

ABSTRACT
Alcohol-related liver disease (ALD) is a major cause of liver disease and represents a global burden, as treatment options are scarce. Whereas 90% of ethanol abusers develop alcoholic fatty liver disease (AFLD), only a minority evolves to steatohepatitis and cirrhosis. Alcohol increases lipogenesis and suppresses lipid-oxidation implying steatosis, although the key role of intestinal barrier integrity and microbiota in ALD has recently emerged. Bacteroides thetaiotaomicron (Bt) is a prominent member of human and murine intestinal microbiota, and plays important functions in metabolism, gut immunity, and mucosal barrier. We aimed to investigate the role of Bt in the genesis of ethanol-induced liver steatosis. Bt DNA was measured in feces of wild-type mice receiving a Lieber-DeCarli diet supplemented with an increase in alcohol concentration. In a second step, ethanol-fed mice were orally treated with living Bt, followed by analysis of intestinal homeostasis and histological and biochemical alterations in the liver. Alcohol feeding reduced Bt abundance, which was preserved by Bt oral supplementation. Bt-treated mice displayed lower hepatic steatosis and triglyceride content. Bt restored mucosal barrier and reduced LPS translocation by enhancing mucus thickness and production of Mucin2. Furthermore, Bt up-regulated Glucagon-like peptide-1 (GLP-1) expression and restored ethanol-induced Fibroblast growth factor 15 (FGF15) down-regulation. Lipid metabolism was consequently affected as Bt administration reduced fatty acid synthesis (FA) and improved FA oxidation and lipid exportation. Moreover, treatment with Bt preserved the mitochondrial fitness and redox state in alcohol-fed mice. In conclusion, recovery of ethanol-induced Bt depletion by oral supplementation was associated with restored intestinal homeostasis and ameliorated experimental ALD. Bt could serve as a novel probiotic to treat ALD in the future.
KEYWORDS: Alcohol-related liver disease, Bacteroides thetaiotaomicron, microbiota, steatosis, intestinal barrier, mitochondria
Introduction
The excessive alcohol introit represents a social, political, and health-care issue worldwide. In Europe and North America, alcohol-related liver disease (ALD) is the main cause of liver disease and severe alcoholic hepatitis (AH), implying high morbidity and mortality rates.1–3 Chronic alcohol consumption provokes the development of hepatic steatosis in 90% of alcoholics, while 10–35% of these evolve to alcoholic steatohepatitis (ASH) and finally only 10–20% develop cirrhosis.4,5 Therefore, ethanol drinking can be seen as the first step in the genesis of ALD, which is still reversible. The enzymatic metabolism of ethanol diminishes the NAD+/NADH ratio with consequent enhancement of fatty acid (FA) synthesis and suppression of FA oxidation (FAO).6,7 Accordingly, studies demonstrated that alcohol assumption enhances the expression of genes involved in lipogenesis, such as Fatty acid synthase (FASN) and Stearoyl-CoA desaturase-1 (SCD-1), while β-oxidation is dampened likely due to inhibition of Peroxisome proliferator-activated receptor-α (PPAR-α) activity.8–12 However, the pathogenesis of ALD is very complex, and recently the key role of intestinal homeostasis has emerged. In fact, ethanol ingestion provokes microbiota perturbations and disruption of barrier function, which lead to translocation of pathogen-associated molecular pattern molecules (PAMPs) (e.g. lipopolysaccharide, LPS) and activation of Kupffer cells with consequent inflammation and oxidative stress in the liver.13,14 Moreover, some studies have showed that intervention in microbiota seems to protect against ethanol-induced liver injury.15–17 Bacteroides thetaiotaomicron (Bt) is a gram-negative anaerobe and a prominent member of intestinal microbiota in humans and mice. Bt constitutes 6% of all intestinal bacteria and 12% of all Bacteroides in humans, and exerts important functions in mucosal barrier, immunity, and nutrient metabolism.18,19 It has been reported that the commensal Bt crucially drives the mucosal barrier development during weaning and also protects from hyperpermeability in an inflammatory contest.20,21 Since an impaired intestinal barrier reflects an important pathogenetic player in ALD, we have hypothesized a loss of this commensal after alcohol assumption. Moreover, a recent study associated Bt depletion with obesity, while Bt administration protected high-fat fed mice against obesity by promoting lipolysis in adipose tissue.22 Therefore, considering the potential role of Bt in modulation of lipid metabolism and its importance in barrier function maintenance, we investigated whether Bt intestinal abundance is impaired by alcohol assumption and whether its supplementation implies metabolic beneficial effects in the liver.
Results
Ethanol reduced Bt amount in vivo and in vitro
In order to investigate whether ethanol impacts the Bt abundance in the intestine, mice were fed an ethanol-containing diet (Lieber-DeCarli diet, EtOH-fed) for 15 days (Figure 1a). Ethanol feeding resulted in increased ALT levels and liver triglyceride accumulation with steatosis development, hallmarks of ALD (Figure 1 b-e).
Figure 1.
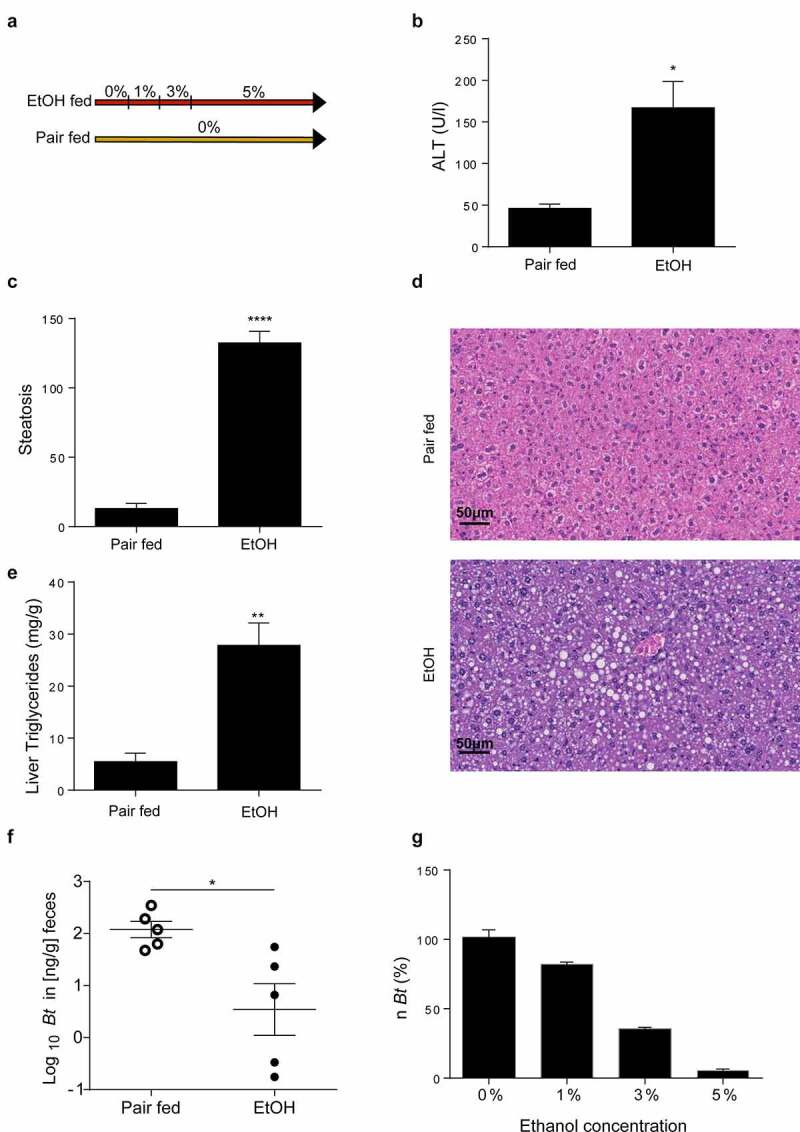
Ethanol depletes Bt. (a) Design of Lieber-DeCarli model. (b) Serum ALT levels (Pair fed = n6; EtOH = n10). (c, d) Histological determination of hepatic steatosis with representative pictures of H&E staining (n = 5 per group). (e) Liver triglyceride content (Pair fed = n5; EtOH = n6). (f) Quantification of Bt DNA in feces measured by qPCR (n = 5 per group). (g) Quantification of Bt number (expressed in percentage) in LYBHI medium supplemented with different ethanol concentrations. Data are expressed in mean ± SEM; *p < .05, **p < .01, ***p < .001, ****p < .0001 according to two-tails student’s t-test.
