

Abstract
Mammalian salivary glands synthesize and secrete saliva via a vast interconnected network of epithelial tubes attached to secretory end units. The extensive morphogenesis required to establish this organ is dependent on interactions between multiple cell types (epithelial, mesenchymal, endothelial, and neuronal) and the engagement of a wide range of signaling pathways. Here we describe critical regulators of salivary gland development and discuss how mutations in these impact human organogenesis. In particular, we explore the genetic contribution of growth factor pathways, nerve-derived factors and extracellular matrix molecules to salivary gland formation in mice and humans.
FUNCTION AND MORPHOGENESIS—AN OVERVIEW
The salivary glands synthesize and secrete saliva, a viscous fluid essential for digestion, vocalization, taste, remineralization, and overall oral health and well-being.1 There are multiple salivary glands located in similar positions in mice and humans, including the major salivary glands (1 pair each of submandibular, parotid, and sublingual) that produce 90% of saliva, as well as 600–1000 minor glands. Each of the major glands differs in regards to the composition of saliva produced due to differences in acinar (secretory) cell type.2 The parotid gland secretes proteinacious saliva by serous acini, the sublingual gland, composed almost exclusively of mucous acini, produces mucin-rich saliva, and the submandibular gland produces a mostly mixed saliva from both acinar cell types. Saliva output from these glands is tightly controlled through the parasympathetic and sympathetic branches of the autonomic nervous system.3–5 In general, parasympathetic and sympathetic nerves control different secretory processes.6 Parasympathetic nerves primarily stimulate water secretion, in part, through transmembrane water channels including aquaporin 5 (AQP5)7 as well as paracellular pathways8,9 by activating acetylcholine muscarinic receptors (CHRM1 and CHRM3) on the acini.10,11 Sympathetic nerves, on the other hand, govern the secretion of digestive proteins such as amylase by activating β-adrenergic receptors on the acini.12,13 Once secreted, saliva flows through a series of water-impermeable ducts (intercalated, striated, and excretory) that create a hypotonic solution via ion absorption for delivery to the oral cavity.14
In order to produce the volume of saliva required for daily living (0.5–1 L in humans), yet be constrained to the craniofacial complex, salivary glands need to maximize space and surface area.15 To achieve this, mouse and human salivary glands establish an interconnected network of secretory acini and ducts through the process of epithelial branching morphogenesis (Figure 1(a) and (b)). Although this process has been described for all salivary glands, the ability to culture the mouse embryonic submandibular gland (SG) ex vivo has resulted in the vast majority of our knowledge being derived from this organ and as such, this review will focus on its development. SG formation is initiated by the thickening of the oral epithelium that invaginates into a condensed mesenchyme containing an endothelial plexus (6–8 weeks in humans and embryonic day (E) 11.5 in mice).17–19 This single epithelial bud then undergoes rounds of branching morphogenesis, defined by multiple cycles of cleft formation, expansion of end buds (pre-acini), and duct tubulogenesis19,20 which involves the elongation of ducts via KRT19+ duct cell proliferation, condensation of KRT19+ duct progenitor cells at the midline, fusion of microlumen to form contiguous lumen and finally lumen expansion (Figure 1(a)).21 Innervation of the epithelium occurs at the onset of development: at E12 in mice neural crest-derived neurons coalesce at the primary duct to form a parasympathetic ganglia.22,23 As epithelial branching ensues (E13), axons extend along the epithelium to envelop the newly forming end buds. Terminal differentiation of end buds into secretory acini is apparent by 19–24 weeks in humans and by E16 in mice and this is followed by further growth and differentiation until a mature organ capable of nerve-stimulated secretion forms (occurring at birth for humans and by post-natal day 6 for mice; Figure 1(b)).17,19 Such a complex series of morphogenic steps suggests that multiple intrinsic and extrinsic signaling pathways are tightly regulated in a spatiotemporal fashion. Below we will discuss how the formation of a viable organ is affected by alterations in these pathways.
FIGURE 1 |.
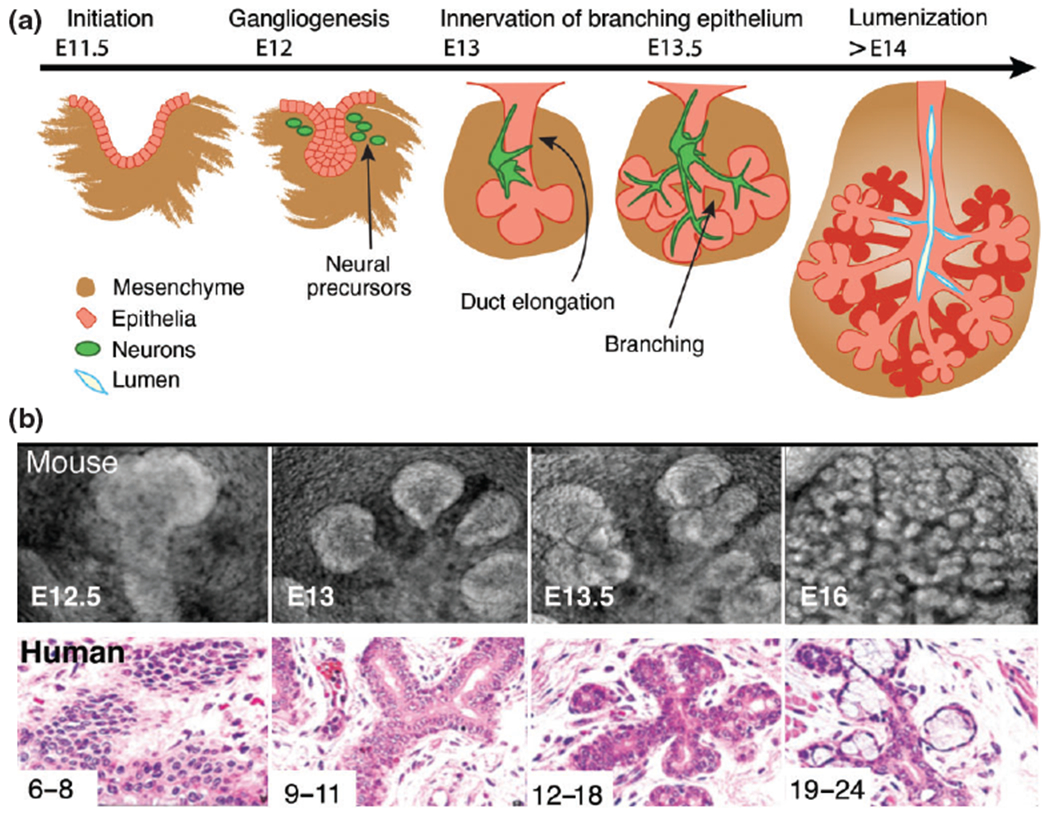
Mouse and human salivary glands develop through the process of branching morphogenesis. Schematic (a) and histological (b) representations of SG development. (b) Mouse and human SG development. Human development is measured in weeks and mouse development in days. E = embryonic day. (Reprinted with permission from Ref 16. Copyright 2010 John Wiley and Sons)
SIGNALING SYSTEMS REGULATING SALIVARY GLAND FORMATION
Fibroblast Growth Factor Family
Fibroblast growth factor (FGF)-FGFR signaling is essential for multiple branching organs including the lungs, pancreas, prostate, and salivary glands. Many of the 23 FGF family members, as well as 3 of the 5 tyrosine kinase FGF receptors (FGFR1, 2, and 3), are expressed in the developing SG and are differentially distributed between the epithelium and mesenchyme.24 In humans, mutations in FGF10 and FGFR2 result in SG phenotypes (Table 1). Patients with haploinsufficiency of FGF10 suffer from one of two overlapping conditions: autosomal dominant aplasia of the lacrimal and salivary glands (ALSG)38 or autosomal dominant lacrimo-auriculo-dento-digital (LADD)39 syndrome. Both are marked by salivary gland aplasia/hypoplasia, xerostomia, increased dental maladies, and oral infections; however, mutations that cause LADD syndrome are thought to be more disruptive to cell signaling than the FGF10 gene mutations that cause ASLG and patients often display various craniofacial, digital, and genitourinary defects.39 These mutations thus highlight the essential role of FGF signaling in salivary gland formation.
TABLE 1 |.
Human Diseases and Associated Mutations with Corresponding Mouse Models
| Disease | Associated Mutations | Human Features | Mouse Model SG Phenotypes |
|---|---|---|---|
| Cystic fibrosis | CFTR | Defective electrolyte, fluid, and mucus secretion; mucus build-up in lungs and digestive system | Cftr-null mice exhibit reduced lumenization of ductal system21 |
| Ehlers-Danlos syndrome | COL3A1, COL4 | Hypermobility, hyper-elastic skin | ? |
| Hyphohidrotic ectodermal dysplasia | EDA, EDAR, EDARRAD | Absent or hypoplastic teeth, hair, sweat glands, sebaceous glands, lacrimal glands, mammary glands, mucous glands of the bronchial, esophageal and colonic mucosa, and salivary glands | Eda, Edar and Nfkbia deficient SG are hypoplastic25,26 |
| Desbuquois dysplasia type 2 | xylosyltransferase 1 (XYLT1) | Characterized by short stature, joint laxity, and advanced carpal ossification. | ? |
| Aplasia of the lacrimal and salivary glands (ALSG) | FGF10 | Salivary gland aplasia/hypoplasia, xerostomia, dental maladies, oral infections | Fgf10-heterozygous mice recapitulate human phenotype27 (Entesarian et al.28–30) |
| Lacrimo-auriculo-dento-digital syndrome (LADD) | FGF10, FGFR2 | Salivary gland aplasia/hypoplasia; craniofacial, digital, and genitourinary abnormalities | Fgf10 and Fgfr2-heterozygous mice recapitulate human phenotype27–30 |
| Kallmann-like idiopathic hypogonadotropic hypogonadism | FGF8, FGFR1 | Anosmia, hyposmia, pubertal defects | Fgf8-null SG initiate but do not develop28 |
| Pfeiffer/Apert/Crouzon/Beare-Stevenson syndrome | FGFR1, FGFR2, (gain-of-function) | Craniosynostosis, facial dysmorphia | Spry1/2-deficient mice have hypoplastic glands with no ganglion or ducts31 |
| Glomerulopathy | Fibronectin (FN1) | Hypertension, defects leading to renal failure | ? |
| Hirschsprung’s disease | GDNF, NRTN, RET, GFRA1 | Agangliogenesis and blockage of the enteric system | Nrtn and Gfra2 deficient mice have reduced salivary function32–34 |
| Erbb3 SG are hypoplastic35 | |||
| NRG1 | |||
| Hereditary gelsolin amyloidosis | Gelsolin (GSN) | Cranial nerve dysfunction; SG atrophy and hypofunction | ? |
| Epidermolysis bullosa | ITGA3 | Compromised barrier functions in kidney, lung, and skin | Itga3; Itga6 SG are hypoplastic and poorly differentiated36 |
| Alagille syndrome | JAG1, NOTCH2 | Heart, liver, face, vertebrae, and eye abnormalities; decreased liver bile ducts | mice with constitutively active Notch4 have immature hyperplastic ducts37 |
| Holoproencephaly, Greig cephalopolysyndactyly syndrome, Pallister-Hall syndrome, Meckel syndrome | SHH, GLI3, Cilia-related genes | Brain/facial abnormalities; polydactyl; kidney and other organ defects | Shh-null are hypoplastic and poorly differentiated28 |
| Treacher collins | TCOF1 | Neurocristopathy resulting in craniofacial abnormalities and hypoplastic cranial ganglia; SG atrophy and hypofunction | ? |
| Hereditary angiopathy with nephropathy, aneurysms, and muscle cramps (HANAC) | COL4a1/2 | Ocular, cerebral, renal and muscular defects | ? |
Understanding the role of FGF signaling in salivary gland development and disease has been greatly aided by mouse models that replicate the phenotypes observed in LADD/ALSG. As in humans, the salivary glands of mice heterozygous for Fgf10 or Fgfr2b (the splice isoform receptor for FGF1, 7, and 10) are hypoplastic and display reduced branching morphogenesis.28 In murine embryos deficient in Fgf10 or Fgfr2b, the phenotype is severe, with salivary gland development stalled at the initial bud stage, indicating FGF10/FGFR2b signaling is essential for early morphogenic events.27–30 In addition to FGF10 and FGFR2b, other FGF ligands and receptors also regulate SG development. Fgf8 hypomorphic and Fgfr2c heterozygous murine embryos (FGFR2c is the receptor for FGF8) exhibit hypoplastic SGs, and conditional ablation of Fgf8 in the ectoderm results in ontogenic arrest followed by involution.28 Humans with FGF8 (or FGFR2) mutations, as found in patients with Kallmann-like idiopathic hypogonadotropic hypogonadism, have numerous craniofacial anomalies including cleft lip and palate, but whether SGs are also affected remains to be determined.
