

Abstract
Blockade of the serotonin 5-HT2A G protein-coupled receptor (5-HT2AR) is a fundamental pharmacological characteristic of numerous antipsychotic medications, which are FDA-approved to treat schizophrenia, bipolar disorder, and as adjunctive therapies in major depressive disorder. Meanwhile, activation of the 5-HT2AR by serotonergic psychedelics may be useful in treating neuropsychiatric indications, including major depressive and substance use disorders. Serotonergic psychedelics and other 5-HT2AR agonists, however, often bind other receptors, and standard 5-HT2AR antagonists lack sufficient selectivity to make well-founded mechanistic conclusions about the 5-HT2AR-dependent effects of these compounds and the general neurobiological function of 5-HT2ARs. This review discusses the limitations and strengths of currently available “selective” 5-HT2AR antagonists, the molecular determinants of antagonist selectivity at 5-HT2ARs, and the utility of molecular pharmacological and computational methods in guiding the discovery of novel unambiguously selective 5-HT2AR antagonists.
Keywords: 5-HT2A, GPCR, antipsychotic, antagonist, selectivity
Graphical Abstract
1. Introduction
Serotonin (5-hydroxytryptamine, 5-HT) is an endogenous signaling molecule that regulates nearly all neurocognitive functions by activating at least 14 genetically-encoded 5-HT receptors, along with dozens of variants generated by post-transcriptional editing or alternative splicing (1, 2). Thirteen genetically encoded 5-HT receptors are G protein-coupled receptors (GPCRs) that, owing to their plasma membrane localization and extensive control over intracellular signaling, are highly accessible and versatile drug targets. Approximately one-third of drugs approved by the United States Food and Drug Administration target GPCRs and ~4% bind to 5-HT2A receptors—arguably the most studied 5-HT receptor (3, 4).
Early antipsychotic medications, known to block dopamine D2-like GPCRs in the striatum, were coincidentally instrumental in the discovery of 5-HT2ARs in the cortex. For example, radioligand binding studies with rat brains showed that both spiperone (Figure 1) and haloperidol bound striatal dopamine receptors, whereas only spiperone had high affinity for a population of cortical receptors labeled by the promiscuous (i.e., nonselective) serotonergic compound lysergic acid diethylamide (LSD), suggesting that spiperone-bound 5-HT receptors (5). Electrophysiology experiments then showed that at least two 5-HT receptors in the brain modulate neuronal firing (6). LSD was subsequently reported to bind both receptors in the rat frontal cortex: those demonstrating higher affinity for [3H]5-HT or [3H]spiperone, termed 5-HT1R and 5-HT2R, respectively (7). The discovery of distinct 5-HT receptors naturally led to investigations into their behavioral and physiological functions, creating demand for selective antagonists to block 5-HT2R-mediated effects (8).
Figure 1:
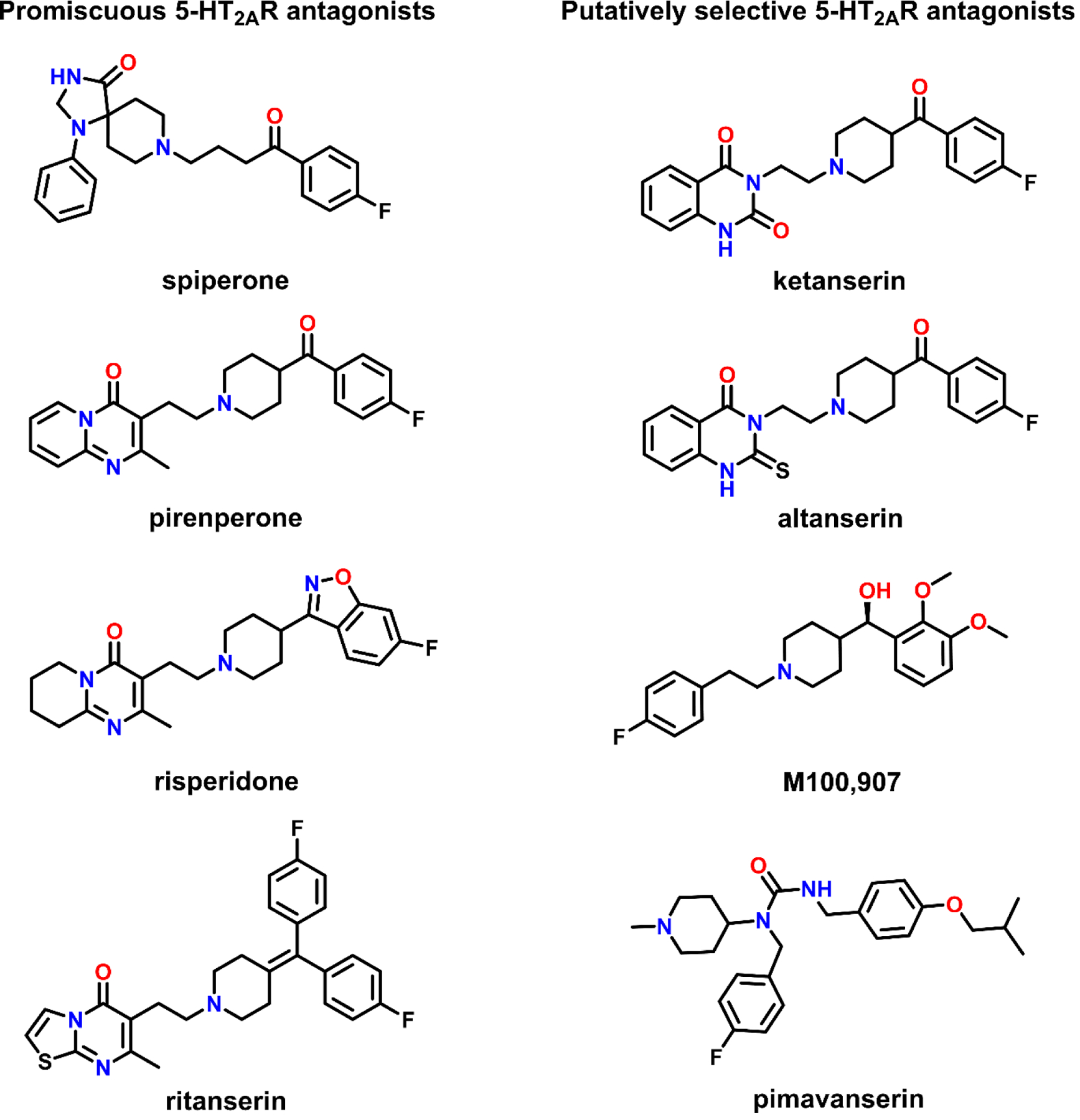
Structures of antagonists, with varying degrees of selectivity, commonly employed to probe the 5-HT2AR.
Among the first behavioral activities to be associated with the 5-HT2R was the ability of laboratory animals to discriminate the subjective effects elicited by serotonergic psychedelics, including LSD, mescaline, and 2,5-dimethoxy-4-methyl-amphetamine, which could be blocked by the 5-HT2R antagonists pirenperone and ketanserin (described below) (9–12). Soon after this, 5-HT was found to effectuate phosphoinositide turnover in the rat cerebral cortex, and the potencies of 5-HT2R antagonists to block this effect were correlated with their affinity for these sites (13). The 5-HT2R was later designated as the 5-HT2AR after the discovery of 5-HT2B and 5-HT2CRs (14–20), classified together based on extensive similarities in their ligand binding affinities, signal transduction mechanisms, and structural homology (60–70% amino acid identity within structurally conserved regions).
Decades have passed since the seminal discoveries described above, and antagonism of 5-HT2ARs has become a mainstay of antipsychotic drug action. In fact, so called selective 5-HT2AR antagonists can effectively treat dimensions of psychosis (21–25), without untoward side effects associated with the off-target binding of more promiscuous antipsychotic agents. Additionally, there is new widespread interest in identifying mechanisms of rapid, persistent neuronal and behavioral plasticity elicited by psychedelics that share activation of 5-HT2ARs, but diverge in their activities at other targets (26–30). The delineation of such mechanisms inherently relies on the use of unambiguously selective 5-HT2AR antagonists to block these effects, yet the utility of currently available antagonists is limited by their selectivity, pharmacological characterization, and structural diversity.
Central to this review, defining the fundamentally vague pharmacological term “selective” remains a formidable challenge. For example, a ligand may bind target A with 10-fold greater potency than targets B and C and be defined as “selective” for target A, albeit, the ligand affinities at targets D and E may not have been considered. In other words, a ligand may be referred to in the literature as “selective” irrespective of the number of off-target proteins against which it has been compared. Thus, it is critical to define under what context a ligand may be “selective”. In this review, we discuss the discovery and relative selectivity of putatively selective 5-HT2AR antagonists. We define here ligands that block 5-HT-elicited Gαq/11-signaling via 5-HT2ARs as “antagonists” owing to a paucity of data at alternative G protein or β-arrestin1/2 signaling pathways. Likewise, there is limited evidence of physiological, therapeutic, or structural distinctions between neutral antagonists and inverse agonists, leading us to refer to neutral antagonists and inverse agonists alike as “antagonists”, unless otherwise specified. We also discuss structural mechanisms underlying selective antagonist binding to 5-HT2ARs, and the potential of molecular modeling and molecular dynamics to advance the design of unambiguously selective 5-HT2AR antagonists.
2. Putatively selective 5-HT2AR antagonists
2.1. Spiperone (spiroperidol)
Spiperone, a butyrophenone discovered by Janssen Pharmaceuticals, was developed as an antipsychotic and binds central dopamine (31) and 5-HT (32) receptors with high affinity. After its affinity for 5-HT receptors was reported, several other antipsychotics were also found to bind 5-HT receptors (5, 7). Spiperone was later found to have >500-fold higher affinity at human 5-HT2ARs than the highly homologous 5-HT2B and 5-HT2CRs (33, 34), making it the first commercially-available compound for clearly labeling and distinguishing 5-HT2ARs from 5-HT2B and 5-HT2CRs. However, its high affinity at dopamine D2-like receptors, along with its moderate (Ki = 30–300 nM) to high (Ki ≤ 30 nM) affinity at α1A-, α1B-, α1D-adrenergic, 5-HT1A, and 5-HT7Rs (Table 1) limits its use for interrogating the physiological and behavioral role of 5-HT2ARs (9).
Table 1:
Binding affinities of various 5-HT2AR antagonists at relevant off-target GPCRs.
| Spiperone | Ketanserin | M100,907 | Pimavanserin Altanserin | Ritanserin | Pirenperone | Risperidone | ||
|---|---|---|---|---|---|---|---|---|
| GPCR | Affinity (Ki, nM) | |||||||
| 5-HT 2A | 1.2 a,e,g | 1.13 a,e,g | 0.47 a,e,g | 0.24 a,e,g,(80) | 0.3 b,e,g,(170) | 0.22 a,e,g | 1.1 a,e,h,(171) | 0.3 a,e,g,(80) |
| 5-HT 2B | 1,114a,e,g | 234a,e,g,(172) | 261a,e,h | 437a,e,g,(80) | - | 0.14a,e,h | 61a,e,h,(171) | 15.5a,e,g,(80) |
| 5-HT 2C | 923a,e,g | 88a,e,g | 100a,e,g | 2.8a,e,g,(80) | 6b,e,g,(170) | 0.24a,e,g | 77a,e,h,(171) | 19a,e,g,(80) |
| 5-HT 1A | 17a,e,g | >1,000a,e,h | >1,000a,e,h | >1,000a,e,g,(78) | >1,000b,f,h,(173) | 309a,e,g | 485b,f,g | 296a,e,g |
| 5-HT 1B | >1,000b,f,g | >1,000a,e,g | >1,000a,e,g | >1,000a,e,g,(78) | >1,000b,f,h,(173) | 194a,e,h | >1,000b,f,g | 53.6a,e,g |
| 5-HT 1D | >1,000a,e,h | 111a,e,g,(174) | >850d,f,h,(58) | >1,000a,e,g,(78) | - | 36.8a,e,h | - | 29a,e,g |
| 5-HT 1E | >1,000a,e,h,(175) | >1,000a,e,h,(175) | >850h,j,(58) | >1,000a,e,g,(78) | - | >1,000a,e,h | - | >1,000a,e,h |
| 5-HT 1F | >1,000a,e,h,(176) | >1,000a,e,h,(176) | >850h,j,(58) | >1,000a,e,g,(78) | - | - | - | >1,000a,e,h,(177) |
| 5-HT 3 | >1,000c,f,g | >1,000c,f,g | >1,000a,e,g | >1,000a,e,g,(78) | - | >1,000b,f,g,(178) | - | >1,000b,e,g,(177) |
| 5-HT 4 | - | >1,000e,(179) | >1,000b,f,g,(58) | >1,000a,e,g,(78) | - | >1,000a,e,g | - | - |
| 5-HT 5A | >1,000a,e,h | >1,000b,e,h | >1,000a,e,h | - | - | 77a,e,h | - | 206a,e,h |
| 5-HT 6 | >1,000b,f,g | >1,000a,e,h | >1,000a,e,h | >1,000a,e,g,(78) | >1,000a,e,h,(170) | 67a,e,h | - | >1,000a,e,h |
| 5-HT 7 | 110a,e,h | 794a,e,h,(180) | 226b,f,g,(58) | >1,000a,e,g,(78) | 15a,e,g,(170) | 29.6a,e,g | 6.5a,e,h,(181) | 4.8a,e,g |
| H 1 | 272c,f,g,(43) | 1.8c,f,g,(43) | >1,000a,b,e,g,(57, 68) | >1,000a,e,g,(80) | 20b,f,g,(173) | <40b,f,g,(95) | - | 18.2a,e,g,(80) |
| H 2 | - | - | - | >1,000i,j,(78) | - | 4.5a,e,g | - | 120a,e,g |
| α 1A | 20a,e,g,(182) | 28b,f,g,k,(35) | 128a,e,g,(67) | >1,000i,j,(78) | 4.55b,f,g,k,(173) | 80.7a,e,g | 20b,f,g,k,(183) | 5a,e,g |
| α 1B | 3a,e,g,(182) | 425a,e,g,(67) | >1,000i,j,(78) | 223a,e,g | 9a,e,g | |||
| α 1D | 8a,e,g,(182) | - | >1,000i,j,(78) | 86.5a,e,g | - | |||
| α 2A | 135b,f,g,k,(182) | - | >1,000a,e,g,(67) | >1,000i,j,(78) | 99b,f,g,k,(96) | 95.8a,e,g | 20b,f,g,k,(183) | 16.5a,e,g |
| α 2B | 199a,e,g,(184) | 271a,e,h | >1,000i,j,(78) | >1,000a,e,g | 8.5a,e,g,(177) | |||
| α 2C | - | >1,000a,e,g,(67) | - | 269a,e,g | 1.3a,e,h | |||
| D 1 | 220a,e,g,(185) | 190a,e,g,(185) | >1,000a,e,g | >1,000i,j,(78) | - | 344a,e,g | - | 249a,e,g |
| D 2 | 0.28a,e,g,(80) | >1,000b,f,g | >1,000a,e,g | >1,000a,e,g,(78) | 62b,f,g,(173) | >1,000a,e,g | - | 4.35a,e,g |
| D 3 | 0.14a,e,g,(80) | - | >1,000b,e,g,(58) | >1,000i,j,(78) | - | 57.7a,e,g | - | 12.2a,e,g |
| D 4 | 1.39a,e,g | - | 540a,e,g,(58) | >1,000i,j,(78) | - | 51.5a,e,g | - | 4.7a,e,g |
| D 5 | >1,000a,e,g,(185) | >1,000a,e,g,(185) | >1,000a,e,g | - | - | 163a,e,g | - | 290a,e,g |
| sigma | 632b,f,g,k,(186) | - | 87b,f,g,k,(58) | >1,000i,j,(78) | - | - | - | - |
Note: Unless otherwise noted by reference (in parenthesis), Ki values represent PDSP certified values or an average obtained from the NIMH Psychoactive Drug Screening Program database, https://pdspdb.unc.edu/pdspWeb/; search conducted 01/18/2022. Ki values were derived from ahuman, (when available), brat/mouse, cguinea pig, or dcow receptors expressed in eheterologous cell systems or fnative tissue, using gantagonist or hagonist radioligands. Some values have undefined iradioligands jspecies or kreceptor subtypes. ‘-’ denotes ligand affinities that are, to the best knowledge of the authors, not reported in the literature.
2.2. Ketanserin (R 48,648)
Since its discovery by Janssen Pharmaceuticals in 1981 (35), the quinazoline derivative ketanserin is among the most widely used tools for probing 5-HT2AR function in preclinical research (26–28, 36), and the sole antagonist used to delineate the 5-HT2AR-dependent effects of serotonergic psychedelics in humans (37–41). Although ketanserin was the first 5-HT2AR antagonist discovered that lacks high affinity for other serotonin and dopamine receptors, it is less appreciated that it has high affinity at several aminergic receptors, including α1A-, α1B-, α1D-adrenergic, and histamine H1 receptors (35, 42–44), as well as, moderate affinity at α2B-adrenergic and 5-HT2C receptors (Table 1). These off-target activities limit the utility of ketanserin as a specific tool for assessing 5-HT2AR activity. The off-target activity of ketanserin at adrenergic and histaminic receptors is particularly confounding because α1-adrenergic receptors colocalize with 5-HT2AR transcripts, and α1-, α2-adrenergic and histamine H1 receptors share signal transduction mechanisms with the 5-HT2AR and modulate the excitability of serotonergic neurons in the dorsal raphe nucleus (45–49).