Abbreviations: Bt, Bacteroides thetaiotaomicron.
Importantly, we note that ethanol feeding induced a significant reduction in Bt compared to pair-fed mice (figure 1f). This was in line with in-vitro study, where Bt growth was inhibited by increasing ethanol concentrations (Figure 1g).
Bt supplementation ameliorated hepatic steatosis
In the next step, ethanol-fed mice were treated with Bt by oral gavage (Figure 2a). Bacterial supplementation was effective to recolonize Bt in EtOH-fed mice (Figure 2b). Notably, although the two ethanol fed groups showed the same alcohol assumption (Supplementary Fig.1), Bt-treated mice presented a grade of hepatic steatosis significantly lower (Figure 2c, d) compared to vehicle treated mice. Moreover, this observation was corroborated by the significant reduction of triglycerides (TG) content in the liver of EtOH-fed Bt-treated mice (Figure 2e). Meanwhile, no difference was found in serum ALT levels, and Bt only mildly reduced IL1β expression and neutrophilic infiltration in the liver (Supplementary Fig.2).
Figure 2.
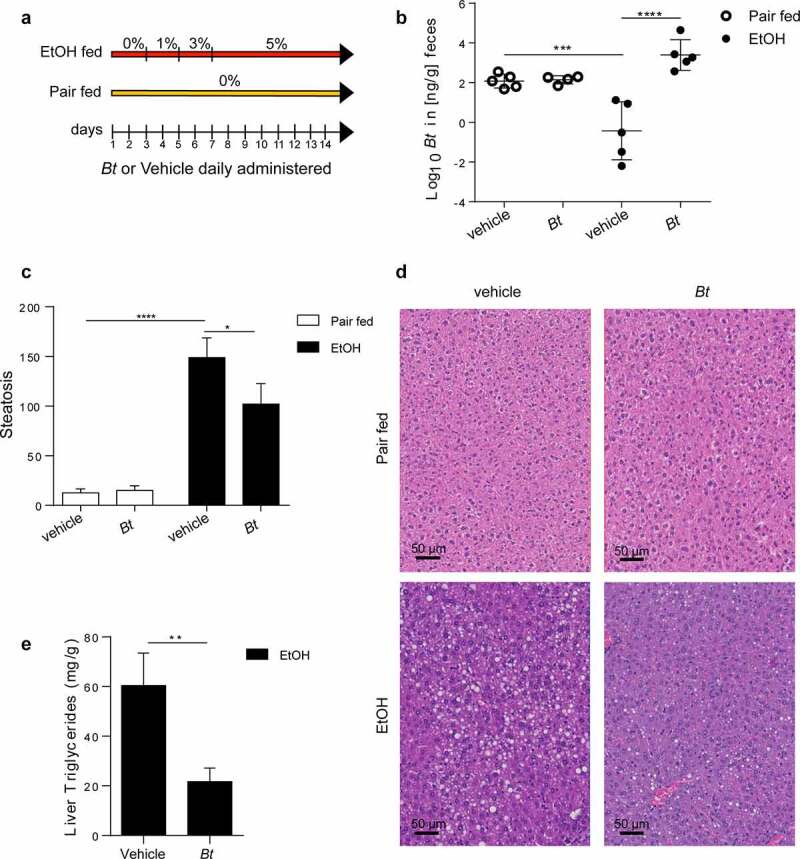
Bt supplementation ameliorates alcoholic fatty liver disease. (a) Experimental design. (b) Quantification of Bt DNA in feces measured by qPCR (Pair fed groups = n4-5; EtOH groups = n5). (c, d) Histological determination of hepatic steatosis with representative pictures of H&E staining (Pair fed groups = n6; EtOH groups = n9). (e) Liver Triglyceride content in EtOH fed mice (n = 9–10). Data are expressed in mean ± SEM; *p < .05; **p < .01; ***p < .001, ****p < .0001 according to one-Way ANOVA followed by post hoc analysis (Bonferroni test) or two-tails student’s t-test.
Abbreviations: Bt, Bacteroides thetaiotaomicron.
Bt preserved mucosal barrier function
Treatment with Bt partially restored the ethanol-induced mucus disruption as shown by higher mucus thickness in PAS-stained colon sections (Figure 3a, b). Although, Bt administration was not associated with altered number of goblet cells in EtOH-fed mice, we could observe a significant increase in goblet cell diameter (Figure 3c-e). It is important to note that lower levels of circulating LPS were detectable in EtOH-fed Bt-treated mice (figure 3f) compared to EtOH-vehicle, suggesting a restored gut barrier function. To further elucidate the role of Bt in mucosal preservation in the colon, we found that Mucin1 (Muc1) was transcriptionally downregulated, while Mucin2 (Muc2) expression was enhanced by Bt, corroborated by Muc2 immunofluorescence (Figure 4a, b). Muc2 is the main component of mucus layer and probably Bt promoted mucus production by increasing muc2. Therefore, ethanol decreased the total mucus amount compared to Bt, which restored the layer of mucus by acting mostly on muc2 production. Although mRNA levels do not show different muc2 expression between pair fed and ethanol, the muc2 histological staining clearly shows the lack of mucin2 in the ethanol vehicle mice. However, we recognize that several other types of mucins could be involved. The integrity of epithelial tight junctions is also important in preserving the barrier function, thus its alteration is crucial in ethanol-related leaky gut.14,23,24 Therefore, we evaluated the expression profile of tight junction proteins, finding a significant down regulation of Claudin 1 (Cldn1) transcription in Bt-treated mice (Figure 4c). Cldn1 over-expression in colon inflammation has been associated with higher MMP9 transcription and ERK phosphorylation to induce Notch activity with the consequent muc2 suppression.25 Accordingly, we found that Bt administration reduced MMP9 expression, ERK phosphorylation, and Hes1 expression (transcriptional marker of Notch activity) (Figure 4d-f). In addition, MMP9 was upregulated by ethanol (Figure 4d).
