

Summary
Neuroendocrine prostate cancer (NEPC) is a lethal subtype of prostate cancer, with a 10% five-year survival rate. However, little is known about its origin and the mechanisms governing its emergence. Our study characterized ADPC and NEPC in prostate tumors from 7 patients using scRNA-seq. First, we identified two NEPC gene expression signatures representing different phases of trans-differentiation. New marker genes we identified may be used for clinical diagnosis. Second, integrative analyses combining expression and subclonal architecture revealed different paths by which NEPC diverges from the original ADPC, either directly from treatment-naïve tumor cells or from specific intermediate states of treatment-resistance. Third, we inferred a hierarchical transcription factor (TF) network underlying the progression, which involves constitutive regulation by ASCL1, FOXA2, and selective regulation by NKX2-2, POU3F2, and SOX2. Together, these results defined the complex expression profiles and advanced our understanding of the genetic and transcriptomic mechanisms leading to NEPC differentiation.
Subject areas: Cancer systems biology, Cancer, Transcriptomics
Graphical abstract
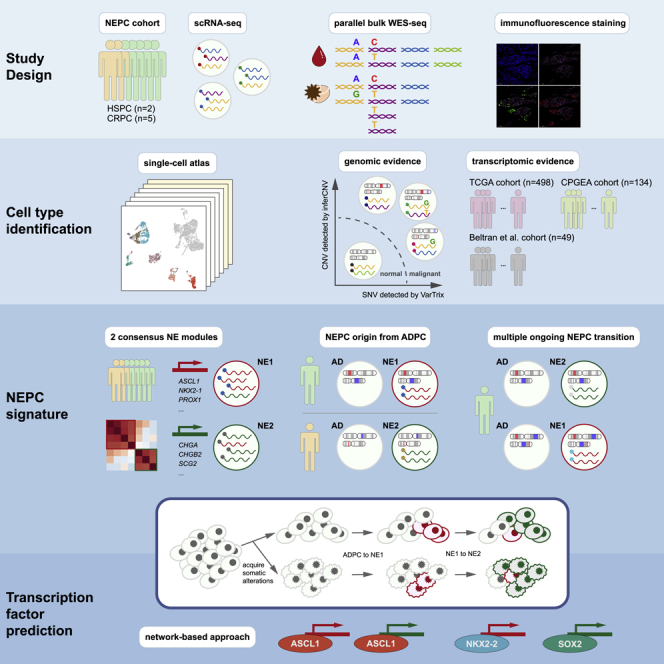
Highlights
-
•
Single-cell RNA sequencing revealed two distinct transcriptional programs of NEPC
-
•
Cell-level clonal evolution analysis extended the divergent model of ADPC to NEPC
-
•
Screening of NEPC-specific transcription factors through network-based approaches
Cancer systems biology; Cancer; Transcriptomics
Introduction
As neuroendocrine prostate cancer (NEPC) was first described more than four decades ago (Wenk et al., 1977), the disease has been estimated to cause approximately 25% of the nearly 34,000 cases of lethal prostate cancer each year in the United States owing to its rapid tumor growth, high vascularity, and early metastatic dissemination (Jemal et al., 2011). A fundamental problem is the inability to diagnose NEPC correctly. The Prostate Cancer Foundation proposed a diagnosis guideline for NEPC in 2013, which is being used in parallel with the guideline updated in 2016 by WHO (Epstein et al., 2014; Priemer et al., 2016). Both guidelines recommend a diagnosis based on pathologists' expertise which can be quite variable and not quantitative. Meanwhile, the sensitivity of existing biomarkers, CHGA, SYP, and NCAM1, is barely satisfactory, varying from 41.5% to 84.0% (Zhang et al., 2020). The advent of next-generation sequencing (NGS) makes a comprehensive depiction of the molecular characteristics of NEPC possible. Beltran et al. (2016) proposed a molecular classifier with integrated genomic, transcriptomic, and epigenetic features to improve the diagnosis of NEPCs. This was followed by an independent, larger cohort study (Aggarwal et al., 2018), in which a novel NE signature was developed based on transcriptomic clustering. However, the current gene panels from these studies showed poor consistency, challenging the translational values of these genes as biomarkers. A possible reason for the inconsistency is the variable prevalence of NE-marker positive cells, from ADPC with focal NE differentiation (NED), ADPC with diffused NED, to pure small cell carcinoma (smCC, or high-grade NE) (Fine, 2018). It was reported that 50–60% of NEPC presents an overlapping expression pattern with ADPC (Conteduca et al., 2019; di Sant'Agnese, 1992), making it challenging to use bulk sequencing techniques to accurately decompose the transcriptome of NEPC from that of ADPC. Furthermore, the trans-differentiation process of NEPC is highly dynamic (Yamada and Beltran, 2021) and difficult to capture by bulk sequencing.
Besides its elusive molecular features, whether NEPC arises from the pre-existing normal NE cells (primary tumor model) or transdifferentiated ADPC (lineage-plasticity model) is also an issue of debate (Rickman et al., 2017). The latter theory hypothesizes that the ADPC differentiates to acquire androgen receptor (AR)-independent phenotypes, e.g., becoming NE cells. About 10–40% of castration-resistant ADPC (CRPC-Adeno) displayed NE phenotype (CRPC-NE) (Nadal et al., 2014; Zhang et al., 2020), suggesting that the lineage-plasticity can be a strategy to circumvent the pressure of androgen-deprivation treatment (ADT). In contrast, ample studies reported focal NED in 30%-100% of ADPC before initiating any treatment (Fine, 2018). Genetic-based evidence (Beltran et al., 2016), including shared TMPRSS2-ERG rearrangement between NE tumor cells and their AD companions (Guo et al., 2011), supports that NEPC is derived from transdifferentiated ADPC. Zaidi et al. identified a stem-like luminal epithelial cell population featuring Sca1+ and Psca + that could develop into Ascl1+ or Pou2f3+ populations in mice (Zaidi et al., 2021).
The molecular mechanism behind acquiring an NE phenotype, however, is still incompletely understood. Current studies suggest that genomic alterations, abnormal activation of specific transcription factors (TFs), and epigenomic dysregulation all play important roles in the NED process. RB1 loss, TP53 loss/mutation, and PTEN loss have been recognized as recurrent molecular events in NEPC (Beltran et al., 2016). Their combined effects in facilitating NED were confirmed by recent studies. For example, the lineage plasticity of ADPC was induced by the loss of RB1 and PTEN, and boosted by the loss of the TP53 gene to acquire NE features (Ku et al., 2017), based on which Park et al. further demonstrated that additional overexpression of c-Myc could lead to high-grade smCC in mouse models (Park et al., 2018). Lee et al. found N-Myc overexpression and PTEN loss are sufficient to induce NED (Lee et al., 2016). The N-Myc overexpression and RB1 loss together could drive NEPC development (Brady et al., 2021). As RB1 and TP53 alterations were found in many but not all NEPCs, some disputed them being indispensable in generating an NE phenotype (Lin et al., 2014). FOXA1 has recently been recognized as most frequently mutated in prostate cancer (Li et al., 2020). FOXA1 mutation at arginine 219 was enriched in NEPC and capable of activating the NED program (Adams et al., 2019). Its clinical importance requires future illumination.
Pioneer TFs, such as ASCL1 (Fraser et al., 2019), FOXA2 (Park et al., 2017), and POU3F2 (Bishop et al., 2017) are upregulated in NEPC and could induce NE-related gene expression. Although more NED-related TFs have been gradually revealed, how different TFs cooperate in the NED process and at what specific stages they act have not been fully clarified.
