

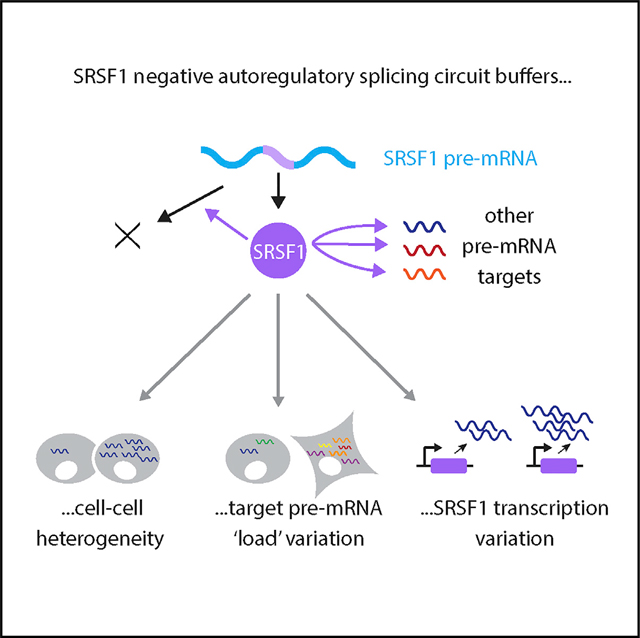
SUMMARY
Non-core spliceosome components are essential, conserved regulators of alternative splicing. They provide concentration-dependent control of diverse pre-mRNAs. Many splicing factors direct unproductive splicing of their own pre-mRNAs through negative autoregulation. However, the impact of such feedback loops on splicing dynamics at the single-cell level remains unclear. Here, we developed a system to quantitatively analyze negative autoregulatory splicing dynamics by splicing factor SRSF1 in response to perturbations in single HEK293 cells. We show that negative autoregulatory splicing provides critical functions for gene regulation, establishing a ceiling of SRSF1 protein concentration, reducing cell-cell heterogeneity in SRSF1 levels, and buffering variation in transcription. Most important, it adapts SRSF1 splicing activity to variations in demand from other pre-mRNA substrates. A minimal mathematical model of autoregulatory splicing explains these experimentally observed features and provides values for effective biochemical parameters. These results reveal the unique functional roles that splicing negative autoregulation plays in homeostatically regulating transcriptional programs.
In brief
Negative autoregulatory feedback is a prevalent feature of splice regulation. Ding et al. show that the splice regulator SRSF1 uses negative autoregulatory splicing to maintain constant levels independent of variations in its own transcription and the “load” of pre-mRNA transcripts. A minimal mathematical model can explain these capabilities.
Graphical Abstract
INTRODUCTION
More than 90% of a typical eukaryotic genome undergoes alternative splicing, producing multiple mRNA isoforms and expanding proteome diversity (Merkin et al., 2012; Barbosa-Morais et al., 2012; Pan et al., 2008; Wang et al., 2008; Nilsen and Graveley, 2010; Ule and Blencowe, 2019; Chaudhary et al., 2019; Liu et al., 2017). Missplicing can cause diverse physiological effects and lead to disease (Scotti and Swanson, 2016; Faustino and Cooper, 2003; Kalsotra and Cooper, 2011; Urbanski et al., 2018; Bonnal et al., 2020; Thomas et al., 2020). Alternative splicing is controlled by many distinct components (Black, 2003; Lee and Rio, 2015; Wilkinson et al., 2020), including non-core spliceosome components (Jangi and Sharp, 2014), the splicing code (Barash et al., 2010; Culler et al., 2010), RNA secondary structures (McManus and Graveley, 2011), RNA polymerase speed (Fong et al., 2014), and epigenetic regulation (Luco et al., 2011).
Of these regulators, non-core spliceosome components play a unique role by modulating splicing activity in a concentration-dependent manner, and maintaining a stable milieu of their concentration (i.e., splicing activity) is critical (Black, 2003; Wang and Burge, 2008; Shin and Manley, 2004; Gehring and Roignant, 2021). Most splicing regulators fall into two main, conserved families, serine-arginine rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs), and are found across diverse tissue types and species (Manley et al., 1996; Dreyfuss et al., 1993; Zahler et al., 1992; Baralle and Giudice, 2017), ranging from Schizosaccharomyces pombe (Shepard and Hertel, 2009) to Arabidopsis (Kalyna et al., 2006). SR or hnRNP proteins modulate alternative splicing of large and diverse sets of target genes (Long and Caceres, 2009; Zhou and Fu, 2013; Wang and Manley, 1995; Leclair et al., 2020), and are implicated in diverse disease processes (Geuens et al., 2016; Anczuków and Krainer, 2016; Bonnal et al., 2020). Maintaining splicing factor homeostasis is thus critical for cellular function.
The control of splicing factor level is commonly achieved through negative autoregulatory splicing (Kalsotra and Cooper, 2011; Lareau et al., 2007; Ni et al., 2007). Specifically, splicing factors alternatively splice their own pre-mRNA to unproductive isoforms, either containing premature termination codons (Wollerton et al., 2004) or introducing new junctions in the 3′ untranslated region (UTR) (Sureau et al., 2001), to trigger degradation by RNA surveillance pathways (Maquat, 2004). The overexpression of splicing factors promotes unproductive splicing (Sureau et al., 2001), leading to negative autoregulation. Previous work investigated many aspects of negative autoregulation, including associated highly or ultra-conserved sequence motifs (Lareau et al., 2007; Ni et al., 2007) and related nonsense-mediated mRNA decay (NMD) triggered by this regulatory mode (Hug et al., 2016; Ni et al., 2007). Nevertheless, the basic question of how autoregulation plays out dynamically at the single-cell level remains unclear.
In other contexts, negative autoregulatory transcriptional feedback is known to speed response times and promote robustness to perturbation (Becskei and Serrano, 2000; Elowitz et al., 2002; Rosenfeld et al., 2002; El-Samad, 2021). However, negative splicing regulation potentially has unique features compared to transcriptional feedback. For instance, rather than operating at a fixed number of DNA binding sites, splicing factors can operate at diverse “loads” of pre-mRNA substrates from their own and other target genes in the cell (Figure 1B). Due to the effects of stochastic gene expression (Raj and van Oudenaarden, 2008; Bohrer and Larson, 2021), total substrate amounts can vary between cell states or over time. This provokes the question of what role splicing autoregulation may play in enabling homeostatic control of splicing factor levels and accelerating responsiveness to changes in substrate.
Figure 1. Splicing factors negatively autoregulate their own synthesis by promoting unproductive splicing of their own transcripts and also operate under variable loads of substrate pre-mRNA produced by other genes.
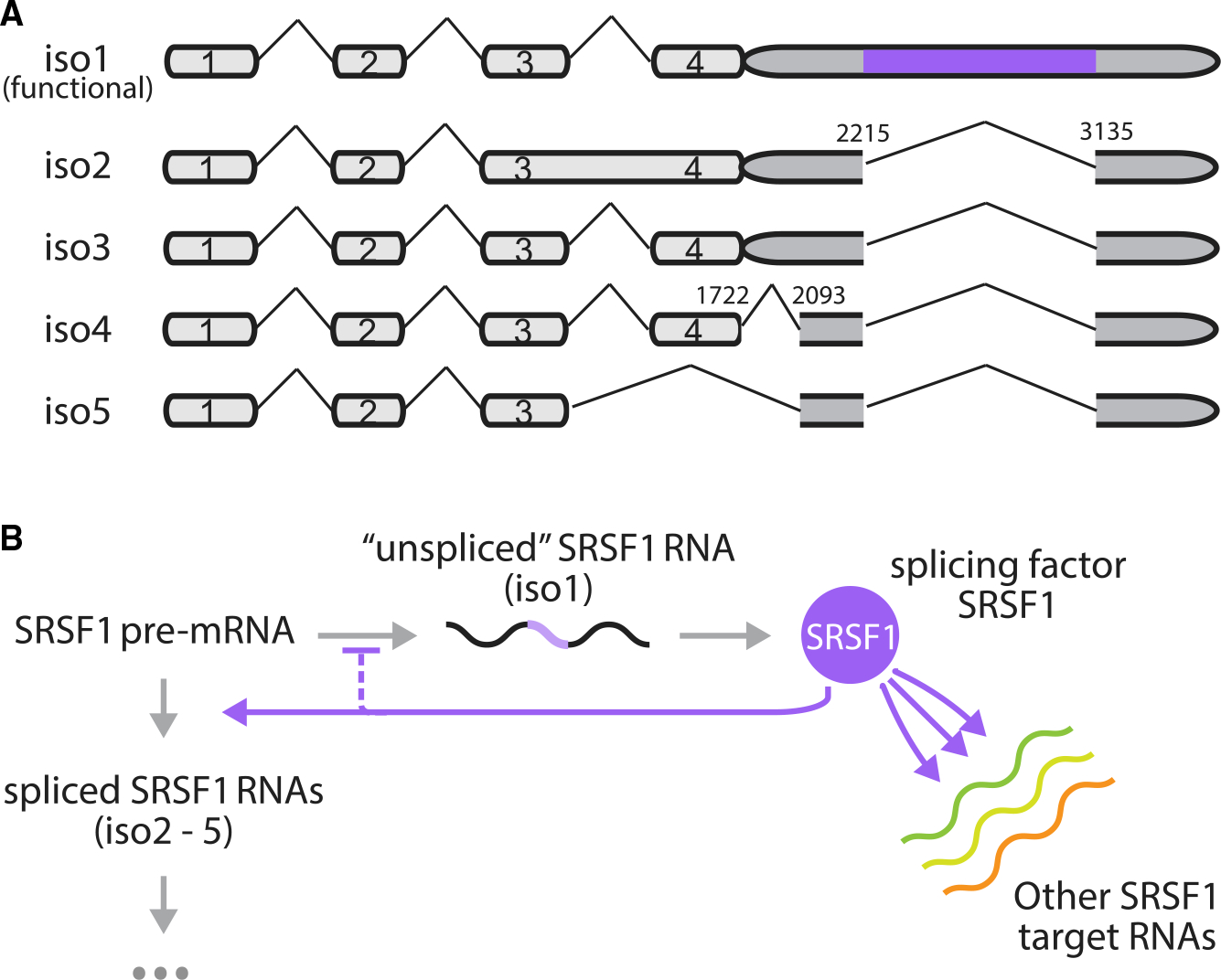
(A) Five isoforms of SRSF1 were observed in HEK293 cells. Each pre-mRNA molecule can be spliced to remove introns in the 3′ UTR (without light purple isoforms, isoforms 2–−5) or left unspliced (with light purple isoforms, isoform 1). Intron removal can lead to degradation through RNA surveillance pathways, while transcripts with retained introns in the 3′ UTR (isoform 1) are translated to produce more splicing factor SRSF1.
(B) Feedback occurs when splicing factors enhance intron removal from their own pre-mRNA, thus negatively regulating their own expression. Apart from their own transcripts, splicing factors additionally act on transcripts produced by other genes. The relative abundance of substrate pre-mRNAs can affect the allocation of splicing factors among transcripts, thereby influencing the dynamics of splicing negative autoregulation.
To address these questions, we developed a system to dynamically and quantitatively investigate negative autoregulatory splicing at the single-cell level. We focused specifically on SRSF1, the protein product of the serine/arginine-rich splicing factor 1 (SRSF1) gene, which is widely expressed in distinct cell types and regulates the splicing pattern of many important genes (Karni et al., 2007; Li and Manley, 2005; Anczuków et al., 2012; Sanford et al., 2009; Paz et al., 2021). Isoform 1 of the SRSF1 transcript, retaining an intron in its 3′ UTR (Figure 1A), is functional (produces SRSF1 protein). We call this the “unspliced” isoform in Figure 1B because it retains a specific intron in its 3′ UTR region, although other introns between exons 1 and 4 of isoform 1 remain spliceable (Figure 1A). To differentiate them from isoform 1, we collectively refer to the other isoforms (Figure 1A iso 2–5) as “spliced” (Figure 1B). These other isoforms are unproductive (do not produce SRSF1 protein).
By tracking SRSF1 accumulation using time-lapse videos with a machine learning-based image analysis system and analyzing SRSF1 levels together with flow cytometry and qRT-PCR/PCR, we found that negative autoregulatory splicing can buffer SRSF1 concentration, achieve ~50% less cell-cell heterogeneity in both expression and response rate upon perturbation of its own pre-mRNA levels, and enable adaptation to total substrate load at both a single-cell level and across 53 human tissue types. We then demonstrated how negative splicing autoregulation can maintain its own dynamics and how it adapts to perturbation in a minimal model. Together, these results quantitatively explain the single-cell dynamics of negative splicing autoregulation and reveal its functional role and impact as a regulatory circuit.
