

Abstract
Purpose:
Up to 30% of patients with Brugada Syndrome (BrS) carry loss-of-function (LoF) variants in the cardiac sodium channel gene SCN5A. Recent studies suggested that the SCN5A protein product NaV1.5 can dimerize and exert dominant negative effects. Here we sought to explore the generality of NaV1.5 dominant negative effects and their clinical severity.
Methods:
We identified 35 LoF variants (<10% of wildtype [WT] peak current) and 15 partial LoF variants (10-50% of WT peak current) that we assessed for dominant negative effects. SCN5A variants were studied in HEK293T cells alone or in heterozygous co-expression with WT SCN5A using automated patch clamp. To assess clinical risk, we compared the prevalence of dominant negative vs. putative haploinsufficient (frameshift/splice/nonsense) variants in a BrS consortium and the gnomAD population database.
Results:
In heterozygous expression with WT, 32/35 LoF and 6/15 partial LoF variants showed reduction to <75% of WT-alone peak current, demonstrating a dominant negative effect. Individuals with dominant negative LoF variants had an elevated disease burden compared to individuals with putative haploinsufficient variants (2.7-fold enrichment in BrS cases, p=0.019).
Conclusions:
Most SCN5A missense LoF variants exert a dominant negative effect. This class of variant confers an especially high burden of BrS.
Introduction
Brugada Syndrome (BrS) is a clinical arrhythmia syndrome with characteristic EKG changes in the absence of underlying structural heart abnormalities1. While often asymptomatic or clinically unrecognized, sudden cardiac death (SCD) due to ventricular tachyarrhythmia can be the sentinel manifestation. Up to 30% of BrS patients have heterozygous loss-of-function (LoF) variants in the cardiac sodium channel gene SCN5A, which encodes the channel protein NaV1.52. A recent evaluation by ClinGen asserted that SCN5A was the only gene with strong evidence for Mendelian associations with BrS3. LoF SCN5A variants are also associated with other arrhythmias including sick sinus syndrome4 and progressive cardiac conduction disease5.
Hundreds of LoF variants within SCN5A have been reported in the literature and in ClinVar across multiple variant classes including missense, nonsense, splice-altering, and frameshift/premature truncation2,6–8. SCN5A encodes a channel with 4 transmembrane domains, each consisting of 6 transmembrane segments9. NaV1.5 has traditionally been thought to function as a monomer; however, a recent study indicated that NaV1.5 can form dimers with coupled intracellular trafficking and/or gating at the plasma membrane10. Similar to variants in established multimeric proteins that can generate dominant negative effects, several missense SCN5A variants with dominant negative effects on trafficking or coupled gating at the cell surface have been reported in vitro and in vivo11–13. However, the dominant negative behavior of most of the approximately 40 functionally-validated LoF missense variants in SCN5A has not been tested14. Moreover, the degree of dominant negative effects among partial LoF missense variants has not been evaluated.
Variable penetrance is a hallmark of pathogenic BrS variants, and the extent to which distinct pathogenic mechanisms (e.g., dominant negative vs haploinsufficiency) contribute to this effect is unknown. Large cohort studies and variant curation efforts provide datasets of SCN5A variants associated with BrS cases2,6,8. In addition, large population cohorts such as gnomAD provide sets of individuals more likely representing putative controls15. Together, these datasets enable the comparison of BrS disease risk among different variant classes.
Here, we study the prevalence of the dominant negative effect among SCN5A LoF and partial LoF missense variants. We use case and control cohorts to test the relative BrS disease risk of dominant negative missense variants compared with other variant classes.
Methods
Selection of Variants.
Variants for this study were selected from previously published functionally characterized variants6,14. Variants with peak currents <10% compared to WT were considered LoF, and variants with peak currents between 10-50% compared to WT were considered partial LoF. A full list of variants in this study is presented in Supplementary Table 1.
SCN5A Mutagenesis.
The SCN5A variant plasmids were mutagenized using a previously described “zone” system14. Briefly, SCN5A individual zones on small plasmids were mutagenized using the QuikChange Lightning Multi kit (Agilent) with primers designed using the online QuikChange Primer Design tool. Primers used in this study are listed in Supplementary Table 2. The variant-containing zone was then subcloned by restriction digestion into a plasmid containing an AttB:SCN5A:IRES:mCherry-blasticidinR plasmid14,16,17. The entire sequence of the zone containing the variant was confirmed by Sanger sequencing. In a previous study of 82 variants generated by this approach, 0/82 plasmids had any additional SCN5A variants outside the target zone14. All analyses used the most common SCN5A transcript in the adult heart, including the adult isoform of exon 6 and a deletion of the alternatively spliced Gln1077 residue (ENST00000443581). As per convention, all variants are named in accordance with the full 2,016 amino acid form (ENST00000333535).
Description of Cell Lines:
All experiments used Human Embryonic Kidney HEK293T “negative selection” landing pad (LP) cells as previously described (gift of Kenneth Matreyek)14,16,17. The AttB/AttP LP allows a single integration event per cell and a consistent level of target gene expression (Figure 1A). Homozygous experiments were carried out in LP cells (Figure 1A). Plasmids carrying SCN5A variants were transfected along with transposase and integrated into the LP site to allow stable expression. We termed these lines LP-SCN5A.
Figure 1. Measurement of loss-of-function homozygous and heterozygous peak current.