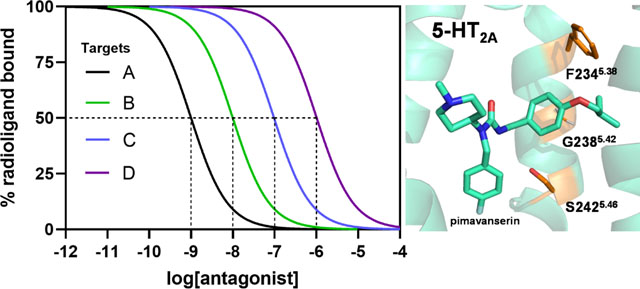
To demonstrate that ketanserin interacts with high affinity at sites independent of the 5-HT2AR, we present an autoradiography experiment wherein mouse brain slices were incubated with 1 nM [3H]ketanserin in the presence or absence of 1 μM M100,907 (Figure 2), another putatively selective 5-HT2AR antagonist (discussed below). Our results demonstrate that [3H]ketanserin prominently labels sites in the striatum that are not specific to 5-HT2-type receptors, consistent with earlier work in rat brain using other 5-HT2-type receptor antagonists (50, 51). Dopaminergic, adrenergic, or histaminergic antagonists were also ineffective at blocking striatal [3H]ketanserin binding (50, 51). Further experiments using rat brains (52), bovine chromaffin granule membranes (53), or heterologous cell systems (54) demonstrate that [3H]ketanserin binds the vesicular monoamine transporter 2 with moderate affinity (Ki, Kd = 22–540 nM). Therefore, studies investigating the effects of 5-HT2AR agonists should use caution when interpreting results obtained with ketanserin as a pharmacological tool since its pharmacodynamic effects are incompletely understood.
Figure 2:
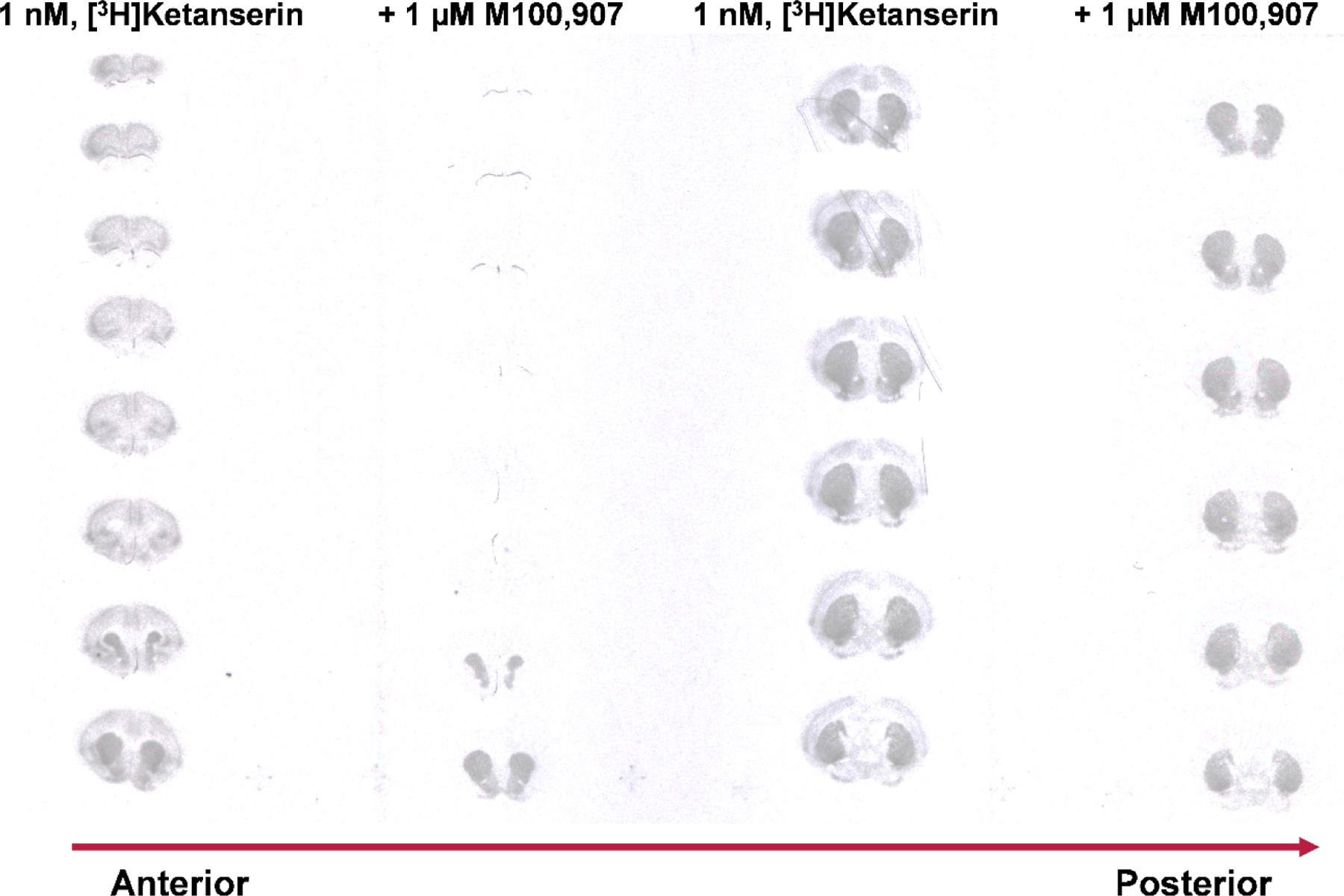
[3H]Ketanserin binds with high affinity (1 nM) a large population of targets in the striatum that are not 5-HT2-type receptors. Note that co-incubation with the putatively selective 5-HT2AR inverse agonist, M100,907 (1 μM, a concentration chosen to occupy and block the vast majority of 5-HT2ARs, in order to reveal off-target binding of [3H]ketanserin), blocked binding of [3H]ketanserin in the cortex, but not the striatum. [3H]ketanserin (42.5 Ci/mmol) autoradiography (10-week film exposure) of coronal brain sections from FVB.129P2-Pde6b+ Tyrc-ch/AntJ (sighted FVB) mice.
2.3. M100,907 (MDL100,907, Volinanserin)
Driven by work suggesting that 5-HT2AR antagonists elicit antipsychotic-like behavioral effects with a low risk of extrapyramidal symptoms compared to antipsychotics that primarily block D2Rs, researchers at Hoechst Marion Roussel in the early 1990s developed the piperidine derivative M100,907 (Figure 1) for the treatment of schizophrenia (55, 56). Until this time, 5-HT2AR antagonists demonstrated only modest selectivity (< 30-fold) over 5-HT2C, α1-adrenergic, H1, and dopamine D2 receptors. Thus, Hoechst Marion Roussel broke ground by reproducibly showing that M100,907 had sub-nanomolar affinity at 5-HT2ARs, with >100-fold selectivity over these off-target GPCRs, along with numerous other receptors and ion channels (57, 58).
An important caveat when considering the selectivity of M100,907 is that much of the work characterizing its binding profile is derived using a variety of species, tissue sources, and radioligands (57, 58). Thus, the apparent ligand affinity may be impacted by differences in the ligand binding pocket between species (59–61), plasma membrane composition (62), and/or radiolabeling discrepancies in native tissue (e.g., [3H]spiperone binds non-selectively to several dopamine and serotonin receptors, Table 1). Moreover, GPCRs exist in a dynamic equilibrium of conformational states often determined by the prevalence of guanine nucleotides and G proteins (63, 64). Agonists tend to recognize these conformational states with variable affinity, making radioligand-based affinity measurements sensitive to differences in the conformation stabilized by the radioligand and competitor ligand. For example, 5-HT has much higher affinity for [3H]5-HT-labeled than [3H]ketanserin-labeled 5-HT2ARs (Ki = 3.8 and 144 nM, respectively) (65). Accordingly, the off-target binding of M100,907 is less certain than one might assume by glancing at available data in Table 1, and since competitive radioligand displacement data provides little information on ligand functional activity, some skepticism is warranted regarding the selectivity of M100,907 at 5-HT2ARs (see ref. (66) for review on defining ligand activity).
Among the currently known off-targets of M100,907 are sigma, 5-HT2C, α1A-, and α2B-adrenergic receptors, with lower affinity at the latter receptors than is observed for ketanserin (Table 1). While selectivity over H1Rs has been challenged (67), most work supports that M100,907 is selective at binding 5-HT2A over H1Rs. (57, 58, 68). Autoradiography studies further support 5-HT2AR engagement by [3H]M100,907 at 0.4 nM in rat brain, exhibiting dense labeling in the frontal cortex, motor cortex, and claustrum (51). Rodent studies indicate that M100,907 does not antagonize apomorphine induced-climbing or phenylephrine lethality, indices of in vivo D2 and α1-adrenergic receptor engagement, respectively, and may only engage 5-HT2CRs at elevated doses (58). Notably, some studies report M100,907 is not a potent modulator of mouse locomotor activity (55, 58, 69), while others report robust locomotor suppressing effects (70, 71), suggesting that different animal strains and/or test conditions are involved in the confounding results reported for M100,907 behavioral pharmacology.
In preclinical studies, M100,907 exhibited promising antipsychotic-like behavioral effects in models of psychomotor stimulation and sensorimotor gating deficits, with a favorable side-effect profile (55, 58, 72, 73). Positron emission tomography (PET) studies in humans confirmed that M100,907 exhibited high levels of apparent 5-HT2AR occupancy in the cortex (74) for over 24 hours, far outlasting its presence in plasma (75). However, clinical trials for schizophrenia were discontinued due to a failure to meet endpoints (76). In summary, M100,907 appears to offer advantages over ketanserin for evaluating 5-HT2AR function in vivo. However, the lack of broad receptor panel screening data with a consistent tissue-source suggests exercising caution when interpreting data reported for M100,907.
2.4. Pimavanserin (ACP-103, Nuplazid®)
Since their development, so-called “atypical” antipsychotic medications (e.g., clozapine) have been noted to demonstrate distinct therapeutic and side-effect profiles compared to their “typical” counterparts (e.g., haloperidol), which were defined primarily by potent dopamine D2R antagonist activity. While differences in D2R occupancy at clinically effective doses or polypharmacology (e.g., partial agonism of 5-HT1ARs) likely contribute to these differences, scientists at Acadia Pharmaceuticals identified functionally distinct effects at the 5-HT2AR as being, at least in part, responsible for atypical antipsychotic therapeutic outcomes (68). Key to this was the development of a sensitive, high-throughput chemical genomics platform (Receptor Selection and Amplification Technology™; R-SAT™) enabling reliable detection of 5-HT2AR constitutive activity (i.e., ligand-independent G protein activation), historically difficult to detect through 5-HT2ARs. By screening numerous antipsychotics via the R-SAT™ platform, it was found that nearly all atypical antipsychotics, as well as spiperone, ketanserin, and M100,907, demonstrated inverse agonist activity (i.e., a concentration-dependent reduction in constitutive activity) at 5-HT2ARs (68). This finding invigorated a high-throughput screening (HTS) campaign that identified several hits for potent 5-HT2AR inverse agonism (77). After thorough selectivity screening and iterative rounds of lead optimization, pimavanserin (ACP-103, Figure 1) was identified as a novel putatively selective 5-HT2AR inverse agonist with preclinical efficacy in multiple models of psychosis, and was devoid of untoward motor side effects (78). Notably, pimavanserin exhibits Gαq-mediated inverse agonist activity in clonal cells expressing wild type (WT) 5-HT2ARs or constitutively activated 5-HT2AR variants (79, 80). A recent study using human and mouse brain tissue, however, reported that pimavanserin may act as a neutral antagonist and inverse agonist of Gαq/11- and Gαi1-mediated signal transduction, respectively, in vivo, apparently via 5-HT2ARs (81). Nevertheless, although the therapeutic relevance of inverse agonism at 5-HT2ARs is uncertain, pimavanserin is approved to treat hallucinations and delusions in Parkinson’s disease patients (22, 82). Pimavanserin also has shown efficacy in clinical trials to treat dementia-related psychosis (23, 24), negative symptoms of schizophrenia (25), and major depression (83).
A broad affinity profiling screen of over 75 GPCRs, ion channels, transporter proteins, and enzymes, indicated that pimavanserin had >1,000-fold selectivity for 5-HT2ARs over all other targets except for 5-HT2CRs (~12-fold), where it also acts as an inverse agonist (78, 80). Pimavanserin also was found to bind L-type calcium channels with moderate to low affinity (Ki = 310 nM) (78). The off-target activity at 5-HT2CRs is especially noteworthy for researchers investigating mechanisms sensitive to midbrain dopamine release, which might be oppositely influenced by 5-HT2A and 5-HT2CRs (67, 84–86); see refs. (87) and (88) for investigations into the effects of 5-HT2CRs on midbrain dopamine release.
Several studies report that pimavanserin may not impact motor function in humans (22, 83, 89, 90), likely due to its nil affinity at D2 and H1Rs (77, 91). In rodents, however, pimavanserin causes robust locomotor suppression (78, 80). This activity appears to be at odds with the role of 5-HT2-type receptors in modulating rodent locomotor activity. For example, 5-HT2AR activation enhances locomotor activity whereas 5-HT2CR activation suppresses locomotor activity in mice (84). Conversely, antagonism of 5-HT2ARs normalizes the locomotor enhancing effects of 5-HT2CR antagonists (85). Therefore, a potent and selective 5-HT2A/5-HT2CR inverse agonist such as pimavanserin might be expected to have neutral effects on locomotor activity in mice. A PET study in pigs also questions the selectivity of pimavanserin, which was not competitively displaced from the thalamus by unlabeled pimavanserin or ketanserin (92), suggesting the parent compound or a metabolite exhibits off-target binding, high non-specific binding, or a slow dissociation rate. Nevertheless, of the ligands discussed here, pimavanserin is the most thoroughly characterized putatively selective 5-HT2A-preferring 5-HT2A/5-HT2CR antagonist/inverse agonist available.
2.5. Other 5-HT2AR antagonists
In addition to spiperone and ketanserin (35, 93), scientists at Janssen Pharmaceuticals also discovered several other highly potent 5-HT2AR antagonists with some selectivity for 5-HT2ARs, including altanserin, pirenperone, risperidone, and ritanserin (9, 94–97) (Figure 1). However, while these ligands demonstrate high affinity at, and some selectivity for, 5-HT2ARs, neither is likely to engage 5-HT2ARs selectively at behaviorally active doses due to their high affinity at off-target receptors (Table 1). Altanserin, a PET ligand for labeling 5-HT2ARs in vivo (98), is a possible exception to this shortcoming, although its in vitro characterization is sparse. Nevertheless, studies with altanserin (99, 100) and pirenperone (9–11, 101) have informed our understanding of 5-HT2AR pharmacology, while structural biology studies with risperidone (79) and ritanserin (102) have begun clearing a path to the rational design of novel and selective 5-HT2AR antagonists (described below).
3. Molecular determinants of polypharmacology and subtype selectivity at 5-HT2A GPCRs
3.1. Polypharmacology of aminergic ligands targeting 5-HT2ARs
The 5-HT2AR orthosteric binding pocket—where the endogenous ligand 5-HT binds—shares significant homology with numerous aminergic GPCRs (receptors for neurotransmitters with basic amine moieties, e.g., acetylcholine, dopamine, histamine, norepinephrine, serotonin), and this overlap is critical to understanding ligand polypharmacology. Within transmembrane domain 3 (TM3) sits the amino acid residue D3.32 (Ballesteros-Weinstein numbering system (103)), which is conserved in the binding pocket of all aminergic receptors. This conserved residue is negatively charged and serves to anchor the positively charged amine moiety present in most aminergic receptor ligands within the binding pocket via an ionic bond (Figure 3). Accordingly, point mutation of D3.32 to uncharged residues drastically diminishes the binding affinity of most agonists and antagonists at aminergic GPCRs, including 5-HT2ARs (104–107).
Figure 3: Molecular determinants of nonselective binding at aminergic receptors.
