


Abstract
Deep learning techniques have significantly advanced the field of protein structure prediction. LOMETS3 (https://zhanglab.ccmb.med.umich.edu/LOMETS/) is a new generation meta-server approach to template-based protein structure prediction and function annotation, which integrates newly developed deep learning threading methods. For the first time, we have extended LOMETS3 to handle multi-domain proteins and to construct full-length models with gradient-based optimizations. Starting from a FASTA-formatted sequence, LOMETS3 performs four steps of domain boundary prediction, domain-level template identification, full-length template/model assembly and structure-based function prediction. The output of LOMETS3 contains (i) top-ranked templates from LOMETS3 and its component threading programs, (ii) up to 5 full-length structure models constructed by L-BFGS (limited-memory Broyden–Fletcher–Goldfarb–Shanno algorithm) optimization, (iii) the 10 closest Protein Data Bank (PDB) structures to the target, (iv) structure-based functional predictions, (v) domain partition and assembly results, and (vi) the domain-level threading results, including items (i)–(iii) for each identified domain. LOMETS3 was tested in large-scale benchmarks and the blind CASP14 (14th Critical Assessment of Structure Prediction) experiment, where the overall template recognition and function prediction accuracy is significantly beyond its predecessors and other state-of-the-art threading approaches, especially for hard targets without homologous templates in the PDB. Based on the improved developments, LOMETS3 should help significantly advance the capability of broader biomedical community for template-based protein structure and function modelling.
Graphical Abstract
Graphical Abstract.
LOMETS3 is a new generation meta-server that integrates deep learning techniques with multiple threading alignments for template-based multi-domain protein structure prediction and function annotation.
INTRODUCTION
The rapid progress of deep learning-based protein structure prediction (1–4), especially the recently developed end-to-end training by AlphaFold2 (5), has dramatically advanced the field of protein structure prediction (6). Nevertheless, the template-based modelling (TBM) (7–9), which builds models from homologous structures identified from the Protein Data Bank (PDB) and has dominated the field for many decades (10), remains an important approach to protein structure prediction. Compared to the deep learning-based approaches, which are built on models that are largely a black box to both users and developers, one advantage of TBM is that the modelling procedure is transparent with clear template resource and sequence–structure alignments, which are important for users to interpret the modelling results and extract functional insights from them. Meanwhile, for many targets with reliable template alignments from the PDB, TBM can build models of comparable and even higher accuracy than the deep learning approaches (11). For this reason, TBM is still contained as a necessary module in many state-of-the-art deep learning approaches and shows important effects on improving the accuracy of final models based on such pipelines (4,5,12). On the other hand, deep learning has also been integrated in many of the threading approaches to increase template recognition and alignment accuracy (13–16).
Given the ongoing importance of the TBM, we developed a meta-threading approach, LOMETS (17), which collects and combines template alignments from multiple individual threading programs for TBM-based protein structure prediction. Since it takes advantages of pooled and complementary information from multiple threading algorithms, LOMETS outperforms individual threading programs in both alignment accuracy and template coverage. Due to the robustness of the results and user-friendly server design, LOMETS has been widely used and successfully completed over 30 000 modelling requests submitted by around 15 000 users between 2007 and 2018. As an improved version, LOMETS2 (18) integrated profiles from deep multiple sequence alignment (MSA) with a new deep learning contact-based threading approach, CEthreader (13), and generated much more accurate templates and models than the previous LOMETS server. Consequently, the number of completed jobs per year by LOMETS2 has increased by 69%, compared with the previous version of server. Given the rapid development of deep learning techniques in the field, however, the previous LOMETS and LOMETS2 servers no longer represent the state of the art, because a number of newly developed, advanced deep learning threading algorithms have yet to be incorporated. In addition, the template ranking and selection in LOMETS/LOMETS2 were mainly based on consensus, which may not work well when combining alignments from different profile and deep learning threading approaches. Third, all existing threading methods, including LOMETS and LOMETS2, are unable to effectively handle multi-domain proteins, although many submitted sequences from the community contain multiple domains. Therefore, an upgraded version of the server integrating new deep learning technology and with the ability to model multi-domain sequences is urgently needed.
In this work, we developed a significantly improved server, LOMETS3, which introduces five major extensions: (i) a new set of deep learning threading approaches is integrated with advanced profile-based threading programs that constitute a comprehensive set of state-of-the-art threading programs; (ii) a new deep learning program, DeepPotential (19), is extended to generate multiple geometric descriptor predictions and used to assist ranking and selection of templates; (iii) the server now adopts a newly developed domain partition/assembly module for modelling multi-domain protein sequences; (iv) a limited-memory Broyden–Fletcher–Goldfarb–Shanno algorithm-based (L-BFGS) protein folding system has been introduced to create models guided by DeepPotential and threading template spatial restraints; and (v) the COFACTOR (20) pipeline in combination with LOMETS3 threading templates and structural analogues is applied to predict protein functions. Large-scale benchmark and blind tests showed that these new developments have significantly improved the overall accuracy for template detection, structure modelling and function prediction, relative to earlier LOMETS versions or other comparable structure prediction servers.