In this study, we identified 19,605 malignant cells from samples of seven patients. Using Consensus Non-Negative Matrix Factorization (cNMF) clustering, we identified two distinct signatures of NEPC, representing the early and late transition programs, respectively. The genomic analysis confirmed the trans-differentiation of NEPC from AD origin in six patients. Notably, we found that NE could be derived from AD cells of different pathological stages, and the process is orchestrated by lineage-specific TFs, such as ASCL1 (common TF), NKX2-2 (NE1-specific), and POU3F2 (NE2-specific).
Results
scRNA-seq and cell typing of non-malignant and prostate tumors
Prostate cancer is a disease of high intra-tumor heterogeneity. To investigate the distinct cell populations and their transcriptomic programs within prostate tumors, we performed scRNA-seq on two hormone-sensitive prostate cancer (HSPC) and two CRPC samples. We also downloaded scRNA-seq data of three CRPC samples from GEO (GSE137829) (Table S1). Five (P2, P4, P5, P6, P7) out of the seven samples came from prostate cancer tissues, and the remaining two (P1, P3) came from metastatic lymph nodes (Figure 1A). Following surgery, tumor samples were rapidly preprocessed for single-cell RNA sequencing by the Drop-seq or 10x Genomics platforms. After stringent filtering, 36,036 high-quality cells with a median of 6,397 mapped reads and 2,064 mapped genes were kept for cell-type annotation and further analysis (Table S2). We first hypothesized that the VIM+/EPCAM-clusters (n = 16,233) (Figure S1) are primarily normal mesenchymal cells and assigned cell types using canonical markers (Figures S2A–S2D). Secondly, using 15,465 normal cells as the normal diploid reference, we inferred copy number variations (CNV) of the EPCAM+/VIM-cells (n = 20,209) and predicted 19,605 malignant cells with non-diploid genomic regions and 604 normal epithelial cells lacking such manifestations. Cell type assignments of each sample were summarized in Figures 1A and Table S3. The putative malignant cells scored much higher than normal cells in tumor gene expression using gene panels from both TCGA and CPGEA prostate cancer cohorts (Cancer Genome Atlas Research, 2015; Li et al., 2020) (Wilcoxon test, p < 0.0001) (Figures 1B and Table S4). For P1, P3, and P6, we used parallel WES data from tumor and paired blood samples to identify somatic single-nucleotide alterations (SNAs) and short insertions/deletions (InDels). 91.9% of cells with somatic mutations detected were in the malignant cell populations (Figures 1A and Table S5, hypergeometric test, p < 0.0001). Taken together, these results indicated that scRNA-seq analysis identified the malignant cell populations from heterogeneous samples.
Figure 1.

Overview of 36,036 single cells from seven prostate cancers
(A) UMAP of the 36,036 cells profiled in seven patients. Two HSPC, three CRPC, and two pure small cell prostate cancers were depicted here. Different cell types were color-coded. For samples with parallel WES data (P1, P3, and P6), cells with at least one mutant read detected were colored black; P4, P5, and P7 come from published data (GSE137829) (also see Figure S1, S2, Tables S1, S2, and S3, and S5).
(B) Distribution of cancer score in TCGA and CPGEA, respectively (STAR Methods) for cells categorized as malignant or normal, data are represented as boxplot (Wilcoxon rank-sum test, ∗∗∗∗: p ≤ 0.0001) (also see Table S4).
Distinct RNA signature of two subtypes of neuroendocrine prostate cancer
Single-cell profiles of malignant cells highlighted intra-tumoral expression heterogeneity of this malignant compartment (Figure S2E). We used the cNMF method (Kotliar et al., 2019) to decompose mixed expression profiles of single cells into a linear combination of biologically interpretable gene expression programs (GEPs). 71 GEPs were deconvolved from seven samples (Figure 2A and Table S6). Despite the apparent inter-patient heterogeneity of malignant phenotypes, correlation clustering illustrated cross-sample similarity between GEPs, which further clustered into nine consensus modules. Gene ontology (GO) enrichment analysis of over-represented genes in each module revealed its biological relevance, including conventional, "housekeeping" modules such as cell cycle, RNA/protein synthesis, and cell stress, as well as less-understood immune-related, interferon-related, and extracellular matrix-related modules (Table S6). Interestingly, two related but distinct modules were enriched with NE-related GO terms (Figure 2B); thus, we termed them NE1 and NE2. The NE1 module consisted of GEPs from P3, P5, P6, and P7, while the NE2 module consisted of GEPs from P1, P2, P3, and P5 (Figure 2A). Genes over-represented in NE1 module (n = 32) and NE2 module (n = 60) were summarized in Table S7. Based on the activities of NE module-related GEPs, we defined NE cells in P1, P2, P3, P5, P6, and P7 (Figure S2F). To confirm the specificity of the NE-associated genes, we applied our NE1 and NE2 module to an external dataset which included 15 CRPC-NEPCs and 34 CRPC-ADPCs, all confirmed by pathologists (Beltran et al., 2016). An NE module was also generated by combining NE1 and NE2 modules using the same approach for validation purpose (Figure S3A). GO enrichment analysis results of the combined NE module confirmed that both NE1-related GO terms (positive regulation of neural precursor cell proliferation), and NE2-related GO terms (modulation of chemical synaptic transmission) were present (Figures 2B and S3B). Single sample gene set enrichment analysis (ssGSEA) revealed that CRPC-NEPCs had much higher scores for all three modules compared with CRPC-ADPCs. We also compared the ssGSEA scores with the "integrated NEPC scores" from the Beltran et al. study, and the two independently developed scoring systems showed good consistency (Figure S3C). These results strongly support the generalizability of our two NE modules which represented the early and late state of the NED process, respectively.
Figure 2.

cNMF algorithm reveals distinct expression signatures
(A) Pairwise correlation clustering of intra-tumoral GEPs. 71 GEPs were derived by cNMF from seven tumors and formed ten consensus modules, whose biological significance was predicted by GO analysis (top). GEPs in two NE modules were annotated (right) (also see Table S6).
(B) GO analysis of genes over-represented in NE1 and NE2, respectively (hypergeometric distribution test adjusted by FDR <5%) (also see Figure S3, Table S7).
(C) UMAP plot of malignant cells from P3. Cells are colored by their NE subtype (right) and the min-max normalized activity score of the corresponding GEPs (left, middle).
(D) Volcano plot of the differentially expressed genes (DE-Gs) between NE1 and NE2 cells of P3 (Wilcoxon rank-sum test adjusted by FDR <5%). Blue, upregulated in NE1; Red, upregulated in NE2; gray, | log 2-fold change <1 | or adjusted P-value > 0.05. Expression of marker genes of note, ASCL1 and CHGB, for NE1 and NE2, respectively, were shown in the UMAP plot. TPM, edtranscript per million (normalized value). (also see Figure S4, Table S8).
We noticed two cases, P3 and P5, which contained GEPs in both NE1 and NE2 (Figure 2A). We then focus on these two samples to further explore the relationship between NE1 and NE2. In P3, GEP7, which represents NE1, and GEP3, which represents NE2, were active in different clusters (Figure 2C). Differential expression analysis identified 109 genes upregulated in NE1 and 164 upregulated in NE2 (adjusted p value < 0.05, | log 2-fold change | ≥ 1), as illustrated in the volcano plot (Figures 2D and S4). Among those DE-Gs, NE1 expressed a high level of ASCL1, which is a TF known to initiate the NEPC trans-differentiation (Vias et al., 2008), consistent with the GO annotation of NE1. In contrast, NE2 cells expressed low to the undetectable level of ASCL1, and instead expressed high level of secretory protein genes including CHGA, CHGB, and ENO2, representing the functional status of terminally differentiated NEPC (Figures 2D and Table S8). To validate the observed expression patterns of ASCL1 and CHGB in single-cell data, we performed in situ immunofluorescence (IF) co-staining in P3 (Figure S5) and found expression of ASCL1 and CHGB were mutually exclusive at the individual cell level, whereas the expression of HOXB13, a prostate-specific marker, was ubiquitous.