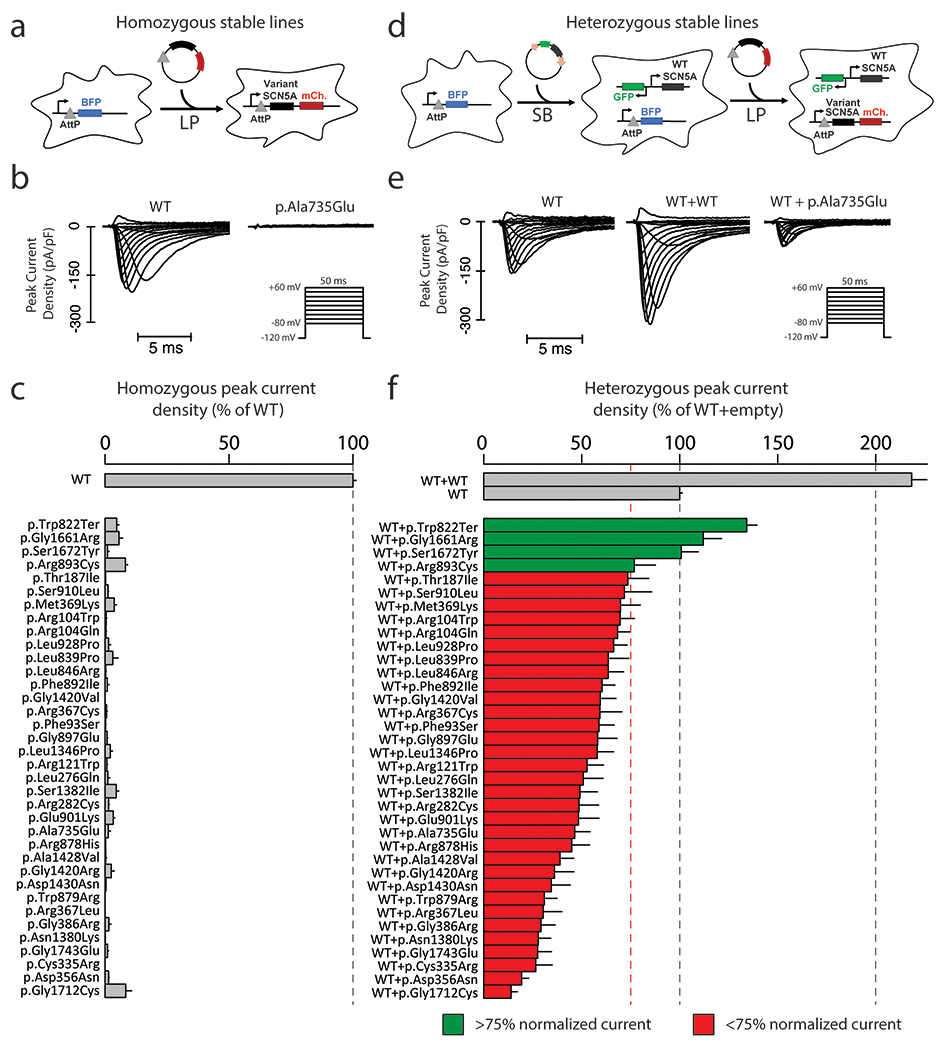
a For homozygous experiments, 1 copy of SCN5A were inserted into engineered HEK293 LP cells. The Landing Pad (LP) comprises an AttP and BFP locus, and allows insertion of a single insert per cell.
b Representative raw peak current densities in a WT and p.Ala735Glu cell. Inset: voltage protocol used.
c Measurement of homozygous peak current density in 35 SCN5A missense variants and one nonsense variant (normalized to WT). Mean ± standard errors. 11-67 cells were studied per variant.
d For heterozygous experiments, we used a combination of LP and SB systems. First, a SB plasmid bearing a WT copy of SCN5A was randomly inserted into the genome. A clone of these cells was identified that has an equal level of NaV1.5 in patch clamp experiments to typical LP expression. Next, a second copy of SCN5A bearing WT or variant was incorporated through the LP system.
e Representative raw peak current densities in a single transfected WT, dually integrated WT+WT, and WT+p.Ala735Glu cell.
f Peak current density measurements for 35 SCN5A missense variants and one nonsense variant in expression with WT SCN5A (normalized to single WT). Mean ± standard errors. 27-164 cells were studied per variant.
For heterozygous expression, we first generated LP cells stably expressing WT SCN5A in a non-LP site using the Sleeping Beauty (SB) transposon system and identified a clone of this cell line with peak sodium current (INa) equivalent to that observed with WT SCN5A in the LP site (Figure 1D). We then generated cell lines with SCN5A variants transfected into the LP site, thereby allowing us to express the two SCN5A alleles at equivalent levels and assess dominant negative effects. These lines are referred to as LP-SB-SCN5A.
