

Abstract
Background:
Allergic contact dermatitis (CD) is a chronic inflammatory skin disease caused by type-1 biased adaptive immunity for which there is an unmet need for antigen (Ag)-specific immunotherapies. Exposure to skin sensitizers stimulates secretion of the proinflammatory neuropeptides substance P (SP) and hemokinin 1 (HK1), which signal via the neurokinin-1 receptor (NK1R) to promote the innate and adaptive immune responses of CD. Accordingly, mice lacking the NK1R develop impaired CD. Nonetheless, the role and therapeutic opportunities of targeting the NK1R in CD remain to be elucidated.
Objective:
To develop an Ag-specific immunosuppressive approach to treat CD by skin co-delivery of hapten and NK1R antagonists integrated in dissolvable microneedle arrays (MNA).
Methods:
In vivo mouse models of contact hypersensitivity and ex-vivo models of human skin were used to delineate the effects and mechanisms of NK1R-signaling and the immunosuppressive effects of the contact sensitizer-NK1R-antagonists-MNA in CD.
Results:
We demonstrate in mice that CD requires NK1R signaling by SP and HK1. Specific deletion of the NK1R in keratinocytes and dendritic cells, but not in mast cells prevented CD. Skin co-delivery of hapten- or Ag-NK1R-antagonist-MNA inhibited neuropeptide-mediated skin inflammation in mouse and human skin, promoted deletion of Ag-specific effector T cells and increased regulatory T cells, which prevented CD onset and relapses locally and systemically in an Ag-specific manner.
Conclusions:
Our findings demonstrate that immune-regulation by engineering localized skin neuroimmune networks can be used to treat cutaneous diseases that like CD, are caused by type-1 immunity.
Keywords: Contact dermatitis, contact hypersensitivity, microneedle arrays, keratinocytes, neuropeptides, neurokinin-1 receptor, neurokinin-1 receptor antagonists, hapten, immunosuppressive therapies
Graphical Abstract
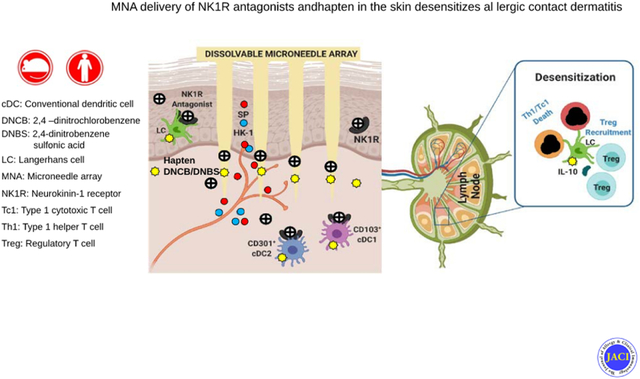
CAPSULE SUMMARY
We developed an immunosuppressive approach for the prevention and treatment of allergic contact dermatitis by skin co-delivery of hapten or protein Ag and neurokinin-1 receptor antagonists integrated in dissolvable microneedle arrays.
INTRODUCTION
Allergic contact dermatitis (CD) is a common chronic inflammatory skin disease caused by exposure to contact sensitizers and sustained by Ag-specific T cells1. Due to an increased exposure to skin sensitizers in domestic and working environments, the incidence of CD has escalated across age and gender differences and is currently the second highest cause of work-related diseases in the USA. Recent studies have shown that the prevalence of CD is up to 18% and affects 10.6 % of the working population1, 2. The management of CD includes preventing exposure to skin sensitizers and administration of corticosteroids or biologics1, 3. Current immunotherapies to treat severe CD are administered systemically and lack antigen (Ag)-specificity, which could result in generalized immunosuppression. Hence, there is an unmet need for Ag-specific treatments that target the physiopathology of CD.
Neuroinflammatory responses have a relevant role in the physiopathology of CD. Skin exposure to hapten or Ag initiates secretion of substance P (SP) and hemokinin 1 (HK1), which are proinflammatory neuropeptides that bind with high affinity to the neurokinin 1 receptor (NK1R), a G protein-coupled receptor that recruits Gαq/11 protein subunits to activate intracellular signaling that supports innate and adaptive immunity4–9. As a result, NK1R signaling increases the severity of chronic inflammatory disorders that, like CD, are mediated by CD4 T helper 1 (Th1) and CD8 T cytotoxic (Tc1) cells. Accordingly, mice lacking the NK1R develop impaired CD4, 6, 7, 10–13.
We hypothesized that inhibition of the proinflammatory function of SP and HK1 by NK1R antagonists, delivered simultaneously with contact sensitizers into a localized area of the skin, would enable a physiopathology-based and Ag-specific immunosuppressive approach to treat type-1 biased CD. To be feasible, this strategy requires further understanding of the role of the NK1R in skin-initiated type-1 immunity and an efficient and reliable system to co-deliver NK1R antagonists and hapten or Ag to the cutaneous microenvironment.
Recent studies have shown that besides signaling via the NK1R, SP and HK1 bind Mas-related G protein-coupled receptors (Mrgpr). Activation of mouse MrgprB2 or human MRGPRX2 by SP in sensory neurons and mast cells (MC) promotes itching, inflammatory pain and adaptive immunity14, 15. These observations challenged our current understanding on the biology of the NK1R and its endogenous ligands in the innate and adaptive immunity of CD, areas of research that require in vivo models in which the NK1R can be deleted in a cell type-specific fashion.
Here we demonstrate that type-1 biased skin contact hypersensitivity (CHS) requires simultaneous NK1R signaling by SP and HK1 but does not require signaling the MrgprB2. We used cell-type specific deletion of the NK1R in mice to identify the skin cells that could be targets of NK1R agonists, including keratinocytes, conventional dendritic cells (cDC) and MC, and we described their downstream regulatory effects on the T cell response that causes CHS. Based on these data, we developed an immunosuppressive approach that utilizes dissolvable multicomponent microneedles arrays (MNA) to co-deliver NK1R antagonists and hapten or Ag to a circumscribed area of the skin. These MNA are delivery platforms that incorporate biologics in a biodegradable matrix. When the microneedles penetrate the skin they dissolve and release their cargo with anatomic precision and dosage control16–19. In mice, we found that co-delivery of NK1R antagonists and hapten or Ag by MNA prevented the development of CD and desensitized the already established disease, locally and systemically in Ag-specific fashion. Importantly, the translational aspects of our results in mice were validated in ex-vivo models of human skin.
METHODS
Supplemental information can be found in the Methods section of the Online Repository.
Study Approval
Mice
Male or female mice (7–12 week old) were randomly selected for the experiments in accordance with the National Institutes of Health scientific rigor policy. Mice were maintained in the pathogen-free animal facility of the University of Pittsburgh School of Medicine that provides with around the clock husbandry and veterinary services. Animal care and handling were performed in accordance with institutional guidelines and the procedures approved by IACUC protocol number 19014279.
Human tissue and blood samples
Human skin samples from cosmetic plastic surgeries were provided by the Health Science Tissue Bank, from the Department of Pathology of the University of Pittsburgh, School of Medicine. Skin samples procured anonymously and without identifiers were used according to Institutional Review Board (IRB) outlines (IRB approval number 0501138). Human blood samples from healthy donors were obtained anonymously and without identifiers from the Central Blood Bank (University of Pittsburgh, Pittsburgh, PA. USA) and used according to IRB guidelines (IRB approval number 0602095). Both human skin and blood samples are classified as non-human subjects.
Induction and assessment of cutaneous CHS
For induction of CHS by topical application of sensitizers, mice were treated once with either 2,4-dinitrochlorobenzene (DNCB) (1% in 4:1 v/v acetone / olive oil), oxazolone (1% in 4:1 v/v acetone / olive oil) (both from Sigma-Aldrich) or vehicle, on the shaved abdominal skin. Topical application of 100 μg of DNCB was used in comparative studies with DNBS-MNA. When indicated, the NK1R antagonists L733060 (10 nmols) or RP67580 (10 nmols), or the NK1R agonists (SarSP) (10−9 M), SP (10−9 M) or HK1 (10−8 M) (all from TOCRIS), were injected i.d. immediately before topical application of the sensitizer. Five days after sensitization, mice were challenged with DNCB (0.5% in 4:1 v/v acetone / olive oil) or with oxazolone (0.5% in 4:1 v/v acetone / olive oil) on the dorsal side of the right ear, and with vehicle on the left ear (control). Ear thickness increase was measured before and after treatment using a digital mini-micrometer (Mitutoyo). Ear thickness was assessed 24 h following sensitization to evaluate the innate immune response and 24, 48 and 72 h after elicitation to evaluate the adaptive immune response. In some experiments, ear thickness was analyzed up to 120 h following elicitation. Values are expressed as percentage in ear thickness increase obtained by the formula: (thickness of treated ear – thickness of untreated ear) / (thickness of untreated ear) × 100. As negative controls, mice were treated with vehicle and challenged with DNCB (0.5%).
For induction of CHS with MNA, mice were sensitized on the right ear by application of one MNA loaded with DNBS (50 μg) (Sigma-Aldrich) or OVA (100 μg) (Sigma-Aldrich) alone or with the NK1R antagonists RP67480 (10 nmols) or L733060 (10 nmols) (TOCRIS). Elicitation was done by topical application of DNCB (0.5% in 4:1 v/v acetone / olive oil) or OVA injected i.d (50 μg in 50 μl of PBS per injection site), 5 days after sensitization. Mice untreated, sensitized with empty-MNA, and then elicited with topical DNCB, were included as controls.
For induction of CHS in hu-Lang-DTR mice, depletion of LC was achieved by administration of diphtheria toxin (500 ng, i.p., once), 72 h before sensitization20. Controls included WT mice and hu-Lang-DTR mice not exposed to diphtheria toxin. LC depletion was confirmed by fluorescence microscopy on epidermal sheets dissected from the ear skin, fixed in cold acetone, and stained with goat-anti human CD207 Ab (R&D Systems) and biotin-IAb Ab, followed by Cy3-conjugated anti-goat IgG and Cy2-streptavidin (Jackson ImmunoResearch).
Statistics
Statistical analysis was performed using the GraphPad Prism v8 software (San Diego, CA). Results are expressed as mean ± 1SD. Comparisons of two means were done by two-tailed Student’s t-test. Comparison of multiple means on a single data set was done by 1-way ANOVA followed by ad hoc comparison between means by Student-Newman-Keuls test. A “p” value < 0.05 was considered significant.
RESULTS
Efficient CHS requires NK1R, SP and HK1
The role of the NK1R and its natural agonists, the neuropeptides SP and HK1, in adaptive cellular immunity of CD was investigated in a model of CHS to DNCB in wild type (WT), NK1RKO, Tac1KO (SP-deficient), Tac4KO (HK1-deficient) and Tac1/Tac4KO and MrgprB2KO mice. DNCB was chosen because is a potent hapten that induces CHS in mice and has the potential to sensitize humans21.
Mice were sensitized with topical DNCB on the skin (abdomen) and elicited 5 days later on the dorsal skin of the right ear. Compared to WT, NK1RKO mice showed significantly reduced CHS as determined by lower ear thickness increase and less mononuclear leukocyte infiltrate at the elicitation site (Fig 1A and B and Fig E 1A and B). To rule out the possibility that low secretion of SP or HK1 in a global NK1RKO mice was causing deficient CHS22, we induced CHS in NK1RKO mice injected i.d. with the agonist [Sar9Met(O2)11] SP (SarSP) during sensitization23. In these conditions, NK1RKO mice did not show a significant increase in ear thickness or the histological changes of CHS compared to NK1RKO mice sensitized without SarSP (Fig 1A and B and Fig E 1A and B). The CHS response in NK1RKO mice remained minimal up to 5 days after elicitation, indicating that absence of NK1R prevents, instead of delaying CHS (Fig 1A). Comparative studies between MrgprB2KO and WT mice showed that lack of MrgprB2 does not decrease the CHS to DNCB (Fig E 1C)
Fig 1. Efficient CHS requires NK1R, SP and HK1.
