

Abstract
Cancer cells respond to various stressful conditions through the dynamic regulation of RNA m6A modification. Doxorubicin is a widely used chemotherapeutic drug that induces DNA damage. It is interesting to know whether cancer cells regulate the DNA damage response and doxorubicin sensitivity through RNA m6A modification. Here, we found that doxorubicin treatment significantly induced RNA m6A methylation in breast cancer cells in both a dose- and a time-dependent manner. However, protein arginine methyltransferase 5 (PRMT5) inhibited RNA m6A modification under doxorubicin treatment by enhancing the nuclear translocation of the RNA demethylase AlkB homolog 5 (ALKBH5), which was previously believed to be exclusively localized in the nucleus. Then, ALKBH5 removed the m6A methylation of BRCA1 for mRNA stabilization and further enhanced DNA repair competency to decrease doxorubicin efficacy in breast cancer cells. Importantly, we identified the approved drug tadalafil as a novel PRMT5 inhibitor that could decrease RNA m6A methylation and increase doxorubicin sensitivity in breast cancer. The strategy of targeting PRMT5 with tadalafil is a promising approach to promote breast cancer sensitivity to doxorubicin through RNA methylation regulation.
Keywords: PRMT5, RNA m6A methylation, ALKBH5, DNA repair, doxorubicin, tadalafil, breast cancer, ALKBH7, nuclear translocation, BRCA1
Graphical abstract
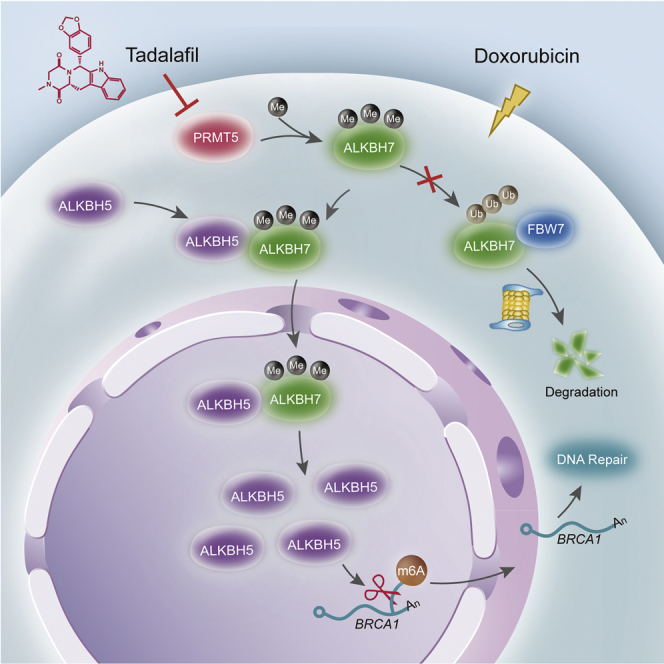
Ying et al. provide evidence that PRMT5 decreases doxorubicin efficacy through enhancing the nuclear translocation of the RNA demethylase ALKBH5 for DNA repair in breast cancer cells. Targeting PRMT5 with the approved drug tadalafil is a promising approach to promote breast cancer sensitivity to doxorubicin through RNA methylation regulation.
Introduction
Methylation at the 6 position of adenosine (N6-methyladenosine, m6A) in RNA is abundant in approximately 25% of RNA transcripts and is involved in the regulation of RNA splicing, translocation, and stability.1 RNA m6A methylation is reversibly regulated by methyltransferase complex (METTL3, METTL14, and the cofactor WTAP), and demethylases (fat mass and obesity-associated protein [FTO] and AlkB homolog 5 [ALKBH5]).1,2 Importantly, RNA m6A methylation and its modulators play vital roles in cancer progression.
Cancer cells have to confront and adapt to different stressful conditions, such as DNA damage, hypoxia, and low nutrient levels.3,4 Especially during the cancer therapeutic regimen, the response of cancer cells to various chemotherapeutic stresses will determine their survival or death fate or even further drug resistance and progression. It has been reported that RNA m6A modification is involved in the short-term UV irradiation-induced DNA damage response.5 Our recent work found reduced m6A methylation in colorectal cancer cells with continuous differentiation signaling.6 Thus, further elucidation of the mechanism that cancer cells respond to chemotherapeutic stresses through regulating RNA m6A modification will help to identify novel targets for cancer therapy.
Protein arginine methyltransferase 5 (PRMT5) is highly expressed in multiple types of cancer and plays important roles in cancer cell proliferation and the maintenance of stem cell properties.7, 8, 9 PRMT5 is reported to regulate homologous recombination (HR) and DNA repair.10 Our previous work found that PRMT5 decreased the breast cancer cell response to doxorubicin.11 Since doxorubicin as a chemotherapeutic drug functions by inducing serious genotoxic stress and DNA damage,12 we hypothesized that PRMT5 decreases the cancer cell response to doxorubicin by increasing the DNA damage repair (DDR) ability. Here, we found that doxorubicin treatment clearly induced RNA m6A methylation in breast cancer cells and tumor tissues. However, PRMT5 inhibited doxorubicin-induced RNA m6A methylation by enhancing the nuclear translocation of ALKBH5 with the partner AlkB homolog 7 (ALKBH7). ALKBH5 removed m6A methylation from BRCA1 mRNA, maintained BRCA1 stability and function, and thereby reduced cell sensitivity to doxorubicin. With unbiased drug screening, we identified tadalafil as a novel PRMT5-targeting inhibitor that produced a synergistic effect with doxorubicin to impede breast cancer growth.
Results
PRMT5 inhibits doxorubicin-induced RNA m6A methylation
To examine whether RNA m6A is involved in DNA damage induced by doxorubicin, we compared RNA m6A methylation levels between breast cancer patient samples with and without neoadjuvant doxorubicin therapy using a dot-blot m6A assay. There was a dramatic increase in RNA m6A methylation in the doxorubicin-treated cancer samples compared with the control samples (Figure 1A, Table S1). We further examined RNA m6A in patient-derived xenografts (PDXs) of breast cancer treated with or without doxorubicin (Figures 1B, S1A, and S1B). Doxorubicin increased the expression of γ-H2AX, a marker for DNA damage, and enhanced RNA m6A levels. Additionally, in breast cancer cells, doxorubicin induced a significant increase in RNA m6A methylation and γ-H2AX expression in a time- and a dose-dependent manner (Figures 1C and S1C). These results showed that doxorubicin induced DNA damage and activated RNA m6A modification.
Figure 1.

PRMT5 inhibits doxorubicin-induced RNA m6A methylation
(A) Left: dot-blot assay of RNA m6A methylation in breast cancer patient samples with (n = 15) or without (n = 18) neoadjuvant doxorubicin (DOX) therapy. Right: semiquantification of the data shown at the left. (B) Semiquantification of the data from a dot-blot assay of RNA m6A methylation in breast cancer tissue samples from a PDX mouse model treated with or without DOX. (C) Dot-blot assay of RNA m6A methylation in MDA-MB-231 and T47-D breast cancer cells treated with DOX for the duration and dose indicated. (D) Dot-blot assay of RNA m6A methylation in MDA-MB-231, MCF-7, and T47-D breast cancer cells overexpressing PRMT5 treated with doxorubicin as indicated. MDA-MB-231, 0.4 μg/mL DOX for 24 h; MCF-7 and T47-D, 0.5 μg/mL DOX for 24 h. (E) Dot-blot assay of RNA m6A methylation in wild-type and prmt5-knockout MEFs treated with 0.4 μg/mL DOX for 24 h. An anti-m6A antibody was used for dot blots with 200 ng of mRNA, and methylene blue staining was used as the loading control in (A), (C), (D) and (E).
