

Abstract
PCR-based assays have become the benchmark for detecting pathogens of poultry and other livestock; however, these techniques are limited in their ability to detect multiple infecting agents, provide limited genetic information on the pathogen, and, for RNA viruses, must be reviewed frequently to assure high sensitivity and specificity. In contrast, untargeted, high-throughput sequencing can rapidly detect all infecting agents in a sample while providing genomic sequence information to allow more in-depth characterization of viruses. Although next-generation sequencing (NGS) offers many advantages, one of its primary limitations is low sensitivity to pathogens given the abundance of host and other non-target sequences in sequencing libraries. We explored methods for improving the sensitivity of NGS to detect respiratory and enteric viruses in poultry from RNA extracts of swab samples. We employed commercial and custom-designed negative enrichment strategies to selectively deplete the most abundant rRNA reads from the host and non-target bacteria; host RNA was diminished from up to 40% of total reads to as low as 3%, and the total number of reads assigned to abundant bacterial classes were reduced greatly. Our treatment resulted in up to a 700-fold increase in the number of viral reads, detection of a greater number of viral agents, and higher average genome coverage for pathogens. Depletion assays added only 2 h to the NGS library preparation workflow. Custom depletion probe design offered significant cost savings (US$7–12 per sample) compared to commercial kits (US$30–50 per sample).
Keywords: detection, next-generation sequencing, poultry, RNA virus
Poultry production is vital to the food supply and economy of developed countries and is a major contributor to the socioeconomic development of developing countries. One of the biggest threats to the global poultry industry is loss of animals and decreased egg production associated with infection by RNA viruses, including influenza A virus (IAV; Alphainfluenzavirus influenzae) and Newcastle disease virus (NDV; Avian orthoavulavirus 1), among others.2,21 For example, the 2014–2015 highly pathogenic avian IAV epidemic affecting U.S. poultry was estimated to have direct control costs of ~$950 million to eradicate (USDA 2016, https://www.ers.usda.gov/publications/pub-details/?pubid=86281). Accurate and efficient detection of infectious agents in poultry flocks is needed to inform decisions about biosecurity measures and control, as well as to properly target vaccination programs to maintain poultry health. However, the diversity of RNA viruses affecting poultry is large, and there are often coinfecting agents that can complicate disease diagnosis.3,4,23 Limitations associated with traditional techniques, such as reverse-transcription PCR, increase the likelihood that some viral infections in poultry go undetected or are not fully characterized.
PCR and serology-based assays have long been the benchmark for detecting respiratory and enteric viruses in poultry and other livestock.22,24 Although multiplex PCR assays are available for the detection of multiple pathogens, these methods usually require additional time and labor for proper validation.14,25 Therefore, although positive or negative results are obtained rapidly from conventional assays, these assays are limited in their ability to detect multiple infecting agents, must be frequently optimized for RNA viruses to maintain sensitivity and specificity (given the high mutation rate of RNA viruses), and provide limited or no genetic information on the pathogen. Additionally, detection of a viral pathogen using conventional approaches is often followed by virus isolation, serologic identification, and targeted molecular sequencing to fully characterize the isolate, including determining serotype, genotype, pathotype, and molecular epidemiologic analysis. 13 This follow-up is labor- and time-intensive and requires specialized laboratory and biosafety containment facilities. Thus, molecular-based tests continue to evolve to overcome the challenges associated with conventional testing.
Advances in next-generation sequencing (NGS) technologies have ushered in a new era of viral detection, and an ever-increasing number of studies employ untargeted NGS as a detection tool.6,7,18,19 In these methods, bulk RNA or DNA extracts are sequenced on high-throughput sequencing instruments, and genome sequences are evaluated to determine the evolutionary history, pathogenicity, and novelty of a particular infecting agent. In contrast to conventional methods, untargeted NGS can rapidly detect multiple infectious agents in a sample while also providing genomic sequence information. Although NGS offers many advantages over traditional tests, one of its primary limitations is low sensitivity to pathogens because of the abundance of host and other non-target sequences in libraries prepared from bulk RNA or DNA extracts.18,20 These non-target sequences often constitute 95–99% of final sequencing libraries, with the identity and contribution of these “background” sequences varying according to sample type. 20
Two major strategies have been employed to improve sensitivity of untargeted NGS for viral detection. One option is a positive selection strategy in which libraries already prepared from bulk extracts are enriched for target sequences. 15 This involves designing oligonucleotide baits complementary to sequences of interest. Many baits can be combined for detection of multiple pathogens, and, if enough baits are included, targeted or full genomic regions of interest can be selected. Similar to conventional PCR assays, the major disadvantage of this strategy is that it requires advance knowledge of the target sequence and is limited to detecting only known pathogens, although there is room to capture some sequence variation because of the “off-target” coverage inherent in these techniques. There is also a high initial cost associated with bait enrichment kits, and long hybridization times for baits (although improving) can add 24–36 h to library preparation workflows.