How does FGF signaling control salivary gland morphogenesis? Early studies using ex vivo cultures of mouse SG affirmed that FGF signaling promotes end bud (pre-acinar) development. Addition of exogenous FGF7 or 10 (both ligands of FGFR2b that are produced by the mesenchyme) to SG organ cultures increased end bud formation via ERK1/2 or PI3K signaling pathways, indicating that these are the major signaling pathways through which FGFs drive morphogenesis.40 Conversely, blockade of FGF signaling by inhibition of FGFR1b or 2b (expressed exclusively by the epithelium) reduces the number of end buds to result in a more ductal phenotype (Figure 2).40 Thus, FGF10/FGFR2b signaling acts centrally on the end buds to increase acinar proliferation and reduce lumen/duct formation.41–43 More recently, Lombaert et al.41 proposed that acinar progenitor cells are centers of FGF activity. Using ex vivo SG models, they determined that FGF7 and FGF10 expand putative progenitor cells marked by MYC, SOX9, and KIT in the end buds.41 Furthermore, upregulation of KIT pathways in KIT+ KRT14+ SOX10+ distal progenitor cells activates the AKT and PI3K pathways to further upregulate FGFR2b targets, increasing progenitor cell proliferation and end bud formation (Figure 2(b)).41
FIGURE 2 |.
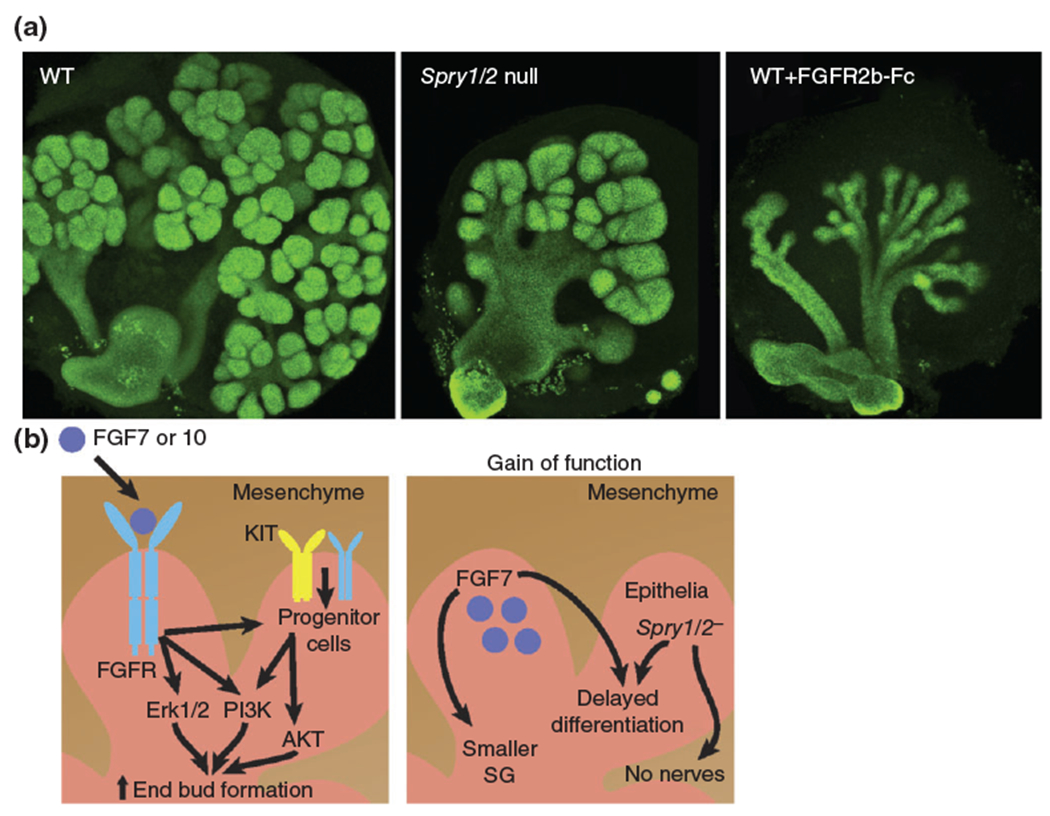
Dysregulation of FGF signaling results in aberrant branching morphogenesis. (a) E13 SGs [wild type and SG deficient in the Sprouty (Spry1) genes 1 and 2] were cultured for 24 h in the presence or absence of recombinant FGFR2b-Fc (10 μg/mL). Epithelium was immunostained for E-cadherin. (b) Left panel: FGF signaling regulates progenitor cells, and epithelial branching. Right panel: Increased FGF signaling [e.g., overexpression of FGF7 in the epithelium or ablation of negative regulators of FGF signaling (Spry1 and 2) in vivo] reduces SG growth, delays differentiation and impairs gangliogenesis. Epithelium is labeled pink, mesenchyme brown.
Given the importance of FGF signaling to epithelial organ morphogenesis, it is surprising that there has been limited analysis of human disorders arising from gain of function mutations in FGFR1 and 2, e.g., Pfeiffer syndrome, Apert syndrome, Crouzon syndrome, and Beare-Stevenson syndrome. Analysis of these patients has focused almost exclusively on craniosynostosis as well as facial dysmorphia, with little to no analysis of branching organs. However, studies in mice have demonstrated that too much FGF signaling disrupts formation of the SG as well as other epithelial organs including the tooth44 and genitalia.45 Targeted overexpression of FGF7 in the SG epithelium with the Krt14 promoter results in smaller salivary glands with delayed differentiation.46 More recently, Knosp et al.31 demonstrated that ablation of epithelial Sprouty (Spry) 1 and 2, antagonists of FGF signaling, in the SG impairs epithelial morphogenesis with the gland appearing as an endbud-like structure with no defined ductal system (Figure 2(a)).31 Moreover, the parasympathetic ganglion innervating the epithelium was absent and epithelial progenitor cells marked by keratin 5 (KRT5) were reduced, suggesting that FGF signaling regulates gangliogenesis and progenitor cells (discussed below; Figure 2(b)). Thus, based on these findings, it would be of significant interest to analyze SGs and other branching organs in humans with activating mutations in the FGF pathway.
If FGF signaling is a global regulator of SG morphogenesis, how is FGF signaling itself regulated? One important and often overlooked component necessary for FGF signaling are heparan sulfate proteoglycans (HSPG) located in the basement membrane or at the cell surface that serve as coreceptors for FGF ligands.47 HSPGs are composed of a protein core with one or more covalently attached unbranched HS glycosaminoglycan chains that consist of a sugar backbone of alternating units of N-acetylglucosamine (GlcNAc) and glucuronic acid (GlcA). Numerous studies have shown that FGF10/FGFR2b signaling is HS dependent. Binding of FGF10 and FGFR2b to HS moieties promotes ligand-receptor dimerization required for signaling.48 Importantly, the HS chain size and pattern of O-sulfation modulates FGF10 signaling and causes morphogenic differences in SG development. Cleavage of HS chains by the endoglycosidase heparanase increases FGF10 mediated branching in ex vivo SG cultures36 and HS with 2-O-sulfation and either an N-or 6-O-sulfate induces end bud expansion, whereas HS with 6-O- and N-sulfation or 6-O-sulfation alone induces duct elongation.49 Recently, Patel et al.43 showed that 3-O-sulfated-HS (3-O-HS) has a specific role in FGF10 mediated expansion of epithelial progenitor cells in the end buds.43 The authors demonstrate that the modifying enzyme, 3-O-sulfotransferase 3 (HS3ST3) is highly expressed in KIT+ progenitor cells in the end buds where it produces 3-O-S rich HS capable of binding and stabilizing FGF10/FGFR2b complexes that in turn increase cell proliferation and Hs3st3 expression. Whether this mechanism also occurs in vivo and is necessary for SG development was not determined, although siRNA knockdown studies suggested Hs3st3 was required for progenitor cell expansion. A number of mutations in genes encoding the enzymes involved in GAG synthesis (e.g., xylosyltransferase 1 (XYLT1)50; Table 1) and HS modifying enzymes (e.g., NDST1)51 as well as sulfate transporters (SLCO5A1)52 occur in humans, but whether these cause abnormal salivary gland formation are yet to be assessed.
Together, these loss and gain of function studies strongly suggest that a balance in FGF signaling is required for epithelial morphogenesis and progenitor cell maintenance. However, a number of questions remain. For example, we know that many FGFs coregulate each other’s expression to create a spatially connected network of signaling molecules. For example, FGF8 signaling modulates mesenchymal FGF10,29 upregulating epithelial FGF1 production to induce epithelial cell proliferation via FGFR1 and 2.40 Given this relationship, one could predict that such a network is compromised in humans heterozygous for FGF mutations, consequently producing the spectrum of phenotypic variation observed in these patients. However, it is also possible that other pathways negatively or positively regulated by FGFs (e.g., EGF, WNTs) may be adversely affected (see next sections). Regardless, there is an essential need to decipher the mechanisms by which perturbations in FGF signaling dysregulate salivary morphogenesis.
Epidermal Growth Factor Family
The epidermal growth factor (EGF) receptor family is a vital component of mammalian development, being involved in a diversity of functions including cell proliferation, migration, differentiation, survival, and apoptosis (reviewed by Ref 53). Since mutations in the EGF family are a leading cause of cancer, most research has focused on cancers rather than developmental defects and accordingly, the phenotype of SGs in patients with mutations in EGF signaling beyond salivary gland carcinomas is not known. However, studies in mice deficient in EGF family members have demonstrated a clear role for these molecules in developmental processes, including SG morphogenesis.
