

Abstract
SETD2 is the sole histone methyltransferase responsible for H3K36me3, with roles in splicing, transcription initiation and DNA damage response. Homozygous disruption of SETD2 yields a tumor suppressor effect in various cancers. However, SETD2 mutation is typically heterozygous in DLBCL. Here we show that heterozygous SETD2 deficiency results in GC hyperplasia, increased competitive fitness, with reduced DNA damage checkpoint activity and apoptosis, resulting in accelerated lymphomagenesis. Impaired DNA damage sensing in Setd2 haploinsufficient GCB and lymphoma cells associated with increased AICDA induced somatic hypermutation, complex structural variants, and increased translocations including those activating MYC. DNA damage was selectively increased on the non-template strand and H3K36me3 loss was associated with greater RNAPII processivity and mutational burden, suggesting that SETD2 mediated H3K36me3 is required for proper sensing of cytosine deamination. Hence, Setd2 haploinsufficiency delineates a novel GCB context specific oncogenic pathway involving defective epigenetic surveillance of AICDA mediated effects on transcribed genes.
Keywords: DLBCL, SETD2, germinal center
INTRODUCTION
Diffuse large B cell lymphomas (DLBCL) are aggressive and heterogeneous tumors arising from B cells transiting the germinal center (GC) humoral immune response (1). GCs are dynamic and transient anatomical structures which develop in secondary lymphoid organs following T cell dependent antigen activation of mature B cells (2). Germinal center B (GCB) cells called centroblasts (CB) undergo rapid rounds of clonal expansion, somatic hypermutation (SHM) to diversify the immunoglobulin (Ig) locus, after which they become post-replicative centrocytes (CC) that compete for T cell help based on B cell receptor affinity for cognate antigen, with selected cells exiting the GC reaction and transitioning to become either memory B cells or antibody producing plasma cells (2). As such, GCB cells are specialized for tolerance of DNA damage (1) and DLBCLs characteristically manifest a high burden of somatic mutations and genomic structural lesions.
SHM is initiated by the deamination of cytosines on uracils at single stranded DNA (ssDNA), by the enzyme, Activation induced cytosine deaminase (AICDA), leading to breaks and nicks on DNA which get repaired with the introduction of mutations to increase the antigen binding affinity (3–5). Although AICDA is targeted to the immunoglobulin heavy chain (IgH) locus, it can act at other actively transcribed and accessible regions of the genome, resulting in abundant off-target mutations (6). AICDA-mediated mutagenesis during the GC reaction occurs due to the rapid proliferative rate of these cells, and many replicating GCB cells undergo genomic damage induced apoptosis (7). In order to survive, GCB cells partially attenuate DNA damage sensing mechanisms and prevent checkpoint engagement that would otherwise impair the process of SHM, for example by the BCL6 transcriptional repressor that attenuates the actions of ATR (8). This scenario makes GCB cells highly prone to malignant transformation, with a majority of B cell tumors including DLBCLs originating from this process (1). Further compounding this effect, many DLBCLs manifest evidence of undergoing further AICDA induced mutagenesis and are composed of highly proliferative cells reflecting their GC origin (9,10).
From the genetic perspective, a dominant theme of somatic mutations in DLBCL is highly recurrent mutations in chromatin modifier genes and transcription factors (11,12). This is critically linked to the extensive and rapid waves of epigenetic reprogramming experienced by B cells as they transit the GC reaction and undergo a variety of phenotypic transitions. Much of this process is controlled by dynamic activation and repression of gene enhancers and promoters by EZH2, CREBBP, KMT2D, EP300 and TET2 (13–18), all of which are mutated at high frequencies in DLBCL (11,12). The outcome of perturbation in their function was shown to include disruption of normal homeostatic interactions within the immune microenvironment, disrupting immune surveillance and impaired exit from the GC reaction (19). Collectively these findings point to disruption in both epigenetic programming of gene regulatory elements and genomic instability as hallmark characteristics of DLBCLs.
Among the highly recurrent DLBCL-associated somatic mutations in chromatin modifiers, those affecting histone methyltransferase SETD2 stand out from the others as being at the cross-roads between epigenetic regulation of transcriptional activation and elongation as well as DNA damage sensing (20). Notably, SETD2 mutations in DLBCL are especially highly recurrent in patients with African ancestry, who also experience inferior clinical outcomes (21). Mechanistically, SETD2 is the sole histone methyltransferase that can trimethylate H3K36, a mark that is primarily localized to the coding region of actively transcribed genes (22). Within gene bodies, SETD2 was shown to interact with phosphorylated C-terminal domain of RNA polymerase II (RNAPII), to deposit H3K36me3 at actively transcribed genes (23). H3K36me3 was found to be involved in recruitment of RNA splicing machinery through recruitment of MRG15 and ZMYND11 and preventing spurious transcription initiation by binding DNMT3B (24–26). Other studies suggest that SETD2 mediated H3K36me3 contributes to sensing of DNA damage through various pathways including mismatch repair (MMR) and recruitment of LEDGF and subsequent activation of homologous recombination (HR) (27,28).
Homozygous loss of function of SETD2 occurs in a variety of solid tumors and leukemias and lead to defective DNA damage repair, and impaired transcription through a variety of potential mechanisms (20,29–31). SETD2 mutations in solid cancers occur as missense and nonsense mutations at roughly equal proportions, with no obvious hotspot but a preference of mutations within the SET domain (20). In early B cell development, homozygous SETD2 deficiency results in defective VDJ recombination due to aberrant end joining of DNA breaks (32,33). However, in DLBCLs, SETD2 is mostly affected by heterozygous missense mutations without loss of heterozygosity, where its role in the highly specialized context of the GC reaction is unknown. Herein, we set out to investigate the role of SETD2 in the humoral immune response and how reduced dosage through heterozygous loss of function could contribute to the malignant transformation of these highly specialized B cells.
RESULTS
Setd2 haploinsufficiency induces GC hyperplasia and dark zone polarization.
A survey of publicly available genomic profiling datasets (n=1917 DLBCLs) revealed the presence of missense (94%) and nonsense (6%) mutations of SETD2 in 5% of cases overall, which was similar in all cohorts (11,34,35) (Figure1A). Of these, ~60% of missense mutations scored as being likely deleterious, including introduction of bulky residues like phenylalanine, introduction of amino acids with opposing ionic charge and hydrophobic residues in place of charged residues (Figure1A–B). In a cohort of 574 DLBCL patients with structural variants, there was heterozygous loss of SETD2 in 7% of patients, with rare cases showing homozygous deletion (Suppl.Fig1A). In contrast, homozygous deletion or loss of heterozygosity is reported to range between 42 to 100% in solid tumors with frequent SETD2 mutations, and 27% in AML and ALL (36–39). Based on the LymphGen classification, we observe SETD2 mutations were most abundant in the ST2 subtype of DLBCL (Suppl.Fig1B), which mostly have GCB like transcriptional profiles (40). Examining SETD2 gene expression in murine and human splenic naive B (NB) and GCB cell populations, we observed robust gene expression (Suppl.Fig1C–D).
Figure 1. Setd2 haploinsufficiency induces GC hyperplasia and dark zone polarization.
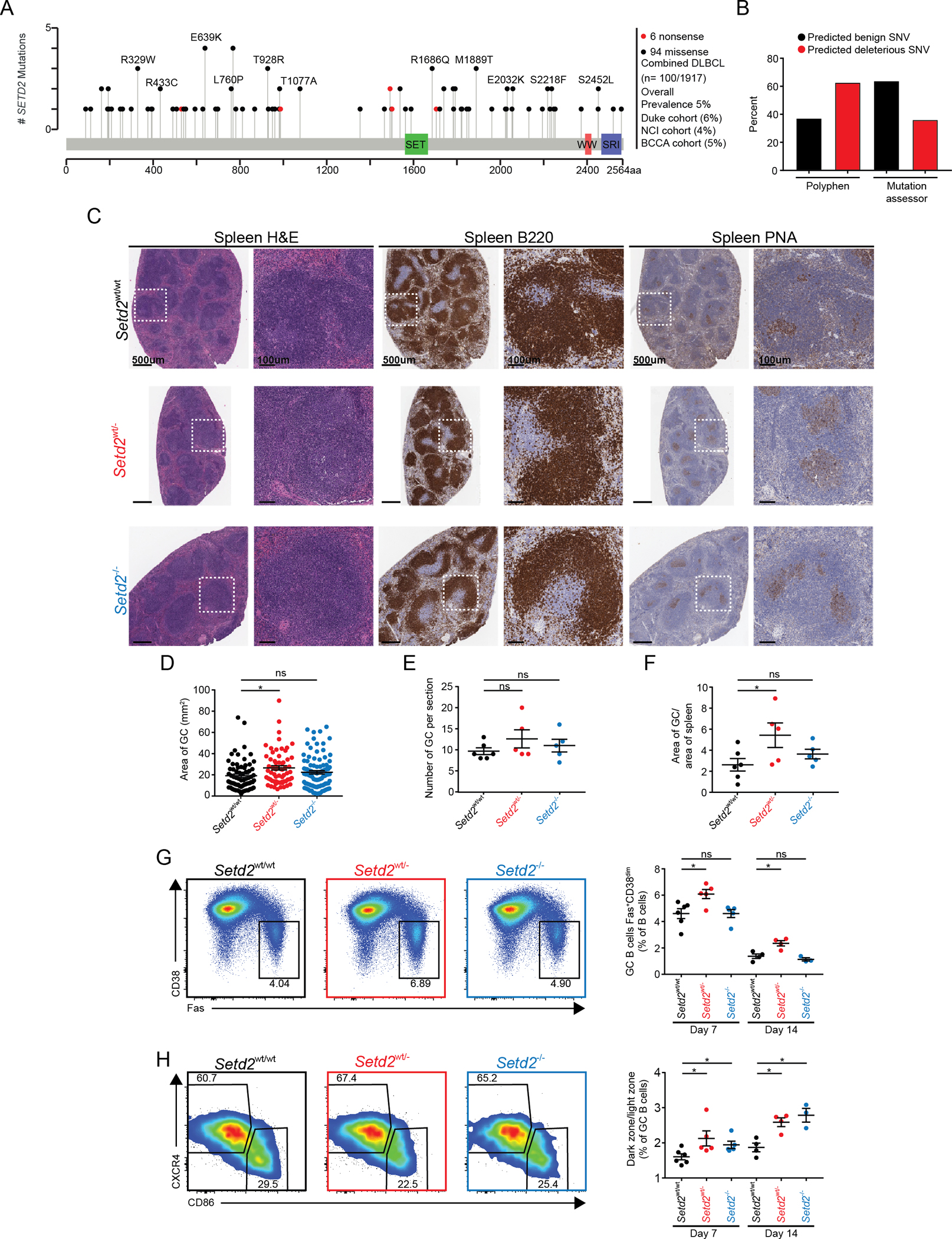
A) Lolliplot of SETD2 mutations from primary DLBCL patients in NCI, Duke and BCCA cohorts (11,34,35).
B) Predicted disruption of SETD2 mutation based on bioinformatics program Polyphen or mutation assessor (102,103).
C-F) Representative histologic sections and quantification of formalin-fixed, paraffin-embedded spleens from Setd2wt/wt (n=6), Setd2wt/− (n=5) and Setd2−/− (n=5) mice. Sections were stained with H&E and antibodies specific for B220 and PNA. Quantification of GC area (D), number of GC (E) and normalized area of GC/total spleen (F).
G) Representative flow cytometry plot and quantification of (B220+Fas+CD38lo) GCB cells from Setd2wt/wt (n=6), Setd2wt/− (n=5) and Setd2−/− (n=5) mice day 7 and 14 after immunization with SRBC.
H) Representative flow cytometry plot and quantification of centroblasts (CXCR4hiCD86lo) and centrocytes (CXCR4loCD86hi) in the GCB cells from Setd2wt/wt (n=6), Setd2wt/− (n=5) and Setd2−/− (n=5) mice day 7 and 14 after immunization with SRBC.
Values represent mean ± SEM. P values were calculated using an unpaired two-tailed t test; ns, not significant; *, p<0.05 (B-F).
To understand the impact of Setd2 loss of function in GCB cells, we crossed mice bearing a floxed Setd2 exon 3 allele with the Cd19-cre strain, expressing cre recombinase in pre-B cells (30,41). Cd19wt/creSetd2wt/wt (Setd2wt/wt), Cd19wt/creSetd2wt/− (Setd2wt/−) and Cd19wt/creSetd2−/− (Setd2−/−) mice (Suppl.Fig1E) were immunized with the T cell dependent antigen, sheep red blood cells (SRBC) to induce GC development and sacrificed seven days later, when GCs are fully formed. Routine histological staining of spleens (H&E and B220 staining) revealed intact tissue architecture and normal-appearing B cell follicles in Setd2wt/− and Setd2−/− animals (Figure1C). Notably, immunostaining with the GC-specific lectin, peanut agglutinin (PNA) revealed enlarged GCs in Setd2wt/−, but not Setd2−/− mice (Figure1C–F). Flow cytometry analysis similarly revealed significantly increased abundance of GCB cells in Setd2wt/− but not Setd2−/− animals, both at day 7 as well as day 14 post-immunization when GCs are resolving (Figure1G, Suppl.Fig1F). We confirmed allele dose-dependent reduction of Setd2 by Q-PCR in sorted GCB cells (Suppl.Fig1G). Further analysis of GCB cell populations revealed an increased ratio of centroblasts to centrocytes in both Setd2wt/− and Setd2−/− mice, indicating perturbations of GC polarity may explain the hyperplastic phenotype (Figure1H). We found no perturbation in any other mature B cell populations or memory B cells in Setd2wt/− mice (Suppl.Fig1H–N), although mature B cells were significantly reduced in Setd2−/− mice, consistent with previous reports (32). Finally, attempts to generate SETD2 knockout human DLBCL cells failed to yield any homozygous clones in 4/5 cell lines (Suppl.Fig.1O–P). Given that SETD2 mutations are recurrently heterozygous in DLBCLs (Figure1A, Suppl.Fig1A), haploinsufficiency resulted in a distinct and more clearly pre-neoplastic phenotype than homozygous deletion, and that homozygous loss is deleterious to DLBCL cells, we subsequently focused specifically on the Setd2wt/− setting.
Setd2wt/− GCB cells manifest superior fitness due to reduced apoptosis.
