

Abstract
The retrovirus HIV‐1 integrates into the host genome and establishes a latent viral reservoir that escapes immune surveillance. Molecular mechanisms of HIV‐1 latency have been studied extensively to achieve a cure for the acquired immunodeficiency syndrome (AIDS). Latency‐reversing agents (LRAs) have been developed to reactivate and eliminate the latent reservoir by the immune system. To develop more promising LRAs, it is essential to evaluate new therapeutic targets. Here, we find that CBX4, a component of the Polycomb Repressive Complex 1 (PRC1), contributes to HIV‐1 latency in seven latency models and primary CD4+ T cells. CBX4 forms nuclear bodies with liquid–liquid phase separation (LLPS) properties on the HIV‐1 long terminal repeat (LTR) and recruits EZH2, the catalytic subunit of PRC2. CBX4 SUMOylates EZH2 utilizing its SUMO E3 ligase activity, thereby enhancing the H3K27 methyltransferase activity of EZH2. Our results indicate that CBX4 acts as a bridge between the repressor complexes PRC1 and PRC2 that act synergistically to maintain HIV‐1 latency. Dissolution of phase‐separated CBX4 bodies could be a potential intervention to reactivate latent HIV‐1.
Keywords: CBX4, HIV‐1 latency, nuclear body, phase separation, SUMOylation
Subject Categories: Chromatin, Transcription & Genomics; Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications & Proteolysis
Polycomb group protein CBX4 recruits EZH2 on the HIV‐1 promoter and SUMOylates EZH2 within CBX4 bodies, which promotes HIV‐1 latency via a liquid–liquid phase separation‐dependent mechanism.

Introduction
The acquired immunodeficiency syndrome (AIDS) is incurable because of human immunodeficiency virus type 1 (HIV‐1) latency (Chun et al, 1997; Finzi et al, 1997; Wong et al, 1997). HIV‐1 proviruses are temporally silenced within resting memory CD4+ T cells and rapidly reactivated upon CD4+ T cell activation. These latently infected CD4+ T cells compose the major latent HIV‐1 reservoirs. The persistent treatment of combined antiretroviral therapy (cART) suppresses HIV‐1 replication effectively. However, the HIV‐1 viremia quickly rebounds upon cART interruption, which prompts that the treatment has to be lifelong (Siliciano et al, 2003). Thus, many functional cure strategies have been proposed to achieve the long‐term suppression of HIV‐1 replication and the permanent remission of HIV‐1 viremia without cART, which include the “shock and kill” strategy, the “block and lock” strategy, stem cell transplantation, CRISPR/Cas9 gene editing, immune checkpoint blockade, and control antigen‐driven proliferation (Davenport et al, 2019; Ahlenstiel et al, 2020; Cohn et al, 2020). All of these strategies rely on a more comprehensive elucidation of the establishment and maintenance of HIV‐1 latency.
Multiple molecular mechanisms are involved in HIV‐1 latency (Mbonye & Karn, 2017; Khoury et al, 2018; Janssens et al, 2021). Both chromatin landscape and transcriptional control play key roles in the establishment and maintenance of HIV‐1 latency. Most of the widely used latency‐reversing agents (LRAs), which expose HIV‐1‐infected cells to immune surveillance, target chromatin landscape modifiers and transcriptional factors (Spivak & Planelles, 2018; Ait‐Ammar et al, 2020). In the early stage of HIV‐1 latency, the expression and the activity of the viral trans‐activator of transcription (Tat) and many cellular transcription factors including NF‐κB, Sp1, AP‐1, NFAT, TFIIH, CDK9 and Cyclin T1 are specifically decreased (Nabel & Baltimore, 1987; Perkins et al, 1993; Kinoshita et al, 1998; Yang et al, 1999; Kim et al, 2006; Budhiraja et al, 2013). Simultaneously, many transcription suppressors including LSF, YY1, CTIP2, DSIF, NELF, PML and TRIM28 are enriched on the HIV‐1 long terminal repeat (LTR) (Coull et al, 2000; Ping & Rana, 2001; He & Margolis, 2002; Marban et al, 2007; Lusic et al, 2013; Ott & Verdin, 2013; Ma et al, 2019). The 5’ end of HIV‐1 LTR contains the HIV‐1 promoter and drives viral gene transcription. Subsequently, many suppressive epigenetic proteins are recruited to the HIV‐1 promoter to shape the inactive chromatin landscape. Active marks acetyls on histone H3 Lysine 9 (H3K9), H3K27 and H4K20 are removed by Histone Deacetylases 1 (HDAC1) and HDAC2 (Marban et al, 2007). Deacetylated H3K9 is methylated by histone methyltransferases SUV39H1, G9a and GLP, which shape the suppressive marks H3K9me2 and H3K9me3 on the HIV‐1 LTR (Chéné et al, 2007; Imai et al, 2010; Ding et al, 2013). Particularly, the transcription suppressor PML protein can orchestrate the G9a methyltransferase to bind to the latent HIV‐1 promoter and shape the H3K9me2‐containing facultative heterochromatin on the proviral DNA (Lusic et al, 2013; Ott & Verdin, 2013). H3K9me2 and H3K9me3 are further maintained by heterochromatin protein 1α (HP1α), HP1β and HP1γ (Chéné et al, 2007). Similarly, deacetylated H3K27 is methylated by EZH2, which is the major subunit of the Polycomb repressive complex 2 (PRC2), resulting in the formation of suppressive mark H3K27me3 on the HIV‐1 LTR (Friedman et al, 2011). Suppressive mark H3K27me3 is further maintained by PRC1 subunits including chromobox protein homolog 2 (CBX2), CBX4, CBX6, CBX7 and CBX8 (Khan et al, 2018). Suppressive mark H4K20me1 is methylated by SMYD2 and maintained by L3MBTL1 (Boehm et al, 2017). DNA methylation on the HIV‐1 LTR, which is catalyzed by DNMT1 and maintained by MBD2, is also found to mediate HIV‐1 latency (Blazkova et al, 2009; Kauder et al, 2009; Palacios et al, 2012; Trejbalová et al, 2016; preprint: Verdikt et al, 2021).
In recent years, many membrane‐less condensates have been found to participate in cellular and viral processes. These non‐membrane‐enclosed compartments are composed of specific proteins or RNAs with liquid–liquid phase separation (LLPS) characteristics (Hyman et al, 2014). BRD4, MED1, OCT4 and GCN4 are found to link super‐enhancers (SEs) and gene activation via LLPS (Boija et al, 2018; Sabari et al, 2018). HP1, SUV39H1, and TRIM28 are found to form LLPS droplets to link heterochromatic H3K9me3 and gene suppression (Larson et al, 2017; Strom et al, 2017; Sanulli et al, 2019; Wang et al, 2019). Specific protein modifications including SUMOylation and hyperphosphorylation also contribute to LLPS (Banani et al, 2016; Lu et al, 2018; Guo et al, 2019). The PML nuclear body, which is also one of the HIV‐1 latency contributors, is one of the first exemplary nuclear bodies shown to be formed through phase separation (Lusic et al, 2013; Banani et al, 2016). Further biochemical analysis indicates that PML forms LLPS nuclear bodies through SUMOylation‐mediated SUMO‐SIM multivalent interactions (Banani et al, 2016; Corpet et al, 2020). Viral processes also have been found to be associated with LLPS. Both measles virus (MeV) and vesicular stomatitis virus (VSV) have been found to form inclusion bodies with LLPS properties (Heinrich et al, 2018; Zhou et al, 2019). Pan‐retroviral nucleocapsids form LLPS condensates to reposition the viral genomic RNA (Monette et al, 2020). SARS‐CoV‐2 nucleocapsids together with viral RNAs form gel‐like condensates to facilitate viral particle assembly (Carlson et al, 2020; Wang et al, 2021). The phosphorylation of SARS‐CoV‐2 nucleocapsids transforms these condensates into LLPS droplets to facilitate viral transcription (Carlson et al, 2020; Savastano et al, 2020). Cellular NLRP6 undergoes LLPS upon RNA virus infection to promote inflammasome activation (Shen et al, 2021). We recently found that the histone chaperone CAF‐1 leads the formation of LLPS nuclear bodies to promote HIV‐1 latency (Ma et al, 2021). The LLPS of CAF‐1 bodies influences the fate of many CAF‐1‐recruited epigenetic modifiers, and plays a key role in maintaining HIV‐1 latency.
Although many transcriptional condensates and heterochromatic condensates which have LLPS characteristics have been found to spatiotemporally regulate cellular or viral processes, the precise function and the molecular basis of these liquid‐like droplets have not been fully elucidated. Recently, two reports showed that the Polycomb group (PcG) protein CBX2 phase‐separates into liquid‐like nuclear puncta, which coalesces into the PRC1 (Plys et al, 2019; Tatavosian et al, 2019). However, whether the LLPS properties of CBX2 condensates or PRC1 condensates play roles in cross‐talking with PRC2 and regulating H3K27me3‐contaning facultative heterochromatin (fHC) is still not defined. Here, we found that the PcG protein CBX4 is enriched on the HIV‐1 LTR and suppresses HIV‐1 expression and promotes HIV‐1 latency in seven cellular models (J‐Lat 6.3, 8.4, 9.2, 10.6, 15.4, Mix and A2) and primary CD4+ T cells. CBX4 forms phase‐separated nuclear condensates which get close to or co‐localize with HIV‐1 proviruses. Further proteomics and biochemical analysis revealed that CBX4 recruits both SUMO4 and EZH2 to CBX4 bodies, and SUMOylates EZH2 utilizing its SUMO E3 ligase activity, which significantly enhances the H3K27 methyltransferase activity of EZH2. Our results indicated that CBX4 bridges PRC1 and PRC2 by recruiting and SUMOylating EZH2 within CBX4 LLPS bodies, which coordinates both PRC1 and PRC2 to establish and maintain H3K27me3‐containing fHC and HIV‐1 latency.
Results
CBX4 contributes to HIV‐1 latency
The Polycomb repressive complex 1 (PRC1) and PRC2 have been found to mediate HIV‐1 latency by establishing and maintaining the H3K27me3 modification on the HIV‐1 promoter (Friedman et al, 2011; Khan et al, 2018). However, the major mediator has not been systematically investigated. To this end, we constructed a custom siRNA library which targeted the major subunits of PRC1 and PRC2. We knocked down each subunit of PRC1 and PRC2 by separately transfecting these siRNAs into the TZM‐bl cell line which harbored an integrated copy of luciferase gene under the control of the HIV‐1 promoter (Platt et al, 1998). The expression level of luciferase reflected the activity of the HIV‐1 promoter. We found that the knockdown of both the PRC1 subunit CBX4 and the PRC2 subunit EZH2 significantly upregulated the HIV‐1 promoter activity (Fig 1A, and Appendix Fig S1A). The enhancement of HIV‐1 promoter activity was much higher upon depleting CBX4 than depleting EZH2. The enhancement was more significant upon co‐treating with HIV‐1 Tat or TNFα (Fig 1B and C). The overexpression of CBX4 suppressed the basal level of HIV‐1 promoter activity and rescued HIV‐1 repression in a dose‐dependent manner (Fig 1D). We also found that CBX4 was highly expressed in unstimulated naïve CD4+ T cells and resting memory CD4+ T cells (Fig 1E). PHA activation in CD4+ T cells resulted in CBX4 downregulation.
Figure 1. CBX4 contributes to HIV‐1 latency.
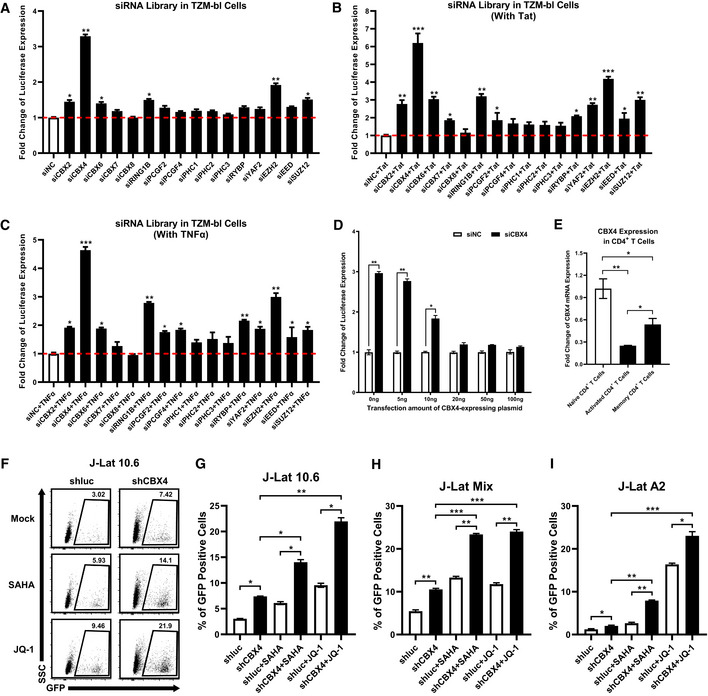
-
AAn siRNA library targeting major PcG proteins was transfected into TZM‐bl cells. Three different siRNAs targeting each gene were transfected as a mixture. Forty‐eight hours post transfection, cells were harvested and lysed, followed by measuring the luciferase activity of each group. Fold changes of luciferase expression were calculated for each gene by normalizing to the negative control siNC. The red dashed line represents the base line.
-
BDifferent siRNAs targeting each gene were transfected as in (A). Twenty‐four hours post siRNA‐transfection, cells in each group were transfected with equal amounts of Tat‐expressing plasmids. Another 24 h later, cells were proceeded to luciferase assay.
-
CsiRNAs targeting different genes were transfected into TZM‐bl cells as in (A). About 24 h later, cells were treated with TNFα. Another 24 h later, cells were proceeded to luciferase assay.
-
DThe endogenous CBX4 in TZM‐bl cells were knocked down by three different siRNAs targeting 3’UTR of CBX4 mRNA, or treated with siNC. Different gradients of CBX4 construct were co‐transfected. The expression of luciferase from each group was measured and normalized to the siNC group which was not co‐transfected with CBX4 construct.
-
ETotal RNAs from naïve CD4+ T cells, PHA‐stimulated CD4+ T cells and resting memory CD4+ T cells were extracted and proceeded to RT‐qPCR to quantitate the relative expression of CBX4. The expression of CBX4 in PHA‐stimulated and resting memory CD4+ T cells was normalized to naïve CD4+ T cells.
-
F, GThe percentages of GFP‐positive J‐Lat 10.6 cells in shluc and shCBX4 groups. LRAs SAHA and JQ‐1 were used as supplements. The representative flow cytometry figure of each group is shown in (F). The overall statistical results are shown in (G).
-
H, IOne heterogeneous latency model J‐Lat Mix and one monoclonal model J‐Lat A2 were treated as in (F). The reactivation efficiencies of each group are indicated by the percentages of GFP‐positive cells.
Data information: Data are presented as mean ± SEM in biological triplicate. P‐values in (A–C) were calculated by one‐way ANOVA with Dunnett's multiple comparisons test. P‐values in (D) were calculated by two‐way ANOVA with Sidak's multiple comparisons test. P‐values in (E–I) were calculated by one‐way ANOVA with Tukey's multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001.
Previous reports have shown that the PRC2 component EZH2 contributed to HIV‐1 latency by catalyzing the H3K27me3 modification on the HIV‐1 promoter (Friedman et al, 2011; Tripathy et al, 2015; Nguyen et al, 2017). However, how PRC1 components, especially CBX4, contributed to HIV‐1 latency was not defined. Thus, we also investigated whether CBX4 contributed to HIV‐1 latency. We knocked down CBX4 in the monoclonal HIV‐1 latency cell line J‐Lat 10.6 which harbored an integrated full‐length HIV‐1 pseudotyped provirus (Jordan et al, 2003). The expression of GFP which was inserted into the HIV‐1 Nef gene open reading frame (ORF) reflected the reactivation of latent HIV‐1. We found that the knockdown of CBX4 upregulated HIV‐1 expression (Fig 1F and G, and Appendix Fig S1B–D). The HIV‐1 reactivation was more significant when supplemented with latency‐reversing agents (LRAs) including HDAC inhibitor SAHA and BRD4 inhibitor JQ‐1. Both SAHA and JQ‐1 have been widely used to reactivate latent HIV‐1 and have no influence on the activity of PcG proteins (Spivak & Planelles, 2018; Ait‐Ammar et al, 2020). These results were well repeated in other monoclonal HIV‐1 latency cell lines including J‐Lat 6.3, 8.4, 9.2 and 15.4 (Fig EV1, EV2, EV3, EV4, EV5, and Appendix Fig S1B–D). We also knocked down CBX4 in a heterogeneous latency cell line J‐Lat Mix which was a mixture of latently infected Jurkat cells. Similarly, the depletion of CBX4 reactivated HIV‐1 pseudotyped provirus (Fig 1H, and Appendix Fig S1E–G). The reactivation was more significant upon co‐treating with SAHA and JQ‐1. To verify whether CBX4 influenced the HIV‐1 promoter activity directly, rather than interplaying with other viral proteins, we also knocked down CBX4 in the monoclonal latency cell line J‐Lat A2 which harbored an integrated HIV‐1 pseudotyped mini‐genome (Jordan et al, 2003). J‐Lat A2 harbored only Tat and GFP ORFs which were under the control of the HIV‐1 promoter. We found that the depletion of CBX4 also upregulated HIV‐1 expression in J‐Lat A2 (Fig 1I, and Appendix Fig S1H–J). The reactivation was more significant upon co‐treating with SAHA and JQ‐1. The latent HIV‐1 reactivation induced by knocking down CBX4 in these cell lines was well confirmed by measuring the relative HIV‐1 mRNA expression (Fig EV1, EV2, EV3, EV4, EV5). These results indicated that CBX4 contributed to HIV‐1 latency by targeting the HIV‐1 promoter.
Figure EV1. CBX4 depletion reactivates latent HIV‐1.