RESULTS
SRSF1 maintains homeostasis through negative splicing feedback
We set out to investigate negative autoregulatory splicing by engineering two HEK293 cell lines expressing the SRSF1 gene with or without autoregulation. We site specifically and stably integrated a single ectopic copy of SRSF1, using either genomic sequence (guide DNA [gDNA], autoregulated) or cDNA sequence (unregulated), under the control of a doxycycline (dox)-inducible cytomegalovirus (CMV) promoter (Figure 2A, top). Because SRSF1 protein is essential for cellular physiology (Li and Manley, 2005; Wang and Manley, 1995), the endogenous genomic copy of SRSF1 was left intact in both cell lines. To distinguish ectopic and endogenous SRSF1, we fused a fluorescent protein, Citrine, at the 5′ end of the ectopic copies (Figure 2A, top). The addition of Citrine did not affect SRSF1 RNA or protein functions (Figures S1A and S1B). Importantly, the splicing pattern of Citrine-fused SRSF1 remained similar to that of the endogenous copy, with 5 isoforms (Figure S1A), including the productively translated functional isoform 1 as well as the unproductive isoforms 2–4 (Sun et al., 2010). Like endogenous SRSF1 protein, ectopic Citrine-fused SRSF1 can downregulate total SRSF1 RNA expression by promoting unproductive splicing to isoforms 2–4 (Figure S1B).
Figure 2. Negative splicing autoregulation establishes a ceiling for SRSF1 protein concentration in response to its own pre-mRNA substrate perturbations.
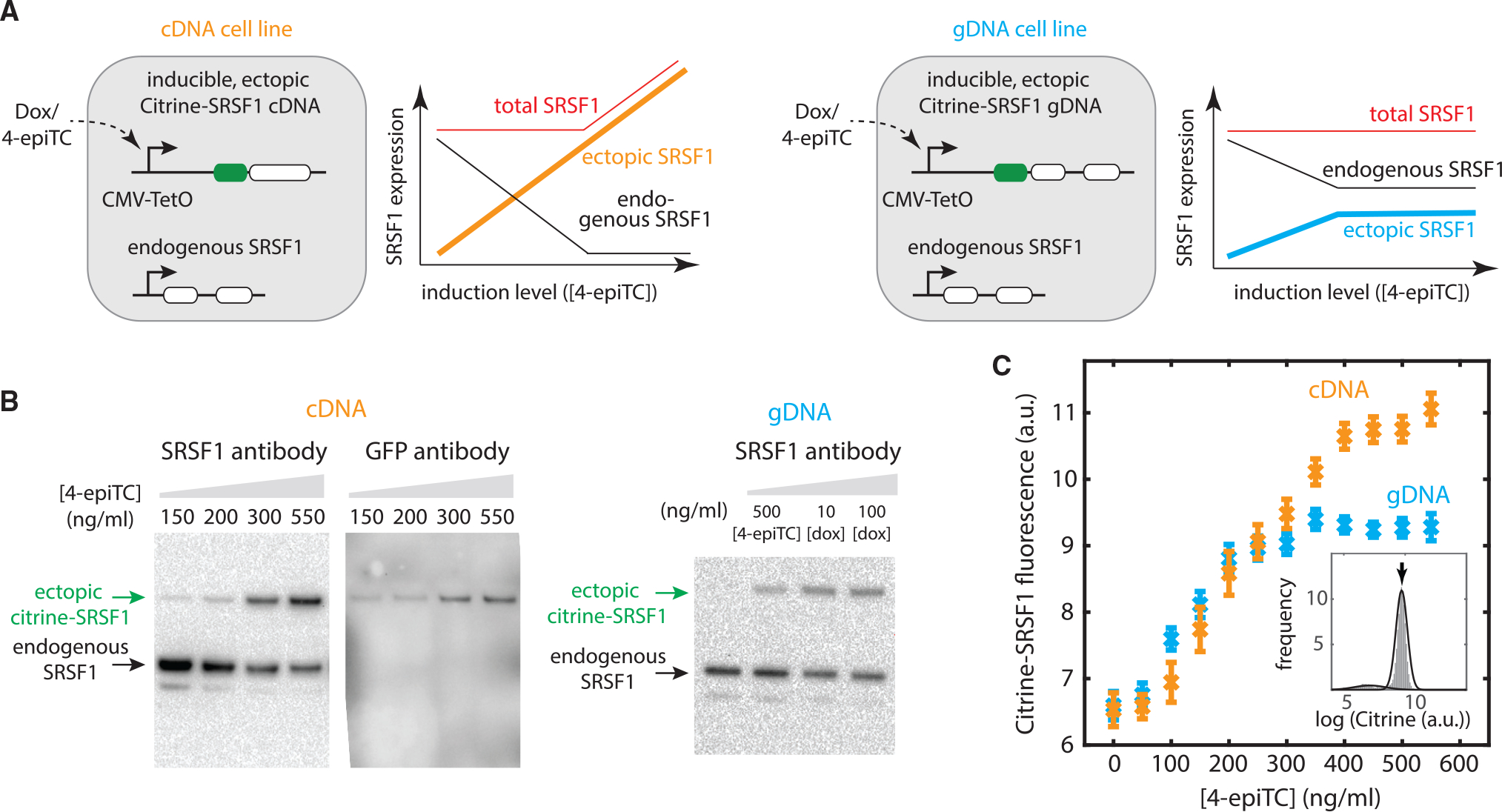
(A) We designed two cell lines: one transfected with a Citrine fused SRSF1 cDNA (i.e., unregulated, with no intron, shown in orange), the other transfected with a Citrine-fused genomic SRSF1 DNA (i.e., autoregulated, shown in blue), both under an inducible Tet-On CMV promoter and stably integrated into the fixed locus of Flp-In T-REx HEK293 cell lines (top). (Bottom) Expected outcomes (schematic): For the cDNA version, increasing ectopic SRSF1 protein level should downregulate endogenous SRSF1 production via splicing feedback (black curve). When the endogenous copy saturates its ability to buffer SRSF1 overexpression, the total SRSF1 level overshoots (red curve). By contrast, for the gDNA cells, due to the negative splicing autoregulation of both the ectopic copy (blue curve) and endogenous copy (black curve), the total SRSF1 level should remain constant (red curve), across a broader range of induction levels.
(B) Western blot shows that the endogenous SRSF1 level decreases with the increasing expression of the ectopic copy. We induced cDNA cells at different 4-epiTC (an analog of doxycycline [dox], with weaker affinity) concentration for 24 h. Anti-SRSF1 antibody (ab133689) staining shows 2 bands: the top band indicates the ectopic copy with fused Citrine (verified by staining Citrine using anti-GFP monoclonal antibody [right]), the bottom band indicates the endogenous copy. Western blot of gDNA cells (induced at different 4-epiTC/dox concentration for 24 h) is also shown as a comparison.
(C) Flow cytometry data shows that ectopic SRSF1 reaches a ceiling (blue curve) with negative splicing autoregulation (i.e., gDNA version), but not with the cDNA version. The 2 cell lines (A) were induced at different 4-epiTC concentration for >24 h and analyzed by flow cytometry. Mean expression levels were extracted from Gaussian fits (Figure S3) to represent the ectopic SRSF1 level. Error bars represent the standard error of the mean from 9 experimental replicates.
Having established the SRSF1(gDNA) and SRSF1(cDNA) cell lines, we next investigated quantitatively how SRSF1 modulates its own expression. In both cell lines, autoregulatory feedback on the endogenous SRSF1 gene is expected to maintain constant SRSF1 protein levels across a modest range of ectopic expression (Figure 2A, lower panels). However, this buffering effect should saturate in the cDNA cell line once endogenous SRSF1 is fully depleted (black curve), leading to increased total SRSF1 levels (orange curve). By contrast, for SRSF1(gDNA) cells, both SRSF1 copies (ectopic and endogenous) are autoregulatory. Therefore, the total SRSF1 expression should remain the same (red curve), with a stable “ceiling” of total ectopic SRSF1 (blue curve).
Consistent with these expectations, the buffering effect from negative autoregulatory splicing could be observed in both cell lines. Inducing ectopic SRSF1 with different concentrations of dox or the weaker affinity inducer 4-epiTC (4 epimers tetracycline, an analog of dox) produced a broad range of ectopic SRSF1 protein expression (Figures 2B and 2C). Concomitant with the increase in ectopic SRSF1 levels, we observed a decrease in endogenous SRSF1 levels in both SRSF1(cDNA) and SRSF1(gDNA) cell lines (western blot in Figures 2B and S2). We also observed a saturation of the buffering effect of endogenous copy in SRSF1(cDNA) cells: At ~100 ng/mL dox induction, all of the endogenous pre-mRNAs were spliced to unproductive isoforms 2–4 by 50 h (Figure S1B). These results, which are consistent with previous work done in HeLa cells (Sun et al., 2010), show that the autoregulatory feedback loop is active and can buffer variations in SRSF1 expression levels.
To obtain a more quantitative view of this behavior, we used single-cell flow cytometry to analyze SRSF1 protein levels in individual cells (Figure 2C). We induced ectopic SRSF1 across a range of levels, fit the resulting data to a simple log-normal distribution with background (Figure S3), and extracted the mean log expression level for each condition. As expected, the Citrine-SRSF1 protein level in SRSF1(cDNA) cells increased monotonically with induction level (orange curve). By contrast, SRSF1(gDNA) cells reached a ceiling at an induction level of 300 ng/mL 4-epiTC (blue curve). The levels of ectopic and endogenous SRSF1 were quantified via gel band intensity in western blot (Figure S2). By correcting for relative protein size (487 amino acids [aa] versus 248 aa), we found the level of endogenous SRSF1 protein is ~3 times that of fully induced ectopic SRSF1 expression.
Together, these results provide a system in which autoregulated and unregulated SRSF1 can be compared quantitatively, showing that negative autoregulatory splicing buffers SRSF1 concentrations at the steady state.
A deep learning system allows label-free single-cell tracking in time-lapse videos
We next sought to use this system to study the dynamics of negative autoregulatory splicing in individual cells in response to the induction of the ectopic constructs, in time-lapse videos. To achieve this requires identifying (segmenting) nuclei in each video frame, tracking their movements over time, and quantifying the changes in total abundance of Citrine-SRSF1 in each individual cell. Segmentation can be challenging to achieve using the Citrine fluorescence signal alone because fluorescence levels are initially too low to reliably label nuclei. To circumvent this issue, we adapted and trained the GoogLeNet deep learning system (Christiansen et al., 2018; Szegedy et al., 2015) to segment nuclei from differential interference contrast (DIC) images (Figure 3A). To train the network, we acquired ~150 paired DIC (Figure 3A, left) and fluorescence (Figure 3A, right) images of the same cells (STAR Methods), under conditions of strong nuclear Citrine expression, in which Citrine clearly labeled nuclei. After training, the network was able to segment cell nuclei from DIC images regardless of Citrine fluorescence level (Figure 3B). It also functioned across DIC images varying in brightness and contrast (Figure 3B). After segmentation, we applied a previously described cell tracking algorithm to follow individual cells over the ~40-h duration of each time-lapse video (Bintu et al., 2016; Singer et al., 2014). Finally, we extracted dynamic traces of Citrine fluorescence from each cell. Together, this deep learning-enabled protocol provides a simple and general method for label-free single-cell fluorescence tracking, avoiding the need to integrate an additional constitutively expressed fluorescent protein into the cell, minimizing cell engineering, and reducing phototoxicity during imaging.
Figure 3. Deep-learning network enables tracking of SRSF1 accumulation in individual cells over time.