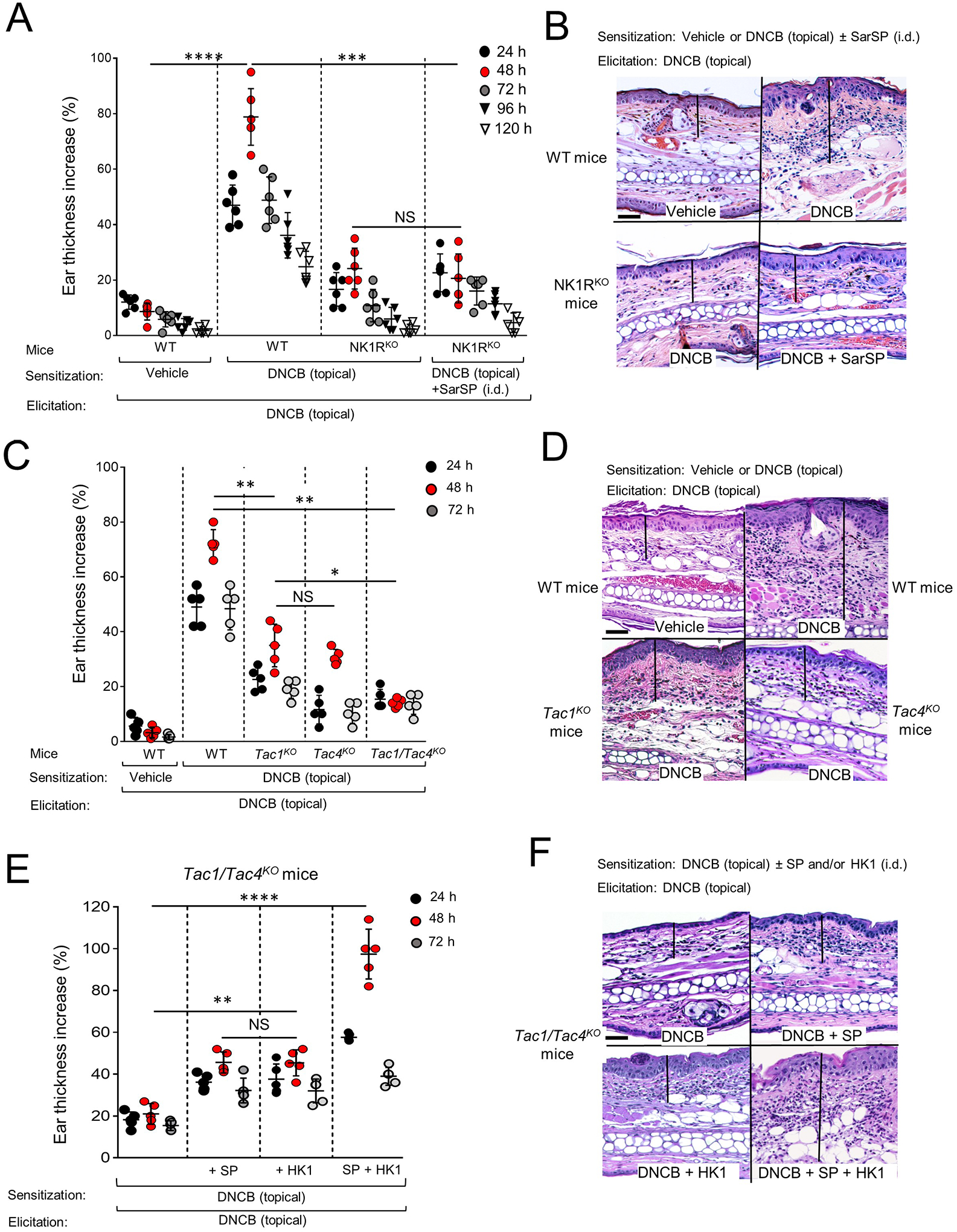
A) Skin CHS to DNCB by ear thickness increase in WT and NK1RKO B6 mice. B) Microscopic images of the ears of mice in (A) showing epidermal-dermal thickness (lines), hyperkeratosis, parakeratosis, acanthosis and leukocyte inflammatory infiltrate. C) Skin CHS to DNCB, in WT, Tac1KO, Tac4KO and Tac1/Tac4KO B6 mice. D) Representative microscopic images of mouse ears in (C). E) CHS to DNCB in Tac1/Tac4KO B6 mice injected i.d. with SP or HK1, or SP and HK1 during sensitization. F) Microscopic images of the ears of mice in (E) showing epidermal-dermal thickness (lines) and leukocyte infiltrate. In (A), (C) and (E), representative experiment of 3. (A) Six mice per group, (B) and (C) 5 mice per group. Means ± 1 SD analyzed by 1-way ANOVA followed by Student-Newman-Keuls test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. NS: not significant. Statistic comparisons among values obtained 48 h following ear skin elicitation are depicted. In (B), (D) and (F), images are from samples obtained 48 h after elicitation. H&E, X200, scale bars, 20μm.
Similar CHS experiments in Tac1KO (SP-deficient), Tac4KO (HK1-deficient) or Tac1/Tac4KO mice demonstrated that lack of either SP or HK1 reduces significantly CHS to DNCB (Fig 1C and D and Fig E 2A and B). The capability of Tac1/Tac4KO mice to develop CHS to DNCB was partially restored when either SP or HK1 was injected i.d. at the sensitization site immediately before topical application of DNCB, and the CHS response was fully restored following co-administration of both neuropeptides (Fig 1E and F and Fig E 2C and D). Thus, skin CHS to DNCB requires signaling of the NK1R by SP and HK1.
Skin co-delivery of hapten and NK1R antagonists integrated in MNA abrogates CHS
Development of an immunosuppressive approach based on skin delivery of hapten and NK1R antagonists to prevent the neuroinflammation triggered by hapten penetration through the skin, requires a system that administers simultaneously reliable doses of the biologics to a circumscribed skin area eliciting minimal or no inflammation, properties that are characteristic of MNA16 (Fig 2A). When the MNA is applied to the skin, the microneedles dissolve within 5–10 min (Fig 2B). Release of the MNA cargo in the skin layers was analyzed using MNA tip-loaded with the fluorochromes Alexa Fluor-488 and Alexa Fluor-555 as surrogates for the NK1R antagonist and the hapten applied to the ears of WT mice. Live-animal imaging by fluorescence microscopy revealed that the MNA delivers both cargos simultaneously and efficiently throughout the skin layers of mouse ears (Fig 2C and D). Following, we tested if the MNA substrate per se was proinflammatory and if the hapten and NK1R antagonists co-delivered by MNA were functional. We compared in a dose response experiment, the cutaneous CHS induced by sensitization with topical DNCB (100 μg) to that induced by MNA loaded with 25, 50 and 100 μg of 2,4 dinitrobenzene sulfonic acid (DNBS) (DNBS-MNA), selected as the water-soluble analog of DNCB suitable for MNA loading. After elicitation with topical DNCB, mice sensitized with DNBS-MNA loaded with either 50 or 100 μg of hapten developed a significantly superior CHS than that of mice sensitized with topical DNCB (Fig 2E). Therefore, MNA loaded with 50 μg of DNBS were selected for the rest of the experiments. Importantly, MNA without cargo (empty MNA) did not induce skin inflammation.
Fig 2. Generation and testing of MNA integrating hapten and NK1R antagonists.
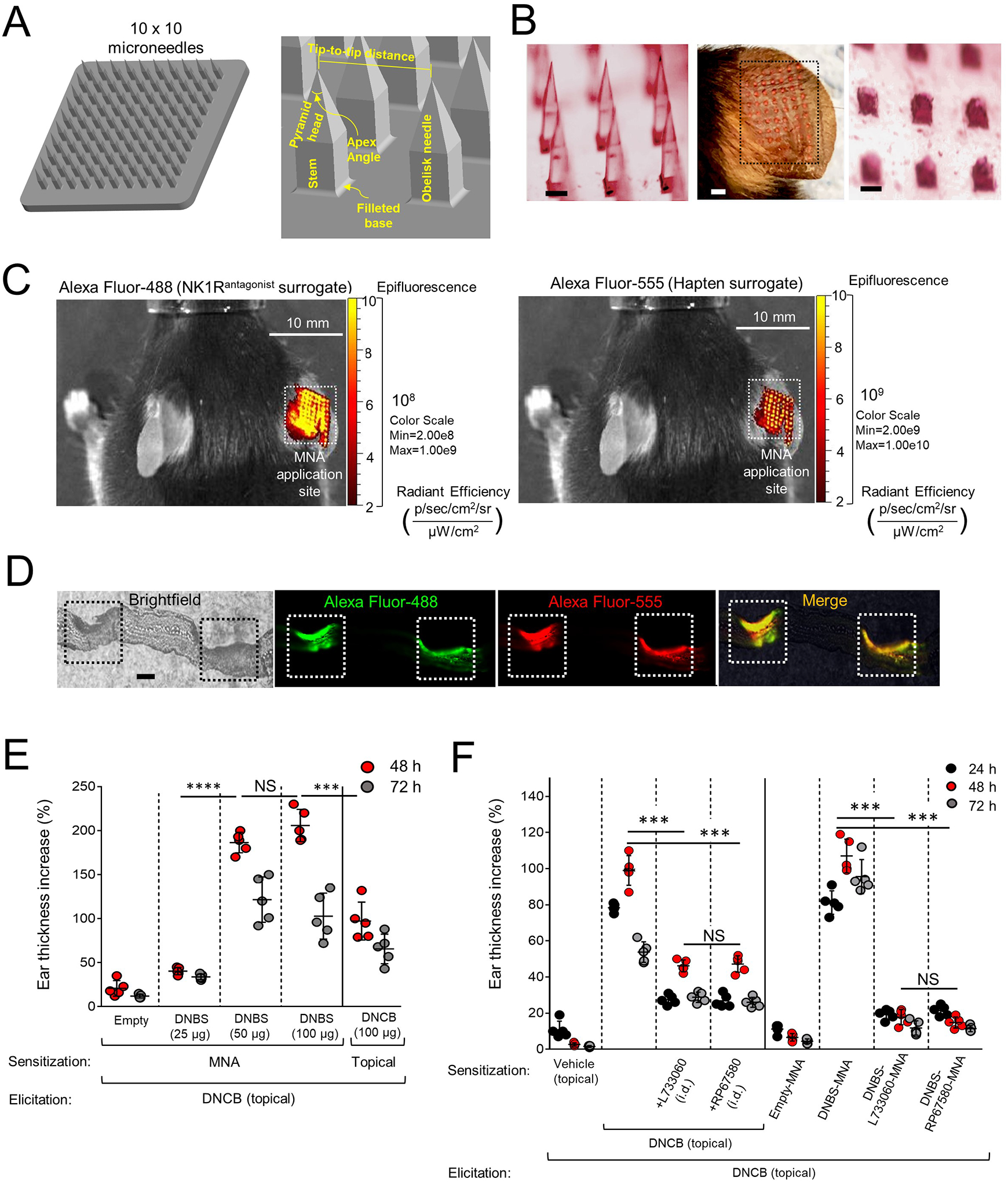
A) Three-dimensional design of MNA and geometric parameters. B) Stereomicroscopy images of the microneedles before (left) and after (right) application to mouse ears. Scale bars, 200μm. Center, MNA-delivered cargo traces (dotted line square) on the mouse ear. Scale bar, 1mm. C) Live-animal fluorescence images of a mouse treated with MNA on the ear skin, showing delivery of Alexa Fluor-488 (left) and Alexa Fluor-555 (right). D) Cryosection of a mouse ear skin showing (left) two areas of MNA penetration (squares), and the corresponding images by fluorescence microscopy showing delivery of Alexa Fluor-488 (green) and Alexa Fluor-555 (red) to skin layers. Scale bar, 40μm X100. E) CHS induced by sensitization with MNA loaded with DNBS versus DNCB applied topically. Each dot represents one mouse. F) Comparative CHS in mice sensitized with topical DNCB plus NK1R antagonists injected i.d. or with DNBS-NK1Rantagonists-MNA. In (E) and (F), a representative experiment of 3 is shown, 5 mice per group. Means ± SD analyzed by one-way ANOVA followed by Student-Newman-Keuls test. ***p < 0.001, ****p < 0.0001, NS: not significant. Statistic comparisons among values obtained 48 h following ear skin elicitation.
We investigated if sensitization with MNA containing DNBS and NK1R antagonists (DNBS-NK1Rantagonist-MNA) affects skin CHS. Because in mice SP and HK1 exert their agonist effects by binding the NK1R and MrgprB2, we compared the effects of RP67580 an antagonist that inhibits specifically the mouse NK1R with those of L733060 that blocks both NK1R and MrgprB2.10, 15, 24–28. The doses of the NK1R antagonists (10 nmols per MNA) were based on previous publications6, 24, 25, 27, 29–31. Co-administration of NK1R antagonist at the sensitization site, incorporated with hapten in MNA or injected i.d. reduced CHS (Fig 2F and Fig E 3A–C). This effect was more pronounced when the NK1R antagonists and DNBS were co-administered via MNA, and both RP67480 and L733060 exerted similar CHS inhibitory effects (Fig 2F and Fig E 3A–C). Thus, the MNA co-deliver NK1R antagonists and hapten efficiently to the skin during sensitization, which prevents cutaneous CHS.
