas9-Flag-Blast | This study | N/A |
| pINDUCER20 | Addgene | Cat. No.: 44012 |
| pIND20-sgRNA-eRNA-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-UBE2E2(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-UBE2E2(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-PRRX2(eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-P2RY2(sgRNA)-bGHpA | This study | N/A |
| pIND20-P2RY2(eRNA)-bGHpA | This study | N/A |
| pIND20-P2RY2(sgRNA)-P2RY2(eRNA)-bGHpA | This study | N/A |
| pIND20-SEMA3C(sgRNA)-bGHpA | This study | N/A |
| pIND20-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-SEMA3C(sgRNA)-SEMA3C(eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-FERM-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-FERM-bGHpA | This study | N/A |
| pIND20-SEMA3C(sgRNA)-FERM-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(ΔFERM eRNA)-bGHpA | This study | N/A |
| pIND20-UBE2E2(sgRNA)-UBE2E2(ΔFERM eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(MT eRNA)-bGHpA | This study | N/A |
| pIND20-PRRX2(sgRNA)-PRRX2(WT.AS eRNA)-bGHpA | This study | N/A |
| pcDNA3.1(+)-PRRX2(WT)eRNA | This study | N/A |
| pcDNA3.1(+)-PRRX2(MT)eRNA | This study | N/A |
| pcDNA3.1(+)-PRRX2(WT.AS)eRNA | This study | N/A |
| pcDNA3.1(+)-UBE2E2(eRNA) | This study | N/A |
| pcDNA3.1(+)-SEMA3C(eRNA) | This study | N/A |
| pcDNA3.1(+)-GFP | This study | N/A |
| pET19b-6xHis-BCAS2 | This study | N/A |
| pCMV-VSV-G | Addgene | Cat. No.: 8454 |
| psPAX2 | Addgene | Cat. No.: 12260 |
| PAdVAntage | Promega | Cat. No.: TB207 |
| Software and algorithms | ||
| FastQC | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Star | Dobin et al. (2013) |
https://github.com/alexdobin/ STAR |
| BWA | Li and Durbin (2009) | http://bio-bwa.sourceforge.net/ |
| StringTie | Pertea et al. (2016) | https://ccb.jhu.edu/software/stringtie/ |
| Cappable-seq | Ettwiller et al. (2016) | https://github.com/Ettwiller/TSS |
| BEDtools | Quinlan and Hall, (2010) | https://bedtools.readthedocs.io/en/latest/ |
| SAMtools | Li et al. (2009) | http://www.htslib.org/ |
| BEDops | Neph et al. (2012) | https://bedops.readthedocs.io/ |
| Deeptools | Ramirez et al. (2016) | https://deeptools.readthedocs.io/en/develop/ |
| SparK | Kurtenbach and Harbour (2019) | https://github.com/harbourlab/SparK |
| Subread | Liao et al., (2013) | http://subread.sourceforge.net/ |
| DESeq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MEME Suite | Bailey et al. (2009) | https://meme-suite.org/meme/index.html |
| DAVID Bioinformatics Resources | Huang da et al. (2009) | https://david.ncifcrf.gov/ |
| RNAfold | Lorenz et al., (2011) | http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi |
| Scanfold | Andrews et al. (2018) | https://github.com/moss-lab/ScanFold |
| Proteome Discoverer | ThermoFisher | Cat. No.: OPTON-30812 |
| msmsTests | Gregori et al. (2020) | https://www.bioconductor.org/packages/release/bioc/html/msmsTests.html |