A second and perhaps more promising approach to increase sensitivity of NGS for pathogen detection is to deplete abundant, non-target sequences.9,27 For many sample types, a large portion of the bulk RNA or DNA extract is dominated by a few host sequences or non-target bacterial reads. To enrich extracts, biotinylated baits can be used to pull out non-target sequences, or standard oligonucleotide baits can be added to generate DNA-RNA hybrids that are selectively degraded enzymatically. These strategies are usually employed prior to library preparation and add only 1–2 h to total library preparation times. There are several readily available commercial kits for depletion of host or bacterial backgrounds using these or similar methods; however, the cost per sample to apply these kits usually doubles the cost of library preparation. These kits are also optimized for only a handful of animal model systems.
We explored the ability of untargeted NGS to detect respiratory and enteric RNA viruses in poultry from RNA extracts of swab and tissue samples. To decrease costs associated with sample enrichment, we developed a custom primer set to selectively deplete the most abundant host-specific rRNA reads (18S, 28S, mitochondrial) and bacteria (16S, 23S). We focused our comparison on differences in viral sequence yield from samples treated with our custom depletion assay, commercial enrichment kits, and in untreated controls. We tested a variety of sample types of clinical relevance for poultry, including oropharyngeal swabs (OPs) and cloacal swabs (CLs), and various tissues preserved on FTA cards.
Materials and methods
Samples
Poultry samples from Tanzania were collected in 2019 as part of ongoing surveillance for NDV and avian IAV in the country. OPs and CLs were taken from apparently healthy chickens in live-bird markets using sterile cotton-tipped plastic swab sticks. Following proper restraint of the bird, the mouth was opened, and a swab was carefully inserted in the choanal slit and swabbed repeatedly in circular motions. The swabs were immediately deposited into a sterile cryovial containing virus transport media (BBI brain heart infusion agar; Becton Dickinson) and were stored in a cool box before being stored in liquid nitrogen. A similar procedure was followed when collecting CLs.
Clinical samples were also collected from apparently healthy and diseased chickens from commercial farms in South America and Mexico in 2020. These samples were obtained from different flocks (breeders, broilers, layers) and included respiratory tissue samples (lung, trachea, choanae), immune tissue samples (spleen, bursa), and enteric samples (CLs). Briefly, from each flock or house, pools of 25 swabs per flock were spotted on each spotting area of a Whatman indicating classic FTA card (MilliporeSigma)—each card with 4 sample areas—translating into 100 swabs per card. After drying at room temperature (15–25°C) for ≥2 h, each FTA card was enclosed individually in a double leakproof zip-lock plastic bag with Whatman desiccant packets (GE Healthcare) and stored at −20°C.
Total RNA extraction and initial virus characterization
For Tanzania swab samples, total RNA was extracted from 50 μL of each CL or OP sample from each bird (MagMAX-96 AI/ND viral RNA isolation kit, Ambion; KingFisher magnetic particle processor, Thermo Fisher) without the addition of carrier RNA. RNA extract concentration was measured (Qubit fluorometer; Thermo Fisher) via an RNA high sensitivity kit. Samples were tested by reverse-transcription real-time PCR (RT-rtPCR) targeting the NDV large polymerase gene (L-test) using the forward primer L+12170 (5′-ACAGCTGGGAATCTCCAACA-3′), reverse primer L-12282 (5′-CTTTGAGAATCATTGGATATGTGA-3′), and probe L+12212 (5′-CAGATGACATTTACCCCTGCATCTCT-3′). The L-test RT-rtPCR was performed in 25-µL reaction volumes comprised of 8 µL of total RNA, 12.5 µL of 2× buffer, 0.5 µL of the forward and reverse primers (20 pmol/µL), 0.5 µL of the probe (6 pmol/µL), 1 µL of AgPath enzyme mix (Ambion), and sterile nuclease-free water. The test included an initial RT step (10 min at 45°C and 10 min 95°C) and PCR steps of 40 cycles (10 s at 95°C, 30 s at 57°C, and 10 s at 72°C). All RT-rtPCR tests were performed on a 7500 fast real-time PCR system (Applied Biosystems). NDV LaSota strain and non-template (nuclease-free water) were used as positive and negative controls, respectively. All samples and controls were run in duplicate. Samples with cycle threshold (Ct) values of <40 were considered as potential positives and recorded.