MATERIALS AND METHODS
Pipeline of LOMETS3 server
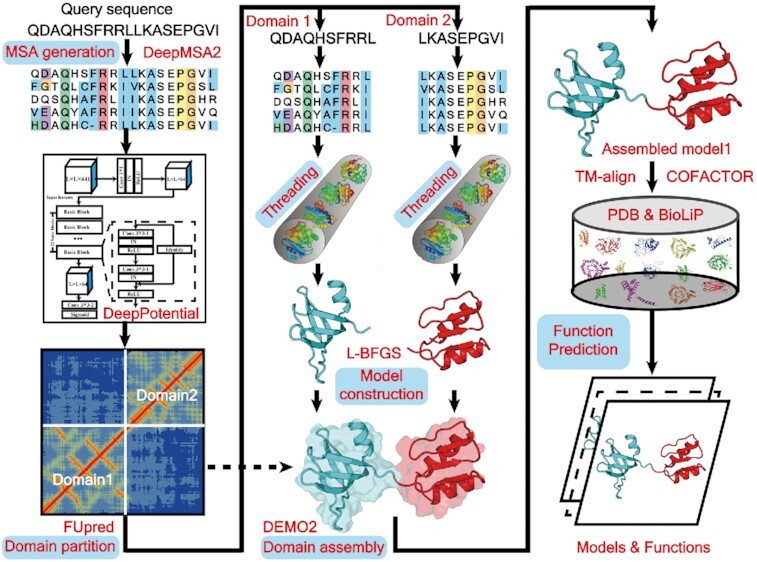
LOMETS3 is an automated meta-threading protein structure prediction pipeline for both single- and multi-domain proteins (Figure 1). For a full-length target (Figure 1A), deep MSAs are generated by iterative sequence homology searches through multiple metagenome sequence databases (see Supplementary Text S1 for details). Then, the MSAs are fed into DeepPotential (see Supplementary Text S1 for details) to predict contact map, and into the threading pipeline to detect full-length templates. FUpred (21) and ThreaDom (22) are used for domain boundary prediction based on the predicted contact map and threading templates, respectively (see Supplementary Text S2 for details). Next, following the domain splitting process, the domain-level sequences are passed into DeepPotential and the basic threading unit again for domain-level threading and model construction. Individual domain-level templates and models are then assembled into ‘full-length’ templates and full-length models by DEMO2 (23) using the deep learning-predicted inter-domain distance restraints from DeepPotential (see Supplementary Text S2 for details). FG-MD (24) and FASPR (25) are used to refine the global topology and re-pack the side-chain conformation for both domain-level models and full-length models. Finally, structural analogues are detected by TM-align (26) by matching the first LOMETS3 model to all structures in the PDB library, where functional insights [including Gene Ontology (GO) term, Enzyme Commission (EC) number and ligand binding interaction] are derived from the BioLiP (27) database based on the structural analogues. Furthermore, COFACTOR is extended by integrating the LOMETS3 threading templates associated with structural analogues to predict protein functions (see Supplementary Text S3 for details).
Figure 1.

Flowchart of the LOMETS3 server pipeline for protein structure and function prediction. (A) Procedure for automated multi-domain threading, domain-level structure assembly and function prediction. (B) Fundamental threading unit for template detection and function annotation of a single-domain target (embedded as needed in multi-domain threading, as shown).
In Figure 1B, we display in detail the basic threading unit that is designed for modelling single-domain proteins or isolated domains segmented from multi-domain proteins. In this unit, the deep MSAs are feed into five contact-based [CEthreader (13), DisCovER (14), EigenTHREADER (15), Hybrid-CEthreader (13) and MapAlign (16)] and six profile-based [FFAS3D (28), HHpred (29), HHsearch (8), MRFsearch (30), MUSTER (9) and SparksX (31)] threading programs (see Supplementary Text S4 for details) to identify structural templates from the PDB library. The MSAs are also used by DeepPotential to predict residue–residue contacts, distances and hydrogen bond restraints. These predicted spatial restraints along with the profile alignment score from original threading programs are used to re-rank the templates detected by the six profile-based threading programs (see Equation S9 in Supplementary Text S5). The top-ranked templates from each threading programs are pooled and sorted by a new scoring function that combines the alignment Z-score, program-specific confidence scores and the sequence identity to the query (Equation S17 in Supplementary Text S5), for template output. In addition, up to five full-length models are constructed by L-BFGS optimization using the distance restraints predicted by DeepPotential and calculated from top threading template alignments (see Supplementary Text S6 for details).
New developments in LOMETS3
Compared to the previous LOMETS servers, the following major updates have been made.
Extension of MSA construction
An updated deep MSA approach, DeepMSA2, was developed to create deep sequence profiles from more extensive metagenome sequence databases than the previous MSA pipeline (Supplementary Figure S1). The deep profiles are then used in all component threading methods.
Updating of contact-based and profile-based threading methods
More than half of the threading programs in the previous LOMETS2 server were renewed and/or replaced by state-of-the-art methods, including deep learning techniques (Supplementary Text S4). Furthermore, a new scoring function, which combines residue–residue distances, contacts and hydrogen bond geometries predicted from DeepPotential, as well as profile alignment scores, is used to re-rank the templates for profile-based threading methods.
Introduction of an L-BFGS system for full-length structure modelling
An L-BFGS folding system is introduced to quickly construct full-length structure models for target sequences based on spatial restraints predicted by DeepPotential and deduced from top threading templates. Furthermore, a new refinement pipeline based on FG-MD and FASPR is used to refine and re-pack the side-chain conformation of the models.
Addition of domain partition and assembly module
FUpred and ThreaDom are used for domain boundary prediction based on the predicted contact map and threading template alignments, respectively. The individual domain-level models and templates are then assembled into full-length models and templates by DEMO2 using deep learning-predicted inter-domain distance restraints.
Inclusion of function annotation and prediction
Structure-based function annotations (including GO term, EC number and ligand binding information), derived from the top LOMETS3 threading templates and protein structure analogues with the closest structural similarity to LOMETS3 models, are reported. Furthermore, COFACTOR is extended by integrating the LOMETS3 threading templates associated with structural analogues to predict three categories of protein functions. Given the newly developed components, the output page of the LOMETS3 server is completely redesigned to facilitate the display of function annotations and predictions.