Skin delivery of hapten and NK1R antagonists by MNA restrains hapten-induced inflammatory environment in mouse and human skin
We investigated in mice and in an ex vivo model of human skin if co-delivery of hapten and NK1R antagonists via MNA affects the skin inflammation triggered by hapten penetration. WT mice were sensitized on the ear skin with DNBS-MNA, DNBS-RP67580-MNA, DNBS-L733060-MNA or empty-MNA (control). Twenty-four h later, DNBS-MNA application induced a robust ear thickness increase and leukocyte infiltrate at the sensitization site, effects that were reduced in mice exposed to either DNBS-NK1Rantagonist-MNA (Fig 3A–D). Next, we investigated if skin application of DNBS-NK1Rantagonist-MNA affects the synthesis and secretion of IL-1β and IL-6, which are relevant for the inflammatory response of CD32–36. Sensitization with DNBS-MNA increased pro-IL-1β and IL-6 mRNA contents and IL-1β and IL-6 protein concentrations at the skin treated site, which did not occur or was substantially reduced when mice were sensitized with DNBS-NK1Rantagonist-MNA (Fig 3E and F).
Fig 3. DNBS and NK1R antagonists integrated in MNA restrain hapten-induced inflammation in mouse and human skin.
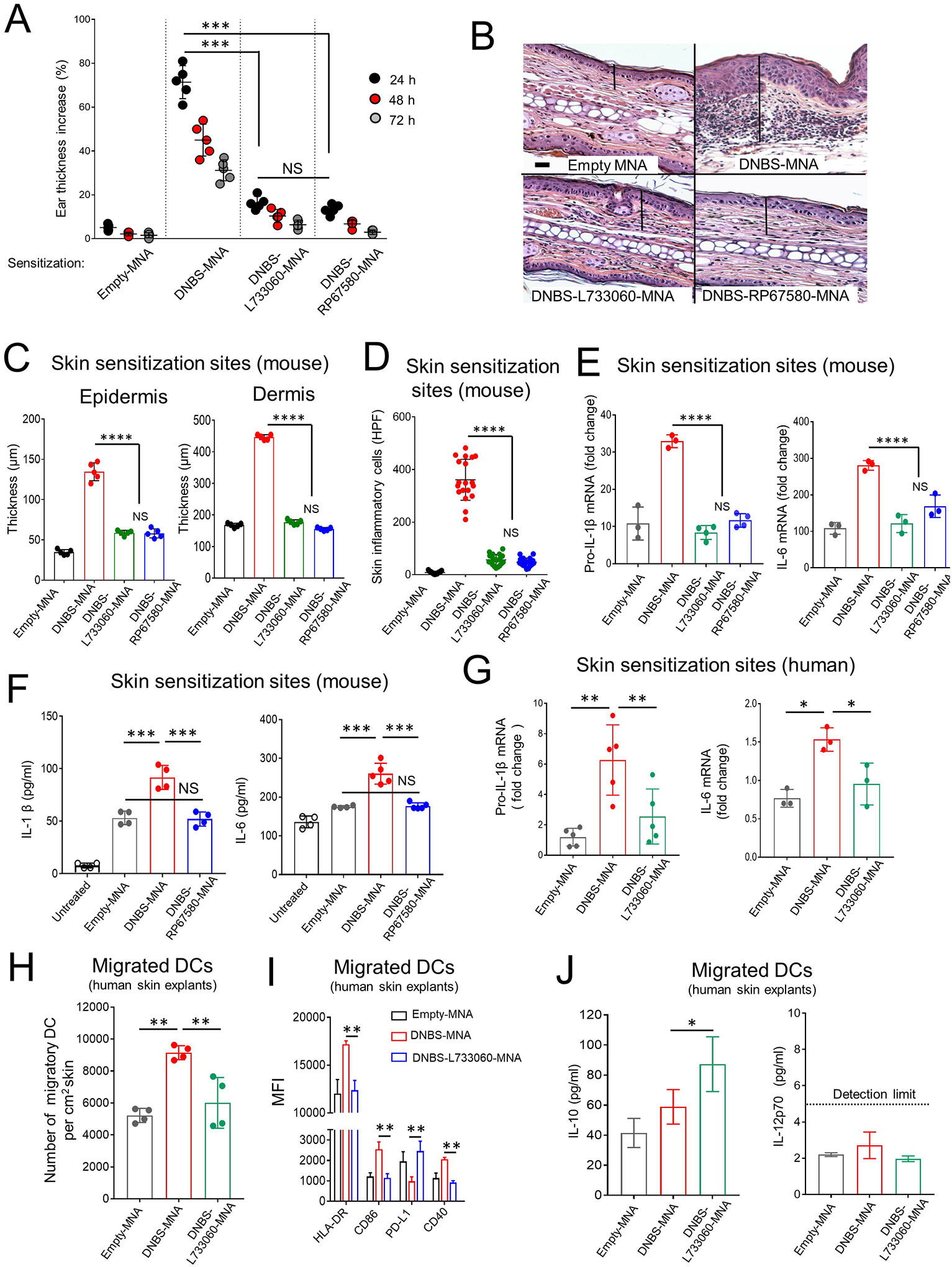
A) Inflammation in WT B6 mice (by the ear thickness) after MNA sensitization. One experiment of 5. Five mice per group. B) Microscopic images of the ears of mice in (A) showing epidermal-dermal thickness (lines) and leukocyte infiltrate. H&E, X200, scale bars 20μm. C) Epidermal and dermal thickness of mouse ears in (A). Five mice / experimental group. D) Quantification of inflammatory cells in the skin sections of mice in (A). Means±1SD of 20 microscopic high-power fields (HPF, X40) per skin section. A representative experiment of 3. Five mice per group. E) Content of IL-1β and IL-6 transcripts in the ears of mice. F) Content of IL-1β and IL-6 in the ears of mice. In (E) and (F) One experiment of 3. G) IL-1β and IL-6 transcripts in human skin treated with MNAs. Each dot represents a skin sample from a different donor. A representative experiment of 3. H) Quantification of DC migrated from the skin samples in (G). One experiment of 3. I) Expression of HLA-DR, CD86, PD-L1 and CD40 on DC mobilized from the human skin shown in (G). J) Concentration of IL-10 and IL-12p70 in supernatants of the migratory DC in (G). Means ± 1 SD analyzed by 1-way ANOVA followed by Student-Newman Keuls test. *p < 0.05, **p < 0.01. ***p < 0.001, NS: not significant.
To analyze if the inhibitory effects of NK1R antagonists on hapten induced inflammation also occur in human skin, we applied DNBS-MNA, DNBS-L733060-MNA, or empty-MNA on the epidermal side of human skin organ cultures composed of epidermis and dermis (4 cm2 size, 1 MNA per cm2). The NK1R antagonist L733060 was selected because it blocks exclusively the human NK1R without affecting the function of the human MRGPR2X receptor that is also signaled by SP and HK115, 26. Six h after MNA application, human skin samples treated with DNBS-L733060-MNA contained less pro-IL-1β and IL-6 mRNA compared to skin treated with DNBS-MNA (Fig 3G). Thus, in mouse and human skin, the NK1R is key for triggering synthesis of pro-inflammatory IL-1β and IL-6 and inflammation at the sensitization site, and these phenomena are minimized by co-administration of hapten and NK1R antagonists via MNA.
Our findings indicate that skin co-delivery of hapten and NK1R antagonists decreases the inflammatory milieu that promotes maturation of immunogenic DC within the skin and their subsequent mobilization to skin-dLN34, 37. Thus, we next analyzed if blockade of NK1R signaling during hapten sensitization affects the mobilization out of the skin and the maturation of human skin DC, which express the NK1R (Fig E 4A and B). Forty-eight h after application of DNBS-L733060-MNA to the epidermis of human skin organ cultures, the DC mobilized out of the skin decreased in numbers (Fig 3H), exhibited lower expression of HLA-DR, CD86 and CD40 and increased PD-L1 surface content (Fig 3I and Fig E 4C). Additionally, we detected higher concentrations of IL-10 in 24 h culture supernatants of purified DC migrated from human skin organ cultures treated with DNBS-NK1Rantagonist-MNA than in supernatants of control DC mobilized from human skin explants treated with DNBS-MNA (Fig 3J). IL-12p70 was undetectable in the same DC culture supernatants, regardless of the skin treatment (Fig 3J). Thus, co-administration of hapten and NK1R antagonists via MNA impairs the maturation and migration out of the skin of human DC.
Skin CHS requires NK1R signaling in keratinocytes and DC
Different subsets of skin cells participate in the sensitization and effector phases of CHS38–41. In mice, the NK1R or its mRNA were detected in skin keratinocytes, MC (dermal-resident and generated from peritoneal progenitors), epidermal and dermal DC, skin DC homed in skin-draining lymph nodes (skin-dLN), and bone marrow-derived DC generated in culture (Fig E 5A–D). Since skin keratinocytes, DC and MC have been shown to be involved in the innate and adaptive immune responses that causes CD6, 42–45, we analyzed on each of these cell subsets the role of NK1R signaling on the sensitization and effector phases of CHS. We used B6 NK1Rfl/fl mice in which Cre driven recombination causes a deletion of exon 2 of the Tac1r locus that contains the initial ATG start codon and encodes a fragment of the 7tm_1 PF00001) domain of the NK1R, which is critical for agonist binding and the functional structure of the receptor. NK1Rfl/fl mice were crossed with K14-Cre, CD11c-Cre or Mcpt5-Cre mice to delete the functional NK1R in keratinocytes, DC or MC, respectively (Fig E 5E). NK1Rfl/fl, K14-Cre, CD11c-Cre, Mcpt5-Cre mouse parent lines developed skin CHS to DNCB similar to WT mice (Fig E 5F). K14-NK1Rfl/fl, CD11c-NK1Rfl/fl and Mcpt5-NK1Rfl/fl mice and respective control littermates were sensitized on the ear skin with DNBS-MNA or empty-MNA, and then elicited with topical DNCB. NK1R deletion in keratinocytes, but not in DC or MC reduced the inflammatory response induced by DNBS-MNA sensitization, based on decreased ear thickness and leukocyte infiltrate, as compared to littermate controls (Fig 4A and Fig E 5G and H). Accordingly, mouse keratinocytes stimulated in vitro with the NK1R agonist SarSP increased the content of pro-IL-1β and IL-6 mRNA (Fig 4B). Likewise, the quantity of pro-IL-1β and IL-6 mRNA in keratinocytes isolated from the ear skin of K14-NK1Rfl/fl mice sensitized with DNBS-MNA was less than that detected in keratinocytes isolated from the ear skin of control littermates exposed to DNBS-MNA (Fig 4C).
Fig 4. Skin CHS requires NK1R signaling in keratinocytes and DC.