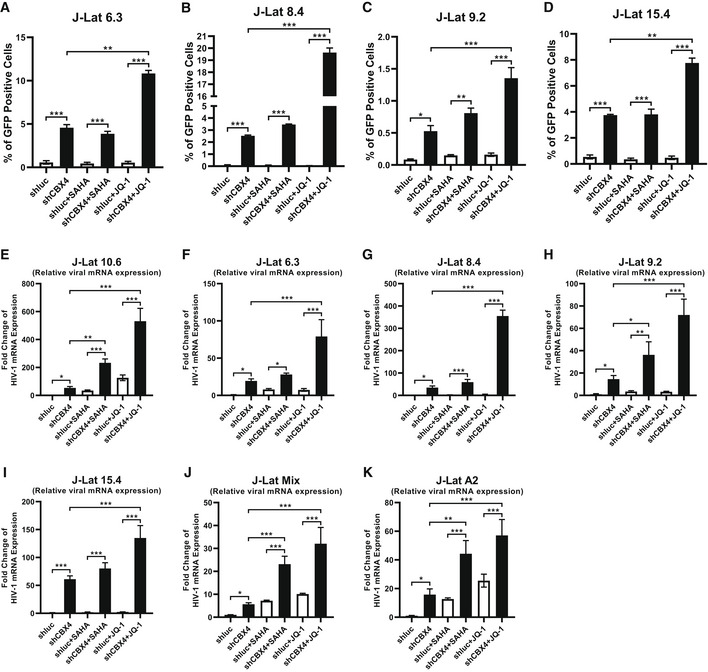
-
A–DMonoclonal latency models including J‐Lat 6.3, 8.4, 9.2 and 15.4 were treated as in Fig 1F. The reactivation efficiencies of each group are indicated by the percentages of GFP‐positive cells.
-
E–KJ‐Lat 10.6, 6.3, 8.4, 9.2, 15.4, Mix and A2 were treated as in Fig 1F. Relative HIV‐1 mRNA expression within each group was quantified and normalized to the shluc group in each cell line.
Data information: Data are presented as mean ± SEM in biological triplicate. P‐values were calculated by one‐way ANOVA with Tukey's multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure EV2. CBX4 bodies colocalize with latent HIV‐1 proviruses and EZH2.
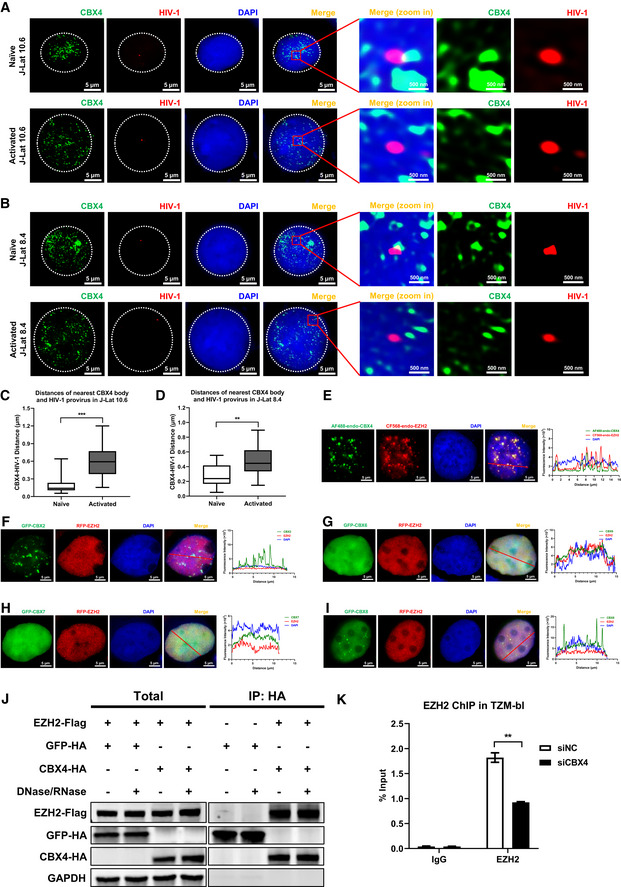
-
A, BImmunoFISH images of the pseudotyped HIV‐1 genomic DNA and CBX4 bodies in J‐Lat 10.6 and 8.4. Naïve cells were treated with DMSO. Activated cells were treated with TNFα. The right images show amplified regions.
-
C, DDistances of the nearest CBX4 body and the HIV‐1 provirus in both naïve and activated cells were measured with Imaris software. In each group, 15 cells were randomly imaged and measured the CBX4‐HIV‐1 distances. The quantification of distances was evaluated in both J‐Lat 10.6 (C) and 8.4 (D) cells.
-
EThe endogenous CBX4 and EZH2 in HEK293T cells were captured with rabbit anti‐CBX4 antibody and mouse anti‐EZH2 antibody respectively. These cells were further incubated with AF488‐conjugated anti‐rabbit IgG antibody and CF568‐conjugated anti‐mouse IgG antibody. DAPI indicated DNA. The co‐localization was evaluated by line scan profile and shown on the right. The red arrow indicates the profiled position.
-
F–IGFP‐tagged CBX2, CBX6, CBX7 and CBX8 were co‐overexpressed with RFP‐tagged EZH2. SIM imaging was performed for each combination. Line scan profiles are shown on the right of each panel. Red arrows in merged images indicate the positions where line scans are profiled.
-
JHA‐tagged CBX4 and GFP were co‐overexpressed with Flag‐tagged EZH2. HA‐tagged proteins were immunoprecipitated (IP) with anti‐HA beads. In both groups, the IP reactions were supplemented with DNase/RNase or left untreated. Both total and IP samples were immunoblotted (IB) with anti‐HA and anti‐Flag antibodies.
-
KChIP assays with antibodies against IgG and EZH2 were performed in siNC and siCBX4 TZM‐bl cells.
Data information: Scale bars in (A), (B) and (E–I) represent 5 μm. Scale bars in amplified images within (A) and (B) represent 500 nm. All the samples were imaged to obtain at least three sets of images. Data in (C) and (D) represent distances in biological replicates (n = 15 for C and D). The central bands of the boxplots represent median values of distances. The heights of the boxes represent the interquartile ranges of distances. The boundaries of the upper whiskers and the lower whiskers represent the maximum distances and the minimum distances, respectively. Data in (K) are presented as mean ± SEM in biological triplicate. P‐values in (C), (D) and (K) were calculated by Student's t‐test. **P < 0.01, ***P < 0.001.
Figure EV3. Proteins in CBX4 bodies were internally diffused.
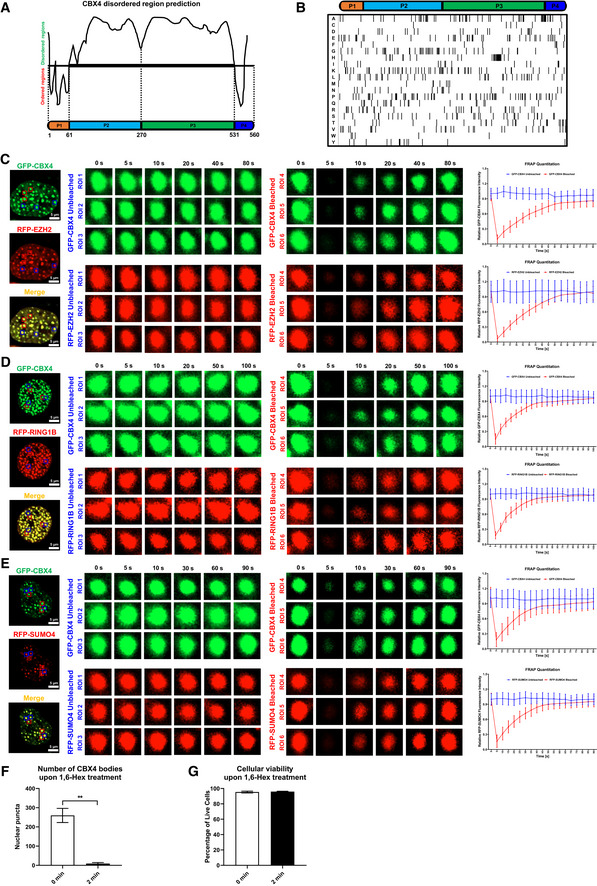
-
AThe schematic of CBX4 disorder regions. P1 represents the N‐terminal Chromodomain (CD). P2 and P3 represent two sub‐regions of IDR. P3 represents C‐terminal CBox domain.
-
BComposition analysis of CBX4 protein. The lower table indicates the distribution of 20 kinds of amino acids.
-
C–EFRAP images of CBX4 bodies and recruited proteins including EZH2 (C), RING1B (D) and SUMO4 (E). RFP‐tagged EZH2, RING1B and SUMO4 were co‐overexpressed with GFP‐tagged CBX4 in HEK293T cells. Live cell images were captured every 5 s. In each combination, six co‐localization bodies were proceeded to FRAP. Three bodies (ROI 1, ROI 2 and ROI 3) were left untreated. Another three bodies (ROI 4, ROI 5 and ROI 6) were bleached with strong 488 and 561 nm laser power. The right histograms show relative fluorescence intensities of unbleached and bleached CBX4 and its partners in each time point.
-
FThe numbers of CBX4 bodies before 1,6‐Hex treatment (0 min) and after 1,6‐Hex treatment (2 min) were calculated.
-
GCellular viabilities before 1,6‐Hex treatment (0 min) and after 1,6‐Hex treatment (2 min) were evaluated by measuring the percentages of amine‐reactive fluorescent dye non‐permeant cells.
Data information: Scale bars in (C–E) represent 5 μm. All the samples were imaged to obtain at least three sets of images. Data are presented as mean ± SEM in biological triplicate. P‐values in (F) and (G) were calculated by Student’s t‐test. **P < 0.01.
Figure EV4. The LLPS of CBX4 depends on the β‐sheet motifs of CBox.
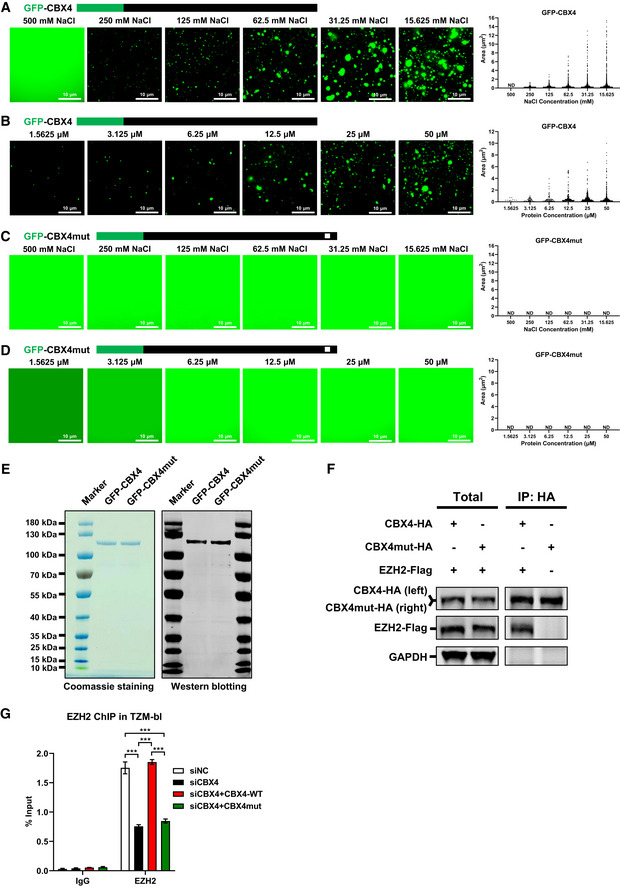
-
AAbout 25 μM of in vitro purified GFP‐CBX4 proteins were incubated with gradient droplet formation buffers. GFP‐CBX4 droplets appeared in 250, 125, 62.5, 31.25 and 15.625 mM NaCl. The right subpanel represents statistical analysis of droplet areas and numbers of GFP‐CBX4 within different NaCl concentrations.
-
BDifferent concentrations of in vitro purified GFP‐CBX4 proteins were incubated with 75 mM NaCl droplet formation buffer. The right subpanel represents statistical analysis of droplets areas and numbers of GFP‐CBX4 within different protein concentrations.
-
C, DThe in vitro purified GFP‐CBX4mut proteins (25 μM) were incubated with different droplet formation buffers which contained different NaCl concentrations (C). Different concentrations of GFP‐CBX4mut proteins were incubated with 75 mM NaCl droplet formation buffer (D). The right subpanel within each panel represents the statistical analysis of droplets areas and numbers of GFP‐CBX4mut within different conditions.
-
EThe purities of in vitro purified GFP‐CBX4 and GFP‐CBX4mut proteins were analyzed by both Coomassie blue staining and Western blotting against GFP.
-
FHA‐tagged CBX4 and CBX4mut were co‐overexpressed with Flag‐tagged EZH2 respectively. HA‐tagged proteins were IP with anti‐HA beads. Both total and IP samples were IB with anti‐HA and anti‐Flag antibodies.
-
GTZM‐bl cells were treated with siCBX4 targeting 3’UTR of CBX4 mRNA or siNC. Another two groups of siCBX4‐treated cells were re‐overexpressed with wild‐type CBX4 and LLPS‐deficient CBX4mut respectively. ChIP assays with antibodies against IgG and EZH2 were performed in these cells.
Data information: Scale bars in (A–D) represent 10 μm. All the samples were imaged to obtain at least three sets of images. Data in (G) are presented as mean ± SEM in biological triplicate. P‐values in (G) were calculated by two‐way ANOVA with Tukey's multiple comparisons test. ***P < 0.001.
Figure EV5. CBX4 SUMOylates EZH2 with SUMO1, SUMO2 and SUMO4.
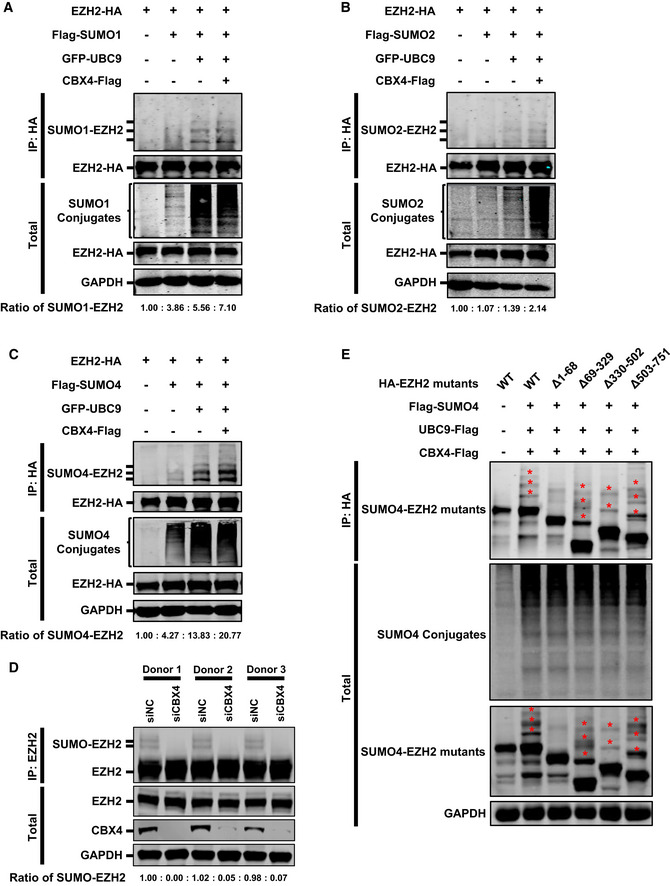
- HA‐tagged EZH2 was co‐overexpressed with Flag‐tagged SUMO1, GFP‐tagged UBC9 or Flag‐tagged CBX4. EZH2 was IP with anti‐HA beads. Both total and IP samples were IB with anti‐HA, ‐Flag, and ‐GAPDH antibodies. The expression ratios of SUMO1‐EZH2 are marked below the panel.
- Target proteins were overexpressed, IP and IB as in (A), except that Flag‐tagged SUMO molecule was changed with SUMO2. The expression ratios of SUMO2‐EZH2 are marked below the panel.
- Target proteins were overexpressed, IP and IB as in (A), except that Flag‐tagged SUMO molecule was changed with SUMO4. The expression ratios of SUMO4‐EZH2 are marked below the panel.
- Primary CD4+ T cells from three healthy donors were transfected with siNC or siCBX4 utilizing 4D‐Nucleofector System. About 24 h post transfection, the endogenous EZH2 within each group was IP with anti‐EZH2 antibodies. Both total and IP samples were IB with anti‐EZH2, anti‐CBX4 and anti‐GAPDH antibodies. The expression ratios of SUMO‐EZH2 within each group are marked below the panel.
- HA‐tagged EZH2 and EZH2 truncation mutants were co‐overexpressed with Flag‐tagged SUMO4, Flag‐tagged UBC9 and Flag‐tagged CBX4. EZH2 and corresponding mutants were IP with anti‐HA beads. Both total and IP samples were IB with antibodies against HA, Flag and GAPDH. The asterisks represent SUMOylated EZH2 mutants.
CBX4 contributes to H3K27me3 modification of HIV‐1 promoter
Both PRC1 and PRC2 are multi‐component complexes which link many chromatin modifications, especially H3K27me3 and H2AK119Ub (Guo et al, 2021). To investigate whether CBX4 influenced the epigenetic status of the HIV‐1 promoter, we evaluated the alterations of histone modifications upon CBX4 knockdown. We firstly observed that CBX4 proteins were highly enriched on the HIV‐1 LTR in TZM‐bl cells, especially the promoter region and nucleosome assembly sites (Fig 2A). The knockdown of CBX4 resulted in the loss of CBX4 proteins on the HIV‐1 LTR (Fig 2A, and Appendix Fig S2A–D). Then, we found that the H3K27me3 modification on the HIV‐1 LTR was significantly decreased upon CBX4 knockdown (Fig 2B). As the main modifier of H3K27me3 was EZH2, this result suggested that CBX4 depletion might influence the distribution and function of EZH2 on the HIV‐1 LTR. Conversely, phosphorylated RNAP II (Pho‐Pol II) proteins were significantly enriched on the HIV‐1 LTR upon knocking down CBX4, which indicated active transcriptional activity of the HIV‐1 promoter (Fig 2C). Besides, we found that the H2AK119Ub modification on the HIV‐1 LTR was slightly downregulated, whereas the H3K27Acetyl on the HIV‐1 LTR was unchanged (Fig 2D and E). The H2AK119Ub modification, which was catalyzed by RING1B, also has been found to contribute to HIV‐1 latency (Yoon et al, 2014; Khan et al, 2018). The downregulation of H2AK119Ub on the HIV‐1 LTR upon knocking down CBX4 indicated that CBX4 might recruit RING1B and establish the suppressive H2AK119Ub modification on the HIV‐1 LTR. While the active mark H3K27Acetyl was mainly deacetylated by HDAC1 and HDAC2 on the HIV‐1 LTR (Marban et al, 2007). The stabilization of H3K27Acetyl on the HIV‐1 LTR upon CBX4 depletion indicated that CBX4 might not cross‐talk with histone acetylation and corresponding deacetylases. We also conducted chromatin immunoprecipitation (ChIP) assays in J‐Lat 10.6. Similarly, we observed significant downregulation of CBX4 and H3K27me3, and significant upregulation of Pho‐Pol II on the HIV‐1 LTR upon CBX4 knockdown (Fig 2F and G, and Appendix Fig S2E–H). The absence of heterochromatin modification and the presence of transcription machinery often indicate higher chromatin accessibility. The integration sites, the removement of specific host factors and the presence of LRAs have been found to alter the chromatin accessibility of the HIV‐1 LTR which contains the HIV‐1 promoter (Conrad et al, 2017; Dupont et al, 2021; Jefferys et al, 2021; Einkauf et al, 2022). Thus, we conducted ATAC‐Seq in J‐Lat 10.6 cells to directly probe the chromatin accessibility of the HIV‐1 LTR upon CBX4 knockout (Buenrostro et al, 2015). Both the HIV‐1 genome and the human genome were analyzed for accessibility. All the sequencing reads were aligned to both HIV‐1 reference genome (K03455.1) and human reference genome (GRCh38). Only reads which contained both HIV‐1 5’LTR and human integration junction DNA were sorted and normalized to the total mapped reads. The tag peaks were generated utilizing these reads and visualized by Igvtools. We found that the transposable tag density, which indicated the accessible region, was significantly increased on the HIV‐1 LTR upon knocking out CBX4 (Fig 2H, and Appendix Fig S2I and J). Overall, our results indicated that CBX4 was enriched on the HIV‐1 LTR and contributed to H3K27me3 modification.
