

Abstract
In this paper we characterize the function of Xylosyltransferase 2 (XylT2) in different tissues to investigate the role XylT2 has in the proteoglycan (PG) biochemistry of multiple organs. The results show that in all organs examined there is a widespread and significant decrease in total XylT activity in Xylt2 knock out mice (Xylt2−/−). This decrease results in increased organ weight differences in lung, heart, and spleen. These findings, in addition to our previous findings of increased liver and kidney weight with loss of serum XylT activity, suggest systemic changes in organ function due to loss of XylT2 activity. The Xylt2−/− mice have splenomegaly due to enlargement of the red pulp area and enhanced pulmonary response to bacterial liposaccharide. Tissue glycosaminoglycan composition changes are also found. These results demonstrate a role of XylT2 activity in multiple organs and their PG content. Because the residual XylT activity in the Xylt2−/− is due to xylosyltransferase 1 (XylT1), these studies indicate that both XylT1 and XylT2 have important roles in PG biosynthesis and organ homeostasis.
Keywords: Proteoglycans, Glycotransferase, Glycosaminoglycans, Extracellular matrix, Disaccharide analyses, Genetic modifier, Organ homeostasis, Proteoglycans, Xylosyltransferase
Introduction
Proteoglycans (PGs) are found on the cell surface, in secretory granules, and in the extracellular matrix (ECM) [1]. At the cell surface, these biopolymers are fundamental to cellular processes including enhancement of receptor binding of cytokines and growth factors, and augmentation of enzyme-substrate interactions important in coagulation and lipid catabolism [2–7]. In the ECM, PGs maintain basement membrane integrity, augment growth factor and cytokine sequestration, and create morphogen gradients [3, 8–10]. PGs consist of one or more glycosaminoglycan (GAG) chains covalently linked to a core protein. In the two major families of PGs, chondroitin/dermatan sulfates (CS/DS) and heparin/heparan sulfates (HP/HS), the GAG chains are attached to the non-reducing terminal glucuronic acid residue of the tetrasaccharide linkage region on the core-protein, GlcA β1–3 Galβ1–3Gal β1–4 Xyl β1–O-Ser (where GlcA is glucuronic acid. Gal is galactose, Xyl is xylose and Ser is serine). XylT catalyzes the initial enzymatic reaction in the assembly of GAG - core protein linkage region. This addition of a Xyl to a designated Ser is hypothesized to be rate limiting for PG biosynthesis [11, 12].
XylT (uridine diphosphate (UDP)-D-xylose: protein-D-xylosyltransferase, E.C. 2.4.2.26) initially studied over 50 years ago was one of the first glycosyltransferases isolated [13, 14]. Genomic analyses have identified two XylT family members in mammals and both are known to possess XylT activity [15–19]. Given this universal and multifunctional activity for PGs, it is surprising that the total loss of XylT2 is not essential to cellular survival [15, 20, 21] nor is it lethal in vivo [22] in mice or humans [23]. Survival has been hypothesized to be due to the expression of XylT1. In humans, our original description of XylT2 deficiency in humans causing Spondylo-ocular syndrome [23] has been confirmed by others as well [24–27]. The degree to which different tissues depend on XylT2 for PG assembly is unknown and how XylT2 deficiency alters the amounts and biochemical profile of PGs is also unknown. Interestingly, multiple diseases with extensive ECM remodeling (e.g. osteoarthritis, and pseudoxanthoma elasticum) have been found to correlate with genetic polymorphisms in XYLT2 [28]. These variants are not causative of these conditions and are not known to cause XylT2 deficiency, but may phenotypically impact or modify ECM homeostasis in these conditions. Furthermore, loss of XylT2 activity in mice induces cyst development in the liver and many similarities with polycystic kidney disease leading to the hypothesis that PG levels may be modifiers of primary mutations [22].
In this paper the organs from the Xylt2−/− mice were examined to investigate the role of XylT2 in the PG biochemistry of multiple organs. We found widespread decreases in total XylT activity due to loss of XylT2 indicating that XylT2 is the predominant XylT in most tissues. Significant changes were found in the heart, spleen, and lungs where the heart is 17% heavier, the spleen weight is 91% higher, and the wet lung weight is 29% increased.
The changes in the spleen were due to an alteration in red pulp to white pulp content and in the lung these changes correlated with an increased response to lipopolysaccharide. Surprisingly, decreases in XylT activity are present in the brain and heart but there was no change in the weights for these organs. In the brain there were no other changes observed; however in the heart, there was an overall decrease in un-sulfated to low sulfated disaccharides.
Materials and methods
XylT activity assays
XylT activity was measured as previously described [29] using the bikunin peptide (QEEEGSGGGQKK) (Bio-Synthesis, Lewisville, TX) as the acceptor substrate [30]. One unit of enzymatic activity represents the incorporation of 1 μmol xylose/min into the acceptor peptide. Each isoform has a slightly different Km for this acceptor substrate where XylT1 is twice as high. Nonetheless this acceptor substrate still can be easily utilized by XylT1 under our assay conditions as published [29]. All reactions were done in duplicate or triplicate. Briefly, To measure XylT activity, tissue lysates were incubated with 1.13 μM UDP-[14C]-D-xylose 150–250 mCi/mol (Perkin Elmer, Waltham, WA), 7.46 μM UDP-D-xylose (CarboSource, University of Georgia, Atlanta, GA) and 160 μM acceptor peptide in a total reaction volume of 100 μL containing 25 mM 2-(4-morpholino)-ethane sulfonic acid, pH 6.5, 25 mM KCl, 5 mM KF, 5 mM MgCl2,5 mM MnCl2 for 1 h at 37 °C. The reaction was stopped and precipitated using BSA as a carrier and 10% trichloroacetic acid/4% phosphotungstic acid [30]. Samples were centrifuged and the precipitate was washed with trichloroacetic acid and resuspended 1 N NaOH followed by determination of radioactivity. For each tissue sample, the average activity represents the average of 3 independent values for brain of each genotype; while for other tissues, the average activity represents 5–6 independent samples.
Animals
Xylt2−/− mice were generated as previously described, and these are a complete knockout line with total loss of Xylt2 function [22]. All mice were housed in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The Institutional Animal Care and Use Committees of all institutions approved all animal procedures and experiments. Numbers of animals used for each analyses are indicated in figure legends. Unless otherwise indicated the mice were females backcrossed to 129SvEvTac. All controls were litter mates.
Splenic morphometrics, staining, and fluorescence activated sorting.
For splenic morphometrics, spleens were harvested and fixed in 4% paraformaldehyde and trimmed longitudinally specifically for morphometric analyses. Paraffin embedded splenic tissue was sectioned at 7 μm thickness and stained with hematoxylin and eosin. Images of spleen were captured using 1X magnification on a Nikon E800 Eclipse fluorescent microscope. Splenic follicular and total splenic areas used to calculate percentage of follicular areas were measured using Image J software version 1.45. Standard reticular staining was performed on 7 μm sections from paraffin embedded tissue. For Western blots, proteins were isolated and prepared as previously described [22] and detected with anti-decorin antibody polyclonal serum [31] (a kind gift from Larry Fisher, National Institutes of Health, Bethesda, MD). For flow cytometry of splenic cells, cells were isolated from whole spleens that were macerated in Dulbecco’s modified Eagle medium (DMEM) (Life Technologies, Inc.) cell culture media containing 5% serum followed by collection of the cellular-rich supernatant. Red blood cells (RBCs) in the supernatant were lysed with RBC lysis buffer containing NH4Cl (0.4 M), NH4HCO3 (0.03 M) and ethylenediaminetetracetic acid (EDTA) (0.3 μM) and debris removed by filtering through a 40 μM filter. Cell pellet was re-suspended in phosphate-buffered saline (PBS) containing 1% BSA buffer for flow analysis. Cells were stained with rat anti-mouse CD16/CD32 for Fc receptor block (BD Biosciences, San Jose, CA) followed by anti-mouse F480-fluorescein isothiocyanate (FITC), CD3-PE and CD19-APC antibodies and the corresponding isotype controls (eBiosciences, San Jose, CA). Cells were analyzed using C6 Accuri Flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software.
