


Abstract
Bacteria and bacteriophages co-evolve in a constant arms race, wherein one tries and finds newer ways to overcome the other. Phage resistance poses a great threat to the development of phage therapy. Hence, it is both essential and important to understand the mechanism of phage resistance in bacteria. First identified in Mycobacterium smegmatis, the gene mpr, upon overexpression, confers resistance against D29 mycobacteriophage. Presently, the mechanism behind phage resistance by mpr is poorly understood. Here we show that Mpr is a membrane-bound DNA exonuclease, which digests DNA in a non-specific manner independent of the sequence, and shares no sequence or structural similarity with any known nuclease. Exonuclease activity of mpr provides resistance against phage infection, but the role of mpr may very well go beyond just phage resistance. Our experiments show that mpr plays a crucial role in the appearance of mutant colonies (phage resistant strains). However, the molecular mechanism behind the emergence of these mutant/resistant colonies is yet to be understood. Nevertheless, it appears that mpr is involved in the survival and evolution of M. smegmatis against phage. A similar mechanism may be present in other organisms, which requires further exploration.
INTRODUCTION
The emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) pathogens or ‘superbugs’ has become a major concern for human health. With the failure of conventional drugs and antibiotics to deal with such pathogens, there is an exigent need to look for alternative therapies and treatment methods. In this regard, the use of bacteriophages, viruses that infect and kill bacteria, appears to be the most viable option (1,2). Phage therapy offers many advantages over conventional medicines, such as a high degree of specificity against pathogenic bacteria and no known severe drawbacks or side effects on the human body (3). While considering phage therapy as an option against drug-resistant bacteria, the arms race between bacteria and bacteriophages (4,5) that has been continuing for millions of years and how the viral pressure has pushed bacteria to develop mechanisms against bacteriophages cannot be neglected (6). This arms race has resulted in the development of a magnitude of bacterial defence mechanisms that target different stages of the phage life cycle (7,8) and equally astounding means by which phages evade bacterial defence mechanisms and infect them (9). This has led to an equilibrium where both phages and bacteria coexist in the same environment without undergoing dramatic changes in their population or extinction events (10). These defence mechanisms may very well impede the widespread use of phage therapy.
The several different mechanisms that the bacteria employ to resist phages include preventing phage attachment by modifying the cell surface receptor (11) or by masking the receptor (12). Bacteria are also known to produce protein (12,13), lipoprotein (14), exopolysaccharide (EPS) (15) and modified EPS (16) to block phage adsorption. Bacteria can resist phage infection by preventing phage DNA entry using Superinfection exclusion (Sie) systems (17), digestion of phage nucleic acid via restriction-modification system (18–21) and CRISPR-cas systems. The CRISPR-cas has been associated with the immune system of bacteria that targets foreign DNA, including plasmids and phage DNA (22–25). Abortive infection is a very unique strategy for defence against phages, which results in death of the infected cell, thus inhibiting the multiplication and propagation of the phage (17). Another interesting bacterial defence mechanism is toxin–antitoxin system; it encodes for a toxin protein capable of interfering with vital biochemical processes arresting cell growth, and an antitoxin that neutralizes the toxin (26). Thus, while continuing with the development of phage therapy, it also becomes crucial to gather a deep understanding of the processes and mechanisms that bacteria employ to resist bacteriophages.
D29 mycobacteriophage is a double-stranded DNA (dsDNA) virus that infects and kills several species of mycobacteria, including the pathogenic Mycobacterium tuberculosis. In M. smegmatis, which is largely used as a model organism for M. tuberculosis, a host gene mpr (for multicopy phage resistance), upon overexpression, has been suggested to play a key role in resistance against phage infection (27). It was also observed that normal infection was achieved upon electroporation of D29 phage DNA in the cell, thus eliminating the possibility that mpr may be involved in some downstream pathway (phage DNA replication, phage gene expression, viral assembly, or host restriction-modification system) to resist phage infection. Moreover, Mpr overexpression was also found to be very toxic to the cell, and this toxicity could be linked to the first 60 amino acids of Mpr (27). Although the exact mechanism behind phage resistance by mpr is not fully understood, the study concluded that the phage resistance phenotype results from blocking of phage DNA entry in the cell. We here present a detailed characterization of Mpr and show that it is a membrane-bound DNA exonuclease. We further provide evidence that suggests that Mpr is required for the emergence and establishment of phage-resistance in M. smegmatis and thus plays an important role in the survival of M. smegmatis. We believe that our study will help in the modulation of phage therapy efforts for mycobacterial infections.
MATERIALS AND METHODS
Bacterial strains, media and growth conditions:
Escherichia coli strain XL1-Blue (Stratagene) was used for all the cloning, and BL21(DE3) (NEB) for protein isolation. Escherichia coli was grown in LB broth (Difco) at 37°C in the presence of either 50 μg/ml kanamycin or 100 μg/ml ampicillin (Sigma-Aldrich), as required, with constant shaking at 200 rpm or on LB agar plates. M. smegmatis mc2155 (MsmWT) was used for localization, phage infection, and knockout studies. Plasmid constructs generated in this study (Table 1) were used to transform various cells, which were selected on the required antibiotic. MsmWT was grown in Middlebrook 7H9 (Difco) liquid broth supplemented with 2% glucose and 0.05% Tween 80, at 37°C with constant shaking at 200 rpm or on Middlebrook 7H9 solid media supplemented with 2% glucose with incubation at 37°C. Whenever required, 25 μg/ml of kanamycin was used for the M. smegmatis culture.
Table 1.
List of constructs used in the study
| S. No. | Name of plasmid | Backbone | Promoter | Type | Reporter tag | Source |
|---|---|---|---|---|---|---|
| 1. | pET21b -mpr | pET21b | T7 promoter | replicon | 6xHis | This study |
| 2. | pET21b-Δ26mpr | pET21b | T7 promoter | replicon | 6xHis | This study |
| 3. | pET21b-Δ32mpr | pET21b | T7 promoter | replicon | 6xHis | This study |
| 4. | pET21b-Δ50mpr | pET21b | T7 promoter | replicon | 6xHis | This study |
| 5. | pMSQSCHS-ΔTMmpr | pMSQSCHS | T7 promoter | replicon | 6xHis | This study |
| 6. | pMSQSNHS-TMGFP | pMSQSNHS | T7 promoter | replicon | 6xHis | This study |
| 7. | pMSQSCHS-ΔCmpr | pMSQSCHS | T7 promoter | replicon | 6xHis | This study |
| 8. | pMSQSCHS-GFP | pMSQSCHS | T7 promoter | replicon | 6xHis | This study |
| 9. | pET21b-M1 | pET21b-mpr | T7 promoter | replicon | 6xHis | This study |
| 10. | pET21b-M2 | pET21b-mpr | T7 promoter | replicon | 6xHis | This study |
| 11. | pET21b-M3 | pET21b-mpr | T7 promoter | replicon | 6xHis | This study |
| 12. | pET21b-M6 | pET21b-mpr | T7 promoter | replicon | 6xHis | This study |
| 13. | pET21b-M8 | pET21b-mpr | T7 promoter | replicon | 6xHis | This study |
| 14. | pET21b-M9 | pET21b-mpr | T7 promoter | replicon | 6xHis | This study |
| 15. | pET21b-M10 | pET21b-mpr | T7 promoter | replicon | 6xHis | This Study |
| 16. | pMA-mprHis | pMA-His | acetamidase | replicon | 6xHis | This study |
| 17. | pMA-mprGFP | pMA-GFP | acetamidase | replicon | GFP | This study |
| 19. | pMA-Δ50GFP | pMA-GFP | acetamidase | replicon | GFP | This study |
| 20. | pMA-ΔTMGFP | pMA-GFP | acetamidase | replicon | GFP | This study |
| 21. | pMA-His | pMA-His | acetamidase | replicon | 6xHis | Lab material |
| 22. | pMIA-GST | pMA-GST | acetamidase | Integration | GST | This study |
| 23. | pMIA-mpr-His | pMA-His | acetamidase | Integration | 6xHis | This study |
| 24. | pMIA-ΔCmpr | pMA-GFP | acetamidase | Integration | GFP | This study |
| 25. | pMIM-mpr | pMIA-His | mpr | Integration | 6xHis | This study |
| 26. | pMIM-GFP | pMIA-GFP | mpr | Integration | GFP | This study |
| 27. | pMIM-Δ26mpr | pMIM-mpr | mpr | Integration | 6xHis | This study |
| 28. | pMIM-ΔCmpr | pMIM-mpr | mpr | Integration | GFP | This study |
| 29. | pMM-GFP | pMA-GFP | mpr | replicon | GFP | This study |
| 30. | pMM-mpr | pMH-His | mpr | replicon | 6xHis | This study |
| 31. | pMH-GFP | pMH-GFP | hsp60 | replicon | GFP | Lab material |
| 32. | pSD5b | pSD5b | *NP | replicon | lacZ | Lab material |
| 33. | pSD5bPrmpr | pSD5b | mpr | replicon | lacZ | Lab material |
*NP, no promoter present in the plasmid.
Reagents
DNA modifying enzymes (such as Antarctic phosphatase, T4 polynucleotide kinase, and restriction enzymes, etc.) were procured from New England Biolabs (NEB) and high-fidelity DNA polymerase, Phusion was procured from Thermo. Gel extraction/PCR cleanup kit were procured from Favorgen. All the enzymes and kits were used as per the manufacturer's instructions. All other reagents were obtained from Sigma Aldrich.
PCR and cloning
PCR reactions were carried out using Phusion high fidelity DNA polymerase (NEB) followed by phosphorylation of the amplicon using T4 Polynucleotide Kinase. For vector backbone preparation, it was digested using respective restriction enzymes followed by cleanup and dephosphorylation by Antarctic phosphatase enzyme as per protocol provided by the supplier. pMSQSCHS and pMSQSNHS (28) were digested with SmaI restriction enzyme, and for pMA-His/pMA-GFP (29), EcoRV or HpaI restriction enzyme sites were used for C-terminal and N-terminal tag cloning, respectively. The ligation of the vector backbone and insert was carried out using T4 DNA ligase at 22°C for 2 h or 16°C overnight incubation, followed by transformation of E. coli XL1 blue cells. A small amount of bacterial colony was re-suspended in 10 μl of MilliQ water and heated at 100°C for 5 min to perform a colony PCR. The samples were then spun for 5 min at room temperature (RT), the resulting supernatant was used in the PCR reaction. Primer sequences used in the study are shown in Table 2.
Table 2.
