

Abstract
Stringent regulation of IgE antibody production is critical for constraining allergic responses. This review discusses recent advances in understanding cell-intrinsic and extrinsic mechanisms that regulate the genesis and fate of IgE B cells. B cell-intrinsic regulation of IgE is orchestrated by the IgE B Cell Receptor (BCR). Through its antigen-independent signaling and low surface expression, the IgE BCR drives IgE B cells to differentiate into short-lived plasma cells and/or undergo apoptosis, restricting IgE-expressing cells from entering long-lived compartments. The pivotal extrinsic regulators of IgE responses are T follicular helper cells (TFH). TFH produce IL-4 and IL-21, which, respectively, are the major activating and inhibitory cytokines for IgE class-switching. Other newly identified T follicular subsets also contribute to IgE regulation. Although IgE responses are normally constrained, recent studies suggest that specific conditions can induce the formation of IgE responses with enhanced affinity or longevity, effectively ‘breaking the rules’ of IgE regulation.
Introduction
The production of IgE antibodies specific for allergens is a major component of allergic sensitization and a key contributor to disease pathogenesis. However, in the majority of immune responses, IgE production is highly constrained. The process of class switch recombination (CSR) to IgE in B cells appears to be tightly controlled, leading to the generation of only small numbers of IgE lymphocytes [1]. Careful studies of these rare IgE lymphocytes in mice reported that IgE B cells were unusual in that a large proportion differentiated into IgE plasma cells (PCs, we note that here we use this term broadly to encompass all antibody-secreting cells including plasmablasts and plasma cells), the majority of which were short-lived [2–7]. IgE B cells also appeared in small numbers and only transiently in germinal centers (GCs), a site for efficient antibody affinity maturation and the generation of long-lived PCs and memory B cells [2–7]. Increased numbers of IgE GC B cells were observed in mice deficient for the master regulator of PC differentiation, Blimp-1, suggesting that normally the predisposition toward PC fate contributes to the reduced representation of IgE B cells in the GC [3]. These studies established the outcomes of IgE regulation: rarity due to limited generation of IgE B cells, low GC participation, high rates of short-lived PC formation, and absence from long-lived compartments. Recently, the mechanisms underlying these features of IgE responses have begun to be illuminated. This review will explore these advances, focusing on examples of B cell-intrinsic regulation through the B cell receptor (BCR) and B cell-extrinsic regulation by T-cell-derived cytokines. We then discuss how escape from these regulatory mechanisms may promote allergy.
Intrinsic regulation of IgE B cell responses
Antigen-independent signaling of the IgE BCR drives premature PC differentiation of IgE B cells
Cell culture studies revealed that the predisposition of IgE B cells for PC differentiation was driven by intrinsic signals from the IgE BCR. Initially, it was observed that when purified murine naïve B cells were cultured with stimuli that mimic T cell help (ligation of CD40 and IL-4) to induce class switch recombination (CSR) to IgE, a large proportion of the IgE cells that formed exhibited a PC phenotype [3,8,9]. Culture studies of human tonsil B cells also showed a higher frequency of PCs among IgE cells compared to IgG1 cells [10]. While these observations could have been due to increased IgE CSR in cells already poised to undergo PC differentiation, or conversely increased PC differentiation in cells poised to undergo IgE CSR, recent studies found that the expression of the IgE BCR itself predisposes IgE B cells for PC fate [8,9]. Specifically, ectopic expression of the IgE BCR in activated B cells drove PC differentiation, whereas the ectopic expression of BCRs of most other isotypes, such as IgG1, did not, with a partial effect observed for IgA. Typically, antigen-induced BCR signaling promotes PC differentiation [11], making it remarkable that ectopic expression of the IgE BCR promoted PC differentiation in an antigen-independent manner.
The above findings suggested that the IgE BCR exhibits autonomous signaling that differs from the tonic signaling of other BCRs. Indeed, evidence was obtained for increased phosphorylation of proximal signaling adapters and increased expression of genes induced by BCR signaling, such as Nur77, in B cells expressing the IgE BCR [8,9]. Notably, however, the antigen-independent signaling from the IgE BCR was substantially weaker than when the BCR was stimulated with antigen [9]. In addition, although expression of the IgE BCR promoted PC differentiation in activated B cells [8,9], it did not do so in naïve B cells [12]. We propose that chronic weak signaling from the IgE BCR synergizes with extrinsic activation signals, such as from T cell help, to promote increased PC differentiation. Consistent with autonomous signaling from the IgE BCR promoting PC differentiation, genetic and pharmacological disruption of a variety of proximal signaling adapters led to reduced IgE PC differentiation in cultured B cells [8,9]. Interestingly, heterozygous mutations in Syk or Cd19 resulted in substantial reductions in IgE PC differentiation, indicating that small changes in the magnitude of BCR signaling can have significant functional consequences.
Based on the observations in cell culture discussed above and previous in vivo observations suggesting that premature PC differentiation hindered IgE GC B cell responses, it was predicted that perturbations of BCR signaling would lead to reduced IgE PC differentiation and enhanced IgE GC B cell responses in vivo. An increase in IgE GC B cells was indeed observed in mice with perturbations of various BCR signaling pathways [8,9]. The effect of these perturbations on in vivo IgE PC responses, however, was less clear, as both decreases and increases in the absolute number of IgE PCs were reported [8,9]. Specifically, one group found that in Blnk-deficient mice, early IgE PC responses were reduced, yet later IgE PC responses were increased and sustained IgE production was observed up to 100 days after immunization [8]. Ig variable region sequencing four weeks after immunization showed similar somatic mutations and selection in IgE and IgG1 PCs [8]. These findings suggest that Blnk-deficiency led to elevated IgE GC B cell responses that in turn resulted in the increased export of long-lived IgE PCs at later timepoints. However, in another study, an increase in the absolute number of IgE PCs was observed early in the immune response in mice with mutations in Blnk, Cd19, or Syk [9]. Analysis of Syk heterozygous cells indicated that the majority of early IgE PCs were not somatically mutated, in contrast to co-isolated IgE GC B cells [9]. These data suggest that early increases in IgE PCs observed in the context of BCR signaling perturbations did not originate from the GC, but rather represented unexplained increases in extrafollicular IgE PC responses. As BCR signaling is known to inhibit CSR [13,14], one possibility to account for these findings is that IgE CSR was increased due to the impaired BCR signaling. Further investigation is therefore needed to elucidate the contributions of BCR signaling to in vivo IgE PC responses.