Disaccharide analyses
Tissues were harvested from mice euthanized with an overdose of isoflurane anesthesia. Tissues were washed in PBS to remove blood, blotted dry, and weighed. Subsequently tissues were flash frozen in liquid nitrogen. GAG purification was performed using previously published methods [32–34]. For each independent sample, brain, liver and kidney tissues were from one animal each, but other tissues were pools of 3 animals. This was required since more tissue was needed for the smaller and less dense organs. Briefly, tissues were frozen in liquid nitrogen and smashed in liquid nitrogen then delipidated with acetone in a pre-weighed tube. Acetone was decanted and residual acetone was removed with isopropyl ether. After ether removal by vacuum, dried tissue was weighed. Tissue was then resuspended in 100 mg tissue/ml pronase digestion buffer (0.1 M Tris-HCl, pH 8; 2 mM CaCl2; 1% Triton X-100). Pronase was added at 0.8 mg/ml final concentration [33] and digestion of the tissue was performed over 24 h with agitation at 55° C. After inactivating the pronase, tissues were digested with benzonase in 2 mM MgCl2 at 37° C for 2 h followed by enzyme inactivation. The supernatant was then applied to DEAE-Sephacel equilibrated in 20 mM Tris-HCl, 0.1 M NaCl, pH 7.5 and washed in the same buffer and eluted in 20 mM Tris-HCl, 2 M NaCl, pH 7.5. GAGs were released from the core proteins with β-elimination using 0.2 N NaOH, 1.0% NaBH4 at 42° C over-night followed by neutralization with glacial acetic acid to pH 7.0. GAGs were precipitated with 100% ethyl alcohol and pellet was washed with 85% ethyl alcohol containing 2% sodium acetate then dried and resuspended in ultrapure water. GAG concentration was determined by orcinol reaction as described [35]. Measurements were done in duplicate at absorbance of 670 nm.
Disaccharide analyses were performed as described [36]. Briefly, for HS disaccharide analysis, purified GAGs (3–7 μg, as determined by orcinol assay) were digested with heparin lyase I, II, and III at 10 mU each in 100 mM sodium acetate/0.1 mM calcium acetate pH 7.0 at 37 °C for 12 h after which the enzymes were inactivated at 100 °C for 5 min, the reaction mixture dried, resuspended in water, and 0.1–0.7 μg injected onto a Dionex DX-600 HPLC with AS-50 auto-sampler and RF2000 fluorescence detector (Thermo Scientific, Sunnyvale, CA), and a temperature controlled post column reactor. Post column solvent was a mixture of 1% 2-cyanoacetamide and 250 mM NaOH. For CS disaccharide analysis, a similar amount of GAG as used in HS analysis was digested in 50 mM Tris-Cl/50 mM sodium acetate pH 8.0 using chondroitinase ABC at 10 mU. The resulting CS disaccharides were similarly analyzed. HS and CS standards for the analyses were purchased from Sigma. All values represent analyses of 4–5 independent samples.
Polyacrylamide gel electrophoresis (PAGE) analysis
Tissue samples were crushed with dry ice into very fine homogenized powder using a mortar and pestle. Fats were removed by washing the tissues with a chloroform/methanol mixture of (2:1, 1:1, 1:2 (v/v)) each left overnight. Defatted samples were desiccated and resuspended in water and proteolyzed at 55 °C with 1% Actinase E at 20 mg/ml for 18 h. After the proteolysis, dry urea and dry CHAPS were added to each sample to give a final concentration of 2% CHAPS and 8 M urea. The resulting cloudy solutions were clarified by passing through a syringe filter containing a 0.2 μm membrane. A Vivapure MAXI Q H spin column was equilibrated with 3 ml of 8 M urea containing 2% CHAPS (pH 8.3). The clarified filtered samples were loaded onto and run through the Vivapure MAXI QH spin columns at 500×g. The columns were first washed with 3 ml of 8 M urea containing 2% CHAPS at pH 8.3 then washed five-times with 5 ml of 200 mM NaCl. GAG was released from the spin column by washing three-times with 1 ml of 16% NaCl. Methanol was added to 80% by volume and the mixture was equilibrated at 4 °C for 18 h. The resulting precipitated GAGs were recovered by centrifugation (2500×g) for 15 min dissolved in 1.0 ml of water and stored frozen for further analysis.
Polyacrylamide gel electrophoresis (PAGE) was used to analyze the molecular weight and polydispersity of each sample. To each lane 5 μg of isolated GAGs was subjected to electrophoresis against a standard composed of heparin oligosaccharides prepared enzymatically from bovine or porcine lung heparin, the gel was visualized with Alcian blue.
Statistics
Quantitative data, including enzyme assays, morphometric measurements, are based on at least 3–5 samples as detailed in the methods. Quantitative data is representative of means ± SDs. All statistical analyses were statistical differences between two groups and were determined by Student’s t test with equaled variance. P < 0.05 was considered significantly different. Analytical software was JMP software version 6 and 12.
Results
XylT2 activity is widespread
Previous studies showed that despite the ubiquitous expression of Xylt2 in mouse tissues [17, 22] and XYLT2 in multiple human cell types [15, 37], mice deficient in XylT2 (Xylt2−/−) survived to adulthood albeit with liver and renal abnormalities [22] and humans deficient also survive [23–27] but develop spondylar-ocular syndrome. In addition, XylT2 was recently shown to be important to adipose tissue homeostasis [38]. In light of these studies, the remaining XylT1 activity was hypothesized to be critical for organ function, organ development, and survivability of the Xylt2−/− mice. By studying the residual XylT activity in the XylT2 deficient animals that arises exclusively from Xylt1, the contribution of XylT1 to organ homeostasis in the mutants could be determined. Subsequently, XylT activity in multiple organs of Xylt2+/+ and Xylt2−/− mice was examined. All organs showed significant decreases in total XylT activity (Fig. 1a). The liver showed the largest decrease in total XylT activity with a 95% reduction and the brain with the least at 40%. The difference in XylT activity between wild type and mutant XylT2 deficient mice demonstrates the relative contribution of XylT1 and XylT2 activity to organ PG biosynthesis. The results show that XylT1 is widespread but XylT2 appears to be the predominant source of XylT activity in most tissues examined (Fig. 1a).
Fig. 1.