In P5, NE cells had three independent GEPs expressed (P5_GEP9, P5_GEP5, P5_GEP14). These NE cells were annotated as NE1, NE2-1, NE2-2 based on the association we made between GEPs and NE modules (Figure 2A), that is, P5_GEP9 belongs to the NE1 module, and P5_GEP5 and P5_GEP14 belong to NE2 module; we then used the differential activation of P5_GEP5 and P5_GEP14 (Figure 3A) to annotate NE2-1 and NE2-2, respectively. This order is associated with the antagonistic changes in the expression levels of CHGB and ASCL1, further corroborating our findings in P3 (Figures 3A and 2D); thus, it is likely that single-cell RNA-seq might have captured cells representing the transitions across the main stages of NEPC trans-differentiation. Indeed, splicing directions inferred from previously published velocity analysis of the same sample supported the hypothesis of NE1-NE2 transition (Dong et al., 2020) (Figure 3A). Pseudo-time trajectory analysis using Monocle2 further extracted the state-specific signatures of NEPC (Cao et al., 2019) (Figure 3B). Cells transitioning from NE-1 to NE2-1 overexpressed ASCL1. In the transition process associated with NE2-1, genes such as IGFBP1, TREM1, CGA, and KLK12 were also upregulated. The NE2-2 program represented the final state of NED, when CHGA, CHGB, and TFF1 were highly expressed (Figure 3B). CHGA has been used as a biomarker for pathological diagnosis but only exhibited a sensitivity of 57.4% in a recent study (Epstein et al., 2014; Zhang et al., 2020). The low sensitivity is likely owing to CHGA’s sparse expression at the early stage of NED when ASCL1 is overexpressed. The dynamic nature of the trans-differentiation process highlights the need to identify marker genes that capture the changing expression landscape at different stages of NEPC, which will provide critical knowledge for clinical diagnosis.
Figure 3.

NE trans-differentiation in P5
(A) The min-max normalized activity score of one NE1 GEP and two NE2 GEPs were colored. UMAP Plots were arranged in the assumed chronological order of differentiation. Expression of marker genes of note, ASCL1 and CHGB, for NE1 and NE2, respectively, were shown. In the right panel, the arrows denote the transition direction inferred from velocity analysis by Dong et al. (2020). TPM, transcript per million (normalized value).
(B) Dynamic expression along the trajectory identified 100 genes that vary significantly over trans-differentiation pseudo-time (likelihood ratio test of nested models adjusted by FDR <5%). Cells were arranged in column by pseudo-time series. Hierarchical clustering of these genes at row via Ward D2’s method recovered three nonredundant groups that covary over the trajectory. Cluster analysis indicated large shifts in gene expression occurred as NE progenitor progressed toward maturation. TPM, transcript per million (scaled value).
(C) inferCNV heatmap with hierarchical clustering of P5. The oncogenes or tumor suppressor genes in Cancer Gene Census were depicted in CNV regions. The top panel indicates a lack of CNV events in reference cells. The bottom panel is the malignant cells. Different types of NE cells were annotated on the sidebar (also see Figure S6).
The genetic origin of neuroendocrine prostate cancer
Somatic alterations of specific genes, including TP53 and RB1, have been recognized as NED-related, underscoring the importance of genetic drivers in NED. Both large-scale CNV inference by inferCNV from scRNA-seq data and CNV calling by GATK from bulk WES-seq confirmed inter-sample heterogeneity at the genetic level as well as frequently observed alterations in prostate cancer, such as 8q gain and 13q loss (Figures S6A and S6B). Based on inferCNV results, RB1 deletion was found in all samples except for P5 (not detected), whereas only one patient (P4) had a clonal TP53 deletion (Figure S6C). In addition, only a single synonymous G > A mutation at chr17: 7,676,071 in P6 was identified (Table S5). The result may support the viewpoint that the alteration of RB1 and TP53, although contributing to higher aggressiveness of NEPC, is not mandatorily required by NED (Rickman et al., 2017).
Having determined the overall CNV landscape of the tumor cell population, we set out to investigate genetic differences between NEPC and ADPC and whether they contribute to NED. In P5, large-scale CNV segments were found consistent across cells (Figure 3C), suggesting shared CNV events between NE and AD cells in this patient, and supporting the lineage-plasticity model. In P3, in addition to the main clone featuring chr2 gain, there was a subclone featuring an additional 12q gain and 15q loss. This subclone contained almost all NE1 cells and a subpopulation of AD cells (Figures 4A and 4B). In contrast, other AD cells and almost all NE2 cells existed outside the subclone (Figures 4A and 4B). Taken together, we concluded the NE2 shared the same origin with the clonal adenocarcinoma characterized by 2p, 7p, 11q, 19q gain, and 6p, 13p loss. The clonal AD continued to acquire gain of 12q and loss of 15q, and this subclone further differentiated into subclonal AD and NE1 (Figure 4D), suggesting that the NEs in the same patient could be derived independently from ADs of different stages.
Figure 4.

Clonal evolution of P3
(A) inferCNV heatmap with hierarchical clustering of P3. The oncogenes or tumor suppressor genes in Cancer Gene Census were depicted in CNV regions. The top panel indicates a lack of CNV events in reference cells. The bottom panel is the malignant cells. Different types of NE cells were annotated on the sidebar. Genes of note in NEPC studies were highlighted in red (also see Figure S6).
(B) Cells are colored according to the amplification status of chr2 and chr12 in the UMAP embedding (also see Figure S5).
(C) Evolution model of P3 extrapolated from inferCNV results.
P1 presented with low PSA (the alias of KLK3 in clinical practice) (8.63 ng/ul) and enlargement of inguinal lymph nodes with whole-body bone metastasis. The patient was diagnosed with treatment-naïve prostate cancer by needle biopsy. Pathological evaluation revealed no positive neuroendocrine markers, with high Gleason scores (4 + 5) (Table S1). However, scRNA-seq data of this patient discovered a cluster of NE2 cells that expressed ASCL1 and CHGB (Figure 5A). The low count of NE2 cells explained the false-negative result from the pathological test. This result further underscored the spatial heterogeneity of prostate cancer and the limitation of needle biopsy-based pathology. Genetic variation analysis revealed that the NE cells and the clonal AD cells shared the same origin. However, we also recovered a subclone of AD cells harboring a gain of 3q (Figure 5B). Although all AD cells expressed AR, as expected, the KLK3 gene expression was in perfect correlation with the gain of chromosome 3q (Figures 5B and 5C). This newly evolved AD subclone explained the low level but positive test of PSA screen in the clinic. Taken together, we constructed a tumor evolution model for P1. The tumor was initiated by MYC amplification and RB1 losses, which produced the clonal ADs and NEs. Following the gain of 3q, a subclone started expressing PSA (Figure 5D). In this patient, we found NE could derive from KLK3 negative and treatment naive adenocarcinoma cells.
Figure 5.

Clonal evolution of P1
(A) UMAP plot of malignant cells from P1. The relative expression of NE2 GEP was colored. Expression of marker genes of note, ASCL1 and CHGB, for NE1 and NE2, respectively, were shown in the UMAP plot. TPM, transcript per million (normalized value).
(B) inferCNV heatmap with hierarchical clustering of P1. The oncogenes or tumor suppressor genes in Cancer Gene Census were depicted in CNV regions. The top panel indicates a lack of CNV events in reference cells. The bottom panel is the malignant cells. NE types, chr3 amplification, and KLK3 expression levels were annotated on the sidebar (also see Figure S6). TPM, transcript per million (normalized value).
(C) Cells were colored by chr3 amplification status (left) and expression levels of KLK3 (middle) and AR (right) in the UMAP embedding.
(D) Evolution model of P1 extrapolated from inferCNV results.
We also analyzed other samples with identified NE cells (Figure S7). Consistent with P1, P3, and P5, NE cells in P2 and P6 also had AD counterparts that shared the same clonality.
