

SUMMARY
The standard of care is unsuccessful to treat recurrent and aggressive soft-tissue sarcomas. Interventions aimed at targeting components of the tumor microenvironment have shown promise for many solid tumors yet have been only marginally tested for sarcoma, partly because knowledge of the sarcoma microenvironment composition is limited. We employ single-cell RNA sequencing to characterize the immune composition of an undifferentiated pleiomorphic sarcoma mouse model, showing that macrophages in the sarcoma mass exhibit distinct activation states. Sarcoma cells use the pleiotropic cytokine macrophage migration inhibitory factor (MIF) to interact with macrophages expressing the CD74 receptor to switch macrophages’ activation state and pro-tumorigenic potential. Blocking the expression of MIF in sarcoma cells favors the accumulation of macrophages with inflammatory and antigen-presenting profiles, hence reducing tumor growth. These data may pave the way for testing new therapies aimed at re-shaping the sarcoma microenvironment, in combination with the standard of care.
Graphical abstract
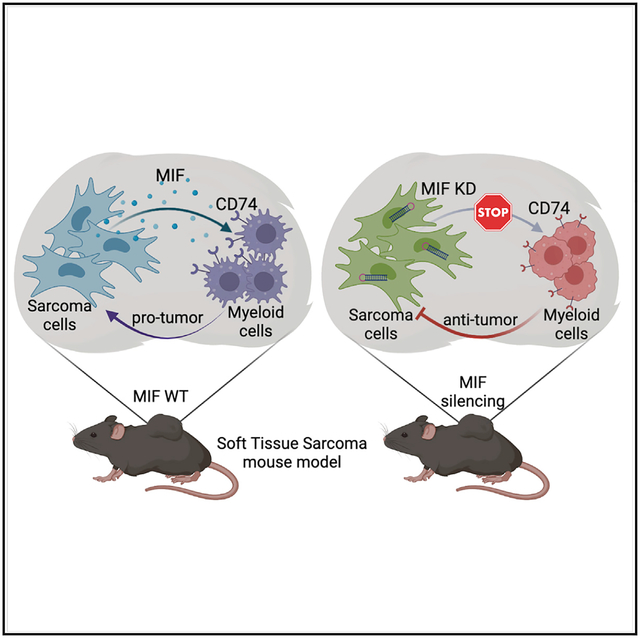
In brief
Macrophages are the main immune compartment of sarcoma. Tessaro et al. report that sarcoma cells interact with macrophages in specific transcriptional states through the soluble factor MIF. MIF signaling biases macrophage functional state and pro-tumorigenic potential. Blocking these interactions leads to differential enrichment of macrophage states and tumor reduction.
INTRODUCTION
Soft-tissue sarcomas (STSs) are tumors of heterogeneous histology that originate from cells of mesenchymal lineage (Cancer Genome Atlas Research Network, 2017). Sarcoma subtypes with complex genetics, such as undifferentiated pleiomorphic sarcoma (UPS), myxofibrosarcoma (MFS), and leiomyosarcoma (LMS) are recurrent and aggressive in adults and often metastatic to the lungs (Eisinger-Mathason et al., 2013; Kelleher and Viterbo, 2013). Conventional therapies, such as chemotherapy and radiotherapy, are often non-curative for the recurrent disease (Grunewald et al., 2020). Novel therapies, including approaches of immunotherapy (e.g., immune checkpoint inhibitors targeting PD1/PDL1 to reinvigorate exhausted T cells; Cogdill et al., 2017), have so far provided mixed results in sarcomas (Linch et al., 2014; Toulmonde et al., 2018). Scarcity of comprehensive characterizations of the critical components of the sarcoma microenvironment is one of the main complications that hinder the discovery of new and more effective therapies. Thus, it is necessary to identify sarcoma cell-extrinsic contributions, both cellular and molecular, responsible for the progression of these aggressive tumors.
Myeloid cells are the most abundant immune components of sarcomas as well as many epithelial tumors, in which they become tumor-associated macrophages (TAMs). While in principle, macrophages could have anti-tumorigenic properties, the tumor microenvironment critically re-shapes the activities of TAMs such that they positively contribute to tumor growth by creating an immunosuppressive microenvironment (Cheng et al., 2021), favoring angiogenesis and by releasing metabolites that have recently emerged as drivers of drug-resistance mechanisms (El-Kenawi et al., 2021). In this respect, interventions aimed at blocking either the myeloid-cell-recruiting cytokine CSF1 or its receptor CSF1R, which are expressed by tumor and myeloid cells, respectively, have been demonstrated in several carcinomas to block recruitment and survival of TAMs (Cassetta and Pollard, 2018). Recently, pexidartinib, an anti-CSF1R inhibitor, has been tested in clinical trials for multiple solid tumors and preclinically in sarcoma (Fujiwara et al., 2021).
Whether TAMs are critical for soft-tissue sarcoma progression and therapeutic response has been only marginally investigated. Here, we sought to determine the mechanisms used by the sarcoma cells to elicit the pro-tumorigenic potential of the TAMs using an immune-competent murine model that recapitulates genetic alterations found in the complex-karyotype sarcomas in patients. We reported a comprehensive single-cell RNA sequencing (scRNA-seq) characterization of sarcoma-associated macrophages and their expression programs, including the mechanisms of interaction with the tumor cells. By executing functional studies in vitro and in vivo, we identified specific microenvironmental elements of the tumor mass that contribute to the TAMs’ pro-tumorigenic functions. These mechanisms represent possible new clinical targets for myeloid-related interventions, in combination with conventional therapies and additional immunotherapies.
RESULTS
Myeloid cells promote tumor growth in a soft-tissue sarcoma mouse model
Recent reports showed that STSs present diverse immune infiltration levels, ranging from highly infiltrated to immune excluded (Petitprez et al., 2020). Furthermore, regardless of degree of infiltration, we found that the bulk RNA-seq profile of STS samples in the The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network, 2017) is dominated by monocyte and macrophage signatures, compared with lymphocytes (T, B, and NK) and other immune cells of the tumor mass (Figure S1A). To corroborate these results, we collected three distinct subtypes of human soft-tissue sarcoma and performed flow cytometry, confirming that monocytes and macrophages represented the largest immune compartments in sarcoma (Figures 1A and S1B).
Figure 1. Macrophages promote sarcoma growth.

(A) Flow cytometry of the main immune populations infiltrating STS, gated in CD45+ (n = 3 independent human sarcomas). DDLPS, dedifferentiated liposarcoma; GIST, gastrointestinal stromal tumor; LMS, leiomyosarcoma.
(B) Schematic representation of the UPS mouse models used (left) and analysis of immune cells that populate the murine sarcoma mass (right).
(C) Schematic representation of the treatment schedule administered to test the effect of anti-CSF1R on sarcoma growth.
(D) Relative proportion of macrophages (CD45+ F4/80+) infiltrating tumors treated with IgG control or anti-CSF1R.
(E) Relative tumor weight after treatment with IgG control or anti-CSF1R (left) and relative % of dsRED+ tumor cells in flow cytometry (right).
(F) CD8+ and CD4+ T cells in proportion to all CD45+ cells infiltrating tumors treated with IgG control or anti-CSF1R.
(G) Schematic representation of the in vivo experiments to test the role of macrophages in promoting sarcoma growth. Bone-marrow-derived macrophages (BMDMs) were generated by macrophage colony stimulating factor (M-CSF) stimulation in vitro and then co-injected with tumor cells. The relative tumor weights (tumor cells alone versus co-injected with BMDMs) are shown on the right.
Unless otherwise indicated, results are presented as mean ± SEM, and results are compared by Student’s t test. *p < 0.05 and **p < 0.01. ns, not significant. See also Figure S1.
To characterize the functional contribution of myeloid cells to sarcoma progression, we employed an immune-competent mouse model of UPS (Guarnerio et al., 2015), which recapitulates genetic aberrations found in patients (Cancer Genome Atlas Research Network, 2017). Accordingly, sarcoma cells were generated by Tp53 knockout and Ccne1 overexpression in mouse mesenchymal stromal cells (Figure 1B), representing two genetic lesions concomitantly found in 14% of human STS, with higher prevalence in UPS, MFS, and LMS (Cancer Genome Atlas Research Network, 2017). In this mouse model, CD45+ immune cells composed 24% ± 4% of the tumor mass. As with human sarcoma samples, monocytes and macrophages were the main components of the CD45+ compartment. Monocytes (CD11b+F4/80−) represented around 40% and macrophages (CD11b+F4/80+) nearly 50% of tumor-infiltrating immune cells. T, B, and NK cells were proportionally scarcer, constituting <10% (Figures 1B and S1C).
To functionally characterize the role of myeloid cells, we employed anti-CSF1R antibodies, which have been evaluated in tumors of multiple histology to block myeloid cell recruitment and survival (Cassetta and Pollard, 2018). We treated the subcutaneous sarcomas, starting at ~0.8 cm diameter, with four doses of anti-CSF1R monoclonal antibody or of immunoglobulin G (IgG) control (Figure 1C). Anti-CSF1R treatment reduced CD11b+F4/80+ macrophages in the tumor mass (Figure 1D) and importantly also reduced tumor burden (Figures 1E and S1D), suggesting that myeloid cells may play pro-tumorigenic functions. No differences were observed in the percentage of T cells present (Figure 1F).
