

Abstract
Findings of early cerebral amyloid‐β deposition in mice after peripheral injection of amyloid‐β‐containing brain extracts, and in humans following cadaveric human growth hormone treatment raised concerns that amyloid‐β aggregates and possibly Alzheimer’s disease may be transmissible between individuals. Yet, proof that Aβ actually reaches the brain from the peripheral injection site is lacking. Here, we use a proteomic approach combining stable isotope labeling of mammals and targeted mass spectrometry. Specifically, we generate 13C‐isotope‐labeled brain extracts from mice expressing human amyloid‐β and track 13C‐lysine‐labeled amyloid‐β after intraperitoneal administration into young amyloid precursor protein‐transgenic mice. We detect injected amyloid‐β in the liver and lymphoid tissues for up to 100 days. In contrast, injected 13C‐lysine‐labeled amyloid‐β is not detectable in the brain whereas the mice incorporate 13C‐lysine from the donor brain extracts into endogenous amyloid‐β. Using a highly sensitive and specific proteomic approach, we demonstrate that amyloid‐β does not reach the brain from the periphery. Our study argues against potential transmissibility of Alzheimer’s disease while opening new avenues to uncover mechanisms of pathophysiological protein deposition.
Keywords: Alzheimer’s disease, brain, multiple reaction monitoring immuno‐mass spectrometry, prion‐like, seeding
Subject Categories: Molecular Biology of Disease, Neuroscience
Stable isotope‐labeled, brain‐derived amyloid‐β does not enter the brain after peripheral injection into mice. These findings argue against transmissibility of amyloid‐β aggregates and consequently Alzheimer’s disease between individuals.

Introduction
Cerebral deposition of amyloid‐β (Aβ), a cleavage product of the amyloid precursor protein (APP), is thought to be integral to the pathogenesis of Alzheimer’s disease (AD; Hardy & Higgins, 1992; Selkoe, 2001). Aβ can form aggregates that expose their surface structure to soluble monomeric Aβ peptides leading to conformational adaption and growing agglomerates (Cohen et al, 2013; Meisl et al, 2014). It was shown in pre‐depositing APP‐transgenic (tg) mice that intraperitoneal (i.p.), intravenous, intramuscular, and intraocular administration of Aβ‐containing brain extract from aged APPtg donor mice or AD patients accelerate Aβ deposition in the meninges and brain, suggesting that Aβ or its so‐called seeds may be transported from the periphery to the brain (Eisele et al, 2010, 2014; Burwinkel et al, 2018; Morales et al, 2021). In the absence of proven transmissibility (Eisele, 2013; Irwin et al, 2013; Asher et al, 2020), these findings were denoted as prion‐like properties of Aβ (Jucker & Walker, 2013). Similarly, early cerebral Aβ deposition was reported in around 50% of the individuals in cohorts that had received treatment with cadaveric human growth hormone (c‐hGH) during childhood and died of iatrogenic Creutzfeldt‐Jakob disease or other causes at a young age (Jaunmuktane et al, 2015; Ritchie et al, 2017; Cali et al, 2018). Certain c‐hGH batches used to treat these patients were shown to contain Aβ and to accelerate Aβ deposition in the brains of APP‐knockin mice after intracerebral injection (Purro et al, 2018). These findings raised concern that Aβ pathology and possibly AD may be transmissible between humans. However, another study found evidence of cerebral β‐amyloidosis in only 1 of 24 iatrogenic Creutzfeldt‐Jakob disease cases following c‐hGH treatment even though Aβ was present in various hGH batches used for the treatment (Duyckaerts et al, 2018).
In general, it remains contentious whether cerebral β‐amyloidosis was induced by seeding from the periphery or by other causes. Underlying neuroendocrine disorders in c‐hGH recipients (Adams et al, 2016; Feeney et al, 2016) or inflammatory stimuli in the Aβ‐containing brain extract injected into mice could also exacerbate Aβ pathology as shown for peripheral injection of lipopolysaccharides in APPtg mice (Wendeln et al, 2018). Moreover, Aβ from donor mice is not biochemically or histologically differentiable from endogenous Aβ in injected host mice, leaving the interpretation that Aβ indeed entered the brain from the periphery speculative. To overcome this key limitation, we combined stable isotope labeling of donor brains and highly sensitive multiple reaction monitoring immuno‐mass spectrometry (MRM‐IMS). This approach enabled us to investigate whether brain‐derived, labeled Aβ reaches the brain from the periphery.
Results and Discussion
Labeling of donor brains
To generate brain extracts containing labeled Aβ that can be differentiated from endogenous Aβ in injected host mice without altering its chemical or conformational characteristics, we fed APPtg mice (APP/PS1, a model featuring early onset of Aβ pathology at 6–8 weeks of age; Radde et al, 2006) a diet containing the fully 13C‐substituted version of lysine (13C‐Lys, 6 additional neutrons; Kruger et al, 2008), starting at 50 or 75 days of age with different feeding durations (Table 1). We assessed the labeling efficiency and concentration of Aβ by mass spectrometry (MS) and immunoassay, respectively (Fig 1A). The percentage of 13C‐Lys‐labeled Aβ increased with both longer feeding duration (50 vs. 125 days of feeding, P < 0.0001) and earlier commencement of feeding (start at 50 vs. 75 days of age, P < 0.0001, Fig 1B–D, Table 1). Correspondingly, the absolute concentration of 13C‐Lys‐labeled Aβ increased with longer feeding duration. Interestingly, the absolute concentration of unlabeled Aβ was 5.0 ± 1.3 ng/mg brain when feeding started at 75 days of age, and 1.7 ± 0.3 ng/mg brain when commenced at 50 days of age, independent of the total feeding time. This implies that approximately 3 ng Aβ/mg brain were newly generated between the ages of 50 and 75 days and that insoluble Aβ that had been generated when labeling started was not subject to dynamic turnover (Fig 1E). Additionally, overall labeling efficiency was assessed by MS in four mice labeled from 50–150 days of age, revealing 97.2 ± 0.2% overall 13C‐Lys‐labeling of the brain proteome (Dataset EV1).
Table 1.
13C‐Lys‐labeling of APPtg donor brains for 13C‐Lys APPtg brain extract generation.
| Animal ID | Age (days) | 13C‐Lys diet (days) | ng Aβ42/mg brain | Ratio 13C‐Lys/unlabeled Aβ17‐28 | %13C‐Lys Aβ17‐28 | Brain extract cohort |
|---|---|---|---|---|---|---|
| 19290 | 125 | 75–125 | 11.9 | 1.4 | 57.6 | 1 |
| 19291 | 175 | 75–175 | 10.5 | 2.9 | 74.5 | 1 |
| 19293 | 175 | 75–175 | 21.7 | 2.4 | 70.2 | 1 |
| 19299 | 175 | 75–175 | 20.7 | 2.8 | 73.5 | 1 |
| 19300 | 175 | 75–175 | 20.1 | 2.7 | 72.9 | 1 |
| 19644 | 100 | 50–100 | 4.0 | 2.2 | 68.7 | 1 |
| 19646 | 100 | 50–100 | 5.2 | 2.5 | 71.8 | 1 |
| 19651 | 100 | 50–100 | 6.0 | 2.3 | 70.0 | 1 |
| 19696 | 100 | 50–100 | 5.3 | 2.5 | 71.4 | 1 |
| 19698 | 100 | 50–100 | 5.9 | 2.2 | 68.8 | 1 |
| 27279 | 175 | 50–175 | 12.6 | 8.8 | 89.8 | 2 |
| 27303 | 175 | 50–175 | 16.7 | 7.0 | 87.5 | 2 |
| 27324 | 175 | 50–175 | 18.9 | 7.2 | 87.8 | 2 |
| 27333 | 175 | 50–175 | 11.1 | 5.7 | 85.0 | 2 |
| 27335 | 175 | 50–175 | 13.1 | 8.0 | 88.9 | 2 |
| 27341 | 175 | 50–175 | 13.9 | 8.2 | 89.1 | 2 |
| 27380 | 175 | 50–175 | 13.9 | 8.3 | 89.3 | 2 |
| 27409 | 175 | 50–175 | 13.9 | 7.3 | 87.9 | 2 |
| 27410 | 175 | 50–175 | 16.1 | 7.0 | 87.5 | 2 |
| 35073 | 125 | 50–125 | 6.4 | 3.8 | 79.3 | 3 |
| 35074 | 125 | 50–125 | 6.5 | 4.1 | 80.4 | 3 |
| 36356 | 150 | 50–150 | 13.1 | 5.6 | 84.8 | 4 |
| 36359 | 150 | 50–150 | 13.3 | 5.4 | 84.3 | 4 |
| 36371 | 150 | 50–150 | 14.8 | 6.1 | 85.9 | 4 |
| 36373 | 150 | 50–150 | 10.8 | 5.8 | 85.3 | 4 |
Animal ID number, age at death, age during which 13C‐Lys‐labeled diet was fed, amount of Aβ42 per mg brain, ratio between 13C‐Lys Aβ17‐28 and unlabeled Aβ17‐28, labeling efficiency in %, cohort for 13C‐Lys APPtg brain extract injection.
Figure 1. 13C‐Lys‐labeling of donor brains.
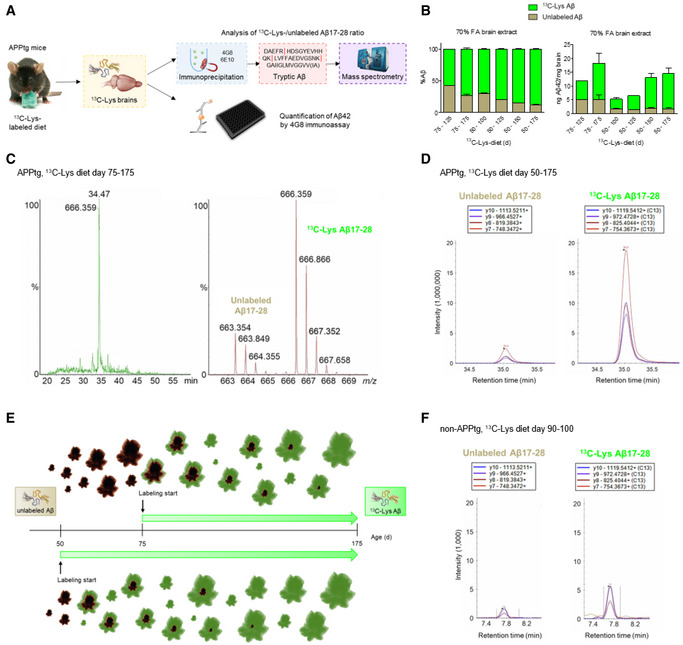
- Labeling strategy (image of mouse with diet provided by Cambridge Isotope Laboratories, Inc.) and quantification of 13C‐Lys‐labeled and unlabeled Aβ.
- Relative (left) and absolute (right) amount of 13C‐Lys‐labeled and unlabeled Aβ in APPtg mouse brains after labeling from 75–125 days (n = 1), 75–175 days (n = 4), 50–100 days (n = 5), 50–125 days (n = 2), 50–150 (n = 4) or 50–175 days (n = 9) of age. Error bars show mean ± SD.
- Representative chromatogram and mass spectra of doubly charged ions of unlabeled Aβ17‐28 (663.3 m/z) and 13C‐Lys Aβ17‐28 (666.3 m/z) in an APPtg brain after labeling from 75–175 days. %‐ ion intensity, m/z‐ mass‐to‐charge. Data were acquired in data‐independent analysis mode (MSE).
- Representative chromatograms of unlabeled Aβ17‐28 (left) and 13C‐Lys Aβ17‐28 (right) in an APPtg brain after labeling from 50–175 days. The intensities of four transitions (y10, y9, y8, and y7) were assessed. Data were acquired by MRM in nanoflow mode.