Multiple members of the EGF family are expressed during SG development. These include the EGF receptors: EGFR (ERBB1), ERBB2, and ERBB3, and the ligands: EGF, transforming growth factor α (TGFa), heparin binding EGF (HBEGF), and neuregulin (NRG). These molecules are differentially distributed between the epithelial and mesenchymal compartments (Figure 3(a)), as well as within the epithelium, e.g., EGFR is enriched in the epithelial ducts, and ERBB3 in the end buds.54 Recent studies in mouse mutants have shown that SG morphogenesis is dependent on ERBB signaling: embryos deficient in Egfr, ErbB3, and Nrg1 type III have reduced SG epithelial branching.35,55,21 However, despite the availability of these mutants, only Egfr deficient animals have been directly characterized. Ablation of Egfr results in reduced epithelial cell proliferation and aberrant duct development with delayed differentiation.56 Consistent with a role in duct morphogenesis, Knox et al.23 recently demonstrated that HBEGF activation of EGFR modulates ductal morphogenesis by controlling progenitor cell differentiation and expansion along the keratin 19 (KRT19+) ductal lineage.23 Furthermore, EGFR transactivation is required for parasympathetic nerve mediated proliferation of KRT5+ epithelial progenitor cells,23 providing an explanation for reduced proliferation in the absence of EGFR. However, the mechanism by which EGFR signaling promotes acinar formation is unclear. One possibility is that loss of EGFR prevents formation of ERBB3/EGFR dimers, where ERBB3 is a dead kinase that requires dimerization with ERBB2 or EGFR for signal transduction, thereby limiting acinar formation. This notion is supported by the reduced branching observed in ErbB3 null SG35 and by ex vivo studies where exogenous NRG1 (the ligand for ERBB3) increases end bud number in epithelial rudiment cultures,54 presumably through an ERBB3/PI3K dependent pathway.57
FIGURE 3 |.
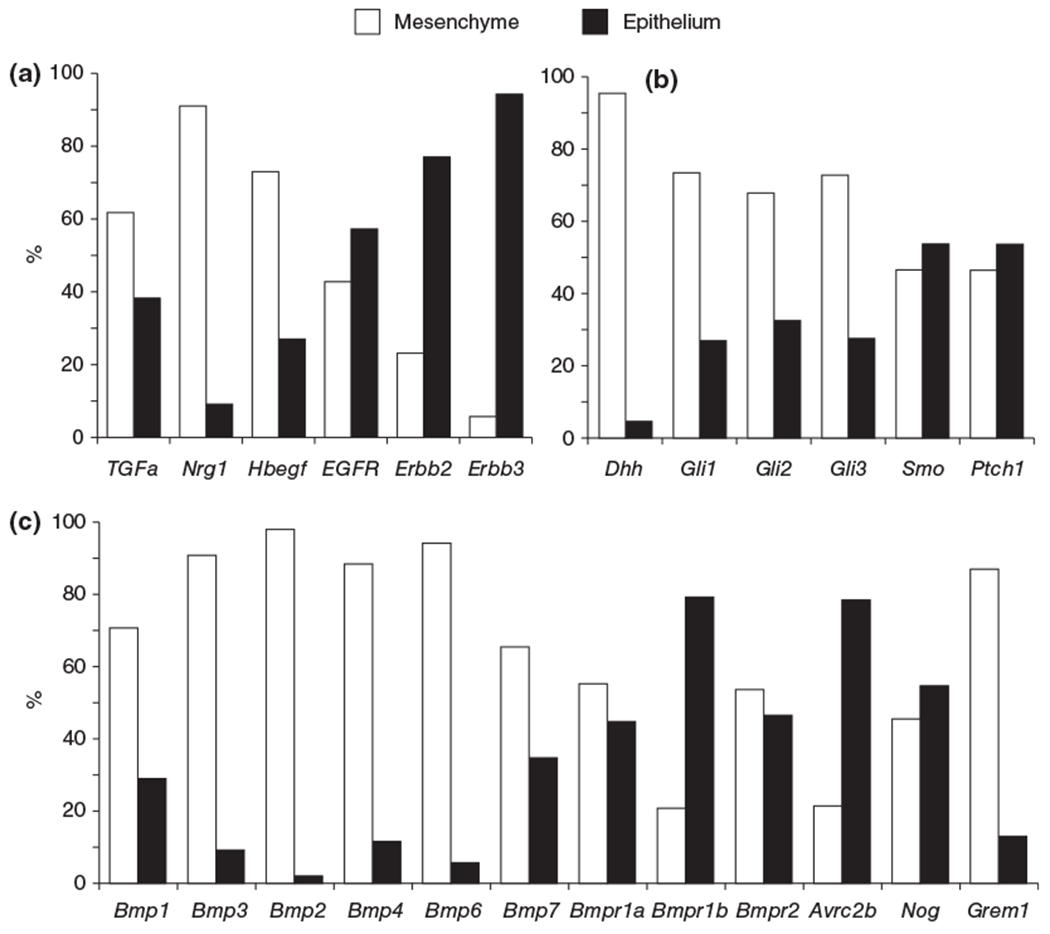
Differential gene expression in the developing SG. Expression of Egf (a), Hh (b), and Bmp (c) family members in epithelial and mesenchymal compartments of the SG at embryonic day 13. Expression patterns were derived from a pre-existing microarray database available online (http://sgmap.nidcr.nih.gov/sgmap/sgexp.html). Data are presented as a percentage of total expression (epithelium + mesenchyme) of the SG.
Although no human mutations in genes that encode the ERBB receptors have been identified as causative for developmental diseases, heterozygous mutations in NRG1 have recently been linked to Hirschsprung’s disease,58 a disorder characterized by aganglionic colon as well as other cardiac and kidney defects. Whether salivary gland morphogenesis or innervation (ERBB3 signaling is necessary for Schwann cell survival) is also affected in these patients remains to be investigated. In addition, we speculate that the spectrum of SG phenotypes found in those patients with LADD/ALSG may be due, in part, to alterations in ERBB signaling from reduced FGF signaling. While gland initiation and early branching is independent of ERBB signaling and dependent on FGF, ERBB mediated acinar expansion has been postulated to be a result of priming by FGF signaling. Ex vivo studies by Nitta et al.59 show that FGF signaling renders epithelial pre-acinar cells at E13 competent for branching in response to EGF ligands.59 This scenario would suggest that in the SG of patients with LADD/ALSG, EGF signaling is not induced during morphogenesis and as such may exacerbate SG aplasia.
WNT Signaling
WNT signaling contributes to embryonic patterning, cell proliferation, cell fate, migration, polarization, and differentiation in multiple organisms and organ systems. Not surprisingly, mutations in WNT genes and their downstream targets have been implicated in a large number of human genetic disorders including those characterized by craniofacial abnormalities, e.g., cleft lip with or without palate,60 as well as neural, kidney, and limb defects,61,62 among many others. Characterization of SGs in humans or mice with similar genetic mutations has not been performed. However, pharmacological approaches using the ex vivo culture system has provided information on the functions of WNT signaling during SG morphogenesis.
The developing SG expresses a large contingent of WNT ligands, receptors and downstream signaling molecules and antagonists in the epithelial and mesenchymal compartments.31 Despite this abundance, WNT signaling itself is strictly spatially and temporally regulated during SG development: AXIN2, a WNT antagonist and marker of canonical WNT activity, first appears at E11.5–E12.5 in the condensed mesenchyme near the endbuds and within the parasympathetic ganglia25,31,42 and becomes restricted to the developing ductal system by E14.25,42 Such confined locations suggest WNT signaling regulates specific morphogenic roles. Consistent with this, Patel et al.42 showed using ex vivo organ culture and pharmacological inhibitors that WNT signaling promotes duct development through the coordination of canonical and non-canonical WNT pathways. Canonical WNT/β-catenin signaling inhibits end bud formation and non-canonical WNT signaling activated by WNT5b upregulates expression of the transcription factor TFCP2L1 to drive duct formation (ducts are disrupted in Tfcp2l1 null SG).63 How does bud formation occur given this WNT-mediated antagonism? The authors show that to maintain a WNT-free end bud, FGF signaling represses expression of Wnt5b and increases expression of the endogenous WNT inhibitor secreted related frizzled protein 1 (SFRP1), which sequesters WNT proteins. How WNTs overcome FGF signals to promote duct formation within the proximal region of the bud is not known. However, it is tempting to speculate that WNTs interact with other duct promoting signaling pathways (e.g., HBEGF/EGFR) to downregulate FGF signaling.
As indicated above, during early SG development (<E13) WNT signaling is restricted to the mesenchyme, but its function in this region was not known. Recently, Knosp et al.31 revealed that mesenchymal WNT signaling is associated with and is essential for the formation of the parasympathetic ganglion.31 In order to promote gangliogenesis in the SG, epithelial cells of the invaginating epithelium secrete WNTs that in turn bind and activate Frizzled (Fzd) receptors on the neurons, resulting in neuronal proliferation and aggregation to form a ganglion. WNT expression was also shown to be dependent on FGF antagonism and ERBB signaling. If FGF signaling becomes overactivated, as in the case of Spry1/2 mutants, or ERBB signaling is inhibited, synthesis of WNTs is prevented leading to impaired WNT signaling and gangliogenesis. The authors further demonstrated that the pancreas of Spry1/2 null embryos also had reduced WNT signaling and fewer intrinsic ganglia, suggesting WNTs regulate gangliogenesis within other developing organs.
How will aberrant WNT signaling affect human SG development? The interaction between FGF and WNT pathways suggests that FGF loss or gain of function mutations would likely result in altered WNT signaling and thus changes in ductal and ganglia morphogenesis. Therefore, it would be of significant interest to determine whether aberrant WNT signaling occurs in individuals with LADD/ALSG or FGF gain of function mutations (e.g., Apert syndrome). This balance in FGF-WNT signaling may be a factor to consider during future therapeutic interventions in patients with these disorders.
Hedgehog Signaling
Hedgehog (HH) signaling has long been tied to development. In humans, congenital brain, limb, cochlear, neural crest, and craniofacial defects are present in individuals harboring mutations in hedgehog proteins [e.g., Sonic hedgehog (SHH), Desert hedgehog (DHH), Indian hedgehog (IHH)], HH receptors [Smoothened (SMO), Patched (PTCH)], downstream mediators (e.g., GLI1–3), or the primary cilia that relay the HH signals. Such diseases include holoprosencephaly, Greig cephalopolysyndactyly syndrome, Pallister-Hall syndrome, and Meckel syndrome.64,65 Mouse models for each of the molecules within the Hh pathway have been made, and have been used to analyze the role of this signaling system in the morphogenesis of multiple organs. However, despite the availability of these mice, and prevalence of these diseases, characterization of the SG in both patients and mice are lacking. As such, we describe the limited studies that have been performed to date in the developing SG.
HH family ligands DHH and SHH (but not IHH) and receptors PTCH1 and SMO as well as downstream effectors (GLI1–3) are expressed during SG development. Shh and Ptch1 are enriched in the epithelium, Dhh in the mesenchyme, and Smo is located in both the mesenchyme and epithelium (Figure 3(b)).25 Activator Gli1, and activators/repressors Gli2 and 3 are also differentially expressed, with Gli1, Gli2 and Gli3 being distributed between the epithelium and mesenchyme, indicating HH signaling could occur in both compartments (Figure 3(b)).66 Despite Shh being lowly expressed,25 Shh deficient SG are hypoplastic and epithelial cells remain undifferentiated.66 To determine if failure of the glands to develop was due to a direct effect on SGs or due indirectly to perturbation of the craniofacial compartment (all tissues are adversely affected in the Shh-null embryo), SGs were treated ex vivo with cyclopamine, an inhibitor of SMO. Cyclopamine treatment significantly reduced epithelial branching,25,66 thus confirming the Hh pathway to directly modulate SG development.