To further assess pro-tumorigenic properties of the macrophages, we performed functional assays in vitro and in vivo. First, we investigated whether the sarcoma cell “secretome” could influence the activation status of tumor-naive macrophages and increase their expression of Arg1 and Nos2 (iNOS), which have been associated with the capacity of myeloid cells to play immune-suppressive and pro-tumorigenic roles (Grzywa et al., 2020). Accordingly, we generated tumor-naive macrophages from bone marrow hematopoietic progenitors and cultured them in medium conditioned by sarcoma cells and their secreted factors. Like the macrophages isolated from the tumor mass (Figure S1E), bone-marrow-derived macrophages (BMDMs) increased the expression of both Arg1 and Nos2 in the presence of tumor-conditioned medium (Figure S1F). Then, we measured in vivo the macrophages’ impact on the tumor growth by injecting tumor cells alone or together with BMDM. When co-injected with BMDM, the tumor cells generated larger tumors (Figure 1G).
Together, these data show that myeloid cells are the main immune cells of the UPS mouse model, mirroring proportions observed in human UPS. In the tumor mass, the myeloid cells appeared to be triggered by the tumor-conditioned microenvironment to acquire pro-tumorigenic properties, and depleting them was able to restrain tumor growth.
scRNA-seq reveals diverse transcriptional modules in myeloid cells
Despite reducing tumor burden, anti-CSF1R as a monotherapy has not sufficed to eradicate sarcoma. However, it has been recently reported that subtypes of myeloid cells differentially respond to CSF1R blockade and that myeloid cells expressing pro-angiogenic and immunosuppressive factors are the least responsive (Zhang et al., 2020). To identify whether the immune microenvironment of STSs contains such diverse myeloid subtypes or states, we employed scRNA-seq of the sarcoma mouse models. Sarcomas were collected when they reached ~1.5 cm diameter, digested, enriched for CD45+ immune cells, hashed, and pooled together for droplet-based scRNA-seq. After computationally removing dead cells and doublets, we identified clusters of cells encompassing the major immune cell types and sarcoma cells (Figures 2A and S2A).
Figure 2. Single-cell transcriptomic profiling of the sarcoma-associated myeloid cells.
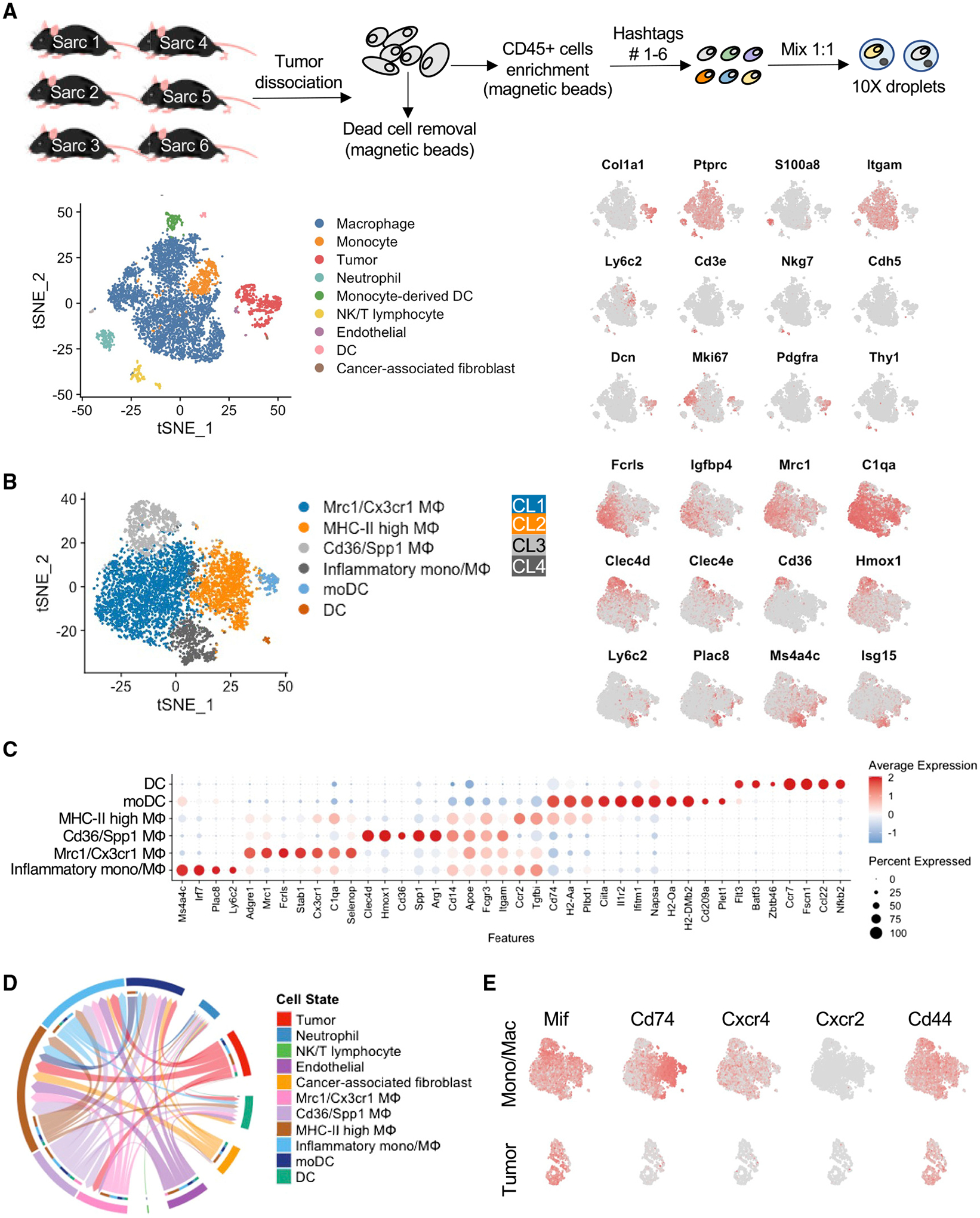
(A) Schematic representation of the experimental design for scRNA-seq analysis (top panel). t-distributed stochastic neighbor embedding (t-SNE) depicting the main immune cell types of the sarcoma mass of six murine p53KOCcne1+ UPS tumors (left bottom panel). Markers of major immune clusters are overlaid on the t-SNE (right bottom panel).
(B) t-SNE derived from sub-clustering of mononuclear phagocytes only (left panel). CL1–4 refer to clusters 1–4 as described in the text. Key myeloid genes broadly or differentially expressed by these cell subtypes are overlaid on the t-SNE (right panel). See also Table S1.
(C) Expression of top marker genes differentially expressed by the myeloid cell clusters.
(D) Circos plot of intercellular MIF signaling between all cell types in murine UPS tumors, with directionality of signaling indicated by arrowheads. The primary sources of Mif signaling are sarcoma cells (red arrows), stromal cells, and Cd36/Spp1 MΦ, while the primary targets of Mif signaling are cells expressing Cd74 along with Cxcr4 and/or Cd44: inflammatory mono/MΦ, MHC class II high MΦ, Mrc1/Cx3cr1 MΦ, and monocytic dendritic cells (moDCs).
(E) Expression of Mif and its receptor and co-receptor genes Cd44, Cd74, Cxcr2, and Cxcr4 by tumor or myeloid cells (right panel).
Six cell clusters of myeloid origin were identified (Figures 2B and 2C); among them, four clusters encompassed monocytes and macrophages in distinct activation states. Cluster 1 was composed of TAMs expressing Adgre1, Mrc1, Cxc3r1, and Fcrls (Figures 2B and 2C; Mrc1/Cx3cr1 Mφ), an expression profile identified in many tumors, including lung cancer (Zilionis et al., 2019) and several other epithelial tumors (Cheng et al., 2021). Consistent with previous reports, these cells expressed high levels of complement (C1qa, C1qb, and C1qc), which have been reported to function pro-tumorigenically by triggering tumor cell growth and metastasis potential (Afshar-Kharghan, 2017). In addition to its role in the complement cascade, C1q can function in the absence of the other C1 subunits as a bridging molecule from macrophages to apoptotic cells, thus mediating efferocytosis, which does not engage the classical complement pathway (Pulanco et al., 2017). These macrophages also expressed high levels of Sirpa, receptor of the “don’t eat me” molecule Cd47, which is highly expressed in the sarcoma cells (data not shown). Cluster 2 macrophages (Figures 2B and 2C; MHC-II high Mφ) expressed major histocompatibility complex class II (MHC class II) and other genes relevant to antigen presentation (H2-Ab1, H2-Aa, Cd74, and Ciita). Cluster 3 macrophages (Figures 2B and 2C; Cd36/Spp1 Mφ) expressed Clec4d, Spp1, and Cd36. Myeloid cells of similar profile have been observed in multiple human tumor histologies, often associated with hypoxic, angiogenic, and lipid-processing signatures (Cheng et al., 2021). Accordingly, cluster 3 macrophages expressed high levels of hypoxia-related (Hmox1, Bnip3, and Egr2) and lipid-metabolism-associated genes in addition to Cd36, such as Lipa, Fabp4, and the lipoprotein lipase (Lpl), whose expression in a subset of macrophages has been correlated with shorter survival in non-small cell lung cancer (Podgornik et al., 2013). Expression of metalloproteinases (Mmp9, Mmp12, and Mmp19) and cathepsin-encoding genes (Ctsb, Ctsd, and Ctsl)—associated with remodeling of the extracellular matrix (Qi et al., 2022)—were also enhanced in Cd36/Spp1 Mφ. Myeloid cells expressing SPP1 have been noted in multiple human cancers for their immunosuppressive properties (Cheng et al., 2021); accordingly, cluster 3 macrophages also expressed high levels of immune-suppressive genes Lgals3 (encoding galectin-3) and Gpnmb, which is upregulated in macrophages upon transforming growth factor β (TGF-β) stimulation (Nickl et al., 2021) and underlies tumorigenic properties of the so-called “M2”-polarized macrophages, by promoting cancer stem cell maintenance and metastasis via CD44 and interleukin-33 (IL-33) (Liguori et al., 2021).