Epigenetic-driven clonal evolution
The cases we examined so far suggest that these very heterogeneous ADs with different genetic alterations could evolve to obtain a similar NE molecular phenotype. We, therefore, hypothesized that there exist epigenetic drivers for this transformation. Thus, we applied pySCENIC to identify the underlying gene co-expression network using the scRNA-seq data. This analysis discovered coherent activities of different TFs involved in the NE program, which represent potential drivers of the epigenetic programming during NED and are decoupled from genetic alternations. They included NE1-specific NKX2-2, NE2-specific POU3F2, and SOX2, as well as ASCL1 and FOXA2, which are common between NE1 and NE2 cells (Figure 6A). One of the common TFs for NE, ARX, was the subject of investigation by several studies for its role in pancreatic neuroendocrine tumors (pNET). ARX is associated with non-functional pNET recurrence (Cejas et al., 2019) and normal development of endocrine cells (Friedman-Mazursky et al., 2016). Another common TF for NE, PBX1, is highly expressed in neural progenitor cells of pigs (Schwartz et al., 2005) and mouse embryos (Roberts et al., 1995), but its function remained understudied in humans. Our results also recapitulated previous findings. For example, the NE2-specific POU3F2 upregulates the expression of NE2-specific SOX2 (Bishop et al., 2017). Similarly, in an ex vivo study, ASCL1 was shown to upregulate the expression of SOX2 (Sinha et al., 2018). Taken together, our study revealed two axes of the evolution of NEPC from ADPC. On one axis, genetic events define the clonal evolution of ADPC as the tumors develop from HSPC to CRPC; on the other axis, a hierarchy of TFs, or epigenetic drivers, defines the trans-differentiation of ADPC to NEPC. Importantly, our data suggest that the epigenetic reprogramming of ADPC to NEPC can happen at multiple stages along ADPC’s own evolutionary trajectory, partially disentangling the complex and heterogeneous relationship in NEPC (Figure 6B).
Figure 6.

TF regulation of NEPC
(A) Heatmap showed three types of TF (Common, NE1 specific, and NE2 specific) predicted by pySCENIC. The black block represents turn "on" status while the white block represents turn "off" status.
(B) The summarized model of transcriptional regulation and genetic evolution of NEPCs in this study.
Discussion
The single-cell sequencing technology empowers us to define the heterogeneous subpopulations of neuroendocrine cancer cells mixed with ADPC. In this study, we isolated NE cells from seven NEPC samples in two independent cohorts and found two related but distinct programs (NE1 and NE2) which might take part in different phases of NE trans-differentiation. NE1 included 32 genes, in which ASCL1, NKX2-2, and PROX1 are known to play critical roles in cell differentiation and cell fate determination. NE2 included 60 genes, such as CHGA, CPE, SCG3, and SYT13. We observed the co-existence of NE1 and NE2 GEPs in both cohorts. Several candidate genes that we identified in this study, including SEC11C, PROX1, SCG3, and PEG10, have been investigated previously in NEPC studies (Aggarwal et al., 2018; Beltran et al., 2016). It is worth noting that, possibly owing to the mixture of NEPC and ADPC, two landmark studies that defined NEPC gene signatures only had a single gene (DNMT1) in common (Aggarwal et al., 2018; Beltran et al., 2016). Akamatsu et al. described the gene expression dynamics associated with the trans-differentiation from ADPC to NEPC using a patient-derived xenograft model and reported the upregulation of PEG10, CHGA, NKX2-2, ASCL1 at the NEPC stage (Akamatsu et al., 2015). This study also reported high expression of ASCL1 at the early stage of NEPC and the apparent loss of expression of ASCL1 in the late stage of NEPC. These results are consistent with our findings. However, contrary to our study, Akamatsu et al. reported consistent expression of CHGA regardless of the stages of NEPC. Dong et al. identified three different NE modules (Dong et al., 2020), but did not further elucidate the relationship among them. Their velocity analysis revealed the process of ADPC differentiating toward NEPC. By re-analyzing Dong et al. data and our data, we discovered NE gene modules representing different stages of NEPC, thus providing a more meaningful array of candidate genes for both mechanistic studies and biomarkers for diagnosis.
In our study, P7 was found to have high NEUROD1 and MYC levels and low ASCL1 levels (Figure 6A). First shown in small cell lung cancer, the mutual exclusiveness of NEUROD1 and ASCL1 has been revealed by bulk RNA-seq analysis on multiple tissue origins (Cejas et al., 2020; Poirier et al., 2013). It was indicated that NEUROD1, instead of acting as an initiator itself, is more likely to function together with MYC after initial NE oncogenesis, to which ASCL1 is more essential (Rudin et al., 2019). A recent study discovered the co-existence of NEUROD1 and ASCL1 in NEPC and proposed a similar model in which ASCL1 clones originated from AD cells and subsequently by subclonal evolution evolved to the NEUROD1 state (Cejas et al., 2020).
Furthermore, by inferring CNV alterations, we identified distinct subpopulations of NEPC and ADPC and their evolutionary relationship. We confirmed that the origin of NEPC is ADPC in six out of seven patients. Our study is the first to report that NEPC could be derived from different stages of ADPC with different background genomic events. Recent investigations, including those utilizing genomic engineering models, have focused on the importance of the double deletion of TP53 and RB1 in the development of NEPC. However, only one patient in our cohort had both a clonal TP53 and an RB1 deletion (Figure S6, Table S5). Previous reports also found that NEPC existed in HSPC samples, but the significance of NEPC in HSPC remains controversial (Aprikian et al., 1994; Casella et al., 1998; Krijnen et al., 1997). Our study confirmed that ADPC in HSPC could, indeed, transdifferentiate into NEPC. Notably, the transcription/epigenetic mechanism of this transition shares a strong commonality with the transition in patients with CRPC. These findings could expand the scope of the investigation into NEPC evolution.
Our study also joins an effort to investigate the TFs network governing NEPC trans-differentiation. We identified program-specific TFs and common TFs in NE1 and NE2. ASCL1 and FOXA2, which have been identified as crucial regulators in NED (Fraser et al., 2019; Park et al., 2017), appeared to dominate early lineage switch. More interestingly, the ASCL1 expression decreases in the late stage, while the downstream target gene network is consistently activated. These early lineage determining factors are often called pioneer factors as they could remodel the chromatin to a more permissive state that allows subsequent TF bindings. The time-dependent expression of NKX2-2, SOX2, and POU3F2 may determine the diverse NEPC phenotypes within the prostate tumors. Incidentally, Sato et al. reported that the cooperation of SOX2 and POU3F2 drives neural lineage in lung squamous cell carcinoma (Sato et al., 2019).
Our study has several limitations. Validating the genetic changes and TF modules remains a challenge, especially in heterogeneous primary patient samples. The CNV inferred by scRNA-seq may have low sensitivity and specificity. The results were validated by parallel bulk-WES-based CNV analysis on three samples and were in concordance. Nevertheless, there is evident disagreement in results in the MHC region (chr6:29,677,984-33,485,635); that is, the deletion reported by inferCNV was not sufficiently supported by the WES data. However, the MHC locus has high gene density; thus, genes in this region violate the "random distribution assumption" that the inferCNV algorithm makes. Further investigation found generally low expression of MHC genes in malignant cells (data not shown), which has been reported as a mechanism of immune evasion (Cornel et al., 2020). Thus, the inferCNV prediction of MHC deletion is likely a false positive.
Another limitation is the sample size. A gold standard in cancer genomics studies is to establish recurrence. Every patient is different, and every tumor is different. They always harbor complex differences in both genomic and epigenomic backgrounds. Owing to this complexity, the power of our current study is likely low. Our key interpretations await replication in much larger patient cohorts.