GC hyperplasia could be caused by either increased proliferation or reduced rates of apoptosis. To explore proliferation effects, we immunized Setd2wt/wt and Setd2wt/− mice and seven days later injected with 5-ethynyl-2′-deoxyuridine (EdU) 120 minutes prior to harvesting splenocytes. We found no difference in EdU+ incorporation, consistent with similar rate of cells transiting S-phase (Figure2A, Suppl.Fig2A). We crossed our Setd2 mice with the cell cycle reporter strain, Rosa26-Fucci2a, which marks cells with different fluorescent signals based on their cell cycle status (42) and did not observe any differences in cell cycle distribution in Setd2wt/− vs Setd2wt/wt GCB cells (Figure2B, Suppl.Fig2B). A high fraction of GCB cells undergo apoptosis (7), and accordingly we observed that 20% of Setd2wt/wt cells stained as AnnexinV+DAPI− (Figure2C), whereas the fraction of apoptotic GCB cells was significantly reduced in Setd2wt/− mice. A similar result was obtained by staining GCB cells with cleaved caspase 3 (Figure2D). We also performed immunohistochemistry stains for cleaved caspase 3 in the spleens of immunized Setd2wt/− and Setd2wt/wt mice and again observed significant reduction in the abundance of apoptotic cells within GCs (delineated by PNA staining, Figure2E). Therefore, GC hyperplasia is mainly attributed to decreased apoptosis and not changes in proliferation or cell cycle dynamics.
Figure 2. Setd2wt/− GCB cells manifest superior fitness due to reduced apoptosis.
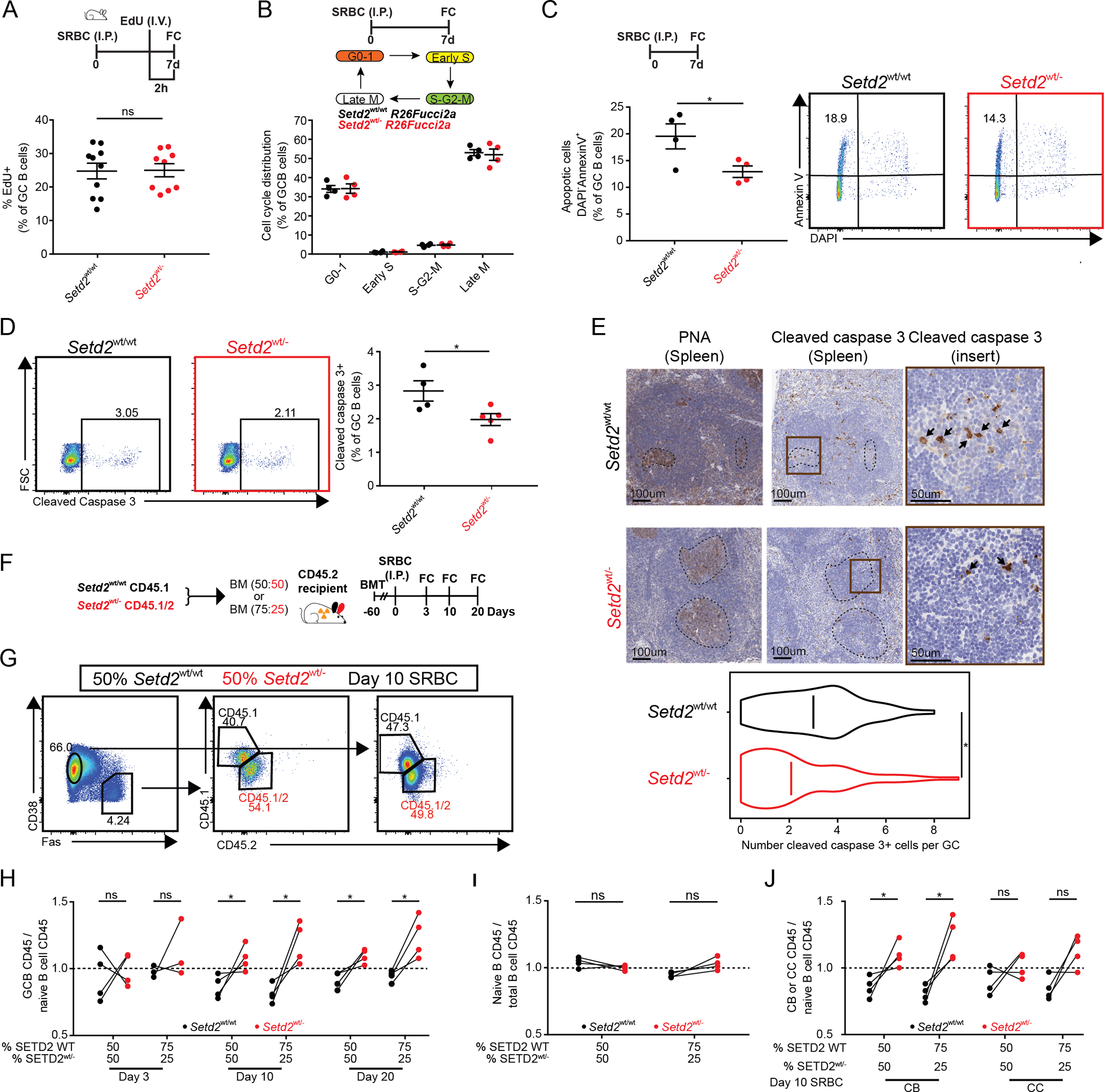
A) Setd2wt/wt (n=10) and Setd2wt/− (n=9) mice were immunized with SRBC and injected with 1mg EdU two hours before sacrifice on day 7. Representative flow cytometry plots (Suppl.Fig2A) and quantification of EdU+ GCB cells are shown.
B) Representative flow cytometry plots (Suppl.Fig2B) and quantification (B) of GCB cells from Setd2wt/wt (n=4) and Setd2wt/− (n=4) FUCCI2a cell cycle reporter mice immunized with SRBC and sacrificed on day 7.
C-E) Representative flow cytometry plots and quantification of AnnexinV+/DAPI− (C) and cleaved caspase 3+ (D) GCB cells from Setd2wt/wt (n=4) and Setd2wt/− (n=4) mice immunized with SRBC and sacrificed on day 7. Representative histologic sections and quantification (E) of formalin-fixed, paraffin-embedded spleens stained with PNA and Caspase 3 antibodies to quantify apoptotic cells within the GC.
F-J) Setd2wt/wt CD45.1+ and Setd2wt/− CD45.1/2+ BM were mixed at either 1:1 or 3:1 ratio and injected into CD45.2+ mice (n=4), immunized with SRBC and euthanized at day 3, 10 and 20 (F). Samples were analyzed by normalizing the percentage of CD45.1+ cells in the target population to their parental CD45.1+ B cells and equivalent normalization with CD45.1/2+ populations. Each pair of connected dots represents a mouse (n=4). Flow cytometry gating strategy of one sample (G). Quantification of GCB cells (Fas+CD38lo) compared to naive B cells (H). Quantification of non GCB cells (Fas−CD38+) compared to total B cells (I). Quantification of centroblasts (CXCR4hiCD86lo) and centrocytes (CXCR4loCD86hi) compared to naive B cells (J).
Values represent mean ± SEM. P values were calculated using an unpaired (A-D) or paired (H-J) two-tailed t test; ns, not significant; *, p<0.05.
To test whether the improved survival of Setd2wt/− GCB cells can confer a clear fitness advantage, we performed mixed bone marrow chimera experiments, where Cd45.1+;Setd2wt/wt or Cd45.1/2+;Setd2wt/− bone marrow (BM) cells were mixed at 50:50 or 75:25 ratios and transplanted into lethally irradiated Cd45.2+ syngeneic recipients. After engraftment, mice were immunized with SRBC and sacrificed 3, 10 or 20 days later (Figure2F). Analysis of GCB cells at the different timepoints showed no advantage for Setd2wt/− over Setd2wt/wt at the initiation of the GC reaction (day 3, Figure2G–H) after normalization to their respective naïve B cell populations by flow cytometry. However, a clear and significant competitive advantage for Setd2wt/− GCB cells did emerge by day 10 and persisted until the end of the GC reaction (day 20, Figure2G–H). Notably, this advantage was also observed even when Setd2wt/wt cells were transduced at the higher (75:25) ratio to Setd2wt/− cells. In contrast there was no competitive advantage for Setd2wt/− naive B cells normalized to total B cell populations at any ratio (Figure2I). Moreover, the advantage for Setd2wt/− GCB cells was most significant among centroblasts, the highly proliferative fraction of GCB cells that undergo somatic hypermutation (Figure2J). Consistent with the above findings related to proliferation and apoptosis, the competing Setd2wt/− GCB cells displayed lower levels of cleaved caspase 3 but no differences in the proliferation marker PCNA compared to Setd2wt/wt GCB cells in the same mice (Suppl.Fig2C–D). These data suggest that Setd2wt/− confers a fitness advantage to GCB cells, associated with reduced rates of apoptotic cell death.
Setd2 haploinsufficiency impairs DNA damage sensing.
Much of the apoptosis occurring in GCB cells is attributed to the DNA damage generated by both AICDA induced hypermutation and via stress from rapid replication (7). Therefore, we wondered whether the improved survival of Setd2wt/− GCB cells was linked to impaired DNA damage sensing, which can occur when there is reduction in SETD2 mediated H3K36 trimethylation (43). Along these lines, performing flow cytometry in spleens of SRBC immunized mice, we detected significantly decreased levels of the early DNA damage marker, γH2AX in Setd2wt/− GCB cells (Figure3A). Repeating this experiment in mice injected with EdU two hours prior to euthanasia, showed that the reduction in γH2AX was not observed in actively dividing EdU+ GCB cells, suggesting the defect occurs after DNA replication (Figure3B). ssDNA damage sensing that occurs during SHM triggers phosphorylation of CHK1, which we observed to also be significantly reduced in Setd2wt/− GCB cells (44) (Figure3C).
Figure 3. Setd2 haploinsufficiency impairs DNA damage sensing.
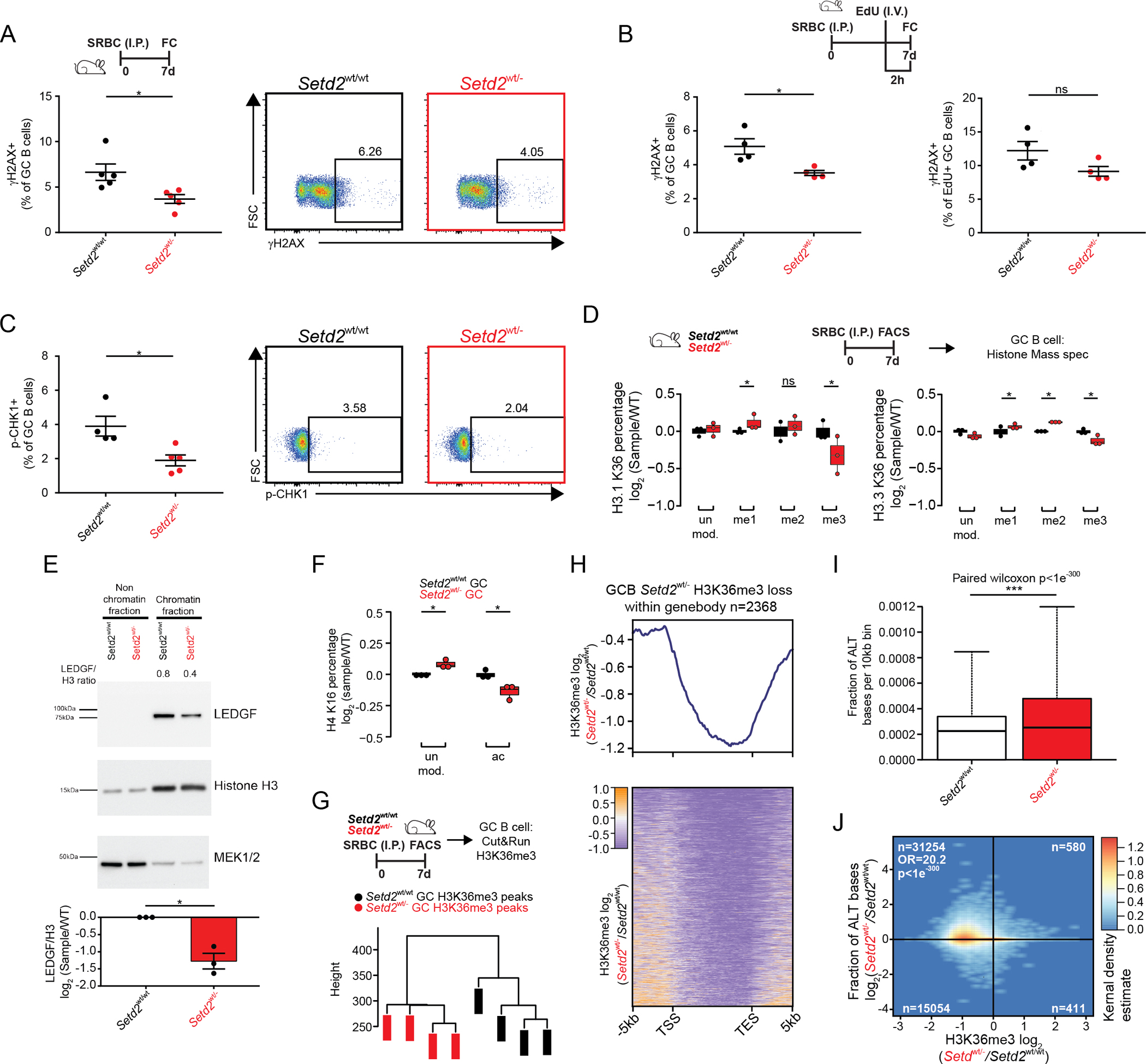
A-C) Representative flow cytometry plots and quantification of γH2AX (A-B) or p-CHK1 (C) stained GCB cells from Setd2wt/wt (n=5) and Setd2wt/− (n=5) mice immunized with SRBC and sacrificed on day 7. Mice were injected with EdU two hours prior to sacrifice (B).
D) Normalized abundance of H3.1 and H3.3 K36 quantified by liquid chromatography separation and mass spectrometry of histones from SRBC immunized mice (n=3).
E) Chromatin fractionation and western blot of LEDGF, Histone H3 and MEK1/2 from GCB cells.
F) Normalized abundance of H4K16 and H4K16ac quantified by liquid chromatography separation and mass spectrometry of histones from SRBC immunized mice (n=3).
G) Hierarchical clustering of H3K36me3 bound peaks by CUT&RUN (n=4)
H) Heatmap and normalized density plot of H3K36me3 bound gene bodies.
I) Abundance of alternative bases identified per 10kb bin in H3K36me3 bound CUT&RUN reads.
J) Correlation plots comparing ratio of alternative bases compared to change in H3K36me3 peaks within gene bodies of Setd2wt/wt and Setd2wt/− GCB cells.