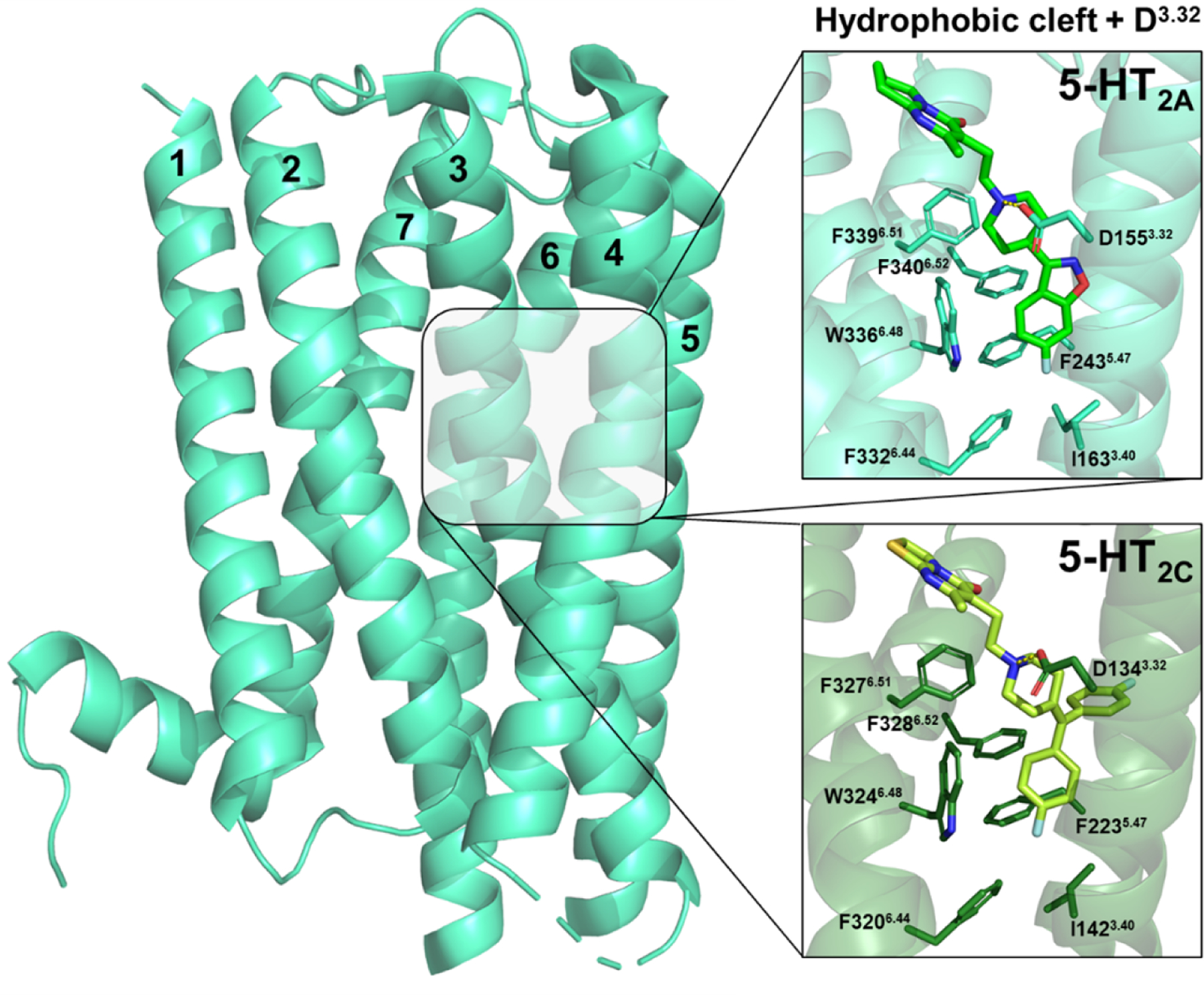
Structures of a risperidone-bound 5-HT2AR (light green, PDB: 6A93) and ritanserin-bound 5-HT2CR (dark green, PDB: 6BQH) show aromatic and hydrophobic interactions with conserved residues forming a hydrophobic cleft deep in the binding pocket, and an ionic bond between the ligand’s protonated amine and the negatively charged side chain of D3.32. The whole 5-HT2AR is shown (left) for perspective, with each transmembrane domain numbered.
The side chains of I3.40, F5.47, F6.44, W6.48, F6.51, and F6.52 form a highly conserved hydrophobic cleft at the base of the orthosteric binding pocket (Figure 3) and contribute to promiscuous antagonist binding at 5-HT1B, 5-HT2A, 5-HT2C, D2, and H1Rs (79, 108). Interestingly, flexible-structured antagonists are more sensitive to manipulations in this region of the binding pocket, likely because they extend deeper than more rigid-structured antagonists such as those of the ergoline class (109, 110). For example, the affinity of flexible ligands such as risperidone and pimavanserin is attenuated by >900-fold at point mutated W336L6.48 (tryptophan to leucine) and F339A6.51 (phenylalanine to alanine) 5-HT2ARs (79). Similarly, the affinity of ritanserin at highly homologous 5-HT2CRs with W324L6.48 or F327L6.51 point mutations is attenuated by >10,000- and ~100-fold, respectively (102). In contrast, the affinity of the relatively structurally-rigid ergoline antagonist mesulergine (Figure 4) is attenuated by only 7-fold at W324L6.48 5-HT2CRs (102). Recently, the side chains of F2435.47 and F3406.52 within the 5-HT2AR were found capable of expanding the binding pocket to accommodate various antagonist structural moieties (111). As such, mutation of either position to alanine or leucine attenuates the affinity (~5–46-fold) of many antagonists, including: spiperone, ketanserin, pimavanserin, risperidone, aripiprazole, and cariprazine (79, 104, 111). Although the affinity of ketanserin at F340A6.52 5-HT2ARs was not different from WT 5-HT2ARs (111).
Figure 4:
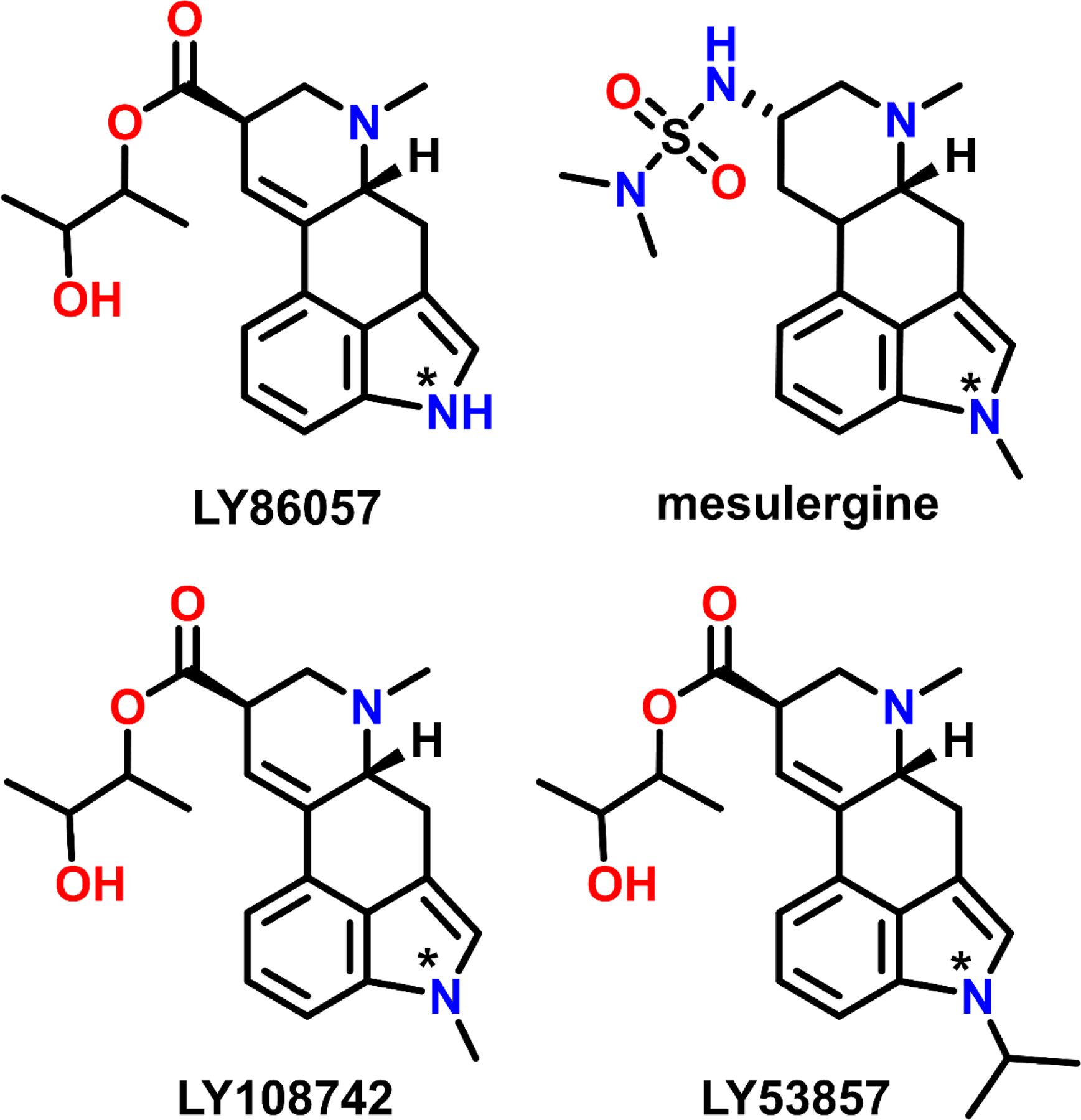
Structures of ergoline-based 5-HT2AR antagonists variably substituted at the N(1) position (denoted by *).
A subset of residues in the hydrophobic cleft forms the P5.50–I3.40–F6.44 motif, a molecular ensemble wherein side-chain rearrangements are understood to mediate activation of class A GPCRs (112), along with other key, conserved motifs, including CWxP, E/DRY, and nPxxY (113). Accordingly, I3.40 and F6.44 are critical for agonist function and antagonist binding at 5-HT2A and 5-HT2CRs (79, 102, 104, 114). Indeed, the agonist activity of 5-HT is abolished at I163A3.40 5-HT2ARs, as is the affinity of mesulergine (79). Moreover, the affinity of pimavanserin and risperidone is attenuated at F332L6.44 5-HT2ARs (10- and 17-fold, respectively) (79), and of ritanserin at I142F3.40 and F320L6.44 5-HT2CRs (20- and 9-fold, respectively) (102).
3.2. Molecular determinants of subtype-selectivity at 5-HT2ARs
Perhaps the most intuitive mechanism of achieving selectivity is the formation of ligand-receptor contacts with amino acid residues that differ across target and off-target receptors. Identifying such residues allows medicinal chemists to rationally design ligands that interact with these residues to realize a degree of selectivity. By combining site-directed mutagenesis, molecular modeling, and structural biology techniques, several non-conserved residues in the 5-HT2AR have been identified and offer significant insight into the design of subtype-selective ligands.
3.2.1. S2425.46
The most thoroughly characterized 5-HT2AR amino acid residue, S2425.46 (A2255.46 and A2225.46 in 5-HT2B and 5-HT2CRs, respectively), has been implicated in species differences in ligand affinity (60, 61, 115), subtype-selective binding (60, 61, 115), binding kinetics (116, 117), and biased signaling (106). Importantly, S5.46 diverges between humans (serine) and rodents (alanine) and is sufficient to explain species differences in the affinity of ergoline-based antagonists, tryptamine-based agonists (60, 115, 116, 118), as well as a 2-aminotetralin-based agonist (61). Such interspecies variations could be due to limited evolutionary pressure to conserve sequence identity for a non-deleterious mutation (119). Although, drug development programs designing ligands that engage interspecies variant residues (intentionally or not) would necessitate the development of humanized animal models.
The species-dependent affinity of 5-HT2AR ligands was first shown by Kao et al. (60) using the N(1)-methyl ergoline antagonist mesulergine, which has a lower affinity at cloned human 5-HT2ARs (S2425.46, Kd = 174 nM) compared to 5-HT2ARs in the rat cortex (A2425.46, Kd = 8.52). Notably, the high affinity of mesulergine at human S242A5.46 point mutated 5-HT2ARs (Kd = 3 nM) recapitulated its affinity at WT rat 5-HT2ARs (60). Johnson et al. (115) extended these findings to various N(1)-substituted ergolines. For example, LY86057, LY108742, and LY53857 differ only in their substitution of the N(1)-position (i.e., −H, −CH3, and −CH[CH3]2, respectively; Figure 4), yet LY86057 (−H) had a higher affinity at human 5-HT2ARs than LY108742 (−CH3) or LY53857 (−CH[CH3]2). In contrast, LY108742 (−CH3) and LY53857 (−CH[CH3]2) had higher affinity at rat 5-HT2ARs than LY86057 (−H). However, these trends were reversed when rat 5-HT2ARs were point mutated to recapitulate human 5-HT2ARs (A242S5.46); e.g., the affinity of LY86057 (−H) better resembled its affinity at human 5-HT2ARs, whereas the affinity of LY108742 (−CH3) and LY53857 (−CH[CH3]2) depreciated by ~4-fold. Such relationships likely stem from a combination of factors, including the availability of favorable electrostatic interactions between the indole N(1)-H and the side chain oxygen of S5.46, as well as steric tolerance and opportunities for hydrophobic interactions between the indole N(1)-alkyl moieties and the side chain of A5.46.
The residue at position X5.46 also diverges between 5-HT2A (serine) and the highly homologous 5-HT2B and 5-HT2CRs (alanine). In an elegant study, Almaula et al. (116) tested the hypothesis that S2425.46 mediates subtype-selective binding to human 5-HT2A over human 5-HT2CRs using exchange mutations to generate S242A5.46 5-HT2ARs and A222S5.46 5-HT2CRs. Ergolines with an N(1)-H moiety (i.e., ergonovine, LSD, and lisuride) that preferentially bound WT 5-HT2ARs over WT 5-HT2CRs, demonstrated lower affinity at S242A5.46 5-HT2ARs, and higher affinity at A222S5.46 5-HT2CRs. In contrast, ergolines with an N(1)-CH3 moiety (i.e., mesulergine) that bound preferentially to 5-HT2CRs over 5-HT2ARs, demonstrated higher affinity at S242A5.46 5-HT2ARs, and lower affinity at A222S5.46 5-HT2CRs. These findings suggest that the affinity of ergolines at 5-HT2A and 5-HT2CRs depends on hydrogen bonding as well as steric and hydrophobic interactions between the N(1)-substituent and the side chains of S/A5.46.
While the side chain of S/A5.46 explains differences in the structure-activity relationships (SAR) of ergolines between human and mouse or rat 5-HT2ARs, and between human 5-HT2A and 5-HT2CRs, the effect of S5.46 vs. A5.46 does not reliably extend to other chemotypes. For example, tryptamine derivatives are not reliably impacted by the S/A5.46 side chain (115, 120). Similarly, we and others have shown that the affinity of various 5-HT2AR antagonists is not different at S242A5.46 5-HT2ARs, including, ketanserin, pimavanserin, risperidone, and certain 2-aminotetralins (79, 80, 115, 116, 121). Taken together, S2425.46 is not a general determinant of subtype-selective antagonist binding at 5-HT2ARs over 5-HT2B and 5-HT2CRs.
3.2.2. G2385.42
The residue G5.42 is unique to 5-HT2-type receptors and is occupied by larger residues including alanine, cysteine, serine, or threonine in other aminergic GPCRs (4). The importance of this to drug design was highlighted in the crystal structure of a ritanserin-bound 5-HT2CR, which indicates that one of the 4-fluorophenyl groups on ritanserin contacts the backbone of TM5 at G2185.42 (102). Given that ritanserin demonstrates some selectivity to bind 5-HT2-type receptors over other aminergic receptors, Peng and coworkers tested the hypothesis that G2185.42 was in part responsible for the preferential binding of ritanserin to 5-HT2-type receptors. They generated a G218S5.42 5-HT2CR to mimic the corresponding S1995.42 residue in 5-HT1ARs—for which ritanserin has low affinity (Ki = 309 nM, Table 1). Ritanserin showed 60-fold lesser affinity at G218S5.42 5-HT2CRs, supporting the hypothesis that G2185.42 contributes to its preferential binding at 5-HT2-type receptors. This hypothesis was further supported by a complementary experiment with clozapine, which exhibits appreciable affinity (Ki < 100 nM) for at least 26 receptors including the 5-HT1AR and has an embedded 4-benzylidenepiperazine core like ritanserin. The affinity of clozapine, however, was attenuated by a mere 2–3-fold at G218S5.42 5-HT2CRs (102, 122, 123).
The role of G5.42 in mediating selective binding at 5-HT2-type receptors has since been extended to the 5-HT2AR by Kimura et al. (79) and was recently replicated and expanded upon by our lab (80). Using a combination of molecular modeling and site-directed mutagenesis, this body of work suggests that the selectivity of pimavanserin to bind 5-HT2A/5-HT2CRs stems from the isobutoxybenzyl moiety occupying a cavity between TM4 and TM5 afforded by the small side chain of G5.42 (79, 80). Generation of a G238S5.42 5-HT2AR confirmed this binding pose, as pimavanserin showed a near-complete loss in affinity and antagonist activity at the receptor variant (79, 80). Moreover, we used novel 2-aminotetralin derivatives to demonstrate that this effect was driven by the size of the ligand motif extending into the cavity between TM4 and TM5, and this was predictive of ligand affinity over several other aminergic GPCRs (80). In contrast, risperidone, which has high affinity at multiple aminergic receptors and binds to 5-HT2ARs in an extended conformation spatially distinct from the side cavity, was only modestly affected by the G238S5.42 mutation (79, 80).
Certain lipids are known to potentiate 5-HT-mediated G protein activation through the 5-HT2AR (124, 125). Very recently, the structural basis of this effect was delineated using crystal structures of 5-HT2ARs bound to the agonists 5-HT, psilocin, lisuride, or LSD (126). In each structure, the lipid monoolein snaked into the cavity adjacent to G2385.42, between TM4 and TM5. The lipids monoolein, oleamide, oleoylethanolamide, and 2-oleoyl glycerol were all shown to modestly activate WT, but not G238S5.42 5-HT2ARs, suggesting that G2385.42 also serves as a structural determinant of lipid-mediated G protein signaling through 5-HT2ARs. It remains unknown how the lipid environment impacts antagonist affinity or selectivity at 5-HT2ARs.