In conclusion, in this study, we have used scRNA-seq profiling to establish a transcriptomic map of NEPC trans-differentiation. We identified two NE programs with related but distinct gene expression patterns and TF networks. The dynamics of NE gene expression and NEPC’s relationship with ADPC provide instrumental knowledge in designing more informed diagnosis strategies in clinical practice. Future studies should focus on replicating these results in larger cohorts and on testing mechanistic hypotheses based on them.
Limitations of the study
In this research, the discovery of two distinct transcriptional profiles of NEPC was based on small sample size. Although external validation has been made on independent cohorts, the results may not be a comprehensive presentation of all NEPC phenotypes. The clonal evolution analysis at a single-cell resolution based mainly on CNV profiles inferred from RNA-seq data. Although parallel bulk WES-seq was applied for validation, experiments like fluorescence in situ hybridization (FISH) and parallel single-cell whole-genome sequencing (scWGS-seq) are optimal for evidence at the cell level. Besides, the NE1-and NE2-specific TFs were identified through computational approaches, and further validation through biology experiments on NEPC disease models, including cell lines, organoids, and animals, are indispensable.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal to Achaete-scute homolog 1 | Abcam | Cat#ab211327 |
| Rabbit monoclonal to Chromogranin A | Abcam | Cat#ab68271; RRID:AB_11154750 |
| Rabbit monoclonal to HOXB13 | Abcam | Cat#ab201682 |
| Biological samples | ||
| Human prostate cancer tissues | Changhai Hospital | N/A |
| Critical commercial assays | ||
| GEXSCOPETM Single-Cell RNA Library Kit | Singleron Biotechnologies | N/A |
| Chromium Next GEM Single Cell 3′ Kit v3.1 | 10X Genomics | PN-1000269 |
| Deposited data | ||
| P1 | This paper; Genome Sequence Archive for Human (GSA-Human) | GSA-Human: HRA002145 |
| P2 | This paper; GSA-Human | GSA-Human: HRA002145 |
| P3 | This paper; GSA-Human | GSA-Human: HRA002145 |
| P4 | GEO | GEO: GSE137829 |
| P5 | GEO | GEO: GSE137829 |
| P6 | This paper; GSA-Human | GSA-Human: HRA002145 |
| P7 | GEO | GEO: GSE137829 |
| TCGA cohort | GDC Data Portal | GDC: TCGA-PRAD |
| CPGEA cohort | GSA-Human | GSA-Human: HRA000099 |
| Beltran et al. cohort | cBioPortal | cBioPortal: Neuroendocrine Prostate Cancer (Multi-Institute, Nat Med, 2016) |
| source code | Code Ocean | Code Ocean: 4157786 |
| Software and algorithms | ||
| Cellranger | 10X Genomics | v5.0.0 |
| Drop-seq tools | github.com/broadinstitute/Drop-seq | v2.3.0 |
| R | cran.r-project.org | v4.1.1 |
| Seurat | cran.r-project.org | v4.0.5 |
| CellMixS | bioconductor.org | v1.10.0 |
| ClusterProfiler | bioconductor.org | v4.2.2 |
| cNMF | github.com/dylkot/cNMF | v1.1 |
| inferCNV | github.com/broadinstitute/inferCNV | v1.3.3 |
| VarTrix | github.com/10XGenomics/vartrix | v1.1.19 |
| Monocle | bioconductor.org | v2.16.0 |
| pySCENIC | github.com/aertslab/pySCENIC | v0.10.3 |
| Fastp | Miniconda3 | v0.23.0 |
| bwa-mem | Miniconda3 | v0.17.12 |
| Limma | bioconductor.org | v3.50.1 |
| Genome Analysis Toolkit (GATK) | gatk.broadinstitute.org | v4.2.0.0 |
| GSVA | bioconductor.org | v1.42.0 |
| ggplot2 | cran.r-project.org | v3.3.5 |
| Tidyverse | cran.r-project.org | v1.3.1 |
| Corrplot | cran.r-project.org | v0.92 |
Resource availability
Lead contact
Further information and requests should be directed to the lead contact, Jing Li (ljing@smmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
P1 is a 66-year-old male diagnosed as HSPC before sampling, the clincal TNM (cTNM) stage is cT3bN1M1b with the PSA level of 8.63 ng/ml; P2 is a 65-year-old male diagnosed as HSPC before sampling, the cTNM stage is cT1cNxMx with the PSA level of 6.39 ng/ml; P3 is a 73-year-old male diagnosed as CRPC before sampling, the cTNM stage is cT3bN1M0 with the PSA level of 71.64 ng/ml; P6 is a 43-year-old male diagnosed as CRPC before sampling, the cTNM stage is cT3bN1M1b with the PSA level of 147.71 ng/ml. Detailed patient information could be found in Table S1. Informed consents were obtained from all subjects and the experiments with human subjects were approved by Changhai Hospital (CHEC2019-012).
Method details
Experimental design
Prostate cancer tissues were sampled from patients whose MRI-scan had a manifestation of prostate cancer disproportionate with the PSA level. The fresh biopsies were stored in GEXSCOPE™ Tissue Preservation Solution (Singleron Biotechnologies) at 4 °C and shipped to the processing lab within 48 hours for single-cell RNA sequencing. Subsequent pathological test confirmed a partial or complete neuroendocrine phenotype. Informed consents were obtained from all subjects. The experiments with human material were approved by Changhai Hospital (CHEC2019-012). Sample information of patients was summarized in Table S1.
Single cell RNA library construction and sequencing
Single-cell suspensions with 1×105 cells/mL in concentration in PBS (HyClone) were prepared. The P3 library was constructed according to Singleron GEXSCOPETM protocol by GEXSCOPETM Single-Cell RNA Library Kit (Singleron Biotechnologies) (B et al., 2019). Libraries of other samples were constructed by Chromium Next GEM Single Cell 3′ Kit v3.1 (10X Genomics). Individual libraries were diluted to 4nM and pooled for sequencing. Pools were sequenced on Illumina HiSeq XTen with 150 bp paired-end reads to obtain a sequencing depth of approximately 6.5K reads/cell and a 70–80% saturation level.
Generation of the raw count matrix
The pair-end sequencing data of libraries constructed by both Dropseq and 10X genomic technique consisted of cell barcode, unique molecular identifier (UMI) barcode in Read 1 and the 3' end of the transcript in Read 2. Thus the same logic was applied to generate the raw count matrix. The adaptors and poly A tails were trimmed and the reads were mapped against GRCh38 (release 98) and annotated with the genome annotation file (GENCODE V32). Reads with identical cell barcode and UMI were collapsed. Besides, the raw count matrix of P4, P5, and P7 were downloaded from GEO database with the accession number GSE137829 (Dong et al., 2020).
Quality control
We used the Seurat package to read in single-cell counting matrics. Basic cell quality control includes 1. Library Complexity: The number of non-zero expressed genes (features) in each cell; 2. Library Size: The number of unique transcripts (counts) in each cell; 3. Mitochondrial Percentage: The percentage of transcripts aligned to the mitochondrial genome (MT percent) in each cell. Cells with low library complexity and/or library size could fail in the subsequent annotation analysis. A generally high mitochondria percentage indicates active metabolic activities, but an unusual proportion of mitochondrial genes suggests cell stress or death. Therefore, cells with features <500, or counts <1000, or MT percent >20% are excluded from downstream analysis as disqualified cells.
Single-cell data analysis
We used the "Seurat" package for the fundamental single-cell data analysis. The SCTransform function was applied to normalize the single-cell expression value against sequencing depth. The resulting residuals were used to determine the high variable genes (HVGs, n = 3000). The residuals of HVGs were z-scaled to give each HVG the same weight. We selected the top 50 principal components (RunPCA) for non-supervised clustering and visualization. To perform non-supervised clustering, we built the shared nearest neighbor (SNN) graph (FindNeighbours) for the module optimization algorithm (Louvain algorithm) to decide the sub-clusters (FindClusters). The "resolution" parameter, which determines the number of sub-clusters, was adjusted in a semi-supervised way to achieve a common-sense between the subcluster and the cell marker-defined population. We used the uniform manifold approximation and projection (UMAP) technique with default parameters for visualization (RunUMAP).