Tendency towards apoptosis in IgE lymphocytes and the transience of IgE responses
In addition to its role in promoting PC differentiation, the IgE BCR has been implicated in IgE B cell apoptosis, although this remains controversial [4,8,9,15]. In vitro-differentiated mouse IgE B cells were observed to be more apoptotic than IgG1 B cells in some cell culture assays [8,15]. The ectopic expression of the IgE BCR in cultured primary B cells and a cell line was also reported to promote apoptosis [8,15]. However, in another study of mouse B cells, these findings could not be reproduced [9]. Specifically, similar rates of apoptosis were observed in IgE and IgG1 B cells differentiated in vitro, or after ectopic expression of the IgE versus IgG1 BCRs in primary B cells and cell lines. In addition, a recent study of cultured human tonsil B cells also concluded that IgE and IgG1 B cells exhibited similar rates of apoptosis [16].
The reasons for these discrepancies in findings remain unclear, but may be related to technical details of the assays. One approach used in cell culture studies was to induce apoptosis by withdrawing pro-survival signals, including CD40 stimulation, IL-4, and in some cases BAFF [8,9,15]. In this context, antigen-independent signaling of the IgE BCR through the Syk→BLNK→JNK/p38 axis, or alternatively the sequestration of Hax1 from the mitochondria by the IgE BCR intracellular tail, was proposed to promote apoptosis [8,15]. One possibility is that antigen-independent signaling of the IgE BCR mimics stimulation of the IgM BCR, which induces apoptosis in the absence of other signals such as ligation of CD40 [17,18]. However, the antigen-independent signaling of the IgE BCR is substantially weaker than direct BCR stimulation and may more closely reflect the chronic stimulation of the BCR in autoreactive B cells. An alternative model is that rather than being a stronger source of pro-apoptotic signals the IgE BCR is a deficient source of pro-survival signals. B cells require membrane BCR expression and resulting tonic signaling via PI3K for survival [19]. The IgE BCR may be deficient in producing these survival signals, either due to its low surface expression (reviewed in depth below) or due to qualitative impairments in its pro-survival signaling. We speculate that specific differences in the culture conditions and the nature of the cell lines used in the above studies may be responsible for the different outcomes observed regarding the IgE BCR and apoptosis. It is unclear the degree to which these findings in cell culture are applicable to IgE B cells in vivo, where a milieu of complex signals are present. One attempt to address this question has been through the analysis of GC B cells ex vivo, however again conflicting findings have emerged. Specifically, one group found that IgE GC B cells exhibited higher rates of apoptosis than IgG1 GC B cells [4], whereas another group could not reproduce these findings [9]. Nevertheless, the low amount of apoptosis of GC B cells detected ex vivo is a tiny fraction of the apoptosis that occurs in vivo, where it is thought that up to half of all GC B cells undergo apoptosis every 6 h [20]. Thus, more studies are needed to determine the extent to which the IgE BCR may promote the apoptosis of IgE B cells in vivo versus promoting their differentiation into short-lived PCs or otherwise rendering them less competitive within the GC, as we discuss further below.
Although conflicting evidence has been obtained regarding the predisposition of IgE B cells towards apoptosis, greater consensus has been achieved regarding the short lifespan of the majority of IgE PCs. For instance, in a model of IgE and IgG1 hypergammaglobulinemia, the overwhelming majority of IgE PCs were proliferating, unlike other PCs, suggesting that the IgE PCs were predominantly short-lived plasmablasts [15]. Indeed, markedly increased numbers of IgE PCs relative to IgG1 PCs were observed in Bcl2 transgenic mice after immunization, indicating that IgE PCs are normally restrained by poor survival [3]. Less data exists on human IgE PCs, but culture studies of human tonsil B cells also showed elevated rates of apoptosis among IgE PCs [16]. More research is needed to understand how IgE PCs are constrained to short-lived fates and what drives their apoptosis, as well as how long-lived IgE PCs might arise pathologically in allergy.
Self-limited surface expression of the IgE BCR on IgE B cells
Another unusual feature of the IgE BCR is its low surface expression on GC B cells [4,9] (Figure 1). IgE GC B cells exhibited reduced antigen-dependent BCR signaling compared with IgG1 GC B cells when stimulated ex vivo [8]. In vivo, IgE GC B cells captured, processed, and presented fewer antigen peptide MHC complexes compared with IgG1 GC B cells [9]. As a result, IgE GC B cells may compete poorly for T cell help in the GC, which is critical for selection. Consistent with this model, IgE GC B cells progressed more slowly through the cell cycle than IgG1 GC B cells [9]. Other evidence of enfeebled function of the IgE BCR comes from studies of mice whose B cells were pre-class-switched to either IgE (IgHε/ε) or IgG1 (IgHγ1/γ1) before development [12]. In the context of a pre-rearranged light chain, IgHε/ε B cells had massively reduced surface BCR expression and failed to mount an antigen-specific B cell response when immunized, whereas their IgHγ1/γ1 counterparts did.
Figure 1: Proposed mechanisms and consequences of low surface BCR expression on IgE B cells.