List of oligos used in the study. Restriction enzyme site, wherever present, is bold-faced
| S. No. | Primer name | Primer sequence |
|---|---|---|
| 1. | mpr-For | ATGACCACTCCTCAGCCGTATCC |
| 2. | mpr-Rev | CGGCGTCAGGTTCACCGTGACC |
| 3. | mprRTfor | CGACGACAAGAAGGACACCA |
| 4. | mprRTrev | ACCACGAACTCGAACTTCCC |
| 5. | Mpr_del26_for | GCGAAGAAGAAGCGCAAATGGC |
| 6. | 32Nter | AAGAAGAAG GCTAGC TGGCCGGCGATCGTCG |
| 7. | 49Nter | GCGCCAGTCG GCTAGC AACGACGACAAGAAGGACACCAC |
| 8. | Mpr_dCter_rev | CATCATCAT CATATG GGCCGGCGTGGTGTCCTTCTTG |
| 9. | Del_Nter for | AACGACGACAAGAAGGACACCACG |
| 10. | Del_tm_for_new | AGCCCGCGAAGAAGAAGCGCAAAAACGACGACAAGAAGGACACCACG |
| 11. | Del_tm_rev_new | CGTGGTGTCCTTCTTGTCGTCGTTTTTGCGCTTCTTCTTCGCGGGCT |
| 12. | Mpr_prom_for | GAGTAG TCTAGA GCTCCGCCCACTTCTCGTGC |
| 13. | mpr_prom_rev2 | ATATATATATAT GCATGC GCCGGGATACGGCTGAGG |
| 14. | GFPFor-rapid | ATGAGCAAGGGCGAGGAGCTGTTC |
| 15. | GFPRev-rapid | CTTGTACAGCTCGTCCATGCCGAG |
| 16. | Mpr_prom1_rev | CATTGTGATCGTTCCTTCATGTCTTGC |
| 17. | AKK_M1_rev | CCATTTGCGCTTCGCCGCCGCAGGCTGC |
| 18. | KRK_M2_rev | CGCTGGCCATGCGGCCGCCTTCTTCGCG |
| 19. | WPA_M3_rev | CGACGATCGCTGCCGCTTTGCGCTTCTTCTTCG |
| 20. | ALM_M6_rev | CCCCACGATCGCCGCCGCGATCACACC |
| 21. | SIF_M8_rev | CTTGTCGTCGTTGGCGGCCGCTCCCACGATCATC |
| 22. | NDD_M9_rev | GGTGTCCTTCTTGGCGGCGGCGAAGATCGACC |
| 23. | KKD_M10_rev | CGGTGTGGTGGCCGCCGCGTCGTCGTTGAAG |
| 24. | pMV_rev_XhoI | TGCTCGAGTCTCGAGCCTGGCAGTCGATCGTACG |
| 25. | mpr-rev | TGCTCGAGT GCGGCCGC CGGCGTCAGGTTCACCGTG |
Protein expression and purification
CDS of mpr and Δ26mpr were cloned between NdeI and XhoI restriction sites while CDS of Δ32mpr and Δ50mpr were cloned between NheI and NotI in pET21b vector. CDS of ΔCmpr and ΔTMmpr were cloned in pMSQSCHS plasmid and TMGFP was cloned in pMSQSNHS (28) were transformed in E. coli BL21(DE3). Bacterial culture was grown in LB broth containing ampicillin at 37°C until the culture optical density at 600 nm (OD600) reached 0.6. The culture was induced by the addition of IPTG to a final concentration of 1 mM. Post-induction, culture was incubated at 37°C for 3 h, and then proceeded for protein purification using Ni-NTA affinity chromatography as described previously (28). Finally, purified proteins were dialysed against storage buffer (50 mM Tris–Cl pH 8.0, 200 mM NaCl, 5 mM β-mercaptoethanol and 40% glycerol). After dialysis, proteins were checked on SDS-PAGE and quantified using Bradford reagent (BIORAD).
Nuclease activity
To prepare linear dsDNA templates for nuclease activity, pMV261 plasmid (4488 bp) was digested with PvuII, PstI or SpeI restriction enzyme to yield blunt, 3′ overhang, or 5′ overhang ends, respectively, followed by ethanol precipitation. 200 ng of the template DNA was subjected to nuclease activity of Mpr (4 μM) in the presence of Buffer T (10×; 50 mM Tris–Cl pH 8.0, 100 mM NaCl, 1 mM DTT, 0.1 mM EDTA and 10 mM MgCl2) and incubated at 37°C for 60 min; Buffer T was used for all the nuclease experiments. The digestion reaction was terminated by adding 20 mM EDTA and loaded on 0.8% agarose gel. The exonuclease assay was also carried out with radiolabelled DNA. Briefly, the blunt end DNA was radiolabelled using polynucleotide kinase and γ32PATP. The radiolabelling at the 3′ end was carried out by digesting the DNA with SpeI followed by end-filling using α32PdCTP and Klenow (exo–) followed by cleanup with Monarch® PCR & DNA Cleanup Kit (NEB). The radiolabelled DNA was subjected to nuclease activity, and the reaction was resolved on PEI Cellulose F TLC plates (Merck) with 1.5 M KH2PO4 (pH 3.4) to separate the released 32P-labelled nucleoside monophosphate; this method was derived from the radioactivity-based assay available to examine the synthesis of secondary messenger molecules of bacteria on TLC (30). The TLC plate was further dried and exposed to a phosphor screen, and the autoradiogram was recorded on Typhoon FLA9000 phosphor imager (GE Healthcare).
Circular dichroism-based analysis
The CD profiles of Mpr and Δ50Mpr were recorded on a Jasco J-815 spectropolarimeter (Jasco, Japan). The proteins were dialyzed against CD buffer (50 mM Sodium phosphate buffer, pH 8.0, 50 mM NaCl and 1 mM dithiothreitol). Wavelength scans were recorded at an optical path length of 1 mm with a final protein concentration of 50 μM. An average of three scans was taken at 25°C, and the buffer subtracted values were converted into molar ellipticity.
Size exclusion chromatography
The native oligomeric state of Mpr was analysed by performing size exclusion chromatography on a Superdex75 10/300 GL column (GE Healthcare). 200 μg of the protein samples in 50 mM Tris–Cl pH 8.0, 5 mM β-mercaptoethanol, 200 mM NaCl and 5% glycerol were loaded on to the column, and the elution of the proteins was monitored at 280 nm. The peaks were compared against the calibration proteins [cytochrome c (12.5 kDa), carbonic anhydrase (29 kDa), albumin (66 kDa) and alcohol dehydrogenase (150 kDa)] (Sigma Aldrich).
Microscopy imaging
MsmWT cells expressing Mpr and its derivatives fused with N-terminal GFP were visualized on Axio Imager.M2 microscope (Zeiss, Jena, Germany) under a 100× oil-immersion objective. The cultures were induced with 0.2% acetamide at 37°C for 3 h with constant shaking. Next, 0.2 ml of the induced bacterial culture was stained with FM4-64 (N-(3-Triethylammoniumpropyl)-4-(6-(4-(diethylamino) phenyl) hexatrienyl) pyridinium dibromide) membrane staining dye as per the manufacturer's instructions. Samples were washed thrice with PBST (phosphate buffered saline with Tween-20), resuspended in 0.1 ml of PBST followed by spotting on 1.0% agarose pad. The agarose pad was then placed on microscopy slide and imaged. Imaging was carried out by using GFP (488 nm) and FM4-64 (506 nm) filters.
Construction of mpr genetic knockout and complementation
Construction and confirmation of mpr genetic knockout were carried out by employing the strategy as described before (31–33). Briefly, upstream and downstream 500bp fragments of mpr were PCR amplified from MsmWT genomic DNA, whereas hygr cassette was PCR amplified from pVV16 vector (obtained through BEI resources, NIAID, NIH: NR-13402). AES (allelic exchange substrate) was then constructed using overlapping PCR with primers listed in Table 2. Linear AES DNA fragments were then electroporated in MsmWT cells containing pJV53 (kind gift from Graham Hatfull, University of Pittsburgh, USA; Addgene plasmid #26904). Plasmid curing of pJV53 was performed as described (34). Plasmid-cured cells were further verified by sequencing of the targeted region. For the complementation of phenotype, pMA-His vector was modified; here, acetamidase gene promoter was removed, and mpr promoter-containing region (1 kb upstream of mpr) with mpr was cloned between XbaI and EcoRV restriction enzyme sites to yield pMM-mpr. For control, pMA-GFP vector was modified to pMM-GFP, wherein acetamidase gene promoter was replaced with mpr promoter between XbaI and EcoRV restriction enzyme sites. Both of the vectors were further modified and mycobacterial origin of replication was replaced with the mycobacterial genome integration cassette from pMV361 (35) between XbaI and PciI, creating pMIM-mpr and pMIM-GFP constructs.
Western blot analysis
Western blot analysis was carried out using appropriate antibodies (anti-Mpr, anti-His, anti-GFP or anti-GST). This was followed by probing with either anti-mouse IgG DyLight 680-conjugated secondary antibody (Thermo Scientific) or anti-rabbit IgG DyLight 800 conjugated secondary antibody (Invitrogen), as required. The blots were scanned on an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE).
Real-time PCR
To monitor the relative expression of mpr in both phage-infected and uninfected conditions, RNA was isolated from cells after 50 min of infection using RT-PCR RNeasy Mini Kit (Qiagen) and cDNA synthesis, and qPCR were performed. Relative expression level was normalized against the expression of the internal control gene, rpoB. Primers used in the RT-PCR are listed in the Table 2.
β-Galactosidase activity assay
The assay was performed as described elsewhere (36). Briefly, M. smegmatis culture was induced at OD600 ∼0.6. After 3 h of induction, 1 ml culture was harvested. The cell pellet was resuspended in 800 μl of Z-buffer (60 mM Na2HPO4·2H2O, 40 mM NaH2PO4·2H2O, 10 mM KCl, 10 mM MgSO4, 50 mM β-mercaptoethanol). Further, 100 μl chloroform and 1% SDS was added and the sample was incubated at 35°C for 5 min. 200 μl of ONPG (stock concentration = 4 mg/ml) was added and the sample was further incubated at 35°C. Once the sample started to develop pale yellow colour, the reaction was stopped by addition of 500 μl of 1 M Na2CO3 and time was noted. The sample was centrifuged at 10 000 r.p.m. for 3 min and OD420 and OD550 were measured. The activity in Miller units was calculated by the following formula:
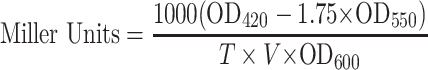 |
where, OD420 and OD550 are from reaction mixture, OD600 reflects the cell density in the washed cell suspension, T is the time of the reaction in minutes and V is the volume of culture used in the assay in ml.