Figure 3.
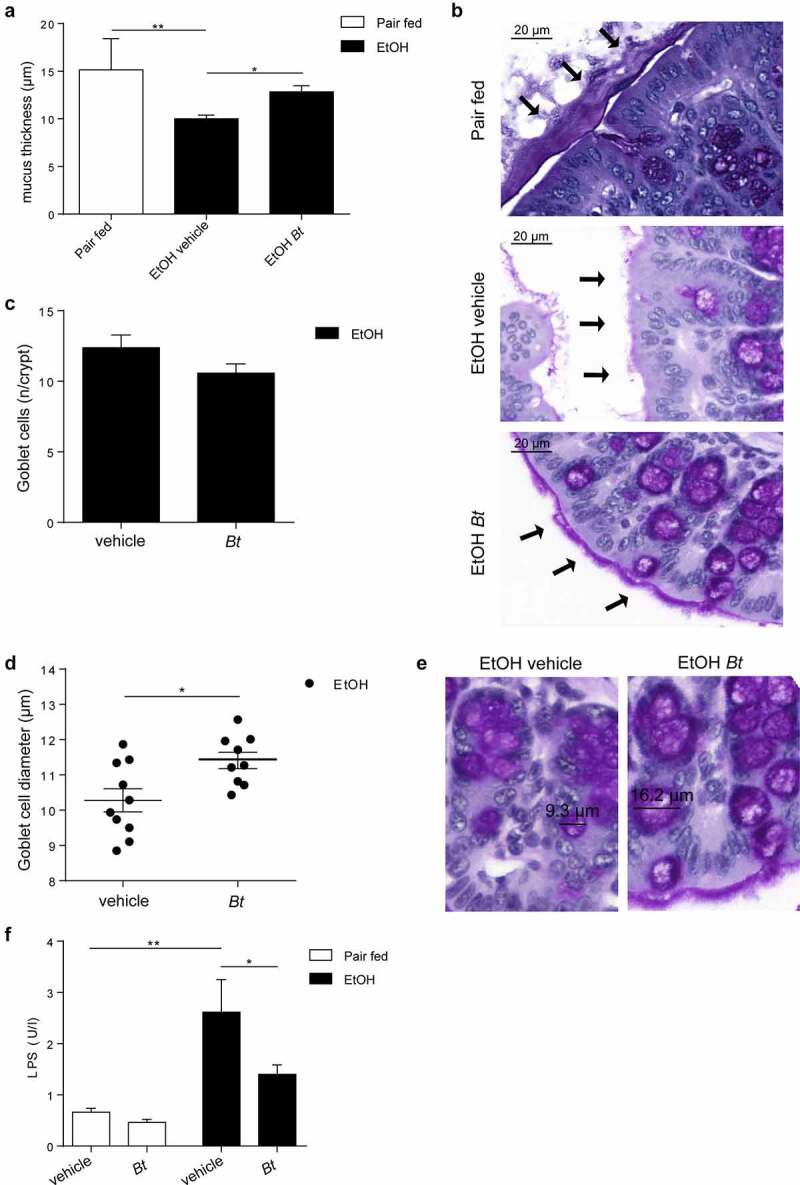
Bt recovery restores mucus production in the colon. (a, b) Quantification of mucus thickness in colon sections stained with periodic acid–Schiff reaction (Pair fed = n4; EtOH groups = n9-10). (c) Number of goblet cells per crypt and (d, e) goblet cells diameter identified by periodic acid–Schiff reaction. (f) Serum LPS concentration (Pair fed groups = n6; EtOH groups = n6-8). Data are expressed in mean ± SEM; *p < .05; **p < .01; ***p < .001, ****p < .0001 according to one-Way ANOVA followed by post hoc analysis (Bonferroni test) or two-tails student’s t-test.
Abbreviations: Bt, Bacteroides thetaiotaomicron, LPS, lipopolysaccharide.
Figure 4.
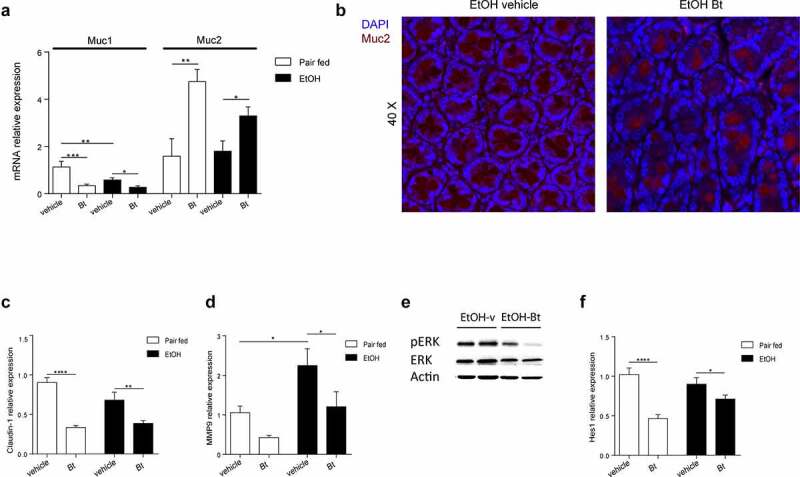
Bt promotes mucin2 production via downregulation of claudin1-Notch axis (a) Colonic expression of muc1 and muc2 fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n6; EtOH groups = n10). (b) Muc2 immunoreactivity (red) with representative confocal microscope image of murine colon in EtOH-fed mice. DAPI, blue. (c-d) Colonic expression of Claudin1 and MMP9 fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n5-6; EtOH groups = n9-10). (e) Representative pictures of protein levels of actin, ERK and pERK determined by western blot analysis (EtOH groups = n5). (f) Colonic expression of Hes1 fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n5-6; EtOH groups = n9-10). Data are expressed in mean ± SEM; *p < .05; **p < .01; ***p < .001, ****p < .0001 according to one-Way ANOVA followed by post hoc analysis (Bonferroni test).
Abbreviations: Muc1, mucin1; muc2, mucin2; DAPI, 4′,6-diamidino-2-phenylindole; MMP9, matrix metallopeptidase 9; ERK, extracellular signal-regulated kinase 1/2; pERK, phosphorylated ERK. Hes1, hairy and enhancer of split-1; Bt, Bacteroides thetaiotaomicron.
The increased expression of Reg3γ and Reg3β lectins also underlined an immunological effect of Bt administration in EtOH fed mice (Supplementary Fig 3).
