

Abstract
Background:
As we move away from the traditional chemotherapy era to targeted therapy, the validity of old assessment paradigms associated with therapeutics are being raised in the context of immunotherapy. The old paradigm required elaborating on the toxicity assessment, with no expectation of efficacy in early phase trials. Safety data from Phase 1 and 2 studies with many immunotherapeutics show limited toxicities and draw attention to the need to demonstrate efficacy in the early evaluation of new agents.
Methods:
Literature searches indicate that molecular oncology mechanistic-based agents are being linked with molecular disease status and clinical benefit. Biomarkers and other endpoints are being employed to accomplish this. Perspectives for a meaningful context of integrating biomarkers and clinical trial design are reviewed.
Results:
The design and conduct of clinical trials have not been fully adjusted to the new era of personalized oncology, and so we are in transition. A part of this transition is the management of expectations and trial designs that need to be considered relative to preclinical experience in the development of therapeutics. For example, pathological complete response is now considered a surrogate marker for favorable prognosis in breast cancer patients who are treated in the neoadjuvant setting. This surrogate marker is tied to novel agents’ mechanistic characteristics with no preclinical counterpart.
Conclusion:
The old paradigm considers patients equal with similar chances to respond to treatments, but the new paradigm considers patient’s heterogeneity, a major fact that informs the design of clinical trials. By linking every treatment to a mechanism of action and to the presence of a specific biomarker, new trials are going to have more subjects who are likely to respond to the treatment.
Keywords: Breast cancer, clinical benefit, clinical trials, immunotherapy, pathological complete response, patient advocacy, process management, proof of concept, tumor response
1. INTRODUCTION/BACKGROUND
There is a push to find and develop effective targeted treatments that are based on the changes in the molecular biology of tumor cells [1]. Such therapeutics are mechanistic-based, blocking biologic transduction pathways regulating cell death [1]. Rapid advances in the understanding of tumor biology and the underlying mechanisms of cancer offer an opportunity for the integration of mechanistic-based therapeutics and identification of populations that will benefit from such therapeutics. This emphasis is also manifested in the increasing interest by the US Food and Drug Administration (FDA) to rapidly approve targeted cancer drugs.
Targeted therapy, while typically thought of as being directed toward a target molecule or pathway, also defines a particular population. Preclinical information, focusing on the mode(s) of action of a therapeutic and its results in experimental models, should help build hypotheses concerning potential efficacy, as it translates to humans [2]. This integrative process should define the intended target patient population, but often it does not [3]. Moreover, milestones and expectations of translatable efficacy, first established in the therapeutic discovery phase, are often not met. The failed endpoint necessitates definition and management of coincident expectations from preclinical studies through proof of concept (PoC) studies [2, 4]. PoC trials typically use an early-efficacy endpoint, rather than a clinical response, as the primary endpoint [5]. Do these endpoints have preclinical homologues that contribute to go/no-go decisions in the development process? In this review, we discuss various perspectives associated with defining and managing expectations in the transition of PoC studies to clinical development that is relevant to breast cancer populations.
1.1. Setting the Stage: New Guidance that Links Mechanism with Target Population
Go/no-go decisions are fundamental to modern therapeutic development, and they require analysis from many fronts [6]. The appropriateness of a target and management of key milestones in preclinical assessments of disease and outcomes comes first [2]. Formal clinical milestones, such as the end of Phase 1, Phase 2a PoC, and Phase 2b, also have intermediate milestones and expectations that need to be managed [7].
Phase 2a PoC clinical trials are commonly conducted as pilot studies to evaluate the efficacy and safety of a therapeutic in a selected patient population [8]. The efficacy objective is to assess the effectiveness of a study treatment in that patient population. This necessitates linking mechanistic actions of the therapeutic with clinical benefit. In this context, new guidance from the FDA has defined a clinical setting for breast cancer studies and the surrogate endpoint, pathological complete response (pCR). This guidance provides opportunities to evaluate the clinical benefit of novel therapeutics in breast cancer patients in a shorter timeframe.
The approval of pertuzumab for use in the neoadjuvant setting is evidence that the FDA is committed to supporting the accelerated-approval pathway [9]. The breakthrough therapy process, in general, is evidence of the commitment by the FDA to facilitate clinical development decisions [10]. This designation includes all of the Fast Track program features, as well as more intensive FDA guidance, on an efficient therapeutic development program. The emerging world of N-of-one medicine highlights a need for new approaches to cancer treatments that are pinpointed at an ever-smaller patient population. But the efficacy of therapeutics is beyond the FDA’s control, and efficacy is dependent on trial design and defining and managing expectations between preclinical studies and early Phase 1 studies [2].
1.2. The Backdrop of Clinical Development
The backdrop of determining clinical benefit is still etched in process models of clinical development (Fig. 1), requiring target discovery and validation, PoC clinical trials, and clinical development, leading to approval [11]. Perhaps the most important part of the process is the PoC stage that links Phase 1 and Phase 2 studies [12]. Phase 2a PoC trials, designed to provide an initial test of a particular therapy in a defined population, are the single most important determinants of research-and-development efficiency in the therapeutic-development process. While Phase 2a clinical-trial design is often viewed as a mini-version of a Phase 3 trial, it is not possible to use the clinical endpoints required for approval in a “first efficacy” study. Choosing the right patient population to test safety and efficacy in a Phase 2a study becomes paramount when deciding to move forward. Consequently, surrogate endpoints (e.g., pCR), which are tied to tumor biology and patient population, present themselves as a logical means to glimpse potential efficacy and warrant further clinical development.
Fig. (1).

The initial steps in the clinical development process for a new therapeutic provides a bridge between the basic science, originally suggesting that a disease-causing pathway could be targeted therapeutically, and the definitive studies that convince regulatory agencies that the therapy can beneficially influence outcomes in a patient population.
It cannot be stressed enough that successful management of the research-to-development interface (Fig. 2) requires robust decision-making in the selection of targets, target-patient characteristics, and endpoints.
Fig. (2).

An important element of therapeutic development is the use of small trials that are underpinned with a deep understanding of tumor molecular pathology that guides trial development, along with a strong sense of the therapeutics’ safety profile.
2. DEFINING EXPECTATIONS
There are special positions for Phase 1 and Phase 2 (2a and 2b) clinical trials in therapeutic development. Phase 1 clinical trials are the gateway between scientific research and clinical medicine. Phase 2a trials are “pilot” trials that evaluate efficacy (and safety) in selected patient populations for the disease or condition to be treated, diagnosed, or prevented [8]. Therapeutic PoC studies rely on Phase 2a studies but without a large number of patients. The expectation is that early Phase 2 studies will provide evidence of a reasonable likelihood of efficacy in pivotal Phase 3 studies and that Phase 2 studies will help set the design for the next clinical trial.
There are technical factors that lend to successful development of a therapeutic (Fig. 3) [13]. Expectations include defining the strength and quality of target validation (the “right target”), demonstrating target engagement (the “right tissue”), defining safety margins (the “right safety”), developing patient stratification plans (the right patient), and defining the medical value proposition (the right commercial potential) [13]. While all of these factors are linked clearly, development of therapeutics requires efficacy.
Fig. (3).