FACS analysis
MsmWT cells transformed with pMA-mprHIS plasmid were grown in MB 7H9 liquid medium supplemented with 2% glucose and 0.05% tween 80. When OD600 reached 0.7, the cultures were induced with 0.2% acetamide at 37°C with constant shaking. Next, 0.5 ml of the induced bacterial culture was stained with DAPI for 30 min followed by washing with PBST (phosphate-buffered saline with Tween-20) and again staining with sytox green; dyes were used as per the manufacturer's instructions. Samples were washed thrice with PBST and resuspended in 0.2 ml of PBST. Cells samples were then subjected to BD FACS Aria III with BD FACS Diva software. Detection was carried out using sytox green (488 nm) and DAPI (450 nm) filters.
RESULTS
Mpr is a membrane-bound protein
We initiated our study with Mpr (MSMEG_1236) protein by carrying out a detailed in silico analysis of the protein sequence. SMART web server analysis of the Mpr sequence shows that it has one 18 amino acids long transmembrane (TM) region (33rd to 50th aa), which is followed by a 13 amino acid low complexity region (63rd to 75th aa) and a 129-amino acid long DUF4352 domain (83rd to 211st aa) (Figure 1A). Further enquiry with Pfam database (37) suggests that there are 25 domain architectures in which DUF4352 is present in nature. The most abundant architecture (∼87%) comprises only of DUF4352 with no other domain present, suggesting that DUF4352 mostly functions independently (Figure 1B). Data from web servers such as PsiPred (38), Protter (39), Hmmtop (40), RHYTHM (41), CCTOP (42) and TOPCONS (43) strongly suggest that Mpr protein harbours a single-pass transmembrane region that spans from 33rd to 50th residue with the C-terminal region containing the DUF4352 located in the periplasmic space (Figure 1C). In order to experimentally verify the membrane localization of Mpr, CDS of the full-length mpr gene, as well as its truncations (Δ50mpr and ΔTMmpr), were cloned in pMA-GFP vector (29) with an N-terminal GFP tag. The clones were transformed and expressed in M. smegmatis followed by imaging under a fluorescence microscope. The Mpr protein is found to be colocalized with FM4-64 dye (a lipophilic dye, known to label the membrane) at the periphery of M. smegmatis (Figure 2A), which confirms membrane localization of the protein. The truncated versions viz. Δ50Mpr and ΔTMMpr are found to be localized in the cytosol (Figure 2B and C), showing that the N-terminal region is indispensable for membrane localization of Mpr. It was interesting to find that the deletion of first 26 residues (Δ26Mpr) and the deletion of the C-terminal region (ΔCMpr) does not affect the membrane localization of Mpr (Figure 2D and E), which suggests that Mpr is able to anchor to the membrane without any signal sequence. However, full-length Mpr and its truncations (Δ26Mpr and ΔCMpr) that carry intact TM region show protein aggregation in the membrane affecting membrane integrity (shown with red arrows in Figure 2A, D, and E), causing toxicity in cell and also leading to loss of plasmid (shown with white arrows in Figure 2D and E). The data thus show that Mpr indeed is a membrane protein and that upon expression in M. smegmatis, it localizes to the membrane. The data also suggest that mpr overexpression affects membrane integrity and causes toxicity in the cell; the same was further explored and verified by monitoring the growth of M. smegmatis expressing Mpr (Supplementary Figure S1).
Figure 1.

In silico analysis of Mpr. (A) Domain organization of Mpr is shown. TM region is (green block) flanked both sides by five charged residues (light blue block) followed by a low complexity region (orange block) and lastly a DUF4352 domain (yellow oval), which contributes to 60% of the protein. Respective sizes of different regions are depicted by the blue lines. (B) Domain architecture of DUF4352 and its architectural abundance as a single domain protein across different DUF4352 domain containing proteins is presented. (C) Localization and membrane topology prediction of Mpr by Protter. The intra and extra-cellular regions are marked. DUF4352 domain is shown in the extracellular region. Two residues, K32 and N51, which form the boundary of the transmembrane region are shown.
Figure 2.
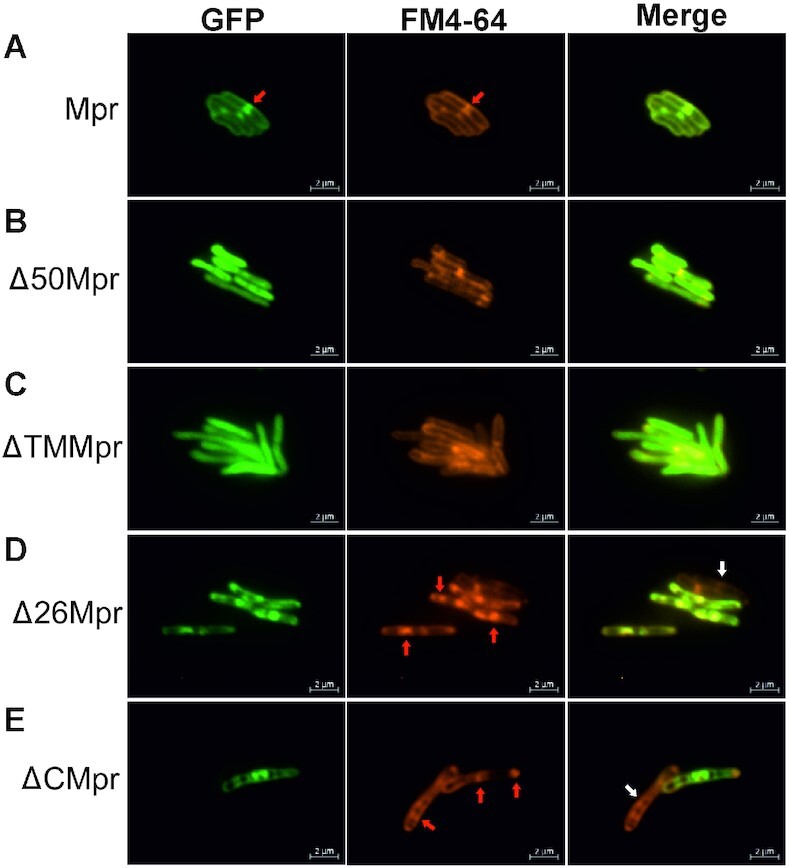
Fluorescent microscopy images of Mpr and its truncations. Respective gene/truncations are cloned with N-terminal GFP tag and expressed in MsmWT. (A) Mpr is observed to colocalize with FM4-64, suggesting membrane localization in vivo. (B) Deletion of first 50 amino acids (Δ50Mpr) renders protein unable to anchor to membrane. (C) loss of TM region (ΔTMMpr) restricts the protein in cytosol. (D) Deletion of first 26 amino acids (Δ26Mpr) does not affect localization of Mpr to the membrane. (E) N-ter region after deletion of C-terminal region (ΔCMpr) still localizes to the membrane. The truncations containing intact TM region show toxicity and cause aggregation in membrane affecting membrane integrity (shown with red arrow). Due to toxicity caused by protein expression, cells most likely lose plasmid and hence, no GFP expression is observed (shown with white arrow). Localization of protein is shown under GFP column. FM4-64 (specific for membrane localization) is used as a reference. Merge of GFP and FM4-64 channel shows relative localization of the target protein. Scale is shown at the bottom right of each image.
Mpr is a Mg2+dependent 3′ → 5′ DNA exonuclease.
Mpr overexpression has been shown to cause phage resistance in M. smegmatis (27). Since Mpr is a membrane protein, we hypothesized that Mpr either blocks the entry of phage DNA or degrades the DNA as it enters the cytoplasm; either of the scenarios will be bypassed if the phage DNA enters the bacterium via electroporation leading to successful phage infection as shown in an earlier study (27). Hence, Mpr was tested for nuclease activity on both linear blunt-end dsDNA as well as supercoiled plasmid DNA. It was observed that while the linear blunt-end dsDNA is readily digested by Mpr, supercoiled plasmid remains intact in the presence of the protein (Figure 3A). This clearly suggests that Mpr acts as an exonuclease. The nuclease activity of Mpr with linear DNA molecules having staggered ends (either 5′ or 3′ overhangs) was also examined to assess the specificity of Mpr towards a particular type of DNA end. Interestingly, Mpr is able to digest both the types of DNA ends equally efficiently (Figure 3B), suggesting that Mpr does not have end-specificity. Furthermore, the exonuclease activity of Mpr remains unaffected by DNA methylation since Mpr can digest linear unmethylated dsDNA (PCR product) and also methylated dsDNA template (prepared by plasmid DNA isolated from XL1-Blue cells) (Figure 3B). With given data, it can be concluded that Mpr protein of M. smegmatis is an exonuclease that is able to digest linear DNA molecules and that its activity is methylation independent.
Figure 3.
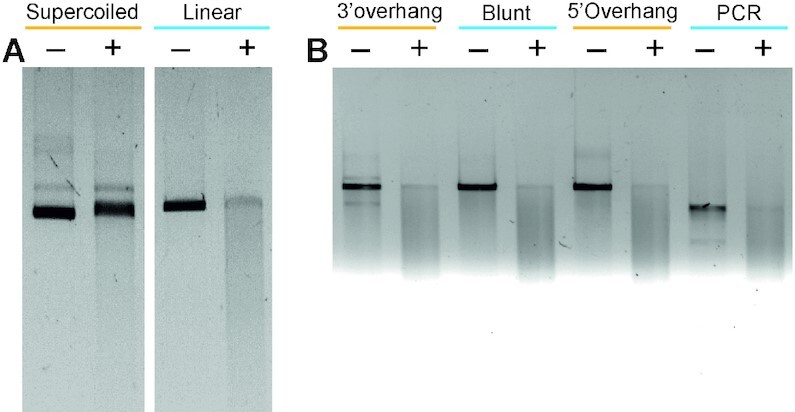
Exonuclease activity of Mpr. Agarose gel images showing the nuclease activity of Mpr are presented. (A) Exonuclease activity of Mpr. Supercoiled dsDNA and linear dsDNA are treated in the absence (–) or presence (+) of Mpr. (B) Nuclease activity of Mpr against different types of DNA ends in the absence (–) or presence (+) of Mpr is shown; ‘PCR’ represents the blunt-end dsDNA produced in a PCR reaction. Relevant information is shown in the figure.
Nucleases are generally known to be dependent on Mg2+ for their activity and it can be readily inhibited by chelating the Mg2+ by EDTA (44–46). Therefore, the Mg2+ dependence of Mpr for its nuclease activity was tested. Purified Mpr protein was dialysed against EDTA to remove any Mg2+ ions present with the protein and used to examine its nuclease activity in the presence and absence of MgCl2 and EDTA. Mpr displayed no activity in the absence of Mg2+ (Figure 4A). Further, the addition of EDTA readily inhibits the nuclease activity of Mpr. All of these data strongly suggest that Mpr is a Mg2+ dependent exonuclease.