HH signaling plays an essential role in duct development in multiple other organs including the prostate, sebaceous glands, mammary gland, and lung, however, in these organs HH signaling is restricted to the mesenchyme (suggesting an indirect action on epithelial morphogenesis), whereas in the SG it occurs primarily in the epithelium.67–70 During lumen formation, signal mediators SMO and GLI3 are enriched in ductal cells and terminal bud cells neighboring the lumen, suggesting that SHH activity may be important for lumen development.66 In support of this, E14 SGs cultured for 72 h with exogenous SHH exhibit fully formed, large lumens compared to control, establishing the Hh pathway as a regulator of lumen formation.71 Moreover, in the adult mouse, overexpression of Hh pathway effector GLI1 in KRT5+ epithelial cells also results in larger lumens, expansion of ductal structures, and loss of acini, reinforcing the significance of the Hh pathway in the salivary ducts.72 Though abnormalities in SG have not yet been reported in individuals with mutations in SHH, these studies suggest that lumen/ductal defects and aplasia are likely affected.
There is also evidence that the Hh pathway interacts with FGF pathways to modulate SG development. FGF8 is a putative target of the Hh pathway through GLI3, and both FGF8 and SHH are able to positively upregulate each other.73,29 Moreover, FGF8 rescues the branching defects in cyclopamine-treated glands.66 Whether SHH signaling is largely operating to upregulate FGF8 or if SHH and FGF8 have redundant downstream effects remains to be determined. Regardless, from these data we would predict that patients with ALSG/LADD could be experiencing reduced HH activity during development due to a lack of stimulation from FGFs, representing another barrier to morphogenesis.
Ectodysplasia/EDAR Signaling
Another critical signaling molecule essential for epithelial morphogenesis is Ectodysplasia (EDA). Mutations in EDA, its cognate receptor EDAR, or adaptor molecule EDARRAD cause hypohidrotic ectodermal dysplasia (HED), a syndrome variably characterized by absent or hypoplastic teeth, hair, sweat glands, sebaceous glands, lacrimal glands, mammary glands, mucous glands of the bronchial, esophageal and colonic mucosa and SGs (Table 1).74,75 As the EDA pathway is highly conserved among mammals, mouse models for these mutations replicate human phenotypes allowing for robust interrogation of this pathway and its effects on tissues.76–78
Both loss and gain of function studies in genetic mouse models have revealed a critical role for the EDA pathway in salivary gland ductal and acinar development. In Eda and Edar null adults, the SGs are hypoplastic with a reduced number of ductal and acinar structures.79,80 Similarly, in Eda and NF-kappaB inhibitor α (Nfkbia) deficient mouse embryos, where NF-kB is the downstream signaling target, SG lumen size and end bud number are reduced.25,80 In contrast, overexpression of EDA in the epithelium using the Krt14 promoter (Krt14Cre-Eda-A1), increases embryonic SG end buds and lumen size25 and in transgenic mice with a high copy number of the wild type Edar locus adult SG display increased epithelial branching.81 The established relationship between EDA and WNT in other organs suggested WNT and EDA pathways might interact to control morphogenesis.82 However, despite EDA and WNT being linked during early SG development (<E13) where suppression of WNT signaling in the mesenchyme reduces Eda expression, after E13 EDA and WNT pathways appear to function independently. Indeed, WNT and EDA/NF-kB activities do not colocalize in the developing SG during duct formation/lumenization: at E12 WNT activity localizes to mesenchyme and shifts to the ducts by E14, while NF-kB signals localize to mesenchyme and shift to the end buds by E14.25,42 This end bud location of NF-kB activity may be counter-intuitive for a pathway involved in duct development. However, lumenization also occurs within the distal portions of the end buds to connect the secretory acini with the ductal system.41 So how does EDA/EDAR control lumenization? A prime candidate for propagating the EDA/EDAR signal is SHH as Shh transcripts and NF-kB activity are in the same location and importantly, in a systems wide analysis of gene regulation in Eda null mice, Shh was found to be heavily downregulated, while addition of recombinant EDA-A1 to skin cultures upregulated Shh expression.83,84 Subsequently, exogenous SHH peptide was shown to rescue branching morphogenesis of ex vivo cultures of Eda deficient SG,25,26 although a thorough analysis of the ducts was not performed. However, based on these outcomes, we predict the EDA/EDAR/Hh signaling pathway is disrupted in patients with HED causing reduced SG branching and lumenization and consequently, poor saliva production.
The localization of NF-kB activity to the end buds and the reduced epithelial branching observed in Eda/Edar//Nfkbia mouse mutants, also suggests EDA signaling regulates acinar development, and as such, may interact with the FGF pathway. Support for such an interaction comes from studies in the developing tooth, where FGF10 partially restores morphogenesis and stimulates the development of additional tooth cusps in cultured Eda deficient molars.85 Additionally, FGF20 was shown to be a major downstream effector of Eda and affects EDA-regulated characteristics of tooth morphogenesis, including the number, size and shape of teeth.86 In SG, addition of recombinant FGF8, a target of EDA signaling, was unable to rescue branching morphogenesis in Eda null SG,26 however, no other FGF signaling molecules were examined. As such, it remains to be determined how the EDA pathway controls acinar formation in SG and whether the SG of humans with HED also show aberrant duct and/or acinar morphogenesis.
BMP/TGF-β Family
The bone morphogenetic proteins (BMPs) are members of the transforming growth factor-β (TGF-β) superfamily and are important in embryonic patterning and the development of many different organ systems including the eye, heart, and kidney.87 BMP-mediated signaling can have a wide variety of effects depending on its spatiotemporal activity and the cell type affected, regulating processes such as cell migration, differentiation, proliferation, and apoptosis.87 Like other members of the TGF-β superfamily, BMP ligands signal through type I and type II transmembrane serine/threonine kinase receptors (BMPR2, ACVR2A and 2B, BMPR1A and 1B, ACVR1A) to activate SMAD-dependent canonical pathways as well as non-canonical, SMAD-independent signaling pathways (e.g., MAPK, PI3K/AKT, PKC, Rho-GTPases). There are also multiple inhibitors of BMP including Noggin, Chordin, and Gremlin-1 that tightly regulate its signaling. Although many human disorders are associated with mutations in BMPs, BMP antagonists or the molecules functioning downstream of BMP signaling pathway, whether these mutations affect SG morphogenesis in humans is not known. However, studies in the mouse have highlighted the importance of this pathway for SG development.
Of the 20 known BMP ligands, the developing SG expresses Bmp 1–4, 6, and 7 as well as the receptors Bmpr1a/b, Bmpr2, and Acvr1a/2a/2b. These factors are differentially expressed between the mesenchyme and epithelium, e.g., Bmp7 is expressed in the epithelium and mesenchyme whereas Bmp4 is enriched in the mesenchyme (Figure 3(c)),24 and suggests they have different roles in SG development. However, despite the availability of mouse mutants for each of these BMP ligands and receptors as well as for the BMP antagonists and downstream signaling molecules (e.g., Smads), only the SG of Bmp7 deficient murine embryos have been studied in vivo. Consistent with its role in epithelial organ development,88 Bmp7 null E17 SG exhibit aberrant morphogenesis with fewer end buds than wild type controls,89,88 suggesting BMP7 regulates acinar formation in a non-redundant fashion. In support of this finding, addition of exogenous BMP7 to ex vivo SG cultures increases end bud formation and rescues branching of FGFR1b-inhibited glands.24 However, not all BMPs promote SG morphogenesis. Similar to what has been observed in the kidney90 and prostate,91 BMP4 has inhibitory activity in the SG: addition of BMP4 acts antagonistically on ex vivo SG cultures to reduce end bud number and branching.24 Given these contrasting functions of BMP4 and 7, how are their actions coordinated? The answer may reside in FGF signaling. Using pharmacological inhibitors, Hoffman et al.24 found FGFR1b signaling induces expression of branch-promoting BMP7 and suppresses branch-inhibiting BMP4.24 Thus, FGF signaling may be a master regulator of BMPs to tightly control salivary morphogenesis. However, as BMP signaling also interacts with many other pathways, including the WNT pathway where the two promote or inhibit one another’s expression depending on the system and developmental timing,92 it is likely BMP actions are governed by many more pathway interactions.
Another member of the TGF-β superfamily, TGF-β1, is also expressed in the developing gland, but clear inferences on its function are more difficult to conclude.93 Exogenous addition to explants/cell culture or overexpression of TGF-β1 in mice is associated with phenotypes of acinar loss and impeded acinar differentiation, elongated ducts, dysplasia, and/or fibrosis94–96; however, studies examining mice deficient in Tgfb1 or Tgfbr1 (TGF-β receptor type 1) found no SG developmental defects in the absence of inflammation.97,98 Therefore, the role of TGF-β signaling in normal development is still under investigation.
Despite extensive investigations into BMP signaling in mice, very few human mutations have been discovered in BMP signaling pathways that are directly causative of disease, and no investigations of salivary gland development or function have been reported. As such, further studies are required to understand how loss, or gain, of function mutations in the BMP family or in other families that affect BMP signaling regulate human (and mouse) SG morphogenesis.
Glial Cell Line-Derived Neurotrophic Growth Factor Family
As highlighted in the Introduction, the salivary gland requires innervation from the peripheral nervous system for development, function, and adult homeostasis. Thus, not surprisingly, if innervation is perturbed, SG formation, function, and homeostasis are adversely affected. How is innervation of the gland achieved during development? Loss and gain of function studies in mice have demonstrated that the Glial cell line-derived neurotrophic growth factor (GDNF) family is essential for establishing and maintaining cranial nerve populations, including those of the submandibular parasympathetic ganglion, and also play a crucial role in epithelial innervation. Mice deficient in the ligand neurturin (NRTN), which is expressed by SG epithelium, or its receptor complex GFRa2/RET, expressed by the nerves, have smaller parasympathetic submandibular ganglia that continue to atrophy over time resulting in lower salivary function.32–34 As would be expected for molecules that function in organ targeting by peripheral nerves, innervation of the SG is also substantially reduced in these mice. Other GDNF family ligands besides NRTN may also be involved in SG innervation as genetic ablation of Gdnf or its receptor Gfra1 results in moderate neuronal loss99,100; however, sufficient compensation by NRTN and GFRa2 likely reduces the impact of individual mutations in these factors on innervation.