Cluster 4 (Figures 2B and 2C; inflammatory Mono/Mφ) included both inflammatory monocytes (Ly6c2 positive) and macrophages (Ly6c2 negative), expressing high levels of interferon-related genes, including Ifit1, Oasl1, Irf7, Cxcl9, Cxcl10, and Isg15. Cells exhibiting this signature have been described by previous investigations as “freshly” recruited myeloid cells in the tumor mass and associated with anti-tumorigenic properties (Medrano et al., 2017).
Finally, of dendritic cell subsets, one expressed conventional dendritic cell (DC) markers Batf3, Zbtb46, and Irf8 as well as a signature associated with an “activated” state (Fscn1, Ccr7, Ccl22, and Nfkb2)—previously described in the context of in vitro lipopolysaccharide (LPS) stimulation (Zanoni et al., 2009) and later in human lung cancer (Zilionis et al., 2019). Other dendritic cells expressed Cd209a while lacking Flt3, consistent with the profile of monocyte-derived DCs. This subset of DCs also expressed high levels of antigen-presentation- and interferon-related genes (Figures 2B and 2C).
Having identified diverse populations of sarcoma-infiltrating myeloid cells with potentially pro- or anti-tumorigenic functions, we next investigated whether and how tumor cells could regulate the functions of macrophages, possibly by changing the balance of macrophage subtypes to enhance pro-tumorigenic behaviors. Accordingly, we identified ligand-receptor pairs that are respectively expressed on tumor cells and four different subtypes of macrophages—and which thereby represent possible mechanisms of interaction or recruitment between tumor and specific macrophage subtypes and states (Figures 2A and 2B). Among the strongest interacting partners were Mif-(Cd74+Cd44), Mif-(Cd74+Cxcr4) (Figure 2D), and App-Cd74 (Figure S2B), where Cd44 and Cxcr4 are co-receptors of Cd74. Cd74, encoding the cognate receptor for MIF, was strongly expressed in both the antigen-presenting and inflammatory MΦ clusters identified by scRNA-seq, while its expression was diminished in the Cd36/Spp1 MΦ cluster (Figure 2C). In contrast, two MIF co-receptor genes Cxcr4 and Cd44 were expressed uniformly but less abundantly across myeloid cells, while the alternative co-receptor Cxcr2 was not expressed (Figure 2E).
Because Cd74 is variably expressed within myeloid cells, we hypothesized that myeloid cells could be differentially affected by CD74’s ligands MIF or amyloid beta precursor protein (APP), which are both soluble molecules expressed abundantly by sarcoma cells (Figure S2B). We therefore investigated whether expression of MIF or APP in tumor cells could alter the immune makeup of the microenvironment, affect tumor progression, or both.
Deciphering tumor-myeloid interactions supports MIF as sarcoma-promoting factor
Involvement of MIF and APP in promoting STSs has never been investigated. Likewise, limited information exists on the capacity of MIF and APP to shape the cellular composition and functional orientation of the tumor microenvironment (TME). Analyzing bulk RNA-seq data from TCGA STS samples, expression levels of MIF, but not of APP, significantly correlated with patient prognosis, with patients presenting higher levels of MIF, showing reduced disease-free survival (Figures 3A and S3A). In addition, MIF is endogenously expressed in multiple mouse and human sarcoma cell lines (Figure 3B), and its mRNA and protein expression levels increase upon cell exposure to hypoxic conditions (Figures 3C and S3B). Based on these assessments, we tested whether tumor-expressed MIF could promote sarcoma. To investigate its role in vivo, Mif was silenced in the p53KOCcne1+ murine sarcoma cells by employing, in two parallel experiments, either CRISPR-Cas9 tools (to achieve knockout [MIF-KO]) or short hairpins RNAs (shRNAs) (to achieve knockdown [MIF-KD]) (Figure 3D). In both KO and KD cases, Mif-silenced sarcoma cells grew significantly smaller tumors than Mif-wild type (WT) (Figures 3E and 3F), suggesting that expression of Mif by the sarcoma cells is critical for tumor growth.
Figure 3. MIF silencing in the tumor cells reduces sarcoma growth.
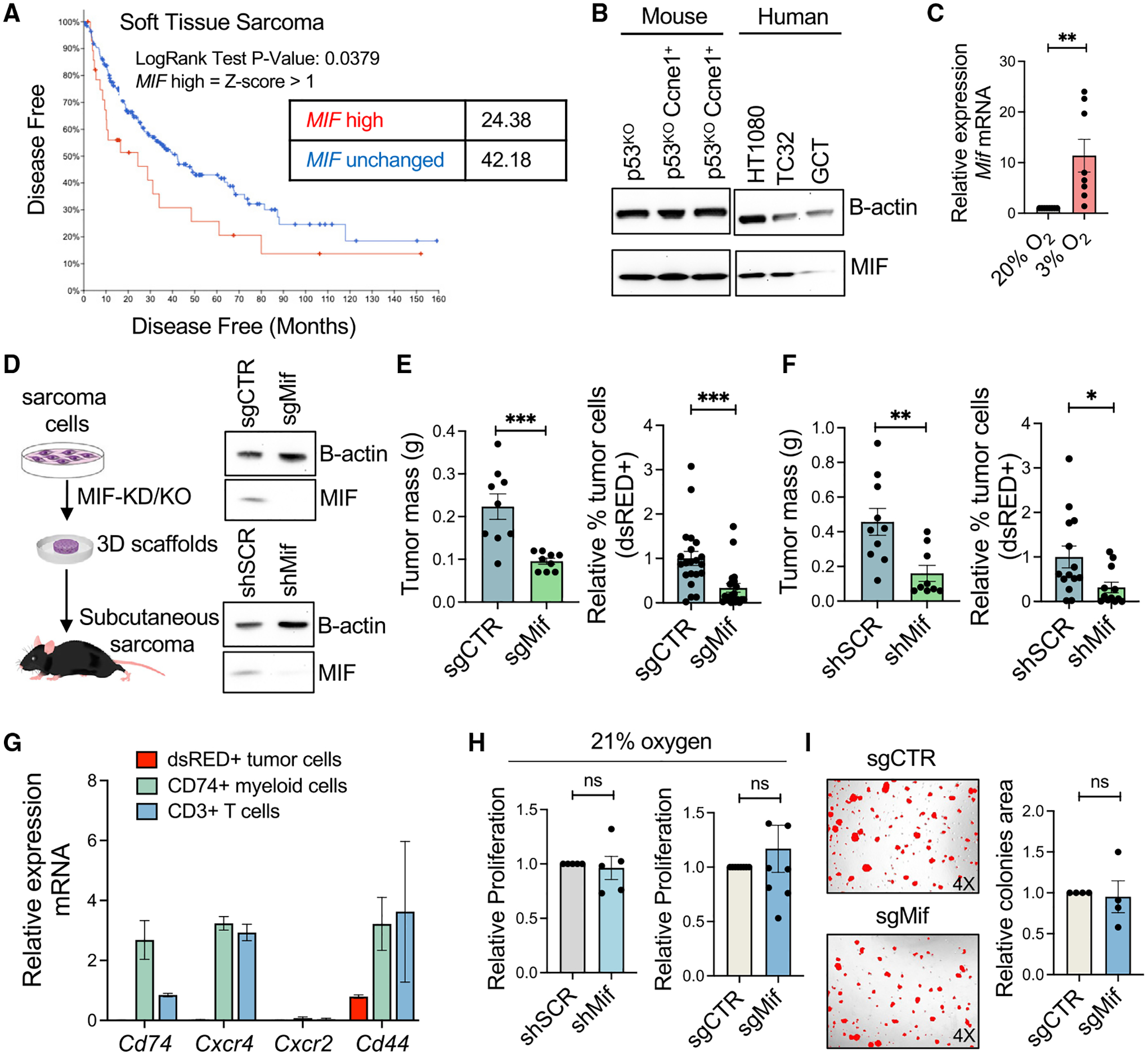
(A) Survival analysis of human soft tissue sarcoma in the TCGA, stratified by MIF mRNA expression level.
