

SUMMARY
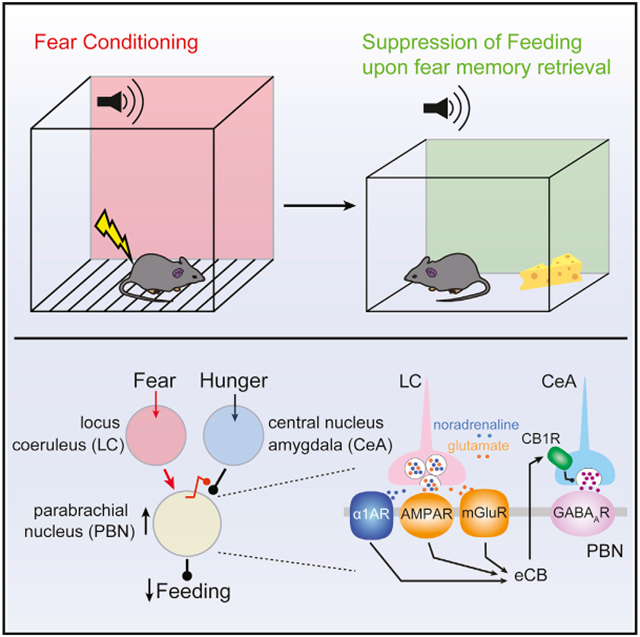
The circuit mechanisms underlying fear-induced suppression of feeding are poorly understood. To help fill this gap, mice were fear conditioned, and the resulting changes in synaptic connectivity among the locus coeruleus (LC), the parabrachial nucleus (PBN), and the central nucleus of amygdala (CeA)—all of which are implicated in fear and feeding—were studied. LC neurons co-released noradrenaline and glutamate to excite PBN neurons and suppress feeding. LC neurons also suppressed inhibitory input to PBN neurons by inducing heterosynaptic, endocannabinoid-dependent, long-term depression of CeA synapses. Blocking or knocking down endocannabinoid receptors in CeA neurons prevented fear-induced depression of CeA synaptic transmission and fear-induced suppression of feeding. Altogether, these studies demonstrate that LC neurons play a pivotal role in modulating the circuitry that underlies fear-induced suppression of feeding, pointing to new ways of alleviating stress-induced eating disorders.
Graphical Abstract
In Brief
Yang et al. demonstrate that locus coeruleus neurons orchestrate fear-induced suppression of feeding by directly activating the parabrachial nucleus neurons and inducing long-term depression of their inhibitory inputs from the amygdala. This dual regulation by locus coeruleus neurons is mediated by co-release of noradrenaline and glutamate.
INTRODUCTION
Stress regulates feeding but does so in a way that depends on a range of factors (Block et al., 2009; Kivimäki et al., 2006; Maniam and Morris, 2012; Pecoraro et al., 2004; Torres and Nowson, 2007; Ulrich-Lai et al., 2015; Yau and Potenza, 2013). When stress increases feeding, brain dopaminergic reward circuitry is engaged (Meye and Adan, 2014). But when stress suppresses feeding, the brain circuitry involved is far from resolved.
One potential node in this circuitry is the locus coeruleus (LC). Noradrenergic LC neurons help orchestrate the neural circuitry controlling both stress and feeding (Sara and Bouret, 2012; Wellman, 2000). The activity of LC neurons and the release of noradrenaline (NA) in the brain are elevated in stress disorders (George et al., 2013; Geracioti et al., 2001; O’Donnell et al., 2004; Olson et al., 2011; Southwick et al., 1993). In animal models of fear conditioning (FC), LC neurons enhance fear memory consolidation and impair fear memory extinction (Bush et al., 2010; Dębiec et al., 2011; Hatfield and McGaugh, 1999; Kabitzke et al., 2011; Liang et al., 1990; Mueller et al., 2008; Sara, 2009; Tully et al., 2007). The effects of LC neurons on feeding are more nuanced. For example, activation of alpha 1-adrenergic receptors (α1ARs) suppresses feeding, whereas activation of beta-adrenergic receptors (βARs) stimulates feeding (Grossman, 1960; Leibowitz, 1988; Morien et al., 1993; Rieg and Aravich, 1994; Wellman et al., 1993; Yeh, 1999), suggesting that the actions of NA on feeding are region or circuit specific. However, whether LC neurons directly regulate feeding is unknown.
Other nuclei known to be involved in both stress and feeding are the central nucleus of amygdala (CeA) and the parabrachial nucleus (PBN). The CeA is widely considered the brain’s stress and fear center (Janak and Tye, 2015; Mahan and Ressler, 2012). Recent studies showed the CeA also regulates feeding (Cai et al., 2014; Douglass et al., 2017; Hardaway et al., 2019; Ip et al., 2019). PBN neurons relay various noxious stimuli to terminate feeding (Campos et al., 2016, 2018; Carter et al., 2013). They are also activated by foot shocks during FC, and silencing them impairs FC (Campos et al., 2018; Han et al., 2015; Sato et al., 2015).
Both the LC and the PBN neurons are reported to be innervated by the CeA neurons (Jia et al., 2005; McCall et al., 2015; Moga and Gray, 1985; Tjoumakaris et al., 2003; Van Bockstaele et al., 1996, 1998). Whether these three nominally interconnected nuclei coordinate their activity to control feeding in response to stress remains an open question. The studies described here were designed to fill that gap by focusing on one type of stress that is induced by fear and to test the hypothesis that fear-induced activation of LC neurons suppresses feeding by modulating the activity of PBN neurons and synaptic transmission from the CeA. To that end, mice were subjected to a FC protocol that reliably suppressed feeding in fasted mice. Next, electrophysiological, optogenetic, and chemogenetic approaches were used to dissect the circuitry mediating the fear-induced suppression of feeding. These studies revealed that LC neurons synapsed on PBN neurons and co-released both NA and glutamate. Co-release directly suppressed feeding by exciting PBN neurons and indirectly suppressed feeding by inducing a heterosynaptic form of endocannabinoid (eCB)-mediate long-term depression (LTD) of inhibitory CeA synapses on the PBN neurons. Preventing the induction of this plasticity blunted fear memory retrieval-induced suppression of feeding in mice, demonstrating the role of this tripartite circuit in controlling behavior.
RESULTS
Retrieval of a fear-induced memory reduced feeding
To study how fear-induced stress affects feeding, mice were subjected to a classical conditioning protocol in which a tone was paired with a trailing electrical foot shock (Figure S1A). Control mice were only exposed to the tone. One hour after conditioning, mice were given access to food for 3 h (Figure S1B). FC animals and control animals (tone) did not differ in food consumption in this situation, regardless of whether feeding occurred during the light or dark cycle (Figure S1B, left or right), although conditioning induced freezing more robustly during the active dark cycle (Figure S2A). To determine whether FC affected the drive or motivation to eat, mice were fasted before FC. In contrast to a previous report (Verma et al., 2016), fasted mice exhibited less freezing during FC (Figure S2B). To avoid fasting-induced effects on FC, mice were FC before fasting and subsequent feeding. In this paradigm, FC did not alter food consumption (Figures S1C and S1D). Altering food palatability did not change this outcome (Figure S1E). Altogether, these results show that homeostatic and hedonic feeding in FC mice did not differ from feeding in mice exposed to tone alone.
Even though FC had no effect on food consumption, remembering a fear-inducing event might. To test this hypothesis, mice were subjected to tone-associated FC or simply tone alone (as described earlier). After conditioning, mice were fasted for 18 h before being given access to food. To induce recollection of the foot shock, the conditioned tone was presented when mice were allowed to feed (Figure 1A, upper panel, tone-induced suppression of feeding [TISF]; Figures S2C-S2F). In this paradigm, FC mice consumed significantly less chow and fewer grain pellets than control mice (Figure 1B). Although freezing duration was not affected by food palatability (Figure S2E), total food consumption in this paradigm was normalized by giving mice access to highly palatable chocolate pellets (Figure S1F).
Figure 1. Fear memory retrieval reduced food consumption after fasting.

(A) Cartoons showing protocols of tone-induced suppression of feeding (B) and retrieval-induced suppression of feeding (C).
(B and C) FC mice consumed significantly less chow and fewer grain pellets during (B) and after (C) fear memory retrieval.
Statistics: *p ≤ 0.05, ***p ≤ 0.001, Mann-Whitney U test. Also see Figures S1 and S2 and Table S1.
To determine whether a retrieved fear memory had a lasting effect on feeding, mice were FC, fasted, and then exposed to a single fear-associated tone before being given access to food (Figure 1A, lower panel, retrieval-induced suppression of feeding [RISF]; Figures S2G-S2T). As when the fear-associated tone was present during feeding, FC mice consumed fewer grain pellets than control mice after presentation of the conditioned tone (Figure 1C). Again, increasing the palatability of the food normalized consumption (Figure S1G). Analysis of movement in the open field revealed that FC and control mice approached food at same rate, but FC mice took longer to start eating once the food was reached (Figures S2L-S2N). Interestingly, in both TISF and RISF, FC mice showed less overall movement and more time around the food in the presence of chocolate pellets (Figures S2F, S2O, and S2R). Altogether, these results demonstrate that recalling a fear-inducing event, but not FC per se, reduced consumption of all but the most palatable foods.
LC neurons suppressed feeding by co-releasing NA and glutamate at PBN synapses
The LC and neighboring PBN have been implicated in fear and feeding (Palmiter, 2018; Sara and Bouret, 2012; Wellman, 2000). To test whether they are synaptically coupled, LC neurons were electrically stimulated while recording from the lateral PBN neurons in ex vivo brain slices (Figure 2A). Lateral PBN neurons were studied, because they were separated from LC and medial PBN by the superior cerebellar peduncle, allowing them to be reliably sampled. Surprisingly, LC stimulation evoked a robust glutamatergic excitatory postsynaptic current (EPSC) in PBN neurons that was blocked by the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX, 5 μM) (Figures 2B, 2C, S3A, and S3B). LC stimulation, but not stimulation at neighboring sites, also induced a persistent inward current (PIC) similar to that evoked by activation of α1ARs in other cell types (Grenhoff et al., 1995) (Figure 2B, green trace; Figures S3C and S3D). Application of α1AR-specific antagonist HEAT (2-[[beta-(−4-hydroxyphenyl)ethyl]aminomethyl]-1-tetralone, 2 μM) blocked the PIC (Figure 2B). In current-clamp recordings, LC stimulation evoked excitatory postsynaptic potentials (EPSPs) and action potentials in PBN neurons, and these responses were blocked by a combination of DNQX and HEAT (Figures S3E-S3H).
Figure 2. LC neurons co-released NA and glutamate to excite PBN neurons and suppress feeding.

(A) Electrical stimulation of LC while recording lateral PBN neurons.
(B and C) 20 Hz electrical stimulation of LC induced both EPSCs (black traces and vertical rectangle in B and enlargement in C) and PICs (green traces) that were blocked by the α1AR-specific antagonist HEAT.
(D) Injection of AAV-hSyn-Con/Fon-ChR2-eYFP into the LC of TH-flp;vGlut2-cre mice.
(E–G) TH immunostaining of brain sections from (D) showing vGlut2 and TH double-positive neurons (eYFP+) in LC. (F) The triangle labels a LC neuron with strong eYFP expression but low TH immunoreactivity (TH-IR), and the star labels a LC neuron with weak eYFP expression but a strong TH-IR signal. (G) White arrowheads label TH-IR and eYFP double-positive fibers, red arrowheads label TH-IR-positive but eYFP-negative fibers, and green arrowheads label eYFP-positive but TH-IR-negative fibers.
(H and I) Recording of lateral PBN neuron while optically stimulating LC input showed both EPSCs and PICs blocked by the α1AR antagonist HEAT.
(J and K) Bilateral expression of hM3D(Gq)-mCherry in LC neurons.
(L–N) Bath application of CNO (3 μM) increased LC firing in ex vivo recordings.
(O) Systemic injection of CNO (1 mg/kg) did not affect feeding in wild-type (WT) mice (left) but inhibited feeding in TH-Cre mice with hM3D(Gq) expressing in LC neurons (right).
(P) Cartoon showing circuitry mechanisms of fear-induced suppression of feeding.
Scale bars: 200 ms in (B); 10 ms in (C) and (H); 500 ms in (I); 0.5 min in (O); 50 pA in (B), (C), (H), and (I); 20 mV in (O); 200 μm in (E) and (K); 20 μm in (F), (G), and (L). Dashed line in (O): −40 mV. Statistics: ***p ≤ 0.001, ****p < 0.0001, Wilcoxon test (N), Mann-Whitney U test (O). Also see Figure S3 and Table S1.