Taken together, published experimental and computational work supports the conclusion that G5.42 contributes to the observed 5-HT2AR selectivity of structurally distinct antagonists. However, this model incompletely describes 5-HT2AR selectivity, as pimavanserin demonstrates high selectivity over 5-HT2B and modest selectivity over 5-HT2CRs despite the presence of G5.42 in both receptors. To better understand the molecular and structural features governing selective binding at 5-HT2ARs over other 5-HT2-type receptors, a more detailed understanding of GPCR structure is required.
3.2.3. Amino acid ensembles governing 5-HT2AR selectivity
Apart from single amino acid differences, amino acid ensembles can change the 3-dimensional (3D) architecture of binding pockets. A sophisticated model of this was recently put forth by Kimura et al. in detailing the first 5-HT2AR crystal structures (79). Structural comparisons of a risperidone-bound 5-HT2AR, ergotamine-bound 5-HT2BR, and ritanserin-bound 5-HT2CR indicated that the length and conformation of extracellular loop 2 (ECL2) was nearly identical between 5-HT2A and 5-HT2CRs yet was 4–6 residues longer in the 5-HT2BR due to a shortened TM4. In addition, divergent amino acid residues near the extracellular ends of TM4 and TM5 within the 5-HT2-type receptors were proposed to contribute to the formation of a unique side chain rotamer of F5.38, located one helical turn closer to ECL2 than G5.42, in the 5-HT2AR compared to that in 5-HT2B and 5-HT2CRs.
In the risperidone-bound 5-HT2AR crystal structure, a hydrophobic interaction between F2134.63 and F2345.38 appeared to orient the side chain rotamer of F2345.38 toward the extracellular end of TM4, leading Kimura et al. (79) to propose that a 5-HT2AR-specific rotamer of F2345.38 increases the volume of the side pocket formed by G2385.42 to realize a “side-extended cavity” (Figure 5). In contrast, the side chains of F2175.38 and F2145.38 in 5-HT2B and 5-HT2CRs, respectively, oriented toward the cytosol, effectively reducing the side-cavity’s volume. The orientation of F5.38 within 5-HT2B and 5-HT2CRs appeared to originate from steric restrictions with K1934.63 in 5-HT2BRs, and I1924.63 or D2115.35 in 5-HT2CRs (79), potentially explaining the observed selectivity of pimavanserin to bind 5-HT2ARs over 5-HT2B and 5-HT2CRs. Notably, although this work represents the most extensive mutagenesis investigation of the 5-HT2AR to date, no mutations of 5-HT2AR were made at positions X4.63 or X5.35 to suggest that a side chain rotamer of F5.38 could be perturbed to impact the affinity of antagonists with subtype-selectivity at 5-HT2ARs.
Figure 5: Putative molecular determinants of subtype-selective binding at 5-HT2-type receptors.
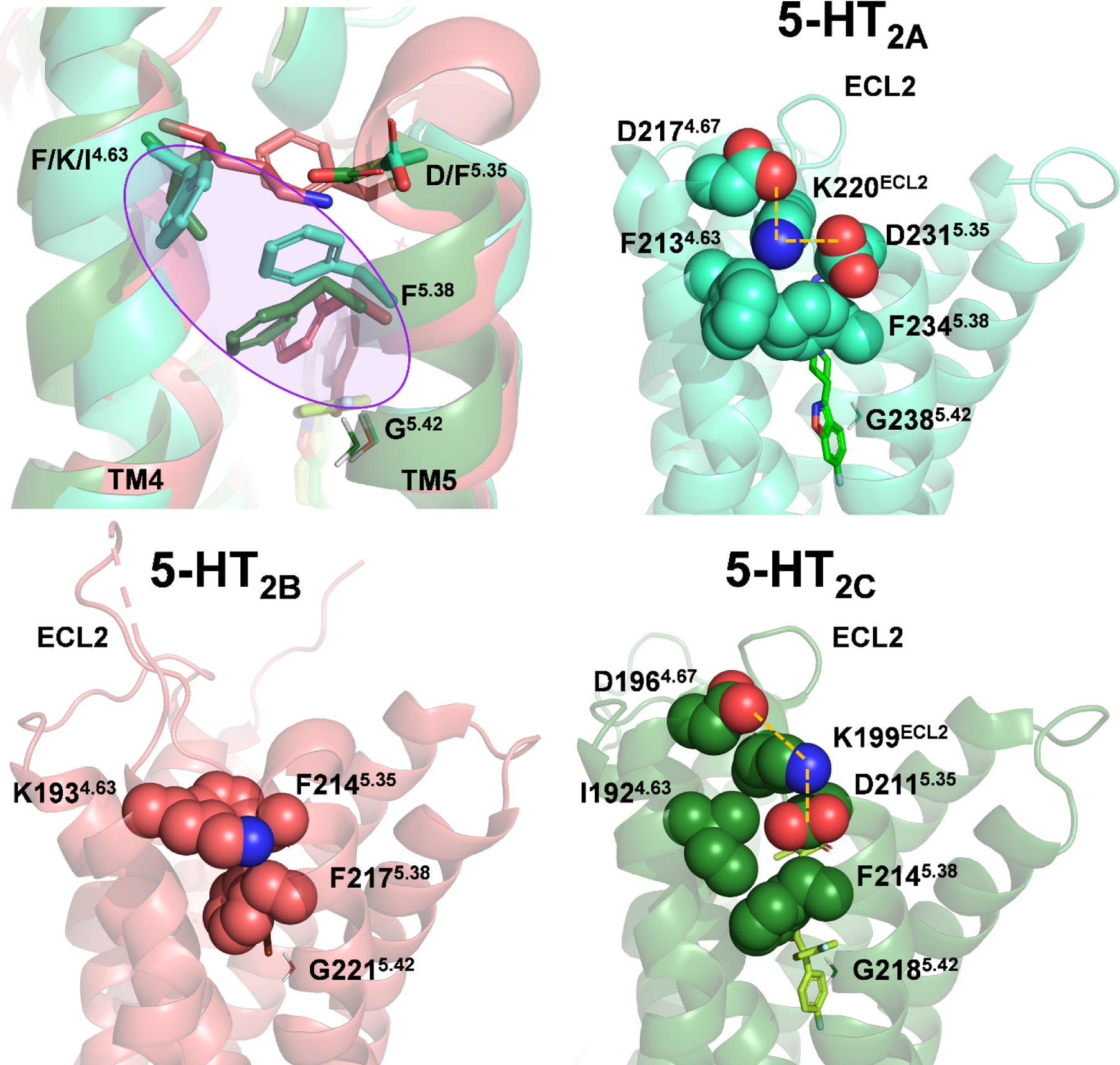
Superimposition of 5-HT2-type receptors suggests that a “side extended cavity” is formed in the 5-HT2AR (light blue color) by a unique rotamer of F5.38, making hydrophobic interactions with the non-conserved residue F4.63 (purple circle, top left). Structures of the 5-HT2AR (PDB: 6A93, top right), 5-HT2BR (PDB: 6DS0, bottom left), and 5-HT2CR (PDB: 6BQH, bottom right) indicate that ECL2 of the 5-HT2BR exhibits greater conformational freedom compared 5-HT2A and 5-HT2CRs, wherein KECL2 is electrostatically constrained (orange dashed lines). The side chain of G5.42 is shown for reference to the side cavity.
To test the role of interactions between F2134.63 and F2345.38 in mediating subtype-selective antagonist binding at 5-HT2ARs, our lab point-mutated F2134.63 within the 5-HT2AR to the structurally equivalent residue in 5-HT2BRs (K4.63) to mimic the side chain rotamer of F5.38 found in 5-HT2BR crystal structures. We then assessed the affinity of several 5-HT2AR antagonists with varying selectivity, including risperidone and pimavanserin, at point-mutated F213K4.63 5-HT2ARs (80). However, no difference in antagonist potency (pKb) was observed for any ligand compared to WT 5-HT2ARs. These results suggest that F2134.63 alone does not meaningfully contribute to facilitating selective antagonist binding to 5-HT2ARs.
Ligand binding is a dynamic process wherein ligands and amino acid residues sample discreet interaction profiles and conformational states across a probability distribution (see ref. (112) for review). Accordingly, receptor crystal structures are static representations of a dynamic system that may not reliably indicate structurally or pharmacologically meaningful interactions. Therefore, we used molecular modeling to identify residues that could dynamically modulate the rotamer of F2345.38 in 5-HT2ARs. Our simulations identified the side chain of D2315.35 (F2145.35 in 5-HT2B and D2115.35 in 5-HT2CRs), located one helical turn above F2345.38, as a candidate for influencing the F2345.38 rotamer (80). However, D231F5.35 5-HT2ARs were functionally null in response to 5-HT, and specific binding was not detected with [3H]spiperone, [3H]ketanserin, or [3H]mesulergine (80). Notably, the absence of a fluorescent tag precluded us from confirming proper membrane localization of D231F5.35 5-HT2ARs.
Careful inspection of published 5-HT2-type receptor crystal structures indicates that the D231F5.35 mutation could have destabilized the conformation of ECL2, potentially leading to a misfolded receptor and, subsequently, inadequate membrane-trafficking. For example, the crystal structure of a risperidone-bound 5-HT2AR shows the formation of an electrostatic cage around K220ECL2, formed by the negatively charged side chains of D2174.67 and D2315.35, which may limit the conformational freedom of, and therefore stabilize, ECL2 (Figure 5). Similar interactions are apparent in the crystal structure of a ritanserin-bound 5-HT2CR, where the positively charged side chain of K199ECL2 is enclosed by the negatively charged side chains of D1964.67 and D2115.35 (Figure 5). Thus, the absence of KECL2 near TM4 in the 5-HT2BR, and the presence of F2145.35, may promote the conformational freedom observed in 5-HT2BR ECL2 (127, 128), and contribute to the ability of some antagonists to bind 5-HT2A and 5-HT2CRs with higher affinity. Moreover, residues on the extracellular side of D3.32 are less conserved across 5-HT2-type receptors than those on the intracellular side of D3.32, suggesting that antagonist selectivity could involve interactions with residues near ECL2. Although, steric interactions with M2185.39 in the 5-HT2BR do not appear to sufficiently explain the low affinity of pimavanserin since it exhibits ~2-fold higher affinity at V235M5.39 5-HT2ARs (80).
Concerning the modest selectivity of pimavanserin to bind 5-HT2A over 5-HT2CRs, careful consideration of its affinity at, and selectivity over, 5-HT2CRs is warranted. Using molecular modeling techniques identical to those described elsewhere (80), we show that the binding pose of pimavanserin in 5-HT2CRs is nearly identical to that reported for 5-HT2ARs, even insofar as F5.38 adopts a raised rotamer conformation (Figure 6). Notably, the only non-conserved residues present in the binding pockets of 5-HT2A and 5-HT2CRs are S/A5.46 and I/V4.56 (80). Although mutagenesis studies do not support a role for S/A5.46 in mediating subtype selectivity of pimavanserin (79, 80), pimavanserin does exhibit 4-fold increase in potency at I206V4.56 5-HT2ARs (79), suggesting that steric tolerance at this position may enhance affinity at 5-HT2CRs.
Figure 6: Proposed binding mode of pimavanserin at 5-HT2A (light green) and 5-HT2CRs (dark green).
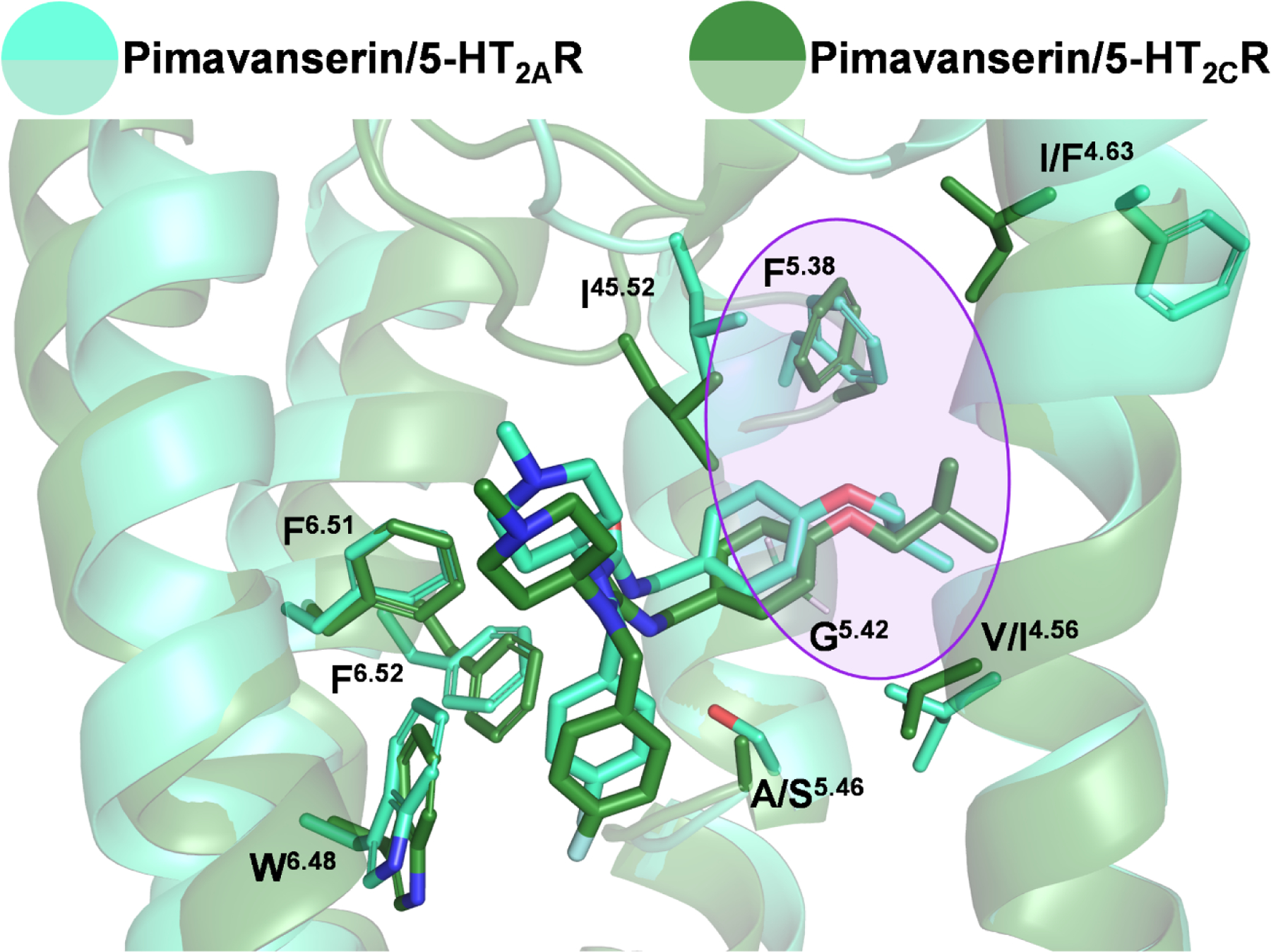
Molecular docking and molecular dynamics simulations indicate that pimavanserin engages nearly identical amino acid residues within the binding pocket of 5-HT2A and 5-HT2CRs. The primary determinant of selectivity, the extension of the isobutoxybenzyl moiety into the side-extended cavity between TM4 and TM5 (purple circle), as allowed by the small side chain of G5.42, is conserved in both receptors and is accompanied by a raised rotamer conformation of F5.38 in both receptor subtypes.
Insight into the ability of ligands to discriminate between 5-HT2A and 5-HT2CRs comes from a recent report detailing the crystal structure of a spiperone-bound D2R that proposes a mechanism for the ability of spiperone to bind D2Rs and 5-HT2ARs selectively over 5-HT2CRs (129). Although the D2R lacks a side-extended cavity, it does possess a spatially distinct extended binding pocket. Located between TM2 and TM3, the extended binding pocket—comprised of V872.57, W902.60, V912.61, L942.64, W10023.50(ECL1), F1103.28, V1113.29, and C18245.50(ECL2)—accommodates the phenyl ring of spiperone. Notably, the side chains of W902.60 in D2Rs and V1302.60 in 5-HT2ARs allow the side chain rotamers of F1103.28 and W1513.28 to flip and expose the extended binding pocket. In 5-HT2CRs, however, the bulkier side chain of L1092.60 may restrict W1303.28 from flipping, thereby impeding the formation of an extended binding pocket (129). These findings were supported by the observation that the affinity of spiperone at W90L2.60 D2Rs was more severely attenuated (~20-fold) than at F110W3.28 D2Rs (~11-fold) (129), although the affinity of spiperone at W90V2.60 D2Rs was not reported. These findings are supported by medicinal chemistry work noting that replacing the phenyl ring on spiperone with smaller lipophilic substituents (e.g., −H, −CH3, −CH2CH3, or − CH[CH3]2) negatively impacted affinity at 5-HT2A, 5-HT2C, and D2Rs (130). In conclusion, these structural differences may contribute, in part, to the observed selectivity of spiperone to bind D2 and 5-HT2ARs over 5-HT2CRs.
3.3. Alternative approaches for designing 5-HT2AR selective antagonists
Most aminergic ligands incorporate a basic amine moiety that, upon protonation, can form a strong ionic bond with D3.32 in the orthosteric binding pocket (105, 131). Such an anchoring strategy can be helpful in generating high affinity ligands, although protonated amines are also significant contributors to ligand promiscuity. In fact, screening studies have found that 20–50% of positively charged novel chemical entities bind to aminergic receptors with an IC50 < 10 μM, and serotonin receptors are among the most common targets (132, 133). Therefore, the design of non-basic ligands can be an effective strategy for developing novel, selective ligands.