- Illustration of 13C‐Lys incorporation into Aβ depending on the starting point of labeling in APPtg mice. The later labeling starts, the higher is the amount of already generated, insoluble Aβ that is not metabolized anymore and consequently, not available for label incorporation. The earlier labeling starts, the higher is the fraction of newly produced Aβ incorporating the label.
- Representative chromatograms of unlabeled Aβ17‐28 (left) and 13C‐Lys Aβ17‐28 (right) in a non‐APPtg brain after labeling from 90–100 days. Note the 1,000‐fold difference in intensity magnitude between APPtg and non‐APPtg brains. Data were acquired by MRM in microflow mode.
Source data are available online for this figure.
Brain homogenates were pooled into four cohorts (Table 1) and diluted to generate 13C‐Lys APPtg brain extracts with 2.9–6.2 ng total Aβ42/µl and 2.3–5.3 ng 13C‐Lys Aβ42/µl (Table EV1). To generate labeled negative controls (13C‐Lys non‐tg extracts), non‐APPtg mice received the 13C‐Lys‐labeled diet for 10 days. We chose this labeling duration as the average life time of brain proteins had been reported to be 9 days (Price et al, 2010). Mass spectrometry analysis of four mice revealed 83.3 ± 2.9% overall 13C‐Lys‐labeling of the brain proteome (Dataset EV2) and detected minute signals of both unlabeled and 13C‐Lys Aβ17‐28 with an intensity 3log10 lower (1/1,000) than in APPtg mice (Fig 1F), representing endogenous murine Aβ.
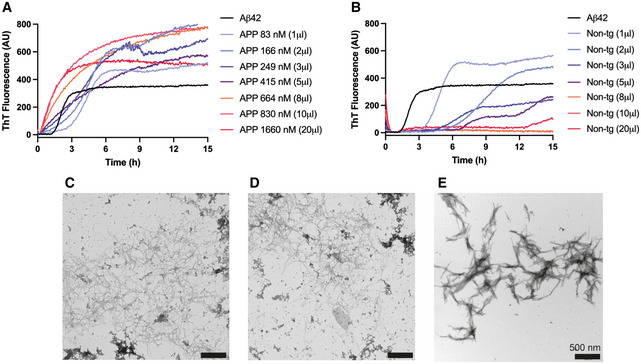
Moreover, unlabeled brain extracts for Aβ quantification in the periphery following i.p. injection were generated from APPtg and non‐APPtg mice fed on an unlabeled standard diet throughout their lifespan. Analysis by immunoassay showed Aβ42 concentrations ranging from 5.1 to 8.5 ng/µl in unlabeled APPtg extracts (Tables EV1 and EV2), while Aβ42 concentrations in non‐tg extracts were below the detection limit. Additionally, we isolated Aβ42 fibrils from unlabeled APPtg extract and tested their in vitro seeding activity by thioflavin T (ThT) fluorescence assay. Aβ42 fibrils extracted from APPtg brain extract exerted a concentration‐dependent seeding effect on Aβ42 aggregation (Fig EV1A) whereas isolate generated from non‐tg brain extract delayed Aβ42 aggregation kinetics (Fig EV1B). Seeded Aβ42 fibrils presented a heterogenous morphorlogy (Fig EV1C and D) while Aβ42 fibrils aggregated without the presence of seeds displayed a more homogenous morphology (Fig EV1E).
Figure EV1. Seeding activity of Aβ42 fibrils extracted from APPtg brain extract.
-
AThT fluorescence aggregation kinetics of recombinant Aβ42 in the presence of extracted fibrils from APPtg brain extract at different concentrations from 83 to 1,660 nM, exhibiting a concentration‐dependent seeding effect.
-
BThT fluorescence aggregation kinetics in the presence of equal amounts of extract from non‐tg brain extract, revealing no seeding activity but a delaying effect on the aggregation kinetics.
-
C, DTEM images showing seeded Aβ42 fibrils at the aggregation kinetic end points using 166 nM seeds (C), and 1,660 nM seeds (D). The fibril morphology is heterogenous.
-
EIn vitro Aβ42 fibrils aggregated without the presence of seeds. The fibril morphology is more homogenous compared to the seeded Aβ42 fibrils with brain extract.
Data information: In (A) and (B), the same curve is shown for Aβ42 aggregated without fibril extract. In (C–E), scale bars correspond to 500 nm. Source data are available online for this figure.
The concept of protein tracking by labeling‐based proteomics has been successfully employed by a recent study featuring ex vivo labeling of the mouse blood plasma proteome to investigate blood–brain transport of plasma proteins (Yang et al, 2020). Our in vivo labeling approach not only generated labeled amyloidogenic brain material for peripheral injection of host mice but also revealed information on the deposition dynamics of Aβ in the brains of APPtg donor mice. The earlier labeling commences, the lower the amount of insoluble Aβ that is not subject to dynamic turnover, and the higher the label incorporation into Aβ, which highlights the importance of an early start of therapies promoting Aβ clearance. This observation is supported by a recent study combining stable isotope labeling with mass spectrometry imaging to monitor plaque growth in an APP‐knockin mouse model and showing that label incorporation into Aβ is highest when labeling starts before plaque onset (Michno et al, 2021).
Detection limit of MS methodology
Considering the possibilities that Aβ may enter the brain in very low amounts or does not reach the brain at all, we sought to use a highly sensitive MS methodology capable of detecting minute quantities of 13C‐Lys‐labeled Aβ in brain samples. Aggregates of insoluble, full‐length Aβ present in APPtg mouse brains are particularly prone to loss in chromatography columns and machinery (Yoo et al, 2018). Thus, we combined an approach extracting both soluble and insoluble Aβ from APPtg mouse brains (Allue et al, 2016) and trypsin digestion generating more readily detectable Aβ fragments (Bateman et al, 2006). First, we performed trypsin digestion of synthetic Aβ42 and analyzed the resulting peptides. The Aβ17‐28 peptide, which contains Lys at position 28, was detected with the highest intensity (Fig 2A) in accordance with previous findings (Bateman et al, 2006; Kim et al, 2014) and thus, was selected as a surrogate to measure total Aβ levels. Next, we analyzed the fragmentation pattern of unlabeled Aβ17‐28 (0 additional neutrons) and 13C6 15N2‐lysine (13C15N‐Lys) Aβ17‐28 (eight additional neutrons), an internal standard well differentiable from both unlabeled and 13C‐Lys Aβ17‐28 (six additional neutrons), revealing several fragment ions of which the y ions y4‐y10 were detected with the highest intensity (Fig 2B). These ions were further analyzed by MRM for unlabeled, 13C‐Lys and 13C15N‐Lys Aβ17‐28, and the transitions with the highest intensity—y10, y9, y8, and y7—were selected for subsequent measurements. To assess the sensitivity of the MRM approach, we subjected decreasing quantities of 13C15N‐Lys Aβ42 to trypsin digestion followed by MRM analysis, revealing a detection limit of 25 amol in the injected volume (Fig 2C). Subsequently, we evaluated the quality of our sample processing by extracting soluble and insoluble Aβ from APPtg mouse brains, spiking decreasing amounts of 13C15N‐Lys Aβ42 into the insoluble fraction, and subjecting the samples to immunoprecipitation, trypsin digestion, and MRM analysis. Our 13C15N‐Lys‐labeled Aβ standard was still clearly detectable at the level of 50 amol in the injected volume despite all sample‐processing steps and the presence of an excess of unlabeled endogenous Aβ (Fig 2D).
Figure 2. Detection limit of mass spectrometry methodology.
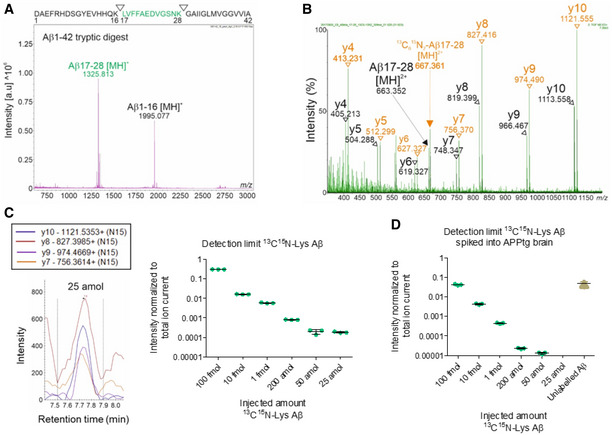
- MALDI‐TOF mass spectrum of trypsin‐digested, synthetic Aβ1‐42 (10 pmol). The Aβ17‐28 [MH]+ fragment was detected with the highest intensity. Measurements in reflectron positive mode. The sequence of Aβ1‐42 with the tryptic cleavage sites is indicated at the top.
- MS/MS spectrum of unlabeled Aβ17‐28 and 13C15N‐Lys Aβ17‐28 synthetic peptides in the 50 fmol range. m/z of precursors ions (m/z 663.34 for Aβ17‐28 [MH]2+, m/z 667.36 for 13C15N‐Lys Aβ17‐28 [MH]2+) and their derived fragments are indicated as a series of y ions (y4‐y10). The peptides were analyzed in data‐independent acquisition MSE mode.
- Decreasing quantities of synthetic 13C15N‐Lys Aβ1‐42 were trypsin‐digested and analyzed by MRM. The intensities of 4 transitions (y10, y9, y8, and y7) were assessed for 13C15N‐Lys Aβ17‐28, showing a detection limit level of 25 amol. Measurements in technical triplicates, normalization to total ion current. Error bars show mean ± SD.
- Decreasing quantities of synthetic 13C15N‐Lys Aβ1‐42 were spiked into the insoluble fraction of APPtg brains, immunoprecipitated, trypsin‐digested, and analyzed by MRM. Assessment of the intensities of y10, y9, y8, and y7 showed a detection limit level of 50 amol for 13C15N‐Lys Aβ17‐28 and detected an excess of brain‐derived unlabeled Aβ17‐28. Measurements in technical triplicates, normalization to total ion current. Error bars show mean ± SD.
Source data are available online for this figure.
Intraperitoneally injected Aβ is detectable in the periphery long after injection
Having generated 13C‐Lys‐labeled brain extracts and set up a reliable MS methodology for Aβ detection at attomolar levels, we now investigated the spatiotemporal distribution of peripherally injected Aβ. To this aim, we administered an i.p. injection of 200 µl 13C‐Lys APPtg brain extract or 13C‐Lys non‐tg brain extract into 50‐day‐old APPtg (APP/PS1 mice; Radde et al, 2006) and harvested the liver, spleen, mesenteric lymph nodes, and brain at 1, 3, 5, 7, 10, 25, 50, 75, or 100 days after injection as well as peritoneal cells at 1–10 days after injection (Fig 3A, Table 2). All experimental samples were spiked with known femtomolar quantities of 13C15N‐Lys Aβ42 prior to immunoprecipitation, serving as a quality control for sample processing and an internal standard.
Figure 3. Intraperitoneally injected 13C‐Lys Aβ is detectable in the periphery long after injection.
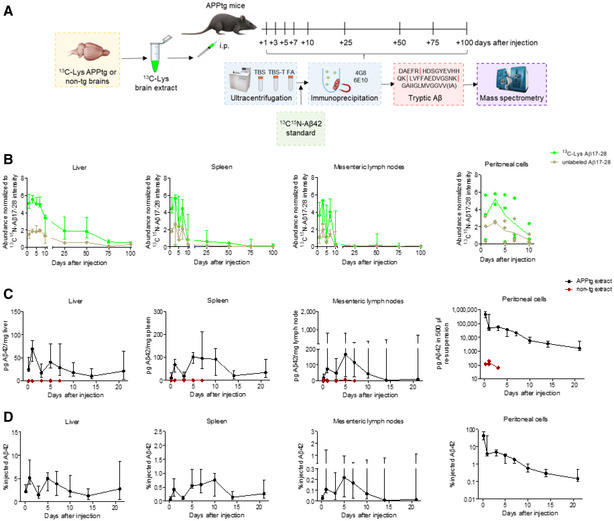
- Experimental design. i.p., intraperitoneal.