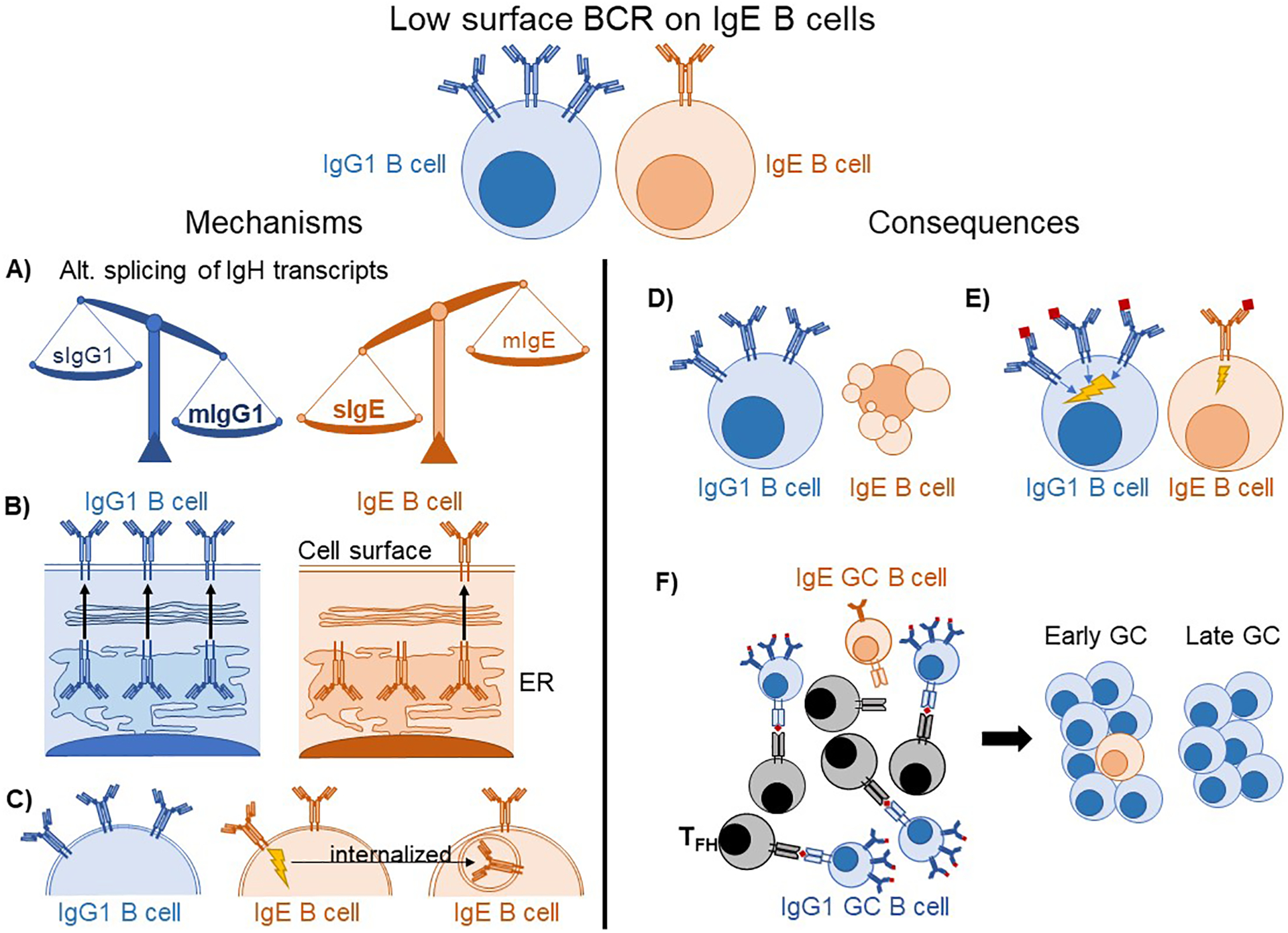
(A–C) Potential mechanisms explaining low IgE BCR surface expression. (A) IgG1 B cells favor the mIg spliceoform of IgH, whereas IgE B cells favor the sIg spliceoform, resulting in fewer mIgE transcripts available for the translation of IgE BCR molecules. (B) The IgE BCR is retained in the ER after translation rather than being exported to the cell surface. (C) Antigen-independent signaling of the IgE BCR leads to its internalization, reducing surface BCR levels. (D–F) Putative consequences of low BCR expression on IgE B cells. (D) IgE B cells undergo apoptosis due to insufficient BCR expression. (E) IgE B cells have weaker antigen-dependent BCR signaling than IgG1 B cells. (F) IgE GC B cells are less able to capture and present antigens to GC TFH, causing them to be progressively out-competed over time.
There are multiple non-mutually exclusive models that might explain the low surface BCR expression in IgE GC B cells (Figure 1). One model is that these cells have low expression of transcripts encoding the IgE BCR. Surface and membrane forms of each Ig isotype are expressed through alternative splicing and polyadenylation. IgG1 B cells primarily express the membrane form of the BCR (mIgG1) whereas IgG1 PCs primarily express the secreted form (sIgG1). IgE B cells, however, are unusual in that they express relatively higher amounts of sIgE than mIgE, and both sIgE and mIgE are greatly upregulated in IgE PCs [4,12,21]. The poor expression of mIgE in IgE B cells may in part be due to an inefficient polyadenylation sequence downstream of the M2 exon [21], although a more classical polyadenylation sequence is located further downstream.
A second model is that mIgE is retained in the endoplasmic reticulum (ER). IgE, unlike IgG1, requires assembly with Igα/β to reach the cell surface [9,22], which could be limiting in some contexts. Recent structural analysis revealed the mIg isotype-specific (migis) segment, a region unique to each isotype membrane BCR that protrudes from the plasma membrane into the extracellular space, was a critical determinant of whether the BCR must associate with Igα/β to reach the cell surface [9]. The IgE migis region was also found to be critical for the antigen-independent signaling activity of the IgE BCR [8,9], leading us to speculate that some characteristics of its association with Igα/β mimic an antigen-binding state. The migis of IgE was also reported to promote enhanced interaction with CD19 [8]. Interestingly, humans and primates have two splice variants of the IgE BCR, one isoform of which encodes a longer M1’ segment of the extracellular membrane proximal domain (EMPD) that includes the migis. This long isoform was associated with retention of the IgE BCR in the ER [23]. Changes in the relative expression of the long versus short isoforms have been reported during IgE PC differentiation in human tonsil B cell cultures [10].
A third possible model to explain low surface IgE BCR expression is that constitutive, antigen-independent signaling induces its internalization, analogous to the downmodulation of surface IgM in autoreactive B cells [24]. Evidence for increased internalization of mIgE was provided by incubating cells with fluorescently-labeled antibodies at different temperatures followed by flow cytometric analysis [15]. Taken together, multiple mechanisms may result in low surface BCR expression on IgE B cells, leading to functional consequences (Figure 1) that impair the ability of these cells to compete in GCs.