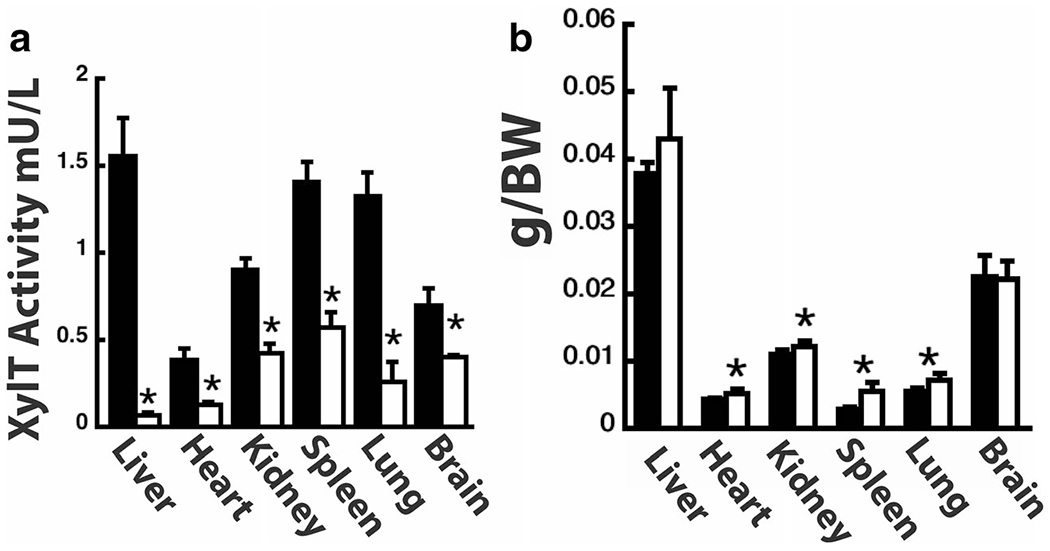
XylT2 deficiency affects multiple organs. a shows XylT activity in organs indicated. n = 5–6 mice, 5–8 month old male for each group. Black Bars Xylt2+/+ mice, Xylt2−/− mice, white bars. (b) Organ weights are changed in Xylt2−/− mice. n = 5 mice for each group, 2–3 month old females. Asterisk indicates significant difference of at least p ≤ 0.05
XylT2 deficiency affects multiple organs
Previous published experiments showed that liver and kidney weights in Xylt2−/− mice were significantly increased 50% and 30%, respectively, as compared to controls [22]. A systematic characterization or organ weights was performed in the Xylt2−/− mice to further investigate the effects of XylT2 deficiency on other organs. Previously, liver and kidney weights were found increased and progressive with age [22]. From the findings here and previous findings, the transition for the changes in these organs occurs at 3–4 months. When we look at 2–3 month animals, we see a significant increase of 10% in the kidney and a trend although not significant in liver weights. Interestingly, there was little change in organ weight for Xylt2−/− brain although it had substantial decreases in XylT activity. However, there was a significant 91% increase in spleen weight, a 29% increase in wet lung lobe weight, and a 17% increase heart weight in the mutants (see Fig. 1b). To further characterize the overall PG changes between organs, GAGs were isolated from PGs and size fractionated on polyacrylamide gels to determine if any overall change in PG GAG chain length had occurred in the Xylt2−/− mice. These analyses showed no obvious change in PG GAG chain length between organs (data not shown). Therefore, in addition to the liver and kidney, loss of XylT2 activity has significant effects on organ weights but no change on overall GAG chain length. To more closely examine GAG changes, disaccharide analysis for GAG chain chemical structural changes was performed.
Brain and kidney PG GAGs in the Xylt2−/− mice are not significantly affected
Isolated PGs from brain and kidney were examined using enzymatic digestion of their GAG chains followed by disaccharide analyses to determine how much of either HS and CS PGs were present in these tissues. As can be seen in Fig. 2a the GAGs derived from total brain PGs (nmoles/g brain dry weight) were unchanged and there are no measureable differences between HS and CS (see Fig. 2b) (HS Xylt2+/+ 382 ± 119, Xylt2−/− 339 ± 68) (CS Xylt2+/+ 1107 ± 33, Xylt2−/− 1134 ± 177). In the Xylt2−/− kidneys there is a trend, although not significant (p > 0.05), of a decrease in GAG derived from total kidney PG (nmoles/g kidney dry weight) (Fig. 2c) where the change affects HS more than CS (HS Xylt2+/+ mice 2603 ± 697, Xylt2−/− mice 2030 ± 319) (CS Xylt2+/+, 579 ± 284, Xylt2−/− 403 ± 110).
Fig. 2.
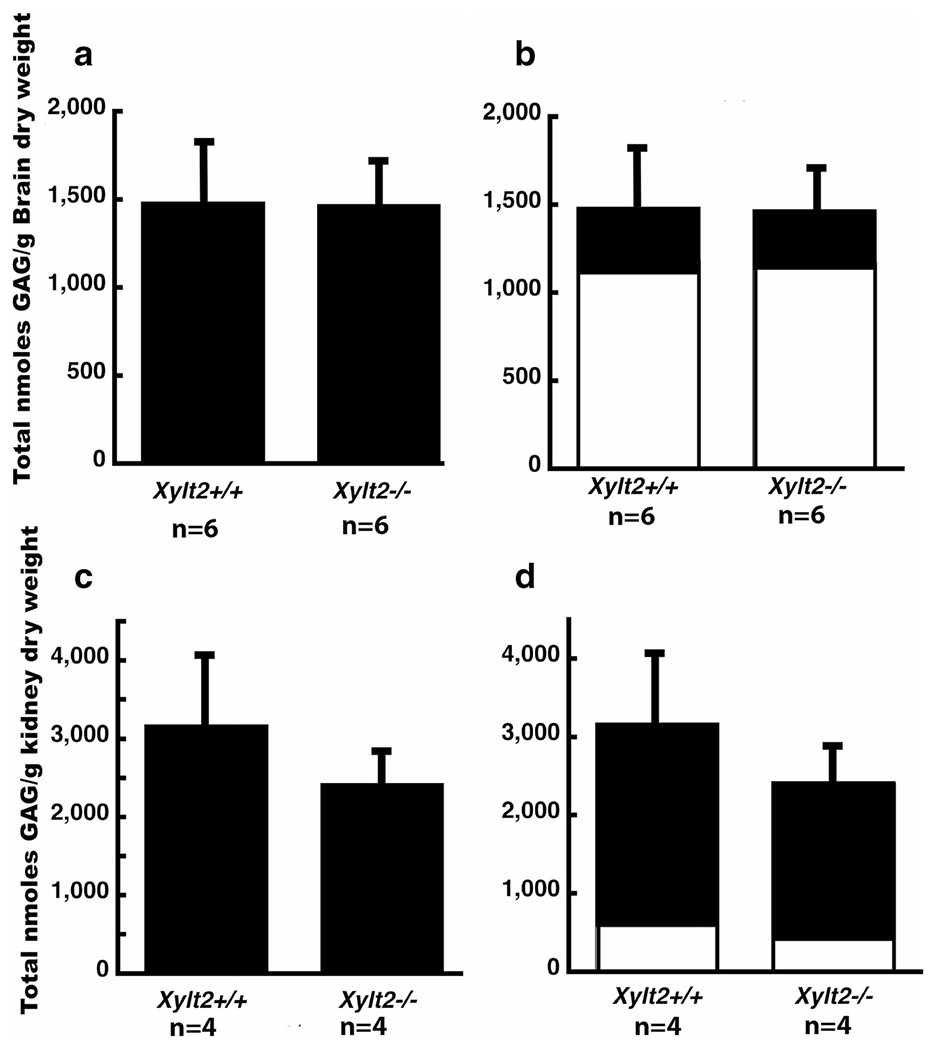
Brain and kidney heparan sulfate and chondroitin sulfate proteoglycans. a shows total brain proteoglycan GAG comparison between Xvlt2+/+ and Xvlt2−/− mice. b shows distribution of HS (dark section) and CS (white section). Error bars are those for total proteoglycans. c shows total proteoglycans in kidney, d shows distribution between HS (black section) and CS (white section). Error bars are those for total proteoglycans
Heart PG GAGs are significantly affected in Xylt2−/− mice
Firstly, the analysis shows that the predominant GAG in the heart is HS. Secondarily, XylT2 deficiency significantly decreases HS GAGs derived from heart PGs by 38% (HS Xylt2+/+ mice 631 ± 66, Xylt2−/− mice 392 ± 61, p = 0.01)(Fig. 3a and b). There is a decrease in total CS but this decrease was not significant (Xylt2+/+ mice 79 ± 44, Xylt2−/− mice 32 ± 6). The decrease in total HS was associated with a decrease in a number of specific disaccharide structures (Fig. 3c). A significant reduction of unsulfated disaccharides, corresponding to UA-GlcNAc (where UA is unsaturated uronic acid and GlcNAc is N-acetylglucosamine) was observed. Moreover, significant decreases were also observed in UA-GlcNS (where GlcNS is N-sulfo-glucosamine), UA2S-GlcNAc (where UA2S is 2-O-sulfo unsaturated uronic acid), and UA2S-GlcNS. No other HS disaccharides showed significant changes. Notably, those disaccharides with a 6-O-sulfo group-containing residues were not affected. The decrease in total CS (Fig. 3d) was associated with a similar decrease in UA-GalNAc, UA-GalNAc4S and UA2S-GalNAc6S (where GalNAc is N-acetylgalactosamine, 4S is 4-O-sulfo and 6S is 6-O-sulfo); however, these decreases were not significant. UA2S-GalNAc and UA2S-GalNAc-6S showed non-significant changes (data not shown) but levels are extremely low in the heart even in the Xylt2+/+ mice, so accurate detection may be difficult.
