

Abstract
Pre-clinical humanized mouse models are a powerful tool to evaluate immunotherapies. NSG-SGM3 mice reconstituted with human stem cells (huSGM3) develop pronounced human myeloid cells due to transgenic expression of stem cell factor, granulocyte-macrophage colony-stimulating factor, and interleukin-3 (IL-3) compared with the widely used humanized NSG (huNSG) model. We assessed in vivo generation of CD19-CAR T cells in huSGM3 mice upon single intravenous injection of the T cell-specific lentiviral vectors (LVs) CD4-LV and CD8-LV. While in vivo CAR T cell generation was clearly detectable in individual mice, generation appeared less efficient than previously observed for huNSG mice. Especially for the CD4-LV group, this correlated with increased IL-15 and decreased GM-CSF levels, indicating activation of monocytes and macrophages. Co-culture assays identified macrophages as a potential barrier for gene transfer. Refining CD4-LV and CD8-LV with a less immunogenic surface by using modified packaging cells substantially improved the transduction of lymphocytes in vitro in the presence of macrophages, as well in vivo in huSGM3 mice. Notably, two mice that developed less CAR T cells showed high interferon-α or -β levels before vector injection. Our data emphasize the relevance of innate immune responses for in vivo generation of CAR T cells, which can be overcome by vector surface engineering.
Key words: macrophage, in vivo gene delivery, shielding, T cell targeted lentiviral vectors, humanized mouse model, innate immune response, CAR T cells
Graphical abstract
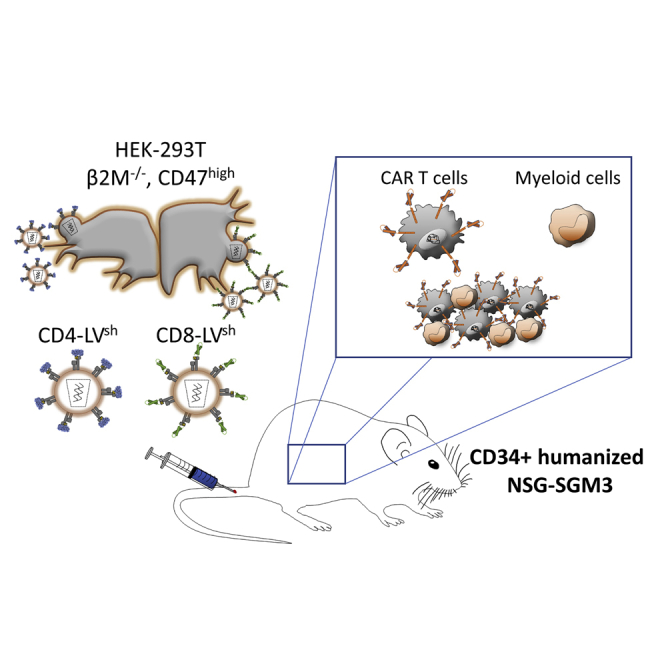
Human macrophages were identified as a road block for in vivo CAR T cell generation with CD4 and CD8 T cell-targeted lentiviral vectors in a mouse model with human myeloid cells in physiologically relevant numbers. Phagocytosis shielding of the vectors improved CAR T cell generation in this model.
Introduction
Due to their crucial role in adaptive immunity and anti-tumor surveillance, T lymphocytes are prime targets in cancer gene therapy. One outstanding approach is equipping T cells with a tumor antigen-specific chimeric antigen receptor (CAR) facilitating TCR-independent anti-tumoral response through its intracellular signaling domains upon target binding. Meanwhile, their potential as a therapeutic regimen has been most prominently demonstrated by the approval of five CAR T cell products directed against CD19 and B cell maturation antigen, thus covering B cell malignancies and multiple myeloma, as well as many more products in clinical trials.1, 2, 3, 4, 5
However, adoptive CAR T cell therapy is highly individualized, based on time- and work-consuming manufacturing process, which limits its broad application in the clinics.6 Genetic modification of T cells is most frequently accomplished by γ-retroviral or lentiviral vectors (LVs), the latter pseudotyped with the vesicular stomatitis virus glycoprotein G (VSV-G). Due to their high transduction activity, VSV-LVs are also under investigation for in vivo gene delivery. In this regard, reducing immunogenicity of the vector particles is an important issue. Recently, this was achieved by excluding the human major histocompatibility complex class I (MHC-I) molecule from the vector particle surface and, instead, increasing the content of human CD47 to prevent phagocytosis. Such phagocytosis-shielded VSV-LVs improved liver gene transfer in nonhuman primates by about 3-fold.7
While manufacturing under GMP conditions is well established for VSV-LVs, they lack cell specificity for T lymphocytes, thus posing a potential risk for CAR gene delivery into lymphoma cells.8 In this regard, our group recently demonstrated selective and functional transfer of a CD19-CAR into CD8+ or CD4+ T cells using CD8-LVs or CD4-LVs, which enter T cells via the respective T cell co-receptor.9,10 These studies provided proof-of-principle for generation of CAR T cells directly in vivo by single intravenous vector administration into CD34+ cord blood humanized NSG (huNSG) mice. Since in vivo gene modification in patients faces many obstacles regarding immune-modulated clearance, targeted LVs for in vivo CAR T cell generation must be evaluated in models that resemble the complex human immune system more closely. Especially, the innate immune system has a key role in the first-line defense against pathogens. For instance, the phagocytic activity of myeloid cells, in particular macrophages, is of most importance at clearing foreign particles.
A constraint of the huNSG model is the limited crosstalk between murine cytokines and human hematopoiesis after CD34+ humanization. Especially, development of mature human myeloid cells is restricted in this system and requires human granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-3 (IL-3) for differentiation of human hematopoietic stem cells (HSCs) into myeloid progenitor cells.11 This has been achieved by in vivo administration of human GM-CSF, IL-4, and Flt-3/Flk-2 in huNSG mice with a boosted human myeloid cell engraftment.12 As further improvement human GM-CSF and IL-3 are transgenically provided in NOG-EXL, MISTRG, and NSG-SGM3 (SGM3) immunodeficient mouse strains, resulting in physiologically relevant human myeloid cell engraftment upon CD34+ humanization.13, 14, 15, 16 Of particular interest is the huSGM3 model, which is a further development of the parental huNSG model and expresses, in addition, human stem cell factor, a critical cytokine for HSC maintenance.17 For instance, the higher proportion of monocytes in this model was shown to be the key point in inducing clinically relevant CAR T cell-associated cytokine release syndrome and neurotoxicity during therapy.18 Therefore, this model resembles a humanized immune system with clinical relevance.