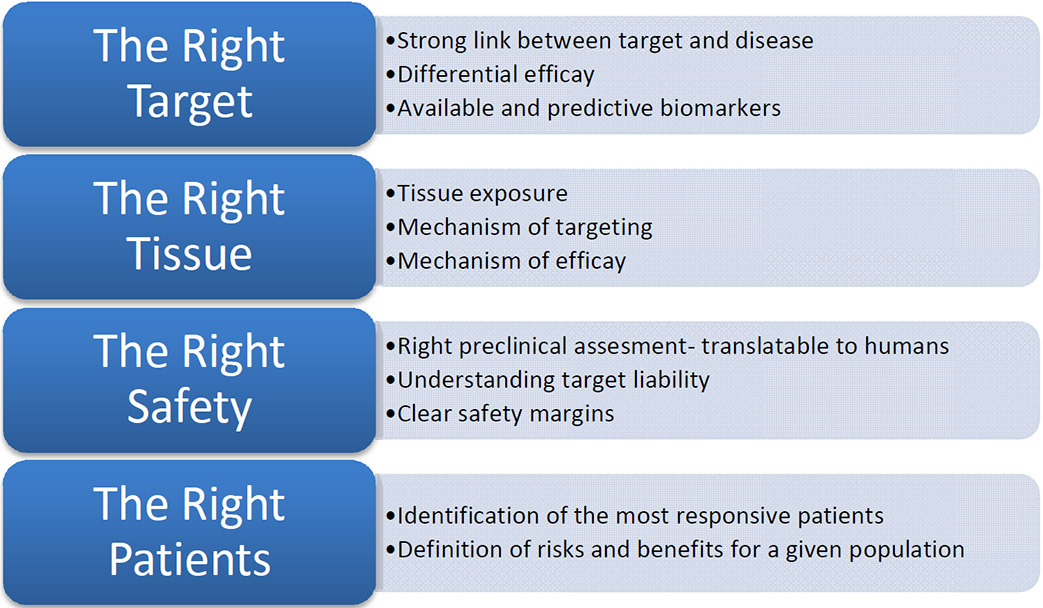
Summary of the key features of an expectation/management framework that can be used to describe a discovery and development project (adapted and used with permission from [13].
2.1. Managing Target Selection and Population
Target selection and the mechanism of efficacy can dictate the target population. Managing expectations of efficacy, starting from preclinical studies and moving through clinical development, is a challenging task, especially in the context of the choices of endpoints used to determine efficacy [2, 14]. It seems obvious that the availability of human-based data that links a target to a disease can drive confidence in target selection.
Major advances in cancer treatment are linked to treatment strategies reflective of the clinical setting, where improved survival is seen with the addition of targeted therapies [15]. Breast cancer treatments were developed by first testing for safety and signs of efficacy in cohorts of patients with advanced disease, with the intent to later test promising drugs in patients who are in earlier disease stages to increase cure rates and improve outcomes for the entire population of women at risk (Fig. 4, left panel) [16].
Fig. (4).

Overall and/or disease-free survival is historically evaluated as the outcome(s) in a randomized Phase 3 trial. Traditional trial concepts on the left emphasized population-based outcomes. Personalized medicine concepts on the right, which are reflective of molecular oncology, focus on disease-related outcomes. This latter concept provides a way to determine what is driving the growth of cancer in an individual patient and to ultimately match the patient with the right targeted therapy.
While this approach has led to therapeutics, we are transitioning to an era of molecular oncology whereby pathways are linked to disease in subpopulations (Fig. 4 right panel). While randomized controlled trials (RCTs) remain the standard for establishing efficacy of new therapies, increased effort is required for the appropriate subpopulation to be found. In this era, patients still define a useful therapy as one that allows them to live longer and helps them to live better [16].
Interestingly, while the primary endpoint of oncology RCTs has shifted away from response rate to survival and other time-to-event endpoints [17], N-of-one trials and the use of surrogate endpoints to suggest clinical benefit are some of the drawbacks of this perspective [18]. Consequently, in the era of molecular oncology, the challenge is to identify which subset of patients is likely to benefit from a novel therapy on the basis of tumor-level molecular characteristics and then determine the best trial design [18].
2.2. Rethinking Randomized Trials
RCTs are considered a necessary element to validate efficacy, eliminating bias in how patients are allocated to treatment options [19]. The FDA often requires randomized trials for the approval of drugs. However, several oncology drugs have been approved on the basis of objective endpoints without a randomized trial [20, 21]. Experiments typically involve a trade-off between internal validity, the ability to trace causal inferences to the intervention, and external validity, indicating the generalizability of the results [22].
RCTs can be large and complex [23] or large and simple [24]. To account for heterogeneity among participants, RCTs must be quite large to achieve statistical significance. RCTs, therefore, provide trends of a large group, making applicability of results to individual patients problematic. Therefore, RCTs can be criticized for not providing enough guidance as to which individuals would benefit from a treatment: the basis for personalized medicine [25-27].
Tumors that express or greatly rely on the therapeutically targeted molecular pathway might be more likely to regress completely than tumors that fail to express the molecular target [28]. Knowing that a target is expressed, patients can be screened prior to trial enrollment, and this will enrich the study population with patients whose tumors are more likely to respond. Nevertheless, it might be unethical to exclude HER2-negative patients from treatment because we know that some patients respond to trastuzumab, despite not having a HER2 expression pattern on their tumor cells [29]. The case might be the same for PDL-1-targeted therapy because of the same type of observations [29, 30]. How accurately can the target be measured?
3. THE DRIVE FOR EFFICACY
Although the widespread impact of new therapeutics has relied, up till now, on clinical trial concepts associated with population-based assessment of a treatment, we are transitioning from population-based to personalized oncology [14, 16]. As noted by Maitland and Schilsky [16], cancer investigators are facing two concurrent and sometimes competing challenges: 1) conducting clinical trials of new, potentially more effective, therapeutic interventions more quickly than ever before [31] and 2) transforming the infrastructure and design of clinical trials from those suited for the era of population oncology to those necessary in the era of personalized oncology [32].
3.1. Precision Oncology and Efficacy
The concept of precision/personalized medicine is the framework for integrating molecular biology with genetic information to define mechanisms of action. There are pros and cons of the concept [33-35]. Cancer therapeutic development continues to lead the way in exploiting the precise molecular pathology that drives the progression of individual cancers [36]. The heterogeneity of cancer cells and their environment is, however, a challenge to targeted therapies [37]. While targeted therapies have been among the most recent approaches to treating cancer, the vast changes in the genetics of tumors via mutations reduce the effectiveness of targeted therapies and lead to disease progression.
Understanding cancer as a disease starts with identifying crucial environmental forces and corresponding adaptive cellular strategies (Darwinian selection), driven by environmental selection forces that interact with individual cellular adaptive strategies (Fig. 5). Proliferating cells - including those in tumors - require mitochondrial respiration. Proliferative cells certainly undergo Darwinian evolution that is powered by genetic instability. The emergence of drug resistance, for example, can result from a cancer cell being under "selective pressure" of chemotherapy, which changes the "adaptive landscape," whereby resistant populations of cancer cells can invariably evolve. This underlying biology also negatively impacts the necessary evidence to enroll patients with a specific molecular signature in a trial that will enable them to achieve event-free survival. This heterogeneity, expressed between subpopulations, extends to intratumoral differences affecting pathways that might impede the discovery of clinically useful biomarkers as predictive of response to treatment [38-40].