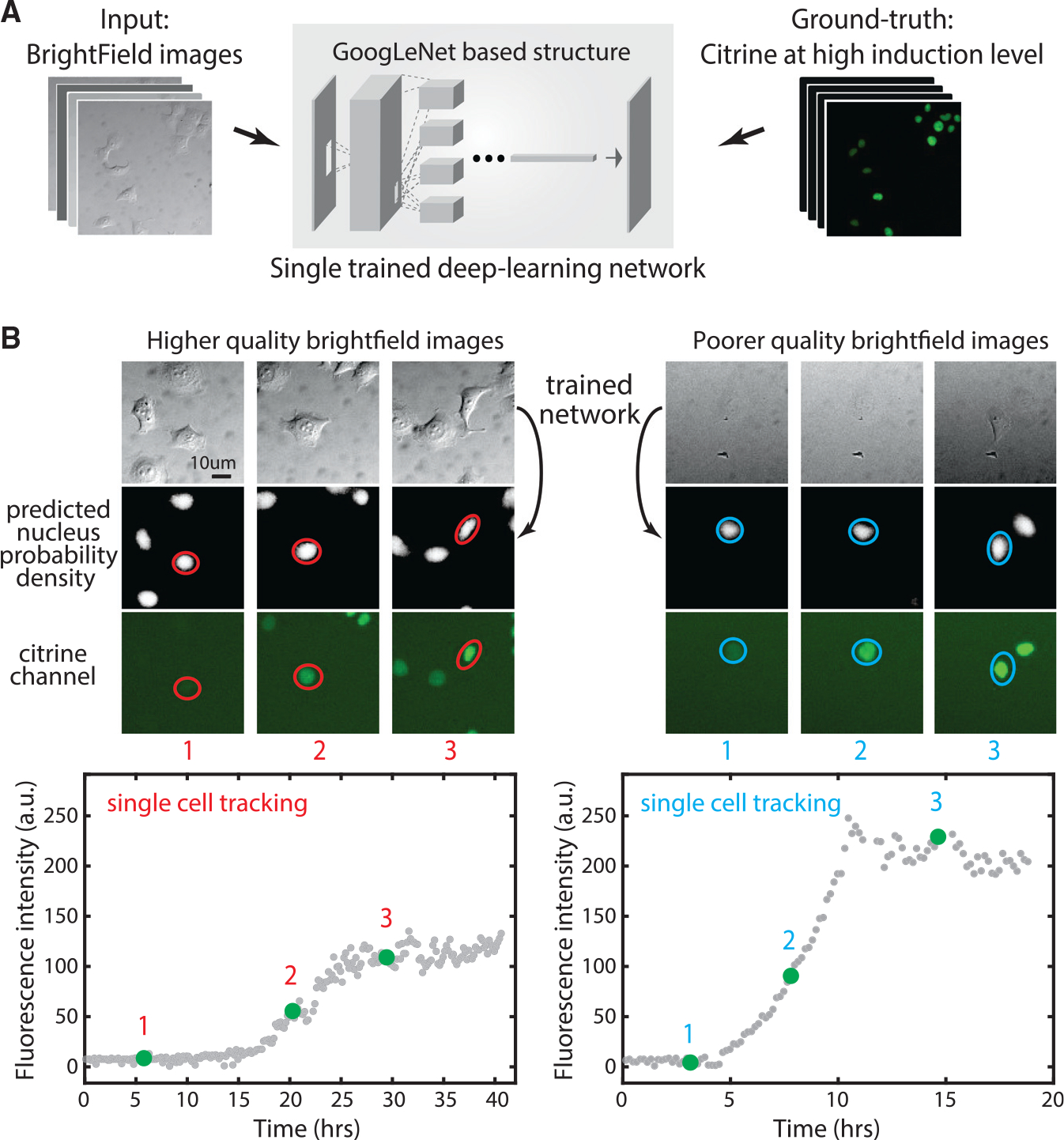
(A) We trained a single deep learning network using ~150 bright-field (DIC) images with their correspondent Citrine signal as the ground truth for learning nuclear location.
(B) The trained network predicts nucleus images (center row) from bright-field images (top row) for 2 different example time-lapse single-cell traces (left and right panels). Note the similarity of predicted fluorescence and actual fluorescence on later images, where visible. Time points are indicated by red/blue numbers (compare with real-time tracking video curves at the bottom). Two videos show diverse bright-field background and contrast, but our trained network works on both. Red circles represent cell segmentation based on deep learning predicted nuclear probability. Left and right traces are the SRSF1(cDNA) cell line induced at t = 0 with 200 ng/mL 4-epiTC or 100 ng/mL dox, respectively.
Negative autoregulatory splicing reduces cell-cell heterogeneity in SRSF1 levels and responsiveness
Equipped with the label-free segmentation and tracking system, we investigated the dynamics of negative autoregulatory splicing by comparing Citrine-SRSF1 dynamics in SRSF1(gDNA) cells to those of unregulated SRSF1(cDNA) cells. Specifically, we induced both cell lines at time 0 under low (200 ng/mL 4-epiTC) and high (100 ng/mL dox) induction levels, recorded DIC and fluorescence images over time, and reconstructed ~200 traces of single-cell Citrine signals for each cell line at each induction level.
Negative autoregulation at the transcriptional level was previously shown to accelerate response times and to reduce cell-cell heterogeneity (illustrated in Figure 4A) (Alon, 2006; Rosenfeld et al., 2002). Time-lapse videos revealed that negative autoregulatory splicing has a similar impact: SRSF1(gDNA) cells reached steady-state levels faster than SRSF1(cDNA) cells (Figure 4B).
Figure 4. Splicing negative autoregulation reduces cell-cell heterogeneity in both the level and response rate of SRSF1 protein.
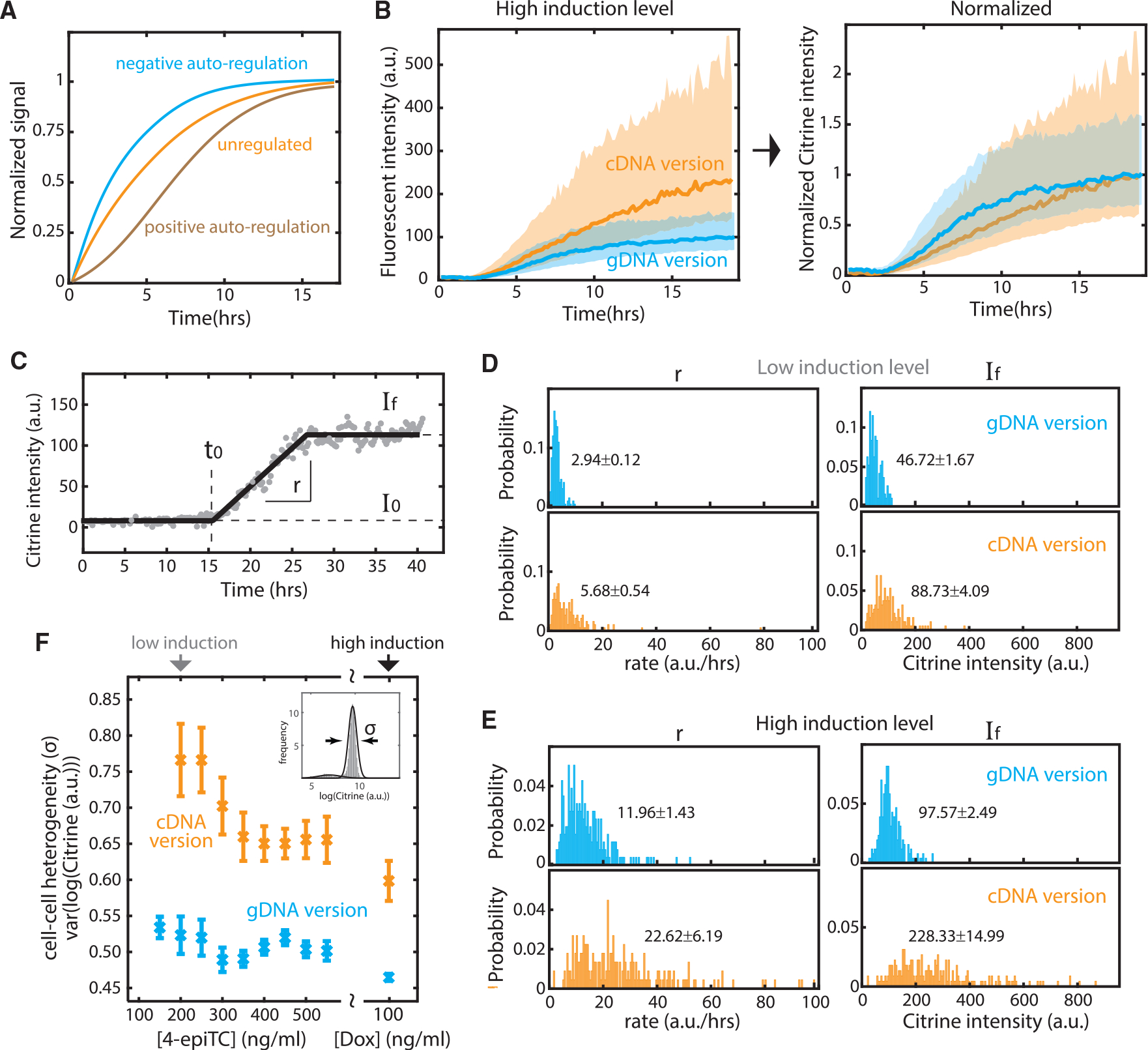
(A) Negative feedback accelerates response time, in comparison to unregulated or positive feedback (schematic).
(B) Negative autoregulatory splicing speeds response rate and reduces cell-cell heterogeneity at high induction level (100 ng/mL dox). (Left) Solid curves are the median of 257 SRSF1(cDNA) and 223 SRSF1(gDNA) single-cell traces. The shade represents the standard deviation of the mean. (Right) The curves are normalized to final expression.
(C) We fit 4 parameters to characterize time-lapse video traces. I0 represents background Citrine intensity and auto-fluorescence. If represents the final (steady-state) Citrine level. t0 denotes the time point when Citrine signal (i.e., ectopic SRSF1 level) surpasses background. r represents the response speed (slope) from I0 to If. The example video trace is the same as in Figure 3 (left).
(D) Distributions of rate and final intensity for 191 gDNA and 188 cDNA traces with 200 ng/mL 4-epiTC added at t0 (see I0 and t0 distribution in Figure S4).
(E) Similar distributions for gDNA and cDNA traces with 100 ng/mL dox added at t0 (see I0 and t0 distribution in Figure S4). The labeled text denotes the median and the standard error of the mean. At both induction levels, the autoregulatory (gDNA) system exhibits a tighter distribution of final equilibrium SRSF1 levels and response rates. The negative splicing feedback loop thus reduces cell-cell heterogeneity both in final level and dynamics.
(F) Flow cytometry data confirms that the autoregulatory (gDNA) system exhibits lower cell-cell heterogeneity across a wide range of induction levels. As in Figure 2C, we induced gDNA and cDNA cells for >24 h, fit the Citrine intensity with a Gaussian curve (Figure S3), and used the standard deviation parameter from the Gaussian fit to represent the variance of ectopic SRSF1 level between cells. Error bars represent the standard error of the mean from 9 experimental replicates.
Furthermore, these traces (Figures 4B and S4A) also showed that negative autoregulatory splicing reduces cell-cell heterogeneity. SRSF1(gDNA) cells exhibited a tighter distribution of temporal traces compared to SRSF1(cDNA) cells. To quantitatively examine cell-cell heterogeneity at different induction levels, we compared SRSF1(gDNA) and SRSF1(cDNA) curves with the distribution of four parameters extracted from each video trace (Figure 4C): I0 and If, characterizing the initial and final fluorescence levels respectively; t0, the time at which the rising signal occurred; and r, the slope of the rise. At low induction levels (Figure 4D), the standard deviation of r and If relative to their median values increased from 0.04 to 0.036 in SRSF1(gDNA) cells to 0.10 and 0.046 in SRSF1(cDNA) cells, respectively. Similarly, at high induction levels (Figure 4E), variability in r and If increased from 0.12 to 0.026 in SRSF1(gDNA) cells to 0.27 and 0.066 in SRSF1(cDNA) cells. We also analyzed a broader range of 4-epiTC concentrations by flow cytometry (Figure S3), and confirmed that negative autoregulatory splicing reduces cell-cell heterogeneity of If across a wide range of induction levels (Figure 4F).
The delay before activation, t0, varied systematically with the induction level, decreasing from ~13 h at low induction levels to ~3 h at the highest induction levels (Figures S4B and S4C). Three hours is comparable to the total expected time required to synthesize mature SRSF1 protein (Milo et al., 2010). The longer 13-h delay may reflect the bursty nature of transcription, which can produce extended intervals between transcriptional bursts (Larsson et al., 2019; Raj et al., 2006), and variable 4-epiTC/dox induction strength and absorption efficiency between cells. This could also explain why we did not observe an accelerated response time for the SRSF1(gDNA) circuit at low induction levels (compare Figures S4A and 4B). Notably, t0 did not differ between the SRSF1(gDNA) and SRSF1(cDNA) cell lines (Figures S4B and S4C), suggesting that it is controlled by factors independent of the splicing regulatory circuit. Similarly, the heterogeneity and level of background signal I0 remained the same for SRSF1(gDNA) and SRSF1(cDNA) cells regardless of induction level (Figures S4B and S4C), suggesting that the sum of autofluorescence and promoter leakage was similar between the two circuits.