Here, we investigate the huSGM3 mouse model for in vivo CAR T cell generation with CD4-LV and CD8-LV. We demonstrate that CAR T cell generation in the presence of this complex humanized immune system is less efficient compared with the previously reported huNSG model. However, in vivo transduction can be improved by shielding the targeted vectors against phagocytosis.
Results
T cell subtype-specific in vivo CAR T cell generation in CD34+ huNSG-SGM3 mice
To generate CD19-CAR T cells in vivo, huSGM3 mice received similar transducing units of either CD4-LV, CD8-LV, or a mixture of both vector suspensions (MIX) intravenously (Table S1). In addition, mice received human IL-7 shortly before and after vector administration to provide minimal stimulation of the T cells and enhance vector transduction as demonstrated previously (Figure 1A).19 Blood was assessed weekly for the presence of CAR T cells as well as cytokine levels in the plasma.
Figure 1.

CAR T cell generation in huSGM3 mice
(A–D) CD34+ cord blood humanized NSG-SGM3 (huSGM3) were injected intravenously (i.v.) with CD4-LV, CD8-LV, a mix of both (MIX), or PBS (Control). Mice received human IL-7 at 1 and 4 days before and 1 and 3 days after vector administration by i.v. or subcutaneous (s.c.) injection (A). Kinetic of CAR+ T cells and their CD19+ target cells in blood are shown as a CAR+ signal of CD8+ T cells (B), CD4+ T cells (C), and normalized B cells of CD3 cells (D) for each mouse. Dotted lines show the cutoff for determining reduction of B cells in mice. n = 4 (MIX), 8 (CD4-LV), 9 (CD8-LV), and 12 (Control) in two independent experiments. dpi, days post injection.
CAR T cells, detected via myc-tag, emerged in blood within 2–5 weeks after LV injection as detected by flow cytometry (FACS). Three out of four MIX mice and three out of nine CD8-LV mice developed detectable CAR T cells (Figure 1B). The maximum level reached was 11% CAR+ T cells in peripheral blood. After CD4-LV injection, in contrast, only for one mouse a detectable CAR signal was measured on day 10 (Figures 1B and 1C). While in MIX mice CAR T cells were detected in both, CD4+ and CD8+ T cell subpopulations, CD8-LV mice developed only CD8+ CAR T cells, demonstrating the specificity of the targeted LVs. Functional activity of the generated CD19-CAR T cells was confirmed by a strong decrease of CD19+ B cells in blood. While for most animals B cell depletion coincided with high CAR T cell levels, it also occurred in three out of eight mice of the CD4-LV group for which pronounced CAR T cells had not been detected via FACS (Figure 1D). The B cell aplasia was permanent for all respective CD8-LV mice and three MIX-treated mice. However, B cell levels recovered within the first 3 weeks in one MIX mouse.
Further analysis of two major lymphocyte residing organs, bone marrow, and spleen, revealed reduced frequencies of B cells in spite of low CAR T cell signals at the cellular level. In general, CAR T cell signals in the organs were within the background of the control group, except for the bone marrow of one CD4-LV mouse (Figure S1A). However, a prominent reduction of B cells was observed for some mice. In spleen, three MIX, three CD4-LV, and four CD8-LV mice showed reduced B cell levels below 20% of all viable CD3 cells (Figure 2A). Two MIX and two CD8-LV mice exhibited a complete B cell eradication in the spleen on day 41 and 70. Most of these mice also showed a strong reduction of B cells in the bone marrow below 6% (Figure 2A). qPCR analysis of cells from bulk spleen and bone marrow samples revealed significant vector copy numbers (VCNs) in the vector-treated groups over the control (Figure 2B). Interestingly, highest VCNs were observed for the CD4-LV group, although the mice of this group had shown the least prominent indication for CAR T cells with respect to CAR T cell detection in blood (Figure 2B). This was significant in the spleen compared with the control. Co-cultivation of splenocytes isolated from these mice with irradiated CD19+ Raji tumor cells resulted in significant CAR T cell expansion in the respective T cell subtype in the case of four mice of the CD8-LV group, three of the CD4-LV group, and two of the MIX group (Figure S1B). This proves that CAR T cells, although below the detection level in FACS analysis, were present and functional in the spleens of some mice, including those of the CD4-LV group, supporting the qPCR data.
Figure 2.

B cells and VCNs in spleen and bone marrow
B cell frequencies and vector copy numbers (VCNs) of the CAR gene in the indicated organs.
(A) B cell levels in the spleen and bone marrow are shown for the day of final analysis determined by FACS.
(B) VCN per cell of the CAR transgene was measured by qPCR for the respective organ. The dotted lines represent the upper 95% confidence interval of the control group. Individual mice are plotted with mean and standard deviation of the group. n = 4 (MIX), 7–8 (CD4-LV), 9 (CD8-LV), and 12 (Control) in two independent experiments. Statistics were determined by non-parametric Kruskal-Wallis ANOVA with Dunn’s multiple comparisons test and indicated significant p values compared with the Control.
Analysis of plasma cytokines revealed no severe elevation of T cell-related pro-inflammatory cytokines, such as TNF-α and IL-2, in the vector-treated mice compared with the control group, although IL-2 was significantly increased in the CD4-LV group over the control, with one mouse reaching about 700 pg/mL in blood (Figure 3). Interestingly, the CD4-LV group, which showed the least robust CAR T cell development, exhibited a cytokine pattern typical for monocyte- and macrophage-associated activities. IL-1β, IL-15, and GM-CSF, as well as the anti-inflammatory IL-10, showed a distinct expression pattern in CD4-LV mice. IL-1β was about 2- to 3-fold increased compared with the other groups and IL-10 was 2-fold increased over the control. In addition, the pro-inflammatory cytokine IL-15, which is related to myeloid activity, was significantly increased (3- to 25-fold) and GM-CSF was 2-fold decreased in CD4-LV mice compared with all the other groups (Figure 3). This could potentially be due to GM-CSF consumption by activated monocytes upon differentiation, but will require further investigations.20,21
Figure 3.

Plasma cytokines in huSGM3
Plasma cytokines of huSGM3 mice on day 17 after vector administration were measured using a bead-based multi-analysis kit. Concentrations for the respective cytokines are shown for each mouse with mean and standard deviation within the group. n = 4 (MIX), 7 (CD4-LV), 8 (CD8-LV), and 11 (Control). Statistics were determined by non-parametric Kruskal-Wallis ANOVA with Dunn’s multiple comparisons test and indicated significant p values.