Benchmark datasets
Three benchmark datasets have been constructed for testing the performance of LOMETS3.
Dataset-1: single-domain protein dataset
Six hundred fourteen non-redundant single-domain proteins were collected from the PDB with pairwise sequence identity <30%. Based on the quality of final threading templates and the consensuses of component threading templates (see details in Supplementary Text S5), the dataset can be classified into two groups: 403 easy targets and 211 hard targets.
Dataset-2: multi-domain protein dataset
The multi-domain dataset contains 408 non-redundant multi-domain proteins. Based on the domain types, this dataset can be further divided into 348 continuous domain proteins and 60 discontinuous domain proteins. Here, a discontinuous domain is defined as containing two or more segments from separate regions of the query sequence, while a continuous domain contains only one continuous segment. Based on the number of domains, our dataset contains 334 two-domain proteins, 50 three-domain proteins and 24 multi-domain proteins with more than three domains. Here, we take the SCOPe (32) domain boundary definition for those proteins as ground truth when assessing the domain partition accuracy. Based on the target types, 321 of these proteins are classified as easy targets and 87 as hard targets.
Dataset-3: function annotation dataset
In order to assess the performance of LOMETS3-driven function predictions, we extracted all ground truth function annotation information for 614 single-domain targets and 408 multi-domain targets from BioLiP database. In total, there are 688 proteins containing at least one type of function annotations, where 573 proteins are annotated with a total of 4061 GO terms [507 proteins have 1728 molecular function (MF) terms, 468 proteins have 1550 biological process (BP) terms and 302 proteins have 783 cellular component (CC) terms], 659 proteins are annotated with four-digit EC numbers and 296 proteins have annotated binding to 403 ligands.
During benchmarking of our threading method, a sequence identity cut-off (<30%) between the query protein and template sequences was enforced to remove any potential bias caused by homologous templates.
RESULTS
Comparison between LOMETS3 and LOMETS2 on single-domain proteins
In Supplementary Table S1, we list a comparison of threading results obtained by LOMETS3 and LOMETS2 for the 614 single-domain proteins from Dataset-1. Overall, the average TM-scores of the first templates identified by LOMETS3 were 0.656, 0.725 and 0.525 for all, easy and hard targets, which are 5%, 2% and 14% higher than those of the first templates from LOMETS2, corresponding to P-values of 1.7E−28, 6.9E−10 and 2.3E−19, respectively (one-tailed Student’s t-test). Figure 2A shows a head-to-head TM-score comparison of the full-length models by the two servers, where LOMETS3 generates higher TM-score models in 550 out of 614 cases (90%). On average, the TM-scores of the LOMETS3 full-length models are 0.814, 0.837 and 0.768, which were 22%, 11% and 55% higher than those by LOMETS2, corresponding to P-values of 4.3E−86, 6.6E−48 and 9.7E−36, for all, easy and hard targets, respectively (one-tailed Student’s t-test). Furthermore, LOMETS2 only generated 105 correct models with TM-score >0.5 (33) for the 211 hard targets, while LOMETS3 can construct nearly twice the correct models (202), achieving success for nearly all targets in this category.
Figure 2.

Comparison between LOMETS3 and LOMETS2 on the 614 single-domain and 408 multi-domain proteins of our benchmarking datasets. (A) TM-scores of the full-length models from LOMETS3 versus those from LOMETS2 on single-domain proteins, where red crosses and blue circles correspond to hard and easy targets, respectively. (B) An illustrative example from pertussis toxin subunit 4 (PDB ID: 1prtD), showing models by L-BFGS, MODELLER and LOMETS3 (blue) overlaid with the experimental structure (yellow). (C) TM-scores of the final assembled models by LOMETS3 versus those built by MODELLER based on the full-chain threading templates. (D) An illustrative example from bifunctional polynucleotide phosphatase (PDB ID: 3zvmA), showing the structure superposition of the LOMETS3 template/model of domain 1 (red) and domain 2 (cyan) with the experimental structure (yellow). The dashed lines are the unaligned regions of the templates.
Several factors have contributed to the significant improvements of LOMETS3 over LOMETS2. First, five state-of-the-art contact-based threading methods have been integrated into LOMETS3, while LOMETS2 was dominated by profile-based programs with only one contact-based threading program, CEthreader, included. As shown in Supplementary Figure S2, the contact-based threading group generally performed better than the profile-based threading group on template identification, especially on the 211 hard targets, where the Hybrid-CEthreader program, which uses a hierarchical procedure to speed up the eigendecomposition and alignment process of CEthreader (Supplementary Text S4), shows the highest performance for both easy and hard targets. Thus, the integration of the contact-based threading methods is particularly important to improve the overall template recognition ability of LOMETS3.
Second, a new scoring function combining profile information and spatial restraints from deep learning has been utilized to re-rank the templates for profile-based threading methods, which can significantly improve the quality of the top templates selected for these programs. As shown in Supplementary Table S2, the TM-score of the first templates after re-ranking is generally higher than that of the original templates obtained by the profile-based threading programs, where the difference is particularly prominent for the hard targets, with TM-score improved by 18–44%. Here, one concern for template re-ranking is that it may reduce the complementariness of the profile-based programs with contact-based program, since both have now used DeepPotential restraints. As shown in Supplementary Table S3, however, the average TM-score of the first templates identified by LOMETS3 was significantly higher than that by the best component threading method, Hybrid-CEthreader, with a P-value of 3.8E−13 (one-tailed Student’s t-test), demonstrating that the template ranking scheme in LOMETS3 is able to take advantage of the complementation between contact-based and profile-based component threading programs.