Values represent mean ± SEM. P values were calculated using an unpaired (A-D, F) or paired (E) two-tailed t test, paired Wilcoxon (I) or Fisher’s exact test (J); ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
The DNA damage sensing actions of SETD2 are linked to its generation of H3K36me3, which can subsequently recruit DNA repair proteins such as LEDGF (45). Therefore, to examine the global effect of Setd2 haploinsufficiency on chromatin states, we performed unbiased histone post-translational modification mass spectrometry on purified Setd2wt/− or Setd2wt/wt GCB cells. We observed significant reduction in the abundance of H3K36me3 in both replication dependent H3.1 and replication independent H3.3 isoforms in Setd2wt/− GCB cells (Figure3D), with modest reciprocal increases in H3K36me1 and to a lesser extent in H3K36me2. This prompted us to measure the abundance of LEDGF in the chromatin and non-chromatin cell fractions, which revealed a significant reduction in the abundance of chromatin-bound LEDGF in Setd2wt/− GCB cells (Figure3E). H3K36me3-dependent LEDGF loading at sites of DNA damage recruits TIP60, which mediates H4K16 acetylation (46). Along these lines our histone mass spectrometry analysis revealed significant reduction on H4K16ac in Setd2wt/− GCB cells with reciprocal gain of unmodified H4K16 (Figure3F).
Finally, we performed H3K36me3 CUT&RUN in Setd2wt/− or Setd2wt/wt GCB cells (Figure3G) to further validate the mass spectrometry data and ascertain whether H3K36me3 loss was associated with specific genes. Unsupervised analysis focused on gene bodies, where most H3K36me3 is located (27), revealed clearly distinct profiles in Setd2wt/− GCB cells (Figure3G). We then performed a supervised analysis focused on gene bodies with significant H3K36 trimethylation. This analysis revealed 2368 differentially methylated gene bodies in Setd2wt/− GCB cells, almost all having reduction of H3K36me3 in Setd2wt/− GCB cells (Figure3H, Suppl.Fig3). To determine whether this reduction in H3K36me3 might lead to accrual of DNA damage, we measured the relative abundance of single nucleotide variants (SNV) occurring within differentially H3K36 trimethylated gene bodies and observed significantly increased abundance of SNVs (p<1e−300) and highly significant correlation between H3K36me3 reduction and gain of SNVs (p<1e−300, Figure3I–J).
H3K36me3 loss is associated with increased SHM and off target AICDA mutations.
The dominant mechanism of mutagenesis in GCB cells is AICDA mediated SHM (47). Indeed, sequencing analysis of the immunoglobulin JH4 variable region locus in GCB cells revealed a significantly higher abundance of mutations in the Setd2 haploinsufficient state (Figure4A). Moreover, Setd2wt/− JH4 alleles often contained higher numbers of point mutations per allele (Figure4B), and a significant increase in the proportion of non-canonical AICDA mediated A>T mutations (Figure4C). Linking this finding to Setd2 deficiency, we observed a marked reduction in H3K36me3 at the JH4 locus (Figure4D). In addition to SHM, AICDA also mediates class switch recombination in GCB cells, an effect that was intact in Setd2wt/− resting B cells induced to undergo class switch recombination (CSR) ex-vivo (Suppl.Fig4A–B).
Figure 4. H3K36me3 loss is associated with increased SHM and off target AICDA mutations.
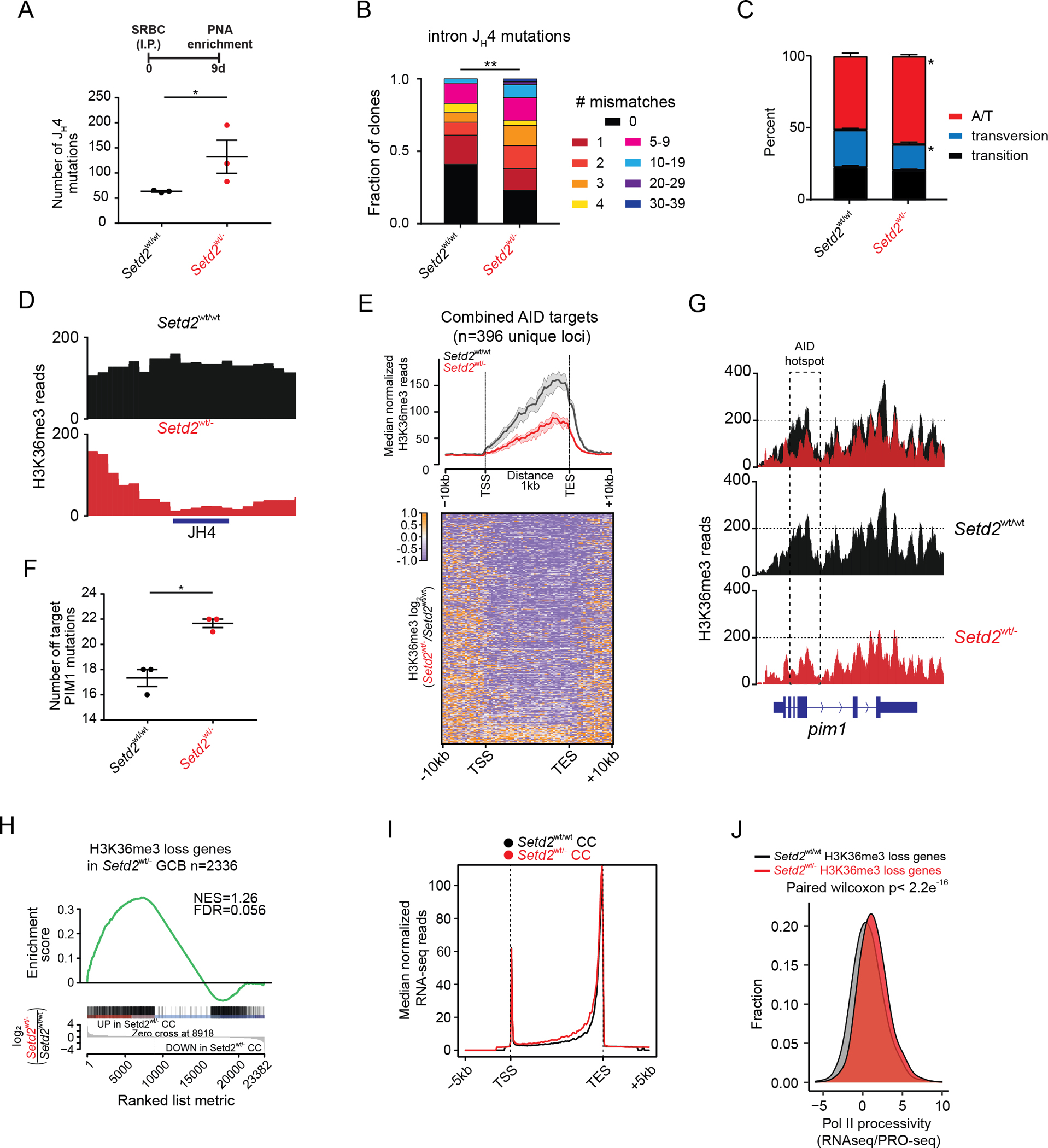
A-C) SHM burden at Ig intron JH4 from Setd2wt/wt (n=3) and Setd2wt/− (n=3) represented as total number (A), proportion of all clones (B) or mutation type (C).
D) H3K36me3 reads across the JH4 locus.
E) Heatmap and normalized density plot of H3K36me3 bound AICDA targets from Setd2wt/wt and Setd2wt/− GC with 95% confidence interval.
F) Mutation burden at Pim1 locus from Setd2wt/wt (n=3) and Setd2wt/− (n=3) GCB cells.
G) H3K36me3 reads across the Pim1 locus.
H) GSEA of Setd2wt/wt Vs Setd2wt/− GC RNA-seq genes against genes losing H3K36me3 in Setd2wt/− GC.
I) Normalized density plot of RNA-seq signal in genes losing H3K36me3 in Setd2wt/wt and Setd2wt/− GC.
J) Density plot of RNAPII processivity score, calculated as RNA-seq signal divided by PRO-seq gene body signal in Setd2wt/wt and Setd2wt/− GC.
Values represent mean ± SEM. P values were calculated using an unpaired two-tailed t test (A, C, F), Wilcoxon ranked-sum test (B) or paired Wilcoxon (J); ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
In addition to on-target Ig loci, AICDA is known to induce off-target mutagenesis at accessible chromatin throughout the genome, contributing to development of lymphomas (6). Certain genes have been shown to be more susceptible to AICDA mutagenesis (6,48), and indeed we observed that these canonical murine and human AICDA off-target genes including critical DLBCL oncogenes featured a significant reduction of gene body H3K36me3 (Figure4E, Suppl.Fig4C). One of the genes known to be most affected by AICDA off-target genes is PIM1, a highly prevalent lymphoma oncogene (49). Sequencing regions of Pim1 known to be affected by AICDA revealed a significantly higher burden of somatic mutations in Setd2wt/− GCB cells (Figure4F). Examining H3K36me3 across the Pim1 locus revealed a clear reduction of this histone mark across the coding region in Setd2wt/− GCB cells, including the 5’ region most affected by somatic hypermutation (6) (Figure4G).
H3K36me3 is normally deposited by SETD2 in actively transcribed genes, suggesting a link between transcriptional activation and H3K36me3. However, RNA-seq performed in Setd2wt/wt or Setd2wt/− centroblasts and centrocytes showed lack of distinct expression profiles and virtually no differentially expressed genes between genotypes (Suppl.Fig4D). Unexpectedly, we did observe significant enrichment for induction of expression at genes that lose H3K36me3 in Setd2wt/− GCB cells (Figure4H). Mapping RNA-seq reads to gene bodies suggest increased reads could be linked to RNAPII processivity, as reads were diffusely distributed across these loci (Figure4I). Similarly, we performed ATAC-seq in these cells and did not observe significant changes in chromatin accessibility, although there was a minor trend towards increased reads in gene bodies that lost H3K36me3 (Suppl.Fig4E–F). To gain further insight into this mechanism we performed nascent transcription PRO-seq (50), calculated a RNAPII processivity score by dividing RNA-seq/PRO-seq counts per gene, and observed an overall increase of RNAPII processivity in Setd2wt/− GCB cells (Figure4J). This suggest that loss of H3K36me3 perturbs the role of RNAPII in transcription coupled detection of DNA damage thus leading to increased processivity.
Setd2 haploinsufficiency results in accelerated lymphomagenesis.
The fitness effect of Setd2 haploinsufficiency along with its hypermutator phenotype pointed to a likely haploinsufficient tumor suppressor role. To explore such effects, we crossed the Setd2-floxed mice with the Rosa26lox-stop-lox-BCL2-IRES-GFP strain (51) for conditional expression of the canonical lymphoma oncoprotein BCL2 and GFP in B cells, generating Cd19wt/cre;Rosa26-BCL2GFP;Setd2wt/− (Setd2/BCL2), Cd19wt/cre;Rosa26-BCL2GFP;Setd2wt/wt (BCL2), Cd19wt/cre;Rosa26wt/wt;Setd2wt/− (Setd2) and Cd19wt/cre;Rosa26wt/wt;Setd2wt/wt (cre). BCL2 was selected as cooperating oncogene since it is upregulated in both GCB and activated B cell (ABC) DLBCLs (40). Exploring a publicly available set of patients with RNA-seq performed in isolated tumor cells and mutational profiles, we confirmed that BCL2 is generally highly expressed in SETD2 mutant patients (Suppl.Fig5A). A cohort of 40 mice per genotype was generated through bone marrow transplantation to lethally irradiated C57BL/6 recipients, which were then immunized three times to stimulate GC formation. To explore whether Setd2 haploinsufficiency could accelerate lymphomagenesis, we sacrificed animals at a timepoint prior to mice manifesting overt disease (182 days post bone marrow transplantation, Figure5A).
Figure 5. Setd2 haploinsufficiency results in accelerated lymphomagenesis.
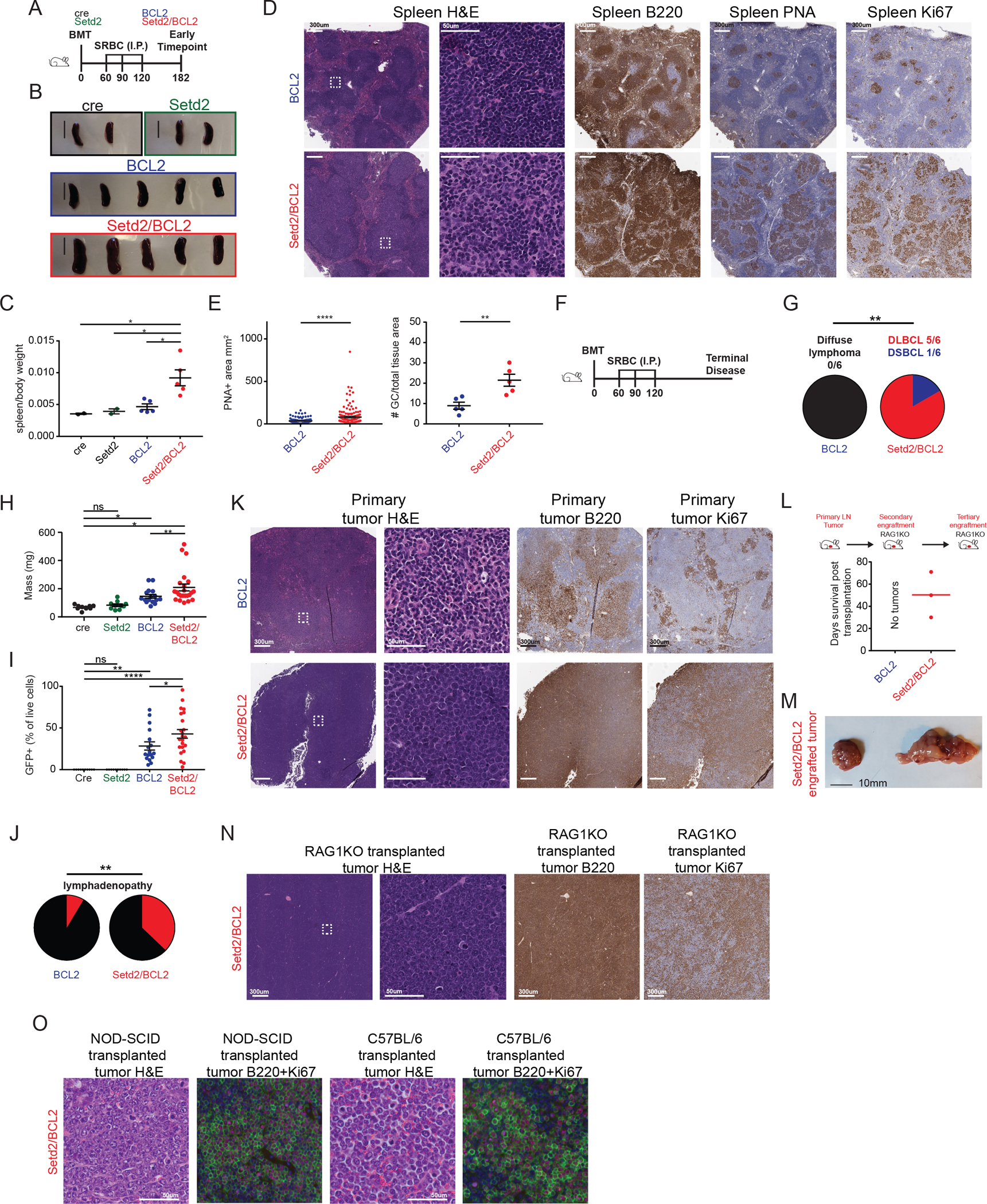
A) Experimental scheme and timeline for (B)-(E).