For the South America–Mexico FTA card samples, 24 three-mm discs were punched out from each FTA card (6 discs per spotted area) using disposable biopsy punches (Robbins Instruments) and incubated in TE buffer (10 mM Tris-HCl, 0.1 mM EDTA; pH 8.0) for 25 min at room temperature to elute nucleic acids. Total RNA was extracted from the TE eluate using the MagMAX protocol as described above. RNA extract concentration was measured on a Qubit fluorometer via an RNA high sensitivity kit. The RNAs from the FTA samples were not subjected to PCR screening prior to sequencing because these samples were part of a shotgun sequencing workflow only.
Methods for host and bacterial depletion
To remove abundant non-target host and bacterial sequences, aliquots of RNA extracts from the samples described above were pretreated with 4 different enrichments (Fig. 1). These enrichments included 2 sets of custom-designed probes in an RNase H–based depletion protocol, as well as 2 commercial kits: 1) the NEB mammalian rRNA depletion kit (designated as MAMribo; NEBNext rRNA depletion kit, human/mouse/rat, New England Biolabs), and 2) a chicken-specific host rRNA removal kit (designated as CXribo; riboPOOL kit, siTOOLS Biotech). Untreated controls for each sample were also included. RNA aliquots for the MAMribo depletion kit were treated according to a standard protocol (Protocol for use with NEBNext rRNA depletion kit (human/mouse/rat) (E6310, E6350), https://www.neb.com/protocols/2017/04/06/nebnext-rrna-depletion-kit-human-mouse-rat-with-rna-sample-purification-beads; New England Biolabs). Briefly, 12-µL aliquots of RNA (2–10 ng/µL) were hybridized with DNA probes by incubating at 95°C for 5 min, cooling gradually to 22°C, and incubated for an additional 5 min at 22°C. RNA-DNA hybrids were degraded by incubating with RNase H at 37°C for 30 min, and DNase-I was used in another 30-min digestion to remove excess DNA probe. RNA was purified (AMPure RNAClean XP beads; Beckman Coulter) at 2.2× volume.
Figure 1.

Overview of methods and RNA pretreatments applied to different samples.
Aliquots of RNA (14 µL; 2–10 ng/µL) for the CXribo treatment were incubated with biotinylated DNA probes complementary to chicken 18S, 23S, and mitochondrial rRNA at 68°C for 10 min and gradually cooled to 37°C. Magnetic beads were used to separate and discard probe-bound RNA, and a final purification step using AMPure RNAClean XP beads at 2.2× volume was carried out.
We also designed a set of custom DNA primers specific to our chicken host and from abundant bacterial reads in previously sequenced swab samples from Tanzania (Suppl. Table 1). Chicken probes included sequences for host 18S, 28S, and mitochondrial rRNA, and were tiled in non-overlapping sequences of ~120 bp covering the entire target sequence. For the bacterial sequences, both conserved and group-specific bacterial 16S and 23S primers were developed. Conserved primers were based on highly expressed regions of Escherichia coli 16S/23S (mapped from prior runs) that were outside the hypervariable regions of those genes. For group-specific bacterial primers, we identified commonly detected bacteria in earlier non-depleted NGS runs, created multiple sequence alignments by phylum, and a consensus sequence was generated (Lasergene; DNAstar). From the consensus sequence, non-overlapping probes of ~120 bp based on the reverse complement of ribosomal sequences were selected and synthesized (Ultramer DNA oligos; IDT) from conserved regions. No probes were targeted to the V3-V4 highly variable region, and a portion of this 16s rRNA region was purposely avoided to allow a fragment large enough (~400 bp) to still identify abundant bacteria. Combinations of these primers within an RNase H–based depletion step (Suppl. Table 2) were applied to RNA extracts of 2–10 ng/µL to remove non-target rRNA. Custom A treatment included all host primers and the conserved bacterial primers. Custom B treatment included all of the primers in custom A and group-specific primers for Pseudomonadota (formerly Proteobacteria), Bacteroides, Firmicutes, Acinetobacter, and Actinobacteria. Custom A and B treatments were applied to different subsets of samples and compared to untreated controls (Fig. 1). To further gauge the effectiveness of our custom depletion assay, we compared RNA extracts from samples treated with our custom A probe set to those treated with the commercial kits described above.