Given previous work concluding that LC neurons lack the machinery for glutamate release (Stornetta et al., 2002), the apparent co-release of glutamate and NA by LC neurons was unexpected. A potential pitfall of our experiments is that electrical stimulation might inadvertently activate axons passing through the LC, in addition to the LC neurons. As a first step toward a more definitive assessment of the possibility of co-transmission, an intersectional genetic strategy was used to determine whether tyrosine hydroxylase (TH) and the type 2 vesicular glutamate transporter (vGlut2) were co-expressed by LC neurons. To this end, Ai65 mice, which express tdTomato in cells co-expressing flippase (Flp) and Cre recombinase (Cre), were crossed with a bitransgenic line of mice expressing Flp under control of the TH promoter and Cre under control of the vGlut2 promoter. In these triple-transgenic mice, tdTomato should be present only in cells that expressed both TH and vGlut2 (Figure S3I). No tdTomato cells were observed in neuronal populations that expressed either vGlut2 or TH only (data not shown). Surprisingly, many LC neurons expressed tdTomato in these mice (Figures S3J-S3L). However, tdTomato-expressing neurons also were seen in the PBN, despite TH immunoreactivity being largely absent from this region in adult mice (Figures S3J and S3K). This mismatch suggests that a subset of PBN neurons expressed TH at some point in development, but not in adulthood. To address adult co-expression of vGlut2 and TH, an adeno-associated virus (AAV) carrying a plasmid that expressed eYFP only in the presence of both Flp and Cre (hSyn-Con/Fon-hChR2(H134R)-eYFP) was injected into the LC of young adult bitransgenic mice (TH-Flp, vGlut2-Cre) (Figure 2D). In these mice, there were many eYFP-expressing neurons in the LC but essentially no eYFP-expressing neurons in the PBN (Figures 2E-2G), confirming the co-expression of TH and vGlut2 in mature LC neurons.
Although suggestive, these experiments do not prove that LC neurons co-release NA and glutamate. To provide a more definitive test of this hypothesis, AAV-EF1a-DIO-hChR2(H134R)-eYFP was stereotaxically injected into the LC of TH-Cre mice, leading to ChR2 expression in TH-expressing LC neurons (Figures S3M-S3U). In these mice, eYFP-labeled LC axons fibers were seen within the PBN (Figures S3V-S3Y). Optical stimulation of LC axons in ex vivo brain slices evoked EPSCs in PBN neurons that were partially blocked by the glutamate receptor antagonist DNQX (5 μM) (Figures 2H and 2I). Thus, in agreement with the inference from the intersectional genetic experiments, at least a subset of LC neurons released glutamate. An optical stimulus train evoked a DNQX-sensitive current and a slower inward current resembling the PIC described earlier that was attributable to α1AR activation (Figures 2H and 2I, green trace). To avoid concerns about ectopic ChR2 expression and to unequivocally assess LC-derived effects, the site of optical stimulation was moved from the PBN to the neighboring LC proper. Optical stimulation of this site at 20 Hz evoked reliable spiking in LC neurons and EPSCs in PBN neurons near the stimulation frequency (Figures S3Z and S3AA). These results provide compelling support for the hypothesis that a subset of LC neurons co-releases NA and glutamate at PBN synapses.
LC excitation of PBN neurons establishes a means by which fear-induced LC activation might terminate feeding (Campos et al., 2016, 2018; Carter et al., 2013). To address this point, chemogenetic methods were used. First, AAV-DIO-hM3D(Gq)-mCherry was injected bilaterally in the LC of TH-Cre mice (Figures 2J and 2K). Next, the efficacy of the excitatory DREADD was confirmed in ex vivo slice recordings (Figures 2L-2N). Lastly, the effect of systemic CNO treatment (to activate LC neurons) on feeding was examined. Although CNO (1 mg/kg) administration did not affect feeding in mice without DREADD expression, it significantly suppressed feeding in fasted mice expressing the excitatory DREADD in LC neurons (Figure 2O), confirming the role of LC neurons in the suppression of feeding (Figure 2P).
FC depressed CeA synapses on PBN neurons
As noted earlier, another potential node in the circuitry controlling fear-induced suppression of feeding is the CeA. To assess its role in the suppression of feeding by fear memory retrieval, the circuitry linking the CeA with the LC and PBN was examined. At the outset, optogenetic approaches were used to assess the functional connectivity of the CeA neurons with those in the LC (McCall et al., 2015; Van Bockstaele et al., 1998). An AAV carrying a hSyn-ChR2(H134R)-eYFP expression construct was stereotaxically injected into the CeA of mice (Figures 3A and 3B). Consistent with previous reports, eYFP fluorescence following this injection was found not in the LC proper but rather in the peri-LC region, where the dendrites of LC neurons are commonly found (Tjoumakaris et al., 2003; Van Bockstaele et al., 1996, 1998) (Figures 3C, S4A, and S4B). Despite this overlap, optogenetic stimulation of CeA axons did not evoke either inhibitory postsynaptic currents (IPSCs) (0/38 cells from 5 animals) (Figures 3D and 3E) or EPSCs (data not shown) in LC neurons. To provide a positive control, neurons in the ventrolateral periaqueductal gray (vlPAG), which are known to receive CeA input (Tovote et al., 2016), were examined. Optical stimulation of CeA axons consistently evoked IPSCs in vlPAG neurons (4/5 cells from 3 animals) (Figure S4C), demonstrating that this approach reliably activated CeA axons. These results suggest that CeA neurons do not directly innervate LC neurons in mice.
Figure 3. FC depressed CeA synapses on PBN neurons.

(A) Schematic showing injection of AAV-ChR2(H134R)-eYFP into the CeA of WT mice.
(B and C) Representative coronal brain sections showing the injection site with ChR2(H134R)-eYFP expression in the CeA (B) and the CeA terminals expressing ChR2(H134R)-eYFP in PBN, but not in LC (C). The CeA also projects to the lateral habenula (LHb).
(D–G) Representative recordings of CeA IPSC (postsynaptic neuron held at 0 mV, black trace) in PBN (F), but not in LC (E). The IPSC was blocked by GABAzine (held at 0 mV, blue trace). A summary of the percentages of cells responding to CeA stimulation is shown in (G).
(H–J) Depression of CeA synapses (ChR2 in H and I and Chronos in J) on PBN after FC (H and J) and after FC-consolidation-reconsolidation (I). Example traces are shown in the upper panels.
(K and L) Representative traces (K) and summary of paired-pulse ratio (PPR) of CeA-Chronos IPSCs in PBN showing an increase of PPRs in FC mice (L).
Scale bars: 1 mm in (B); 200 μM in (C); 200 pA and 20 ms in (E), (F), and (H)–(J); 50 pA and 20 ms in (K). Statistics: **p ≤ 0.005, Mann-Whitney U test. BLA, basolateral amygdala; LPBN, lateral PBN; MPBN, medial PBN; scp, superior cerebellar peduncle; 4V, fourth ventricle. Also see Figure S4 and Table S1.
CeA also has been reported to innervate the PBN (Douglass et al., 2017; Jia et al., 2005; Moga and Gray, 1985). In contrast to LC, optical stimulation of CeA axons in ex vivo brain slices reliably evoked IPSCs in both lateral and medial PBN neurons (Figures 3F, 3G, and S4D). In current-clamp recordings, these inhibitory inputs were large enough to silence spiking in PBN neurons (Figure S4E). Altogether, these results show that γ-aminobutyric acid (GABA)-ergic CeA neurons implicated in FC innervate PBN neurons, but not neighboring LC neurons.
To determine whether FC affected the functional connectivity of the CeA with the PBN, CeA neurons were infected with AAV-hSyn-ChR2(H134R)-eYFP, and then 3–4 weeks later, mice were subjected to FC using either one- or three-day protocols (Figures S2U and S2V). These protocols induced pronounced freezing during fear memory acquisition and retrieval (Figures S2U-S2W). In ex vivo brain slices from FC mice, CeA-evoked IPSCs in lateral PBN neurons were significantly smaller than those evoked in control mice exposed to tone alone (Figures 3H and 3I). To determine whether the reduction in IPSC amplitude was associated with a change in release probability, CeA neurons were infected with an AAV carrying a hSyn-Chronos-EGFP expression construct, rather than one carrying ChR2; Chronos allows axons to be stimulated at frequencies relevant for assessing changes in release probability (Klapoetke et al., 2014). Chronos-evoked IPSCs in lateral PBN neurons were also significantly smaller in FC mice than in control mice (Figure 3J). Consistent with a presynaptic reduction in release probability, the pair-pulse ratio (PPR) at CeA synapses on lateral PBN neurons was significantly increased in slices from FC mice compared with controls (Figures 3K and 3L). These results suggest FC induced a form of plasticity at CeA synapses on lateral PBN neurons that was accompanied by a decrease in presynaptic release probability.
LTD of CeA synapses on PBN was mediated by eCBs
CeA neurons express cannabinoid type 1 receptors (CB1Rs) (Kamprath et al., 2011; Metna-Laurent et al., 2012), making it possible that these receptors, which are widely implicated in presynaptic plasticity, mediated the fear-induced depression of CeA synapses. As a first step toward testing this hypothesis, a selective CB1R agonist Win55,212-2 (WIN, 5 μM) was bath applied to ex vivo brain slices while recording optically evoked IPSCs in PBN neurons. In naive mice with CeA injections of either AAV-hSyn-ChR2(H134R)-YFP or AAV-hSyn-Chronos-GFP, WIN induced a robust and long-lasting reduction in IPSC amplitude (Figures 4A and 4B). Moreover, the LTD at CeA synapses on PBN neurons was associated with an increased PPR (Figure 4C), implicating a presynaptic, CB1R-dependent mechanism, as described in many other brain regions (Castillo et al., 2012).
Figure 4. Activation of CB1Rs induced eCB-LTD at CeA synapses on lateral PBN after FC.

(A and B) WIN induced LTD of CeA IPSCs in PBN (postsynaptic neurons held at −50 mV) in naive animals. Summary in (B).
(C) WIN increased PPR of CeA IPSCs in PBN.
(D) WIN-induced LTD of CeA IPSCs in PBN was occluded in the FC mice.
(E–H) Rimonabant injection before FC (E) and fear retrieval (G) did not affect fear learning or retrieval but blocked depression of CeA IPSCs in PBN (uninjected controls of tone and FC were from historical data in Figures 3H and 3I but at 100% LED intensity).
Scale bars: 20 ms in (A), (C), and (D); 100 pA in (A); 50 pA in (C) and (D). Statistics: *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001, ****p ≤ 0.0001, Wilcoxon test (C), Kruskal Wallis with Dunn’s correction (F and H). Also see Table S1.
To determine whether the plasticity at CeA synapses induced by FC was mediated by CB1Rs, two sets of experiments were performed. First, an occlusion experiment was performed. WIN was bath applied to ex vivo brain slices from FC mice while monitoring the CeA-evoked IPSCs in PBN neurons. In contrast to the situation in naive mice, WIN failed to affect CeA-evoked IPSCs in slices from FC mice (Figure 4D), suggesting CB1R-dependent LTD had already been induced. Next, rimonabant, a brain penetrant and inverse agonist of CB1Rs, was systematically administrated (3 mg/kg) 2 h before FC. Rimonabant injection per se did not affect fear memory acquisition or retrieval (Figures 4E and 4G). However, in rimonabant-treated FC mice, the amplitudes of the CeA-evoked IPSCs in lateral PBN neurons were indistinguishable from those in controls in both one-day and three-day FC protocols (Figures 4F and 4H). Thus, CB1Rs appeared to be responsible for the fear-induced LTD at CeA synapses on PBN neurons.
LC neurons heterosynaptically control eCB-LTD induction
The postsynaptic generation of eCBs requires a combination of depolarization and activation of G-protein-coupled receptors (GPCRs) coupled through Gq proteins to phospholipase C isoforms (Hashimotodani et al., 2007). Thus, the induction of eCB-LTD at inhibitory GABAergic CeA synapses on PBN neurons must be controlled heterosynaptically. Neurons in the LC are obvious candidates for this role in FC. To determine whether they alone could meet the signaling requirements for the induction of eCB-LTD at CeA synapses, LC neurons were activated with a patterned electrical stimulus train intended to mimic the activity produced by foot shocks (Martins and Froemke, 2015). Indeed, LC stimulation, but not stimulation at neighboring sites, induced robust LTD at CeA synapses on PBN neurons (Figures 5A and S5A-S5F). Moreover, the induced LTD was blocked by the CB1R antagonist AM251 (Figure 5B).
Figure 5. LC induced eCB-LTD at CeA synapses on lateral PBN.

(A and B) Electrical stimulations of LC-induced LTD of CeA IPSCs (A), which was blocked by a CB1R antagonist (4 μM AM251, B).
(C) Injection of AAV-Syn-ChrimsonR-tdTomato into the CeA and AAV-EF1α-DIO-ChR2-eYFP into the LC of TH-cre animals for recordings of both CeA and LC inputs in the lateral PBN neurons with different optical stimulations.