- Temporal MRM analysis of 13C‐Lys Aβ17‐28 and unlabeled Aβ17‐28 in the liver, spleen, mesenteric lymph nodes, and peritoneal cells after 13C‐Lys‐APPtg brain extract i.p. injection (n = 3 mice per time point and tissue at 1–10 days, except for n = 2 mice for peritoneal cells at 3 days; n = 4 mice per time point and tissue at 25–100 days, except for n = 3 mice for spleen at 100 days). Measurements in technical triplicates, normalization to 13C15N‐Lys Aβ17‐28 intensity as a spiked internal standard and additionally to organ weight (mg) for lymph nodes. Error bars show median ± interquartile range.
- Temporal 4G8 immunoassay quantification of Aβ42 in the liver, spleen, mesenteric lymph nodes, and peritoneal cells after unlabeled APPtg (n = 8 mice at 5 h to 7 days, except for n = 5 mice for peritoneal lavage, n = 5 mice at 10–14 days, n = 4 mice at 21 days) or non‐tg brain extract injection (n = 2 mice at 5 h and 3 days, n = 3 mice at 1 day, n = 1 mouse at 5 and 7 days, except for peritoneal cells where n = 1 mouse at 5 h and 3 days, and n = 2 mice at 1 day). Error bars show median ± interquartile range.
- Normalization of APPtg extract group data presented in (C) to organ weight (mg) and injected Aβ amount (range 1,027–1,426 ng). Error bars show median ± interquartile range.
Source data are available online for this figure.
Table 2.
13C‐Lys brain extract injection groups.
| n | Genotype | Injected 13C‐Lys brain extract | Labeling duration brain donors (days) | Time point post‐injection (days) | Age (days) |
|---|---|---|---|---|---|
| 3 | APPtg | APPtg, cohort 1 | 50, 100 | 1 | 51 |
| 3 | APPtg | non‐tg | 10 | 1 | 51 |
| 3 | APPtg | APPtg, cohort 1 | 50, 100 | 3 | 53 |
| 3 | APPtg | non‐tg | 10 | 3 | 53 |
| 3 | APPtg | APPtg, cohort 1 | 50, 100 | 5 | 55 |
| 3 | APPtg | non‐tg | 10 | 5 | 55 |
| 3 | APPtg | APPtg, cohort 1 | 50, 100 | 7 | 57 |
| 3 | APPtg | non‐tg | 10 | 7 | 57 |
| 3 | APPtg | APPtg, cohort 1 | 50, 100 | 10 | 60 |
| 3 | APPtg | non‐tg | 10 | 10 | 60 |
| 4 | APPtg | APPtg, cohort 2 | 125 | 25 | 75 |
| 4 | APPtg | non‐tg | 10 | 25 | 75 |
| 4 | APPtg | APPtg, cohort 2 | 125 | 50 | 100 |
| 4 | APPtg | non‐tg | 10 | 50 | 100 |
| 4 | APPtg | APPtg, cohort 2 | 125 | 75 | 125 |
| 4 | APPtg | non‐tg | 10 | 75 | 125 |
| 3 | APPtg | APPtg, cohort 1 | 50, 100 | 100 | 150 |
| 1 | APPtg | APPtg, cohort 2 | 125 | 100 | 150 |
| 4 | APPtg | non‐tg | 10 | 100 | 150 |
| 6 | APPtg | APPtg, cohort 4 | 100 | 1 | 51 |
| 6 | APPtg | non‐tg | 100 | 1 | 51 |
| 3 | non‐APPtg | APPtg, cohort 3 | 75 | 1 | 51 |
n = number of animals, genotype of injected animals, injected brain extract, number of days during which the donors of brains used for extract generation had received the 13C‐Lys diet, sacrifice time point after injection, age at death.
Analysis of the liver, spleen, mesenteric lymph nodes, and peritoneal cells from 13C‐Lys APPtg brain extract‐injected mice by MRM‐IMS revealed detectable 13C‐Lys Aβ17‐28 and unlabeled Aβ17‐28 at abundance ratios comparable to the 13C‐Lys/unlabeled Aβ ratios observed in the APPtg donor brains (Fig 3B). The abundance of both peptides was highest at 1–3 days after i.p. injection and decreased over time in all investigated tissues, with low levels still detectable at 100 days in the liver, at 75 days in the spleen and at 50 days in the mesenteric lymph nodes. In the liver, Aβ‐positivity was also recognizable by immunohistochemistry in perisinusoidal macrophages (Browicz‐Kupffer cells) for up to 10 days after injection of 13C‐Lys APPtg brain extract but not 13C‐Lys non‐tg brain extract (Fig EV2, images shown for 1 day after injection).
Figure EV2. Immunopositivity for Aβ in liver cells (macrophages) after intraperitoneal Aβ injection.
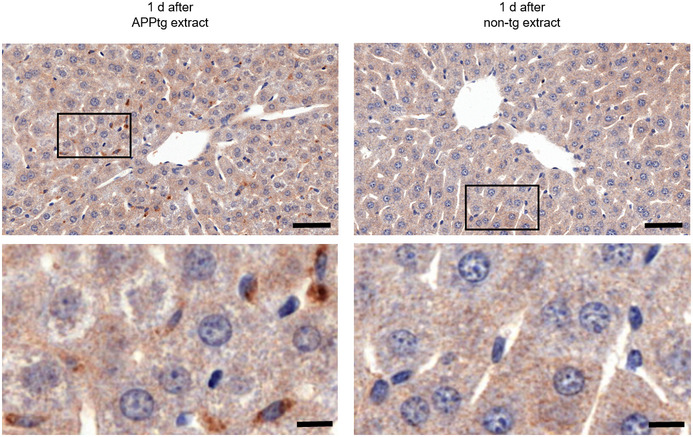
Representative images of liver sections (anti‐Aβ staining, clone 4G8) at 1 day after intraperitoneal 13C‐Lys APPtg or non‐tg brain extract injection, bottom row insets of delineated areas. Aβ‐positivity is detectable in mice injected with APPtg brain extract but not in mice injected with non‐tg brain extract. Scale bars correspond to 50 µm (top row) and 10 µm (bottom row), respectively.
To quantify the amount of Aβ reaching peripheral tissues after i.p. injection, we administered 200 µl of unlabeled APPtg brain extract or non‐tg brain extract into 50‐day‐old APPtg mice and harvested the liver, spleen, mesenteric lymph nodes, and peritoneal cells at 5 h, 1, 3, 5, 7, 10, 14, or 21 days after injection as well as blood at the time points 5 h to 7 days after injection. With immunoassay, we found Aβ42 in the liver, spleen, mesenteric lymph nodes, and peritoneal cells of mice injected with APPtg brain extract until 21 days after injection (Fig 3C and D). In the liver, spleen, and mesenteric lymph nodes, Aβ42 levels reached two peaks at 1 and 5 days after injection, with the liver accumulating most of the injected Aβ and the mesenteric lymph nodes the least (Fig 3D). In the peritoneal cells consisting predominantly of lymphocytes and to a lesser extent of macrophages and mesothelial cells, Aβ42 levels were highest at 5 h after injection, with about half of the injected Aβ still being detectable, and subsequently decreased over time (Fig 3D). The decrease in Aβ observed over time in the liver and lymphoid tissues, suggests that Aβ does not replicate in the periphery and is in accordance with a previous study reporting that peripheral expression of murine or human Aβ is not required for accelerating cerebral β‐amyloidosis after i.p. injection of APPtg brain extracts (Eisele et al, 2014).
In blood plasma, Aβ42 levels were comparable between mice injected with APPtg brain extract and those injected with non‐tg brain extract at all time points (Fig EV3A). To further trace i.p. administered Aβ in blood, we isolated mononuclear blood cells, that is, lymphocytes and monocytes from mice injected with APPtg brain extract or non‐tg brain extract at 5 h to 7 days after injection. We found higher levels of Aβ42 in the APPtg brain extract‐injected mice compared to non‐tg brain extract‐injected mice at 5 h and 1 day after injection, whereas Aβ42 was only detectable in single individuals thereafter (Fig EV3B). The finding that injected Aβ is present in mononuclear blood cells but not in blood plasma during the first day after injection is in further concordance with the study by Eisele et al (2014). However, we cannot rule out that injected Aβ is present in blood plasma at levels below the detection limit of the immunoassay or that binding of Aβ to soluble transport proteins such as low‐density lipoprotein receptor‐related protein‐1 (Sagare et al, 2007) leads to epitope masking, thereby reducing Aβ detection in the immunoassay.
Figure EV3. Intraperitoneally injected Aβ is not detectable in plasma but in mononuclear blood cells shortly after injection.
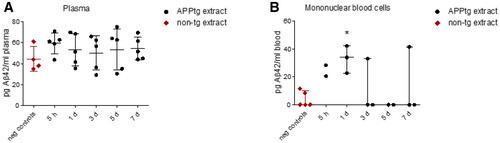
- Temporal and comparative 4G8 immunoassay quantification of Aβ42 in blood plasma after unlabeled APPtg or non‐tg brain extract injection (for APPtg extract, n = 5 mice per time point after injection; for non‐tg extract, mice (n = 1 at time points 5 h and 3 days, n = 2 at 1 day) were pooled and referred to as negative controls). Data are shown as mean ± SD.
- Temporal and comparative 4G8 immunoassay quantification of Aβ42 in mononuclear blood cells after unlabeled APPtg or non‐tg brain extract injection (for APPtg extract, n = 2 mice at time points 5 h and 5 days, and n = 3 mice at time points 1, 3, and 7 days; for non‐tg extract, mice (n = 1 per time point) were pooled and referred to as negative controls). Mann‐Whitney U‐test * P = 0.0325, APPtg extract‐injected vs. negative controls. Normalization to blood volume. Error bars show median ± interquartile range.
Source data are available online for this figure.
Taken together, these data suggest that Aβ injected into the peritoneum first reaches the liver and spleen via mononuclear blood cells, and the mesenteric lymph nodes via lymphatic drainage. The second peak observed in the liver, spleen, and lymph nodes may be attributable to clearance of Aβ‐loaded immune cells from the peritoneum.
Labeled Aβ is detectable in the brain after peripheral injection of both 13C‐Lys APPtg and 13C‐Lys non‐tg brain extract due to incorporation of 13C‐Lys
Having traced i.p. injected Aβ in the periphery, we now sought to answer the central question whether Aβ entered the brain. Analysis of the soluble brain fraction by MRM‐IMS did not detect 13C‐Lys Aβ17‐28 at any time point after injection of 13C‐Lys APPtg brain extract while unlabeled Aβ17‐28 was present in low abundance at all time points (Fig 4A). Surprisingly, we observed 13C‐Lys Aβ17‐28 already at 1 day after injection in the insoluble brain fraction of both groups of mice, injected either with 13C‐Lys APPtg brain extract or with 13C‐Lys non‐tg brain extract (Fig 4B). The abundance of 13C‐Lys Aβ17‐28 was significantly higher in the 13C‐Lys APPtg brain extract‐injected group compared to the 13C‐Lys non‐tg brain extract‐injected group between 1 and 25 days after injection (Fig 4B). Unlabeled Aβ17‐28 was detected in the insoluble brain fraction with an abundance 1,000‐fold higher than that of 13C‐Lys Aβ17‐28, steadily increasing over time without differences between the experimental groups (Fig 4C). In accordance with this, we did not find differences between groups at any time point when quantifying total cerebral Aβ load by immunoassay at 1–100 days after injection (Fig 4D), and immunohistochemistry at 25–100 days after injection (Fig EV4A). Similarly, we did not observe group differences in microglial activation as assessed by immunohistochemistry 25–100 days after injection (Fig EV4B). To evaluate potential long‐term effects, we administered an i.p. injection of 200 µl unlabeled APPtg or non‐tg brain extract into 50‐day‐old APPtg mice and harvested the brain at 240 days after injection. Quantification of total cerebral Aβ load by immunoassay did not reveal differences between groups (Fig EV5). These results indicate that cerebral β‐amyloidosis and concurrent neuroinflammation are not accelerated by i.p. injection of Aβ‐containing brain extract in the APPtg model used in our study.