Implications for IgE memory
Long term IgE production could theoretically arise from either long-lived IgE PCs or from quiescent memory B cells. The peculiar features of the IgE BCR described above suggest that IgE B cells are unlikely to be capable of differentiating into quiescent memory B cells. Indeed, clear direct evidence of IgE memory B cells in normal murine immune responses is lacking [4,25], and rigorous attempts at their detection in humans have failed [26]. Instead, the increased production of IgE that occurs during recall responses has been proposed to originate from IgG1 memory B cells [4,25,27,28]. A recent elegant study characterized the role of distinct subpopulations of IgG1 memory B cells in low and high affinity IgE recall responses [28]. Providing evidence that recall IgE responses involve sequential CSR from IgG1 memory B cells, mice in which the extracellular domains of IgG1 were swapped with those of IgE, thereby making the IgG1 BCR more like the IgE BCR, had defective IgE recall responses [25]. Interestingly, however, the intracellular tail of the IgE BCR appears to be required for optimal IgE antibody production in recall responses, suggesting that this molecular region may be important for the function of the IgE BCR in activated IgE B cells and/or PCs [29,30], as has been observed for the IgG1 BCR [31]. A major remaining question is what types of intrinsic or extrinsic signals determine whether memory IgG1 B cells undergo CSR to IgE in a recall response, and whether other types of memory B cells may also contribute. In addition, further assessment is needed of the contribution of sequential CSR to IgE production in IgE-mediated allergic diseases.
Extrinsic regulation of IgE B cell responses
TFH as the major direct extrinsic regulators of IgE responses
IL-4 has long been appreciated as critical to IgE responses [32]. In classical studies of CD4 T cell differentiation in cell culture, TH1 cells and TH2 cells were found to produce IFN-γ versus IL-4, which inhibited or promoted IgE production, respectively [33]. Consequently, it was presumed for many years that TH2 cells interacted with B cells and were the cellular source of IL-4 that induced IgE CSR. However, over the past two decades, T follicular helper cells (TFH) have emerged as the major subset of T cells that provide help to B cells [34]. Contemporaneously, early IL-4 reporter studies found that IL-4 production in the lymph node was almost exclusive to TFH [35,36]. It was later found that eliminating TFH-derived IL-4 abolished IgE production whereas deletion of the TH2 master transcription factor GATA3 had a negligible impact on IgE production [37,38]. Recently, an impressive division of labour between TH2 and TFH cells was revealed in mouse models of allergic airway inflammation [39]. TH2 cells were positioned in the lung and produced local IL-4, IL-5, and IL-13, while TFH localized to the mediastinal lymph node where they drove IgE antibody production. Together, this work has established TFH as the major cellular source of IL-4 for IgE CSR (Figure 2).
Figure 2: The opposing effects of IL-4 and IL-21 on IgE CSR are tuned by the strength of CD40 ligation.
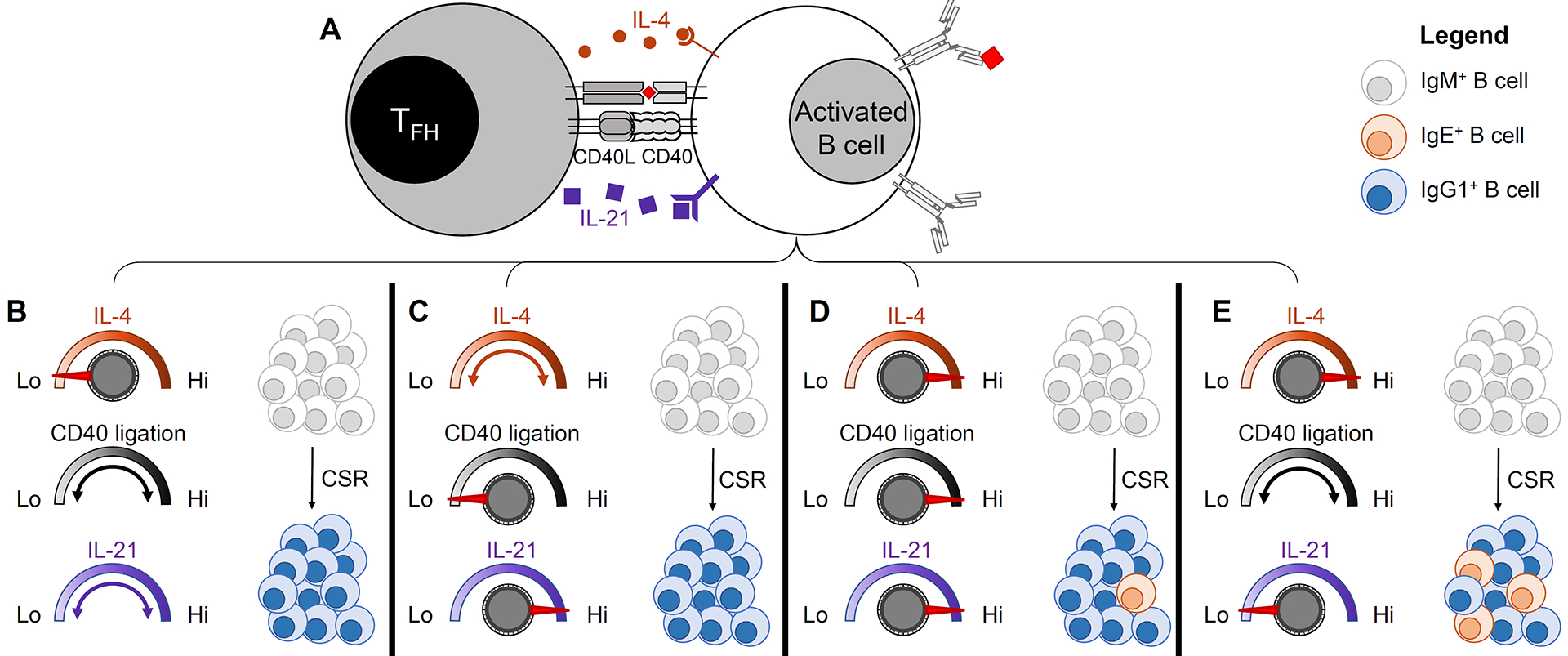
(A) Cognate interaction between a T follicular helper cell (TFH) and an activated B cell. The B cell has presented antigen to the T cell and is receiving T cell help in the form of IL-4, CD40 ligation, and IL-21. (B–E) represent potential outcomes of the T-B interaction. (B) IgE CSR is disfavored in the context of low amounts of IL-4, regardless of the strength of CD40 ligation and the amount of IL-21. (C) IgE CSR is disfavored in the context of high amounts of IL-21 when CD40 ligation is weak, regardless of the amount of IL-4 present. (D) Some IgE CSR may occur despite high amounts of IL-21 in the context of high amounts of IL-4 and strong CD40 ligation. (E) IgE CSR is strongly promoted in the context of high amounts of IL-4 and minimal amounts of IL-21, across a range of the strength of CD40 ligation.