B-C) Spleens at early time point necropsy (B) and quantification of normalized spleen weight (C).
D-E) Representative histologic sections and quantification of formalin-fixed, paraffin-embedded spleens from BCL2 and Setd2/BCL2 mice (D). Sections were stained with H&E and antibodies specific for B220, PNA and Ki67. Quantification of GC area and normalized area of GC/total spleen (E).
F) Experimental scheme and timeline for (G)-(K).
G) Lymphoma penetrance of mice at terminal timepoint. DSBCL=diffuse small B cell lymphoma
H) Spleen mass of mice at terminal disease.
I) Flow cytometry quantification of tumor burden as measured by GFP+ percent from organs of mice at terminal disease.
J) Pie chart representing the presence of lymphadenopathy in terminal time point BCL2 and Setd2/BCL2 mice.
K) Representative histologic sections of formalin-fixed, paraffin-embedded lymph nodes from BCL2 and Setd2/BCL2 mice at terminal disease. Sections were stained with H&E and antibodies specific for B220 and Ki67.
L) Experimental scheme and quantification of days for tumor growth in serially transplanted RAG1KO mice (n=3).
M) Serially transplanted Setd2/BCL2 tumors from RAG1KO mice.
N-O) Representative histologic sections of formalin-fixed, paraffin-embedded tumors from Setd2/BCL2 mice transplanted into RAG1KO (N), NOD-SCID or C57BL/6 (O) mice. Sections were stained with H&E and antibodies specific for B220 (green) and Ki67 (pink). Values represent mean ± SEM. P values were calculated using one-way ANOVA (C, H-I), unpaired two-tailed t test (E) or Fisher’s exact test (G, J); ns, not significant; *, p<0.05; **<0.01; ***, p<0.001; ****, p<0.0001.
Gross examination of spleens revealed marked splenomegaly in Setd2/BCL2 as compared to BCL2, Setd2 or cre mice (Figure5B–C). Histological evaluation of splenic sections from Setd2/BCL2 animals showed disruption of splenic architecture by a heterogeneous B cell population with variable nuclear size and amount of cytoplasm. There was striking expansion and disruption of GCs as shown by PNA and Ki67 staining in Setd2/BCL2 vs BCL2 spleens, consistent with the effect of Setd2 haploinsufficiency inducing GC hyperplasia and impairing apoptosis (Figure5D). In marked contrast, spleens in BCL2 mice retained their follicular structure, although these were enlarged and somewhat distorted. There were enlarged but well delimited GCs within these follicular structures, with B cells appearing more uniform and consisting of tightly packed centrocytic cells. GCB cells occupied three-fold greater area in Setd2/BCL2 vs BCL2 mice (Figure5E). Flow cytometry from these spleens showed significantly greater abundance of B cells in both Setd2/BCL2 and BCL2 mice as compared to Setd2 and controls, almost 100% of which were GFP+ (Suppl.Fig5B–D), whereas T cells did not express GFP. GC expansion in the spleen of Setd2/BCL2 animals was confirmed by flow cytometry (Suppl.Fig5E). Examination of other lymphoid tissues revealed massive enlargement of lymph nodes in Setd2/BCL2 vs BCL2 mice, again with completely effaced architecture by heterogeneous expanded areas of proliferative GCB cell (Suppl.Fig5F–H). There was also extensive perivascular infiltration of B220+ lymphoma cells into other tissues including the kidney and liver in Setd2/BCL2 mice (Suppl.Fig5I–J). Overall, these data indicate that Setd2 haploinsufficiency results in significant acceleration and dissemination of lymphomagenesis and yields a distinctive proliferative GC phenotype in these tumors.
Despite this, plotting overall survival yielded no difference between Setd2/BCL2 vs BCL2 mice (p=0.4703, log-rank mantel-cox test, Suppl.Fig5K). However, mortality in BCL2 mice was not associated with overt and widespread aggressive lymphoma, but instead was most likely due to thrombotic microangiopathy resulting in severe renal disease, as previously reported in BCL2 transgenic mice (Supp.Fig5L–N) (52,53). In contrast, Setd2/BCL2 mice showed 100% diffuse lymphoma penetrance and accordingly significantly greater tumor burden as compared to BCL2 mice, as manifested by greater spleen weights, greater fraction of GFP+ and B220+ tumor cells within spleens (Figure5F–I, Suppl.Fig5O–P). Moreover, Setd2/BCL2 displayed significantly greater frequency of major lymphadenopathy (37% Vs 9%) defined as lymph nodes greater than 100mg in weight (Figure5J). Histologic analysis of end stage Setd2/BCL2 lymphoid tissues showed total effacement of normal architecture by diffuse sheets of large and highly proliferative B cells, consistent with full transformation into DLBCL (Figure5K). In contrast, the spleens in terminal BCL2 mice contained infiltrates of smaller and heterogeneous cells, with only small islands of highly proliferative cells (Figure5K). B cell receptor profiling of terminal Setd2/BCL2 and BCL2 lymphomas revealed that Setd2/BCL2 lymphomas were more likely to consist of dominant clonal populations, whereas BCL2 lymphomas were highly polyclonal (Suppl.Fig5Q–R). Gene expression profiles of GFP+ tumor cells (54) indicated that these lymphomas were distributed across the board as GCB, ABC and unclassified based on the cell of origin classification system (Suppl.Fig5S). Intraperitoneal adoptive transfer of tumor cells from Setd2/BCL2 mice readily led to their engraftment (3/3) in RAG1KO mice within 30–70 days, leading to their death due to abdominal lymphomas that retained the histologic appearance of the original primary B cell lymphoma (Figure5L–N). These tumors could additionally be engrafted into tertiary RAG1KO, NOD-SCID and C57BL/6 (syngeneic immunocompetent) mice (Figure5O). In contrast, none of the terminal BCL2 lymphomas engrafted in RAG1KO recipients. Collectively, the data show that Setd2 deficiency leads to the formation of highly malignant and invasive high grade DLBCLs with superior fitness to initiate lymphomas in recipient mice.
SETD2 lymphomas display a high abundance of clustered AICDA signature mutations skewed to non-template strand DNA.
The evidence for reduced sensing of DNA damage induced during the GC reaction led us to consider whether Setd2 haploinsufficiency might contribute to lymphomagenesis by enhancing mutagenesis and genetic heterogeneity, which are linked to increased tumor fitness (55). To address this question, we performed whole genome sequencing in the sorted GFP+ tumor cells from three Setd2/BCL2 and three BCL2 terminal mice, using as germline control the DNA from GFP negative bone marrow cells from the original donor animals. We noted a significant increase in the global abundance of SNVs in Setd2/BCL2 tumors (+/−1800 vs 900 per tumor), as well as SNVs located in gene exons (+/−12 vs 3 per tumor, Figure6A–B) and the frequency of regional clustering of point mutations (Figure6C). Focusing on coding mutations, we identified 30 genes with exonic non-synonymous mutations in Setd2/BCL2 lymphomas that included canonical AICDA off-target mutation genes (6), such as Pax5 and Cd83, compared to only five exonic mutations in BCL2 lymphomas (Figure6D, Suppl.Table1). Known AICDA off target genes featured more frequent and abundant SNVs in Setd2/Bcl2 tumor cells as compared to BCL2 only mice (Suppl.Fig6A). Along these lines, the mutation profiles of Setd2/BCL2 lymphomas were significantly enriched for canonical (C>T) and non-canonical (T>C) AICDA type mutations (Figure6E).
Figure 6. SETD2 lymphomas display a high abundance of clustered AICDA signature mutations skewed to non-template strand DNA.
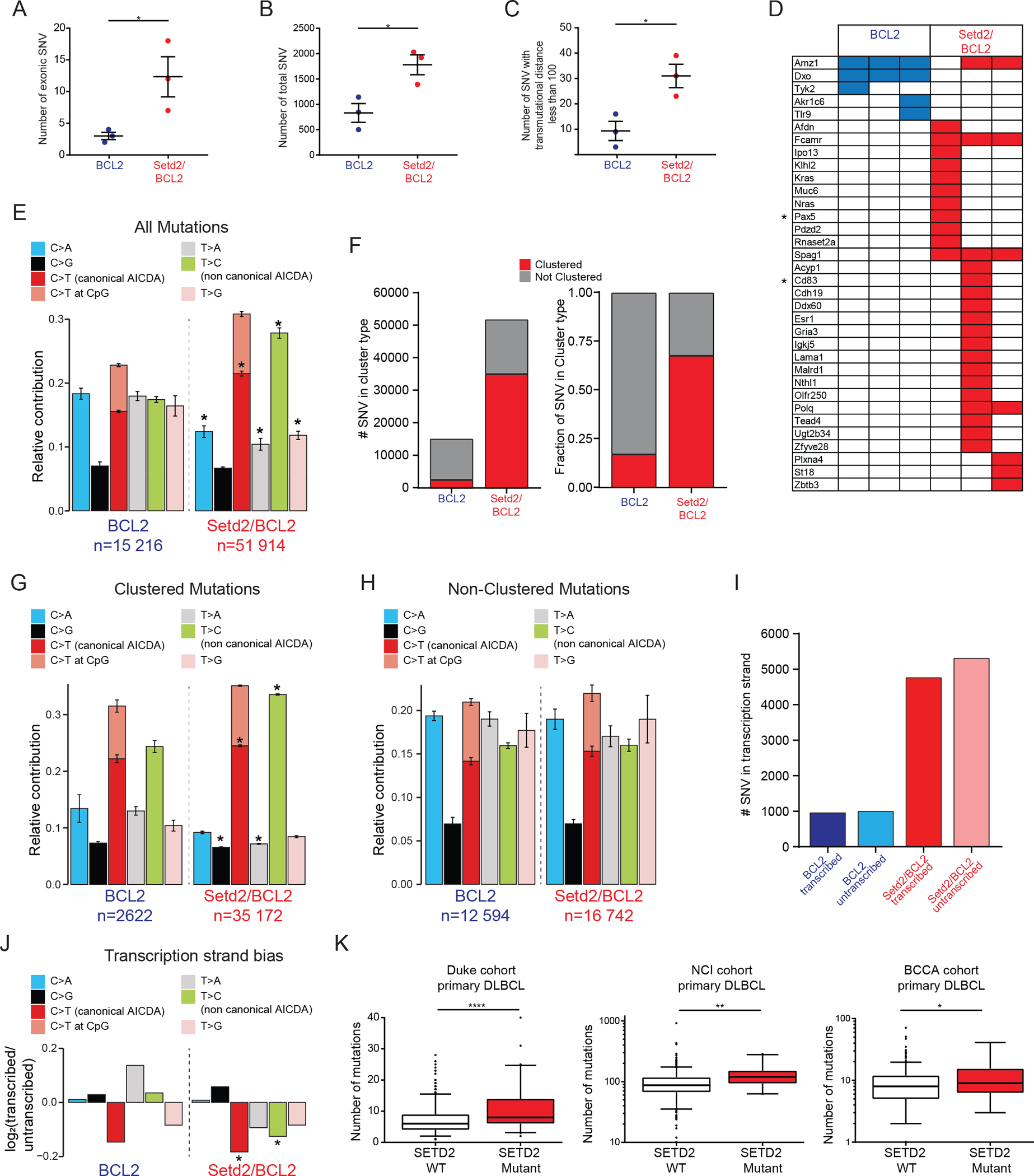
A-B) Exonic (A) and total (B) SNV number identified in BCL2 (n=3) and Setd2/BCL2 (n=3) tumors.
C) Number of clustered SNV within 100bp.
D) Table of exonic SNV, * represent AICDA targets.
E-H) SNV from BCL2 (n=3) and Setd2/BCL2 (n=3) tumors grouped according to SNV type across the whole genome (E), number and proportion of clustered mutations (F), proportion of clustered (G) and non-clustered (H) mutations according to SNV type.
I-J) Number (I) and relative change (J) in SNV in BCL2 (n=3) and Setd2/BCL2 (n=3) tumors according to transcriptional strand bias.
K) Number of exonic SNV in SETD2 WT or mutated primary DLBCL patients from the Duke, NCI and BCCA cohort (11,34,35).
Values represent mean ± SEM (A-C) or SD (E, G-H); median for (K). P values were calculated using unpaired two-tailed t test (A-C, E, G-H), Poisson asymmetry test (J) or one-way ANOVA with Kruskal-Wallis H test (K); ns, not significant; *, p<0.05; **<0.01; ***, p<0.001; ***, p<0.0001.
Further analysis revealed that the increase in mutation burden in Setd2/BCL2 lymphomas was mainly due to highly clustered SNVs (<1kb), which represent a significantly greater fraction of differential point mutations in these tumors (Figure6F). Even though much fewer in number, canonical and non-canonical AICDA associated mutations tend to be more clustered in BCL2 than the other base substitution types, yet this difference was still far greater in the Setd2/BCL2 lymphomas, where such mutations were far more abundant (Figure6G). In contrast, non-clustered mutations had comparable mutation profiles in both Setd2/BCL2 and BCL2 lymphomas (Figure6H). Off-target AICDA mutations are more abundant at highly transcribed genes (6), raising the possibility that this SETD2-associated clustered mutation pattern might reflect DNA damage occurring on ssDNA during RNAPII elongation, where the non-template strand is more vulnerable to attack by AICDA. Indeed, we observed significant over-representation of non-template strand AICDA mutations occurring in Setd2/BCL2 lymphomas, which were also far more abundant than in BCL2 lymphomas (Figure6I). Moreover, there was also greater skewing of clustered mutations to the non-template DNA strand especially at canonical and non-canonical AICDA associated mutations in Setd2/BCL2 tumors (Figure6J, Suppl.Fig6B–C). Finally, examining three independent cohorts of human DLBCL patients showed a significantly increased burden of SNVs in SETD2 mutant vs SETD2 wild type lymphomas (Figure6K). These findings suggest that SETD2 is important for repair of AICDA induced DNA damage and loss of function leads to genomic instability.