Phagocytosis-shielded vectors improve in vitro T cell transduction in the presence of macrophages
Taking into account the different engraftment efficiency of human myeloid cells between the huNSG and the huSGM3 mouse models, we recapitulated the in vivo T cell transduction in a defined in vitro model. We established a co-culture system of human T cells with in vitro polarized macrophages to determine the effect of the myeloid population on CD4-LV- and CD8-LV-mediated transduction. Monocytes were isolated from human peripheral blood and polarized into macrophages with GM-CSF. Characterization of these macrophages revealed high expression of CD209 and CD206, typical markers associated with phagocytosis (Figure S2).22 In the co-culture we tested the ratio 1:1 or the ratio reflecting human T cells and human myeloid cells found in human peripheral blood (10%) and blood of huNSG mice (4%). Comparison of the efficiencies for CAR T cell generation revealed reductions of 27% for CD4-LV and 57% for CD8-LV in the highest macrophage ratio. This was dose dependent as with increasing numbers of macrophages the transduction efficiencies decreased (Figure 4A).
Figure 4.

Reduced T cell transduction by macrophages is ameliorated by LV shielding
(A) Normalized transduction of T cells co-cultivated with the indicated percentages of macrophages using CD4-LV (blue) or CD8-LV (green) produced in conventional packaging cells.
(B) Comparison of conventional (blank bars) and shielded LVs (stripped bars) in transducing T cells in a 1:1 co-culture with (+) or without (O) macrophages. Mean with standard deviation from three to seven donors performed in four different experiments in technical triplicates. Statistics were determined by two-way ANOVA with Turkey’s (A) or Bonferroni’s (B) multiple comparisons test and indicated significant p values.
In the next step, we aimed to improve T cell transduction. For this purpose we took advantage of the recently described phagocytosis-shielded LVs.7 Shielding is achieved through producing LVs in cells lacking surface exposed MHC-I and overexpressing the human inhibitor of phagocytosis CD47, thus obtaining vector particles that are both MHC free and have high amounts of CD47 in their envelope (CD47high).7,23 These MHC-free and CD47high LVs showed efficient transduction of the liver following intravenous administration to non-human primates and improved resistance toward professional phagocytes compared with unmodified parental LV.7 Accordingly, we produced shielded CD4-LVsh and CD8-LVsh by transfecting the modified HEK293T packaging cell line with the required plasmids for LV production. Interestingly, the shielded vectors reached significantly increased transduction rates in the co-culture setting over their conventional counterparts. On average, transduction rate increased about 2-fold for CD4-LVsh and about 3-fold for CD8-LVsh (Figure 4B).
Phagocytosis-shielded LVs improve in vivo CAR T cell generation
To study if the results from the co-culture model had any in vivo relevance, we compared CD4-LVsh and CD8-LVsh with their conventionally produced counterparts upon administration into huSGM3 mice to generate in vivo CD19-CAR T cells. We focused on just these two vector groups to investigate the improvement for each vector type separately and simplify experimental groups. The same experimental setup was used as before and the vectors were normalized for the numbers of injected particles to compare the influence of shielding and non-shielding within the targeted receptor group (Table S1). Although, the injected CD8-LV in here had 2- to 5-fold lower titer than the vector stock in the previous experiment in Figure 1, comparable transducing units were injected between the CD8-LV and CD8-LVsh group in this experiment (Table S1). To analyze dose correlations, two additional groups were included in which mice received double doses of CD4-LVsh and CD8-LVsh, respectively.
Analysis of CAR T cell kinetics in blood revealed a fast appearing peak of CD4 CAR+ T cells on day 10 in the CD4-LV group, which dropped 1 week later. Mice having received CD4-LVsh showed about 2-fold higher CAR T cell levels. All mice, except one mouse of the single and double dose, respectively, developed clearly detectable levels of CAR T cells with high specificity for CD4+ T cells. At the peak, they reached up to 2.2% CAR+ of CD4+ T cells and resembled up to 104 CAR T cells per mL blood (Figures 5A and S3). Two out of five mice from the conventional CD4-LV group reached detectable CAR T cells in blood ranging up to 1% CAR+ CD4 T cells and 4 × 103 CAR T cells per mL blood (Figures 5A and S3). In contrast, in the CD8-LV group none of the single-dose-injected vector groups showed clear evidence for the presence of CD8+ CAR T cells in blood. Only after administration of the double dose of CD8-LVsh four out of five mice developed CAR T cells, specifically in CD8+ T lymphocytes. The CD8+ CAR T cells arose 1 week later than the CD4+ CAR T cells in the CD4-LV group and reached about 1% frequency. In one mouse, the CD8+ CAR T cells expanded further to 4.5%, corresponding to 5 × 103 CAR T cells per mL blood (Figures 5A and S3). Of note, CAR signals in the blood were detected exclusively in the respective T cell subset without detectable signal in the CD3– population (Figures S4A and S4B).
Figure 5.

In vivo CAR T cell generation using shielded LVs
(A and B) huSGM3 mice were injected i.v. with the indicated vectors as a single or a double dose (2×). As control, PBS was injected into the mice. Kinetics of CAR+ T cells (A) in blood are shown as percentage CAR+ of respective T cell subtype and (B) normalized CD19+ cells of the CD3 population. Dotted lines show the cutoff for determining the reduction of B cells in mice. Mice determined as CAR– in blood are depicted with gray symbols and black connecting lines. n = 5 (CD4-LV), 6 (CD4-LVsh), 5 (CD4-LVsh (2×)), 4 (CD8-LV), 5 (CD8-LVsh), 5 (CD8-LVsh (2×)), and 4 (Control) in one experiment. dpi, days post injection.
Furthermore, we determined the monocyte level in the blood of huSGM3 mice before starting the experiment. On average, 1.6 × 103 CD14+ cells per mL blood were found in the periphery, representing about 0.4% of human CD45+ cells (Figure S5A). However, some mice showed noticeable higher levels with up to 7 × 103 cells per mL blood. Interestingly, CD14+ cell counts showed a tendency to reversely correlate with the number of in-vivo-generated CAR T cells in the CD4-LV group (Figure S5B).
In line with the appearance of CAR T cells, a strong decrease of the initial B cell levels was observed in all CD4-LV groups starting from day 10. This was valid not only for mice with detectable CAR T cells in the blood but also for mice with absent or no clear CAR signals. Three mice in the conventional CD4-LV group, all mice in the single-dose CD4-LVsh group and four mice in the double dose group showed substantially reduced or even completely eradicated B cell levels (Figure 5B). In agreement with the lower CAR T cells levels, the kinetics and the extent of B cell depletion was much less pronounced in the groups having received CD8-LVs. Within this group, the time point of B cell reduction correlated well with the first appearance of CAR T cells on day 17. Mainly mice which received the double dose of CD8-LVsh showed B cell reduction, of which one mouse exhibited a moderate reduction to below 40% and three mice displayed a strong reduction down to 2% or 14%. Notably, B cells were reduced also in one mouse of the single-dose CD8-LVsh group, but in none of the mice having received the conventional CD8-LV (Figure 5B).