(B) Western blot verification of MIF protein expression in mouse and human sarcoma cells.
(C) qRT-PCR analysis of Mif mRNA expression levels in normoxic (20% O2) or hypoxic (3% O2) culture conditions.
(D) Schematic representation of the generation of MIF-KO and KD sarcoma models and assessment of MIF KO or KD by western blot.
(E) Relative growth in vivo of MIF-WT (sgCTR) and MIF-KO (sgMif) sarcoma cells, represented as tumor mass at day 21 (left panel) and relative proportion of total digested tumor that was sarcoma cells (right panel).
(F) Relative growth in vivo of MIF-WT (shSCR) and MIF-KD (shMif) sarcoma cells, represented as tumor mass at day 21 (left panel) and relative proportion of total digested tumor that was sarcoma cells (right panel).
(G) Relative expression of Cd74, Cxcr2, Cd44, and Cxcr4 in dsRed+ tumor cells, CD74+ myeloid cells, and CD3+ T cells. All cells were FACS sorted from whole mouse tumors.
(H) Relative proliferation in vitro of MIF-WT (shSCR) and MIF-KD (shMif) sarcoma cells (left) and of MIF-WT (sgCTR) and MIF-KO (sgMif) sarcoma cells (right).
(I) Relative capacity of spheroid formation for the MIF-WT (sgCTR) and MIF-KO (sgMif) sarcoma cells in vitro.
Unless otherwise indicated, results are presented as mean ± SEM and results are compared by Student’s t test. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S3.
We first investigated whether MIF’s effect was cell autonomous in the sarcoma cells. MIF can display autocrine activity, sustaining the proliferation of tumor cells that express both the MIF receptor CD74 and one of its co-receptors CXCR2, CXCR4, and CD44 (Penticuff et al., 2019). scRNA-seq data already excluded that sarcoma cells express Cd74, Cxcr4, or Cxcr2; this was corroborated by fluorescence-activated cell sorting (FACS) whole tumors and probing the MIF receptor genes by qRT-PCR. They were not detected in the sarcoma cells but only in the tumor-infiltrating myeloid and lymphoid cells (Figure 3G).
However, MIF was recently reported to function intracellularly and independently of any of these receptors as a nuclease regulating DNA replication (Wang et al., 2021), thus potentially promoting tumor growth in another cell-intrinsic manner. Accordingly, we compared proliferation rates of MIF-KO/KD and MIF-WT sarcoma cells in vitro. Mif silencing did not reduce cell proliferation, whether seeded in normal growth conditions (medium containing 10% FBS and normoxia) (Figure 3H) or in hypoxic conditions (3% O2) (Figure S3C). Similarly, no proliferation defects were observed for the Mif-silenced sarcoma cells when grown as spheroids (Figure 3I) or when cultured with limited growth factors (1% FBS) (Figure S3D). Furthermore, comparing expression profiles of MIF-KO/KD and MIF-WT cells by RNA-seq of FACS-purified tumors (n = 4 each group), few genes were differentially expressed between the MIF-KO/KD sarcoma cells and their respective controls (108 genes up and 80 genes down in MIF-KD tumors, out of 15,303 filtered genes, at 5% false discovery rate [FDR]), with no downregulation in pathways related to cell cycle, making it unlikely that the reduction in tumor size could be due to cell-autonomous growth defects following MIF KD (Figure S3E). In the case of the KO experiment, the sgMif cells even showed slight (though non-significant) upregulation in cell cycle pathways compared with cells from the larger sgCTR tumors (Figure S3F), further diminishing the possibility that the reduced size of sgMif tumors could be explained by a reduction in proliferative capacity.
Having excluded a cell-autonomous role for MIF in promoting tumor growth, we investigated whether tumor MIF could change the TME content. In flow cytometry, we observed that MIF-silenced tumors were globally more infiltrated by CD45+ cells, compared with MIF-WT tumors (Figure 4A). More specifically, there was enrichment in monocytes and CD4+ T cells. No differences were observed for macrophages and CD8+ T cells (Figures 4B and 4C). Then, we analyzed the transcriptional states of the myeloid cells across conditions by scRNA-seq (Figure S4A). Treating the four macrophage clusters described in Figure 2 as reference states, all cells identified as macrophages within MIF-KO or MIF-WT tumors were scored en masse for expression of the marker genes of each of the four “reference” macrophage subtypes. Although overall macrophage proportion had been similar in MIF-WT and MIF-KO/KD tumors, their transcriptional profile was markedly distinct, depending on MIF status of the tumor of origin. Macrophages from MIF-KO tumors exhibited greatest similarity, on average, to the MHC-II high MΦ cluster (Figure 4D) identified previously (Figure 2). In line with this, qRT-PCR of FACS-sorted myeloid cells (CD11b+) from MIF-WT and KO tumors showed that silencing Mif in the sarcoma cells favored the accumulation of inflammatory and antigen-presenting macrophages in the tumor (Figures 4D–4F). On the contrary, macrophages from the MIF-WT tumors were comparatively more similar to Cd36/Spp1 MΦ (Figure 4D) and compared with macrophages from MIF-KO tumors, upregulated pathways related to lipid processing, formation of extracellular matrix, and membrane-extracellular matrix (ECM) interactions (Figure 4E)—functions which have been ascribed to SPP1+ macrophages that predicted poor prognosis in colorectal cancer (Qi et al., 2022).
Figure 4. MIF silencing in the tumor cells re-shapes the myeloid microenvironment.
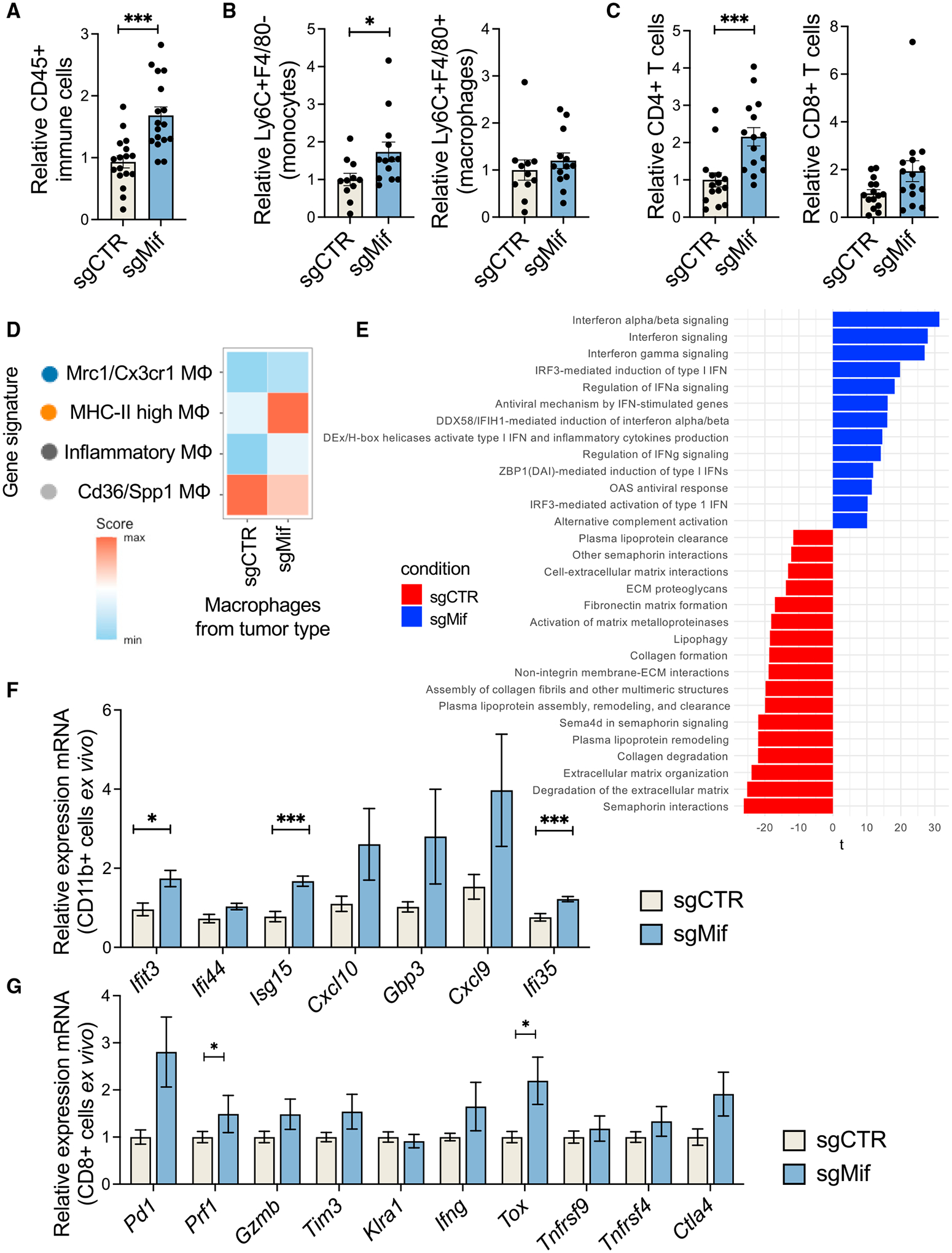
(A) Relative proportions in flow cytometry of immune cells (CD45+) infiltrating MIF-WT (sgCTR) and MIF-KO (sgMIFif) sarcomas.