Figure 4. Insoluble 13C‐Lys Aβ is detectable in the brain after peripheral injection of both 13C‐Lys APPtg and 13C‐Lys non‐tg brain extract due to incorporation of 13C‐Lys.
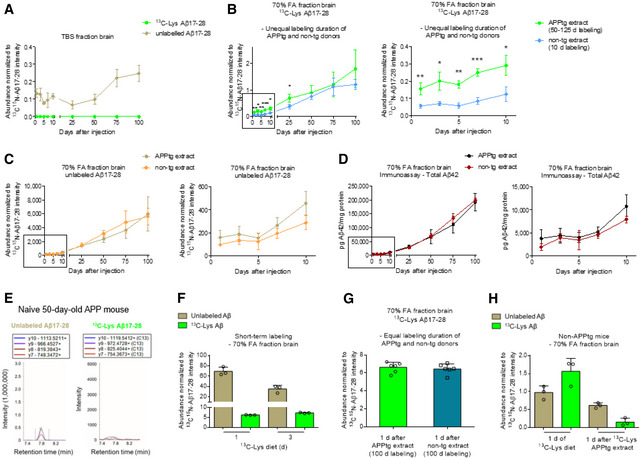
-
ATemporal MRM analysis of unlabeled Aβ17‐28 and 13C‐Lys Aβ17‐28 in the soluble brain fraction after 13C‐Lys APPtg brain extract i.p. injection (n = 3 mice per time point at 1–10 days, n = 4 mice per time point at 25–100 days).
-
B, CComparative MRM analysis of 13C‐Lys Aβ17‐28 (B) and unlabeled Aβ17‐28 (C) in the insoluble brain fraction after 13C‐Lys‐APPtg brain extract (donors labeled for 50–125 days) and 13C‐Lys‐non‐tg brain extract (donors labeled for 10 days) i.p. injection (n = 3 mice per group and time point at 1–10 days, n = 4 mice per group and time point at 25, 50, 100 days, n = 3 mice in the APPtg extract group and n = 4 in the non‐tg extract group at 75 days), right side zoom for time points 1–10 days. Unpaired two‐tailed t‐test *P < 0.05 (P = 0.0129 at 3 days, P = 0.0156 at 10 days, P = 0.0109 at 25 days), **P < 0.01 (P = 0.0091 at 1 day, P = 0.0011 at 5 days), ***P < 0.001 (P = 0.0006 at 7 days), 13C‐Lys APPtg extract‐injected vs. 13C‐Lys non‐tg extract‐injected for each time point.
-
DComparative 4G8 immunoassay quantification of Aβ42 in the insoluble brain fraction after 13C‐Lys APPtg or 13C‐Lys non‐tg brain extract i.p. injection (n = 3 mice per group and time point at 1–10 days, n = 4 mice per group and time point at 25–100 days), right side zoom for time points 1–10 days. Normalization to protein concentration.
-
ERepresentative chromatograms of unlabeled Aβ17‐28 (left) and 13C‐Lys Aβ17‐28 (right) in the brain of an unlabeled 50‐day‐old APPtg mouse not injected with labeled brain extract. Note the absence of 13C‐Lys Aβ17‐28.
-
FMRM analysis of unlabeled and 13C‐Lys‐labeled Aβ17‐28 in APPtg mouse brains after 13C‐Lys‐labeled diet for 1 or 3 days (n = 3 mice per time point).
-
GComparative MRM analysis of 13C‐Lys Aβ17‐28 in the insoluble brain fraction after 13C‐Lys APPtg and 13C‐Lys non‐tg brain extract (all donors labeled for 100 days) i.p. injection (n = 6 mice per group).
-
HMRM analysis of unlabeled and 13C‐Lys‐labeled Aβ17‐28 in non‐APPtg mouse brains after 13C‐Lys‐labeled diet for 1 days (n = 3 mice) or at 1 days after 13C‐Lys‐APPtg brain extract i.p. injection (n = 3 mice).
Data information: In (A–C) and (F–H), measurements in technical triplicates, normalization to 13C15N‐Lys Aβ17‐28 intensity as a spiked internal standard and brain weight (mg). All error bars show mean ± SD.
Source data are available online for this figure.
Figure EV4. Peripheral injection of APPtg brain extract has no influence on the accumulation of Aβ plaques or microglial activation.
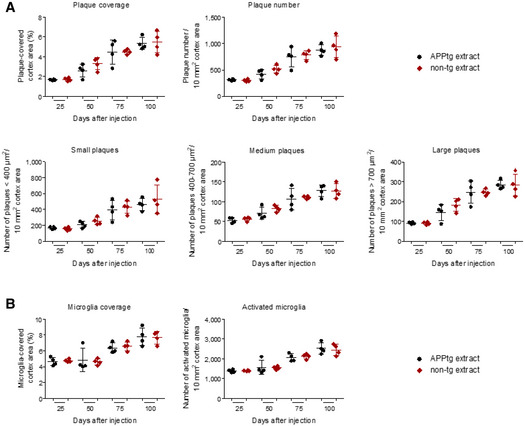
- Analysis of cortical Aβ plaque load, plaque number, and plaque size distribution in anti‐Aβ (clone 4G8)‐stained brain sections from mice injected with 13C‐Lys APPtg or 13C‐Lys non‐tg extract (n = 4 mice per time point and group). Data are presented as mean ± SD.
- Analysis of cortical coverage and number of activated microglia in anti‐IBA1‐stained brain sections from mice injected with 13C‐Lys APPtg or 13C‐Lys non‐tg extract (n = 4 mice per time point and group). Error bars are shown as mean ± SD.
Source data are available online for this figure.
Figure EV5. Intraperitoneal injection of APPtg brain extract does not induce exacerbation of β‐amyloidosis 240 days after injection.
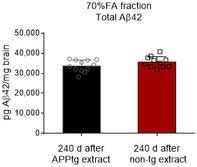
Comparative 4G8 immunoassay quantification of Aβ42 in the insoluble brain fraction after unlabeled APPtg or non‐tg brain extract injection (n = 12 mice in the APPtg extract group, n = 11 mice in the non‐tg extract group). Normalization to brain weight. Data are presented as mean ± SD.
Source data are available online for this figure.
To explain the presence of increasing amounts of 13C‐Lys Aβ17‐28 in the 13C‐Lys non‐tg brain extract‐injected group and to investigate whether the higher 13C‐Lys Aβ17‐28 abundance in the 13C‐Lys APPtg brain extract‐injected group was due to brain penetration of Aβ or due to different labeling durations in APPtg and non‐APPtg brain donor mice, we performed additional experiments. First, we probed for the presence of naturally occurring 13C‐Lys Aβ17‐28 in the insoluble brain fraction of unlabeled APPtg mice at the age of 50 days. We did not detect 13C‐Lys Aβ17‐28 in unlabeled mice (Fig 4E), thereby excluding naturally occurring 13C‐Lys Aβ17‐28 as an explanation and implying that metabolized 13C‐Lys from labeled extract had been incorporated into endogenous Aβ. To confirm this, we fed the 13C‐Lys‐labeled diet to 50‐day‐old APPtg mice for 1 or 3 days and analyzed the insoluble brain fraction by MRM‐IMS. We detected 13C‐Lys Aβ17‐28 accounting for an 8% of total Aβ17‐28 already after 1 day of 13C‐Lys‐labeled diet (Fig 4F), demonstrating rapid incorporation of 13C‐Lys into endogenous Aβ. Next, we fed non‐APPtg mice the 13C‐Lys labeled diet for 100 days to have the same labeling duration as for APPtg donor mice, resulting in 96.5 ± 0.2% overall 13C‐Lys‐labeling of the brain proteome (Dataset EV3). We administered an i.p. injection of 200 µl 13C‐Lys APPtg brain extract or 13C‐Lys non‐tg brain extract generated from cohorts of donor mice with equal labeling duration into 50‐day‐old APPtg mice and harvested the brain at 1 day after injection (Table 2). Analysis of the insoluble brain fraction by MRM‐IMS revealed the presence of 13C‐Lys Aβ17‐28 at a comparable abundance in both groups, mice injected with 13C‐Lys APPtg brain extract and those injected with 13C‐Lys non‐tg brain extract (Fig 4G). Taken together, these results indicate that the mice rapidly incorporated metabolized 13C‐Lys, which is an essential amino acid, from the donor extracts into newly generated endogenous Aβ, whereas Aβ present in the APPtg donor extract did not enter the brain. Finally, non‐APPtg mice injected with 13C‐Lys APPtg brain extract (Table 2) showed substantially lower abundance of 13C‐Lys Aβ17‐28 at 1 day after injection (0.15 ± 0.09 relative to 13C15N‐Lys Aβ17‐28 abundance) compared to non‐APPtg mice after 1 day of 13C‐Lys diet (1.56 ± 0.29 relative to 13C15N‐Lys Aβ17‐28 abundance; Fig 4H), supporting the idea of a dose‐dependent incorporation of 13C‐Lys into murine Aβ rather than brain penetration of injected human Aβ.
Using in vivo stable isotope labeling of brain‐derived Aβ and highly sensitive targeted MRM‐IMS, we provide evidence that peripherally injected Aβ does not penetrate the brain but may be metabolized along with other labeled proteins, with the label being incorporated into newly generated Aβ. Thus, our findings argue against transmission of Aβ pathology via Aβ aggregates entering the brain from the periphery.
However, our data pointed to cerebral incorporation of metabolized 13C‐Lys from the donor extract into endogenous Aβ from 1 day after injection in both APPtg mice and non‐APPtg mice, which only express murine APP and produce substantially lower Aβ amounts compared to APPtg mice. Incorporation of labeled amino acids into Aβ plaques has previously been observed in an AD patient 8 days after injection of 13C‐labeled leucine (Wildburger et al, 2018). We speculate that 13C‐Lys‐containing proteins and peptides from the injected donor extract may be metabolized in the periphery, resulting in release of 13C‐Lys – an essential amino acid – into the bloodstream. From there, 13C‐Lys may be transported into the brain and used for de novo production of Aβ. While we observed incorporation of 13C‐Lys into insoluble Aβ, we did not detect 13C‐Lys Aβ in the soluble brain fraction at any time point. This discrepancy is most likely attributable to the proportion of soluble to insoluble Aβ in our brain samples, with soluble unlabeled Aβ being detected with an abundance 1,000–10,000‐fold lower than insoluble unlabeled Aβ. Given that this low abundance of soluble unlabeled Aβ was already close to the limit of detection and that insoluble 13C‐Lys Aβ was detected with an abundance 1,000‐fold lower than insoluble unlabeled Aβ, the abundance of soluble 13C‐Lys Aβ would be below the limit of detection in our study.