Generation of Cell Lines:
Cells were cultured at 37°C in humidified 95% air/5% CO2 incubator in “HEK media”: Dulbecco’s Eagle’s medium supplemented with 10% fetal bovine serum, 1% non-essential amino acids, and 1% penicillin/streptomycin. Stable integration of a WT SCN5A into LP-cells was achieved using an optimized SB transposon system18 using the pSBbi-GN plasmid (a gift from Eric Kowarz, Addgene #60517), which contains SB transposon sequences for genomic integration flanking a promoter upstream of GFP and a second promoter upstream of a multiple cloning site (MCS) for expression of a gene of interest. A NotI restriction site was first cloned into the multiple cloning site using Gibson assembly (New England Biolabs). Then, WT SCN5A was cloned into the MCS by NotI digestion (New England Biolabs). Next, 1 ug of pSBbiGN-SCN5A and 100 ng of pCMV(CAT)T7-SB100, a plasmid expressing SB transposase (a gift from Zsuzsanna Izsvak, Addgene #34879), were cotransfected into the cells19, using FuGENE 6 (Promega) following manufacturer’s instructions. At day 7 post-transfection, GFP+ cells were sorted by fluorescence-activated cell sorting (FACS), and individual colonies were picked and re-analyzed by analytical flow cytometry to identify clones expressing varying levels of GFP (and thus varying levels of NaV1.5). Clones were then tested by SyncroPatch automated patch clamping (see below) to identify a clone expressing an equal peak sodium current as results from typical integration of a single copy of wild-type Nav1.5 into the AttB/AttP landing pad.
For homozygous patch clamp experiments, LP cells were transfected with an AttB-SCN5A variant:IRES:mCherry-BlasticidinR plasmid and studied as previously described14. For heterozygous patch clamp experiments, LP-SB-SCN5A cells were transfected using similar methods. For all cell lines, cells were transfected with FuGENE 6 or Lipofectamine 2000 following manufacturer’s suggested protocols using an AttB-containing SCN5A:IRES:mCherry:blasiticidinR plasmid and a plasmid bearing Bxb1 recombinase; cells underwent negative selection for 6 days with 1 ug/mL doxycycline (to induce promoter expression; Sigma), 100 ug/mL blasticidin S (to kill cells not expressing the blasticidin-resistant plasmid; Sigma), and 10 nM AP1903 (to kill un-integrated cells expressing the AP1903-sensitive caspase gene; MedChemExpress) in HEK media. At the end of selection, cells were assessed by analytical flow cytometry to assess percentage of mCherry-positive, BFP-negative cells (LP integration of SCN5A variant) and GFP-positive cells (SB integration of SCN5A).
Automated Patch Clamping.
Electrophysiology data were collected with the SyncroPatch 384PE automated patch clamping device (Nanion) using the same cell preparation and solutions as previously reported14. Peak currents are reported at −20 mV after a 200 msec pulse from a resting potential of −120 mV; peak sodium current is presented as the mean of data obtained in ≥8 cells/variant (homozygous experiments) or ≥27 cells/variant (heterozygous experiments). Voltage of half activation, voltage of half inactivation, time of 50% recovery from inactivation, and late current at 200 ms were obtained using previously published protocols14. As previously described, cells with values greater than 2.5 standard deviations from the mean were removed in an automated process14. For these additional parameters, only variants with data collected from >10 cells were included.
Case-control analysis.
We performed case-control analyses to test the penetrance of different classes of variants. We used BrS case counts from a recent International BrS Genetics Consortium and putative controls from gnomAD; the frequency of these variants is presented in Supplementary Table 18,15. We use gnomAD as putative controls; although phenotypes are not available for gnomAD participants, the vast majority of these individuals should not have Brugada Syndrome. All gnomAD counts were taken from gnomAD v2.1.1 transcript ENST00000333535.4. A cut-off minor allele frequency of 2.5e-5 was used to designate ultra-rare variants, as previously suggested20. To test the severity of each disease class (i.e., missense vs. indel vs. splice/frameshift/nonsense), we compared the relative number of cases versus controls by variant, drawing from the BrS consortium and gnomAD. Frameshift, splice, and nonsense variants at amino acid position > 1800 (post-transmembrane domain IV) were excluded due to the possibility that these variants may not be full LoF. We calculated the odds ratio associated with each variant class according to the formula (a/b)/(c/d), where a = BrS cases with variant, b = BrS cases without variant, c = gnomAD controls with variant, and d = gnomAD controls without variant. Since the allele number varied for different variants in gnomAD, the average allele number was calculated over all relevant variant types (missense, frameshift, nonsense, and splice site) and divided by 2 to obtain a count of sequenced gnomAD participants to use in odds ratio calculations, following a previously published approach8.
Data Analysis.
SyncroPatch 384PE data were analyzed as previously reported14. Peak current densities were calculated by dividing peak current at −20 mV by cell capacitance. For homozygous experiments, peak current densities were normalized to peak current densities observed in cells expressing WT plasmid. For heterozygous experiments, peak current densities were normalized to that observed in LP-SB-SCN5A cells, i.e., those expressing a single WT allele. As described below, WT+WT cells displayed ~200% peak INa compared to LP-SB-SCN5A cells. Heterozygote (WT+variant) cells displaying <75% of peak INa compared to LP-SB-SCN5A cells were designated as exerting a dominant negative effect. Statistical comparisons were made using two-tailed Fisher’s exact tests, implemented in R Studio (version 1.3.1093). R code used for all analyses is available upon request.
Methods for the Western blotting, supplemental case-control analyses, and structural modeling are in the Supplemental methods.
Results
Homozygous and Heterozygous Measurements of LoF Variants
We generated 37 LP-SCN5A stable lines (1 SCN5A allele expressed/line), each expressing LoF variants or the nonsense variant p.Trp822Ter (Figures 1A and Table S1)16,17. Representative traces for WT and p.Ala735Glu are shown in Figure 1B. We recorded peak INa at −20 mV: 35/37 missense variants exhibited a peak current density <10% compared to WT (Figure 1C – only LoF shown; Table S1). The remaining 2 variants (previously reported to be LoF) showed >10% peak current when compared to WT and were studied separately with other partial LoF variants. One LoF variant, p.Arg893Cys, was previously detected in patients with BrS but has not been previously assessed by patch clamping2.