Sequencing and analysis
Treated and untreated RNA extracts from each sample were amplified via sequence-independent, single-primer amplification (SISPA) as described previously. 6 First-strand cDNA was synthesized in a 20-μL reaction mixture with 5 μL of viral nucleic acids from each sample, 100 pmol of primer K-8N (GACCATCTAGCGACCTCCACNNNNNNNN), SuperScript IV reverse transcriptase (Thermo Fisher), and dNTPs (10 μmol) following the manufacturer’s instructions. To convert the first-strand cDNA into double-stranded cDNA (dsDNA), 20 μL of the first-strand cDNA was heated to 95°C for 3 min and then cooled to 4°C in the presence of 10 pmol of primer K-8N, and 10 μmol dNTPs in 1× Klenow reaction buffer (NEB). Afterwards, 1 µL of Klenow fragment were added and incubated at 37°C for 60 min (final volume, 25 μL). After conversion into dsDNA by Klenow polymerase (NEB), the products were purified (Agencourt AMPure XP DNA beads; Beckman Coulter). The purified dsDNA of the Klenow reaction was subsequently used as a template for PCR amplification. Sequence-independent PCR amplification was conducted with 5 μL of the double-stranded cDNA template in a final reaction volume of 50 μL, which contained 1× Phusion HF buffer, 200 μM deoxynucleoside triphosphate (dNTP), 10 μM primer K (GACCATCTAGCGACCTCCAC), and 0.5 U Phusion DNA polymerase (NEB). The PCR cycling was performed as follows: 98°C for 30 s, followed by 35 cycles of 98°C for 10 s, 55°C for 30 s, and 72°C for 1 min, with a final extension at 72°C for 10 min. PCR products were purified using Agencourt AMPure XP DNA beads. For quantification of the ds cDNA, the Qubit dsDNA HS assay (Invitrogen) was performed according to the manufacturer’s instructions.
Sequencing libraries were prepared from SISPA PCR products (Nextera Flex kit; Illumina); paired-end sequencing (2 × 250 bp) of the pooled libraries was performed (MiSeq platform, 500-cycle MiSeq reagent kit v.2; Illumina) according to the manufacturer’s instructions. Pre-processing of the raw sequencing data was completed within the Galaxy platform1,8 using publicly available tools (https://usegalaxy.org), and fastq sequences were deposited in GenBank under Bioproject PRJNA773734. Raw sequence reads were quality assessed using FASTQC, and residual adaptor sequence and low-quality bases were trimmed using Cutadapt. 17 Forward and reverse reads were merged using PEAR v.0.9.6.0 (https://cme.h-its.org/exelixis/web/software/pear/), and individual read counts were normalized using the “normalize by khmer abundance” script within Galaxy. Normalized reads were de novo assembled with MIRA v.3.4.1. 5 De novo contigs were used to identify virus presence via BLASTn (https://blast.ncbi.nlm.nih.gov/Blast.cgi) of contigs, and raw read mapping with the Burrows–Wheeler alignment tool (BWA-MEM) 16 was employed to enumerate the viral agents present and to quantify the contribution of host reads in each treatment. After quantifying host and viral reads, a paired t-test was used to evaluate statistical differences in host/viral read relative abundance between the different pretreatments. A metagenomic approach was used to confirm that custom B treatment reduced relative abundance of the bacterial phyla targeted for depletion. For this meta-analysis, non-host reads were assigned a taxonomic label and tallied using Kraken 2 26 within the Galaxy platform. The custom python script, “combine_kreports.py”, from Krakentools was used to merge/average taxonomy tables for treatment replicates.