Since PRMT5 regulates the DNA damage response in leukemia cells,10 we further analyzed this effect on breast cancer cells treated with doxorubicin. Consistent with our previous study,11 we found that overexpressed PRMT5 reversed the γ-H2AX expression induced by doxorubicin in MDA-MB-231 and T47D breast cancer cells. We subcutaneously injected MDA-MB-231 cells with PRMT5-overexpressing or control cells into nude mice and treated the mice with doxorubicin. Breast cancer tumors with PRMT5 overexpression exhibited a reduced, weaker response to doxorubicin, showing increased proliferation activity and an enhanced DNA repair capacity detected with staining for Ki-67 and replication protein A 32-kDa subunit (RPA32), respectively (Figures S1D–S1G). These data suggest that PRMT5 may increase the DDR ability of cancer cells, neutralizing the effect of doxorubicin. Therefore, we wondered whether PRMT5 regulates RNA m6A methylation induced by doxorubicin. In different breast cancer cells, the higher expression of PRMT5 was able to offset the methylation of RNA m6A induced by doxorubicin (Figure 1D). Furthermore, using established mouse embryonic fibroblasts (MEFs), we observed that prmt5-knockout MEFs exhibited enhanced RNA m6A methylation compared with wild-type MEFs under doxorubicin treatment (Figures 1E and S1H, and prmt5-knockout MEFs were more sensitive to doxorubicin, with decreased proliferation and increased γ-H2AX expression (Figures S1I–S1L). Taken together, our results showed that PRMT5 decreased doxorubicin sensitivity in breast cancer cells, decreased RNA m6A methylation, and increased DDR, which prompted us to examine whether PRMT5 regulates RNA m6A on DNA repair genes.
PRMT5 regulates m6A methylation of BRCA1 mRNA for DNA repair
To identify PRMT5-mediated transcriptome-wide RNA m6A methylation patterns for the doxorubicin response, we performed immunoprecipitation (IP) of m6A-methylated RNA followed by sequencing (MeRIP-seq) in MDA-MB-231 cells. As shown in Figures 2A and S2A, the m6A peaks were enriched mainly near start and stop codons and characterized by the canonical GGAC motif. Compared with vehicle, doxorubicin induced an increase in m6A peaks mainly at stop codons; however, PRMT5 overexpression in cancer cells decreased these peaks induced by doxorubicin (Figure S2B). Gene Ontology (GO) term analysis revealed that RNA m6A methylation increase induced by doxorubicin treatment and PRMT5 overexpression resulted in enrichment of genes related to the DNA damage response and repair (Figures S2C and S2D). The GO terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation analysis identified BRCA1 signaling as the target of m6A methylation induced by doxorubicin but inhibited by PRMT5 (Figure S2D). BRCA1 is the critical regulator of HR repair of damaged DNA to maintain genome stability,13 and PRMT5 increased BRCA1 mRNA expression in MDA-MB-231 cells (Figure S2E). Because RNA m6A methylation modulates RNA stability by regulating mRNA degradation,14 we measured BRCA1 RNA stability under doxorubicin treatment in breast cancer cells treated with the cyclin-dependent kinase (CDK) inhibitor flavopiridol to block global mRNA transcription.15 Doxorubicin decreased BRCA1 mRNA stability, but PRMT5 reversed this effect (Figure 2C). Given that PRMT5 decreased m6A methylation, the data indicated that PRMT5 might maintain BRCA1 RNA expression through inhibition of the m6A methylation of BRCA1 and thereby promote DNA repair. Furthermore, we knocked down BRCA1 expression by siRNAs in PRMT5-overexpressing and control cells and treated the cells with doxorubicin. PRMT5 overexpression reduced the percentage of comet tail DNA (transfer DNA (T-DNA))-positive cells, which represented the cells with damaged DNA. However, this reduction was reversed by knocking down BRCA1 expression (Figure 2D). Simultaneously, knocking down BRCA1 expression in PRMT5-overexpressing cells recovered the cell sensitivity to doxorubicin (Figures S2F and S2G). Thus, PRMT5 may reduce the doxorubicin sensitivity of breast cancer cells by improving BRCA1-mediated DDR.
Figure 2.

PRMT5 decreases the m6A methylation of BRCA1 mRNA to increase BRCA1 stability
(A) The m6A peak enrichment was analyzed in each group as indicated and characterized by the canonical GGAC motif. (B) The m6A abundances in BRCA1 mRNA transcripts in different groups were detected by m6A-seq. (C) MDA-MB-231 cells were treated with 0.5 μg/mL DOX for 24 h and then treated with vehicle or 3.2 μM flavopiridol for 6 h. BRCA1 mRNA levels were measured by RT-qPCR, and the flavopiridol/vehicle (F/V) ratio of BRCA1 mRNA was determined (mean ± SEM; n = 3). (D) A comet assay was performed with MDA-MB-231 cells with or without PRMT5 overexpression and BRCA1-specific siRNA treated with 0.4 μg/mL DOX for 24 h. Right: quantification of the comet tail that represents broken DNA strands (T-DNA%). ##p < 0.01 versus MDA-MB-231-PRMT5-siNC, ∗∗p < 0.01 versus MDA-MB-231-siNC, and ∗∗∗p < 0.001 versus MDA-MB-231-siNC. NC, negative control. (E) A relative luciferase assay was performed to analyze the BRCA1 mRNA 3′-UTR with intact or mutant m6A sites. (F) RT-PCR analysis was performed following m6A-IP of the BRCA1 mRNA 3′-UTR in MDA-MB-231 cells with PRMT5 overexpression treated with 0.4 μg/mL doxorubicin for 24 h. BRCA1-149 and BRCA1-1268 represent two different m6A sites in the BRCA1 mRNA 3′-UTR. ∗∗p < 0.01, and ∗∗∗p < 0.001. (G) RT-PCR analysis was performed following m6A-IP of the BRCA1 3′-UTR in wild-type and prmt5-knockout MEFs treated with 0.4 μg/mL doxorubicin. ∗p < 0.05.
To further validate whether the regulation of BRCA1 by PRMT5 is related to m6A methylation, we analyzed the structure of BRCA1 mRNA and found two potential m6A methylation sites at adenosines 149 and 1,268 in the 3′ untranslated region (UTR) (Figures S2H). As expected, PRMT5 upregulated the luciferase activity of wild-type BRCA1 3′-UTR reporter constructs, but mutations in these two m6A sites substantially decreased the construct luciferase activity (Figure 2E). We further conducted m6A IP followed by gene-specific RT-PCR analysis of BRCA1 with primers flanking the two potential m6A sites (BRCA1-149 and BRCA1-1268), using hypoxanthine phosphoribosyltransferase 1 (HPRT1) as the internal control.16 Doxorubicin induced a relatively high abundance of m6A methylation on BRCA1 mRNA at adenosines 149 and 1,268 in the BRCA1 3′-UTR, whereas the methylation at these sites was inhibited by PRMT5 (Figure 2F). Using a similar method, we observed more enrichment of m6A methylation on BRCA1 mRNA in prmt5-knockout cells treated with doxorubicin compared with wild-type MEFs (Figure 2G). Taken together, our data demonstrate that PRMT5 decreases m6A methylation of BRCA1 mRNA to increase its stability and thereby improve DDR.