Analysis of the B cell levels in bone marrow revealed stronger activities of in-vivo-generated CAR T cells by the shielded LV stocks. B cell depletion was highly pronounced in all CD4-LV groups but only significant for the shielded CD4-LVs (Figure 6A). Moreover, the reductions mediated by the shielded CD8-LVs were statistically significant for both vector doses compared with mice having received conventional CD8-LV (Figure 6A).
Figure 6.

B cell depletion and VCNs in spleen and bone marrow
(A) B cell levels in spleen and bone marrow at final analysis determined by FACS gated on CD19+ of human CD3 cells.
(B) VCN of the CAR transgene measured by qPCR in enriched T cells from spleen. Dotted lines represent the upper standard deviation of the control group. X indicates datapoint below the axis. Individual mice are shown as data points with mean and standard deviation for n = 5 (CD4-LV), 6 (CD4-LVsh), 5 (CD4-LVsh ([2×)), 4 (CD8-LV), 5 (CD8-LVsh), 5 (CD8-LVsh (2×)), and 4 (Control) in one experiment. Statistics were determined by one-way ANOVA with Dunnett’s multiple comparisons test and indicated significant p values.
In spleen, both the single and double dose of CD4-LVsh showed a similar B cell reduction, which differed only slightly from that obtained with the conventional group. With the CD8-LVs, the shielded vector group clearly showed a stronger B cell depletion compared with the conventional group in a dose-dependent manner (Figure 6A). In line with the spleen data, VCNs of human T cells revealed successful CAR gene integration. At identical particle dose, CD4-LVsh-injected mice exhibited more than 2-fold higher VCNs compared with mice of the conventional CD4-LV group (Figure 6B). A similar tendency was obtained for the CD8-LVs, which resulted in more than 3-fold higher VCN when shielded particles were injected. Furthermore, samples from spleens of mice that received the 2 times higher dose of CD8-LVsh contained 10 times higher VCNs (Figure 6B). Importantly, flow cytometry analysis of the spleen showed no off-target transduction in the CD33+ myeloid cell population (Figure S4C). The expansion assay of splenocytes with irradiated CD19+ tumor cells confirmed these results. Only with cells from mice having received the shielded LVs, a significant fraction of co-cultures resulted in successful CAR T cell expansion (Figure S6).
In-vivo-generated CD4 CAR T cells exhibit distinct cytokine responses
In line with the initial mouse study described above, we observed pronounced changes of T cell- and myeloid-associated cytokines in plasma for the CD4-LV, but not for the CD8-LV groups. IL-2 and TNF-α, which are mainly secreted by CAR T cells, increased in plasma of some CAR+ mice of the CD4-LVsh groups. Highest values were observed on day 17 after vector application (Figure S7). A similar kinetic was observed for IL-1β and IL-15, reaching up to 7- and 9-fold elevated levels over the control (Figures 7 and S7). Notably, this pattern was not observed for the conventionally produced CD4-LV (Figures 7 and S7). A common observation for all CD4-LV groups was a significant 2-fold decrease of GM-CSF by day 17 (Figures 7 and S8).
Figure 7.

Kinetic of plasma cytokines in CD4-LV-injected huSGM3 mice
Plasma cytokines of huSGM3 mice injected with the indicated LVs were determined by bead-based multi-analysis kit. The concentrations for each cytokine over time are shown for each mouse. Mice determined as CAR– in blood are depicted in gray with black connecting lines. n = 5 (CD4-LV), 6 (CD4-LVsh), 5 (CD4-LVsh [2×]), and 4 (Control) from one experiment. dpi, days post injection.
Analysis of cytokines relevant for the antiviral defense revealed a slight increase for interferon-α (IFN-α) and IFN-β in mice of the CD4-LVsh group (Figure 7). Notably, mice no. 11 and no. 34 in the single- and double-dose CD4-LVsh groups showed a reverse kinetic for these cytokines compared with the initial levels before vector administration. Starting with over 200 pg/mL of IFN-α and over 600 pg/mL IFN-β on day −4, levels declined to background until day 17 (Figure 7). Interestingly, exactly these mice had not developed robust CAR T cell levels in blood, suggesting the antiviral defense as a possible explanation.
Discussion
In this study, we investigated in vivo CAR T cell generation in the highly sophisticated human immune system of the huSGM3 mouse model using targeted CD4-LV and CD8-LV. We could improve in vivo CAR T cell generation by shielding the vector surface with a less immunogenic envelope, while maintaining target specificity for CD4 and CD8 T cells.
Our results demonstrate an overall successful target-specific T cell transduction after systemic administration of the vectors resulting in reduced endogenous target cells. Using flow cytometry analysis of the blood, we could trace the kinetic of CAR T cells and their target cells. Although some mice developed less than 1 × 103 detectable CAR T cell per mL blood, most showed a pronounced reduction and depletion of B cells. Therefore, B cell reduction seems to be a more sensitive readout than CAR T cell detection in the blood by FACS. This was also true for CAR T cells residing in the bone marrow and spleen, where FACS signals for CAR T cells were ambiguous. Resting T cell status and CAR down-modulation are potential explanations.24 However, B cell levels, qPCR, and expansion assays of splenocytes co-cultivated with CD19+ tumor cells proved the presence of CD19– CAR T cells even several weeks after vector injection.
An important finding of this study is that CAR T cell generation was overall lower in huSGM3 mice than in huNSG mice. While comparing data from different publications, there is a clear tendency that, after single injection of CD4-LV and CD8-LV, less huSGM3 mice developed detectable CAR T cells than huNSG mice (Table 1). The NSG-SGM3 mouse model supports a better human hematopoiesis, with a more pronounced myeloid population, than the parental NSG strain when humanized with CD34+ stem cells from human umbilical cord blood.15,16,25 Therefore, we suspected the myeloid system to be causative for the lower in vivo transduction efficiency in huSGM3 mice, in particular macrophages, which are highly active in clearing foreign particles. Indeed, in co-cultures of T cells with increasing amounts of intercepting macrophages, we detected reduced T cell transduction for both CD4-LV and CD8-LV. Notable effects were only significant in the artificial 1:1 ratio, likely because of the excess of vector particles in the culture, masking the reducing transduction effect at lower ratios of macrophages.
Table 1.