Figure 2. CBX4 contributes to H3K27me3 modification of HIV‐1 promoter.
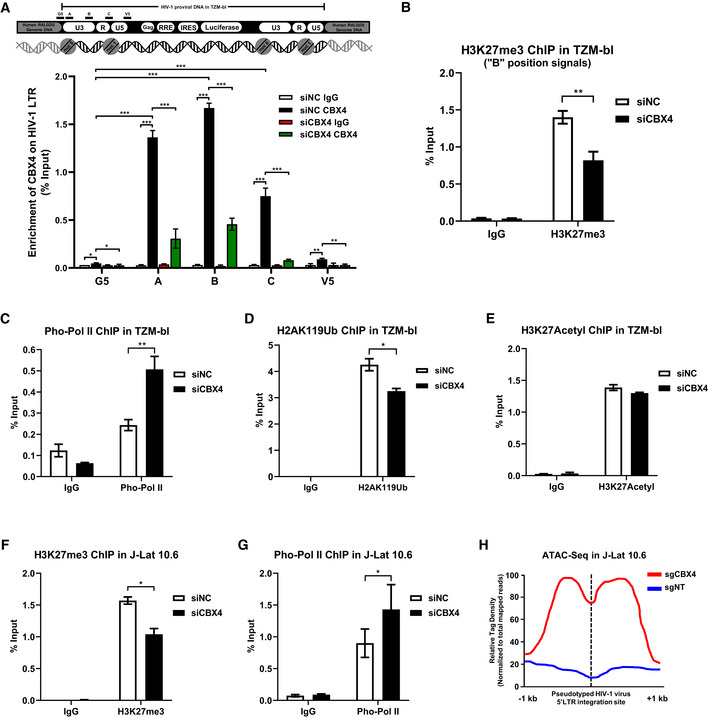
-
AThe upper schematic represents the pseudotyped HIV‐1 provirus and corresponding integration site in TZM‐bl cells. The HIV‐1 mini‐genome was integrated in the intron of human RALGDS gene. Five pairs of ChIP‐qPCR primers were designed on the HIV‐1 LTR, which are indicated above the backbone. G5: Cellular DNA and viral 5’LTR junction; A: Nucleosome (Nuc) 0 assembly site; B: Nuc‐free region; C: Nuc 1 assembly site; V5: Viral 5’LTR and gag leader sequence junction. ChIP assays with antibodies against IgG and CBX4 were performed in both siNC‐treated and siCBX4‐treated TZM‐bl cells. All the ChIP‐qPCR DNA signals were normalized to Input. The lower statistical graph represents the ChIP‐qPCR results.
-
B–EChIP assays with antibodies against H3K27me3, Pho‐Pol II, H2AK119Ub and H3K27Acetyl were performed in TZM‐bl cells as in (A). Only “B” position signals are shown and normalized to Input.
-
F, GChIP assays with antibodies against H3K27me3 and Pho‐Pol II were performed in J‐Lat 10.6 cells as in (A). ChIP‐qPCR DNA signals on position “B” were normalized to Input.
-
HATAC‐Seq was performed in WT and CBX4‐knockout J‐Lat 10.6 cells. The relative tag densities of the pseudotyped HIV‐1 5’LTR integration site in each group were calculated. The highest tag density was set as 100. Data represent 2 kb ranges of tag densities centered the 5’LTR integration site. The integration site is indicated with dashed line.
Data information: Data are presented as mean ± SEM in biological triplicate. P‐values in (A) were calculated by two‐way ANOVA with Tukey's multiple comparisons test. P‐values in (B–G) were calculated by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
CBX4 recruits PcG proteins and forms nuclear bodies
To investigate the mechanisms of CBX4‐mediated epigenetic suppression of HIV‐1, we utilized mass spectrometry (MS) to identify CBX4‐enriched proteins. We overexpressed CBX4 in TZM‐bl cells and separated CBX4‐co‐immunoprecipitated proteins by SDS‐PAGE. Distinct gel bands, which contained CBX4‐enriched proteins, as well as control gel bands were proceeded to in‐gel digestion, followed by nanoscale LC‐MS/MS to characterize digested peptides. All the peptides were annotated by PEAKS Studio to identify corresponding proteins. Finally, we identified 701 candidate proteins which were enriched by CBX4 (Fig 3A). Apart from classical PRC1 components including RNF1 and RNF2, we also found that CBX4 enriched PRC2 core component EZH2 and SUMO paralog SUMO4. We speculated that CBX4 might orchestrate both EZH2‐mediated H3K27me3 and SUMO4‐mediated SUMOylation to contribute to HIV‐1 latency. To investigate the distribution of CBX4 within cells, we visualized CBX4 proteins with super‐resolution Structured Illumination Microscopy (SIM). We found that CBX4 formed hundreds of cellular bodies within nuclei (Fig 3B). We also evaluated the sizes of these CBX4 bodies within 3D‐Stack images. The diameters of CBX4 bodies ranged from 100 to 1,500 nm (Fig 3C, and Appendix Fig S3A–C). Besides, we measured the sphericities of CBX4 bodies. All the CBX4 bodies formed spherical shapes with a median sphericity of 0.9 (Fig 3D). We knocked down the endogenous CBX4 by siRNA targeting 3’UTR of CBX4 mRNA and expressed equal amounts of GFP‐CBX4. We found that GFP‐tagged CBX4 also formed many nuclear bodies, the size and distribution of which were similar to the endogenous CBX4 bodies (Appendix Fig S3D and E). To further exclude the possibility that CBX4 condensation might be caused by artificially high protein concentrations, we expressed equal amounts of GFP‐CBX4 in HEK293T cells and used CF568‐conjugated antibodies to capture both endogenous and exogenous CBX4 proteins. We found that GFP‐CBX4 bodies still co‐localized with CF568‐CBX4 condensates (Appendix Fig S3F and G). We next investigated the positions of CBX4 bodies and HIV‐1 proviruses in different stimulation conditions utilizing immunolabeling‐based fluorescence in situ hybridization (ImmunoFISH) assay (Lusic et al, 2013; Marini et al, 2015). We found that the HIV‐1 genomic DNA was close to or co‐localized with CBX4 bodies in naïve J‐Lat 10.6 and 8.4 cells (Figs 3E, and EV2A and B). Upon TNFα stimulation, the HIV‐1 genomic DNA was away from CBX4 bodies (Figs 3E and EV2A–D). These results are in line with what was observed previously for PML nuclear bodies and latent HIV‐1 proviruses (Lusic et al, 2013; Ott & Verdin, 2013). The latent HIV‐1 provirus resides in close proximity to PML bodies. While TPA treatment results in the progressive displacement of the HIV‐1 provirus from the PML bodies. Our above results indicated that CBX4 bodies were enriched on HIV‐1 proviruses in latent status. In activation status, HIV‐1 proviruses were disassociated with CBX4 bodies.
Figure 3. CBX4 recruits PcG proteins and forms nuclear bodies.
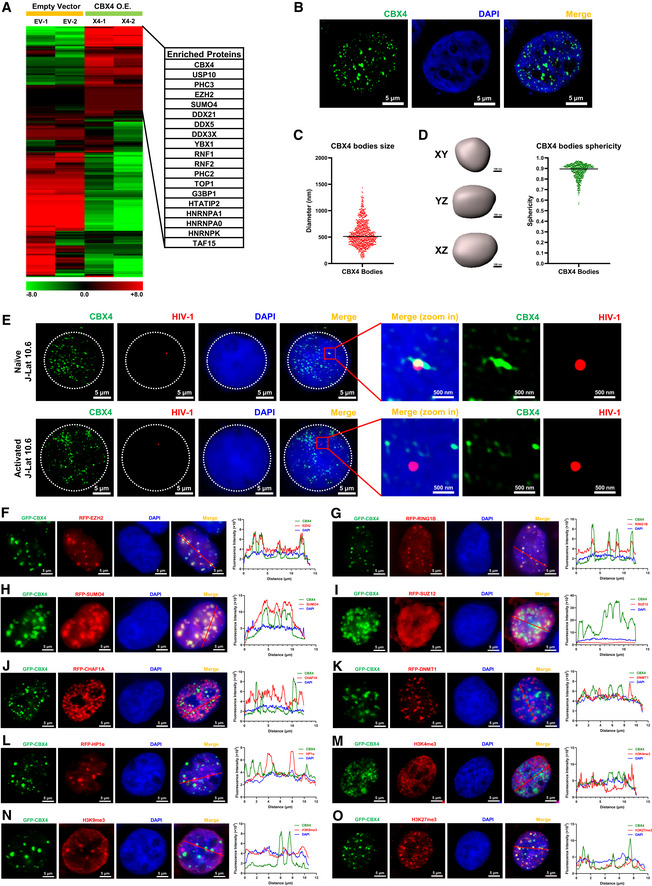
-
ATZM‐bl cells were transfected with empty vectors and CBX4‐expressing plasmids, followed by the enrichment of CBX4‐bound proteins. The heatmap represents CBX4‐enriched proteins. Representative genes are shown in table aside the heatmap. The heatmap scale represents fold changes in gene expression (min: −8 fold; max: 8 fold).
-
BSuper‐resolution SIM image of CBX4 in HEK293T cells. DAPI was used to dye DNA which is colored in blue. FITC‐tagged antibodies were used to label endogenous CBX4 which is colored in green.
-
CThe distribution of CBX4 bodies diameters. The black line indicates the median value of diameters which was 512 nm. Data were collected from three cells of three independent samples.
-
DRendered 3D shapes of CBX4 bodies. Three panels represent XY, YZ and XZ planes of one CBX4 body. The right scatter plot represents the distribution of CBX4 bodies sphericities. The black line indicates the median value of sphericities which was 0.9. Data were collected from three cells of three independent samples.
-
EImmunoFISH images of the pseudotyped HIV‐1 genomic DNA and CBX4 bodies in J‐Lat 10.6. Naïve cells were treated with DMSO. Activated cells were treated with TNFα.
-
F–LRFP‐tagged EZH2, RING1B, SUMO4, SUZ12, CHAF1A, DNMT1 and HP1α were co‐overexpressed with GFP‐tagged CBX4 in HEK293T cells. SIM imaging was performed for each combination. The line scan profiles of SIM images which show co‐localization and non‐co‐localization between CBX4 and its partners are shown on the right of each panel. The red arrows in merged images indicate the positions where the line scans are profiled.
-
M–OGFP‐tagged CBX4 was overexpressed in HEK293T cells, followed by treating with AF568‐tagged antibodies against H3K4me3, H3K9me3 and H3K27me3. CBX4 and each histone modification are imaged with SIM. Line scan profiles are shown on the right of each panel. Red arrows in merged images indicate the positions where line scans are profiled.
Data information: Scale bars in (B) and (E–O) represent 5 μm. Scale bars in (D) represent 100 nm. Scale bars in amplified images of (E) represent 500 nm. All the samples were imaged to obtain at least three sets of images.
To verify whether the CBX4 body co‐localized with its enriched proteins, especially PcG and SUMO proteins which were identified by MS, we co‐overexpressed GFP‐tagged CBX4 and RFP‐tagged candidates in HEK293T cells. We found that CBX4 significantly co‐localized with EZH2 (Fig 3F). To exclude the possibility that the co‐localization might be caused by protein overexpression, we used AF488‐conjugated antibodies and CF568‐conjugated antibodies to image the endogenous CBX4 and the endogenous EZH2 in physiological conditions. Similarly, The endogenous EZH2 was also co‐localized with the endogenous CBX4 (Fig EV2E). Conversely, chromobox protein homologs CBX2, CBX6, CBX7 and CBX8, all of which were PRC1 subunits, did not co‐localize with EZH2, although both CBX2 and CBX8 were able to form nuclear bodies (Figs EV2, EV3, EV4, EV5). Our co‐immunoprecipitation assay also showed that EZH2 was recruited by CBX4 in DNA‐ and RNA‐independent manner (Fig EV2J). Besides, the knockdown of CBX4 significantly downregulated EZH2 on the HIV‐1 LTR (Fig EV2K). CBX4 also co‐localized with RING1B and SUMO4 (Fig 3G and H). However, CBX4 did not co‐localize with SUZ12 which was another major component of PRC2 (Fig 3I). To verify whether CBX4 bodies might cross‐talk with other canonical cellular bodies, we also co‐overexpressed CBX4 with other body components. We found that CBX4 did not co‐localize with CAF‐1 bodies, DNMT1 bodies or HP1α bodies (Fig 3J–L). We also evaluated the relative positions of CBX4 bodies and chromatin compartments. We found that CBX4 bodies did not co‐localize with H3K4me3‐contating euchromatin (EC) or H3K9me3‐containing constitutive heterochromatin (cHC) (Fig 3M and N). CBX4 bodies co‐localized with H3K27me3‐containing facultative heterochromatin (fHC) (Fig 3O). Taken together, our above results indicated that CBX4 formed nuclear bodies on the HIV‐1 proviruses and recruited PcG and SUMO proteins to CBX4 bodies.
CBX4 bodies are phase‐separated condensates
In recent years, many proteins which harbored intrinsically disordered regions (IDRs) have been found to form liquid–liquid phase‐separated (LLPS) condensates (Brangwynne et al, 2015). Through IDR prediction, we found that 86.61% of CBX4 protein sequence was disordered (Fig EV3A and B). Thus, we speculated that CBX4 bodies might also be LLPS condensates. Firstly, we performed the fluorescence recovery after photobleaching (FRAP) assay on GFP‐tagged CBX4 bodies within live cells. To exclude the possibility that CBX4 condensation might be caused by artificially high protein concentrations, we expressed equal amounts of GFP‐CBX4 in HEK293T cells compared with the amounts of endogenous CBX4 proteins. We found that the bleached CBX4 bodies recovered fluorescence intensities quickly (Fig 4A). We also utilized strong laser power to bleach only half of one CBX4 body. We found that the bleaching not only quenched the fluorescence intensities of bleached parts but also impaired the intensities of unbleached parts (Appendix Fig S4A), while the fluorescence intensities of both parts quickly recovered. These results indicated that CBX4 bodies were internally diffused and able to diffuse across boundary. Our previous data have shown that CBX4 recruited EZH2, RING1B and SUMO4 to CBX4 bodies (Fig 3F–H). We speculated that these partners might also be internally diffused and diffuse across boundary. We co‐overexpressed RFP‐tagged EZH2, RING1B and SUMO4 with GFP‐tagged CBX4 in HEK293T cells respectively. About 24 h post transfection, we used strong 488 and 561 nm laser power to bleach CBX4 bodies. We found that both RFP‐tagged proteins and GFP‐CBX4 proteins within bleached CBX4 bodies quickly recovered fluorescence intensities, while the fluorescence intensities of unbleached CBX4 bodies were almost unchanged (Fig EV3, EV4, EV5). Besides the internal diffusion properties, we also observed that two CBX4 bodies were able to fuse into one bigger CBX4 body (Fig 4B). One CBX4 body was also able to split into two smaller CBX4 bodies (Fig 4C). Internal diffusion, diffusion across boundary, fusion and fission were golden criteria of LLPS condensates (Alberti et al, 2019). Our data shown above were consistent with these criteria, which indicated that CBX4 bodies were characterized by LLPS condensates. The 1,6‐hexanediol compound has been widely used to disrupt phase‐separated condensates (Sabari et al, 2018). We treated GFP‐tagged CBX4‐expressing cells with 1,6‐hexanediol and found that CBX4 bodies were gradually dissolved upon treatment, which indicated that CBX4 bodies were sensitive to 1,6‐hexanediol (Figs 4D, and EV3F and G). We also purified GFP‐tagged CBX4 proteins in vitro. We found that CBX4 proteins were able to form droplets along with the decrease of NaCl concentration (Fig EV4A and E). CBX4 proteins also formed more and bigger droplets at higher protein concentrations (Fig EV4B). Overall, these results indicated that CBX4 bodies were phase‐separated nuclear condensates.
Figure 4. CBX4 bodies are phase‐separated condensates.