We then tested each LoF variant in heterozygous expression using engineered cell lines (WT+variant) (Figure 1D). Figure 1E shows representative traces of cells expressing WT, WT+WT, and WT+p.Ala735Glu (an example dominant negative variant). Figure 1F presents peak INa for the same 35 LoF variants presented in Figure 1C. WT+WT cells expressed peak INa of 218.4±7.7% relative to WT alone in LP-SB-SCN5A cells, i.e., those expressing a single WT allele. By contrast, 32/35 of the WT+variant cell lines showed <75% peak INa compared to LP-SB-SCN5A cells, indicating a dominant negative effect. The heterozygous dominant negative variants displayed a gradient of effect, from 13.9±3.3% to 74.4±5.4% of WT alone. Two previously studied dominant negative variants, p.Arg104Trp and p.Arg121Trp21, both also exhibited dominant negative effects in this study (69.6±7.3% and 52.7±8.4% of WT, respectively). While p.Trp822Ter, p.Gly1661Arg, p.Ser1672Tyr, and p.Arg893Cys had LoF peak currents in homozygous experiments, they did not exhibit a dominant negative effect (Table S1).
Prior to each SyncroPatch experiment, we assessed expression of each variant using flow cytometry (Supplementary Figure 1A–D). We observed consistently high expression of our reporter gene mCherry sharing the same promoter as the SCN5A variants in homozygous and heterozygous cells (Supplementary Figure 1E). In an orthogonal experiment, we tested the expression of selected variants in homozygous expression by Western blotting (Supplementary Figure 2). Consistent with the flow cytometry measurements, we observed uniform expression of mCherry by Western blot; however, we observed somewhat variable expression of NaV1.5 across 8 tested variants. This result could indicate a diversity of mechanisms of LoF, including a subset arising from variable rates of protein degradation.
Homozygous and Heterozygous Measurements of Partial LoF Variants
We also studied the prevalence of dominant negative effects in 15 partial LoF variants using LP-SB-SCN5A lines. We first confirmed that variant peak currents were 10%-50% compared to WT in homozygous expression with LP-SCN5A cells. (Figure 2A). The set of 15 variants included two variants (p.Arg282His and p.Gly1740Arg) previously reported to be LoF but measured as >10% peak INa in our system22,23. Figure 2B shows a gradient of INa, with partial LoF variants showing a greater range of effect in heterozygous expression than those of LoF variants (24.7±5.6% to 231.6±10.8%). 6/15 partial LoF variants had a dominant negative effect whereas the remaining 9 variants all exceeded normalized WT peak current.
Figure 2. Some partial loss-of-function variants have a dominant negative effect.
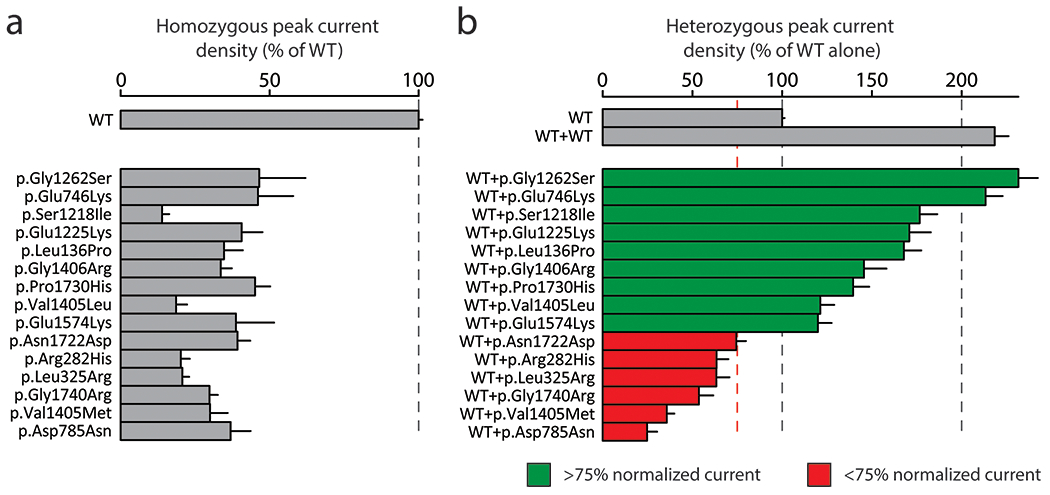
a Measurement of homozygous peak current density in 15 partial LoF SCN5A variants (normalized to WT). Mean ± standard errors. 8-36 cells were studied per variant.
b Measurement of heterozygous peak current density in 15 partial LoF SCN5A variants (normalized to WT). Mean ± standard errors. 27-53 cells were studied per variant.
Coupled gating in heterozygous expression
In addition to assessing peak sodium current, we also examined other parameters of channel function to measure the extent of coupled gating, a phenomenon where the LoF allele alters the gating properties of the WT allele. These parameters required additional experimental protocols and quality control filters, so these parameters were not comprehensively obtained in all variants studied; only variants with data from >10 qualifying cells are presented. We examined voltage of half activation among the missense variants investigated above (representative raw data shown in Figure 3A and 3B). 16/50 variants (14 LoF and 2 partial LoF) showed a >10 mV shift in the voltage of half activation, suggesting widespread coupled gating affecting this parameter (Figure 3C). We did not observe widespread changes for other parameters beyond voltage of half activation. No variants were shown to induce a shift in voltage of half inactivation >10 mV (Figure 3D). One variant (p.Gly1406Arg) had a 1.71-fold change in recovery from inactivation when compared to WT; the other 34 qualifying variants had <50% shifts in recovery from inactivation (Figure 3E). No variants induced late current >1% when co-expressed with WT (Figure 3F). Due to the very low or absent peak currents in homozygous LoF variants, it was not feasible to assess parameters other than peak current in homozygous expression.