In accordance with the MS data not pointing to brain penetration of Aβ, we did not observe an acceleration of cerebral Aβ deposition in APP/PS1 mice injected with APPtg brain extracts, not even at 8 months after injection, the incubation period reported in previous peripheral injection studies (Eisele et al, 2014; Morales et al, 2021). The APPtg brain extracts used for i.p. injection in our experiments had Aβ concentrations of 2.9–8.5 ng total Aβ42/µl. This is comparable to previous peripheral injection studies using APPtg brain extracts with Aβ concentrations of 10–20 ng/µl and demonstrating that extract diluted 10‐fold (i.e., containing 1–2 ng Aβ/µl) still accelerated cerebral Aβ deposition (Eisele et al, 2010, 2014) or estimating the amount of intraperitoneally injected Aβ to be 171.8 ng (corresponding to an Aβ concentration of 1.7 ng/µl; Morales et al, 2021). Thus, insufficiently high levels of injected Aβ can be excluded as a potential confounder in our study.
Our findings imply that accelerated cerebral β‐amyloidosis previously observed after peripheral injection of Aβ‐containing brain extract or blood into APPtg mouse models (Eisele et al, 2010, 2014; Burwinkel et al, 2018; Morales et al, 2020, 2021) was caused by other factors present in the donor material rather than by seeding from the periphery. Indeed, a peripheral injection of lipopolysaccharides exacerbated Aβ pathology in a similar fashion as an i.p. injection of APPtg brain extract in APP23 mice (Wendeln et al, 2018). Moreover, it has been shown that exposing young mice to the systemic milieu or plasma of aged mice resulted in cognitive impairment and decreased synaptic plasticity and vice versa (Villeda et al, 2011). However, we did not observe accelerated cerebral β‐amyloidosis in our study. This discrepancy with previous studies may be explained by differences in the rate of Aβ deposition in the different APPtg models used as host mice. While APP23 and Tg2576 mice used in previous studies start depositing Aβ at 6–7 months (Sturchler‐Pierrat et al, 1997) and 8–10 months (Kawarabayashi et al, 2001), respectively, APP/PS1 mice used in our study already show first Aβ deposits at 1.5 months (Radde et al, 2006), potentially masking any accelerated plaque formation caused by still unknown factors in the donor material. In contrast to our findings arguing against brain penetration of Aβ, a recent study demonstrated that human Aβ derived from APPtg mice entered and accumulated in the brain of non‐APPtg mice after 12 months of parabiosis (Bu et al, 2018). However, parabiosis models cause severe pain and distress that might promote sterile inflammatory factors facilitating the entrance of Aβ into the brain. As most of the Aβ was found in or near blood vessels, it cannot be discarded that these findings are the result of a loss of blood‐brain barrier integrity due to the severity of the experimental model.
Our study implying that Aβ does not penetrate the brain from the periphery provides evidence against the intensely discussed hypothesis that human transmission of Aβ seeds and potentially AD may occur by medical procedures not directly involving the central nervous system (Jucker & Walker, 2015; Huynh & Holtzman, 2018; Purro et al, 2018; Asher et al, 2020; Lauwers et al, 2020; Lau et al, 2021). Further studies are warranted to confirm our findings and investigate alternative mechanisms (e.g. inflammatory factors) that may have induced cerebral β‐amyloidosis and other AD‐related pathologies after injection of c‐hGH or Aβ‐containing brain material, thereby gaining new insights into the pathogenesis of AD. In addition to arguing against the potential transmissibility of AD, our proteomic approach combining in vivo stable isotope labeling and highly sensitive MRM‐IMS provides a framework for deciphering deposition dynamics of proteins associated with proteopathic diseases.
Materials and Methods
Animals
APP/PS1 transgenic (APPtg) mice co‐expressing human amyloid precursor protein with the KM670/671NL mutation, and L166P‐mutated presenilin 1 under the control of the Thy1 promotor were provided by the University of Tübingen (Radde et al, 2006) and maintained on a C57BL/6J background. APPtg mice and their non‐transgenic littermates were housed in groups in open cages at 21–22°C under a 12/12‐h light/dark circle with free access to food and water. All animal experiments were in accordance with the guidelines of the European Directive 2013/63/EU and Norwegian national laws and were formally approved by regional authorities (reference number 11909). Data is reported in accordance with the ARRIVE guidelines.
Brain donor mice
Male and female APPtg donor mice (n = 25) received a commercial 13 6C6‐lysine (13C‐Lys)‐labeled diet (EURISO‐TOP GmbH, Saarbrücken, Germany) from 75–175 days (n = 4), 75–125 days (n = 1 sacrificed at 125 instead of 175 days due to skin lesions), 50–100 days (n = 5), 50–125 days (n = 2), 50–150 days (n = 4) or 50–175 days (n = 9) of age. Male and female non‐APPtg donor mice aged 90–98 days (n = 19) received the diet for 10 days until the age of 100–108 days. An additional cohort of female non‐APPtg donor mice was fed on the diet from 50–150 days of age (n = 4). Mice were sacrificed by cervical dislocation and intracardially perfused with cold phosphate‐buffered saline (PBS). The brain was removed without cerebellum and lower brainstem, snap‐frozen, and stored at −80°C.
Assessment of concentration and labeling efficiency of Aβ
Thawed brains were individually homogenized for 30 s with four 2.8 mm ceramic beads (PreCellys®, Peqlab, Germany) using a SpeedMill PLUS homogenizer (AnalytikJena AG, Germany). Brain homogenates from APPtg mice (~15 mg per sample) were mixed with 10 µl cold 70% formic acid (FA)/mg brain, sonicated for 2 × 30 s (Branson Digital Sonifier SFX 150, 3.2 mm microtip, amplitude 70%, pulses of 1 s) on ice, incubated overnight at 4°C, and centrifuged at 16,000 × g for 90 min at 4°C. Supernatants were neutralized with 19 volumes of neutralization solution (1 M Tris, 0.5 M Na2HPO4, 0.05% NaN3), and further diluted for electrochemiluminescence‐linked immunoassay. Overall Aβ42 levels were quantified using the V‐PLEX Plus Aβ42 Peptide (4G8) Kit (detection limit 0.156 pg/ml; quantification limit 2.50 pg/ml) and a MESO QuickPlex SQ120 imager (Mesoscale Discovery, USA) according to the manufacturer’s instructions.
To determine the percentage of 13C‐Lys Aβ, brain homogenates from APPtg mice (~6–11 mg per sample) were mixed with 200 µl of cold 70% FA, sonicated on ice, and centrifuged at 30,000 × g for 5 min at 4°C. Supernatants were neutralized with 12 ml of PBS/2 M Tris (1:1 ratio) solution, pH 8.45, and immunoprecipitated overnight with monoclonal anti‐Aβ antibodies, clones 4G8 and 6E10 (BioLegend™) cross‐linked to magnetic beads (2.5 µg of each antibody covalently coupled to 50 µl of Protein G Dynabeads, Invitrogen; Thermo Scientific™). Bound Aβ peptides were eluted from the beads with 2 × 50 µl of 5% FA and digested overnight with sequencing grade trypsin (Promega) in 20% acetonitrile (ACN)/50 mM ammonium bicarbonate (AMBIC). The tryptic Aβ peptides were desalted and concentrated through C18 reversed phase (RP) ZipTips (Millipore™), eluted thrice from the ZipTips with 50% ACN/0.1% trifluoroacetic acid (TFA), and dried under vacuum. Brain homogenates from four non‐tg mice were processed identically.
Tryptic digests from the APPtg mice that had received the labeled diet from 75–125, 75–175, or 50–100 days of age were re‐suspended in 5% ACN and injected into a nanoAcquity UPLC system equipped with a trapping column (5 μm Xbridge C8 RP, 180 μm × 20 mm), followed by an analytical C8 RP column (1.7 µm BEH C8, 75 µm × 250 mm). The digests were separated with a linear gradient of 3–85% of solution B (0.1% FA/ACN) for 60 min at a flow rate of 250 nl/min and a stable column temperature of 35°C. All samples were run in triplicates, with three blanks between runs. The tryptic samples were analyzed in data‐independent analysis mode (MSE) on a Synapt G2‐S mass spectrometer (Waters™) by alternating between low collision energy (6 V) and high collision energy ramp in the transfer compartment (20–45 V) and utilizing 1 s cycle time. The separated peptides were detected online with a mass spectrometer operated in positive, resolution mode in the range of m/z 50–2,000 amu. Human [Glu1]‐fibrinopeptide B (Sigma Aldrich, St. Louis, MO, USA) at 150 fmol/µl in 25% ACN/0.1% FA solution at a flow rate of 300 nl/min applied every 30 s, was used for lock mass correction. Chromatograms were analyzed with Progenesis QI Proteomics™ using a mouse FASTA library (UniProtKB/Swiss‐Prot reviewed) spiked with the human Aβ sequence (release 2018.08.01 with 16,994 sequences), and using SILAC 13C6 and oxidation (M) as variable modifications with one trypsin‐missed cleavage allowed. Database searches were carried out with the ion accounting algorithm and using the following parameters: default peptide and fragment tolerance, maximum protein mass 250 kDa; minimum fragment ions matches per protein ≥ 7; minimum fragment ions matches per peptide ≥ 3; minimum unique peptide matches per protein ≥ 2; false discovery rate (FDR) < 4%. The extracted ion chromatograms were generated and peak areas for all 13C‐Lys labeled and unlabeled Aβ peptides were obtained using MassLynx v.4.1 software. Both procedures produced a peak table used for subsequent calculations. The ratios of 13C/12C were calculated based on normalized intensities and assessing peak areas of 13C‐Lys‐labeled and unlabeled Aβ17‐28 peptide (detected at [MH]+, [MH]2+ and [MH]3+ charge states).
In another set of experiments, tryptic digests from the APPtg mice that had received the labeled diet from 50–125, 50–150, or 50–175 days of age and non‐APPtg mice were re‐suspended in 5% ACN and injected into a 6500+ QTrap – Eksigent nanoLC system (SCIEX™). Tryptic peptides from APPtg mice labeled from 50–175 days were separated using an ACQUITY UPLC M‐Class Symmetry C18 Trap column (5 µm, 180 µm × 20 mm, Waters™) and an ACQUITY UPLC M‐Class Peptide CSH C18 analytical column (3.5 µm, 75 µm × 250 mm, Waters™) with a 60 min gradient of 3–35% ACN/0.1% FA at a flow of 300 nl/min (nanoflow mode). Tryptic peptides from APPtg mice labeled from 50–125 or 50–150 days and non‐tg mice were separated using a YMC Triart C18 column (1.9 µm; 15 cm × 150 mm; YMC Separation technology, Japan) at a flow of 7 µl/min (microflow mode) and a column heater kept at 30°C with a 15‐min step‐wise gradient method of 3–85% ACN/0.1% FA. The multiple reaction monitoring (MRM) approach was used to analyze the samples, selecting four transitions for 13C‐Lys‐labeled and unlabeled Aβ17‐28 with the highest intensity (y10, y9, y8, and y7). Raw MRM data (as.wiff files) were exported to Skyline (MacCoss Lab Software v.19.1) for peak integration, normalization and abundance calculations.
Assessment of total 13C‐Lys incorporation into the brain
Brain homogenate samples (~11 mg per sample) from APPtg mice (n = 4, fed for 100 days on 13C‐Lys diet) and control non‐APPtg mice (n = 4, fed for 10 days, and n = 4, fed for 100 days on 13C‐Lys diet) were processed utilizing a previously described 3‐step ultracentrifugation method (Allue et al, 2016) generating the soluble Tris‐buffered saline (TBS) fraction, the membrane‐soluble TBS + 1% Triton X‐100 (TBS‐T) fraction, and the insoluble 70% FA fraction but with the modification of using 2 ml of TBS and TBS‐T, respectively, and 200 µl of 70% FA. Protein concentration in TBS fractions was assessed with Bradford assay (Pierce). 10 µg of protein mixture was reduced with 5.6 mM DTT for 5 min at 95°C and then alkylated with 5 mM iodoacetamide for 30 min at RT. Trypsin solution (Promega, Mannheim, Germany) was added in a ratio of 1:50 w/w in 250 mM ammonium bicarbonate and incubated overnight at room temperature with shaking. Tryptic digests were purified using C18‐reverse‐phase ZipTip™ (Millipore). Dried peptide digest was resuspended in 1% trifluoroacetic acid and sonicated in a water bath for 1 min before injection.