Recent work highlights that TFH are also critical negative regulators of IgE CSR. Although IFN-γ was considered to inhibit IgE responses, minimal changes in IgE responses were observed in Ifng gene-targeted mice [40]. Instead, the cytokine IL-21 appears to be a critical negative regulator of IgE in a broad range of immune responses in mice [40]. TFH-derived IL-21 inhibits IgE CSR by binding to the IL-21 receptor on B cells which then transduces signals through the phosphorylation of STAT3 [40] (Figure 2). This finding is consistent with the early studies of IL-21 receptor (IL-21R)-deficient mice [41,61], the observation of high serum IgE in IL-21-deficient and IL-21R-deficient patients [42], and the detection of STAT3 mutations in a significant fraction of human patients with hyper-IgE syndrome (HIES) [43]. However, these findings were in contrast to some previous human cell culture studies, in which IL-21 augmented IgE production [44]. These studies measured secreted IgE as a proxy for IgE CSR, but IgE secretion would also be affected by B cell proliferation and PC differentiation. Indeed, IL-21 potently stimulates both proliferation and PC differentiation of human B cells [44]. Recent studies of both mouse and human B cells additionally found that the effect of IL-21 on IgE CSR depended on the relative strength of CD40 ligation [40]. In particular, IL-21 inhibited IgE CSR in the context of limiting amounts of CD40 stimulation, whereas this effect was abrogated in the context of strong CD40 stimulation [40] (Figure 2). These results suggest that past discordant findings on the inhibitory effects of IL-21 on IgE class-switching are likely related to the strength of CD40 stimulation. In addition to the role of CD40, the relative balance of IL-4 and IL-21 was recently found to be an important determinant of IgE versus IgG1 CSR in both mouse and human B cells [40].
Recent work has identified a specialized subset of TFH cells that, in contrast to most TFH, are capable of expressing the cytokine IL-13 [45–47]. These cells have been variably referred to as TFH2 or TFH13 to denote similarities to TH2 cells or signify IL-13 production. A portion of this IL-13-competent subset was found to co-express GATA3 and Bcl-6 while also producing IL-4 [45–47]. One group reported that the presence of this subset was positively associated with high-affinity IgE titers as detected by ELISA [45]. While intriguing, the interpretation of these findings is complicated by the lack of data on the ratio of high-affinity to total antigen-binding antibody for each sample, as is standard practice. In addition, ELISA measurements of antigen-binding IgE can be significantly impacted by competition with antigen-binding IgG, which is much more abundant [48]. Thus, these assays are sensitive to differences in the amount of antigen-binding IgG in addition to the amount of antigen-binding IgE [48]. The absence of TFH13 cells in Il13Cre Bcl6flox/flox mice was associated with reduced detection of high-affinity IgE by ELISA and reduced passive cutaneous anaphylaxis [45]. This was provided as evidence for a critical role for TFH13 cells and supported the conclusion that IL-13 is involved in high affinity IgE production [45]. Alternatively, these results could also reflect the loss of TFH that were derived from T cells that previously produced IL-13 (e.g. former TH2 cells), or could be related to the finding that the TFH13 cells exhibited reduced production of IL-21 compared with other TFH cells [45]. Notably, IL-21 suppressed sequential CSR of IgG1 GC B cells to IgE ex vivo [2] and immunized IgG1-deficient mice have normal IgE levels but impaired IgE affinity [27]. Further work is needed to determine the precise contributions of IL-13, IL-21, and TFH2/TFH13 cells to IgE production and affinity in allergic immune responses. Future studies would also benefit from Ig variable region sequencing to determine the frequency of high affinity somatic mutations.
In addition to TFH, other T cell subsets regulate IgE production. FoxP3-expressing T regulatory cells (Treg) are also important contributors to the control of IgE responses [49,50]. In general, T follicular regulatory (TFR) cells that co-express Bcl-6 and FoxP3 have been shown to regulate B cell responses [34]. Genetic strategies to deplete TFR cells based on the co-expression of these molecules have paradoxically been reported to result in both augmented and reduced IgE responses [46,51,52]. Interestingly IL-10 made by TFR cells was reported to promote peanut-specific IgE and IgG1 production [51], yet another study found that IL-10-deficiency had a negligible impact upon IgE levels [40]. Meanwhile, in human tonsils, the abundance of an IL-10-producing CD25+ follicular T cell subset was found to inversely correlate with circulating IgE levels, whereas the abundance of FoxP3+ follicular T cells was found to positively correlate with circulating IgE levels [53]. Yet, recent work suggests TFR cells also produce neuritin, a neuropeptide, that inhibits IgE production [52]. An improved understanding of the interacting partners of follicular T cell subsets and the molecular mechanism by which the molecules produced by these cells augment/inhibit IgE CSR may help to reconcile these conflicting findings.
Allergic disease as an outcome of IgE dysregulation
The intrinsic and extrinsic regulatory mechanisms described in this review for constraining IgE responses likely serve to limit the ability of IgE to promote allergic reactions, which in extreme forms, such as systemic anaphylaxis in response to peanuts, can be fatal. This notion is compatible with observations that the regulation of IgE responses results in the limited generation of IgE switched B cells and the transient production of IgE with low to moderate affinity for antigen. However, even in the context of constrained IgE production, IgE antibodies coat the surface of FcεR1-expressing cells where they are retained for extended periods of time [54]. In this setting, high concentrations of antigen may be bound co-operatively by polyclonal IgE antibodies of low-to-moderate affinity, thereby inducing the degranulation of mast cells and/or basophils proximal to the site of exposure. While the potential physiological roles of IgE in healthy immunity remain an area of active discussion [55], it seems likely that IgE evolved to elicit local hypersensitivity responses in this manner. In contrast, the sustained production of allergen-reactive IgE and the production of high-affinity IgE have been reported in some instances of human allergic disease and likely contribute to the tendency to develop systemic anaphylaxis [56]. One possible explanation for the emergence of atypical IgE responses in allergic individuals is that specific allergens or conditions of exposure can ‘break the rules’ of IgE responses and permit high-affinity and/or sustained IgE production, and that these types of exposures associate with the development of allergy. Supporting the plausibility of this notion, it was reported recently that extended repetitive immunization with house dust mite, as a model of continuous aeroallergen exposure in asthma, produced a population of long-lived IgE PCs in the bone marrow that remained for up to 32 weeks without further immunization [57]. While the underlying mechanisms are unclear, this study provides important clues to understand what factors may lead to the differentiation of long-lived rather than short-lived IgE PC during allergic sensitization.