Figure 3. Coupled gating revealed by missense variants in heterozygous expression.
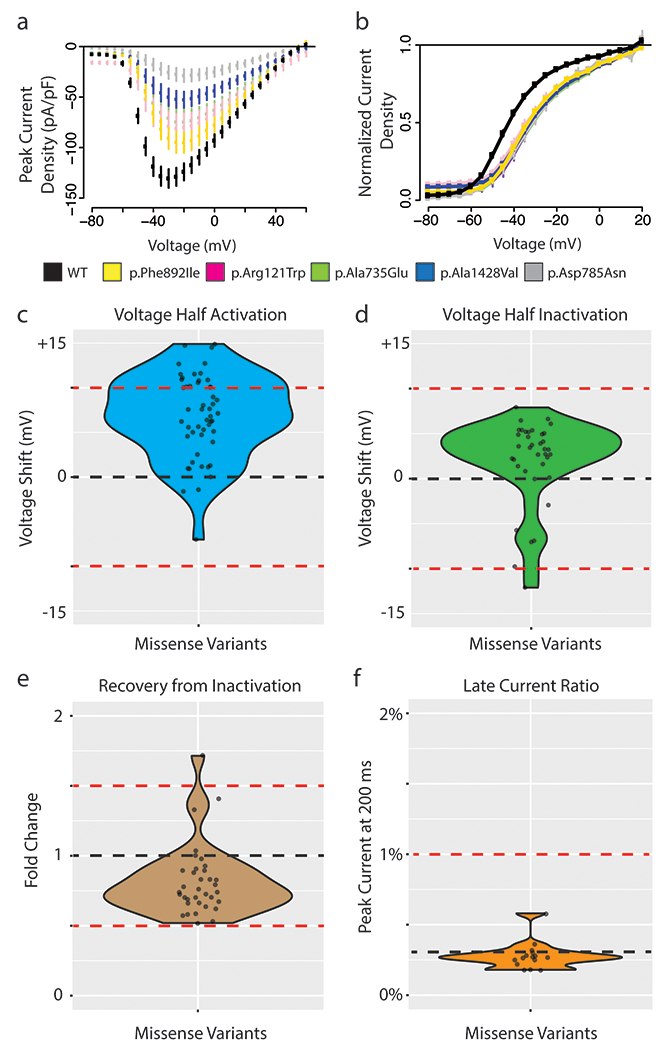
a Current-voltage plot of WT (black) and 5 missense SCN5A variants with large shifts in voltage of half activation: p.Ala735Glu (light green), p.Arg121Trp (pink), p.Asp785Asn (grey), p.Ala1428Val (blue), p.Phe892Ile (gold). The voltage protocol is identical to that used in Figure 1.
b Voltage of half activation curve for WT and 5 missense SCN5A variants (variants and color same as in A). B-D) WT indicated with black line and abnormal cutoffs indicated with red lines. For B-D only variants with at least 10 qualifying cells meeting quality control criteria were analyzed.
c Voltage of half activation shift of all missense variants compared to WT. Thresholds correspond to +10 mV and −10 mV shifts from WT to indicate dominant negative effect.
d Voltage of half inactivation shift of all missense variants compared to WT. Thresholds correspond to +10 mV and −10 mV shifts from WT to indicate dominant negative effect.
e Time of 50% recovery from inactivation measured in fold change for all missense variants normalized to WT. Thresholds of fold change of 1.5 and 0.5 represent strong deviations from WT.
f Late current percentage (% of peak current) measured at 200 ms for all missense variants compared to WT. Threshold at 1% indicates threshold of potential dominant negative effect.
Elevated BrS Risk Among Dominant Negative Variants
Having identified dominant negative missense variants as a variant subclass with functional features predictive of increased clinical penetrance, we aimed to characterize the clinical impact of these variants. We therefore compared the relative frequencies of BrS cases to controls among discrete variant classes (missense, indel, haploinsufficient, dominant negative). This approach was inspired by similar studies comparing variant frequencies in cases from disease consortia to those in large population databases as putative controls8. We restricted our study to rare variants meeting a population frequency of less than 2.5e-05 as previously described20. From these comparisons we could derive odds ratios to quantify the relationship between an exposure (rare SCN5A variant) and BrS.
Case and control counts of heterozygotes of the dominant negative variants described above were interrogated using a published consortia of BrS cases8 and gnomAD, a database of population variation that we considered to contain largely putative controls15 (Figure 4A, Table S3). In Figure 4B we present the odds ratios (ratio of odds in BrS cohort:gnomAD). The LoF missense dominant negative variants had an odds ratio of 323 compared to 11.0 for missense, 24.2 for indel, and 118 for putative haploinsufficient variants (nonsense, splice, frameshift). Thus, the relative risk of dominant negative missense variants compared to haploinsufficient variants is 2.7 (Fisher’s exact test, p = 0.019). All categories were significantly enriched compared to all missense variants (Fisher’s exact test, p<0.05).
Figure 4. Case-Control analysis by variant class.