In the scene of gene expression visualization (FeaturePlot), differential expression analysis (FindMarkers), and gene-set score calculation (AddModuleScore), we opted for the traditional log-normalized values (NormalizeData) rather than residuals as suggested by the Seurat developer.
Cell type annotation
EPCAM is abounded in normal epithelial cells and significantly elevates in tumors (van der Gun et al., 2010). In comparison, VIM is widely used to identify mesenchymal cells and epithelial-mesenchymal transition (EMT) state (Mendez et al., 2010). In the first-pass analysis, sub-clusters were assigned epithelial- or mesenchymal-like by their EPCAM and VIM expression. Considering the epithelial origin of prostate cancer, we assumed mesenchymal-like cells were likely benign cells and further refined their identity in the second-pass analysis. Cells from lymph nodes or in situ lesions are integrated respectively using the CCA integration method in Seurat V3 to remove nested batch effect and to discover rare populations better. The removal of batch effect was evaluated through visualization and the CellMixS method (Lütge et al., 2021). For each cluster, cell types were determined by: 1. observing the expression intensity of curated cell type marker genes (Table S3); 2. Using Seurat FindAllMarkers module with default parameters (adjusted P-value < 0.05, and log2 fold change >0.25, and minimal expression proportion difference >0.25) to identify cluster-specific highly expressed genes. Unknown cells were those without highly expressed markers. Ambiguous cells were those mesenchymal-like cells in the first-pass analysis which formed an EPCAM-enriched cluster in the second-pass; 3. Seurat FindMarkers help discerns subtle differences between clusters of the same lineage.
Clonality analysis
Copy number variation (CNV) profile of single cells was detected by "inferCNV" software (https://github.com/broadinstitute/inferCNV) in each patient. We set "cutoff = 0.1" for 3' library. Normal cell types with at least 15 cells were selected as reference, by which multiple baselines were established to minimize false positive CNV signals. The CNV profiles of epithelial-like cells in the first pass and ambiguous cells in the second pass were queried.
In order to determine the copy state of each chromosome arm. HMM based CNV prediction method implemented in inferCNV was applied. For each cell, a chromosomal arm was considered gain or loss if more than 40% of genes located on this arm have corresponding gain or loss. The percentage of cells harboring copy number altered arm in each sample were then calculated and visualized.
WES and mutation calling
When preparing single-cell libraries for P1, P3, and P6 samples, we kept some tumor tissues and sent them together with the paired whole blood samples for WES sequencing. The whole exon DNA library of P1 was captured using Agilent Human All Exon V6. DNA libraries of P3 and P6 were captured using IDT xGen Exome Research Panel v2. A panel of normal (PON) built on blood samples (n = 20) was also available for P3 and P6. The sequencing library was prepared using the TruSeq Nano DNA HT Sample Prep Kit (Illumina). After quality check using an Agilent Bio-analyzer 2100 and quantified via real-time PCR, the library was sequenced by Illumina Novaseq.
Step by step, raw sequenced reads were quality controlled using fastp (v0.20.1)(Chen et al., 2018), aligned using bwa-mem (v0.17.12)(Li and Durbin, 2009), processed by picard tools (built-in GTAK4) for duplicates marking and base quality score recalibration. The detection of SNVs and InDels followed the GTAK4 best practice using GATK toolkit (v4.2.0.0)(DePristo et al., 2011). The parameter "--f-score-beta" in the "FilterMutectCalls" command was set at 0.8. Only those sites that passed all filters were kept for VarTrix (https://github.com/10XGenomics/vartrix) to validate the assignment of malignancy in the single-cell data.
WES CNV calling
The WES calling followed the GATK4 best practice pipeline (https://gatk.broadinstitute.org/) using GATK toolkit (v4.2.0.0)(DePristo et al., 2011). Read counts were denoised against paired blood normal sample in P1, and against PON in P3 and P6 with GC-bias also corrected simultaneously. The denoised copy ratios were further grouped into segments, where both read counts ratios and germline heterogeneous allelic frequencies information were considered. Sex chromosomes were dropped from the analysis. The parameter "--number-of-changepoints-penalty-factor" in the "ModelSegments" command was tuned to 5 for smooth segmentation. Segments with a mean log2 copy ratio value lower than 0.9 or higher than 1.1 were called as "deletion" or "amplification" respectively.
Putative adenocarcinoma (AD) score of TCGA and CPGEA
The RNA-seq raw count matrix and sample metadata were retrieved from the Chinese Prostate Cancer Genome and Epigenome Atlas (CPGEA) data portal (http://bigd.big.ac.cn/gsa-human/) with the accession number PRJCA001124 (Li et al., 2020) (134 tumors vs. 134 normals). The RNA-seq raw count matrix and sample metadata of The Cancer Genome Atlas (TCGA)-PRAD project were retrieved from the DGC data portal (https://portal.gdc.cancer.gov/) (498 tumors vs. 52 normals). RNA expression data were FPKM normalized, and log2 transformed. To summarize the ADPC signature gene-set, differential expression analysis was carried out between prostate cancer and benign tissue in each cohort using the limma package (Ritchie et al., 2015). Genes highly expressed in tumor tissue with a log 2-fold change >1 and an adjusted P-value < 0.05 were obtained for ADPC signature (Table S4). We thus defined 248 genes in the CPGEA gene-set and 199 in the TCGA gene-set to calculate the malignant scores using AddModuleScore in the Seurat package.
cNMF analysis
Various functional programs, which consist of a group of functionally related genes, help sustain cells' diverse phenotypes. A single cell would have programs defining its specific cell identity and function, and other programs work as "housekeeping" activities. The sequenced single-cell transcriptome is a mixture of various programs with different expression intensities. cNMF algorithm accords with the non-negative nature of the single-cell count matrix. It decomposes the high-dimensional cell (N) × gene (M) matrix into the product of two non-negative low-dimensional matrices, N×k and k×M (K is an integer that represents the number of programs). The k×M matrix represents the gene expression programs (GEPs), which describes genes' contribution to each GEP. The N×k matrix represents the program usages, which describes cells' usage of each GEP.
cNMF iterates the factorization steps 100 times to summarize the best solution for M and N under each k. We selected the optimal value of k ranging from 5 to 15 for each sample by considering the trade-off between Silhouette stability score and Frobenius reconstruction error. 13 GEPs in P1, 12 in P2, 7 in P3, 8 in P4, 14 in P5, 8 in P6, and 9 in P7 was determined. We selected the top 100 genes to represent the GEP. The gene-set score of each GEP was calculated across all malignant cells in each sample. Correlation clustering of the cross-sample GEP scores was performed using "1 - Pearson correlation coefficients" as the distance metric and "ward D2" linkage. After hierarchical clustering, we identified appropriate cutoffs to obtain consensus modules by cutting the clustering tree. The target number of modules was set to 15. Genes from all GEPs in the same module were aggregated and frequencies of their occurrence in samples were calculated. Genes that appeared in at least two samples in a consensus module were considered over-represented and used for GO enrichment analysis. The GO analysis was based on the clusterProfiler R package. We set “minGSSize = 3”, and “adjusted p value cutoff = 0.1”. The results of GO enrichment analysis were then used to annotate the modules.
External validation
An external bulk RNA expression dataset (Beltran et al., 2016) was obtained from cBioPortal (cbioportal.org). The study included 15 CRPC-NEPCs and 34 CRPC-ADPCs, all confirmed by pathologists. We also generated a consensus NE module by combining NE1 and NE2 modules using the same approach (cNMF method). Single-sample gene set enrichment analysis (ssGSEA) score was calculated by GSVA R package on NE1, NE2, and combined NE modules.