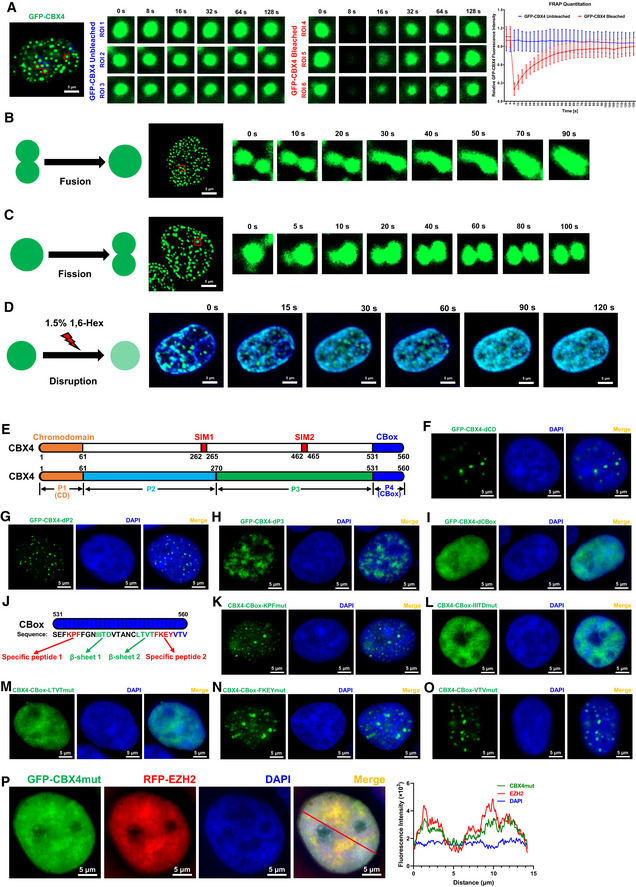
-
AFRAP images of CBX4 bodies to indicate the internal diffusion and diffusion across boundary properties. GFP‐tagged CBX4 was overexpressed in HEK293T cells. These live cells were proceeded to time series imaging 24 h post transfection. Six GFP‐CBX4 bodies are circled and marked as region of interest 1 (ROI 1), ROI 2, ROI 3, ROI 4, ROI 5 and ROI 6. Three CBX4 bodies (ROI 4, ROI 5 and ROI 6) were bleached with strong 488 nm laser pulse. Another three unbleached CBX4 bodies (ROI 1, ROI 2 and ROI 3) were set as control. Images were captured every 4 s. The right histogram shows relative fluorescence intensities of unbleached and bleached CBX4 bodies in each time point.
-
B, CRepresentative images of two CBX4 bodies fusing into one and one CBX4 body splitting into two. Images were captured every 5 s.
-
D1.5% 1,6‐hexanediol (1,6‐Hex) compound was used to treat GFP‐tagged CBX4‐expressing live cells. The DNA was stained with Hoechst and is colored in blue. Images were captured every 5 s.
-
EThe schematic of CBX4 backbone. P1 represents Chromodomain (CD). P2 and P3 represent two sub‐regions of IDR, which harbored two SUMO‐interacting motifs named SIM1 and SIM2. P4 represents the CBox domain.
-
F–IGFP‐tagged CBX4 truncation mutants including CBX4‐dCD, CBX4‐dP2, CBX4‐dP3 and CBX4‐dCBox were overexpressed in HEK293T cells and imaged with SIM.
-
JThe schematic of five motifs within CBox domain, including Specific peptide 1 (KPF), β‐sheet 1 (IIITD), β‐sheet 2 (LTVT), Specific peptide 2 (FKEY) and C‐terminal disordered motif (VTV).
-
K–OGFP‐tagged CBX4 mutants including CBox‐KPFmut, CBox‐IIITDmut, CBox‐LTVTmut, CBox‐FKEYmut and CBox‐VTVmut were overexpressed in HEK293T cells and imaged with SIM. The motif mutants were constructed by mutating these motifs into corresponding numbers of alanine residues.
-
PRFP‐tagged EZH2 was co‐overexpressed with GFP‐tagged CBX4 mutant named CBX4mut which was CBX4‐LTVTmut. SIM images were captured. The line scan profile is shown on the right. The red arrow within merged image indicates the line scan position.
Data information: Data are presented as mean ± SEM in biological triplicate in (A). Scale bars in (A), (B–D), (F–I) and (K–P) represent 5 μm. All the samples were imaged to obtain at least three sets of images.
We next tried to find the key amino acids which mediated the LLPS of CBX4 bodies. The CBX4 protein sequence harbored a big IDR which was between the Chromodomain (CD) and the CBox domain (Fig EV3A). We firstly split the CBX4 protein sequence into four parts (Fig 4E). P1 represented CD. P2 and P3 represented two major sub‐IDRs. P4 represented CBox. We found that the absence of CD (P1) and two sub‐IDRs (P2 and P3) did not dissolve CBX4 bodies, although the size and the number of these bodies have changed slightly (Fig 4F–H). Interestingly, we found that the sole deletion of CBox (P4) completely dissolved CBX4 bodies (Fig 4I). We also constructed many CBX4 mutants which expressed nuclear localization signal (NLS)‐conjugated segments and CBox‐conjugated segments. We found that the CD‐NLS mutant did not form bodies within nuclei; however, the CBox‐conjugated CD‐NLS mutant formed hundreds of nuclear puncta (Appendix Fig S4B and C). Although P2‐NLS mutant formed few and big condensates within nucleoli, the size, the number and the distribution of these bodies have significantly changed (Appendix Fig S4D). The conjugation of CBox on P2‐NLS was able to redistribute these condensates out of nucleoli and reshape them into many small bodies (Appendix Fig S4D). The P3 mutant, which physiologically harbored the CBX4 NLS, did not form nuclear bodies either (Appendix Fig S4E). However, the conjugation of CBox enabled P3 to form nuclear condensates (Appendix Fig S4E). The NLS‐conjugated CBox mutant formed many nuclear puncta which were similar to CBX4 bodies (Appendix Fig S4F). These results indicated that the CBox of CBX4 mediated the LLPS of CBX4 bodies.
To further identify the key amino acids or motif which mediated the LLPS of CBX4, we constructed many CBX4 mutants which harbored limited mutations within CBox. Based on secondary structure prediction, we focused on five motifs on CBox, including Specific peptide 1 (KPF), β‐sheet 1 (IIITD), β‐sheet 2 (LTVT), Specific peptide 2 (FKEY) and the C‐terminal disordered motif (VTV) (Fig 4J). These CBX4 mutants were constructed by mutating these motifs into corresponding numbers of alanine residues. We found that mutations of both β‐sheet 1 (IIITD) and β‐sheet 2 (LTVT) were able to dissolve CBX4 bodies; however, CBX4 bodies still existed upon mutating Specific peptide 1 (KPF), Specific peptide 2 (FKEY) and C‐terminal disordered motif (VTV) (Fig 4K–O). As CBX4‐LTVTmut harbored only four mutations which dissolved CBX4 bodies, we conducted our further investigation utilizing CBX4‐LTVTmut and renamed it as CBX4mut. We purified GFP‐CBX4mut proteins in vitro and conducted in vitro droplet formation assays. We found that GFP‐CBX4mut proteins were unable to form droplets at any NaCl concentration and at any protein concentration (Fig EV4, EV5). Previously, we have found that CBX4 recruited EZH2 to CBX4 bodies, which enabled EZH2 to form similar nuclear bodies. We wondered whether EZH2 still formed nuclear puncta upon mutating CBX4. We co‐overexpressed GFP‐tagged CBX4mut and RFP‐tagged EZH2 and found that both CBX4mut and EZH2 bodies were dissolved (Fig 4P). Besides, we found that CBX4mut was unable to interact with EZH2 and recruit EZH2 on the HIV‐1 LTR (Fig EV4F and G). Taken together, our results indicated that the β‐sheet motifs of CBox determined the LLPS properties of CBX4 bodies and played a key role in recruiting EZH2 to CBX4 bodies.
CBX4 SUMOylates EZH2
Our previous MS data have shown that CBX4 recruited SUMO4 to CBX4 bodies. Many reports also indicated that CBX4 was a SUMO E3 ligase which SUMOylated CtBP, DNMT3A, HNRNPK, PRDM16 and many others (Kagey et al, 2003; Li et al, 2007; Pelisch et al, 2012; Chen et al, 2018). To systematically identify CBX4‐SUMOylated proteins, we conducted global SUMO‐MS assay. We constructed the SUMO4‐Q88R mutant which mimicked yeast SUMO Smt3 to enable efficient identification of SUMO‐acceptor lysines by MS (Hendriks et al, 2014). Then, we co‐overexpressed SUMO4‐Q88R mutant with CBX4 and SUMO E2 UBC9 in HEK293T cells, followed by the enrichment of SUMO4‐conjugated substrates. All these substrates were proceeded to in‐gel digestion, characterized by nanoscale LC‐MS/MS, and annotated by PEAKS Studio. Finally, we identified 1,928 SUMOylated candidates at a significance threshold below 10−7. All these SUMOylated candidates formed a large STRING network at the interaction confidence of 0.7, which could be further clustered into 12 sub‐clusters utilizing k‐means clustering (Fig 5A). Through Gene Ontology (GO) analysis, these candidates could be classified into 21 functional classes, most of which belonged to nucleic acid metabolism proteins, enzymes and translational regulators (Fig 5B). Some chromatin‐binding proteins and chaperones were also among the candidates. Among the highly interconnected STRING networks, we found that many proteins belonged to the mRNA splicing complex, PcG protein complex, NuRD complex and activation of SUMOylation (Fig 5C–F). Remarkably, we found that PcG protein EZH2 was also among the candidates (Fig 5D).
Figure 5. CBX4 SUMOylates EZH2.
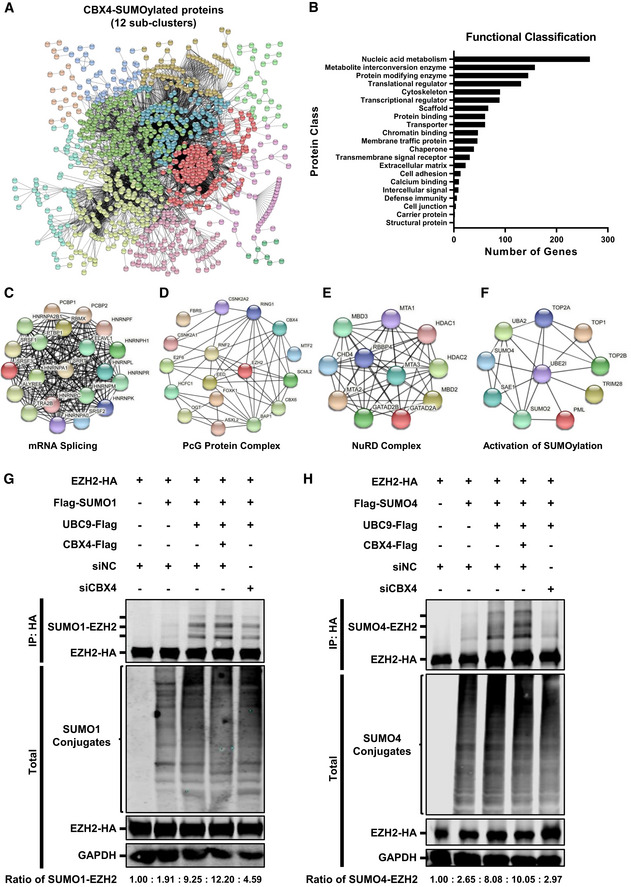
-
ACBX4‐SUMOylated candidates formed a large STRING network, which were further clustered into 12 sub‐clusters utilizing k‐means clustering. These 12 interconnected sub‐clusters are shown in different colors.
-
BSUMOylated proteins were proceeded to Gene Ontology (GO) analysis utilizing PANTHER classification system, which were classified into 21 functional classes.
-
C–FFour highly interconnected STRING networks including mRNA splicing complex, PcG protein complex, NuRD complex and activation of SUMOylation.
-
GHA‐tagged EZH2 was co‐overexpressed with Flag‐tagged SUMO1, Flag‐tagged UBC9 or Flag‐tagged CBX4, and siNC. In the last group, HA‐tagged EZH2 was co‐overexpressed with Flag‐tagged SUMO1, Flag‐tagged UBC9 and siCBX4. EZH2 was IP with anti‐HA beads. Both total and IP samples were IB with antibodies against HA, Flag and GAPDH. The expression ratios of SUMO1‐EZH2 within each group are shown below the figure.
-
HHA‐tagged EZH2 was co‐overexpressed with Flag‐tagged SUMO4, Flag‐tagged UBC9 or Flag‐tagged CBX4, and siNC. In the last group, HA‐tagged EZH2 was co‐overexpressed with Flag‐tagged SUMO4, Flag‐tagged UBC9 and siCBX4. EZH2 was IP with anti‐HA beads. Both total and IP samples were IB with antibodies against HA, Flag and GAPDH. The expression ratios of SUMO4‐EZH2 within each group are shown below the figure.
To verify that EZH2 could be SUMOylated, we co‐overexpressed EZH2 and UBC9 with different SUMO paralogs including SUMO1, SUMO2 and SUMO4. We found that all the three SUMO paralogs were able to conjugate to EZH2, resulting in additional SUMO‐EZH2 Western blotting bands above EZH2 bands (Fig EV5A–C). More importantly, the co‐overexpression of CBX4 with EZH2, UBC9 and SUMO molecules significantly enhanced the SUMOylation of EZH2, resulting in elevated amounts of SUMO‐EZH2 (Fig EV5A–C). Besides, SUMO4‐mediated SUMOylation of EZH2 was more significant compared with SUMO1 and SUMO2. To further verify that EZH2 was SUMOylated by CBX4, we knocked down endogenous CBX4 along with co‐overexpressing EZH2, UBC9 and SUMO molecules. We found that the knockdown of CBX4 significantly decreased the amounts of SUMO1‐EZH2 and SUMO4‐EZH2 (Fig 5G and H, and Appendix Fig S5A and B). Because all the above SUMOylation assays were conducted in an overexpression system, we wondered whether CBX4 could mediate EZH2 SUMOylation in physiological conditions. We isolated primary CD4+ T cells from three healthy donors and transfected these cells with siRNAs targeting CBX4 utilizing 4D‐Nucleofector System. We found that the depletion of CBX4 resulted in a significant decrease of SUMOylated EZH2 (Fig EV5D). We also co‐overexpressed different EZH2 mutants with SUMO4, UBC9 and CBX4 to identify the key SUMOylated region of EZH2. We found that the absence of the N‐terminal of EZH2, ranging from 1 to 68 amino acid residues, disenabled EZH2 SUMOylation (Fig EV5E). Overall, our results indicated that CBX4 utilized SUMO paralogs to SUMOylate EZH2.
LLPS‐deficient CBX4 bodies are unable to suppress HIV‐1 and SUMOylate EZH2
To investigate whether the deficiency of LLPS property of CBX4 body would influence its function on HIV‐1 latency as well as EZH2 SUMOylation, we conducted multiple functional assays with different CBX4 mutants. We performed rescue experiments in TZM‐bl cells and found that the re‐introduction of wild‐type CBX4 in CBX4‐knockdown cells was able to rescue HIV‐1 suppression to the basal level (Fig 6A). The co‐treatment of Tat transfection or TNFα stimulation did not influence wild‐type CBX4‐mediated rescue (Appendix Fig S6A and B). The deletion of Chromodomain (CD) did not influence its suppression on HIV‐1 expression. However, the mutation of two SUMO‐interacting motifs (SIMs), the deletion of CBox and the mutation of LLPS motif within CBox aborted CBX4‐mediated HIV‐1 re‐suppression (Fig 6A, and Appendix Fig S6A–E). These results indicated that both the LLPS property and the SUMO E3 ligase activity of CBX4 contributed to the suppression of HIV‐1 expression. Next, we investigated whether the LLPS of CBX4 influenced its SUMOylation activity. We found that the LLPS‐deficient CBX4mut did not increase the amount of SUMO4‐conjugated EZH2 compared with wild‐type CBX4 (Fig 6B). Subsequently, we evaluated whether CBX4‐mediated EZH2 SUMOylation would influence EZH2‐mediated H3K27me3 modification, and found that the co‐overexpression of SUMO4, UBC9, CBX4 and EZH2 significantly increased the H3K27me3 modification on the HIV‐1 LTR compared with overexpression of EZH2 only (Fig 6C). However, the co‐overexpression of CBX4mut with EZH2, SUMO4 and UBC9 did not increase H3K27me3 signals on HIV‐1 LTR compared with overexpression of EZH2, SUMO4 and UBC9 only (Fig 6C). Besides, the co‐overexpression of SUMO4, UBC9, CBX4 and EZH2 in EZH2‐knockdown cells was able to rescue and enhance global H3K27me3 more significantly than the sole overexpression of EZH2 (Fig 6D). We also conducted in vitro methyltransferase assay to probe the activity of EZH2 in different conditions. In the first group, HEK293T cells were transfected with an empty vector. In the second group, we overexpressed EZH2 in HEK293T cells. In the third group, we co‐overexpressed EZH2 with EED and SUZ12, both of which were major subunits of PRC2. In the fourth and sixth groups, we co‐overexpressed EZH2 with EED, SUZ12, SUMO4, UBC9 and CBX4. In the fifth group, we co‐overexpressed EZH2 catalytic mutant (Y731D, named EZH2mut) with EED, SUZ12, SUMO4, UBC9 and CBX4 (Ernst et al, 2010; Lavarone et al, 2019). Forty‐eight hours post transfection, we enriched EZH2 and EZH2mut proteins and incubated EZH2 or EZH2mut from each group with in vitro purified mononucleosomes and the cofactor S‐adenosyl‐L‐methionine (SAM). We found that the co‐overexpression of EZH2, EED and SUZ12 could potentially enhance EZH2 activity on H3K27me3 modification compared with overexpression of EZH2 only (Fig 6E, and Appendix Fig S6F). Whereas, the co‐overexpression of EZH2 with EED, SUZ12, SUMO4, UBC9 and CBX4 significantly enhanced EZH2‐mediated H3K27me3 modification (Fig 6E). However, Y731D‐mutated EZH2mut was unable to catalyze H3K27me3 modifications on mononucleosomes (Fig 6E). We also incubated EZH2 and CBX4‐SUMOylated EZH2 with H3K27‐mutated mononucleosomes (H3.1 K27 M) and found that no H3K27me3 signals were observed within mononucleosomoes (Fig 6E, and Appendix Fig S6F). Taken together, our results indicated that the LLPS property of CBX4 contributed to the suppression of HIV‐1 expression and the SUMOylation of EZH2. Besides, CBX4‐mediated EZH2 SUMOylation enhanced the methyltransferase activity of EZH2, resulting in elevated H3K27me3 on the HIV‐1 LTR.
Figure 6. LLPS‐deficient CBX4 bodies are unable to suppress HIV‐1 and SUMOylate EZH2.