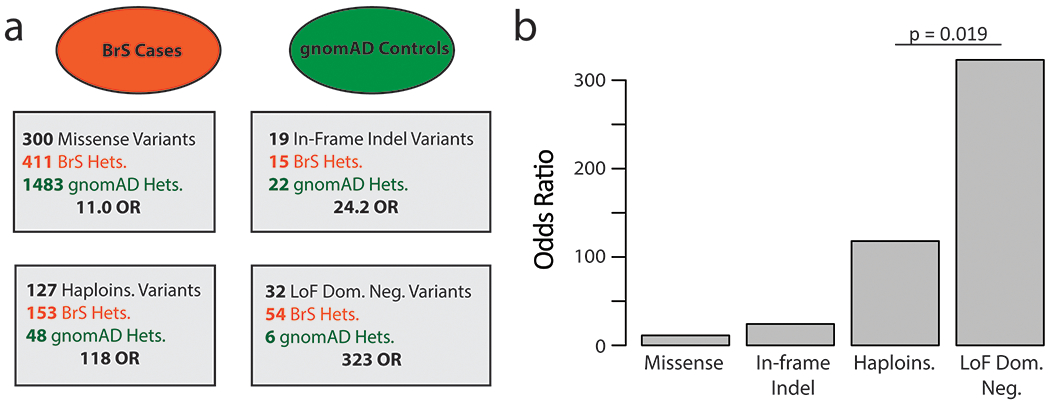
a Case-control breakdown by data source stratified by variant class. BrS cases are shown in red, with putative gnomAD controls shown in green. Heterozygous individuals demarked as Hets. Haploinsufficient (haploins.) indicates nonsense, splice, and frameshift variants. Odds ratios are calculated for each variant class.
b Barplot of BrS odds ratios by variant class.
Next, we performed a matched ancestry contingency analysis using only a subset of gnomAD and the BrS case consortium. We restricted our BrS cases to those from European centers in Walsh et. al. and used a subset of gnomAD (Non-Finnish-European ancestry) as controls. We observed very similar results to the main analysis with odds ratios of 380.6 for dominant negative missense variants, vs 18.10 for all missense, 73.06 for indel, and 126.1 for the putative haploinsufficient variants (Supplementary Figure 4). While we observe a slightly higher enrichment of cases in dominant negative variants compared to haploinsufficient variants in this contingency analysis (3.1 vs 2.7), the p-value increased from 0.01 to 0.09 due to the smaller sample size (two-tailed Fisher’s exact test). We performed an additional analysis further comparing groups of functionally characterized variants in this study (Supplementary Figure S5). The analysis showed that among LoF and partial LoF variants, those with dominant negative effects had a significantly greater odds ratio for BrS than non-dominant negative variants (OR 249 vs 58; Fisher’s exact test, p = 0.0011). To test the sensitivity of our findings to the chosen dominant negative threshold of 0.75 peak current, we recalculated odds ratios at various thresholds (Supplementary Figure 3). We found that the odds ratios were stable across a range of thresholds between 0.60 and 0.80. With a more stringent cut-off (≤0.55), the odds ratios were even higher.
Structural Distribution of Dominant Negative Variants
Dominant negative variants were present throughout the structured transmembrane regions of NaV1.5 and did not predominate in any single hotspot region (Supplementary Figure 6A). Structural modeling further showed that dominant negative variants were distributed throughout the three-dimensional structure of NaV1.5, with apparent enrichment in the S5-S6 linker domains (Supplementary Figure 6B–C).
DISCUSSION
Dominant Negative Effect Among Most Missense LoF SCN5A Variants
This study assessed the dominant negative properties of 50 LoF and partial LoF variants. A large majority of examined LoF variants (32/35) and some partial LoF variants (6/15) showed dominant negative behavior. Dominant negative effects are pervasive throughout biology, especially for multimeric proteins, and involve several distinct mechanisms to compromise WT function24. In the case of SCN5A, the dominant negative effect has been posited to arise by both deficient trafficking to the membrane as well as coupled gating at the cell surface. One study showed that the variants p.Arg104Trp and p.Arg121Trp induced a dominant negative effect primarily through endoplasmic reticulum retention of WT protein due to interactions among the channel alpha-subunits21. Follow up studies with extensive biochemical analyses showed that the dominant negative variant p.Leu325Arg acted through coupled gating at the cell surface11.
Previous research suggested that the residues between 493 and 517 are critical for the dimerization and coupled gating of NaV1.5 at the cell surface, and another study found an enrichment of dominant negative variants at the N-terminus of the protein10,25. We did not observe an enrichment of dominant negative variants among these previously described residues, but rather a broader distribution of variants spanning the four transmembrane domains of the protein (Supplementary Figure 6). Thus, dominant negative effects appear to be a general property of most LoF missense variants in SCN5A, independent of location within the protein. Particularly interesting are examples of disparate effects within close physical proximity, such as the partial LoF variants p.Val1405Met (35.7% peak current in heterozygous expression), p.Val1405Leu (121% peak current), and p.Gly1406Arg (146% peak current). We surprisingly observed a small but non-zero peak current (4.7% of WT) for an early nonsense variant p.Trp822Ter. This variant has an intact Domain I, and therefore may be forming a poorly conducting multimeric channel.