Pseudo-time trajectory analysis
Pseudo-time trajectory analysis was carried out on the P5 sample using Monocle 2. The size factors and dispersions were estimated, and low-quality cells were filtered using the parameter "min_expr = 0.1". We chose the top 100 genes of each GEP of the NE1 and NE2 modules, and generalized them as an input marker genes list for Monocle 2 to infer the trans-differentiation process in a semi-supervised manner. The "DDRTree" method implemented was applied for dimensionality reduction. Differential expression analysis among different NE types was performed, and the top 100 pseudo-time-dependent genes (q value <0.01) were displayed for visualization.
Single-cell regulatory network inference
We used pySCENIC 0.10.3 (Van de Sande et al., 2020) to answer the activation of what transcription factor (TF) regulates the genes that determine the phenotype of each single cell. The primary network between putative TFs (Lambert et al., 2018) and their potential target genes were inferred from the raw count matrix of each sample based on the positive co-expression relationship (pyscenic grn, seed = 1). Then the primary network was pruned to keep only the direct target genes that had corresponding binding motif for the regulator TF (pyscenic ctx). The TF and its direct target formed a regulon. The activity of regulons was calculated and binarized using pyscenic aucell.
To answer what TF is essential in different phages of the NE trans-differentiation. We defined TFs as common, NE1-specific, or NE2-specific. For each NE subcluster in each sample, if a regulon is positive in over 25% of cells, the TF was then considered "positive" in that subcluster. Since there were nine NE subclusters, including four NE1 clusters, and five NE2 clusters, we applied an arbitrary cutoff where a TF was considered "common" if it is positive in more than five out of nine NE subclusters, "NE1-specific" if it is positive in more than two out of four NE1 clusters, and "NE2-specific" if it is positive in more than three out of five NE2 subclusters.
Quantification and statistical analysis
The statistical test method can be found in the figure legends. In Figures 1B, 2D, S4A, and S4B, Wilcoxon rank sum test was performed; In Figure 2B, hypergeometric distribution test was performed; In Figure 3B, likelihood ratio test of nested models. The significance level was defined as p < 0.05, and was adjusted by FDR <5% when multiple comparisons exist. R (version 4.1.1) was utilized for data processing and visualization.
Acknowledgments
Funding
Shanghai Rising-Star Program 20QA1411800 (J.L).
National Natural Science Foundation of China grant 82022055 (J.L).
National Natural Science Foundation of China grant 31871317 (J.H).
National Natural Science Foundation of China grant 32070635 (J.H).
Author contributions
Z.W: Data curation, Formal Analysis, Methodology, Visualization, Writing original draft. T.W: Data curation, Formal Analysis, Visualization, Writing original draft. D.H: Formal Analysis, Validation, Software, Visualization, Writing original draft. B.D: Investigation, Methodology, Visualization, Writing original draft. Y.W: Investigation, Validation. H.H: Formal Analysis, Validation, Writing review and editing. W.Z: Validation, Visualization. B.L: Software, Validation. B.J: Validation. H.S: Data curation. M.Q: Data curation. X.G: Funding acquisition, Resources. D.L: Validation. C.C: Investigation, Supervision. G.W: Supervision. C.X: Funding acquisition, Project administration, Resources, Supervision, Writing review and editing. H.L: Investigation, Software, Writing review and editing, Supervision. J.H: Conceptualization, Investigation, Project administration, Funding acquisition, Writing review and editing. J.L: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Writing review and editing.
Declarations of interests
The authors declare no potential conflicts of interest.
Published: July 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104576.
Contributor Information
Chuanliang Xu, Email: chuanliang_xu@126.com.
Hyung Joo Lee, Email: hyungjoo.lee@pintherapeutics.com.
Jialiang Huang, Email: jhuang@xmu.edu.cn.
Jing Li, Email: ljing@smmu.edu.cn.
Supplemental information
Data and code availability
-
•
Single-cell RNA-seq data have been deposited at GSA-Human and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at Code Ocean and is publicly available as of the date of publication. The capsule number is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adams E.J., Karthaus W.R., Hoover E., Liu D., Gruet A., Zhang Z., Cho H., DiLoreto R., Chhangawala S., Liu Y., et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature. 2019;571:408–412. doi: 10.1038/s41586-019-1318-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal R., Huang J., Alumkal J.J., Zhang L., Feng F.Y., Thomas G.V., Weinstein A.S., Friedl V., Zhang C., Witte O.N., et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol. 2018;36:2492–2503. doi: 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akamatsu S., Wyatt A.W., Lin D., Lysakowski S., Zhang F., Kim S., Tse C., Wang K., Mo F., Haegert A., et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015;12:922–936. doi: 10.1016/j.celrep.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Aprikian A.G., Cordon-Cardo C., Fair W.R., Zhang Z.F., Bazinet M., Hamdy S.M., Reuter V.E. Neuroendocrine differentiation in metastatic prostatic adenocarcinoma. J. Urol. 1994;151:914–919. doi: 10.1016/s0022-5347(17)35121-2. [DOI] [PubMed] [Google Scholar]
- Beltran H., Prandi D., Mosquera J.M., Benelli M., Puca L., Cyrta J., Marotz C., Giannopoulou E., Chakravarthi B.V.S.K., Varambally S., et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop J.L., Thaper D., Vahid S., Davies A., Ketola K., Kuruma H., Jama R., Nip K.M., Angeles A., Johnson F., et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017;7:54–71. doi: 10.1158/2159-8290.CD-15-1263. [DOI] [PubMed] [Google Scholar]
- Brady N.J., Bagadion A.M., Singh R., Conteduca V., Van Emmenis L., Arceci E., Pakula H., Carelli R., Khani F., Bakht M., et al. Temporal evolution of cellular heterogeneity during the progression to advanced AR-negative prostate cancer. Nat. Commun. 2021;12:3372. doi: 10.1038/s41467-021-23780-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J., et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella R., Bubendorf L., Sauter G., Moch H., Mihatsch M.J., Gasser T.C. Focal neuroendocrine differentiation lacks prognostic significance in prostate core needle biopsies. J. Urol. 1998;160:406–410. doi: 10.1097/00005392-199808000-00031. [DOI] [PubMed] [Google Scholar]
- Cejas P., Drier Y., Dreijerink K.M.A., Brosens L.A.A., Deshpande V., Epstein C.B., Conemans E.B., Morsink F.H.M., Graham M.K., Valk G.D., et al. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat Med. 2019;25:1260–1265. doi: 10.1038/s41591-019-0493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejas P., Xie Y., Font-Tello A., Lim K., Syamala S., Qiu X., Tewari A.K., Shah N., Nguyen H.M., Patel R.A., et al. Subtype heterogeneity and epigenetic convergence in neuroendocrine prostate cancer. bioRxiv. 2020 doi: 10.1101/2020.09.13.291328. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conteduca V., Oromendia C., Eng K.W., Bareja R., Sigouros M., Molina A., Faltas B.M., Sboner A., Mosquera J.M., Elemento O., et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer. 2019;121:7–18. doi: 10.1016/j.ejca.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornel A.M., Mimpen I.L., Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers. 2020;12:1760. doi: 10.3390/cancers12071760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Sant'Agnese P.A. Neuroendocrine differentiation in carcinoma of the prostate. Diagnostic, prognostic, and therapeutic implications. Cancer. 1992;70:254–268. doi: 10.1002/1097-0142(19920701)70:1+<254::aid-cncr2820701312>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Dong B., Miao J., Wang Y., Luo W., Ji Z., Lai H., Zhang M., Cheng X., Wang J., Fang Y., et al. Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun Biol. 2020;3:778. doi: 10.1038/s42003-020-01476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J.I., Amin M.B., Beltran H., Lotan T.L., Mosquera J.M., Reuter V.E., Robinson B.D., Troncoso P., Rubin M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014;38:756–767. doi: 10.1097/PAS.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine S.W. Neuroendocrine tumors of the prostate. Mod. Pathol. 2018;31:S122–S132. doi: 10.1038/modpathol.2017.164. [DOI] [PubMed] [Google Scholar]