Although these in vivo studies indicate the importance of NRTN and innervation to epithelial organ function and maintenance (epithelial organ atrophy is observed in the absence of innervation), as well as salivary function, the SG themselves were not examined. However, a recent ex vivo study supports the notion that NRTN is necessary for functional innervation of the developing salivary epithelium. Knox et al.101 show that NRTN expressed by the developing SG end buds is required for guiding GFRa2 expressing parasympathetic nerves toward these end buds (Figure 4).101 This study also highlights the need for bidirectional communication between the branching epithelium and ganglia: NRTN secreted by the epithelium increases secretion of acetylcholine and other nerve-derived factors that maintain/expand epithelial progenitor cells and regulate branching (Figure 4(d); discussed below). Notably, Nrtn transcripts are enriched in KIT+ epithelial progenitor cells, thereby providing an even more finely tuned mechanism to control progenitors and morphogenesis.41 The enrichment of Nrtn mRNA in KIT+ progenitor cells is similar to that observed in the adult SG, where GDNF is expressed by a subset of LIN−CD24+KIT+SCA1+ progenitors.102 The role of GDNF in the adult may be similar to NRTN during development, i.e., expression may be required for maintaining innervation. Indeed, Xiao et al. found that treatment of irradiated adult mice with GDNF expanded KIT+ progenitor cells and rescued SG function,102 but whether this was due to increased innervation was not investigated.
FIGURE 4 |.
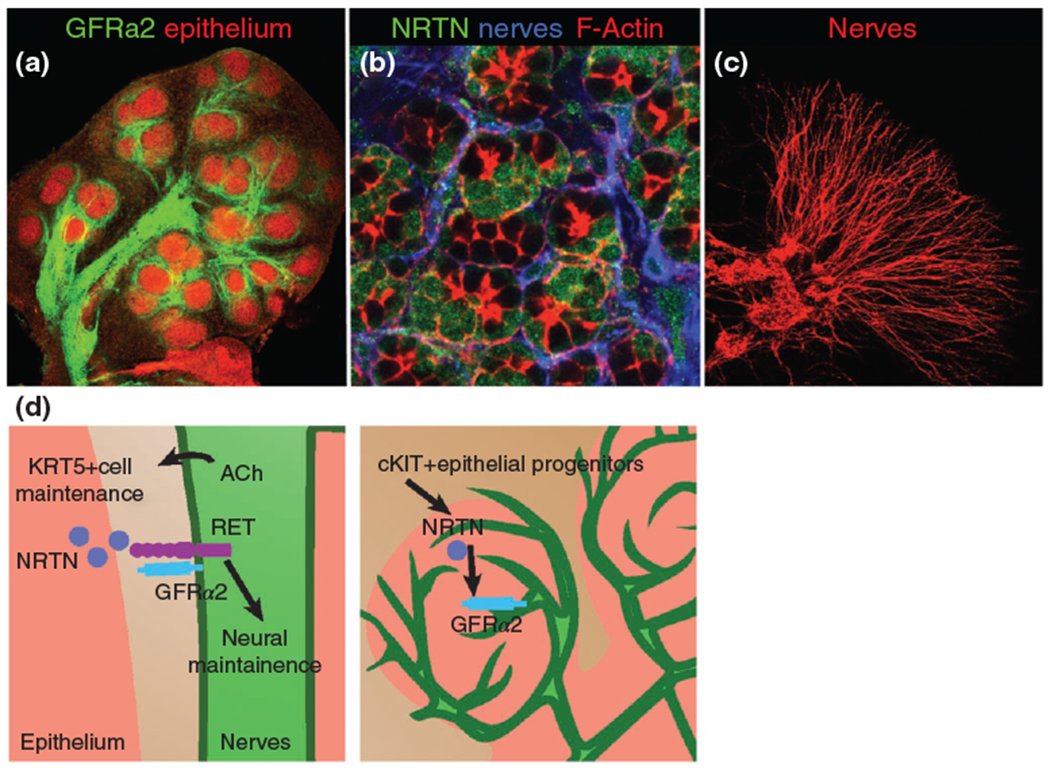
GFRa2 localizes to the nerves and neurturin (NRTN) to the acinar cells of developing salivary glands and NRTN promotes axon outgrowth from parasympathetic ganglia. (a) E13.5 SG labeled with GFRa2 and peanut agglutinin (epithelial marker). (b) Post-natal day 1 SG immunostained for neurturin, acinar cell cytoskeleton (F-actin, red) and neurons (blue). (c) E13 parasympathetic ganglia cultured with NRTN for 48 h shows extensive outgrowth. (d) Left panel: Bidirectional communication between parasympathetic nerves and the SG epithelial progenitor cells. NRTN from epithelium (pink) promotes innervation and increases production of acetylcholine (ACh), which in turn maintains KRT5+ basal progenitor cells. Right panel: NRTN is synthesized by cKIT+ progenitor cells in the end buds.
Morphogenesis of human SG (and other organs) has rarely been assessed in diseases associated with loss of innervation. However, salivary hypofunction can result from familial amyloid neuropathy, in which amyloid aggregates form in nerves and tissues, leading to a degeneration of nerve fibers,103,104 and in familial dysautonomia, which is due to sensory and autonomic dysfunction.105 Furthermore, salivary gland atrophy and dysfunction have been found in patients with hereditary gelsolin amyloidosis, a disease that manifests with late-onset dysfunction of the cranial nerves106 as well as in patients with the neurocristopathy Treacher Collins syndrome.107 Treacher Collins syndrome is caused by mutations in TCOF1, a gene necessary for neural crest cell survival and migration, which results in craniofacial abnormalities including hypoplastic cranial ganglia.108 Future studies are needed to determine if salivary hypofunction in these disorders is a result of poor innervation of the secretory epithelium, aberrant salivary development or tissue atrophy.
Neurotransmitter Signaling: Acetylcholine and Vasoactive Intestinal Peptide
Parasympathetic nerves of the autonomic nervous system have historically been investigated in a functional context and typically in post-natal or adult tissues. In the adult, these nerves function to regulate organ homeostasis in an unconscious manner. For example, stimulation of parasympathetic nerves innervating the smooth muscle cells of the intestine increases motility to enable digestion. However, recent in vivo and ex vivo studies in which parasympathetic nerves have been removed23 or reduced21 have indicated that they also play an essential role in developmental processes. Here we will discuss two of the many secreted factors, acetylcholine (ACh) and vasoactive intestinal peptide (VIP) that have specific functions in gland development.
ACh is a neurotransmitter synthesized by peripheral and central nervous system and was the first neuronal derived factor characterized in the developing SG.22 In the salivary gland, ACh binds three of the 5 G-protein-coupled muscarinic receptors CHRM1–3, where CHRM1/CHRM3 are epithelial and parasympathetic nerves express CHRM2. CHRM1 and 3 mediate saliva secretion from adult acinar cells, with CHRM3 eliciting the majority of secretory function.10,11 In addition to its secretory role, CHRM1 serves in the maintenance of epithelial progenitor cells in developing and adult SG as inhibition of Ach release or Chrm1 knockdown reduces the pool of undifferentiated KRT5+ basal progenitors.23,31 ACh/CHRM1-mediated maintenance of progenitors is also dependent on HBEGF/EGFR: ACh/CHRM1 signaling transactivates EGFR through release of HBEGF by MMPs. However, in the absence of CHRM1 activation, HBEGF/EGFR drives differentiation along the KRT19+ ductal lineage,23 indicating that a connection between nerve and growth factor signaling is required for maintaining the KRT5+ progenitor cell population. Based on these findings, it is possible that patients with reduced innervation, as found in familial amyloid neuropathy and Hirschsprung’s disease, also have reduced progenitor cell pools required for tissue morphogenesis and homeostasis.
While ACh facilitates progenitor proliferation, VIP and its receptor VIPR1 play an instrumental role in salivary morphogenesis and more specifically, tube formation. Nedvetsky et al.21 found VIP to regulate each of these processes, in part through a cAMP/PKA pathway.21 Notably, the authors showed VIP to mediate lumen expansion via cystic fibrosis transmembrane regulator (CFTR), a cAMP-activated ion channel that localizes to the luminal surfaces of ductal epithelia. This was further supported in vivo using Cftr deficient mice, which exhibited aberrant lumen expansion and ductal tubulogenesis.21 Mutations in CFTR are known to cause cystic fibrosis, a disease characterized by defective electrolyte, fluid, and mucus secretion. Though morphological abnormalities of the SG have not been rigorously assessed in CF patients, it is known that Na+/Cl− ion concentrations are also higher in the saliva of CF patients, and salivary flow is often reduced.109–112 Thus, in cystic fibrosis patients, saliva secretion may be affected by not only inherent transport defects in CFTR, but also structural defects stemming from disrupted VIP signaling during development.
BASEMENT MEMBRANE PROTEINS: COLLAGENS, FIBRONECTIN, LAMININS
Mutations in basement membrane and extracellular matrix (ECM) proteins and their integrin receptors result in a plethora of diseases including Ehlers-Danlos syndrome (collagen III or IV), dystrophic epidermolysis bullosa (collagen VII) and glomerulopathy (fibronectin) that affect multiple organs. Typically, mouse models of ECM/basement membrane mutations have focused on the skin, vasculature and kidney; but how these specific mutations affect development of other organs has not been explored. However, there is substantial evidence indicating collagens, fibronectin and laminins, and their integrin receptors, are required for SG development.
Early studies of SG development established that collagens are required for epithelial branching morphogenesis: degradation of COL1 and 3 reduced epithelial cleft formation and consequently branching.113–115 More recently, Rebustini et al.116 demonstrated that bioactive fragments of collagen IV, produced by MMP cleavage, are also necessary for epithelial branching.116 Cleavage of collagen IV by MT2-MMP releases bioactive NC1 domains (COL4a1 and 2) that bind and activate β1 integrins. Downstream AKT activation then increases epithelial proliferation via both FGFR and HBEGF-mediated mechanisms, linking the basement membrane to growth factor-mediated epithelial proliferation. Whether patients with collagen IV mutations (e.g., Alports disease or hereditary angiopathy with nephropathy, aneurysms, and muscle cramps (HANAC), Table 1), or mutations in MMPs that cleave collagens, have defects in salivary branching remains to be investigated.
Fibronectin (FN) has similarly been shown to be integral to branching morphogenesis. FN promotes cell adhesion, migration, and signaling and is widely expressed in developing tissues, including the SG. In SGs, wedges of FN translocate inward as clefts form between randomly motile epithelial cells, which is accompanied by loss of the cell–cell adhesion molecule E-cadherin in cells adjacent to the FN with FN also regulating the accumulation of other matrix components (e.g., collagen III).117 Ex vivo studies have demonstrated that FN is necessary for SG development: anti-FN antibody or siRNA or anti-integrin-α5/6 (FN receptors) inhibits cleft formation to reduce epithelial branching.117–119 How does FN regulate epithelial morphogenesis? Onodera et al.117 recently established a mechanism whereby FN induces expression of a cleft localized factor, BTBD7, that increases the cell-scattering gene Snail2 and suppresses E-cadherin levels, thereby altering cell morphology and reducing cell–cell adhesion required for cleft formation and branching. Thus, FN appears to be a master regulator of cleft formation controlling intracellular networks to establish and propagate clefts for new bud formation. This would also suggest that compromised FN function, as occurs in glomerulopathy, would impede SG morphogenesis.