Taken together, these results demonstrate that negative autoregulatory splicing can speed response rates and reduce cell-cell heterogeneity in response rate and expression level. These effects are similar to those of other well-known negative autoregulatory feedback loops (Becskei and Serrano, 2000; Rosenfeld et al., 2002, 2007). Despite the similarity, negative autoregulatory splicing has a feature potentially distinct from other types of negative feedback—its ability to simultaneously affect a large and variable load of target pre-mRNAs (Figure 1B).
Negative autoregulatory splicing modulates response of SRSF1 level to splicing load
In principle, SRSF1 negative autoregulatory splicing could operate in two opposite regimes that differ in their response to increased substrate (pre-mRNA) load (Figure 5A). In “robust” mode, the feedback strength would be independent of the load level. Despite the competition between distinct pre-mRNA substrates, SRSF1 protein is abundant in the cell, and SRSF1 production maintains a constant concentration in the cell. Alternatively, in an “adaptive” mode, the feedback loop would modulate its negative autoregulatory strength, tuning the SRSF1 level in response to total substrate load. In this case, increased substrate load, like a sponge, would dilute the available SRSF1 protein per substrate molecule, reducing the feedback strength, generating more functional SRSF1 isoform 1, and thereby producing more SRSF1 protein.
Figure 5. Splicing negative autoregulation modulates its feedback strength in response to variable total substrate load.
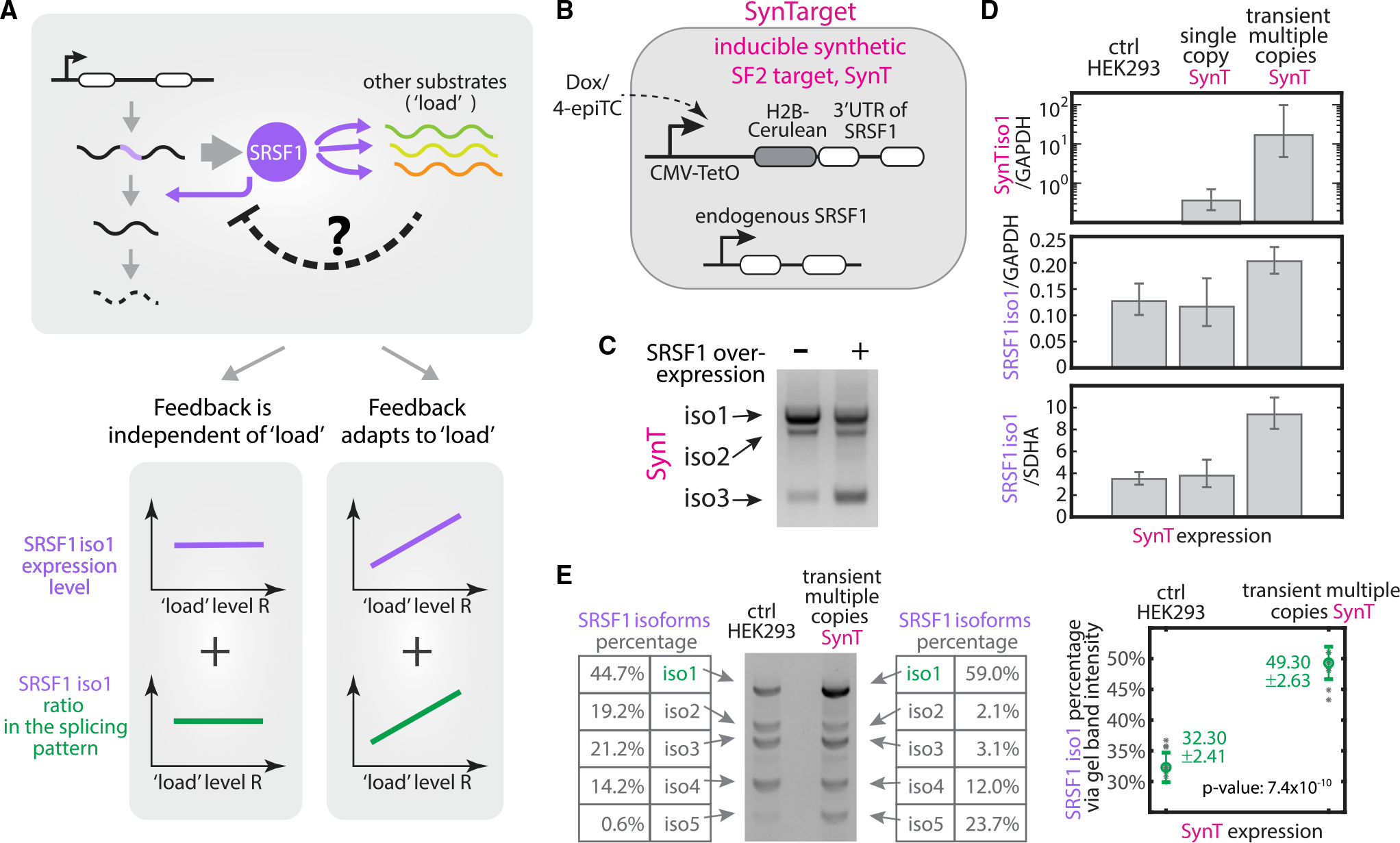
(A) Two possible outcomes in response to total substrate load. (Left) Robust feedback scheme: The total SRSF1 level (purple line) and its splicing pattern (green line) remain constant across a broad range of substrate levels. The amount of SRSF1 involved in negative feedback is independent of load level. (Right) Adaptive feedback scheme: more SRSF1 is produced (purple and green curves) via weakening negative autoregulatory splicing (i.e., dashed negative arrow in the top gray box), as increased substrate load titrates away available SRSF1 in the cell.
(B) The inducible synthetic SRSF1 target (SynTarget [SynT]) cell line contains H2B-Cerulean fused with the spliceable 3′ UTR of SRSF1. This synthetic gene is expressed under a Tet-On CMV promoter and stably integrated at the Flp-In locus in a T-REx HEK293 cell line.
(C) SynT is a splicing target of SRSF1. We used RT-PCR and gel-imaged 3 isoforms of SynT cells with 100 ng/mL dox (left lane) and of SynT cells with transiently transfected SRSF1(cDNA) plasmid in 100 ng/mL dox (right lane). We found that SRSF1 overexpression promotes the splicing of SynT, increasing the expression of short isoforms.
(D) We induced SynT at different levels and quantified the concentration of SynT isoform 1 (top row) and the functional SRSF1 isoform 1 (bottom 2 rows) by qRT-PCR (see qPCR qualification in Figure S6). We found that SRSF1 levels remained unchanged by expression from a single copy of SynT (center column, by inducing the stably integrated SynT with 100 ng/mL dox), but increased ~50% when multiple SynT copies were induced in the same cell (right column, by transiently transfecting SynT plasmid with 100 ng/mL dox). qPCR results were verified by normalizing to 2 housekeeping genes, GAPDH and SDHA, respectively. The data represent the exponential logarithmic mean of normalized qPCR reads. Error bars represent the minimum and maximum values over 3–10 experimental replicates.
(E) SRSF1 splice isoform pattern changes in response to increased total substrates. We quantified SRSF1 isoforms using RT-PCR and analyzed the gel band intensity by Bio-Rad ChemiDoc Image Lab 6.0 band analyzer. Two gel band examples are presented—one from HEK293 control (left), the other from transient multiple copies of SynT (right). Multiple copies of SynT trigger ~50% more SRSF1 isoform 1 (i.e., functional unspliced isoform) through splicing. The data represent the median of gel band intensity percentage reads and error bars represent the standard deviation over 6–7 experimental replicates.
To discriminate between these two regimes and investigate whether and how negative autoregulatory splicing responds to perturbations of load, we introduced a synthetic target (SynT) of SRSF1 in HEK293 cells (Figure 5B) under a dox-inducible CMV promoter. SynT contains the spliceable 3′ UTR of SRSF1, fused with a fluorescent protein, H2B-cerulean, at the 5′ end. It generates three isoforms (Figure S5), with the same splicing junction as SRSF1 isoforms (Figures 1A and S1A). The overexpression of SRSF1 protein (through transiently transfecting the cDNA version of SRSF1, as in Figure S1B) altered the splicing pattern of SynT toward more spliced isoforms (Figure 5C), consistent with SynT acting as a SRSF1 target.
To determine the response of SRSF1 splicing to various SynT loads, we need to quantify the splicing pattern of SRSF1, as well as the expression level of the only functional isoform of SRSF1, isoform 1, across different SynT induction levels (Figure 5A, bottom). The splicing pattern can be quantified using RT-PCR (Figures 5C and S5) by amplifying all of the isoforms at once with a single primer set targeting the 5′ and-3′ end of the gene (STAR Methods). The amount of SRSF1 isoform 1 can be quantified using qRT-PCR by a primer set (Figure S1A) specifically targeting isoform 1 without amplifying isoforms 2–5 (Figure S6A). The quantitative calibration of the designed qPCR primers is verified in the dilution curve shown in Figure S6B.
We then tested the response of SRSF1 splicing to two different ectopic expression levels of SynT: A site-specifically integrated single copy of SynT (Figure 5D, single-copy SynT), and transiently transfected multiple copies of the SynT that together produce ~20–100 times more SynT Figure 5D, transient multi-copy SynT). The latter ensures that the ectopic SynT load effectively competes with the large number of endogenous SRSF1 target substrates (Das and Krainer, 2014). As shown in Figure 5D (middle and lower panels), we observed no significant change in SRSF1 isoform 1 levels in response to the single-copy perturbation. By contrast, SRSF1 isoform 1 levels increased ~2-fold in response to transient, multi-copy SynT transfection.
Negative autoregulatory splicing contributed to this increased SRSF1 isoform 1 level. As shown in Figure 5E, when the SynT level increased, a higher fraction of SRSF1 pre-mRNA remained unspliced, producing more functional isoform 1. Based on gel band intensity quantification, the ratio of isoform 1 increased approximately 1.53 times, consistent with the readout from Agilent Bioanalyzer 2100 (Figure S7). This result suggests that the overexpression of SRSF1 substrates can reduce SRSF1 availability, impeding the unproductive self-splicing of SRSF1, and thereby increasing the level of functional SRSF1 isoform 1. Notice that the isoform 1 ratio increment (Figure 5E) is smaller than the increased SRSF1 isoform 1 level (Figure 5D), indicating the existence of other regulation layers on SRSF1 production, apart from the negative autoregulatory splicing (see more in the Discussion section).
SynT is a synthetic target. We further checked whether SRSF1 splicing similarly responds to variations in the load presented by its endogenous targets. Specifically, we obtained a list of SRSF1 target genes from the cross-linking and immunoprecipitation with high-throughput sequencing (CLIP-seq) database POSTAR2 (Zhu et al., 2019), and the corresponding transcript levels (transcripts per million, TPM) of both SRSF1 and its CLIP-based target genes from GTEx (www.gtexportal.org) across 53 human tissue types. In addition to SRSF1, we included two other splicing factors, hnRNPA1 and PTBP1, with conserved autoregulatory negative splicing feedback loops (Ni et al., 2007; Wollerton et al., 2004). In contrast, we picked six RNA binding proteins not known to regulate splicing of their own mRNAs: UPF1, RNA helicase, and ATPase (involved in mRNA nuclear export and surveillance); TAF15, TATA-binding protein-associated factor (involved in RNA polymerase II transcription); RBM10, RNA-binding motif (involved in splicing regulation); RTCB, RNA ligase; MBNL2, RNA splicing regulator; and CELF2, RNA splicing regulator. The abundance of the three negatively autoregulated splicing factors positively correlated with that of their targets (Figures 6A, S8A, and S9A), while the six other RNA regulators did not present any clear correlation (Figures 6B, S8B, and S9B). This lack of correlation occurred across multiple CLIP-seq data background-extraction methods (STAR Methods). The quantitative correlation coefficients are listed in Figure S11. Note that these data do not rule out roles for other potential regulatory interactions that could also contribute to these correlations (see Discussion).
Figure 6. The adaptation function of splicing negative autoregulation responds to variations in endogenous targets load.