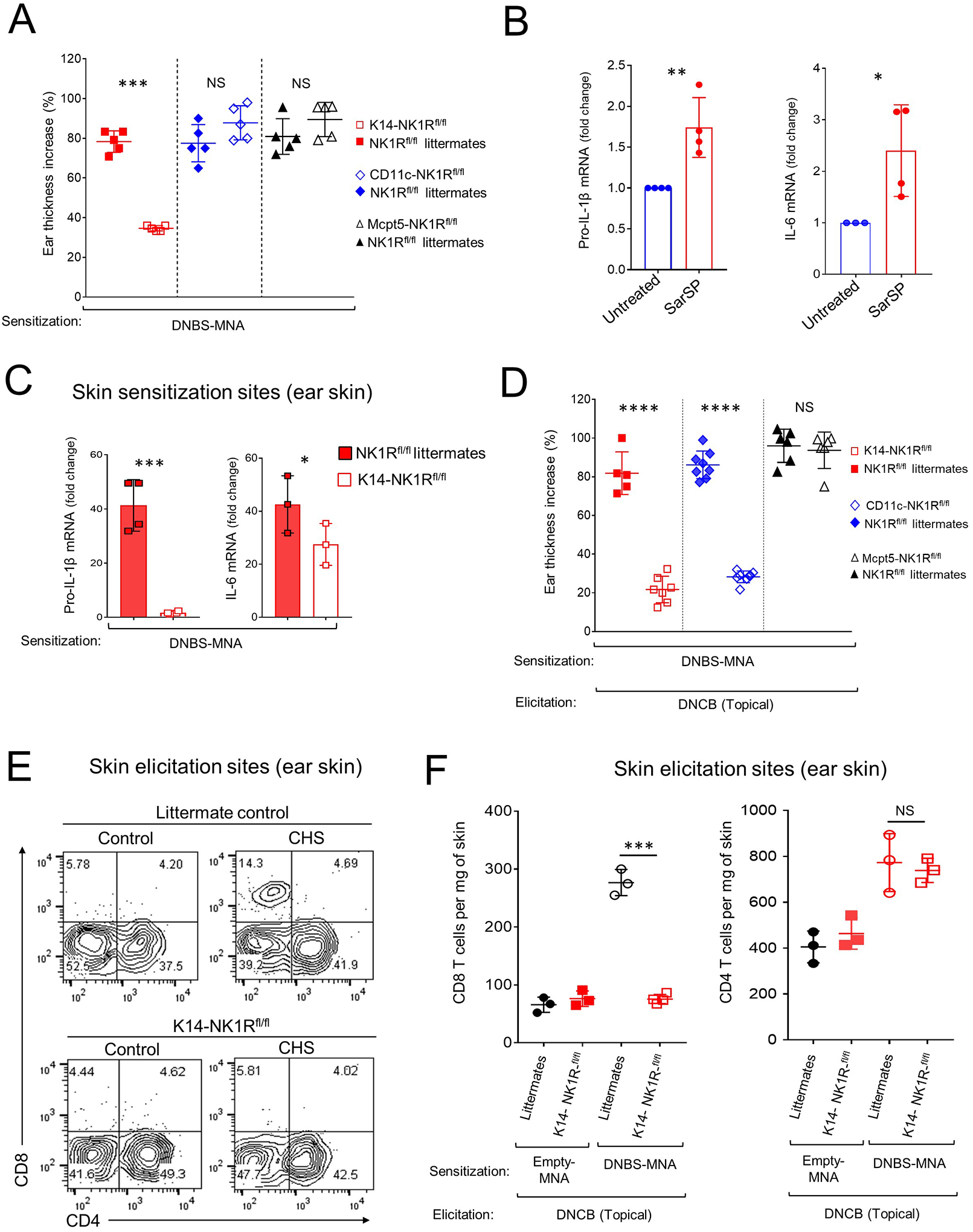
A) Inflammatory response, by ear thickness increase, in K14-NK1Rfl/fl, CD11c-NK1Rfl/fl, or Mcpt5-NK1Rfl/fl and littermate controls. One experiment of 3. Five mice per experimental group. B) IL-1β and IL-6 mRNA in COCA cells cultured alone or with SarSP. C) IL-1β and IL-6 mRNA content in the ears of K14-NK1Rfl/fl mice and littermate controls treated with DNBS-MNA. One experiment of 3. Four mice per experimental group. D) CHS effector response in K14-NK1Rfl/fl, CD11c-NK1Rfl/fl, and Mcpt5-NK1Rfl/fl mice treated with MNA. One experiment of 3. Five mice per group. E) Quantification of CD4 and CD8 T cells in the ears of K14-NK1Rfl/fl mice and littermate controls in (D) 48 h after elicitation. Numbers are percentages of cells per quadrant. F) Quantification of T cells in the ears of mice in (E). One experiment of 3. Means ± 1 SD analyzed by 1-way ANOVA followed by Student-Newman-Keuls test. *p < 0.05, **p < 0.01, *** p < 0.001, **** p < 0.0001. NS: not significant. In (A) statistic comparisons among values obtained 24 h following ear skin elicitation are depicted. In (D) statistic comparisons among values obtained 48 h following ear skin elicitation are depicted.
Mice sensitized with DNBS-MNA on the ear skin and with deletions of functional NK1R in keratinocytes or DC, but not in MC, developed a decreased effector phase of skin CHS against topical DNCB, compared to littermate controls (Fig 4D and Fig E 5I and J). Under the same conditions, mice lacking functional NK1R in keratinocytes exhibited fewer effector CD8 T cells at the elicitation site (Fig 4E and F). Thus, NK1R expression in keratinocytes is critical for the early inflammatory response induced by hapten sensitization and NK1R expression in keratinocytes and DC is key for development of the effector phase of CHS.
Effect of co-delivery of hapten and NK1R antagonists on mouse skin DC
Our findings indicate that NK1R antagonists restrain the pro-inflammatory skin microenvironment necessary for the activation of DC with potent T-cell stimulatory function required for the elicitation of CHS. Therefore, we investigated if NK1R antagonists affect migration, maturation and Ag transport of skin-resident DC. For these experiments, we used a mouse model of skin CHS to chicken ovalbumin (OVA) after validating that skin application of MNA loaded with OVA (OVA-MNA) promote potent CHS, which is significantly decreased when NK1R antagonists and OVA are co-delivered by the MNA (Fig E 6A). To trace the migration of cutaneous DC exclusively from the skin area treated with the MNA, the experiments were done in mice expressing ubiquitously the Kikume Green-Red (KikGR) fluorescent photoconvertible protein46. The skin area sensitized with MNA was exposed to violet light immediately before MNA application, to shift the fluorescence of the photoconvertible skin cells from green to red. In this model, the photoconverted (red) DC that migrate from the skin area treated with MNA are distinguished from the non-photoconverted (green) DC that traffic from untreated skin or from DC that reside constitutively in LN. Using this model, we tested if co-delivery of OVA and NK1R antagonists via MNA alters the traffic of cutaneous DC to skin-dLN. Since the NK1R antagonists RP67580 and L733060 exerted similar effects on CHS to OVA (Fig E 6A), the following experiments were done with the selective mouse NK1R antagonist RP67580. OVA-MNA, OVA-RP67580-MNA or empty-MNA were applied to the ears of KikGR mice. Two days after MNA application, photoconverted DC mobilized from the MNA treated ear were identified by flow cytometry in the auricular skin-dLN as CD11cPos MHC class-IIHigh cells, which included CD207Pos CD103Neg Langerhans cells (LC), dermal conventional CD103Pos (cDC1) and dermal conventional CD301bPos (cDC2) dendritic cells (Fig 5A and Fig E 6B). Treatment with OVA-MNA increased the number of LC and both dermal DC subsets in skin-dLN compared to skin application of empty-MNA. Addition of RP67580 to the OVA-MNA applied on the skin decreased the number of dermal cDC1 and cDC2 detected in skin-dLN, without altering significantly the number skin LC found in skin-dLN (Fig 5B). It also decreased expression of HLA-DR, CD86 and CD40 and augmented PD-L1 expression in photoconverted skin LC and dermal DC mobilized from the sensitization area to skin-dLN (Fig E 6C).
Fig 5. Effect of cutaneous co-delivery of hapten and NK1R antagonists on mouse skin DC.
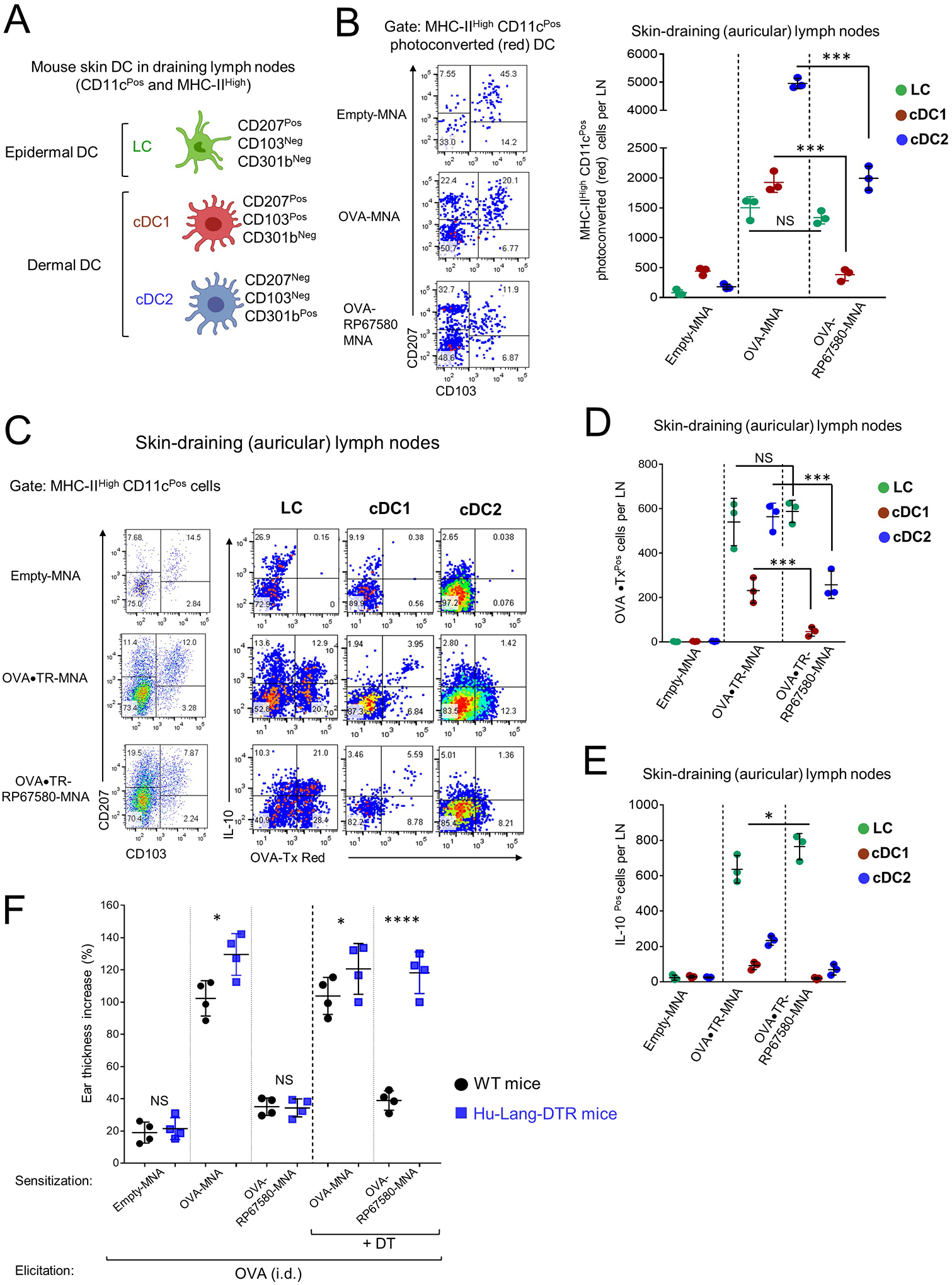
A) Populations of mouse skin DCs analyzed by FACS in skin-dLN. B) Quantification of percentages (left) and numbers (right) of skin DCs homing in skin-dLN of KikGR mice treated with MNAs. One experiment of 4. C) Transport of OVA•TR and IL-10 content in DCs mobilized to skin-dLN of IL-10-EGFP-reporter mice treated with MNAs. In (B and C) Numbers in dot plots are percentages of cells per quadrants. D and E) Numbers of skin DC expressing OVA•TR (D) or with IL-10 content (E), in skin-dLN of mice. One experiment of 3. F) Effect of LC depletion on the inhibition of CHS to OVA assessed by ear thickness. One experiment of 3. Four mice per experimental group. Means ± 1 SD were analyzed by 1-way ANOVA followed by comparison between all means by Student-Newman-Keuls test. *p < 0.01, ***p < 0.001, ****p < 0.0001. NS: not significant.
Next, we investigated if NK1R blockade during sensitization affects transport of Ag by cutaneous DC mobilized to skin-dLN. To track the Ag applied to the skin, we used MNA loaded with OVA conjugated to the fluorochrome Texas Red (OVA•TR-MNA) alone or plus RP67580 (OVA•TR-RP67580-MNA). Since IL-10 has been associated with the immunosuppressive capacity of DC20, 47, 48, these experiments were done in IL-10-enhanced green fluorescent protein (EGFP) reporter B6 mice in which EGFP expression is driven by the endogenous IL-10 promoter. OVA•TR-MNA, OVA•TR-RP67580-MNA or empty-MNA were applied to the ears of IL-10-EGFP mice, and cell suspensions of skin-dLN were analyzed by flow cytometry 2 days later. Following MNA treatment, OVA•TR was transported to skin-dLN mainly by skin LC and dermal cDC2 and to a less extent by dermal cDC1 (Fig 5C and D). Addition of RP67580 to the OVA•TR-MNA reduced the number of cDC1 and cDC2 with OVA•TR detected in skin-dLN without affecting significantly the number of migrating LC bearing OVA•TR in skin-dLN (Fig 5C and D). Following treatment with OVA-MNA, LC were the main subset of skin migratory DC containing IL-10 in skin-dLN and the number of LC containing OVA•TR and IL-10 in skin-dLN increased in mice treated with OVA•TR-RP67580-MNA (Fig 5E).