Bt modulated GLP1 and FGF15 expression
Intriguingly, Bt modulated intestinal hormone production as Bt enhanced GLP-1 expression and restored the ethanol-induced FGF15 downregulation (Figure 5a, b), in the colon and ileum, respectively. The serum levels of GLP-1 and FGF-15 corroborated these findings (Figure 5a, b). Supposing the role of the intestinal FXR in FGF15 dysregulation, the expression of other FXR targets was measured (i.e., IBABP, Ost-α, and Ost-β), underlying a decreased expression in EtOH-vehicle mice and an increased expression (significant for Ost-α and Ost-β) in EtOH-Bt mice (Figure 5a). These findings highlight the intestinal FXR dysregulation in ALD, while the administration of Bt improved FXR function probably by modulating bile acid metabolism or by the production of unknown FXR activators. Accordingly, in the liver, the expression of CYP7A1, a major bile acid synthesis modulator typically downregulated by FXR and FGF15-FGFR4, was significantly up-regulated in EtOH-fed mice and normalized in Bt-treated mice (supplementary Fig.4).
Figure 5.
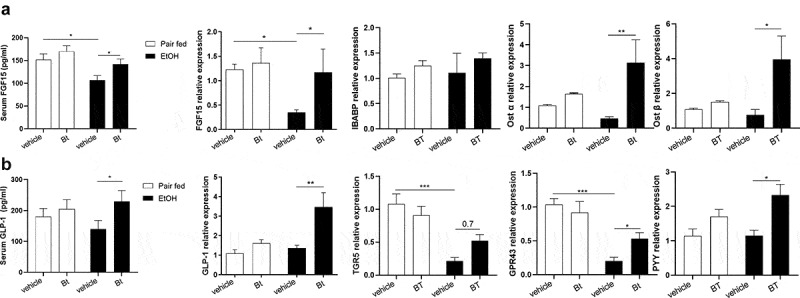
Bt modulated GLP1 and FGF15 expression. (a) Serum levels of FGF15 and ileal expression of FGF15, IBABP, Ost-α and Ost-β fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n4-6; EtOH groups = n6-10). (b) Serum levels of GLP-1 and colonic expression of GLP-1, TGR5, GPR43 and PYY fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n4-6; EtOH groups = n6-10). Data are expressed in mean ± SEM; *p < .05; **p < .01; ***p < .001, according to one-Way ANOVA followed by post hoc analysis (Bonferroni test).
Abbreviations: FGF15, fibroblast growth factor 15; IBABP, ileal bile acid-binding protein; Ost-α/β, Organic solute transporter α/β; GLP-1, Glucagon-like peptide-1; G protein-coupled bile acid receptor 1; GPR43, G-protein-coupled receptor 43; Peptide YY; Bt, Bacteroides thetaiotaomicron.
When analyzing the GLP-1 induction pathways in the colon, we found that GPR43, a SCFA receptor, and TGR5, a bile acid receptor, were both downregulated in EtOH-fed mice and restored by Bt treatment (Figure 5b). GPR43 and TGR5 induce prompt GLP-1 and PYY production;26 accordingly, also PYY was up-regulated in EtOH-Bt mice (Figure 5b). The modulation of both receptors underlines that bile acid homeostasis together with bacterial SCFA could contribute to GLP-1 production in Bt-treated mice.
Bt improved hepatic metabolism
Considering the results described above, we have hypothesized that Bt improved hepatic steatosis via the recovery of intestinal homeostasis. Interestingly, we found that the liver expression of FGFR4 was up-regulated in EtOH-Bt mice (Figure 6a), while GLP-1 receptor (GLP1R) was up-regulated in EtOH-vehicle and EtOH-Bt mice (supplementary Fig.5). Analyzing the main lipid metabolism regulators, we found that Bt significantly counteracted the reduction of AMPK activity, and the increase of SREBP-1c induced by alcohol (Figure 6b). In particular, Bt restored levels of AMPK phosphorylation and normalized total amount and nuclear translocation of SREBP-1c (Figure 6b). AMPK has been described as a target of FGFR4 and GLP1R signaling, and is considered a cellular energetic sensor, able to suppress FA synthesis and stimulate FA oxidation (FAO) to preserve ATP storage.27 SREBP-1c is a transcriptional factor of genes involved in FA synthesis; accordingly, FASN and SCD-1 expression was reduced in Bt-treated mice compared to EtOH vehicle mice (Figure 6c, d). Overall, these findings suggest that alcohol stimulates FA synthesis by inhibiting AMPK and enhancing SREBP-1c probably by affecting FGF-15 and GLP-1 production. Bt administration prevents this by restoring the gut-liver axis. Other AMPK targets are PPAR-α, PGC-1α, and probably PGC-1β, whose primary function is to induce mitochondrial biogenesis, OXPHOS and FAO.28 Interestingly, we found that ethanol reduced the expression of all major mitochondrial biogenesis markers such as PPAR-α, PGC-1 α, PGC-1β, and TFAM, while Bt significantly restored their expression (Figure 7a). Accordingly, Bt recovered protein levels of mitochondrial respiratory chain (RC) complexes I, II, and V, and consequently the cellular bioenergetics improved as shown by ATP levels, which were reduced in EtOH-vehicle and normal in EtOH-Bt (Figure 7b). The expression of CPT1a and VDAC1 levels, whose coupled activity is essential for transport of acyl-CoA esters and fuel FAO,29 confirmed that FAO was inhibited in ethanol-fed mice and stimulated in Bt supplemented ones (Figure 7c). Moreover, PGC-1β is also involved in VLDL exportation.28 Part of lipid accumulation in ALD is due to lower lipid exportation.30 Accordingly, we found that DGAT1, target of PGC1β involved in VLDL trafficking, was upregulated in EtOH-Bt mice and serum triglycerides were lower in EtOH-vehicle mice and normal in EtOH-Bt (Figure 7d). Moreover, PGC-1β is known to stimulate the antioxidant response and interestingly, we found that Bt restored the SOD activity, PRDX3- and −5 expression, reducing serum lipoperoxidation markers, such as MDA- and HNE-protein adducts (Figure 7e). Taken together, the recovery of Bt intestinal abundance in ethanol fed mice permits to counteract the hepatic bioenergetic aberrations induced by alcohol, likely by gut-liver axis rewiring.
Figure 6.