PRMT5 promotes the arginine methylation of ALKBH7 to regulate the RNA m6A modification and doxorubicin sensitivity
To understand the molecular mechanism by which PRMT5 regulates the m6A methylation of BRCA1 mRNA, we explored the downstream effectors of PRMT5 using mass spectrometry analysis. We identified ALKBH7 as a potential PRMT5-interacting protein (Figure 3A). ALKBH7 belongs to the ALKBH family and predominantly localizes in the mitochondria, where it is involved in alkylation and oxidation-induced programmed necrosis.17, 18, 19 We validated that PRMT5 bound with ALKBH7 in the context of doxorubicin treatment by IP (Figure 3B). Notably, both the human and the mouse ALKBH7 proteins contain the consensus PRMT5-recognized methylation motif RGRR (Figure S3A). We transfected HA-tagged ALKBH7 and FLAG-tagged PRMT5 into HEK293 cells and then pulled down ALKBH7 with an anti-HA tag antibody. We detected the obvious symmetric dimethylarginine (SDMA) methylation of ALKBH7 catalyzed by PRMT5 (Figure S3B). However, with either PRMT5 with the R368 mutation (PRMT5 R368A, defective methyltransferase activity) or mutant ALKBH7 with three arginine sites mutated to lysine (ALKBH73RK), ALKBH7 dramatically lost SDMA methylation (Figures 3C and S3B), suggesting that ALKBH7 is a substrate of PRMT5. We further confirmed this finding with an in vitro methylation assay to evaluate the ability of purified PRMT5 to catalyze the methylation of ALKBH7.20 To optimize the expression and purification conditions of mutant ALKBH73RK, 20 amino acid residues of signal peptides in the N terminus were deleted. As shown in Figure 3D, in the presence of recombinant FLAG-tagged PRMT5, histone H4, the methyl donor S-adenosylmethionine (SAM), and His-tagged wild-type ALKBH7 (ALKBH7-WT) or mutant ALKBH73RK, PRMT5 dramatically catalyzed the arginine methylation of histone H4 (positive control; Figure 3D, left) and ALKBH7-WT. However, the arginine methylation of mutant ALKBH73RK by PRMT5 was obviously decreased (Figure 3D, right), demonstrating that these three arginine sites were methylation targets of PRMT5 in ALKBH7.
Figure 3.

PRMT5 promotes the arginine methylation of ALKBH7 to regulate RNA m6A modification of BRCA1 and DOX sensitivity
(A) Silver staining of proteins associated with PRMT5. MDA-MB-231 cells stably expressing FLAG-PRMT5 were treated with DOX and immunoprecipitated with an anti-FLAG antibody, and the MDA-MB-231 cells with same treatment were set for control (Ctrl). Samples were resolved by SDS-PAGE and visualized by silver staining. The bands of interest were cut out and analyzed with a mass spectrometer. (B) IP of FLAG-PRMT5 and MYC-ALKBH7 in PRMT5-or ALKBH7-overexpressing MDA-MB-231 breast cancer cells treated with or without 0.4 μg/mL DOX for 24 h. (C) Wild-type (WT) ALKBH7 and mutant ALKBH73RK with arginine-to-lysine mutation were used for protein methylation detection. HEK293 cells were transfected with HA-ALKBH7, HA-ALKBH73RK or FLAG-PRMT5 as indicated. IP with anti-HA or IgG antibodies and western blotting with an anti-symmetric dimethylarginine (SDMA) antibody were performed to assess the methylation of ALKBH7 at the 194RGRR site. (D) In vitro methylation assay with recombinant FLAG-tagged PRMT5, unlabeled S-adenosylmethionine (SAM), and the substrates His-tagged WT or methylation site-mutant (3RK) ALKBH7. The same blot was stripped and re-probed with an anti-ALKBH7 antibody, showing the amount of substrate in each reaction. H4 was used as a positive control. (E) Protein stability analysis by western blotting of MDA-MB-231 cells stably expressing WT ALKBH7 or ALKBH73RK treated with 100 μg/mL cycloheximide (CHX) for the indicated times. (F) HEK293 cells transfected with V5-ubiqutin, FBW7α, MYC-ALKBH7, and MYC-ALKBH73RK as indicated and then treated with 100 μM MG132 for 6 h. ALKBH7 and ALKBH73RK were immunoprecipitated from whole-cell lysates with an anti-MYC antibody. The ubiquitination of ALKBH7 and ALKBH73RK in the immunoprecipitants and levels of ALKBH7, ALKBH73RK, and FBW7α in whole-cell lysates (Input) were determined by western blotting as indicated. (G) Proliferation analysis (MTT) of PRMT5 overexpression with ALKBH7-specific siRNA knockdown in MDA-MB-231 cells treated with 0.4 μg/mL DOX for the indicated times. (H) Dot-blot assay of RNA m6A methylation in MDA-MB-231 cells overexpressing PRMT5 with ALKBH7-specific siRNA transfection treated with 0.4 μg/mL DOX for 24 h i, RT-PCR analysis following m6A-IP of the BRCA1 mRNA 3′-UTR in MDA-MB-231 cells overexpressing PRMT5 with ALKBH7-specific siRNA transfection treated with DOX for 24 h ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To examine the function of ALKBH7 arginine methylation, we measured ALKBH7 protein stability. Remarkably, arginine mutation to either lysine (ALKBH73RK) or alanine (ALKBH73RA) reduced the protein half-life of ALKBH7(Figures 3E and S3C), suggesting that arginine methylation of ALKBH7 is essential for its protein stability. MDA-MB-231 cells containing ALKBH73RK were treated with the proteasome inhibitor MG132, lysosome inhibitor leupeptin, or neddylation inhibitor MLN4924, but only MG132 treatment reversed ALKBH7 expression (Figure S3D), suggesting that the ubiquitin-proteasome pathway might be responsible for non-arginine-methylated ALKBH7 degradation. Using bioinformatic prediction, we identified F-box and WD repeat domain-containing 7 alpha (FBW7α) as the potential E3 ligase responsible for ALKBH7 degradation (Figure S3E). We transfected ALKBH7-WT, ALKBH73RK, and FBW7α into HEK-293T cells, treated the cells with cycloheximide (CHX), and found that FBW7α decreased ALKBH7 and ALKBH73RK stability (Figures S3F and S3G). Furthermore, MYC-tagged ALKBH7 was pulled down from cells transfected with V5-tagged ubiquitin (V5-UB), FBW7α, and ALKBH7-WT or ALKBH73RK, followed by western blotting with an anti-V5 tag antibody; this showed that ALKBH73RK exhibited more ubiquitin binding than ALKBH7-WT (Figure 3F), suggesting that arginine methylation was required to prevent ubiquitin-mediated degradation of ALKBH7. All data suggested that PRMT5 maintained ALKBH7 protein stability through methylating its arginine to avoid FBW7α-mediated ubiquitination.
