

Abstract
The clinical outcome and disease severity in coronavirus disease 2019 (COVID-19) are heterogeneous, and the progression or fatality of the disease cannot be explained by a single factor like age or comorbidities. In this study, we used system-wide network-based system biology analysis using whole blood RNA sequencing, immunophenotyping by flow cytometry, plasma metabolomics, and single-cell-type metabolomics of monocytes to identify the potential determinants of COVID-19 severity at personalized and group levels. Digital cell quantification and immunophenotyping of the mononuclear phagocytes indicated a substantial role in coordinating the immune cells that mediate COVID-19 severity. Stratum-specific and personalized genome-scale metabolic modeling indicated monocarboxylate transporter family genes (e.g., SLC16A6), nucleoside transporter genes (e.g., SLC29A1), and metabolites such as α-ketoglutarate, succinate, malate, and butyrate could play a crucial role in COVID-19 severity. Metabolic perturbations targeting the central metabolic pathway (TCA cycle) can be an alternate treatment strategy in severe COVID-19.
Keywords: COVID-19, similarity network fusion, personalized genome-scale metabolic model
Graphical abstract
Ambikan et al. used blood cell transcriptomics, immunophenotyping, plasma metabolomics, and single-cell-type metabolomics of monocytes to identify the system-level metabolic rewiring in COVID-19 patients. Integrative omics improved the clinical definition of the risk group of COVID-19 severity. The personalized and group-specific metabolic models indicated the essential role of transporters and metabolites of central metabolism (TCA cycle) in COVID-19 severity. This can lead to an alternate treatment strategy through metabolic perturbations of central metabolism in severe COVID-19.
Introduction
Coronavirus disease 19 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), presents mild to moderate symptoms in most of the infected patients. Several studies, including ours, have indicated altered systems-level metabolic disruption in SARS-CoV-2-infected patients compared with the healthy individuals, where dysregulated amino acid, central carbon, and energy metabolisms were observed as hallmarks of the disease (Krishnan et al., 2021; Shen et al., 2020; Wu et al., 2020). Although many patients with severe COVID-19 have underlying comorbidities like obesity, diabetes, and cardiovascular disease related to metabolic syndrome (MetS), there is a high disparity in COVID-19 severity and mortality. The interplay between metabolic and signaling molecular effectors driving disease severity has not been systematically examined. Further, in addition to comorbidities, the high physiological heterogeneity has not been considered when addressing the mechanisms of population-level predisposition to the severe phenotype.
Viruses are obligatory parasites and entirely rely on their hosts for replication. This reliance is evidenced by experimental findings showing significant metabolic flux disruption in the host cells. This is further highlighted by observed variations in the cell-specific viral replication and production, leading to changes in the host metabolism (Yu et al., 2011). Virus infection leads to significant metabolic alterations in the host, such as altered glycolysis rates and changes in ATP production rates (Aller et al., 2018). Therefore, the changes in energy metabolism can be seen as an evolving property of the combined host-virus metabolic system. This could be related to changes in host cellular demands arising from viral production, increased demand from immune cell activation, and damage due to toxicities (e.g., oxidative damage) (Molenaar et al., 2009). Even though the primary site of infection is the upper respiratory tract, SARS-CoV-2 can invade several organs, tissues, and cells of the body (Delorey et al., 2021). Therefore, a systems-level characterization of metabolic and signaling dysregulation can comprehensively and quantitatively capture the implications of COVID-19 infection throughout the body.
Systems biology approaches have shown promise in systematically elucidating complex disease-underlying mechanisms by integrating different layers of omics information. The application of the methods, including machine learning, biological network analyses, and multi-omics data integration, has allowed us to generate multi-omics integrated networks to characterize the host response to SARS-CoV-2 (Appelberg et al., 2020). Interrogating these integrated networks permits identifying and understanding the sequence of specific biological elements coordinately altered in pathological conditions. Further, comprehensive metabolic characterizations allow us to simulate system-level metabolic fluxes through genome-wide metabolic modeling in human cells and tissues (Robinson et al., 2020). Combining biological network analysis and genome-scale metabolic modeling (GSMM) as multi-omics integrative approaches can attain holistic and temporally dynamic characterizations of complex rearrangement in response to viral infection and can computationally predict potential therapeutic targets. Importantly, these characterizations permit a group-specific and personalized characterization of global metabolism in healthy and progressive disease states.
In the present study, we employ data-driven multi-omics, whole blood transcriptomics, and plasma metabolomics (Krishnan et al., 2021) characterization and clustering of patient data through the construction of integrated networks. Further, the multidimensional stratification of patients through similarity network fusion (SNF) enables addressing population heterogeneity and redefining the clinical classification of COVID-19 severity. We also develop a GSMM encompassing both host and viral metabolic demands that permit metabolic characterization at a personalized level and attain a data-driven patient stratification. Subsequently, a combination of metabolic flux prediction, topological network analysis, reporter metabolite analysis, and in silico target prediction of the personalized networks permits identifying commonalities and patient-specific targets and hubs. Finally, we measure the intracellular metabolites that are predicted to be the key regulators during SARS-CoV-2 infection in the specific cell type identified by digital cell quantification (DCQ), immunophenotyping, and single-cell RNA sequencing (scRNA-seq) data (obtained from Zhang et al., 2020) from COVID-19 patients. We combine integrated network analysis and personalized GSMM for COVID-19 patients at the metabo-transcriptomic levels that could pave the way for personalized treatment strategies dependent on the patient’s metabolic state.
Results
Study design and patient cohorts
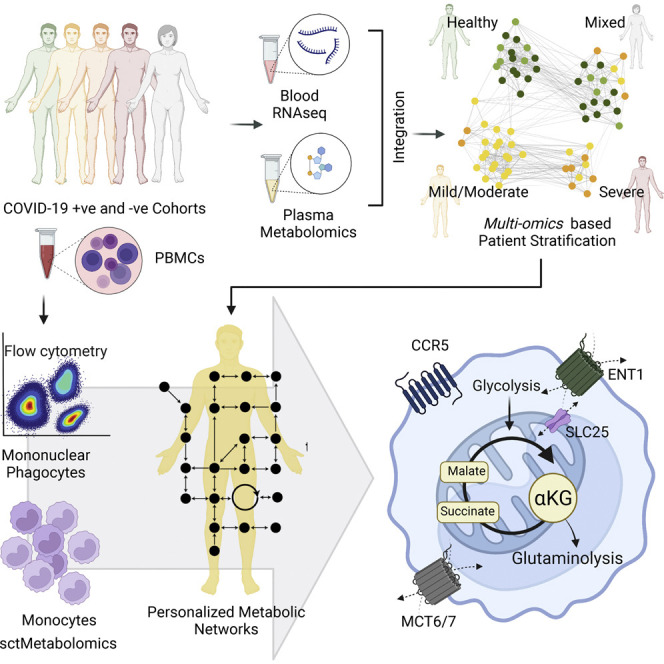
Based on the oxygen (O2) requirement and the clinician’s decision, we clinically categorized COVID-19 patients into hospitalized-mild (n = 26, O2 requirement < 4 L/min) and hospitalized-severe (n = 11, O2 requirement > 4 L/min). The patient characteristics were presented elsewhere (Krishnan et al., 2021). Additional COVID-19 PCR-negative samples were also collected (healthy control [HC], n = 31) and tested for SARS-CoV-2 antibody levels. The serology identified 10 participants as SARS-CoV-2 antibody-positive whose samples were termed as convalescent. RNA sequencing (RNA-seq) was performed on the whole blood samples obtained from SARS-CoV-2-infected and -noninfected patients. We used DCQ to quantify the different cell types, followed by differential analysis between clinically identified groups after adjusting for the high abundant cell types. Immunophenotyping using flow cytometry further validated the changes in mononuclear phagocytes (MNPs). To improve the classification of clinical COVID-19 severity, transcriptomics and metabolomics data were integrated using SNF, leading to the re-classification of the patient groups into healthy, mild/moderate, and severe. SNF-classified patient groups led to context-specific GSMM both at personalized and group levels. The flux balance analysis (FBA) was performed to study the metabolic rearrangement in deeper resolution and identify the essential genes and metabolites associated with SNF-defined COVID-19 severity in groups. Previously published scRNA-seq data (Zhang et al., 2020) was re-analyzed to examine the expression of the essential genes that are associated with disease severity in specific cell types. Finally, single-cell-type metabolomics analysis (sctMetabolomics), targeting TCA-cycle intermediates, confirmed the metabolic rearrangement in the monocytes. The complete study design is presented in Figure 1 .
Figure 1.

The study design and project description
The key methods are marked with bold texts. The dotted arrow indicates the leading experiments. The figure is created with biorender.com.
Distinct changes in immune cell types in hospitalized-severe patients
The transcriptomics data was generated from whole blood samples; therefore, the gene expression variation may be due to altered gene regulation in cell types whose abundances differed between patient groups. Thus, we have performed DCQ that computationally estimates the cell type proportions from bulk blood RNA-seq data. DCQ uses a cell-specific expression profile of 18 blood cell types obtained from the Human Protein Atlas (Uhlen et al., 2017) and a deconvolution algorithm adapted from the method estimation of proportions of immune and cancer cells (EPIC) (Racle and Gfeller, 2020). Proportions of 18 blood cell populations in all the samples were computed and analyzed for groupwise differences. Kruskal-Wallis’s test identified four cell types, namely classical monocytes (CM), neutrophils, mucosa-associated invariant T (MAIT) cells, and γδ-T cells, which differed among the patient groups (adj. p < 0.05; Figure 2 A). The pairwise analysis identified that in both hospitalized-mild and -severe groups, neutrophils and CM displayed significantly increased abundance in COVID-19 patients compared with HC, whereas γδ-T cells, memory B cells, and MAIT cells had lower abundance in severe COVID-19 patients compared with HC (adj. p < 0.05; Figure 2B). The pairwise co-expression landscape of marker genes of specific cell types showed a preserved correlation between the signature genes in the immune cell types in HC (Figure 2C) but a dysregulated correlation in convalescent (Figure 2D), mild (Figure 2E), and severe (Figure 2F) patients. Specifically, the associations between marker genes of T cells and dendritic cells (DC) and the associations between granulocytes (neutrophils/eosinophils) and DC or T cells were altered, indicating an impaired interaction between these cell types during COVID-19 infection at a system level (Figure S1A) that may lead to an inability of the innate immune cells to elicit an effective adaptive immune response in severe COVID-19 patients. The GSEA performed on the lost marker genes associations indicated significant changes in PI3K-Akt signaling, chemokine signaling pathway, NF-kB, and MAPK signaling pathways (Figure S1B).
Figure 2.