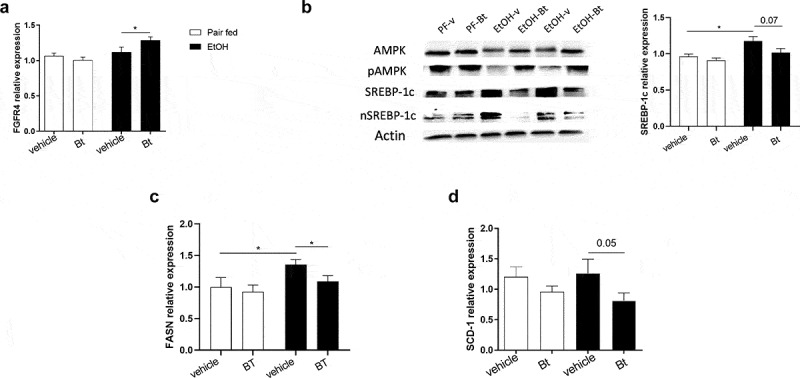
Bt reduced hepatic FA synthesis (a) Liver expression of FGFR4 fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n5-6; EtOH groups = n9-10). (b) Representative pictures of protein levels of actin, AMPK, pAMPK, SREBP-1c, and nuclear SREBP-1c, determined by western blot analysis and expression of SREBP-1c determined by qPCRin the liver (for western blot: Pair fed groups = n3; EtOH groups = n5; for qPCR: Pair fed groups = n5-6; EtOH groups = n9-10). (c, d) Liver expression of FASN and SCD-1 fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n4; EtOH groups = n8-10). Data are expressed in mean ± SEM; *p < .05, according to one-Way ANOVA followed by post hoc analysis (Bonferroni test).
Abbreviations: FA, fatty acids; FGFR4, Fibroblast Growth Factor Receptor 4; AMPK, 5’ AMP-activated protein kinase; pAMPK, phosphorylated AMPK; SREBP-1c, Sterol-regulatory element-binding protein-1C; n SREBP-1c, nuclear SREBP-1c; FASN, Fatty acid synthase; SCD-1, Stearoyl-CoA desaturase; Bt, Bacteroides thetaiotaomicron.
Figure 7.
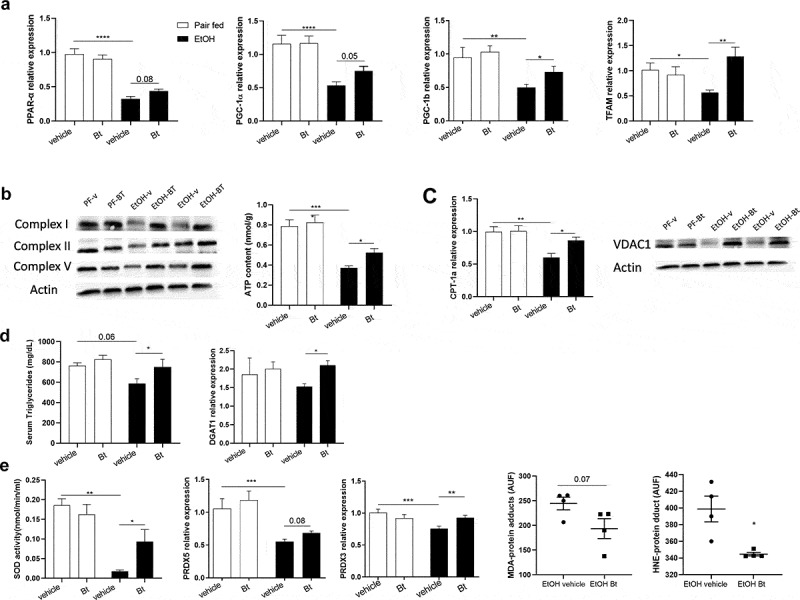
Bt improved hepatic mitochondrial fitness and function (a) Liver expression of PPAR-α, TFAM, PGC-1α, PGC-1β fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n4-5; EtOH groups = n6-7). (b) Representative pictures of protein levels of actin, respiratory chain complexes I, II and V determined by western blot analysis (Pair fed groups = n3; EtOH groups = n5), and and hepatic ATP content (Pair fed groups = n4; EtOH groups = n5). (c) Liver expression of CPT-1a fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n4; EtOH groups = n5); and representative pictures of protein levels of actin and VDAC1determined by western blot analysis (Pair fed groups = n3; EtOH groups = n5). (d) Serum levels of Triglycerides (Pair fed groups = n5; EtOH groups = n9), and liver expression of DGAT1 fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n4-5; EtOH groups = n9-10). (e) Hepatic SOD enzymatic activity (Pair fed groups = n4; EtOH groups = n4) and expression of PRDX-3 and −5 fold over Pair fed vehicle group and determined by qPCR (Pair fed groups = n5-6; EtOH groups = n9); and serum MDA- and HNE- protein adducts (Pair fed groups = n4; EtOH groups = n4). Data are expressed in mean ± SEM; *p < .05, **p < .01; ***p < .001, ****p < .0001 according to one-Way ANOVA followed by post hoc analysis (Bonferroni test).
Abbreviations: PPAR-α, Peroxisome Proliferator Activated Receptorα; PGC-1α/β, peroxisome proliferator-activated receptor-gamma coactivator-1α/β; TFAM, Transcription Factor A, mitochondrial; CPT-1a, Carnitine Palmitoyltransferase 1a; VDAC1, Voltage-dependent anion-selective channel 1; DGAT1, Diacylglycerol O-Acyltransferase 1; SOD, Superoxide dismutase; PRDX-3/5, Peroxiredoxin 3/5; MDA, Malondialdehyde; HNE, 4Hydroxynonenal; Bt, Bacteroides thetaiotaomicron.
Discussion
The alterations of intestinal microbiota have been described as the driving force of ALD development,13,31,32 and in some experimental models, oral supplementation with probiotics, bacteria, and some immunomodulatory molecules attenuated liver alcohol-related liver16,33,34 injury. Ethanol ingestion provokes leaky gut and bacterial overgrowth through disruption of mucus layer and tight junction, and downregulation of antimicrobial peptides, such as Reg3b and Reg3g lectins.35,36 Bt is a gram-negative anaerobe and a prominent member of intestinal bacteria both in humans and mice. Several studies demonstrated Bt capacity to modulate host functions such as immunity, mucosal barrier, and nutrients metabolism.18,21 In this study, we have further demonstrated that Bt drastically decreased in chronically alcohol fed mice and its growth in culture medium is strongly inhibited by ethanol supplementation. Therefore, we investigated whether the recovery of Bt intestinal abundance in Lieber-DeCarli fed mice could protect the liver from alcohol exposure. The daily oral administration of Bt was effective in restoring alcohol-induced depletion. Interestingly, although the two ethanol-exposed groups showed the same quantity of ethanol in the serum, highlighting equal absorption and metabolization, Bt-treated mice presented several benefits. In particular, hepatic steatosis and lipid accumulation were significantly reduced. Perhaps, the shortness of Lieber-DeCarli model does not allow us to appreciate the potential effects of Bt on inflammation and liver injury, but only its benefits in alcoholic fatty liver disease (AFLD).