Another possibility is that, rather than ‘breaking the rules’ of IgE regulation, allergic responses are initiated at tissue sites where these rules are less enforced. As discussed above, TFH-derived IL-21 appears to be a major negative regulator of IgE CSR. A tissue microenvironment with a low abundance of IL-21-producing TFH may therefore be more favorable for IgE CSR. This is reminiscent of a hypothesis that potent allergens are those which induce IgE responses outside of GCs [58] and reports of local IgE CSR in mucosal tissues [59]. Supporting the plausibility of this idea, abundant IgE clones were recently identified in the stomach and duodenum of peanut-allergic individuals [60]. The numbers of these clones correlated with serum IgE, implying they represent an important fraction of IgE-producing cells in these individuals. Additionally, the clones were highly restricted to individual tissue sites, suggesting that they derived from local activation events rather than emigrating from lymph nodes, where IL-21 producing TFH would be more abundant. Determining the extent and mechanism by which specific tissue niches could support B cell activation and promote IgE class-switching, and whether this differs in allergic versus non-allergic individuals, would be of great interest in future studies.
Conclusion
Recent work has provided key insights into the regulatory mechanisms that limit the initial generation of IgE B cells and control their fate. The IgE BCR has emerged as a critical B cell-intrinsic regulator of IgE responses through its antigen-independent signaling and low surface expression, predisposing IgE B cells towards short-lived PC differentiation and/or apoptosis, and limiting the ability of IgE B cells to compete in GCs. Collectively, these pathways largely prevent IgE lymphocytes from developing into long-lived memory B cells and PCs. Many of the unique features of the IgE BCR have been associated with one particular structural domain: the EMPD, yet much remains to be learned about how this region interacts with other parts of the IgE molecule and associated coreceptors.
While intrinsic regulation governs the fate of IgE-expressing cells, extrinsic regulation controls their formation. Recent work identifies TFH as the critical source of IL-4 for IgE CSR. TFH are also powerful negative regulators of IgE CSR due to their production of IL-21. IgE responses also appear to be regulated by TFR cells and an IL-13 producing TFH subtype may be associated with the production of high-affinity serum IgE. Finally, recent work has raised the question of whether unique settings of allergen sensitization (e.g. particular allergens, conditions of exposure or tissue sites) can induce IgE responses with qualitatively distinct properties, such as enhanced affinity or longevity, that defy the norms of IgE production and may thereby facilitate allergic pathogenesis.
Acknowledgments
Relevant research in this laboratory on IgE B cells was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01AI130470 and R21AI154335), the Pew Charitable Trusts (C.D.C.A. is a Pew Scholar in the Biomedical Sciences), the Weston Havens Foundation, the UCSF Sandler Asthma Basic Research Center, and the UCSF Cardiovascular Research Institute. A.K.W.V. was supported by a Doctoral Foreign Study Award from the Canadian Institutes of Health Research (DFD-170769). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Declaration of Interests
C.D.C.A. is on the Scientific Advisory Board for Walking Fish Therapeutics. C.D.C.A.’s spouse is an employee and shareholder of Bristol Myers Squibb. The authors declare no other competing financial interests.
References
- 1.Geha RS, Jabara HH, Brodeur SR: The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol 2003, 3:721–732. [DOI] [PubMed] [Google Scholar]
- 2.Erazo A, Kutchukhidze N, Leung M, Christ AP, Urban JF Jr., Curotto de Lafaille MA, Lafaille JJ: Unique maturation program of the IgE response in vivo. Immunity 2007, 26:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Z, Sullivan BM, Allen CD: Fluorescent in vivo detection reveals that IgE(+) B cells are restrained by an intrinsic cell fate predisposition. Immunity 2012, 36:857–872. [DOI] [PubMed] [Google Scholar]
- 4.He JS, Meyer-Hermann M, Xiangying D, Zuan LY, Jones LA, Ramakrishna L, de Vries VC, Dolpady J, Aina H, Joseph S, et al. : The distinctive germinal center phase of IgE+ B lymphocytes limits their contribution to the classical memory response. J Exp Med 2013, 210:2755–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Talay O, Yan D, Brightbill HD, Straney EEM, Zhou M, Ladi E, Lee WP, Egen JG, Austin CD, Xu M, et al. : Reply to “On the differentiation of mouse IgE+ cells”. Nat Immunol 2012, 13:623–624.22713817 [Google Scholar]
- 6.Talay O, Yan D, Brightbill HD, Straney EE, Zhou M, Ladi E, Lee WP, Egen JG, Austin CD, Xu M, et al. : IgE(+) memory B cells and plasma cells generated through a germinal-center pathway. Nat Immunol 2012, 13:396–404. [DOI] [PubMed] [Google Scholar]
- 7.Talay O, Yan D, Brightbill HD, Straney EEM, Zhou M, Ladi E, Lee WP, Egen JG, Austin CD, Xu M, et al. : Addendum: IgE+ memory B cells and plasma cells generated through a germinal-center pathway. Nat Immunol 2013, 14:1302–1304. [DOI] [PubMed] [Google Scholar]
- 8.Haniuda K, Fukao S, Kodama T, Hasegawa H, Kitamura D: Autonomous membrane IgE signaling prevents IgE-memory formation. Nat Immunol 2016, 17:1109–1117. [DOI] [PubMed] [Google Scholar]; ** This report established that the IgE BCR exhibits antigen-independent signaling that promotes PC differentiation and apoptosis of IgE B cells through specific BCR signaling pathways. These features of the IgE BCR depended on the IgE migis region. Deficiency in Blnk permitted the generation of long-lived IgE PC and memory B cell responses. See also Yang et al. 2016.
- 9.Yang Z, Robinson MJ, Chen X, Smith GA, Taunton J, Liu W, Allen CD: Regulation of B cell fate by chronic activity of the IgE B cell receptor. eLife 2016, 5:e21238. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study showed that the IgE BCR exhibits antigen-independent signaling that promotes PC differentiation, dependent on the IgE migis region. In contrast to Haniuda et al. 2016, increased rates of apoptosis due to IgE BCR expression were not observed. Low surface IgE BCR expression was found to contribute to reduced antigen uptake and presentation by IgE GC B cells, which had slower cell cycle times than their IgG1 counterparts.