Results
We compared 20 OPs and 5 CLs from Tanzania using the MAMribo and CXribo depletion kits, our custom A primer set (Fig. 1; Suppl. Table 1; Table 1), and by sequencing untreated libraries. Our full complement of custom depletion primers (custom B, Suppl. Table 1) was used to selectively deplete host and bacterial rRNA from 28 samples of various tissue types from Mexico and South America and 14 CLs from Tanzania (Fig. 1). Compared to untreated controls, rRNA depletion significantly reduced host reads from up to 15% of total reads to <3% with custom A treatment, MAMribo, and CXribo kits (Fig. 2; paired t-test, p < 0.05). This level of host reduction is comparable to those observed in other host systems using similar methods with fold enrichment from 5–10× following removal of host and carrier RNA via RNase H–based protocols.9,11,12,27
Table 1.
Viral enrichment method comparison, limited to Tanzania swab samples for which all treatments were tested. Fold enrichment is relative to untreated controls.
| Pretreatment strategy | Cost per sample, US$ | Additional time, h | Average % total reads mapped to host | Fold enrichment in viral reads |
|---|---|---|---|---|
| MAMribo depletion | 45 | 1–2 | 0.8 | 10.6× |
| CXribo depletion | 33 | 1–2 | 1.2 | None |
| Custom A probe set | 7 | 1–2 | 2.8 | 66× |
| Custom B probe set | 12 | 1–2 | 37.8 | 106× |
CXribo = riboPOOL kit; siTOOLS biotech; MAMribo = NEBNext rRNA depletion kit, human/mouse/rat; New England Biolabs.
Figure 2.
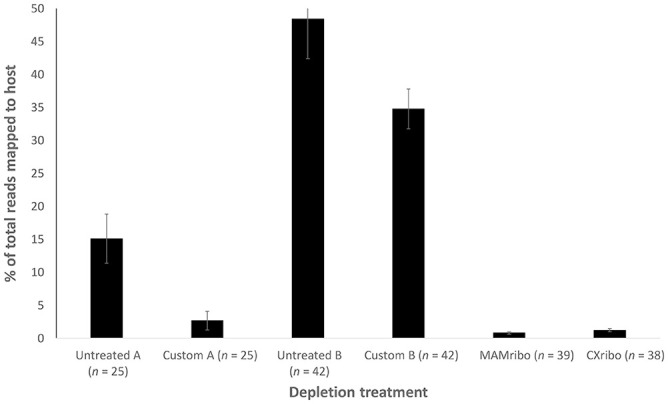
Average percentage of total reads mapping to host in each depletion treatment. All treatments significantly reduced host reads relative to untreated controls (t-test, p < 0.01). Error bars are ±SE. Untreated A and custom A are comparing 20 oral and 5 cloacal swabs. Untreated B and custom B include 14 cloacal swabs and 28 tissue samples. MAMribo (NEBNext rRNA depletion kit, human/mouse/rat; New England Biolabs) and CXribo (riboPOOL kit; siTOOLS Biotech) were tested on all 20 oral and 19 cloacal swabs (1 CXribo-treated sample did not have sufficient read depth for comparison). See also data presented in Table 1.
Sensitivity to virus was dramatically improved using the custom probe sets compared to untreated controls and both commercial kits. This was true for detection of NDV, which predominated in OPs and CLs from Tanzania, infectious bronchitis virus (Avian coronavirus), and picornaviruses, which were abundant in choanal or lung tissues, and infectious bursal disease virus, which was present in some bursal tissues (Suppl. Table 3). Improvement in sensitivity was reflected in viral richness and diversity across samples and in the number of near-complete viral genome sequences recovered (Suppl. Table 3). There were 9 instances in which no virus was detected in untreated samples, but significant viral reads were obtained via custom depletion (Suppl. Table 3). Although detection ability was similar between the custom approach and MAMribo kit, the custom A primer combination increased normalized virus read count up to 10× more than the MAMribo and 70× more than what was observed in untreated swab samples and CXribo-treated aliquots (Fig. 3A). Inclusion of additional bacterial probes (custom B) enhanced virus yield further, up to 100× in swabs (Fig. 3A) and 700-fold in tissue samples (Fig. 3B). This resulted in the recovery of near-complete (>80%) genomes for 33 viral pathogens from 31 different samples (Suppl. Table 3). Only a single MAMribo-treated sample yielded a near complete genome for NDV; no near-complete genomes were obtained from CXribo-treated and untreated samples (Suppl. Table 3).