Comparison of in vivo CAR T cell generation in huNSG and huSGM3 mice
| Mouse model | Vector | No. of animals | aCAR+ mice | bB cell depletion |
|---|---|---|---|---|
| huNSG | cCD4-LV | 8 | 7 (88%) | 7 (88%) |
| dCD8-LV | 10 | 7 (70%) | 8 (80%) | |
| huSGM3 | eCD4-LV | 13 | 6 (46%) | 5 (39%) |
| eCD8-LV | 13 | 8 (62%) | 4 (31%) | |
| fCD4-LVsh | 6 | 6 (100%) | 5 (83%) | |
| fCD4-LVsh (2×) | 5 | 4 (80%) | 4 (80%) | |
| fCD8-LVsh | 5 | 2 (40%) | 1 (20%) | |
| fCD8-LVsh (2×) | 5 | 4 (80%) | 4 (80%) |
Mice are stated as CAR+ when two out of five independently assessed parameters (flow cytometry of blood, spleen, and bone marrow, qPCR, and expansion assay) were positive for the presence of CAR.
B cell depletion was determined if at least two out of three organs exhibited reduced B cell levels.
Data from Agarwal et al.10
Data from Pfeiffer et al.9
Data from Figure 5.
To reduce the immunogenicity of our vectors and refine in vivo transduction in the huSGM3 model, we produced the T cell-targeted vectors in the optimized packaging cell line β2M−/−, CD47high HEK293T. This modified cell line had been shown to improve in vivo gene transfer of VSV-G pseudotyped LVs into liver of non-human primates by inhibiting macrophage-induced phagocytosis through the SIRPα receptor.7 Indeed, CD4-LVsh and CD8-LVsh refined with a less immunogenic surface could recover transduction efficiency of T cells in the co-culture with macrophages in vitro.
Remarkably, CD4-LVsh and CD8-LVsh also improved in vivo gene delivery in the huSGM3 model and outperformed conventionally produced LVs while maintaining high T cell target specificity. This was observed for CD4-LVsh in the periphery. Analysis of VCN and B cell levels in lymphocyte residing organs confirmed improved gene transfer activities for CD8-LVsh as well. For CD8-LV, CAR T cell generation was less efficient in the huSGM3 mouse experiment shown in Figure 5 than in Figure 1. This was most likely due to a less-potent vector stock administered (Table S1). Of note, increasing CD4-LVsh and CD8-LVsh vector dosages led to an even more pronounced and effective in vivo CAR T cell generation in cell counts in the blood suggesting that saturation for in vivo CAR T cell generation was not yet reached. In agreement with previous observations in PBMC-engrafted NSG mice, in-vivo-generated CAR T cells appear weeks earlier in peripheral blood of CD4-LV than of CD8-LV-injected mice.10 Here, we confirm this for fully humanized mice.
The reduced CAR T cell generation in huSGM3 mice was especially pronounced for CD4-LV. A possible explanation could be binding of CD4-LVs to CD4 on monocytes and macrophages, which might lead to sequestration of vector particles and accordingly reduced in vivo gene delivery.26 That we did not observe transduction of CD14+ cells despite vector particle binding is not surprising, since CD4-LV lacked the accessory viral protein Vpx, which is required to bypass antiviral restriction factors, such as SAMHD1 in myeloid cells, especially at low vector particle numbers, as is the case in vivo.27 This explanation is well in agreement with the substantial improvement that CD4-LVsh showed in the huSGM3 model, likely due to CD47 displayed on LVsh, which in particular reduces phagocytic activity and therefore LV clearance. Furthermore, CD4 ligation on monocytes induces activation and differentiation toward macrophages.26,21,28 Therefore, it is possible that particularly CD4-LV induces a pronounced innate associated immune response that restricts gene delivery in the huSGM3 model. This was in agreement with the plasma cytokine patterns particularly observed for CD4-LV-injected mice. Elevated IL-15 and especially reduced GM-CSF around day 17 underline an ongoing immune response of the innate immune system. This late cytokine increase might fit to a priming-associated immune response, potentially directed against the mouse-derived anti-CD19 scFv antibody FMC63, which has been shown in patients to elicit anti-CAR immune responses.29, 30, 31 Moreover, we observed a tendency of initially high monocyte counts in the blood of huSGM3 mice to correlate with low CAR T cell generation in vivo of some mice in the CD4-LV group, underlining the potential influence of the myeloid system on in vivo CAR T cell generation.
An aspect of the non-obese diabetic, severe combined immunodeficiency (NOD-SCID) strain derived NSG-SGM3 model is the inter-species cross-reactivity of murine SIRPα, including reactivity with human CD47. This is one of the reasons why such high engraftment of human hematopoietic cells can be achieved in NSG models, since human leukocytes ubiquitously express CD47 and thus are less prone to be cleared by the murine innate system.32 Thus, CD47 present on shielded LVs may not only interact with human myeloid cells but also with murine SIRPα-positive murine phagocytes, for instance in liver. This adds another advantage of the current model, as this is reflecting a human-like situation where the LVs are not only interacting with myeloid cells of the peripheral system but also organ residing cells.
Another interesting feature, which we observed in few mice, was elevated levels of human IFN-α and IFN-β before LV administration. Strikingly, these mice did not develop pronounced levels of CAR T cells in the periphery, most likely due to early immune clearance of the vectors. Similar innate immune responses toward systemically injected VSV-LV in immunocompetent mice with increased antiviral IFN-α and IFN-β in the plasma were reported before and shown to originate from plasmacytoid dendritic cells.33 Brown et al.33 showed that the IFN-α response after VSV-LV administration limited in vivo GFP gene delivery and was improved in mice incapable of responding toward IFN-α and IFN-β. Indeed, huSGM3 mice were described to contain human plasmacytoid dendritic cells.34 Accordingly, application of transient IFN-α- and IFN-β-neutralizing antibody or respective receptor blocker might be beneficial before systemic LV injection to reduce the initial antiviral immune response.
This study has further advanced the novel strategy of in vivo generation of CAR T cells with T cell-targeted LVs by demonstrating the validity of this strategy in huSGM3 mice, which reconstitute a sophisticated human immune system. Our data confirm that this model provides many advantages in recapitulating key points of the human immune system, thereby making it an ideal preclinical model for testing immunological therapies. Here, we observe some road blocks in a more physiological human immune system for T cell-targeted in vivo gene delivery, which could not be detected in simpler models. Innate immune responses against the vectors limit in vivo CAR T cell generation but can be overcome by phagocytosis-shielded LVs. These modifications allowed improved in vivo CAR T cell generation in the huSGM3 mouse model and highlight the potential to refine CAR T cell therapy, or T cell gene therapy in general, with targeted LVs.