Figure 4.
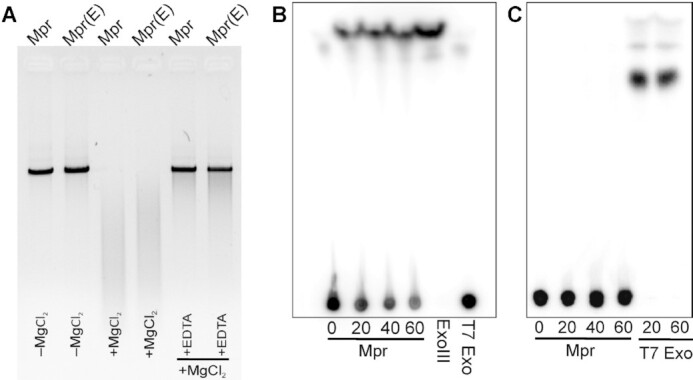
Nuclease property of Mpr. (A) Agarose gel image showing magnesium dependence of Mpr for its nuclease activity against blunt end linear dsDNA is presented. Mpr(E) depicts Mpr dialysed against EDTA. Digestion reaction in the presence (+) or absence (–) of MgCl2 or EDTA is labelled. (B) TLC plate image showing nuclease activity of Mpr against 3′ radiolabelled dsDNA is presented. T7 Exo and ExoIII are used as negative and positive controls, respectively. (C) TLC plate image showing nuclease activity of Mpr against 5′ radiolabelled dsDNA is presented. T7 Exo is used as positive control. Nuclease activity at different time interval is shown in mins.
After establishing the exonuclease activity, directionality of nuclease activity by Mpr was explored. It is widely known that exonucleases digest the linear DNA in either 5′→ 3′ or 3′→ 5′ direction or both. The examples include T7 exonuclease (47), Exonuclease III and Exonuclease V (48). To check the direction of the Mpr exonuclease activity, linear dsDNA was prepared and radiolabelled with 32P at either 3′ or 5′. T7 Exonuclease (digests in 5′→ 3′) and Exonuclease III (digests in 3′→ 5′) with known directionality for linear DNA digestion were used as controls. The release of radiolabelled nucleotide could be resolved on TLC only after treating the 3′ end labelled DNA with Mpr, which clearly suggests that Mpr is a 3′→ 5′ exonuclease (Figure 4B); similar digestion could also be observed with the Exonuclease III which is known to digest in 3′ to 5′ direction. Furthermore, such release of free nucleotide could not be observed on TLC with 5′ end radiolabelled DNA, which could be readily digested with the T7 exonuclease (Figure 4C).
The exonuclease activity lies in the N-terminus of Mpr and the DUF4352 domain is dispensable for nuclease activity in vitro
Several truncations of the protein were generated to understand which region of Mpr protein is essential for its nuclease activity. 26, 32 and 50 residues were deleted from the N-terminus of Mpr thus yielding Δ26Mpr, Δ32Mpr and Δ50Mpr, respectively. ΔCMpr that carried residues 1–60 aa from the N-terminus of Mpr followed by GFP was also generated. All the proteins were purified and the nuclease activity assay was carried out against a linear dsDNA. Surprisingly, it was observed that the first 60 residues are sufficient to show the nuclease activity, and that the DUF4352 domain is dispensable for the nuclease activity of Mpr in vitro (Figure 5). Furthermore, the first 26 residues are also not essential for the nuclease activity. We, therefore, conclude that the exonuclease activity of Mpr lies within the region corresponding to 27th to 60th amino acids.
Figure 5.
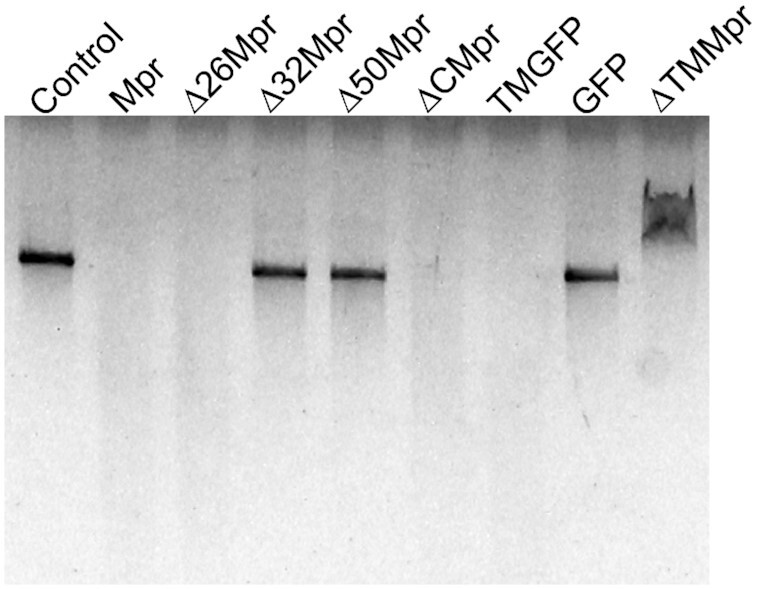
Nuclease activity of Mpr and its truncations. Agarose gel image showing nuclease activity of full length Mpr and its truncations on blunt end linear dsDNA is presented. Δ32Mpr, Δ50Mpr and ΔTMMpr are not able to digest DNA, whereas Mpr, Δ26Mpr, ΔCMpr and TMGFP can digest linear DNA. ΔTMMpr is able to bind to dsDNA, but is unable to digest it, resulting in mobility shift on agarose gel. Dialysis buffer is used as a control; GFP is used as a nonspecific control protein.
To further dissect the exonuclease activity-harbouring region in Mpr, two more truncations namely ΔTMMpr and TMGFP were created. The ΔTMMpr was constructed by deleting the transmembrane (TM) region, which spans from 33rd to 50th amino acids, from the full-length protein leaving the flanking charged residues intact. In TMGFP, the sequence spanning from 27th to 60th amino acids carrying the TM region along with the flanking charged residues were installed at the N-terminus of GFP. Exonuclease activity assays carried out with both the truncated proteins clearly show that the TMGFP protein, and not the ΔTMMpr, is able to digest linear dsDNA. Taken together, the data strongly supports the presence of exonuclease activity in the TM region of the Mpr protein. It is also interesting to note that ΔTMMpr is able to bind to dsDNA but is unable to digest it, as shown by upward mobility shift of the template DNA on agarose gel (Figure 5), indicating that the charged residues flanking the TM region are crucial for DNA binding, whereas the TM region itself harbours the exonuclease activity.
To conclude that the nuclease activity of Mpr lies in the TM region, and is not a result of a contaminating protein being co-purified, alanine scanning mutagenesis of the TM region, including the charged residues on either side, was performed. The entire 30aa (27th to 56th residue) carrying TM region and flanking charged residues were substituted with alanine as a group of three residues per mutant, thus generating a total of 10 triple alanine mutants, namely M1 to M10 (Figure 6A). Out of these mutants, M4 (IVG), M5 (GVI) and M7 (IVG) were omitted, considering these residues are hydrophobic in nature and highly unlikely to be involved in nuclease activity. M1, M2, M9, and M10 represent mutants in the flanking charged regions, and M3, M6 and M8 represent mutants in the TM region. Site-director mutagenesis (SDM) was performed as described previously (49) using respective SDM primers listed in Table 2. The mutants were transformed in E. coli BL21 (DE3) cells, followed by induction and protein purification on Ni-NTA chromatography. 4 μM of protein sample was used to digest 200 ng of linear dsDNA in buffer T at 37°C for 60 min. Mutants M2, M6 and M9 are unable to efficiently digest the template DNA, whereas Mpr, M1, M3, M8 and M10 are able to digest template DNA efficiently (Figure 6B). This clearly shows that the TM region indeed harbours nuclease activity of Mpr. Loss of function mutants also eliminate the possibility that the nuclease activity shown by Mpr is a result of co-purification of a protein having nuclease activity. SDS PAGE analysis of Mpr and its mutants did not show any significant difference in protein purification profile (Figure 6C).
Figure 6.

Nuclease activity of Mpr with alanine-substituted mutants. (A) Schematic representation of the TM region of Mpr; charged residues flanking the TM region are depicted in red. Residues mutated to alanine are highlighted in bold; respective positions of residues are mentioned. (B) Agarose gel image showing nuclease activity of Mpr and its mutants is presented; dialysis buffer is used as control. (C) Coomassie-stained SDS-PAGE gel image of purified Mpr and its mutants is presented; first lane corresponds to protein ladder with size of various protein bands marked for reference.
The N-terminal region of Mpr is largely unstructured
To further understand the structural and biophysical aspect of Mpr, MFDP2 (50) was used to predict the disordered region of the protein. The result from MFDP2 web server suggests that 44.19% (first 95 residues) of Mpr is disordered, whereas the C-terminal region of Mpr, which is predicted to house the DUF4352 domain, is structured (Figure 7A). To experimentally verify it, circular dichroism (CD) analysis of full-length Mpr as well as Δ50Mpr was performed. CD data clearly show that the full-length protein has a significant amount of random coil, whereas the Δ50Mpr protein has a more ordered structure with both α helix and β sheet (Figure 7B). This suggests the presence of a large disordered region in the N-terminus of Mpr, which also correlates with the data predicted by MFDP2 webserver. We also examined the oligomerization of Mpr protein by subjecting both full-length as well as Δ50Mpr proteins to size exclusion chromatography (SEC). SEC is based on the shape and size (hydrodynamic radius) of the eluted macromolecule (51). The data show that both full-length as well as Δ50Mpr proteins elute close to their apparent dimeric molecular mass of 38.5 and 34.5 kDa, respectively (Figure 7C); this is not uncommon as some exonucleases have been shown to function as dimers (52).
Figure 7.

Structural and biophysical properties of the protein. (A) Prediction of disordered content in Mpr by MFDp2. (B) Molar ellipticity plot of Mpr and Δ50Mpr as recorded by circular dichroism spectroscopy. (C) Size exclusion chromatography of Mpr and Δ50Mpr. ‘Standard’ represents the known molecular weight proteins that are used for calibration; peaks corresponding to specific molecular weights are marked. Relevant information is shown in the figure.