In addition to decreased peak current, we observed that 16/50 variants also influenced voltage of half activation when measured in heterozygous expression with WT. This finding is consistent with the concept of coupled gating at the cell surface, and reflects the influence of the LoF allele on properties of the WT allele of the protein, possibly through a multi-channel complex11. These shifts in V1/2 activation in a loss of function direction combine with reduced peak currents to result in additional reduction of channel function in heterozygous expression. V1/2 activation was the only additional property that varied substantially from WT NaV1.5 activity, as we did not observe large differences in voltage of half inactivation, recovery from inactivation, or late current.
While dominant negative effects were very common among the variants studied, there were LoF variants and partial LoF variants that did not diminish peak current. These results further support the idea that there are yet to be recognized channel-channel interactions that may or may not induce dominant negative behavior. In experiments assessing protein expression, we found a range of relative expression of NaV1.5 compared to the mCherry reporter by Western blot. We did not observe a strong correlation between degree of dominant negative effects and relative protein expression (Supplementary Figure 2). This may indicate a diversity of underlying mechanisms including protein degradation, altered gating, or aberrant trafficking.
Previous studies10,11,21 have experimentally demonstrated at least two mechanisms for individual dominant negative variant effects: coupled gating and deficient trafficking. Our observation of 16 variants that modulate voltage of half activation indicate that these variants may act through coupled gating. However, at this point the mechanism of the remaining dominant negative variants is unclear. Through Western blot data, we observe a range of protein expression in selected variants; these remaining variants may act by causing a failure to traffic to the plasma membrane or may traffic properly and reduce channel function at the plasma membrane.
Increased BrS Risk of Dominant Negative Variants
Our results indicate that dominant negative SCN5A variants are significantly more likely to cause manifest Brugada Syndrome than other missense variants. Previous work has established that homozygous peak current of SCN5A variants is the strongest in vitro electrophysiological predictor of each variant’s BrS risk6,26. Since dominant negative missense variants cause an especially low peak current, we hypothesized that dominant negative variants would confer an especially high risk for BrS. Importantly, our expanded catalog of 38 dominant negative SCN5A variants enabled us to calculate cohort-based estimates of disease risk of this class of variants. Using gnomAD and a recently published cohort of BrS cases8,15, we demonstrated that dominant negative variants are highly overrepresented in cases vs controls when compared to other variant classes, with a striking odds ratio of 323 for dominant negative LoF missense variants. In contrast, other variant classes have lower odds ratios of 11 (all missense variants) or 118 (putative haploinsufficient frameshift/nonsense/splice site variants). Thus, the relative risk of BrS among dominant negative LoF missense variants compared to putative haploinsufficient variants is 2.7. Previous studies have shown that truncating and functionally inactive missense variants cause a more severe phenotype than partially active missense variants, but the penetrance of dominant negative variants had not yet been extensively studied27. Our results indicate that the penetrance of dominant negative missense variants is higher than penetrance of other variant classes. One potential explanation for the different disease penetrance among variant classes is that nonsense mediated decay (NMD) removes aberrant transcripts for splice-altering and nonsense variants, preventing their interaction with WT NaV1.5. Given the data presented here, dominant negative missense variants should arouse high clinical suspicion for BrS risk when detected in patients.
Current evidence suggests that peak NaV1.5 current is highly predictive of clinical BrS penetrance6,26. Our finding of a higher BrS penetrance for dominant negative loss of function variants compared to other classes (putative haploinsufficient and non-dominant negative missense variants) further supports a model in which total NaV1.5 peak current (adjusting for dominant negative effects) is correlated with disease severity.
High-throughput Electrophysiological Assays to Study Dominant Negative Effects
High-throughput automated patch clamping has emerged as a tool for rapidly assessing functional consequences of ion channel genetic variation28. This technique has been used to assess pathogenicity of variants in KCNQ129, SCN5A14, and KCNH230. Here, we present the most extensive evaluation of heterozygous NaV1.5 expression to date using this platform, studying 51 total variants with 27-164 cells per heterozygous measurement. Heterozygous measurements are already common for the cardiac potassium channels KCNQ1 and KCNH2; this study suggests that heterozygous studies may also be necessary for LoF SCN5A variants in future studies. This work shows that high-throughput automated patch-clamp can help establish molecular mechanisms of disease.
Limitations
Results from heterologous expression in HEK293T cells may not fully recapitulate behavior in native cardiomyocytes in human hearts. In particular, contributions such as polygenic modifiers, as has been previously observed in BrS31, may not be fully captured by this non-native system. Two common alternative splicing events impact SCN5A splicing (p.Gln1077 deletion/insertion and fetal/adult exon 6); only the most common splice isoform in the adult heart was examined in this study. The gnomAD population database does not have available phenotypic information, so a small fraction of individuals included in gnomAD may in fact have BrS.
Conclusions
Most LoF missense variants in SCN5A have a dominant negative effect. These missense dominant negative variants have a 2.7-fold increased risk of BrS when compared to haploinsufficient variants. These results may help refine prediction of BrS risk in dominant negative variant heterozygotes.
Supplementary Material
Acknowledgements
This research was funded by NIH grants K99 HG010904 (AMG), R01 HL149826 (DMR), T32GM007347 (MJO and AM), AHA grants AHA 20PRE35180088 (AM) and 20POST35220002 (BL), and a Heart Rhythm Society Clinical Research Award in Honor of Mark Josephson and Hein Wellens (YW). We thank Victoria Parikh for helpful discussions and Kenneth Matreyek for suppling the LP-negative HEK293 cell line. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). SyncroPatch 384PE experiments were performed in the Vanderbilt High-Throughput Screening (HTS) Core Facility. The HTS Core receives support from the Vanderbilt Institute of Chemical Biology and the Vanderbilt Ingram Cancer Center (P30 CA68485).