Material and methods
Vector production and characterization
Detailed protocols for production and characterization of CD4-LV and CD8-LV stocks were recently published.35 Both vector stocks contained the transfer plasmid pS-CD19-CAR-W coding for a second-generation CD19-CAR with a CD28 and CD3z signaling domain under control of the SFFV promotor. In brief, in 2.5 × 107 HEK293T cells or β2M−/−, CD47high HEK293T cells were seeded in a T175 flask 24 h before cotransfection with 14.4 μg of packaging plasmid pCMVdR8.9, 15.1 μg of transfer plasmid pS-CD19-CAR-W and 0.93 μg of pH-CD4-DARPin (29.2), 4.63 μg of pFcΔ30 for CD4-LVs, or 0.9 μg of pCAGGS-NiV-Gd34-CD8, 4.49 μg of plasmid NiV-F pCAGGS-NiVFd22 for CD8-LVs. After 2 days the vector particles released in the supernatant were collected, concentrated, and purified through a 20% sucrose cushion (4,500 rpm, 24 h at 4°C). The supernatant was discarded, the pellet was resuspended in 60 μL of PBS, and the vector stocks were stored at −80°C. Gene transfer activity of the vector stocks were determined by transduction of 8 × 104 activated human PBMCs or 4 × 104 A301 (CD4+) and J76S8ab (CD8+) cell line in a serial dilution of the vector stocks. PBMCs were transduced via spinfection (at 850 × g, 90 min and 32°C). Transducing units were calculated based on the linear range of the gene transfer activity after 3 days in the CD4+ (CD4-LV) or CD8+ (CD8-LV) cell populations. To determine particle numbers, nanoparticle tracking analysis was performed at the NanoSight NS300 (Malvern Panalytical).
Primary cells and cell lines
Human PBMCs were isolated from freshly sampled human blood, purchased from the German blood donation center (DRK-Blutspendedienst, Frankfurt am Main) by Histopaque gradient centrifugation. For purified T cells, a negative selection using the magnetic PAN T cell isolation Kit (Miltenyi Biotech) was performed. Primary cells were cultured in T cell medium (TCM) made out of RPMI 1640 medium (Biowest), 10% fetal bovine serum (FBS) (Biochrom AG), 2 mM glutamine (Sigma-Aldrich), 0.5% penicillin/streptomycin, 25 mM HEPES (Sigma-Aldrich), and further supplemented with 25 U/mL human IL-7 (Miltenyi Biotech) and 50 U/mL human IL-15 (Miltenyi Biotech). For T cell activation, plates were coated with 1 μg/mL anti-human CD3 mAb (clone: OKT3, Miltenyi Biotech) and 3 μg/mL anti-human CD28 mAb (clone: 15E8, Miltenyi Biotech) were added to the cell culture medium for 72 h. For generating primary macrophages, monocytes from PBMCs were labeled with CD14 (REA599, APC, Miltenyi Biotech) antibody, isolated using a magnetic anti-APC-Microbead Kit (Miltenyi Biotech) and polarized for 5–6 days in TCM supplemented with 50 ng/mL recombinant human GM-CSF (Leukine Sanofi-Aventis). Nalm-6, Raji, and A301 cells were cultivated in RPMI 1640 medium supplemented with 10% FBS and 2 mM glutamine (RPMI complete). J76S8ab cells were cultivated in RPMI complete further supplemented with 1 mM sodium pyruvate (Gibco) and 1 mM non-essential amino acids (Gibco). HEK293T and β2M−/−, CD47high HEK293T cells were cultivated in DMEM (Gibco) supplemented with 10% FBS and 2 mM glutamine. All cells were cultivated at 37°C, 5% CO2, and 90% humidity.
Macrophage-T cell transduction assay
A total of 8 × 104 activated T cells was seeded together with the respective ratio of macrophages in a flat-bottom 96-well plate in TCM supplemented with 25 U/mL human IL-7 (Miltenyi Biotech) and 50 U/mL human IL-15 (Miltenyi Biotech). Co-culture was transduced with the indicated LVs normalized for vector particles centrifuged at 850 × g, 90 min, 32°C, and co-culture was subsequently cultivated in 200 μL total volume until day 3 for flow cytometry analysis.
In vivo CAR T cell generation
All animal experiments were performed in accordance with the regulations of the German animal protection law and the respective European Union guidelines.
Humanized CD34+ cord-blood-engrafted NSG-SGM3 (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ, ID:013,062) or NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ, ID: 005,557) mice were purchased from Jackson Laboratory. Human CD45+ cells and B cell and T cell ratio were analyzed in the blood by flow cytometry before the start of the experiment and animals were randomly allocated to the vector and control groups. Investigators were not blinded to group allocation. Data of mice, which had to be sacrificed due to strong, donor related, weight loss within the first 8 days after experiment start, were excluded. Mice were injected subcutaneously or intravenously (i.v.) with 200 ng human IL-7 (Peprotech) 4 and 1 day before and 1 and 3 days after vector injection. In the huSGM3 experiment in Figure 1 only conventionally produced LVs, 2.4 × 1011 particles of CD4-LV, 1.2 or 2 × 1011 particles of CD8-LV or 1.6 × 1011 CD4-LV and 7.6 × 1010 CD8-LV particles (MIX) were injected i.v.. For the experiment in Figure 5, conventional and phagocytosis-shielded LVs, 8.7 × 1010 particles of CD4-LV or CD4-LVsh, 1.4 × 1011 particles of CD8-LV or CD8-LVsh, or PBS as control was injected i.v.. For the higher dose group (2×) a double amount of LVsh was administered. Mice were regularly monitored for their general health status. CAR T cell (via myc-tag) and B cell levels (via CD19) were determined in the blood of the mice weekly by flow cytometry analysis. On the final day blood, spleen, and bone marrow were harvested for further analysis.
Preparation of single-cell suspension from organs and expansion assay
Blood, spleen, and bone marrow were collected from each mouse. Single-cell suspensions of spleen were obtained by mincing the organ through a 70 μm cell strainer. Bone marrow were harvested by opening ends of bones and centrifugation at 4,600 × g for 3 min in perforated 0.5 mL tubes inside a fresh 1.5 mL tube containing RPMI medium. Cell suspension afterward was singularized by a 70 μm cell strainer. Isolated single-cell suspension of each organ and the blood were washed with PBS and subsequently incubated in BD Pharm Lyse buffer (BD Biosciences) for 10 min for erythrocyte lysis. Isolated single-cell suspensions of the organs were used for flow cytometry analysis, T cell enrichment, VCN determination, and expansion assay experiments. Expansion assay was performed with 1 × 105 fresh isolated splenocytes and CD19+ irradiated Raji or Nalm6 cells at an effector:target ratio of 1:1 in RPMI complete supplemented with 50 U/mL human IL-2 (Miltenyi Biotech) or 25 U/mL human IL-7 (Miltenyi Biotech) with 50 U/mL human IL-15 (Miltenyi Biotech) and a medium change two times a week.