Finally, the newly developed L-BFGS folding system further improved the quality of the full-length models by integrating spatial restraints from DeepPotential with the threading templates. As shown in Supplementary Table S4, even using the same set of templates, LOMETS3 with L-BFGS generated models with TM-score (0.814) 17.1% higher than that for models built on MODELLER (34), an approach used in LOMETS2 that did not use deep learning restraints. Meanwhile, the results showed that the template alignment information also helps in the full-length model construction, where the models by L-BFGS with DeepPotential but without using templates have the TM-score (0.797) significantly lower than that with using templates, with P-value = 6.6E−55. In Figure 2B, we showed an illustrative example from pertussis toxin subunit 4 (PDB ID: 1prtD), where L-BFGS + DeepPotential generated a model with a poor TM-score of 0.34, due to an incorrect distance map prediction from DeepPotential (Supplementary Figure S3A). However, the model built by MODELLER with threading information had an improved TM-score of 0.61, indicating that better spatial restraints can be extracted from the LOMETS3 threading templates. As a result, the LOMETS3 server generates the best model with a TM-score of 0.69, due to optimal combination of spatial restraints from deep learning and threading alignments (Supplementary Figure S3B).
Performance of LOMETS3 on multi-domain proteins
A new domain partition and assembly module has been developed and optimized for LOMETS3, which allows the server to deal effectively with the threading and domain assembly of multi-domain proteins for the first time. As shown in Figure 1, the multi-domain modelling procedure consists of three steps of domain boundary prediction, domain-level template recognition, and full-length model construction and assembly.
First, we examine the domain boundary prediction accuracy of LOMETS3, which is a combination of two complementary predictors: FUpred and ThreaDom. As shown in Supplementary Figure S4, the average domain boundary distance score (35) of LOMETS3, which is defined based on the distance between the predicted and the true domain boundaries along the sequence, was 8% and 16% higher than those of FUpred and ThreaDom, and 76% and 191% higher than those of two third-party deep learning-based domain partition methods, ConDo (36) and DoBo (37), respectively. To examine the effect of domain-level template recognitions, we list in Supplementary Table S5 a TM-score comparison of the full-chain threading templates versus the templates assembled from the domain-level threading templates. It is shown that the average TM-scores of the assembled templates are 0.65 and 0.55 for individual domain and full-chain sequence, respectively, which are 30% and 8% higher than those obtained by the traditional full-chain threading approaches, demonstrating the utility of the domain-level threading and assembly procedure for multi-domain proteins.
In Figure 2C, we list a head-to-head TM-score comparison of the full-length models obtained by LOMETS3 based on domain partition and assembly versus that obtained by MODELLER built on the full-chain threading templates, a protocol similar to LOMETS2 but using LOMETS3 threading programs. Out of the 408 cases, the domain-based threading/assembly approach achieved a higher TM-score in 341 cases, while the full-chain threading did so only in 67 cases. Overall, the average TM-score of the domain-based assembled models is 23% higher than that by full-chain threading (P-values = 5.4E−45, one-tailed Student’s t-test).
Figure 2D shows an example from bifunctional polynucleotide phosphatase (PDB ID: 3zvmA), which is a two-domain protein. For full-chain threading, the first template has a poor TM-score of 0.29, whereas the full-length model built by MODELLER has a TM-score of 0.33. When using the domain threading/assembly approach, the full-chain template assembled from the domain-level templates has a TM-score = 0.66. After the L-BFGS simulation, LOMETS3 obtained a final model of TM-score = 0.91. Thus, we see that both domain split/assembly and DeepPotential-guided structural simulations contribute to the significantly improved model quality for the multi-domain protein structure modelling.
Comparison between LOMETS3 and other structure modelling servers
To examine the quality of LOMETS3 with other approaches, we compared LOMETS3 with two widely used protein structure modelling servers, including a threading-based method, HHpred (29), and a deep learning-based method, trRosetta (38). We note that the comparison with other fully automated servers is essential to fairly test the performance that is likely to be achieved by LOMETS3 and other comparable methods for average, non-expert users. For LOMETS3 and HHpred, the same sequence identity cut-off (<30%) between the query protein and template sequences was utilized to exclude the homologous templates. Figure 3A and B shows the TM-score of the three methods on the 614 single-domain and 408 multi-domain proteins of our Dataset-1 and Dataset-2, respectively. Overall, the average TM-score of LOMETS3 was 8% (10%) and 26% (17%) higher than those of trRosetta and HHpred on the single-domain (multi-domain) targets, respectively. Notably, for the 211 single-domain hard targets (87 multi-domain hard targets), LOMETS3 performed 8% (12%) better than trRosetta, although trRosetta was designed for deep learning-based template-free modelling. These results demonstrated the advancement of the hybrid TBM and deep learning approached adopted by the LOMETS3 server.
Figure 3.

The comparison between LOMETS3 and other structural modelling servers. The TM-scores of full-length models predicted by LOMETS3, HHpred and trRosetta for (A) single- and (B) multi-domain proteins for our benchmarking datasets. (C) Case study of four targets for which LOMETS3 generated better models than other servers in CASP14, where the LOMETS3 models (blue) and the second-best server models (red) are overlaid with the experimental structures (yellow).