Relevant were the effects on intestinal health, which may explain the improvement of hepatic disease. Bt partially restored mucus layer in EtOH-fed mice by increasing muc2 production. Wrzosek et al reported that in a gnotobiotic model Bt is involved in goblet cell differentiation and induction of mucins expression such as muc2.37 Moreover, a recent study showed that in colitis, goblet cell differentiation and muc2 production are dampened by overexpression of cludin1, which in turn induces MMP9 upregulation and ERK phosphorylation with consequent activation of Notch signaling.25 Interestingly, Bt supplementation reduced the expression of cldn1 and MMP9, ERK phosphorylation and Hes1 expression (marker of Notch signaling activation), suggesting a potential mechanism by which intestinal bacteria promote mucin production and limit inflammation. Preventing mucus disruption and enhancing antimicrobial peptides expression, Bt significantly reduced LPS translocation. Beyond the barrier function, Bt showed impact on enterokines production, as it induced a significant increase of GLP1 and restored the ethanol-induced FGF15 reduction. Some studies highlighted the importance of these two hormones (i.e. GLP1, FGF15) in gut liver axis and how bacterial metabolites can modulate their expression promoting or reducing hepatic steatosis in NAFLD.38 According to our observation Xie G et al described that chronic alcohol assumption decreased FGF15 intestinal production in rats39 and recently, the FXR/FGF15 axis was proposed as a potential treatment in ALD.40 It is known that alcohol exposure increases intestinal bile acids absorption and hepatic bile acid production with consequent overload in the gut, liver and serum.39,41 In the intestines, bile acids activate FXR which in turn promotes transcription of FGF15/19, which are secreted in the portal circulation. FGF15/19 bind FGFR4 in the liver, suppressing CYP7A1 and bile acid synthesis.31,42 Here we found that the expression of FXR targets, IBABP, and in more extent Ost-α and Ost-β, decreased in alcohol-exposed mice and was restored in Bt-treated mice, highlighting that Bt can prevent the intestinal FXR inhibition occurring in ethanol fed mice. Moreover, the CYP7A1 downregulation in EtOH-Bt mice confirmed that the modulation of bile acid metabolism might represent one of the protective mechanisms of Bt. GLP-1 is an incretin secreted after a meal ingestion by intestinal L-cells, which are primarily located in the distal ileum and colon. Human and animal studies associated treatments with GLP1 receptor agonists to amelioration of hepatic fat accumulation, insulin resistance and increase of FAs β-oxidation in NAFLD,43–47 while in ALD only an effect to reduce ethanol intake was demonstrated both in rodents and humans.48 GLP-1 colonic expression can be modulated by bile acids (i.e. deoxycholic and lithocholic acid) via TGR5, or by bacterial derived short chain fatty acids (SCFAs) via GPR43.38,49 In our experiments we found that ethanol reduced expression of both TGR5 and GPR43, which were restored by Bt supplementation. Therefore, the GLP-1 production in Bt-treated mice was likely promoted either by SCFAs and bile acids profile change. Interestingly, analyzing the hepatic metabolic profile we showed that Bt administration efficiently prevented the ethanol-induced metabolic abnormality. In particular, Bt recovered the ethanol-induced AMPK inhibition, with the consequent normalization of SREBP-1c levels, a known transcriptional factor of FA synthesis genes such as FASN and SCD-1, which accordingly were downregulated in Bt-treated mice. Moreover, Bt contrasted the ethanol-induced downregulation of mitochondrial biogenesis markers such as PPAR-α, PGC-1α, PGC-1β and TFAM. It is known that the simultaneous activation of these factors stimulates OXPHOS and FAO.28 In accordance, we found that Bt prevented the RC complexes I, II and V depletion and stimulated FAO. Moreover, the higher antioxidant activity in Bt-treated mice might represent an effect of PGC-1 β activation. Interestingly, the entire liver bioenergetic profile could be due to the effect of gut-liver axis perturbations induced by alcohol exposure, and Bt improved hepatic metabolism by rewiring intestinal homeostasis. In fact, it is demonstrated that the FGF15/FGFR4 signaling induces AMPK activity, reducing SREBP1c and FA synthesis gene expression.50 PPARα can also be activated by FGFR4 activity.51 On the other hand, AMPK and PPAR-α can be activated by the GLP1R signaling as well, while FASN and SCD-1 are typically downregulated.43 Notably, PGC1β liver expression seems to be modulated by fasting, nutrients, and other unknown gut-derived molecules,28,52 underling once more the importance of gut-liver axis and the impact of Bt on it. However, it has also been supposed that AMPK activity could enable PGC-1β expression.28
In conclusion, ALD development depends on multiple factors and probably the gut-liver axis dysregulation, in terms of bile acid metabolism, enterokine production, and LPS translocation, alters the hepatic capability to regulate the cellular bioenergetics. This results in lipid metabolism abnormality, mitochondrial dysfunction, and oxidative stress. Therefore, it would be conceivable to hypothesize that, as for NASH, a multiple target therapeutic strategy should be tested in ALD, taking into account the possibility of intervening simultaneously on bile acid system, FGF15 and GLP-1 signaling, intestinal barrier, liver mitochondria, and others. The limit of our study is the lack of “loss of function” studies; hence, further investigations will have to clarify the impact of any mechanism involved in the microbiota-intestine-liver axis. However, we have demonstrated that by modulating ethanol-induced microbiota alterations by restoring the intestinal abundance of Bt, it is possible to obtain pleiotropic effects by simulating a multiple target therapy. This candidates Bt as a potent novel probiotic to treat patients with ALD.
Materials and methods
Mouse experiments
An experimental model of ALD was used to study the impact of ethanol introit on intestinal Bt and whether oral supplementation with Bt could exert beneficial effects. All experiments were performed according to ethical principles and legal laws. 8- to 10-week-old female wild-type (C57BL/6) mice were fed with a Lieber-DeCarli diet containing up to 5% alcohol for 15 days (EtOH-fed) or Lieber-DeCarli diet alone (pair-fed), as shown in Figure 1a and previously described.33,53
In order to study the potential properties of Bt to protect the liver against ethanol introit, the mice were treated every day (from day 1 to day 14) with Bt (3 × 109 bacteria/200 μl phosphate-buffered saline (PBS)) or vehicle (PBS) by oral gavage. Mice were weighed every other day, and drinking amounts were monitored daily. At day 15 mice were sacrificed by being anaesthetized with xylazine (5 mg/kg) and ketamine (100 mg/kg), and blood, liver, and intestinal samples were collected.
Quantification of Bt in mice feces.
DNA extraction from feces was performed by DNeasy PowerSoil Kit (QIAGEN, Hilden, Germany) according to manufacturer’s protocol. Following this, the amount of Bt DNA was quantified by qPCR with SybrGreen (Eurogentec, Köln, Germany) on MXPro3000 Cycler (Agilent Technology, Waldbronn, Germany). The primers for Bt DNA detection were based on 16rDNA gene sequences: forward-GGCAGCATTTCAGTTTGCTTG; reverse-GGTACATACAAAATTCCACACGT. The cycle was performed using 30 ng of fecal DNA, primer concentrations at 250 nM and 60°C as annealing temperature. The standard curve was obtained by serial dilutions of bacterial DNA extracted from Bt colonies, and the cycle threshold of each sample was compared with the standard. The quantity of Bt DNA in the feces was expressed in Log10 ng/g.