Flow cytometry analysis
Staining for flow cytometry analysis was performed in PBS supplemented with 2% FBS. Primary cells were blocked with human FcR blocking reagent (Miltenyi Biotech) and mouse-derived samples were additionally blocked with mouse FcR-blocking reagent (Miltenyi Biotech). The following anti-human antibodies were used for FACS staining: CD45 (2D1, BV510, BioLegend), CD3 (BW264/56, PerCP, Miltenyi Biotec; HIT3a, BV605, BD Biosciences), CD4 (VIT-4, FITC, Miltenyi Biotech; RPA-T4, PE-CF594, BD Biosciences, VIT-4, VioBlue, Miltenyi Biotech), CD8 (BW135/80, FITC, Miltenyi Biotech; RPA-T8, BV786, BD Biosciences; BW135/80, APC, Miltenyi Biotech), Myc for CAR detection (9B11, PE, Cell Signaling Technology; SH1-26e7.1.3, FITC, Miltenyi Biotech), CD19 (LT19, PE-Vio770, Miltenyi Biotech; HIB19, Alexa Fluor 700, Thermo Fisher), CD14 (REA599, APC, Miltenyi Biotech), CD206 (DCN228, VioBlue, Miltenyi Biotech), and CD209 (REA617, PE, Miltenyi Biotech). To determine the viability, Fixable Viability Dye (eFluor 780, Thermo Fisher Scientific) was used. Stained samples were fixated in 1% PFA and measured at LSR Fortessa (BD Biosciences) or at the MACS Quant Analyzer10 (Miltenyi Biotec) and data were analyzed using FlowJo v.10.1 (BD Biosciences) or FCS Express 6 (De Novo Software). The percentage of human cells was identified as the living single-lymphocyte population that expresses human CD45. Cells within the human CD45 gate were further gated for CD3+, CD19+, and CD3–/CD19– populations. Within the CD3 gate, cells were gated for human CD8 and CD4 cells and CAR+ cells. To determine absolute counts in the blood, defined volumes of 4 μm CountBright Plus absolute counting beads (Invitrogen) were added to the samples before flow cytometry measurement.
VCN
VCN was determined on genomic DNA, isolated from bone marrow and spleen suspension cells from huSGM3 mice using the DNeasy Blood and Tissue Kit (QIAGEN) following the manufacturer’s protocol. Quantification was performed by TaqMan-based qPCR assay using the LightCycler 480 Probes Master, the LightCycler 480 Instrument II, and the appropriate LightCycler software for data analysis (all Roche). Transgene detection was performed using woodchuck hepatitis post transcriptional element (WPRE)-specific probe (5′-Cy5-CGCCGCCTGCCTTGCCCGCT-BHQ2-3′) and primers (fwd: 5′-CACCACCTGTCAGCTCCTTT-3′; and rev: 5′-GGACGATGATTTCCCCGACA-3′). For internal reference human albumin-specific probe (5′-6FAM-ACGTGAGGAGTATTTCATTACTGCATGTGT-BHQ1-3′) and primers (fwd: 5′-CACACTTTCTGAGAAGGAGAGAC-3′; and rev: 5′-GCTTGAATTGACAGTTCTTGCTAT-3′) were used. Simultaneously on the same plate, albumin and WPRE were measured as single reactions for samples as well as for the standard (plasmid) in duplicates (1× 95°C for 5 min; 45× [95°C for 10 s and 60°C for 40 s]). In each case, a linear standard curve was used to calculate the amount of human cells (albumin) and vector integration (WPRE). The VCN was then calculated as a ratio (copies WPRE/copies albumin) for human cells carrying the WPRE.
Cytokine assay
Plasma was isolated from the blood of mice by two-step centrifugation. First, the tubes were centrifuged at 300 × g for 5 min at RT, followed by a second centrifugation of the supernatant at 16,000 × g for 5 min. Samples were stored at −80°C until analysis with a customized 13-plex cytokine Legendplex kit (BioLegend). Samples were measured at the MACS Quant Analyzer10 (Miltenyi Biotec) and analyzed with the Legendplex software (v.8.0).
Statistical analysis
Data were analyzed using the GraphPad Prism 8 software (GraphPad Software). Statistical differences were assessed as indicated in the figure legend by using one-way, two-way, or Kruskal-Wallis ANOVA test with Dunnett’s, Bonferroni’s, Turkey’s, or Dunn’s multiple comparisons test. Correlation was assessed by Pearson correlation analysis. Differences were considered significant at p < 0.05.
Acknowledgments
We thank Manuela Gallet and Gundula Braun for their excellent work performing the qPCR and producing the lentiviral vectors. This work was supported by grants from the Bundesminsterium für Gesundheit (ZMV I 1 - 25 18 FSB 404) and the Deutsche Krebshilfe (70114099) to C.J.B. and the LOEWE Center Frankfurt Cancer Institute (FCI) funded by the Hessian Ministry for Higher Education, Research and the Arts [III L 5 - 519/03/03.001 - (0015)] to F.B.T.
Author contributions
N.H., S.A., and F.B.T. designed and performed the experiments. N.H. and F.B.T. evaluated the data. M.M. and A.C. contributed unique reagents. C.J.B. and F.B.T. conceived and designed the study, acquired grants, and supervised the work. N.H., C.J.B., and F.B.T. wrote the manuscript and all authors contributed to its completion.
Declaration of interests
C.J.B. is listed as inventor on patents covering T cell-targeted lentiviral vectors. A.C. is inventor on patents related to MHC-negative lentiviral vectors. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.06.004
Supplemental information
Data availability
All authors declare that data are available within the article or the supplemental information files.