Our findings indicate that delivery of Ag and NK1R antagonist to the skin via MNA reduces the number of dermal DC transporting Ag to skin-dLN, without affecting the quantity of LC bearing allergen and IL-1020, 48. Thus, we tested if LC have a role in the inhibitory effect of the NK1R antagonists in hu-Lang-DTR mice, a model that allows selective depletion of epidermal LC by administration of diphtheria toxin (DT)20. Three days after DT injection, when epidermal LC were depleted (Fig E6 D), hu-Lang-DTR mice were sensitized with OVA-MNA, OVA-RP67580-MNA, or empty-MNA on the ear skin and elicited with OVA injected i.d., LC depletion abolished the inhibitory effect of the NK1R antagonist on the effector phase of CHS (Fig 5F).These results demonstrate that LC are required for the regulatory effect of NK1R antagonists.
Skin co-delivery of hapten and NK1R antagonists impairs the T cell response that causes CHS to DNCB
Since NK1R antagonists reduce the effector phase of skin CHS, we tested if co-delivery of hapten plus NK1R antagonists via MNA affects the T cell response that causes the disorder. We used two complementary relevant mouse models of CHS, one against DNBS that is mediated by a polyclonal T cell response, and the other against OVA in which the Ag-specific T cell response can be tracked with TCR transgenic OT-II CD4 and OT-I CD8 T cells.
In the CHS model against DNBS, following sensitization of WT B6 mice with DNBS-NK1Rantagonist-MNA, analysis by flow cytometry revealed a higher percentage of activated (CD44High) CD4 and CD8 T lymphocytes undergoing cell death (Fig 6A) and a higher percentage of CD4 FoxP3 Treg (Fig 6B) in skin-dLNs, compared to those detected in skin-dLNs of mice treated with DNBS-MNA. Next, we investigated if the increase in the CD4 Foxp3 Treg in skin-dLN of mice treated with DNBS-NK1Rantagonist-MNA was due to proliferation or recruitment of natural occurring Treg (nTreg), or de novo differentiation of induced Treg (iTreg). To address that, untreated CD45.1 congenic B6 mice were injected i.v. with equal numbers of nTreg (CD4Pos Foxp3-EGFPPos) labelled with Violet Proliferation Dye (VPD) and naïve CD4 T cells (Foxp3-EGFPNeg CD25Neg) labelled with Far-Red Proliferation Dye (RPD), both FACS-sorted from spleens of CD45.2 congenic DEREG mice (Fig E 7A). Because in DEREG mice the DT-receptor-EGFP fusion protein is under control of the endogenous foxp3 promoter49, this strategy allowed analyzing simultaneously in skin-dLN the recruitment and proliferation of VPD labelled nTreg and the generation of iTreg from RPD-labeled naïve CD4 T cells expressing de novo Foxp3 EGFP (Fig E7 A). One day after T cell transfer, mice were sensitized on the ear with DNBS-MNA or DNBS-NK1Rantagonist-MNA. Four days later, proliferation and cell number of nTreg and iTreg in skin-dLN was assessed by flow cytometry. The results indicate that the increase in Treg numbers in skin-dLN of mice sensitized with DNBS-NK1R antagonist–MNA was due to recruitment of non-proliferating nTreg (Fig E 7B).
Fig 6. Co-delivery of hapten and NK1R antagonists via MNA impairs the CHS-T cell response to DNCB.
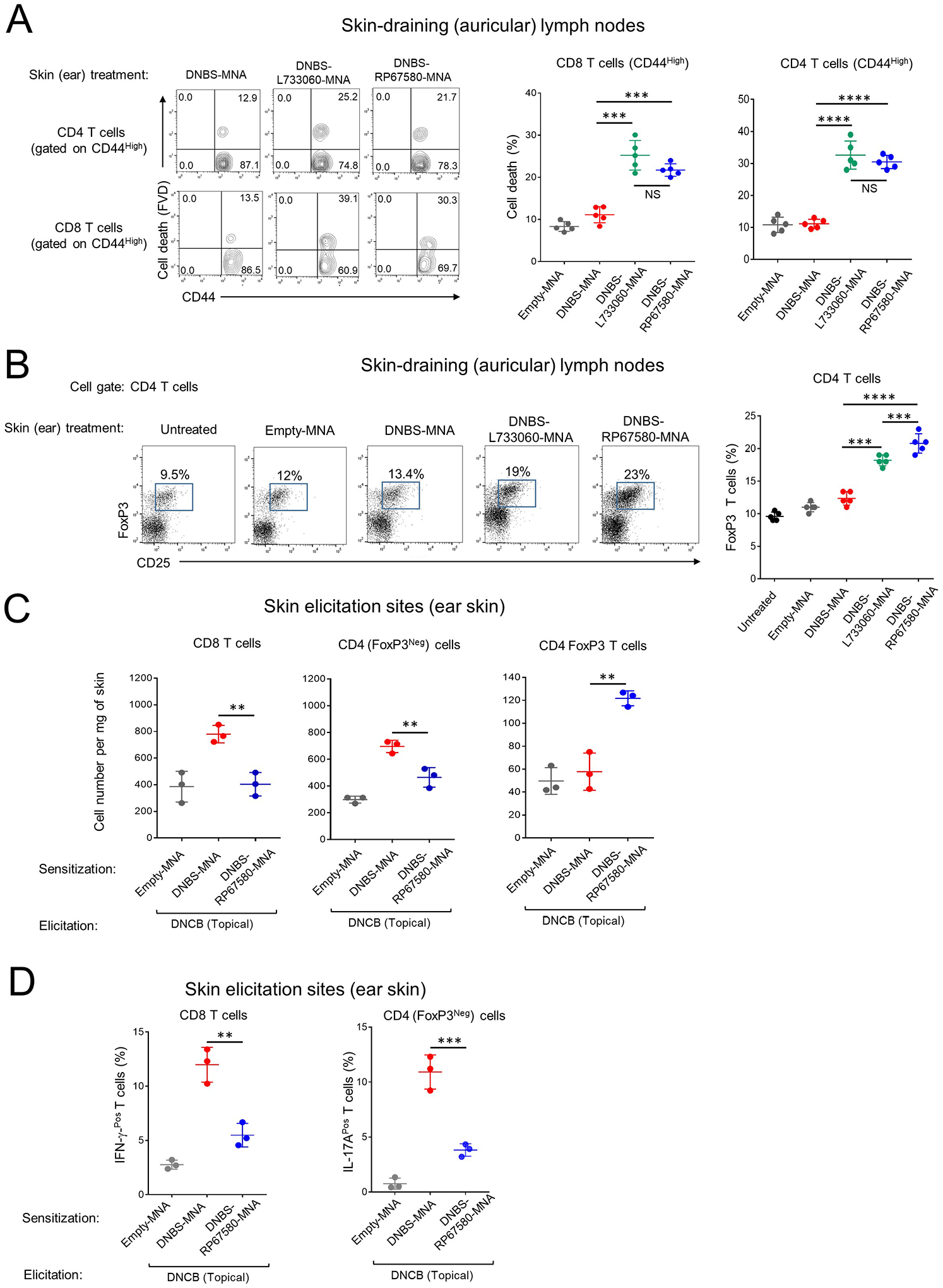
A) Quantification of cell death in activated CD4 and CD8 T cells in skin-dLN of mouse ears treated with MNA. One experiment of 4. Each dot represents a sample including 4 auricular LN from 2 mice. Numbers in contour plots are percentages of cells per quadrant. B) Percentages of Treg cells in skin-dLN of mice treated with MNAs. In the diagram on the right, each dot represents a sample containing 4 auricular LN pooled from 2 mice. One experiment of 3. C) Quantification of CD8, CD4 (Foxp3Neg) and Treg cells in the ear skin of mice sensitized with MNA and elicited with topical DNCB. D) Percentage of CD4 (FoxP3Neg) and CD8 T cells expressing IL-17 or IFN-γ at the elicitation site of mice in (C). One experiment of 3. Each dot represents a sample of skin pooled from 2 ears from the same mouse. Means ± 1 SD were analyzed by 1-way ANOVA followed by Student-Newman-Keuls test. **p < 0.01, ***p < 0.001, ****p < 0.0001. NS: not significant.
Following, we analyzed in the CHS model against DNBS if cutaneous co-delivery of hapten and NK1R antagonists affects the number of effector T cells at the skin elicitation site (Fig 6C and Fig E 7C and D). Mice were treated on the ear skin with DNBS-RP67580-MNA, DNBS-MNA or empty-MNA, and elicited with topical DNCB. Forty-eight h later, the elicitation sites of mice treated with DNBS-RP67580-MNA contained fewer CD8 and CD4 T cells with decreased secretion of IL-17A and IFN-γ, respectively (Fig 6C and D and Fig E 7D) and more CD4 Foxp3 Treg, than the skin area of mice sensitized with MNA without the antagonist (Fig 6C and Fig E 7C). Thus, blockade of the NK1R during hapten sensitization of the skin results in death of activated T cells and recruitment of nTreg in skin-dLN, and fewer effector T cells secreting IL-17A and IFN-γ and more CD4 Foxp3 Treg at the skin elicitation site.
Skin co-delivery of OVA and NK1R antagonists impairs the CHS-effector response in an Ag-specific fashion
The ability of the immunosuppressive MNA to abrogate the T cell response in an Ag-specific manner was validated in the mouse model of CHS to OVA. Since the NK1R antagonists RP67580 and L733060 inhibited to a similar extent skin CHS to OVA (Fig E 6A), the experiments were done with MNA loaded with RP67580. WT B6 mice (Thy1.2 congenic) were injected i.v. with CFSE-labeled OT-II naïve CD4 T cells specific for OVA323–339 -IAb or with naïve OT-I CD8 T cells against OVA257–264 (SIINFEKL)-H2Kb (in this system OT cells are Thy1.1). The next day, mice were treated on the ear with OVA-MNA, OVA-RP67580-MNA or control empty-MNA. Three days (for OT-I) or 4 days (for OT-II) after treatment we analyzed by flow cytometry the absolute number, proliferation, activation, cell death and content of IFN-γ and IL-17A in OT-I and OT-II cells in skin-dLN. Treatment with OVA-RP67580-MNA did not affect OT-I or OT-II cell proliferation (by CFSE dilution) or activation (based on CD44High expression) but increased the percentage of cell death (by FVD incorporation) (Fig 7A and B). Proliferating OT cells from mice treated with OVA-RP67580 MNA underwent similar number of cell cycles than those OT cells from mice treated with OVA-MNA, which indicates that treatment with OVA-NK1Rantagonist-MNA did not induce T-cell anergy (Fig 7C). In addition compared to mice skin sensitized with OVA-MNA, sensitization with OVA-RP67580-MNA reduced the content of IFN-γ and IL-17A in dividing OT cells in skin-dLNs (Fig 7D and E). In accord with the results obtained in the CHS to DNCB, we did not detect de novo generation of OT-II iTreg in mice treated with OVA-RP67580-MNA (Fig 7F). Thus, the model of CHS to OVA revealed that skin delivery of Ag plus NK1R antagonists via MNA leads to cell deletion and reduced IFN-γ-content in Ag-specific T cells in skin-dLNs.
Fig 7. Co-administration of NK1R antagonists and Ag impairs mouse CHS to OVA and decreases the T cell-stimulatory function of human skin DC.