Digital cell quantification (DCQ) identified severity-specific signature in COVID-19
(A) Bubble plot describing DCQ results of all samples in each patient cohorts (HC [n = 21], convalescent [n = 10], hospitalized-mild [n = 26], and hospitalized-severe [n = 11]). Bubble size and color gradient are relative to median cell proportions calculated in each cohort for various cell types. Kruskal-Wallis test results between the cohorts are labeled by asterisks (adj. p < 0.05).
(B) Boxplot of cell proportion estimated in all cohort samples represented in (A). Asterisks represent a significant change between cohorts computed by the Mann-Whitney U test (adj. p < 0.05).
(C–F) Network visualization of co-expression among marker genes of various cell types in samples of healthy control (HC) cohort (C), convalescent cohort (D), hospitalized-mild cohort (E), and hospitalized-severe cohort (F). Each node in the network represents marker genes of the corresponding cell type, and node size is relative to the mean expression value (TPM). Edge denotes a significant Spearman correlation (adj. p < 0.001) between marker genes in HC, convalescent, and hospitalized-mild cohorts while adjusted p < 0.2 in hospitalized-severe.
Immunophenotyping of blood MNPs associated with COVID-19 severity
The loss in the co-expression of the marker genes between the T cells and myeloid cells observed in the transcriptomics data prompted us to characterize these populations in peripheral blood mononuclear cells (PBMCs) of COVID-19 mild and severe patients. We performed immunophenotyping of MNPs consisting of monocytes and DCs and innate regulatory cells such as myeloid-derived suppressor cells (MDSCs) and low-density granulocytes (LDG). We used CD4+ and CD8+ T cells from our earlier study (Krishnan et al., 2021). The gating strategy and markers used to define the different cell populations of MNPs are provided (Figure S2; Table S1), and representative contour plots for HC, mild, and severe patients are shown in Figure 3 A. Integrating all analyzed markers and projection of the gated populations into the UMAP space revealed differences in several cell populations between HC and COVID-19 patients (Figures 3B and S3A), indicating significant alterations in the phenotype of the MNP compartment during SARS-CoV-2 infection. Differences in relative frequencies of all populations in all individuals in each group are illustrated in Figures 3C and S3B. A decrease in G-MDSC and an increase in M-MDSC (albeit very low levels) and LDG frequencies were observed in COVID-19 patients, with differences being more pronounced in patients with severe disease (Figure 3C). In the DC and monocyte populations, significant decreases in relative frequencies between HC and COVID-19 patients were noted for plasmacytoid DC (pDC), CD16+ non-classical monocytes, which also includes monocyte-derived DCs (NCM/MD-DC), and total CD16− classical DC (cDC). On the contrary, the frequencies of the CD303− DC5 population (Villani et al., 2017) and of the CM were increased in COVID-19 patients compared with HC. We also observed a significant decrease in CD16+CD141lowCD1c− DC4 population in mild COVID-19 patients compared with HC (Figure 3C). Although the total CD16− cDCs were decreased, no difference in the relative frequency of CD141+ DC1 or CD1c+ DC2/DC3 populations (Figure S3B) was observed. In line with the DCQ, the association between the T cells (CD4+ and CD8+) and some of the MNPs (e.g., pDC) was altered (Figure 3D). We then assessed the expression of the chemokine (C-C motif) receptor type 2 (CCR2), CX3 chemokine receptor 1 (CX3CR1), and C-C chemokine receptor type 5 (CCR5) in the MNPs, using flow cytometry (Figures 3E–3G). Our analysis showed an increase in the frequency of CCR5+ CM, IM, NCM/MD-DC, DC4/NCM cells, and DC1 cells, especially in the mild COVID-19 patients (Figure 3G). The frequency of CCR5+ G-MDSCs was significantly decreased in COVID-19 patients in a severity-dependent way. Similarly, CCR2+ IM, NCM/MD-DC, DC4, and DC5 were increased in frequency, and CCR2+ DC1 cells were decreased in COVID-19 patients, compared with HC (Figure 3G). We also observed a significant decrease in the frequency of CX3CR1+ pDC (both in mild and severe COVID-19), and in CM (in severe COVID-19 patients) and NCM/MD-DC (in mild COVID-19 patients), compared with HC (Figures 3G and S3C). The median fluorescence intensity (MFI) of the chemokine receptors was also altered in COVID-19 patients, with altered CCR5 expression more predominant in mild patients and altered CCR2 expression in all COVID-19 patients, compared with HC (Figures 3E and S3D). These data highlight essential changes in the MNP landscape in the blood of COVID-19 patients, including a phenotype consistent with chemokine-mediated migration of MNPs (particularly of DCs) in the infected lung and additionally defective interaction of MNP with lymphocytes, which probably prevent efficient viral clearance.
Figure 3.

Immunophenotyping of blood MNPs associated with COVID-19 severity
(A) Contour plots of the different MNP populations in HC (n = 9), mild (n = 16), and severe (n = 8) COVID-19 patients.
(B) UMAPs on concatenated files of MNPs in HC, mild, and severe COVID-19 patients.
(C) Boxplots showing the relative frequencies of MNP populations in HC, mild, and severe COVID-19 patients. p values determined by Mann-Whitney U test, ∗ p < 0.05, ∗∗ p < 0.001.
(D) Spearman correlation analysis between the myeloid cells and CD4+/CD8+ T cells (∗ p < 0.05).
(E) Bubble plots of the different chemokine receptor expressions in MNP. The bubble size denotes cells expressing the receptor, whereas the color denotes the median fluorescence intensity (MFI).
(F) MFI of chemokine receptors CCR5, CCR2, and CX3CR1 in the UMAP described in (B).
(G) Boxplots showing relative frequencies of MNPs expressing the chemokine receptors. p values were determined by the Mann-Whitney U test, ∗ p < 0.05, ∗∗ p < 0.001.
System-level whole blood transcriptomics signature differentiates COVID-19 patients
To identify the system-level host response following the SARS-CoV-2 infection, we performed a transcriptomic profile of the total RNA isolated from whole blood by RNA-seq. A UMAP projection of the data showed a clear separation of the COVID-19 patients from HC and convalescent controls but failed to efficiently distinguish between hospitalized-mild and -severe patients (Figure 4 A). Differential gene expression (DGE) analysis (adj. p < 0.05 and log2-fold change ≥1.5) identified 581 genes that were differentially regulated between the HC and COVID-19 patients (Figure 4B; Data S1) and 154 genes differentially regulated between hospitalized-mild and -severe patients (Figure 4C; Data S1). While comparing the HC with the hospitalized-mild and -severe groups, 445 and 1,068 genes were significantly different between the two groups, respectively (Data S1). The hierarchical clustering analysis (HCA) based on the significantly regulated genes also identified distinct patient clustering but with a heterogeneity among the COVID-19 patients (Figure 4D). The gene set analysis between the HC and the COVID19 patients identified distinct upregulations of pathways related to antiviral response (e.g., NOD-like receptor signaling, RIG-I-like signaling, and TNF signaling pathways), metabolism (e.g., fructose mannose metabolism, oxidative phosphorylation (OXPHOS), and pentose phosphate pathway), and pathways like C-type lectin receptor signaling pathway, complement, and coagulation cascades that are related to thromboembolism (Figure 4E). Interestingly, a comparison between mild and severe patients’ profiles showed that pathways like PI3K-Akt, mTOR, AMPK, and HIF-1 signaling were distinctly upregulated in severe patients (Figure 4E). These pathways are the master regulators of central carbon and energy metabolism (e.g., TCA cycle, OXPHOS, and pyruvate metabolism) and are regulated by the SARS-CoV-2 replication (Appelberg et al., 2020; Codo et al., 2020; Krishnan et al., 2021). The gene set analysis result for group-specific (mild and severe) comparisons with HC is provided (Figures 6 and S4A).
Figure 4.

System-wide transcriptomics profile in COVID-19 patients identified dysregulated immune and metabolic pathways
(A) Sample distribution using normalized transcriptomics data (log2 counts per million) of all protein-coding genes through UMAP, colored by cohort.
(B) Volcano plot visualizing gene expression changes in SARS-CoV-2-infected individuals (mild [n = 26] and severe [n = 11] in comparison with healthy individuals [HC, n = 21]). The top five significantly regulated genes are labeled.
(C) Volcano plot visualizing gene expression changes in SARS-CoV-2-infected individuals with severe COVID-19 (n = 11) in comparison with mild disease (n = 26). The top five significantly regulated genes are labeled.
(D) Heatmap visualizing expression pattern (Z score transformed log2 counts per million) and log2 scaled fold change values of significantly regulated genes in pairwise comparisons (adj. p < 0.05 and log2-fold change > 1.5). Column annotation represents patient groups, and pairwise differential expression comparisons and rows represent genes.
(E) Heatmap visualizing significantly regulated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (adj. p < 0.05) in SARS-CoV-2-infected individuals (mild [n = 26] and severe [n = 11] in comparison with healthy individuals [HC, n = 21]) and COVID-19 patients with severe disease (n = 11) in comparison with patients with mild disease (n = 26). The color scale represents negative log10 scaled adjusted p values of different directionality regulation classes. The non-directional p values are computed from the gene statistics disregarding the direction of expression. Mix-direction down and mix-direction up p values were computed by considering the segment of the gene statistics that are downregulated and upregulated, respectively. Distinct-directional p values were calculated using expression direction along with gene statistics. The distinct-direction up p values is exclusively influenced by the upregulation of genes. By contrast, distinct-direction down p values is influenced by downregulation of genes and not influenced by upregulation and downregulation together.
Figure 6.

Flux balance analysis (FBA) and sctMetabolomics identified COVID-19 and severity-specific dysregulated metabolic reaction
(A) Representative diagram of selected transport reactions found to have different flux in SNF-1 (mild/moderate, n = 22) and SNF-3 (severe, n = 10) than SNF-4 (healthy, n = 17). The diagram shows the cellular location of the responses, transporter genes, and the transported metabolic products.
(B) UMAP clustering of cells and their associated cell types generated from the scRNA-seq data published by Zhang et al. (2020). Expression (log expression > 2) of SLC25A1, SLC25A10, and SLC25A11 in various cell types is shown in subsequent UMAPs.
(C) Single-cell-type metabolomics (sctMetabolomics) of the monocyte population identified increased α-ketoglutarate (α-KG), citrate, and malate (HC [n = 12] and COVID-19 [n = 18]).
(D) Heatmap of metabolic reactions showed different flux among all the samples identified from individual genome-scale modeling and flux balance analysis. The color scale represents the metabolic flux measurements (mmol/h/gDCW) derived from FBA. Severity-specific (reactions having different flux in more than half of the samples in SNF-3 [severe, n = 10] compared with more than half of the samples in SNF-1 [mild/moderate, n = 22]; labeled in red) and COVID-19-specific (reactions having different flux in more than half of the samples in SNF-1 [mild/moderate, n = 22] and SNF-3 [severe, n = 10] compared with more than half of the samples in SNF-4 [healthy, n = 17]) reactions are labeled.