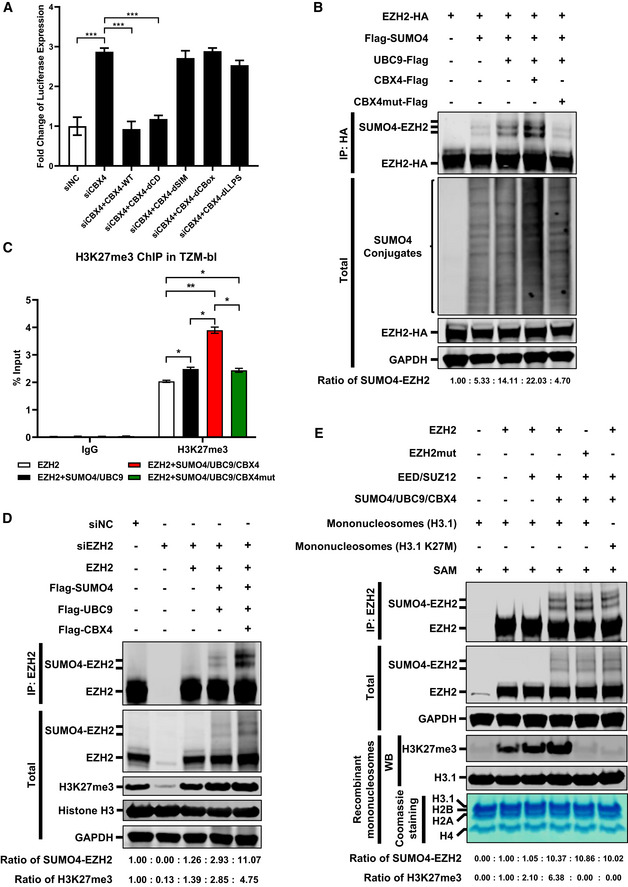
- The endogenous CBX4 was knocked down by siRNA targeting 3’UTR of CBX4 mRNA, followed by the overexpression of wild‐type CBX4 and CBX4 mutants including CBX4‐dCD, CBX4‐dSIM, CBX4‐dCBox and CBX4‐dLLPS. The expression of luciferase within each group was measured and normalized to the siNC group.
- Two micrograms (2 μg) of HA‐tagged EZH2 were co‐overexpressed with 4 μg of Flag‐tagged SUMO4, 250 ng of Flag‐tagged UBC9, 250 ng of Flag‐tagged CBX4 or CBX4mut within 6 cm dishes. The CBX4mut was the LLPS‐deficient CBX4. EZH2 was IP with anti‐HA beads. Both total and IP samples were IB with antibodies against HA, Flag and GAPDH. The expression ratios of SUMO4‐EZH2 are marked below the panel.
- Two micrograms (2 μg) of EZH2 was overexpressed in TZM‐bl cells within 6 cm dishes. In the second group, 2 μg of EZH2 was co‐overexpressed with 4 μg of SUMO4 and 250 ng of UBC9. In the third group, 2 μg of EZH2 was co‐overexpressed with 4 μg of SUMO4, 250 ng of UBC9 and 250 ng of CBX4. In the last group, 2 μg of EZH2 was co‐overexpressed with 4 μg of SUMO4, 250 ng of UBC9 and 250 ng of CBX4mut. ChIP assays with antibodies against IgG and H3K27me3 were performed for each group. Only “B” position signals are shown and normalized to input.
- The endogenous EZH2 in TZM‐bl cells within 6 cm dishes was knocked down by siRNAs targeting 3’UTR of EZH2 mRNA, followed by the overexpression of 2 μg of EZH2, 4 μg of Flag‐tagged SUMO4, 250 ng of Flag‐tagged UBC9 or 250 ng of Flag‐tagged CBX4. EZH2 was IP with anti‐EZH2 antibodies. IP samples were IB with anti‐EZH2 antibodies. Total samples were IB with anti‐EZH2, anti‐H3K27me3, anti‐Histone H3 and anti‐GAPDH antibodies. The expression ratios of SUMO4‐EZH2 and H3K27me3 are marked below the panel.
- In the first group, HEK293T cells within 6 cm dishes were transfected with empty vectors. In the second group, 3 μg of EZH2 was overexpressed. In the third group, 3 μg of EZH2 was co‐overexpressed with 2 μg of EED and 2 μg of SUZ12. In the fourth and sixth group, 3 μg of EZH2 was co‐overexpressed with 2 μg of EED, 2 μg of SUZ12, 2 μg of SUMO4, 500 ng of UBC9 and 2 μg of CBX4. In the fifth group, 3 μg of EZH2mut (Y731D) was co‐overexpressed with 2 μg of EED, 2 μg of SUZ12, 2 μg of SUMO4, 500 ng of UBC9 and 2 μg of CBX4. Forty‐eight hours post transfection, EZH2 and EZH2mut were IP with anti‐EZH2, followed by the incubation with 2 μg of mononucleosomes (H3.1) and 20 μM of the cofactor S‐adenosyl‐L‐methionine (SAM) to measure the methyltransferase activity of EZH2. The IP reaction in the sixth group was incubated with 2 μg of mononucleosomes (H3.1 K27M) and 20 μM of SAM. Both total and IP samples were IB with anti‐EZH2, GAPDH, H3K27me3 and H3.1 antibodies. The purities of recombinant mononucleosomes which included H3.1, H2B, H2A and H4 were verified by Coomassie blue staining. The expression ratios of SUMO4‐EZH2 and H3K27me3 are marked below the panel.
Data information: Data in (A) and (C) are presented as mean ± SEM in biological triplicate. P‐values in (A) were calculated by one‐way ANOVA with Tukey's multiple comparisons test. P‐values in (C) were calculated by two‐way ANOVA with Tukey's multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001.
CBX4 depletion reactivates latent HIV‐1 in cells from HIV‐1‐infected individuals
To evaluate whether CBX4 contributed to HIV‐1 latency in primary CD4+ T cells, we conducted multiple experiments in primary CD4+ T cell latency models and resting CD4+ T cells isolated from HIV‐1‐infected individuals. Firstly, we investigated whether CBX4 could delay HIV‐1 entering into latency. We infected PHA‐activated CD4+ T cells with wild‐type HIV‐1. The wild‐type HIV‐1 model used here harbored the full‐length HIV‐1 genome. All the viral genes including Envelope and Nef were intact. A GFP ORF was in frame with Nef and spaced by a P2A sequence to indicate the intensity of HIV‐1 expression. One day post HIV‐1 infection, these CD4+ T cells were divided into two groups and infected with shluc and shCBX4 respectively. The percentages of GFP‐positive cells and the amounts of intracellular HIV‐1 mRNAs were measured every three days. We found that the depletion of CBX4 significantly delayed HIV‐1 entering into latency, resulting in much higher percentages of HIV‐1 expression cells as well as higher amounts of viral RNAs upon knocking down CBX4 (Fig 7A, and Appendix Fig S7A–D). We also conducted ChIP assays utilizing cells on Day 6 post infection. We found that both H3K27me3 and H2AK119Ub modifications on the HIV‐1 LTR were significantly decreased upon knocking down CBX4 in primary CD4+ T cells (Fig 7B and C). Next, we evaluated whether CBX4 contributed to the maintenance of HIV‐1 latency. Activated CD4+ T cells were infected with wild‐type HIV‐1 followed by the infection of shluc and shCBX4 lentiviruses. Another group of cells were infected with HIV‐1 only and treated as mock sample. Two weeks post infection, one part of HIV‐1‐infected mock cells were reactivated by αCD3/αCD28/IL‐2. One part of shluc‐ and shCBX4‐infected cells were co‐stimulated with LRA JQ‐1. We found that the absence of CBX4 significantly upregulated the expression of latent HIV‐1, which was indicated by both the percentages of GFP‐positive cells and the amounts of intracellular HIV‐1 RNAs (Fig 7D, and Appendix Fig S7E–H). The combination of CBX4 knockdown and JQ‐1 treatment upregulated HIV‐1 expression more significantly compared with separate intervention. To verify whether CBX4 contributed to HIV‐1 latency in resting CD4+ T cells isolated from HIV‐1‐infected individuals, we also knocked down CBX4 in these cells and measured the amount of intracellular HIV‐1 RNAs. We found that the depletion of CBX4 was able to reactivate considerable amount of HIV‐1 RNAs in Patient 1 samples, while the absence of CBX4 only induced slight increase of intracellular HIV‐1 RNAs in samples from Patient 2 and Patient 3 (Fig 7E–G, and Appendix Fig S7I–L). Other possible mechanisms besides CBX4 might also contribute to HIV‐1 silencing in different patient samples, which could result in patient‐dependent variabilities of HIV‐1 reactivation. Overall, our results indicated that CBX4 contributed to HIV‐1 latency in primary CD4+ T cells, and the depletion of CBX4 reactivated latent HIV‐1 in cells isolated from HIV‐1‐infected individuals.
Figure 7. CBX4 depletion reactivates latent HIV‐1 in cells from HIV‐1‐infected individuals.
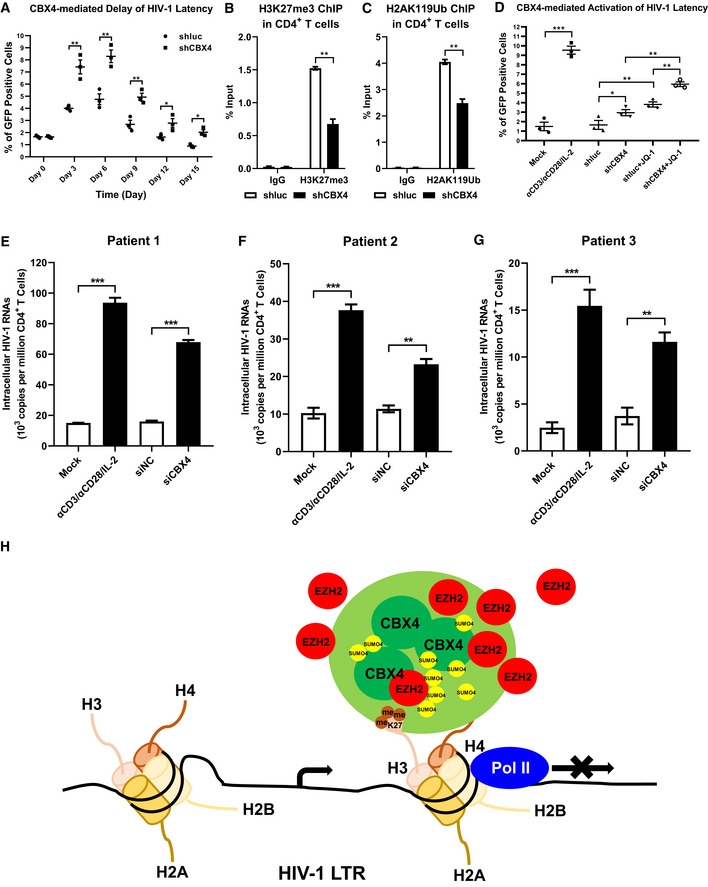
-
APHA‐activated primary CD4+ T cells were infected with wild‐type HIV‐1 viruses which harbored a GFP ORF after Nef gene, followed by infecting with shluc and shCBX4 lentiviruses. The percentages of GFP‐positive cells, which were HIV‐1‐expressing cells, were monitored every three days.
-
B, CPrimary CD4+ T cells were treated as in (A). On Day 6 post infection with HIV‐1 viruses and shRNA lentiviruses, ChIP assays with antibodies against H3K27me3 and H2AK119Ub were conducted in both shluc and shCBX4 groups.
-
DPHA‐activated CD4+ T cells were infected with wild‐type HIV‐1 viruses as in (A). One group of HIV‐1‐infected CD4+ T cells were left untreated. One group of HIV‐1‐infected cells were infected with shluc. Another group of HIV‐1‐infected cells were infected with shCBX4. Two weeks later, one part of HIV‐1‐infected CD4+ T cells were reactivated by αCD3/αCD28/IL‐2. One part of shluc‐ and shCBX4‐infected cells were co‐stimulated with LRA JQ‐1. GFP‐positive cells in each group were measured by flow cytometry to indicate reactivated HIV‐1‐infected cells.
-
E–GPrimary CD4+ T cells, which contained latent HIV‐1‐infected cells, were isolated from three HIV‐1‐infected individuals. One group of cells were untreated. One group of cells were activated with αCD3/αCD28/IL‐2. Another two groups of cells were transfected with siNC and siCBX4 utilizing 4D‐Nucleofector System. Three days post transfection, the amounts of intracellular HIV‐1 RNAs within each group were quantitated by RT‐qPCR and represented as 103 copies per million CD4+ T cells.
-
HThe schematic of CBX4 body‐mediated HIV‐1 latency. Detailed information for the schematic is given in the Discussion section.
Data information: Data are presented as mean ± SEM in biological triplicate. P‐values in (A–C) were calculated by two‐way ANOVA with Sidak's multiple comparisons test. P‐values in (D–G) were calculated by one‐way ANOVA with Tukey's multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
CBX4 was originally identified as a PcG protein involved in proto‐oncogene suppression (Satijn et al, 1997). Then, it was further identified as a SUMO E3 ligase which conjugated SUMO molecules to many cellular proteins (Kagey et al, 2003). In recent years, CBX4 was found to participate in epidermal stem cells differentiation and senescence as well as tumor angiogenesis (Luis et al, 2011; Li et al, 2014; Wang et al, 2016; Hu et al, 2020). Our study reported here showed that CBX4 contributed to HIV‐1 latency by forming LLPS nuclear bodies and SUMOylating EZH2. Based on our data, we proposed a model of CBX4‐mediated HIV‐1 latency (Fig 7H). CBX4 was enriched on the HIV‐1 LTR and formed nuclear bodies with LLPS characteristics. The LLPS of CBX4 relied on its CBox domain. CBX4 bound to H3K27me3 through its Chromodomain (CD), which maintained the H3K27me3 on the HIV‐1 LTR. Besides, CBX4 recruited EZH2 and SUMO4 to CBX4 bodies, and SUMOylated EZH2 with SUMO4 molecules. The SUMOylation of EZH2 enhanced the H3K27me3 catalytic activity, which further maintained H3K27me3‐containing heterochromatin around the HIV‐1 promoter. The formation of CBX4 bodies and H3K27me3‐containing heterochromatin on the HIV‐1 LTR also disenabled RNAP II to enrich on the HIV‐1 promoter, resulting in transcriptional suppression. Our data indicated that CBX4 bridged PRC1 and PRC2 by recruiting and SUMOylating EZH2, resulting in synergistically maintaining HIV‐1 latency.
In recent years, the Polycomb repressive complexes PRC1 and PRC2 have been found to shape the 3D genomic architecture by forming facultative heterochromatin (fHC), mediating chromatin looping and inducing chromatin compaction (Schoenfelder et al, 2015; Eagen et al, 2017; Ogiyama et al, 2018; Oksuz et al, 2018; Boyle et al, 2020). The condensed PRC‐related nuclear compartments act as hubs to suppress the expression of recruited genes simultaneously, which significantly enhances the efficiency of various biological processes including inducing differentiation, safeguarding cellular identity, maintaining proliferation, modulating cellular plasticity, and many others. However, most of these studies focused on RING1B and EZH2 which are major subunits of PRC1 and PRC2, respectively. RING1B‐mediated H2AK119Ub and EZH2‐mediated H3K27me3 are two remarkable signatures of condensed compartments. Our study showed that CBX4 is able to form liquid‐like nuclear condensates, and recruit both RING1B and EZH2 to CBX4 bodies. CBX4 could be the major driving force of coalescing PcG proteins and seeding the suppressive heterochromatin center. We cannot rule out the possibility that other PcG proteins may form similar suppressive hubs. We have found that both CBX2 and CBX8 form nuclear puncta. Other reports also showed that CBX2 is of LLPS characteristics (Plys et al, 2019; Tatavosian et al, 2019). However, we found that CBX2 or CBX8 is unable to recruit EZH2 to its nuclear bodies. CBX2 and CBX8 bodies may represent other forms of PRC1. Future work should focus on the functions of different chromobox proteins‐containing Polycomb repressive complexes.
Our data also showed that CBX4 bodies could be modification centers which contain multiple modification proteins. CBX4‐recruited protein RING1B is a ubiquitin E3 ligase which monoubiquitylates Lysine 119 of Histone H2A (H2AK119Ub) (Tamburri et al, 2020). Another CBX4‐recruited protein EZH2 is a methyltransferase which catalyzes H3K27 trimethylation (van Mierlo et al, 2019). Both RING1B‐mediated H2AK119Ub and EZH2‐mediated H3K27me3 are typical functions of PRC1 and PRC2 respectively, which collaborate to repress gene expression. Besides, CBX4 bodies could also be SUMOylation centers. CBX4 itself is a SUMO E3 ligase which SUMOylates many cellular proteins (Kagey et al, 2003; Li et al, 2007; Pelisch et al, 2012; Chen et al, 2018). Our investigation also showed that CBX4 recruits both SUMO4 and EZH2 to CBX4 bodies and SUMOylates EZH2 with SUMO4 molecules, which significantly enhances the H3K27me3 catalytic activity of EZH2 and upregulates H3K27me3 on the HIV‐1 LTR. We believe that CBX4‐mediated EZH2 SUMOylation could bridge PRC1 and PRC2. Previous reports have shown that PRC2 subunit EZH2‐mediated H3K27me3 can act as docking sites for CBX4 of PRC1 (Guo et al, 2021). Thus, the H3K27me3 modification may serve as the initial signal for the accumulation of CBX4 on the HIV‐1 LTR. Our data show that CBX4 can mediate positive feedback to PRC2 by SUMOylating EZH2 to enhance the H3K27me3 activity of EZH2. The collaboration of CBX4 and EZH2 can orchestrate both PRC1 and PRC2, which significantly maintains the H3K27me3‐containing fHC on target genomic DNA including the HIV‐1 promoter. Other proteins may recruit CBX4 on the HIV‐1 LTR directly. YY1 can recruit PRC1 to its DNA binding site by physically interacting with YAF2 and CBX proteins (Basu et al, 2013). Moreover, YY1 has been found to bind to the HIV‐1 LTR directly and contribute to HIV‐1 latency (Coull et al, 2000; Bernhard et al, 2013). Interestingly, SUV39H1 proteins, which catalyze H3K9me3 modifications on the HIV‐1 LTR, can also methylate CBX4 and recruit PRC1 to cellular gene promoters, which represses the expression of target genes (Sewalt et al, 2002; Yang et al, 2011). We hypothesize that CBX4‐containing PRC1 suppressive complex may be recruited by SUV39H1 to the latent HIV‐1 promoter as well. CpG islands (CGIs) have been found to act as polycomb response elements (PREs) and be recognized by both PRC1 and PRC2 (Ku et al, 2008). Another report shows that KDM2B can recognize non‐methylated DNA in CGIs and recruit PRC1 to these regions (Farcas et al, 2012). We hypothesize that CBX4 and corresponding PRC1 may also be recruited by KDM2B to the HIV‐1 LTR and the transcription start site which harbor two CGIs (Blazkova et al, 2009; Kauder et al, 2009). Future work should focus on the exact mechanisms of how CBX4 is recruited to the HIV‐1 promoter.