Non-basic ligands comprise the most subtype selective 5-HT2-type receptor antagonists available. For example, RS-127445 is a 5-HT2BR antagonist that does not possess a basic amine moiety and is not positively charged at physiological pH. Yet, it displays >1,000-fold selectivity over 5-HT1A, 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2C, 5-HT3, 5-HT5, 5-HT6Rs, and 100 other receptors and ion channels, determined by Cerep screening (134). Successfully generating a selective 5-HT2BR antagonist is an impressive accomplishment since ligand affinity at 5-HT2BRs may predict pharmacological promiscuity, with some surveys finding that ~50% of positively charged compounds interact with 5-HT2BRs (132, 133). Similarly, indoline urea derivatives such as SB 242084, SB 243213, and SB 206553 lack basic amines and are among the most selective antagonists targeting 5-HT2B and/or 5-HT2CRs (135–137). Thus, rational strategies to identify non-basic 5-HT2AR ligands hold promise for discovering novel subtype-selective ligands. One such approach was taken to discover 6-(1-ethyl-3-(quinolin-8-yl)-1H-pyrazol-5-yl)pyridazin-3-amine, a non-basic subtype-selective 5-HT2CR agonist, identified through screening a compound library against D134A3.32 5-HT2CRs—an active site receptor variant that is functionally responsive to neutral, but not positively charged, agonists (138). Analogous point-mutated 5-HT2ARs might also be useful in identifying selective non-basic inverse agonists in platforms like R-SAT™ (68).
Results from recent molecular docking studies provide an alternative approach to achieving subtype-selective binding. Docking studies with SB 242084 suggest that it may interact with the same conserved amino acid residues in each of the 5-HT2-type receptors, but that its selectivity stems from its ability to make closer, more numerous hydrophilic contacts in the 5-HT2CR, e.g., with D1343.32, S1383.36, and Y3587.43 (117). Moreover, ligand affinity is defined by the relationship between the association rate (kon) of a free ligand to an unbound receptor, forming a binary complex, and the dissociation rate (koff) of the bound ligand from the binary complex (Kd = koff/kon). Conceptually, it is helpful to consider koff as reflecting the stability of a ligand-receptor complex, with more stable complexes having lower koff values, and hence, higher affinity. Given this, it is interesting to speculate that closer hydrophilic contacts between SB 242084 and 5-HT2CR residues may promote a uniquely stable ligand-receptor complex with a low koff value. Further computational and experimental investigations to this end would provide a more textured understanding of how ligands are stabilized in 5-HT2-type receptors.
4. Computational approaches to assist in drug design targeting 5-HT2ARs
4.1. Ligand-based drug discovery: QSAR
In the pre-structural biology era, 3D quantitative structure-activity relationships (3D-QSAR) were commonly used in drug discovery programs targeting 5-HT receptors. Among the most popular methods is comparative molecular field analysis (CoMFA), which correlates the biological activity (e.g., binding affinity) of a series of compounds with molecular features governing non-covalent ligand-receptor interactions, such as electrostatic and steric properties. Compared to conventional QSAR models based on the Hansch analysis, CoMFA models are similarly capable of predicting biological activity yet can incorporate structurally diverse analogs typically omitted from traditional QSAR analyses (139).
The CoMFA method to predict the activity of novel ligands is based on a training set of experimental data. For example, 3D-QSAR studies on a series of 3-(1,2,5,6-tetrahydropyridin-4-yl)indole derivatives targeting 5-HT2ARs, yielded a model that accurately predicted the affinity of six novel ligands based on a training set composed of 45-compounds (139). Moreover, CoMFA models modified to account for hydrogen bonding and hydrophobic interactions have been generated to predict electrostatic, steric, and hydrophobic determinants of affinity for a series of conformationally restricted butyrophenones at rat 5-HT2A and D2Rs (140).
4.2. Use of homology modeling and molecular docking to delineate mechanisms of receptor inactivation and ligand affinity
Several GPCR crystal structures were solved in the early 2000s, including rhodopsin (141) and human β2-adrenergic receptors (142). They were widely used to build homology models of 5-HT receptors to better understand the molecular mechanisms governing receptor function and ligand affinity and selectivity. Using a 5-HT2AR model based on the crystal structure of bovine rhodopsin in combination with molecular modeling, site-directed mutagenesis, and functional assays, Shapiro et al. (143) investigated the molecular mechanism of 5-HT-mediated 5-HT2AR activation. Their findings were consistent with the hypothesis that an ionic bond between R1733.50 and E3186.30, on the intracellular face of the receptor, serves as an essential interaction that is disrupted upon receptor activation (143). The side chain of R1733.50 has also been implicated in resolving the inverse agonist activity of constitutively activated C322K6.34 5-HT2AR variants, supported by molecular docking and molecular dynamics simulations with a model of the 5-HT2AR based on metarhodopsin (144, 145).
A homology model of the 5-HT2AR based on rhodopsin helped predict the relative contribution of S2395.43 and S2425.46 in the binding of tryptamine and phenylalkylamine-based agonists. Using a combination of site-directed mutagenesis and radioligand binding to confirm model predictions, Braden and Nichols demonstrated that a hydrogen bond could form between S2395.43 and tryptamines substituted at the 4- or 5-position with hydrogen-bond acceptors, as well as phenylalkylamines substituted at the 4-position (121). Moreover, phenylalkylamine binding was predicted to involve a hydrogen bond between the 2- and 5-position substituents and S1593.36 and S2395.43, respectively. This prediction was supported over a decade later by the structure of a 5-HT2AR bound to the phenylalkylamine derivative N-(2-hydroxybenzyl)-2,5-dimethoxy-4-cyanophenylethylamine, demonstrating the formation of a unique hydrogen bond between the C(2)-OH moiety and the side chain of S1593.36 (106). In contrast, the side chain of S1593.36 does not appear to be involved in the binding of LSD or ketanserin at 5-HT2ARs (106, 120).
4.3. Virtual high-throughput screening
Traditional drug discovery programs often rely on experimental HTS with large compound libraries in biological assays, and known active ligands, to identify leads for SAR and guide drug development (146). However, while these methods have proved fruitful, they barely scratch the surface of exploring the chemical space of drug-like molecules—routinely cited to be >1020—thus limiting the pace of medication development (147). Compared to experimental HTS, virtual HTS, pairing large computational libraries of compounds and molecular docking, is a more economical screening approach to drug discovery (148–151). Several studies have used virtual HTS with models of 5-HT2-type receptors to identify possible novel ligands (152, 153), with some predicted to demonstrate subtype-selective binding (154). Importantly, these investigations have led to the discovery of experimentally validated novel ligands with high affinity at (155), or selectivity for (156, 157), particular 5-HT2-type receptors.
With the increasing abundance of experimental 5-HT2AR structures, 13 at the time of writing (79, 106, 111, 126), coupled with the availability of free ultra-large ligand libraries, including the ZINC database (http://zinc20.docking.org)—an expanding library of over a billion compounds—the discovery of novel ligands with new scaffolds is increasingly accessible. Virtual HTS and ultra-large library docking have proven fruitful in identifying novel and potent receptor modulators (158–161). It seems increasingly likely that these methods will assume a more prominent role in drug discovery campaigns searching for novel 5-HT2AR ligands as we advance. To predict the functional activity of novel ligands as agonists or antagonists, and especially functionally selective (biased) agonists, however, requires additional computational methods capable of tracking global changes in receptor structure and receptor interactions with signaling proteins.
4.4. Molecular dynamics as a method to predict functionally relevant receptor conformations
Both structural and dynamic factors determine protein function. To better understand the dynamic and temporal aspects of protein function, molecular dynamic (MD) simulations are an integral and powerful computational approach. For example, one microsecond-long MD simulations using a 5-HT2AR homology model bound to psychedelic or non-psychedelic 5-HT2AR agonists identified distinct conformations of intracellular loop 2 (ICL2) characteristic of psychedelic agonists (162). These findings suggest that psychedelic 5-HT2AR agonists could stabilize discreet receptor conformational states relevant to intracellular signaling cascades, as suggested by experimental work (163). Moreover, MD simulations have been used to predict that the side chains of S2425.46 and N3436.55 in the 5-HT2AR contribute to biased agonism, a prediction later supported by experiment (164).
When designed to encompass a sufficient temporal window, MD simulations may also distinguish agonist from antagonist ligands at the 5-HT2AR. This was shown by Shan et al., using 350 nanosecond MD simulations of a 5-HT2AR model, based on rhodopsin and β2AR crystal structures, in complex with a full agonist, a partial agonist, and an antagonist of canonical 5-HT2AR signaling (i.e., 5-HT, LSD, and ketanserin, respectively) (165). The simulations indicated that ligands of varying functional activity differentially influenced well-documented microswitches of GPCR activation and stabilized conformational states of the receptor that lead to distinct deformations in the surrounding plasma membrane. Using a complementary approach, we have aimed to expand the work described elsewhere (143) to characterize 5-HT2AR dynamics with one microsecond MD simulations of an apo-5-HT2AR, methiothepin-bound 5-HT2AR (antagonist-state), and an ergotamine-bound 5-HT2AR (agonist-state). We confirmed that a stable ionic bond (~2 Å) between R1733.50 and E3186.30 locks the apo-5-HT2AR in an inactive state. This bond is also present in the inactive methiothepin-bound 5-HT2AR, and broken (~4 Å) upon conformational transitions to the active state in the ergotamine-bound 5-HT2AR between 300–700 nanoseconds (Figure 7). Similar findings have been presented independently for the 5-HT2BR (166).
Figure 7:
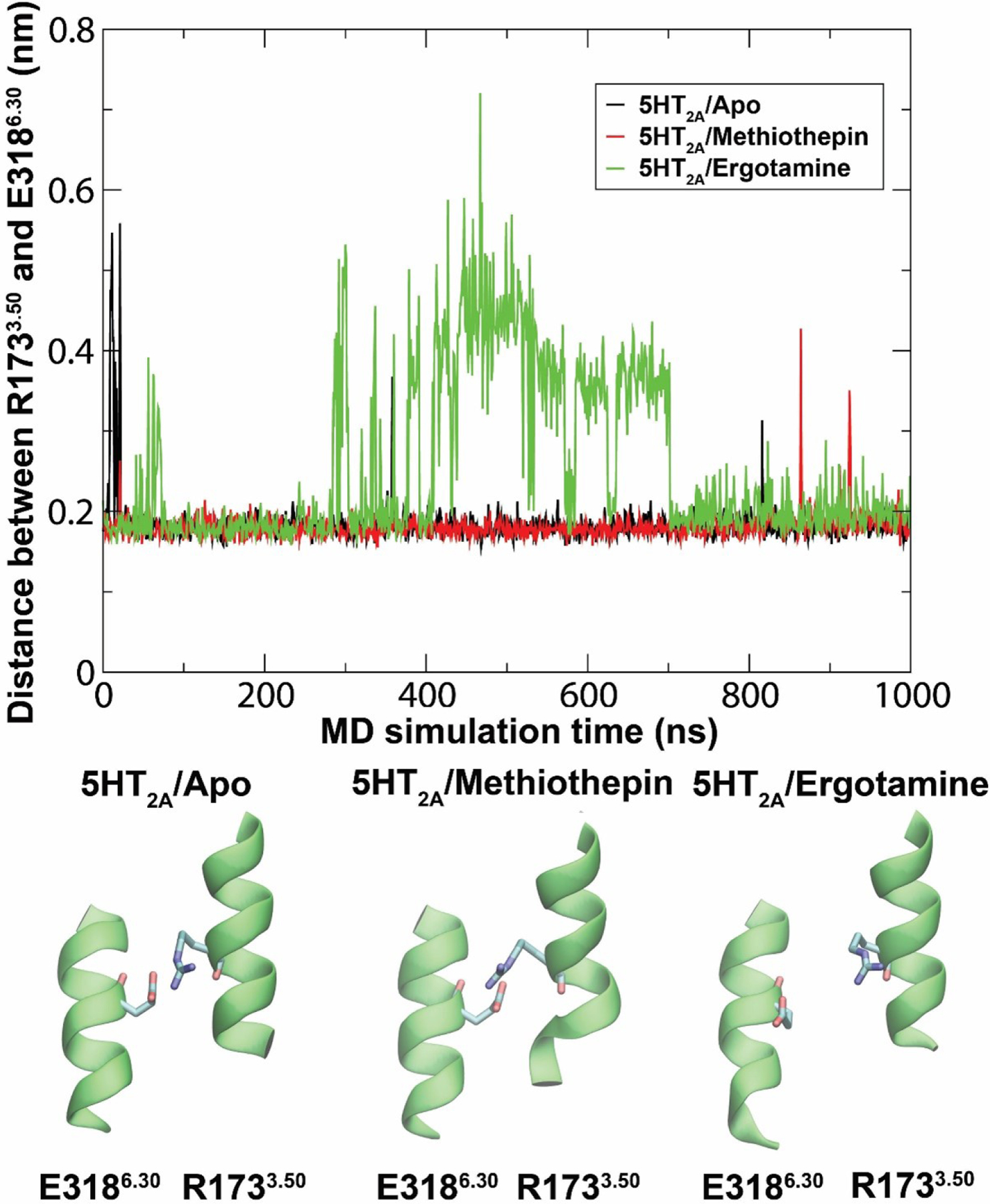
Molecular dynamics simulation results of an unbound, antagonist-, and agonist-bound 5-HT2AR (APO, methiothepin, and ergotamine, respectively). Ionic lock (E3186.30—R1733.50) distances are shown as a function of simulation time.
5. Considerations and conclusions
The development of selective antagonists for clinical and preclinical purposes will likely continue, although the methods used to discover them are destined to evolve. Historically, the discovery of putatively selective 5-HT2AR antagonists relied on experimental screening and lead optimization. Yet, sparingly few compounds have withstood the test of rigorous receptor profiling in vitro, and the number of chemically diverse antagonists with robust selectivity for 5-HT2ARs remains low. Moving forward, it would be incumbent for medicinal chemists and pharmacologists reporting on selective ligands to clearly describe the context used to define a ligand as “selective”. As an example, a ligand may be defined as selective if it has >100-fold higher affinity at its target GPCR, based on in vitro affinity tests at a pre-defined set of off-target GPCRs within the target’s receptor family, e.g., “compound X is selective for 5-HT2A relative to 5-HT2B, and 5-HT2CRs based on in vitro affinity assays using antagonist radiolabels.” Moreover, novel compounds should be characterized alongside reference ligands in orthogonal assays to account for affinity differences due to assay conditions and facilitate fair comparison across literature reports (80, 167). Authors should then note that the compound may only be useful—without further characterization—as an in vitro tool.
Discerning ligand selectivity with confidence in vivo poses a distinct challenge, due to a need to quantify ligand occupancy at on- and off-target binding sites across multiple tissues, doses, and time points following administration. In addition, the pharmacokinetic parameters used to determine occupancy will likely vary between routes of administration (i.e., intraperitoneal, subcutaneous, oral, or intravenous), and thus would require further experimentation to validate selectivity following dosing across routes. These experiments may be seen as cost-prohibitive, especially for academic labs developing medication candidates, but these data ultimately would help prevent misinterpretation of results and provide the broader scientific community (e.g., behavioral pharmacologists, neurophysiologists, etc.) critical information to guide the selection, and dosing, of tool compounds for in vivo use.
In the absence of detailed pharmacokinetic profiling, there is the modus operandi of titrating antagonist dose to prevent behaviorally disruptive or off-target effects while achieving a specific behavioral outcome. However, there can be problems with this approach if the behavioral outcome under evaluation is affected by multiple targets. For example, the head-twitch response in mice is associated with activation of the 5-HT2AR, yet is also modulated through numerous other mechanisms (163, 168, 169). Without the pharmacodynamic and pharmacokinetic information described above, we cannot make firm conclusions about selective receptor engagement in vivo or about the role of the GPCR target in the measured outcome. For example, we have shown that M100,907—which has ~160-fold in vitro selectivity for mouse 5-HT2A over 5-HT2CRs—significantly suppresses the head-twitch response in adult, male C57BL/6J mice elicited by 1 mg/kg (±)-2,5-dimethoxy-4-iodo-amphetamine (s.c.), when administered at 0.0025 mg/kg (s.c.) (71), though, at this dose, only ~35% of the head-twitch response is blocked. Meanwhile at 0.025 mg/kg and 0.25 mg/kg M100,907, ~85% and 100% of the head-twitch response is blocked, respectively. Without evaluating receptor occupancy at these doses, it is not possible to conclude what dose (if any) of M100,907 selectively blocks central 5-HT2ARs that elicit the head-twitch response.
Selective knockdown of 5-HT2AR with genetic tools in combination with autoradiography would also clarify the in vivo selectivity of a ligand following administration at a defined dose, route, timestep. For example, [3H]M100,907 could be administered at various doses to 5-HT2AR knockout mice together with another, structurally unique 5-HT2AR antagonist, and receptor occupancy could be used to determine the dose at which it binds off-targets, considering potential issues of non-specific binding.
There is also the option of combining ligands in an experiment to interrogate mechanism. For example, SB 242084, a putatively selective neutral antagonist of 5-HT2CRs might be co-administered with pimavanserin to mask the potent inverse agonist activity of pimavanserin at 5-HT2CRs. However, SB 242084 would block activity-dependent signaling through 5-HT2CRs, like pimavanserin, without preserving its native signaling capacity in response to synaptic release of 5-HT. Moreover, ligand combinations increase the possibility of confounding pharmacokinetic and pharmacodynamic interactions in vivo, e.g., competition for common metabolic enzymes might increase the plasma concentration of each ligand, and parent compound metabolites, and their pharmacological activities, are often unknown.