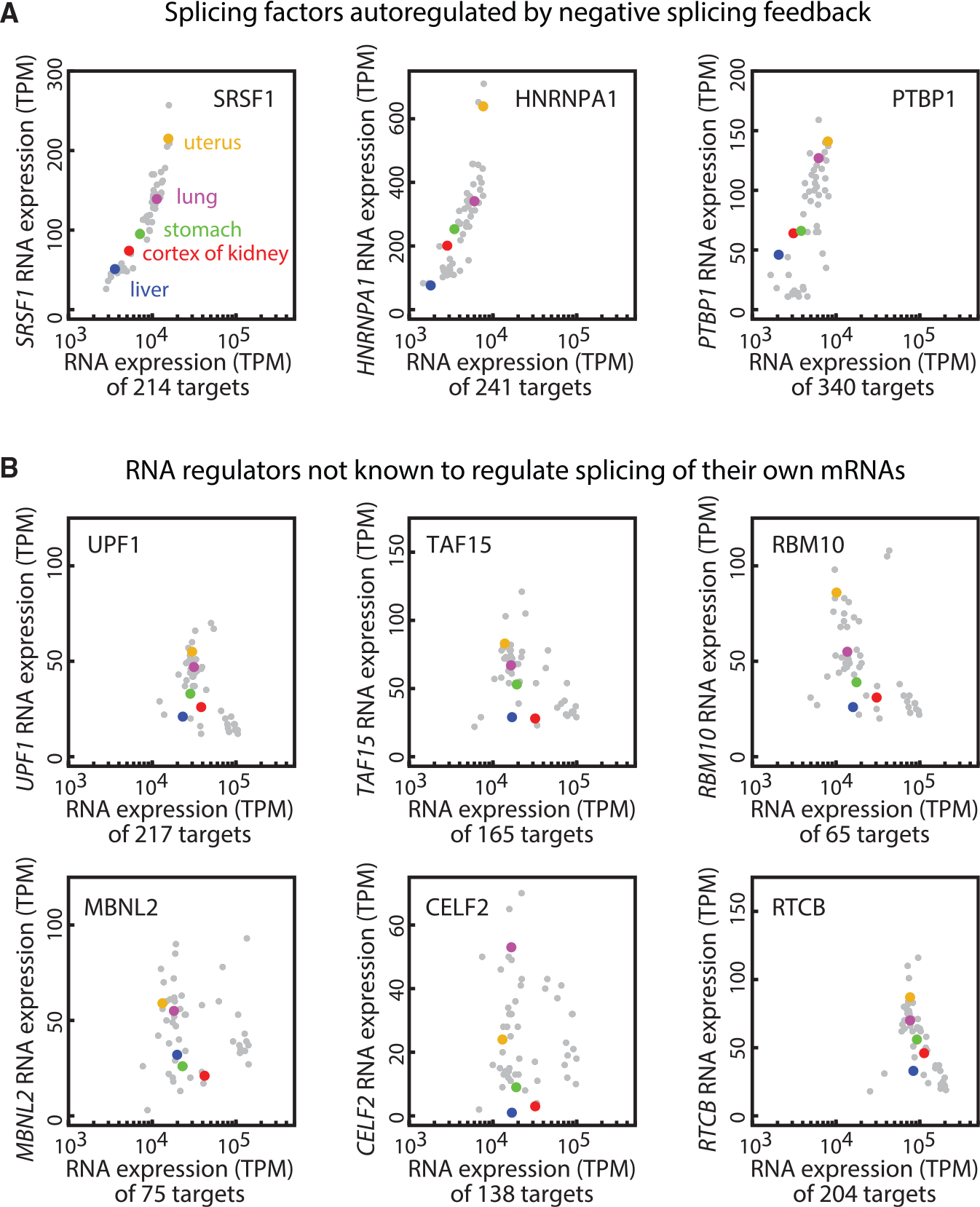
(A) The expression level of splicing factors positively correlates with their target expression across 53 human tissue types (gray dots), in which 5 example tissues (uterus, lung, cortex of kidney, stomach, liver) are labeled in distinct colors. The splicing factor RNA expression levels (TPM) were extracted from the GTEx database. The respective target genes are selected based on POSTAR2, specifically, the top 2% “binding site records” in each CLIP database (STAR Methods). The 3 splicing factors SRSF1, hnRNPA1, and PTBP1 are autoregulated via negative splicing feedback.
(B) The RNA regulation proteins not known to regulate splicing of their own mRNAs do not show correlative patterns as in (A).
Taken together, these results indicate that substrate load can modulate negative autoregulatory splicing feedback. This load-adaptive feedback scheme helps ultra-conserved splicing factors to adapt their own protein expression levels to variable substrate levels across diverse tissues (Hanamura et al., 1998; Lareau et al., 2007; Zahler et al., 1992).
A mathematical model explains the dynamics and function of negative autoregulatory splicing
Having shown experimentally that negative autoregulatory splicing accelerates SRSF1 response times and enables their adaptation to substrate load, we sought to understand how these features arise from the autoregulatory architecture. We developed a mathematical model describing the dynamics of SRSF1 with and without feedback, and fit the model to SRSF1 dynamics observed in time-lapse videos of the SRSF1(gDNA) and SRSF1(cDNA) cell lines.
We formulated differential equations describing the dynamics of unspliced pre-mRNA u, functional mRNA isoform m, and protein p from the endogenous (subscript en) and ectopic (subscript ec) copies (Figure 7A). For both sets of equations, we assumed the same constant production rate (α) for either of the two ectopic constructs, controlling production of either mec for SRSF1(cDNA) or uec for SRSF1(gDNA). We also assumed that the translation of p is linearly related to the corresponding mRNA concentration m, at rate g, and that all of the species undergo first-order degradation with rate constant β.
Figure 7. A minimal mathematical model describes splicing regulation with and without negative feedback.
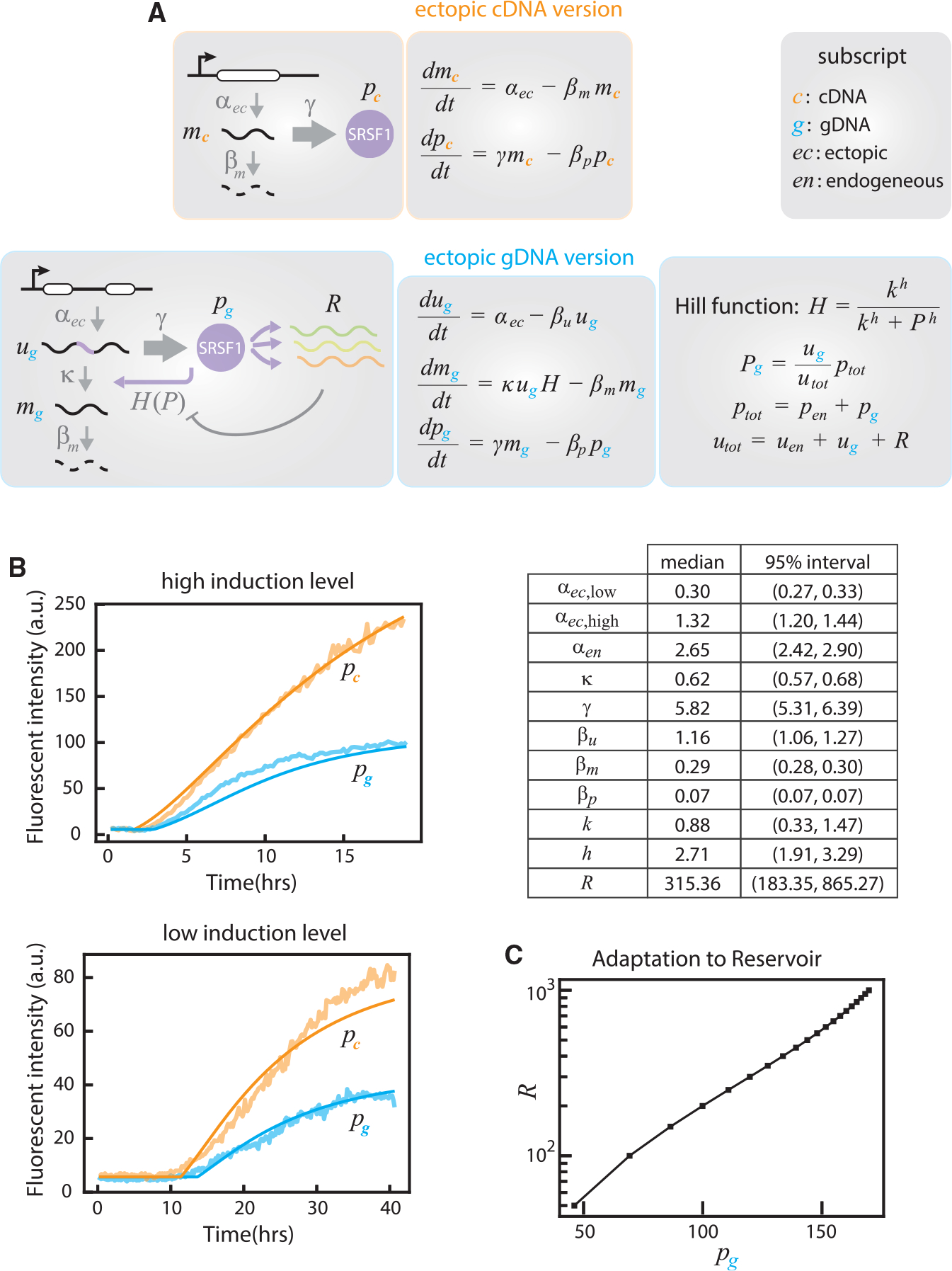
(A) Differential equation sets for the regulation of ectopic SRSF1(cDNA) (without feedback) and SRSF1(gDNA) (with feedback) cell lines. Unspliced pre-mRNA, functional mRNA, and SRSF1 protein are labeled u, m, and p, respectively. Subscripts are defined at left. As each ectopic copy is stably integrated into the same genomic locus of HEK293 cells, they share the same transcription rate (α), translation rate (γ), and degradation rate (β) between cell lines. Note that gDNA system incorporates a Hill function H(P) to represent the dependence of splicing activity on SRSF1 protein. For details, see STAR Methods.
(B) Fits of median time-lapse traces from Figures 4B and S4A to the equations in (A). Fitted curves are shown with smooth solid lines. Bestfit parameter estimates are shown in the table and Figure S12.
(C) Negative splicing autoregulation adapts to load. Using the fitted parameters, the model predicts a positive correlation between target load and SRSF1 level, p, similar to experimental observations (Figure 5).
We define two sets of equations, with and without feedback. In the case of SRSF1(cDNA), the functional isoform mec is not subject to negative autoregulatory splicing. Since the endogenous components do not have any regulatory effect, modeling the dynamics of mec and pec is sufficient (Figure 7A, ectopic cDNA version; STAR Methods). In contrast, the SRSF1(gDNA) cell line features negative feedback affecting the splicing of uec to mec via both the endogenous and ectopic SRSF1 proteins (Figure 7A, ectopic gDNA version and endogenous copy; STAR Methods). Since SRSF1 also acts on many other target genes, collectively denoted R, we reasoned that the level of SRSF1 available for autoregulatory splicing would be reduced by competition with R. Therefore, we modeled negative feedback by a Hill function H(p), where the effective protein level acting on SRSF1 transcripts depends on the abundance of those transcripts relative to those of the reservoir. The resulting differential equations are summarized in Figure 7A; more details on the derivation are given in STAR Methods.
We then fit the model parameters to the averaged traces in Figure 4B using a Bayesian inference framework (Figure 7B; STAR Methods). The data comprise ectopic SRSF1 protein levels for the SRSF1(cDNA) and SRSF1(gDNA) lines at both low and high induction levels (Figures 4B and S4). Shared biochemical parameters (e.g., translation and degradation rates) are fit jointly across all of the conditions. Parameters specific to the negative feedback case are κ, the efficiency of splicing to the unproductive isoform; k, the substrate concentration that produces half-maximum splicing activity; h, the Hill coefficient; and R, the reservoir level (STAR Methods). Only the ectopic production rate was allowed to change between low and high induction levels.
We computed probability distributions for each parameter (Figure S12) and identified median values and confidence intervals (Figure 7B, table). These values were approximately consistent with independent parameter estimates. In particular, the ratio between production rates of the ectopic and endogenous SRSF1 copy in SRSF1(gDNA) cells (i.e., αec versus αen) matched that obtained from the western blot in Figure S2. Similarly, the fit degradation rate values generate half-lives of approximately 35 min for u, 2.4 h for m, and 10 h for p, broadly consistent with values in other studies (Milo and Phillips, 2015; Moulton et al., 2014). Finally, the fitted value of k suggests that 62% of transcripts are spliced to isoform 1, generally consistent with the percentage seen in multi-copy SynT transfection (Figure 5E).