- Fraser J.A., Sutton J.E., Tazayoni S., Bruce I., Poole A.V. hASH1 nuclear localization persists in neuroendocrine transdifferentiated prostate cancer cells, even upon reintroduction of androgen. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-55665-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman-Mazursky O., Elkon R., Efrat S. Redifferentiation of expanded human islet beta cells by inhibition of ARX. Sci. Rep. 2016;6 doi: 10.1038/srep20698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.C., Dancer J.Y., Wang Y., Aparicio A., Navone N.M., Troncoso P., Czerniak B.A. TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum. Pathol. 2011;42:11–17. doi: 10.1016/j.humpath.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA. Cancer. J. Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Kotliar D., Veres A., Nagy M.A., Tabrizi S., Hodis E., Melton D.A., Sabeti P.C. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife. 2019;8:e43803. doi: 10.7554/eLife.43803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnen J.L., Bogdanowicz J.F., Seldenrijk C.A., Mulder P.G., van der Kwast T.H. The prognostic value of neuroendocrine differentiation in adenocarcinoma of the prostate in relation to progression of disease after endocrine therapy. J. Urol. 1997;158:171–174. doi: 10.1097/00005392-199707000-00054. [DOI] [PubMed] [Google Scholar]
- Ku S.Y., Rosario S., Wang Y., Mu P., Seshadri M., Goodrich Z.W., Goodrich M.M., Labbé D.P., Gomez E.C., Wang J., et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T. The human transcription factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- Lee J.K., Phillips J.W., Smith B.A., Park J.W., Stoyanova T., McCaffrey E.F., Baertsch R., Sokolov A., Meyerowitz J.G., Mathis C., et al. N-myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer. Cell. 2016;29:536–547. doi: 10.1016/j.ccell.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Xu C., Lee H.J., Ren S., Zi X., Zhang Z., Wang H., Yu Y., Yang C., Gao X., et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature. 2020;580:93–99. doi: 10.1038/s41586-020-2135-x. [DOI] [PubMed] [Google Scholar]
- Lin D., Wyatt A.W., Xue H., Wang Y., Dong X., Haegert A., Wu R., Brahmbhatt S., Mo F., Jong L., et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74:1272–1283. doi: 10.1158/0008-5472.CAN-13-2921-T. [DOI] [PubMed] [Google Scholar]
- Lütge A., Zyprych-Walczak J., Brykczynska Kunzmann U., Crowell H.L., Calini D., Malhotra D., Soneson C., Robinson M.D. CellMixS: quantifying and visualizing batch effects in single-cell RNA-seq data. Life Science Alliance. 2021;4 doi: 10.26508/lsa.202001004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez M.G., Kojima S., Goldman R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB. J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal R., Schweizer M., Kryvenko O.N., Epstein J.I., Eisenberger M.A. Small cell carcinoma of the prostate. Nat. Rev. Urol. 2014;11:213–219. doi: 10.1038/nrurol.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.W., Lee J.K., Sheu K.M., Wang L., Balanis N.G., Nguyen K., Smith B.A., Cheng C., Tsai B.L., Cheng D., et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science. 2018;362:91–95. doi: 10.1126/science.aat5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.W., Lee J.K., Witte O.N., Huang J. FOXA2 is a sensitive and specific marker for small cell neuroendocrine carcinoma of the prostate. Mod. Pathol. 2017;30:1262–1272. doi: 10.1038/modpathol.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier J.T., Dobromilskaya I., Moriarty W.F., Peacock C.D., Hann C.L., Rudin C.M. Selective tropism of Seneca Valley virus for variant subtype small cell lung cancer. J. Natl. Cancer. Inst. 2013;105:1059–1065. doi: 10.1093/jnci/djt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priemer D.S., Montironi R., Wang L., Williamson S.R., Lopez-Beltran A., Cheng L. Neuroendocrine tumors of the prostate: emerging insights from molecular data and updates to the 2016 world health organization classification. Endocr. Pathol. 2016;27:123–135. doi: 10.1007/s12022-016-9421-z. [DOI] [PubMed] [Google Scholar]
- Rickman D.S., Beltran H., Demichelis F., Rubin M.A. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat Med. 2017;23:664–673. doi: 10.1038/nm.4341. [DOI] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts V.J., van Dijk M.A., Murre C. Localization of Pbx1 transcripts in developing rat embryos. Mech. Dev. 1995;51:193–198. doi: 10.1016/0925-4773(95)00364-9. [DOI] [PubMed] [Google Scholar]
- Rudin C.M., Poirier J.T., Byers L.A., Dive C., Dowlati A., George J., Heymach J.V., Johnson J.E., Lehman J.M., MacPherson D., et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat. Rev. Cancer. 2019;19:289–297. doi: 10.1038/s41568-019-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Yoo S., Kong R., Sinha A., Chandramani-Shivalingappa P., Patel A., Fridrikh M., Nagano O., Masuko T., Beasley M.B., et al. Epigenomic profiling discovers trans-lineage SOX2 partnerships driving tumor heterogeneity in lung squamous cell carcinoma. Cancer Res. 2019;79:6084–6100. doi: 10.1158/0008-5472.CAN-19-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P.H., Nethercott H., Kirov I.I., Ziaeian B., Young M.J., Klassen H. Expression of neurodevelopmental markers by cultured porcine neural precursor cells. Stem Cell. 2005;23:1286–1294. doi: 10.1634/stemcells.2004-0306. [DOI] [PubMed] [Google Scholar]
- Sinha S., Nyquist M.D., Corella A., Coleman I., Nelson P.S. Abstract LB-199: role of ASCL1 in neuroendocrine prostate cancer progression. Cancer Res. 2018;78 LB-199-LB-199. [Google Scholar]
- Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q., et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- van der Gun B.T.F., Melchers L.J., Ruiters M.H.J., de Leij L.F.M.H., McLaughlin P.M.J., Rots M.G. EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis. 2010;31:1913–1921. doi: 10.1093/carcin/bgq187. [DOI] [PubMed] [Google Scholar]
- Vias M., Massie C.E., East P., Scott H., Warren A., Zhou Z., Nikitin A.Y., Neal D.E., Mills I.G. Pro-neural transcription factors as cancer markers. BMC Med Genomics. 2008;1:17. doi: 10.1186/1755-8794-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk R.E., Bhagavan B.S., Levy R., Miller D., Weisburger W. Ectopic ACTH, prostatic oat cell carcinoma, and marked hypernatremia. Cancer. 1977;40:773–778. doi: 10.1002/1097-0142(197708)40:2<773::aid-cncr2820400226>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Yamada Y., Beltran H. Clinical and biological features of neuroendocrine prostate cancer. Curr. Oncol. Rep. 2021;23:15. doi: 10.1007/s11912-020-01003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S., Zhao J.L., Chan J.M., Roudier M.P., Wadosky K.M., Gopalan A., Karthaus W.R., Choi J., Lawrence K., Chaudhary O., et al. Multilineage plasticity in prostate cancer through expansion of stem–like luminal epithelial cells with elevated inflammatory signaling. bioRxiv. 2021 doi: 10.1101/2021.11.01.466599. Preprint at. [DOI] [Google Scholar]
- Zhang Q., Han Y., Zhang Y., Liu D., Ming J., Huang B., Qiu X. Treatment-emergent neuroendocrine prostate cancer: a clinicopathological and immunohistochemical analysis of 94 cases. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.571308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq data have been deposited at GSA-Human and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at Code Ocean and is publicly available as of the date of publication. The capsule number is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.