Fig. 3.
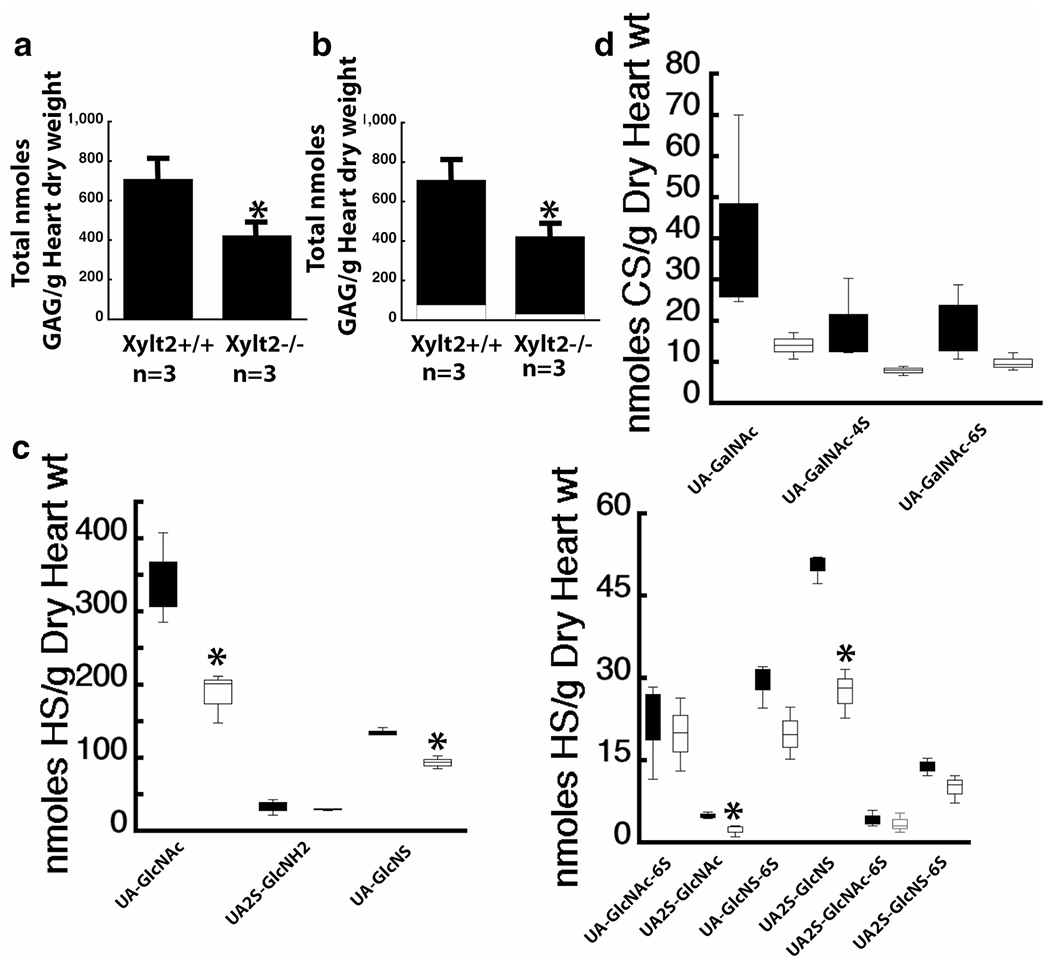
XylT2 deficiency significantly decreases HS in heart. a shows heart total GAGs are significantly decreased where the significant decrease is in HS. b shows the distribution of total PGs between HS, black section, and CS, white section. c shows the disaccharide distribution of the significant decreases in Xylt2−/− mice HS, white bars versus the Xylt2+/+, Black bars. Left panel shows high concentration disaccharides and right panel shows low concentration disaccharides. (d) shows the CS disaccharide distribution of Xylt2+/+ mice black bars and Xylt2−/− mice, white bars. Asterisks are significant differences of p ≤ 0.05
Spleen PG GAGs are significantly affected which induces splenic structural changes.
The increase in splenic weight is associated with an overall significant reduction in GAG (p = 0.0120) of 35% with significant decreases in CS levels. The greater decrease is in CS with 40% (Fig. 4a and b) (HS Xylt2+/+ mice 495 ± 124, Xylt2−/− mice 356 ± 36, p = 0.07; CS Xylt2+/+ mice 744 ± 182, Xylt2−/− mice 448 ± 80 nmoles/g dry organ weight, p = 0.03). Interestingly, CS predominates in this organ normally. The decrease in splenic HS was reflected in significant decreases in UA-GlcNS and UA-GlcNS6S, and a strong trend in UA-GlcNAc although not significant (supplemental Fig. 1). This appears to spare mostly 2-O-sulfated disaccharides (Fig. 4c). The decrease in CS was similarly reflected in decreases in all disaccharides but only significant for UA-GalNAc, UA-GalNAc4S and UA2S-GalNAc, seemingly sparing 6-O-sulfated disaccharides (Fig. 4d). Furthermore, the impact of XylT2 deficiency on splenic core protein modifications is demonstrated in decorin western blots that show the splenic stroma decorin is lacking GAG CS since core protein detection is unaffected by chondroitinase digestion, supporting the disaccharide findings (Fig. 4e). Besides the stroma, the spleen consists of the white pulp containing mostly T and B cells and monocytes and the red pulp area that contains mostly RBCs, splenic sinuses, and macrophages/monocytes. To explain the higher splenic weight, we hypothesized that one or both areas were somehow increased due to loss of GAGs. Therefore, we investigated the distribution of macrophages, T cells and B cells in the white pulp by fluorescent activated sorting using F4/80, CD3, and CD19 which are markers for these cells, respectively (Fig. 4f). All cell populations were found to be similar to Xylt2+/+ mice. Next, we did morphometric analyses on the spleens to measure the ratio of white pulp follicular area to total splenic area and found interestingly this ratio was significantly less in the Xylt2−/− mice (Fig. 4g). Given no difference in Xylt2−/− white pulp cellular counts exists, then the decreased follicular to total splenic area in the Xylt2−/− mice demonstrates an expanded red pulp area rather than a decreased white pulp areas is the reason for the increased splenic weight in the Xylt2−/− mice. Since we have observed fibrosis in other organs in the Xylt2−/− mice [22], we stained the spleens for collagen I with Sirius red that showed no difference between Xylt2−/− and controls (data not shown). The structural integrity of the red pulp depends on its reticular fiber network that consists of primarily collagen III. Furthermore, decorin is important to collagen fibrillogenesis. Therefore, we examined reticular fibers in the spleen using a reticular stain that binds to these collagen fibrils. We found normal variation of periarterial distribution of reticular fibers in the Xylt2+/+ mice, but in most Xylt2−/− mice there was consistently more layers of reticular staining positive material around splenic arteries as compared to Xylt2+/+ mice (Fig. 4h) suggesting a periarterial increase in collagen III in the Xylt2−/− mice.