Fig. (5).

(A) Cancers are heterogeneous and dynamic entities. Under host and treatment pressures, Darwinian evolution drives these cancers to lose multicellularity traits and develop multidrug resistance. Heterogeneity of a tumor cannot be conceived without heterogeneity of its host. The interaction of the host and the tumor results in cancer disease. (B) Immune heterogeneity is illustrated by the different strategies used by the tumor to achieve immune evasion. In well differentiated tumors, immune evasion occurs by shedding unfavorable antigens to the tumor (active immune editing), which allows it to “drive under the radar screen” of the immune system. At an advanced stage, with the acquisition of genomic instability and development of multiple neo-antigens, immune suppression by the tumor becomes the main mechanism of immune evasion.
The fundamental fact that cancer cells evolve to resist targeted therapies is commonly ignored in the design of treatment strategies. In an effort to develop patient- specific, long-term therapeutic strategies, resistance needs to be anticipated by developing "adaptive therapies" prior to the emergence of resistance. Nevertheless, success in targeted therapy is exemplified by high-profile drugs, such as Herceptin (trastuzumab), Gleevec (imatinib), Tarceva (erlotinib), and Avastin (bevacizumab).
3.2. Patient Advocacy and Efficacy
In 2015, the FDA approved 30 drugs for cancer (http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm), which is more than three times the number they approved in 2009. Ipilimumab (Yervoy; Bristol-Myers Squibb, New York, NY), approved by the FDA for the treatment of metastatic melanoma, provides a survival benefit (over and above the standard-treatment arms) of 3.7 months in previously treated patients and 2.1 months in previously untreated patients [41-43]. The cost, $120,000 for 4 doses, is comparable to the cost of other oncology treatments [41]. Ipilimumab is important because about 20% of patients who use this therapy achieved a long-lasting complete remission that can be equal to a cure of a disease that in the past was invariably fatal. However, identifying those patients upfront and administering the drug to them becomes the challenge.
Patient advocacy, with the realization of treatment costs, followed up by discussions associated with the Moonshot Initiative, has contributed to the drive for faster approvals. It is presumed that faster FDA approval will lower treatment costs because development costs should be lower. In short, the US drug-approval process is fuelled by competing concerns: the significant financial investment that drug manufacturers make throughout the clinical trial and approval processes, the need for drugs to be safe and effective, and the need to speed a drug to market in cases where there are gaps in treatments [44].
3.3. Endpoints and Efficacy
A relevant issue in clinical trials is the selection of an appropriate endpoint. Endpoints used in oncology trials can be grouped into two general categories: patient-centered endpoints and tumor-centered endpoints [45]. The FDA first approved ipilimumab on the basis of an improvement in overall survival (OS), compared with the gp100 vaccine, in patients with advanced melanoma [42]. Although ipilimumab was approved based on OS, objective response rate (ORR) was initially the primary endpoint. The primary endpoint was amended to OS because of Phase 2 results and data from an ongoing, blinded Phase 3 study. The totality of the data from two Phase 3 studies led to ipilimumab being approved based on an extension of survival, despite its failure to significantly raise ORR (i.e., the initial primary endpoint).
OS is the most meaningful clinical endpoint in cancer research and ultimate metric for clinical benefit of a therapeutic [27]. Studies with a hard clinical endpoint, such as survival, are not always feasible for establishing the efficacy or risk of an intervention [46]. So, how is clinical benefit defined in the absence of a hard clinical marker? Biomarkers, under appropriate conditions, are proposed to substitute for hard clinical markers [47]. An appropriate biomarker is defined as “a characteristic that is objectively measured and evaluated as an indication of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [47]. Biomarkers for clinical efficacy can be divided into several groups, including natural-history markers, biological-activity markers, and surrogate markers. Surrogate markers are events of a more intermediate nature and are associated more with biological mechanisms than survival [48].
3.4. Surrogate Markers - pCR as a Special Case
The FDA has defined efficacy as a clinically meaningful benefit, which can be codified under their accelerated-approval process. Some time ago, the FDA started to think about surrogate markers as a substitute for clinical endpoints to define and correlate efficacy of treatments [47-49]. Surrogate markers can include patterns, such as response rates, time-to-disease-relapse, or molecular or genetic changes in blood or tissue samples. These metrics reflect patient milestones that researchers can use as stand-ins to predict longer-term patient results, such as survival.
Recent discussions about pCR being used as a potential endpoint to support accelerated approval have started to change the landscape. Studies have demonstrated that pCR is the most significant prognostic factor for patients with breast cancer who are treated with neoadjuvant therapy [50, 51]. Patients with breast cancer who experience a pCR with neoadjuvant chemotherapy can have significant improvements in both disease-free survival (hazard ratio [HR] 0.48, 95% CI: 0.37-0.63) and OS (HR 0.48, 95% CI: 0.33-0.69), compared with patients with residual invasive disease [52].
Does pCR mean the same thing in all types of breast cancer? pCR might be considered by some to represent a valid surrogate endpoint for long-term outcomes, such as progression-free survival (PFS) and OS, the usual primary trial endpoints in adjuvant or metastatic-disease settings [50, 53]. However in luminal type, pCR does not have the same predictive power of long-term clinical outcomes, such as disease-free survival (DFS) and OS [53]. Expressed in other terms, when pCR succeeds in predicting long-term outcomes it reflects the sensitivity of the primary tumor cells and the disseminated tumor cells to the tested therapy. In tumor types where pCR does not predict long-term outcomes, the eradication of the primary tumor does not correlate with the eradication of disseminated tumor cells because these cells are different from the primary cells. Surrogate biomarkers are often used as proxies since they define intermediary mechanisms of disease processes and are considered as indicators of a biological or pathological process or a therapeutic response [54, 55]. ORR is the portion of patients with a tumor size reduction of a predefined amount for a minimum time period. However, promising early results measured by response rates often do not translate into long-term benefits, such as longer PFS or OS [56].
Long-term follow-up of subjects in trials is clearly needed [57]. Short-term trials cannot address long-term risks and benefits, and small studies cannot reliably assess treatment differences in clinical survival outcomes [58]. Historically, single-arm trials with objective tumor responses were viewed as sufficient Phase 2 evidence of biologic activity [58], and most Phase 3 trials were based on the efficacy data from these relatively small single-arm Phase 2 trials [58].
Herein might be a cause for Phase 3 trial failures. This is the reason why the FDA mandated the conduct of traditional confirmatory trials that would take years to mature for all drugs that benefited from the pCR-based accelerated approval process, such as pertuzumab.
4. TARGET POPULATIONS IN BREAST CANCER
Gene-expression profiling has led to the molecular classification of breast cancer characterized by four intrinsic subtypes: basal-like, HER2-positive, luminal A, and luminal B [59, 60]. Among the estrogen receptor (ER)-positive types of breast cancer, the luminal A subtype has been shown to exhibit good clinical outcomes with endocrine therapy, whereas luminal B represents the more complicated subtype, diagnostically as well as therapeutically. For luminal A breast cancers, the addition of chemotherapy to endocrine therapy generally provides little benefit [61].