Polycomb repressive complexes‐mediated HIV‐1 latency has guided us to develop novel latency‐reversing agents (LRAs). However, until now, only few LRAs targeting PcG proteins have been tested to reactivate latent HIV‐1. Besides, all of these LRAs target PRC2 components. DZNep, GSK126, GSK‐343 and EPZ‐6438, which target EZH2, have been found to downregulate H3K27me3 on the HIV‐1 LTR (Friedman et al, 2011; Matsuda et al, 2015; Nguyen et al, 2017). However, the reduction of H3K27me3 is not specific to HIV‐1. All the global H3K27me3 levels are downregulated, resulting in potential toxicity. Recently, EED226 and A‐395, which target EED of PRC2, are found to reactivate latent HIV‐1 by decreasing H3K27me3 on the HIV‐1 LTR in latency model cell lines (Turner et al, 2020). Whether these two compounds reactivate latent HIV‐1 in primary CD4+ T cells from HIV‐1‐infected individuals needs to be further evaluated. Our study showed that the CBX4 protein of PRC1 promotes HIV‐1 latency by coalescing PRC1 and PRC2 in both latency model cell lines and HIV‐1‐infected primary CD4+ T cells. Developing LRAs targeting CBX4 can reactivate latent HIV‐1 and bypass the potential toxicity of directly inhibiting EZH2. We believe that specifically degrading CBX4 proteins or dissolving phase‐separated CBX4 bodies could be safe and efficient therapeutic interventions to massively reactivate latent HIV‐1.
Materials and Methods
Study participants
Blood samples from healthy donors were obtained from Shenzhen Blood Center. We did not have any interaction with these healthy participants or protected information. Thus, no informed consent was required.
Blood samples from chronically HIV‐1‐infected donors were obtained from the Department of Infectious Diseases, Guangzhou 8th People's Hospital. The Ethics Review Boards of Sun Yat‐sen University and Guangzhou 8th People's Hospital approved this study. All the participants were given written informed consent with the approval of both Ethics Committees. The enrollment of HIV‐1‐infected participants was based on the following criteria: under combined antiretroviral therapy (cART) treatment, long‐term suppression of HIV‐1 viremia, undetectable plasma HIV‐1 RNAs (less than 50 copies per milliliter) and high CD4+ T cells number (more than 350 cells per square milliliter), and all the above requirements have been guaranteed for at least 6 months.
Cell lines and primary cultures
Jurkat cells, HEK293T cells and HeLa cells were obtained from ATCC. TZM‐bl cells were obtained from NIH AIDS Reagent Program. J‐Lat 6.3, 8.4, 9.2, 10.6, 15.4 and A2 cells, which were constructed in Dr. Eric Verdin (The Buck Institute for Research on Aging, Novato, CA, USA) Laboratory, were obtained from Dr. Robert F. Siliciano (Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA) Laboratory. Adherent cells including HEK293T, HeLa and TZM‐bl were cultured in DMEM supplemented with 1% penicillin‐streptomycin (ThermoFisher) and 10% FBS (ThermoFisher). Suspension cells including Jurkat, J‐Lat 6.3, 8.4, 9.2, 10.6, 15.4 and A2 were cultured in RPMI 1640 supplemented with 1% penicillin‐streptomycin and 10% FBS.
Peripheral blood mononuclear cells (PBMCs) and primary CD4+ T cells were isolated and purified from study participants and cultured in RPMI 1640 supplemented with 1% penicillin‐streptomycin and 10% FBS.
All cells were cultured in a sterile incubator at 37°C with 5% CO2. All cells have been tested for mycoplasma utilizing PCR‐based assays and confirmed to be mycoplasma‐free.
Microbe strains
Escherichia coli DH5α (Vazyme), HB101 (Takara), BL21 (Vazyme) and Stbl3 (ThermoFisher) were grown in LB medium with the corresponding antibiotic at 37°C. DH5α was used to propagate most of the protein‐coding constructs including wild‐type CBX4 and corresponding CBX4 mutants. HB101 and Stbl3 were used to propagate lentivirus‐based constructs including shluc, shCBX4, sgNT and sgCBX4 to avoid homologous recombination. BL21 was used to express and purify prokaryon‐expressed proteins including GFP‐CBX4 and GFP‐CBX4mut proteins.
The HIV‐1 infectious clone pNL4‐3 was obtained from NIH AIDS Reagent Program. All the modified clones including pNL4‐3‐P2A‐EGFP (NPG) and pNL4‐3‐ΔEnv/ΔNef‐d2EGFP were constructed from pNL4‐3. These HIV‐1‐related clones were amplified and purified from HB101 to avoid homologous recombination, and transfected to HEK293T cells to package corresponding viruses in Biosafety Level 3 (BSL‐3) facility.
SiRNA library screening
A custom siRNA library which targeted major subunits of PRC1 and PRC2, and negative control siNC were transfected into TZM‐bl cells. Three different siRNAs targeting the same gene were synthesized and transfected as a mixture. Forty‐eight hours post transfection, these transfected cells were lysed with passive lysis buffer (Promega) for 30 min while shaking. Cell lysates were clarified by centrifuging. The supernatant which contained luciferase proteins was proceeded to measure the amounts of luciferase which were indicated by relative luciferase units. Fold changes of luciferase expression in each group were normalized to the siNC group.
shRNA‐mediated knockdown
shRNA constructs targeting CBX4 (shCBX4‐CDS: 5’‐GACGCATCGTGATCGTGAT‐3’; shCBX4‐3UTR: 5’‐GACTCTACTCCCGTTTGTA‐3’) were generated by introducing core sequences into pLKO.3G‐RFP. The pLKO.3G‐RFP backbone was constructed by replacing GFP ORF within pLKO.3G with RFP ORF. shRNA construct targeting luciferase mRNA (shluc: 5’‐ACCGCCTGAAGTCTCTGATTAA‐3’) was used as negative control. These shRNA lentiviruses were packaged by transfecting 10 cm‐dish HEK293T cells with 3 μg of VSV‐G glycoprotein‐expressing vectors, 6 μg of lentiviral packaging constructs psPAX2 and 6 μg of shRNA constructs utilizing Lipofectamine 2000 (ThermoFisher) according to the manufacturer’s instructions. Forty‐eight hours post transfection, shRNA lentiviruses within the supernatant were concentrated with PEG6000. Target cells including J‐Lat cell lines and primary CD4+ T cells were spin‐infected with shluc and shCBX4 respectively. LRAs including SAHA and JQ‐1 were added to each group forty‐eight hours post infection. The infection efficiencies were indicated by the percentages of RFP‐positive cells. The knockdown efficiencies were confirmed by both RT‐qPCR and Western blot. The reactivation efficiencies for GFP‐tagged HIV‐1 were indicated by the percentages of GFP‐positive cells as well as the amounts of HIV‐1 RNAs (HIVTotRNA Forward Primer: 5’‐CTGGCTAACTAGGGAACCCACTGCT‐3’ and HIVTotRNA Reverse Primer: 5’‐GCTTCAGCAAGCCGAGTCCTGCGTC‐3’). The reactivation efficiencies for clinical samples were indicated by the amounts of intracellular HIV‐1 RNAs. Cellular viabilities upon CBX4 knockdown by siRNAs or shRNAs were evaluated by measuring the percentages of amine‐reactive fluorescent dye non‐permeant cells with Zombie Violet Fixable Viability Dye (BioLegend).
CRISPR‐CAS9‐sgRNA‐mediated knockout
sgRNA construct targeting CBX4 (sgCBX4: 5’‐GAGGCAGTTCGCGGTGACGT‐3’) was generated by introducing the core sequence into lentiCRISPRv2.0 backbone. sgRNA targeting dummy guide (sgNT: 5’‐ACGGAGGCTAAGCGTCGCAA‐3’) was used as negative control. These CRISPR/CAS9 sgRNA lentiviruses were packaged as shRNA lentiviruses. J‐Lat 10.6 cells were infected with sgNT and sgCBX4 respectively. Forty‐eight hours post infection, infected cells were treated with 1 μg/ml puromycin (Sigma‐Aldrich) to eliminate uninfected cells. Three days post treatment, cells were transferred into fresh RPMI 1640. The knockout efficiencies were confirmed by Western blot. The reactivation efficiencies were indicated by the percentages of GFP‐positive cells. Cellular viabilities upon CBX4 knockout by sgRNA were evaluated by measuring the percentages of amine‐reactive fluorescent dye non‐permeant cells with Zombie Violet Fixable Viability Dye (BioLegend).
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) assays were performed according to the manufacturer’s instruction (CST). Briefly, four million cells in each group were crosslinked with 1% formaldehyde (Sigma‐Aldrich) and lysed with Buffer A (CST) supplemented with DTT and protease inhibitor cocktail (PIC). The nuclei were pelleted and digested with micrococcal nuclease (CST) in Buffer B (CST), followed by sonication with three sets of 20‐s pulses at 40% amplitude. The supernatant contained digested chromatin was clarified by centrifuging. One‐tenth of the supernatant was proceeded to DNA purification to determine the size distribution and concentration of digested DNA.
For each IP reaction, approximately 10 μg of chromatin was diluted into 500 μl of ChIP Buffer supplemented with PIC. Ten microliters of diluted chromatin was used as the input sample. ChIP antibodies against normal rabbit IgG (CST, 2729), CBX4 (Abcam, ab242149), H3K27me3 (Abcam, ab6002), Pho‐RNAP II (Abcam, ab5095), H2AK119Ub (CST, 8240), H3K27Acetyl (Abcam, ab4729) and EZH2 (Abcam, ab191250) were added into each reaction and incubated for at least 6 h at 4°C while rotating. Antibodies‐bound proteins and DNAs were pull down with ChIP Grade Protein G Magnetic Beads (CST) and eluted with ChIP Elution Buffer (CST). These enriched complexes and input samples were proceeded to DNA purification.
Purified DNA fragments in each group were quantitated by Real‐Time Quantitative PCR. Five pairs of ChIP‐qPCR primers were designed on the HIV‐1 LTR, which included G5: Cellular DNA and viral 5’LTR junction; A: Nucleosome (Nuc) 0 assembly site; B: Nuc‐free region; C: Nuc 1 assembly site; V5: Viral 5’LTR and gag leader sequence junction. ChIP‐qPCR DNA signals were normalized to siNC IgG of G5.
ATAC‐Seq
About 30,000 cells of sgNT and sgCBX4 J‐Lat 10.6 cells were used for building ATAC‐Seq library. The ATAC‐Seq library was built with TruePrep DNA Library Prep Kit V2 (Vazyme) according to the manufacturer’s instructions. Briefly, cells were lysed with ice‐cold lysis buffer (10 mM Tris‐HCl buffered at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Igepal CA‐630), followed by centrifuging to remove supernatant. The nuclear pellets were resuspended with transposition reaction mix and incubated for 30 min at 37°C. Transposed DNAs were purified by VAHTS DNA Clean Beads (Vazyme) and PCR‐amplified. The amplified ATAC‐Seq library was purified again with VAHTS DNA Clean Beads. The library qualities were evaluated by Qubit 3.0 Fluorometer (ThermoFisher) and BioAnalyzer 2100 (Aglient), followed by sequencing with HiSeq X Ten (Illumina) under the PE150 protocol. The ATAC‐Seq reads were trimmed, filtered and quality‐controlled by FastQC. All the reads were aligned to HIV‐1 reference genome K03455, M38432 (Version K03455.1) by Bowtie2, followed by rearranging with Samtools. The sequence reads were also aligned to human reference genome GRCh38. Reads which contained both human integration junction sequence and the HIV‐1 5’LTR DNA were specifically sorted out. These reads were visualized as tag peaks in Igvtools. Tag densities of two groups were calculated by normalizing to the total mapped reads. The highest tag density was set as 100. Relative tag densities of 2 kb ranges centered HIV‐1 5’LTR integration sites were calculated and compared with sgNT.
Mass spectrometry (MS)
To identify CBX4‐recruited proteins, HA‐tagged CBX4 was overexpressed in TZM‐bl cells. The control group was transfected with empty vectors. Forty‐eight hours post transfection, cells were lysed with NP‐40 lysis buffer (10 mM Tris‐HCl buffered at pH 7.5, 150 mM NaCl, 0.5% NP‐40, 1% Triton X‐100, 10% Glycerol, 2 mM EDTA, 1 mM NaF, 1 Mm Na3VO4) supplemented with protease inhibitor cocktail (PIC). HA‐tagged CBX4 was IP with anti‐HA beads, followed by SDS‐PAGE to separate CBX4‐enriched proteins. Distinct gel bands, which contained CBX4‐enriched proteins, as well as control gel bands were proceeded to in‐gel digestion. Digested peptides were extracted with the following extraction solutions sequentially: 50% ACN / 5%TFA, 75% ACN / 0.1% TFA, and 100% ACN. The extracts were subjected to vacuum to remove the solvent, followed by desalting and enriching with C18 ZipTip (Millipore). The enriched peptides were dissolved in 0.01% formic acid and proceeded to nanoscale LC‐MS/MS to characterize digested peptides. The generated raw data was annotated by PEAKS Studio to identify corresponding proteins. Significantly enriched proteins were presented as heatmap.
To identify CBX4‐SUMOylated proteins, HA‐tagged CBX4 was co‐overexpressed with E2 UBC9 and Flag‐tagged SUMO4‐Q88R in HEK293T cells. SUMO4‐Q88R mutants were constructed to mimic yeast SUMO Smt3 to enable efficient identification of SUMO‐acceptor lysines by MS. Forty‐eight hours post transfection, SUMO4‐conjugated proteins were IP with anti‐Flag beads and separated by SDS‐PAGE. Distinct gel bands which contained SUMOylated proteins were proceeded to in‐gel digestion. The digested peptides were enriched and characterized as above. CBX4‐SUMOylated candidates were analyzed by STRING to build the network which was further clustered into 12 sub‐clusters with k‐means clustering analysis.
SIM imaging
To prepare samples which were proceeded to the super‐resolution Structured Illumination Microscopy (SIM) imaging, HEK293T cells were plated into Lab‐Tek II chambered cover glass (ThermoFisher) which was pre‐treated with poly‐lysine (Sigma‐Aldrich). Twenty‐four hours later, cells were transfected with GFP‐tagged or RFP‐tagged constructs. Another 24 h later, cells were fixed with 3% paraformaldehyde (Electron Microscopy Sciences) / 0.1% glutaraldehyde (Electron Microscopy Sciences) followed by permeabilized with 0.2% Triton X‐100 (Sigma‐Aldrich). Subsequently, cells were blocked with 10% normal donkey serum (NDS) (Jackson ImmunoResearch) / 0.05% Triton X‐100. Blocked samples were sequentially incubated with specific primary antibodies and fluorescently‐labeled secondary antibodies. 4', 6‐Diamidino‐2‐Phenylindole, Dihydrochloride (DAPI) (ThermoFisher) solution was added to samples to dye DNA.
Prepared samples were imaged on an Eclipse Ti inverted microscope equipped with a CFI Apo TIRF objective (1.49 NA, oil immersion), a NIS‐Elements AR software, an sCMOS camera (Hamamatsu Flash 4.0, 6.5 μm × 6.5 μm pixel size), and four lasers including SIM 405, SIM 488, SIM 561 and SIM 647. Original images were acquired with 512 × 512 resolution and reconstructed to form the SIM image with 1024 × 1024 resolution. The lateral resolution of reconstructed SIM images was 115 nm. While, the axial resolution was 300 nm. For 3D‐Stack SIM imaging, the Z‐step size was set to 0.20 μm per stack. For each focal plane, fifteen images (5 phases, 3 angles, 3D‐SIM mode) were captured with the NIS‐Elements. SIM images were reconstructed and analyzed with the N‐SIM module of the NIS‐Elements AR software (Nikon). Line scans profiles were also conducted with NIS‐Elements AR software. The size, number, distribution and sphericity of CBX4 bodies were analyzed with Imaris software (Version 9.2) (Bitplane).
ImmunoFISH
Immunolabelling‐based fluorescence in situ hybridization (ImmunoFISH) assays were conducted in naïve and TNFα‐activated J‐Lat 10.6 and 8.4 cells. These suspended cells were grown on coverslips pre‐treated with poly‐lysine, followed by fixing with 4% paraformaldehyde/PBS and permeabilizing with 0.5% Triton X‐100/PBS while shaking. Cells were blocked with 4% BSA/PBS, followed by sequentially incubating with rabbit anti‐CBX4 primary antibodies and goat anti‐rabbit FITC‐conjugated secondary antibodies. Cells were further post‐fixed with 4% paraformaldehyde/PBS, post‐permeabilized with 0.5% Triton X‐100/PBS, and soaked in 20% glycerol/PBS for at least 1 h. These cells were proceeded to six rounds of freezing and thawing in liquid nitrogen and glycerol/PBS, followed by incubating with freshly prepared 0.1 N HCl for 10 min. Cells were permeabilized again and stabilized in 2×SSC (Sigma‐Aldrich). Then, cells were digested with RNase A/2×SSC (100 μg/ml) to remove RNAs. The finished samples were soaked in a hybridization solution (50% formamide/2×SSC) at 4°C overnight.
Probes of HIV‐1 DNA were prepared with a FISH Tag DNA kit (ThermoFisher) according to the manufacturer’s recommendations. Briefly, five micrograms of pNL4‐3 plasmids were used for nick translation reaction to enzymatically incorporate amine‐modified nucleotides into the probe templates. The amine‐modified DNA was purified and labeled with Alexa Fluor 594 dye. Fluorescent dye‐labeled DNA was purified and co‐precipitated with Cot‐1 DNA (ThermoFisher) and herring sperm DNA (Sigma‐Aldrich). The DNA pellet which contained probes was dissolved in 10 μl 100% formamide, followed by adding 10 μl 4 × SSC/20% Dextran. The probe solution was denatured at 95°C for 5 min, followed by quickly inserting into ice to cool for 3 min. The prepared probes were stored at −80°C or used directly for hybridization.