(B) Relative proportions in flow cytometry of monocytes (CD45+LyC6+F4/80−) and macrophages (CD45+LyC6−F4/80+) infiltrating MIF-WT (sgSCR) and MIF-KO (sgMif) tumors.
(C) Relative proportions in flow cytometry of CD4+ and CD8+ T lymphocytes infiltrating MIF-WT (sgCTR) and MIF-KO (sgMif) tumors.
(D) Scoring the expression of four macrophage gene signatures (of the previously defined tumor-infiltrating macrophage subtypes; Figure 2) in macrophages coming from MIF-WT (sgCTR) or MIF-KO (sgMif) tumors, using Seurat’s Module Score method.
(E) Pathway analysis of macrophages infiltrating tumors generated by MIF-WT (sgCTR) or MIF-KO (sgMif) tumor cells.
(F) Differential expression of interferon-related genes in FACS-sorted myeloid cells (CD11b+) from the MIF-WT (sgCTR) and MIF-KO (sgMif) tumors.
(G) Differential expression of genes related to activation and exhaustion of T cells in FACS-sorted CD8+ T cells from the MIF-WT (sgCTR) and MIF-KO (sgMif) tumors.
Unless otherwise indicated, results are presented as mean ± SEM and results are compared by Student’s t test. *p < 0.05 and ***p < 0.001. See also Figure S4.
We also corroborated higher and more inflammatory CD4+ T cell content in the tumor mass upon Mif silencing (Figure 4C). These expressed higher levels of interferon-gamma; Rora, which is associated with activated Th17 cells (Castro et al., 2017); and Tbx21 (T-bet), the transcription factor driving differentiation into Th1 cells (Szabo et al., 2000; Figure S4B). CD8+ T cells from the MIF-KO tumors showed higher expression of activation and exhaustion markers Pdcd1, Havcr2 (Tim3), and Tox (Figure 4G), in line with the higher inflammatory content of the tumor. Together, these observations suggest that MIF expressed by tumor cells is critical to promote the formation of a microenvironment that promotes sarcoma growth. MIF targeting could favor the accumulation of anti-tumorigenic immune cells in the tumor mass and thus restrain tumor growth.
DISCUSSION
The microenvironment of sarcoma has been poorly characterized at the single-cell level, especially compared with the epithelial tumors. Therefore, we employed an scRNA-seq to profile the microenvironment in an immune-competent mouse model of UPS that recapitulates genetic defects found frequently in sarcoma patients (Guarnerio et al., 2015). Macrophages were the principal immune infiltrate of the murine tumors, mirroring proportions observed in human soft-tissue sarcomas, including UPS. Macrophages displayed a spectrum of multiple activation states, with numerous possible functions, including immunomodulation, angiogenesis, efferocytosis, and altered metabolic processing. Decades of previous research, based largely on in vitro experiments, suggested two distinct “activation states” that macrophages could polarize into. In the tumor context, anti-tumorigenic and inflammatory properties were ascribed to the so-called M1 macrophages, while M2 macrophages were described to have angiogenic and pro-tumorigenic features (DeNardo and Ruffell, 2019). However, more recently, in vivo single-cell characterizations of the tumor have partially challenged the hypothesis that macrophages exist in dichotomous activation states. Our findings corroborate this notion and demonstrate that macrophages in the sarcoma context show a continuous range of expression profiles and that more than two diametric subtypes can be appreciated (Zilionis et al., 2019). Indeed, many small sets of genes appear to be highly correlated within our and other myeloid single-cell data, suggesting a diverse repertoire of possible transcriptional “modules” for macrophages; furthermore, various combinations of such gene sets can in principle be co-expressed in a single cell. Extending this idea, the amalgam of gene modules that are manifest in a macrophage at a given point in time could be determined by numerous factors, including recent history of cellular activity, present and past conditions in the microenvironment, and interaction with tumor cells and immune cells.
With a mind to the emerging complexity of myeloid functional states, we examined how such heterogeneity might affect and be affected by the progression of STS—whose many subtypes are characterized by heavy myeloid infiltration (Figures S1A and S1B). Employing a murine model, we demonstrated that anti-CSF1R antibodies reduce UPS growth, reinforcing previous observations that the expression of CSF1 and CSF1 receptor is associated with poor prognosis in LMS (Espinosa et al., 2009) and that the depletion of TAMs using the chemotherapeutic agent trabectedin decreases growth in a mouse model of fibrosarcoma (Germano et al., 2013). However, we also noticed that a consistent proportion of TAMs survive the use of anti-CSF1R antibodies. Because TAMs exhibit numerous transcriptional states—corresponding to specific functional programs, such as pro-inflammatory, pro-angiogenic, antigen presenting, and so on—it would be critical to understand whether the anti-TAM therapies deplete macrophages with specific signatures while sparing others, as recently exemplified in a study of colon cancer (Zhang et al., 2020). In this case, new interventions tailored to macrophages in specific activation states that harbor pro-tumorigenic potential, as opposed to therapies that deplete macrophages indiscriminately, may be more successful in restraining tumor growth. Based on our results, in the case of sarcoma, interventions that specifically tackle Spp1/Cd36 TAMs, which appear to drive ECM remodeling and tumor angiogenesis, may be critical.
Finally, this work identifies the MIF/CD74 axis as a critical determinant of TME composition in sarcoma. When MIF was silenced in sarcoma cells, tumor-infiltrating macrophages displayed higher levels of anti-tumorigenic inflammatory factors and antigen presentation and lower levels of pro-angiogenic and ECM-remodeling components, which on the contrary have been reported to favor tumor growth (Cheng et al., 2021). Accordingly, MIF-KO cells formed smaller tumors compared with MIF-WT. Thus, we speculate that MIF released by sarcoma cells could condition macrophage activation and switch their properties from anti-tumorigenic to pro-tumorigenic. MIF targeting (e.g., with the small molecule 4-IPP; Varinelli et al., 2015) may be therapeutically beneficial for cancer patients, including for sarcoma. In this respect, higher expression levels of MIF correlate with worse survival for sarcoma patients (Figure 3A), including the most aggressive sarcoma subtypes, such as LMS, MFS, and UPS. However, although we observed a clear correlation between TAM activation state and tumor growth, a causative mechanism remains to be identified. Consequently, additional studies should functionally characterize whether and how TAMs in different activation states can restrain or promote tumor growth. The main challenges in this respect would be to identify cell-surface markers useful for the targeting of the specific TAM activation states and methods allowing the isolation of such specific TAMs while sufficiently preserving their transcriptional state for functional experiments.
Limitations of the study
The tumor immune microenvironment that occurs following transplantation of tumor cells into recipient mice may be different from the immune microenvironment after tumor coevolution with the immune system in autochthonous tumor models (Wisdom et al., 2020). In addition, tumor cells carrying distinct genetic backgrounds may also elicit different TMEs. Accordingly, while the current study uses a novel model of sarcoma, it will be critical to extend these investigations to additional sarcoma models and assess how changes in the immune microenvironment influence the capability of MIF to condition macrophage activation and switch their properties from anti-tumorigenic to pro-tumorigenic.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jlenia Guarnerio (jlenia.guarnerio@cshs.org).
Materials availability
Plasmids generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
Bulk RNA-seq and scRNA-seq have been deposited in Gene Expression Omnibus and are publicly available (GEO: GSE201615, GSE201618, GSE201616).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57Bl/6J wild type, Cas9-mice and p53KO mice (002101) were purchased from The Jackson Laboratory. Eight-week-old female mice were used as tumor recipients. Animal experiments were performed in accordance with the guidelines of the Cedars-Sinai Medical Center Institutional Animal Care and Use Committee.
Human cell lines
The human 293T cell line for viral preparation and the human sarcoma cell lines (HT1080 and GCT) were purchased from ATCC. TC32 cells were a gift from Dr. Mona Batish at the University of Delaware. Human sarcoma cell lines were grown in DMEM supplemented with glutamine, 10% fetal bovine serum (Gibco), 100 IU/mL penicillin and 100ug/mL streptomycin (Gibco). Cells were cultured in an incubator at 37°C and 5% CO2.