To further elucidate the role of ALKBH7 in PRMT5-mediated DDR and doxorubicin sensitivity, we blocked ALKBH7 expression in PRMT5-overexpressing breast cancer cells by siRNAs. We found that the decreased doxorubicin sensitivity caused by PRMT5 was abolished by ALKBH7 knockdown (Figure 3G). With a comet assay and the Western blot analysis of γ-H2AX and immunofluorescence of RPA32, we found that knocking down ALKBH7 expression boosted the T-DNA-positive cell numbers and increased the γ-H2AX expression and RPA32 expression induced by doxorubicin (Figures S3H–S3J). These results indicate that ALKBH7 might mediate PRMT5 function in DNA repair. Given that PRMT5 inhibited RNA m6A methylation induced by doxorubicin, we asked whether ALKBH7 is involved in the m6A methylation regulated by PRMT5. We observed that ALKBH7 overexpression significantly decreased m6A methylation (Figures S3K and S3L) as a function of PRMT5. ALKBH7 knockdown reversed total RNA m6A methylation and the m6A methylation on BRCA1 mRNA at adenosines 149 and 1,268 in the BRCA1 3′-UTR induced by PRMT5 (Figures 3H and 3I) and reduced BRCA1 mRNA stability and expression (Figures S3M and S3N). Overall, PRMT5 promoted the arginine methylation of ALKBH7 to maintain ALKBH7 protein stability, which is critical for RNA m6A methylation of BRCA1 and doxorubicin sensitivity.
PRMT5 enhances the ALKBH7-mediated nuclear translocation of ALKBH5 required for RNA demethylation
As mentioned above, PRMT5 reduced the RNA m6A methylation induced by doxorubicin through ALKBH7, and it was therefore reasonable to further assess how ALKBH7 regulates RNA m6A demethylation. We first analyzed whether PRMT5 or ALKBH7 alters the expression of the well-known RNA demethylases ALKBH5 and FTO.21, 22, 23, 24 However, neither PRMT5 nor ALKBH7 affected the expression of ALKBH5 or FTO (Figures S4A and S4B). Surprisingly, we found that PRMT5 and ALKBH7 dramatically promoted the nuclear translocation of ALKBH5 with doxorubicin treatment by using an immunofluorescence assay (Figures 4A and S4C). We further confirmed this observation with a nuclear and cytoplasmic fractionation assay and found that ALKBH5 nuclear translocation was remarkably elevated when ALKBH7 was over-expressed. When ALKBH73RK is over-expressed, the upregulation of nucleic ALKBH5 is slight, which corresponds to the lower expression of ALKBH7(Figures 4B and 4C). ALKBH7 knockdown reduced the nuclear translocation of ALKBH5 induced by PRMT5 (Figure 4D), showing that the large amount of ALKBH7, especially that kept through methylation mediated by PRMT5, was required for the PRMT5-induced nuclear translocation of ALKBH5 induced by doxorubicin treatment. We next asked how ALKBH7 regulates the nuclear translocation of ALKBH5. An IP assay clearly showed the interaction of ALKBH7 with endogenous ALKBH5 in total cell lysates of MDA-MB-231 or T47-D cells (Figure S4D). Although ALKBH5 is believed to localize exclusively in the nucleus for RNA m6A demethylation,22,25 our results collectively provided evidence that ALKBH5 was also located in the cytoplasm, while ALKBH7 bound with ALKBH5 and promoted ALKBH5 translocation from the cytoplasm to the nucleus with doxorubicin treatment.
Figure 4.

PRMT5 promotes the nuclear translocation of ALKBH5 to support RNA demethylation mediated by the partner ALKBH7
(A) Immunofluorescence assay of ALKBH5 in MDA-MB-231 cells overexpressing PRMT5 or ALKBH7 treated with 0.4 μg/mL DOX for 24 h as indicated. (B) Nuclear/cytoplasmic fractionation assay assessing ALKBH5 and FTO in MDA-MB-231 cells overexpressing PRMT5 treated with 0.4 μg/mL DOX for 24 h as indicated. Lamin A/C and α-tubulin served as the nuclear and cytosolic controls, respectively. (C) Nuclear/cytoplasmic fractionation assay assessing ALKBH5 in MDA-MB-231 cells overexpressing ALKBH7 or ALKBH73RK treated with 0.4 μg/mL DOX for 24 h as indicated. (D) Nuclear/cytoplasmic fractionation assay assessing ALKBH5 in MDA-MB-231 cells overexpressing PRMT5 with ALKBH7-specific siRNA knockdown treated with 0.4 μg/mL DOX for 24 h as indicated. (E and F) Dot-blot assay of RNA m6A methylation in MDA-MB-231 cells overexpressing PRMT5 with ALKBH5-specific siRNA transfection treated with 0.4 μg/mL DOX for 24 h. (F) RT-PCR analysis following m6A-IP of the BRCA1 mRNA 3′-UTR in MDA-MB-231 cells overexpressing PRMT5 with ALKBH5-specific siRNA treated with 0.4 μg/mL DOX for 24 h ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To further determine whether ALKBH5 is the main downstream effector of PRMT5 and ALKBH7, which regulates RNA m6A modification, we examined the total m6A methylation level in PRMT5-and ALKBH7-overexpressing breast cancer cells with ALKBH5 expression knocked down by siRNAs. The results showed that ALKBH5 knockdown dramatically reversed the m6A level inhibition by PRMT5 and ALKBH7 (Figures 4E and S4E), especially reversing the inhibition of the enrichment of m6A methylation on the 3′-UTR of BRCA1 mRNA mediated by PRMT5 (Figure 4F), and decreased the stability and expression of BRCA1 mRNA (Figures S4F and S4G). Taken together, our results suggest that PRMT5 inhibits RNA m6A methylation probably through enhancing the ALKBH7-mediated nuclear translocation of the RNA demethylase ALKBH5.
PRMT5 positively correlates with nuclear ALKBH5 and predicts relatively poor survival in breast cancer patients
We further evaluated the clinical significance of the regulation of ALKBH5 and BRCA1 by PRMT5 signaling in breast cancer. Both upregulated PRMT5 expression and upregulated BRCA1 (with approximately 4% mutation) expression were correlated with relatively poor survival in breast cancer patients in The Cancer Genome Atlas (TCGA) program (Figures 5A and 5B). There was a positive correlation between PRMT5 and BRCA1 expression using mRNA analysis in the TCGA and immunohistochemical (IHC) analysis of protein levels in the TCGA dataset and our cohort (Figures 5C and 5D; Table S2). Most importantly, we found that PRMT5 was highly correlated with ALKBH7 and nuclear localization of ALKBH5 by IHC analysis of breast cancer samples (Figures 5E–5G). Thus, PRMT5 might be critical in breast cancer progression and the chemotherapeutic response by regulating the nuclear localization of ALKBH5 and maintaining BRCA1 expression. Therefore, PRMT5 is an ideal therapeutic target in breast cancers to improve the response to doxorubicin in the clinic.
Figure 5.