DNA probe solution was diluted with hybridization buffer (50% formamide/10% Dextran/2 × SSC), and denatured at 95°C for 5 min again, followed by quick cooling in the ice. These denatured DNA probes were loaded onto the glass slide and covered with coverslips which contained cell samples prepared above. Edges of coverslips were sealed with rubber cement. The whole slide was denatured at 77.5°C water bath for 6 min and cooled to 37°C gradually. The hybridization was carried out in a box floating at 37°C water bath for 48 h, followed by incubating at 42°C for another 24 h. The rubber cement was removed after hybridization. The coverslips were washed with 2 × SSC, 0.5 × SSC and PBS sequentially. Samples were incubated with DAPI to dye DNA and mounted with antifade mountant (ThermoFisher). The prepared ImmunoFISH samples were imaged with SIM as described above.
Fluorescence recovery after photobleaching (FRAP)
Fluorescence Recovery After Photobleaching (FRAP) assay was conducted to evaluate the internal diffusion and diffusion across boundary characteristics of CBX4 bodies. GFP‐tagged CBX4 was overexpressed in HEK293T cells, followed by live cell imaging with Zeiss LSM 900. Our preliminary experiments have shown that CBX4 proteins could aggregate into hardened condensates upon expressing extremely high amounts of CBX4 proteins in HEK293T cells. These hardened condensates were characterized by both liquid phase and solid phase, resulting in the insensitivity of fluorescence recovery (slower and less recovery). To mimic physiological conditions, HEK293T cells were expressed with equal amounts of GFP‐CBX4 compared with the amounts of endogenous CBX4 proteins. Six CBX4 bodies for each cell were circled as regions of interest (ROIs). Three ROIs were treated as negative control (unbleached regions). The other three ROIs were bleached with strong 488 nm laser power. Live cells were imaged every 4 s. Fluorescence intensities in each time point were recorded and analyzed with Imaris software. The background intensities were subtracted. Fluorescence intensities in each time point were normalized to pre‐bleaching time point. For half‐body FRAP, only half parts of each CBX4 body were bleached with strong laser power. Both bleached and unbleached parts of each CBX4 body were measured for the relative fluorescence intensities at each time point. For two‐color FRAP, RFP‐tagged EZH2, RING1B and SUMO4 were co‐expressed with GFP‐tagged CBX4 in HEK293T cells respectively. These cells were imaged with Zeiss LSM 900. Six co‐localization bodies within each cell were circled as ROIs. Three ROIs were treated as negative control. Three ROIs were co‐bleached with strong 488 and 561 nm laser power. Live cells were imaged every 5 s. Fluorescence intensities in each time point were recorded and analyzed with Imaris software.
Live cell imaging
GFP‐tagged CBX4 proteins were expressed in HEK293T cells and imaged directly with Zeiss LSM 900. HEK293T cells were expressed with equal amounts of GFP‐CBX4 compared with the amounts of endogenous CBX4 proteins. Single cells which expressed GFP‐CBX4 were imaged every 5 s. Two CBX4 bodies which fused into one big CBX4 body were marked and amplified. One CBX4 body which split into two smaller bodies was marked and amplified. For 1,6‐hexanediol experiment, GFP‐CBX4‐expressing HEK293T cells were pre‐treated with Hoechst to dye DNA. These live cells were imaged with Zeiss LSM 900 and captured every 5 s. During the capturing, cells were treated with 1.5% 1,6‐hexanediol to dissolve CBX4 bodies.
In vitro droplet formation assay
The GFP‐CBX4 ORF was cloned into pET28a vector which contained 6 × His tag. The construct was transformed into BL21 (Takara), and amplified in LB with kanamycin while shaking. Upon the OD600 of bacterial solution reached 0.4 to 0.6, isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) (Takara) was added to the solution to induce the expression of GFP‐CBX4 proteins. Eighteen hours later, protein‐expressing bacteria were concentrated in 25 ml of Buffer A (50 mM Tris‐HCl, pH7.5, 500 mM NaCl) supplemented with PIC. Resuspended bacteria were lysed by sonication for 20 min in ice. The sonication was conducted by 5‐s pulse and 5‐s pause in 70% of maximum power. Lysates were clarified and filtered to remove residual bacteria or insoluble components. The His‐tagged GFP‐CBX4 proteins were enriched by Ni‐NTA agarose (QIAGEN), followed by sequentially washing with 30 ml of Buffer B (50 mM Tris‐HCl, pH7.5, 500 mM NaCl, 10 mM Imidazole), 30 ml of Buffer C (50 mM Tris‐HCl, pH7.5, 500 mM NaCl, 20 mM Imidazole) and 30 ml of Buffer D (50 mM Tris‐HCl, pH7.5, 500 mM NaCl, 50 mM Imidazole). Proteins on agarose were eluted with 2 ml of Buffer E (50 mM Tris‐HCl, pH7.5, 500 mM NaCl, 100 mM Imidazole), 2 ml of Buffer F (50 mM Tris‐HCl, pH7.5, 500 mM NaCl, 200 mM Imidazole) and 2 ml of Buffer G (50 mM Tris‐HCl, pH7.5, 500 mM NaCl, 500 mM Imidazole). The eluates which contained GFP‐CBX4 proteins were combined, concentrated and buffer‐exchanged with Buffer H (50 mM Tris‐HCl, pH7.5, 500 mM NaCl, 10% Glycerol, 1 mM DTT). The LLPS‐deficient GFP‐CBX4mut proteins were purified as above. The purities of GFP‐CBX4 and GFP‐CBX4mut proteins were verified by both Coomassie blue staining and Western blotting.
The in vitro purified GFP‐CBX4 or GFP‐CBX4mut proteins were used for in vitro droplet formation. Briefly, proteins were added into different droplet formation buffers which contained different gradients of NaCl. The Buffer I (50 mM Tris‐HCl, pH7.5, 10% Glycerol, 1 mM DTT) was used to dilute NaCl. Droplet formation buffers included Buffer J (50 mM Tris‐HCl, pH7.5, 250 mM NaCl, 10% Glycerol, 1 mM DTT), Buffer K (50 mM Tris‐HCl, pH7.5, 125 mM NaCl, 10% Glycerol, 1 mM DTT), Buffer L (50 mM Tris‐HCl, pH7.5, 62.5 mM NaCl, 10% Glycerol, 1 mM DTT), Buffer M (50 mM Tris‐HCl, pH7.5, 31.25 mM NaCl, 10% Glycerol, 1 mM DTT), Buffer N (50 mM Tris‐HCl, pH7.5, 15.625 mM NaCl, 10% Glycerol, 1 mM DTT). Protein solutions with different NaCl concentrations and different protein concentrations were prepared simultaneously and incubated for 10 min to facilitate protein droplet formation within 8‐well confocal plates. Samples were imaged with Nikon Confocal A1. Laser 488 nm was used to capture droplets. The scan size was 1,024 × 1,024 utilizing CFI Apo TIRF objective (1.49 NA, oil immersion).
SUMOylation assay
Two micrograms (2 μg) of HA‐tagged EZH2 was co‐overexpressed with 4 μg of Flag‐tagged SUMO molecules, 250 ng of E2 UBC9, and/or 250 ng of E3 CBX4/CBX4mut in HeLa cells within each 6 cm dish. The SUMO molecules used in this study included SUMO1, SUMO2 and SUMO4. Forty‐eight hours post transfection, cells were lysed with NP‐40 buffer containing 2 M NEM which was used to prevent deSUMOylation. HA‐tagged EZH2 was IP with anti‐HA beads. Both total and IP samples were IB with antibodies against HA, Flag and GAPDH. The SUMOylated EZH2 would be 20, 40 or 60 kD larger than wildtype EZH2. In experiments to evaluate EZH2 SUMOylation status upon CBX4 knockdown, the endogenous CBX4 was knocked down by siCBX4, followed by the co‐overexpression of EZH2, SUMO molecules and UBC9.
In vitro methyltransferase assay
The in vitro methyltransferase assays were conducted to evaluate the activities of wild‐type EZH2 and SUMOylated EZH2. Six groups of HEK293T cells were prepared. In the first group, cells within 6 cm dishes were transfected equal amounts of empty vectors. In the second group, cells were transfected 3 μg of EZH2 constructs. In the third group, cells were co‐overexpressed with 3 μg of EZH2, 2 μg of EED and 2 μg of SUZ12. In both fourth and sixth groups, cells were co‐overexpressed with 3 μg of EZH2, 2 μg of EED, 2 μg of SUZ12, 2 μg of SUMO4, 500 ng of UBC9 and 2 μg of CBX4. In the fifth group, cells were co‐overexpressed with 3 μg of EZH2mut (Y731D), 2 μg of EED, 2 μg of SUZ12, 2 μg of SUMO4, 500 ng of UBC9 and 2 μg of CBX4. Forty‐eight hours post transfection, cells were harvested and lysed with NP‐40 buffer. EZH2 and corresponding enriched proteins were IP with anti‐EZH2, followed by incubating the protein‐bound beads with 2 μg of mononucleosomes (H3.1) and 20 μM of the cofactor S‐adenosyl‐L‐methionine (SAM). While in the sixth group, the protein‐bound beads were incubated with 2 μg of mononucleosomes (H3.1 K27M) and 20 μM of the cofactor SAM. The reaction was conducted at 30°C for 1 h, followed by adding loading buffer to quench the reaction. The samples were boiled at 100°C for 10 min. EZH2, Histone H3.1 and H3K27me3 were IB with corresponding antibodies. The purity of mononucleosomes which contained H3.1, H2B, H2A and H4 was verified by Coomassie blue staining.
HIV‐1 latency model construction
The pNL4‐3‐ΔEnv/ΔNef‐d2EGFP pseudotyped HIV‐1 construct was used to build Jurkat latency model. HEK293T cells were co‐transfected with 3 μg of VSV‐G constructs and 9 μg of pseudotyped virus constructs. Forty‐eight hours post transfection, pseudotyped viruses within supernatant were concentrated by PEG6000. About 1 × 106 Jurkat cells were infected with pseudotyped viruses. Forty‐eight hours post infection, GFP‐negative cells were sorted by fluorescence‐activated cell sorting (FACS) and cultured for another week to recover. Sorted cells were activated by TNFα, followed by the sorting of GFP‐positive cells. The GFP‐positive cells were cultured for two weeks to recover. Two weeks later, if there were still many GFP‐positive cells, the GFP‐negative cells were sorted out. After several rounds of cell sorting, these infected cells would be totally GFP‐negative. While upon TNFα stimulation, over 50% of these GFP‐negative cells would be reactivated and turned to GFP‐positive. We named this Jurkat latency cell line as J‐Lat Mix which was used for evaluating the contribution of CBX4 on HIV‐1 latency.
The NL4‐3‐P2A‐EGFP (NPG) wildtype HIV‐1 construct was used to build primary CD4+ T cell latency model. The wild‐type virus constructs were transfected to HEK293T cells directly without co‐transfecting VSV‐G to package viruses. Viruses were proceeded to infect PHA‐activated primary CD4+ T cells. During two‐week resting, AZT was added to the culture medium to prevent persistent HIV‐1 infection. Two weeks later, most of these infected cells would be GFP‐negative, which were latently‐infected CD4+ T cells.
HIV‐1 intracellular RNA quantitation
Intracellular total RNAs from clinical samples, which were transfected with siNC or siCBX4 utilizing 4D‐Nucleofector System (Lonza), were extracted with TRIzol reagent (ThermoFisher) according to the manufacturer’s instruction, followed by the treatment of DNase (Promega) to remove residual DNAs. HIV‐1‐specific RNAs were firstly reverse‐transcribed by a reverse primer (5’‐GCTTCAGCAAGCCGAGTCCTGCGTC‐3’). The qPCR reactions were performed to quantitate reverse‐transcribed HIV‐1 cDNAs with the primer pair including HIVTotRNA Forward Primer (5’‐CTGGCTAACTAGGGAACCCACTGCT‐3’) and HIVTotRNA Reverse Primer (5’‐GCTTCAGCAAGCCGAGTCCTGCGTC‐3’). An in vitro transcribed HIV‐1 RNA was used as the external control. The Ct values of each sample were converted to mass and further converted to HIV‐1 RNA copies. The final expression of intracellular HIV‐1 RNAs was represented as 103 copies of viral RNA per million CD4+ T cells for each sample.
Data and statistical analysis
Statistical details for experiments, which included statistical tests, sample numbers, mean values, median values, standard errors of the mean (SEM) and P‐values, have been clearly described in the main text, figure legends and methods. Statistical analyses, which contained Student’s t‐test, one‐way ANOVA with Tukey's multiple comparisons test, two‐way ANOVA with Sidak's multiple comparisons test, and two‐way ANOVA with Tukey's multiple comparisons test, were conducted with Graphpad Prism 8 or Microsoft Excel. The network analysis and k‐means clustering analysis were conducted with STRING. Triplicate data were presented as mean ± SEM. A value of P < 0.05 was considered to be statistically significant and represented as asterisk (*). Value of P < 0.01 was considered to be more statistically significant and represented as double asterisks (**). Value of P < 0.001 was considered to be the most statistically significant and represented as triple asterisks (***).
Author contributions
Liyang Wu: Data curation; Validation; Investigation; Visualization; Methodology. Ting Pan: Data curation; Validation; Investigation; Visualization; Methodology. Mo Zhou: Data curation; Validation; Investigation. Tao Chen: Data curation; Investigation. Shiyu Wu: Data curation; Investigation. Xi Lv: Validation; Investigation; Visualization. Jun Liu: Formal analysis; Visualization; Methodology. Fei Yu: Validation; Investigation. Yuanjun Guan: Investigation; Visualization. Bingfeng Liu: Investigation; Methodology. Wanying Zhang: Investigation; Methodology. Xiaohui Deng: Validation; Investigation. Qianyu Chen: Validation; Investigation; Visualization. Anqi Liang: Validation; Investigation; Visualization. Yingtong Lin: Investigation; Visualization. Lilin Wang: Validation; Investigation; Visualization. Xiaoping Tang: Resources; Validation. Weiping Cai: Resources; Validation. Linghua Li: Resources; Validation. Xin He: Investigation; Visualization. Hui Zhang: Conceptualization; Supervision; Funding acquisition; Writing ‐ original draft; Writing ‐ review & editing. Xiancai Ma: Conceptualization; Data curation; Supervision; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing ‐ original draft; Writing ‐ review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization, HZ and XM; Methodology, LiyW, TP, MZ, TC, SW, XL, JL, FY, YG, BL, WZ, XD, QC, AL, YL, and XM; Software, JL and XM; Validation, LiyW, TP, MZ, TC, and XM; Formal Analysis, LiyW, MZ, and XM; Investigation, LiyW, TP, MZ, TC, SW, XL, JL, FY, YG, BL, WZ, XD, QC, AL, YL, and XM; Resources, FY, YG, BL, WZ, LilW, XT, WC, LL, and XH; Data Curation, LiyW, TP, MZ, and XM; Writing‐Original Draft, HZ and XM; Writing‐Review & Editing, LiyW, TP, HZ, and XM; Visualization, LiyW, TP, MZ, TC, and XM; Supervision, HZ and XM; Funding Acquisition, TP, HZ, and XM.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (82102385) to XM. This work was also supported by the Important Key Program of NSFC (81730060, 92169201), the Exchange Program of NSFC (82150710553), the Special 2019‐nCoV Program of NSFC (82041002), the Emergency Key Program of Guangzhou Laboratory (EKPG21‐24), and the Special Research and Development Program of Guangzhou (202008070010) to HZ. This work was also supported by National NSFC (81971918), Shenzhen Science and Technology Program (Grant No. JSGG20200225150431472 and JCYJ20200109142601702), the Pearl River S&T Nova Program of Guangzhou (201806010118) and the Fundamental Research Funds for the Central Universities of Sun Yat‐sen University (19ykpy164 and 2021qntd43) to TP.
EMBO reports (2022) 23: e53855.
Contributor Information
Hui Zhang, Email: zhangh92@mail.sysu.edu.cn.
Xiancai Ma, Email: maxc6@mail.sysu.edu.cn.
Data availability
The ATAC‐Seq data from this publication have been deposited to the Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra/PRJNA797956) and assigned the identifier (PRJNA797956). The MS data from this publication have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD031011) via the iProX partner repository (https://www.iprox.cn//page/project.html?id=IPX0003941000) and assigned the identifier (PXD031011) (Ma et al, 2018). Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Dr. Xiancai Ma (maxc6@mail.sysu.edu.cn). Plasmids sequences for CBX4 and corresponding mutants will be made available upon request. Purified CBX4 proteins for in vitro experiments will be provided upon execution of a material transfer agreement (MTA) with inquiries directed to Dr. Xiancai Ma.