In a recent study, DeepMind released AlphaFold2 (5), which was found to be the highest-performing structure prediction method in the 14th Critical Assessment of Structure Prediction (CASP14) experiment. On the 614 single-domain and 408 multi-domain proteins of our Dataset-1 and Dataset-2, AlphaFold2 achieved impressive TM-scores of 0.921 and 0.841, respectively. These are considerably higher than those of LOMETS3 that has average TM-scores of 0.814 and 0.669, respectively, after excluding close homologous templates. Interestingly, we found that AlphaFold2 has 4% targets with a lower TM-score than LOMETS3 on the 1022 benchmark proteins in our dataset (see examples in Supplementary Figure S5). However, without experimental structures, it is uncertain in which cases LOMETS3 can perform better than AlphaFold2. Thus, how to identify those cases in practice to allow the user to pick a LOMETS3 model instead of an AlphaFold2 model is the subject of future research. Note that the AlphaFold2 algorithm also includes a threading component based on HHsearch. To examine the effectiveness of LOMETS3, we modified AlphaFold2 by replacing the threading component with LOMETS3, and compared it with default AlphaFold2 based on HHsearch threading. As shown in Supplementary Table S6, we found that the AlphaFold2 with LOMETS3 had a consistently higher TM-score than AlphaFold2 using the default HHsearch on the 614 single-domain proteins, showing a consistent improvement (P-value = 4.5E−18, Student’s t-test). These results demonstrate the usefulness and complementarity of the LOMETS3 compared to the state-of-the-art end-to-end deep learning algorithm, both in terms of providing better predictions for some targets and in terms of improving Alphafold2 performance over an even wider variety of structures.
An early version of LOMETS3, which did not contain the automated domain partition/assembly module and template re-ranking algorithm, participated (as ‘Zhang-TBM’) in the CASP14 experiment. It was ranked as the fifth structure prediction algorithm (Supplementary Figure S6), following three other Zhang Lab servers and ‘BAKER-ROSETTASERVER’ in the automated server section of CASP14 (https://www.predictioncenter.org/casp14/zscores_final.cgi?gr_type=server_only). Figure 3C presents four representative targets for which LOMETS3 generated a better model than all other servers, excluding the ‘Zhang-Server’ and ‘QUARK’ servers (both of which used LOMETS3 as a component module). These four targets belong to ‘FM’, ‘FM/TBM’ or ‘TBM-hard’ categories, which are roughly consistent with hard targets defined by LOMETS3. The highlighted areas represent the regions where LOMETS3 models are closer to the native structure than the second-best server models, which result in the overall higher TM-score. These results demonstrated again the advantage of LOMETS3 on modelling the non-homologous targets when coupling TBM with deep learning potentials.
Performance of LOMETS3 on protein function prediction
Protein functions in LOMETS3 are predicted by an extended version of the COFACTOR (20) pipeline, which integrates LOMETS3 threading templates and structural analogues that searched through the BioLiP protein function database for identifying functional information. Function annotations provided by LOMETS3 include GO, EC and ligand binding sites (LBS), where GO is further categorized into three sub-aspects of MF, BP and CC.
To examine the quality of LOMETS3 function prediction, we compared LOMETS3 with an AlphaFold2-based function annotation pipeline, which detects functional templates by structurally matching the AlphaFold2 model to all proteins in the PDB/BioLiP library based on TM-align search, and derives functional annotations from the BioLiP database based on the structural analogues. The function prediction performance is assessed by the F1-score, which is defined as the harmonic average between precision and recall.
Figure 4A shows the F1-scores of the two methods on the 1728 MF, 1550 BP and 783 CC GO terms of our Dataset-3. When calculating the F1-score for GO terms, all parent terms of the annotated GO terms are also considered as annotations for each target. We found that the extended combination of LOMETS3 and COFACTOR outperforms AlphaFold2 on all three sub-aspects of GO terms. Similarly, Figure 4B lists the comparison results on the 659 EC numbers of Dataset-3. Each EC number is a code of four numbers, where the first digit defines the general type of reaction catalysed by the enzyme and ranges from 1 to 6, and each subsequent digit indicates a subclass of the previous digit. The data again show a significant advantage of the LOMETS3 over the AlphaFold2 EC annotation. In particular, the F1-score for the first digit of EC number for LOMETS3 is 0.98, while that for AlphaFold2 is just 0.88. In Figure 4C, we compare the two methods on 403 binding ligands of Dataset-3. The F1-score of LOMETS3 is also considerably higher than that of AlphaFold2 (0.28 versus 0.20). These results suggest that LOMETS3 can provide high-quality function predictions beyond those obtained even from the highest accuracy available structural models, probably because of the combination of multiple sources of functional modelling pipelines in LOMETS3/COFACTOR (see Supplementary Text S3).
Figure 4.

Comparison between LOMETS3 and AlphaFold2 on the function prediction. (A) The F1-scores of the two methods on the 1728 MF, 1550 BP and 783 CC GO terms of Dataset-3. (B) The F1-scores of the two methods on the 659 EC numbers of Dataset-3. Each EC number is a code of four numbers, where the first digit defines the general type of reaction catalysed by the enzyme and ranges from 1 to 6, and each subsequent digit indicates a subclass of the previous digit. (C) The F1-scores of the two methods on 403 binding ligands of Dataset-3.
WEB SERVER
Server input
The input to the LOMETS3 server is a single-chain amino acid sequence in FASTA format. After making a submission, a URL link with a random job ID will be created and allows the user to check the results and keep the data privacy. The user is encouraged (but not required) to provide an email address when submitting a job. LOMETS3 server will automatically send a notification email with a link to the Results page upon job completion. Three advanced options are provided to allow users to (i) either keep all templates or remove the homologous templates (sequence identity >30%) for benchmarking, (ii) either use the automated domain partition or not, and (iii) either predict protein functions or not. Here, the second option is mainly designed in case the user knows that the input sequence is a single-domain protein. By selecting ‘run threading without domain partition’ option, the system will ignore the domain partition step and only run one-round full-chain threading, which can avoid false-positive domain prediction and make the job complete faster. If the third option (‘function prediction’) is selected, the job will need an additional 1–10 h to finish.
Server output
The user will receive the results within 24 h after submitting a job if the length of deposited sequence is <350 residues (Supplementary Figure S7). Generally, a larger protein takes longer time, and a multi-domain protein usually require twice the runtime of a single-domain protein with the same protein size. Additionally, the actual server response time is equal to the sum of the job pending time and running time. Thus, if too many sequences are accumulated in the queue, the job pending time may be longer.