There is an important practical consideration for neoadjuvant therapy. While the accelerated approval of pertuzumab in the neoadjuvant setting relied on pCR as a surrogate endpoint for clinical benefit, FDA guidance on the generality of using pCR as an endpoint applies only to targeted agents that were developed for high-risk patients. In breast cancer, the triple negative (TN) and luminal B breast cancers typically are rapidly dividing and have higher expression levels of Ki67, which may explain their chemosensitivity, in keeping with the idea that chemotherapy is most effective at killing cells that are rapidly dividing. Luminal A cancers represent a significant continuing challenge because patients who present with a high tumor burden and with four or more positive lymph nodes have a largely unsatisfactory outcome with endocrine therapy [62]. The benefit from chemotherapy is either very minimal or nonexistent in patients with luminal A cancers. The effectiveness of endocrine therapy is limited by de novo or intrinsic resistance (i.e., existing before any treatment is given) and acquired resistance during treatment (i.e., resistance that develops during therapy after an initial period of response). About 50% of patients with metastatic disease do not respond to initial endocrine treatment [63]. Inevitably, patients with ER-positive advanced breast cancer will become refractory to endocrine therapy, and they will actually have a worse overall long-term survival than same-stage TN patients [62].
4.1. Molecular Pathways of Efficacy
Clinical trials in breast cancer have provided insight on the molecular characteristics of breast cancer and treatment strategies [53]. Treatment combinations targeting receptor signaling that block the crosstalk between pathways and eliminate escape routes have been proven highly effective in clinical outcomes [63]. Results of recent clinical studies, while partly supporting this approach, also highlight the need to better identify a priori the appropriate patients whose tumors are most likely to benefit from specific co-targeting strategies.
Since monitoring biologic disease processes increasingly employs biomarkers, various task forces and agencies have assessed guidelines in the hope that improvements in biomarker studies will enhance the efficiency of investigational drug development [64-68]. Although we have yet to show which levels of biological information are most informative to predict outcome, an integrated molecular characterization of tumors will likely offer the most comprehensive view for individualized therapy approaches [69] as shown in Fig. (5). Signaling pathways themselves might be biomarkers [70]. Fey et al. showed that constructing a pathway that reproduces the all-or-nothing, switch-like activation of the stress-activated kinase JNK could be used to stratify neuroblastoma patients [70].
The use of either biomarkers or surrogate markers in the early phases of therapeutic development is generally noncontroversial from a regulatory viewpoint. Reliance on informative markers obtained early in the development process might be problematic in clinical development if the markers in preclinical models are vastly different than in the targeted population.
4.2. Linking Target with Patient Population
Glycosylation is the stepwise procedure of covalent attachment of oligosaccharide chains to proteins or lipids, and alterations in this process have been associated with malignant transformation [71]. Glycans are involved in a number of processes relevant to carcinogenesis, including regulation of growth factors/growth factor receptors, cell-cell adhesion and motility, as well as immune system modulation. Expression analyses of glycan-related genes reveal that mRNA levels for many of the glycan-related genes differ significantly between normal and malignant breast tissue [72]. Immunohistochemical staining also indicates this differential expression [73]. The totality of glycan expression on cancer cells defines them as tumor associated carbohydrate antigens (TACAs) [74].
It is postulated that antibodies reactive with TACAs are part of immune-surveillance mechanisms [75, 76] and have apoptotic activity that affect metastases [77]. TACAs define a right target (Fig. 3) because of their tissue distribution and functional importance in tumor biology [71, 74]. These glycan signatures are linked to signaling cascades [78]. We have taken advantage of the nature of antibodies to bind to the right target by developing carbohydrate mimetic peptides (CMPs) that will induce antibodies with proapoptotic activity toward cancer cells, much like natural antibodies [79]. We therefore expect to develop such antibodies, given the right choice of CMP, with characteristics that allow manufacturing success for clinical testing [2]. This peptide is a pan-immunogen, synthesized with the Pan T cell peptide PADRE, and is able to induce antibody responses to multiple TACAs expressed on multiple tumor phenotypes in mice and humans.
Our preclinical studies indicate that CMP immunizations are effective in mouse models of cancer, which indicate that we are targeting the right tissue [79-83]. In addition, antibodies that bind to a broad spectrum of TACAs can reduce tumor-cell dissemination by multiple mechanisms, including blocking the adhesion of metastatic cells to adhesion molecules, mediating cell death via complement pathways, and generally functioning as regulatory molecules to thwart signaling processes that underlie migration, autocrine, and paracrine activities that grant immune privilege to cancer [75].
Glycan expression on human and murine tissues can similarly react with various lectins [73, 83]. In the context of safety profile, using tissue destruction or immune pathology as a metric, we observed that immunization with CMPs in general do not destroy normal tissue or induce antibodies that could lead to neuronal damage in experimental animals [82, 83], indicating that they have the right safety level with immunogen dosing ranges of 100-500 ug. In a Phase 1 clinical trial where metastatic breast cancer subjects were immunized with P10s CMP, we found the immunogen to be safe and tolerable, with no adverse response of grade 3 tumors and subjects to have the same immunogen concentration as the experimental animals [84].
In terms of defining the “right patients”, we are using in vitro assessments to better understand combination therapies and immunogen-induced antibodies [84]. We have observed that immunogen-induced antibodies can sensitize tumor cells for cell death through apoptotic mechanisms [84]. Consequently, the induction of such antibodies links the right target, the right tissue, and the right safety with mechanisms of efficacy that are translatable from preclinical models to humans. The observation that the induced antibodies can sensitize tumor cells to more efficient killing by taxanes [84] has led us to start a Phase 2 study in the neoadjuvant setting with estrogen-positive, HER2-negative patients with a change in pCR as the surrogate endpoint (see below). Immunization with CMPs offers a new direction in the treatment of cancer that links innate and adaptive responses to target TACAs [85].
4.3. Linking Mechanism with Efficacy
We are using the rationale of Fig. (6) in our neoadjuvant Phase 2 trial with the P10s CMP. Results from our early-stage Phase 1 trial with the P10s immunogen indicate that the CMP induces antibodies with GD2/GD3/GM2 reactivity, much like it did in our preclinical studies in mice [82]. More broadly, we believe our results suggest that targeting TACAs by active specific immunization, as a means to perturb signaling, offers new opportunities to target cancer beyond the conventional lytic killing of cancer cells by the immune system. We also observe that P10s-induced antibodies play a role in sensitizing tumor cells for more efficient activity of taxanes [84]. Gene-expression profiling suggests that Bcl-2 is upregulated in luminal A tumor cells, which may explain why these tumors are resistant to cytotoxic chemotherapy. Reduced sensitivity to various chemotherapeutic drugs is well known to be mediated by high levels of the anti-apoptotic protein Bcl-2 [86] and associated with a low Ki67 expression profile, typical for luminal A. Consequently, patients with luminal A tumors do not derive much benefit from chemotherapy, and pCRs are low in patients within this subgroup who are treated with neoadjuvant chemotherapy.
Fig. (6).