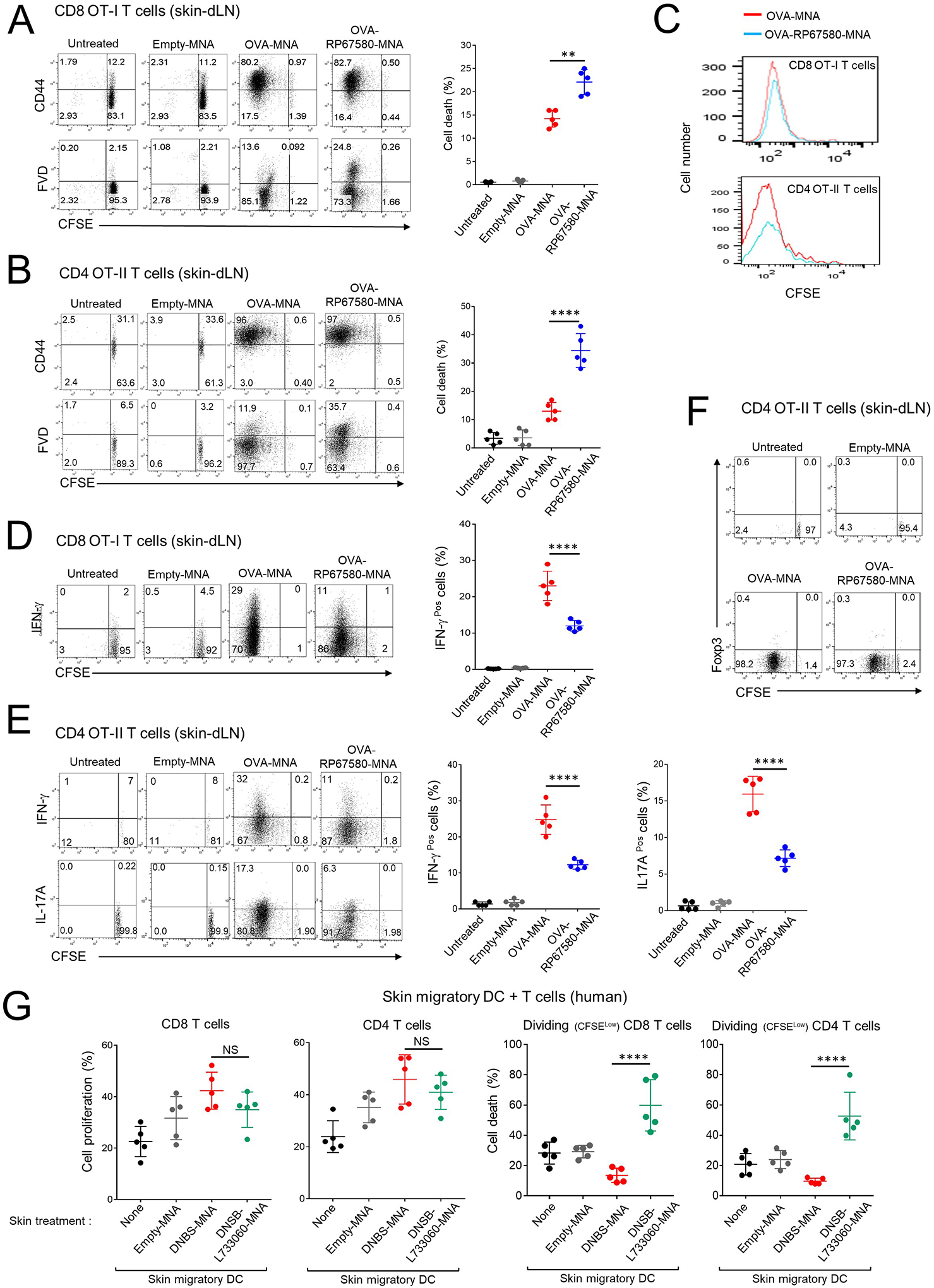
A and B) Cell death in proliferating CD8 OT-I (A) or OT-II cells (B) in skin-dLN of mice sensitized with MNAs. C) Side-by-side comparison of the proliferation of CD8 OT-I and CD4 OT-II cells shown in (A) and (B). D) Content of IFN-γ in the CD8 OT-I cells shown in (A). E) Content of IFN-γ and IL-17 in the CD4 OT-II cells shown in (B). F) Percentages of Treg in CD4 OT-II cells of the mice in (B). G) Proliferation and cell death of human CD4 and CD8 T cells stimulated with allogeneic DCs migrated from human skin treated with MNA. In the dot diagrams on the right of (A), (B), (D) and (E), one experiment of 3. In dot plots of (A), (B), (D), (E) and (F), numbers are percentages of cells per quadrant. In (G), each dot is a sample of responding T cells from a different mixed leukocyte culture. In (A), (B), (D), (E) and (G), means ± 1 SD were analyzed by 1-way ANOVA followed by Student-Newman-Keuls test. **p < 0.01, ****p< 0.0001.
Cutaneous co-delivery of hapten and NK1R antagonists by MNA limits the T-cell stimulatory function of human skin DC
To assess the clinical applicability of our approach, we compared the T cell stimulatory function of DC mobilized out of human epidermal-dermal organ cultures (4 cm2 surface area) treated with DNBS-MNA, DNBS-L733060-MNA or empty-MNA (1 MNA per cm2, 5 explants per donor). Forty-eight h after MNA application, DC harvested from skin cultures of each donor were used as stimulators of CFSE-labelled allogeneic T cells in 5 day–mixed leukocyte cultures that were analyzed by flow cytometry. DC mobilized from skin explants treated with DNBS-L733060-MNA induced similar proliferation but increased percentage of cell death in dividing CD4 and CD8 T cells, as compared to that elicited by DC migrated from explants treated with DNBS-MNA (Fig 7G and Fig E 8A). In both situations, proliferating T cells underwent similar number of cell cycles, which indicates that treatment with DNBS-L733060-MNA did not cause T cell anergy (Fig E 8B). Thus, DC mobilized from human skin treated with hapten in presence of NK1R antagonists trigger proliferation of T cells unfit for survival.
Hapten and NK1R antagonists integrated in MNA desensitize established CD
We tested if co-delivery of DNBS and NK1R antagonists to the skin desensitizes established CHS and prevents its relapses. To address that, skin CHS to DNCB was first induced in WT B6 mice by topical DNCB sensitization on the skin (abdomen) followed on day 6, by cutaneous elicitation (right ear) with topical DNCB. Skin CHS to DNCB was confirmed in each mouse. Sixteen days after (day 22), mice were treated on the elicitation site (right ear) with (i) DNBS-L733060-MNA or DNBS-RP67580-MNA, (ii) L733060-MNA or RP67580-MNA (no hapten), (iii) DNBS-MNA (positive control) or empty-MNA (Fig 8A). A week later (day 29), mice were re-challenged with topical DNCB on the homolateral (right) ear to evaluate the effect of the treatment on the local immune response, or on the contralateral (left) ear to assess the effect on the systemic response (Fig 8A). Co-administration of hapten with either NK1R antagonist via MNA prevented the relapse of local (Fig 8B) and systemic (Fig 8C) CHS to DNCB. These effects were superior to those observed when CHS desensitization was induced by application of MNA loaded with NK1R antagonists alone (Fig 8B and C).
Figure 8. Co-delivery of hapten and NK1R antagonists in the skin desensitizes CD locally and systemically in hapten-specific fashion.
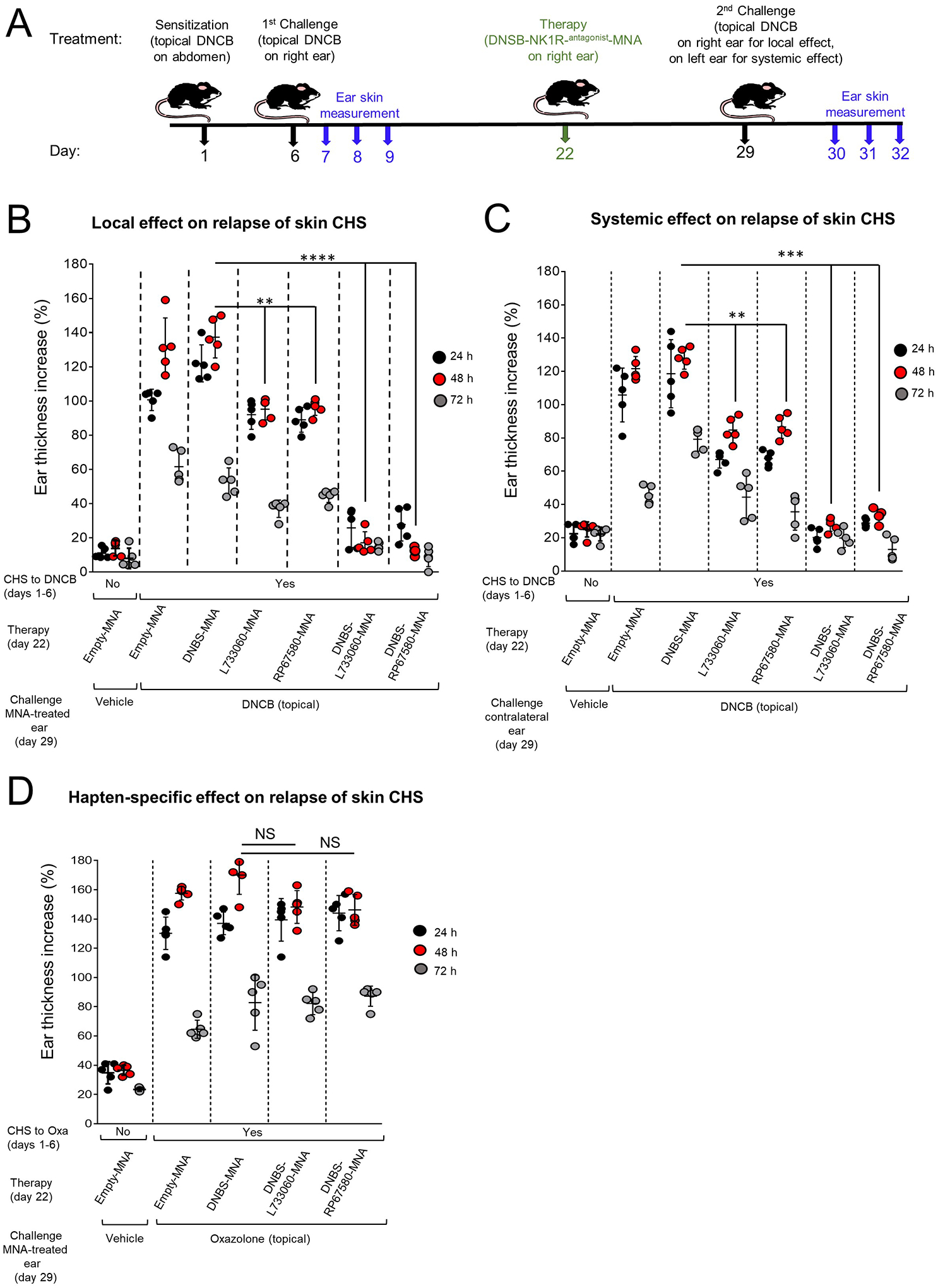
A) Timeline describing the experiments of induction and desensitization of CHS in mice. B and C) Reduction of established CHS in mice, treated with MNA on day 22 and re-challenged on day 29, with topical DNCB on the treated homolateral (right) ear (B), or on the contralateral (left) ear (C). D) Mice with established skin CHS to oxazolone, treated with DNBS-NK1Rantagonist-MNA on the homolateral (right) ear on day 22 and re-challenged with topical oxazolone on the same ear on day 29. In (B), (C) and (D), a representative experiment of 3 is shown. Five mice per group. Each dot represents one mouse. Means ± 1 SD were analyzed by 1-way ANOVA followed by Student-Newman-Keuls test. **p < 0.01, ***p < 0.001, ****p < 0.0001. NS: not significant.
Next, we tested if desensitization by DNBS-NK1Rantagonist-MNA is hapten-specific in WT B6 mice that had developed CHS against oxazolone induced by skin (abdomen) sensitization and confirmed by cutaneous elicitation with topical oxazolone (right ear). After 16 days (day 22), mice were treated on the skin (right ear) with (i) DNBS-L733060-MNA or DNBS-RP67580-MNA or (ii) DNBS-MNA or empty-MNA. A week later (day 29), mice were re-challenged with topical oxazolone on the skin (right ear). MNA loaded with DNBS and either NK1R antagonist were unable to desensitize CHS to oxazolone (Fig 8D). Thus, co-delivery of hapten and NK1R antagonists into the skin results in hapten-specific desensitization of established CHS and prevents CD relapses locally and systemically.