(D) Expression of ChrimsonR-tdTomato in the CeA.
(E–G) Expression of ChR2-eYFP in the LC (F) and LC-eYFP and CeA tdTomato fibers in the lateral PBN (G).
(H and I) Optical stimulation of LC-induced LTD of CeA IPSCs (H), which was blocked by AM251 (I).
Scale bars: 40 ms and 100 pA in (A), (B), (H), and (I); 200 μm in (D) and (E); 20 μm in (F) and (G). Also see Figures S5 and S6.
To verify that LC stimulation per se was sufficient to induce eCB-LTD at CeA synapses on PBN neurons, a dual opsin strategy was used. First, AAV-hSyn-ChR2(H134R)-YFP or AAV-Syn-ChrimsonR-tdTomato was injected into the CeA (Figures S6A-S6E). In ex vivo brain slices from these mice, it was verified that ChrimsonR was activated by both 470 or 615 nm pulses (although 615 nm was more effective) (Figures S6B-S6E) (Klapoetke et al., 2014). Next, LC neurons were induced to express ChR2 by injecting AAV-EF1a-DIO-ChR2(H134R)-eYFP into TH-Cre mice, and lateral PBN neurons were recorded following stimulation by 470 or 615 nm pulses in LC. EPSCs in lateral PBN neurons were evoked only by 470 nm pulses (Figure S6F). Lastly, the ability of LC stimulation to modulate CeA-evoked responses in PBN neurons was assessed in ex vivo slices (Figures 5C-5G and S6G-S6T). After baseline recording of CeA IPSCs in PBN neurons evoked with 615 nm pulses, LC axons were repetitively (5×) stimulated with a short 20 Hz train (1 s duration; 1 min inter-stimulation interval, mimicking the activity produced by foot shocks) of 470 nm pulses. After this induction protocol, CeA-evoked IPSCs in PBN neurons were recorded again by switching back to 615 nm pulses. LC stimulation induced a robust LTD at CeA synapses on PBN neurons (Figure 5H). As expected, antagonizing CB1Rs with AM251 blocked LTD induction (Figure 5I). These results confirm that LC stimulation is sufficient to induce eCB-LTD at CeA synapses on PBN neurons.
To initially assess the potential role of co-transmission in the LC-induced eCB-LTD, pharmacological approaches were taken. Consistent with a central role for adrenergic signaling in the LC-induced LTD, bath application of NA (10 μM) to ex vivo brain slices induced long-lasting depression of CeA-evoked IPSCs in PBN neurons held at −50 mV (to mimic the effect of ionotropic receptor activation) (Figure 6A); as expected, the CB1R antagonist AM251 (4 μM) blunted the effects of NA on the amplitude of CeA IPSCs in PBN neurons (Figure 6B). The α1AR agonist phenylephrine (10 μM), but not the βAR agonist isoproterenol (10 μM), mimicked the effect of NA on synaptic transmission (Figures 6C and 6F). As expected, this LTD was blocked by the CB1R antagonist AM251 (Figure 6F). Consistent with a role for metabotropic glutamate receptors (mGluRs), bath application of the type I mGluR (mGluR-I) agonist, (S)-3,5-dihydroxyphenylglycine (DHPG, 100 μM) produced LTD at CeA synapses on PBN neurons when they were held at −50 mV; as expected, this LTD was also blocked by the CB1R antagonist AM251 (Figures 6D-6F). So, with depolarization of PBN neurons, either α1AR or mGluR signaling was sufficient to induce CB1R-dependent LTD at CeA synapses.
Figure 6. Activation of α1ARs and mGluR-I induced LTD at CeA synapses on lateral PBN.

(A and B) NA (10 μM, A) induced LTD of CeA IPSCs, which was blocked by AM251 (B).
(C) Activation of α1ARs with phenylephrine (Phe, 10 μM) induced LTD of CeA IPSCs.
(D and E) Activation of mGluR-I with DHPG (100 μM, D) induced LTD of CeA IPSCs, which was blocked by AM251 (E).
(F) Summary showing activation of α1ARs with Phe and mGluR-I with DHPG, but not βARs with isoproterenol, induced eCB-LTD at CeA synapses on PBN.
Scale bars: 40 ms and 100 pA in (A)–(E). Statistics: *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001, Kruskal Wallis with Dunn’s correction (left), Mann-Whitney U test (middle and right). Also see Table S1.
Next, the necessity of co-transmission for LC-induced LTD was tested. As a first step toward making this determination, the impact of antagonizing both α1ARs and mGluRs was examined. Indeed, antagonizing α1ARs with HEAT (2 μM), mGluR5s with 2-methyl-6-(phenylethynyl)pyridine (MPEP, 10 μM), and mGluR1s with 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt, 25 μM) fully blocked the LTD induced by LC stimulation (Figures S5G-S5L). To test for the necessity of α1ARs, HEAT (2 μM) was bath applied before LC electrical stimulation. Antagonizing α1ARs significantly diminished LTD induction at CeA synapses (Figures 7A and 7C). In contrast, antagonizing mGluR5s and mGluR1s with the combination of MPEP and CPCCOEt did not significantly reduce the median LTD (Figures 7B, 7C, and S7H-S7J).
Figure 7. NA and glutamate co-released by LC synergized LTD at CeA synapses on lateral PBN.

(A–C) LC electrical-stimulation-induced LTD of CeA IPSCs was significantly diminished by the α1AR antagonist HEAT (2 μM, A), but not by the combination of mGluR1 and mGluR5 antagonists MPEP (10 μM) and CPCCOEt (25 μM) (MP+CP, B). Summary in (C).
(D) Scatterplot of EPSC and PIC amplitudes of LC synapses on PBN (n = 30 cells from 22 animals). Highlights are PBN neurons that received high glutamate but low NA release or receptor activation.
(E) Two example PBN neurons with strong EPSCs but weak PICs. MP+CP blocked LTD, but HEAT did not.
(F–H) In a subset of PBN neurons with weak PICS (<20 pA), MP+CP blocked LTD (F). In a subset of PBN neurons with strong EPSCs (>80 pA), HEAT failed to block LTD (G). Summary in (H).
(I) Example images showing the variability of TH and eYFP (TH+vGlut2) expression in TH-flp;vGlut2-cre animals injected with AAV-hSyn-Con/Fon-ChR2-eYFP into the LC.
(J–M) Activation of mGluR-I with DHPG (100 μM, J) or α1ARs with Phe (10 μM, K) induced labile eCB-LTD, whereas activation of both receptors with DHPG+Phe (L) induced static eCB-LTD of CeA IPSCs. Summary in (M).
(N) Cartoon showing LC activates both α1ARs and mGluR-I in PBN neurons, which synergize to induce eCB-LTD at the CeA synapses on PBN.
Scale bars: 40 ms, 100 pA in (A), (B), (F), (G), and (J)–(L); 10 μm in (I). Statistics: *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001, ****p ≤ 0.0001, Kruskal Wallis with Dunn’s correction. Also see Figure S7 and Table S1.
These results were surprising given the evidence for co-transmission and the sufficiency of mGluR signaling in the induction of LTD at CeA synapses. To better understand the role of glutamatergic co-transmission, the relative amplitudes of the LC-evoked α1AR-dependent PIC and the AMPA-receptor-mediated EPSC were plotted for a sample of PBN neurons (Figure 7D). Most PBN neurons had a relatively large evoked PIC, consistent with a dominant role for NA signaling in LTD induction. However, in a subset of PBN neurons, the LC-evoked glutamatergic EPSC was relatively large (more than −80 pA) and the PIC was relatively small (less than −20 pA) (Figure 7D, red rectangle highlight). This suggests that in this type of PBN neuron, the relative roles of adrenergic and glutamatergic signaling in LTD induction might be reversed. In PBN neurons with small adrenergic PICs (less than −20 pA), antagonizing mGluR signaling disrupted LC-induced LTD induction at CeA synapses; in contrast, in PBN neurons with large glutamatergic EPSCs (more than −80 pA), antagonizing α1ARs did not disrupt LC-induced LTD induction at CeA synapses (Figures 7E-7H and S7). These results show that in most PBN neurons, α1AR signaling plays a critical role in LTD induction at CeA synapses; however, in a subset of PBN neurons, glutamatergic signaling is necessary and sufficient to induce LTD (Figure S7).
The apparent heterogeneity in PBN LTD may arise from a variation in the properties of presynaptic LC neurons. In bitransgenic mice (TH-Flp, vGlut2-Cre) injected with Con/Fon-eYFP virus, many LC neurons strongly expressed eYFP (indicating co-release), but there was considerable variability in immunoreactivity for TH (Figure 2F, triangle, and Figure 7I), suggesting mosaicism in the properties of LC neurons.
A more basic question is why LC neurons would co-release NA and glutamate. One possibility is that the intracellular signaling pathways of α1AR and mGluR1/5 complement one another in a way that has a functional consequence. One intriguing idea is that co-activation of these receptors controls the transition from labile to static (or persistent) LTD (Atwood et al., 2014). To test this hypothesis, α1AR or mGluR agonists were washed out and the CB1R inverse agonist AM251 was applied after synaptic transmission had been depressed for 15 min. Surprisingly, the depression induced by either DHPG or phenylephrine alone was labile and reversed in the presence of AM251 (Figures 7J and 7K). However, when α1AR and mGluR1/5 agonists were co-applied—mimicking glutamate and NA co-release—the LTD at CeA synapses became static (Figures 7L and 7M). Thus, co-transmission may gate the stability of LTD at CeA synapses and the duration of the suppression in feeding caused by LC activation (Figure 7N).
CeA CB1Rs are necessary for fear-induced suppression of feeding
Our results suggest that LC-dependent eCB-LTD at CeA synapses on PBN neurons reduced feeding induced by retrieval of a fearful memory. If this was the case, silencing LC neurons or blocking LTD induction should disrupt the ability of fear retrieval to suppress feeding. To silence LC neurons during FC, chemogenetic methods were used. First, AAV-DIO-hM4D(Gi)-mCherry was injected bilaterally in the LC of TH-Cre mice (Figure 8A). Next, the efficacy of the inhibitory DREADD was confirmed in ex vivo slice recordings (Figures 8B and 8C). Lastly, the effect of systemic CNO treatment (to inhibit LC neurons) during FC on RISF was examined (Figures S8A-S8E). CNO (1 mg/kg) administration prevented the suppression of feeding induced by fear memory retrieval (Figure 8D), confirming the role of LC neurons in the RISF.
Figure 8. Silencing LC neurons and knocking down CB1Rs specifically in CeA blocked reduction of feeding induced by fear memory retrieval; knocking down CB1Rs also blocked loss of weight induced by Phenylephrine.

(A) Bilateral injection of AAV-hSyn-DIO-hM4D(Gi)-mCherry into the LC of TH-cre mice.
(B and C) Bath application of CNO (3 μM) inhibits LC neuron firing in ex vivo recordings.
(D) Systemic injection of CNO (1 mg/kg) blocked retrieval-induced suppression of feeding. Dashed lines: median of historical data in WT mice in Figure 1C.
(E) Bilateral injection of AAV-Cre-tdTomato into the CeA of CB1Rlox/lox mice.
(F and G) Cre injection significantly reduced CB1R mRNA expression in the CeA (F), but not BLA (G). FC per se did not affect CB1R expression in both CeA and BLA (WT).
(H and I) Knocking down CB1Rs in CeA significantly blocked tone-induced suppression of feeding (H) and retrieval-induced suppression of feeding (I). Dashed lines: medians of historical data in WT mice in Figures 1B and 1C.
(J) Single injection of 10 mg/kg, but not 1 or 0.1 mg/kg, of Phenylephrine-induced weight loss ten days afterward in WT mice.
(K) Knocking down CB1Rs in CeA significantly diminished Phenylephrine-induced weight loss.
Scale bars: 200 μm in (A) and (E). 30 s and 100 pA in (C). Statistics: *p ≤ 0.05, **p ≤ 0.005, ****p ≤ 0.0001, Wilcoxon test (B), Mann-Whitney U test (D, H, and I), Kruskal Wallis with Dunn’s correction (F and G). Also see Figure S8 and Table S1.
To disrupt LTD induction, the brain penetrant and CB1R inverse agonist rimonabant was administrated systemically (3 mg/kg) 2 h before FC (Figures S8F-S8K). Rimonabant significantly increased the consumption of grain pellets by FC mice (Figure S8G). The problem with this pharmacological strategy is that the systemic effects of rimonabant on feeding are complex (McLaughlin et al., 2003; Soria-Gómez et al., 2014). To more directly test our hypothesis, a genetic strategy was used. CB1Rs in CeA neurons were specifically downregulated by injecting an AAV carrying a Cre-tdTomato expression construct into the CeA of CB1R floxed (CB1Rlox/lox) mice. Confocal microscopy was used to confirm infection of CeA neurons (Figure 8E) and quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) methods were used to verify loss of mRNA coding for CB1Rs in CeA (Figure 8F). Importantly, the local AAV injection did not result in the knockdown mRNA for CB1Rs in the neighboring basolateral amygdala (Figure 8G). In mice injected with AAV-expressing Cre, but not those receiving a control vector, TISF was significantly blunted (Figure 8H). Similarly, CeA knockdown of CB1R expression significantly diminished the suppression of feeding induced by retrieval of a fearful memory (Figure 8I). The effect of disrupting CB1R signaling in CeA neurons was specific to feeding, because freezing was not significantly different in treated and control mice (Figures S8L-S8S).