In order to further demonstrate the presence of living Bt in the intestine of mice treated with oral gavage, mice feces were cultured on blood agar plate, in anaerobic condition, and a qPCR was performed from the growing colonies, showing the presence of Bt DNA.
Cultivation of Bt
Bt (DSM 2079) was purchased by DSMZ (Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures) and cultured on blood agar (Biomerieux, Marcy l’Etoile, France) at 37°C under anaerobic conditions with GENbox and GENbox anaer systems (Biomeriux, Marcy l’Etoile, France).
Statistical analysis
GraphPad PRISM v8 (La Jolla, California, USA) was used for statistical analysis. Unpaired two-tailed Student’s t-test and one-way analysis of variance followed by post hoc Bonferroni test were used when appropriate. The results are shown as mean ± standard error of mean (SEM). Data were considered statistically significant when p < .05.
Supplementary Material
Funding Statement
This study is supported by the excellence initiative VASCage (Centre for Promoting Vascular Health in the Ageing Community), an R&D K-Centre (COMET program – Competence Centers for Excellent Technologies) funded by the Austrian Ministry for Transport, Innovation and Technology, the Austrian Ministry for Digital and Economic Affairs and the federal states Tyrol, Salzburg and Vienna.
Abbreviations
ALD, alcohol-related liver disease; AFLD, alcoholic fatty liver disease; Bt, Bacteroides thetaiotaomicron; AH, alcoholic hepatitis; ASH, alcoholic steatohepatitis; FAs, fatty acids; FAO, fatty acid oxidation; FASN, fatty acid synthase; SCD-1, stearoyl-CoA desaturase-1; PPAR-α, peroxisome proliferator-activated receptor-α; PAMPs, pathogen-associated molecular patterns; LPS, lipopolysaccharides; EtOH, ethanol-fed; PBS, phosphate-buffered saline; ALT, alanine aminotransferase; FGF15, fibroblast growth factor 15; IBABP, ileal bile acid-binding protein; Ost-α/β, Organic solute transporter α/β; GLP-1, Glucagon-like peptide-1; G protein-coupled bile acid receptor 1; GPR43, G-protein-coupled receptor 43; Peptide YY; AMPK, 5’ AMP-activated protein kinase; SREBP-1c, Sterol-regulatory element-binding protein-1C; PGC-1α/β, peroxisome proliferator-activated receptor-gamma coactivator-1α/β; TFAM, Transcription Factor A, Mitochondrial; CPT-1a, Carnitine Palmitoyltransferase 1a; VDAC1, Voltage-dependent anion-selective channel 1; DGAT1, Diacylglycerol O-Acyltransferase 1; SOD, Superoxide dismutase; PRDX-3/5, Peroxiredoxin 3/5; MDA, Malondialdehyde; HNE, 4Hydroxynonenal; Muc1, mucin1; Muc2, mucin2; Cldn1, claudin1; MMP9, matrix metallopeptidase 9; Hes1, hairy and enhancer of split-1; GLP1R, GLP-1 receptor; FGFR4, FGF receptor 4.
Data availability statement
The data that support the findings of this study are available in repository 4TU.ResearchData at https://doi.org/10.4121/16825366.v1.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2089006
References
- 1.Mathurin P, Bataller R.. Trends in the management and burden of alcoholic liver disease. J Hepatol. 2015;62:S38–14. doi: 10.1016/j.jhep.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol. 2013;59:160–168. doi: 10.1016/j.jhep.2013.03.007 [DOI] [PubMed] [Google Scholar]
- 3.WHO . Global status report on alcohol and health 2018.
- 4.Lefkowitch JH. Morphology of alcoholic liver disease. Clin Liver Dis. 2005;9:37–53. doi: 10.1016/j.cld.2004.11.001 [DOI] [PubMed] [Google Scholar]
- 5.Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet. 1995;346:987–990. doi: 10.1016/S0140-6736(95)91685-7 [DOI] [PubMed] [Google Scholar]
- 6.Abenavoli L, Masarone M, Federico A, Rosato V, Dallio M, Loguercio C, Persico M. Alcoholic hepatitis: pathogenesis, diagnosis and treatment. Rev Recent Clin Trials. 2016;11:159–166. doi: 10.2174/1574887111666160724183409 [DOI] [PubMed] [Google Scholar]
- 7.Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J Gastroenterol. 2014;20:17756–17772. doi: 10.3748/wjg.v20.i47.17756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou SL, Gordon RE, Bradbury M, Stump D, Kiang CL, Berk PD. Ethanol up-regulates fatty acid uptake and plasma membrane expression and export of mitochondrial aspartate aminotransferase in HepG2 cells. Hepatology. 1998;27:1064–1074. doi: 10.1002/hep.510270423 [DOI] [PubMed] [Google Scholar]
- 9.Zhong W, Zhao Y, Tang Y, Wei X, Shi X, Sun W, Sun X, Yin X, Sun X, Kim S. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am J Pathol. 2012;180:998–1007. doi: 10.1016/j.ajpath.2011.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200 [DOI] [PubMed] [Google Scholar]
- 11.Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–28004. doi: 10.1074/jbc.M302140200 [DOI] [PubMed] [Google Scholar]
- 12.Berk PD, Zhou S, Bradbury MW. Increased hepatocellular uptake of long chain fatty acids occurs by different mechanisms in fatty livers due to obesity or excess ethanol use, contributing to development of steatohepatitis in both settings. Trans Am Clin Climatol Assoc. 2005;116:335–344. discussion 345. [PMC free article] [PubMed] [Google Scholar]
- 13.Tilg H, Mathurin P. Altered intestinal microbiota as a major driving force in alcoholic steatohepatitis. Gut. 2016;65:728–729. doi: 10.1136/gutjnl-2015-311014 [DOI] [PubMed] [Google Scholar]
- 14.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94:200–207. doi: 10.1111/j.1572-0241.1999.00797.x [DOI] [PubMed] [Google Scholar]
- 15.Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA, Schnabl B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grander C, Adolph TE, Wieser V, Lowe P, Wrzosek L, Gyongyosi B, Ward DV, Grabherr F, Gerner RR, Pfister A. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut. 2018;67:891–901. doi: 10.1136/gutjnl-2016-313432 [DOI] [PubMed] [Google Scholar]
- 17.Ferrere G, Wrzosek L, Cailleux F, Turpin W, Puchois V, Spatz M, Ciocan D, Rainteau D, Humbert L, Hugot C. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J Hepatol. 2017;66:806–815. doi: 10.1016/j.jhep.2016.11.008 [DOI] [PubMed] [Google Scholar]
- 18.Zocco MA, Ainora ME, Gasbarrini G, Gasbarrini A. Bacteroides thetaiotaomicron in the gut: molecular aspects of their interaction. Dig Liver Dis. 2007;39:707–712. doi: 10.1016/j.dld.2007.04.003 [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. A genomic view of the human-bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029 [DOI] [PubMed] [Google Scholar]
- 20.Resta-Lenert S, Barrett KE. Probiotics and commensals reverse TNF-alpha- and IFN-gamma-induced dysfunction in human intestinal epithelial cells. Gastroenterology. 2006;130:731–746. doi: 10.1053/j.gastro.2005.12.015 [DOI] [PubMed] [Google Scholar]