While DUF4352 is important for in vivo function of mpr, the protein itself may not be important in phage resistance, under normal conditions
To understand the role of mpr in vivo, protein coding sequence of full-length mpr and N-terminus region (first 60aa of mpr) were cloned in a mycobacterial genome integration vector under acetamidase gene promoter (since mpr overexpression causes toxicity (27), use of replicon system was avoided and integration system was preferred). pMIA series of vectors are derivatives of pMA series, where the origin of replication is replaced with integration cassette from pMV361 plasmid. These vectors offer two restriction sites EcoRV and HpaI, on either side of the tag that provides C-terminal and N-terminal tag, respectively. While the full-length mpr was cloned in pMIA-His vector so as to yield pMIA-Mpr-His with C-terminal His tag, the N-terminal 60 residues coding sequence was cloned in pMIA-GFP vector at EcoRV restriction site to obtain pMIA-ΔCmpr expressing N-terminal 60 amino acids of Mpr fused with a C-terminus GFP. A non-specific protein GST (pMIA-GST) is used as a control for phage infection assay. Phage infection was performed after 3 h of induction by acetamide using double agar overlay plaque assay (53). No plaques were observed in the cells overexpressing mpr gene, suggesting mpr could resist D29 phage infection as has been shown previously (27,54) and normal plaques were observed in GST. However, surprisingly, cells expressing the ΔCMpr protein do not resist D29 infection and show normal plaque morphology (Figure 8A), even though ΔCMpr protein is found to be functionally active in vitro and is able to digest linear dsDNA (Figure 5). This suggests that DUF4352 is critical for in vivo function of Mpr.
Figure 8.
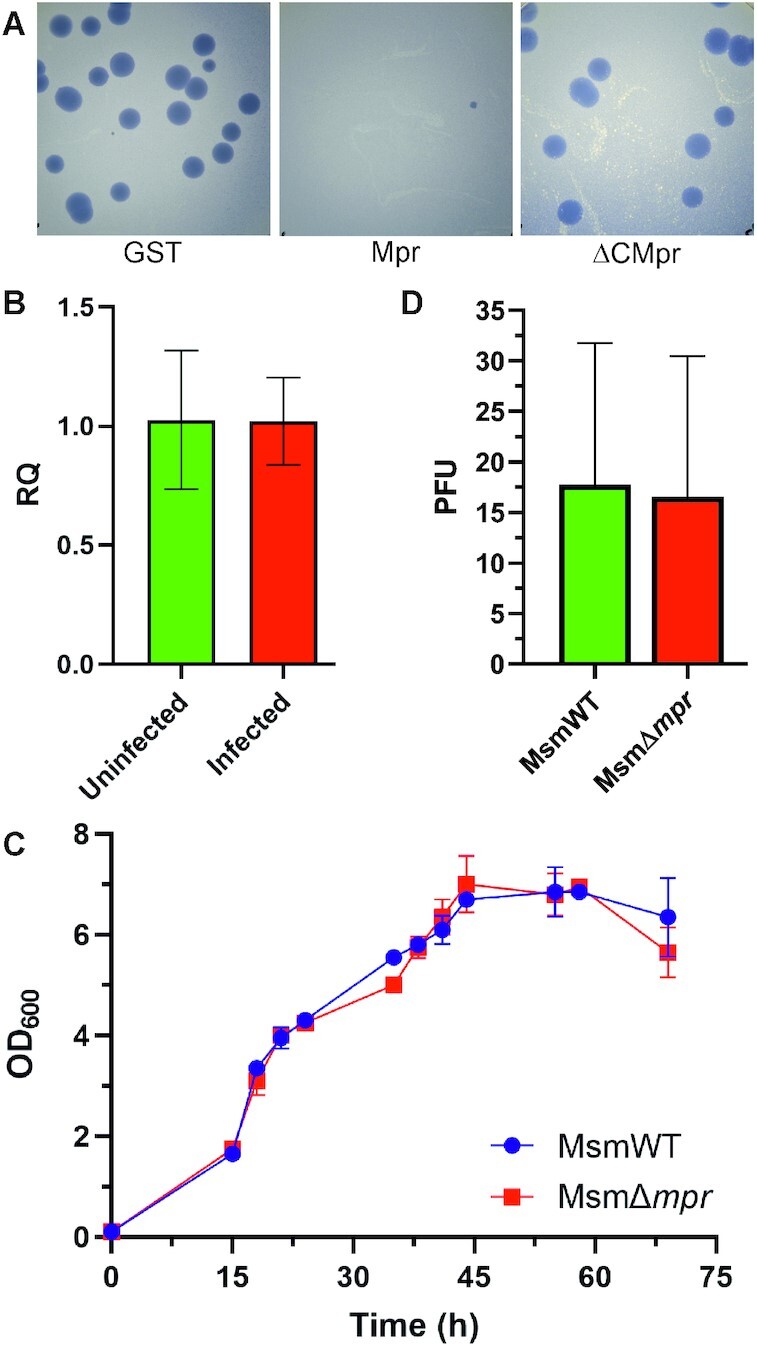
Phage resistance by Mpr. (A) Agar plate images showing phage resistance from a single copy of target gene (GST, mpr, or ΔCmpr) expressed under acetamidase promoter from an integrative system using double agar overlay plaque assay are shown. (B) RT-PCR of mpr under uninfected and phage-infected conditions. (C) Growth curve of MsmΔmpr vs MsmWT. (D) Graph showing PFU count in MsmWT and MsmΔmpr is shown.
mpr upon overexpression has been shown to confer resistance against phage infection but even so, wildtype M. smegmatis cells carrying the full-length mpr gene are very much susceptible to phage infection. To answer this conundrum, RT-PCR was performed under phage infection condition to examine if mpr overexpresses during phage infection. Interestingly, RT-PCR of M. smegmatis carried out 50 min after D29 phage infection shows no overexpression of mpr gene in wildtype cells (Figure 8B). The result correlates with the fact that M. smegmatis wildtype cells (MsmWT) are susceptible to D29 phage infection and further indicates that mpr plays some other comprehensive role in vivo. Hence to examine the significance of Mpr in M. smegmatis biology, the mpr gene from M. smegmatis genome was deleted and a knockout of mpr (MsmΔmpr) was generated. MsmWT and MsmΔmpr growth was monitored in MB7H9 liquid medium supplemented with 0.05% tween 80 and 2% glucose by measuring the optical density of the culture at 600 nm (OD600) at an interval of 3 h. The growth profile of MsmΔmpr is found to be indifferent from that of the MsmWT (Figure 8C), which suggests that mpr is a non-essential gene at least under normal growth conditions. To further check if deletion of mpr affects phage susceptibility, a D29 phage infection with MsmΔmpr and MsmWT was performed using double agar overlay method, which surprisingly shows no significant change in PFU (Figure 8D), indicating that mpr is not a surface receptor for D29 phage. Additionally, mpr does not play an essential role against phage infection under natural conditions.
mpr promoter is tightly regulated and shows no expression under any growth phase
To better understand the biological relevance of mpr and its role in vivo, it was important to determine if mpr is expressed during any of the growth phases of the cell and if yes, what is the level of expression? Levels of expression are tightly regulated and maintained in the cell, as over or reduced expression of genes may affect physiological conditions in the cell, resulting in a mutant phenotype (55). To ascertain expression from mpr promoter, 1kb upstream of mpr gene was cloned in pSD5b vector, a promoter-less mycobacterial shuttle vector having lacZ as a reporter gene (56), giving rise to pSD5bPrmpr vector. β-galactosidase assay was performed at 20, 44, and 68 h intervals corresponding to log, stationary, and decline phase of M. smegmatis; empty pSD5b vector was used as negative control and rel promoter (pR300lac), which is known to give weak constitutive expression in M. smegmatis (30) was used as a positive control. Expression from mpr promoter is found to be less than 20 Miller units (MU), which is nearly 4–5-fold less as compared to weak rel promoter, suggesting a tight regulation under mpr promoter (Figure 9A). We next checked the mpr expression during different growth phases of wild type M. smegmatis. For this purpose, western blotting was performed with 24, 48 and 72 h cultures. Mpr could not be detected in any of the growth phases (Figure 9B), and such levels of Mpr are not able to resist phage infection as evident from susceptibility of MsmWT against phage D29. Mpr has been shown to resist phage infection, when expressed under hsp60 promoter (27). To have a better understanding of such level of expression, western blot analysis was carried out. For this purpose, mpr promoter was cloned upstream of GFP in pMH-GFP vector backbone (29) by replacing hsp60 promoter between DraI and EcoRV, creating pMM-GFP; the pMH-GFP was used as a reference. The amount of GFP expression from mpr promoter was found to be roughly 100-fold less as compared to that from hsp60 promoter (Figure 9C). It is to be noted that this level of expression is from a replicon system having an origin of replication from pAL5000 plasmid and having a copy number of 3–5 (57). In wildtype M. smegmatis cell, there is only one copy of mpr gene, effectively reducing the amount of Mpr present in the cell. Again, these levels of mpr expression are not sufficient to resist phage infection, as evident from the susceptibility of wildtype cells to phage infection. Moreover, overexpression of mpr is toxic to cell (Figure 2 and Supplementary Figure S1). To check if mpr can resist phage infection with higher level of expression from its endogenous promoter, the gene was cloned along with its promoter in pMH-His vector (a replicon system) between DraI and EcoRV sites (29), creating pMM-Mpr. Phage infection was carried out in M. smegmatis having either pMM-Mpr or pMM-GFP (control). Interestingly, even with 3–5-fold more expression (achieved because of the copy number of plasmid), mpr–overexpressing cells were unable to resist phage infection effectively and turbid plaques were observed (Figure 9D). This again suggests that mpr expression under its endogenous promoter is not sufficient to confer resistance against phage infection.
Figure 9.