Fig. 4.
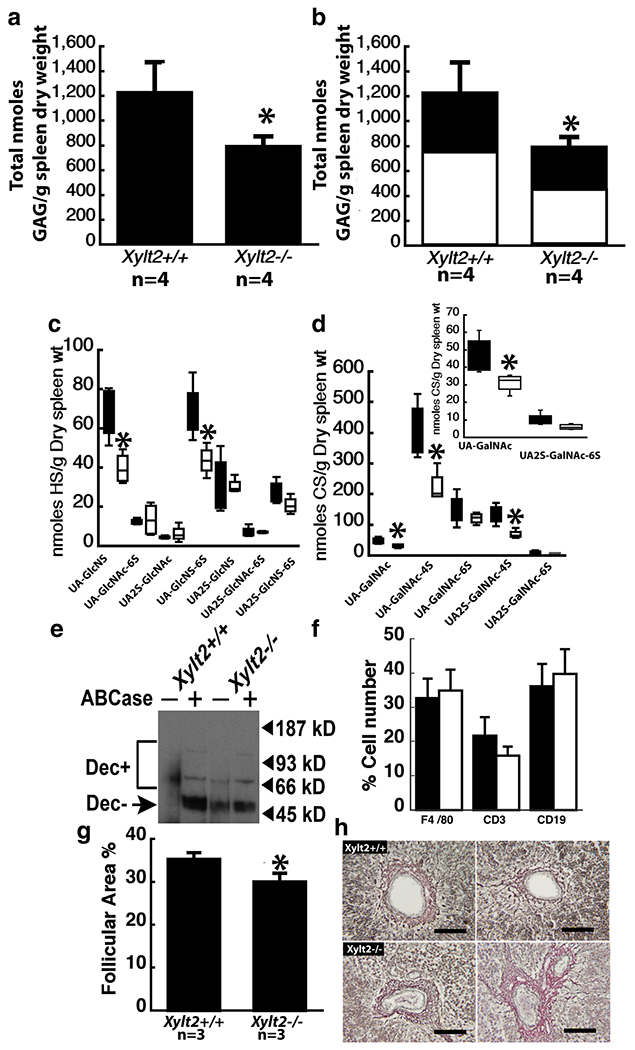
Xylt2 deficiency results in splenic changes in both HS and CS. a shows a significant decrease in splenic total proteoglycans in the Xylt2−/− mice. b shows that both HS, black section, and CS, white section, are decreased. c shows the HS disaccharide distribution in the spleens. Black bars are Xylt2+/+ mice and white bars are Xylt2−/− mice. d shows the significant decreases in CS disaccharides in the Xylt2−/− spleens. Bars are as in c. Inset is lower concentration disaccharides. e Western blot of splenic protein extracts with decorin antibody. Dec + indicates the smear of decorin core protein with intact CS. Dec-indicates decorin core proteins without CS. Plus and minus signs above the lanes indicate digestion or no digestion with chondroitinase ABC. Lanes 1 and 2 are Xylt2+/+ splenic protein extracts and lanes 3 and 4 are Xylt2−/− splenic extracts. f Fluorescent activated sorting of splenic cells. Bars are as in c. Xylt2+/+, n = 13, Xylt2−/−, n = 4. g Morphometric analyses of splenic follicular area as a percentage to the total splenic area. h Reticular staining of spleens. Upper panels are Xylt2+/+ spleens and lower panels are Xylt2−/− spleens. Scale bars 50 um. Asterisks are significant differences of p ≤ 0.05
Lung PGs are significantly decreased and result in abnormal response to LPS
The increase in lung weight is correlated with a significant decrease of 58% in GAGs derived from total lung PGs (Fig. 5a). This total GAG decrease is due to significant decreases of 62% for HS and 50% for CS GAGs (Fig. 5b) (HS Xylt2+/+ mice 2451 ± 635, Xylt−/− mice 941 ± 206, p = 0.004; CS Xylt2+/+ mice 1015 ± 242, Xylt2−/− mice 506 ± 327, p = 0.05). Similarly, nearly all disaccharides for both HS and CS were significantly decreased as well (Fig. 5c). In fact, the HS disaccharide UA2S-GlcNH2 was not detected in the Xylt2−/− mice and may reflect low sensitivity of detection. The only disaccharide unaffected was UA2S-GlcNAc-6S which is in very low amounts even in the Xylt2+/+ mice; therefore, a difference may go undetected. Lung tissues from patients with chronic obstructive pulmonary disease (COPD) of which emphysema can be a component have reduced PGs [39, 40]. However, we do not see any pathological changes associated with alveolar septal loss in the Xylt2−/− mice. Histologically, the lungs from some Xylt2−/− mice possessed a perivascular infiltration of mononuclear inflammatory cells (data not shown) but this was not in all animals. However looking for expression of markers (see supplemental methods) that would recruit these cells to the lung environment shows lung tissue from the Xylt2−/− mice has significantly increased Ccl2 levels and increasing trends in several other mononuclear inflammatory markers (Fig. 5e) suggesting the Xylt2−/− mouse lungs have an environment of slightly increased inflammation correlating with the occasional mice with perivascular inflammatory cell infiltration. To investigate this further, we challenged the mice with bacterial lipopolysaccharide (LPS) to mimic acute lung bacterial infection (see supplemental methods). These experiments showed an exaggerated response to LPS with significant higher levels of hemorrhage and inflammatory cell infiltrate in the Xylt2−/− mice (Fig. 5f).
Fig. 5.