Typtic digests were analyzed in nano‐LC‐Thermo Q Exactive Plus Orbi‐Trap MS as described by us before (Gholizadeh et al, 2021). 1 µg of peptides in each biological sample was separated by Easy‐nLC system (Thermo Scientific) equipped with a reverse‐phase trapping column Acclaim PepMapTM 100 (C18, 75 μm × 20 mm, 3‐μm particles, 100 Å; Thermo Scientific), followed by an analytical Acclaim PepMapTM 100 RSLC reversed‐phase column (C18, 75 µm × 250 mm, 2‐µm particles, 100 Å; Thermo Scientific). The injected sample analytes were trapped at a flow rate of 2 µl/min in 100% of solution A (0.1% FA). After trapping, the peptides were separated with a linear gradient of 120 min comprising 96 min from 3 to 30% of solution B (0.1% FA/80% ACN), 7 min from 30 to 40% of solution B, and 4 min from 40 to 95% of solution B. The resolution was set to 140,000 for MS scans and 17,500 for the MS/MS scans. Full MS scans were acquired in 350–1,400 m/z range, and the 10 most abundant precursor ions were selected for fragmentation with 30 s dynamic of exclusion time. Ions with 2+, 3+and 4+ charge states were selected for MS/MS analysis. Secondary ions were isolated with a window of 1.2 m/z. The MS automatic gain control target was set to 3 × 106 counts, whereas the MS/MS automatic gain control target was set to 1 × 105. Dynamic exclusion was set with a duration of 20 s. The Normalized Collision Energy was set to 28 kJ/mol. Each sample run was followed by two empty runs to wash out any remaining peptides from the previous runs.
The raw files were analyzed by Proteome Discoverer (PD, version 2.4.0.305, Thermo Scientific). The identification of proteins was performed against the cptac2_mouse_hcd_selected mouse spectral library (24‐11‐2014, updated on 08‐07‐2018 from https://chemdata.nist.gov/; compatible with MSPepSearch node in PD) and UniProt mouse protein database (release 08‐2020 with 17,033 entries spiked with human AβPP sequence), a tolerance level of 10 ppm for MS and 0.02 Da for MS/MS, and with two tryptic missed cleavages allowed. The carbamidomethylation of cysteines was set as a fixed modification, whereas 13C6‐Lys, the oxidation of methionines and deamidation of asparagines and glutamines were set as variable modifications. A minimum length of six amino acids (one peptide per protein) was required for each peptide hit. The 13C/12C ratios were calculated taking into account the ratios of normalized intensities for identified proteins, detected as heavy (13C6‐Lys) and light forms (12C6‐Lys). The efficiency of 13C‐Lys labeling was assessed by utilizing the following equation: SILAC ratio = [ratio (H/L) × 100]/[ratio (H/L) + 1]. Only proteins detected at FDR < 0.01 at all levels of protein confidence with ≥ 2 unique peptides were taken into account in the quantitative analyses.
Preparation of brain extracts
To generate 13C‐Lys‐labeled APPtg brain extracts, 13C‐Lys APPtg brain homogenates were pooled into four cohorts, diluted with 1 µl PBS per mg brain, and sonicated for 3 × 30 s on ice. Negative control 13C‐Lys non‐tg brain extracts were prepared identically. Brain extracts were aliquoted and stored at −80°C until use.
Additional cohorts of unlabeled APPtg and non‐tg brain extracts originating from 100 to 175‐day‐old APPtg (n = 59, Table EV2) and 115 to 175‐day‐old non‐APPtg donor mice (n = 34), which had received a standard laboratory diet throughout their entire life, were generated in an identical manner. Samples of 13C‐Lys‐labeled and unlabeled APP brain extracts were treated with nine volumes of 70% FA and overall Aβ42 levels were measured as described above.
Fibril extraction from brain extracts
Fibrils were extracted using a modified method from a recent study (Kollmer et al, 2019). Unlabeled APPtg brain extract (300 µl) was diluted with an equal volume of Tris calcium buffer (20 mM Tris, 138 mM NaCl, 2 mM CaCl2, 0.1% NaN3, pH 8) and centrifuged at 21,000 g for 30 min. The pellet was re‐suspended in 1 ml Tris calcium buffer and 50 μl of Collagenase (5 mg/ml) and 10 μl of DNAse I were added. The solution was vortexed, incubated overnight at 37°C, and centrifuged at 21,000 g for 30 min. The pellet was dissolved in 1 ml wash buffer (50 mM Tris, 10 mM EDTA, pH 8) and 40 μl of 10% SDS was added. The solution was incubated at 37°C for 30 min and centrifuged. This step was repeated twice and two further washing steps of the pellet were performed in wash buffer without SDS. Finally, the pellet containing fibrils was dissolved in 50 μl of 20 mM sodium phosphate buffer, 0.2 mM EDTA pH 8.0, and used for further experiments. Unlabeled non‐tg brain extract was processed identically as control.
Dot blot assay
The concentration of Aβ in fibril extract was estimated from dot blot using Aβ42 monomer as reference. Fibril extract generated from APPtg brain extract was sonicated and spotted on a nitrocellulose membrane in different volumes (1, 2, 3, and 4 µl, respectively). 4 µl of Aβ42 in different concentrations (1, 2, 3, 6, and 8 µM, respectively) was spotted as standard. The membrane was air‐dried, blocked with blocking buffer (intercept® blocking buffer, LI‐COR, Lincoln, USA) and incubated overnight at 4°C with primary anti‐Aβ antibody (6E10, anti‐mouse, 1:5,000, BioLegend™). The membrane was washed thrice with PBS‐T, followed by incubation with secondary anti‐mouse antibody (IRDye 800 CW, 1:20,000, LI‐COR) for 60 min at 25°C. The membrane was washed thrice with PBS‐T and developed using Odyssey® DLx Imaging System. Blot analysis was performed for estimation of Aβ concentration using ImageJ.
Thioflavin T fluorescence assay
To test the seeding activity of fibrils extracted from APPtg brain extract, a Thioflavin T (ThT) fluorescence assay was performed. 20 µl of sodium phosphate buffer (20 mM, 0.2 mM EDTA, pH 8.0) containing 3 µM Aβ42 and 10 µM ThT was added to 384‐well plates along with different volumes (1, 2, 3, 5, 8, 10, and 20 µl, respectively) of isolate generated form APPtg or non‐tg brain extracts. Excitation filter at 440 nm and emission filter at 480 nm were selected and fluorescence was recorded at quiescent conditions at 37°C using a fluorimeter (FLUOStar Galaxy from BMG Labtech, Offenberg, Germany). The fluorescence data were plotted with respect to time after subtracting the minimum fluorescence value for each sample from all data points.
Transmission electron microscopy
To visualize the morphology of seeded Aβ42 fibrils at the aggregation kinetic end points, transmission electron microscopy (TEM) was performed. 5 µl of aggregated samples from 384‐well plates were spotted on a 400 mesh formvar/carbon‐coated copper grid. Samples were washed twice with 10 µl of MQ water and stained with 1% uranyl formate. TEM (FEI Tecnai 12 Spirit BioTWIN, operated at 100 kV) images were recorded using a 2 × 2 k Veleta CCD camera (Olympus Soft Imaging Solutions, GmbH, Münster, Germany). For each sample, 10–15 images were taken at a magnification of 20,000×–43,000×.
Detection limit of mass spectrometry methodology
Peptides Aβ1‐40 and Aβ1‐42 (Sigma‐Aldrich), Aβ17‐28, 13 6C6 15 7N2‐lysine (13C15N‐Lys) Aβ17‐28 and 13C15N‐Lys Aβ1‐42 (all amino acid‐analyzed; JPT Peptide Technologies GmbH, Berlin‐Germany™) were solubilised in 1,1,1,3,3,3‐hexafluoroisopropanol (Sigma‐Aldrich). The peptides were digested in 40% ACN/0.2 M AMBIC overnight at 37°C using trypsin at 1:1 stoichiometric ratio, and subsequently desalted and concentrated using C18 RP ZipTips. Digested Aβ1‐40 and Aβ1‐42 (10 pmol) were analyzed by MALDI‐TOF in reflectron positive mode (m/z 1,000–5,000 Da on UltrafleXtreme, Bruker™) using α‐hydroxycinnamic acid as a matrix, and the well‐detectable Aβ17‐28 peptide was selected for further experiments. Peptides Aβ17‐28 and 13C15N‐Lys Aβ17‐28 were analyzed at 50 fmol in data‐independent acquisition MSE mode on a C8 RP nano‐column (1.7 µm BEH C8, 75 µm × 250 mm) with a linear gradient of 3–85% of solution B (0.1% FA/ACN), for 60 min at a flow rate of 250 nl/min and stable column temperature of 35°C. Subsequently, MRM analyses were carried out, initially detecting seven MRM transitions for each labeling status of the tryptic peptide (y10, y9, y8, y7, y6, y5, and y4 for unlabeled, 13C‐Lys and 13C15N‐Lys internal standard) achieving a cycle time of 1.5 s. The four transitions with highest intensity (y10, y9, y8, and y7) were selected for further studies. Decreasing amounts of 13C15N‐Lys Aβ1‐42 were trypsin‐digested and concentrated to find the limit of detection (100 fmol down to 25 amol in the final injection volume). Samples were re‐suspended in 5% ACN and 4 µl injected each time into a 6500+ QTrap‐ Eksigent system operating in microflow mode as described above.
Brains from APPtg mice were processed by the 3‐step ultracentrifugation method (Allue et al, 2016) described above but with the modification of using 2 ml of each solvent. The 70% FA fraction was spiked with 13C15N‐Lys Aβ1‐42 at decreasing concentrations to find the limit of detection (100 fmol down to 25 amol in the final injection volume). Spiked samples were neutralized, immunoprecipitated, digested and analyzed by MRM as described above.
Intraperitoneal injection and short‐term labeling experiments
For all injection experiments, mice were randomly assigned to experimental groups assuring that littermates were equally distributed between groups. Sample sizes were informed by prior literature using similar peripheral injection experiments with unlabeled brain material and our previous experience.
Male 50‐day‐old APPtg mice were injected i.p. with 200 µl of 13C‐Lys APPtg brain extract (donors labeled for 50, 100 or 125 days) or 13C‐Lys non‐tg brain extract (donors labeled for 10 days). Mice were sacrificed by ketamine/xylazine overdose (400 mg/kg ketamine, 40 mg/kg xylazine) and intracardially perfused with 20 ml of cold PBS 1, 3, 5, 7, 10, 25, 50, 75, and 100 days after injection (n = 3–4 per time point and extract). Peritoneal lavage with 2 × 5 ml cold PBS was performed before intracardial perfusion at the time points 1–10 days. The brain, mesenteric lymph nodes, the spleen, and the liver were removed. Both brain hemispheres were snap‐frozen at time points 1–10 days while one hemisphere was snap‐frozen and the other one stored in 4% paraformaldehyde at time points 25–100 days. Peripheral organs were snap‐frozen after removal of a small segment that was stored in 4% paraformaldehyde. The recovered cell suspension from the peritoneal lavage was centrifuged at 300 g for 8 min at room temperature. The cell pellet was re‐suspended in 500 µl PBS and snap‐frozen. All frozen samples were stored at −80°C.