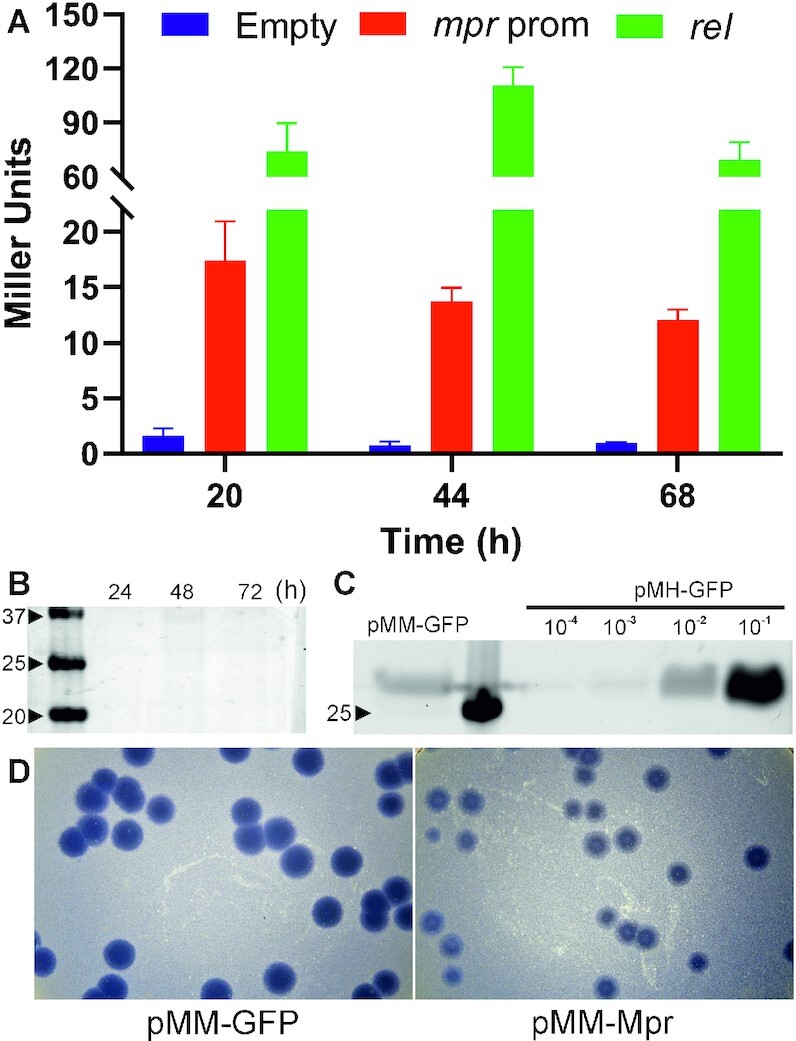
Analysis of expression under mpr promoter. (A) β-Galactosidase assay to check expression under mpr promoter during different growth phases of M. smegmatis. Empty pSD5b vector is used as a negative control and rel promoter (weak constitutive promoter) is used as a positive control. (B) Western blot analysis of mpr expression from wildtype M. smegmatis during log (24 h), stationary (48 h) and decline phase (72 h); anti-Mpr antibody is used to detect Mpr expression (C) Western blot analysis of GFP expression under mpr promoter (pMM-GFP) as compared to hsp60 promoter (pMH-GFP). hsp60 promoter is used as a reference (both the cultures were grown at 37°C and no heat shock or induction was given); anti-GFP antibody is used to detect GFP. (D) Phage infection in wildtype cells expressing mpr (pMM-Mpr) from its endogenous promoter in a replicon system (copy number 3–5); wildtype cells expressing GFP (pMM-GFP) from mpr promoter are used as control.
mpr plays a key role in appearance of mutant colonies that are resistant to phage infection
In order to further understand the significance of Mpr, both MsmWT and MsmΔmpr cells were infected with a high titer of D29 phage (MOI ≥ 1000) using double agar overlay plaque assay and incubated at 37°C. On the next day, it was observed that all the MsmWT cells were lysed by phage infection and the agar plate was found to be clear. The infected culture plates were further incubated for 4–5 days, and new colonies started to appear. The experiment resulted in the appearance of bacterial colonies with several fold higher numbers of colonies being observed in the wildtype cells compared to the knockout (Figure 10A and B). Since these colonies appeared on a D29 phage-seeded plate, it implied that the appeared colonies were resistant to D29 phage.
Figure 10.
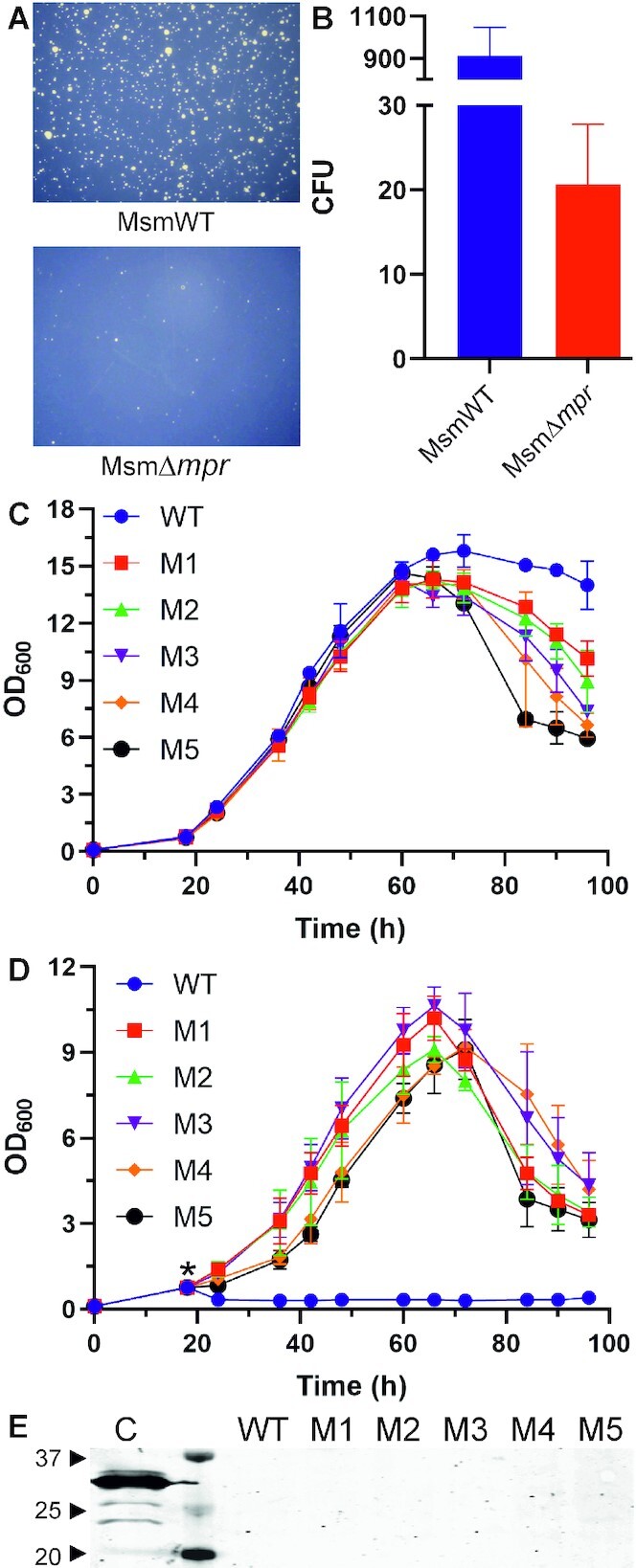
Phage resistant strains. (A) Emergence of phage resistant colonies in D29 infected MsmWT and MsmΔmpr culture after prolonged incubation on agar plate is shown. (B) Graphical representation of number of resistant colonies in MsmWT as compared to MsmΔmpr is shown. Single mutant colony was isolated by using serial dilution and secondary culture was grown in MB 7H9 media supplemented with OADC till 0.8 OD600 (normalized). Growth curve was performed and OD600 was observed at interval of 6 h under uninfected (C) and infected (D) conditions. ‘*’ in panel D represents phage addition into the culture. (E) Western blot analysis for the expression of Mpr in the infected mutant cultures is shown. Cell lysate (C) of the Mpr overexpressing E. coli cells from an expression vector is used as a control. WT represents the uninfected wildtype M. smegmatis, whereas M1 to M5 are the five different mutants. The lane between the control and the WT is the protein ladder; a few molecular weight bands are marked (in kDa). Anti-Mpr antibody is used to detect Mpr expression.
To assess if the phage resistance is because of a change in the proteome (resulting in a transient resistance) or the genome (establishing stable resistance in the population), the colonies were first patched on MB agar in the absence of phage and grown over several generations. Next, the mutant colonies were grown in MB 7H9 liquid medium supplemented with Tween 80 and glucose, in the absence of phage. The log phase cultures were washed with PBST and serially diluted followed by plating on MB agar plate to obtain single isolated colonies. Five of these mutant colonies (M1 to M5) were randomly selected and tested for D29 phage resistance in liquid culture with wildtype M. smegmatis as control. The OD600 of the culture was normalized to 0.8 and observed at an interval of 6 h in the presence and absence of phage D29. The mutants showed a similar growth profile to wildtype in the log phase but differentiated in stationary and decline phase in the uninfected condition (Figure 10C). In the infected condition, however, wildtype showed steep decline in OD600 while mutants resisted the infection and showed a continuous rise in OD600 during log phase, followed by a short stationary phase and then a decline phase (Figure 10D). Furthermore, we checked Mpr expression in these mutants that are challenged with phage. Interestingly, no expression of Mpr was detected (Figure 10E); lysate from E. coli cells overexpressing Mpr was used as positive control. This shows that the phenotypic changes are stable and permanent, and not because of change in proteome, thus validating that the emergence of phage-resistant colonies in the presence of mpr is definitely a result of genetic changes occurring in the bacterial genome.
To further validate the role of mpr in the appearance of mutant colonies, complementation of phenotype in the mpr knockout was carried out. Here, in order to match the expression and regulation of mpr to that in the wildtype bacterium, the mpr gene was cloned with its endogenous promoter region in a mycobacterial integration system (pMIM-Mpr), while GFP (pMIM-GFP) was used as control. MsmWT (WT::GFP) when challenged with phage D29 showed the emergence of mutant colonies, whereas MsmΔmpr complemented with GFP (Δmpr::GFP) did not show emergence of such mutant colonies. Interestingly, complementation of MsmΔmpr with mpr (Δmpr::mpr) showed recovery of the phenotype and number of colonies that appeared were comparable to wildtype (Figure 11A). The data thus obtained from the complementation study strongly indicates the critical role of mpr in the emergence of mutant colonies.
Figure 11.