Lastly, to verify that the LC circuitry controlling the CeA synapses affected feeding, mice were given systemic injections of the α1AR agonist phenylephrine. As predicted by the ability of α1AR signaling to suppress feeding, a single 10 mg/kg injection caused significant weight loss (Figure 8J). However, in mice lacking CeA CB1Rs, the effect of phenylephrine on body weight was significantly blunted (Figure 8K), suggesting that LC control of CeA synapses and feeding was not limited to fear-inducing situations.
DISCUSSION
Our studies demonstrate that there is a tripartite circuit controlling the ability of fear to suppress feeding. Five key observations were made. First, it was shown that a tone associated with foot shock was able to suppress feeding in fasted mice. Second, LC neurons excited PBN neurons known to mediate the suppression of feeding. The excitation of PBN neurons was evoked by the co-release of NA and glutamate. Third, FC induced a CB1R-dependent form of presynaptic LTD at inhibitory synapses formed by GABAergic CeA neurons on PBN neurons. Fourth, the conditioning-induced LTD at CeA synapses was mediated heterosynaptically by LC neurons. Lastly, the ability of FC stimuli to suppress feeding was blunted by either silencing LC neurons or deleting CeA CB1Rs, establishing a causal link between the tripartite circuit and the fear-memory-induced suppression of feeding. Altogether, these results suggest that in response to real fearful events, LC neurons directly excite PBN neurons to transiently suppress feeding and enable defensive or escape behaviors. However, LC activation also has lasting effects that are mediated by attenuation of inhibitory CeA control of PBN. This synaptic attenuation may be maladaptive, because it may blunt the ability of amygdalar networks to promote feeding during fear retrieval, when no tangible threat exists. These studies shed new light on the neural mechanisms underlying the ability of fear to suppress eating, even when hungry—opening new therapeutic avenues for disorders like post-traumatic stress disorder (PTSD) and anorexia.
LC neurons co-released NA and glutamate to excite PBN neurons and suppress feeding
The closeness of LC and PBN neurons makes it difficult to interpret responses to local electrical stimulation. Using unambiguous genetic tools, our experiments revealed that LC neurons co-released NA and glutamate to excite PBN neurons. Although contrary to the inference drawn from a previous study that relied upon in situ hybridization, which may not be sensitive enough to detect vGlut2 in LC neurons (Stornetta et al., 2002), three observations supported the conclusion that LC neurons released glutamate (in addition to NA). First, intersectional genetic methods revealed that a subset of LC neurons co-expressed vGlut2 and TH in adult mice. Second, optogenetic activation of LC neurons evoked EPSCs in PBN neurons that were blocked by AMPA receptor antagonists. Third, in a subset of PBN neurons, LC-induced LTD at CeA synapses was blocked by group 1 mGluR antagonists.
The co-release of NA and glutamate from LC neurons directly excited PBN neurons, creating a means by which threatening events could suppress feeding and enable escape or defensive behaviors. Chemogenetic activation of LC neurons, mimicking the response to fear-inducing events, suppressed feeding in fasted animals that had not been FC. Our observations are consistent with recent work showing that activation of local GABAergic neurons in LC promotes feeding (Marino et al., 2020). Interestingly, PBN neurons also make excitatory, glutamatergic synapses on LC neurons (Liu et al., 2015), suggesting that the suppression of feeding might be augmented by a positive feedback loop between LC and PBN neurons.
FC attenuated CeA coupling to PBN
The CeA has been implicated in the regulation of both fear-associated behavior and feeding (Cai et al., 2014; Douglass et al., 2017; Hardaway et al., 2019; Ip et al., 2019; Janak and Tye, 2015). Still, how the CeA might link fear and feeding is just beginning to be understood. It is likely that different subcircuits in the CeA are involved. CeA neurons that express protein kinase Cδ (PKCδ) are inhibited by conditional stimulus after FC (Ciocchi et al., 2010; Haubensak et al., 2010). Moreover, activation of PKCd-expressing CeA (CeAPKCδ) neurons suppresses feeding, whereas activation of CeA neurons that do not express PKCδ (CeAPKCδ−) does not (Cai et al., 2014). In contrast, fasting activates a subset of CeA neurons (Wu et al., 2014). This latter group of neurons expresses some combination of the serotonin receptor 2a (Htr2a) and the Drd1 dopamine receptor (Douglass et al., 2017; Kim et al., 2017). The CeAPKCδ− GABAergic neurons are robustly connected to PBN neurons that suppress feeding, creating a means by which hunger can promote food consumption (Douglass et al., 2017). In contrast, the CeAPKCδ GABAergic neurons do not directly innervate PBN neurons but instead inhibit CeAPKCδ− neurons driven by hunger (Cai et al., 2014). Optogenetic stimulation of presumptive CeAPKCδ− neurons evoked robust GABAergic IPSCs in both medial and lateral PBN neurons.
FC attenuated the strength of this coupling by inducing a CB1R-dependent LTD on CeA terminals in the PBN. This conclusion was based upon the ability of CB1Rs to produce a classical presynaptic LTD at CeA synapses on PBN neurons in ex vivo tissue from unconditioned mice, the inability of CB1Rs to modulate CeA transmission in tissue from conditioned mice, and the normalization of CeA-evoked responses in FC animals by systemic treatment with the CB1R inverse agonist rimonabant. Moreover, knocking down CB1Rs in CeA neurons significantly reduced the suppression of feeding by fear retrieval, arguing that the presynaptic LTD made a substantive contribution to the network activity controlling feeding.
LC neurons induced eCB-LTD at CeA synapses on PBN neurons
Several lines of evidence point to the LC as being responsible for the CB1R-dependent LTD at CeA synapses on PBN neurons. First, the LC is activated during FC (Martins and Froemke, 2015). Second, it was shown here that electrical or optogenetic activation of LC neurons induced a heterosynaptic, CB1R-dependent form of LTD at CeA synapses on PBN neurons. These experiments took advantage of dual opsin methods to selectively activate LC axons while recording from PBN neurons. However, a caveat in these experiments was that CeA axons also were activated during the induction of LTD. Whether presynaptic activity in CeA axons is required for the LTD induction is not clear. Previously, presynaptic activity was found to be necessary for eCB-LTD induction at excitatory synapses (Singla et al., 2007). It is also unclear whether the LC-induced LTD was induced in all types of PBN neurons. LTD was evident in the region of the PBN invested with LC axons and lacking strong immunoreactivity for calcitonin gene-related peptide (CGRP) (Figures S3R-S3Y), but CGRP-expressing PBN neurons (Campos et al., 2016; Carter et al., 2013) were not explicitly examined. Additional work will be required to determine whether LTD is induced in this subtype of PBN neurons.
The induction of LTD at CeA synapses by LC neurons largely depended on release of NA and activation of postsynaptic PBN α1ARs. The induction of this form of LTD has two core requirements (Castillo et al., 2012; Hashimotodani et al., 2007). First, the postsynaptic membrane must be depolarized enough to open the Cav1 Ca2+ channels. Second, Gq-linked GPCRs need to be activated to stimulate the phospholipase C metabolism of membrane lipids. LC release of NA and activation of Gq-linked α1ARs met both of these requirements, because their activation triggered a depolarizing PIC that was of sufficient magnitude to cause spiking in PBN neurons. This excitatory effect was similar to that seen in dopaminergic neurons following α1AR activation (Grenhoff et al., 1995). Although the mechanisms mediating the depolarization remain to be resolved, Ca2+-stimulated opening of TRP channels is one possibility (Launay et al., 2002).
In addition to engaging α1ARs, many LC neurons co-released glutamate, resulting in activation of both ionotropic receptors and mGluRs on PBN neurons—well-known drivers of activity-dependent eCB-LTD (Castillo et al., 2012; Hashimotodani et al., 2007). The ability of co-release-mediated signaling in PBN neurons to both drive spiking and depress inhibitory GABAergic input from CeA provides an elegant mechanism by which LC activation in response to fear or threatening events could exert both transient and sustained effects on feeding. The lack of effect of FC on body weight argues that the lasting depression of CeA synaptic transmission did not alter the relationship between hunger and feeding in the absence of fear-inducing stimuli.
In addition to providing an ancillary means of inducing LTD at CeA synapses on PBN neurons, co-release of glutamate by noradrenergic LC neurons may serve another important function at CeA synapses on PBN neurons. eCB-dependent, presynaptic LTD can be labile or static (Atwood et al., 2014). Termination of presynaptic CB1R signaling with AM251 15 min after initiating induction led to reversal of the LTD produced by bath application of either α1AR or mGluR agonists. Hence, both signaling pathways induced a labile form of eCB-LTD when stimulated on their own. However, when both receptors were engaged, the eCB-LTD was resistant to AM251 and was static, persisting for as long as the recording was held. Although the precise mechanism governing the transition from labile to static remains to be determined, it is tempting to speculate that it is a threshold phenomenon governed by presynaptic CB1Rs. If the generation of eCBs by combined α1AR/mGluR stimulation exceeds that possible by stimulating either one alone, then that transition threshold might be reached. It is also possible that this convergence enables the participation of other synaptic inputs in the induction of LTD at CeA synapses. Regardless, our results establish a biological logic for co-transmission that has largely been lacking at other synapses in which co-transmission has been documented. Although our studies are the first to demonstrate co-release of glutamate and NA by LC neurons, adrenergic neurons in the nucleus of the solitary tract (NTS) also have been reported to co-release glutamate (Roman et al., 2016), as have dopaminergic neurons innervating the striatum (Poulin et al., 2018). The biological logic of co-release by these other cell types remains to be determined but could follow that of LC neurons at CeA synapses.
The lack of demonstrable CeA connectivity with LC
Although several recent studies have suggested that CeA neurons directly innervate LC neurons (Bouret et al., 2003; Schwarz et al., 2015; Van Bockstaele et al., 1996, 1998), optical stimulation of CeA axons failed to evoke a response in most LC neurons in our hands, in agreement with earlier work (Aston-Jones et al., 1986). There are two obvious interpretations of this discrepancy. One is that CeA neurons are polysynaptically coupled, but not monosynaptically coupled, to LC neurons. Several potential polysynaptic pathways link CeA and LC. For example, CeA neurons innervate PBN neurons, which in turn innervate LC neurons (Liu et al., 2015). Although the previously reported electrophysiological coupling could be accounted for in this way, it is more difficult to reconcile the anatomical data using monosynaptic rabies virus mapping (msRVm) (Schwarz et al., 2015). Although it is possible that the starter populations in these experiments included neurons outside the LC, another explanation is that CeA neurons make synaptic contact with LC neurons but that these connections are functionally insignificant. A limitation of the msRVm approach is that it cannot be used to judge the functional strength of a connection. In principle, msRVm does not distinguish between a presynaptic neuron that make one synapse with a starter cell and a neuron that makes a thousand. It also is possible that CeA neurons innervating LC rely upon a transmitter whose postsynaptic effects would not have been apparent in our experiments. CeA neurons release various neuromodulators that could fall into this category, but CeA neurons typically co-release GABA and produced robust GABAergic IPSCs in neighboring PBN neurons, making this explanation unlikely. Determining which of these possibilities is true will require additional study.
Summary and translational implications
Altogether, our studies have identified a novel neural mechanism that controls fear-induced suppression of feeding. Upon fear or threatening stimuli, LC neurons co-release both NA and glutamate to depolarize PBN neurons to transiently suppress feeding (Figure 2P). Co-release also induced a heterosynaptic form of presynaptic, CB1R-dependent LTD at CeA synapses on PBN neurons (Figure 7N). Following the retrieval of a fearful memory, this LTD appears to blunt the ability of hunger-driven activity in CeA neurons to inhibit PBN neurons and promote feeding. Deletion of CB1Rs selectively from CeA neurons prevented fear retrieval from affecting food consumption in hungry mice.