PRMT5 positively correlates with nuclear ALKBH5 and predicts relatively poor survival in breast cancer patients
(A) The relationship between PRMT5 expression and human breast cancer patient survival in TCGA datasets was analyzed. (B) The relationship between BRCA1 expression and human breast cancer patient survival in TCGA datasets was analyzed. (C) The relationship between PRMT5 and BRCA1 gene expression in breast cancer patients was analyzed, and the correlation coefficient R was calculated by the Pearson method. (D) The relationship between PRMT5 and BRCA1 protein expression in breast cancer patients was analyzed, and the correlation coefficient R was calculated by the Pearson method. (E) The relationship between PRMT5 and ALKBH5 nuclear expression in breast cancer patients was analyzed. (F) The relationship between PRMT5 and ALKBH7 protein expression in breast cancer patients was analyzed. (G) Accumulated nuclear ALKBH5, BRCA1, and ALKBH7 proteins were observed in human breast cancer tissue with high PRMT5 expression by IHC compared with breast cancer tissue with low PRMT5 expression.
Approved drug tadalafil is a novel PRMT5-targeting inhibitor
Currently, there are several reported potent and selective inhibitors targeting PRMT5, such as EPZ015666, JNJ-64619178, and PJ-68.26, 27, 28 However, thus far, only JNJ-64619178 and PF-06939999 have been assessed in phase I clinical trials, indicating that a long time is required for final clinical application. Herein, we successfully re-positioned potential PRMT5 inhibitors from 1,813 approved drugs by use of virtual screening and molecular docking to the crystal structure of the PRMT5 protein (Figure 6A).29 With rigorous selection, tadalafil was selected as a potential PRMT5 inhibitor for further bioanalysis (Figure 6B), which demonstrated the desired binding affinity with the PRMT5/MEP50 complex using surface plasmon resonance (SPR) analysis (Figure 6C). Furthermore, tadalafil inhibited the symmetric methylation of the well-known PRMT5 downstream targets H4R3me2s and SDMA in a dose- and time-dependent manner (Figures S5A and S5B).
Figure 6.

Approved drug tadalafil is identified as a novel PRMT5-targeting inhibitor
(A) Structure-based virtual screening of PRMT5 inhibitors among 1,813 approved drugs. (B) Molecular structures and binding modules of the re-positioned approved drug tadalafil docked in the 3UA4 crystal structure of the PRMT5 protein. (C) SPR analysis to show the binding affinity of tadalafil with the purified PRMT5/MEP50 protein complex. Kd = 2.265 × 10−5 M. (D) In vitro methylation assay with recombinant FLAG-tagged PRMT5 and unlabeled SAM. The substrate was wild-type His-tagged ALKBH7, and the conditions were treatment with or without 100 μM tadalafil. The same blot was stripped and re-probed with an anti-ALKBH7 antibody, showing the amount of substrate in each reaction. H4 was used as a positive control. (E) Tadalafil-mediated inhibition of the arginine methylation of ALKBH7 by PRMT5. MDA-MB-231 cells with or without PRMT5 overexpression were treated with 100 μM tadalafil for 24 h. Then, cell lysates were immunoprecipitated with an anti-MYC antibody and detected with anti-SDMA and anti-ALKBH7 antibodies. (F) Western blot analysis of ALKBH7 expression in MDA-MB-231 cells treated with tadalafil (100 μM) for 24 h and CHX (100 μg/mL) as indicated before harvest.
To obtain further evidence of tadalafil functioning as a specific PRMT5 inhibitor, we performed in vitro methylation assays for PRMT5 with the substrates recombinant His-tagged ALKBH7 and histone H4 in the presence of SAM and tadalafil. Tadalafil clearly inhibited the methylation of ALKBH7 and histone H4 catalyzed by PRMT5 (Figure 6D) in vitro, confirming that tadalafil blocked the arginine methylation activity of PRMT5. We also found that tadalafil significantly inhibited ALKBH7 methylation induced by PRMT5 in breast cancer cells and decreased the protein stability of ALKBH7 (Figures 6E and 6F). Thus, we were the first to identify the approved drug tadalafil as a novel PRMT5-targeting inhibitor, which might have extensive potential to improve the doxorubicin sensitivity of breast cancer.
Tadalafil exhibits a synergetic effect with doxorubicin on breast cancer
To evaluate whether tadalafil increases doxorubicin sensitivity by targeting PRMT5, a cell proliferation assay was performed, and tadalafil significantly increased the inhibitory effect of doxorubicin on MDA-MB-231 breast cancer cells with wild-type BRCA1 but not on HCC1937 cells with mutant BRCA1 (Figures S5C and S5D), indicating that the synergistic effect of tadalafil with doxorubicin is most likely dependent on BRCA1 status. To test whether tadalafil takes effect dependently on PRMT5 inhibition, we blocked PRMT5 or PDE5 (phosphodiesterase type 5, the originally designed target of tadalafil) with siRNA and analyzed the drug response respectively. Interestingly, only PRMT5 knockdown, but not PDE5 inhibition, could abolish the synergistic effect of tadalafil with doxorubicin (Figures S5E–S5G), suggesting that tadalafil functions to improve the doxorubicin effect mainly by targeting PRMT5. Surprisingly, tadalafil also enhanced the sensitivity of MDA-MB-231 cells to the PARP (poly adenosine diphosphate-ribose polymerase) inhibitor olaparib (Figure S5H), which indicates the application potential of olaparib combined with the PRMT5 inhibitor tadalafil in breast cancer patients with wild-type BRCA1.
Moreover, the combination of tadalafil and doxorubicin clearly reduced orthotopic tumor growth in athymic nude mice (Figure 7A), with enhanced DNA damage measured by evaluating γ-H2AX and decreased cancer cell proliferation measured by Ki67 staining (Figures S6A and S6B). After treatment with gradually increasing concentrations of doxorubicin for up to 6 months, MDA-MB-231 cells acquired resistance to doxorubicin.30 Similar to our previous finding, PRMT5 expression was increased in breast cancer cells with doxorubicin resistance (Figure S6C)11, and we further analyzed whether tadalafil re-sensitized the resistant cells to doxorubicin treatment. Tadalafil dramatically increased the sensitivity of resistant cells to doxorubicin in vitro (Figure 7B) and deterred tumor growth when combined with doxorubicin therapy in a MDA-MB-231-ADR xenograft mouse model (Figures 7C and S6D). Importantly, the synergistic effect of tadalafil and doxorubicin on PDX mouse models of breast cancer with wild-type BRCA1 was remarkable (Figures 7D and S6E), which further supports the conclusion that tadalafil can increase doxorubicin sensitivity for breast cancer treatment.
Figure 7.