The LOMETS3 Results page consists of nine sections, including (i) summary of the user input, including sequence and predicted secondary structure and solvent accessibility (Supplementary Figure S8A), (ii) spatial restraints predicted by the deep learning method DeepPotential (Supplementary Figure S8B), (iii) domain partition results and summary of individual domain threading results (Figure 5A), (iv) summary of the top 10 domain-level assembled template alignments (Figure 5B), (v) summary of the top 10 full-chain threading templates (Figure 5C), (vi) up to 5 full-length models (Figure 5D), (vii) the top 10 closest structural analogues identified from the PDB, along with the associated function annotations (Figure 5E), (viii) function predictions by COFACTOR with LOMETS3 threading templates and structural analogues (Figure 5F), and (ix) the top templates from the component threading programs and the associated function annotations (Supplementary Figure S9). In the following, we go into some details on sections (iii)–(ix), which are new additions to the LOMETS3 server. The example presented in Figure 5 and Supplementary Figure S9 comes from the bifunctional polynucleotide phosphatase (PDB ID: 3zvmA) run with ‘remove templates sharing >30% sequence identity with target’ and ‘run domain partition and assembly’ options selected.
Figure 5.

Illustration of the LOMETS3 server output, including (A) domain partition results and summary of individual domain threading results, (B) summary of the top 10 domain-level assembled template alignments, (C) summary of the top 10 full-chain threading template alignments identified by LOMETS3, (D) up to 5 full-length models, (E) top 10 closest structure analogues in PDB and the associated function annotations, and (F) function predictions from COFACTOR with LOMETS3 threading templates and structural analogues.
Figure 5A shows the domain partition results of FUpred and LOMETS3 (section iii). Three images, including contact map associated with the predicted domain boundary, FUpred continuous domain scoring function and discontinuous domain scoring function, are listed in the FUpred results panel. In particular, the different domains are depicted by different colours in the contact map in order to highlight the domain partition results. Following that, a table that summarizes the LOMETS3 domain partition is shown, and the table contains the number of domains, the boundary and length of each domain, and the sequence of each domain. In each row, users can click the image or the hyperlink of the domain to check the modelling results of each domain in an additional output page, which contains sections (i), (ii), (v)–(vii) and (ix). Section (iii) is optional for multi-domain proteins. If the target is defined as ‘easy’ and the alignment coverages of top five ranked templates are all >0.8, the target will be modelled without domain partition since the full-chain threading can generate reliable models directly.
Figure 5B and C lists the top 10 assembled templates (section iv) and full-chain threading templates (section v) identified by LOMETS3, respectively. Here, the presented templates are assembled from the top-ranked domain-level templates by DEMO2, and thus section (iv) is only available for multi-domain targets. For each template in sections (iv) and (v), the left panel displays the template’s PDB ID (only in section v), contact map overlapping score (CMO), mean absolute distance error (MAE), sequence identity to the query, alignment coverage, normalized Z-score and the threading algorithm. Two PDB IDs and corresponding hyperlinks are provided, where the domain-level PDBs are templates used in the LOMETS3 library, and the full-length PDBs are linked to the original RCSB PDB. CMO is the contact map overlapping score (see Equation S13 in Supplementary Data) between template contact map and predicted query contact map, and MAE is the mean absolute distance error (see Equation S11 in Supplementary Data) between template distance map and predicted query distance map. The normalized Z-score (see Equation S9 in Supplementary Data) shows the significance of the template alignment, where the normalized Z-score ≥1 indicates a good alignment by the corresponding threading program. The right panel of sections (iv) and (v) shows query–template alignments by LOMETS3 associated with the predicted secondary structure and solvent accessibility. In the bottom of section (v), 3D structures of the templates, together with the threading methods, are provided.
Figure 5D shows up to five full-length models generated by LOMETS3 (section vi). The user can drag, rotate or zoom in on the structure showed in the figures, and the PDB-formatted files storing the model coordinates can be downloaded from the links under the figures.
Figure 5E presents the top 10 closest protein structures in the PDB to the LOMETS3 first model, along with functional annotations derived from those structures (section vii). The table provides detailed information on the structural analogues identified by TM-align, including the template ID, TM-score, root-mean-square deviation (RMSD), sequence identity and alignment coverage. In addition, links for downloading the superposed structures and pairwise sequence alignments are provided in the same table. Below the table, the function annotations for those aligned structural analogues are shown, where the functions obtained from the BioLiP database (27) include three aspects: GO terms, EC numbers and LBS. In detail, MF, BP, CC GO terms, four-digit EC number, three-letter ligand code and the LBS information are listed in this table, and hyperlinked to QuickGO (39) Gene Ontology, the ExPASy (40) ENZYME database or the BioLiP database, as appropriate. We note that the results of section (vii) can be different from those of sections (iv) and (v), because the structural analogues in section (vii) are detected based on predicted model structures through TM-align, while the templates in sections (iv) and (v) are detected based only on the query sequence via the LOMETS3 threading unit.