Here, we conclude that M100,907 and pimavanserin, on balance, are the most authenticated putatively selective 5-HT2AR antagonists available. Nevertheless, M100,907 and pimavanserin, as well as all other 5-HT2AR-selective antagonists, have off-target activities that need to be considered when used to elucidate biological mechanisms. Thus, we echo a recommendation made nearly forty-years ago (8) that mechanistic investigation of 5-HT2ARs by pharmacological antagonism should utilize two structurally distinct antagonists with different off-target activities, albeit, feasibility of conducting the same experiments twice with different antagonists is limited by increased costs.
For the reasons detailed above, we define “selective” empirically within a context, meaning that if a ligand can be administered such that it acts on its intended target without acting on pre-defined off-targets, then it is selective. We acknowledge the shortcomings of this definition, because of the “unknown unknowns” phenomenon, i.e., there are tens of thousands of proteins, and ~800 GPCRs, expressed in mammalian tissue, and a ligand could have activity at any one of them. Because this is a critical issue, we believe that medicinal chemists and pharmacologists should convene to establish parameters for defining a ligand as “selective” with the goal of providing the general scientific community information to choose dose ranges for in vivo studies in particular species.
The design of novel unambiguously selective 5-HT2AR antagonists will require expertise spanning various fields. Structural biology studies could help by delineating the structure of 5-HT2-type receptors bound to subtype-selective antagonists from multiple chemotypes to identify points of contact reliably involved in subtype-selective binding. Notably, structural biology studies often detail a single receptor conformational state, leaving unanswered questions about the dynamic nature of the receptor. Therefore, a library of diverse ligand-receptor structures could improve the efficiency of ultra-large library docking and prediction of ligand function in silico, which will likely lead to the next generation of selective 5-HT2AR antagonists. Nevertheless, the proper implementation of MD simulations in the context of drug discovery is still a challenge, and computational approaches supported by experimental evidence will provide the most compelling models for ligand-receptor interactions.
Funding and acknowledgments:
Some of the original research described in this review was funded by the National Institute on Drug Abuse (NIDA) grants R01DA047130 and R01DA030989 (R.G.B.), National Institute of Neurological Disorders and Stroke (NINDS) grant R15 NS118352-01 (C.E.C), U.S. Department of Defense grants CDMRP W81XWH-17-1-0322 (R.G.B.) and CDMRP W81XWH-17-1-0329 (C.E.C), and the Northeastern University David Bear Scholar Fund (A.B.C.). The computational work shown in figures 6 and 7 was completed using the Discovery cluster, supported by Northeastern University’s Research Computing team.
Abbreviations
- LSD
lysergic acid diethylamide
- R-SAT™
receptor selection and amplification technology™
- TM
transmembrane domain
- ECL
extracellular loop
- ICL
intracellular loop
- Q-SAR
Quantitative structure-activity relationships
- CoMFA
comparative molecular field analysis
- MD
molecular dynamics
- HTS
high throughput screening
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: The authors declare no conflicts of interest.
CRediT author statement
Austen B. Casey: Writing - Original Draft, Conceptualization, Visualization. Meng Cui: Writing - Review & Editing, Resources. Raymond G. Booth: Writing - Review & Editing, Funding acquisition. Clinton E. Canal: Writing - Review & Editing, Conceptualization, Funding acquisition, Resources, Supervision.
References:
- 1.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, et al. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997; 387(6630): 303–8. [DOI] [PubMed] [Google Scholar]
- 2.Isles AR. Htr2c Splice Variants and 5HT2CR-Mediated Appetite. Trends Endocrinol Metab. 2017; 28(8): 542–4. [DOI] [PubMed] [Google Scholar]
- 3.Sriram K, Insel PA. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol. 2018; 93(4): 251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pandy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpsoe K, Hauser AS, et al. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 2018; 46(D1): D440–D6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leysen JE, Niemegeers CJ, Tollenaere JP, Laduron PM. Serotonergic component of neuroleptic receptors. Nature. 1978; 272(5649): 168–71. [DOI] [PubMed] [Google Scholar]
- 6.McCall RB, Aghajanian GK. Serotonergic facilitation of facial motoneuron excitation. Brain Res. 1979; 169(1): 11–27. [DOI] [PubMed] [Google Scholar]
- 7.Peroutka SJ, Snyder SH. Multiple serotonin receptors: differential binding of [3H]5-hydroxytryptamine, [3H]lysergic acid diethylamide and [3H]spiroperidol. Mol Pharmacol. 1979; 16(3): 687–99. [PubMed] [Google Scholar]
- 8.Bradley PB, Engel G, Feniuk W, Fozard JR, Humphrey PP, Middlemiss DN, et al. Proposals for the classification and nomenclature of functional receptors for 5-hydroxytryptamine. Neuropharmacology. 1986; 25(6): 563–76. [DOI] [PubMed] [Google Scholar]
- 9.Colpaert FC, Niemegeers CJ, Janssen PA. A drug discrimination analysis of lysergic acid diethylamide (LSD): in vivo agonist and antagonist effects of purported 5-hydroxytryptamine antagonists and of pirenperone, a LSD-antagonist. J Pharmacol Exp Ther. 1982; 221(1): 206–14. [PubMed] [Google Scholar]
- 10.Glennon RA, Young R, Rosecrans JA. Antagonism of the effects of the hallucinogen DOM and the purported 5-HT agonist quipazine by 5-HT2 antagonists. Eur J Pharmacol. 1983; 91(2–3): 189–96. [DOI] [PubMed] [Google Scholar]
- 11.Glennon RA, Titeler M, McKenney JD. Evidence for 5-HT2 involvement in the mechanism of action of hallucinogenic agents. Life Sci. 1984; 35(25): 2505–11. [DOI] [PubMed] [Google Scholar]
- 12.Hirschhorn ID, Winter JC. Mescaline and lysergic acid diethylamide (LSD) as discriminative stimuli. Psychopharmacologia. 1971; 22(1): 64–71. [DOI] [PubMed] [Google Scholar]
- 13.Conn PJ, Sanders-Bush E. Serotonin-stimulated phosphoinositide turnover: mediation by the S2 binding site in rat cerebral cortex but not in subcortical regions. J Pharmacol Exp Ther. 1985; 234(1): 195–203. [PubMed] [Google Scholar]
- 14.Foguet M, Hoyer D, Pardo LA, Parekh A, Kluxen FW, Kalkman HO, et al. Cloning and functional characterization of the rat stomach fundus serotonin receptor. EMBO J. 1992; 11(9): 3481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foguet M, Nguyen H, Le H, Lubbert H. Structure of the mouse 5-HT1C, 5-HT2 and stomach fundus serotonin receptor genes. Neuroreport. 1992; 3(4): 345–8. [DOI] [PubMed] [Google Scholar]
- 16.Kursar JD, Nelson DL, Wainscott DB, Cohen ML, Baez M. Molecular cloning, functional expression, and pharmacological characterization of a novel serotonin receptor (5-hydroxytryptamine2F) from rat stomach fundus. Mol Pharmacol. 1992; 42(4): 549–57. [PubMed] [Google Scholar]
- 17.Loric S, Launay JM, Colas JF, Maroteaux L. New mouse 5-HT2-like receptor. Expression in brain, heart and intestine. FEBS Lett. 1992; 312(2–3): 203–7. [DOI] [PubMed] [Google Scholar]
- 18.Pazos A, Hoyer D, Palacios JM. The binding of serotonergic ligands to the porcine choroid plexus: characterization of a new type of serotonin recognition site. Eur J Pharmacol. 1984; 106(3): 539–46. [DOI] [PubMed] [Google Scholar]
- 19.Conn PJ, Sanders-Bush E, Hoffman BJ, Hartig PR. A unique serotonin receptor in choroid plexus is linked to phosphatidylinositol turnover. Proc Natl Acad Sci U S A. 1986; 83(11): 4086–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humphrey PP, Hartig P, Hoyer D. A proposed new nomenclature for 5-HT receptors. Trends Pharmacol Sci. 1993; 14(6): 233–6. [DOI] [PubMed] [Google Scholar]
- 21.Bugarski-Kirola D, Arango C, Fava M, Nasrallah H, Liu IY, Abbs B, et al. Pimavanserin for negative symptoms of schizophrenia: results from the ADVANCE phase 2 randomised, placebo-controlled trial in North America and Europe. Lancet Psychiatry. 2021. [DOI] [PubMed] [Google Scholar]
- 22.Cummings J, Isaacson S, Mills R, Williams H, Chi-Burris K, Corbett A, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet. 2014; 383(9916): 533–40. [DOI] [PubMed] [Google Scholar]
- 23.Ballard C, Banister C, Khan Z, Cummings J, Demos G, Coate B, et al. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol. 2018; 17(3): 213–22. [DOI] [PubMed] [Google Scholar]
- 24.Tariot PN, Cummings JL, Soto-Martin ME, Ballard C, Erten-Lyons D, Sultzer DL, et al. Trial of Pimavanserin in Dementia-Related Psychosis. N Engl J Med. 2021; 385(4): 309–19. [DOI] [PubMed] [Google Scholar]
- 25.Bugarski-Kirola D, Arango C, Fava M, Nasrallah H, Liu IY, Abbs B, et al. Pimavanserin for negative symptoms of schizophrenia: results from the ADVANCE phase 2 randomised, placebo-controlled trial in North America and Europe. Lancet Psychiatry. 2022; 9(1): 46–58. [DOI] [PubMed] [Google Scholar]
- 26.Shao LX, Liao C, Gregg I, Davoudian PA, Savalia NK, Delagarza K, et al. Psilocybin induces rapid and persistent growth of dendritic spines in frontal cortex in vivo. Neuron. 2021; 109(16): 2535–44 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hesselgrave N, Troppoli TA, Wulff AB, Cole AB, Thompson SM. Harnessing psilocybin: antidepressant-like behavioral and synaptic actions of psilocybin are independent of 5-HT2R activation in mice. Proc Natl Acad Sci U S A. 2021; 118(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ly C, Greb AC, Cameron LP, Wong JM, Barragan EV, Wilson PC, et al. Psychedelics Promote Structural and Functional Neural Plasticity. Cell Rep. 2018; 23(11): 3170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hibicke M, Landry AN, Kramer HM, Talman ZK, Nichols CD. Psychedelics, but Not Ketamine, Produce Persistent Antidepressant-like Effects in a Rodent Experimental System for the Study of Depression. ACS Chem Neurosci. 2020; 11(6): 864–71. [DOI] [PubMed] [Google Scholar]
- 30.de la Fuente Revenga M, Zhu B, Guevara CA, Naler LB, Saunders JM, Zhou Z, et al. Prolonged epigenomic and synaptic plasticity alterations following single exposure to a psychedelic in mice. Cell Rep. 2021; 37(3): 109836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fields JZ, Reisine TD, Yamamura HI. Biochemical demonstration of dopaminergic receptors in rat and human brain using [3H]spiroperidol. Brain Res. 1977; 136(3): 578–84. [DOI] [PubMed] [Google Scholar]
- 32.Creese I, Snyder SH. 3H-Spiroperidol labels serotonin receptors in rat cerebral cortex and hippocampus. Eur J Pharmacol. 1978; 49(2): 201–2. [DOI] [PubMed] [Google Scholar]
- 33.Bonhaus DW, Bach C, DeSouza A, Salazar FH, Matsuoka BD, Zuppan P, et al. The pharmacology and distribution of human 5-hydroxytryptamine2B (5-HT2B) receptor gene products: comparison with 5-HT2A and 5-HT2C receptors. Br J Pharmacol. 1995; 115(4): 622–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoyer D, Srivatsa S, Pazos A, Engel G, Palacios JM. [125I]LSD labels 5-HT1C recognition sites in pig choroid plexus membranes. Comparison with [3H]mesulergine and [3H]5-HT binding. Neurosci Lett. 1986; 69(3): 269–74. [DOI] [PubMed] [Google Scholar]
- 35.Leysen JE, Awouters F, Kennis L, Laduron PM, Vandenberk J, Janssen PA. Receptor binding profile of R 41 468, a novel antagonist at 5-HT2 receptors. Life Sci. 1981; 28(9): 1015–22. [DOI] [PubMed] [Google Scholar]
- 36.Preller KH, Herdener M, Pokorny T, Planzer A, Kraehenmann R, Stampfli P, et al. The Fabric of Meaning and Subjective Effects in LSD-Induced States Depend on Serotonin 2A Receptor Activation. Curr Biol. 2017; 27(3): 451–7. [DOI] [PubMed] [Google Scholar]
- 37.Preller KH, Burt JB, Ji JL, Schleifer CH, Adkinson BD, Stampfli P, et al. Changes in global and thalamic brain connectivity in LSD-induced altered states of consciousness are attributable to the 5-HT2A receptor. Elife. 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraehenmann R, Pokorny D, Vollenweider L, Preller KH, Pokorny T, Seifritz E, et al. Dreamlike effects of LSD on waking imagery in humans depend on serotonin 2A receptor activation. Psychopharmacology (Berl). 2017; 234(13): 2031–46. [DOI] [PubMed] [Google Scholar]
- 39.Vollenweider FX, Vollenweider-Scherpenhuyzen MF, Babler A, Vogel H, Hell D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport. 1998; 9(17): 3897–902. [DOI] [PubMed] [Google Scholar]
- 40.Knudsen GM. The Neurobiological Effect of 5-HT2AR Modulation. ClinicalTrials.gov 2020. https://www.clinicaltrials.gov/ct2/show/NCT03289949?term=ketanserin&draw=2&rank=6. [Google Scholar]
- 41.Liechti ME. Role of the Serotonin 5-HT2A Receptor in Mescaline-induced Altered States of Consciousness (MDR). ClinicalTrials.gov; 2021. https://www.clinicaltrials.gov/ct2/show/NCT04849013?term=ketanserin&draw=2&rank=8. [Google Scholar]
- 42.Hoyer D, Vos P, Closse A, Pazos A, Palacios JM, Davies H. [3H]ketanserin labels 5-HT2 receptors and alpha 1-adrenoceptors in human and pig brain membranes. Naunyn Schmiedebergs Arch Pharmacol. 1987; 335(3): 226–30. [DOI] [PubMed] [Google Scholar]