Figure 3.
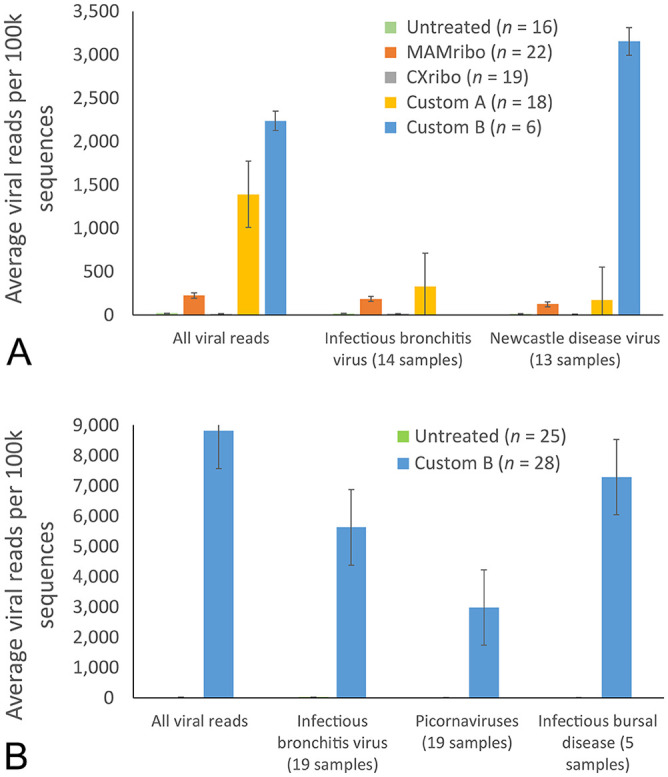
A. Average number of viral reads per 100k sequences for Tanzania oral and cloacal swabs. Virus sensitivity was significantly higher in custom treatments compared to untreated controls and treatment with MAMribo (NEBNext rRNA depletion kit, human/mouse/rat; New England Biolabs) and CXribo (riboPOOL kit; siTOOLS Biotech). B. Average number of viral reads per 100k sequences for Mexico–Peru–Guatemala tissue samples preserved on FTA cards. Untreated controls are plotted but not visible because average virus read count per 100k sequences was <20. Custom treatment increased virus sensitivity up to 700-fold compared to untreated controls. Sample sizes in parentheses do not represent total number of samples evaluated, only those with virus present.
For swab samples with corresponding Ct values, depletion lowered the threshold for detection of NDV. In untreated samples, Ct values of 21–22 were required to detect NDV via NGS, whereas Ct values of 26–27 yielded a significant number of sequence reads in custom A and MAMribo-treated libraries. Custom A treatment showed the highest sensitivity across any of the given Ct values (Fig. 4). There were only 4 custom B–treated samples with corresponding Ct values (data not plotted on Fig. 4). The lowest value was 17.5 for sample SM71, and the NDV read count from this sample was 19,013 per 100k. Highest observed Ct was 21.5 from sample M573, and the read count for NDV was 1,327 per 100k.
Figure 4.
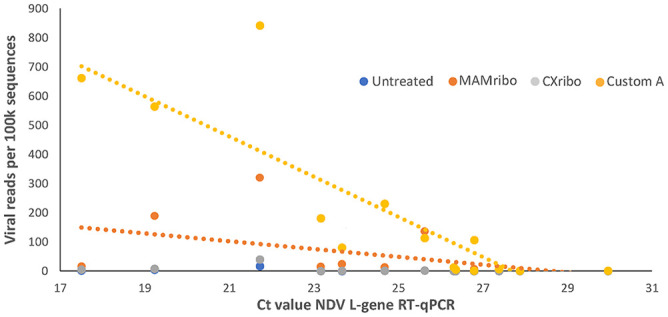
Relationship between Newcastle disease virus (NDV) L-test gene RT-qPCR Ct value and viral read yield under different RNA depletion treatments. Shown are oral and/or cloacal samples from Tanzania for which there were viral reads present and measurable Ct values (n = 14). Ct of 26–27 was the threshold for obtaining significant reads, and custom depletion yielded the greatest number of viral reads at a given Ct value. Dotted lines are lines of best fit for data points. MAMribo = NEBNext rRNA depletion kit, human/mouse/rat, New England Biolabs; Cxribo = riboPOOL kit, siTOOLS Biotech.