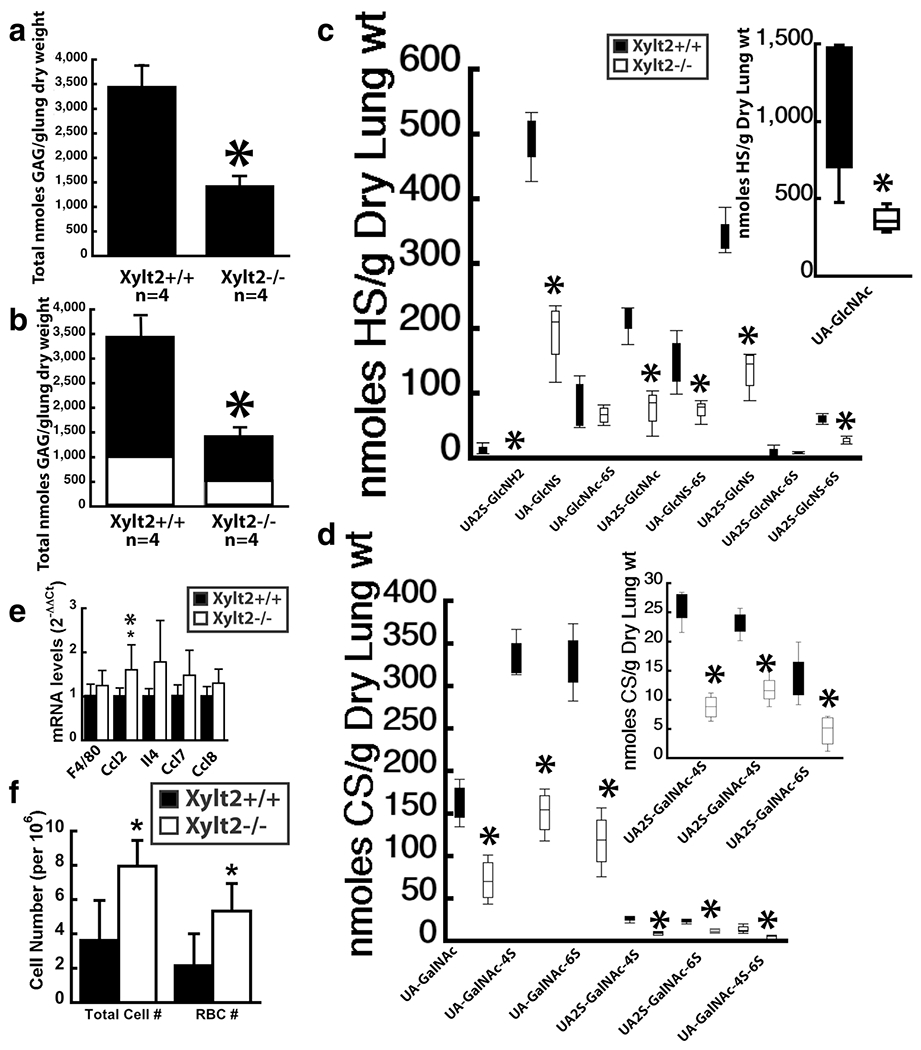
Lung proteoglycans are significantly altered in the Xylt2−/− mice resulting in inflammatory changes. a shows significant decrease in total lung PGs. b shows both HS (black section) and CS (white section) are significantly decreased. c HS disaccharide distribution in Xylt2+/+ mice (black bars) versus the Xylt2−/− mice (white bars). Inset shows values of UA-GlcNAc levels since they are extremely high. d shows CS disaccharide distribution in the lung. e shows mRNA expression of inflammatory markers in lung, n = 4–6 mice per group (f) Cell counts in bronchiolar lavage fluid from LPS challenged mice, n = 5–6 mice per group. All asterisks are p ≤ 0.05
Discussion
In this paper and another [22] we find that XylT2 deficiency has substantial impact on total XylT activity throughout many tissues where the liver has the greatest decrease and brain the least. Previously, we have shown that liver and kidney weight are impacted with XylT2 deficiency [22]. In addition, this paper shows there are significant changes in the organ weights of the spleen, heart, and lung. These results reveal the dependence that various organs have on XylT2 activity for GAG assembly on PGs. Furthermore, despite substantial loss of XylT activity in the brain there is not a significant change in organ weight or total HS and CS in this tissue. Loss of XylT2 function and decreases in total XylT activity does correlate with previous expression analyses. In all, loss of XylT2 function results in changes in total organ XylT activity, weight, disaccharide levels, and disaccharide distribution in multiple organs.
We show that the predominant PG in the heart is HS. In the heart of the Xylt2−/− mice, there is a significant decrease in total HS disaccharides, particularly those containing N-sulfo glucosamine residues or 2-O-sulfo uronic acid. But nonsulfated disaccharides are also reduced significantly. Interestingly, those disaccharides with a 6-O-sulfo glucosamine group-containing residues are spared. Heart CS is low to begin with and changes in disaccharide levels in the Xylt2−/− mice were not significant. These results indicate that a significant amount of the HS disaccharides in this organ are dependent on XylT2. There was a trend of decreasing total CS and of decreases in CS disaccharides but these were not significant. This suggests that XylT1 may be more responsible for CS and XylT2 for HS assembly in this organ.
Our results demonstrate that the spleen is a CS dependent tissue, and that XylT2 activity is important to splenic structural homeostasis, however both HS and CS are impacted in this organ. The fact that a substantial increase in the red pulp area associates with increased reticular fiber staining suggests that loss of HS and CS has caused stromal tissue instability leading to a compensatory increased reticular tissue production. The Xylt2−/− mice do have liver fibrosis [22]. Therefore, the splenic changes could be exacerbated secondary to portal vein hypertension due to liver fibrosis as seen in polycystic kidney patients [41]. Furthermore, with the extensive loss of GAGs from decorin, defective collagen III fibrillogenesis leading to abnormalities in reticular fibers should not be dismissed. Our previous data suggests that cells rely on one XylT isoform over another and that XylT2 predominates in many cells [15] and Roch et al., had similar findings [37]. In the spleens of the Xylt2−/− mice, HS glucosamine disaccharides that contain 2-O-sulfo uronic acids were spared suggesting these GAGs arose from XylT1 activity and not XylT2 since XylT2 is missing in the Xylt2−/− mice. For splenic CS disaccharides in the Xylt2−/− mice, a similar observation is found for 6-O-sulfated disaccharides. The mechanistic details for this are complex and left up to speculation.
In the lung, there are significant changes grossly, biochemically, and functionally. Nearly all disaccharides are affected with no selective decrease in any specific disaccharide. Surprisingly, given the large decrease in XylT activity and change in weight, the lung histologically is relatively normal. However, when the Xylt2−/− mice are challenged with intratracheal LPS, they have an exaggerated inflammatory cell infiltration and hemorrhage. The loss of GAGs in the Xylt2−/− may create changes in the surfactant, ECM, and alveolar basement membrane that result in increased baseline permeability of the blood-air barrier or lead to increased damage to this structure with LPS exposure. The GAGs are important to the integrity of these structures and their homeostasis [42] and when absent this may lead to a greater instability with a higher susceptibility to vascular leakage and leukocyte diapedesis. In addition, GAGs and the pulmonary glycocalyx has an important homeostatic role that is disturbed with trauma [43]. In the case of the Xylt2−/− mice, reduced GAGs in the glycocalyx may facilitate LPS access to the cell surface TLR4 receptor its known to activate. Thus, PG and GAG levels could be modifiers of disease in this organ. We have hypothesized previously that PG levels may be genetic modifiers of single gene disorders [22] and given the wide range of effects imposed by tissue PGs, their levels could very easily be genetic modifiers of polygenic disorders. Decreased PGs/GAGs could arise from decreased GAG assembly, increased degradation due to trauma and infection, or an imbalance of turnover and replacement. All of these processes could be influenced by genetic background leading to changes in GAG levels and significantly impacting disease severity.