Laminins are heterotrimeric (α, β, γ) glycoproteins that are a major component of basement membranes and are essential for embryonic implantation, induction, and maintenance of cell polarity, tissue morphogenesis, and organogenesis. Many of the laminin isoforms are expressed in the SG and display developmentally regulated expression patterns during development.36 The involvement of laminins in SG development has been revealed through a combination of in vivo and ex vivo studies. Function blocking antibodies or inhibitory peptides against laminin α1 or γ1 inhibited branching in SG cultures120–122 and mice deficient in laminin α5 (lama5) had reduced SG branching morphogenesis, with the complete loss of the sublingual gland.36 How does laminin regulate branching? Intriguingly, Rebustini et al.36 reveal LAMA5 to regulate epithelial morphogenesis through promoting FGF signaling: activation of the LAMA5 receptor integrin α3 β1, which is necessary for basement membrane formation and epithelial organization,123 increases FGFR mediated proliferation, which in turn upregulates lama5. Given mutations in integrin α3 (ITGA3) can result in diseases characterized by compromised barrier functions in kidney, lung, and skin (e.g., epidermolysis bullosa) it would be interesting to determine if SG are also compromised. Furthermore, these data suggest LAMA5 may be affected in patients that have defective FGF signaling (e.g., ASLG/LADD), adding to the severity of the disease.
CONCLUSION
As this review indicates, there are many common signaling pathways that regulate initiation and/or progression of salivary gland morphogenesis (Figure 5). These pathways operate in distinct spatiotemporal patterns to establish acinar and ductal growth, development of ganglia, and progenitor cell survival/proliferation. Yet, it is also clear from mouse studies that these molecular pathways act within complex signaling networks that will require a systematic approach to elucidate how they affect morphogenic processes. There also remains a large gap in our understanding on how different cell types impact others during development. For example, endothelial cells form a vascular plexus very early in gland development, yet the function of endothelial cells at these stages and the timing of maturation of the plexus into functional vessels is not known. Moreover, despite extensive investigations on epithelial–mesenchymal interactions and their influence on development, we know very little about the identity of the cells in the neural crest-derived mesenchymal compartment or whether these cells are also adversely affected by disease. As such, there are many more questions than answers; however, with advances in human genetics and the ever-increasing number of mouse models we will no doubt greatly increase our knowledge of how signaling pathways and cells establish tissue architecture and function during salivary gland formation.
FIGURE 5 |.
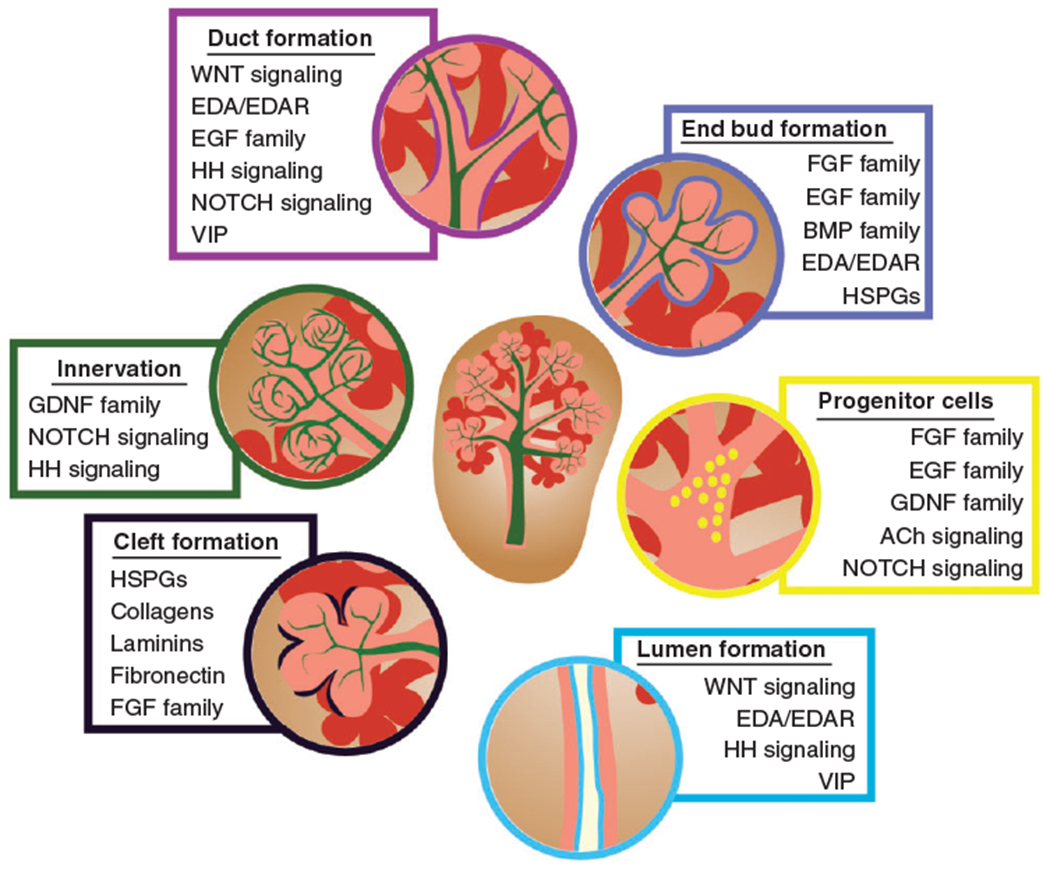
Developmental processes, cell interactions, and signaling pathways involved in salivary gland organogenesis.
Footnotes
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1.Pedersen AM, Bardow A, Jensen SB, Nauntofte B. Saliva and gastrointestinal functions of taste, mastication, swallowing and digestion. Oral Dis 2002, 8:117–129. [DOI] [PubMed] [Google Scholar]
- 2.Martinez-Madrigal F, Micheau C. Histology of the major salivary glands. Am J Surg Pathol 1989, 13:879–899. [DOI] [PubMed] [Google Scholar]
- 3.Garrett JR. The proper role of nerves in salivary secretion: a review. J Dent Res 1987, 66:387–397. [DOI] [PubMed] [Google Scholar]
- 4.Langley JN. On the physiology of the salivary secretion: part I. The influence of the chorda tympani and sympathetic nerves upon the secretion of the sub-maxillary gland of the cat. J Physiol 1878, 1:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ludwig C Neue Versuch Ober die Beihdlfe der Nerven zu der Speichel-sekretion. Mitt Naturforsch Ges 1850, 53–54:210–239. [Google Scholar]
- 6.Proctor GB, Carpenter GH. Regulation of salivary gland function by autonomic nerves. Auton Neurosci 2007, 133:3–18. [DOI] [PubMed] [Google Scholar]
- 7.Ma T, Song Y, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J Biol Chem 1999, 274:20071–20074. [DOI] [PubMed] [Google Scholar]
- 8.Murakami M, Shachar-Hill B, Steward MC, Hill AE. The paracellular component of water flow in the rat submandibular salivary gland. J Physiol 2001, 537:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakahari T, Morimatsu S, Imai Y. Inhibition of salivary fluid secretion by occlusion of the intercellular space. Eur J Morphol 1998, 36(Suppl):107–111. [PubMed] [Google Scholar]
- 10.Gautam D, Heard TS, Cui Y, Miller G, Bloodworth L, Wess J. Cholinergic stimulation of salivary secretion studied with M1 and M3 muscarinic receptor single- and double-knockout mice. Mol Pharmacol 2004, 66:260–267. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura T, Matsui M, Uchida K, Futatsugi A, Kusakawa S, Matsumoto N, Nakamura K, Manabe T, Taketo MM, Mikoshiba K. M(3) muscarinic acetylcholine receptor plays a critical role in parasympathetic control of salivation in mice. J Physiol 2004, 558:561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson LC, Garrett JR, Proctor GB. Advantages of burst stimulation for inducing sympathetic salivary secretion in rats. Q J Exp Physiol 1988, 73:1025–1028. [DOI] [PubMed] [Google Scholar]
- 13.Asking B Sympathetic stimulation of amylase secretion during a parasympathetic background activity in the rat parotid gland. Acta Physiol Scand 1985, 124:535–542. [DOI] [PubMed] [Google Scholar]
- 14.Baum BJ. Principles of saliva secretion. Ann N Y Acad Sci 1993, 694:17–23. [DOI] [PubMed] [Google Scholar]
- 15.Catalan MA, Nakamoto T, Melvin JE. The salivary gland fluid secretion mechanism. J Med Invest 2009, 56(Suppl):192–196. [DOI] [PubMed] [Google Scholar]
- 16.Ianez RF, Buim ME, Coutinho-Camillo CM, Schultz R, Soares FA, Lourenço SV. Human salivary gland morphogenesis: myoepithelial cell maturation assessed by immunohistochemical markers. Histopathology 2010, 57:410–417. [DOI] [PubMed] [Google Scholar]
- 17.Knox SM, Hoffman MP. Salivary Gland Development and Regeneration. Wiley-Blackwell; 2008. [Google Scholar]
- 18.Lourenco SV, Kapas S. Integrin expression in developing human salivary glands. Histochem Cell Biol 2005, 124:391–399. [DOI] [PubMed] [Google Scholar]
- 19.Patel VN, Rebustini IT, Hoffman MP. Salivary gland branching morphogenesis. Differentiation 2006, 74:349–364. [DOI] [PubMed] [Google Scholar]
- 20.Borghese E The development in vitro of the submandibular and sublingual glands of Mus musculus. J Anat 1950, 84:287–302. [PMC free article] [PubMed] [Google Scholar]
- 21.Nedvetsky PI, Emmerson E, Finley JK, Ettinger A, Cruz-Pacheco N, Prochazka J, Haddox CL, Northrup E, Hodges C, Mostov KE, et al. Parasympathetic innervation regulates tubulogenesis in the developing salivary gland. Dev Cell 2014, 30:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coughlin MD. Early development of parasympathetic nerves in the mouse submandibular gland. Dev Biol 1975, 43:123–139. [DOI] [PubMed] [Google Scholar]
- 23.Knox SM, Lombaert IM, Reed X, Vitale-Cross L, Gutkind JS, Hoffman MP. Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science 2010, 329:1645–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffman MP, Kidder BL, Steinberg ZL, Lakhani S, Ho S, Kleinman HK, Larsen M. Gene expression profiles of mouse submandibular gland development: FGFR1 regulates branching morphogenesis in vitro through BMP- and FGF-dependent mechanisms. Development 2002, 129:5767–5778. [DOI] [PubMed] [Google Scholar]
- 25.Haara O, Fujimori S, Schmidt-Ullrich R, Hartmann C, Thesleff I, Mikkola ML. Ectodysplasin and Wnt pathways are required for salivary gland branching morphogenesis. Development 2011, 138: 2681–2691. [DOI] [PubMed] [Google Scholar]
- 26.Wells KL, Mou C, Headon DJ, Tucker AS. Recombinant EDA or sonic Hedgehog rescue the branching defect in Ectodysplasin A pathway mutant salivary glands in vitro. Dev Dyn 2010, 239:2674–2684. [DOI] [PubMed] [Google Scholar]
- 27.De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 2000, 127:483–492. [DOI] [PubMed] [Google Scholar]
- 28.Jaskoll T, Abichaker G, Witcher D, Sala FG, Bellusci S, Hajihosseini MK, Melnick M. FGF10/FGFR2b signaling plays essential roles during in vivo embryonic submandibular salivary gland morphogenesis. BMC Dev Biol 2005, 5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaskoll T, Witcher D, Toreno L, Bringas P, Moon AM, Melnick M. FGF8 dose-dependent regulation of embryonic submandibular salivary gland morphogenesis. Dev Biol 2004b, 268:457–469. [DOI] [PubMed] [Google Scholar]
- 30.Ohuchi H, Hori Y, Yamasaki M, Harada H, Sekine K, Kato S, Itoh N. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem Biophys Res Commun 2000, 277:643–649. [DOI] [PubMed] [Google Scholar]
- 31.Knosp WM, Knox SM, Lombaert IM, Haddox CL, Patel VN, Hoffman MP. Submandibular parasympathetic gangliogenesis requires sprouty-dependent wnt signals from epithelial progenitors. Dev Cell 2015, 32:667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heuckeroth RO, Enomoto H, Grider JR, Golden JP, Hanke JA, Jackman A, Molliver DC, Bardgett ME, Snider WD, Johnson EM Jr, et al. Gene targeting reveals a critical role for neurturin in the development and maintenance of enteric, sensory, and parasympathetic neurons. Neuron 1999, 22:253–263. [DOI] [PubMed] [Google Scholar]
- 33.Rossi J, Herzig KH, Voikar V, Hiltunen PH, Segerstrale M, Airaksinen MS. Alimentary tract innervation deficits and dysfunction in mice lacking GDNF family receptor α2. J Clin Invest 2003, 112:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rossi J, Tomac A, Saarma M, Airaksinen MS. Distinct roles for GFRα1 and GFRα2 signalling in different cranial parasympathetic ganglia in vivo. Eur J Neurosci 2000, 12:3944–3952. [DOI] [PubMed] [Google Scholar]