Data-driven patient stratification results in the re-classification of COVID-19 subgroups
Clinical categorization of COVID-19 patients relies on their oxygen requirement. However, our previous study identified that O2 need at hospitalization did not predict mortality (Saccon et al., 2021). Moreover, the HCA analysis identified heterogeneity among the significantly dysregulated genes in COVID-19 patients (Figure 4D). Therefore, we sought to improve the classification of clinical COVID-19 severity definitions as driven from a molecular point of view through a data-driven integrative approach capable of handling the high patient-patient molecular heterogeneity. We integrated information from both transcriptomics and metabolomics through SNF, which has been shown to capture both shared and complementary information from different data sources and produce clinically relevant subtypes in multiple studies (Wang et al., 2014). Through SNF, we observed that while individual omics did not show evident similarity patterns (Figures 5 A and 5B), the integrative characterization of the samples pointed to four clusters (Figure 5C; Table S2). Among these, we observed that many HC (52.4%, 11/21;) samples shared high similarities with convalescent (60%, 6/10) samples (SNF cluster 4; SNF-4) (Figures 5C–5E), in line with our observations at transcriptomic-only level (Figure 4A). Furthermore, 50% of hospitalized-severe samples shared high similarity with hospitalized-mild samples (SNF-3). In addition, we observed that SNF-2 included samples from all four groups, including HC (38%, 8/21), convalescent (40%, 4/10), hospitalized-mild (6.9%, 2/29), and hospitalized-severe (27%, 3/11) samples, suggesting that homogeneous features exist in each layer of data type. Furthermore, SNF-1 was dominated by hospitalized-mild samples (69.0%, 20/29) (Figures 5C and 5D). Based on the plasma inflammation profile identified by the interleukins (IL) IL6, IL7, IL8, IL10, IL15, and IL18 (Figure 5F)—measured using the Olink Immuno-oncology panel (Krishnan et al., 2021)—we defined SNF-1 as COVID-19 mild/moderate, SNF-3 as COVID-19 severe, and SNF-4 as HC. In subsequent analyses, we used these newly defined clusters except for SNF-2 due to its mixed populations that may share common molecular features.
Figure 5.

Integrated clustering of transcriptomics and metabolomics using similarity network fusion (SNF) shows novel stratifications for patients based on molecular data
(A) Heatmap showing sample to sample similarity between all samples in each patient cohorts (HC [n = 21], convalescent [n = 10], hospitalized-mild [n = 26], and hospitalized-severe [n = 11]) for transcriptomics data. Row annotation denotes patient cohort.
(B) Heatmap showing sample to sample similarity between all samples used in (A) for metabolomics data. Row annotation denotes patient cohort.
(C) Sample to sample similarity of SNF-derived patient clusters. Column annotation denotes SNF clusters and associated clinical categories.
(D) Network fusion diagram of the four integrated patient clusters. Node color indicates the clinical category of the samples; edges indicate a similarity >0.01. (HC [dark green], convalescent [light green], hospitalized-mild [yellow], and hospitalized-severe [orange])
(E) Sankey plot depicting association of clinical categories, and new clusters defined based on molecular data for each sample.
(F) Mean-scaled interleukins (IL) of plasma inflammation profile identified by the IL6, IL7, IL8, IL10, IL15, and IL18 to define the severity of the data-driven clusters.
(G) Community characterization results obtained from topology analysis of the association network between genes and metabolites among samples of SNF-1, 3, and 4. Node size is relative to the centrality (degree) of the feature (gene/metabolite). Node colors denote log2-fold change values of the feature in SNF-3 (severe, n = 10) compared with SNF-1 (mild/moderate, n = 22). Each edge denotes a significant Spearman correlation (adj. p < 0.00001).
(H) Significantly enriched pathways (adj. p < 0.01) in the most central community found in (F) (community 1) based on average node centrality (degree).
Multi-omics network topology analysis identifies severity-specific mechanisms
Integrated networks based on transcriptomics and metabolomics data were generated to identify severity-specific omics hallmarks and associated mechanisms. Topological network analysis (adj. p < 10−5, Spearman rank correlation) among features from samples of SNF-1, 3, and 4 was used to create a network, followed by a community identification (Traag et al., 2019). The analysis highlighted nine communities (c1–c9) of co-expressed genes and metabolites (Figure 5G). The expression direction of genes and metabolites in SNF-3 (severe) compared with SNF-1 (mild/moderate) was mapped to the network to define severity associations at the feature community level. Out of the total genes in c1, 42% of them were found significantly (adj. p < 0.05) upregulated in SNF-3 (severe) compared with SNF-1 (mild/moderate), and thus it was termed a severity-specific community. Among the rest of the genes in c1, 6.6% were upregulated in SNF-1. We employed centrality analysis to identify and rank the importance of genes and metabolites based on how connected they were to the network (i.e., degree centrality). We observed (Data S2) that c1 was the most central community, thus showing that the most connected molecular elements tended to be associated with coordinated expression changes related to COVID-19 severity state. Among the top 5% central elements in the network (542 nodes), 290 genes/metabolites were found in c1 and included the glycolytic genes ADP-dependent glucokinase (ADPGK), phosphoglycerate mutase 1 (PGAM1), and the TCA-cycle and OXPHOS gene succinate dehydrogenase complex iron-sulfur subunit B (SDHB). We observed that mannose, a biomarker for COVID-19 (Krishnan et al., 2021), was also found in c1. Enrichment analysis showed that c1 was associated with NOD-like receptor signaling, chemokine signaling, Fc gamma R-mediated phagocytosis and platelet activation pathways, and other pathways such as NF-κ B, Notch, RIG-I, HIF-1, and FoxO signaling (Figure 5H). The enrichment analysis of the other communities (c2–c9) is given (Figure S5). These observations indicate that substantial signaling alterations in the immune response are closely linked with central metabolism through mannose, ADPGK, PGAM1, and SHDB, all of which are associated with disease severity.
Metabolic modeling highlights host metabolic dysregulation in COVID-19
Since prior omics analysis has identified metabolic and signaling pathway dysregulation in COVID-19 (Figure 4E), we have used context-specific GSMM to find how the metabolic reactions are impacted upon COVID-19 and their cellular localization further to increase the resolution of perception on metabolic rearrangement. We have generated group-specific GSMMs based on the newly defined SNF clusters by integrating transcriptomics data and constrained the nutrient uptake of the models based on metabolomics data. Each model accounted for the intracellular energy requirements for the growth of host cells (see STAR Methods). Models were deemed feasible if they passed 57 biological functions (Agren et al., 2014). After FBA and based on metabolic flux distributions, we identified 100 reactions differentially active (with or without flux) or with opposite reaction directions among SNF clusters (Data S3). Further, we identified 15 transporter reactions with unique flux in SNF-1 (mild/moderate) and SNF-3 (severe) compared with SNF-4 (HC) (Table S3). Among the transporter reactions, there were reactions involved in the transport of TCA-cycle intermediates such as cis-aconitate, α-ketoglutarate, succinate, malate, and fumarate between cytosol and mitochondria. Out of these 15 reactions, 13 had flux only in SNF-1 (mild/moderate) and SNF-3 (severe), indicating active transport of the metabolites in COVID-19 patients, which then potentially feed the TCA-cycle pathway (Figure 6 A). We used previously published single-cell transcriptomics data (Zhang et al., 2020), which characterized patient groups similar to ours. The members of the mitochondrial carrier family (SLC25) (SLC25A1 and SLC25A11) were highly expressed (log expression > 2) in more cells in the monocyte populations (Figures 6B and S6). As we observed that signatures of monocytes were enriched both in the DCQ (Figure 2) and in immunophenotyping analyses (Figure 3) and metabolism in monocytes plays an important role in regulating innate immune function (Pence and Yarbro, 2019), we performed sctMetabolomics targeting TCA-cycle intermediates in monocytes. We observed increased cellular levels of α-ketoglutarate, malate, and citrate in monocytes in COVID-19 samples (Figure 6C). To further handle the individual heterogeneity underlying the molecular profiles of all patients, we generated personalized GSMMs to characterize COVID-19-specific and severity-specific metabolic alterations. FBA identified 274 metabolic reactions with distinct fluxes across patients (Figure 6D; Data S3), many of which recapitulated the above-described group-specific observations. Reactions that had flux in the same direction in more than or half of the samples in SNF-1 and SNF-3 and opposite or no flux in more than or half of the samples in SNF-4 were regarded as COVID-19-specific reactions. By contrast, reactions that had flux in the same direction in more than or half of the samples in SNF-3 and opposite or no flux in more than or half of the samples in SNF-1 were regarded as COVID-19 severity-specific reactions. The analysis provided 16 reactions as COVID-19 specific and 10 reactions as COVID-19 severity specific (Figure 6D; Data S3).
FBA-derived network topology analysis identified essential genes and metabolites in COVID-19
The results above indicated substantial rearrangement of intracellular metabolic fluxes concomitant with COVID-19 and its severity. To further explore the biological implications of the metabo-transcriptomic alterations while still considering the biologically driven characterization provided by the predicted flux distributions, we performed a topological analysis of the personalized and group-specific metabolic networks. These weighted networks were created using reactions that displayed divergent metabolic fluxes, where nodes were reactants, products, and the associated enzymatic genes (Data S3), and edge weight was scaled down measurement of the reaction fluxes. Further, network communities were identified, and centrality was calculated to prioritize nodes (Data S3). We observed that 7 genes and 51 metabolites showed very high centrality (Figure 7 A; betweenness > mean across samples; 83.9 for genes and 22 for metabolites). These included the monocarboxylate transporter SLC16A6 and nucleoside transporter SLC29A1 and metabolites such as α-ketoglutarate in the cytosol, succinate and malate in mitochondria and cytosol, and butyrate in the cytosol and extracellular space, which were identified as uniquely essential for COVID-19 samples. Topology analysis of SNF-3 (severe) networks also identified α-ketoglutarate as the most central among other intermediate products of the TCA cycle such as cis-aconitate, malate, fumarate, succinate, and mitochondrial transporter genes like SLC25A1 and SLC25A11 (Figure 7B). The reactions associated with SLC25A1 and SLC25A11 displayed fluxes (Figure 6D) only in SNF-1 (mild/moderate) and SNF-3 (severe) but not in any other SNF cluster, further reinforcing the rearrangement of malate and α-ketoglutarate metabolism observed at sctMetabolomic level (Figures 6A and 6B). Moreover, gene essentiality analysis identified mitochondrial genes as essential in COVID-19 patients (Figure S7), including NADH: ubiquinone oxidoreductase core subunit S2 (NDUFS2) and NADH: ubiquinone oxidoreductase subunit B3 (NDUFB3) in OXPHOS, glycine N-methyltransferase (GNMT) in glycine metabolism, 5-oxoprolinase, ATP-hydrolysing (OPLAH) in glutathione metabolism, and amidohydrolase domain containing 1 (AMDHD1) in histidine metabolism, all of which were part of the central community c1 (Figure 5G). To find the cell types where the top central genes are mostly expressed, we used the published scRNA-seq data (Zhang et al., 2020). COVID-19-specific essential gene SLC16A6 was mostly expressed in monocytes and monocyte-derived DCs (Figures 7C and S8), whereas SLC29A1 was mostly expressed in NCM and proT cells (Figures 7C and S8).