In principle, this insight should provide a potential path for the development of pharmacotherapies to treat patients for whom traumatic memories disrupt eating, as in PTSD or anorexia (Carmassi et al., 2015; Mitchell et al., 2012). CeA CB1Rs are the obvious target for this kind of approach. However, CB1Rs are widely expressed in the brain and shape a range of behaviors, in addition to feeding (Lutz et al., 2015). Systemically administered CB1R agonists stimulate and antagonists reduce food consumption, particularly of highly palatable food (Arnone et al., 1997; Di Marzo et al., 2001; Jo et al., 2005). Even within the PBN, infusion of CB1R agonists increases the consumption of highly palatable food, possibly by modulating hypothalamic synapses on CGRP-expressing neurons (Campos et al., 2016; DiPatrizio and Simansky, 2008). These considerations and the problematic clinical experience with rimonabant make CB1Rs an untenable target for a systemic drug (Sam et al., 2011).
An alternative target is the α1ARs expressed by PBN neurons. Systemic administration of an α1AR agonist induces weight loss (Morien et al., 1993; Yeh, 1999). Our findings directly implicate the LC innervation of PBN in this effect, complementing early work implicating the paraventricular hypothalamus and medial raphe nucleus (Silva et al., 2017; Wellman et al., 1993). Because LC and PBN neurons are activated by various fearful stimuli, but not painful stimuli (Aston-Jones et al., 1991; Palmiter, 2018), it is likely that the mechanisms described here extend to other fear-producing situations. Thus, an α1AR antagonist should attenuate stress-induced maladaptive suppression of feeding. The α1AR antagonist prazosin has been shown to reduce nightmares of PTSD patients (Ahmadpanah et al., 2014; Raskind et al., 2013), suggesting that this class of drug might have broader utility in treating the consequences of trauma.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be addressed to Lead Contact, D. James Surmeier (j-surmeier@northwestern.edu).
Materials availability
Request for CB1Rlox/lox line should be addressed to Dr. Eric Delpire (eric.delpire@vanderbilt.edu); request for vGlut2-cre; TH-2A-Flp and vGlut2-cre; TH-2A-Flp; Ai65 lines should be addressed to Dr. Rajeshwar Awatramani (r-awatramani@northwestern.edu); request for all other materials should be addressed to D. James Surmeier (j-surmeier@northwestern.edu).
Data and code availability
All datasets contributing to this study is summarized in Table S1. Raw data are available with reasonable request to the Lead Contact D. James Surmeier (j-surmeier@northwestern.edu). No custom code, software, or algorithm was used in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Adult (8-12 weeks) mice were used for this study. All surgeries for stereotaxic injections of recombinant adeno-associated virus (rAAV) carrying ChR2, Chronos, ChrimsonR, hM3D(Gq), hM4D(Gi) or Cre recombinase were carried out in 8-9 weeks old mice. All fear conditioning and feeding behaviors were performed in 12 weeks old mice. C57BL/6J (JAX #00064) mice were used for expression of hSyn-hChR2(H134R)-YFP, Syn-Chronos-GFP, and behaviors. CRF-Cre (JAX #011087) and TH-Cre (JAX #008601) were used for expression of DIO-hChR2(H134R)-eYFP, DIO-hM3D(Gq)-mCherry or DIO-hM4D(Gi)-mCherry. vGlut2-Cre (JAX#016963) and TH-Flp (Poulin et al., 2018) were crossed to generate TH-Flp;vGlut2-Cre mice, which were used for expression of Con/Fon-eYFP. These mice were further crossed with Ai65 (JAX#021875) to generate TH-Flp;vGlut2-Cre;Ai65 mice (Dr. Rajeshwar Awatramani). CB1Rlox/lox mice (Dr. Eric Delpire), generated as previously described (Marcus et al., 2020), were used for expression of CMV-Cre-tdTomato or CMV-control-tdTomato (from Virovek) and behaviors. Males were used for all behavior experiments, including fear conditioning and feeding (Figures 1, 2J-2O, 3H-3L, 4D-4H, and 8). Both males and females were used for all other experiments addressing the circuit mechanisms (Figures 2A-2I, 3A-3G, 4A-4C, 5, 6, and 7). We did not see a clear difference between sexes, thus data from both sexes were combined. All animals were housed and handled according to the guidelines established by the Northwestern University Animal Care and Use Committee, the National Institutes of Health and the Society for Neuroscience.
Fear conditioning
Fear conditioning training (fear memory acquisition) and retention tests (fear memory retrieval) were performed at the Northwestern University Behavioral Phenotyping Core. Animals were allowed to acclimate to the behavior room for at least 30 minutes before all behavior. For the training, a rat-sized Coulbourn arena (Coulbourn Rat Arena: E63-20) with mouse shock floor (H10-11R/M-TC-SF) was used. The rat-sized arena increases baseline ambulation to make it easier to detect freezing and the mouse shock floor provides properly sized rails for mouse. A precision animal shocker (H13-15) was used to deliver the shocks. For the retention test, a clean open rat cage without lid and bedding placed in a 56 × 56 × 30 cm gray PVC open field arena (Phenome Technologies, Inc. Lincolnshire, IL) was used. Training and retention tests were both performed in a light- and sound-attenuating chamber (Industrial Acoustics Corp.). The Coulbourn arena with mouse shock floor for training and rat cage within the open field arena for retention test provide different environmental contexts and floors. Other than that, training was performed with white light and curtain up; the retention test was performed with red dim light and curtain down. Between individual tests, arenas and rat cages were cleaned to prevent odor influences from the previous test. The activity of each mouse was recorded with CCD camera vertically mounted above. Shock and sound delivery and camera recordings were remotely controlled by the experimenter with FreezeFrame 5 software (Actimetrics, Wilmette, IL). For each experiment, mice were gently placed in the center of the arena or rat cage at the beginning of the test. Mice were allowed to explore the environment for three minutes before the recording started. Each recording consists of three minutes baseline, fear conditioning training or retention test, and one-minute end recording. Fear conditioning training consists of 20 s tone (2900 Hz, 80 dB) that co-terminates with 1 s of shock (0.6 mA) and 40 s inter-stimulation-interval. The retention test is a single 60 s tone (2900 Hz, 80 dB). Also see details in Figures S2U-S2V. Times that mice freeze and move were analyzed with Actimetrics’ FreezeFrame software. Percentage of time that mice freeze (time of freeze/total time) was reported for baseline and during tone. In some experiments, the motion index was also analyzed (See Figures S2C, S2F, S8I, S8K, S8M, and S8O).
METHOD DETAILS
Feeding
All feedings, except TISF that was done in a clean rat cage and RISF that was done in an open field arena, were done in a fresh mouse cage without bedding and with food in a food cup in a corner under where home cage food used to be. Mice were habituated and familiarized with the feeding setting and food types for at least two consecutive days before the experiment.
For light-cycle feeding measurement, mice were either fear-conditioned or treated with tone alone at 9:00 a.m. in the fear conditioning arena as described above. Mice were then transferred to fresh cages with no food provided. One hour later, feeding was performed in another fresh cage with food in a food cup on the floor and water bottle above the lid as in home cages for three hours. Both fear conditioning and feeding were performed under white light.
For dark-cycle feeding measurement, mice were either fear-conditioned or treated with tone alone at 6:00 p.m., and food intake was monitored during a three hours period starting one hour after fear conditioning. All contexts were the same as light-cycle feeding except under dim red light.
For post-fast feeding measurement, mice were either fear-conditioned or treated with tone alone at 9:00 a.m., then transferred to a fresh cage with no food provided on the same day either at 3:00 p.m. for 18 hours fasting or 12:00 a.m. for 9 hours fasting. After fasting, on the second day at 9:00 a.m., animals were allowed to feed as described above. Foods were weighed at 10 min, 30 min, 1 hour, and 3 hours after feeding.
For the tone-induced suppression of feeding (TISF) test, mice were fear-conditioned and fasted as described above. After fasting, on the second day at 9:00 a.m., mouse was transferred to a clean rat cage without bedding placed within a light- and sound-attenuating chamber as described above for retention test under dim red light. In a corner of the rat cage, a food cup with food was placed on the floor, allowing the mouse to feed for 10 minutes. During this 10 minutes feeding, four tones were delivered for fear memory retrieval, each 60 s, with 90 s inter-stimulation interval (also see Figures 1A and S2C).
For the retrieval-induced suppression of feeding (RISF) test, mice were first treated the same as for the TISF test, except when transferred to a clean rat cage on the second day, no food cup was placed on the floor of the cage. After 90 s baseline recording, a 60 s tone was delivered for fear memory retrieval. Immediately after fear memory retrieval, the mouse was transferred to a clean open field arena in another light- and sound-attenuating chamber as described above for the retention test but under white light and with food in a food cup placed in the center of the arena, allowing the mouse to feed for 10 minutes (also see Figures 1A and S2G).
Note that fear memory retrieval was under the same context in all experiments except that for the retention test, in which the mice were not fasted and no food provided during tone delivery, and for the TISF test, in which mice were fasted and food was provided during tone delivery, and for the RISF test, in which mice were fasted but no food provided during tone delivery (also see Figure S2W). For both TISF and RISF, mice were fasted to ensure that a readily measurable amount of food was consumed during the observation period.
For CNO injection, either wild-type mice or TH-cre mice were injected with either hM3D(Gq) or hM4d(Gi) virus 3-4 weeks before experiments. For hM3D(Gq) injected mice, one day before feeding, mice were transferred to a fresh cage with no food provided at 3:00 p.m. for 18 hours fasting. After fasting, on the second day at 8:30 a.m., mice were i.p. injected with either saline or CNO (1 mg/kg) then allowed to feed standard chow as described above at 9:00 a.m for three hours. For hM4d(Gi) injected mice, CNO (1 mg/kg) was injected 0.5 h before fear conditioning. Mice were then fasted for 18 hours from 3 p.m. to 9 a.m. before an exposure to a brief tone and feeding in an open arena as described above for RISF.
Following are foods used:
Standard Chow (Teklad #7912): Protein 19.1%, Fat 5.8%, Carbohydrate 44.3%, Calorie 3.10 kCal/g; Grain Pellet (Bio-Serv #F0163): Protein 21.3%, Fat 3.8%, Carbohydrate 54% (Monosaccharides 105 g/kg, Disaccharides 140 g/kg, Polysaccharides 278 g/kg), Calorie 3.35 kCal/g; Chocolate Pellet (Bio-Serv #F05301): Protein 18.4%, Fat 5.5%, Carbohydrate 59.1% (Monosaccharides 268 g/kg, Disaccharides 310 g/kg, Polysaccharides 12.8 g/kg), Calorie 3.60 kCal/g. All foods are similar in energy density.
In all feeding experiments, food was weighed before and after feeding. In rare cases where food got wet by urine or water, food was dried and weighed the second day.
Brain slice preparation
Mice were anesthetized with a ketamine/xylazine mixture, followed by a transcardial perfusion with ice-cold, carbogen-saturated cutting solution containing 185 mM Sucrose, 2.5 mM KCl, 25 mM NaHCO3, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 10 mM MgCl2, and 25 mM glucose, pH 7.3 (315–320 mOsm L−1). After perfusion, mice were decapitated and brains removed rapidly, followed by sectioning in an ice-cold carbogen-saturated cutting solution using a vibratome (VT1000S, Leica Microsystems). Either coronal or horizontal slices (220 μm thick) were incubated in carbogen-saturated artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgCl2, and 25 mM glucose, pH 7.3 (315–320 mOsm L−1) at 32–34°C for 30 min, then at room temperature for another 30 min before electrophysiological recordings. Slices were then transferred to a small-volume (< 0.5 ml) recording chamber that mounted on a fixed-stage, upright microscope (BX51, Olympus America). Electrophysiological recordings were performed at 32–34°C. The chamber was superfused with carbogen-saturated ACSF running through an in-line heater (SH-27B with TC-324B controller, Warner Instruments). For AM251 pre-treatment, slices were pre-incubated for 60 min with AM251 containing ACSF and then placed in a recording chamber with continuous drug superfusion.