References
- 1.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y., et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/nejmoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/nejmoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W., et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020;382:1331–1342. doi: 10.1056/nejmoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abramson J.S., Palomba M.L., Gordon L.I., Lunning M.A., Wang M., Arnason J., Mehta A., Purev E., Maloney D.G., Andreadis C., et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–852. doi: 10.1016/s0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 5.Munshi N.C., Anderson L.D., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/nejmoa2024850. [DOI] [PubMed] [Google Scholar]
- 6.Hartmann J., Schüßler-Lenz M., Bondanza A., Buchholz C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017;9:1183–1197. doi: 10.15252/emmm.201607485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milani M., Annoni A., Moalli F., Liu T., Cesana D., Calabria A., Bartolaccini S., Biffi M., Russo F., Visigalli I., et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci. Transl. Med. 2019;11:eaav7325. doi: 10.1126/scitranslmed.aav7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruella M., Xu J., Barrett D.M., Fraietta J.A., Reich T.J., Ambrose D.E., Klichinsky M., Shestova O., Patel P.R., Kulikovskaya I., et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018;24:1499–1503. doi: 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfeiffer A., Thalheimer F.B., Hartmann S., Frank A.M., Bender R.R., Danisch S., Costa C., Wels W.S., Modlich U., Stripecke R., et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018;10:e9158. doi: 10.15252/emmm.201809158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agarwal S., Hanauer J.D.S., Frank A.M., Riechert V., Thalheimer F.B., Buchholz C.J. In vivo generation of CAR T cells selectively in human CD4+ lymphocytes. Mol. Ther. : the journal of the American Society of Gene Therapy. 2020;28:1783–1794. doi: 10.1016/j.ymthe.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dougan M., Dranoff G., Dougan S.K. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity. 2019;50:796–811. doi: 10.1016/j.immuni.2019.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Q., Khoury M., Chen J. Proceedings of the National Academy of Sciences of the United States of America. Vol. 106. 2009. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice; pp. 21783–21788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito R., Takahashi T., Katano I., Kawai K., Kamisako T., Ogura T., Ida-Tanaka M., Suemizu H., Nunomura S., Ra C., et al. Establishment of a human allergy model using human IL-3/GM-CSF-transgenic NOG mice. J. Immunol. 2013;191:2890–2899. doi: 10.4049/jimmunol.1203543. [DOI] [PubMed] [Google Scholar]
- 14.Rongvaux A., Willinger T., Martinek J., Strowig T., Gearty S.V., Teichmann L.L., Saito Y., Marches F., Halene S., Palucka A.K., et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014;32:364–372. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wunderlich M., Chou F.-S., Link K.A., Mizukawa B., Perry R.L., Carroll M., Mulloy J.C. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24:1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Billerbeck E., Barry W.T., Mu K., Dorner M., Rice C.M., Ploss A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-granulocyte-macrophage colony-stimulating factor-and interleukin-3-expressing NOD-SCID IL2Rγ(null) humanized mice. Blood. 2011;117:3076–3086. doi: 10.1182/blood-2010-08-301507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broudy V.C. Stem cell factor and hematopoiesis. Blood. 1997;90:1345–1364. doi: 10.1182/blood.v90.4.1345.1345_1345_1364. [DOI] [PubMed] [Google Scholar]
- 18.Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P., et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 19.Frank A.M., Braun A.H., Scheib L., Agarwal S., Schneider I.C., Fusil F., Perian S., Sahin U., Thalheimer F.B., Verhoeyen E., Buchholz C.J. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood advances. 2020;4:5702–5715. doi: 10.1182/bloodadvances.2020002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamilton J.A. GM-CSF-Dependent inflammatory pathways. Front. Immunol. 2019;10:2055. doi: 10.3389/fimmu.2019.02055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhen A., Krutzik S.R., Levin B.R., Kasparian S., Zack J.A., Kitchen S.G. CD4 ligation on human blood monocytes triggers macrophage differentiation and enhances HIV infection. J. Virol. 2014;88:9934–9946. doi: 10.1128/jvi.00616-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulz D., Severin Y., Zanotelli V.R.T., Bodenmiller B. In-depth characterization of monocyte-derived macrophages using a mass cytometry-based phagocytosis assay. Sci. Rep. 2019;9:1925. doi: 10.1038/s41598-018-38127-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milani M., Annoni A., Bartolaccini S., Biffi M., Russo F., Di Tomaso T., Raimondi A., Lengler J., Holmes M.C., Scheiflinger F., et al. Genome editing for scalable production of alloantigen-free lentiviral vectors for in vivo gene therapy. EMBO Mol. Med. 2017;9:1558–1573. doi: 10.15252/emmm.201708148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W., Qiu S., Chen J., Jiang S., Chen W., Jiang J., Wang F., Si W., Shu Y., Wei P., et al. Chimeric antigen receptor designed to prevent ubiquitination and downregulation showed durable antitumor efficacy. Immunity. 2020;53:456–470.e6. doi: 10.1016/j.immuni.2020.07.011. e6. [DOI] [PubMed] [Google Scholar]
- 25.Coughlan A.M., Harmon C., Whelan S., O'Brien E.C., O'Reilly V.P., Crotty P., Kelly P., Ryan M., Hickey F.B., O'Farrelly C., Little M.A. Myeloid engraftment in humanized mice: impact of granulocyte-colony stimulating factor treatment and transgenic mouse strain. Stem Cell. Dev. 2016;25:530–541. doi: 10.1089/scd.2015.0289. [DOI] [PubMed] [Google Scholar]
- 26.Stewart S.J., Fujimoto J., Levy R. Human T lymphocytes and monocytes bear the same Leu-3(T4) antigen. J. Immunol. 1986;136:3773–3778. [PubMed] [Google Scholar]
- 27.Hrecka K., Hao C., Gierszewska M., Swanson S.K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M.P., Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong M.E., Jaworowski A., Hearps A.C. The HIV reservoir in monocytes and macrophages. Front. Immunol. 2019;10:1435. doi: 10.3389/fimmu.2019.01435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turtle C.J., Hanafi L.-A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. The Journal of clinical investigation. 2016;126:2123–2138. doi: 10.1172/jci85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brudno J.N., Lam N., Vanasse D., Shen Y.-W., Rose J.J., Rossi J., Xue A., Bot A., Scholler N., Mikkilineni L., et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020;26:270–280. doi: 10.1038/s41591-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner D.L., Fritsche E., Pulsipher M.A., Ahmed N., Hamieh M., Hegde M., Ruella M., Savoldo B., Shah N.N., Turtle C.J., et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021;18:379–393. doi: 10.1038/s41571-021-00476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takenaka K., Prasolava T.K., Wang J.C.Y., Mortin-Toth S.M., Khalouei S., Gan O.I., Dick J.E., Danska J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007;8:1313–1323. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- 33.Brown B.D., Sitia G., Annoni A., Hauben E., Sergi L.S., Zingale A., Roncarolo M.G., Guidotti L.G., Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007;109:2797–2805. doi: 10.1182/blood-2006-10-049312. [DOI] [PubMed] [Google Scholar]
- 34.Maser I.-P., Hoves S., Bayer C., Heidkamp G., Nimmerjahn F., Eckmann J., Ries C.H. The tumor milieu promotes functional human tumor-resident plasmacytoid dendritic cells in humanized mouse models. Front. Immunol. 2020;11:2082. doi: 10.3389/fimmu.2020.02082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weidner T., Agarwal S., Perian S., Fusil F., Braun G., Hartmann J., Verhoeyen E., Buchholz C.J. Genetic in vivo engineering of human T lymphocytes in mouse models. Nat. Protoc. 2021;16:3210–3240. doi: 10.1038/s41596-021-00510-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All authors declare that data are available within the article or the supplemental information files.