Figure 7.

Network-based essential gene and metabolite analysis reveal the role of transporter genes and TCA-cycle intermediates in COVID-19
(A) The essential genes and metabolites are identified based on topology analysis of the network created using metabolites and associated genes of selected reactions in each sample. The heatmap visualizes the centrality measurements (betweenness) of genes and metabolites in each sample network. Top column annotation denotes sample classification by SNF clustering. The bottom column annotation denotes original cohorts.
(B) Topology analysis results of the network created using metabolites and associated genes of selected reactions in SNF cluster 3 (severe, n = 10). The figure shows two communities identified. The color gradient denotes the centrality of the communities (betweenness). Node size is relative to the centrality (betweenness) of each feature.
(C) UMAP clustering of cells and their associated cell types generated from scRNA-seq data published by Zhang et al. (2020). Expression of top essential genes identified in (A), in various cell types, is shown in subsequent UMAPs.
Discussion
This study combined system-level transcriptomics, DCQ, blood cell immunophenotyping of MNPs, integrated network analysis, and generated personalized and group-specific GSMM of COVID-19 patients to provide comprehensive systems-level characterizations in SARS-CoV-2 infection. The transcriptomics data identified distinct upregulation of pathways related to the antiviral response and metabolism in the COVID-19 patients. While comparing the mild and severe COVID-19 patients’, pathways that are master regulators of central carbon and energy metabolism (e.g., TCA cycle, OXPHOS, pyruvate metabolism, etc.), the PI3K-Akt, mTOR, AMPK, and HIF-1 signaling were distinctly upregulated, which are also known to be regulated during SARS-CoV-2 infection (Appelberg et al., 2020; Krishnan et al., 2021). Based on DCQ, in both mild and severe COVID-19 patients, a disease pathogenicity-specific pattern in neutrophils and CM was determined. The homeostatic association between the immune cells like T cells and DCs was dysregulated, further supported by the immunophenotyping of MNPs and its association with the T cells (CD4+ and CD8+). Due to the high molecular heterogeneity displayed by patients, we performed a sample stratification through SNF, based on both metabolomic and transcriptomic data. Using the personalized GSMM and SNF cluster-driven mild/moderate and severe COVID-19 groups, the FBA identified the transporter reactions that fuel TCA-cycle intermediates, such as the cis-aconitate, α-ketoglutarate, succinate, malate, and fumarate, that may play an essential role in COVID-19, which was further supported by sctMetabolomics of the monocytes. Gene and metabolite prioritization analysis indicated that monocarboxylate transporters (MCTs or SLC16 gene family) like SLC16A6 or nucleoside transporter genes like SLC29A1 that are expressed in the monocytes and metabolites such as α-ketoglutarate, succinate, malate, and butyrate could play a crucial role in COVID-19.
The DCQ predicted an increase in the neutrophils and CM in COVID-19 patients, which corroborated earlier findings (reviewed in Reusch et al., 2021). Following SARS-CoV-2 infection, an elevated number of both mature and immature neutrophils were observed in the nasopharyngeal epithelium (Chua et al., 2020), lung (Liao et al., 2020), and blood (Giamarellos-Bourboulis et al., 2020; Guan et al., 2020; Lourda et al., 2021). Elevation of neutrophil counts can lead to excessive formation of neutrophil extracellular traps (NETs), an extracellular network of chromatin, oxidant enzymes, and microbicidal proteins released by the neutrophils to control infection (Zuo et al., 2020), potentially contributing to hyperinflammation in COVID-19 patients. Moreover, a myeloid-driven atypical cytokine storm, mediated by the classical proinflammatory monocytes and neutrophils, could be the critical mediator of COVID-19 disease severity (Vanderbeke et al., 2021). The decrease in the number of circulating unconventional T cells, γδ-T cells, and MAIT cells in severe COVID-19 patients compared with HC was in line with previous studies (Lei et al., 2020; Parrot et al., 2020). Given that both cell types are important for antiviral responses, decreases in the blood could indicate their recruitment to tissue and increased cell death (Shi et al., 2021). Interestingly, the co-expression network of the cell-specific markers indicated a gradual loss (convalescent > mild > severe) of the association between marker genes of the T cells and DCs, as well as between marker genes of granulocytes and other immune cells at the system level. These findings are indicative of dysregulated crosstalk between T cells and DCs (Hivroz et al., 2012) and granulocytes with other immune cells (Lourda et al., 2021), which could explain the insufficient control of the viral replication (Saichi et al., 2021) leading to severe COVID-19. Interestingly, the COVID-19 convalescent samples also showed the dysregulated crosstalk, further supporting a recent study where it has been shown that mild SARS-CoV-2 infection can leave an inflammatory imprint in the myeloid cells and can affect the macrophage effector functions (Bohnacker et al., 2022).
Several studies, including ours, have reported alterations in myeloid cells in blood and tissues of COVID-19 patients—with increased levels of CM, M-MDSC, and LDG (Cabrera et al., 2021) and a decrease in NCM, pDC, and DC1 cells—with these changes linked to platelet activation and degranulation, activation of the coagulation cascade, and overall disease severity (Agrati et al., 2020; Chilunda et al., 2021; Falck-Jones et al., 2021; Krishnan et al., 2021; Lourda et al., 2021; Schulte-Schrepping et al., 2020; Trombetta et al., 2021). In our study, using DCQ and immune profiling, we report similar changes in the MNP profile, and in addition to that, we also observed altered chemokine receptor expression in different MNP cells. Chemokines play a crucial role in modulating the immune landscape during viral infections by regulating innate and adaptive immune responses (Khalil et al., 2021). Systemic changes in chemokines’ levels and receptors play critical roles in disease pathogenesis and progression (Majumdar and Murphy, 2020). Chemokine receptor signaling controls emigration from the bone marrow, cell survival, and migration into inflamed sites. Changes indicate the dynamic nature of an ongoing immune response to viral infection. Changes in MDSC cell subsets could have significant impacts on immune responses to SARS-CoV-2 (i.e., decreased T cell responses, antigen-presenting cell responses, etc.) (Emsen et al., 2022; Koushki et al., 2021). The fate of circulating monocytes during infection and inflammation is determined by differential expression of specific cell surface receptors, including CCR2, CX3CR1, and CCR5, which are known to modulate monocyte differentiation, migration, function, and survival (Chen et al., 2013b; Gschwandtner et al., 2019; Käufer et al., 2018; Mehta and Reilly, 2012). SARS-CoV-2 has been shown to upregulate CCR2, CCR5, and CCR1 expression in the human thoracic dorsal root ganglion (Khalil et al., 2021) and could directly or indirectly affect the levels of these receptors also on myeloid cells. In this study, we observed significantly increased levels of CCR2 and CCR5 on all monocyte subsets and in some DC subsets in the blood of COVID-19 patients, which are in line with the high levels of their ligands in the blood of COVID-19 patients (Kvedaraite et al., 2021; Patterson et al., 2021a) and attraction to the inflamed lung. SARS-CoV-2-infected lung macrophages have also been shown to express the chemokine ligand 5 (CCL5), and the CCL5/CCR5 axis disruption by CCR5 blockade reduced inflammation, restored T cell lymphocytopenia, and reduced SARS-CoV-2 viremia in COVID-19 patients (Patterson et al., 2021b). Of note, the significant decrease in pDC frequency in the blood of COVID-19 patients and the downregulation of CX3CR1 in the remaining pDC in blood could possibly be explained by the binding of CX3CL1 (fractalkine) to CX3CR1. Fractalkine is a potent lung chemoattractant of immune cells and was previously found to be elevated in the serum of COVID-19 patients (Tong et al., 2020). Since DCs were reported in COVID-19 lungs (Liao et al., 2020), it is possible that DC disappearance from the circulation is related to their influx in lymph nodes and lung tissue to initiate and assist in the adaptive immune responses against SARS-CoV-2.
Another feature observed in COVID-19 disease is the altered plasma metabolic profile indicating a shift towards amino acid and fatty acid metabolism, energy, and lipid metabolism (Shen et al., 2020; Thomas et al., 2020; Wu et al., 2020). Previously, we and others reported that SARS-CoV-2 modulated the AKT/mTOR/HIF-1 pathway that is a central regulator of glycolysis and other central metabolic pathways (Appelberg et al., 2020; Chen et al., 2020; Codo et al., 2020). We have also shown that the inhibition of the glycolysis, glutaminolysis, and AKT/mTOR/HIF-1 pathways inhibited the SARS-CoV-2 replication, as SARS-CoV-2 utilizes these pathways for its efficient replication (Appelberg et al., 2020; Krishnan et al., 2021). The solute carrier (SLC) transporters are membrane-associated proteins that mediate the influx and efflux of metabolites (Schumann et al., 2020). Thus, SLCs play a significant role in maintaining glycolytic balance within a cell and significantly influence the development of metabolic diseases (Zhang et al., 2019b). In our previous study, immunophenotyping identified increased expression of the carbohydrate transporter GLUT1 (SLC2A1) in CD8+ T cells, IM, and NCM and amino acid transporter xCT (SLC7A11) in all monocytes in COVID-19 patients compared with HC, highlighting the role of metabolite transporters in COVID-19 disease (Krishnan et al., 2021). Moreover, the alterations of the transporters were more prominent in the monocytic population, which could potentially contribute to mediating the disease severity by modulating its impaired immune function through metabolic alterations. In the present study, instead of explicitly looking into specific transporters, we tried to comprehensively identify the transporters uniquely associated with COVID-19. For this purpose, we have used metabolic flux-prediction models by integrating the blood transcriptomics and plasma metabolomics data and determined that SLC transporters, such as SLC29A1 (ENT1; equilibrative nucleoside transporter 1) and SLC16A6 (MCT7; monocarboxylate transporter 7), were uniquely essential in COVID-19 samples. The SLC29A1 is a nucleoside transporter and is critical for nucleotide biosynthesis. The SLC16 gene family, comprising 14 members of the MCT family, plays an important role in energy metabolism as it catalyzes the rapid transport of lactate and pyruvate across the cell membrane, which are essential components for glycolysis (reviewed in Halestrap, 2013). Although no study reported the role of the SLC transporter earlier, a co-expression analysis performed in microarray analysis data of influenza-infected pediatric patients reported upregulated expression of SLC16A6 (Zarei Ghobadi et al., 2019), suggesting its potential role in respiratory viral infections. The SLC16A6 has been reported to act as a transporter of ketone bodies like β-hydroxybutyrate out of the liver (Newman and Verdin, 2017) that is again internalized by tissues other than the liver and is utilized in the TCA cycle in mitochondria or fatty acid synthesis in the cytoplasm (Sheraj et al., 2021). A more recent study also reported SLC16A6 as a taurine transporter (Higuchi et al., 2022), indicating its role in energy metabolism. It is also possible that transporters such as SLC16A6, which was differentially upregulated in COVID-19, can also regulate the intracellular and extracellular levels of α-keto acids like α-ketoglutarate, which are essential components of the TCA cycle. We have also identified mitochondrial transporters such as SLC25A1 (CIC; citrate/isocitrate carrier) and SLC25A11 (OCG; mitochondrial 2-oxoglutarate/malate carrier) along with TCA-cycle intermediates such as α-ketoglutarate, citrate, succinate, fumarate, and malate differentially represented either in the cytosol or extracellular space. The prominent expression of SLC25A11 and SLC25A1 together with the upregulation of intracellular malate and α-ketoglutarate observed at the sctMetabolomic level is further supported by our GSMMs. Further, metabolite essentiality analysis identified butyrate, a microbiome-derived four-carbon short-chain fatty acid, as an essential metabolite in severe COVID-19, which plays an essential role in energy metabolism and intestinal homeostasis (Liu et al., 2018). Recently our integrative analysis of cytokines, metabolites, and microbiome features suggested a potential role of microbial-derived immunoregulatory processes in fatal outcomes in COVID-19 due to the failure of the negative feedback mechanism that should confine the cytokine storm (Albrich et al., 2022). Interestingly, MCTs are also involved in the butyrate transportation (Chang et al., 2014). The role of SLC transporters in the regulation of energy metabolism has been studied extensively for cancer but requires further attention in the context of viral infections that affect the metabolic pathways.