Mouse mesenchymal stromal cells isolation, maintenance, and in vivo tumorigenesis
Subcutaneous sarcomas were generated as previously described (Guarnerio et al., 2015). Briefly, long bones were collected from p53KO mice, crushed and digested with collagenase II (1 mg/mL) for 1 h at 37°C on a shaker. Recovered cells were stained and FACS-sorted (CD45−CD31−Ter119−Sca1+PDGFRα+) to obtain mesenchymal stem cells and cultured in complete MesenCult medium (STEMCELL Technologies). MSCs were maintained in a humidified chamber with 5% CO2 and 1% O2, with half of medium changed every 3 days. After 7 days in culture at 1% O2, cells formed visible CFU-F colonies; after this point cells were periodically split at 80% confluency. To generate sarcoma cells, mesenchymal cells were transduced for the stable expression of Ccne1 and red fluorescent protein (see below for plasmid generation). The stable cells were assessed by RTqPCR and/or western blot, expanded in vitro and then used for in vivo tumorigenesis assays. Experiments aimed at measuring in vivo tumorigenesis (subcutaneous tumors) were carried out following the protocol previously described (Guarnerio et al., 2015). Briefly, 3D scaffolds (5 mm × 2 mm discs) made with reticulated polycarbonate polyurethane urea matrix (CS1-0502-25, Biomerix Corp/DSM Biomedical) were seeded with MSCs at a concentration of 1 × 105 cells/scaffold. Cells were allowed to adhere to the scaffolds for a minimum of 6 h. Scaffolds were then implanted subcutaneously into mouse flanks, and tumors were harvested 3 weeks after implantation. After isolation from primary recipient mice, sarcoma cells were expanded in culture, carried in DMEM supplemented with glutamine, 10% fetal bovine serum, 100 IU/mL penicillin and 100ug/mL streptomycin (Gibco), and maintained at 37°C and 5% CO2. Cells were then implanted into secondary recipients for sarcoma generation and the experiments presented in the manuscript.
In specific experiments, tumor-bearing mice were treated with an anti-CSF1R blocking antibody (BioXCell BE0213), or with control IgG (BioXCell BE0089). Mice were treated with 200 μg antibody every other day for eight days.
Bone marrow derived macrophage isolation and in vivo experiments
Bone marrow-derived macrophages were generated by isolating total hematopoietic cells from mouse bone marrow and stimulated in vitro with recombinant mouse M-CSF (Biolegend) for 6 days. New M-CSF was added every other day with fresh medium. In specific experiments, BMDM were treated with 50% conditioned medium collected from tumor cell cultures; RNA was then isolated from the treated cells and analyzed. In selected experiments, BMDM were injected together with the sarcoma cells into the recipient mice.
METHOD DETAILS
Harvesting of mouse and human sarcoma cells ex vivo
Sarcomas were grown in mice until approximately 1 × 1 × 1 cm in size. Tumors were resected and enzymatically digested. Where applicable, tumor cells were FACS-sorted based on the expression of the fluorescent marker dsRED.
Human sarcoma samples were minced and then enzymatically and mechanically digested using the 37C_h_TDK_2 protocol on the Miltenyi gentleMACS with Tumor Dissociation Kit for Human cells (Miltenyi Biotec, Auburn, CA). Cell suspensions were washed in DPBS with 0.04% BSA and filtered through 70 μm strainers (Bioland Scientific LLC, Paramount, CA). Red blood cells were lysed with ACK buffer prior to staining with the following surface antibodies: anti-CD45 Pacific Blue, anti-CD11b APC, anti-CD163 PE, anti-CD3 FITC, anti-CD4 PE, anti-CD8 APC, anti-CD56 APC Cy7, anti-CD19 FITC (all purchased from Biolegend).
Cell proliferation assay
Cell proliferation was measured as previously described (Guarnerio et al., 2015). Briefly, cells were plated at low confluency in 12-well plates (2,000 cells per well) and allowed to proliferate for 5 days. Cell viability was measured by crystal violet staining (Sigma Aldrich, 0.1% in 20% methanol) of adherent cells after 10 min fixation with 10% formalin. After washing twice and air-drying, stained cells were washed with 10% acetic acid to solubilize the crystal violet, and OD600 values were measured with a spectrophotometer. Spheroid formation assay was carried out by seeding 1 × 104 sarcoma in DMEM containing 2% Matrigel and 10% FBS. The number of colonies was scored 7 days later, and area quantification was completed using ImageJ.
Generation of retrovirus, lentivirus, knockdown, knockout and overexpressing cells
The retroviral vector pCMMP-MCS-IRES-mRFP (Addgene #36972) was used for the overexpression of Ccne1 gene. The gene was amplified from the cDNA of mouse mesenchymal cells and cloned into the retroviral vector by using the Gibson Assembly kit (New England Biolabs). The expression of the transgene was assessed by RTqPCR. The shRNAs were cloned into the pLKO.1 lentiviral vector (Addgene #10879), following Addgene instructions. The shRNA sequences were designed according to the following program provided by the Broad Institute GPP Web Portal: https://portals.broadinstitute.org/gpp/public/seq/search.
CRISPR sgRNAs were cloned in a Cas9-Puromycin expressing lentiviral vector (Addgene #98290). The CRISPR sgRNA sequences were designed according to the following program provided by the Broad Institute: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design.
All the viral particles were produced in 293T cells, which were co-transfected with the specific viral vector and packaging-expressing plasmids: pECO for the retroviral vectors, and VSV-G, REV and d8.74 for the lentiviral vectors. Transfection of the cells was performed by using Lipofectamine 3,000 diluted in Opti-MEM, according to manufacturer instructions. Transfection medium was changed 8 h after transfection, and the lentiviral particles were collected 24 and 48 h after transfection. Viral supernatant was used with 10ug/mL polybrene (TR-1003-G, Sigma Aldrich) to infect the sarcoma cells, which were seeded at a confluence of 50% the day prior to transduction. Sarcoma cells were incubated overnight with the viral supernatant, washed with PBS and then supplemented with complete medium. Antibiotic selection (puromycin 2ug/mL) was performed at least 72 h post-infection.
RNA extraction, PCR and RT-qPCR
Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions. RNA was directly reverse-transcribed using the RETROscript System (Life Technologies) according to manufacturer instructions. 5–10 ng of RNA were used for each PCR reaction. Quantitative PCRs were carried out using SYBR Green master mix and StepOnePlus real-time PCR system (Applied Biosystems).
Bulk RNA sequencing and analysis
Library preparation and mRNA-sequencing of the mouse sarcoma cells silenced for the expression of Mif was conducted at the Cedars-Sinai Center for Bioinformatics and Functional Genomics. Quality control of FASTQ files was performed with FastQC and adaptor sequences were trimmed with Trimmomatic. Transcript abundances were quantified using the mm10 transcriptome and salmon (Patro et al., 2017), then aggregated to gene-level counts with tximeta (Love et al., 2020). Genes were pre-filtered for those genes expressed (at least 10 counts) in at least 3 samples, and then tested for differential expression using DESeq2 (Love et al., 2014). p values were adjusted using the Benjamini-Hochberg procedure. For pathway analysis, gene lists ranked by p value were used as input to Gene Set Variation Analysis as implemented in the GSVA R package (Hanzelmann et al., 2013).
Flow cytometry
Cells were analyzed using LSRII (BD, Pharmingen) and sorted using FACS-ARIA II (BD, Pharmingen). The following antibodies were used: anti-CD45 FITC, anti-CD31 FITC, anti-Ter119 FITC, anti-Sca1 Pacific Blue, anti-PDGFRα PE, anti-CD3 APC, anti-CD8 FITC, anti-CD45 Pacific Blue, anti-CD4 PE, anti-NK1.1 APC-cy7, anti-B220 APC-cy7 (all purchased from Biolegend). Tumors were digested to single cell suspension enzymatically and filtered twice through 70 μm filters. Red blood cells were lysed with ACK solution (Gibco), washed twice with PBS, and then stained with the fluorophore-conjugated antibodies for 15 min at room temperature. The excess of unbound antibodies was washed out before acquisition in flow cytometry.
Western blot
Protein lysates were prepared with ice-cold RIPA buffer (Boston Bio Products) supplemented with protease and phosphatase inhibitors (Roche). Protein lysates were separated using SDS-polyacrylamide gel electrophoresis (pre-cast gels, Thermo Fisher) and transferred to a nitrocellulose membrane. After blocking the membrane with 5% milk in PBST (PBS with 0.1% Tween20, Sigma), the membranes were incubated overnight at 4°C with anti-MIF (Cell Signaling) and anti-Beta Actin (Bethyl) antibodies, diluted in PBST with 5% BSA. The blots were washed 3 times and then incubated with secondary antibodies (anti-Rabbit HRP, Thermo Fisher), diluted in PBST with 5% milk. Finally, the membranes were incubated with ECL substrate (Pierce) for 1 min and exposed for signal detection with the iBright Imager (Thermo Fisher).
Single-cell RNA sequencing and analysis
For each tumor, sarcoma cells were seeded onto 3D scaffolds as described above, implanted subcutaneously into sex- and age-matched mice via a small incision on the shoulder, and allowed to grow for 3 weeks. Tumors were harvested, minced and then enzymatically and mechanically digested using the 37C_m_TDK_2 protocol on the Miltenyi gentleMACS with Tumor Dissociation Kit for Mouse (Miltenyi Biotec, Auburn, CA). Cell suspensions were washed in DPBS with 0.04% BSA and filtered through 70 μm strainers (Bioland Scientific LLC, Paramount, CA). Red blood cells were lysed with ACK buffer. To enrich the cell suspension for immune cells, samples were stained with anti-CD45 antibody conjugated to magnetic particles using the CD45 EasySep selection kit (Stemcell Technologies, Vancouver, BC), and separated into positive and negative fractions using a magnet. CD45-enriched cell suspensions were each tagged with a unique TotalSeq-A Hashtag reagent (BioLegend, San Diego, CA)—an antibody-conjugated oligo barcode. Final cell suspensions were washed 3 times in PBS, filtered through 40 μm Bel-Art FlowMi strainers (Bel-Art/SP Scienceware, Wayne, NJ), counted, and pooled into a single sample at a concentration of 1,000 cells/uL. The cell suspension was loaded into the Chromium Controller (10x Genomics, Pleasanton, CA) for cell lysing and mRNA capture, with the Chromium Single Cell 3’ Reagent Kit (v3 chemistry). Sequencing libraries were prepared using the same kit according to standard protocol and sequenced by the Cedars Sinai Genomics Core on the NovaSeq 6,000 (Illumina, San Diego, CA) at 400M reads per library. Sequencing data were demultiplexed, and then converted to read counts using the Cell Ranger (10X Genomics) pipeline.