Integration of mechanism-based rationale for Phase 2 studies. The future of oncologic therapeutic development lies in using predictive biomarkers to identify subsets of patients who will benefit from particular therapies. Taxanes function through apoptotic pathways mediated by focal-adhesion kinase, for example. Tumor cells can be sensitized for more efficient killing by taxanes, which in turn can lead to better tumor response. This is manifested in increased pCR rates. ER-estrogen receptor; PR’-progesterone receptor; TN-Triple Negative; CR-complete regression; PR-partial regression.
If there were a way to increase pCR in the luminal A subpopulation, will pCR for this subpopulation become predictive of survival and would standard of care change? Based on meta-analyses, the effect by HR status is dependent on tumor grade [50]. Patients with grades 1 and 2 who reached pCR still did not do well. This manuscript suggests that killing all cancer cells in the primary tumor bed does not matter because patients who had a pCR did not do better than those who did not reach a pCR. In other words, eradicating all primary tumor cells in ER-positive grades 1 and 2 breast cancer is not parallel with eradication of disseminated tumor cells. It is possible that the indolent nature of these cancers leads to early clonal evolution with a major genomic drift that separates the disseminated tumor cells from the primary-tumor-bed cells by causing different sensitivities to cytotoxic agents. In this case, increasing the number of pCRs for the sake of increasing pCR numbers is unlikely to increase survival. Yet, the fact that pCR rates that are associated with grade 3 tumors, and other tumor subtypes, can reflect survival leads to a “what if” one can change grade 1 and grade 2 tumor types to be responsive, as grade 3 tumor types are.
The concept that antibodies can sensitize low-grade tumors (of the luminal A type) to the efficacious action of chemotherapeutics on grade 3 cancers can possibly change treatment paradigms for this subpopulation. Targeting this subpopulation for future clinical therapy development should include:
Generally, endocrine therapy alone would be recommended for luminal A, which carries an excellent ten-year breast cancer-specific survival rate. However, tumor dormancy and late recurrences beyond 10 years are characteristic of luminal A.
-
TACAs are involved in tumor-cell dissemination and in dormancy [87]. Tumor cells exhibit striking changes in cell-surface glycosylation, as a consequence of dysregulated glycosyltransferases and glycosidases. Circulating tumor cells can evolve with the expression of different TACA moieties that affect distant tumor-cell dissemination and organ colonization. These disseminated tumor cells carry the same indolent nature as the primary tumor, but since they are evolving independently from the primary tumor and are undergoing different environmental pressures, they may acquire different genetic/epigenetic changes.
In particular, an increase in the expression of certain sialylated glycans is a prominent feature of many transformed cells [87]. Altered sialylation has long been associated with metastatic cell behaviors, including invasion and enhanced cell survival; however, there is limited information regarding the molecular details of how distinct sialylated structures or sialylated carrier proteins regulate cell signaling to control responses, such as adhesion/migration or resistance to specific apoptotic pathways [87]. We have shown that a breast cancer cell line that targets the bone had higher levels of α2-6-linked sialic acid [88], whereas upregulation of ST6GaLNAcV, which adds α2-6 sialic acid to gangliosides, directs breast cancer metastasis to the brain [89].
The optimal manner and duration of endocrine therapy in either pre- or postmenopausal women is still debated; extension of endocrine treatment beyond 5, or even 10, years has received support from clinical studies [90], but the most effective endocrine therapy agent requires ongoing study to better define treatment algorithms for luminal A breast cancer.
If we can overcome Bcl-2 resistance in luminal A cancers, we can optimize this approach for other breast cancer subtypes. Because diminished Bcl-2 expression in cancer confers increased sensitivity to cytotoxic chemotherapy, it is possible that breast cancer patients with endocrine-resistant disease could achieve significant therapeutic benefit from cytotoxic agents [91, 92].
In breast cancer, a relationship between host defense mechanisms and prognosis was proposed some time ago [93]. Subjects with tumors densely infiltrated with CD8+ T cell, a critical component of antitumor immunity, tend to have a better prognosis, especially in rapidly proliferating tumors [94]. Denkert et al. [95] showed that the presence of tumor-associated lymphocytes in breast cancer is a new and independent predictor of response to anthracycline/taxane. CD8+ cytotoxic T lymphocytes are a key component of Tumor Infiltrating Lymphocytes (TILs) associated with chemo-response [96]. Without chemo-response, TILs might not be functional for luminal A patients.
PD-L1 upregulation, which is more frequent in basal breast cancers, is associated with increased T-cell cytotoxic immune response [97]. Upregulation of PD-L1 is associated with better survival and chemotherapy response and is also associated with increased pCR after neoadjuvant chemotherapy [97]. Consequently, reactivation of dormant tumor-infiltrating lymphocytes by PDL1-inhibitors could represent a promising strategy in PD-L1-upregulated basal breast cancer.
5. THE POLITICS AND FASHIONS OF THERAPEUTICS
Predicting clinical efficacy is one of the greatest bottlenecks in therapeutic development. Biomarkers continue to appeal as an essential part of clinical development strategies. Useful biomarkers that offer information on mechanisms that can be tied to the clinical benefit of particular individuals undergoing treatment are viewed as the future of medicine. In principle, this approach offers an alternative to conventional therapeutic development for large populations (one size fits all), and they promise that correlative relationships will lead to safer therapies and faster approval rates.
Biomarker development now relies on the FDA Critical Path Opportunities list, which has potential to speed up the development and approval of new drugs. Interestingly, while biomarkers are increasingly popular, adding such biomarkers might actually increase the burden of evaluation in clinical trials. In addition, there is often a gap in the ability to predict a drug candidate’s performance early and with a large degree of certainty, due in part to a mismatch in managing expectations in the early phases of evaluation [2]. Calculating small differences in gene expression, for example, requires a large number of patients. Biomarker qualification is the preferred terminology for the evidentiary procedure of causally linking a biomarker to a biological process. This is a lengthy process that requires prospective and or retrospective analysis of prospectively conducted clinical trials and large population screening, which is at odds with the push for smaller and faster trials.
Biomarkers are argued to be predictors of drug efficacy that help to reach conclusions faster than evaluating conventional clinical endpoints. Safety profiles in preclinical and human studies, along with biomarker evaluation that are associated with mechanisms, can certainly lend themselves to the decision-making process to move forward with clinical development. Emphasizing mechanistic studies at the preclinical stage with robust biomarker assays should increase prospects of successful outcomes in the clinic. Interestingly, the old markers of Ki-67, Bcl-2, and the various caspases might work well in validating if a patient will or will not respond to a therapeutic that triggers signaling pathways associated with cell survival. Mechanism-based therapies under clinical evaluation in oncology can directly induce apoptosis by targeting molecular components of apoptosis regulatory pathways [98]. Application of informative, validated biomarkers of apoptosis in clinical trials of anti-cancer therapies is therefore urgently required, as noted some time ago [99].
Recent studies using explant tissue samples from patients suggest that low levels of activated caspase-3 predict favorable response to 5FU-based chemotherapy in advanced colorectal cancer (CRC) [100]. Using tissue microarrays, the authors examined caspase-3 activation and individual patient response. Their results demonstrated that caspase-3 activity induces cell proliferation in patient tumors, suggesting a yinand-yang effect of caspase-3 in relation to 5FU therapy. Further analysis suggested a role for Cox-2 inhibition that could be mediated by aspirin [100]. It was concluded that measurement of caspase-3 in the tumor or of caspase-3/7 activities in the serum might enable reliable stratification of patients based on response, and it might also allow for identification of patients likely to respond poorly to 5FU-based therapy [100]. However, it was also concluded that analysis in larger cohorts and long-term studies are required to further validate caspase-3 or caspase-3/7 activities as a biomarker for early prediction of treatment outcome or disease progression in CRC patients [100].