The system-wide metabolomics profile in COVID-19 patients with different disease states identified several biomarkers and shed mechanistic insight into SARS-CoV-2 infection. Even though metabolomics is closer to the phenotype, the metabolomics-only approach limits identifying the alterations of the metabolites or the metabolic pathways that can be influenced by gender, age, ethnicity, comorbidities, diet, etc. Application of GSMM by integrating the host transcriptomics and metabolomics and the virus biomass function can provide information about the altered metabolic reactions. Earlier seminal studies in the context of SARS-CoV-2, based on host-virus GSMM of human alveolar macrophages (Renz et al., 2020), human airway epithelial cells (Bannerman et al., 2021), and liver cancer cells (Huh7) (Yaneske et al., 2021), reported that the role of biosynthesis and degradation of amino acids, glycolytic shift, and energy metabolism are essential for progressive viral replication. As the SARS-CoV-2 was not detected in the blood cell populations in our cohort, we therefore hypothesized that the systemic metabolic alterations are most likely because of the bystander effect of the infection due to the inflammatory conditions. The specific metabolic environment may differ from the metabolic alteration during productive replication in the infecting cells.
In addition to using GSMMs for metabolic flux characterization and reporter metabolites of different COVID-19 subgroups, the generation of personalized metabolic models enabled the identification and prioritization of genes and metabolites at a patient-specific level in our study. We did a comprehensive characterization of COVID-19 at a personalized level integrating transcriptomic data combining information from biologically agreeable metabolic flux predictions and topological network analysis. Our data indicated a system-level altered flux in the mitochondrial TCA-cycle intermediates that can explain the dysregulated central metabolic pathways in COVID-19 and could potentially be linked with the glycolytic modulation by mTOR/HIF-1 signaling and mitochondrial dysfunction (Krishnan et al., 2021). We also reported that by blocking two critical central metabolic pathways, glycolysis and glutaminolysis, viral replication and production were inhibited (Krishnan et al., 2021). Although systemic GSMM was not reported in other respiratory diseases caused by viruses, in our recent study to understand the natural control of HIV-1 infection, we observed the regulation of similar metabolic pathways, but the difference in the metabolic reactions potentiates a disease-specific contextualization of the metabolic flux (Ambikan et al., 2022). A viral disease-specific systemic GSMM atlas for other emerging and re-emerging viruses is underway.
The study has limitations that merit comments. Firstly, the RNA-seq data were generated from whole blood samples, which can cause bias in DGE analysis due to different blood cell populations. To overcome the bias, we computationally estimated the cell type proportions in each sample through DCQ. We used the information to adjust the bias to a minimum possible while performing DGE analysis. Second, although the DCQ analysis adapted from the original EPIC algorithm has worked here because of high sequence depth and sequence quality, it may not work in data with low sequence depth samples. It is recommended to cross-validate the finding with the original EPIC algorithm or by other phenotypic methods like flow cytometry. Third, the definition of hospitalized-mild and hospitalized-severe patient groups was based on oxygen consumption, and clinicians’ definitions may leave a great deal of ambiguity as observed in the HCA. However, one of the strengths of our study was that we grouped the samples using high-throughput transcriptomics and metabolomics data by employing the multi-omics SNF methodology. The new patient groups were exclusively built on samples’ multi-omics profiles, which brings high confidence than the clinical definition of the COVID-19 severity. Further, the group level GSMM and FBA were performed on the new patient groups (SNF) to minimize the ambiguity in the clinical definition, which may have caused erroneous results otherwise. Finally, the GSMMs was created by considering the whole blood as a single biological system. This poses an additional challenge as different cells perform metabolic functions in a slightly different manner. Ideally, the models should have been explicitly created for each cell type, but due to technological challenges, it was impossible to generate single-cell-type metabolomics data to accurately constrain the model’s nutrient uptake of the system in various conditions, which is an essential factor in the procedure. Future developments enabling scRNA-seq integration in metabolic models to overcome the high sparsity that characterizes this data type will allow disentangling of the distinct metabolic flux distributions displayed by different cell types. However, to validate the findings observed, we developed and applied sctMetabolomics for the critical cell type, i.e., monocytes, further strengthening our GSMM observation.
In conclusion, with a combination of multi-modal systems-wide transcriptomics, DCQ, and immunophenotyping of MNPs, we observed an essential role in coordinating the immune cells in COVID-19 severity. Novel data-driven patient stratification recapitulated many clinical properties previously known but further enabled us to uncover the potential mechanical consequences of COVID-19 infection in immune cells. The systems biology approaches, including network topology analysis and personalized GSMM, sctMetabolomics of monocytes, and gene mapping in published scRNA-seq data, displayed a progressive dysregulation of the central metabolic pathway (TCA cycle) concomitant with disease severity. Alterations in central carbon and energy metabolism, TCA-cycle intermediates like malate and α-ketoglutarate, and expression of metabolite transporters in monocytes were associated with disease severity at individual and group levels. These observations prompt the metabolic modulation of monocytes in specific COVID-19 patient groups and potentiate personalized targets for treatment in severe patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | ThermoFisher Scientific | Cat#L34957 |
| HLADR-PECY7-L243 | BD Bioscience | Cat#335830; RRID:AB_2868691 |
| CD3- PECF594-UCHT1 | BD Bioscience | Cat#562280; RRID:AB_11153674 |
| CD19-PECF594-HIB19 | BD Bioscience | Cat#562294; RRID:AB_11154408 |
| CD56-PECF594-B159 | BD Bioscience | Cat#562289; RRID:AB_11152080 |
| CD33-FITC-WM-53 | BD Bioscience | Cat#564588; RRID:AB_2738856 |
| CD11c-BUV737-B-LY6 | BD Bioscience | Cat#741827; RRID:AB_2871162 |
| CD14-PE-M5E2 | BD Bioscience | Cat#555398; RRID:AB_395799 |
| CD16-APCH7-3G8 | BD Bioscience | Cat#560195; RRID:AB_1645466 |
| CD1C-BV421-F10/21A3 | BD Bioscience | Cat#565050; RRID:AB_2744319 |
| CD141-BV711-1A4 | BD Bioscience | Cat#563155; RRID:AB_2738033 |
| CD303-BV786-V24-785 | BD Bioscience | Cat#748000; RRID:AB_2872461 |
| CCR2-BV650-1D9 | BD Bioscience | Cat#747849; RRID:AB_2872311 |
| CCR5-PECY5-2D7 | BD Bioscience | Cat#556889; RRID:AB_396529 |
| CX3CR1-BV605-2A9-1 | BD Bioscience | Cat#744488; RRID:AB_2742268 |
| CD11B-BUV395-ICRF44 | BD Bioscience | Cat#563839; RRID:AB_2716869 |
| CD15-AF700-HI98 | Biolegend | Cat#301920; RRID:AB_2728239 |
| CD66B-AF647-G10F5 | BD Bioscience | Cat#561645; RRID:AB_10894001 |
| Biological Samples | ||
| Human blood from COVID-19 patients and COVID-19 negative samples | This paper and (Krishnan et al., 2021) | |
| Critical Commercial Assays | ||
| Global Metabolomics (HD4) | Metabolon. Inc., US | HD4 |
| Olink Immuno-Oncology Panel | Olink, Sweden | Immuno-Oncology Panel |
| EasySep human monocyte isolation kit | STEMCELL Technologies, US | Cat#19359 |
| Deposited Data | ||
| Transcriptomics | This paper | SRA: PRJNA828431 |
| Metabolomics | Krishnan et al. (2021) | 10.6084/m9.figshare.13336862 |
| Proteomics | Krishnan et al. (2021) | 10.6084/m9.figshare.13336862 |
| scRNASeq | Zhang et al. (2020) | Kindly obtained from the authors |
| Software and Algorithms | ||
| PIANO v2.2.0 | Väremo et al. (2013) | https://doi.org/10.18129/B9.bioc.piano |
| GSEAPY v0.9.16 | Subramanian et al. (2005) | https://pypi.org/project/gseapy/0.10.8/ |
| Estimating the Proportions of Immune and Cancer cells (EPIC) | Racle and Gfeller (2020) | https://github.com/GfellerLab/EPIC |
| CellMarker | Zhang et al. (2019a) | http://bio-bigdata.hrbmu.edu.cn/CellMarker/ |
| PanglaoDB | Franzén et al. (2019) | https://panglaodb.se/ |
| ggplot2 v3.3.2 | Wickham (2016) | https://ggplot2.tidyverse.org/ |
| ComplexHeatmap v2.2.0 | Gu et al. (2016) | https://doi.org/10.18129/B9.bioc.ComplexHeatmap |
| Cytoscape v3.6.1 | Shannon et al. (2003) | https://cytoscape.org/ |
| InteractiVenn | Chen and Boutros (2011) | http://www.interactivenn.net/ |
| RAVEN toolbox v2.4.0 | Wang et al. (2018) | https://github.com/SysBioChalmers/RAVEN |
| COBRA Toolbox v3.0 | Heirendt et al. (2019) | https://opencobra.github.io/ |
| RUVSeq v1.28.0 | Risso et al. (2014) | https://doi.org/10.18129/B9.bioc.RUVSeq |
| Original source code | This paper | https://dx.doi.org/10.5281/zenodo.6706499 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ujjwal Neogi (ujjwal.neogi@ki.se).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cohort description
In this study, we included two cohorts of hospitalized-mild (n=26) and severe (n=11) COVID-19 patients and COVID-19 PCR negative samples (n=31). The cohorts' clinical and demographic parameters were described elsewhere (Krishnan et al., 2021). The disease pathogenesis was defined based on oxygen consumption. The study was approved by the Regional Ethics Committee in Stockholm, Sweden, and performed in accordance with the Declaration of Helsinki.