Tadalafil produces a synergetic effect with DOX to impede breast cancer growth
(A) Xenograft mouse model of breast cancer treated with tadalafil and DOX. MDA-MB-231 cells were orthotopically implanted into the mammary fat pad (subcutaneously) of female athymic nude mice (n = 5–7/group). The mice were treated with tadalafil (2 mg/kg body weight) every day via gavage and DOX (2 mg/kg body weight) once a week via tail vein injection after the tumor volume had reached 100–150 mm3. Tumor volume was measured every 3 days, and the mice were euthanized 30 days after injection. ∗p < 0.05 and ∗∗p < 0.01. (B) Proliferation analysis of MDA-MB-231 cells with DOX resistance (MDA-MB-231-ADR) treated with 0.4 μg/mL DOX and 0, 25, 50, or 100 μM tadalafil for 24 h. (C) Tumor growth curves of MDA-MB-231-ADR cells orthotopically implanted into athymic nude mice that were treated with tadalafil and DOX as described in (A) (n = 5–7/group). (D) Two patient-derived xenograft (PDX) mouse models of breast cancer, UM1 and UM2. The models were treated with tadalafil and DOX as mentioned above (n = 5–7/group). Tumor volume was measured every 3 days, and the mice were euthanized 30 days after injection. (E) Dot-blot assay of RNA m6A methylation in breast cancer tumors harvested from PDX mouse models as indicated in (D) (n = 5–7/group). (F) RT-PCR analysis following m6A-IP of the BRCA1 mRNA 3′-UTR in MDA-MB-231 cells treated with 0.4 μg/mL DOX with or without 100 μM tadalafil for 24 h.
Finally, we asked whether tadalafil promotes doxorubicin sensitivity by regulating ALKBH5 localization and RNA m6A methylation. Strikingly, tadalafil inhibited the nuclear localization of ALKBH5 induced by doxorubicin (Figures S7A and S7B). Tadalafil treatment increased RNA m6A methylation in both breast cancer cells and tumor cells isolated from PDX-model mice (Figures 7E and S7C) and further increased the enrichment of m6A methylation on the BRCA1 3′-UTR when combined with doxorubicin treatment (Figure 7F). Tadalafil combined with doxorubicin further decreased BRCA1 mRNA stability and expression (Figures S7D and S7E) and increased the number of T-DNA-positive cells and expression of cleaved caspase-3, suggesting that tadalafil strengthens doxorubicin-mediated DNA damage and tumor cell apoptosis (Figures S7F and S7G). Thus, we identified the approved drug tadalafil as a novel PRMT5 inhibitor that increases doxorubicin sensitivity in breast cancer by regulating ALKBH5 nuclear localization and RNA m6A methylation.
Discussion
The stress response regulation of cancer cells to various drugs is critical for the life-and-death decisions that determine the ultimate tumor efficacy. Doxorubicin is one of the most important chemotherapeutic drugs for breast cancer, mainly by inducing DNA damage.31 However, the complete molecular mechanism of the doxorubicin-mediated DNA damage response remains unclear.32 Herein, we first reported that doxorubicin induced RNA m6A methylation in vitro and in vivo, suggesting that breast cancer cells regulated doxorubicin-mediated DNA damage through RNA m6A methylation. However, further study is needed to understand the detailed mechanism.
A number of studies have confirmed the important functions of RNA m6A methylation and its modulators in cancer proliferation and invasion.1,16,33 It was found that the RNA demethylase ALKBH5 was highly expressed in breast cancer tissues and that the inhibition of ALKBH5 expression significantly inhibited the invasion and metastasis of breast cancer cells.34,35 To date, ALKBH5 is believed to localize predominantly in the nucleus for RNA m6A demethylation.22,25 However, the mechanism that controls the cellular localization of ALKBH5 is unknown. Here, we have provided the first evidence that ALKBH5 translocates from the cytoplasm to the nucleus through an interaction with ALKBH7 in the context of doxorubicin treatment, suggesting that RNA m6A methylation regulated by the dynamic localization of ALKBH5 may be crucial for the doxorubicin-induced DNA damage response.
Although PRMT5 is believed to be important for preserving genome integrity,9,36 the molecular machinery by which PRMT5 regulates the DNA damage response is not completely understood. Notably, we identified ALKBH7 as a novel PRMT5 substrate with arginine methylation, which is critical for preserving ALKBH7 protein stability. It is believed that ALKBH7 does not have RNA demethylase activity and is localized predominantly the mitochondria.17, 18, 19 Herein, we have provided evidence that ALKBH7 is located in both the cytoplasm and the nucleus and supports ALKBH5 nuclear translocation for RNA m6A demethylation, suggesting the complicated function of ALKBH7 with changes in subcellular localization under different stress conditions.
Although PRMT5 is a well-recognized druggable cancer target,26,37,38 no PRMT5 inhibitors have been approved for clinical use. Tadalafil was primarily approved for the treatment of erectile dysfunction (ED) and pulmonary arterial hypertension, functioning as a PDE5 inhibitor.39 Our work is the first to identify tadalafil as a novel PRMT5 inhibitor by confirming its synergistic efficacy with doxorubicin in inhibiting breast cancer cells and even its compentency of reversing doxorubicin resistance, providing a feasible strategy to promote doxorubicin sensitivity for the treatment of breast cancer.
In summary, our present study has identified a novel mechanism by which PRMT5 inhibits the doxorubicin-induced RNA m6A methylation of BRCA1 by regulating the nuclear translocation of ALKBH5 and has provided a promising approach to elevate doxorubicin sensitivity in breast cancer through the PRMT5 inhibitor tadalafil.
Materials and methods
Cell lines and culture
The MDA-MB-231 cell line was obtained from the American Type Culture Collection (HTB-26) and grown in L-15 medium (HyClone, SH30525.01) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (Gibco, 15140). Cells were free of mycoplasma (IDEXX STAT-Myco). The T47-D and MCF-7 cell lines were obtained from ATCC (HTB-133 and HTB-22) and grown in DMEM supplemented with 10% FBS, 0.005 mg/mL insulin (Sigma, I0516), and 1% penicillin-streptomycin. The HCC1937 cell line was obtained from ATCC (CRL-2336) and grown in RPMI 1640 medium supplemented with 10% FBS.
Animals
Female BALB/c nude mice (5–6 weeks old) were purchased from the Animal Center of Fourth Military Medical University (Xi’an, China) and raised under pathogen-free conditions. All animal care and experiments were approved by the Institutional Animal Care and Use Committee of Fourth Military Medical University. For the subcutaneously transplanted tumor model, wild-type, PRMT5-overexpressing or doxorubicin-resistant MDA-MB-231 cells (5 × 106 per mouse, n = 5–7 for each group) were diluted in 100 μL of phosphate-buffered saline (PBS) plus 100 μL of Matrigel (BD Biosciences) and subcutaneously injected into female nude mice to investigate tumor growth. When all tumor volumes reached 100 mm3, the mice were randomly assigned and treated with the indicated drugs. In the experiment, doxorubicin was administered once a week via intravenous tail vein injection at 2 mg/kg body weight, and tadalafil was administered daily via oral gavage at 2 mg/kg body weight. Tumor volume was measured every 3 days using a digital caliper and calculated using the formula V = 1/2 × (diameter) × (smaller diameter)2. The mice were euthanized 27 days after injection. Tumor growth curves and harvested tumors are displayed as indicated.
Human subjects
All tumor collection and analysis protocols were approved by the medical ethics committee of Fourth Military Medical University, with informed consent provided by the participants.
Cell proliferation assay
For cell proliferation assays, cells were seeded in 96-well plates at a concentration of 8,000–10,000 cells per well in quintuplicate. Cell proliferation was assessed with MTT (0.5 mg/mL, Sigma) following the manufacturer’s instructions. Briefly, the cells were seeded in 96-well plates, and MTT was added at the indicated time points. After incubation at 37°C for 4 h, the reaction was stopped by adding DMSO (150 μL, Sigma). The absorbance at 490 nm was measured using a microplate reader (Bio-Rad).
m6A quantification
The total m6A content was measured in 200 ng of mRNA extracted from cells by use of a m6A RNA methylation quantification kit (cat. no. P-9005, Epigentek) according to the manufacturer’s instructions.