- 21.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881 [DOI] [PubMed] [Google Scholar]
- 22.Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, Shi J, Zhao S, Liu W, Wang X. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med. 2017;23:859–868. doi: 10.1038/nm.4358 [DOI] [PubMed] [Google Scholar]
- 23.Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol. 1999;276:G965–74. doi: 10.1152/ajpgi.1999.276.4.G965 [DOI] [PubMed] [Google Scholar]
- 24.Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pope JL, Bhat AA, Sharma A, Ahmad R, Krishnan M, Washington MK, Beauchamp RD, Singh AB, Dhawan P. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of notch-signalling. Gut. 2014;63:622–634. doi: 10.1136/gutjnl-2012-304241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greiner TU, Backhed F. Microbial regulation of GLP-1 and L-cell biology. Mol Metab. 2016;5:753–758. doi: 10.1016/j.molmet.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249 [DOI] [PubMed] [Google Scholar]
- 28.Piccinin E, Villani G, Moschetta A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat Rev Gastroenterol Hepatol. 2019;16:160–174. doi: 10.1038/s41575-018-0089-3 [DOI] [PubMed] [Google Scholar]
- 29.Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem. 2011;286:25655–25662. doi: 10.1074/jbc.M111.228692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu M, Yin H, Mitra MS, Liang X, Ajmo JM, Nadra K, Chrast R, Finck BN, You M. Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology. 2013;58:1953–1963. doi: 10.1002/hep.26589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, Puchois V, Martin JC, Lepage P, Le Roy T. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. 2016;65:830–839. doi: 10.1136/gutjnl-2015-310585 [DOI] [PubMed] [Google Scholar]
- 32.Cassard AM, Gerard P, Perlemuter G. Microbiota, liver diseases, and alcohol. Microbiol Spectr. 2017;5. doi: 10.1016/j.chom.2016.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sangineto M, Grabherr F, Adolph TE, Grander C, Reider S, Jaschke N, Mayr L, Schwärzler J, Dallio M, Moschen AR. Dimethyl fumarate ameliorates hepatic inflammation in alcohol related liver disease. Liver Int. 2020;40:1610–1619. doi: 10.1111/liv.14483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li F, Duan K, Wang C, McClain C, Feng W. Probiotics and alcoholic liver disease: treatment and potential mechanisms. Gastroenterol Res Pract. 2016;2016:5491465. doi: 10.1155/2016/5491465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Fouts DE, Starkel P, Hartmann P, Chen P, Llorente C, DePew J, Moncera K, Ho SB, Brenner DA. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe. 2016;19:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartmann P, Chen P, Wang HJ, Wang L, McCole DF, Brandl K, Stärkel P, Belzer C, Hellerbrand C, Tsukamoto H. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology. 2013;58:108–119. doi: 10.1002/hep.26321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wrzosek L, Miquel S, Noordine ML, Bouet S, Joncquel Chevalier-Curt M, Robert V, Philippe C, Bridonneau C, Cherbuy C, Robbe-Masselot C. Bacteroides thetaiotaomicron and faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013;11:61. doi: 10.1186/1741-7007-11-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou D, Fan JG. Microbial metabolites in non-alcoholic fatty liver disease. World J Gastroenterol. 2019;25:2019–2028. doi: 10.3748/wjg.v25.i17.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie G, Zhong W, Li H, Li Q, Qiu Y, Zheng X, Chen H, Zhao X, Zhang S, Zhou Z. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J. 2013;27:3583–3593. doi: 10.1096/fj.13-231860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, Wang L, Alnouti Y, Fouts DE, Stärkel P. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology. 2018;67:2150–2166. doi: 10.1002/hep.29676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manley S, Ding W. Role of farnesoid X receptor and bile acids in alcoholic liver disease. Acta Pharm Sin B. 2015;5:158–167. doi: 10.1016/j.apsb.2014.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001 [DOI] [PubMed] [Google Scholar]
- 43.Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43:173–181. doi: 10.1002/hep.21006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, Anania FA. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51:1584–1592. doi: 10.1002/hep.23569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma S, Mells JE, Fu PP, Saxena NK, Anania FA. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One. 2011;6:e25269. doi: 10.1371/journal.pone.0025269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svegliati-Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, Faraci G, Pacetti D, Vivarelli M, Nicolini D. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31:1285–1297. doi: 10.1111/j.1478-3231.2011.02462.x [DOI] [PubMed] [Google Scholar]
- 47.Tushuizen ME, Bunck MC, Pouwels PJ, van Waesberghe JH, Diamant M, Heine RJ. Incretin mimetics as a novel therapeutic option for hepatic steatosis. Liver Int. 2006;26:1015–1017. doi: 10.1111/j.1478-3231.2006.01315.x [DOI] [PubMed] [Google Scholar]
- 48.Jerlhag E. Alcohol-mediated behaviours and the gut-brain axis; with focus on glucagon-like peptide-1. Brain Res. 2020;1727:146562. doi: 10.1016/j.brainres.2019.146562 [DOI] [PubMed] [Google Scholar]
- 49.Federico A, Dallio M, R DIS, Giorgio V, Miele L. Gut microbiota, obesity and metabolic disorders. Minerva Gastroenterol Dietol. 2017;63:337–344. doi: 10.23736/S1121-421X.17.02376-5 [DOI] [PubMed] [Google Scholar]
- 50.Markan KR, Potthoff MJ. Metabolic fibroblast growth factors (FGFs): mediators of energy homeostasis. Semin Cell Dev Biol. 2016;53:85–93. doi: 10.1016/j.semcdb.2015.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cui G, Martin RC, Jin H, Liu X, Pandit H, Zhao H, Cai L, Zhang P, Li W, Li Y. Up-regulation of FGF15/19 signaling promotes hepatocellular carcinoma in the background of fatty liver. J Exp Clin Cancer Res. 2018;37:136. doi: 10.1186/s13046-018-0781-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin J, Yang R, Tarr PT, Wu PH, Handschin CS, Li S, Yang S, Pei L, Uldry M, Tontonoz P. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120:261–273. doi: 10.1016/j.cell.2004.11.043 [DOI] [PubMed] [Google Scholar]
- 53.Grabherr F, Grander C, Adolph TE, Wieser, V, Mayr, L, Enrich, B, Macheiner, S, Sangineto, M, Reiter, A, Viveiros, A. Ethanol-mediated suppression of IL-37 licenses alcoholic liver disease. Liver Int. 2018;38:1095–1101. doi: 10.1111/liv.13642 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in repository 4TU.ResearchData at https://doi.org/10.4121/16825366.v1.