The fitted parameters can provide deeper insight into the biology of SRSF1 regulation. The Hill coefficient value h ~ 2–3 suggests ultra-sensitivity of SRSF1 activity. The estimated reservoir level R of nearly 300 suggests approximately several hundred additional SRSF1 target transcripts (with relatively high abundances) per SRSF1 transcript in this cell type, also consistent with typical values in a variety of cell types (Figures 6A, S8A, and S9A). Finally, using the model to compute the steady-state ectopic protein level as a function of R reveals that SRSF1 level adapts to target load (Figure 7C), consistent with experimental results (Figures 6A, S8A, and S9A).
DISCUSSION
Negative autoregulation is a prominent feature of many splicing regulatory systems. To understand its functional impact, we constructed three HEK293 cell lines, with inducible gDNA (autoregulated), cDNA (unregulated), and SynT (synthetic SRSF1 targets) and quantitatively investigated the dynamic function of negative autoregulatory SRSF1 splicing in individual cells. By combining deep learning-based single-cell video analysis (Figure 3) with flow cytometry, qRT-PCR, and RT-PCR, we found that negative autoregulatory splicing can stabilize SRSF1 levels independent from its own transcription strength, reduce cell-cell heterogeneity in both SRSF1 levels and response times (Figure 4), and adapt to changes in target load (Figures 5, S8, and S9). Although we focused on SRSF1, the approach presented here can be extended to study other splicing factors.
A particularly interesting aspect of negative splicing autoregulation, compared to better-known negative autoregulatory transcriptional feedback, is its ability to homeostatically control splicing activity in response to changes in total substrate load. In contrast to a transcriptional circuit facing a relatively fixed number of binding sites, splicing factors operate on variable amounts of substrate, including their own pre-mRNAs as well as other target genes (Figure 1B). Thus, to maintain a constant splicing activity, negative autoregulatory splicing buffers SRSF1 effective concentration against variations in both SRSF1 pre-mRNAs level (Figures 2 and 4) as well as their target levels (Figures 5 and 6). Noticeably, even though both SRSF1 and their target SynT overexpression increases absolute SRSF1 levels, they produce distinct and opposite effects on SRSF1 splicing patterns (Figures S1B and S5D), with more spliced SRSF1 isoforms produced when SRSF1(cDNA) was overexpressed (Figure S1B) and more unspliced SRSF1 isoforms when SynT was overexpressed (Figure 5E). These counterintuitive results confirm that the cell maintains subsaturating SRSF1 levels.
In addition to the experimental data, mathematical modeling provided further insights into SRSF1 autoregulation. First, it shows that experimentally observed effects can arise from basic aspects of splicing and transcriptional regulation more generally, by phenomenologically incorporating Hill functions into the differential equations listed in Figure 7A to describe the concentration dynamics of involved RNA (spliced, unspliced) and protein molecules. Second, the model provides a unified understanding of the sometimes counterintuitive responses of SRSF1 to different perturbations driven by autogenous splicing and feedback (i.e., SRSF1 overexpression in Figure 7B, versus perturbations in other target substrates, the amount of reservoir in Figure 7C). Finally, the model identified biochemical parameters, such as the half-lives of SRSF1 protein and its spliced/unspliced RNAs, the co-transcriptional splicing efficiency, as well as the amount of target substrates, that recapitulate experimental observations and can help to inform a more quantitative understanding of splicing dynamics.
Negative autoregulatory splicing is not the only factor affecting SRSF1 expression. Our experimental data and mathematical modeling do not rule out more complex mechanisms. For instance, the ratio of isoforms 2–5 in SRSF1 splicing pattern varies when we perturb the SRSF1 level (Figure S1B) or SRSF1 substrate load (Figure 5E), indicating that SRSF1 concentration (either overexpression or depletion) is not the only factor affecting SRSF1 splicing. In addition, a previous study (Sun et al., 2010) revealed translational level regulation on SRSF1 expression. Further molecular analysis will be necessary to fully characterize mechanistic aspects of autoregulation.
This analysis does not explicitly incorporate the non-uniform spatial distribution of SRSF1 in the nucleus. SRSF1 concentrates in interchromatin granule clusters called speckles (Lamond and Spector, 2003; Misteli et al., 1997) that exhibit dynamic structures (Misteli et al., 1997). Recent work suggests that the spatial distribution of splicing factors correlates with active transcription hubs in the nucleus (Ding and Elowitz, 2019; Quinodoz et al., 2018). Because most splicing occurs co-transcriptionally (Bentley, 2014; Das et al., 2007; Rosonina and Blencowe, 2002), negative autoregulatory splicing should, in principle, feedback on the local SRSF1 concentration within a subnuclear neighborhood, rather than the global concentration averaged over the nucleus as a whole. It thus remains unclear how splicing factors balance their local and global concentrations in the nucleus. In the future, it will be interesting to develop a more complete analysis of negative autoregulatory splicing that includes SRSF1 subcellular spatial distribution and may provide an integrated view of how cells maintain constant effective SRSF1 concentrations despite heterogeneity in their subnuclear spatial distributions.
More generally, negative autoregulatory splicing has the potential to affect diverse cellular functions. For instance, splice regulators could impact cell physiology by regulating “poison” exons, among other mechanisms (Thomas et al., 2020; Leclair et al., 2020). Splice regulation could also be useful in synthetic circuits in which microRNAs are expressed from introns (Strovas et al., 2014). These natural and synthetic systems both suggest the importance of further investigation into the actual and potential roles of splicing feedback systems.
Limitations of the study
Splicing is a complicated process. Both our experiments and mathematical models focus only on the negative autoregulatory aspect of splicing (with transcriptional control). They do not analyze the roles for other regulatory mechanisms, such as NMD regulation, translational regulation, and epigenetic regulation. A complete understanding of splice regulation will eventually need to account for multiple regulatory systems. In addition, we focused here on quantifying the dynamics of SRSF1 autoregulation in the HEK293 cell line. Cell lines and cell types differ in the expression of splicing components, and autoregulatory splicing dynamics could correspondingly vary between different cell types or in more complex contexts, such as specific tissues.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests regarding resources and reagents can be directed to Michael Elowitz (melowitz@caltech.edu).
Materials availability
Cell lines generated in this study are available from the lead contact upon request.
Data and code availability
Data generated in this study are available from the lead contact upon request.
All data reported in this paper will be shared by the lead contact upon request.
Original codes are deposited and publicly accessible. DOI is listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Human Fibronectin | Oxford Biomedical Research | SKU:GP40 |
| 4-epi tetracycline hydrochloride | Sigma Aldrich | 37918 |
| doxycycline | Ding et al., 2019 | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Lipofectamine LTX plasmid transfection reagent | ThermoFisher | A12621 |
| RNeasy Mini Kit | Qiagen | 74106 |
| iScript cDNA Synthesis Kit | Bio-Rad Laboratories | 1708891 |
| AccuPrime™ PfxSuperMix | ThermoFisher | 12344–040 |
| iQ SYBR Green Supermix | Bio-Rad Laboratories | 1708880 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Flp-In™ T-Rex™ HEK293 cells | ThermoFisher | R78007 |
| gDNA cells: FLP-in T-Tex 293 + CMV-TO- Citrine-EndoSFRS1 | This paper | N/A |
| cDNA cells: FLP-in T-Tex 293 + CMV-TO- Citrine-cDNA of SFRS1 | This paper | N/A |
| synT cells: FLP-in T-Tex 293 + CMV-TO- H2B-Ceru-3'UTR | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| RT-PCR SRSF1 isoforms forward -ACATC GACCTCAAGAATCGCCGC | IDT DNA | N/A |
| RT-PCR SRSF1 isoforms reverse -ATCCA GTGAGCCCTCTCCAA | IDT DNA | N/A |
| RT-PCR SynT isoforms forward- CACATG AAGCAGCACGACTT | IDT DNA | N/A |
| RT-PCR SynT isoforms reverse - ATCCA GTGAGCCCTCTCCAA | IDT DNA | N/A |
| SRSF1 isoform1 gene-specific primer-TCA TCCTCCCTATCCTATCCACA | IDT DNA | N/A |
| RT-qPCR SRSF1 isoform1 forward - GCA GAGGATCACCACGCTAT | IDT DNA | N/A |
| RT-qPCR SRSF1 isoform1 reverse - GCC AAGGTTTAAAAAGCAAAGCA | IDT DNA | N/A |
| RT-qPCR SynT isoform 1 forward - CGG CATGGACGAGCTGTA | IDT DNA | N/A |
| RT-qPCR SynT isoform 1 reverse - AGTTC ACACAAACCAGGGCA | IDT DNA | N/A |
| GAPDH isoform1 gene-specific primer - AGT GATGGCAT GGACTGTGG | IDT DNA | N/A |
| RT-qPCR GAPDH forward - GGTGTGAACC ATGAGAAGTATGA | IDT DNA | N/A |
| RT-qPCR GAPDH reverse - GAGTCCTTCC ACGATACCAAAG | IDT DNA | N/A |
| SDHA isoform1 gene-specific primer - CTC CAGTGCTCCTCAAAGGG | IDT DNA | N/A |
| RT-qPCR SDHA forward - AGAGGGAGGC ATTCTCATTAAC | IDT DNA | N/A |
| RT-qPCR SDHA reverse - ACCGAGACAC CACATCTCTA | IDT DNA | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| SRSF1(gDNA): CMV-TO-Citrine-EndoSFRS1 | This paper | N/A |
| SRSF1(cDNA): CMV-TO-Citrine-cDNA of SFRS1 | This paper | N/A |
| SynT: CMV-TO-H2B-Ceru-3’UTR | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Automated acquisition software Metamorph | Molecular Devices | N/A |
| Easyflow | Y. Antebi | https://github.com/AntebiLab/easyflow |
| Fiji | Schindelin et al., 2012 | https://imagej.net/Fiji |
| Cell tracking | Y. Antebi | https://github.com/AntebiLab/EasyTrack |
| MATLAB | MathWorks | N/A |
| The code and database for tissue-specific analysis (related to Figures 6 and S8–S10) | This paper | https://doi.org/10.5281/zenodo.6522983 |
| The code for the mathematical modeling section (related to Figures 7 and S11) | This paper | https://doi.org/10.5281/zenodo.6525681 |
| The deep-learning image analysis code | Darvin Yi | https://github.com/yidarvin/FirstAid |
|
| ||
| Other | ||
|
| ||
| 24-well 10 mm diameter glass No. 1.5 coverslip plates | MatTek Corp | P24G-1.5–10-F |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plasmids
SRSF1(gDNA), or SRSF1(cDNA), or SynT was cloned into a Flp-In™ pFRT expression vector (Life Technologies) under an inducible CMV-TO promoter. SRSF1(gDNA) incorporated the same sequence as the endogenous SRSF1 gene (based on gDNA sequencing), including the 3’UTR part. SRSF1(cDNA) incorporated the sequence of mRNA of endogenous SRSF1 (without the 3’UTR site) with the BGH polyadenylation sequence in the pFRT vector. SynT contained H2B-Citrine fused with the 3’UTR site of the endogenous SRSF1 gene.
Cell lines
Flp-In™ T-Rex™ HEK293 cells (Life Technologies, we did not test for mycoplasm) were cultured following the manufacturer’s protocol. For transfection, the cells were pre-plated in 24-well plates with 80% confluency. We added 800–1000 ng of plasmid (SRSF1(gDNA) or SRSF1(cDNA) or SynT) using the Lipofectamine LTX plasmid transfection reagent (ThermoFisher Scientific) and changed the culture media to Opti-MEM™ Reduced Serum Medium (ThermoFisher Scientific). Cells were left in the incubator overnight, then trypsinized (using 0.25% Trypsin-EDTA (Thermo Fisher Scientific)) into new 6-well plates with complete culture media the next day. These cells were then cultured for 1–2 weeks with 100 ug/ml Hygromycin. The surviving transfected cells were subcloned by limiting dilution.
METHODS DETAILS
Time-lapse microscopy imaging and data analysis
24-well 10 mm diameter glass No. 1.5 coverslip plates (MatTek Corp.) were coated with 5 ug/ml Human Fibronectin (Oxford Biomedical Research, Rochester Hills, MI) in PBS buffer for 1hr at room temperature. Fibronectin was then aspirated, and 4,000 – 10,000 cells were plated in the coated 24-well plate with complete cell media. The plate was manually swayed (Hui and Bhatia, 2007) to uniformly spread the cells, and left in the incubator for 2–3 h before imaging. Details of microscopy have been described previously (Nandagopal et al., 2019). For each movie, 40–60 stage positions were picked manually, and YFP and differential interference contrast (DIC) images were acquired every 10 min with an Olympus 20x objective using automated acquisition software (Metamorph, Molecular Devices, San Jose, CA). The details of cell tracking algorithm was in (Bintu et al., 2016; Singer et al., 2014).