Further male and female 50‐day‐old APPtg mice were injected i.p. with 200 µl of unlabeled APPtg brain extract (sacrificed at 5 h, and 1, 3, 5, 7, 14, and 21 days after injection; n = 4–8 per time point) or non‐tg extract as negative controls (sacrificed at 5 h, and 1, 3, 5 and 7 days after injection; n = 1–3 per time point). The liver, spleen, mesenteric lymph nodes, peritoneal cells (at each time point), and EDTA‐anticoagulated whole‐blood samples from the caudal vena cava (at time points 5 h to 7 days) were collected. Ten microliters of resuspended peritoneal cells were air‐dried overnight on Superfrost® Plus slides at room temperature. Slides were fixed in methanol for 5 min and subjected to Giemsa staining to identify the composition of the peritoneal cells. Blood samples were centrifuged at 2,000 g for 15 min at 4°C and plasma was collected. In a separate experiment, EDTA‐anticoagulated blood was taken by intra‐cardiac puncture (n = 2–3 mice per time point at 5 h to 7 days after APPtg extract injection, n = 1 per time point at 5 h to 7 days after non‐tg extract injection) and mononuclear cells were isolated from blood samples using Ficoll‐Paque PREMIUM 1.084 (GE Healthcare) according to the manufacturer’s instructions. All samples were snap‐frozen and stored at −80°C.
Moreover, male and female 50‐day‐old APPtg mice were injected i.p. with 200 µl of unlabeled APPtg brain extract (n = 12) or non‐tg brain extract (n = 11) and sacrificed as described above at 240 days after injection. The brain was harvested and one hemisphere was snap‐frozen and the other one stored in 4% paraformaldehyde. All frozen samples were stored at −80°C.
Additional male 50‐day‐old APPtg mice either were injected i.p. with 200 µl of 13C‐Lys APPtg or non‐tg brain extract (all donors labeled for 100 days) and sacrificed 1 day after injection (n = 6 per group) or received the 13C‐Lys‐labeled diet following overnight fasting and were sacrificed after 1 or 3 days of labeling (n = 3 per time point). Age‐matched non‐APPtg mice either were injected i.p. with 200 µl of 13C‐Lys APPtg brain extract and sacrificed 1 day after injection (n = 3) or received the 13C‐Lys‐labeled diet following overnight fasting and were sacrificed after 1 day of labeling (n = 3). Mice were sacrificed as described above and the brain was harvested. Both hemispheres were snap‐frozen and stored at −80°C.
Detection of labeled and unlabeled Aβ by multiple reaction monitoring immuno‐mass spectrometry
The liver and spleen from mice injected with 13C‐Lys APPtg brain extract were homogenized on ice. Liver and spleen homogenates (20 mg per sample), and mesenteric lymph nodes were mixed with 200 µl of cold 70% FA, sonicated on ice, and centrifuged at 25,000 × g for 90 min at 4°C. The supernatants were spiked at 200 µl with 1.25 fmol of 13C15N‐Lys Aβ1‐42 and neutralized in 12 ml of PBS/2 M Tris solution. Peritoneal cell suspensions (cell pellet in 500 µl of PBS) were incubated with 1 ml of 100% FA and processed as the other peripheral tissues. The supernatant was spiked at 1.5 ml with 1.25 fmol of 13C15N‐Lys Aβ1‐42 and neutralized in 50 ml of PBS/2 M Tris solution.
Brains from mice either injected with 13C‐Lys APPtg or non‐tg brain extracts or labeled for 1 or 3 days as well as from unlabeled APPtg mice at 50 days of age (n = 2) were processed as described in detection limit of mass spectrometry methodology and the resulting fractions were stored at −80°C. The TBS fraction from mice injected with 13C‐Lys APPtg brain extract was spiked at 200 µl with 1.25 fmol of 13C15N‐Lys Aβ1‐42 in 20 µl of 70% FA and neutralized with 1.2 ml of PBS/2 M Tris (1:1 ratio) solution, pH 8.45. The 70% FA fraction from all mice was spiked at 200 µl with 10 fmol of 13C15N‐Lys Aβ1‐42 and neutralized in 12 ml of PBS/2 M Tris solution.
Spiked, neutralized samples were immunoprecipitated as described above. Eluted Aβ peptides were neutralized to pH 8.5 with 1 M AMBIC and enzymatically digested by the addition of 2.5 µg of trypsin in 50% acetonitrile. The digested peptide solution was dried, re‐suspended in 0.1–1% TFA to bring to pH < 4, concentrated and desalted twice, utilizing C18 RP ZipTip cartridges (50 cycles of sample binding and Aβ peptide elution by 10 × 10 µl of 70% ACN). The resulting eluate was dried and stored at −20°C until analysis. Subsequent MRM analysis was carried out by detecting the 4 transitions with the highest intensity (y10, y9, y8 and y7) for each labeling status of the tryptic peptide (unlabeled, 13C‐Lys and 13C15N‐Lys internal standard), with a dwell time of 100 ms, and achieving a cycle time of 1.26 s. Samples were re‐suspended in 25 µl of 5% ACN and analyzed in technical triplicates by time points, injecting 4 µl each into a 6500+ QTrap ‐ Eksigent system (SCIEX™) operating in microflow mode. Tryptic peptides were separated using a YMC Triart C18 column as described above. To monitor the system performance, MS Qual/Quant QC Mix, (MRM calibration standard, MSQC1; Sigma‐Aldrich) was injected every 15 samples. Two blank runs followed each sample.
Raw MRM data (as.wiff files) were exported to Skyline (MacCoss Lab Software v.19.1) for peak integration, normalization and abundance calculations. Abundance was reported as the ratio between the Aβ17‐28 peptide or 13C‐Lys Aβ17‐28 peptide and the 13C15N‐Lys Aβ17‐28 peptide (global standard). For the brain and lymph nodes, abundance of the Aβ17‐28 peptide and 13C‐Lys Aβ17‐28 peptide was additionally normalized to tissue weight. Investigators were blinded to group allocation during all data acquisitions and analyses.
Quantification of Aβ42 levels in peripheral and brain samples
The liver and spleen from mice injected with unlabeled APPtg or non‐tg brain extracts were homogenized for 1 min. Liver and spleen homogenates (~20–25 mg per sample), and mesenteric lymph nodes were treated with 10 µl of cold 70% FA/mg tissue. Mononuclear blood cell pellets were mixed with 100 µl of cold 70% FA. The samples were processed and Aβ42 levels analyzed as described for brain homogenates. Aβ42 concentrations were calculated as pg Aβ42/mg tissue for tissue samples and normalized to total organ weight and amount of injected Aβ42, yielding percent injected Aβ42. For mononuclear blood cells, Aβ42 concentrations were calculated as pg Aβ42/ml blood. Re‐suspended peritoneal cells were treated with two volumes of cold formic acid (minimum of 98%), sonicated for 2 × 30 s on ice, and incubated overnight at 4°C. Peritoneal cell samples were centrifuged at 25,000 × g for 60 min at 4°C. Aβ42 levels were analyzed as described above, calculated as pg Aβ42/500 µl cell suspension, and normalized to amount of injected Aβ42. Blood plasma was assayed for Aβ42 without additional treatment.
Brains from mice injected with unlabeled APPtg or non‐tg brain extracts were processed as described in detection limit of mass spectrometry methodology. Aβ42 levels were analyzed in the 70% FA fraction from mice injected with 13C‐Lys APPtg or non‐tg brain extracts or unlabeled APPtg or non‐tg brain extracts as described for brain homogenates. Aβ42 concentrations were normalized to protein concentrations measured with a spectrophotometer (ScanDrop, Analytik Jena GmbH, Germany) for samples from mice injected with 13C‐Lys brain extracts or to brain weight for samples from mice injected with unlabeled brain extracts.
The investigator was blinded to group allocation during all analyses.
Immunohistochemistry
Formalin‐fixed hemispheres and segments of livers were embedded in paraffin and sliced on a microtome (4 µm). Slices were stained using a BOND‐III automated immunostaining system (Leica Biosystems GmbH, Germany). Coronal brain sections (at approximately −1.4 and −2.5 mm relative to bregma) and liver sections were stained for Aβ (anti‐human Aβ clone 4G8; 1:4,000, BioLegend™) as previously described by us (Steffen et al, 2017; Paarmann et al, 2018). Additionally, brain sections were stained for microglia (IBA1, 1:1,000, Wako, 019‐19741). After staining, tissue sections were digitized at 230 nm resolution using a Pannoramic MidiII slide scanner (3DHistotech, Hungary).
Brain sections were analyzed by an investigator blinded to group allocation, using artificial intelligence in DeePathology STUDIO™ software as previously described by us (Bascuñana et al, 2021; Möhle et al, 2021). Briefly, the software was trained separately to recognize Aβ plaques and activated microglia, respectively. Once the training was completed, stained brain sections were analyzed automatically by DeePathology software after delineating the region of interest including parietal, somatosensory, auditory, and temporal cortices. Results were normalized to 10 mm² cortex area.
Statistical analysis
All statistical analysis was performed using Prism 6 software (GraphPad). Data resulting from Aβ quantification after injection of brain extracts were tested for normality by Kolmogorov–Smirnov test. Normality assumptions were rejected if P < 0.05. Unpaired two‐tailed t‐tests were used to compare mass spectrometry, immunoassay, and immunohistochemistry data of 13C‐Lys APPtg extract‐injected and 13C‐Lys non‐tg extract‐injected groups at each time point after injection. Variance was comparable between groups. Plasma Aβ concentration was analyzed by one‐way ANOVA. Levels of unlabeled Aβ in mononuclear blood cells were compared between negative controls and unlabeled APPtg extract‐injected mice at 1 day after injection by Mann–Whitney U‐test. Values of P < 0.05 were considered statistically significant.
Author contributions
Mirjam Brackhan: Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Giulio Calza: Formal analysis; Validation; Investigation; Visualization; Methodology. Kristiina Lundgren: Investigation; Methodology. Pablo Bascuñana: Formal analysis; Investigation; Methodology. Thomas Brüning: Investigation; Methodology. Rabah Soliymani: Formal analysis; Investigation; Methodology. Rakesh Kumar: Investigation; Visualization. Axel Abelein: Formal analysis; Investigation; Visualization; Writing—review and editing. Marc Baumann: Data curation; Supervision; Validation; Writing—original draft. Maciej Lalowski: Formal analysis; Supervision; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Jens Pahnke: Conceptualization; Resources; Data curation; Formal analysis; Supervision; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
MBr designed and performed mouse experiments, biochemical and molecular analyses, analyzed data, wrote manuscript. GC designed and performed the IP‐MALDI‐MS, MSE measurements, established MRM analysis pipelines and analyzed MS data. KL performed the IP‐MRM and SILAC experiments. PB performed morphological analyses, wrote manuscript. TB performed tissue processing and analyses. RS provided MSE and SILAC analysis. RK performed measurements about seeding activity. AA analyzed measurements about seeding activity. MBa provided the MS facility support, provided methodological study discussions. ML designed, supervised, and analyzed MS data, edited manuscript. JP developed the project, acquired the funding, designed experiments, wrote manuscript.
Disclosure and competing interests statement
Jens Pahnke declares competing financial interests related to the publication of this study, including being scientific advisor for DeePathology Ltd., Ra’anana, Israel. Maciej Lalowski is president of the Finnish Proteomics Society and member of the FEBS Fellowships Committee.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Iván Eiriz for skillful assistance with the mouse colonies; Markus Krohn, Luisa Möhle, and Kristin Paarmann for fruitful discussions; Elisabeth Nyberg for establishing early phase measurements of Aβ in non‐tg brains; Magdalena Łuczak (IBCh, PAS‐Poznan) for sharing her expertise on the MS statistical software and analysis of the MRM data; Jörg A. Gsponer (University of British Columbia, Vancouver, Canada) and Martin Fuhrmann (Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE) e.V. Bonn, Germany) for helpful comments on the manuscript.