Method details
Multi-omics analysis
Whole blood was collected in Tempus™ blood RNA tubes. The RNA sequencing was performed using Illumina NovaSeq6000 in S4 mode. The transcriptomics data processing was performed as recently described by us (Appelberg et al., 2020). Differential gene expression analysis was performed using the R/Bioconductor package DESeq2 v1.26.0 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html). Confounding factors such as cell type proportions, BMI, and other possible hidden factors were adjusted while performing differential expression analysis. Bioconductor package RUVSeq v1.28.0 was used to compute the factor of unwanted variation and added to the DESeq2 design matrix (Risso et al., 2014). KEGG gene set enrichment analysis for differentially regulated genes was performed using PIANO v2.2.0 (Väremo et al., 2013) (nperm=500, geneset statistic=mean) and for communities using enrichr module of python package GSEAPY v0.9.16 (Chen et al., 2013a; Subramanian et al., 2005). Metabolomics and proteomics data generation were performed using the Metabolon HD4 (Metabolon Inc, NC, US) and Olink Immuno-Oncology Panel (Olink, Sweden), respectively (Krishnan et al., 2021). Normalized values of interleukins from Olink proteomics data are scaled down using the R function scale, and the mean values of each sample were visualized as boxplots. The single-cell RNAseq (scRNAseq) data was kindly obtained from Zhang et al. (2020), which had similar study groups to the present study.
Digital cell quantification (DCQ)
The deconvolution algorithm adapted from the method Estimating the Proportions of Immune and Cancer cells (EPIC) (Racle and Gfeller, 2020) was employed in the digital cell quantification (DCQ) procedure. The DCQ uses a reference cell type-specific gene expression profile and a list of signature genes specific for the cell types to calculate the cell type proportion. We have used RNA HPA blood cell gene data obtained from the Human Protein Atlas as a reference profile (Uhlen et al., 2017). The data consists of gene-level expression data of 18 blood cell types, and it is based on Human Protein Atlas version 20.1 and Ensembl version 92.38. Signature genes for the 18 blood cell types in the reference profile were downloaded from the online database CellMarker (Zhang et al., 2019a) and PanglaoDB (Franzén et al., 2019). The transcript per million (TPM) transformed gene expression data of all genes from the samples were inputted to EPIC along with reference profile and signature gene list to estimate the cell proportion.
SARS-CoV-2 virus detection in blood
We used two approaches in the study to detect viral RNA as described by us previously (Appelberg et al., 2020). Firstly, the genome alignment method was employed for virus detection. Adapter trimmed and quality filtered (Q>30) transcriptomics reads were aligned against the SARS-CoV-2 virus genome (GenBank: MT093571.1). The alignment percentage (>0%) was used to determine the presence of the virus. TrimGalore v0.6.3_dev (DOI:10.5281/zenodo.5127899) was used for data pre-processing, and STAR v2.7.3a (Dobin et al., 2013) was used for the alignment process. The detection of the viral RNA in the plasma was performed using the primers as described by us previously (Appelberg et al., 2020).
Similarity network fusion and network analysis
The Similarity network fusion (SNF) (Wang et al., 2014)) was based on transcriptomics and metabolomics from 68 individuals. First, we generated similarity matrices using features derived from each platform individually; we filtered out lowly-expressed features (TPM<5) for transcriptomics and features with variance less than 0.01 in both transcriptomics and metabolomics, followed by SNF (K=6, T=20, alpha=0.7). Association networks of transcriptomics and metabolomics data were built by computing pairwise Spearman rank correlations between and among all genes and metabolites after removal of non-expressed (row median TPM<1) and lowly variant (row variance<0.1) genes and metabolites. Top 10% significant correlation (FDR<0.00005) was analyzed in igraph (https://igraph.org) using correlation coefficient as edge weight. Centrality analysis was performed by computing degree and betweenness centralities. Communities were identified by modularity maximization through the Leiden algorithm (Traag et al., 2019). A similar approach was employed for topological analysis after flux distribution prediction.
Genome-scale metabolic model (GSMM), flux balance analysis (FBA), and essentiality analysis
Personalized (individual sample) and group-specific human metabolic models were reconstructed by integrating transcriptomics data on human reference genome-scale metabolic model obtained from Metabolic Atlas (Robinson et al., 2020). Personalized models were generated using the gene expression table of each sample, whereas the average gene expression value was used for group-specific model generation. Task-driven Integrative Network Inference for Tissues (tINIT) algorithm was used for the metabolic model reconstruction (Agren et al., 2012; Agren et al., 2014; Robinson et al., 2020). The threshold expression of TPM 1 was used for the process. Firstly, the metabolic reference model was converted to closed-form by adding boundary metabolites. After model reconstruction, biological feasibility was examined by checking their capability to perform essential metabolic tasks that are known to occur in all cell types. A list of 57 metabolic tasks (Agren et al., 2014) which are expected to be carried out by cells to be viable was used for the purpose. Matlab implementation of tINIT algorithm was used for the entire analysis (https://github.com/SysBioChalmers/Human-GEM). To emulate the energetic demands construed by virus growth onto host cells, SARS-CoV2 specific viral biomass objective function (VBOF) was incorporated into models created for COVID-19 positive samples to account for the host metabolic requirement for the virus. A unidirectional pseudo-reaction consisting of the products of VBOF and ATP hydrolysis as reactants was used as the objective function. For the remaining models, ATP hydrolysis was used as the objective function for flux balance analysis (FBA). FBA was also performed without VBOF function and ATP hydrolysis as the objective function to study the influence of VBOF in the model. The addition of VBOF did not show any change in FBA results. Matlab function solveLP implemented in the RAVEN toolbox v2.4.0 (Wang et al., 2018) was used for FBA. Essentiality analysis was performed in COBRA Toolbox v3.0 utilizing a single-gene deletion function (Heirendt et al., 2019) and the same objective functions used for FBA. The exchange reactions in the model were constrained using plasma metabolomics data as a reference. We considered that all exchange reaction fluxes are proportionally affected by the availability of extracellular metabolites, i.e. that transport reactions are limited by metabolite abundance, effectively approaching first-order kinetics with respect to extracellular metabolites. We computed log2 fold changes between each group against the control group, and reaction bounds were constrained proportionally to the computed increase or decreases. The bounds were validated based on literature data (Thiele et al., 2020).
Network topology analysis was performed on the set of reactants, products, and enzymatic genes for all reactions found to have flux from FBA. Networks were analyzed in igraph using the absolute value of flux scaled between 0 and 1 as edge weight. The centrality analysis was performed by calculating betweenness and degree centralities. Communities were identified using the Leiden algorithm (Traag et al., 2019).
Visualization
R package ggplot2 v3.3.2 (Wickham, 2016) was used to create bubble plots, scatter plots, and boxplots. R/Bioconductor package ComplexHeatmap v2.2.0 (Gu et al., 2016) was used to create all the heatmaps. Network diagrams were drawn in Cytoscape ver 3.6.1 (Shannon et al., 2003). Venn diagrams were generated using the online tool InteractiVenn (Chen and Boutros, 2011).
Flow cytometry
Peripheral blood mononuclear cells (PBMCs) were subjected to flow cytometry analysis. All samples were stained with live/dead fixable aqua dye (Invitrogen), and cell surface markers (listed in key resources table) were detected by incubating cells with relevant antibodies for 20 min at room temperature in a flow cytometry buffer. All cells were fixed with 2% paraformaldehyde before acquiring on a BD FACS Symphony flow cytometer (BD Bioscience) and data were analyzed and compensated with FlowJo 10.8 (TreeStar Inc). The gating strategy is given in Figure S2. Dimensionality reduction was performed with the UMAP FlowJo plugin v3.1.
Single cell-type metabolomics analysis (sctMetabolomics) of TCA-cycle metabolites
Monocytes were purified from stored PBMCs by using a magnetic-based EasySep human monocyte isolation kit (STEMCELL Technologies, US) as per the manufacturer's guidelines with specific modifications of the amount of specified reagents used based upon the starting cell numbers that yielded a purity of more than 85% and viability of more than 90% (Figure S9). Following isolation of the monocytes, they were washed three times in PBS at room temperature for downstream metabolomics analysis. Absolute quantification of the TCA-cycle intermediates was measured by gas chromatography coupled to triple-quadrupole analyzers operating in tandem mass-spectrometer (GC-QQQ-MS) at Swedish Metabolomics Centre, Umea,
For GCMS analysis, an 11-point calibration curve (cis-aconitic acid, a-keto-glutaric acid, citric acid, fumaric acid, glucose, glucose 6-phosphate, isocitric acid, lactic acid, malic acid, shikimic acid, succinic acid, sucrose, urea) spanning from 25-2500 pg/μL was prepared by serial dilutions and spiked with internal standards: Fumaric acid (13C4), L-Malic acid (13C4), D-Glucose (13C6), α-ketoglutaric acid (13C4), Succinic acid (D4), citric acid (D4), Sucrose (13C12) at a final concentration of 350 pg/μL. For metabolite extraction 275μL of ice-cold extraction mixture of 80%MeOH, was added to each sample. The metabolites were extracted using a mixer mill set to a frequency 30 Hz for 3 min, with 1 tungsten carbide bead added to each tube. Obtained extracts were centrifuged at 14000 r.p.m (18 620g) for 10 min. 260 μL of the supernatant was transferred into GC-vials, spiked with 1050 pg of each GCMS internal standard, and evaporated until dryness under a stream of nitrogen.
Derivatization was performed according to Gullberg et al. (2004). In detail, 10μL of methoxyamine (15 μg/μL in pyridine) was added to the dry sample that was shaken vigorously for 10 minutes before being left to react at room temperature. After 16 hours 10 μL of MSTFA was added, and the sample was shaken and left to react for 1 hour at room temperature. 10 μL of methyl stearate (1050 pg/μL in heptane) was added before analysis.