Likewise, Ki-67 expression in breast cancer tissue may be an effective predictive factor of response to neoadjuvant chemotherapy [101]. Moreover, Ki-67 was found to be a useful predictive factor for pCR, especially in patients with ER-negative and HER2-positive breast cancer [101]. Validating pathways associated with pCR is warranted in order to more accurately use the neoadjuvant setting for drug approval studies.
CONCLUSION
Moving from the blind approach used in traditional oncology treatment to more targeted and personalized therapy will require major adjustment and invention of novel conceptual and practical frameworks for testing new drugs. The old paradigm considers patients, who have similar chances to respond to treatments, as equals, while the new paradigm considers patient heterogeneity a major fact that informs the design of clinical trials. In the new paradigm, only patients who carry the biomarker in their cancer will be eligible for the planned study, which allows an expectation for high-response rate at an early phase of development using smaller trials. Here again, an expectation of response is justified if the patients are selected based on the presence of the target. Once safety is documented the process should move quickly forward to efficacy studies.
The case for pCR as a clinical end point is unique. The mechanistic basis of pCR lies in the interaction between survival and cell-death pathways associated with tumor cell survival/death decisions. When survival pathways are inhibited and the cells are “pushed” over the cliff of apoptosis, complete eradication of cancer cells ensues, which defines pCR. The importance of pCR in breast cancer, which is now seen by the FDA as a potential solid surrogate endpoint for long-term survival, stems from the fact that it is considered sufficient evidence by the FDA for accelerated approval of new drugs. In our experience, targeting TACA associated with survival/apoptotic pathways reinforces apoptotic activity in a way that is similar to chemotherapeutics. But not all tumors respond in this way. This suggests that old biomarkers, associated with apoptosis, need further understanding and validation as predictors of efficacy.
ACKNOWLEDGEMENTS
This work was supported by a Clinical Translational Award from the Department of Defense Breast Cancer Program (W81XWH-06-1-0542) to TKE. Also supported by the UAMS Translational Research Institute (TRI), UL1TR000039 through the NIH National Center for Research Resources and National Center for Advancing Translational Sciences and the UAMS Center for Microbial Pathogenesis and Host Inflammatory Responses, P20 GM103625. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or UAMS. Light editing was provided by DeAnn Hubberd of UAMS.
LIST OF ABBREVIATIONS
- CMP
Carbohydrate Mimetic Peptide
- CRC
Colorectal Cancer
- DFS
Disease Free Survival
- ER
Estrogen Receptor
- FDA
Food and Drug Administration
- HR
Hazard Ratio
- ORR
Objective Response Rate
- OS
Overall Survival
- pCR
Pathological Complete Response
- PFS
Progression-Free Survival
- PoC
Proof of Concept
- PR
Partial Response
- PR’
Progesterone Receptor
- RCT
Randomized Controlled Trials
- TACA
Tumor Associated Carbohydrate Antigen
- TILs
Tumor Infiltrating Lymphocytes
- TN
Triple Negative
Footnotes
CONFLICT OF INTEREST
Drs. Kieber-Emmons, Monzavi-Karbassi, and UAMS have a financial interest in the technology discussed in this publication. These financial interests have been reviewed and approved in accordance with the UAMS conflict of interest policies.
REFERENCES
- [1].Perez-Herrero E, Fernandez-Medarde A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015; 93: 52–79. [DOI] [PubMed] [Google Scholar]
- [2].Kieber-Emmons T, Pennisi A, Lane A, et al. Defining and managing expectations for early immunotherapy cancer trials. Rev Rec Clin Trials 2015; 10(1): 47–60. [DOI] [PubMed] [Google Scholar]
- [3].Butler D Translational research: Crossing the valley of death. Nature 2008; 453(7197): 840–2. [DOI] [PubMed] [Google Scholar]
- [4].Preskorn SH. The role of proof of concept (POC) studies in drug development using the EVP-6124 POC study as an example. J Psychiatr Pract 2014; 20(1): 59–60. [DOI] [PubMed] [Google Scholar]
- [5].Chen C, Sun L, Li CL. Evaluation of early efficacy endpoints for proof-of-concept trials. J Biopharm Stat 2013; 23(2): 413–24. [DOI] [PubMed] [Google Scholar]
- [6].Pritchard JF, Jurima-Romet M, Reimer ML, Mortimer E, Rolfe B, Cayen MN. Making better drugs: Decision gates in non-clinical drug development. Nat Rev Drug Discov 2003; 2(7): 542–53. [DOI] [PubMed] [Google Scholar]
- [7].Potter WZ. Optimizing early Go/No Go decisions in CNS drug development. Expert Rev Clin Pharmacol 2015; 8(2): 155–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhong B Single-arm phase IIA clinical trials with go/no-go decisions. Contemp Clin Trials 2012; 33(6): 1272–9. [DOI] [PubMed] [Google Scholar]
- [9].Prowell TM, Pazdur R. Pathological complete response and accelerated drug approval in early breast cancer. N Engl J Med 2012; 366(26): 2438–41. [DOI] [PubMed] [Google Scholar]
- [10].Sherman RE, Li J, Shapley S, Robb M, Woodcock J. Expediting drug development--the FDA's new "breakthrough therapy" designation. N Engl J Med 2013; 369(20): 1877–80. [DOI] [PubMed] [Google Scholar]
- [11].Orloff J, Douglas F, Pinheiro J, et al. The future of drug development: advancing clinical trial design. Nat Rev Drug Discov 2009; 8(12): 949–57. [DOI] [PubMed] [Google Scholar]
- [12].Schmidt B Proof of Principle studies. Epilepsy Res 2006; 68(1): 48–52. [DOI] [PubMed] [Google Scholar]
- [13].Cook D, Brown D, Alexander R, et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov 2014; 13(6): 419–31. [DOI] [PubMed] [Google Scholar]
- [14].Beckman RA, Chen C. New evidence-based adaptive clinical trial methods for optimally integrating predictive biomarkers into oncology clinical development programs. Chin J Cancer 2013; 32(5): 233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Burney IA, Al-Moundhri MS. Major advances in the treatment of cancer: What does a Non-Oncologist need to know? Sultan Qaboos Univ Med J 2008; 8(2): 137–48. [PMC free article] [PubMed] [Google Scholar]
- [16].Maitland ML, Schilsky RL. Clinical trials in the era of personalized oncology. CA Cancer J Clin 2011; 61(6): 365–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Booth CM. Evaluating patient-centered outcomes in the randomized controlled trial and beyond: Informing the future with lessons from the past. Clin Cancer Res 2010; 16(24): 5963. [DOI] [PubMed] [Google Scholar]
- [18].Lai TL, Lavori PW. Innovative clinical trial designs: Toward a 21st-Century health care system. Stat Biosci 2011; 3(2): 145–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Viera AJ, Bangdiwala SI. Eliminating bias in randomized controlled trials: importance of allocation concealment and masking. Fam Med 2007; 39(2): 132–7. [PubMed] [Google Scholar]
- [20].Tsimberidou AM, Braiteh F, Stewart DJ, Kurzrock R. Ultimate fate of oncology drugs approved by the US food and drug administration without a randomized Trial. J Clin Oncol 2009; 27(36): 6243–50. [DOI] [PubMed] [Google Scholar]