We had originally describe XylT2 deficiency in humans responsible for spondylo-ocular syndrome [23] this has been confirmed by others [24–27]. Although some variability exists between patients, possibly due to the causative mutation and genetic background of individuals, there are some consistent phenotypic findings suggesting that the tissues affected are dependent on XylT2 activity. These patients commonly experience facial dysmorphism, cataracts, detached retina, osteoporosis, platyspondyly, long bone fractures, and reduced height. Other abnormalities not consistently assessed, or if they were are variable in patients, are developmental delay, hearing defects, and sternum defects [25]. Since extensive biochemical analyses of tissues are prohibitive in humans, the value of the Xylt2−/− mice is apparent in assessing GAG changes in tissues as a result of loss of XylT2. The Xyt2−/− mice, appear relatively normal under standard conditions except for those tissues that have been extensively and systematically phenotyped such as the liver, kidney, and adipose tissue [22, 44]. These tissues/organs have not been extensively assessed in patients or the age of the patients precludes a final assessment of these defects. At present, no gross fractures have been observed in the Xylt2−/− mice. As skeletal weight bearing in mice is different than in humans, a lack of fractures in the mice is not a reliable clinical parameter, but an extensive phenotyping of the bone tissue is under way as is for retina and ocular defects.
Previously we have shown in cultured cells of various origins that very few cell types express both XylT1 and XylT2 [15]. However, tissue expression analyses shows that most organs express both but the liver, lung, and heart seemingly are XylT2 dependent [17, 22]. In this paper we further show the loss of XylT2 activity impacts these three organs dramatically in regard to total XylT activity in the Xylt2−/− mice, and that spleen and brain are significantly affected. It needs to be noted that we had previously found an age-specific organ dysfunction in the liver and kidney and that there was no compensatory gain in Xylt1 expression in these organs [22]. In this paper, we demonstrate that the Xylt2−/− spleen is abnormal too;however, the lung is normal until challenged suggesting there is some homeostatic threshold either of glycosylation or physiology or both maintained in this organ of the Xylt2−/− mice despite the decrease in XylT activity and changes in glycosylation. As for the liver and kidney, this is maintained by residual XylT1 activity and likely not an increase in Xylt1 expression. Only until challenged is this threshold compromised. Therefore, the disaccharide alterations we see in the Xylt2−/− mice, due to residual XylT1 activity, could be providing some functional glycosylation threshold that maintains organ homeostasis. We don’t believe this is due to an increase in Xylt1 gene expression. In all, these findings support the concept that cells can express both Xylt1 and Xylt2 but usually one predominates possibly based on availability of acceptor and acceptor affinity. Also these findings suggest that cells within a tissue cannot fully replace or compensate the full loss of XylT2 activity by utilizing XylT1. Given the cellular heterogeneity in an organ, XylT activity needs further study at the cellular level. For example, since some sulfation modifications are preserved in the Xylt2−/− mice, our results indicate that some cells within those tissues depend on XylT1 to initiate the assembly of those GAGs, but knowing which cells these are and if these changes are compensatory is unknown and needs further study. Lastly, this paper’s findings suggest and support the view that changes in GAG levels may be important modifiers of disease.
Supplementary Material
Acknowledgements
This work was supported by Oklahoma Center for Advancement of Science and Technology, Oklahoma Center for Adult Stem Cell Research, and National Institutes of Health DK087989. We are thankful to the University of California at San Diego’s Glycoanalytics Core facility as well for expertise in disaccharide analyses. We are also very thankful for the helpful statistical advice of Ms. Binu Sharma.
Abbreviations
- CS
Chondroitin sulfate
- DMEM
Dulbecco’s modified Eagle medium
- ECM
extracellular matrix
- EDTA
ethylenediaminetetracetic acid
- FITC
fluorescein isothiocyanate
- GAG
glycosaminoglycan
- HP
heparin sulfate
- HS
Heparan sulfate
- LPS
bacterial lipopolysaccharide
- PAGE
Polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- PG
proteoglycans
- RBCs
red blood cells
- XylT2
xylosyltransferase 2
- XylT1
xylosyltransferase 1
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10719-020-09945-9) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare no conflict of interests.
Ethical approval All mice were housed in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International: The Institutional Animal Care and Use Committees of all institutions approved all animal procedures and experiments.
References
- 1.Esko JD, Kimata K, and Lindahl U: Proteoglycans and Sulfated Glycosaminoglycans. In: Essentials of glycobiology. pp. xxix, 784 p. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (2009) [PubMed] [Google Scholar]
- 2.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M: Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem 68, 729–777 (1999) [DOI] [PubMed] [Google Scholar]
- 3.Esko JD, Selleck SB: Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem 71, 435–471 (2002) [DOI] [PubMed] [Google Scholar]
- 4.Gustafsson M, Boren J: Mechanism of lipoprotein retention by the extracellular matrix. Curr. Opin. Lipidol 15(5), 505–514 (2004) [DOI] [PubMed] [Google Scholar]
- 5.Mahley RW, Ji ZS: Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res 40(1), 1–16 (1999) [PubMed] [Google Scholar]
- 6.Perrimon N, Bernfield M: Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 404(6779), 725–728 (2000) [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg RD, Shworak NW, Liu J, Schwartz JJ, Zhang L: Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J Clin Invest 99(9), 2062–2070. (1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hacker U, Nybakken K, Perrimon N: Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 6(7), 530–541 (2005) [DOI] [PubMed] [Google Scholar]
- 9.Iozzo RV: Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 6(8), 646–656 (2005) [DOI] [PubMed] [Google Scholar]
- 10.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH: Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U. S. A 100(4), 1885–1890 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kearns AE, Vertel BM, Schwartz NB: Topography of glycosylation and UDP-xylose production. J. Biol. Chem 268(15), 11097–11104 (1993) [PubMed] [Google Scholar]
- 12.Stoolmiller AC, Horwitz AL, Dorfman A: Biosynthesis of the chondroitin sulfate proteoglycan. Purification and properties of xylosyltransferase. J Biol Chem. 247(11), 3525–3532 (1972) [PubMed] [Google Scholar]
- 13.Iozzo RV: Proteoglycan Protocols. Humana Press, Totowa, NJ: (2001) [Google Scholar]