Electrophysiology
Conventional tight-seal (> 2 GΩ) whole-cell patch-clamp recordings were made on visually identified (60 × , 0.9 NA water-immersion objective) LC and PBN neurons based on location (LC locates near the floor of the fourth ventricle, LPBN & MPBN separated by superior cerebellar peduncle), size (LC: a cluster of neurons with large somatic compartment, typical soma diameter ~20 μm), somatodendritic morphology (LC: generally fusiform or triangular soma with 3-6 primary dendrites; PBN: medium-to small-sized cells with more rounded soma) and firing (LC regular spiking between 1 and 4 Hz). Signals were filtered at 1–4 kHz and digitized at 5–20 kHz with a Digidata 1400 (Molecular Devices). For current-clamp recordings, the amplifier bridge circuit was adjusted to compensate for electrode resistance and monitored. For both cell-attached and whole-cell current-clamp recordings, internal solution containing 135 mM KMeSO4, 5 mM KCl, 5 mM HEPES, 0.05 mM EGTA, 10 mM Na2PCr (phosphocreatine disodium), 2 mM ATP-Mg and 0.5 mM GTP-Na, the pH adjusted to 7.3 (290–300 mOsm L−1) was used. For all cell-attached or whole-cell recordings of LC intrinsic firing with CNO application, synaptic blockers (10 μM DNQX, 50 μM D-AP5, 10 μM GABAzine, 1 μM CGP55845) were bath applied. For whole-cell voltage-clamp recordings of IPSCs or EPSCs, internal solution containing 135 mM CsMeSO3, 3.3 mM QX-314-Cl, 10 mM HEPES, 0.05 mM EGTA, 8 mM phosphocreatine-di(tris), 4 mM ATP-Mg and 0.3 mM GTP-Na, the pH adjusted to 7.3 (290–300 mOsm L−1) was used. The liquid junction potential in our recording ACSF using this internal solution was 7 mV and not corrected for. For IPSCs recordings, AMPA and NMDA antagonists (10 μM DNQX, 50 μM D-AP5) were bath applied, except where EPSCs were also recorded (experiments in characterizing the CeA inputs to PBN and LC, and LC inputs induced LTD at the CeA synapses on PBN). For EPSCs recordings, GABAA and GABAB antagonists (10 μM GABAzine, 1 μM CGP55845) were bath applied except where IPSCs were also recorded (LC inputs induced LTD at the CeA synapses on PBN).
All optical-stimulated IPSCs or EPSCs were evoked by either ChR2/Chronos stimulation using 470-nm wavelength or ChrimsonR stimulation using a 615-nm wavelength with 1 ms light pulses (CoolLED pE-100). For LTD and PPR recordings, the light power was adjusted in each recording to evoke a baseline IPSCs between 200 - 400 pA at - 50 mV. In experiments where WIN was applied in fear-conditioned mice, baseline IPSCs between 100 and 200 pA at −50 mV were also used. Series resistance was monitored by a hyperpolarizing step (−5mV, 100 ms) with each sweep. The series resistances were typically <20 MΩ, and data were discarded if the value changed by more than 20% during the recordings.
There were three protocols for the LTD experiments. In the first protocol, PBN neurons were voltage-clamped at −50 mV while CeA axons were optically stimulated. CeA-IPSCs were recorded every 30 s and the average of two consecutive IPSCs was calculated and its amplitude plotted (one data point for every minute in Figures 4A, 4B, 4D, 6A-6E, and 7J-7L). In the second protocol, CeA and LC axons were alternatingly stimulated in successive 30 s intervals during LTD induction; in the interval when CeA axons were stimulated, PBN neurons were held at −50 mV and when LC axons were stimulated, PBN neurons were held at −70 mV. Before and after LTD induction, two consecutive CeA-IPSCs (one every 30 s) were averaged and the amplitude plotted (one data point for every minute); during LTD induction segment, the amplitude of a single CeA-IPSC (every minute) was plotted (one data point for every minute in Figures 5A, 5B, 5H, 5I, 7A, 7B, 7E-7G, S5E, S5K, S7C, and S7F). In the third protocol, PBN neurons were in current clamp during LC stimulation (the LTD induction period). CeA-IPSCs were measured at −50 mV (in voltage clamp) before and after LC stimulation. The average of every two consecutive recordings was calculated and its amplitude plotted (one data point for every minute in Figures S7H, S7I, S7M, and S7N). See more details of LC optical-/electrical-stimulation below.
For the LC optical-stimulation induced LTD experiment, before recording CeA-IPSCs with 615-nm wavelength stimulation of ChrimsonR, single 470-nm wavelength optical stimulations of ChR2 were delivered and EPSCs recorded. Stimulation intensity was adjusted. Only cells with clear EPSCs (> 10 pA at −70 mV) were used for LTD experiments. After baseline recording of CeA-IPSCs at −50 mV, cells were voltage-clamped at −70 mV for 20 Hz optical-stimulation of LC for one second, with one-minute inter-stimulation interval for five times. In the middle of each 20 Hz optical-stimulation of LC, cells were voltage-clamped back at −50 mV for recordings of CeA-IPSCs (Figures 5H and 5I).
The electrical-stimulated EPSCs were evoked by electrical stimulation using a concentric bipolar electrode (FHC #30203) and an isolated current stimulator (100 μs, Digitimer Model DS3). For the LC electrical-stimulation induced LTD experiments, before recording CeA-IPSCs, single electrical stimulations were delivered and EPSCs were recorded. Stimulation intensity was adjusted. Only cells with clear EPSCs (> 10 pA at −70 mV) were used for LTD experiments. After baseline recording of CeA-IPSCs at −50 mV, cells were either voltage-clamped at −70 mV or current-clamped at 0 pA for 20 Hz electrical-stimulation of LC for one second, with one-minute inter-stimulation interval for five times. For voltage-clamp recordings, in the middle of each 20 Hz electrical-stimulation of LC, cells were voltage-clamped back at −50 mV for recordings of CeA-IPSCs (Figures 5A, 5B, 7A, 7B, 7E-7G, S5E, S5K, S7C, and S7F). For current clamp recordings, cells were current-clamped throughout LC stimulation (Figures S7H, S7I, S7M, and S7N).
For paired-pulse recordings, the ratio of the amplitudes of second IPSC (IPSC2) over the first IPSC (IPSC1) was calculated. The second IPSC typically rose before the first IPSC decayed to baseline, initially a curved was fitted to the decay of the first IPSC for the accurate estimation of the amplitude of the second IPSC. Because the first IPSC had decayed back by within 2 pA by the time of second IPSC peak, the amplitude estimated of the second IPSC was not corrected for residual current from the first IPSC for all recordings.
For most recordings of IPSCs, EPSCs, or action potentials, both coronal and horizontal slices were used. For comparing CeA-IPSCs between Tone and FC animals, only LPBe neurons in horizontal sections (the fourth section in Figure S4B) that showed the strongest fluorescence signals and were anatomically distinct and easy to identify were patched to keep consistency between animals. For LC electrical stimulations, we found coronal sections better preserved both LC and PBN in the same slices and easier to record LC-EPSCs in PBN neurons.
Viral vectors
100 nL of AAV2/9-hSyn-hChR2(H134R)-eYFP, AAV9-Syn-Chronos-GFP, AAV9-Syn-ChrimsonR-tdTomato, 200 nL of AAV9-EF1a-DIO-hChR2(H134R)-eYFP (UNC vector core) were injected into either CeA or LC for optical stimulations. 200 nL of AAV8-hSyn-DIO-hM3D(Gq)-mCherry or AAV8-hSyn-DIO-hM4D(Gi)-mCherry (UNC vector core) was injected into LC of TH-Cre animals for chemogenetic manipulation. 150 nL of AAV5-hSyn-Con/Fon-hChR2(H134R)-eYFP-WPRE (UNC vector core) was injected into LC of TH-Flp;vGlut2-Cre animals for intersectional genetic labeling of vGlut2 and TH co-expressing neurons. 200 nL of AAV9-CMV-Cre-2A-tdTomato, AAV9-U6-Control-shRNA-CMV-tdTomato-WPRE-bGHpA (customer made from Virovek) were injected into CeA of CB1Rlox/lox animals for knocking down CB1R expression specifically in CeA. The shRNA in the control vector was a scrambled construct that had been used as a control in another context. It was used as a control here for viral infection and expression of tdTomato.
Stereotaxic injection
Adult mice (8-9 weeks) were anaesthetized with isoflurane (induction 3%, maintenance 2%) delivered continuously with 100% medical O2. Throughout the surgery, mice were placed under a heating pad to avoid hypothermia. Mice were injected subcutaneously with Metacam (0.1mg/ml) and placed in a Kopf stereotaxic frame apparatus with head position to obtain a flat skull between bregma and lambda. A craniotomy was performed using micro-drill to expose brain tissue and recombinant adeno-associated virus (AAV) virus was injected with a glass micropipette (VWR) directly into the CeA region (1.00 mm posterior to bregma; 2.78 mm lateral to midline; and 3.90 mm deep from the cortical surface) or LC region (4.90 mm posterior to bregma; 1.10 mm lateral to midline; and 3.25 mm deep from the cortical surface). After three-four weeks post-surgery for ChR2, ChrimsonR, hM3D(Gq) or hM4D(Gi) expression and four weeks for Cre expression, mice were used for behavior training and/or slice preparation for recordings and/or tissue collection for quantification of CB1R mRNA levels and/or fixation for immunostaining and imaging.
Immunohistochemistry
Mice were transcardially perfused with 4% paraformaldehyde (PFA) in PBS (1X, pH 7.4). Brains were removed and postfixed in 4% PFA for overnight at 4°C. Thin brain sections (40-50 μm-thick, except for the triple-crossed mice that were sliced at 25 μm-thick) were obtained using a Leica vibratome VT1200S. Brain slices after electrophysiological recordings (220 μm-thick) from the following two conditions were also fixed in 4% PFA for overnight at 4°C for immunostaining: 1). When the cells were filled with biocytin for staining and labeling of the cells patched; 2). When a live imaging was taken showing the placement of stimulating electrode and location of cells being patched, then immunostaining showing TH expression and location of LC neurons were performed. Sections were permeabilized with 0.3% Triton X-100 in PBS, rinsed with PBS for three times, blocked in 10% normal goat serum followed by incubation with a mouse monoclonal antibody against tyrosine hydroxylase (TH) (Immunostar #22941, except for the triple-crossed animals that were stained with sheep polyclonal anti-TH from Pel-Freez #P60101-150, both with a working dilution of 1:1000), goat anti-CGRP (abcam#ab36001, 1:1000 dilution), or rabbit anti-GFP (ThermoFisher#A11122, 1:1000 dilution) for overnight at 4°C. Staining was visualized with goat anti-mouse secondary antibody conjugated to Alexa 594, goat anti-sheep Alexa 488, donkey anti-goat Alexa 350, or goat anti-rabbit Alexa 488 (ThermoFisher: #A11032, A11015, A21081, A11034, all 1:1000 dilution) and Cy5-Streptavidin (ThermoFisher: #434316, 1:200 dilution) for biocytin either two hours at room temperature or overnight at 4°C. Images were acquired with an Olympus FluoView FV10i self-contained confocal microscope (equipped with four (405/473/559/635nm) diode lasers and two objectives (10x and 60x)), with the exception of the triple-crossed animals that were acquired with an Olympus SZX-12 fluorescence stereomicroscope with a digital camera (Q image Retiga 2000R), a fluorescence slide scanner (Olympus) and a 20x objective. All images were stored at 12-bit image depth at a resolution of 1024x1024 pixels.
Quantitative PCR analysis of cannabinoid receptor1 (CB1R)
RNA was isolated from the amygdala tissues collected by microdissection of the interested areas from 300 μm brain slices using RNAeasy mini kit (QIAGEN). The RNA was reverse transcribed with SuperScript VILO cDNA Synthesis Kit (Invitrogen). Quantitative real-time PCR was performed using ABI StepOnePlus Real-Time PCR System with Taqman PCR Master Mix (Applied Biosystems, Forster City, CA). The abundance of different transcripts was assessed by Taqman quantitative PCR in triplicates. Taqman probes were used for PCR amplification of Crn1 (#Mm01212171_s1), and Gapdh (#Mm99999915_g1)) genes. Experimental Ct values were normalized to Gapdh values using the formula: ΔCt = Ct (Crn1) −Ct (gapdh). An averaged ΔCt value of CeA from animals treated with tone alone was calculated as ΔCt(reference). The final expression levels were shown as 2^[ΔCt(reference)- ΔCt(sample)] as fold change of mRNA expression level.