Figure 5F provides an example of the results of function predictions by the LOMETS3 built on the extended COFACTOR pipeline (section viii). The first three panels show the consensus GO prediction results in three aspects of the MF, BP and CC. The predicted GO terms are displayed together with their parent terms as a directed acyclic graph, where each GO term is highlighted with purple through red colours representing CscoreGO values on a scale from 0.4–0.5 to 0.9–1.0, respectively. Only the GO terms with CscoreGO ≥ 0.4 are displayed, although the full prediction result is available for download from a link in the right table. Furthermore, the right table lists predicted GO terms, along with CscoreGO and common names for those terms. The fourth panel shows the top five EC number prediction results, each displayed alongside the template structure with predicted active sites. The right table lists the predicted EC number, CscoreEC, PDB ID, TM-score, RMSD, sequence identity, coverage and predicted active sites, respectively. The last panel shows the ligand binding prediction results. For each template, the positions of the LBS residues are highlighted. The right table lists the three-digit ligand code and predicted binding site, together with CscoreLB, PDB ID, TM-score, RMSD, sequence identity, coverage and local similarity score. The predicted ligand binding complex can be downloaded from the links in the right table.
Supplementary Figure S9 displays the templates and corresponding function annotations from the component threading programs (section ix). In each program, up to 10 templates associated with the pairwise alignment information (template ID, CMO, MAE, Z-score, etc.) and the function annotations (GO, EC and LBS) are displayed.
CONCLUSION
We have proposed LOMETS3 as a significant extension of the previous LOMETS2 meta-threading server for template-based protein structure prediction and function annotation. In LOMETS3, a balanced set of start-of-the-art threading programs, including five deep learning contact-based and six profile-based methods, is integrated for multiple template identifications, and a new deep learning method, DeepPotential, was extended to create high-quality contact maps to guide the deep learning threading alignments. Meanwhile, the entire set of spatial potentials, including contact and distance maps and hydrogen-bonding potential, is used to re-rank and select templates from the profile-based threading alignments, in order to improve the accuracy of the later programs. To provide atomic models, which are usually needed for detailed function annotation and virtual screening, a gradient-based L-BFGS folding system has been developed to construct atomic full-length models guided by the restraints from deep learning models and threading alignments. In addition, protein functions are predicted from COFACTOR using a combination of LOMETS3 threading templates and structural analogues. For the first time, a domain split and assembly protocol was introduced to allow for the LOMETS3 server to effectively handle multi-domain protein structure prediction.
Large-scale benchmark tests indicated that the new implementations have significantly improved the quality and functionality of the LOMETS3 server relative to earlier LOMETS versions. On the 614 single-domain proteins in our benchmarking dataset, for example, the average TM-scores for the first template/full-length model are 5%/22% higher than those obtained by the previous LOMETS2. The new LOMETS3 server also outperforms the widely used third-party servers HHpred and trRosetta, with a TM-score increase by 26% and 8%, respectively; this advantage is mainly attributed to the efficient coupling of multiple template recognition and deep learning potentials in LOMETS3. Although LOMETS3 underperforms the record-holding method AlphaFold2, there are cases for which LOMETS3 generated higher TM-scores than AlphaFold2 (Supplementary Figure S5). Meanwhile, the incorporation of LOMETS3 threading alignments into AlphaFold2 can further improve the performance, indicating the usefulness and complementarity of LOMETS3 with the state-of-the-art deep learning approaches. For multi-domain proteins, the domain-level template threading and assembly process generated full-length models with a TM-score 23% higher than the traditional full-chain threading, demonstrating the effectiveness of the new protocol for multi-domain protein structure modelling. A former version of LOMETS3 was also tested (as ‘Zhang-TBM’) in the CASP14 and ranked as the second-best server, after excluding the other Zhang Lab servers that used the LOMETS3 models as the starting point in their pipelines. Furthermore, we tested the quality of LOMETS3 function predictions by comparing with an AlphaFold2-based function annotation pipeline. The benchmark results indicate that LOMETS3 outperforms AlphaFold2 model-based pipeline on protein function predictions, in terms of GO term, EC number and LBS prediction.
As an online server, in addition to high-quality modelling results, it is also important to consider the convenience of use, transparency of modelling process and ease of interpretability of the modelling results. In this regard, we have optimized the server interface that includes multiple advanced options to control homology cut-off and domain partition and assembly process. The output page contains now nine carefully designed sections to display detailed modelling results ranging from domain boundary prediction, deep learning potentials, domain- and chain-level threading alignments, and atomic full-length atomic models, as well as structure-based function annotations and predictions, including GO, EC and LBS predictions. These new web interface and output designs help significantly improve the transparency and interpretability of the LOMETS3 modelling results.
Despite the success, there is still room for further improvement of LOMETS3 in the future. For instance, the current deep learning information is mainly derived from DeepPotential, which was trained on a residual convolutional neural network system, and may not work well for the targets with few homologous sequences. A new approach utilizing attention-based MSA transformer coupled with microbiome-targeted metagenome MSA collections (41) might help in modelling such targets. Meanwhile, improved deep learning models will also help improve the accuracy of domain splitting and domain orientation assembly for large multi-domain proteins (e.g. >1000 residues), which are still challenging to LOMETS3 when the targets lack full-length template structures. Efforts along these lines will continue to enhance LOMETS as one of the most robust and useful TBM platforms to serve the broader biological community.
DATA AVAILABILITY
The web server and benchmark dataset are available at https://zhanglab.ccmb.med.umich.edu/LOMETS/.
Supplementary Material
ACKNOWLEDGEMENTS
This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (ACI-1548562).
Contributor Information
Wei Zheng, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Qiqige Wuyun, Department of Computer Science and Engineering, Michigan State University, East Lansing, MI 48824, USA.