- 10.Ramadani F, Bowen H, Upton N, Hobson PS, Chan YC, Chen JB, Chang TW, McDonnell JM, Sutton BJ, Fear DJ, et al. : Ontogeny of human IgE-expressing B cells and plasma cells. Allergy 2017, 72:66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shinnakasu R, Kurosaki T: Regulation of memory B and plasma cell differentiation. Curr Opin Immunol 2017, 45:126–131. [DOI] [PubMed] [Google Scholar]
- 12.Tong P, Granato A, Zuo T, Chaudhary N, Zuiani A, Han SS, Donthula R, Shrestha A, Sen D, Magee JM, et al. : IgH isotype-specific B cell receptor expression influences B cell fate. Proc Natl Acad Sci U S A 2017, 114:E8411–E8420. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study generated mice pre-class switched to IgE or IgG1. By crossing these mice to a strain with a pre-arranged light chain, it was demonstrated that IgE B cells had altered sIg/mIg IgH transcript ratios and were refractory to immunization.
- 13.Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, Rickert RC: Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity 2006, 25:545–557. [DOI] [PubMed] [Google Scholar]
- 14.Jabara HH, Chaudhuri J, Dutt S, Dedeoglu F, Weng Y, Murphy MM, Franco S, Alt FW, Manis J, Geha RS: B-cell receptor cross-linking delays activation-induced cytidine deaminase induction and inhibits class-switch recombination to IgE. J Allergy Clin Immunol 2008, 121:191–196 e192. [DOI] [PubMed] [Google Scholar]
- 15.Laffleur B, Duchez S, Tarte K, Denis-Lagache N, Peron S, Carrion C, Denizot Y, Cogne M: Self-Restrained B Cells Arise following Membrane IgE Expression. Cell Rep 2015, 10:900–909. [DOI] [PubMed] [Google Scholar]
- 16.Ramadani F, Bowen H, Gould HJ, Fear DJ: Transcriptional Analysis of the Human IgE-Expressing Plasma Cell Differentiation Pathway. Front Immunol 2019, 10:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsubata T, Wu J, Honjo T: B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature 1993, 364:645–648. [DOI] [PubMed] [Google Scholar]
- 18.Finkelman FD, Holmes JM, Dukhanina OI, Morris SC: Cross-linking of membrane immunoglobulin D, in the absence of T cell help, kills mature B cells in vivo. J Exp Med 1995, 181:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K: PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, et al. : The microanatomic segregation of selection by apoptosis in the germinal center. Science 2017, 358:eaao2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karnowski A, Achatz-Straussberger G, Klockenbusch C, Achatz G, Lamers MC: Inefficient processing of mRNA for the membrane form of IgE is a genetic mechanism to limit recruitment of IgE-secreting cells. Eur J Immunol 2006, 36:1917–1925. [DOI] [PubMed] [Google Scholar]
- 22.Venkitaraman AR, Williams GT, Dariavach P, Neuberger MS: The B-cell antigen receptor of the five immunoglobulin classes. Nature 1991, 352:777–781. [DOI] [PubMed] [Google Scholar]
- 23.Vanshylla K, Opazo F, Gronke K, Wienands J, Engels N: The extracellular membrane-proximal domain of membrane-bound IgE restricts B cell activation by limiting B cell antigen receptor surface expression. Eur J Immunol 2018, 48:441–453. [DOI] [PubMed] [Google Scholar]; * These authors investigated the short and long spliceoforms of human mIgE in vitro and found that the long isoform of mIgE was preferentially retained in the ER and had diminished expression on the plasma membrane.
- 24.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. : Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988, 334:676–682. [DOI] [PubMed] [Google Scholar]
- 25.Turqueti-Neves A, Otte M, Schwartz C, Schmitt ME, Lindner C, Pabst O, Yu P, Voehringer D: The Extracellular Domains of IgG1 and T Cell-Derived IL-4/IL-13 Are Critical for the Polyclonal Memory IgE Response In Vivo. PLoS Biol 2015, 13:e1002290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jimenez-Saiz R, Ellenbogen Y, Bruton K, Spill P, Sommer DD, Lima H, Waserman S, Patil SU, Shreffler WG, Jordana M: Human BCR analysis of single-sorted, putative IgE(+) memory B cells in food allergy. J Allergy Clin Immunol 2019, 144:336–339 e336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong H, Dolpady J, Wabl M, Curotto de Lafaille MA, Lafaille JJ: Sequential class switching is required for the generation of high affinity IgE antibodies. J Exp Med 2012, 209:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He JS, Subramaniam S, Narang V, Srinivasan K, Saunders SP, Carbajo D, Wen-Shan T, Hidayah Hamadee N, Lum J, Lee A, et al. : IgG1 memory B cells keep the memory of IgE responses. Nat Commun 2017, 8:641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmitt MER, Lutz J, Haase P, Bosl MR, Wienands J, Engels N, Voehringer D: The B-cell antigen receptor of IgE-switched plasma cells regulates memory IgE responses. J Allergy Clin Immunol 2020, 146:642–651 e645. [DOI] [PubMed] [Google Scholar]
- 30.Achatz G, Nitschke L, Lamers MC: Effect of Transmembrane and Cytoplasmic Domains of IgE on the IgE Response. Science 1997, 276:409–411. [DOI] [PubMed] [Google Scholar]
- 31.Wienands J, Engels N: The Memory Function of the B Cell Antigen Receptor. Curr Top Microbiol Immunol 2016, 393:107–121. [DOI] [PubMed] [Google Scholar]
- 32.Finkelman FD, Holmes J, Katona IM, Urban JF Jr., Beckmann MP, Park LS, Schooley KA, Coffman RL, Mosmann TR, Paul WE: Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol 1990, 8:303–333. [DOI] [PubMed] [Google Scholar]
- 33.Coffman RL: Origins of the T(H)1-T(H)2 model: a personal perspective. Nat Immunol 2006, 7:539–541. [DOI] [PubMed] [Google Scholar]
- 34.Crotty S: T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50:1132–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reinhardt RL, Liang HE, Locksley RM: Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 2009, 10:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King IL, Mohrs M: IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J Exp Med 2009, 206:1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harada Y, Tanaka S, Motomura Y, Harada Y, Ohno S, Ohno S, Yanagi Y, Inoue H, Kubo M: The 3’ enhancer CNS2 is a critical regulator of interleukin-4-mediated humoral immunity in follicular helper T cells. Immunity 2012, 36:188–200. [DOI] [PubMed] [Google Scholar]
- 38.Liang HE, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM: Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat Immunol 2011, 13:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi T, Iijima K, Dent AL, Kita H: Follicular helper T cells mediate IgE antibody response to airborne allergens. J Allergy Clin Immunol 2017, 139:300–313 e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Z, Wu CM, Targ S, Allen CDC: IL-21 is a broad negative regulator of IgE class switch recombination in mouse and human B cells. J Exp Med 2020, 217:e20190472. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study reconciled past conflicting results in mice and humans to identify IL-21 as the major cytokine suppressor of IgE CSR. Furthermore, it was shown that TFH are the primary source of IL-21, and that IL-21 inhibits IgE CSR in B cells by signaling through the IL-21 receptor and STAT3. IgE CSR was found to be regulated by the relative amounts of IL-21 versus IL-4, and the strength of CD40 signaling.