Ligands and protein crystal structures
Structures of the 1,813 approved drugs were collected by PubChem Compound ID. The 3D structures were generated using the chemistry toolbox Open Babel, version 2.4.0. The PRMT5 structures were downloaded from the Protein Data Bank (PDB)12 with PDB IDs 3UA3 and 3UA4.11
Virtual screening
Virtual screening was performed with AutoDock Vina. The default docking parameters were used. The area around Glu499 and Glu508 was taken as the target area for docking. The center point was set to (72.75–1.43, 13.06), and the docking space was set to a cube with a side length of 30.
In vitro protein methylation assays
A recombinant PRMT5/MEP50 complex (31521, Active Motif) was incubated with 1.5 μg of recombinant His-ALKBH7-WT (81132, Active Motif) or His-ALKBH7-3RK proteins (prokaryotic expression and purification in our laboratory). Methylation reactions were carried out using methylation buffer (50 mM Tris-HCl, [pH 8.5], 5 mM MgCl2, and 4 mM DTT) containing 100 μM unlabeled S-(5'-adenosyl)-L-methionine chloride dihydrochloride (SAM; A7007, Sigma) for 2 h at 30°C. The histone H4 protein (31493, Active Motif) was introduced as a positive control. Reactions were stopped by adding 5× SDS loading buffer (Beyotime Biotechnology) and boiling the samples for 5 min. The methylation reaction products were separated by SDS-PAGE, transferred to PVDF membrane, and analyzed by western blotting using anti-SDMA, anti-PRMT5, and anti-ALKBH7 antibodies separately.
Surface plasmon resonance (SPR)
Interactions between the recombinant PRMT5/MEP50 and tadalafil were analyzed using the Biacore T200 system at 25°C, as described before.40 Briefly, recombinant PRMT5/MEP50 was immobilized on a sensor chip (CM5), using an amine coupling kit (GE Healthcare). Final PRMT5/MEP50-immobilized levels were typically 1 mg/mL. Subsequently, tadalafil was injected as analytes at various concentrations and using PBS-P (10 mM phosphate buffer with 2.7 mM KCl and 137 mM NaCl, 0.05% Surfactant P20, pH 4.5) as running buffer. For binding studies, analytes were applied at corresponding concentrations in running buffer at a flow rate of 30 μL/min with a contact time of 60 s and a dissociation time of 60 s. Chip platforms were washed with running buffer and 50% DMSO. Data were analyzed with the Biacore evaluation software (T200 version 1.0) by curve fitting using a 1:1 binding model.
Quantification and statistical analysis
Data are presented as the mean ± SEM or SD. Differences in the mean between two groups were assessed for significance with a two-tailed Student’s t test by using GraphPad Prism 6.0. Kaplan-Meier survival curves were analyzed using the log rank test. The Pearson correlation test was used to assess relationships between variables in tumor tissue samples.
Data availability
The sequencing data obtained in this study were uploaded to the Gene Expression Omnibus (GEO) database under the accession number GSE155657.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 82073202, 81972710, 81902677), the State Key Laboratory of the Cancer Biology Project (CBSKL2017Z08 and CBSKL2017Z11), the Natural Science Basic Research Program of Shaanxi (2020JM-312), and the Fund for Distinguished Young Scholars of ShaanXi province (2019JC-22).
Author contributions
Y.W., Z.W., B.Y., M.W., Z.G., and N.H. performed the experiments. L.H. and W.Z. screened the PRMT5 inhibitors in the approved drug library. H.Z., J.Y., Z.W., T.W., and J.Y. collected and analyzed the breast cancer patient samples. S.L. and C.S. provided the PDX mouse model. C.C. and L.Y. supervised the research and discussed the data. R.L. and J.Z. conceived, designed, and supervised the research and analyzed the data. W.Y. and Z.J. wrote the manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.03.003.
Contributor Information
Wenxia Zhou, Email: zhouwx@bmi.ac.cn.
Rui Ling, Email: lingrui0105@163.com.
Jian Zhang, Email: biozhangj@fmmu.edu.cn.
Supplemental information
Document S2. Figures S1–S7 and Tables S1–S2
References
- 1.Lan Q., Liu P.Y., Haase J., Bell J.L., Huttelmaier S., Liu T. The critical role of RNA m(6)A methylation in cancer. Cancer Res. 2019;79:1285–1292. doi: 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- 2.Chen X.Y., Zhang J., Zhu J.S. The role of m(6)A RNA methylation in human cancer. Mol. Cancer. 2019;18:103. doi: 10.1186/s12943-019-1033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo A., Gong Y., Kim H., Chen Y. Proteome dynamics analysis identifies functional roles of SDE2 and hypoxia in DNA damage response in prostate cancer cells. NAR Cancer. 2020;2:zcaa010. doi: 10.1093/narcan/zcaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckley A.M., Lynam-Lennon N., O'Neill H., O'Sullivan J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat. Rev. Gastroenterol. Hepatol. 2020;17:298–313. doi: 10.1038/s41575-019-0247-2. [DOI] [PubMed] [Google Scholar]
- 5.Xiang Y., Laurent B., Hsu C.H., Nachtergaele S., Lu Z., Sheng W., Xu C., Chen H., Ouyang J., Wang S., et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–576. doi: 10.1038/nature21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S., Fan X., Zhu J., Xu D., Li R., Chen R., Hu J., Shen Y., Hao J., Wang K., et al. The differentiation of colorectal cancer is closely relevant to m6A modification. Biochem. Biophys. Res. Commun. 2021;546:65–73. doi: 10.1016/j.bbrc.2021.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Chiang K., Zielinska A.E., Shaaban A.M., Sanchez-Bailon M.P., Jarrold J., Clarke T.L., Zhang J., Francis A., Jones L.J., Smith S., et al. PRMT5 is a critical regulator of breast cancer stem cell function via Histone methylation and FOXP1 expression. Cell Rep. 2017;21:3498–3513. doi: 10.1016/j.celrep.2017.11.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamiya H., Kim H., Klymenko O., Kim H., Feng Y., Zhang T., Han J.Y., Murao A., Snipas S.J., Jilaveanu L., et al. SHARPIN-mediated regulation of protein arginine methyltransferase 5 controls melanoma growth. J. Clin. Invest. 2018;128:517–530. doi: 10.1172/JCI95410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu D., Gur M., Zhou Z., Gamper A., Hung M.C., Fujita N., Lan L., Bahar I., Wan Y. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 2015;6:8419. doi: 10.1038/ncomms9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamard P.J., Santiago G.E., Liu F., Karl D.L., Martinez C., Man N., Mookhtiar A.K., Duffort S., Greenblatt S., Verdun R.E., Nimer S.D. PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell Rep. 2018;24:2643–2657. doi: 10.1016/j.celrep.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z., Kong J., Wu Y., Zhang J., Wang T., Li N., Fan J., Wang H., Zhang J., Ling R. PRMT5 determines the sensitivity to chemotherapeutics by governing stemness in breast cancer. Breast Cancer Res. Treat. 2018;168:531–542. doi: 10.1007/s10549-017-4597-6. [DOI] [PubMed] [Google Scholar]