Xiaogen Zhou, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Yang Li, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Peter L Freddolino, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA; Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Yang Zhang, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA; Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences [GM136422, S10OD026825]; National Institute of Allergy and Infectious Diseases [AI134678]; National Science Foundation [IIS1901191, DBI2030790, MTM2025426]. Funding for open access charge: National Institute of General Medical Sciences [GM136422].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang S., Sun S., Li Z., Zhang R., Xu J.. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol. 2017; 13:e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mortuza S.M., Zheng W., Zhang C., Li Y., Pearce R., Zhang Y.. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions. Nat. Commun. 2021; 12:5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zheng W., Zhang C., Li Y., Pearce R., Bell E.W., Zhang Y.. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods. 2021; 1:100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D.et al.. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021; 373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pearce R., Zhang Y.. Toward the solution of the protein structure prediction problem. J. Biol. Chem. 2021; 297:100870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bowie J.U., Lüthy R., Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991; 253:164–170. [DOI] [PubMed] [Google Scholar]
- 8. Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005; 21:951–960. [DOI] [PubMed] [Google Scholar]
- 9. Wu S., Zhang Y.. MUSTER: improving protein sequence profile–profile alignments by using multiple sources of structure information. Proteins. 2008; 72:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008; 18:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pereira J., Simpkin A.J., Hartmann M.D., Rigden D.J., Keegan R.M., Lupas A.N.. High-accuracy protein structure prediction in CASP14. Proteins. 2021; 89:1687–1699. [DOI] [PubMed] [Google Scholar]
- 12. Zheng W., Li Y., Zhang C., Zhou X., Pearce R., Bell E.W., Huang X., Zhang Y.. Protein structure prediction using deep learning distance and hydrogen-bonding restraints in CASP14. Proteins. 2021; 89:1734–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zheng W., Wuyun Q., Li Y., Mortuza S.M., Zhang C., Pearce R., Ruan J., Zhang Y.. Detecting distant-homology protein structures by aligning deep neural-network based contact maps. PLoS Comput. Biol. 2019; 15:e1007411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhattacharya S., Roche R., Moussad B., Bhattacharya D. DisCovER: distance- and orientation-based covariational threading for weakly homologous proteins. Proteins. 2022; 90:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buchan D.W.A., Jones D.T.. EigenTHREADER: analogous protein fold recognition by efficient contact map threading. Bioinformatics. 2017; 33:2684–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ovchinnikov S., Park H., Varghese N., Huang P.S., Pavlopoulos G.A., Kim D.E., Kamisetty H., Kyrpides N.C., Baker D. Protein structure determination using metagenome sequence data. Science. 2017; 355:294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu S., Zhang Y.. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007; 35:3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng W., Zhang C., Wuyun Q., Pearce R., Li Y., Zhang Y.. LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res. 2019; 47:W429–W436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y., Zhang C., Zheng W., Zhou X., Bell E.W., Yu D.J., Zhang Y.. Protein inter-residue contact and distance prediction by coupling complementary coevolution features with deep residual networks in CASP14. Proteins. 2021; 89:1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang C., Freddolino P.L., Zhang Y.. COFACTOR: improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 2017; 45:W291–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng W., Zhou X., Wuyun Q., Pearce R., Li Y., Zhang Y.. FUpred: detecting protein domains through deep-learning-based contact map prediction. Bioinformatics. 2020; 36:3749–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xue Z., Xu D., Wang Y., Zhang Y.. ThreaDom: extracting protein domain boundary information from multiple threading alignments. Bioinformatics. 2013; 29:i247–i256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou X., Hu J., Zhang C., Zhang G., Zhang Y.. Assembling multidomain protein structures through analogous global structural alignments. Proc. Natl Acad. Sci. U.S.A. 2019; 116:15930–15938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang J., Liang Y., Zhang Y.. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011; 19:1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang X., Pearce R., Zhang Y.. FASPR: an open-source tool for fast and accurate protein side-chain packing. Bioinformatics. 2020; 36:3758–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y., Skolnick J.. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005; 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., Roy A., Zhang Y.. BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2013; 41:D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu D., Jaroszewski L., Li Z., Godzik A.. FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics. 2014; 30:660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meier A., Söding J.. Automatic prediction of protein 3D structures by probabilistic multi-template homology modeling. PLoS Comput. Biol. 2015; 11:e1004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma J., Wang S., Wang Z., Xu J.. MRFalign: protein homology detection through alignment of Markov random fields. PLoS Comput. Biol. 2014; 10:e1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou H., Zhou Y.. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 2005; 58:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chandonia J.M., Fox N.K., Brenner S.E.. SCOPe: classification of large macromolecular structures in the structural classification of proteins—extended database. Nucleic Acids Res. 2019; 47:D475–D481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu J., Zhang Y.. How significant is a protein structure similarity with TM-score = 0.5?. Bioinformatics. 2010; 26:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Webb B., Sali A.. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics. 2016; 54:5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tress M., Cheng J., Baldi P., Joo K., Lee J., Seo J.H., Lee J., Baker D., Chivian D., Kim D.et al.. Assessment of predictions submitted for the CASP7 domain prediction category. Proteins. 2007; 69:137–151. [DOI] [PubMed] [Google Scholar]
- 36. Hong S.H., Joo K., Lee J.. ConDo: protein domain boundary prediction using coevolutionary information. Bioinformatics. 2019; 35:2411–2417. [DOI] [PubMed] [Google Scholar]
- 37. Eickholt J., Deng X., Cheng J.. DoBo: protein domain boundary prediction by integrating evolutionary signals and machine learning. BMC Bioinformatics. 2011; 12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang J., Anishchenko I., Park H., Peng Z., Ovchinnikov S., Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl Acad. Sci. U.S.A. 2020; 117:1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Binns D., Dimmer E., Huntley R., Barrell D., O’Donovan C., Apweiler R.. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics. 2009; 25:3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000; 28:304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang P., Zheng W., Ning K., Zhang Y.. Decoding the link of microbiome niches with homologous sequences enables accurately targeted protein structure prediction. Proc. Natl Acad. Sci. U.S.A. 2021; 118:e2110828118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The web server and benchmark dataset are available at https://zhanglab.ccmb.med.umich.edu/LOMETS/.