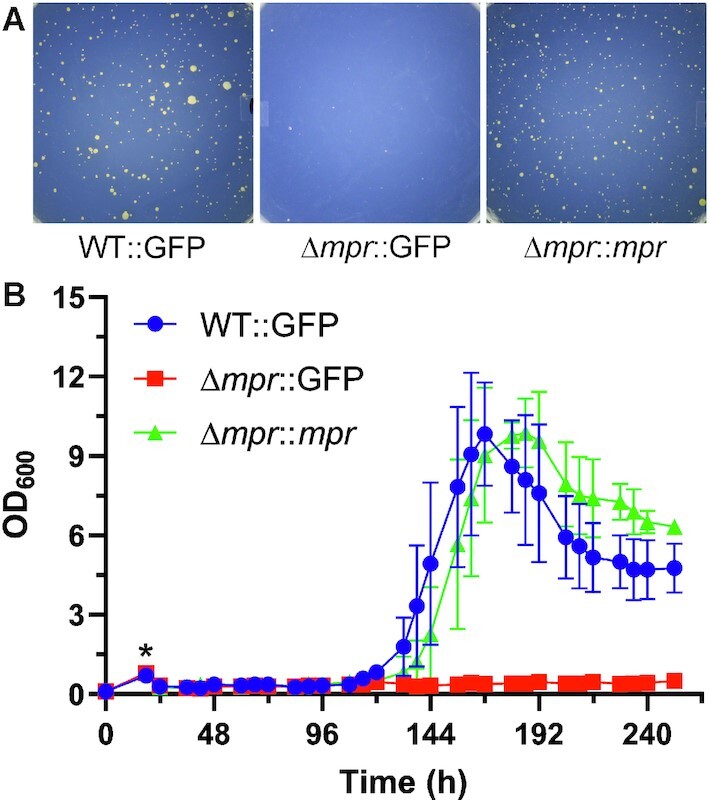
Complementation of phenotype. (A) Agar plate images of the complementation of the phenotype in Δmpr by GFP (Δmpr::GFP) and mpr (Δmpr::mpr) are shown. (B) Growth curve of M. smegmatis wildtype, mpr knockout, and knockout complemented with mpr under phage infection condition is shown. Cultures were allowed to grow till OD600 ∼0.8 (normalized) and infected (‘*’) with high titre phage. OD600 was monitored at every 6 h and plotted.
To understand the population dynamics of emerging mutant colonies in wildtype and mpr knockout cells under phage infection condition, growth profiles of wildtype (WT::GFP), knockout (Δmpr::GFP) and complemented (Δmpr::mpr) bacteria were recorded. The cultures were grown in MB7H9 liquid media supplemented with OADC and glucose till OD600 reached 0.8 followed by infection with high titre phage D29. The growth was monitored by measuring OD600 at an interval of 6 h. All the three cultures showed a decline in OD600 soon after infection, indicating susceptibility to D29 phage. A rise in OD600 was observed from fifth day onward in wildtype culture followed by complemented culture, whereas no rise in OD600 in knockout culture was observed (Figure 11B). The mutants emerged and the cultures followed a typical growth profile with a log phase followed by a stationary and a decline phase. This further validates the critical role of mpr in the emergence of mutant colonies.
DISCUSSION
Phage therapy is being considered as an alternative strategy to combat bacterial infections, especially those being caused by drug-resistant bacteria. However, phage resistance is one phenomenon that presents a serious threat to the widespread use of phage therapy. It is, therefore, important to understand the mechanism(s) involved in phage resistance by bacteria. In this regard, we present here the molecular dissection of Mpr, a 23 kDa protein. Mpr contains only two identifiable regions, viz. transmembrane (TM) region, which is required for membrane insertion, and DUF4352 domain, whose function, as the name suggests domain of unknown function (DUF), is unknown. The microscopy data clearly shows membrane localization of Mpr. Interestingly, deletion of first 26 residues (Δ26Mpr) or deletion of the whole C-terminal (ΔCMpr) region does not affect Mpr localization, suggesting that the membrane localization of Mpr is a signal sequence-independent process. The overexpression of mpr leads to toxicity in cell, as observed by growth curve analysis under mpr overexpression condition (Supplementary Figure S1A). The cause of toxicity was found to be the result of membrane perturbation. The overexpression of membrane localizing proteins resulted in compromised cell membrane, which can be observed as the membrane is now permeable to otherwise nonpermeable sytox green (Supplementary Figure S1B). With the several lines of evidence, the toxicity can be linked to the overexpression of 34 amino acids (27th–60th aa) residues that span the membrane. These 34 aa are critical for membrane localization and accumulation of protein in the membrane upon overexpression, causing toxicity in the cell. The deletion of 61st–136th base pairs (21st–45th aa) eliminates the phage resistance phenotype of Mpr (27); this is part of the TM region of Mpr and its truncation will affect membrane localization of Mpr, and its accumulation in the membrane. Hence, it is understandable that the deletion of 61–136 bp would eliminate the phage resistance phenotype of Mpr.
Since overexpression of mpr causes resistance against phage infection and its localization to the membrane could be established, the possibility of interaction of Mpr with injected phage DNA to resist phage infection could not be ignored and thus exonuclease activity of Mpr was discovered. Upon further investigation, Mpr was found to be novel membrane-bound exonuclease whose catalytic activity resides in 34aa (27th–60th aa) of TM region, and DUF4352 was found to be critical for resistance against phage infection in vivo. With the nuclease activity of Mpr discovered, it was tempting to assume that upon overexpression, Mpr is able to interact with and digest the injected phage DNA, thus providing resistance against phage infection. Besides phage resistance, mpr overexpression also causes toxicity in cell. This presents a conflicting situation, where mpr overexpression is needed to resist phage infection but the same would lead to toxicity in cell, indicating mpr is probably playing a different biological function in M. smegmatis. Moreover, additional copy of gene is insufficient to resist phage infection, again hinting towards a different biological role of mpr in MsmWT.
The biological function of a gene in vivo is determined by the level of expression or amount of protein molecules available in the cell to perform its function, in this case phage resistance. We checked Mpr levels under different growth phases of M. smegmatis and found that mpr is not expressed in detectable amounts during any of the growth phases or is expressed in extremely low (undetectable) amounts. Mpr when expressed from its endogenous promoter in a replicon system (copy number 3–5) still could not effectively resist phage infection even with 3–5-fold higher level of expression (due to copy number of the expression vector), and turbid plaques were observed. Moreover, mpr is not overexpressed during phage infection as well. With all these evidences, we believe that mpr is possibly playing some other biological function in vivo which is explored in this study. Again, phage resistance phenotype is only observed upon ‘overexpression’ of mpr, but to determine its biological function, its endogenous expression levels must be considered which are insufficient to resist phage infection. It also appears logical that mpr would not be overexpressed under normal conditions as mpr overexpression leads to toxicity, which would be disadvantageous to the cell, thus suggesting that mpr expression is tightly regulated in the cell. The same was observed with western blotting analysis of GFP when expressed from mpr promoter (pMM-GFP). A very low level of GFP expression was observed under mpr promoter, which is ∼100-fold less than what is desirable to effectively resist D29 phage infection. Nevertheless, the possibility of mpr overexpression under some particular condition cannot be ruled out. Furthermore, no difference in phage infection or PFU was observed between MsmWT and MsmΔmpr cells. Thus, a different biological function of mpr beyond phage resistance cannot be ignored.
Overexpression of mpr can certainly resist phage infection but the emergence of mutant/phage resistant colonies in the presence of Mpr is a different function altogether, which was not explored in any earlier study. In case of resistance, cells immediately start to grow, resisting the infection, but in case of emergence of mutant colonies, they emerge after a prolong incubation of 4–5 days. The appeared colonies were found to have different morphology as compared MsmWT (Supplementary Figure S2A). The rough phenotype, typical of wildtype cells was lost in the mutants. Most of the mutant colonies displayed a mucoidal and smooth phenotype. In contrast to wildtype, which shows dry and flaky colonies, mutant colonies were sticky in nature. Some mutant colonies also showed concentric circles, which are not observed in wildtype, further suggesting that the appearing colonies are different mutant strains of MsmWT. With all the morphological changes, it appears that the change in phenotype in mutants is mostly associated with a change in cell surface. Furthermore, many mutant colonies were found to have similar morphology, indicating same or similar pathways are affected. To check if phage resistance in mutant cells is a result of compromised phage adsorption, cells were incubated with D29 phage for 30 min followed by removal of the cell-phage complex by centrifugation. PFU of unabsorbed phage in solution was determined by infection with wildtype cells (Supplementary Figure S2B). Interestingly, phage adsorption is found to be affected in all of the mutants, suggesting alteration in cell surface, which is consistent with change in cell morphology in mutants. The emergence of phage-resistant colonies in the presence of mpr is definitely a result of genetic changes occurring in the bacterial genome. Moreover, our data strongly confirm that mpr is responsible for appearance of mutant colonies. It, therefore, appears that Mpr is either directly or indirectly interacting with host DNA or triggering some downstream pathway, possibly stress response pathway, causing mutations in the genome, thus increasing the survivability of M. smegmatis.
It was interesting to see the emergence of mutant/resistant colonies appearing in the presence of Mpr. Although a few colonies appeared in mpr knockout as well, the mutants appearing in the absence of mpr were found to be still susceptible to phage infection, with the exception of one out of seven experiments, in which OD600 for mutant appearing in knockout background reached 3.8 (data not shown). The high level of resistance in mutants appearing in MsmWT as compared to MsmΔmpr suggests that the mechanism underlying in the appearance of mutant colonies in the presence and absence of mpr is different. Interestingly, continuous fluctuation in OD600 (between 0.2 and 0.6) was observed in case of knockout suggesting complex dynamics of appearance of mutants and lysis happening due to phage infection (Supplementary Figure S3). It shows that mutant colonies that are appearing in knockout culture show very low level of resistance against infection and are still susceptible to phage; at the same time, they manage to persist while resisting the infection.
With the growing use of phage in medicine and therapies to control infectious diseases, it is imperative to have a deep understanding of the mechanisms behind phage resistance if we want to exploit phage therapy to its full potential. Lack of profound understanding of phage resistance poses a great threat to the widespread application of phage therapy. Reckless and unaccounted use of antibiotics has led to the ascent of MDR and XDR. In similar lines, a careless and an irresponsible use of phage therapy without an in-depth understanding of phage resistance may have grave consequences and may sound the advent of pathogenic strains that are resistant to not only multiple drugs but also phages (58,59). Phage resistance is a direct threat to phage therapy, the understanding of which is still lacking. We believe that our work will enhance the understanding of phage resistance and will help in further developing phages as therapeutics, while considering phage resistance also as one of the possible problems.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mohit Garg for helping in optimisation of protein isolation protocol. SPS thanks Council of Scientific and Industrial Research, Govt. of India for the Senior Research Fellowship.
Contributor Information
Surya Pratap Seniya, Microbiology and Molecular Biology Laboratory, Department of Biological Sciences, Indian Institute of Science Education and Research (IISER), Bhopal 462066, India.