DISCUSSION
We developed an MNA-based immunosuppressive approach that restrains skin inflammation during hapten-sensitization and impairs the adaptive immune response of type-1 biased CD. Specifically, we targeted the function of proinflammatory neuropeptides that signal via the NK1R, which are released in the skin immediately after exposure to contact sensitizers. To our knowledge, the potential of this Ag-specific therapeutic strategy has not been previously explored likely due to lack of appropriate delivery systems able to simultaneously overcome the cutaneous physical barrier and deliver multiple biologics in the skin without disturbing its quiescent state. In addition, the role of neuropeptide-induced inflammation in the pathogenesis of chronic inflammatory skin disorders is beginning to be elucidated and the understanding of its mechanisms in vivo required genetically-modified mouse models that were not available until recently. In this study, we tested the therapeutic potential of dissolvable multicomponent MNA to co-deliver hapten and NK1R inhibitors efficiently in the skin to hinder the immune response of CD in mice and in an ex vivo human skin system, the later a biologically relevant translational model50.
We described the role that NK1R signaled by its endogenous agonists, SP and HK1, plays in the pathogenesis of CD. Although it has been established that signaling the NK1R by SP promotes CHS, the function of HK1, had not been previously investigated13, 51, 52. Besides, SP and HK1 also bind the mouse MrgprB2 and human MRGPRX214,15. Therefore, the exclusive roles the NK1R in CHS needed further analysis. Here, we demonstrated in mice deficient in NK1R or its endogenous ligands that induction of CHS requires NK1R, HK1 and SP, but not MrgprB2. Indeed, the CHS response was not compromised in MrgprB2KO mice, but was impaired in NK1RKO mice. Likewise in mice, blockade of the NK1R alone by the antagonist RP67580 induced similar immuno-suppressive effects than those caused by blockade of the NK1R and MrgprB2 by the antagonist L733060. In our mouse model, SP and HK1 exerted additive effects on CHS, which could be due to binding to different receptor sites, induction of a distinctive conformation of the receptor, redundancy or partial agonistic function of these tachykinins53–57.
Our results demonstrated that delivery of hapten or Ag integrated in MNA induced robust CHS whereas inclusion of NK1R antagonists in the MNA exerted potent immunoregulatory effects at the sensitization and effector sites of the CHS. During CHS sensitization, proinflammatory neuropeptides trigger the release of IL-1β and IL-6, which activate immunostimulatory skin DC that migrate to skin-dLN and initiate effector type-1 biased T cell responses32–36. In addition, in the context of ACD, IL-1β and IL-6 are required for the induction of IL-17 secretion by activated T cells in the skin and skin-dLN and sustain the inflammatory skin microenvironment that facilitate the recruitment of leukocytes to the skin32. We showed that the NK1R is required for the skin inflammation induced by the hapten of Ag and that MNA administration of hapten and NK1R antagonists minimizes release of IL-1β and IL-6 in the skin treated area and reduced the number of skin DC homed to skin dLN during the sensitization of the CHS. Thus, co-delivery of hapten and NK1R antagonists, integrated in MNA, prevents the skin inflammation necessary for the development of the adaptive immune response that sustains CD. Importantly, similar findings were obtained in an ex vivo model of human skin, which support the translational benefits of this therapeutic approach for future clinical application.
The cell types of the skin that express the NK1R and contribute to CD have not been previously identified. The lack of in vivo models allowing cell type-specific deletion of the NK1R precluded analyzing the relevance of NK1R-signaling in different skin cells that could be the targets of NK1R antagonist-based therapies for CD. We validated that NK1R is expressed by keratinocytes, skin DC and MC, which are all involved in the pathogenesis of CD32, 42, 43. The use of cell type-specific NK1RKO mice revealed that the NK1R-dependent inflammation triggered by the hapten was mediated mainly by keratinocytes, and that the adaptive immune component of CD required expression of functional NK1R in keratinocytes and DC. Our findings indicate that keratinocytes are early targets of hapten-induced neuroinflammation and they provide the ideal skin microenvironment for the activation of immunostimulatory DC. In addition, our data suggest that signaling DC via the NK1R was necessary for their ability to sustain type-1 immunity an observation that was previously inferred by our laboratories4, 5. By contrast, in our model, the absence of functional NK1R in MC did not decrease the severity of CD, which seems to agree with previous observations that MC have a rather regulatory role in CHS against otent sensitizers40, 41, 58.
The DC subsets that are affected by cutaneous inflammation differ on their ability to prime and polarize effector T cell responses. Epidermal LC exert a regulatory function20, 59 whereas CD103Pos cDC1 promote type-1 immunity and CD301bPos cDC2 stimulate type-2 bias60–65. Using the KikGR mouse model to identify and quantify exclusively the DC from the skin treated area we demonstrated that the three DC subsets transported Ag to the skin-dLN. Interestingly, delivery of NK1R antagonists during sensitization led to a pronounced decrease of dermal DC without altering the influx of LC carrying Ag and IL-10 in the skin-dLN. These data indicate that the immunosuppressive effects of NK1R antagonists in CHS depend at least in part, on an imbalance between stimulatory and suppressive skin DC homed in skin-dLN20, 48. In addition, our results from in vivo experiments in which LC were depleted confirmed that LC are necessary for the effect of the NK1R antagonist-based MNA immunosuppressive approach. Experiments beyond the scope of this study will determine the individual roles of specific NK1R-signaling in different subsets of skin DC.
Abrogation of the immune response in CD requires expansion of Treg and deletion of hapten-specific effector T cells7, 66–72. Here, we provide in vivo evidence that the NK1R antagonist-based immunosuppressive approach for CD increased the percentage of nTreg and promoted cell deletion and reduced IL-17A and IFN-γ-content in Ag-specific activated T cells within the skin-dLN. Likewise, this approach reduced the number of effector T cells and increased the quantity of Treg at the skin elicitation site, resulting in defective effector CHS response.
Although our data requires further confirmation in clinical trials, relevant for its clinical applicability, the immunosuppressive MNA developed here prevented the relapse of established CD, locally and systemically, in an Ag-specific fashion. Importantly, our findings in human skin support the translational relevance of this immunosuppressive approach.
In summary, our results validate the concept that systemic immune regulation is achievable through localized control of skin neuro-immune networks to treat CD and likely other skin diseases mediated by type-1 biased immune responses.
Supplementary Material
KEY MESSAGES.
Skin exposure to haptens stimulates secretion of the neuropeptides substance P (SP) and hemokinin-1 (HK1), which signal via the neurokinin-1 receptor (NK1R) to initiate the inflammatory response that promotes the T-cell stimulatory function of skin-resident dendritic cells (DC).
Simultaneous skin delivery of hapten and NK1R antagonists incorporated in dissolvable microneedle arrays prevents hapten-induced skin inflammation, reduces the migration of conventional dermal DC without affecting the migration of epidermal Langerhans cells (LC), and inhibits the adaptive immune response of contact dermatitis.
AKNOWLEDGEMENTS:
We thank Denise Prosser for assistance with Luminex at the UPMC Hillman Cancer Center Cancer Biomarkers Facility, Luminex Core Laboratory, Dr. C.J. Paige and A. Berger for providing NK1RKO, Tac4KO and Tac1/4KO mice, Dr. A. Roers for providing Mcpt5-Cre mice and Dr B. McNeil for providing the MrgprB2KO mice.
Source of Funding.
This work was supported by the NIH grants R01AR068249 and R01AR071277 (to L.D.F and A.T.L.), R01AR074285 (to L.D.F.), R01HL130191 and R01AI148690 (to A.E.M.), R21AR063852, P30AR039750 and P50AR070590 (to N.L.W.), R01 AI145881 (to M.O.), K01AR067250 (to T.L.S.), T32-CA175294 fellowship (to S.C.B.) and P30-CA047904 to the UPMC Hillman Cancer Center Cancer Biomarkers Facility.
ABBREVIATIONS
- Ag
Antigen
- CD
Allergic contact dermatitis
- cDC
Conventional dendritic cells
- CHS
Contact hypersensitivity
- DC
Dendritic cells
- dLN
Draining lymph node
- DNBS
2,4-dinitrobenzene sulfonic acid
- DNCB
2,4-dinitrochlorobenzene
- DT
Diphtheria toxin
- HK1
Hemokinin 1
- HPF
High power-field
- LC
Langerhans cell
- LN
Lymph node
- MC
Mast cells
- MNA
Microneedle arrays
- Mrgpr
Mas-related G protein-coupled receptors
- NK1R
Neurokinin-1 receptor
- OVA
Chicken ovalbumin
- SarSP
[Sar9,Met(O2)11] SP
- SP
Substance P
- Th1
CD4 T helper 1 cells
- Tc1
CD8 cytotoxic T cells
- Treg
Regulatory T cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST: The authors declare that they have not significant conflict of interest.
REFERENCES
- 1.Nassau S, Fonacier L. Allergic Contact Dermatitis. Med Clin North Am 2020; 104:61–76. [DOI] [PubMed] [Google Scholar]
- 2.Chu C, Marks JG Jr., Flamm A. Occupational Contact Dermatitis: Common Occupational Allergens. Dermatol Clin 2020; 38:339–49. [DOI] [PubMed] [Google Scholar]
- 3.Bhatia J, Sarin A, Wollina U, Lotti T, Navarini AA, Mueller SM, et al. Review of biologics in allergic contact dermatitis. Contact Dermatitis 2020; 83:179–81. [DOI] [PubMed] [Google Scholar]
- 4.Janelsins BM, Sumpter TL, Tkacheva OA, Rojas-Canales DM, Erdos G, Mathers AR, et al. Neurokinin-1 receptor agonists bias therapeutic dendritic cells to induce type 1 immunity by licensing host dendritic cells to produce IL-12. Blood 2013; 121:2923–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janelsins BM, Mathers AR, Tkacheva OA, Erdos G, Shufesky WJ, Morelli AE, et al. Proinflammatory tachykinins that signal through the neurokinin 1 receptor promote survival of dendritic cells and potent cellular immunity. Blood 2009; 113:3017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathers AR, Tckacheva OA, Janelsins BM, Shufesky WJ, Morelli AE, Larregina AT. In vivo signaling through the neurokinin 1 receptor favors transgene expression by Langerhans cells and promotes the generation of Th1- and Tc1-biased immune responses. J Immunol 2007; 178:7006–17. [DOI] [PubMed] [Google Scholar]
- 7.Morelli AE, Sumpter TL, Rojas-Canales DM, Bandyopadhyay M, Chen Z, Tkacheva O, et al. Neurokinin-1 Receptor Signaling Is Required for Efficient Ca(2+) Flux in T-Cell-Receptor-Activated T Cells. Cell Rep 2020; 30:3448–65 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinhoff MS, von Mentzer B, Geppetti P, Pothoulakis C, Bunnett NW. Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol Rev 2014; 94:265–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morteau O, Lu B, Gerard C, Gerard NP. Hemokinin 1 is a full agonist at the substance P receptor. Nat Immunol 2001; 2:1088. [DOI] [PubMed] [Google Scholar]
- 10.Yu M, Lee SM, Lee H, Amouzegar A, Nakao T, Chen Y, et al. Neurokinin-1 Receptor Antagonism Ameliorates Dry Eye Disease by Inhibiting Antigen-Presenting Cell Maturation and T Helper 17 Cell Activation. Am J Pathol 2020; 190:125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theoharides TC, Tsilioni I, Bawazeer M. Mast Cells, Neuroinflammation and Pain in Fibromyalgia Syndrome. Front Cell Neurosci 2019; 13:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunin P, Caillon A, Corvaisier M, Garo E, Scotet M, Blanchard S, et al. The tachykinins substance P and hemokinin-1 favor the generation of human memory Th17 cells by inducing IL-1beta, IL-23, and TNF-like 1A expression by monocytes. J Immunol 2011; 186:4175–82. [DOI] [PubMed] [Google Scholar]