Pharmacological reagents and chemicals
Reagents were purchased from Sigma except for GABAzine (SR95531, Tocris#1262), CGP55845 (Tocris#1248), D-AP5 (Tocris#0106), DNQX (Tocris#2312), Noradrenaline (Abcam#ab120717), HEAT (Tocris#0535), Phenylephrine (Tocris#2838), Isoproterenol (Tocris#1747), Win (Tocris#1038), AM251 (Tocris#1117), Rimonabant (Tocris#0923), MPEP (Tocris#1212), CPCCOEt (Tocris#1028), CNO (Hello Bio#HB6149), Cy5-streptavidin (ThermoFisher#434316). For i.p. injection, Rimonabant (Tocris #0923) was first prepared 30 mg/mL stock in DMSO, then diluted in saline with 1% DMSO, 1% Cremophor, 1% Ethanol to make 0.3 mg/mL for injection, finally injected 0.01 mL/g to reach 3 mg/kg injection. Phenylephrine (Tocris #2838) was prepared at 10 mM stock solution in water, then diluted in saline for injection. CNO (HelloBio #HB6149) was prepared 0.1 mg/mL in saline and injected 0.01 mL/1 g to reach 1 mg/kg injection. For slice recordings, all stocks were diluted to final concentrations in ACSF to achieve a final solvent concentration of less than 0.1% v/v
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analyses were performed with Clampex 10.7 and Clampfit 10.7 (Molecular Devices Inc.) and Igor Pro 6.32 (WaveMetrics, Lake Oswego, OR). Cell counting was performed with IMARIS 8.2 (Bitplane). Statistical analyses were conducted in GraphPad Prism 6 (GraphPad Software Inc., CA, USA) (see all statistic analysis in Table S1). Data were scatterplotted and boxplotted with a thick line as median, a box showing first/third quartiles and whiskers representing minimum/maximum values. In other cases, lines with symbols and shadow were used with symbols showing median and shadow representing first/third quartiles or thick lines showing median and thin lines representing first/third quartiles. All data, including outliers, were included in the scatterplot. Outliers were excluded in boxplots and statistical analysis. Outliers were defined as numbers with values that are more than 1.5 times the length of the box away from either the lower (< Q1 - 1.5 × IQR) or upper ((> Q3 + 1.5 × IQR) quartiles. Two-tailed Mann-Whitney test was used for comparison between two groups. Kruskal-Wallis test with Dunn’s correction was used for comparison between multiple groups. For comparison of two paired groups, two-tailed Wilcoxon matched-pairs signed-rank test was used. For LTD data, Mann-Whitney U test was performed comparing the average of all baseline 10 min data with all end 10 min data. If the U test showed significance, we would consider as LTD. If the U test showed non-significance, then the average of baseline 10-min data with the average of 10-min starting from the last 5 min of induction were compared. If significant, we consider as short-term depression (STD) or labile LTD if antagonist was applied (Figures 7J and 7K); if non-significant, we consider as no effect. In this study, we did not observe STD. For comparison between two or more groups with two factors, two-way ANOVA with Bonferroni’s correction for multiple comparisons between each group and each data point in each group was used. “n” indicates cell number, “N” indicates animal number. Data exclusion: one wild-type animal was found dead three days after 10 mg/kg phenylephrine injection with continuous weight loss.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Tyrosine Hydroxylase (TH) mouse monoclonal antibody | Immunostar | 22941; RRID: AB_572268 |
| Tyrosine Hydroxylase (TH) sheep polyclonal antibody | Pel-Freez | P60101-150; RRID: AB_461070 |
| CGRP goat polyclonal antibody | Abcam | 36001; RRID: AB_725807 |
| GFP rabbit polyclonal antibody | ThermoFisher | A11122; RRID: AB_221569 |
| goat anti-mouse Alexa 594 | ThermoFisher | A11032; RRID: AB_2534091 |
| goat anti-sheep Alexa 488 | ThermoFisher | A11015; RRID: AB_2534082 |
| donkey anti-goat Alexa 350 | ThermoFisher | A21081; RRID: AB_2535738 |
| goat anti-rabbit Alexa 488 | ThermoFisher | A11034; RRID: AB_2576217 |
| Bacterial and virus strains | ||
| AAV2/9-hSyn-hChR2(H134R)-eYFP | UNC vector core | N/A |
| AAV9-Syn-Chronos-GFP | UNC vector core | N/A |
| AAV9-EF1a-DIO-hChR2(H134R)-eYFP | UNC vector core | N/A |
| AAV9-Syn-ChrimsonR-tdTomato | UNC vector core | N/A |
| AAV8-hSyn-DIO-hM3D(Gq)-mCherry | UNC vector core | N/A |
| AAV8-hSyn-DIO-hM4D(Gi)-mCherry | UNC vector core | N/A |
| AAV5-hSyn-Con/Fon-hChR2(H134R)-eYFP-WPRE | UNC vector core | N/A |
| AAV9-CMV-Cre-2A-tdTomato | Virovek (Hayward, CA) | N/A |
| AAV9-U6-Control-shRNA-CMV-tdTomato-WPRE-bGHpA | Virovek (Hayward, CA) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| GABAzine | Tocris | 1262 |
| DNQX | Tocris | 2312 |
| D-AP5 | Tocris | 0106 |
| MPEP | Tocris | 1212 |
| CPCCOEt | Tocris | 1028 |
| Win | Tocris | 1038 |
| AM251 | Tocris | 1117 |
| Rimonabant | Tocris | 0923 |
| Noradrenaline | Abcam | Ab120717 |
| Phenylephrine | Tocris | 2838 |
| Isoproterenol | Tocris | 1747 |
| HEAT | Tocris | 0535 |
| Biocytin | Sigma | B4216 |
| CNO | Hello Bio | HB6149 |
| Cy5-streptavidin | ThermoFisher | 434316 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: CRF-cre: B6;FVB-Tg(Crh-cre) 1Kres/J | The Jackson Laboratory | RRID: IMSR_JAX:011087 |
| Mouse: TH-cre: B6.Cg-7630403G23RikTg(Th-cre)1Tmd/J | The Jackson Laboratory | RRID: IMSR_JAX:008601 |
| Mouse: vGlut2-Cre: Slc17a6tm2(cre)Lowl/j | The Jackson Laboratory | RRID: IMSR_JAX:016963 |
| Mouse: TH-2A-Flp | Poulin et al., 2018 | N/A |
| Mouse: vGlut2-cre; TH-2A-Flp | Poulin et al., 2018 | N/A |
| Mouse: Ai65: B6;129S-Gt(ROSA) 26Sortm65.1(CAG-tdTomato)HZe/J | The Jackson Laboratory | RRID: IMSR_JAX:021875 |
| Mouse: vGlut2-cre; TH-2A-Flp; Ai65 | Dr. Rajeshwar Awatramani This paper | N/A |
| Mouse: CB1Rlox/lox | Marcus et al., 2020 | N/A |
| Oligonucleotides | ||
| crn1 | ThermoFisher | Mm01212171_s1 |
| Gapdh | ThermoFisher | Mm99999915_g1 |
| Software and algorithms | ||
| Clampex & Clampfit 10.7 | Molecular Devices Inc. | RRID: SCR_011323 |
| FreezeFrame 5 | Actimetrics | RRID: SCR_014429 |
| Igor Pro 6.32 | WaveMetrics | RRID: SCR_000325 |
| Prism 6 | GraphPad Software Inc. | RRID: SCR_002798 |
| IMARIS 8.2 | Bitplane | RRID: SCR_007370 |
| Other | ||
| Standard chow | Teklad | 7912 |
| Grain pellet | Bio-Serv | F0163 |
| Chocolate pellet | Bio-Serv | F05301 |
Highlights.
Retrieval of fearful memories suppresses feeding in fasted animals
LC neurons co-release noradrenaline and glutamate to suppress feeding
The co-release activates PBN neurons and induces eCB-LTD of amygdalar inputs to PBN
Disruption of eCB signaling alleviates fear-induced suppression of feeding
ACKNOWLEDGMENTS
We thank Kang Chen, Dannielle R. Schowalter, Marisha Alicea, and Bonnie M. Erjavec for genotyping and mouse colony management and Sasha Ulrich for lab management and administrative assistance. We thank Drs. Weixing Shen, Tamara Perez-Rosello, and Asami Tanimura for discussion and comments on the manuscript; Dr. Tristano Pancani for providing the drawing of a mouse in Illustrator; and all members of the Surmeier laboratory for their support. We especially thank Yu Chen for help with RT-PCR and Dannielle R. Schowalter for help with FC behavior experiments. We thank Dr. Jelena Radulovic for help with designing behavior tests and comments on the manuscript and her lab members, particularly Lynn Ren, for comments on the manuscript, and Dr. Craig Weiss, Mary Kando, and the Northwestern University Behavioral Phenotyping Core for help with FC behavior and data analysis. This work is supported in part by NIH Pre-Doctoral Training Grant NUIN-MRS T32 (to B.Y.) and by the DOD and the JPB Foundation (to D.J.S.). The CB1Rlox/lox mouse generation was supported by the Gene-Targeted Mouse Core of the INIAstress Consortium, supported by National Institute on Alcohol Abuse and Alcoholism grant U01 AA013514 (to E.D.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2020.12.023.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmadpanah M, Sabzeiee P, Hosseini SM, Torabian S, Haghighi M, Jahangard L, Bajoghli H, Holsboer-Trachsler E, and Brand S (2014). Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology 69, 235–242. [DOI] [PubMed] [Google Scholar]
- Arnone M, Maruani J, Chaperon F, Thiébot MH, Poncelet M, Soubrié P, and Le Fur G (1997). Selective inhibition of sucrose and ethanol intake by SR 141716, an antagonist of central cannabinoid (CB1) receptors. Psychopharmacology (Berl). 132, 104–106. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Ennis M, Pieribone VA, Nickell WT, and Shipley MT (1986). The brain nucleus locus coeruleus: restricted afferent control of a broad efferent network. Science 234, 734–737. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Chiang C, and Alexinsky T (1991). Discharge of noradrenergic locus coeruleus neurons in behaving rats and monkeys suggests a role in vigilance. Prog. Brain Res 88, 501–520. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Lovinger DM, and Mathur BN (2014). Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 37, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block JP, He Y, Zaslavsky AM, Ding L, and Ayanian JZ (2009). Psychosocial stress and change in weight among US adults. Am. J. Epidemiol 170, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret S, Duvel A, Onat S, and Sara SJ (2003). Phasic activation of locus ceruleus neurons by the central nucleus of the amygdala. J. Neurosci 23, 3491–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush DEA, Caparosa EM, Gekker A, and Ledoux J (2010). Beta-adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front. Behav. Neurosci 4, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Haubensak W, Anthony TE, and Anderson DJ (2014). Central amygdala PKC-δ(+) neurons mediate the influence of multiple anorexigenic signals. Nat. Neurosci 17, 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos CA, Bowen AJ, Schwartz MW, and Palmiter RD (2016). Parabrachial CGRP Neurons Control Meal Termination. Cell Metab. 23, 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos CA, Bowen AJ, Roman CW, and Palmiter RD (2018). Encoding of danger by parabrachial CGRP neurons. Nature 555, 617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmassi C, Antonio Bertelloni C, Massimetti G, Miniati M, Stratta P, Rossi A, and Dell Osso L (2015). Impact of DSM-5 PTSD and gender on impaired eating behaviors in 512 Italian earthquake survivors. Psychiatry Res. 225, 64–69. [DOI] [PubMed] [Google Scholar]
- Carter ME, Soden ME, Zweifel LS, and Palmiter RD (2013). Genetic identification of a neural circuit that suppresses appetite. Nature 503, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, and Hashimotodani Y (2012). Endocannabinoid signaling and synaptic function. Neuron 76, 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SBE, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, et al. (2010). Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468, 277–282. [DOI] [PubMed] [Google Scholar]
- Dębiec J, Bush DEA, and LeDoux JE (2011). Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats—a possible mechanism for the persistence of traumatic memories in PTSD. Depress. Anxiety 28, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Bátkai S, Járai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, and Kunos G (2001). Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 410, 822–825. [DOI] [PubMed] [Google Scholar]
- DiPatrizio NV, and Simansky KJ (2008). Activating parabrachial cannabinoid CB1 receptors selectively stimulates feeding of palatable foods in rats. J. Neurosci 28, 9702–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass AM, Kucukdereli H, Ponserre M, Markovic M, Gründemann J, Strobel C, Alcala Morales PL, Conzelmann K-K, Lüthi A, and Klein R (2017). Central amygdala circuits modulate food consumption through a positive-valence mechanism. Nat. Neurosci 20, 1384–1394. [DOI] [PubMed] [Google Scholar]
- George SA, Knox D, Curtis AL, Aldridge JW, Valentino RJ, and Liberzon I (2013). Altered locus coeruleus-norepinephrine function following single prolonged stress. Eur. J. Neurosci 37, 901–909. [DOI] [PubMed] [Google Scholar]
- Geracioti TD Jr., Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, Schmidt D, Rounds-Kugler B, Yehuda R, Keck PE Jr., and Kasckow JW (2001). CSF norepinephrine concentrations in posttraumatic stress disorder. Am. J. Psychiatry 158, 1227–1230. [DOI] [PubMed] [Google Scholar]
- Grenhoff J, North RA, and Johnson SW (1995). Alpha 1-adrenergic effects on dopamine neurons recorded intracellularly in the rat midbrain slice. Eur. J. Neurosci 7, 1707–1713. [DOI] [PubMed] [Google Scholar]
- Grossman SP (1960). Eating or drinking elicited by direct adrenergic or cholinergic stimulation of hypothalamus. Science 132, 301–302. [DOI] [PubMed] [Google Scholar]
- Han S, Soleiman MT, Soden ME, Zweifel LS, and Palmiter RD (2015). Elucidating an Affective Pain Circuit that Creates a Threat Memory. Cell 162, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardaway JA, Halladay LR, Mazzone CM, Pati D, Bloodgood DW, Kim M, Jensen J, DiBerto JF, Boyt KM, Shiddapur A, et al. (2019). Central Amygdala Prepronociceptin-Expressing Neurons Mediate Palatable Food Consumption and Reward. Neuron 102, 1037–1052.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, and Kano M (2007). Ca(2+)-assisted receptor-driven endocannabinoid release: mechanisms that associate presynaptic and postsynaptic activities. Curr. Opin. Neurobiol 17, 360–365. [DOI] [PubMed] [Google Scholar]