Flow cytometry
Experimental procedures and data analysis for flow cytometry have been described previously (Nandagopal et al., 2019).
Transient transfection
Cells were plated at 50% confluency in 24-well plates and grown to 80% confluency overnight. We added 800–1000 ng of plasmid (SRSF1(gDNA) or SRSF1(cDNA) or SynT) using the Lipofectamine LTX plasmid transfection reagent (ThermoFisher Scientific) and changed the culture media to Opti-MEM™ Reduced Serum Medium (ThermoFisher Scientific). Cells were left in the incubator for 5.5 h, then trypsinized (by 0.25% Trypsin-EDTA (Thermo Fisher Scientific)) off the plates and resuspended in PBS (Phosphate-Buffered Saline buffer, Thermo Fisher Scientific). These cells were centrifuged, and the pellet washed 3x with PBS buffer to remove Trypsin. The final cell pellets were either dissolved for RNA extraction, or frozen and stored at −80C.
RT-PCR
We extracted total RNA using the RNeasy Mini Kit (Qiagen), and used 500 ng - 1 ug RNA to make cDNA using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). 0.1 of cDNA (i.e. 1ul after diluting cDNA 10x) was used in the PCR reaction, using AccuPrime™ Pfx SuperMix (Thermo Fisher Scientific) with an annealing temperature at 62.5 degree for 35 cycles. PCR primers are listed in Table S1. 3–8 ul of the PCR product was then run on a 1–2% Agarose gel. Gel band intensity was analyzed by Bio-Rad ChemiDoc Image Lab 6.0 band analyzer.
PT-qPCR
RNA was extracted as in RT-PCR. We then used 1 ug RNA to make cDNA using the SuperScript™ III First-Strand Synthesis kit (Thermo Fisher Scientific) with all gene-specific primers (SRSF1, SynT, GAPDH, and SDHA isoform1 gene-specific primers). We used gene-specific cDNA for qPCR, rather than random cDNA, to minimize the influence between isoforms with similar sequences. Experimental procedures and data analysis of qPCR were performed as described previously (Nandagopal et al., 2019). All primers are listed in Table S1.
Western blot
Frozen or fresh cell pellets (with 106 cells) were denatured using 200ul SDS loading buffer (1x sodium dodecyl sulfate (Sigma-Aldrich), 1x protease inhibitor, 4 mM EDTA) and heated for 5mins at 68°C in a water bath. The heated cells were then centrifuged at 55,000 rpm for 1hr at 4°C. 30 ul of the supernatant was loaded onto a NuPAGE™ 4–12% Bis-Tris Protein Gel (Thermo Fisher Scientific) and transferred using iBlot™ Transfer Stack to nitrocellulose (Thermo Fisher Scientific) following the manufacturer’s protocol. The blot was then blocked in 1xTBST, 5% dry milk, 2% BSA for 1 h at room temperature, followed by overnight incubation at 4°C with the primary antibody, anti-SRSF1 antibody (ab133689) at 1:1000 and anti-GFP (ab1218) at 1:2000, together. The next day, the blot was washed with 1x TBST three times, then incubated with anti-rabbit and anti-mouse HRP conjugated secondary antibody (GE Healthcare Life Sciences) at 1:2000 for 2hrs at 4°C. The blot was then washed with 1x TBST at room temperature for 1hr and five more times for 3 min. Gel bands were detected using SuperSignal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific) and analyzed using a Bio-Rad ChemiDoc Image Lab 6.0 band analyzer.
QUANTIFICATION AND STATISTICAL ANALYSIS
CLIP-seq data analysis
CLIP-seq database is downloaded from POSTAR2 (Zhu et al., 2019). The TPM data of target gene expression across 53 human tissue types is downloaded from GTEx (www.gtexportal.org).
As shown in Figures S10A and S10B, CLIP-seq has found more than 10,000 targets for most RNPs. With such a big number, when we add up the TPM of all the targets, the sum is directly comparable to total transcriptome (Figures S10C and S10D). To minimize the effect of targets with weak and non-specific binding, we only pick the strongest ones based on “Binding site records” in the CLIP-seq database.
We define two different ways to set up the threshold for binding-strength selection, based on either ‘percentile’ or ‘absolute’ modes. In the ‘percentile’ mode, for each RNP, we pick the target genes with its top 2% (Figure 6) or 5% (Figure S8) “Binding site records”. By changing from 2% to 5%, the number of selected CLIP-seq target genes is doubled. In the ‘absolute’ mode (Figure S9), for each RNP, we pick the threshold of “Binding site records” that can achieve to set the median TPM of selected target genes in between 2% and 5% of total transcriptome expression. In short, ‘percentile’ mode depends on the “Binding site records” distribution of each RNP, while ‘absolute’ mode aims for a comparable TPM sum of selected targets between RNPs.
As shown in Figures S8 and S9, despite the number of selected targets are different across various threshold setting (for instance, hnRNPA1 changed from 241, to 603, then 2049), the data pattern (indicating the adaption function of negative autoregulatory splicing) persists.
This pattern starts to diminish with a higher threshold, where the TPM sum of selected targets is beyond 10% of total transcriptome.
Mathematical model with and without negative splicing feedback
For the cDNA construct, ectopic mRNA transcripts mec are generated with some production rate αec, while protein pec is translated from mRNA with rate γ. Both species are degraded by first-order kinetics with rate constants βm and βp respectively. Therefore, their dynamics are described by the following set of differential equations:
For the gDNA construct with negative autoregulation of splicing, we must consider not only mRNA and protein levels m and p but also unspliced pre-mRNA levels u. Here, ectopic uec is generated with production αec and endogenous transcripts uen with rate αen. These transcripts are spliced productively to mec and men with rate k. This splicing is regulated by negative feedback from the effective SRSF1 protein level P, which we describe phenomenologically by a Hill function
The total protein level comprises both endogenous and ectopic SRSF1, or ptot = pen + pec. However, SRSF1 acts not only on ectopic and endogenous SRSF1 transcripts but also on other target RNAs, or a “reservoir” R. Therefore, the effective protein level for any given target is not the total amount. SRSF1 not only regulates the ectopic and endogenous SRSF1 transcripts but also acts on other target RNAs, or a “reservoir” R. We assume that the effective protein level for a given target is determined by the relative proportion of the target RNA. Defining the total level of unspliced targets by utot = uen + uec, the effective protein level acting on endogenous SRSF1 is
and similar for ectopic transcripts. Pre-mRNAs undergo first-order decay with rate constant βu; other biochemical parameters - translation rate as well as mRNA and protein degradation rates - are shared with the no-feedback model. The dynamics are given by the following set of differential equations:
The equations for the endogenous species are analogously defined:
Model parameters
We fit this model to the averaged movie curves of Figure 4, representing SRSF1 protein levels with and without feedback for both low and high induction levels. For the no-feedback case, the relevant model parameters include the transcription rates of the ectopic construct with low or high induction, the translation rate (γ), and the degradation rates of mRNA (βm) and protein (βp). Since we measure only the ectopic SRSF1 protein, we do not need to model the endogenous SRSF1 here.
Once we introduce regulation at the level of splicing, we must consider the ectopic as well as the endogenous SRSF1. We also include the parameters describing the feedback: k, the ratio of full transcript spliced to the productive isoform; k, the activation coefficient (substrate concentration of half-maximum activity); h, the Hill coefficient; and R, the reservoir level.
In addition to these key model parameters, we fit the starting levels of endogenous SRSF1 species u0, p0, and m0. Based on the analysis of Figure 4, we account for time delays between induction and cellular response, considering two values tlow and thigh for the low- and high-induction conditions. We also account for background noise by setting a floor bg on the predicted output.
Parameter fitting
To determine the model parameters, we performed Bayesian inference using Stan (Carpenter et al., 2017) to sample the probability distributions for each parameter given the experimental data. Bayesian statistics requires specifying priors, or the probability distributions for each parameter based on prior knowledge, and the likelihood, or the probability of observing the data given a set of parameters. Together, they allow inference of the posterior, or the probability distributions for each parameter after observing the experimental data.
For the likelihood, we assumed that experimental values lie in a Gaussian distribution centered at the theoretical value with a standard deviation σ. For the priors, we assumed relatively broad distributions based on biological knowledge and included applicable constraints. These prior distributions for each parameter are summarized in Table S2. All parameters are constrained to be nonnegative; any additional constraints are listed.
For these differential equations, the starting levels of each species can be fit, but the starting time is not permitted to vary in this framework. Therefore, we screened a broad range of choices of time delays, performed an initial fit for all possible combinations, and selected the values that minimized the resulting error. These initial fits were done on 4 chains with 250 iterations per chain (125 warm up, 125 sampling). For time delays, we evaluated a range of 6–9 h in the low-induction case and a range of 0–1 h in the high-induction case, sampling the ranges at intervals of 0.25 h (15 min). To quantify error for each set of time delays, we took the median value across samples for each parameter, simulated the resulting curve, and calculated the sum of squared errors (SSE) between the theoretical and observed values. This procedure yielded an optimal set of time delays of 8.25 h for the low-induction condition and 0.25 h for the high-induction condition. We then reran a more extensive fitting for this set of values, using 4 chains with 2000 iterations per chain (1000 warm up, 1000 sampling). The resulting samples are the basis for all results presented in the main text.
Supplementary Material
Highlights.