- 35.Dyachuk V, Furlan A, Shahidi MK, Giovenco M, Kaukua N, Konstantinidou C, Pachnis V, Memic F, Marklund U, Muller T, et al. Neurodevelopment. Parasympathetic neurons originate from nerve-associated peripheral glial progenitors. Science 2014, 345:82–87. [DOI] [PubMed] [Google Scholar]
- 36.Rebustini IT, Patel VN, Stewart JS, Layvey A, Georges-Labouesse E, Miner JH, Hoffman MP. Laminin α5 is necessary for submandibular gland epithelial morphogenesis and influences FGFR expression through β1 integrin signaling. Dev Biol 2007, 308:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jhappan C, Gallahan D, Stahle C, Chu E, Smith GH, Merlino G, Callahan R. Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev 1992, 6:345–355. [DOI] [PubMed] [Google Scholar]
- 38.Entesarian M, Matsson H, Klar J, Bergendal B, Olson L, Arakaki R, Hayashi Y, Ohuchi H, Falahat B, Bolstad AI, et al. Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nat Genet 2005, 37:125–127. [DOI] [PubMed] [Google Scholar]
- 39.Milunsky JM, Zhao G, Maher TA, Colby R, Everman DB. LADD syndrome is caused by FGF10 mutations. Clin Genet 2006, 69:349–354. [DOI] [PubMed] [Google Scholar]
- 40.Steinberg Z, Myers C, Heim VM, Lathrop CA, Rebustini IT, Stewart JS, Larsen M, Hoffman MP. FGFR2b signaling regulates ex vivo submandibular gland epithelial cell proliferation and branching morphogenesis. Development 2005, 132:1223–1234. [DOI] [PubMed] [Google Scholar]
- 41.Lombaert IM, Abrams SR, Li L, Eswarakumar VP, Sethi AJ, Witt RL, Hoffman MP. Combined KIT and FGFR2b signaling regulates epithelial progenitor expansion during organogenesis. Stem Cell Rep 2013, 1:604–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel N, Sharpe PT, Miletich I. Coordination of epithelial branching and salivary gland lumen formation by Wnt and FGF signals. Dev Biol 2011, 358:156–167. [DOI] [PubMed] [Google Scholar]
- 43.Patel VN, Lombaert IM, Cowherd SN, Shworak NW, Xu Y, Liu J, Hoffman MP. Hs3st3-modified heparan sulfate controls KIT+ progenitor expansion by regulating 3-O-sulfotransferases. Dev Cell 2014, 29:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klein OD, Lyons DB, Balooch G, Marshall GW, Basson MA, Peterka M, Boran T, Peterkova R, Martin GR. An FGF signaling loop sustains the generation of differentiated progeny from stem cells in mouse incisors. Development 2008, 135:377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ching ST, Cunha GR, Baskin LS, Basson MA, Klein OD. Coordinated activity of Spry1 and Spry2 is required for normal development of the external genitalia. Dev Biol 2014, 386:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo L, Yu QC, Fuchs E. Targeting expression of keratinocyte growth factor to keratinocytes elicits striking changes in epithelial differentiation in transgenic mice. EMBO J 1993, 12:973–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64:841–848. [DOI] [PubMed] [Google Scholar]
- 48.Makarenkova HP, Hoffman MP, Beenken A, Eliseenkova AV, Meech R, Tsau C, Patel VN, Lang RA, Mohammadi M. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci Signal 2009, 2:ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel VN, Likar KM, Zisman-Rozen S, Cowherd SN, Lassiter KS, Sher I, Yates EA, Turnbull JE, Ron D, Hoffman MP. Specific heparan sulfate structures modulate FGF10-mediated submandibular gland epithelial morphogenesis and differentiation. J Biol Chem 2008, 283:9308–9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bui C, Huber C, Tuysuz B, Alanay Y, Bole-Feysot C, Leroy JG, Mortier G, Nitschke P, Munnich A, Cormier-Daire V. XYLT1 mutations in Desbuquois dysplasia type 2. Am J Hum Genet 2014, 94:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reuter MS, Musante L, Hu H, Diederich S, Sticht H, Ekici AB, Uebe S, Wienker TF, Bartsch O, Zechner U, et al. NDST1 missense mutations in autosomal recessive intellectual disability. Am J Med Genet A 2014, 164A:2753–2763. [DOI] [PubMed] [Google Scholar]
- 52.Isidor B, Pichon O, Redon R, Day-Salvatore D, Hamel A, Siwicka KA, Bitner-Glindzicz M, Heymann D, Kjellen L, Kraus C, et al. Mesomelia-synostoses syndrome results from deletion of SULF1 and SLCO5A1 genes at 8q13. Am J Hum Genet 2010, 87:95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci 2008, 65:1566–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyazaki Y, Nakanishi Y, Hieda Y. Tissue interaction mediated by neuregulin-1 and ErbB receptors regulates epithelial morphogenesis of mouse embryonic submandibular gland. Dev Dyn 2004, 230:591–596. [DOI] [PubMed] [Google Scholar]
- 55.Jaskoll T, Melnick M. Submandibular gland morphogenesis: stage-specific expression of TGF-α/EGF, IGF, TGF-β, TNF, and IL-6 signal transduction in normal embryonic mice and the phenotypic effects of TGF-β2, TGF-β3, and EGF-r null mutations. Anat Rec 1999, 256:252–268. [DOI] [PubMed] [Google Scholar]
- 56.Haara O, Koivisto T, Miettinen PJ. EGF-receptor regulates salivary gland branching morphogenesis by supporting proliferation and maturation of epithelial cells and survival of mesenchymal cells. Differentiation 2009, 77:298–306. [DOI] [PubMed] [Google Scholar]
- 57.Tan M, Grijalva R, Yu D. Heregulin β1-activated phosphatidylinositol 3-kinase enhances aggregation of MCF-7 breast cancer cells independent of extracellular signal-regulated kinase. Cancer Res 1999, 59:1620–1625. [PubMed] [Google Scholar]
- 58.Tang CS, Ngan ES, Tang WK, So MT, Cheng G, Miao XP, Leon TY, Leung BM, Hui KJ, Lui VH, et al. Mutations in the NRG1 gene are associated with Hirschsprung disease. Hum Genet 2012, 131:67–76. [DOI] [PubMed] [Google Scholar]
- 59.Nitta M, Kume T, Nogawa H. FGF alters epithelial competence for EGF at the initiation of branching morphogenesis of mouse submandibular gland. Dev Dyn 2009, 238:315–323. [DOI] [PubMed] [Google Scholar]
- 60.He F, Chen Y. Wnt signaling in lip and palate development. Front Oral Biol 2012, 16:81–90. [DOI] [PubMed] [Google Scholar]
- 61.Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, Johnson CA. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol Genet 2013, 22:1358–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y, Pawlik B, Elcioglu N, Aglan M, Kayserili H, Yigit G, Percin F, Goodman F, Nurnberg G, Cenani A, et al. LRP4 mutations alter Wnt/β-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am J Hum Genet 2010, 86:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamaguchi Y, Yonemura S, Takada S. Grainyhead-related transcription factor is required for duct maturation in the salivary gland and the kidney of the mouse. Development 2006, 133:4737–4748. [DOI] [PubMed] [Google Scholar]
- 64.Aguilar A, Meunier A, Strehl L, Martinovic J, Bonniere M, Attie-Bitach T, Encha-Razavi F, Spassky N. Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. Proc Natl Acad Sci USA 2012, 109:16951–16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ming JE, Roessler E, Muenke M. Human developmental disorders and the Sonic hedgehog pathway. Mol Med Today 1998, 4:343–349. [DOI] [PubMed] [Google Scholar]
- 66.Jaskoll T, Leo T, Witcher D, Ormestad M, Astorga J, Bringas P Jr, Carlsson P, Melnick M. Sonic hedgehog signaling plays an essential role during embryonic salivary gland epithelial branching morphogenesis. Dev Dyn 2004a, 229:722–732. [DOI] [PubMed] [Google Scholar]
- 67.Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development 1997, 124:53–63. [DOI] [PubMed] [Google Scholar]
- 68.Gu LH, Coulombe PA. Hedgehog signaling, keratin 6 induction, and sebaceous gland morphogenesis: implications for pachyonychia congenita and related conditions. Am J Pathol 2008, 173:752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lamm ML, Catbagan WS, Laciak RJ, Barnett DH, Hebner CM, Gaffield W, Walterhouse D, Iannaccone P, Bushman W. Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev Biol 2002, 249:349–366. [DOI] [PubMed] [Google Scholar]
- 70.Moraes RC, Zhang X, Harrington N, Fung JY, Wu MF, Hilsenbeck SG, Allred DC, Lewis MT. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development 2007, 134:1231–1242. [DOI] [PubMed] [Google Scholar]
- 71.Hashizume A, Hieda Y. Hedgehog peptide promotes cell polarization and lumen formation in developing mouse submandibular gland. Biochem Biophys Res Commun 2006, 339:996–1000. [DOI] [PubMed] [Google Scholar]
- 72.Fiaschi M, Kolterud A, Nilsson M, Toftgard R, Rozell B. Targeted expression of GLI1 in the salivary glands results in an altered differentiation program and hyperplasia. Am J Pathol 2011, 179:2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aoto K, Nishimura T, Eto K, Motoyama J. Mouse GLI3 regulates Fgf8 expression and apoptosis in the developing neural tube, face, and limb bud. Dev Biol 2002, 251:320–332. [DOI] [PubMed] [Google Scholar]
- 74.Clarke A Hypohidrotic ectodermal dysplasia. J Med Genet 1987, 24:659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nordgarden H, Johannessen S, Storhaug K, Jensen JL. Salivary gland involvement in hypohidrotic ectodermal dysplasia. Oral Dis 1998, 4:152–154. [DOI] [PubMed] [Google Scholar]
- 76.Headon DJ, Emmal SA, Ferguson BM, Tucker AS, Justice MJ, Sharpe PT, Zonana J, Overbeek PA. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414:913–916. [DOI] [PubMed] [Google Scholar]
- 77.Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet 1999, 22:366–369. [DOI] [PubMed] [Google Scholar]