- Hatfield T, and McGaugh JL (1999). Norepinephrine infused into the basolateral amygdala posttraining enhances retention in a spatial water maze task. Neurobiol. Learn. Mem 71, 232–239. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong H-W, Deisseroth K, Callaway EM, et al. (2010). Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip CK, Zhang L, Farzi A, Qi Y, Clarke I, Reed F, Shi Y-C, Enriquez R, Dayas C, Graham B, et al. (2019). Amygdala NPY Circuits Promote the Development of Accelerated Obesity under Chronic Stress Conditions. Cell Metab. 30, 111–128.e6. [DOI] [PubMed] [Google Scholar]
- Janak PH, and Tye KM (2015). From circuits to behaviour in the amygdala. Nature 517, 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H-G, Zhang G-Y, and Wan Q (2005). A GABAergic projection from the central nucleus of the amygdala to the parabrachial nucleus: an ultrastructural study of anterograde tracing in combination with post-embedding immunocytochemistry in the rat. Neurosci. Lett 382, 153–157. [DOI] [PubMed] [Google Scholar]
- Jo Y-H, Chen Y-JJ, Chua SC Jr., Talmage DA, and Role LW (2005). Integration of endocannabinoid and leptin signaling in an appetite-related neural circuit. Neuron 48, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabitzke PA, Silva L, and Wiedenmayer C (2011). Norepinephrine mediates contextual fear learning and hippocampal pCREB in juvenile rats exposed to predator odor. Neurobiol. Learn. Mem 96, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamprath K, Romo-Parra H, Häring M, Gaburro S, Doengi M, Lutz B, and Pape H-C (2011). Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology 36, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Zhang X, Muralidhar S, LeBlanc SA, and Tonegawa S (2017). Basolateral to Central Amygdala Neural Circuits for Appetitive Behaviors. Neuron 93, 1464–1479.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivimäki M, Head J, Ferrie JE, Shipley MJ, Brunner E, Vahtera J, and Marmot MG (2006). Work stress, weight gain and weight loss: evidence for bidirectional effects of job strain on body mass index in the Whitehall II study. Int. J. Obes 30, 982–987. [DOI] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, and Kinet JP (2002). TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 109, 397–407. [DOI] [PubMed] [Google Scholar]
- Leibowitz SF (1988). Hypothalamic paraventricular nucleus: interaction between alpha 2-noradrenergic system and circulating hormones and nutrients in relation to energy balance. Neurosci. Biobehav. Rev 12, 101–109. [DOI] [PubMed] [Google Scholar]
- Liang KC, McGaugh JL, and Yao HY (1990). Involvement of amygdala pathways in the influence of post-training intra-amygdala norepinephrine and peripheral epinephrine on memory storage. Brain Res. 508, 225–233. [DOI] [PubMed] [Google Scholar]
- Liu C-Y, Lee M-L, Yang C-S, Chen C-M, Min M-Y, and Yang H-W (2015). Morphological and physiological evidence of a synaptic connection between the lateral parabrachial nucleus and neurons in the A7 catecholamine cell group in rats. J. Biomed. Sci 22, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz B, Marsicano G, Maldonado R, and Hillard CJ (2015). The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci 16, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan AL, and Ressler KJ (2012). Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci. 35, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniam J, and Morris MJ (2012). The link between stress and feeding behaviour. Neuropharmacology 63, 97–110. [DOI] [PubMed] [Google Scholar]
- Marcus DJ, Bedse G, Gaulden AD, Ryan JD, Kondev V, Winters ND, Rosas-Vidal LE, Altemus M, Mackie K, Lee FS, et al. (2020). Endocannabinoid Signaling Collapse Mediates Stress-Induced Amygdalo-Cortical Strengthening. Neuron 105, 1062–1076.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino RAM, McDevitt RA, Gantz SC, Shen H, Pignatelli M, Xin W, Wise RA, and Bonci A (2020). Control of food approach and eating by a GABAergic projection from lateral hypothalamus to dorsal pons. Proc. Natl. Acad. Sci. USA 117, 8611–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins ARO, and Froemke RC (2015). Coordinated forms of noradrenergic plasticity in the locus coeruleus and primary auditory cortex. Nat. Neurosci 18, 1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, and Bruchas MR (2015). CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron 87, 605–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin PJ, Winston K, Swezey L, Wisniecki A, Aberman J, Tardif DJ, Betz AJ, Ishiwari K, Makriyannis A, and Salamone JD (2003). The cannabinoid CB1 antagonists SR 141716A and AM 251 suppress food intake and food-reinforced behavior in a variety of tasks in rats. Behav. Pharmacol 14, 583–588. [DOI] [PubMed] [Google Scholar]
- Metna-Laurent M, Soria-Gómez E, Verrier D, Conforzi M, Jégo P, Lafenêtre P, and Marsicano G (2012). Bimodal control of fear-coping strategies by CB1 cannabinoid receptors. J. Neurosci 32, 7109–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meye FJ, and Adan RAH (2014). Feelings about food: the ventral tegmental area in food reward and emotional eating. Trends Pharmacol. Sci 35, 31–40. [DOI] [PubMed] [Google Scholar]
- Mitchell KS, Mazzeo SE, Schlesinger MR, Brewerton TD, and Smith BN (2012). Comorbidity of partial and subthreshold ptsd among men and women with eating disorders in the national comorbidity survey-replication study. Int. J. Eat. Disord 45, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moga MM, and Gray TS (1985). Evidence for corticotropin-releasing factor, neurotensin, and somatostatin in the neural pathway from the central nucleus of the amygdala to the parabrachial nucleus. J. Comp. Neurol 241, 275–284. [DOI] [PubMed] [Google Scholar]
- Morien A, McMahon L, and Wellman PJ (1993). Effects on food and water intake of the alpha 1-adrenoceptor agonists amidephrine and SK&F-89748. Life Sci. 53, 169–174. [DOI] [PubMed] [Google Scholar]
- Mueller D, Porter JT, and Quirk GJ (2008). Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J. Neurosci 28, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell T, Hegadoren KM, and Coupland NC (2004). Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology 50, 273–283. [DOI] [PubMed] [Google Scholar]
- Olson VG, Rockett HR, Reh RK, Redila VA, Tran PM, Venkov HA, Defino MC, Hague C, Peskind ER, Szot P, and Raskind MA (2011). The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol. Psychiatry 70, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD (2018). The Parabrachial Nucleus: CGRP Neurons Function as a General Alarm. Trends Neurosci. 41, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecoraro N, Reyes F, Gomez F, Bhargava A, and Dallman MF (2004). Chronic stress promotes palatable feeding, which reduces signs of stress: feedforward and feedback effects of chronic stress. Endocrinology 145, 3754–3762. [DOI] [PubMed] [Google Scholar]
- Poulin J-F, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, Chan CS, Dombeck DA, Deisseroth K, and Awatramani R (2018). Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat. Neurosci 21, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskind MA, Peterson K, Williams T, Hoff DJ, Hart K, Holmes H, Homas D, Hill J, Daniels C, Calohan J, et al. (2013). A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am. J. Psychiatry 170, 1003–1010. [DOI] [PubMed] [Google Scholar]
- Rieg TS, and Aravich PF (1994). Systemic clonidine increases feeding and wheel running but does not affect rate of weight loss in rats subjected to activity-based anorexia. Pharmacol. Biochem. Behav 47, 215–218. [DOI] [PubMed] [Google Scholar]
- Roman CW, Derkach VA, and Palmiter RD (2016). Genetically and functionally defined NTS to PBN brain circuits mediating anorexia. Nat. Commun 7, 11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam AH, Salem V, and Ghatei MA (2011). Rimonabant: From RIO to Ban. J. Obes 2011, 432607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ (2009). The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci 10, 211–223. [DOI] [PubMed] [Google Scholar]
- Sara SJ, and Bouret S (2012). Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron 76, 130–141. [DOI] [PubMed] [Google Scholar]
- Sato M, Ito M, Nagase M, Sugimura YK, Takahashi Y, Watabe AM, and Kato F (2015). The lateral parabrachial nucleus is actively involved in the acquisition of fear memory in mice. Mol. Brain 8, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, and Luo L (2015). Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva ESD, Flores RA, Ribas AS, Taschetto AP, Faria MS, Lima LB, Metzger M, Donato J Jr., and Paschoalini MA (2017). Injections of the of the α1-adrenoceptor antagonist prazosin into the median raphe nucleus increase food intake and Fos expression in orexin neurons of free-feeding rats. Behav. Brain Res 324, 87–95. [DOI] [PubMed] [Google Scholar]
- Singla S, Kreitzer AC, and Malenka RC (2007). Mechanisms for synapse specificity during striatal long-term depression. J. Neurosci 27, 5260–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria-Gómez E, Massa F, Bellocchio L, Rueda-Orozco PE, Ciofi P, Cota D, Oliet SHR, Prospéro-García O, and Marsicano G (2014). Cannabinoid type-1 receptors in the paraventricular nucleus of the hypothalamus inhibit stimulated food intake. Neuroscience 263, 46–53. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Krystal JH, Morgan CA, Johnson D, Nagy LM, Nicolaou A, Heninger GR, and Charney DS (1993). Abnormal noradrenergic function in posttraumatic stress disorder. Arch. Gen. Psychiatry 50, 266–274. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Sevigny CP, and Guyenet PG (2002). Vesicular glutamate transporter DNPI/VGLUT2 mRNA is present in C1 and several other groups of brainstem catecholaminergic neurons. J. Comp. Neurol 444, 191–206. [DOI] [PubMed] [Google Scholar]
- Tjoumakaris SI, Rudoy C, Peoples J, Valentino RJ, and Van Bockstaele EJ (2003). Cellular interactions between axon terminals containing endogenous opioid peptides or corticotropin-releasing factor in the rat locus coeruleus and surrounding dorsal pontine tegmentum. J. Comp. Neurol 466, 445–456. [DOI] [PubMed] [Google Scholar]
- Torres SJ, and Nowson CA (2007). Relationship between stress, eating behavior, and obesity. Nutrition 23, 887–894. [DOI] [PubMed] [Google Scholar]
- Tovote P, Esposito MS, Botta P, Chaudun F, Fadok JP, Markovic M, Wolff SBE, Ramakrishnan C, Fenno L, Deisseroth K, et al. (2016). Midbrain circuits for defensive behaviour. Nature 534, 206–212. [DOI] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, and Bolshakov VY (2007). Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc. Natl. Acad. Sci. USA 104, 14146–14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Fulton S, Wilson M, Petrovich G, and Rinaman L (2015). Stress exposure, food intake and emotional state. Stress 18, 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Chan J, and Pickel VM (1996). Input from central nucleus of the amygdala efferents to pericoerulear dendrites, some of which contain tyrosine hydroxylase immunoreactivity. J. Neurosci. Res 45, 289–302. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Colago EE, and Valentino RJ (1998). Amygdaloid corticotropin-releasing factor targets locus coeruleus dendrites: substrate for the co-ordination of emotional and cognitive limbs of the stress response. J. Neuroendocrinol 10, 743–757. [DOI] [PubMed] [Google Scholar]
- Verma D, Wood J, Lach G, Herzog H, Sperk G, and Tasan R (2016). Hunger Promotes Fear Extinction by Activation of an Amygdala Microcircuit. Neuropsychopharmacology 41, 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman PJ (2000). Norepinephrine and the control of food intake. Nutrition 16, 837–842. [DOI] [PubMed] [Google Scholar]
- Wellman PJ, Davies BT, Morien A, and McMahon L (1993). Modulation of feeding by hypothalamic paraventricular nucleus alpha 1- and alpha 2-adrenergic receptors. Life Sci. 53, 669–679. [DOI] [PubMed] [Google Scholar]
- Wu Q, Lemus MB, Stark R, Bayliss JA, Reichenbach A, Lockie SH, and Andrews ZB (2014). The temporal pattern of cfos activation in hypothalamic, cortical, and brainstem nuclei in response to fasting and refeeding in male mice. Endocrinology 155, 840–853. [DOI] [PubMed] [Google Scholar]
- Yau YHC, and Potenza MN (2013). Stress and eating behaviors. Minerva Endocrinol. 38, 255–267. [PMC free article] [PubMed] [Google Scholar]
- Yeh SY (1999). Comparative anorectic effects of metaraminol and phenylephrine in rats. Physiol. Behav 68, 227–234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets contributing to this study is summarized in Table S1. Raw data are available with reasonable request to the Lead Contact D. James Surmeier (j-surmeier@northwestern.edu). No custom code, software, or algorithm was used in this study.