- 13.Scholzen TE, Steinhoff M, Sindrilaru A, Schwarz A, Bunnett NW, Luger TA, et al. Cutaneous allergic contact dermatitis responses are diminished in mice deficient in neurokinin 1 receptors and augmented by neurokinin 2 receptor blockage. FASEB J 2004; 18:1007–9. [DOI] [PubMed] [Google Scholar]
- 14.McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015; 519:237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azimi E, Reddy VB, Shade KC, Anthony RM, Talbot S, Pereira PJS, et al. Dual action of neurokinin-1 antagonists on Mas-related GPCRs. JCI Insight 2016; 1:e89362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bediz B, Korkmaz E, Khilwani R, Donahue C, Erdos G, Falo LD Jr., et al. Dissolvable microneedle arrays for intradermal delivery of biologics: fabrication and application. Pharm Res 2014; 31:117–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim E, Erdos G, Huang S, Kenniston TW, Balmert SC, Carey CD, et al. Microneedle array delivered recombinant coronavirus vaccines: Immunogenicity and rapid translational development. EBioMedicine 2020; 55:102743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korkmaz E, Friedrich EE, Ramadan MH, Erdos G, Mathers AR, Burak Ozdoganlar O, et al. Therapeutic intradermal delivery of tumor necrosis factor-alpha antibodies using tip-loaded dissolvable microneedle arrays. Acta Biomater 2015; 24:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arora A, Prausnitz MR, Mitragotri S. Micro-scale devices for transdermal drug delivery. Int J Pharm 2008; 364:227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bobr A, Olvera-Gomez I, Igyarto BZ, Haley KM, Hogquist KA, Kaplan DH. Acute ablation of Langerhans cells enhances skin immune responses. J Immunol 2010; 185:4724–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hordinsky M, Donati A. Alopecia areata: an evidence-based treatment update. Am J Clin Dermatol 2014; 15:231–46. [DOI] [PubMed] [Google Scholar]
- 22.Sumpter TL, Ho CH, Pleet AR, Tkacheva OA, Shufesky WJ, Rojas-Canales DM, et al. Autocrine hemokinin-1 functions as an endogenous adjuvant for IgE-mediated mast cell inflammatory responses. J Allergy Clin Immunol 2015; 135:1019–30 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niizeki H, Kurimoto I, Streilein JW. A substance p agonist acts as an adjuvant to promote hapten-specific skin immunity. J Invest Dermatol 1999; 112:437–42. [DOI] [PubMed] [Google Scholar]
- 24.Hamity MV, White SR, Hammond DL. Effects of neurokinin-1 receptor agonism and antagonism in the rostral ventromedial medulla of rats with acute or persistent inflammatory nociception. Neuroscience 2010; 165:902–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bang R, Sass G, Kiemer AK, Vollmar AM, Neuhuber WL, Tiegs G. Neurokinin-1 receptor antagonists CP-96,345 and L-733,060 protect mice from cytokine-mediated liver injury. J Pharmacol Exp Ther 2003; 305:31–9. [DOI] [PubMed] [Google Scholar]
- 26.Azimi E, Reddy VB, Pereira PJS, Talbot S, Woolf CJ, Lerner EA. Substance P activates Mas-related G protein-coupled receptors to induce itch. J Allergy Clin Immunol 2017; 140:447–53 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen W, Marvizon JC. Neurokinin 1 receptor activation in the rat spinal cord maintains latent sensitization, a model of inflammatory and neuropathic chronic pain. Neuropharmacology 2020; 177:108253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munoz M, Rosso M, Covenas R. The NK-1 receptor antagonist L-732,138 induces apoptosis in human gastrointestinal cancer cell lines. Pharmacol Rep 2017; 69:696–701. [DOI] [PubMed] [Google Scholar]
- 29.Tauer U, Zhao Y, Hunt SP, Culman J. Are biological actions of neurokinin A in the adult brain mediated by a cross-talk between the NK1 and NK2 receptors? Neuropharmacology 2012; 63:958–65. [DOI] [PubMed] [Google Scholar]
- 30.Zhong B, Ma S, Wang DH. Protease-activated receptor 2 protects against myocardial ischemia-reperfusion injury through the lipoxygenase pathway and TRPV1 channels. Exp Ther Med 2019; 18:3636–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noailles PA, Angulo JA. Neurokinin receptors modulate the neurochemical actions of cocaine. Ann N Y Acad Sci 2002; 965:267–73. [DOI] [PubMed] [Google Scholar]
- 32.Popov A, Mirkov I, Miljkovic D, Belij S, Zolotarevski L, Kataranovski D, et al. Contact allergic response to dinitrochlorobenzene (DNCB) in rats: insight from sensitization phase. Immunobiology 2011; 216:763–70. [DOI] [PubMed] [Google Scholar]
- 33.Yasukawa S, Miyazaki Y, Yoshii C, Nakaya M, Ozaki N, Toda S, et al. An ITAM-Syk-CARD9 signalling axis triggers contact hypersensitivity by stimulating IL-1 production in dendritic cells. Nat Commun 2014; 5:3755. [DOI] [PubMed] [Google Scholar]
- 34.Larregina AT, Morelli AE, Kolkowski E, Sanjuan N, Barboza ME, Fainboim L. Pattern of cytokine receptors expressed by human dendritic cells migrated from dermal explants. Immunology 1997; 91:303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cumberbatch M, Dearman RJ, Kimber I. Langerhans cells require signals from both tumour necrosis factor-alpha and interleukin-1 beta for migration. Immunology 1997; 92:388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi X, Wang L, Clark JD, Kingery WS. Keratinocytes express cytokines and nerve growth factor in response to neuropeptide activation of the ERK1/2 and JNK MAPK transcription pathways. Regul Pept 2013; 186:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morelli AE, Rubin JP, Erdos G, Tkacheva OA, Mathers AR, Zahorchak AF, et al. CD4+ T cell responses elicited by different subsets of human skin migratory dendritic cells. J Immunol 2005; 175:7905–15. [DOI] [PubMed] [Google Scholar]
- 38.Cevikbas F, Steinhoff A, Homey B, Steinhoff M. Neuroimmune interactions in allergic skin diseases. Curr Opin Allergy Clin Immunol 2007; 7:365–73. [DOI] [PubMed] [Google Scholar]
- 39.Choi JE, Di Nardo A. Skin neurogenic inflammation. Semin Immunopathol 2018; 40:249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reber LL, Sibilano R, Starkl P, Roers A, Grimbaldeston MA, Tsai M, et al. Imaging protective mast cells in living mice during severe contact hypersensitivity. JCI Insight 2017; 2 (9): e92900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaudenzio N, Marichal T, Galli SJ, Reber LL. Genetic and Imaging Approaches Reveal Pro-Inflammatory and Immunoregulatory Roles of Mast Cells in Contact Hypersensitivity. Front Immunol 2018; 9:1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin SF, Jakob T. From innate to adaptive immune responses in contact hypersensitivity. Curr Opin Allergy Clin Immunol 2008; 8:289–93. [DOI] [PubMed] [Google Scholar]
- 43.Kaplan DH, Igyarto BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol 2012; 12:114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albanesi C Keratinocytes in allergic skin diseases. Curr Opin Allergy Clin Immunol 2010; 10:452–6. [DOI] [PubMed] [Google Scholar]
- 45.Deckers J, De Bosscher K, Lambrecht BN, Hammad H. Interplay between barrier epithelial cells and dendritic cells in allergic sensitization through the lung and the skin. Immunol Rev 2017; 278:131–44. [DOI] [PubMed] [Google Scholar]
- 46.Tomura M, Hata A, Matsuoka S, Shand FH, Nakanishi Y, Ikebuchi R, et al. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci Rep 2014; 4:6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo Y, Wang S, Liu X, Wen H, Li W, Yao X. Langerhans cells mediate the skin-induced tolerance to ovalbumin via Langerin in a murine model. Allergy 2019; 74:1738–47. [DOI] [PubMed] [Google Scholar]
- 48.van der Burg NMD, Depelsenaire ACI, Crichton ML, Kuo P, Phipps S, Kendall MAF. A low inflammatory, Langerhans cell-targeted microprojection patch to deliver ovalbumin to the epidermis of mouse skin. J Control Release 2019; 302:190–200. [DOI] [PubMed] [Google Scholar]
- 49.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 2007; 8:191–7. [DOI] [PubMed] [Google Scholar]
- 50.Saarnilehto M, Chapman H, Savinko T, Lindstedt K, Lauerma AI, Koivisto A. Contact sensitizer 2,4-dinitrochlorobenzene is a highly potent human TRPA1 agonist. Allergy 2014; 69:1424–7. [DOI] [PubMed] [Google Scholar]
- 51.Scholzen TE, Steinhoff M, Bonaccorsi P, Klein R, Amadesi S, Geppetti P, et al. Neutral endopeptidase terminates substance P-induced inflammation in allergic contact dermatitis. J Immunol 2001; 166:1285–91. [DOI] [PubMed] [Google Scholar]
- 52.Peters EM, Ericson ME, Hosoi J, Seiffert K, Hordinsky MK, Ansel JC, et al. Neuropeptide control mechanisms in cutaneous biology: physiological and clinical significance. J Invest Dermatol 2006; 126:1937–47. [DOI] [PubMed] [Google Scholar]
- 53.Milligan G, Ward RJ, Marsango S. GPCR homo-oligomerization. Curr Opin Cell Biol 2018; 57:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berger A, Paige CJ. Hemokinin-1 has Substance P-like function in U-251 MG astrocytoma cells: a pharmacological and functional study. J Neuroimmunol 2005; 164:48–56. [DOI] [PubMed] [Google Scholar]
- 55.Mou L, Xing Y, Kong Z, Zhou Y, Chen Z, Wang R. The N-terminal domain of human hemokinin-1 influences functional selectivity property for tachykinin receptor neurokinin-1. Biochem Pharmacol 2011; 81:661–8. [DOI] [PubMed] [Google Scholar]
- 56.Nederpelt I, Bleeker D, Tuijt B, AP IJ, Heitman LH. Kinetic binding and activation profiles of endogenous tachykinins targeting the NK1 receptor. Biochem Pharmacol 2016; 118:88–95. [DOI] [PubMed] [Google Scholar]
- 57.Borbely E, Helyes Z. Role of hemokinin-1 in health and disease. Neuropeptides 2017; 64:9–17. [DOI] [PubMed] [Google Scholar]
- 58.Serhan N, Basso L, Sibilano R, Petitfils C, Meixiong J, Bonnart C, et al. House dust mites activate nociceptor-mast cell clusters to drive type 2 skin inflammation. Nat Immunol 2019; 20:1435–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dioszeghy V, Mondoulet L, Laoubi L, Dhelft V, Plaquet C, Bouzereau A, et al. Antigen Uptake by Langerhans Cells Is Required for the Induction of Regulatory T Cells and the Acquisition of Tolerance During Epicutaneous Immunotherapy in OVA-Sensitized Mice. Front Immunol 2018; 9:1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Malissen B, Tamoutounour S, Henri S. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol 2014; 14:417–28. [DOI] [PubMed] [Google Scholar]
- 61.Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med 2010; 207:823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 2011; 35:260–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity 2005; 23:611–20. [DOI] [PubMed] [Google Scholar]
- 64.Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 2013; 39:733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sokol CL, Camire RB, Jones MC, Luster AD. The Chemokine Receptor CCR8 Promotes the Migration of Dendritic Cells into the Lymph Node Parenchyma to Initiate the Allergic Immune Response. Immunity 2018; 49:449–63 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balmert SC, Donahue C, Vu JR, Erdos G, Falo LD, Jr., Little SR. In vivo induction of regulatory T cells promotes allergen tolerance and suppresses allergic contact dermatitis. J Control Release 2017; 261:223–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Long CM, Marshall NB, Lukomska E, Kashon ML, Meade BJ, Shane H, et al. A Role for Regulatory T Cells in a Murine Model of Epicutaneous Toluene Diisocyanate Sensitization. Toxicol Sci 2016; 152:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bruhs A, Schwarz T. Ultraviolet Radiation-Induced Immunosuppression: Induction of Regulatory T Cells. Methods Mol Biol 2017; 1559:63–73. [DOI] [PubMed] [Google Scholar]
- 69.Ouyang Z, Wang X, Meng Q, Feng L, Sun Y, Wu X, et al. Suppression of adenosine monophosphate-activated protein kinase selectively triggers apoptosis in activated T cells and ameliorates immune diseases. Biochem Biophys Res Commun 2017; 487:223–9. [DOI] [PubMed] [Google Scholar]
- 70.Muro R, Nitta T, Kitajima M, Okada T, Suzuki H. Rasal3-mediated T cell survival is essential for inflammatory responses. Biochem Biophys Res Commun 2018; 496:25–30. [DOI] [PubMed] [Google Scholar]
- 71.Kitashima DY, Kobayashi T, Woodring T, Idouchi K, Doebel T, Voisin B, et al. Langerhans Cells Prevent Autoimmunity via Expansion of Keratinocyte Antigen-Specific Regulatory T Cells. EBioMedicine 2018; 27:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity 2012; 36:873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.