- [21].Hatswell AJ, Baio G, Berlin JA, Irs A, Freemantle N. Regulatory approval of pharmaceuticals without a randomised controlled study: analysis of EMA and FDA approvals 1999-2014. BMJ Open 2016; 6(6): e011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shadish WR, Clark MH, Steiner PM, Hill J. Can nonrandomized experiments yield accurate answers? A randomized experiment comparing random and nonrandom assignments [with comments, rejoinder]. J Am Stat Assoc 2008; 103(484): 1334–50. [Google Scholar]
- [23].Sully BG, Julious SA, Nicholl J. A reinvestigation of recruitment to randomised, controlled, multicenter trials: a review of trials funded by two UK funding agencies. Trials 2013; 14: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shuster JJ. Sample size verification for clinical trials. Clin Transl Sci. 2014; 7(1): 60–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ghislain I, Zikos E, Coens C, et al. Health-related quality of life in locally advanced and metastatic breast cancer: methodological and clinical issues in randomised controlled trials. Lancet Oncol 2016; 17(7): e294–304. [DOI] [PubMed] [Google Scholar]
- [26].Pocock SJ, Stone GW. The primary outcome fails - What next? N Engl J Med 2016; 375(9): 861–70. [DOI] [PubMed] [Google Scholar]
- [27].Pocock SJ, Stone GW. The primary outcome is positive - Is that good enough? N Engl J Med 2016; 375(10): 971–9. [DOI] [PubMed] [Google Scholar]
- [28].Schilsky RL. End points in cancer clinical trials and the drug approval process. Clin Cancer Res 2002; 8(4): 935–8. [PubMed] [Google Scholar]
- [29].Ithimakin S, Day KC, Malik F, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res 2013; 73(5): 1635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Carbognin L, Pilotto S, Milella M, et al. Differential activity of nivolumab, pembrolizumab and mpdl3280a according to the tumor expression of programmed death-ligand-1 (pd-l1): Sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS One 2015; 10(6): e0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schork NJ. Personalized medicine: Time for one-person trials. Nature 2015; 520(7549): 609–11. [DOI] [PubMed] [Google Scholar]
- [32].Biankin AV, Piantadosi S, Hollingsworth SJ. Patient-centric trials for therapeutic development in precision oncology. Nature 2015; 526(7573): 361–70. [DOI] [PubMed] [Google Scholar]
- [33].Millner LM, Strotman LN. The future of precision medicine in oncology. Clin Lab Med 2016; 36(3): 557–73. [DOI] [PubMed] [Google Scholar]
- [34].Garofalo A, Sholl L, Reardon B, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med 2016; 8(1): 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morgan G, Aftimos P, Awada A. current-day precision oncology: from cancer prevention, screening, drug development, and treatment - have we fallen short of the promise? Curr Opin Oncol 2016; 28(5): 441–6. [DOI] [PubMed] [Google Scholar]
- [36].Collins I, Workman P. New approaches to molecular cancer therapeutics. Nat Chem Biol 2006; 2(12): 689–700. [DOI] [PubMed] [Google Scholar]
- [37].Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer 2013; 108(3): 479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Longo DL. Tumor heterogeneity and personalized medicine. N Engl J Med 2012; 366(10): 956–7. [DOI] [PubMed] [Google Scholar]
- [39].Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366(10): 883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kirk R Genetics: Personalized medicine and tumour heterogeneity. Nat Rev Clin Oncol 2012; 9(5): 250. [DOI] [PubMed] [Google Scholar]
- [41].Siddiqui M, Rajkumar SV. The high cost of cancer drugs and what we can do about it. Mayo Clin Proc 2012; 87(10): 935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363(8): 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364(26): 2517–26. [DOI] [PubMed] [Google Scholar]
- [44].Ashley D, Thomas D, Gore L, et al. Accepting risk in the acceleration of drug development for rare cancers. Lancet Oncol 2015; 16(4): e190–4. [DOI] [PubMed] [Google Scholar]
- [45].Fiteni F, Westeel V, Pivot X, Borg C, Vernerey D, Bonnetain F. Endpoints in cancer clinical trials. J Visc Surg 2014; 151(1): 17–22. [DOI] [PubMed] [Google Scholar]
- [46].Kluft C Principles of use of surrogate markers and endpoints. Maturitas 2004; 47(4): 293–8. [DOI] [PubMed] [Google Scholar]
- [47].Aronson JK. Biomarkers and surrogate endpoints. Br J Clin Pharmacol 2005; 59(5): 491–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx 2004; 1(2): 189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Stat Med 2012; 31(25): 2973–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014; 384(9938): 164–72. [DOI] [PubMed] [Google Scholar]
- [51].Bonnefoi H, Jacot W, Saghatchian M, et al. Neoadjuvant treatment with docetaxel plus lapatinib, trastuzumab, or both followed by an anthracycline-based chemotherapy in HER2-positive breast cancer: Results of the randomised phase II EORTC 10054 study. Ann Oncol 2015; 26(2): 325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mieog JS, van der Hage JA, van de Velde CJ. Neoadjuvant chemotherapy for operable breast cancer. Br J Surg 2007; 94(10): 1189–200. [DOI] [PubMed] [Google Scholar]
- [53].Pennisi A, Kieber-Emmons T, Makhoul I, Hutchins L. Relevance of pathological complete response after neoadjuvant therapy for breast cancer. Breast Cancer (Auckl) 2016; 10: 103–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wagner JA. Overview of biomarkers and surrogate endpoints in drug development. Dis Markers 2002; 18(2): 41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Aronson JK. Research priorities in biomarkers and surrogate endpoints. Br J Clin Pharmacol 2012; 73(6): 900–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hutchinson L Tumour response, correlates of survival and clinical benefit. Nat Rev Clin Oncol 2015; 12(8): 433. [DOI] [PubMed] [Google Scholar]
- [57].Cuzick J Statistical controversies in clinical research: long-term follow-up of clinical trials in cancer. Ann Oncol 2015; 26(12): 2363–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Korn EL, Sachs MC, McShane LM. Statistical controversies in clinical research: assessing pathologic complete response as a trial-level surrogate end point for early-stage breast cancer. Ann Oncol. 2016; 27(1): 10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wirapati P, Sotiriou C, Kunkel S, et al. Meta-analysis of gene expression profiles in breast cancer: Toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res 2008; 10(4): R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med 2009; 360(8): 790–800. [DOI] [PubMed] [Google Scholar]
- [61].Uchida N, Suda T, Ishiguro K. Effect of chemotherapy for luminal a breast cancer. Yonago Acta Med 2013; 56(2): 51–6. [PMC free article] [PubMed] [Google Scholar]
- [62].Colleoni M, Sun Z, Price KN, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: Results from the international breast cancer study group trials I to V. J Clin Oncol 2016; 34(9): 927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 2011; 62: 233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Dancey JE, Dobbin KK, Groshen S, et al. Guidelines for the development and incorporation of biomarker studies in early clinical trials of novel agents. Clin Cancer Res 2010; 16(6): 1745–55. [DOI] [PubMed] [Google Scholar]