Funding to J.P. was provided by Deutsche Forschungsgemeinschaft/ Germany (DFG 263024513); HelseSØ/ Norway (2019‐054, 2019‐055, 2022‐046); EEA grant/Norway grants (TAČR TARIMAD TO100078); Norges forskningsråd/ Norway (251290 (PROP‐AD), 295910 (PETABC), 327571 (NAPI)).
PROP‐AD was supported through the following funding organizations under the aegis of JPND ‐ www.jpnd.eu (AKA #301228 – Finland, BMBF #01ED1605 – Germany, CSO‐MOH #30000‐12631 – Israel, NFR #260786 – Norway, SRC #2015‐06795 – Sweden). PETABC is supported through the following funding organizations under the aegis of JPND ‐ www.jpnd.eu (NFR #327571 – Norway, FFG #882717 – Austria, BMBF #01ED2106 – Germany, MSMT #8F21002– Czech Republic, VIAA #ES RTD/2020/26 – Latvia, ANR #20‐JPW2‐0002‐04 ‐ France, SRC #2020‐02905 – Sweden). The projects receive funding from the European Union’s Horizon 2020 research and innovation program under grant agreement #643417 (JPco‐fuND).
Funding to M.L. was provided by ERA‐NET NEURON (ERA‐NET MicroSynDep project); AKA 318857 – Finland.
Funding to A.A was provided by Svenska Sällskapet för Medicinsk Forskning (SSMF) (P17‐0047), Svenska Forskningsrådet (Formas) (2020‐01013), Åke Wiberg stiftelse (M20‐0148), Alzheimerfonden (AF‐968052), Petrus och Augusta Hedlunds stiftelse (M‐2020‐1313), Foundation for Geriatric Diseases at Karolinska Instititet (2020‐2267), and Åhlén‐stiftelsen (203087).
EMBO reports (2022) 23: e54405.
Data availability
All raw and processed data are available at OSF PROP‐AD (http://www.doi.org/10.17605/OSF.IO/VNGPM). MS data are deposited at: MassIVE MSV000089339 (https://massive.ucsd.edu/ProteoSAFe/FindDatasets?query=MSV000089339; www.doi.org/10.25345/C57H1DR1J) and PeptideAtlas PASS01757 (http://www.peptideatlas.org/PASS/PASS01757).
References
- Adams HHH, Swanson SA, Hofman A, Arfan Ikram M (2016) Amyloid‐β transmission or unexamined bias? Nature 537: E7–E9 [DOI] [PubMed] [Google Scholar]
- Allue JA, Sarasa L, Izco M, Perez‐Grijalba V, Fandos N, Pascual‐Lucas M, Ogueta S, Pesini P, Sarasa M (2016) Outstanding phenotypic differences in the profile of amyloid‐beta between Tg2576 and APPswe/PS1dE9 transgenic mouse models of Alzheimer's disease. J Alzheimers Dis 53: 773–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher DM, Belay E, Bigio E, Brandner S, Brubaker SA, Caughey B, Clark B, Damon I, Diamond M, Freund M et al (2020) Risk of transmissibility from neurodegenerative disease‐associated proteins: experimental knowns and unknowns. J Neuropathol Exp Neurol 79: 1141–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bascuñana P, Brackhan M, Pahnke J (2021) Machine learning‐supported analyses improve quantitative histological assessments of amyloid‐β deposits and activated microglia. J Alzheimers Dis 79: 597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM (2006) Human amyloid‐beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo . Nat Med 12: 856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu X‐L, Xiang Y, Jin W‐S, Wang J, Shen L‐L, Huang Z‐L, Zhang K, Liu Y‐H, Zeng F, Liu J‐H et al (2018) Blood‐derived amyloid‐β protein induces Alzheimer's disease pathologies. Mol Psychiatry 23: 1948–1956 [DOI] [PubMed] [Google Scholar]
- Burwinkel M, Lutzenberger M, Heppner FL, Schulz‐Schaeffer W, Baier M (2018) Intravenous injection of beta‐amyloid seeds promotes cerebral amyloid angiopathy (CAA). Acta Neuropathol Commun 6: 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali I, Cohen ML, Haїk S, Parchi P, Giaccone G, Collins SJ, Kofskey D, Wang H, McLean CA, Brandel J‐P et al (2018) Iatrogenic Creutzfeldt‐Jakob disease with Amyloid‐β pathology: an international study. Acta Neuropathol Commun 6: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TPJ (2013) Proliferation of amyloid‐β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA 110: 9758–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Sazdovitch V, Ando K, Seilhean D, Privat N, Yilmaz Z, Peckeu L, Amar E, Comoy E, Maceski A et al (2018) Neuropathology of iatrogenic Creutzfeldt‐Jakob disease and immunoassay of French cadaver‐sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol 135: 201–212 [DOI] [PubMed] [Google Scholar]
- Eisele YS (2013) From soluble Aβ to progressive Aβ aggregation: could prion‐like templated misfolding play a role? Brain Pathol 23: 333–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M (2010) Peripherally applied Abeta‐containing inoculates induce cerebral beta‐amyloidosis. Science 330: 980–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Fritschi SK, Hamaguchi T, Obermuller U, Fuger P, Skodras A, Schafer C, Odenthal J, Heikenwalder M, Staufenbiel M et al (2014) Multiple factors contribute to the peripheral induction of cerebral beta‐amyloidosis. J Neurosci 34: 10264–10273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney C, Scott GP, Cole JH, Sastre M, Goldstone AP, Leech R (2016) Seeds of neuroendocrine doubt. Nature 535: E1–E2 [DOI] [PubMed] [Google Scholar]
- Gholizadeh E, Karbalaei R, Khaleghian A, Salimi M, Gilany K, Soliymani R, Tanoli Z, Rezadoost H, Baumann M, Jafari M et al (2021) Identification of celecoxib‐targeted proteins using label‐free thermal proteome profiling on rat hippocampus. Mol Pharmacol 99: 308–318 [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256: 184–185 [DOI] [PubMed] [Google Scholar]
- Huynh TV, Holtzman DM (2018) Amyloid‐β 'seeds' in old vials of growth hormone. Nature 564: 354–355 [DOI] [PubMed] [Google Scholar]
- Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ (2013) Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver‐derived human growth hormone. JAMA Neurol 70: 462–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaunmuktane Z, Mead S, Ellis M, Wadsworth JDF, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard‐Loendt A, Walker AS et al (2015) Evidence for human transmission of amyloid‐β pathology and cerebral amyloid angiopathy. Nature 525: 247–250 [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2013) Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2015) Amyloid‐β pathology induced in humans. Nature 525: 193–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age‐dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: 372–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Ahn HS, Cho SM, Lee JE, Kim Y, Lee C (2014) Detection and quantification of plasma amyloid‐β by selected reaction monitoring mass spectrometry. Anal Chim Acta 840: 1–9 [DOI] [PubMed] [Google Scholar]
- Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, Schmidt M, Sigurdson CJ, Jucker M, Fändrich M (2019) Cryo‐EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer's brain tissue. Nat Commun 10: 4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fässler R, Mann M (2008) SILAC mouse for quantitative proteomics uncovers kindlin‐3 as an essential factor for red blood cell function. Cell 134: 353–364 [DOI] [PubMed] [Google Scholar]
- Lau HHC, Ingelsson M, Watts JC (2021) The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer's disease. Acta Neuropathol 142: 17–39 [DOI] [PubMed] [Google Scholar]
- Lauwers E, Lalli G, Brandner S, Collinge J, Compernolle V, Duyckaerts C, Edgren G, Haïk S, Hardy J, Helmy A et al (2020) Potential human transmission of amyloid β pathology: surveillance and risks. Lancet Neurol 19: 872–878 [DOI] [PubMed] [Google Scholar]
- Meisl G, Yang X, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SIA, Dobson CM, Linse S, Knowles TPJ (2014) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc Natl Acad Sci USA 111: 9384–9389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michno W, Stringer KM, Enzlein T, Passarelli MK, Escrig S, Vitanova K, Wood J, Blennow K, Zetterberg H, Meibom A et al (2021) Following spatial Aβ aggregation dynamics in evolving Alzheimer's disease pathology by imaging stable isotope labeling kinetics. Sci Adv 7: eabg4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhle L, Bascuñana P, Brackhan M, Pahnke J (2021) Development of deep learning models for microglia analyses in brain tissue using DeePathology™ STUDIO. J Neurosci Methods 364: 109371. 10.1016/j.jneumeth.2021.109371 [DOI] [PubMed] [Google Scholar]
- Morales R, Duran‐Aniotz C, Bravo‐Alegria J, Estrada LD, Shahnawaz M, Hu PP, Kramm C, Morales‐Scheihing D, Urayama A, Soto C (2020) Infusion of blood from mice displaying cerebral amyloidosis accelerates amyloid pathology in animal models of Alzheimer's disease. Acta Neuropathol Commun 8: 213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Bravo‐Alegria J, Moreno‐Gonzalez I, Duran‐Aniotz C, Gamez N, Edwards Iii G, Soto C (2021) Transmission of cerebral amyloid pathology by peripheral administration of misfolded Aβ aggregates. Mol Psychiatry 26: 5690–5701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paarmann K, Prakash SR, Krohn M, Möhle L, Brackhan M, Brüning T, Eiriz I, Pahnke J (2019) French maritime pine bark treatment decelerates plaque development and improves spatial memory in Alzheimer's disease mice. Phytomedicine 57: 39–48 [DOI] [PubMed] [Google Scholar]
- Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S (2010) Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci USA 107: 14508–14513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purro SA, Farrow MA, Linehan J, Nazari T, Thomas DX, Chen Z, Mengel D, Saito T, Saido T, Rudge P et al (2018) Transmission of amyloid‐β protein pathology from cadaveric pituitary growth hormone. Nature 564: 415–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S et al (2006) Aβ42‐driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7: 940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie DL, Adlard P, Peden AH, Lowrie S, Le Grice M, Burns K, Jackson RJ, Yull H, Keogh MJ, Wei W et al (2017) Amyloid‐beta accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol 134: 221–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T et al (2007) Clearance of amyloid‐beta by circulating lipoprotein receptors. Nat Med 13: 1029–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766 [DOI] [PubMed] [Google Scholar]
- Steffen J, Krohn M, Schwitlick C, Brüning T, Paarmann K, Pietrzik CU, Biverstål H, Jansone B, Langer O, Pahnke J (2017) Expression of endogenous mouse APP modulates β‐amyloid deposition in hAPP‐transgenic mice. Acta Neuropathol Commun 5: 49. 10.1186/s40478-017-0448-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler‐Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA et al (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease‐like pathology. Proc Natl Acad Sci USA 94: 13287–13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A et al (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln A‐C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A et al (2018) Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556: 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildburger NC, Gyngard F, Guillermier C, Patterson BW, Elbert D, Mawuenyega KG, Schneider T, Green K, Roth R, Schmidt RE et al (2018) Amyloid‐beta plaques in clinical Alzheimer's disease brain incorporate stable isotope tracer in vivo and exhibit nanoscale heterogeneity. Front Neurol 9: 169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AC, Stevens MY, Chen MB, Lee DP, Stähli D, Gate D, Contrepois K, Chen W, Iram T, Zhang L et al (2020) Physiological blood‐brain transport is impaired with age by a shift in transcytosis. Nature 583: 425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Zhang S, Kreutzer AG, Nowick JS (2018) An efficient method for the expression and purification of Aβ(M1‐42). Biochemistry 57: 3861–3866 [DOI] [PMC free article] [PubMed] [Google Scholar]