Meta-omic analysis of non-host reads showed that the custom B primer mix was effective in depleting target sequences. Gammaproteobacteria made up 40% and 35% of the total reads in untreated CLs and tissues, respectively (Fig. 5). Depletion reduced Gammaproteobacteria to 28% of total reads in CLs and 20% in tissues. The Clostridia (Firmicutes) in both untreated tissues and CLs were almost completely removed in treated samples. There was up to a 50% reduction in Bacteroidia and Epsilonproteobacteria in CLs following custom treatment. Similarly, treatment reduced the signal of Bacilli and Betaproteobacteria in tissues by half. Viral reads were 16% of the total community in treated tissues and an average of 4% of the total community in treated CLs. Viral reads did not make up a significant percentage of the community in untreated samples. Overall, bacterial diversity was higher in treated samples, with a greater number of bacterial classes contributing <1% of the total community. Thus, at a given sequence depth, the custom probe set allowed detection of a greater number of bacterial taxa.
Figure 5.

Average percentage of total non-host reads contributed by each bacterial or viral class. Only classes with >1% average contribution to the total community are shown. Viral classes are highlighted in red boxes.
Discussion
Our efficient, low-cost method improved the sensitivity of NGS to detect respiratory and enteric viruses in poultry from RNA extracts of swab samples. Although the initial cost of purchasing probes is significant, the price per reaction for the custom A probes in combination with the RNase H protocol was only $7 per reaction compared to $45 per reaction with the MAMribo kit and $33 per reaction with the CXribo kit. Treatment with additional probes for specific bacterial groups (custom B, Suppl. Table 1; Table 1) added ~$5 per reaction and reduced host reads from up to 48% of total reads in corresponding untreated controls to ~35% of total reads. The apparent lower efficiency of custom B treatment in reducing host reads is likely the result of the amount of host starting material (higher in tissues) and the potential for additional bacterial probes to increase the relative contribution of host reads in the final library; however, application of custom A and B probes on the same sample set would be needed to verify this hypothesis.
Sensitivity to virus was greatly improved utilizing our custom approach as evidenced by the relationship between Ct values and the total viral reads obtained. We obtained 33 near-complete viral genomes across 31 samples using our custom approach, whereas no near-complete genomes were recovered in untreated samples, and only a single MAMribo-treated sample yielded a near-complete viral genome. For samples with corresponding Ct data, significant viral reads were obtained at corresponding Ct values between 26–27, and custom B treatment showed potential to further reduce the threshold of viral detection (more data points are needed to confirm this trend). Prior studies suggest that a Ct of 32 is the approximate cutoff for detection of virus via NGS, and significant viral reads are only obtained at Ct values of 20–25, depending on sample type. 10 This detection ability will vary by sequencing effort or depth, and our results demonstrate that custom depletion is likely to improve detection and pathotyping ability at any sequence depth.
Although another custom RNase H–based depletion protocol has been developed for avian species, 11 we validated our method on a variety of sample types of clinical relevance for poultry. We also focused our comparison on how enrichment strategies affect viral detection and viral read enrichment, whereas prior studies typically focused on efficiency of removal of the non-target RNA as a validation strategy. A virus-focused validation more clearly demonstrates the utility in our method for detecting and recovering genomes for RNA viruses. Finally, our custom method is one of the first negative enrichment strategies to include probes for both host and abundant non-target bacteria, showing that the inclusion of additional probes can greatly improve virus recovery.
Supplementary Material
Acknowledgments
We thank Jesse Gallagher, Dawn Williams-Coplin, Ricky Zoller, Edna Espinoza, Peter Msoffe, and Gaspar H. Chiwanga for technical assistance with this work.