One μL of the derivatized sample was injected by an Agilent 7693 autosampler, in splitless mode into an Agilent 7890A gas chromatograph equipped with a multimode inlet (MMI) and 10 m x 0.18 mm fused silica capillary column with a chemically bonded 0.18 μm DB 5-MS UI stationary phase (J&W Scientific). The injector temperature was 260 °C. The carrier gas flow rate through the column was 1 ml min-1, the column temperature was held at 70 °C for 2 minutes, then increased by 40 °C min-1 to 320 °C and held there for 2 min. The column effluent is introduced into the electron impact (EI) ion source of an Agilent 7000C QQQ mass spectrometer. The thermal AUX 2 (transfer line) and the ion source temperatures were 250 °C and 230 °C, respectively. Ions were generated by a 70 eV electron beam at an emission current of 35 μA and analyzed in dMRM-mode. The solvent delay was set to 2 minutes. The list of MRM transitions is provided (Table S4).
Data were processed using MassHunter Qualitative Analysis and Quantitative Analysis (QqQ; Agilent Technologies, Atlanta, GA, USA) and Excel (Microsoft, Redmond, Washington, USA) software. Methyl stearate was used as the internal standard for compounds lacking a stable isotope internal standard.
Acknowledgments
The authors would like to thank Elisabet Storgärd and Ronnie Ask, Study Nurses, Södersjukhuset for their excellent support with patient recruitment and all the clinicians and nurses who are the frontline warriors fighting against COVID-19. The authors acknowledge support from the National Genomics Infrastructure in Genomics Production Stockholm, funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish Research Council, and SNIC/Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure. We are thankful to the Swedish Metabolomics Centre, Umeå, Sweden, for measuring TCA-cycle intermediates by GC-QQQ-MS and providing the methodology. The study is funded by the Swedish Research Council grants 2021-00993 (U.N.), 2017-01330 (U.N.), 2018-06156 (U.N.), and 2021-03035 (S.G.) and received support from Karolinska Institutet Stiftelser och Fonder grant 2020-01554 (U.N.) and 2020-02153 (S.G.), The Center for Medical Innovation grant CIMED-FoUI-093304 (S.G.), and Åke Wiberg Stiftelse grant M20-0220 (S.G.).
Author contributions
Conceptualization, U.N. and R.B.; methodology, A.T.A., H.Y., S.K., S.S.A., M.S., M.A., C.Z., S.M.P., A.M., R.B., and U.N.; investigation, clinical, H.N., C.J.T., and A.S.; investigation, computational and laboratory, A.T.A., H.Y., S.K., S.S.A., S.G., M.S., M.L., M.A., C.Z., A.M., R.B., and U.N.; visualization, A.T.A., H.Y., S.K., S.S.A., M.L., R.B., and U.N.; funding acquisition, S.G. and U.N.; project administration, C.J.T., A.M., R.B., and U.N.; supervision, C.J.T., A.M., R.B., and U.N.; writing—original draft, A.T.A., H.Y., S.K., S.S.A., S.G., and U.N.; writing—review & editing, M.L., M.S., M.A., C.Z., H.N., S.M.P., A.S., C.J.T., L.O.M., A.M., and R.B.
Declaration of interests
The authors declare no competing interests.
Published: July 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cels.2022.06.006.
Supplemental information
Data and code availability
All of the data generated or analyzed during this study are included in this published article and/or the supplementary materials. Created datasets and code are publicly available. The metabolomics data can be obtained from the https://dx.doi.org/10.6084/m9.figshare.13336862. All the codes are available at https://dx.doi.org/10.5281/zenodo.6706499. Additional Supplemental Items are available from Mendeley Data at https://dx.doi.org/10.17632/5prg4tsz7n.1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Agrati C., Sacchi A., Bordoni V., Cimini E., Notari S., Grassi G., Casetti R., Tartaglia E., Lalle E., D’Abramo A., et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19) Cell Death Differ. 2020;27:3196–3207. doi: 10.1038/s41418-020-0572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agren R., Bordel S., Mardinoglu A., Pornputtapong N., Nookaew I., Nielsen J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012;8:e1002518. doi: 10.1371/journal.pcbi.1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agren R., Mardinoglu A., Asplund A., Kampf C., Uhlen M., Nielsen J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 2014;10:721. doi: 10.1002/msb.145122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrich W.C., Ghosh T.S., Ahearn-Ford S., Mikaeloff F., Lunjani N., Forde B., Suh N., Kleger G.R., Pietsch U., Frischknecht M., et al. A high-risk gut microbiota configuration associates with fatal hyperinflammatory immune and metabolic responses to SARS-CoV-2. Gut Microbes. 2022;14:2073131. doi: 10.1080/19490976.2022.2073131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aller S., Scott A., Sarkar-Tyson M., Soyer O.S. Integrated human-virus metabolic stoichiometric modelling predicts host-based antiviral targets against Chikungunya, Dengue and Zika viruses. J. R. Soc. Interface. 2018;15 doi: 10.1098/rsif.2018.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambikan A.T., Svensson-Akusjärvi S., Krishnan S., Sperk M., Nowak P., Vesterbacka J., Sönnerborg A., Benfeitas R., Neogi U. Genome-scale metabolic models for natural and long-term drug-induced viral control in HIV infection. Life Sci. Alliance. 2022;5 doi: 10.26508/lsa.202201405. e202201405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelberg S., Gupta S., Svensson Akusjärvi S., Ambikan A.T., Mikaeloff F., Saccon E., Végvári Á., Benfeitas R., Sperk M., Ståhlberg M., et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 2020;9:1748–1760. doi: 10.1080/22221751.2020.1799723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman B.P., Júlvez J., Oarga A., Blundell T.L., Moreno P., Floto R.A. Integrated human/SARS-CoV-2 metabolic models present novel treatment strategies against COVID-19. Life Sci. Alliance. 2021;4 doi: 10.26508/lsa.202000954. e202000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnacker S., Hartung F., Henkel F., Quaranta A., Kolmert J., Priller A., Ud-Dean M., Giglberger J., Kugler L.M., Pechtold L., et al. Mild COVID-19 imprints a long-term inflammatory eicosanoid- and chemokine memory in monocyte-derived macrophages. Mucosal Immunol. 2022;15:515–524. doi: 10.1038/s41385-021-00482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera L.E., Pekkarinen P.T., Alander M., Nowlan K.H.A., Nguyen N.A., Jokiranta S., Kuivanen S., Patjas A., Mero S., Pakkanen S.H., et al. Characterization of low-density granulocytes in COVID-19. PLoS Pathog. 2021;17:e1009721. doi: 10.1371/journal.ppat.1009721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P.V., Hao L., Offermanns S., Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA. 2014;111:2247–2252. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Boutros P.C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Zhang Z., Barletta K.E., Burdick M.D., Mehrad B. Heterogeneity of lung mononuclear phagocytes during pneumonia: contribution of chemokine receptors. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013;305:L702–L711. doi: 10.1152/ajplung.00194.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.M., Zheng Y., Yu Y., Wang Y., Huang Q., Qian F., Sun L., Song Z.G., Chen Z., Feng J., et al. Blood molecular markers associated with COVID-19 immunopathology and multi-organ damage. EMBO J. 2020;39:e105896. doi: 10.15252/embj.2020105896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilunda V., Martinez-Aguado P., Xia L.C., Cheney L., Murphy A., Veksler V., Ruiz V., Calderon T.M., Berman J.W. Transcriptional changes in CD16+ monocytes may contribute to the pathogenesis of COVID-19. Front. Immunol. 2021;12:665773. doi: 10.3389/fimmu.2021.665773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- Codo A.C., Davanzo G.G., Monteiro LdB., de Souza G.F., Muraro S.P., Virgilio-da-Silva J.V., Prodonoff J.S., Carregari V.C., de Biagi Junior C.A.O., Crunfli F., et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab. 2020;32:498–499. doi: 10.1016/j.cmet.2020.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorey T.M., Ziegler C.G.K., Heimberg G., Normand R., Yang Y., Segerstolpe Å., Abbondanza D., Fleming S.J., Subramanian A., Montoro D.T., et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. 2021;595:107–113. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsen A., Sumer S., Tulek B., Cizmecioglu H., Vatansev H., Goktepe M.H., Kanat F., Koksal Y., Arslan U., Artac H. Correlation of myeloid-derived suppressor cells with C-reactive protein, ferritin and lactate dehydrogenase levels in patients with severe COVID-19. Scand. J. Immunol. 2022;95:e13108. doi: 10.1111/sji.13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck-Jones S., Vangeti S., Yu M., Falck-Jones R., Cagigi A., Badolati I., Österberg B., Lautenbach M.J., Åhlberg E., Lin A., et al. Functional monocytic myeloid-derived suppressor cells increase in blood but not airways and predict COVID-19 severity. J. Clin. Invest. 2021;131 doi: 10.1172/JCI144734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzén O., Gan L.M., Björkegren J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019;2019:baz046. doi: 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P., et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwandtner M., Derler R., Midwood K.S. More Than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front. Immunol. 2019;10:2759. doi: 10.3389/fimmu.2019.02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullberg J., Jonsson P., Nordström A., Sjöström M., Moritz T. Design of experiments: an efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 2004;331:283–295. doi: 10.1016/j.ab.2004.04.037. [DOI] [PubMed] [Google Scholar]
- Halestrap A.P. The SLC16 gene family—structure, role and regulation in health and disease. Mol. Aspects Med. 2013;34:337–349. doi: 10.1016/j.mam.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Heirendt L., Arreckx S., Pfau T., Mendoza S.N., Richelle A., Heinken A., Haraldsdóttir H.S., Wachowiak J., Keating S.M., Vlasov V., et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019;14:639–702. doi: 10.1038/s41596-018-0098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi K., Sugiyama K., Tomabechi R., Kishimoto H., Inoue K. Mammalian monocarboxylate transporter 7 (MCT7/Slc16a6) is a novel facilitative taurine transporter. J. Biol. Chem. 2022;298:101800. doi: 10.1016/j.jbc.2022.101800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hivroz C., Chemin K., Tourret M., Bohineust A. Crosstalk between T lymphocytes and dendritic cells. Crit. Rev. Immunol. 2012;32:139–155. doi: 10.1615/critrevimmunol.v32.i2.30. [DOI] [PubMed] [Google Scholar]