- 43.Bucholtz EC, Brown RL, Tropsha A, Booth RG, Wyrick SD. Synthesis, evaluation, and comparative molecular field analysis of 1-phenyl-3-amino-1,2,3,4-tetrahydronaphthalenes as ligands for histamine H(1) receptors. J Med Chem. 1999; 42(16): 3041–54. [DOI] [PubMed] [Google Scholar]
- 44.Yoshio R, Taniguchi T, Itoh H, Muramatsu I. Affinity of serotonin receptor antagonists and agonists to recombinant and native alpha1-adrenoceptor subtypes. Jpn J Pharmacol. 2001; 86(2): 189–95. [DOI] [PubMed] [Google Scholar]
- 45.Pan ZZ, Grudt TJ, Williams JT. Alpha 1-adrenoceptors in rat dorsal raphe neurons: regulation of two potassium conductances. J Physiol. 1994; 478 Pt 3: 437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amargos-Bosch M, Adell A, Bortolozzi A, Artigas F. Stimulation of alpha1-adrenoceptors in the rat medial prefrontal cortex increases the local in vivo 5-hydroxytryptamine release: reversal by antipsychotic drugs. J Neurochem. 2003; 87(4): 831–42. [DOI] [PubMed] [Google Scholar]
- 47.Santana N, Mengod G, Artigas F. Expression of alpha(1)-adrenergic receptors in rat prefrontal cortex: cellular co-localization with 5-HT(2A) receptors. Int J Neuropsychopharmacol. 2013; 16(5): 1139–51. [DOI] [PubMed] [Google Scholar]
- 48.Brown RE, Sergeeva OA, Eriksson KS, Haas HL. Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J Neurosci. 2002; 22(20): 8850–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pudovkina OL, Cremers TI, Westerink BH. Regulation of the release of serotonin in the dorsal raphe nucleus by alpha1 and alpha2 adrenoceptors. Synapse. 2003; 50(1): 77–82. [DOI] [PubMed] [Google Scholar]
- 50.Leysen JE, Niemegeers CJ, Van Nueten JM, Laduron PM. [3H]Ketanserin (R 41 468), a selective 3H-ligand for serotonin2 receptor binding sites. Binding properties, brain distribution, and functional role. Mol Pharmacol. 1982; 21(2): 301–14. [PubMed] [Google Scholar]
- 51.Lopez-Gimenez JF, Mengod G, Palacios JM, Vilaro MT. Selective visualization of rat brain 5-HT2A receptors by autoradiography with [3H]MDL 100,907. Naunyn Schmiedebergs Arch Pharmacol. 1997; 356(4): 446–54. [DOI] [PubMed] [Google Scholar]
- 52.Roth BL, McLean S, Zhu XZ, Chuang DM. Characterization of two [3H]ketanserin recognition sites in rat striatum. J Neurochem. 1987; 49(6): 1833–8. [DOI] [PubMed] [Google Scholar]
- 53.Darchen F, Scherman D, Laduron PM, Henry JP. Ketanserin binds to the monoamine transporter of chromaffin granules and of synaptic vesicles. Mol Pharmacol. 1988; 33(6): 672–7. [PubMed] [Google Scholar]
- 54.Erickson JD, Schafer MK, Bonner TI, Eiden LE, Weihe E. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996; 93(10): 5166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sorensen SM, Kehne JH, Fadayel GM, Humphreys TM, Ketteler HJ, Sullivan CK, et al. Characterization of the 5-HT2 receptor antagonist MDL 100907 as a putative atypical antipsychotic: behavioral, electrophysiological and neurochemical studies. J Pharmacol Exp Ther. 1993; 266(2): 684–91. [PubMed] [Google Scholar]
- 56.Dudley M, Ogden A, Carr A, Nieduzak T Kehne J MDL 100,907, (+-α-(2,3-Dimethoxyphenyl)-1-[2-(4-Fluorophenylethyl)]-4-Piperidinemethanol), a Potent, Chiral, 5-HT2 Receptor Antagonist. In: Society for Neuroscience: 1990. [Google Scholar]
- 57.Palfreyman MG, Schmidt CJ, Sorensen SM, Dudley MW, Kehne JH, Moser P, et al. Electrophysiological, biochemical and behavioral evidence for 5-HT2 and 5-HT3 mediated control of dopaminergic function. Psychopharmacology (Berl). 1993; 112(1 Suppl): S60–7. [DOI] [PubMed] [Google Scholar]
- 58.Kehne JH, Baron BM, Carr AA, Chaney SF, Elands J, Feldman DJ, et al. Preclinical characterization of the potential of the putative atypical antipsychotic MDL 100,907 as a potent 5-HT2A antagonist with a favorable CNS safety profile. J Pharmacol Exp Ther. 1996; 277(2): 968–81. [PubMed] [Google Scholar]
- 59.Oksenberg D, Marsters SA, O’Dowd BF, Jin H, Havlik S, Peroutka SJ, et al. A single amino-acid difference confers major pharmacological variation between human and rodent 5-HT1B receptors. Nature. 1992; 360(6400): 161–3. [DOI] [PubMed] [Google Scholar]
- 60.Kao HT, Adham N, Olsen MA, Weinshank RL, Branchek TA, Hartig PR. Site-directed mutagenesis of a single residue changes the binding properties of the serotonin 5-HT2 receptor from a human to a rat pharmacology. FEBS Lett. 1992; 307(3): 324–8. [DOI] [PubMed] [Google Scholar]
- 61.Canal CE, Cordova-Sintjago T, Liu Y, Kim MS, Morgan D, Booth RG. Molecular pharmacology and ligand docking studies reveal a single amino acid difference between mouse and human serotonin 5-HT2A receptors that impacts behavioral translation of novel 4-phenyl-2-dimethylaminotetralin ligands. J Pharmacol Exp Ther. 2013; 347(3): 705–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pucadyil TJ, Chattopadhyay A. Cholesterol modulates ligand binding and G-protein coupling to serotonin(1A) receptors from bovine hippocampus. Biochim Biophys Acta. 2004; 1663(1–2): 188–200. [DOI] [PubMed] [Google Scholar]
- 63.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011; 469(7329): 175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011; 477(7366): 549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sleight AJ, Stam NJ, Mutel V, Vanderheyden PM. Radiolabelling of the human 5-HT2A receptor with an agonist, a partial agonist and an antagonist: effects on apparent agonist affinities. Biochem Pharmacol. 1996; 51(1): 71–6. [DOI] [PubMed] [Google Scholar]
- 66.Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011; 51: 117–44. [DOI] [PubMed] [Google Scholar]
- 67.Pehek EA, Nocjar C, Roth BL, Byrd TA, Mabrouk OS. Evidence for the preferential involvement of 5-HT2A serotonin receptors in stress- and drug-induced dopamine release in the rat medial prefrontal cortex. Neuropsychopharmacology. 2006; 31(2): 265–77. [DOI] [PubMed] [Google Scholar]
- 68.Weiner DM, Burstein ES, Nash N, Croston GE, Currier EA, Vanover KE, et al. 5-hydroxytryptamine2A receptor inverse agonists as antipsychotics. J Pharmacol Exp Ther. 2001; 299(1): 268–76. [PubMed] [Google Scholar]
- 69.Barr AM, Lehmann-Masten V, Paulus M, Gainetdinov RR, Caron MG, Geyer MA. The selective serotonin-2A receptor antagonist M100907 reverses behavioral deficits in dopamine transporter knockout mice. Neuropsychopharmacology. 2004; 29(2): 221–8. [DOI] [PubMed] [Google Scholar]
- 70.Gleason SD, Shannon HE. Blockade of phencyclidine-induced hyperlocomotion by olanzapine, clozapine and serotonin receptor subtype selective antagonists in mice. Psychopharmacology (Berl). 1997; 129(1): 79–84. [DOI] [PubMed] [Google Scholar]
- 71.Canal CE, Booth RG, Morgan D. Support for 5-HT2C receptor functional selectivity in vivo utilizing structurally diverse, selective 5-HT2C receptor ligands and the 2,5-dimethoxy-4-iodoamphetamine elicited head-twitch response model. Neuropharmacology. 2013; 70: 112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carlsson ML. The selective 5-HT2A receptor antagonist MDL 100,907 counteracts the psychomotor stimulation ensuing manipulations with monoaminergic, glutamatergic or muscarinic neurotransmission in the mouse--implications for psychosis. J Neural Transm Gen Sect. 1995; 100(3): 225–37. [DOI] [PubMed] [Google Scholar]
- 73.Padich RA, McCloskey TC, Kehne JH. 5-HT modulation of auditory and visual sensorimotor gating: II. Effects of the 5-HT2A antagonist MDL 100,907 on disruption of sound and light prepulse inhibition produced by 5-HT agonists in Wistar rats. Psychopharmacology (Berl). 1996; 124(1–2): 107–16. [DOI] [PubMed] [Google Scholar]
- 74.Barrett FS, Zhou Y, Carbonaro TM, Roberts JM, Smith GS, Griffiths RR, et al. Human Cortical Serotonin 2A Receptor Occupancy by Psilocybin Measured Using [11C]MDL 100,907 Dynamic PET and a Resting-State fMRI-Based Brain Parcellation. Frontiers in Neuroergonomics. 2022; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grunder G, Yokoi F, Offord SJ, Ravert HT, Dannals RF, Salzmann JK, et al. Time course of 5-HT2A receptor occupancy in the human brain after a single oral dose of the putative antipsychotic drug MDL 100,907 measured by positron emission tomography. Neuropsychopharmacology. 1997; 17(3): 175–85. [DOI] [PubMed] [Google Scholar]
- 76.de Paulis T M-100907 (Aventis). Curr Opin Investig Drugs. 2001; 2(1): 123–32. [PubMed] [Google Scholar]
- 77.Hacksell U, Burstein ES, McFarland K, Mills RG, Williams H. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014; 39(10): 2008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vanover KE, Weiner DM, Makhay M, Veinbergs I, Gardell LR, Lameh J, et al. Pharmacological and behavioral profile of N-(4-fluorophenylmethyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy)phen ylmethyl) carbamide (2R,3R)-dihydroxybutanedioate (2:1) (ACP-103), a novel 5-hydroxytryptamine(2A) receptor inverse agonist. J Pharmacol Exp Ther. 2006; 317(2): 910–8. [DOI] [PubMed] [Google Scholar]
- 79.Kimura KT, Asada H, Inoue A, Kadji FMN, Im D, Mori C, et al. Structures of the 5-HT2A receptor in complex with the antipsychotics risperidone and zotepine. Nat Struct Mol Biol. 2019; 26(2): 121–8. [DOI] [PubMed] [Google Scholar]
- 80.Casey AB, Mukherjee M, McGlynn RP, Cui M, Kohut SJ, Booth RG. A new class of serotonin 5-HT2A /5-HT2C receptor inverse agonists: Synthesis, molecular modeling, in vitro and in vivo pharmacology of novel 2-aminotetralins. Br J Pharmacol. 2021. [DOI] [PubMed] [Google Scholar]
- 81.Muneta-Arrate I, Diez-Alarcia R, Horrillo I, Meana JJ. Pimavanserin exhibits serotonin 5-HT2A receptor inverse agonism for Galphai1- and neutral antagonism for Galphaq/11-proteins in human brain cortex. Eur Neuropsychopharmacol. 2020; 36: 83–9. [DOI] [PubMed] [Google Scholar]
- 82.Stahl SM. Mechanism of action of pimavanserin in Parkinson’s disease psychosis: targeting serotonin 5HT2A and 5HT2C receptors. CNS Spectr. 2016; 21(4): 271–5. [DOI] [PubMed] [Google Scholar]
- 83.Fava M, Dirks B, Freeman MP, Papakostas GI, Shelton RC, Thase ME, et al. A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study of Adjunctive Pimavanserin in Patients With Major Depressive Disorder and an Inadequate Response to Therapy (CLARITY). J Clin Psychiatry. 2019; 80(6). [DOI] [PubMed] [Google Scholar]
- 84.Halberstadt AL, van der Heijden I, Ruderman MA, Risbrough VB, Gingrich JA, Geyer MA, et al. 5-HT(2A) and 5-HT(2C) receptors exert opposing effects on locomotor activity in mice. Neuropsychopharmacology. 2009; 34(8): 1958–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McOmish CE, Lira A, Hanks JB, Gingrich JA. Clozapine-induced locomotor suppression is mediated by 5-HT2A receptors in the forebrain. Neuropsychopharmacology. 2012; 37(13): 2747–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.De Deurwaerdere P, Navailles S, Berg KA, Clarke WP, Spampinato U. Constitutive activity of the serotonin2C receptor inhibits in vivo dopamine release in the rat striatum and nucleus accumbens. J Neurosci. 2004; 24(13): 3235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De Deurwaerdere P, Ramos M, Bharatiya R, Puginier E, Chagraoui A, Manem J, et al. Lorcaserin bidirectionally regulates dopaminergic function site-dependently and disrupts dopamine brain area correlations in rats. Neuropharmacology. 2020; 166: 107915. [DOI] [PubMed] [Google Scholar]
- 88.Cathala A, Devroye C, Maitre M, Piazza PV, Abrous DN, Revest JM, et al. Serotonin2C receptors modulate dopamine transmission in the nucleus accumbens independently of dopamine release: behavioral, neurochemical and molecular studies with cocaine. Addict Biol. 2015; 20(3): 445–57. [DOI] [PubMed] [Google Scholar]
- 89.Meltzer HY, Mills R, Revell S, Williams H, Johnson A, Bahr D, et al. Pimavanserin, a serotonin(2A) receptor inverse agonist, for the treatment of parkinson’s disease psychosis. Neuropsychopharmacology. 2010; 35(4): 881–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ancoli-Israel S, Vanover KE, Weiner DM, Davis RE, van Kammen DP. Pimavanserin tartrate, a 5-HT(2A) receptor inverse agonist, increases slow wave sleep as measured by polysomnography in healthy adult volunteers. Sleep Med. 2011; 12(2): 134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry. 1992; 49(7): 538–44. [DOI] [PubMed] [Google Scholar]
- 92.Andersen VL, Hansen HD, Herth MM, Dyssegaard A, Knudsen GM, Kristensen JL. (11)C-labeling and preliminary evaluation of pimavanserin as a 5-HT2A receptor PET-radioligand. Bioorg Med Chem Lett. 2015; 25(5): 1053–6. [DOI] [PubMed] [Google Scholar]
- 93.Leysen JE, Gommeren W, Laduron PM. Spiperone: a ligand of choice for neuroleptic receptors. 1. Kinetics and characteristics of in vitro binding. Biochem Pharmacol. 1978; 27(3): 307–16. [DOI] [PubMed] [Google Scholar]
- 94.Colpaert FC. Discovering risperidone: the LSD model of psychopathology. Nat Rev Drug Discov. 2003; 2(4): 315–20. [DOI] [PubMed] [Google Scholar]
- 95.Leysen JE, Gommeren W, Van Gompel P, Wynants J, Janssen PF, Laduron PM. Receptor-binding properties in vitro and in vivo of ritanserin: A very potent and long acting serotonin-S2 antagonist. Mol Pharmacol. 1985; 27(6): 600–11. [PubMed] [Google Scholar]
- 96.Janssen PA. Pharmacology of potent and selective S2-serotonergic antagonists. J Cardiovasc Pharmacol. 1985; 7 Suppl 7: S2–11. [DOI] [PubMed] [Google Scholar]
- 97.Kennis JEJV J Novel 1,2-benzisoxadol-3-yl and 1,2-benzisothiazol-3-yl derivatives. 1985.