- 12.Martin M., Romero A., Cheang M.C., Lopez Garcia-Asenjo J.A., Garcia-Saenz J.A., Oliva B., Roman J.M., He X., Casado A., de la Torre J., et al. Genomic predictors of response to doxorubicin versus docetaxel in primary breast cancer. Breast Cancer Res. Treat. 2011;128:127–136. doi: 10.1007/s10549-011-1461-y. [DOI] [PubMed] [Google Scholar]
- 13.Wang H., Bierie B., Li A.G., Pathania S., Toomire K., Dimitrov S.D., Liu B., Gelman R., Giobbie-Hurder A., Feunteun J., et al. BRCA1/FANCD2/BRG1-Driven DNA repair stabilizes the differentiation state of human mammary epithelial cells. Mol. Cell. 2016;63:277–292. doi: 10.1016/j.molcel.2016.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaccara S., Jaffrey S.R. A unified model for the function of YTHDF proteins in regulating m(6)a-modified mRNA. Cell. 2020;181:1582–1595.e18. doi: 10.1016/j.cell.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang C., Samanta D., Lu H., Bullen J.W., Zhang H., Chen I., He X., Semenza G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. U S A. 2016;113:E2047–E2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J., Eckert M.A., Harada B.T., Liu S.M., Lu Z., Yu K., Tienda S.M., Chryplewicz A., Zhu A.C., Yang Y., et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018;20:1074–1083. doi: 10.1038/s41556-018-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu D., Jordan J.J., Samson L.D. Human ALKBH7 is required for alkylation and oxidation-induced programmed necrosis. Genes Dev. 2013;27:1089–1100. doi: 10.1101/gad.215533.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordan J.J., Chhim S., Margulies C.M., Allocca M., Bronson R.T., Klungland A., Samson L.D., Fu D. ALKBH7 drives a tissue and sex-specific necrotic cell death response following alkylation-induced damage. Cell Death Dis. 2017;8:e2947. doi: 10.1038/cddis.2017.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker A.R., Silvestrov P., Muller T.A., Podolsky R.H., Dyson G., Hausinger R.P., Cisneros G.A. ALKBH7 variant related to prostate cancer exhibits altered substrate binding. Plos Comput. Biol. 2017;13:e1005345. doi: 10.1371/journal.pcbi.1005345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bikkavilli R.K., Avasarala S., Van Scoyk M., Karuppusamy Rathinam M.K., Tauler J., Borowicz S., Winn R.A. In vitro methylation assay to study protein arginine methylation. J. Vis. Exp. 2014:e51997. doi: 10.3791/51997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su R., Dong L., Li C., Nachtergaele S., Wunderlich M., Qing Y., Deng X., Wang Y., Weng X., Hu C., et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90–105.e23. doi: 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J., Vagbo C.B., Shi Y., Wang W.L., Song S.H., et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao X., Yang Y., Sun B.F., Shi Y., Yang X., Xiao W., Hao Y.J., Ping X.L., Chen Y.S., Wang W.J., et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z., Chen Y., Sulman E.P., Xie K., Bogler O., et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thalhammer A., Bencokova Z., Poole R., Loenarz C., Adam J., O'Flaherty L., Schodel J., Mole D., Giaslakiotis K., Schofield C.J., et al. Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1alpha (HIF-1alpha) PLoS One. 2011;6:e16210. doi: 10.1371/journal.pone.0016210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu F., Cheng G., Hamard P.J., Greenblatt S., Wang L., Man N., Perna F., Xu H., Tadi M., Luciani L., Nimer S.D. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Invest. 2015;125:3532–3544. doi: 10.1172/JCI81749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan-Penebre E., Kuplast K.G., Majer C.R., Boriack-Sjodin P.A., Wigle T.J., Johnston L.D., Rioux N., Munchhof M.J., Jin L., Jacques S.L., et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015;11:432–437. doi: 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- 28.Li X., Wang C., Jiang H., Luo C. A patent review of arginine methyltransferase inhibitors (2010-2018) Expert Opin. Ther. Pat. 2019;29:97–114. doi: 10.1080/13543776.2019.1567711. [DOI] [PubMed] [Google Scholar]
- 29.Tao H., Yan X., Zhu K., Zhang H. Discovery of novel PRMT5 inhibitors by virtual screening and biological evaluations. Chem. Pharm. Bull (Tokyo) 2019;67:382–388. doi: 10.1248/cpb.c18-00980. [DOI] [PubMed] [Google Scholar]
- 30.Yang J.Y., Ha S.A., Yang Y.S., Kim J.W. p-Glycoprotein ABCB5 and YB-1 expression plays a role in increased heterogeneity of breast cancer cells: correlations with cell fusion and doxorubicin resistance. BMC Cancer. 2010;10:388. doi: 10.1186/1471-2407-10-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andres J.L., Fan S., Turkel G.J., Wang J.A., Twu N.F., Yuan R.Q., Lamszus K., Goldberg I.D., Rosen E.M. Regulation of BRCA1 and BRCA2 expression in human breast cancer cells by DNA-damaging agents. Oncogene. 1998;16:2229–2241. doi: 10.1038/sj.onc.1201752. [DOI] [PubMed] [Google Scholar]
- 32.Yang F., Teves S.S., Kemp C.J., Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta. 2014;1845:84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barbieri I., Tzelepis K., Pandolfini L., Shi J., Millan-Zambrano G., Robson S.C., Aspris D., Migliori V., Bannister A.J., Han N., et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552:126–131. doi: 10.1038/nature24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu L., Wu D., Ning J., Liu W., Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19:326. doi: 10.1186/s12885-019-5538-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C., Zhi W.I., Lu H., Samanta D., Chen I., Gabrielson E., Semenza G.L. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527–64542. doi: 10.18632/oncotarget.11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He W., Ma X., Yang X., Zhao Y., Qiu J., Hang H. A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Res. 2011;39:4719–4727. doi: 10.1093/nar/gkq1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu X., Fernando T., Lossos C., Yusufova N., Liu F., Fontan L., Durant M., Geng H., Melnick J., Luo Y., et al. PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood. 2018 doi: 10.1182/blood-2018-02-831438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y., Chitnis N., Nakagawa H., Kita Y., Natsugoe S., Yang Y., Li Z., Wasik M., Klein-Szanto A.J., Rustgi A.K., Diehl J.A. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 2015;5:288–303. doi: 10.1158/2159-8290.CD-14-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aversa A. Systemic and metabolic effects of PDE5-inhibitor drugs. World J. Diabetes. 2010;1:3–7. doi: 10.4239/wjd.v1.i1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao S., Si L., Tian Z., Jiao P., Fan Z., Meng K., Zhou X., Wang H., Xu R., Han X., et al. Pentacyclic triterpenes grafted on CD cores to interfere with influenza virus entry: a dramatic multivalent effect. Biomaterials. 2016;78:74–85. doi: 10.1016/j.biomaterials.2015.11.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S2. Figures S1–S7 and Tables S1–S2
Data Availability Statement
The sequencing data obtained in this study were uploaded to the Gene Expression Omnibus (GEO) database under the accession number GSE155657.