Footnotes
Ethics Declaration
All data generated from previous studies used in this analysis received IRB/REC approval.
Data Availability
Variant-level datasets generated and analyzed in the current study are available in the supplement. Materials (e.g. “zone” or full-length plasmids), detailed cell-level raw patch clamp data, and R analysis scripts are available from the corresponding author on request.
References
- 1.Mizusawa Y & Wilde AA Brugada syndrome. Circ Arrhythm Electrophysiol 5, 606–616, doi: 10.1161/circep.111.964577 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Kapplinger JD et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46, doi: 10.1016/j.hrthm.2009.09.069 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosseini SM et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 138, 1195–1205, doi: 10.1161/circulationaha.118.035070 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gui J et al. Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One 5, e10985, doi: 10.1371/journal.pone.0010985 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bezzina CR et al. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ Res 92, 159–168, doi: 10.1161/01.res.0000052672.97759.36 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Kroncke BM, Glazer AM, Smith DK, Blume JD & Roden DM SCN5A (Na(V)1.5) Variant Functional Perturbation and Clinical Presentation: Variants of a Certain Significance. Circ Genom Precis Med 11, e002095, doi: 10.1161/circgen.118.002095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landrum MJ et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980–985, doi: 10.1093/nar/gkt1113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh R et al. Enhancing rare variant interpretation in inherited arrhythmias through quantitative analysis of consortium disease cohorts and population controls. Genet Med 23, 47–58, doi: 10.1038/s41436-020-00946-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan X et al. Comparative structural analysis of human Na(v)1.1 and Na(v)1.5 reveals mutational hotspots for sodium channelopathies. Proc Natl Acad Sci U S A 118, doi: 10.1073/pnas.2100066118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clatot J et al. Voltage-gated sodium channels assemble and gate as dimers. Nat Commun 8, 2077, doi: 10.1038/s41467-017-02262-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clatot J et al. Mutant voltage-gated Na(+) channels can exert a dominant negative effect through coupled gating. Am J Physiol Heart Circ Physiol 315, H1250–h1257, doi: 10.1152/ajpheart.00721.2017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller DI et al. Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res 67, 510–519, doi: 10.1016/j.cardiores.2005.03.024 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Doisne N et al. In vivo Dominant-Negative Effect of an SCN5A Brugada Syndrome Variant. Front Physiol 12, 661413, doi: 10.3389/fphys.2021.661413 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glazer AM et al. High-Throughput Reclassification of SCN5A Variants. Am J Hum Genet 107, 111–123, doi: 10.1016/j.ajhg.2020.05.015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443, doi: 10.1038/s41586-020-2308-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matreyek KA, Stephany JJ, Chiasson MA, Hasle N & Fowler DM An improved platform for functional assessment of large protein libraries in mammalian cells. Nucleic Acids Res 48, e1, doi: 10.1093/nar/gkz910 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matreyek KA, Stephany JJ & Fowler DM A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res 45, e102, doi: 10.1093/nar/gkx183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowarz E, Löscher D & Marschalek R Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J 10, 647–653, doi: 10.1002/biot.201400821 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Mátés L et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 41, 753–761, doi: 10.1038/ng.343 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Whiffin N et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med 19, 1151–1158, doi: 10.1038/gim.2017.26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clatot J et al. Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Na(v)1.5 α-subunits. Cardiovasc Res 96, 53–63, doi: 10.1093/cvr/cvs211 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poelzing S et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 114, 368–376, doi: 10.1161/circulationaha.105.601294 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Baroudi G, Napolitano C, Priori SG, Del Bufalo A & Chahine M Loss of function associated with novel mutations of the SCN5A gene in patients with Brugada syndrome. Can J Cardiol 20, 425–430 (2004). [PubMed] [Google Scholar]
- 24.Veitia RA & Birchler JA Dominance and gene dosage balance in health and disease: why levels matter! J Pathol 220, 174–185, doi: 10.1002/path.2623 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Wang Z et al. Calmodulin binds to the N-terminal domain of the cardiac sodium channel Na(v)1.5. Channels (Austin) 14, 268–286, doi: 10.1080/19336950.2020.1805999 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishikawa T et al. Functionally validated SCN5A variants allow interpretation of pathogenicity and prediction of lethal events in Brugada syndrome. Eur Heart J 42, 2854–2863, doi: 10.1093/eurheartj/ehab254 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Meregalli PG et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 6, 341–348, doi: 10.1016/j.hrthm.2008.11.009 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Obergrussberger A, Friis S, Brüggemann A & Fertig N Automated patch clamp in drug discovery: major breakthroughs and innovation in the last decade. Expert Opin Drug Discov 16, 1–5, doi: 10.1080/17460441.2020.1791079 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Vanoye CG et al. High-Throughput Functional Evaluation of KCNQ1 Decrypts Variants of Unknown Significance. Circ Genom Precis Med 11, e002345, doi: 10.1161/circgen.118.002345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng CA et al. High-throughput phenotyping of heteromeric human ether-à-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants. Heart Rhythm 17, 492–500, doi: 10.1016/j.hrthm.2019.09.020 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Bezzina CR et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 45, 1044–1049, doi: 10.1038/ng.2712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Variant-level datasets generated and analyzed in the current study are available in the supplement. Materials (e.g. “zone” or full-length plasmids), detailed cell-level raw patch clamp data, and R analysis scripts are available from the corresponding author on request.