- [65].Paller CJ, Bradbury PA, Ivy SP, et al. Design of phase I combination trials: Recommendations of the clinical trial design task force of the NCI investigational drug steering committee. Clin Cancer Res 2014; 20(16): 4210–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ivy SP, Siu LL, Garrett-Mayer E, Rubinstein L. Approaches to phase 1 clinical trial design focused on safety, efficiency, and selected patient populations: A report from the clinical trial design task force of the national cancer institute investigational drug steering committee. Clin Cancer Res 2010; 16(6): 1726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].LoRusso PM, Boerner SA, Seymour L. An overview of the optimal planning, design, and conduct of phase I studies of new therapeutics. Clin Cancer Res 2010; 16(6): 1710–8. [DOI] [PubMed] [Google Scholar]
- [68].Yang B, Zhou Y, Zhang L, Cui L. Enrichment design with patient population augmentation. Contemp Clin Trials 2015; 42: 60–7. [DOI] [PubMed] [Google Scholar]
- [69].Dietel M, Johrens K, Laffert MV, et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: A review focussing on clinical relevance. Cancer Gene Ther 2015; 22(9): 417–30. [DOI] [PubMed] [Google Scholar]
- [70].Fey D, Halasz M, Dreidax D, et al. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci Signal 2015; 8(408): ra130. [DOI] [PubMed] [Google Scholar]
- [71].Cazet A, Julien S, Bobowski M, Burchell J, Delannoy P. Tumour-associated carbohydrate antigens in breast cancer. Breast Cancer Res 2010; 12(3): 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Potapenko IO, Haakensen VD, Luders T, et al. Glycan gene expression signatures in normal and malignant breast tissue; possible role in diagnosis and progression. Mol Oncol 2010; 4(2): 98–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Korourian S, Siegel E, Kieber-Emmons T, Monzavi-Karbassi B. Expression analysis of carbohydrate antigens in ductal carcinoma in situ of the breast by lectin histochemistry. BMC Cancer 2008; 8: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hakomori S Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci USA 2002; 99(16): 10231–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pashov A, Monzavi-Karbassi B, Kieber-Emmons T. Glycan mediated immune responses to tumor cells. Hum Vaccin 2011; 7(Suppl): 156–65. [DOI] [PubMed] [Google Scholar]
- [76].Monzavi-Karbassi B, Pashov A, Kieber-Emmons T. Tumor-Associated Glycans and Immune Surveillance. Vaccines (Basel) 2013; 1(2): 174–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Vollmers HP, Brandlein S. Natural antibodies and cancer. N Biotechnol 2009; 25(5): 294–8. [DOI] [PubMed] [Google Scholar]
- [78].Hanover JA. Glycan-dependent signaling: O-linked N-acetylglucosamine. FASEB J 2001; 15(11): 1865–76. [DOI] [PubMed] [Google Scholar]
- [79].Monzavi-Karbassi B, Artaud C, Jousheghany F, et al. Reduction of spontaneous metastases through induction of carbohydrate cross-reactive apoptotic antibodies. J Immunol 2005; 174(11): 7057–65. [DOI] [PubMed] [Google Scholar]
- [80].Kieber-Emmons T, Luo P, Qiu J, et al. Vaccination with carbohydrate peptide mimotopes promotes anti-tumor responses. Nat Biotechnol 1999; 17(7): 660–5. [DOI] [PubMed] [Google Scholar]
- [81].Monzavi-Karbassi B, Cunto-Amesty G, Luo P, Shamloo S, Blaszcyk-Thurin M, Kieber-Emmons T. Immunization with a carbohydrate mimicking peptide augments tumor- specific cellular responses. Int Immunol 2001; 13(11): 1361–71. [DOI] [PubMed] [Google Scholar]
- [82].Monzavi-Karbassi B, Hennings LJ, Artaud C, et al. Preclinical studies of carbohydrate mimetic peptide vaccines for breast cancer and melanoma. Vaccine 2007; 25(16): 3022–31. [DOI] [PubMed] [Google Scholar]
- [83].Hennings L, Artaud C, Jousheghany F, Monzavi-Karbassi B, Pashov A, Kieber-Emmons T. Carbohydrate mimetic peptides augment carbohydrate-reactive immune responses in the absence of immune pathology. Cancers (Basel) 2011; 3(4): 4151–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Makhoul I, Hutchins L, Emanuel PD, et al. Moving a carbohydrate mimetic peptide into the clinic: Clinical response of a breast cancer patient after mimotope-based immunotherapy. Hum Vaccin Immunother 2014; 11: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pashov A, Monzavi-Karbassi B, Raghava GP, Kieber-Emmons T. Bridging innate and adaptive antitumor immunity targeting glycans. J Biomed Biotechnol 2010; 2010: 354068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dole M, Nunez G, Merchant AK, et al. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res 1994; 54(12): 3253–9. [PubMed] [Google Scholar]
- [87].Schultz MJ, Swindall AF, Bellis SL. Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev 2012; 31(3-4): 501–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Carcel-Trullols J, Stanley JS, Saha R, et al. Characterization of the glycosylation profile of the human breast cancer cell line, MDA-231, and a bone colonizing variant. Int J Oncol 2006; 28(5): 117–383. [PubMed] [Google Scholar]
- [89].Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009; 459(7249): 1005–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013; 381(9869): 805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Von MG. Breakthrough for neoadjuvant chemotherapy. Breast Cancer 2008; 15(1): 27–30. [DOI] [PubMed] [Google Scholar]
- [92].Von MG, Untch M, Blohmer JU, et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol 2012; 30(15): 1796–804. [DOI] [PubMed] [Google Scholar]
- [93].Di Paola M, Angelini L, Bertolotti A, Colizza S. Host resistance in relation to survival in breast cancer. Br Med J 1974; 4(5939): 268–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Aaltomaa S, Lipponen P, Eskelinen M, et al. Lymphocyte infiltrates as a prognostic variable in female breast cancer. Eur J Cancer 1992; 28A(4-5): 859–64. [DOI] [PubMed] [Google Scholar]
- [95].Denkert C, Loibl S, Noske A, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 2010; 28(1): 105–13. [DOI] [PubMed] [Google Scholar]
- [96].Seo AN, Lee HJ, Kim EJ, et al. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br J Cancer 2013; 109(10): 2705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Sabatier R, Finetti P, Mamessier E, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015; 6(7): 5449–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Taylor K, Micha D, Ranson M, Dive C. Recent advances in targeting regulators of apoptosis in cancer cells for therapeutic gain. Expert Opin Investig Drugs 2006; 15(6): 669–90. [DOI] [PubMed] [Google Scholar]
- [99].Ward TH, Cummings J, Dean E, et al. Biomarkers of apoptosis. Br J Cancer 2008; 99(6): 841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Flanagan L, Meyer M, Fay J, et al. Low levels of Caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis 2016; 7: e2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kim KI, Lee KH, Kim TR, Chun YS, Lee TH, Park HK. Ki-67 as a predictor of response to neoadjuvant chemotherapy in breast cancer patients. J Breast Cancer 2014; 17(1): 40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]