Increased burden of AICDA associated structural genomic lesions in SETD2 mutant lymphomas.
AICDA is known to contribute to structural genomic lesions arising from GCB cells, such as translocations that activate MYC activity (56). Strikingly, an overview of structural variants (SV) revealed significantly greater abundance of translocations, duplications, and deletions in Setd2/BCL2 lymphomas (Figure7A). Using a more complex genome graph-based structural variant analysis method (57), we found more highly complex genomic lesions in Setd2/BCL2 lymphomas including chromoplexy, kataegis and rigma (Figure7B). There was an average of 13 translocations per sample in Setd2/BCL2 and 4 per sample in BCL2 lymphomas. We noted the presence of translocations involving the Ig loci (one in BCL2, and three in Setd2/BCL2 tumors). Notably two thirds of Setd2/BCL2 lymphomas (but 0/3 BCL2 lymphomas) had either a a function (58), Figure7C–D), also known to be mediated through AICDA (59). The Ig locus translocation in the BCL2 tumor was not proximal to any annotated genes. We identified a IgH-Gadd45b translocation in a Setd2/BCL2 tumor, a pro survival factor in GCB cells and tumors (60) (Figure7C). A gene set enrichment analysis of RNA-seq data from murine tumors revealed significant enrichment for canonical MYC target gene sets, and Myc transcript abundance was significantly higher in Setd2/BCL2 lymphomas, suggesting that deregulation of MYC is a common event in Setd2 driven lymphomagenesis (Figure7E–G, Suppl.Fig7A–B). These data again point to Setd2 haploinsufficiency impairing DNA damage sensing, facilitating severe genetic instability due to AICDA, including the development of canonical Myc translocations. Similarly, Tp53 deficiency was shown to facilitate development of Myc translocations and malignant transformation due to the actions of AICDA (61). Notably, SETD2 and TP53 mutations tend to be mutually exclusive in human DLBCL patients, but not in a pan-cancer analysis, suggesting context-dependent phenocopy of SETD2 and TP53 in GCB cells (Figure7H). Finally, examination the NCI cohort of human DLBCL patients with structural variant data indicated that SETD2 mutant or copy number deficient lymphomas also manifested significantly greater burden of copy number loss across the genome (Figure7I, Suppl.Fig7C). Collectively these suggest that Setd2 deficiency can lead to lymphoma fitness by fostering genomic instability through the actions of AICDA, enabling acquisition of numerous canonical AICDA induced genomic lesions that confer selective advantage, such as translocations of the Myc locus.
Figure 7. Increased burden of AICDA associated structural genomic lesions in SETD2 mutant lymphomas.
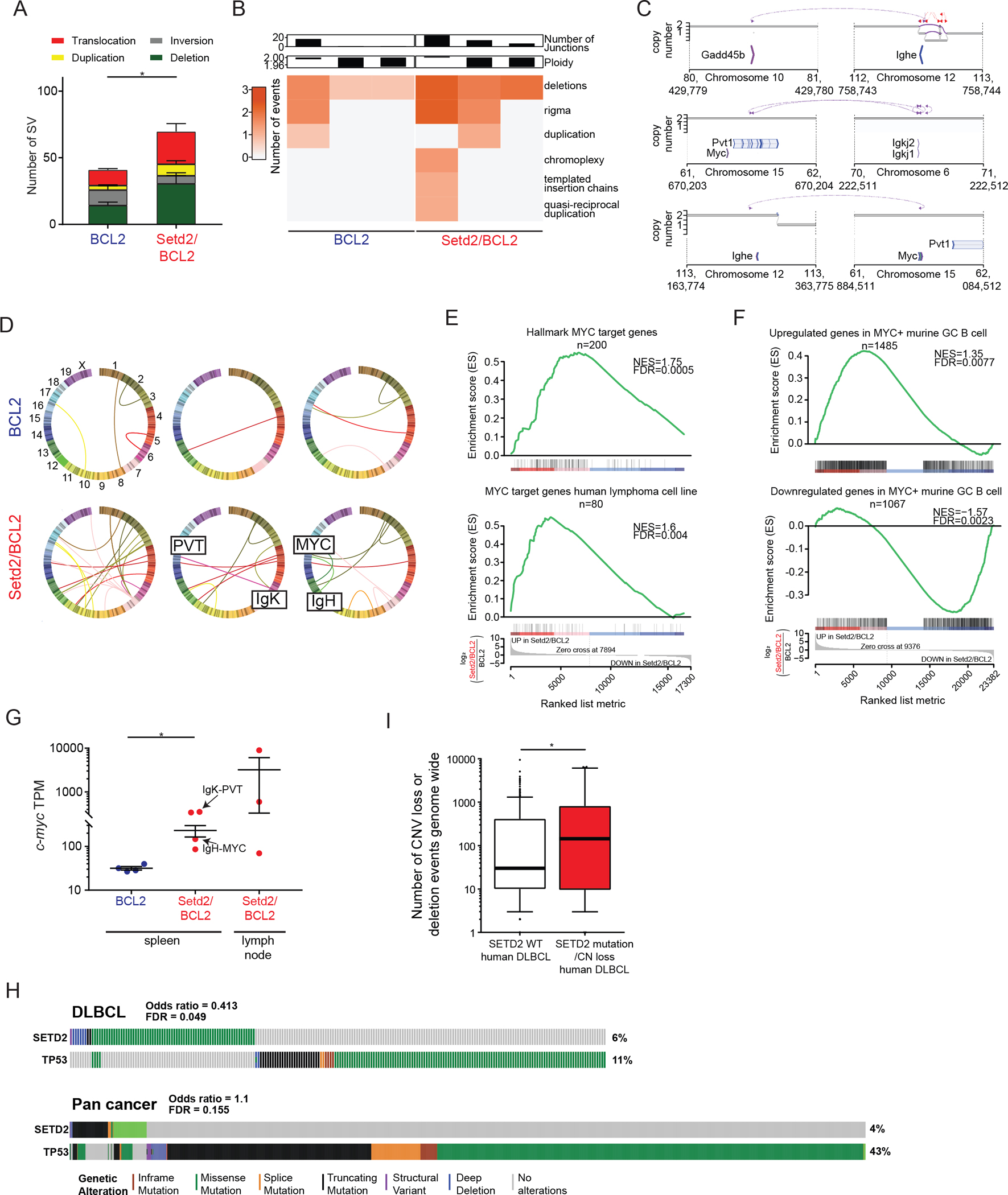
A) Chromosomal alterations from BCL2 (n=3) and Setd2/BCL2 (n=3) tumors.
B) Presence of simple and complex SV events identified using JaBba.
C) Genomic track showing translocation events from the immunoglobulin and c-myc locus. The top segment (purple) shows the rearrangement junction.
D) Circos plots showing translocations in mouse tumors.
E-F) GSEA of BCL2 Vs Setd2/BCL2 tumor RNA-seq genes against human (E) or murine (F) MYC targets.
G) c-myc gene expression from BCL2 (n=4) and Setd2/BCL2 (n=4) tumors.
H) Mutation matrix for TP53 and SETD2 mutations from all cancers and DLBCL cases identified in cBIOPORTAL. Each vertical line represents one patient, with grey lines indicating no known mutation.
I) Copy number loss or deletion genome wide in SETD2 WT or mutated/loss of heterozygosity primary DLBCL patients from the NCI cohort (34).
Values represent mean ± SEM; median for (I). P values were calculated using unpaired two-tailed t test (A, I), one-way ANOVA (G) or Fisher’s exact test (H); ns, not significant; *, p<0.05.
DISCUSSION
Here, we show that SETD2, a histone methyltransferase responsible for depositing H3K36me3 at gene bodies in actively transcribed genes, has unique functions as a haploinsufficient tumor suppressor in GC derived lymphomas. The reason for this is due to a synthetic DNA damage susceptibility scenario that is unique to GCB cells due to the effect of AICDA, a cytosine deaminase that normally induces ssDNA lesions to induce SHM of the immunoglobulin loci, as well as double strand DNA breaks during CSR in B cells entering the GC reaction (3). Upon sensing DNA damage, most cells trigger checkpoint mechanisms to permit DNA damage repair, prior to proceeding through the cell cycle. However, GCB cells uniquely attenuate expression of DNA damage sensing mechanisms such as ATR and TP53 through the actions of the transcriptional repressor BCL6, thus allowing these cells to continuously proliferate and accumulate genetic lesions (8,62). Perturbation of DNA damage sensing in GCB cells disrupts function, for example Atm knockout mice have decreased GC size due to accumulation of DNA damage and increased rates of cell death (63). This scenario may cause GCB cells to be exceptionally dependent on remaining DNA damage sensing and repair mechanisms to repair AICDA induced damage and thus vulnerable to lymphomagenesis arising due to loss of function in repair mechanisms. This is the case in MMR deficient mice, where loss of Msh2 in combination with BCL6 overexpression results in GCB cell lymphomas with signs of genomic instability (64). Hence our data point to SETD2 as a novel and critical DNA damage response mechanism in GCB cells required to restrict their potential for malignant transformation, at least in part through failure to trigger DNA damage-associated apoptosis mechanisms. GCB cells that fail to engage in productive interactions with T cells may also undergo apoptosis (7), and we cannot exclude that such effects could also occur here.
Mechanistically, the effect of SETD2 deficiency seemed to link both to sensing and repair of double and single strand DNA breaks, and hence is relevant to damage occurring during both CSR and SHM (65). Double strand breaks can also occur during AICDA mutagenesis within the GC reaction during SHM (47), which may be reflected by the increased abundance of structural genomic lesions in Setd2 haploinsufficient lymphomas. The finding of increased Myc translocations and MYC target gene activation in Setd2 haploinsufficient lymphomas is reminiscent of the effect of compound H2ax and Tp53 loss of function in GCB cells, which also results in increased incidence of such lesions (61,66). Hence SETD2 mutation may be one of the many mechanisms through which DLBCLs induce aberrant MYC expression. It is striking that loss of one Setd2 allele in the GC context is sufficiently deleterious to mimic biallelic deletion ofTp53. The fact that there were increased complex genomic lesions such as rigma and chromoplexy, speaks to the significant dependency of GCB cells on SETD2 to keep order in the genome in the context of their high mutational risk due to AICDA.
SETD2 has been mechanistically linked to transcription, due to its role in depositing H3K36me3 at actively transcribed genes through its interaction with phosphorylated, elongating RNAPII, enhanced through transcriptional activation signals such as H3.3S31 phosphorylation (23,67). However, it is not clear whether the H3K36me3 mark is required for transcriptional activation. Instead, it has been mostly linked with changes in nucleosomal occupancy and prevention of spurious intragenic transcriptional initiation through its interaction with the FACT complex and DNMT3B, respectively (68,69). Alternatively spliced exons display lower levels of H3K36me3 and loss of SETD2 has been implicated in altering exon usage via recruitment of splicing factors including MRG15, ZMYMD11 and hnRNP proteins (25,26,70), a key process disrupted in SETD2 mutated colorectal cancers (31). The presence of H3K36me3 is linked instead to a greater tendency of DNA damage to induce HR repair thus maintaining the integrity of coding genes (45). This function is especially critical in GCB cells, where the predominant DNA repair mechanism is non homologous end joining (NHEJ) and HR is critical for reducing the abundance of off-target mutations at coding genes (71,72). Indeed, disruption of HR through loss of Brca2 or Xrcc2 led to impaired GC reactions and accumulation of DNA breaks, respectively (71,73). The effect of Setd2 heterozygous loss of function may thread the needle and induce just the right level of impaired DNA damage response during transcription to reduce efficiency of HR and likely MMR without otherwise impairing GCB cell functions. SETD2 is not really sensing but instead may be marking transcribed chromatin at risk for damage to provide a histone code setting that can be recognized by DNA damage proteins such as LEDGF upon DNA damage signaling occurring in cells.
Although SETD2 has been proposed to also mediate putative epigenetic effects relevant to transcriptional regulation (20), our data suggest that its role as a tumor suppressor is primarily through the described AICDA-DNA damage effect. Along these lines we did not observe significant changes in gene expression profiles in Setd2wt/− GCB cells, nor did we observe any significant differential chromatin accessibility by ATAC-seq, whereas such effects on gene expression and chromatin accessibility were highly prominent in renal cell carcinomas with homozygous SETD2 deletion (74). This is in contrast to the impact of somatic mutations of other chromatin modifier proteins such as EZH2, CREBBP, KMT2D and TET2, which result in dramatic perturbation of transcription and repressive chromatin modification profiles (13–18). Moreover, these other mutations are strongly associated with GCB-DLBCLs, whereas SETD2 mutations are not particularly linked to lymphomas with a specific transcriptional signature. These considerations nonetheless do not completely rule out subtle epigenetic effects of SETD2 haploinsufficiency, and future efforts to illuminate such mechanisms are certainly warranted. However, the data point to mutagenesis as the most likely force for natural selection favoring outgrowth of Setd2 deficient GCB cells, through events such as Myc translocations and point mutations of other oncogenes and tumor suppressors. Our finding of substantial DNA base pair variation in the form of AICDA-associated “mutational noise” supports this notion, suggesting that SETD2 induces genetic heterogeneity as a potential metric of GCB cell fitness allowing outgrowth of lymphoma initiating cells (55). Fitness may be further conferred by decreased cell death observed in Setd2 haploinsufficient GCB cells due to suppressed DNA damage checkpoint activation. The combination of increased genetic diversity and decreased apoptosis presents a scenario allowing for malignant transformation to readily occur, and is likely a significant driving force in humans, given the concordance of mutagenic phenotypes in both murine and human SETD2 deficient/mutated lymphomas.
In summary, we provide a novel context-specific tumor suppressor mechanism for Setd2 haploinsufficiency in GCB cells, where it cooperates with GC specific expression of AICDA to impair a newly defined critical role of SETD2 in restricting off target mutagenesis during class-switch recombination and somatic hypermutation. These findings make it tempting to speculate whether or not SETD2 haploinsufficiency could serve as a therapeutic vulnerability for lymphoma patients, by targeting with newly developed SETD2 inhibitors (75). This question is of particular interest and potential impact given the over-representation of SETD2 mutations in lymphoma patients of African ancestry (21), who also manifest inferior clinical outcomes as compared to the Caucasian population. In contrast to solid tumors and leukemias which often feature homozygous loss of function, DLBCLs are virtually always heterozygous, suggesting a critical dependency on the remaining SETD2 allele, which might reflect the importance of this genomic checkpoint mechanism in maintaining viability of these tumors, and providing a potential avenue for precision therapy for SETD2 mutant patients, with special benefit to patients of African ancestry.