Vikas Jain, Microbiology and Molecular Biology Laboratory, Department of Biological Sciences, Indian Institute of Science Education and Research (IISER), Bhopal 462066, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Science and Engineering Research Board, Govt. of India [CRG/2020/004231 to V.J.]. Funding for open access charge: Science and Engineering Research Board, Govt. of India [CRG/2020/004231].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dedrick R.M., Guerrero-Bustamante C.A., Garlena R.A., Russell D.A., Ford K., Harris K., Gilmour K.C., Soothill J., Jacobs-Sera D., Schooley R.T.et al.. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019; 25:730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kortright K.E., Chan B.K., Koff J.L., Turner P.E.. Phage therapy: a renewed approach to combat antibiotic-resistant bacteria. Cell Host Microbe. 2019; 25:219–232. [DOI] [PubMed] [Google Scholar]
- 3. Sulakvelidze A., Alavidze Z., Morris J.G.J.. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001; 45:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Samson J.E., Magadán A.H., Sabri M., Moineau S.. Revenge of the phages: defeating bacterial defences. Nat. Rev. Microbiol. 2013; 11:675–687. [DOI] [PubMed] [Google Scholar]
- 5. Hampton H.G., Watson B.N.J., Fineran P.C.. The arms race between bacteria and their phage foes. Nature. 2020; 577:327–336. [DOI] [PubMed] [Google Scholar]
- 6. Suttle C.A. Marine viruses — major players in the global ecosystem. Nat. Rev. Microbiol. 2007; 5:801–812. [DOI] [PubMed] [Google Scholar]
- 7. Dy R.L., Richter C., Salmond G.P.C., Fineran P.C.. Remarkable mechanisms in microbes to resist phage infections. Annu. Rev. Virol. 2014; 1:307–331. [DOI] [PubMed] [Google Scholar]
- 8. van Houte S., Buckling A., Westra E.R.. Evolutionary ecology of prokaryotic immune mechanisms. Microbiol. Mol. Biol. Rev. 2016; 80:745–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rostøl J.T., Marraffini L.. Ph)ighting phages: how bacteria resist their parasites. Cell Host Microbe. 2019; 25:184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandes S., São-José C.. Enzymes and mechanisms employed by tailed bacteriophages to breach the bacterial cell barriers. Viruses. 2018; 10:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu M., Deora R., Doulatov S.R., Gingery M., Eiserling F.A., Preston A., Maskell D.J., Simons R.W., Cotter P.A., Parkhill J.et al.. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science. 2002; 295:2091–2094. [DOI] [PubMed] [Google Scholar]
- 12. Nordström K., Forsgren A.. Effect of protein a on adsorption of bacteriophages to Staphylococcus aureus. J. Virol. 1974; 14:198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Riede I., Eschbach M.L.. Evidence that TraT interacts with OmpA of Escherichia coli. FEBS Lett. 1986; 205:241–245. [DOI] [PubMed] [Google Scholar]
- 14. Pedruzzi I., Rosenbusch J.P., Locher K.P.. Inactivation in vitro of the Escherichia coli outer membrane protein FhuA by a phage T5-encoded lipoprotein. FEMS Microbiol. Lett. 1998; 168:119–125. [DOI] [PubMed] [Google Scholar]
- 15. Hammad A.M.M. Evaluation of alginate-encapsulated Azotobacter chroococcum as a phage-resistant and an effective inoculum. J. Basic Microbiol. 1998; 38:9–16. [Google Scholar]
- 16. Zaleski P., Wojciechowski M., Piekarowicz A.. The role of Dam methylation in phase variation of Haemophilus influenzae genes involved in defence against phage infection. Microbiology. 2005; 151:3361–3369. [DOI] [PubMed] [Google Scholar]
- 17. Labrie S.J., Samson J.E., Moineau S.. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010; 8:317–327. [DOI] [PubMed] [Google Scholar]
- 18. Pingoud A., Jeltsch A.. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001; 29:3705–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Williams R.J. Restriction endonucleases: classification, properties, and applications. Mol. Biotechnol. 2003; 23:225–244. [DOI] [PubMed] [Google Scholar]
- 20. Tock M.R., Dryden D.T.. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005; 8:466–472. [DOI] [PubMed] [Google Scholar]
- 21. Vasu K., Nagaraja V.. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013; 77:53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Makarova K.S., Grishin N.V., Shabalina S.A., Wolf Y.I., Koonin E.V.. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct. 2006; 1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vestergaard G., Shah S.A., Bize A., Reitberger W., Reuter M., Phan H., Briegel A., Rachel R., Garrett R.A., Prangishvili D.. Stygiolobus rod-shaped virus and the interplay of crenarchaeal rudiviruses with the CRISPR antiviral system. J. Bacteriol. 2008; 190:6837–6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. La M., Ej S.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yaung S.J., Esvelt K.M., Church G.M.. CRISPR/Cas9-mediated phage resistance is not impeded by the DNA modifications of phage t4. PLoS One. 2014; 9:e98811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Page R., Peti W.. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016; 12:208–214. [DOI] [PubMed] [Google Scholar]
- 27. Barsom E.K., Hatfull G.F.. Characterization of a Mycobacterium smegmatis gene that confers resistance to phages L5 and D29 when overexpressed. Mol. Microbiol. 1996; 21:159–170. [DOI] [PubMed] [Google Scholar]
- 28. Singh M.I., Jain V.. Tagging the expressed protein with 6 histidines: rapid cloning of an amplicon with three options. PLoS One. 2013; 8:e63922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seniya S.P., Yadav P., Jain V.. Construction of E. coli—Mycobacterium shuttle vectors with a variety of expression systems and polypeptide tags for gene expression in mycobacteria. PLoS One. 2020; 15:e0230282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jain V., Saleem-Batcha R., China A., Chatterji D. Molecular dissection of the mycobacterial stringent response protein Rel. Protein Sci. 2006; 15:1449–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Kessel J.C., Hatfull G.F.. Recombineering in Mycobacterium tuberculosis. Nat. Methods. 2007; 4:147–152. [DOI] [PubMed] [Google Scholar]
- 32. Dubey A.A., Wani S.R., Jain V.. Methylotrophy in mycobacteria: dissection of the methanol metabolism pathway in Mycobacterium smegmatis. J. Bacteriol. 2018; 200:e00288-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patil V., Jain V.. Insights into the physiology and metabolism of a mycobacterial cell in an energy-compromised state. J. Bacteriol. 2019; 201:e00210-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mao X.-J., Yan M.-Y., Zhu H., Guo X.-P., Sun Y.-C.. Efficient and simple generation of multiple unmarked gene deletions in Mycobacterium smegmatis. Sci. Rep. 2016; 6:22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stover C.K., de la Cruz V.F., Fuerst T.R., Burlein J.E., Benson L.A., Bennett L.T., Bansal G.P., Young J.F., Lee M.H., Hatfull G.F.et al.. New use of BCG for recombinant vaccines. Nature. 1991; 351:456–460. [DOI] [PubMed] [Google Scholar]
- 36. Miller J.H. Experiments in Molecular Genetics. 1972; NY: Cold Spring Harbor Laboratory. [Google Scholar]
- 37. Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J.et al.. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021; 49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buchan D.W.A., Jones D.T.. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019; 47:W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Omasits U., Ahrens C.H., Müller S., Wollscheid B.. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014; 30:884–886. [DOI] [PubMed] [Google Scholar]
- 40. Tusnady G.E., Simon I.. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001; 17:849–850. [DOI] [PubMed] [Google Scholar]
- 41. Rose A., Lorenzen S., Goede A., Gruening B., Hildebrand P.W.. RHYTHM–a server to predict the orientation of transmembrane helices in channels and membrane-coils. Nucleic Acids Res. 2009; 37:W575–W580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dobson L., Reményi I., Tusnády G.E.. CCTOP: a consensus constrained TOPology prediction web server. Nucleic Acids Res. 2015; 43:W408–W412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsirigos K.D., Peters C., Shu N., Käll L., Elofsson A.. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015; 43:W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bickler S.W., Heinrich M.C., Bagby G.C.. Magnesium-dependent thermostability of DNase I. Biotechniques. 1992; 13:64–66. [PubMed] [Google Scholar]
- 45. Hoffmann P.J. Mechanism of degradation of duplex DNA by the DNase induced by herpes simplex virus. J. Virol. 1981; 38:1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cowan J.A. Magnesium activation of nuclease enzymes—the importance of water. Inorg. Chim. Acta. 1998; 275–276:24–27. [Google Scholar]
- 47. Mitsunobu H., Zhu B., Lee S.-J., Tabor S., Richardson C.C.. Flap endonuclease activity of gene 6 exonuclease of bacteriophage T7. J. Biol. Chem. 2014; 289:5860–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lovett S.T. The DNA exonucleases of Escherichia coli. EcoSal Plus. 2011; 4: 10.1128/ecosalplus.4.4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erijman A., Dantes A., Bernheim R., Shifman J.M., Peleg Y.. Transfer-PCR (TPCR): a highway for DNA cloning and protein engineering. J. Struct. Biol. 2011; 175:171–177. [DOI] [PubMed] [Google Scholar]
- 50. Mizianty M.J., Peng Z., Kurgan L.. MFDp2: accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. Intrinsically Disord. Proteins. 2013; 1:e24428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mori S., Barth H.G.. Size Exclusion Chromatography. 2013; Springer Science & Business Media. [Google Scholar]
- 52. Zuo Y. The DNase activity of RNase T and its application to DNA cloning. Nucleic Acids Res. 1999; 27:4077–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kropinski A.M., Mazzocco A., Waddell T.E., Lingohr E., Johnson R.P.. Clokie M.R.J., Kropinski A.M.. Enumeration of bacteriophages by double agar overlay plaque assay. Bacteriophages, Methods in Molecular Biology. 2009; 501:Totowa, NJ: Humana Press; 69–76. [DOI] [PubMed] [Google Scholar]
- 54. Rubin E.J., Akerley B.J., Novik V.N., Lampe D.J., Husson R.N., Mekalanos J.J.. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:1645–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Prelich G. Gene overexpression: uses, mechanisms, and interpretation. Genetics. 2012; 190:841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jain S., Kaushal D., Das Gupta S.K., Tyagi A.K.. Construction of shuttle vectors for genetic manipulation and molecular analysis of mycobacteria. Gene. 1997; 190:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ranes M.G., Rauzier J., Lagranderie M., Gheorghiu M., Gicquel B.. Functional analysis of pAL5000, a plasmid from Mycobacteriumfortuitum: construction of a ‘mini’ Mycobacterium-Escherichia coli shuttle vector. J. Bacteriol. 1990; 172:2793–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hesse S., Rajaure M., Wall E., Johnson J., Bliskovsky V., Gottesman S., Adhya S.. Phage resistance in multidrug-resistant Klebsiellapneumoniae ST258 evolves via diverse mutations that culminate in impaired adsorption. Mbio. 2020; 11:e02530-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Luong T., Salabarria A.-C., Roach D.R.. Phage therapy in the resistance era: where do we stand and where are we going?. Clin. Ther. 2020; 42:1659–1680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.