- 98.Kristiansen H, Elfving B, Plenge P, Pinborg LH, Gillings N, Knudsen GM. Binding characteristics of the 5-HT2A receptor antagonists altanserin and MDL 100907. Synapse. 2005; 58(4): 249–57. [DOI] [PubMed] [Google Scholar]
- 99.Elmenhorst D, Kroll T, Matusch A, Bauer A. Sleep deprivation increases cerebral serotonin 2A receptor binding in humans. Sleep. 2012; 35(12): 1615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fisher PM, Meltzer CC, Price JC, Coleman RL, Ziolko SK, Becker C, et al. Medial prefrontal cortex 5-HT(2A) density is correlated with amygdala reactivity, response habituation, and functional coupling. Cereb Cortex. 2009; 19(11): 2499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Green AR, O’Shaughnessy K, Hammond M, Schachter M, Grahame-Smith DG. Inhibition of 5-hydroxytryptamine-mediated behaviour by the putative 5-HT2 antagonist pirenperone. Neuropharmacology. 1983; 22(5): 573–8. [DOI] [PubMed] [Google Scholar]
- 102.Peng Y, McCorvy JD, Harpsoe K, Lansu K, Yuan S, Popov P, et al. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell. 2018; 172(4): 719–30 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors Methods in Neuroscience. 1995; 25: 366–428. [Google Scholar]
- 104.Shapiro DA, Kristiansen K, Kroeze WK, Roth BL. Differential modes of agonist binding to 5-hydroxytryptamine(2A) serotonin receptors revealed by mutation and molecular modeling of conserved residues in transmembrane region 5. Mol Pharmacol. 2000; 58(5): 877–86. [DOI] [PubMed] [Google Scholar]
- 105.Vass M, Podlewska S, de Esch IJP, Bojarski AJ, Leurs R, Kooistra AJ, et al. Aminergic GPCR-Ligand Interactions: A Chemical and Structural Map of Receptor Mutation Data. J Med Chem. 2019; 62(8): 3784–839. [DOI] [PubMed] [Google Scholar]
- 106.Kim K, Che T, Panova O, DiBerto JF, Lyu J, Krumm BE, et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell. 2020; 182(6): 1574–88 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang CD, Gallaher TK, Shih JC. Site-directed mutagenesis of the serotonin 5-hydroxytrypamine2 receptor: identification of amino acids necessary for ligand binding and receptor activation. Mol Pharmacol. 1993; 43(6): 931–40. [PubMed] [Google Scholar]
- 108.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011; 475(7354): 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013; 340(6132): 610–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell. 2017; 168(3): 377–89 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen Z, Fan L, Wang H, Yu J, Lu D, Qi J, et al. Structure-based design of a novel third-generation antipsychotic drug lead with potential antidepressant properties. Nat Neurosci. 2022; 25(1): 39–49. [DOI] [PubMed] [Google Scholar]
- 112.Latorraca NR, Venkatakrishnan AJ, Dror RO. GPCR Dynamics: Structures in Motion. Chem Rev. 2017; 117(1): 139–55. [DOI] [PubMed] [Google Scholar]
- 113.Zhou Q, Yang D, Wu M, Guo Y, Guo W, Zhong L, et al. Common activation mechanism of class A GPCRs. Elife. 2019; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Y, Canal CE, Cordova-Sintjago TC, Zhu WY, Booth RG. Mutagenesis Analysis Reveals Distinct Amino Acids of the Human Serotonin 5-HT2c Receptor Underlying the Pharmacology of Distinct Ligands. Acs Chemical Neuroscience. 2017; 8(1): 28–39. [DOI] [PubMed] [Google Scholar]
- 115.Johnson MP, Loncharich RJ, Baez M, Nelson DL. Species variations in transmembrane region V of the 5-hydroxytryptamine type 2A receptor alter the structure-activity relationship of certain ergolines and tryptamines. Mol Pharmacol. 1994; 45(2): 277–86. [PubMed] [Google Scholar]
- 116.Almaula N, Ebersole BJ, Ballesteros JA, Weinstein H, Sealfon SC. Contribution of a helix 5 locus to selectivity of hallucinogenic and nonhallucinogenic ligands for the human 5-hydroxytryptamine2A and 5-hydroxytryptamine2C receptors: direct and indirect effects on ligand affinity mediated by the same locus. Mol Pharmacol. 1996; 50(1): 34–42. [PubMed] [Google Scholar]
- 117.Wang YQ, Lin WW, Wu N, Wang SY, Chen MZ, Lin ZH, et al. Structural insight into the serotonin (5-HT) receptor family by molecular docking, molecular dynamics simulation and systems pharmacology analysis. Acta Pharmacol Sin. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller KJ, Wu GY, Varnes JG, Levesque P, Li J, Li D, et al. Position 5.46 of the serotonin 5-HT2A receptor contributes to a species-dependent variation for the 5-HT2C agonist (R)-9-ethyl-1,3,4,10b-tetrahydro-7-trifluoromethylpyrazino[2,1-a]isoindol-6(2H)-o ne: impact on selectivity and toxicological evaluation. Mol Pharmacol. 2009; 76(6): 1211–9. [DOI] [PubMed] [Google Scholar]
- 119.Miller MP, Kumar S. Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet. 2001; 10(21): 2319–28. [DOI] [PubMed] [Google Scholar]
- 120.Almaula N, Ebersole BJ, Zhang D, Weinstein H, Sealfon SC. Mapping the binding site pocket of the serotonin 5-Hydroxytryptamine2A receptor. Ser3.36(159) provides a second interaction site for the protonated amine of serotonin but not of lysergic acid diethylamide or bufotenin. J Biol Chem. 1996; 271(25): 14672–5. [DOI] [PubMed] [Google Scholar]
- 121.Braden MR, Nichols DE. Assessment of the roles of serines 5.43(239) and 5.46(242) for binding and potency of agonist ligands at the human serotonin 5-HT2A receptor. Mol Pharmacol. 2007; 72(5): 1200–9. [DOI] [PubMed] [Google Scholar]
- 122.BL Roth D Contract NO2MH80002. In: Psychoactive Drug Screening Program database. National Institute of Mental Health, 2012. [Google Scholar]
- 123.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively nonselective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004; 3(4): 353–9. [DOI] [PubMed] [Google Scholar]
- 124.Huidobro-Toro JP, Harris RA. Brain lipids that induce sleep are novel modulators of 5-hydroxytrypamine receptors. Proc Natl Acad Sci U S A 1996; 93(15): 8078–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thomas EA, Carson MJ, Neal MJ, Sutcliffe JG. Unique allosteric regulation of 5-hydroxytryptamine receptor-mediated signal transduction by oleamide. Proc Natl Acad Sci U S A 1997; 94(25): 14115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cao D, Yu J, Wang H, Luo Z, Liu X, He L, et al. Structure-based discovery of nonhallucinogenic psychedelic analogs. Science. 2022; 375(6579): 403–11. [DOI] [PubMed] [Google Scholar]
- 127.Liu W, Wacker D, Gati C, Han GW, James D, Wang D, et al. Serial femtosecond crystallography of G protein-coupled receptors. Science. 2013; 342(6165): 1521–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ishchenko A, Wacker D, Kapoor M, Zhang A, Han GW, Basu S, et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc Natl Acad Sci U S A 2017; 114(31): 8223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Im D, Inoue A, Fujiwara T, Nakane T, Yamanaka Y, Uemura T, et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat Commun. 2020; 11(1): 6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Metwally KA, Dukat M, Egan CT, Smith C, DuPre A, Gauthier CB, et al. Spiperone: influence of spiro ring substituents on 5-HT2A serotonin receptor binding. J Med Chem. 1998; 41(25): 5084–93. [DOI] [PubMed] [Google Scholar]
- 131.Kristiansen K, Kroeze WK, Willins DL, Gelber EI, Savage JE, Glennon RA, et al. A highly conserved aspartic acid (Asp-155) anchors the terminal amine moiety of tryptamines and is involved in membrane targeting of the 5-HT(2A) serotonin receptor but does not participate in activation via a “salt-bridge disruption” mechanism. J Pharmacol Exp Ther. 2000; 293(3): 735–46. [PubMed] [Google Scholar]
- 132.Peters JU, Hert J, Bissantz C, Hillebrecht A, Gerebtzoff G, Bendels S, et al. Can we discover pharmacological promiscuity early in the drug discovery process? Drug Discov Today. 2012; 17(7–8): 325–35. [DOI] [PubMed] [Google Scholar]
- 133.Whitebread S, Hamon J, Bojanic D, Urban L. Keynote review: in vitro safety pharmacology profiling: an essential tool for successful drug development. Drug Discov Today. 2005; 10(21): 1421–33. [DOI] [PubMed] [Google Scholar]
- 134.Bonhaus DW, Flippin LA, Greenhouse RJ, Jaime S, Rocha C, Dawson M, et al. RS-127445: a selective, high affinity, orally bioavailable 5-HT2B receptor antagonist. Br J Pharmacol. 1999; 127(5): 1075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wood MD, Reavill C, Trail B, Wilson A, Stean T, Kennett GA, et al. SB-243213; a selective 5-HT2C receptor inverse agonist with improved anxiolytic profile: lack of tolerance and withdrawal anxiety. Neuropharmacology. 2001; 41(2): 186–99. [DOI] [PubMed] [Google Scholar]
- 136.Kennett GA, Wood MD, Bright F, Trail B, Riley G, Holland V, et al. SB 242084, a selective and brain penetrant 5-HT2C receptor antagonist. Neuropharmacology. 1997; 36(4–5): 609–20. [DOI] [PubMed] [Google Scholar]
- 137.Kennett GA, Wood MD, Bright F, Cilia J, Piper DC, Gager T, et al. In vitro and in vivo profile of SB 206553, a potent 5-HT2C/5-HT2B receptor antagonist with anxiolytic-like properties. Br J Pharmacol. 1996; 117(3): 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Carpenter J, Wang Y, Wu G, Feng J, Ye XY, Morales CL, et al. Utilization of an Active Site Mutant Receptor for the Identification of Potent and Selective Atypical 5-HT2C Receptor Agonists. J Med Chem. 2017; 60(14): 6166–90. [DOI] [PubMed] [Google Scholar]
- 139.Agarwal A, Pearson PP, Taylor EW, Li HB, Dahlgren T, Herslof M, et al. Three-dimensional quantitative structure-activity relationships of 5-HT receptor binding data for tetrahydropyridinylindole derivatives: a comparison of the Hansch and CoMFA methods. J Med Chem. 1993; 36(25): 4006–14. [DOI] [PubMed] [Google Scholar]
- 140.Ravina E, Negreira J, Cid J, Masaguer CF, Rosa E, Rivas ME, et al. Conformationally constrained butyrophenones with mixed dopaminergic (D(2)) and serotoninergic (5-HT(2A), 5-HT(2C)) affinities: synthesis, pharmacology, 3D-QSAR, and molecular modeling of (aminoalkyl)benzo- and -thienocycloalkanones as putative atypical antipsychotics. J Med Chem. 1999; 42(15): 2774–97. [DOI] [PubMed] [Google Scholar]
- 141.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000; 289(5480): 739–45. [DOI] [PubMed] [Google Scholar]
- 142.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007; 450(7168): 383–7. [DOI] [PubMed] [Google Scholar]
- 143.Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, Roth BL. Evidence for a model of agonist-induced activation of 5-hydroxytryptamine 2A serotonin receptors that involves the disruption of a strong ionic interaction between helices 3 and 6. J Biol Chem. 2002; 277(13): 11441–9. [DOI] [PubMed] [Google Scholar]
- 144.Egan C, Grinde E, Dupre A, Roth BL, Hake M, Teitler M, et al. Agonist high and low affinity state ratios predict drug intrinsic activity and a revised ternary complex mechanism at serotonin 5-HT(2A) and 5-HT(2C) receptors. Synapse. 2000; 35(2): 144–50. [DOI] [PubMed] [Google Scholar]
- 145.Hossain M, Muntasir HA, Ishiguro M, Bhuiyan MA, Rashid M, Sugihara T, et al. Mechanism of inverse agonist action of sarpogrelate at the constitutively active mutant of human 5-HT2A receptor revealed by molecular modeling. Biol Pharm Bull. 2012; 35(9): 1553–9. [DOI] [PubMed] [Google Scholar]
- 146.Brown DG, Bostrom J. Where Do Recent Small Molecule Clinical Development Candidates Come From? J Med Chem. 2018; 61(21): 9442–68. [DOI] [PubMed] [Google Scholar]
- 147.Walters WP. Virtual Chemical Libraries. J Med Chem. 2019; 62(3): 1116–24. [DOI] [PubMed] [Google Scholar]
- 148.Bailey D, Brown D. High-throughput chemistry and structure-based design: survival of the smartest. Drug Discov Today. 2001; 6(2): 57–9. [DOI] [PubMed] [Google Scholar]
- 149.Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol. 2008; 153 Suppl 1: S7–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011; 7(2): 146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sadybekov AA, Sadybekov AV, Liu Y, Iliopoulos-Tsoutsouvas C, Huang XP, Pickett J, et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gandhimathi A, Sowdhamini R. Molecular modelling of human 5-hydroxytryptamine receptor (5-HT2A) and virtual screening studies towards the identification of agonist and antagonist molecules. J Biomol Struct Dyn. 2016; 34(5): 952–70. [DOI] [PubMed] [Google Scholar]
- 153.Wang Y, Joseph J, Gao Y, Hu B, Geng X, Wu D, et al. Revealing the interaction modes of 5-HT2A receptor antagonists and the Structure-Based virtual screening from FDA and TCMNP database. J Biomol Struct Dyn. 2020: 1–12. [DOI] [PubMed] [Google Scholar]
- 154.Veeramachaneni GK, Thunuguntla V, Bhaswant M, Mathai ML, Bondili JS. Pharmacophore Directed Screening of Agonistic Natural Molecules Showing Affinity to 5HT2C Receptor. Biomolecules. 2019; 9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ahmed A, Choo H, Cho YS, Park WK, Pae AN. Identification of novel serotonin 2C receptor ligands by sequential virtual screening. Bioorg Med Chem. 2009; 17(13): 4559–68. [DOI] [PubMed] [Google Scholar]
- 156.Zhou Y, Ma J, Lin X, Huang XP, Wu K, Huang N. Structure-Based Discovery of Novel and Selective 5-Hydroxytryptamine 2B Receptor Antagonists for the Treatment of Irritable Bowel Syndrome. J Med Chem. 2016; 59(2): 707–20. [DOI] [PubMed] [Google Scholar]
- 157.Kozikowski AP, Cho SJ, Jensen NH, Allen JA, Svennebring AM, Roth BL. HTS and rational drug design to generate a class of 5-HT(2C)-selective ligands for possible use in schizophrenia. ChemMedChem. 2010; 5(8): 1221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science. 2017; 358(6361): 381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lyu J, Wang S, Balius TE, Singh I, Levit A, Moroz YS, et al. Ultra-large library docking for discovering new chemotypes. Nature. 2019; 566(7743): 224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stein RM, Kang HJ, McCorvy JD, Glatfelter GC, Jones AJ, Che T, et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature. 2020; 579(7800): 609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Alon A, Lyu J, Braz JM, Tummino TA, Craik V, O’Meara MJ, et al. Structures of the sigma2 receptor enable docking for bioactive ligand discovery. Nature. 2021; 600(7890): 759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Perez-Aguilar JM, Shan J, LeVine MV, Khelashvili G, Weinstein H. A functional selectivity mechanism at the serotonin-2A GPCR involves ligand-dependent conformations of intracellular loop 2. J Am Chem Soc. 2014; 136(45): 16044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gonzalez-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, et al. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007; 53(3): 439–52. [DOI] [PubMed] [Google Scholar]
- 164.Marti-Solano M, Iglesias A, de Fabritiis G, Sanz F, Brea J, Loza MI, et al. Detection of new biased agonists for the serotonin 5-HT2A receptor: modeling and experimental validation. Mol Pharmacol. 2015; 87(4): 740–6. [DOI] [PubMed] [Google Scholar]
- 165.Shan J, Khelashvili G, Mondal S, Mehler EL, Weinstein H. Ligand-dependent conformations and dynamics of the serotonin 5-HT(2A) receptor determine its activation and membrane-driven oligomerization properties. PLoS Comput Biol. 2012; 8(4): e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim SK, Li Y, Abrol R, Heo J, Goddard WA 3rd. Predicted structures and dynamics for agonists and antagonists bound to serotonin 5-HT2B and 5-HT2C receptors. J Chem Inf Model. 2011; 51(2): 420–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tran KT, Pallesen JS, Solbak SMO, Narayanan D, Baig A, Zang J, et al. A Comparative Assessment Study of Known Small-Molecule Keap1-Nrf2 Protein-Protein Interaction Inhibitors: Chemical Synthesis, Binding Properties, and Cellular Activity. J Med Chem. 2019; 62(17): 8028–52. [DOI] [PubMed] [Google Scholar]
- 168.Canal CE, Morgan D. Head-twitch response in rodents induced by the hallucinogen 2,5-dimethoxy-4-iodoamphetamine: a comprehensive history, a re-evaluation of mechanisms, and its utility as a model. Drug Test Anal. 2012; 4(7–8): 556–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Brandt SD, Kavanagh PV, Twamley B, Westphal F, Elliott SP, Wallach J, et al. Return of the lysergamides. Part IV: Analytical and pharmacological characterization of lysergic acid morpholide (LSM-775). Drug Test Anal. 2018; 10(2): 310–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tan PZ, Baldwin RM, Van Dyck CH, Al-Tikriti M, Roth B, Khan N, et al. Characterization of radioactive metabolites of 5-HT2A receptor PET ligand [18F]altanserin in human and rodent. Nucl Med Biol. 1999; 26(6): 601–8. [DOI] [PubMed] [Google Scholar]
- 171.Wainscott DB, Lucaites VL, Kursar JD, Baez M, Nelson DL. Pharmacologic characterization of the human 5-hydroxytryptamine2B receptor: evidence for species differences. J Pharmacol Exp Ther. 1996; 276(2): 720–7. [PubMed] [Google Scholar]
- 172.Cussac D, Newman-Tancredi A, Quentric Y, Carpentier N, Poissonnet G, Parmentier JG, et al. Characterization of phospholipase C activity at h5-HT2C compared with h5-HT2B receptors: influence of novel ligands upon membrane-bound levels of [3H]phosphatidylinositols. Naunyn Schmiedebergs Arch Pharmacol. 2002; 365(3): 242–52. [DOI] [PubMed] [Google Scholar]
- 173.Leysen JE. Use of 5-HT Receptor Agonists and Antagonists for the Characterization of Their Respective Receptor Sites. In: Drugs as Tools in Neurotransmitter Research. Humana Press, 1989. [Google Scholar]
- 174.Domenech T, Beleta J, Palacios JM. Characterization of human serotonin 1D and 1B receptors using [3H]-GR-125743, a novel radiolabelled serotonin 5HT1D/1B receptor antagonist. Naunyn Schmiedebergs Arch Pharmacol. 1997; 356(3): 328–34. [DOI] [PubMed] [Google Scholar]
- 175.Zgombick JM, Schechter LE, Macchi M, Hartig PR, Branchek TA, Weinshank RL. Human gene S31 encodes the pharmacologically defined serotonin 5-hydroxytryptamine1E receptor. Mol Pharmacol. 1992; 42(2): 180–5. [PubMed] [Google Scholar]
- 176.Adham N, Kao HT, Schecter LE, Bard J, Olsen M, Urquhart D, et al. Cloning of another human serotonin receptor (5-HT1F): a fifth 5-HT1 receptor subtype coupled to the inhibition of adenylate cyclase. Proc Natl Acad Sci U S A 1993; 90(2): 408–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl). 1996; 124(1–2): 57–73. [DOI] [PubMed] [Google Scholar]
- 178.Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, et al. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr. 1998; 178: 440–66. [PubMed] [Google Scholar]
- 179.Gerald C, Adham N, Kao HT, Olsen MA, Laz TM, Schechter LE, et al. The 5-HT4 receptor: molecular cloning and pharmacological characterization of two splice variants. EMBO J. 1995; 14(12): 2806–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Thomas DR, Gittins SA, Collin LL, Middlemiss DN, Riley G, Hagan J, et al. Functional characterisation of the human cloned 5-HT7 receptor (long form); antagonist profile of SB-258719. Br J Pharmacol. 1998; 124(6): 1300–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jasper JR, Kosaka A, To ZP, Chang DJ, Eglen RM. Cloning, expression and pharmacology of a truncated splice variant of the human 5-HT7 receptor (h5-HT7b). Br J Pharmacol. 1997; 122(1): 126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Corradetti R, Mlinar B, Falsini C, Pugliese AM, Cilia A, Destefani C, et al. Differential effects of the 5-hydroxytryptamine (5-HT)1A receptor inverse agonists Rec 27/0224 and Rec 27/0074 on electrophysiological responses to 5-HT1A receptor activation in rat dorsal raphe nucleus and hippocampus in vitro. J Pharmacol Exp Ther. 2005; 315(1): 109–17. [DOI] [PubMed] [Google Scholar]
- 183.Megens AA, Leysen JE, Awouters FH, Niemegeers CJ. Further validation of in vivo and in vitro pharmacological procedures for assessing the alpha 2/alpha 1-selectivity of test compounds: (1). Alpha-adrenoceptor antagonists. Eur J Pharmacol. 1986; 129(1–2): 49–55. [DOI] [PubMed] [Google Scholar]
- 184.Weinshank RL, Zgombick JM, Macchi M, Adham N, Lichtblau H, Branchek TA, et al. Cloning, expression, and pharmacological characterization of a human alpha 2B-adrenergic receptor. Mol Pharmacol. 1990; 38(5): 681–8. [PubMed] [Google Scholar]
- 185.Sunahara RK, Guan HC, O’Dowd BF, Seeman P, Laurier LG, Ng G, et al. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature. 1991; 350(6319): 614–9. [DOI] [PubMed] [Google Scholar]
- 186.Largent BL, Gundlach AL, Snyder SH. Psychotomimetic opiate receptors labeled and visualized with (+)-[3H]3-(3-hydroxyphenyl)-N-(1-propyl)piperidine. Proc Natl Acad Sci U S A. 1984; 81(15): 4983–7. [DOI] [PMC free article] [PubMed] [Google Scholar]