METHODS
Animal Models
The following mouse strains were obtained from The Jackson Laboratory (Bar Harbor, ME, USA): B6 Cd45.2 (C57BL6/6J, RRID:IMSR_JAX:000664), Cd19Cre (B6.129P2(C)-Cd19tm1(cre)Cgn/J, RRID:IMSR_JAX:006785), B6 Cd45.1 (B6.SJL-PtprcaPepcb/BoyJ, RRID:IMSR_JAX:002014), RAG1KO (B6.129S7-Rag1tm1Mom/J, RRID:IMSR_JAX:002216). SETD2fl/fl were a generous gift from Scott Armstrong, Dana-Farber Cancer Institute (30). Rosa26-BCL2-GFP mice for the lymphoma studies were obtained from H. C. Reinhardt (51). The R26-Fucci2a model was developed by I.J. Jackson (42). All mouse experiments were conducted using unbiased age and sex matched specimens. Unless stated otherwise, all animals were 8–16 weeks of age at the time of experimentation. All procedures were approved, and animals maintained according to guidelines established by the Research Animal Resource Center of Weill Cornell Medicine. All mice were monitored until any one of several criteria for euthanizing were met, including severe lethargy and more than 10% body-weight loss in accordance with the Weill Cornell Medicine IACUC–approved animal protocol (protocol #2011–0031).
Germinal center assessment in mice
To induce germinal center formation, age and sex matched mice were immunized intraperitoneally with 0.5mL of 2% sheep red blood cells (SRBC) suspended in PBS (Cocalico Biologicals, 20–1334A).
Bone Marrow transplantation
Bone marrow cells were harvested from the femur and tibia of 8–12 week old donor mice and treated with red blood cell lysis solution (QIAGEN, 158904). 1 million cells were injected into lethally irradiated C57BL/6J host mice (2 doses of 450 rad, Rad source Technologies RS 2000 Biological Research X-ray Irradiator) through the retro-orbital sinus. Transplanted mice were used for experiments 6–8 weeks after transplant.
Flow Cytometry
Mice were sacrificed at the indicated time points, with organs harvested and mononuclear cells purified using Histopaque gradient centrifugation (Atlanta Biologicals, I40650). Single cell suspensions were resuspended in flow buffer (PBS + 2% FBS, 2mM EDTA), blocked with mouse FC block (BD Biosciences, Cat# 553141, RRID:AB_394656) and stained with the following fluorescent labeled anti-mouse antibodies: from BD Biosciences: APC Annexin V (dilution 1:100, 550475, RRID:AB_2868885), APC anti-CD138 (dilution 1:500, 558626, RRID:AB_1645216), APC anti-IgG1 (dilution 1:500, 550083, RRID:AB_393553), BV421 anti-CD95 (dilution 1:500, 562633, RRID:AB_2737690), BV786 anti-B220 (dilution 1:500, 563894, RRID:AB_2738472), FITC anti-B220 (dilution 1:500, 553087, RRID:AB_394617), FITC anti-CD23 (dilution 1:500, 553138, RRID:AB_394653), PerCp-Cy5.5 anti-CD19 (dilution 1:500, 551001, RRID:AB_394004). ThermoFisher Scientific: APC anti-CD4 (dilution 1:500, 17–0041-81, RRID:AB_469319), APC anti-CD38 (dilution 1:500, 17–0381-81, RRID:AB_469381), APC anti-IgM (dilution 1:500, 17–5790-82, RRID:AB_469458), PE anti-CXCR4 (dilution 1:250, 12–9991-82, RRID:AB_891391), PE-Cy7 Streptavidin (dilution 1:500, RRID:AB_10116480), PerCp-Cy5.5 anti-CD45.1 (dilution 1:500, 45–0453-82, RRID:AB_1107003). Biolegend: APC-Cy7 anti-CD38 (dilution 1:500, 102728, RRID:AB_2616968), APC-Cy7 anti-CD45.2 (dilution 1:500, 109824, RRID:AB_830789), BV510 anti-IgD (dilution 1:500, 405723, RRID:AB_2562742), PE-Cy7 anti-CD23 (dilution 1:500, 123420, RRID:AB_1953277), PE-Cy7 anti-CD86 (dilution 1:500, 105014, RRID:AB_439783), PE-Cy7 anti-CD138 (dilution 1:500, 142514, RRID:AB_2562198), PerCp-Cy5.5 anti-CD38 (dilution 1:500, 102728, RRID:AB_2616968), PerCp-Cy5.5 anti-CD95 (dilution 1:500, 152610, RRID:AB_2632905), PerCp-Cy5.5 anti-GL7 (dilution 1:500, 144610, RRID:AB_2562979).
Intracellular staining was performed by first fixing and permeabilizing cells followed by staining of membrane bound targets. Fixation/permeabilization was performed by either BD cytofix/cytoperm (BD Biosciences, 554714, RRID:AB_2869008) then stained with the following fluorescent labeled antibodies Alexa Fluor 647 anti Phospho-Histone H2A.X S139 (Cell Signaling, dilution 1:100, 9720, RRID:AB_10692910), Alexa Fluor 647 anti-active caspase 3 (BD Biosciences, dilution 1:100, 560626, RRID:AB_1727414), PE anti Phospho-CHK1 S345 (Cell Signaling, dilution 1:100, 12268, RRID:AB_2797863).
DAPI (ThermoFisher Scientific, D1306, RRID:AB_2629482) or Ghost Dye Violet 510 (Tonbo Biosciences, 13–0870) was used for the exclusion of dead cells. Data were collected on the FACS Canto II or Fortessa (BD Biosciences) flow cytometry analyzer and analyzed using FlowJo software package (TreeStar, RRID:SCR_008520).
Cell Sorting
Naïve and GC B cells were sorted from the spleens of mice immunized with SRBC for 7 days. Briefly, single cell suspensions were stained with anti-B220, anti-Fas and anti-CD38. Centroblasts and centrocytes were separated from the GC fraction using anti-CXCR4 and anti-CD86 antibodies. DAPI was used to exclude dead cells. Cell sorting was performed in a BD Influx cell sorter in the WCM Flow Cytometry Core Facility. Magnetic bead cell isolation for GC B-cells was performed using the PNA MicroBeads kit (Miltenyi Biotec, #130–110-479) or resting B cells using the anti-43 MicroBeads kit (Miltenyi Biotech, #130–049-801) according to manufacturer’s protocol. Cell purity was confirmed by flow cytometry and all samples had over 90% cell purity of selected populations.
Histology and immunohistochemistry
Mouse organs were fixed with 4% formaldehyde and embedded in paraffin. Tissue processing and staining was performed by the Laboratory of Comparative Pathology (MSKCC). Briefly, 5um sections were deparaffinized and heat antigen retrieval in citrate buffer pH 6.4 and endogenous peroxidase activity blocked with 3% hydrogen peroxide in methanol. Indirect immunohistochemistry was performed with anti-specifies specific biotinylated secondary antibodies followed by avidin horseradish peroxidase or avidin-AP and developed by Vector Blue or DAB color substrates (Vector Laboratories). Sections were counterstained with hematoxylin. Immunofluorescence slides were stained with the following secondary antibodies: Donkey anti-Rat AlexaFluor488 (Invitrogen, A21208, RRID:AB_2535794), Donkey anti-Rabbit AlexaFluor594 (Invitrogen, A21207, RRID:AB_141637). The following antibodies were used: biotin-conjugated anti-B220 (BD Biosciences, 550286, RRID:AB_393581), anti-PNA (Vector Laboratories, B1075, RRID:AB_2313597), anti-CD138 (BD Biosciences, 553712, RRID:AB_394998), anti-cleaved caspase 3 (Asp175) (Cell Signaling Technology, 9661, RRID:AB_2341188) and anti-Ki67 (Cell Signaling Technology, 12202, RRID:AB_2620142). Slides were scanned using a Zeiss Mirax Slide Scanner and photomicrographs were examined using Aperio eSlide Manager (Leica Biosystems). QuPath software was used to quantify GC area.
Genomic DNA and RNA extraction
Genomic DNA was extracted using QuickExtract DNA Extraction Solution (Epicentre, QE09050) or DNeasy Blood & Tissue Kit (QIAGEN, 27106). Total RNA was extracted from cells using TRIzol (ThermoScientific, 15596018). RNA concentration was determined by Qubit Fluorometric Quantification (ThermoScientific) and integrity was verified by Agilent 2100 Bioanalyzer (Agilent Technologies).
CUT&RUN
NB and GC B-cells, at least 300 000 each from Setd2wt/wt and Setd2wt/− mice were sorted by FACS. CUT&RUN was performed following CUTANA protocol v1.5.2 (Epicypher). Briefly, cells were washed, immobilized onto Convanavalin-A beads (Bangs Laboratories, Inc., BP531) and incubated overnight at 4C with 0.01% digitonin and 0.5ug H3K36me3 antibody (Epicypher, 13–0035) or anti-rabbit IgG (antibodies-online Inc., ABIN101961, RRID:AB_10775589). CUT&RUN enriched DNA was purified using Monarch DNA cleanup kit (NEB, T1030S) and 5–10ng used to prepare sequencing libraries with Ultra II DNA library Prep kit (NEB, 9645L). DNA libraries were sequenced on a Hi-Seq 2×150bp (Illumina) at GENEWIZ.
Data was aligned to mm10 genome using BWA-MEM (76,77) and FPKM normalized coverage tracks were generated using the bamCoverage tool from the deepTools framework (78). H3K36me3 peaks were called SICER2 (79) (window size 200bp, redundancy_threshold 1) and genes bound by H3K36me3 were identified as those with >40% of their respective gene bodies covered by called peaks (n=9225 genes). Genes showing gain or loss of H3K36me3 were calculated using normalized read counts within H3K36me3-bound genes using the deepTools multiBigwigSummary tool (FC>1.5, nominal Wilcoxon p<0.05; n=1 gain, n=2368 loss). Read density plots were generated using the deepTools computeMatrix and plotHeatmap tools.
RNA-seq
Library preparation, sequencing and post-processing of raw data was performed at the Epigenomics Core at Weill Cornell Medicine or New York Genome Center. Library preparation using the Illumina TruSeq stranded mRNA Library prep kit (Illumina, 20020594) and sequenced with PE50 paired end-sequencing, performed on an Illumina NovaSeq 6000 sequencer. Sequencing results were aligned to mm10 using STAR and annotated to RefSeq using the R subread package. Differential gene expression was identified using the EdgeR package (80) with thresholds of fold change >1.5 and p<0.01, adjusted for multiple testing using Benjamini-Hochberg correction. Hierarchical clustering was performed using Euclidean distance of log TPM values of genes. Gene set enrichment analysis was performed using the GSEA algorithm (81) and pathways analysis performed using the PAGE algorithm (82).
PRO-seq data preparation and processing
PRO-seq libraries were prepared according to previously described protocols (50). Exceptions include using a run-on Master Mix, with biotinylated nucleotides at 10mM Biotin-11-ATP, 10mM Biotin-11-GTP, 100mM Biotin-11-CTP, and 100mM Biotin-11-UTP; digested RNA by base hydrolysis in 0.2N NaOH on ice was reduced from 8 min to 6min. Briefly, chromatin from 1^6 cells per sample were mixed with 1:10,000 ratio of S2 chromatin and normalized by dividing mouse reads in each sample by the total number of S2 reads in the same sample. Libraries were prepared using adapters that contain a 6bp unique molecular identifier sequence on read1. Libraries were competitively aligned to a genome resulted by merging mm10 assembly with D.melanogaster dm3 genome assembly. Alignment was performed using the proseq2.0 pipeline developed by the Danko lab (https://github.com/Danko-Lab/proseq2.0) using the parameters -PE --RNA5=R2_5prime --UMI1=6.
Downstream analysis was performed in R, using Genomic (83) and BRgenomics1.1.3 (https://mdeber.github.io/index.html). Gene expression was quantified using the GENCODE v20 annotations in mouse and differential expression was quantified using DEseq2 (84). Total number of reads around each TSS or within gene bodies of annotated GENCODE v20 genes was counted with a 200bp window centered on gene start sites, while gene bodies were defined as the entirety of the gene excluding the first and last 300bp from TSS and TES respectively. Raw PRO-seq counts was used as input to DEseq2 along with the mouse/fly ratios as scaling factors.
Omni-ATAC-seq
Omni-ATAC-seq was performed as previously described (85). Nuclei were prepared from 50 000 Setd2wt/wt and Setd2wt/− centroblasts or centrocytes and incubated with 2.5ul transposase (Illumina, 15028212) in a 50ul reaction volume for 30min at 37C. Following purification of transposase fragmented DNA, the library was amplified by PCR and subjected to high throughput sequencing on a Hi-Seq 2×150bp sequencer (Illumina) at GENEWIZ.
WGS
Library preparation, sequencing and post processing of raw data was performed at the New York Genome Center. DNA quality was confirmed by Fragment Analyzer (Advanced Analytics) and all samples had a genomic quality number (GQN) above 8.9. Whole genome sequencing (WGS) libraries were prepared using the Truseq DNA PCR-free Library Preparation Kit in accordance with the manufacturer’s instructions. Briefly, 1ug of DNA was sheared using a Covaris LE220 sonicator (Adaptive Focused Acoustics). DNA fragments underwent bead-based size selection and were subsequently end-repaired, adenylated, and ligated to Illumina sequencing adapters. Final libraries were evaluated using fluorescent-based assays including qPCR with the Universal KAPA Library Quantification Kit and Fragment Analyzer (Advanced Analytics) or BioAnalyzer (Agilent 2100). Libraries were sequenced on an Illumina Novaseq sequencer (v1.5 chemistry) using 2×150bp cycles.