- Käufer C., Chhatbar C., Bröer S., Waltl I., Ghita L., Gerhauser I., Kalinke U., Löscher W. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc. Natl. Acad. Sci. USA. 2018;115:E8929–E8938. doi: 10.1073/pnas.1806754115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil B.A., Elemam N.M., Maghazachi A.A. Chemokines and chemokine receptors during COVID-19 infection. Comput. Struct. Biotechnol. J. 2021;19:976–988. doi: 10.1016/j.csbj.2021.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushki K., Salemi M., Miri S.M., Arjeini Y., Keshavarz M., Ghaemi A. Role of myeloid-derived suppressor cells in viral respiratory infections; hints for discovering therapeutic targets for COVID-19. Biomed. Pharmacother. 2021;144:112346. doi: 10.1016/j.biopha.2021.112346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S., Nordqvist H., Ambikan A.T., Gupta S., Sperk M., Svensson-Akusjärvi S., Mikaeloff F., Benfeitas R., Saccon E., Ponnan S.M., et al. Metabolic perturbation associated With COVID-19 disease severity and SARS-CoV-2 replication. Mol. Cell. Proteomics. 2021;20:100159. doi: 10.1016/j.mcpro.2021.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvedaraite E., Hertwig L., Sinha I., Ponzetta A., Hed Myrberg I., Lourda M., Dzidic M., Akber M., Klingström J., Folkesson E., et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2018587118. e2018587118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L., Qian H., Yang X., Zhang X., Zhang D., Dai T., Guo R., Shi L., Cheng Y., Zhang B., et al. The phenotypic changes of γδ T cells in COVID-19 patients. J. Cell. Mol. Med. 2020;24:11603–11606. doi: 10.1111/jcmm.15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Liu H., Wang J., He T., Becker S., Zhang G., Li D., Ma X. Butyrate: a double-edged sword for health? Adv. Nutr. 2018;9:21–29. doi: 10.1093/advances/nmx009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourda M., Dzidic M., Hertwig L., Bergsten H., Palma Medina L.M., Sinha I., Kvedaraite E., Chen P., Muvva J.R., Gorin J.B., et al. High-dimensional profiling reveals phenotypic heterogeneity and disease-specific alterations of granulocytes in COVID-19. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2109123118. e2109123118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S., Murphy P.M. Chemokine regulation During epidemic coronavirus infection. Front. Pharmacol. 2020;11:600369. doi: 10.3389/fphar.2020.600369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta N.N., Reilly M.P. Monocyte mayhem: do subtypes modulate distinct atherosclerosis phenotypes? Circ. Cardiovasc. Genet. 2012;5:7–9. doi: 10.1161/CIRCGENETICS.111.962647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar D., van Berlo R., de Ridder D., Teusink B. Shifts in growth strategies reflect tradeoffs in cellular economics. Mol. Syst. Biol. 2009;5:323. doi: 10.1038/msb.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J.C., Verdin E. β-hydroxybutyrate: a Signaling Metabolite. Annu. Rev. Nutr. 2017;37:51–76. doi: 10.1146/annurev-nutr-071816-064916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrot T., Gorin J.B., Ponzetta A., Maleki K.T., Kammann T., Emgård J., Perez-Potti A., Sekine T., Rivera-Ballesteros O., Gredmark-Russ S., et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci. Immunol. 2020;5:eabe1670. doi: 10.1126/sciimmunol.abe1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson B.K., Guevara-Coto J., Yogendra R., Francisco E.B., Long E., Pise A., Rodrigues H., Parikh P., Mora J., Mora-Rodríguez R.A. Immune-based prediction of COVID-19 severity and chronicity decoded using machine learning. Front. Immunol. 2021;12:700782. doi: 10.3389/fimmu.2021.700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson B.K., Seethamraju H., Dhody K., Corley M.J., Kazempour K., Lalezari J., Pang A.P.S., Sugai C., Mahyari E., Francisco E.B., et al. CCR5 inhibition in critical COVID-19 patients decreases inflammatory cytokines, increases CD8 T-cells, and decreases SARS-CoV2 RNA in plasma by day 14. Int. J. Infect. Dis. 2021;103:25–32. doi: 10.1016/j.ijid.2020.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pence B.D., Yarbro J.R. Classical monocytes maintain ex vivo glycolytic metabolism and early but not later inflammatory responses in older adults. Immun. Ageing. 2019;16:3. doi: 10.1186/s12979-019-0143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racle J., Gfeller D. EPIC: a tool to estimate the proportions of different cell types from bulk gene expression data. Methods Mol. Biol. 2020;2120:233–248. doi: 10.1007/978-1-0716-0327-7_17. [DOI] [PubMed] [Google Scholar]
- Renz A., Widerspick L., Dräger A. FBA reveals guanylate kinase as a potential target for antiviral therapies against SARS-CoV-2. Bioinformatics. 2020;36:i813–i821. doi: 10.1093/bioinformatics/btaa813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch N., De Domenico E., Bonaguro L., Schulte-Schrepping J., Baßler K., Schultze J.L., Aschenbrenner A.C. Neutrophils in COVID-19. Front. Immunol. 2021;12:652470. doi: 10.3389/fimmu.2021.652470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D., Ngai J., Speed T.P., Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.L., Kocabaş P., Wang H., Cholley P.E., Cook D., Nilsson A., Anton M., Ferreira R., Domenzain I., Billa V., et al. An atlas of human metabolism. Sci. Signal. 2020;13:eaaz1482. doi: 10.1126/scisignal.aaz1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccon E., Bandera A., Sciumè M., Mikaeloff F., Lashari A.A., Aliberti S., Sachs M.C., Billi F., Blasi F., Gabriel E.E., et al. Distinct metabolic profile associated with a fatal outcome in COVID-19 patients during the early epidemic in Italy. Microbiol. Spectr. 2021;9:e0054921. doi: 10.1128/Spectrum.00549-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saichi M., Ladjemi M.Z., Korniotis S., Rousseau C., Ait Hamou Z., Massenet-Regad L., Amblard E., Noel F., Marie Y., Bouteiller D., et al. Single-cell RNA sequencing of blood antigen-presenting cells in severe COVID-19 reveals multi-process defects in antiviral immunity. Nat. Cell Biol. 2021;23:538–551. doi: 10.1038/s41556-021-00681-2. [DOI] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann T., König J., Henke C., Willmes D.M., Bornstein S.R., Jordan J., Fromm M.F., Birkenfeld A.L. Solute carrier transporters as potential targets for the treatment of metabolic disease. Pharmacol. Rev. 2020;72:343–379. doi: 10.1124/pr.118.015735. [DOI] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L., et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheraj I., Guray N.T., Banerjee S. A pan-cancer transcriptomic study showing tumor specific alterations in central metabolism. Sci. Rep. 2021;11:13637. doi: 10.1038/s41598-021-93003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Zhou J., Zhang X., Hu W., Zhao J.F., Wang S., Wang F.S., Zhang J.Y. Single-cell transcriptomic profiling of MAIT cells in patients with COVID-19. Front. Immunol. 2021;12:700152. doi: 10.3389/fimmu.2021.700152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I., Sahoo S., Heinken A., Hertel J., Heirendt L., Aurich M.K., Fleming R.M. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 2020;16:e8982. doi: 10.15252/msb.20198982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T., Stefanoni D., Reisz J.A., Nemkov T., Bertolone L., Francis R.O., Hudson K.E., Zimring J.C., Hansen K.C., Hod E.A., et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. 2020;5:e140327. doi: 10.1172/jci.insight.140327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M., Jiang Y., Xia D., Xiong Y., Zheng Q., Chen F., Zou L., Xiao W., Zhu Y. Elevated expression of serum endothelial cell adhesion molecules in COVID-19 patients. J. Infect. Dis. 2020;222:894–898. doi: 10.1093/infdis/jiaa349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta A.C., Farias G.B., Gomes A.M.C., Godinho-Santos A., Rosmaninho P., Conceição C.M., Laia J., Santos D.F., Almeida A.R.M., Mota C., et al. Severe COVID-19 recovery is associated with timely acquisition of a myeloid cell immune-regulatory phenotype. Front. Immunol. 2021;12:691725. doi: 10.3389/fimmu.2021.691725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F., et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Vanderbeke L., Van Mol P., Van Herck Y., De Smet F., Humblet-Baron S., Martinod K., Antoranz A., Arijs I., Boeckx B., Bosisio F.M., et al. Monocyte-driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID-19 disease severity. Nat. Commun. 2021;12:4117. doi: 10.1038/s41467-021-24360-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väremo L., Nielsen J., Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013;41:4378–4391. doi: 10.1093/nar/gkt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani A.C., Satija R., Reynolds G., Sarkizova S., Shekhar K., Fletcher J., Griesbeck M., Butler A., Zheng S., Lazo S., et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356:eaah4573. doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Mezlini A.M., Demir F., Fiume M., Tu Z., Brudno M., Haibe-Kains B., Goldenberg A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods. 2014;11:333–337. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- Wang H., Marcišauskas S., Sánchez B.J., Domenzain I., Hermansson D., Agren R., Nielsen J., Kerkhoven E.J. RAVEN 2.0: a versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018;14:e1006541. doi: 10.1371/journal.pcbi.1006541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer International Publishing; 2016. ggplot2. [Google Scholar]
- Wu D., Shu T., Yang X., Song J.X., Zhang M., Yao C., Liu W., Huang M., Yu Y., Yang Q., et al. Plasma metabolomic and lipidomic alterations associated with COVID-19. Natl. Sci. Rev. 2020;7:1157–1168. doi: 10.1093/nsr/nwaa086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaneske E., Zampieri G., Bertoldi L., Benvenuto G., Angione C. Genome-scale metabolic modelling of SARS-CoV-2 in cancer cells reveals an increased shift to glycolytic energy production. FEBS Lett. 2021;595:2350–2365. doi: 10.1002/1873-3468.14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Clippinger A.J., Alwine J.C. Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011;19:360–367. doi: 10.1016/j.tim.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei Ghobadi M., Mozhgani S.H., Farzanehpour M., Behzadian F. Identifying novel biomarkers of the pediatric influenza infection by weighted co-expression network analysis. Virol. J. 2019;16:124. doi: 10.1186/s12985-019-1231-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhang Y., Sun K., Meng Z., Chen L. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J. Mol. Cell Biol. 2019;11:1–13. doi: 10.1093/jmcb/mjy052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Y., Wang X.M., Xing X., Xu Z., Zhang C., Song J.W., Fan X., Xia P., Fu J.L., Wang S.Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- Zuo Y., Yalavarthi S., Shi H., Gockman K., Zuo M., Madison J.A., Blair C., Weber A., Barnes B.J., Egeblad M., et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:e138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data generated or analyzed during this study are included in this published article and/or the supplementary materials. Created datasets and code are publicly available. The metabolomics data can be obtained from the https://dx.doi.org/10.6084/m9.figshare.13336862. All the codes are available at https://dx.doi.org/10.5281/zenodo.6706499. Additional Supplemental Items are available from Mendeley Data at https://dx.doi.org/10.17632/5prg4tsz7n.1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.