Read count data from Cell Ranger were imported for use with the Seurat R package (version 3) (Stuart et al., 2019), which was used for all subsequent analysis. HTODemux was used to call sample identities for each cell, and to exclude likely cell doublets bearing more than one hashtag. Low quality cells (with < 200 genes or with mitochondrial gene counts >10%) were removed. Gene expression values were normalized and scaled with SCTransform. Cell nearest neighbors were calculated using the first 30 principal components of the gene expression data. Cell clusters were identified using the Louvain algorithm for modularity optimization, and marker genes were calculated for each cluster. In the MIF-knockout scRNAseq experiment, in order to score macrophage signatures within sgMif and sgCTR macrophages, first a signature of top markers was derived from each macrophage cluster described in Figure 2 (MHC-II high MΦ, Cd36/Spp1 MΦ, Mrc1/Cx3cr1 MΦ, and Inflammatory mono/MΦ). A cluster’s top markers were defined as upregulated genes detected in greater than 50% of that cluster’s cells, with expression average log2 fold-change > 0.7 and adjusted p-val <0.001. Finally, the expression of each of the four signatures was scored across all sgMif macrophages and across all sgCTR macrophages, using the Seurat function AddModuleScore.
Inference of immune cell type proportions from bulk RNA sequencing of human tumors
mRNAseq data (normalized RSEM counts) were downloaded for Soft Tissue Sarcoma patients from the TCGA SARC cohort. This expression matrix was analyzed with MCP-counter (Nirmal et al., 2018) to estimate abundances of 8 immune subpopulations.
Ligand-receptor analysis
CellChat was used to infer intercellular communications based on differentially expressed signaling genes, using the package’s native curated and literature-annotated database of known ligand-receptor pairings—based on the Kyoto Encyclopedia of Genes and Genomes (KEGG), and including multimeric receptors, soluble molecules, and co-stimulatory receptors (Jin et al., 2021). Possible sender and receiver cell types were those obtained from single-cell RNA sequencing of whole mouse tumor. A subset of significant interactions, with tumor cells as sender and myeloid subtypes as receivers, are plotted in Figure S2B.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism 9 software (GraphPad, San Diego, USA). A two-tailed unpaired Student’s t-test was used for comparisons between two groups. In all graphs, each symbol represents an individual sample, and the error bars represent the mean ± standard error of mean. *p < 0.05, **p < 0.01, ***p < 0.001. Differential expression testing of bulk RNA-seq data was performed in DESeq2 using the Wald test and Benjamini-Hochberg multiple-testing correction. Differential expression testing of scRNA-seq data was conducted in Seurat using the Wilcoxan rank sum test and Bonferroni p-value correction.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 PB | Biolegend | Clone 30-F11 Cat: 103126 |
| Anti-mouse CD4 PE | Biolegend | Clone GK1.5 Cat: 100408 |
| Anti-mouse CD8 FITC | Biolegend | Clone 53-6.7 Cat: 100705 |
| Anti-mouse B220 APC/cy7 | Biolegend | Clone RA3-6B2 Cat: 103223 |
| Anti-mouse Ly6C APC/cy7 | Biolegend | Clone HK1.4 Cat: 128026 |
| Anti-mouse F4/80 PE or FITC | Biolegend | Clone BM8 Cat: 123109 |
| Anti-mouse CD94 PE | Biolegend | Clone 18d3 Cat: 105507 |
| Anti-mouse CD3 APC | Biolegend | Clone 17A2 Cat: 100236 |
| Anti-human CD45 PB | Biolegend | Clone 2d1 Cat: 368539 |
| Anti-human CD4 PE | Biolegend | Clone A161A1 Cat: 357403 |
| Anti-human CD8 APC | Biolegend | Clone SKI Cat: 344721 |
| Anti-human CD19 PE/cy7 | Biolegend | Clone H1B19 Cat: 302215 |
| Anti-human CD11b APC | Biolegend | Clone ICRF44 Cat: 301309 |
| Anti-human CD163 PE | Biolegend | Clone GHI/61 Cat: 333605 |
| Anti-human CD56 PE/cy7 | Biolegend | Clone 5.1H11 Cat: 362509 |
| Anti-human CD3 FITC | Biolegend | Clone OKT3 Cat: 317305 |
| InVivo anti-mouse CSF1R neutralizing antibody | BioXCell | Clone AFS98 Cat: BE0213 |
| Anti-MIF | Cell Signaling Technology | Clone E7T1W Cat: 87501 |
| Anti-beta actin | Bethyl | Cat: A300-485A |
| TotalSeq-A0301 anti-mouse Hashtag 1 Antibody 10ug | Biolegend | Cat#155801; RRID: AB_2750032 |
| TotalSeq-A0302 anti-mouse Hashtag 2 Antibody 10ug | Biolegend | Cat#155803; RRID: AB_2750033 |
| TotalSeq-A0303 anti-mouse Hashtag 3 Antibody 10ug | Biolegend | Cat#155805; RRID: AB_2750034 |
| TotalSeq-A0304 anti-mouse Hashtag 4 Antibody 10ug | Biolegend | Cat#155807; RRID: AB_2750035 |
| TotalSeq-A0305 anti-mouse Hashtag 5 Antibody 10ug | Biolegend | Cat#155809; RRID: AB_2750036 |
| TotalSeq-A0306 anti-mouse Hashtag 6 Antibody 10ug | Biolegend | Cat#155811; RRID: AB_2750037 |
| Bacterial and virus strains | ||
| Stable 3 E. Coli | Thermo Fisher | Cat#C737303 |
| Biological samples | ||
| Mouse Sarcoma cells | This paper | N/A |
| Primary sarcoma samples (GIST, LMS, DDLPS) | This paper | N/A |
| BMDM | This paper | N/A |
| Tumor-infiltrating immune cells | This paper | N/A |
| Critical commercial assays | ||
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3, 4 rxns | 10X Genomics | Cat#1000092 |
| Chromium Chip B Single Cell Kit, 16 rxns | 10X Genomics | Cat#1000154 |
| Chromium i7 multiplex kit | 10X Genomics | Cat#120262 |
| Mouse CD45-magnetic beads | Stem Cell Technology | Cat#18945 |
| Tumor dissociation kit, mouse | Miltenyi Biotec | 130-096-730 |
| Deposited data | ||
| Single-cell RNA-sequencing | This paper | GEO: GSE201615 GEO: GSE201618 |
| Bulk RNA-sequencing | This paper | GEO: GSE201616 |
| TCGA PanCancer Atlas - Sarcoma | https://gdc.cancer.gov/about-data/publications/pancanatlas | N/A |
| Experimental models: Cell lines | ||
| HT1080 sarcoma cell lines | ATCC | Cat#CCL-121 |
| TC32 sarcoma cell lines | Laboratory of Dr. Mona Batish | N/A |
| GCT sarcoma cell lines | ATCC | Cat#TIB-223 |
| 293T cells for viral production | ATCC | Cat#CRL-3216 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | Strain #: 000664; IMSR_JAX: 000664 |
| Oligonucleotides | ||
| Primers are listed in Table S2 | Thermo Fisher | N/A |
| shMIF_Fw CCGGCAGAACCGCAAC TACAGTAAGCTCGAGCTTACTGTA GTTGCGGTTCTGIIIIIG | This Paper | N/A |
| sgMIF_Rv: GGAACCGTTCCAGCCCACGT | This Paper | N/A |
| Recombinant DNA | ||
| MCS-IRES-Ccne1 | This Paper | Backbone from Addgene: 36972 |
| pLKO.1-shMIF | This Paper | Backbone from Addgene: 10878 |
| lentiCRISPRv2-sgMIF | This Paper | Backbone from Addgene: 98290 |
| Software and algorithms | ||
| Cell Ranger pipeline v3.1.0 | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest/ | N/A |
| R v4.1.1 | https://cran.r-project.org/ | N/A |
| Rstudio Desktop v2020.09.0 | https://www.rstudio.com/products/rstudio/ | N/A |
| tximport v1.20.0 | https://doi.org/10.18129/B9.bioc.tximport | N/A |
| tximeta v1.10.0 | https://doi.org/10.18129/B9.bioc.tximeta | N/A |
| salmon v1.5.2 | https://combine-lab.github.io/salmon/ | N/A |
| DESeq2 v1.32.0 | https://doi.org/10.18129/B9.bioc.DESeq2 | N/A |
| GSVA v1.40.1 | https://doi.org/10.18129/B9.bioc.GSVA | N/A |
| limma v3.48.3 | https://doi.org/10.18129/B9.bioc.limma | N/A |
| CellChat v1.1.3 | https://github.com/sqjin/CellChat | N/A |
| MCPcounter v1.2.0 | https://github.com/ebecht/MCPcounter | N/A |
| Seurat v4.0.4 | https://satijalab.org/seurat/ | N/A |
| cBioPortal v4.0.3 | https://www.cbioportal.org/ | N/A |
| Other | ||
| Biomerix Biomaterial 3D reticulated polyurethane disc scaffolds, 5 mm × 2 mm | DSM Biomedical | Cat# CS1-0502-25 Rev B; Lot# FC-082521-1 |
Highlights.