Whole Genome data were processed on NYGC automated pipeline. Paired-end 150 bp reads were aligned to the GRCm38 mouse reference using the Burrows-Wheeler Aligner (BWA-MEM v0.7.8) and processed using the GATK best-practices workflow that includes marking of duplicate reads using Picard tools (v1.83, http://picard.sourceforge.net), local realignment around indels, and base quality score recalibration (BQSR) via Genome Analysis Toolkit (GATK v3.4.0). Tumor and normal bam files are processed through NYGC’s variant calling pipeline consisting of MuTect2 (GATK v4.0.5.1) (86), Strelka2 (v2.9.3) (87) and Lancet (v1.0.7) (88) for calling SNVs and short Insertion-or-Deletion (Indels), SvABA (v0.2.1) (89) for calling Indels and SVs, Manta (v1.4.0) (90) and Lumpy (v0.2.13) (91) for calling SVs and FACETS (v0.5.5) (92), EXCAVATOR2 (v1.1.2) (93) and Biseq2 (v0.2.6) (94) for calling Copy-number variants (CNVs). Calls are merged by variant type (SNVs, Multi Nucleotide Variants (MNVs), Indels and SVs). SVs are converted to bedpe format, all SVs below 500bp are excluded and the rest are merged across callers using bedtools (95) pair to pair (slope of 300bp, same strand orientation, and 50% reciprocal overlap). SNVs and Indels with minor allele frequency (MAF) of 1% or higher in either 1000Genomes (phase3) or gnomeAD (r.2.0.1) were removed. Allele counts and frequencies were used to filter the somatic callset. Variants were filtered if: a) the variant allele frequency (VAF) in the tumor sample was less than 0.0001, or b) if the VAF in the normal sample is greater than 0.2, or c) if the depth at position is less than 2 in either the tumor or normal sample. Variants were also filtered if the VAF in the normal sample was greater than the VAF in the tumor sample. Variant rearrangement junctions were identified using SvABA and GRIDSS with standard settings. 1-kb binned read depth was computed and corrected for GC and mappability using fragCounter. Junction-balanced genome graphs were generated from binned read depth and junction calls using JaBbA (57). Rigma are isolated cluster of deletions at a single chromosomal locus, while chromoplexy are multi-way reciprocal rearrangement where three or more breakpoints on distinct chromosomes are rearranged. Genome graphs and corresponding genomic data (e.g., binned coverage, allelic bin counts) were visualized using gTrack.
All mutational signature analyses were performed using R (v4.0.1). The R package, MutationalPatterns (v3.2.0) (96) was run on the High Confidence somatic SNV calls to estimate contributions of known COSMIC mutational signatures (v3) in the tumor sample. Nearest mutation distance (NMD) was computed for SNVs within the same tumor-normal pair and was used to partition SNVs in two groups of clustered (NMD < 1kb) and non-clustered (NMD >= 1kb) mutations. Mutations were evaluated for involvement in transcriptional strands, where SNVs on the opposite strand of gene bodies were classified as transcribed. Strand bias was assessed using a Poisson test of strand asymmetry.
Quantitative real-time PCR
cDNA synthesis from total RNA was performed using Verso cDNA Synthesis kit (ThermoScientific, AB1453B). Gene expression was detected using the Fast SYBR Green Master Mix (ThermoScientific, 4385614) on a QuantStudio6 Flex Real-Time PCR System (ThermoScientific). Gene expression was normalized to HPRT levels, using the ΔΔC(t) method, with results presented as mRNA expression.
JH4 Intron sequencing and assessment of Pim1 somatic mutations
GC B-were isolated using PNA enrichment. DNA was purified using DNA Clean & Concentrator kit (Zymo Research, D4013). For JH4 intron, sequences were amplified from GCB cell gDNA by PCR using JH4 forward primer (5’- GGA ATT CGC CTG ACA TCT GAG GAC TCT GC-3’), JH4 reverse primer (5’- GAC TAG TCC TCT CCA GTT TCG GCT GAA TCC −3’) (97) and Phusion Hot Start II DNA polymerase (ThermoScientific, F549S). PCR conditions (98C 3’)x1, (98C 30”, 72C 1’)x39, (72C 10’)x1. The Pim1 locus was amplified using primers Pim1 forward primer (5’- TTC GGC TCG GTC TAC TCT G-3’) and Pim1 reverse primer (5’- GGA GGG AAA AGT GGG TCA TAC −3’). PCR program (95C 2’)x1, (95C 1’, 65C 1’, 72C 1’)x25, (72C 15’)x1. PCR products were resolved by agarose gel electrophoresis and corresponding band extracted using the QIAquick Gel Extraction Kit (QIAGEN, 28704).
Purified PCR products were cloned into the Zero Blunt TOPO PCR cloning kit (ThermoScientific, 450245) and grown overnight at 37C on Kanamycin agar plates as per manufacturer’s instructions. Bacteria colony sequencing was performed by GENEWIZ, using the T7 universal sequencing primer (5’- TAA TAC GAC TCA CTA TAG GG −3’) for Pim1 locus and JH4 sequencing primer (5’- CCA TAC ACA TAC TTC TGT GTT CC-3’). Mutation mismatch counts were calculated from Sanger sequencing and compared to the consensus sequencing using the tool Basic Local Alignment Search Tool (BLAST).
Immunoseq
Genomic DNA was isolated using the QIAGEN DNeasy Blood & Tissue Kit (QIAGEN, 69504) from murine tumors and 1 ug of genomic DNA per sample was used for sequencing. Sample data was generated using the immunoSEQ Assay (Adaptive Biotechnologies). The somatically rearranged mouse IGH CDR3 was amplified from genomic DNA using a two-step, amplification bias-controlled multiplex PCR approach (98). The first PCR consists of forward and reverse amplification primers specific for every V and J gene segment and amplifies the hypervariable complementarity-determining region 3 (CDR3) of the immune receptor locus. The second PCR adds a proprietary barcode sequence and Illumina adapter sequences. BCR repertoire analyses were performed using the immunoSEQ Analyzer 3.0 (Adaptive Biotechnologies). Sequences were subjected to analysis using IMGT/Vquest software to define all V, D and J genes as well as CDR3 sequences.
Histone mass spectrometry
Mouse B cells were sorted by FACS as described above with 105 cells collected into 2N H2SO4, as described in Camarillo et al (99). Cellular debris was removed by centrifugation at 4000xg for 5min and histones precipitated with trichloroacetic acid at 20% (v/v) overnight at 4C. Histones were pelleted by centrifugation at 10 000xg for 5min, washed with 0.1% HCl in acetone, followed by a wash in acetone and centrifugation at 15 000xg for 5min. Pellets were dried in a fume hood and store at −80C. Derivatization and digestions were performed based on Garcia et al (100). Dried histones were resuspended in 50mM ammonium bicarbonate, sodium hydroxide then propionic anhydride was added to the histone solution and adjusted to pH 8 with additional sodium hydroxide. Samples were incubated at 52C for 1 hour before drying to completion in a SpeedVac concentrator. Propionylated histones were resuspended in 50mM ammonium bicarbonate and digested for 16 hours with 0.5ug trypsin. Digests were dried in a Speedvac concentrator and subjected to a final propionylation as described above.
Samples were resuspended in water with 0.1% TFA and analyzed by nano-LC (Dionex) on a TSQ Quantiva triple quadrupole mass spectrometer (ThermoScientific). Peptides were loaded on a 3 cm × 150 μm trapping column, packed with ProntoSIL C18-AQ, 3μm, 200Å resin (New Objective) in water with 0.1% TFA for 10 min at 2.5 μL/min. The peptides were eluted at 0.30 μL/min from the trapping and PicoChip analytical column, 10 cm × 75 μm packed with ProntoSIL C18-AQ, 3 μm, 200 Å resin (New Objective) over a 45 min gradient from 1 to 35% Nano Pump Solvent B (95% acetonitrile with 0.1% formic acid; Nano Pump Solvent A, water with 0.1% formic acid). Ions were produced by electrospray from a 10 μm emitter tip and introduced into the mass spectrometer with the following settings: collision gas pressure of 1.5 mTorr; Q1 peak width of 0.7 (FWHM); cycle time of 3 s; skimmer offset of 10 V; electrospray voltage of 2.5 kV. All injections were performed in technical triplicate. Targeted analysis of unmodified and various modified histone peptides was performed with transitions specific to each peptide species as described previously. Raw MS files were analyzed with Skyline (v4.1) using Savitzky-Golay smoothing and peak area assignments were manually assessed. The percent relative abundance of each histone PTM was calculated from the total peak areas exported from Skyline.
Chromatin fractionation
Cells were collected and washed twice with PBS. Pellet was resuspended in 200ul of 1X Abcam Working Lysis buffer with protease inhibitor (Abcam, Ab117152) and incubated on ice for 10min followed by 10s vortex. Supernatant was removed and saved for analysis as the soluble fraction. 100ul of 1X Working Extraction Buffer was added to chromatin pellet and resuspended, followed by 10min incubation on ice followed by 10s vortex. Samples were sonicated twice, 20s each then centrifuged at 12 000rpm for 10min at 4C. Chromatin supernatant was transferred to a new tube, followed by 1:1 addition of Chromatin buffer and stored at −80C.
Western blot
Protein concentration was quantified using the BCA protein assay kit (ThermoScientific, 23225), with a BSA standard curve. Samples were resolved by SDS-polyacrylamide gel electrophoresis, transferred to PVDF membranes and probed with primary antibodies LEDGF (1:2000 dilution, ThermoFisher Scientific, MA5–14821, RRID:AB_11009140), Histone H3 (1:2000 dilution, Abcam, Ab1791, RRID:AB_302613), MEK1/2 (1:2000 dilution, Cell Signaling, 8727, RRID:AB_10829473), followed by HRP-conjugated secondary antibodies (1:5000 dilution, Cell Signaling, 7074, RRID:AB_2099233) and detected with chemiluminescence (WBKLS0500, Millipore). Densitometry was performed using ImageJ (101).
In vitro class switch recombination
Purified splenic resting B cells were culture with B cell media containing RPMI, 15% FBS, penicillin/streptomycin, 55nM β-mercaptoethanol, 2mM L-glutamine. Induction of IgG1 switching was performed by the addition of LPS (33μg/mL, Sigma, L4130) and mIL4 (12.5ng/mL, R&D, 404-ML). Induction of IgG3 switching was performed by the addition of LPS (33μg/mL, Sigma, L4130).
In silico prediction of mutation impact
SETD2 SNV identified from human DLBCL cohorts were used as inputs for the prediction tools, Polyphen2 (102) and Mutation Assessor (103). Deleterious SNV were predictions that were possibly or probably damaging (Polyphen2) or neutral, medium, high (Mutation Assessor).
Secondary tumor transplantation
Lymph node tumors from BCL2 and Setd2/BCL2 animals were collected and single cell suspensions prepared followed by SRBC lysis and resuspended in PBS. 10X106 cells were injected into the peritoneal cavity of RAG1KO mice and monitored until any one of the criteria for euthanasia were met.
Cell lines
The DLBCL cell lines OCI-Ly7 and RIVA was grown in Iscove Modified Dulbecco Media (ThermoScientific, 12440061) supplemented with 10% FBS (OCI-Ly7) or 20% FBS (RIVA) and antibiotics; SU-DHL-4, HBL1, SU-DHL-2 was grown in Roswell Park Memorial Institute Media (Corning, 10–040-CV) supplemented with 10% FBS (SU-DHL-4, HBL1) or 20% FBS (SU-DHL-2) and antibiotics. OCI-Ly7 were obtained from the Ontario Cancer Institute (OCI), SU-DHL-4, RIVA from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ), SU-DHL-2 from American Type Culture Collection (ATCC), HBL1 were obtained from Jose A. Martinez-Climent (Centre for Applied Medical Research, CIMA). All cells were grown in a 37C incubator at a 5% CO2 environment. Cell line authentication was performed on all parental and CRISPR KO cell lines at the University of Arizona Genetics Core, using the short tandem repeat (STR) assay and genetic profiles were compared to established cell line profiles. Cell lines were also routinely tested for mycoplasma contamination in the laboratory.
Generation of SETD2 KO cell lines
To generate SETD2 KO cell lines (HBL1, OCI-Ly7, SU-DHL-4, SU-DHL-2), parental cells were transduced with lentivirus expressing doxycycline inducible Cas9 and blasticidin resistance gene (Addgene, #83481) followed by 5 days of blasticidin selection. Cas9 expressing cells were transduced with lentivirus expressing a SETD2 specific sgRNA (SETD2G#1: 5’ AAA GAA ACA ATA GTA GAA GT 3’; SETD2G#2: 5’ AAT CTG ATG AAG ATT CTG TA 3’) and GFP (Addgene, #57822). 4 days post doxycycline induction, GFP+ cells were single cell sorted into 96 well plates and allowed to grow for at least two weeks. For RIVA, parental cells were electroporated with Amaxa Nucleofector Unit and the SF Cell line 4D-Nucleofector X kit (Lonza, PBC2–22500) to incorporate a recombinant Cas9 nuclease (Alt-R S.p. Cas9 Nuclease V3, Integrated DNA Technologies, 1081058), a SETD2 targeting Alt-R CRISPR-Cas9 crRNA (SETD2G#1: 5’ AAA GAA ACA ATA GTA GAA GT 3’; SETD2G#2: 5’ AAT CTG ATG AAG ATT CTG TA 3’ Integrated DNA Technologies) and Alt-R CRISPR-Cas9 tracrRNA (Integrated DNA Technologies, 1075927) using manufacturer’s protocol. Forty-eight hours after electroporation, ATTO550+ single cells were sorted into 96 well plates and allowed to grow for at least two weeks. Clones were screened by PCR amplification of a 500bp region encompassing the CRISPR-Cas9 cleavage site and verified by sanger sequencing at GENEWIZ.
Quantification and statistical analyses
Data were analyzed with GraphPad Prism 7.01 and represented with number of replicates, type of measurement and statistical significance reported in the Figures and Figure Legends. Data is judged to be statistically significant when p<0.05 in a two-sided t-test or one-way ANOVA, with asterisks denoting degree of significance (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001). Fisher exact test were performed for single or multiple pair wise comparisons.
Data availability
RNA-seq, CUT&RUN, PRO-seq, ATAC-seq and WGS data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE189867.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Our findings define a B cell specific oncogenic effect of SETD2 heterozygous mutation, which unleashes AICDA mutagenesis of non-template strand DNA in the germinal center reaction, resulting in lymphomas with heavy mutational burden. GC derived lymphomas did not tolerate SETD2 homozygous deletion pointing to a novel context specific therapeutic vulnerability.
ACKNOWLEDGEMENTS
The authors show their immense gratitude to the late Dr Kristy Richards (Cornell University, WCM) for first conceiving of this project and contributions to lymphoma research. We would also like to acknowledge Dr Surya Seshan (Department of Pathology and Laboratory Medicine, WCM) for support in interpreting histological disease. The work was supported by the following grants awarded to A.M.M. NCI/NIH R35 CA220499, NCI/NIH P01 CA229086–01A1, LLS-SCOR 7012–16, LLS-TRP 6572–19, the Samuel Waxman Cancer Research Foundation, the Follicular Lymphoma Consortium, and the Chemotherapy Foundation. N.L.K. is supported by NIGMS/NIH P41 GM108569. S.A.A is supported by the NIH grants CA176745 and CA066996. C.E.M. would like to thank the Scientific Computing Unit (SCU), XSEDE Supercomputing Resources, as well as the Starr Cancer Consortium (I13–0052), the Vallee Foundation, the WorldQuant Foundation, The Pershing Square Sohn Cancer Research Alliance, the National Institutes of Health (R01CA249054, R35GM138152), and the Leukemia and Lymphoma Society (LLS) grants MCL7001–18, LLS 9238–16, LLS-MCL7001–18. The authors thank the Laboratory of Comparative Pathology, Epigenomics Core and Flow Cytometry Core Facility at Weill Cornell Medicine.