SRSF1 uses negative autoregulatory splicing to maintain its own concentration
This feedback reduces heterogeneity in both SRSF1 levels and response times
Splice autoregulation adapts SRSF1 levels to variation in target transcript load
A minimal mathematical model explains these capabilities
ACKNOWLEDGMENTS
We thank F. Tan for providing the western blot protocol, N. Nandagopal and Y. Antebi for technical assistance, and Life Sciences Editors (Sabbi Lall) for critical feedback on the manuscript. We also thank S. Sun, A.R. Krainer, D. Sprinzak, D. Baltimore, J.G. Ojalvo, and N. Wingreen for discussion and feedback on the project. F.D. was supported by a fellowship from the Schlumberger Foundation. C.J.S. is supported by NIH National Institute of General Medical Sciences training grant no. GM008042, and by a David Geffen Medical Scholarship. The work was funded by the Gordon and Betty Moore Foundation through grant no. GBMF2809 to the Caltech Programmable Molecular Technology Initiative and the Institute for Collaborative Biotechnologies through grant no. W911NF-09–0001 from the US Army Research Office, AWS Machine Learning Research Awards, and the Intel Corporation. The content of the information does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred. M.B.E. is a Howard Hughes Medical Institute investigator. This article is subject to HHMI’s Open Access to Publications policy. HHMI lab heads have previously granted a nonexclusive CC BY 4.0 license to the public and a sublicensable license to HHMI in their research articles. Pursuant to those licenses, the author-accepted manuscript of this article can be made freely available under a CC BY 4.0 license immediately upon publication.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110985.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alon U (2006). An Introduction to Systems Biology: Design Principles of Biological Circuits (Chapman and Hall/CRC; ). [Google Scholar]
- Anczuków O, and Krainer AR (2016). Splicing-factor alterations in cancers. RNA 22, 1285–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, and Krainer AR (2012). The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 19, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, and Frey BJ (2010). Deciphering the splicing code. Nature 465, 53–59. [DOI] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. (2012). The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593. [DOI] [PubMed] [Google Scholar]
- Baralle FE, and Giudice J (2017). Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18, 437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becskei A, and Serrano L (2000). Engineering stability in gene networks by autoregulation. Nature 405, 590–593. [DOI] [PubMed] [Google Scholar]
- Bentley DL (2014). Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 15, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, and Elowitz MB (2016). Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336. [DOI] [PubMed] [Google Scholar]
- Bohrer CH, and Larson DR (2021). The stochastic genome and its role in gene expression. Cold Spring Harb. Perspect. Biol. 13. 10.1101/cshperspect.a040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnal SC, López-Oreja I, and Valcárcel J (2020). Roles and mechanisms of alternative splicing in cancer—implications for care. Nat. Rev. Clin. Oncol. 17, 457–474. [DOI] [PubMed] [Google Scholar]
- Carpenter B, Gelman A, Hoffman MD, Lee D, Goodrich B, Betancourt M, Brubaker M, Guo J, Li P, and Riddell A (2017). Stan: a probabilistic programming language. J. Stat. Softw 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary S, Khokhar W, Jabre I, Reddy ASN, Byrne LJ, Wilson CM, and Syed NH (2019). Alternative splicing and protein diversity: Plants versus animals. Front. Plant Sci. 10, 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen EM, Yang SJ, Ando DM, Javaherian A, Skibinski G, Lipnick S, Mount E, O’Neil A, Shah K, Lee AK, et al. (2018). Silico labeling: predicting fluorescent labels in unlabeled images. Cell 173, 792–803.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culler SJ, Hoff KG, and Smolke CD (2010). Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science 330, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, and Krainer AR (2014). Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. 12, 1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, and Reed R (2007). SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 26, 867–881. [DOI] [PubMed] [Google Scholar]
- Ding F, and Elowitz MB (2019). Constitutive splicing and economies of scale in gene expression. Nat. Struct. Mol. Biol. 26, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Matunis MJ, Piñol-Roma S, and Burd CG (1993). hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 62, 289–321. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, and Swain PS (2002). Stochastic gene expression in a single cell. Science 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- El-Samad H (2021). Biological feedback control-Respect the loops. Cell Syst. 12, 477–487. [DOI] [PubMed] [Google Scholar]
- Faustino NA, and Cooper TA (2003). Pre-mRNA splicing and human disease. Genes Dev. 17, 419–437. [DOI] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu X-D, and Bentley DL (2014). Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 28, 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, and Roignant J-Y (2021). Anything but ordinary–emerging splicing mechanisms in eukaryotic gene regulation. Trends Genet. 37, 355–372. [DOI] [PubMed] [Google Scholar]
- Geuens T, Bouhy D, and Timmerman V (2016). The hnRNP family: insights into their role in health and disease. Hum. Genet. 135, 851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamura A, Cáceres JF, Mayeda A, Franza BR Jr., and Krainer AR (1998). Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA 4, 430–444. [PMC free article] [PubMed] [Google Scholar]
- Hug N, Longman D, and Cáceres JF (2016). Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44, 1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui EE, and Bhatia SN (2007). Silicon microchips for manipulating cell-cell interaction. J. Vis. Exp 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, and Sharp PA (2014). Building robust transcriptomes with master splicing factors. Cell 159, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, and Cooper TA (2011). Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyna M, Lopato S, Voronin V, and Barta A (2006). Evolutionary conservation and regulation of particular alternative splicing events in plant SR proteins. Nucleic Acids Res. 34, 4395–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, and Krainer AR (2007). The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 14, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond AI, and Spector DL (2003). Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4, 605–612. [DOI] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, and Brenner SE (2007). Unproductive splicing of SR genes associated with highly conserved and ultra-conserved DNA elements. Nature 446, 926–929. [DOI] [PubMed] [Google Scholar]
- Larsson AJM, Johnsson P, Hagemann-Jensen M, Hartmanis L, Faridani OR, Reinius B, Segerstolpe Å, Rivera CM, Ren B, and Sandberg R (2019). Genomic encoding of transcriptional burst kinetics. Nature 565, 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclair NK, Brugiolo M, Urbanski L, Lawson SC, Thakar K, Yurieva M, George J, Hinson JT, Cheng A, Graveley BR, et al. (2020). Poison exon splicing regulates a coordinated network of SR protein expression during differentiation and tumorigenesis. Mol. Cell 80, 648–665.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, and Rio DC (2015). Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 84, 291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, and Manley JL (2005). Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122, 365–378. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gonzàlez-Porta M, Santos S, Brazma A, Marioni JC, Aebersold R, Venkitaraman AR, and Wickramasinghe VO (2017). Impact of alternative splicing on the human proteome. Cell Rep. 20, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, and Caceres JF (2009). The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 417, 15–27. [DOI] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, and Misteli T (2011). Epigenetics in alternative pre-mRNA splicing. Cell 144, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley JL, Tacke R, Hogan BLM, Friedman KL, Diller JD, Ferguson BM, Nyland SVM, Brewer BJ, Fangman WL, Saavedra C, et al. (1996). SR proteins and splicing control 1569. Genes Dev. 10, 1569–1579. [DOI] [PubMed] [Google Scholar]
- Maquat LE (2004). Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5, 89–99. [DOI] [PubMed] [Google Scholar]
- McManus CJ, and Graveley BR (2011). RNA structure and the mechanisms of alternative splicing. Curr. Opin. Genet. Dev. 21, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, and Burge CB (2012). Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R, and Phillips R (2015). Cell Biology by the Numbers (Garland Science; ). [Google Scholar]
- Milo R, Jorgensen P, Moran U, Weber G, and Springer M (2010). BioNumbers—the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Cáceres JF, and Spector DL (1997). The dynamics of a pre-mRNA splicing factor in living cells. Nature 387, 523–527. [DOI] [PubMed] [Google Scholar]
- Moulton VR, Gillooly AR, and Tsokos GC (2014). Ubiquitination regulates expression of the serine/arginine-rich splicing factor 1 (SRSF1) in normal and systemic lupus erythematosus (SLE) T cells. J. Biol. Chem. 289, 4126–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandagopal N, Santat LA, and Elowitz MB (2019). Cis-activation in the Notch signaling pathway. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, and Ares M Jr. (2007). Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 21, 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, and Graveley BR (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, and Blencowe BJ (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Paz S, Ritchie A, Mauer C, and Caputi M (2021). The RNA binding protein SRSF1 is a master switch of gene expression and regulation in the immune system. Cytokine Growth Factor Rev. 57, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-order interchromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, and van Oudenaarden A (2008). Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, and Tyagi S (2006). Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4, e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Elowitz MB, and Alon U (2002). Negative autoregulation speeds the response times of transcription networks. J. Mol. Biol. 323, 785–793. [DOI] [PubMed] [Google Scholar]
- Rosenfeld N, Young JW, Alon U, Swain PS, and Elowitz MB (2007). Accurate prediction of gene feedback circuit behavior from component properties. Mol. Syst. Biol. 3, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosonina E, and Blencowe BJ (2002). Gene expression: the close coupling of transcription and splicing. Curr. Biol. 12, R319–R321. [DOI] [PubMed] [Google Scholar]
- Sanford JR, Wang X, Mort M, Vanduyn N, Cooper DN, Mooney SD, Edenberg HJ, and Liu Y (2009). Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 19, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti MM, and Swanson MS (2016). RNA mis-splicing in disease. Nat. Rev. Genet. 17, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, and Hertel KJ (2009). The SR protein family. Genome Biol. 10, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, and Manley JL (2004). Cell signalling and the control of pre-mRNA splicing. Nat. Rev. Mol. Cell Biol. 5, 727–738. [DOI] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, and Elowitz MB (2014). Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol. Cell 55, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strovas TJ, Rosenberg AB, Kuypers BE, Muscat RA, and Seelig G (2014). MicroRNA-based single-gene circuits buffer protein synthesis rates against perturbations. ACS Synth. Biol. 3, 324–331. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhang Z, Sinha R, Karni R, and Krainer AR (2010). SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat. Struct. Mol. Biol. 17, 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A, Gattoni R, Dooghe Y, Stévenin J, and Soret J (2001). SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 20, 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegedy C, Liu W, Jia Y, Sermanet P, Reed S, Anguelov D, Erhan D, Vanhoucke V, and Rabinovich A (2015). Going deeper with convolutions. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, pp. 1–9. [Google Scholar]
- Thomas JD, Polaski JT, Feng Q, De Neef EJ, Hoppe ER, McSharry MV, Pangallo J, Gabel AM, Belleville AE, Watson J, et al. (2020). RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat. Genet. 52, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, and Blencowe BJ (2019). Alternative splicing regulatory networks: Functions, mechanisms, and evolution. Mol. Cell 76, 329–345. [DOI] [PubMed] [Google Scholar]
- Urbanski LM, Leclair N, and Anczuków O (2018). Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 9, e1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Manley JL (1995). Overexpression of the SR proteins ASF/SF2 and SC35 influences alternative splicing in vivo in diverse ways. RNA 1, 335–346. [PMC free article] [PubMed] [Google Scholar]
- Wang Z, and Burge CB (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, and Burge CB (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson ME, Charenton C, and Nagai K (2020). RNA splicing by the spliceosome. Annu. Rev. Biochem. 89, 359–388. [DOI] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, and Smith CWJ (2004). Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol. Cell 13, 91–100. [DOI] [PubMed] [Google Scholar]
- Zahler AM, Lane WS, Stolk JA, and Roth MB (1992). SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 6, 837–847. [DOI] [PubMed] [Google Scholar]
- Zhou Z, and Fu X-D (2013). Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 122, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Xu G, Yang YT, Xu Z, Chen X, Shi B, Xie D, Lu ZJ, and Wang P (2019). POSTAR2: deciphering the post-transcriptional regulatory logics. Nucleic Acids Res. 47, D203–D211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are available from the lead contact upon request.
All data reported in this paper will be shared by the lead contact upon request.
Original codes are deposited and publicly accessible. DOI is listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Human Fibronectin | Oxford Biomedical Research | SKU:GP40 |
| 4-epi tetracycline hydrochloride | Sigma Aldrich | 37918 |
| doxycycline | Ding et al., 2019 | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Lipofectamine LTX plasmid transfection reagent | ThermoFisher | A12621 |
| RNeasy Mini Kit | Qiagen | 74106 |
| iScript cDNA Synthesis Kit | Bio-Rad Laboratories | 1708891 |
| AccuPrime™ PfxSuperMix | ThermoFisher | 12344–040 |
| iQ SYBR Green Supermix | Bio-Rad Laboratories | 1708880 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Flp-In™ T-Rex™ HEK293 cells | ThermoFisher | R78007 |
| gDNA cells: FLP-in T-Tex 293 + CMV-TO- Citrine-EndoSFRS1 | This paper | N/A |
| cDNA cells: FLP-in T-Tex 293 + CMV-TO- Citrine-cDNA of SFRS1 | This paper | N/A |
| synT cells: FLP-in T-Tex 293 + CMV-TO- H2B-Ceru-3'UTR | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| RT-PCR SRSF1 isoforms forward -ACATC GACCTCAAGAATCGCCGC | IDT DNA | N/A |
| RT-PCR SRSF1 isoforms reverse -ATCCA GTGAGCCCTCTCCAA | IDT DNA | N/A |
| RT-PCR SynT isoforms forward- CACATG AAGCAGCACGACTT | IDT DNA | N/A |
| RT-PCR SynT isoforms reverse - ATCCA GTGAGCCCTCTCCAA | IDT DNA | N/A |
| SRSF1 isoform1 gene-specific primer-TCA TCCTCCCTATCCTATCCACA | IDT DNA | N/A |
| RT-qPCR SRSF1 isoform1 forward - GCA GAGGATCACCACGCTAT | IDT DNA | N/A |
| RT-qPCR SRSF1 isoform1 reverse - GCC AAGGTTTAAAAAGCAAAGCA | IDT DNA | N/A |
| RT-qPCR SynT isoform 1 forward - CGG CATGGACGAGCTGTA | IDT DNA | N/A |
| RT-qPCR SynT isoform 1 reverse - AGTTC ACACAAACCAGGGCA | IDT DNA | N/A |
| GAPDH isoform1 gene-specific primer - AGT GATGGCAT GGACTGTGG | IDT DNA | N/A |
| RT-qPCR GAPDH forward - GGTGTGAACC ATGAGAAGTATGA | IDT DNA | N/A |
| RT-qPCR GAPDH reverse - GAGTCCTTCC ACGATACCAAAG | IDT DNA | N/A |
| SDHA isoform1 gene-specific primer - CTC CAGTGCTCCTCAAAGGG | IDT DNA | N/A |
| RT-qPCR SDHA forward - AGAGGGAGGC ATTCTCATTAAC | IDT DNA | N/A |
| RT-qPCR SDHA reverse - ACCGAGACAC CACATCTCTA | IDT DNA | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| SRSF1(gDNA): CMV-TO-Citrine-EndoSFRS1 | This paper | N/A |
| SRSF1(cDNA): CMV-TO-Citrine-cDNA of SFRS1 | This paper | N/A |
| SynT: CMV-TO-H2B-Ceru-3’UTR | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Automated acquisition software Metamorph | Molecular Devices | N/A |
| Easyflow | Y. Antebi | https://github.com/AntebiLab/easyflow |
| Fiji | Schindelin et al., 2012 | https://imagej.net/Fiji |
| Cell tracking | Y. Antebi | https://github.com/AntebiLab/EasyTrack |
| MATLAB | MathWorks | N/A |
| The code and database for tissue-specific analysis (related to Figures 6 and S8–S10) | This paper | https://doi.org/10.5281/zenodo.6522983 |
| The code for the mathematical modeling section (related to Figures 7 and S11) | This paper | https://doi.org/10.5281/zenodo.6525681 |
| The deep-learning image analysis code | Darvin Yi | https://github.com/yidarvin/FirstAid |
|
| ||
| Other | ||
|
| ||
| 24-well 10 mm diameter glass No. 1.5 coverslip plates | MatTek Corp | P24G-1.5–10-F |