- 14.Wilson IB: The never-ending story of peptide O-xylosyltransferase. Cellular and molecular life sciences : CMLS. 61(7–8), 794–809 (2004). 10.1007/s00018-003-3278-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuellar K, Chuong H, Hubbell SM, Hinsdale ME: Biosynthesis of chondroitin and heparan sulfate in Chinese hamster ovary cells depends on xylosyltransferase II. J. Biol. Chem 282(8), 5195–5200 (2007). M611048200 [pii]). 10.1074/jbc.M611048200 [DOI] [PubMed] [Google Scholar]
- 16.Gotting C, Kuhn J, Zahn R, Brinkmann T, Kleesiek K: Molecular cloning and expression of human UDP-d-xylose: proteoglycan core protein beta-d-xylosyltransferase and its first isoform XT-II. J. Mol. Biol 304(4), 517–528 (2000). 10.1006/jmbi.2000.4261 [DOI] [PubMed] [Google Scholar]
- 17.Ponighaus C, Ambrosius M, Casanova JC, Prante C, Kuhn J, Esko JD, Kleesiek K, Gotting C: Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. J. Biol. Chem 282(8), 5201–5206 (2007). 10.1074/jbc.M611665200 [DOI] [PubMed] [Google Scholar]
- 18.Voglmeir J, Voglauer R, Wilson IB: XT-II, the second isoform of human peptide-O-xylosyltransferase, displays enzymatic activity. J. Biol. Chem 282(9), 5984–5990 (2007). 10.1074/jbc.M608087200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunner A, Kolarich D, Voglmeir J, Paschinger K, Wilson IB: Comparative characterisation of recombinant invertebrate and vertebrate peptide O-Xylosyltransferases. Glycoconj. J 23(7–8), 543–554 (2006) [DOI] [PubMed] [Google Scholar]
- 20.Esko JD, Elgavish A, Prasthofer T, Taylor WH, Weinke JL: Sulfate transport-deficient mutants of Chinese hamster ovary cells. Sulfation of glycosaminoglycans dependent on cysteine. J Biol Chem. 261(33), 15725–15733 (1986) [PubMed] [Google Scholar]
- 21.Esko JD, Stewart TE, Taylor WH: Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. U. S. A 82(10), 3197–3201 (1985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Condac E, Silasi-Mansat R, Kosanke S, Schoeb T, Towner R, Lupu F, Cummings RD, Hinsdale ME: Polycystic disease caused by deficiency in xylosyltransferase 2, an initiating enzyme of glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. U. S. A 104(22), 9416–9421 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munns CF, Fahiminiya S, Poudel N, Munteanu MC, Majewski J, Sillence DO, Metcalf JP, Biggin A, Glorieux F, Fassier F, Rauch F, Hinsdale ME: Homozygosity for frame-shift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am. J. Hum. Genet 96(6), 971–978 (2015). 10.1016/j.ajhg.2015.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylan F, Costantini A, Coles N, Pekkinen M, Heon E, Siklar Z, Berberoglu M, Kampe A, Kiykim E, Grigelioniene G, Tuysuz B, Makitie O: Spondyloocular syndrome: novel mutations in XYLT2 gene and Expansion of the phenotypic Spectrum. J. Bone Miner. Res 31(8), 1577–1585 (2016). 10.1002/jbmr.2834 [DOI] [PubMed] [Google Scholar]
- 25.Guleray N, Simsek Kiper PO, Utine GE, Boduroglu K, Alikasifoglu M: Intrafamilial variability of XYLT2-related spondyloocular syndrome. Eur J Med Genet. 62(11), 103585 (2019). 10.1016/j.ejmg.2018.11.019 [DOI] [PubMed] [Google Scholar]
- 26.Taylan F, Yavas Abali Z, Jantti N, Gunes N, Darendeliler F, Bas F, Poyrazoglu S, Tamcelik N, Tuysuz B, Makitie O: Two novel mutations in XYLT2 cause spondyloocular syndrome. Am. J. Med. Genet. A 173(12), 3195–3200 (2017). 10.1002/ajmg.a.38470 [DOI] [PubMed] [Google Scholar]
- 27.Umair M, Eckstein G, Rudolph G, Strom T, Graf E, Hendig D, Hoover J, Alanay J, Meitinger T, Schmidt H, Ahmad W: Homozygous XYLT2 variants as a cause of spondyloocular syndrome. Clin. Genet 93(4), 913–918 (2018). 10.1111/cge.13179 [DOI] [PubMed] [Google Scholar]
- 28.Gotting C, Kuhn J, Kleesiek K: Human xylosyltransferases in health and disease. Cellular and molecular life sciences : CMLS. 64(12), 1498–1517 (2007). 10.1007/s00018-007-7069-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Condac E, Dale GL, Bender-Neal D, Ferencz B, Towner R, Hinsdale ME: Xylosyltransferase II is a significant contributor of circulating xylosyltransferase levels and platelets constitute an important source of xylosyltransferase in serum. Glycobiology. 19(8), 829–833 (2009). cwp058 [pii]). 10.1093/glycob/cwp058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeil U, Wenzel KW: Purification and some properties of UDP-xylosyltransferase of rat ear cartilage. Glycobiology. 10(8), 803–807 (2000) [DOI] [PubMed] [Google Scholar]
- 31.Fisher LW, Stubbs JT 3rd, Young MF: Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop. Scand 266, 61–65 (1995) [PubMed] [Google Scholar]
- 32.Brown AH: Determination of pentose in the presence of large quantities of glucose. Arch. Biochem 11, 269–278 (1946) [PubMed] [Google Scholar]
- 33.Ledin J, Staatz W, Li JP, Gotte M, Selleck S, Kjellen L, Spillmann D: Heparan sulfate structure in mice with genetically modified heparan sulfate production. J. Biol. Chem 279(41), 42732–42741 (2004) [DOI] [PubMed] [Google Scholar]
- 34.Warda M, Toida T, Zhang F, Sun P, Munoz E, Xie J, Linhardt RJ: Isolation and characterization of heparan sulfate from various murine tissues. Glycoconj. J 23(7–8), 555–563 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruckner J: Estimation of monosaccharides by the orcinol-sulphuric acid reaction. Biochem. J 60(2), 200–205 (1955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, Cummings RD, Esko JD: Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem 283(48), 33674–33684 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roch C, Kuhn J, Kleesiek K, Gotting C: Differences in gene expression of human xylosyltransferases and determination of acceptor specificities for various proteoglycans. Biochem. Biophys. Res. Commun 391(1), 685–691 (2010). 10.1016/j.bbrc.2009.11.121 [DOI] [PubMed] [Google Scholar]
- 38.Sivasami P, Poudel N, Munteanu MC, Hudson J, Lovern P, Liu L, Griffin T, Hinsdale ME: Adipose tissue loss and lipodystrophy in xylosyltransferase II deficient mice. Int. J. Obes 43, 1783–1794 (2019). 10.1038/s41366-019-0324-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hallgren O, Nihlberg K, Dahlback M, Bjermer L, Eriksson LT, Erjefalt JS, Lofdahl CG, Westergren-Thorsson G: Altered fibroblast proteoglycan production in COPD. Respir. Res 11, 55 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Straaten JF, Coers W, Noordhoek JA, Huitema S, Flipsen JT, Kauffman HF, Timens W, Postma DS: Proteoglycan changes in the extracellular matrix of lung tissue from patients with pulmonary emphysema. Mod. Pathol 12(7), 697–705 (1999) [PubMed] [Google Scholar]
- 41.O’Brien K, Font-Montgomery E, Lukose L, Bryant J, Piwnica-Worms K, Edwards H, Riney L, Garcia A, Daryanani K, Choyke P, Mohan P, Heller T, Gahl WA, Gunay-Aygun M: Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease. J. Pediatr. Gastroenterol. Nutr. 54(1), 83–89 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park PW: Introduction to the thematic mini-review series on “matrix biology in lung health and disease”. Matrix Biol. 73, 1–5 (2018). 10.1016/j.matbio.2018.07.002 [DOI] [PubMed] [Google Scholar]
- 43.Chignalia AZ, Yetimakman F, Christiaans SC, Unal S, Bayrakci B, Wagener BM, Russell RT, Kerby JD, Pittet JF, Dull RO: The Glycocalyx and trauma: a review. Shock. 45(4), 338–348 (2016). 10.1097/SHK.0000000000000513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sivasami P, Poudel N, Munteanu MC, Hudson J, Lovern P, Liu L, Griffin T, Hinsdale ME: Adipose tissue loss and Lipodystrophy in Xylosyltransferase II deficient mice. International Journal of Obesity (Accepted). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.