Sarcoma-infiltrating macrophages exhibit a range of distinct activation states
Sarcoma cells interact with surrounding macrophages through the MIF-CD74 axis
Blocking tumor MIF favors accumulation of inflammatory and antigen-presenting macrophages
ECM-remodeling Spp1+ macrophages are reduced when MIF signaling is blocked
ACKNOWLEDGMENTS
This work was supported by the NIH/NCI (K99/R00 CA212200 and R01 CA258265 awarded to J.G.), The Sarcoma Foundation of America (grant 2019 SFA 15–19 awarded to J.G.), and the Cedars-Sinai Cancer Center. The graphical abstract was created with BioRender.com.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110977.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afshar-Kharghan V (2017). The role of the complement system in cancer. J. Clin. Invest 127, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2017). Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 171, 950–965.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, and Pollard JW (2018). Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov 17, 887–904. 10.1038/nrd.2018.169. [DOI] [PubMed] [Google Scholar]
- Castro G, Liu X, Ngo K, De Leon-Tabaldo A, Zhao S, Luna-Roman R, Yu J, Cao T, Kuhn R, Wilkinson P, et al. (2017). RORγt and RORα signature genes in human Th17 cells. PLoS One 12, e0181868. 10.1371/journal.pone.0181868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, et al. (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e23. 10.1016/j.cell.2021.01.010. [DOI] [PubMed] [Google Scholar]
- Cogdill AP, Andrews MC, and Wargo JA (2017). Hallmarks of response to immune checkpoint blockade. Br. J. Cancer 117, 1–7. 10.1038/bjc.2017.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, and Ruffell B (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol 19, 369–382. 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger-Mathason TK, Zhang M, Qiu Q, Skuli N, Nakazawa MS, Karakasheva T, Mucaj V, Shay JE, Stangenberg L, Sadri N, et al. (2013). Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 3, 1190–1205. 10.1158/2159-8290.cd-13-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kenawi A, Dominguez-Viqueira W, Liu M, Awasthi S, Abraham-Miranda J, Keske A, Steiner KK, Noel L, Serna AN, Dhillon J, et al. (2021). Macrophage-derived cholesterol contributes to therapeutic resistance in prostate cancer. Cancer Res. 81, 5477–5490. 10.1158/0008-5472.can-20-4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa I, Beck AH, Lee CH, Zhu S, Montgomery KD, Marinelli RJ, Ganjoo KN, Nielsen TO, Gilks CB, West RB, and van de Rijn M (2009). Coordinate expression of colony-stimulating factor-1 and colony-stimulating factor-1-related proteins is associated with poor prognosis in gynecological and nongynecological leiomyosarcoma. Am. J. Pathol 174, 2347–2356. 10.2353/ajpath.2009.081037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Yakoub MA, Chandler A, Christ AB, Yang G, Ouerfelli O, Rajasekhar VK, Yoshida A, Kondo H, Hata T, et al. (2021). CSF1/CSF1R signaling inhibitor Pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates T-cell infiltration in the sarcoma microenvironment. Mol Cancer Ther 20, 1388–1399. 10.1158/1535-7163.mct-20-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, et al. (2013). Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 23, 249–262. 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Grünewald TG, Alonso M, Avnet S, Banito A, Burdach S, Cidre-Aranaz F, Di Pompo G, Distel M, Dorado-Garcia H, Garcia-Castro J, et al. (2020). Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med 12, e11131. 10.15252/emmm.201911131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, and Golab J (2020). Myeloid cell-derived arginase in cancer immune response. Front. Immunol 11, 938. 10.3389/fimmu.2020.00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnerio J, Riccardi L, Taulli R, Maeda T, Wang G, Hobbs RM, Song MS, Sportoletti P, Bernardi R, Bronson RT, et al. (2015). A genetic platform to model sarcomagenesis from primary adult mesenchymal stem cells. Cancer Discov. 5, 396–409. 10.1158/2159-8290.cd-14-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 14, 7. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun 12, 1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher FC, and Viterbo A (2013). Histologic and genetic advances in refining the diagnosis of “undifferentiated pleomorphic sarcoma. Cancers 5, 218–233. 10.3390/cancers5010218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori M, Digifico E, Vacchini A, Avigni R, Colombo FS, Borroni EM, Farina FM, Milanesi S, Castagna A, Mannarino L, et al. (2021). The soluble glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness and metastasis via CD44 and IL-33. Cell. Mol. Immunol 18, 711–722. 10.1038/s41423-020-0501-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linch M, Miah AB, Thway K, Judson IR, and Benson C (2014). Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nat. Rev. Clin. Oncol 11, 187–202. 10.1038/nrclinonc.2014.26. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Soneson C, Hickey PF, Johnson LK, Pierce NT, Shepherd L, Morgan M, and Patro R (2020). Tximeta: reference sequence checksums for provenance identification in RNA-seq. PLoS Comput. Biol 16, e1007664. 10.1371/journal.pcbi.1007664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrano RFV, Hunger A, Mendonça SA, Mendonca SA, Barbuto JAM, and Strauss BE (2017). Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 8, 71249–71284. 10.18632/oncotarget.19531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickl B, Qadri F, and Bader M (2021). Anti-inflammatory role of Gpnmb in adipose tissue of mice. Sci. Rep 11, 19614. 10.1038/s41598-021-99090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirmal AJ, Regan T, Shih BB, Hume DA, Sims AH, and Freeman TC (2018). Immune cell gene signatures for profiling the microenvironment of solid tumors. Cancer Immunol Res 6, 1388–1400. 10.1158/2326-6066.cir-18-0342. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penticuff JC, Woolbright BL, Sielecki TM, Weir SJ, and Taylor JA 3rd. (2019). MIF family proteins in genitourinary cancer: tumorigenic roles and therapeutic potential. Nat. Rev. Urol 16, 318–328. 10.1038/s41585-019-0171-9. [DOI] [PubMed] [Google Scholar]
- Petitprez F, de Reyniès A, Keung EZ, Chen TWW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougoüin A, et al. (2020). B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560. 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- Podgornik H, Sok M, Kern I, Marc J, and Cerne D (2013). Lipoprotein lipase in non-small cell lung cancer tissue is highly expressed in a subpopulation of tumor-associated macrophages. Pathol. Res. Pract 209, 516–520. 10.1016/j.prp.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Pulanco MC, Cosman J, Ho MM, Huynh J, Fing K, Turcu J, and Fraser DA (2017). Complement protein C1q enhances macrophage foam cell survival and efferocytosis. J. Immunol 198, 472–480. 10.4049/jimmunol.1601445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, Ding X, Bao R, Hong L, Jia W, et al. (2022). Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun 13, 1742. 10.1038/s41467-022-29366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, Bompas E, Piperno-Neumann S, Cousin S, Grellety T, et al. (2018). Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: a phase 2 clinical trial. JAMA Oncol. 4, 93–97. 10.1001/jamaoncol.2017.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varinelli L, Caccia D, Volpi CC, Caccia C, De Bortoli M, Taverna E, Gualeni AV, Leoni V, Gloghini A, Manenti G, and Bongarzone I (2015). 4-IPP, a selective MIF inhibitor, causes mitotic catastrophe in thyroid carcinomas. Endocr. Relat. Cancer 22, 759–775. 10.1530/erc-15-0299. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen Y, Wang C, Yang M, Wang Y, Bao L, Wang JE, Kim B, Chan KY, Xu W, et al. (2021). MIF is a 3’ flap nuclease that facilitates DNA replication and promotes tumor growth. Nat. Commun 12, 2954. 10.1038/s41467-021-23264-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisdom AJ, Mowery YM, Hong CS, Himes JE, Nabet BY, Qin X, Zhang D, Chen L, Fradin H, Patel R, et al. (2020). Single cell analysis reveals distinct immune landscapes in transplant and primary sarcomas that determine response or resistance to immunotherapy. Nat. Commun 11, 6410. 10.1038/s41467-020-19917-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, Ronchi AE, Rocchetti M, Mingozzi F, Foti M, Chirico G, et al. (2009). CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature 460, 264–268. 10.1038/nature08118. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, et al. (2020). Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459.e29. 10.1016/j.cell.2020.03.048. [DOI] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50, 1317–1334.e10. 10.1016/j.immuni.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-seq and scRNA-seq have been deposited in Gene Expression Omnibus and are publicly available (GEO: GSE201615, GSE201618, GSE201616).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.