- 41.Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC 3rd, Liu C, Schwartzberg PL, Leonard WJ: A critical role for IL-21 in regulating immunoglobulin production. Science 2002, 298:1630–1634. [DOI] [PubMed] [Google Scholar]
- 42.Kotlarz D, Zietara N, Milner JD, Klein C: Human IL-21 and IL-21R deficiencies: two novel entities of primary immunodeficiency. Curr Opin Pediatr 2014, 26:704–712. [DOI] [PubMed] [Google Scholar]
- 43.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. : Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007, 448:1058–1062. [DOI] [PubMed] [Google Scholar]
- 44.Moens L, Tangye SG: Cytokine-Mediated Regulation of Plasma Cell Generation: IL-21 Takes Center Stage. Front Immunol 2014, 5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, Joseph J, Gertie JA, Xu L, Collet MA, et al. : Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science 2019, 365:eaaw6433. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study provided a detailed analysis of a subset of TFH, dubbed TFH13, that, similar to previously described TFH2 cells, expressed GATA3 and IL-13. In wild-type mice, these cells were only present following particular immunizations with allergens and were associated with high-affinity IgE production and reduced IL-21 levels.
- 46.Clement RL, Daccache J, Mohammed MT, Diallo A, Blazar BR, Kuchroo VK, Lovitch SB, Sharpe AH, Sage PT: Follicular regulatory T cells control humoral and allergic immunity by restraining early B cell responses. Nat Immunol 2019, 20:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim CJ, Lee CG, Jung JY, Ghosh A, Hasan SN, Hwang SM, Kang H, Lee C, Kim GC, Rudra D, et al. : The Transcription Factor Ets1 Suppresses T Follicular Helper Type 2 Cell Differentiation to Halt the Onset of Systemic Lupus Erythematosus. Immunity 2018, 49:1034–1048 e1038. [DOI] [PubMed] [Google Scholar]
- 48.Lehrer SB, Reish R, Fernandes J, Gaudry P, Dai G, Reese G: Enhancement of murine IgE antibody detection by IgG removal. J Immunol Methods 2004, 284:1–6. [DOI] [PubMed] [Google Scholar]
- 49.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ: Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 2008, 29:114–126. [DOI] [PubMed] [Google Scholar]
- 50.Wing JB, Ise W, Kurosaki T, Sakaguchi S: Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity 2014, 41:1013–1025. [DOI] [PubMed] [Google Scholar]
- 51.Xie MM, Chen Q, Liu H, Yang K, Koh B, Wu H, Maleki SJ, Hurlburt BK, Cook-Mills J, Kaplan MH, et al. : T follicular regulatory cells and IL-10 promote food antigen-specific IgE. J Clin Invest 2020, 130:3820–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez-Figueroa P, Roco JA, Papa I, Nunez Villacis L, Stanley M, Linterman MA, Dent A, Canete PF, Vinuesa CG: Follicular regulatory T cells produce neuritin to regulate B cells. Cell 2021, 184:1775–1789.e19. [DOI] [PubMed] [Google Scholar]
- 53.Canete PF, Sweet RA, Gonzalez-Figueroa P, Papa I, Ohkura N, Bolton H, Roco JA, Cuenca M, Bassett KJ, Sayin I, et al. : Regulatory roles of IL-10-producing human follicular T cells. J Exp Med 2019, 216:1843–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jimenez-Saiz R, Chu DK, Mandur TS, Walker TD, Gordon ME, Chaudhary R, Koenig J, Saliba S, Galipeau HJ, Utley A, et al. : Lifelong memory responses perpetuate humoral TH2 immunity and anaphylaxis in food allergy. J Allergy Clin Immunol 2017, 140:1604–1615 e1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palm NW, Rosenstein RK, Medzhitov R: Allergic host defences. Nature 2012, 484:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Croote D, Darmanis S, Nadeau KC, Quake SR: High-affinity allergen-specific human antibodies cloned from single IgE B cell transcriptomes. Science 2018, 362:1306–1309. [DOI] [PubMed] [Google Scholar]
- 57.Asrat S, Kaur N, Liu X, Ben LH, Kajimura D, Murphy AJ, Sleeman MA, Limnander A, Orengo JM: Chronic allergen exposure drives accumulation of long-lived IgE plasma cells in the bone marrow, giving rise to serological memory. Sci Immunol 2020, 5:eaav8402. [DOI] [PubMed] [Google Scholar]
- 58.Aalberse RC, Platts-Mills TA: How do we avoid developing allergy: modifications of the TH2 response from a B-cell perspective. J Allergy Clin Immunol 2004, 113:983–986. [DOI] [PubMed] [Google Scholar]
- 59.Gould HJ, Takhar P, Harries HE, Durham SR, Corrigan CJ: Germinal-centre reactions in allergic inflammation. Trends Immunol 2006, 27:446–452. [DOI] [PubMed] [Google Scholar]
- 60.Hoh RA, Joshi SA, Lee JY, Martin BA, Varma S, Kwok S, Nielsen SCA, Nejad P, Haraguchi E, Dixit PS, et al. : Origins and clonal convergence of gastrointestinal IgE(+) B cells in human peanut allergy. Sci Immunol 2020, 5:eaay4209. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Using next-generation sequencing and GI biopsies, this investigation revealed the presence of IgE PCs in the stomach and duodenum of peanut-allergic patients but not healthy individuals. Further analysis determined that these cells correlated with serum IgE and were derived from local class-switching events.
- 61.Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, Johnson KA, Witek JS, Senices M, Konz RF et al. : IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity 2002, 16:559–569 [DOI] [PubMed] [Google Scholar]