Footnotes
Declaration of conflicting interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: Our research was supported by USDA-NIFA Coordinated Agricultural Project 2015-68004-23131 and by an appointment to the Intelligence Community Postdoctoral Research Fellowship Program at Southeast Poultry Research Laboratory administered by Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy and the Office of the Director of National Intelligence (ODNI).
Supplemental material: Supplemental material for this article is available online.
ORCID iDs: Joshua Parris  https://orcid.org/0000-0001-7040-5482
https://orcid.org/0000-0001-7040-5482
Henry Kariithi
https://orcid.org/0000-0002-9250-9108
References
- 1. Afgan E, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acid Res 2016;44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bagust TJ. Poultry health and disease control in developing countries. In: Poultry Development Review. FAO, 2013:95–100. http://www.fao.org/3/a-i3531e.pdf
- 3. Barzon L, et al. Next-generation sequencing technologies in diagnostic virology. J Clin Virol 2013;58:346–350. [DOI] [PubMed] [Google Scholar]
- 4. Boonham N, et al. Methods in virus diagnostics: from ELISA to next generation sequencing. Virus Res 2014;186:20–31. [DOI] [PubMed] [Google Scholar]
- 5. Chevreux B, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 2004;14:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chrzastek K, et al. Use of sequence-independent, single-primer-amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017;509:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De la Torre DI, et al. Enteric virus diversity examined by molecular methods in Brazilian poultry flocks. Vet Sci 2018;5:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dimitrov KM, et al. A robust and cost-effective approach to sequence and analyze complete genomes of small RNA viruses. Virol J 2017;14:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fauver JR, et al. A reverse-transcription/RNase H based protocol for depletion of mosquito ribosomal RNA facilitates viral intrahost evolution analysis, transcriptomics and pathogen discovery. Virology 2019;528:181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fischer N, et al. Evaluation of unbiased next-generation sequencing of RNA (RNA-seq) as a diagnostic method in influenza virus-positive respiratory samples. J Clin Microbiol 2015;53:2238–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gu H, et al. Novel rRNA-depletion methods for total RNA sequencing and ribosome profiling developed for avian species. Poult Sci 2021;100:101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hasan MR, et al. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J Clin Microbiol 2016;54:919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kapczynski DR, et al. Characterization of the 2012 highly pathogenic avian influenza H7N3 virus isolated from poultry in an outbreak in Mexico: pathobiology and vaccine protection. J Virol 2013;87:9086–9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim LM, et al. Detection of a broad range of class I and II Newcastle disease viruses using a multiplex real-time reverse transcription polymerase chain reaction assay. J Vet Diagn Invest 2008;20:414–425. [DOI] [PubMed] [Google Scholar]
- 15. Lee JS, et al. Targeted enrichment for pathogen detection and characterization in three felid species. J Clin Microbiol 2017;55:1658–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv 1303.3997, 2013. [Google Scholar]
- 17. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011;17:10–12. [Google Scholar]
- 18. Matranga CB, et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol 2014;15:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matranga CB, et al. Unbiased deep sequencing of RNA viruses from clinical samples. J Vis Exp 2016;113:54117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O’Flaherty BM, et al. Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res 2018;28:869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rehan MA, et al. Potential economic impact of Newcastle disease virus isolated from wild birds on commercial poultry industry of Pakistan: a review. Hosts Viruses 2019;6:1–15. [Google Scholar]
- 22. Richt JA, et al. Real-time reverse transcription-polymerase chain reaction assays for the detection and differentiation of North American swine influenza viruses. J Vet Diagn Invest 2004;16:367–373. [DOI] [PubMed] [Google Scholar]
- 23. Seifi S, et al. Natural co-infection caused by avian influenza H9 subtype and infectious bronchitis viruses in broiler chicken farms. Vet Arh 2010;80:269–281. [Google Scholar]
- 24. Spackman E, et al. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol 2002;40:3256–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spackman E, et al. Development of multiplex real-time RT-PCR as a diagnostic tool for avian influenza. Avian Dis 2003;47(Suppl 3):1087–1090. [DOI] [PubMed] [Google Scholar]
- 26. Wood DE, et al. Improved metagenomic analysis with Kraken 2. Genome Biol 2019;20:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeng Z, et al. A reverse transcriptase-mediated ribosomal RNA depletion (RTR2D) strategy for the cost-effective construction of RNA sequencing libraries. J Adv Res 2020;24:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.