Footnotes
CONFLICTS OF INTEREST
C.M. has financial interest in Thorne HealthTech. MI is an advisor to ImmPACT Bio. S.S.D. owns equity in Data Driven Bioscience. C.F. is a consultant for AstraZeneca, BeiGene, BioAscend, Bristol Myers Squibb, Celgene, Curio Sciences, Denovo Biopharma, Epizyme/Incyte, Genentech/Roche, Genmab, MEI Pharmaceuticals, MorphoSys AG, Pharmacyclics/ Janssen, SeaGen and research funding from 4D, Abbvie, Acerta, Adaptimmune, Allogene, Amgen, Bayer, Celgene, Cellectis, EMD, Gilead, Genentech/Roche, Guardant, Iovance, Janssen Pharmaceutical, Kite, Morphosys, Nektar, Novartis, Pfizer, Pharmacyclics, Sanofi, Takeda, TG Therapeutics, Xencor, Ziopharm, Burroughs Wellcome Fund, Eastern Cooperative Oncology Group, National Cancer Institute, V Foundation, Cancer Prevention and Research Institute of Texas: CPRIT Scholar in Cancer Research. S.A.A. has been a consultant and/or shareholder for Neomorph Inc, Imago Biosciences, Vitae/Allergan Pharma, Cyteir Therapeutics, C4 Therapeutics, Accent Therapeutics, and Mana Therapeutics and has received research support from Janssen, Novartis, and Syndax. C.E.M. is a cofounder and board member for Biotia and Onegevity Health as well as an advisor or compensated speaker for Abbvie, Acuamark Diagnostics, ArcBio, Bio-Rad, DNA Genotek, Genialis, Genpro, Karius, Illumina, New England BioLabs, Qiagen, Whole Biome and Zymo Research. A.M.M receives research funding from Epizyme, Janssen, Astra Zeneca, Sanofi, Daiichi Sankyo, has consulted for Epizyme, Constellation, Exo-Therapeutics, Astra Zeneca, BMS, Daiichi Sankyo, Janssen and is on the scientific advisory board of KDAC. No other conflicts of interest are declared.
REFERENCES
- 1.Mlynarczyk C, Fontan L, Melnick A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev 2019;288(1):214–39 doi 10.1111/imr.12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mesin L, Ersching J, Victora GD. Germinal Center B Cell Dynamics. Immunity 2016;45(3):471–82 doi 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 2007;76:1–22 doi 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 4.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nature immunology 2003;4(5):452–6 doi 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 5.Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med 2003;197(10):1291–6 doi 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature 2008;451(7180):841–5 doi 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 7.Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, et al. The microanatomic segregation of selection by apoptosis in the germinal center. Science 2017;358(6360) doi 10.1126/science.aao2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, et al. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nature immunology 2007;8(7):705–14 doi 10.1038/ni1478. [DOI] [PubMed] [Google Scholar]
- 9.Lossos IS, Alizadeh AA, Eisen MB, Chan WC, Brown PO, Botstein D, et al. Ongoing immunoglobulin somatic mutation in germinal center B cell-like but not in activated B cell-like diffuse large cell lymphomas. Proc Natl Acad Sci U S A 2000;97(18):10209–13 doi 10.1073/pnas.180316097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenz G, Nagel I, Siebert R, Roschke AV, Sanger W, Wright GW, et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. J Exp Med 2007;204(3):633–43 doi 10.1084/jem.20062041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017;171(2):481–94 e15 doi 10.1016/j.cell.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011;476(7360):298–303 doi 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013;23(5):677–92 doi 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov 2017;7(1):38–53 doi 10.1158/2159-8290.CD-16-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 2015;21(10):1199–208 doi 10.1038/nm.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov 2018;8(12):1632–53 doi 10.1158/2159-8290.CD-18-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov 2017;7(3):322–37 doi 10.1158/2159-8290.CD-16-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 2015;21(10):1190–8 doi 10.1038/nm.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beguelin W, Teater M, Meydan C, Hoehn KB, Phillip JM, Soshnev AA, et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 2020;37(5):655–73 e11 doi 10.1016/j.ccell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fahey CC, Davis IJ. SETting the Stage for Cancer Development: SETD2 and the Consequences of Lost Methylation. Cold Spring Harb Perspect Med 2017;7(5) doi 10.1101/cshperspect.a026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee MJ, Koff JL, Switchenko JM, Jhaney CI, Harkins RA, Patel SP, et al. Genome-defined African ancestry is associated with distinct mutations and worse survival in patients with diffuse large B-cell lymphoma. Cancer 2020;126(15):3493–503 doi 10.1002/cncr.32866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 2008;27(2):406–20 doi 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 2005;25(8):3305–16 doi 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015;520(7546):243–7 doi 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 25.Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Molecular cell 2014;56(2):298–310 doi 10.1016/j.molcel.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science 2010;327(5968):996–1000 doi 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 2014;21(4):366–74 doi 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013;153(3):590–600 doi 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanu N, Gronroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene 2015;34(46):5699–708 doi 10.1038/onc.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mar BG, Chu SH, Kahn JD, Krivtsov AV, Koche R, Castellano CA, et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood 2017;130(24):2631–41 doi 10.1182/blood-2017-03-775569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan H, Li N, Fu D, Ren J, Hui J, Peng J, et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J Clin Invest 2017;127(9):3375–91 doi 10.1172/JCI94292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu SH, Chabon JR, Matovina CN, Minehart JC, Chen BR, Zhang J, et al. Loss of H3K36 Methyltransferase SETD2 Impairs V(D)J Recombination during Lymphoid Development. iScience 2020;23(3):100941 doi 10.1016/j.isci.2020.100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji Z, Sheng Y, Miao J, Li X, Zhao H, Wang J, et al. The histone methyltransferase Setd2 is indispensable for V(D)J recombination. Nat Commun 2019;10(1):3353 doi 10.1038/s41467-019-11282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 2018;378(15):1396–407 doi 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ennishi D, Mottok A, Ben-Neriah S, Shulha HP, Farinha P, Chan FC, et al. Genetic profiling of MYC and BCL2 in diffuse large B-cell lymphoma determines cell-of-origin-specific clinical impact. Blood 2017;129(20):2760–70 doi 10.1182/blood-2016-11-747022. [DOI] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013;499(7456):43–9 doi 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497(7447):67–73 doi 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017;171(3):540–56 e25 doi 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet 2014;46(3):287–93 doi 10.1038/ng.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020;37(4):551–68 e14 doi 10.1016/j.ccell.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res 1997;25(6):1317–8 doi 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mort RL, Ford MJ, Sakaue-Sawano A, Lindstrom NO, Casadio A, Douglas AT, et al. Fucci2a: a bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle 2014;13(17):2681–96 doi 10.4161/15384101.2015.945381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carvalho S, Vitor AC, Sridhara SC, Martins FB, Raposo AC, Desterro JM, et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 2014;3:e02482 doi 10.7554/eLife.02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schoeler K, Jakic B, Heppke J, Soratroi C, Aufschnaiter A, Hermann-Kleiter N, et al. CHK1 dosage in germinal center B cells controls humoral immunity. Cell Death Differ 2019;26(12):2551–67 doi 10.1038/s41418-019-0318-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sorensen CS, et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol 2012;19(8):803–10 doi 10.1038/nsmb.2314. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Wang Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J Biol Chem 2017;292(28):11951–9 doi 10.1074/jbc.M117.788224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol 2009;30(4):173–81 doi 10.1016/j.it.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 48.Alvarez-Prado AF, Perez-Duran P, Perez-Garcia A, Benguria A, Torroja C, de Yebenes VG, et al. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J Exp Med 2018;215(3):761–71 doi 10.1084/jem.20171738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001;412(6844):341–6 doi 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 50.Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 2016;11(8):1455–76 doi 10.1038/nprot.2016.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knittel G, Liedgens P, Korovkina D, Seeger JM, Al-Baldawi Y, Al-Maarri M, et al. B-cell-specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood 2016;127(22):2732–41 doi 10.1182/blood-2015-11-684183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Egle A, Harris AW, Bath ML, O’Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 2004;103(6):2276–83 doi 10.1182/blood-2003-07-2469. [DOI] [PubMed] [Google Scholar]
- 53.Flumann R, Rehkamper T, Nieper P, Pfeiffer P, Holzem A, Klein S, et al. An Autochthonous Mouse Model of Myd88- and BCL2-Driven Diffuse Large B-cell Lymphoma Reveals Actionable Molecular Vulnerabilities. Blood Cancer Discov 2021;2(1):70–91 doi 10.1158/2643-3230.BCD-19-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cardenas MG, Yu W, Beguelin W, Teater MR, Geng H, Goldstein RL, et al. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest 2016;126(9):3351–62 doi 10.1172/JCI85795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Persi E, Wolf YI, Horn D, Ruppin E, Demichelis F, Gatenby RA, et al. Mutation-selection balance and compensatory mechanisms in tumour evolution. Nat Rev Genet 2021;22(4):251–62 doi 10.1038/s41576-020-00299-4. [DOI] [PubMed] [Google Scholar]
- 56.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell 2008;135(6):1028–38 doi 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hadi K, Yao X, Behr JM, Deshpande A, Xanthopoulakis C, Tian H, et al. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 2020;183(1):197–210 e32 doi 10.1016/j.cell.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tseng YY, Bagchi A. The PVT1-MYC duet in cancer. Mol Cell Oncol 2015;2(2):e974467 doi 10.4161/23723556.2014.974467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 2011;147(1):95–106 doi 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zazzeroni F, Papa S, Algeciras-Schimnich A, Alvarez K, Melis T, Bubici C, et al. Gadd45 beta mediates the protective effects of CD40 costimulation against Fas-induced apoptosis. Blood 2003;102(9):3270–9 doi 10.1182/blood-2003-03-0689. [DOI] [PubMed] [Google Scholar]
- 61.Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015;162(4):727–37 doi 10.1016/j.cell.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004;432(7017):635–9 doi 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 63.Nicolas L, Cols M, Smolkin R, Fernandez KC, Yewdell WT, Yen WF, et al. Cutting Edge: ATM Influences Germinal Center Integrity. J Immunol 2019;202(11):3137–42 doi 10.4049/jimmunol.1801033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gu X, Booth CJ, Liu Z, Strout MP. AID-associated DNA repair pathways regulate malignant transformation in a murine model of BCL6-driven diffuse large B-cell lymphoma. Blood 2016;127(1):102–12 doi 10.1182/blood-2015-02-628164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol 2013;8:79–103 doi 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 66.Gostissa M, Bianco JM, Malkin DJ, Kutok JL, Rodig SJ, Morse HC 3rd, et al. Conditional inactivation of p53 in mature B cells promotes generation of nongerminal center-derived B-cell lymphomas. Proc Natl Acad Sci U S A 2013;110(8):2934–9 doi 10.1073/pnas.1222570110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Armache A, Yang S, Martinez de Paz A, Robbins LE, Durmaz C, Cheong JQ, et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 2020;583(7818):852–7 doi 10.1038/s41586-020-2533-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, et al. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res 2013;41(5):2881–93 doi 10.1093/nar/gks1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017;543(7643):72–7 doi 10.1038/nature21373. [DOI] [PubMed] [Google Scholar]
- 70.Bhattacharya S, Wang S, Reddy D, Shen S, Zhang Y, Zhang N, et al. Structural basis of the interaction between SETD2 methyltransferase and hnRNP L paralogs for governing co-transcriptional splicing. Nat Commun 2021;12(1):6452 doi 10.1038/s41467-021-26799-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, et al. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nature immunology 2010;11(9):820–6 doi 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev 2004;200:115–31 doi 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- 73.Hirth G, Svensson CM, Bottcher K, Ullrich S, Figge MT, Jungnickel B. Regulation of the Germinal Center Reaction and Somatic Hypermutation Dynamics by Homologous Recombination. J Immunol 2019;203(6):1493–501 doi 10.4049/jimmunol.1900483. [DOI] [PubMed] [Google Scholar]
- 74.Xie Y, Sahin M, Sinha S, Wang Y, Nargund AM, Lyu Y, et al. SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes. Nat Cancer 2022;3(2):188–202 doi 10.1038/s43018-021-00316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lampe JW, Alford JS, Boriak-Sjodin PA, Brach D, Cosmopoulos K, Duncan KW, et al. Discovery of a First-in-Class Inhibitor of the Histone Methyltransferase SETD2 Suitable for Preclinical Studies. ACS Med Chem Lett 2021;12(10):1539–45 doi 10.1021/acsmedchemlett.1c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013;1303.3997v1 [Google Scholar]
- 77.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 2016;44(W1):W160–5 doi 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009;25(15):1952–8 doi 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40 doi 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goodarzi H, Elemento O, Tavazoie S. Revealing global regulatory perturbations across human cancers. Molecular cell 2009;36(5):900–11 doi 10.1016/j.molcel.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol 2013;9(8):e1003118 doi 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 2017;14(10):959–62 doi 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31(3):213–9 doi 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 2018;15(8):591–4 doi 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 88.Narzisi G, Corvelo A, Arora K, Bergmann EA, Shah M, Musunuri R, et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol 2018;1:20 doi 10.1038/s42003-018-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res 2018;28(4):581–91 doi 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016;32(8):1220–2 doi 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 91.Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 2014;15(6):R84 doi 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44(16):e131 doi 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.D’Aurizio R, Pippucci T, Tattini L, Giusti B, Pellegrini M, Magi A. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res 2016;44(20):e154 doi 10.1093/nar/gkw695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xi R, Lee S, Xia Y, Kim TM, Park PJ. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res 2016;44(13):6274–86 doi 10.1093/nar/gkw491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–2 doi 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med 2018;10(1):33 doi 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jolly CJ, Klix N, Neuberger MS. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res 1997;25(10):1913–9 doi 10.1093/nar/25.10.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun 2013;4:2680 doi 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 99.Camarillo JM, Swaminathan S, Abshiru NA, Sikora JW, Thomas PM, Kelleher NL. Coupling Fluorescence-Activated Cell Sorting and Targeted Analysis of Histone Modification Profiles in Primary Human Leukocytes. J Am Soc Mass Spectrom 2019;30(12):2526–34 doi 10.1007/s13361-019-02255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, et al. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat Protoc 2007;2(4):933–8 doi 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9(7):671–5 doi 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7(4):248–9 doi 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011;39(17):e118 doi 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, CUT&RUN, PRO-seq, ATAC-seq and WGS data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE189867.