References
- Ahlenstiel CL, Symonds G, Kent SJ, Kelleher AD (2020) Block and lock HIV cure strategies to control the latent reservoir. Front Cell Infect Microbiol 10: 424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait‐Ammar A, Kula A, Darcis G, Verdikt R, De Wit S, Gautier V, Mallon PWG, Marcello A, Rohr O, Van Lint C (2020) Current status of latency reversing agents facing the heterogeneity of HIV‐1 cellular and tissue reservoirs. Front Microbiol 10: 3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gladfelter A, Mittag T (2019) Considerations and challenges in studying liquid‐liquid phase separation and biomolecular condensates. Cell 176: 419–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, Rosen MK (2016) Compositional control of phase‐separated cellular bodies. Cell 166: 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Wilkinson FH, Colavita K, Fennelly C, Atchison ML (2013) YY1 DNA binding and interaction with YAF2 is essential for Polycomb recruitment. Nucleic Acids Res 42: 2208–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard W, Barreto K, Raithatha S, Sadowski I (2013) An upstream YY1 binding site on the HIV‐1 LTR contributes to latent infection. PLoS One 8: e77052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Trejbalova K, Gondois‐Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J et al (2009) CpG methylation controls reactivation of HIV from latency. PLoS Pathog 5: e1000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Jeng M, Camus G, Gramatica A, Schwarzer R, Johnson JR, Hull PA, Montano M, Sakane N, Pagans S et al (2017) SMYD2‐mediated histone methylation contributes to HIV‐1 latency. Cell Host Microbe 21: 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM et al (2018) Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell 175: 1842–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle S, Flyamer IM, Williamson I, Sengupta D, Bickmore WA, Illingworth RS (2020) A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev 34: 931–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Tompa P, Pappu Rohit V (2015) Polymer physics of intracellular phase transitions. Nat Phys 11: 899–904 [Google Scholar]
- Budhiraja S, Famiglietti M, Bosque A, Planelles V, Rice AP (2013) Cyclin T1 and CDK9 T‐loop phosphorylation are downregulated during establishment of HIV‐1 latency in primary resting memory CD4+ T cells. J Virol 87: 1211–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC‐seq: a method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol 109: 21.29.21–21.29.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CR, Asfaha JB, Ghent CM, Howard CJ, Hartooni N, Safari M, Frankel AD, Morgan DO (2020) Phosphoregulation of phase separation by the SARS‐CoV‐2 N protein suggests a biophysical basis for its dual functions. Mol Cell 80: 1092–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Huang L, Pan D, Zhu LJ, Wang Y‐X (2018) Cbx4 sumoylates Prdm16 to regulate adipose tissue thermogenesis. Cell Rep 22: 2860–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chéné ID, Basyuk E, Lin Y‐L, Triboulet R, Knezevich A, Chable‐Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P et al (2007) Suv39H1 and HP1γ are responsible for chromatin‐mediated HIV‐1 transcriptional silencing and post‐integration latency. EMBO J 26: 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun T‐W, Stuyver L, Mizell SB, Ehler LA, Mican JAM, Baseler M, Lloyd AL, Nowak MA, Fauci AS (1997) Presence of an inducible HIV‐1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA 94: 13193–13197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn LB, Chomont N, Deeks SG (2020) The biology of the HIV‐1 latent reservoir and implications for cure strategies. Cell Host Microbe 27: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad RJ, Fozouni P, Thomas S, Sy H, Zhang Q, Zhou M‐M, Ott M (2017) The short isoform of BRD4 promotes HIV‐1 latency by engaging repressive SWI/SNF chromatin‐remodeling complexes. Mol Cell 67: 1001–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet A, Kleijwegt C, Roubille S, Juillard F, Jacquet K, Texier P, Lomonte P (2020) PML nuclear bodies and chromatin dynamics: catch me if you can! Nucleic Acids Res 48: 11890–11912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun J‐M, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM (2000) The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol 74: 6790–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport MP, Khoury DS, Cromer D, Lewin SR, Kelleher AD, Kent SJ (2019) Functional cure of HIV: the scale of the challenge. Nat Rev Immunol 19: 45–54 [DOI] [PubMed] [Google Scholar]
- Ding D, Qu X, Li L, Zhou X, Liu S, Lin S, Wang P, Liu S, Kong C, Wang X et al (2013) Involvement of histone methyltransferase GLP in HIV‐1 latency through catalysis of H3K9 dimethylation. Virology 440: 182–189 [DOI] [PubMed] [Google Scholar]
- Dupont L, Bloor S, Williamson JC, Cuesta SM, Shah R, Teixeira‐Silva A, Naamati A, Greenwood EJD, Sarafianos SG, Matheson NJ et al (2021) The SMC5/6 complex compacts and silences unintegrated HIV‐1 DNA and is antagonized by Vpr. Cell Host Microbe 29: 792–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagen KP, Aiden EL, Kornberg RD (2017) Polycomb‐mediated chromatin loops revealed by a subkilobase‐resolution chromatin interaction map. Proc Natl Acad Sci USA 114: 8764–8769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einkauf KB, Osborn MR, Gao CE, Sun W, Sun X, Lian X, Parsons EM, Gladkov GT, Seiger KW, Blackmer JE et al (2022) Parallel analysis of transcription, integration, and sequence of single HIV‐1 proviruses. Cell 185: 266–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo‐Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A et al (2010) Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42: 722–726 [DOI] [PubMed] [Google Scholar]
- Farcas AM, Blackledge NP, Sudbery I, Long HK, McGouran JF, Rose NR, Lee S, Sims D, Cerase A, Sheahan TW et al (2012) KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 1: e00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R et al (1997) Identification of a reservoir for HIV‐1 in patients on highly active antiretroviral therapy. Science 278: 1295–1300 [DOI] [PubMed] [Google Scholar]
- Friedman J, Cho W‐K, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J (2011) Epigenetic silencing of HIV‐1 by the histone H3 lysine 27 methyltransferase enhancer of zeste 2. J Virol 85: 9078–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille J‐H, Afeyan LK, Zamudio AV, Shrinivas K et al (2019) Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572: 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zhao S, Wang GG (2021) Polycomb gene silencing mechanisms: PRC2 chromatin targeting, H3K27me3 'Readout', and phase separation‐based compaction. Trends Genet 37: 547–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Margolis DM (2002) Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV‐1) by the HIV‐1 repressor YY1 and HIV‐1 activator tat. Mol Cell Biol 22: 2965–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich BS, Maliga Z, Stein DA, Hyman AA, Whelan SPJ, Palese P (2018) Phase transitions drive the formation of vesicular stomatitis virus replication compartments. MBio 9: e02290‐02217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks IA, D'Souza RCJ, Yang B, Verlaan‐de Vries M, Mann M, Vertegaal ACO (2014) Uncovering global SUMOylation signaling networks in a site‐specific manner. Nat Struct Mol Biol 21: 927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Zhang Q, Tang Q, Zhou H, Liu W, Huang J, Liu Y, Wang Q, Zhang J, Zhou M et al (2020) CBX4 promotes the proliferation and metastasis via regulating BMI‐1 in lung cancer. J Cell Mol Med 24: 618–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, Jülicher F (2014) Liquid‐liquid phase separation in biology. Annu Rev Cell Dev Biol 30: 39–58 [DOI] [PubMed] [Google Scholar]
- Imai K, Togami H, Okamoto T (2010) Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV‐1 latency and its reactivation by BIX01294 *. J Biol Chem 285: 16538–16545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens J, Bruggemans A, Christ F, Debyser Z (2021) Towards a functional cure of HIV‐1: insight into the chromatin landscape of the provirus. Front Microbiol 12: 391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferys SR, Burgos SD, Peterson JJ, Selitsky SR, Turner A‐M, James LI, Tsai Y‐H, Coffey AR, Margolis DM, Parker J et al (2021) Epigenomic characterization of latent HIV infection identifies latency regulating transcription factors. PLoS Pathog 17: e1009346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E (2003) HIV reproducibly establishes a latent infection after acute infection of T cells in vitro . EMBO J 22: 1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Melhuish TA, Wotton D (2003) The polycomb protein Pc2 is a SUMO E3. Cell 113: 127–137 [DOI] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E (2009) Epigenetic regulation of HIV‐1 latency by cytosine methylation. PLoS Pathog 5: e1000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Iqbal M, Tariq M, Baig SM, Abbas W (2018) Epigenetic regulation of HIV‐1 latency: focus on polycomb group (PcG) proteins. Clin Epigenetics 10: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G, Darcis G, Lee MY, Bouchat S, Van Driessche B, Purcell DFJ, Van Lint C (2018) The molecular biology of HIV latency. Adv Exp Med Biol 1075: 187–212 [DOI] [PubMed] [Google Scholar]
- Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu S‐Y, Chiang C‐M, Karn J (2006) Recruitment of TFIIH to the HIV LTR is a rate‐limiting step in the emergence of HIV from latency. EMBO J 25: 3596–3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita S, Chen BK, Kaneshima H, Nolan GP (1998) Host control of HIV‐1 parasitism in T cells by the nuclear factor of activated T cells. Cell 95: 595–604 [DOI] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS et al (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ (2017) Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547: 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavarone E, Barbieri CM, Pasini D (2019) Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat Commun 10: 1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhou J, Liu P, Hu J, Jin H, Shimono Y, Takahashi M, Xu G (2007) Polycomb protein Cbx4 promotes SUMO modification of de novo DNA methyltransferase Dnmt3a. Biochem J 405: 369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Xu Y, Long X‐D, Wang W, Jiao H‐K, Mei Z, Yin Q‐Q, Ma L‐N, Zhou A‐W, Wang L‐S et al (2014) Cbx4 governs HIF‐1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 25: 118–131 [DOI] [PubMed] [Google Scholar]
- Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q (2018) Phase‐separation mechanism for C‐terminal hyperphosphorylation of RNA polymerase II. Nature 558: 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis NM, Morey L, Mejetta S, Pascual G, Janich P, Kuebler B, Roma G, Nascimento E, Frye M, Di Croce L et al (2011) Regulation of human epidermal stem cell proliferation and senescence requires polycomb‐ dependent and ‐independent functions of Cbx4. Cell Stem Cell 9: 233–246 [DOI] [PubMed] [Google Scholar]
- Lusic M, Marini B, Ali H, Lucic B, Luzzati R, Giacca M (2013) Proximity to PML nuclear bodies regulates HIV‐1 latency in CD4+ T cells. Cell Host Microbe 13: 665–677 [DOI] [PubMed] [Google Scholar]
- Ma J, Chen T, Wu S, Yang C, Bai M, Shu K, Li K, Zhang G, Jin Z, He F et al (2018) iProX: an integrated proteome resource. Nucleic Acids Res 47: D1211–D1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Chen T, Peng Z, Wang Z, Liu J, Yang T, Wu L, Liu G, Zhou MO, Tong M et al (2021) Histone chaperone CAF‐1 promotes HIV‐1 latency by leading the formation of phase‐separated suppressive nuclear bodies. EMBO J 40: e106632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Yang T, Luo Y, Wu L, Jiang Y, Song Z, Pan T, Liu B, Liu G, Liu J et al (2019) TRIM28 promotes HIV‐1 latency by SUMOylating CDK9 and inhibiting P‐TEFb. eLife 8: e42426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O (2007) Recruitment of chromatin‐modifying enzymes by CTIP2 promotes HIV‐1 transcriptional silencing. EMBO J 26: 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini B, Kertesz‐Farkas A, Ali H, Lucic B, Lisek K, Manganaro L, Pongor S, Luzzati R, Recchia A, Mavilio F et al (2015) Nuclear architecture dictates HIV‐1 integration site selection. Nature 521: 227–231 [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Kobayashi‐Ishihara M, Fujikawa D, Ishida T, Watanabe T, Yamagishi M (2015) Epigenetic heterogeneity in HIV‐1 latency establishment. Sci Rep 5: 7701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbonye U, Karn J (2017) The molecular basis for human immunodeficiency virus latency. Annu Rev Virol 4: 261–285 [DOI] [PubMed] [Google Scholar]
- van Mierlo G, Veenstra GJC, Vermeulen M, Marks H (2019) The complexity of PRC2 subcomplexes. Trends Cell Biol 29: 660–671 [DOI] [PubMed] [Google Scholar]
- Monette A, Niu M, Chen L, Rao S, Gorelick RJ, Mouland AJ (2020) Pan‐retroviral nucleocapsid‐mediated phase separation regulates genomic RNA positioning and trafficking. Cell Rep 31: 107520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel G, Baltimore D (1987) An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326: 711–713 [DOI] [PubMed] [Google Scholar]
- Nguyen K, Das B, Dobrowolski C, Karn J, Goff SP, Rice A, Zack J, Marsden M, Kashanchi F (2017) Multiple histone lysine methyltransferases are required for the establishment and maintenance of HIV‐1 latency. MBio 8: e00133‐00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiyama Y, Schuettengruber B, Papadopoulos GL, Chang J‐M, Cavalli G (2018) Polycomb‐dependent chromatin looping contributes to gene silencing during Drosophila development. Mol Cell 71: 73–88 [DOI] [PubMed] [Google Scholar]
- Oksuz O, Narendra V, Lee C‐H, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA et al (2018) Capturing the onset of PRC2‐mediated repressive domain formation. Mol Cell 70: 1149–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Verdin E (2013) Three rules for HIV latency: location, location, and location. Cell Host Microbe 13: 625–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios JA, Pérez‐Piñar T, Toro C, Sanz‐Minguela B, Moreno V, Valencia E, Gómez‐Hernando C, Rodés B (2012) Long‐term nonprogressor and elite controller patients who control viremia have a higher percentage of methylation in their HIV‐1 proviral promoters than aviremic patients receiving highly active antiretroviral therapy. J Virol 86: 13081–13084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Pozzi B, Risso G, Muñoz MJ, Srebrow A (2012) DNA damage‐induced heterogeneous nuclear ribonucleoprotein K SUMOylation regulates p53 transcriptional activation *. J Biol Chem 287: 30789–30799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ (1993) A cooperative interaction between NF‐kappa B and Sp1 is required for HIV‐1 enhancer activation. EMBO J 12: 3551–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Y‐H, Rana TM (2001) DSIF and NELF interact with RNA polymerase II elongation complex and HIV‐1 tat stimulates P‐TEFb‐mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation *. J Biol Chem 276: 12951–12958 [DOI] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D (1998) Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72: 2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, Kingston RE (2019) Phase separation of Polycomb‐repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev 33: 799–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC et al (2018) Coactivator condensation at super‐enhancers links phase separation and gene control. Science 361: eaar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, Narlikar GJ (2019) HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satijn DP, Olson DJ, Jvd V, Hamer KM, Lambrechts C, Masselink H, Gunster MJ, Sewalt RG, Rv D, Otte AP (1997) Interference with the expression of a novel human polycomb protein, hPc2, results in cellular transformation and apoptosis. Mol Cell Biol 17: 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savastano A, Ibáñez de Opakua A, Rankovic M, Zweckstetter M (2020) Nucleocapsid protein of SARS‐CoV‐2 phase separates into RNA‐rich polymerase‐containing condensates. Nat Commun 11: 6041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Sugar R, Dimond A, Javierre B‐M, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares‐Cadete F, Furlan‐Magaril M et al (2015) Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47: 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewalt RGAB, Lachner M, Vargas M, Hamer KM, Blaauwen JLd, Hendrix T, Melcher M, Schweizer D, Jenuwein T, Otte AP (2002) Selective interactions between vertebrate polycomb homologs and the SUV39H1 histone lysine methyltransferase suggest that histone H3–K9 methylation contributes to chromosomal targeting of polycomb group proteins. Mol Cell Biol 22: 5539–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Li R, Negro R, Cheng J, Vora SM, Fu T‐M, Wang A, He K, Andreeva L, Gao PU et al (2021) Phase separation drives RNA virus‐induced activation of the NLRP6 inflammasome. Cell 184: 5759–5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF (2003) Long‐term follow‐up studies confirm the stability of the latent reservoir for HIV‐1 in resting CD4+ T cells. Nat Med 9: 727–728 [DOI] [PubMed] [Google Scholar]
- Spivak AM, Planelles V (2018) Novel latency reversal agents for HIV‐1 cure. Annu Rev Med 69: 421–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH (2017) Phase separation drives heterochromatin domain formation. Nature 547: 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburri S, Lavarone E, Fernández‐Pérez D, Conway E, Zanotti M, Manganaro D, Pasini D (2020) Histone H2AK119 mono‐ubiquitination is essential for polycomb‐mediated transcriptional repression. Mol Cell 77: 840–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavosian R, Kent S, Brown K, Yao T, Duc HN, Huynh TN, Zhen CY, Ma B, Wang H, Ren X (2019) Nuclear condensates of the polycomb protein chromobox 2 (CBX2) assemble through phase separation. J Biol Chem 294: 1451–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejbalová K, Kovářová D, Blažková J, Machala L, Jilich D, Weber J, Kučerová D, Vencálek O, Hirsch I, Hejnar J (2016) Development of 5‘ LTR DNA methylation of latent HIV‐1 provirus in cell line models and in long‐term‐infected individuals. Clin Epigenetics 8: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy MK, McManamy MEM, Burch BD, Archin NM, Margolis DM, Silvestri G (2015) H3K27 demethylation at the proviral promoter sensitizes latent HIV to the EFFECTS OF VORInostat in ex vivo cultures of resting CD4+ T cells. J Virol 89: 8392–8405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A‐M, Dronamraju R, Potjewyd F, James KS, Winecoff DK, Kirchherr JL, Archin NM, Browne EP, Strahl BD, Margolis DM et al (2020) Evaluation of EED inhibitors as a class of PRC2‐targeted small molecules for HIV latency reversal. ACS Infect Dis 6: 1719–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdikt R, Bouchat S, Pasternak AO, Nestola L, Darcis G, Avettand‐Fenoel V, Vanhulle C, Aït‐Ammar A, Bendoumou M, Plant E et al (2021) Novel role of UHRF1 in DNA methylation‐mediated repression of latent HIV‐1. bioRxiv 10.1101/2021.06.15.448539 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Wang L, Gao Y, Zheng X, Liu C, Dong S, Li RU, Zhang G, Wei Y, Qu H, Li Y et al (2019) Histone modifications regulate chromatin compartmentalization by contributing to a phase separation mechanism. Mol Cell 76: 646–659 [DOI] [PubMed] [Google Scholar]
- Wang S, Dai T, Qin Z, Pan T, Chu F, Lou L, Zhang L, Yang B, Huang H, Lu H et al (2021) Targeting liquid–liquid phase separation of SARS‐CoV‐2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat Cell Biol 23: 718–732 [DOI] [PubMed] [Google Scholar]
- Wang X, Li L, Wu Y, Zhang R, Zhang M, Liao D, Wang G, Qin G, Xu R‐H, Kang T (2016) CBX4 suppresses metastasis via recruitment of HDAC3 to the Runx2 promoter in colorectal carcinoma. Can Res 76: 7277–7289 [DOI] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Günthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD (1997) Recovery of replication‐competent HIV despite prolonged suppression of plasma viremia. Science 278: 1291–1295 [DOI] [PubMed] [Google Scholar]
- Yang L, Lin C, Liu W, Zhang J, Ohgi Kenneth A, Grinstein Jonathan D, Dorrestein Pieter C, Rosenfeld Michael G (2011) ncRNA‐ and Pc2 methylation‐dependent gene relocation between nuclear structures mediates gene activation programs. Cell 147: 773–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Chen Y, Gabuzda D (1999) ERK MAP kinase links cytokine signals to activation of latent HIV‐1 infection by stimulating a cooperative interaction of AP‐1 and NF‐κB *. J Biol Chem 274: 27981–27988 [DOI] [PubMed] [Google Scholar]
- Yoon CH, Jang DH, Kim KC, Park SY, Kim H, Kim SS, Chi SG, Choi BS (2014) Disruption of polycomb repressor complex‐mediated gene silencing reactivates HIV‐1 provirus in latently infected cells. Intervirology 57: 116–120 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Su JM, Samuel CE, Ma D, Dutch RE (2019) Measles virus forms inclusion bodies with properties of liquid organelles. J Virol 93: e00948‐00919 [DOI] [PMC free article] [PubMed] [Google Scholar]