- 78.Srivastava AK, Pispa J, Hartung AJ, Du Y, Ezer S, Jenks T, Shimada T, Pekkanen M, Mikkola ML, Ko MS, et al. The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc Natl Acad Sci USA 1997, 94:13069–13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blecher SR, Debertin M, Murphy JS. Pleiotropic effect of Tabby gene on epidermal growth factor-containing cells of mouse submandibular gland. Anat Rec 1983, 207:25–29. [DOI] [PubMed] [Google Scholar]
- 80.Jaskoll T, Zhou YM, Trump G, Melnick M. Ectodysplasin receptor-mediated signaling is essential for embryonic submandibular salivary gland development. Anat Rec A Discov Mol Cell Evol Biol 2003, 271:322–331. [DOI] [PubMed] [Google Scholar]
- 81.Chang SH, Jobling S, Brennan K, Headon DJ. Enhanced Edar signalling has pleiotropic effects on craniofacial and cutaneous glands. PLoS One 2009, 4:e7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cui CY, Schlessinger D. EDA signaling and skin appendage development. Cell Cycle 2006, 5:2477–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Melnick M, Phair RD, Lapidot SA, Jaskoll T. Salivary gland branching morphogenesis: a quantitative systems analysis of the Eda/Edar/NFkappaB paradigm. BMC Dev Biol 2009, 9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pummila M, Fliniaux I, Jaatinen R, James MJ, Laurikkala J, Schneider P, Thesleff I, Mikkola ML. Ectodysplasin has a dual role in ectodermal organogenesis: inhibition of Bmp activity and induction of Shh expression. Development 2007, 134:117–125. [DOI] [PubMed] [Google Scholar]
- 85.Pispa J, Jung HS, Jernvall J, Kettunen P, Mustonen T, Tabata MJ, Kere J, Thesleff I. Cusp patterning defect in Tabby mouse teeth and its partial rescue by FGF. Dev Biol 1999, 216:521–534. [DOI] [PubMed] [Google Scholar]
- 86.Haara O, Harjunmaa E, Lindfors PH, Huh SH, Fliniaux I, Aberg T, Jernvall J, Ornitz DM, Mikkola ML, Thesleff I. Ectodysplasin regulates activator-inhibitor balance in murine tooth development through Fgf20 signaling. Development 2012, 139:3189–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sieber C, Kopf J, Hiepen C, Knaus P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev 2009, 20:343–355. [DOI] [PubMed] [Google Scholar]
- 88.Zouvelou V, Luder HU, Mitsiadis TA, Graf D. Deletion of BMP7 affects the development of bones, teeth, and other ectodermal appendages of the orofacial complex. J Exp Zool B Mol Dev Evol 2009, 312B:361–374. [DOI] [PubMed] [Google Scholar]
- 89.Jaskoll T, Zhou YM, Chai Y, Makarenkova HP, Collinson JM, West JD, Hajihosseini MK, Lee J, Melnick M. Embryonic submandibular gland morphogenesis: stage-specific protein localization of FGFs, BMPs, Pax6 and Pax9 in normal mice and abnormal SMG phenotypes in FgfR2-IIIc(+/δ), BMP7(−/−) and Pax6(−/−) mice. Cells Tissues Organs 2002, 170:83–98. [DOI] [PubMed] [Google Scholar]
- 90.Piscione TD, Phan T, Rosenblum ND. BMP7 controls collecting tubule cell proliferation and apoptosis via Smad1-dependent and -independent pathways. Am J Physiol Renal Physiol 2001, 280:F19–F33. [DOI] [PubMed] [Google Scholar]
- 91.Lamm ML, Podlasek CA, Barnett DH, Lee J, Clemens JQ, Hebner CM, Bushman W. Mesenchymal factor bone morphogenetic protein 4 restricts ductal budding and branching morphogenesis in the developing prostate. Dev Biol 2001, 232:301–314. [DOI] [PubMed] [Google Scholar]
- 92.Itasaki N, Hoppler S. Crosstalk between Wnt and bone morphogenic protein signaling: a turbulent relationship. Dev Dyn 2010, 239:16–33. [DOI] [PubMed] [Google Scholar]
- 93.Kizu Y, Sakurai H, Katagiri S, Shinozaki N, Ono M, Tsubota K, Saito J. Immunohistological analysis of tumour growth factor β1 expression in normal and inflamed salivary glands. J Clin Pathol 1996, 49:728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hall B, Zheng C, Swaim W, Cho A, Nagineni C, Flanders K, Ambudkur I, Maum B, Kulkarni A. Conditional overexpression of TGF-β1 disrupts mouse salivary gland development and function. Lab Invest. 2010, 90:543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hardman WE, Cameron IL. Colonic crypts located over lymphoid nodules of 1,2-dimethylhydrazine-treated rats are hyperplastic and at high risk of forming adenocarcinomas. Carcinogenesis 1994, 15:2353–2361. [DOI] [PubMed] [Google Scholar]
- 96.Janebodin K, Buranaphatthana W, Ieronimakis N, Hays AL, Reyes M. An in vitro culture system for long-term expansion of epithelial and mesenchymal salivary gland cells: role of TGF-β1 in salivary gland epithelial and mesenchymal differentiation. BioMed Res Int 2013, 2013:815895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McCartney-Francis NL, Mizel DE, Redman RS, Frazier-Jessen M, Panek RB, Kulkarni AB, Ward JM, McCarthy JB, Wahl SM. Autoimmune Sjogren’s-like lesions in salivary glands of TGF-β1-deficient mice are inhibited by adhesion-blocking peptides. J Immunol 1996, 157:1306–1312. [PubMed] [Google Scholar]
- 98.Nandula SR, Amarnath S, Molinolo A, Bandyopadhyay BC, Hall B, Goldsmith CM, Zheng C, Larsson J, Sreenath T, Chen W, et al. Female mice are more susceptible to developing inflammatory disorders due to impaired transforming growth factor β signaling in salivary glands. Arthritis Rheum 2007, 56:1798–1805. [DOI] [PubMed] [Google Scholar]
- 99.Enomoto H, Heuckeroth RO, Golden JP, Johnson EM, Milbrandt J. Development of cranial parasympathetic ganglia requires sequential actions of GDNF and neurturin. Development 2000, 127:4877–4889. [DOI] [PubMed] [Google Scholar]
- 100.Rossi J, Airaksinen MS. GDNF family signalling in exocrine tissues: distinct roles for GDNF and neurturin in parasympathetic neuron development. Adv Exp Med Biol 2002, 506:19–26. [DOI] [PubMed] [Google Scholar]
- 101.Knox SM, Lombaert IM, Haddox CL, Abrams SR, Cotrim A, Wilson AJ, Hoffman MP. Parasympathetic stimulation improves epithelial organ regeneration. Nat Commun 2013, 4:1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xiao N, Lin Y, Cao H, Sirjani D, Giaccia AJ, Koong AC, Kong CS, Diehn M, Le QT. Neurotrophic factor GDNF promotes survival of salivary stem cells. J Clin Invest 2014, 124:3364–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Johansson I, Ryberg M, Steen L, Wigren L. Salivary hypofunction in patients with familial amyloidotic polyneuropathy. Oral Surg Oral Med Oral Pathol 1992, 74:742–748. [DOI] [PubMed] [Google Scholar]
- 104.Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol 2011, 10:1086–1097. [DOI] [PubMed] [Google Scholar]
- 105.Shohat M, Weisz Hubshman M. Familial dysautonomia. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, eds. GeneReviews(R). Seattle, WA: University of Washington; 1993. [Google Scholar]
- 106.Juusela P, Tanskanen M, Nieminen A, Kari K, Suominen L, Uitto VJ, Kiuru-Enari S. Xerostomia in hereditary gelsolin amyloidosis. Amyloid 2013, 20:39–44. [DOI] [PubMed] [Google Scholar]
- 107.Osterhus IN, Skogedal N, Akre H, Johnsen UL, Nordgarden H, Asten P. Salivary gland pathology as a new finding in Treacher Collins syndrome. Am J Med Genet A 2012, 158A:1320–1325. [DOI] [PubMed] [Google Scholar]
- 108.Trainor PA. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am J Med Genet A 2010, 152A:2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ceder O, van Dijken J, Ericson T, Kollberg H. Ribonuclease in different types of saliva from cystic fibrosis patients. Acta Paediatr Scand 1985, 74:102–106. [DOI] [PubMed] [Google Scholar]
- 110.Davies H, Bagg J, Goodchild MC, McPherson MA. Defective regulation of electrolyte and protein secretion in submandibular saliva of cystic fibrosis patients. Acta Paediatr Scand 1991, 80:1094–1095. [DOI] [PubMed] [Google Scholar]
- 111.Goncalves AC, Marson FA, Mendonca RM, Ribeiro JD, Ribeiro AF, Paschoal IA, Levy CE. Saliva as a potential tool for cystic fibrosis diagnosis. Diagn Pathol 2013, 8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kolberg H, Danielsson A, Glitterstam K, Henriksson R, Marklund S. Studies on parotid saliva in cystic fibrosis. Acta Paediatr Scand 1982, 71:321–322. [DOI] [PubMed] [Google Scholar]
- 113.Fukuda Y, Masuda Y, Kishi J, Hashimoto Y, Hayakawa T, Nogawa H, Nakanishi Y. The role of interstitial collagens in cleft formation of mouse embryonic submandibular gland during initial branching. Development 1988, 103:259–267. [DOI] [PubMed] [Google Scholar]
- 114.Grobstein C, Cohen J. Collagenase: effect on the morphogenesis of embryonic salivary epithelium in vitro. Science 1965, 150:626–628. [DOI] [PubMed] [Google Scholar]
- 115.Nakanishi Y, Sugiura F, Kishi J, Hayakawa T. Scanning electron microscopic observation of mouse embryonic submandibular glands during initial branching: preferential localization of fibrillar structures at the mesenchymal ridges participating in cleft formation. J Embryol Exp Morphol 1986, 96:65–77. [PubMed] [Google Scholar]
- 116.Rebustini IT, Myers C, Lassiter KS, Surmak A, Szabova L, Holmbeck K, Pedchenko V, Hudson BG, Hoffman MP. MT2-MMP-dependent release of collagen IV NC1 domains regulates submandibular gland branching morphogenesis. Dev Cell 2009, 17:482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Onodera T, Sakai T, Hsu JC, Matsumoto K, Chiorini JA, Yamada KM. Btbd7 regulates epithelial cell dynamics and branching morphogenesis. Science 2010, 329:562–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Larsen M, Wei C, Yamada KM. Cell and fibronectin dynamics during branching morphogenesis. J Cell Sci 2006, 119:3376–3384. [DOI] [PubMed] [Google Scholar]
- 119.Sakai T, Larsen M, Yamada KM. Fibronectin requirement in branching morphogenesis. Nature 2003, 423:876–881. [DOI] [PubMed] [Google Scholar]
- 120.Kadoya Y, Kadoya K, Durbeej M, Holmvall K, Sorokin L, Ekblom P. Antibodies against domain E3 of laminin-1 and integrin α 6 subunit perturb branching epithelial morphogenesis of submandibular gland, but by different modes. J Cell Biol 1995, 129:521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kadoya Y, Nomizu M, Sorokin LM, Yamashina S, Yamada Y. Laminin α1 chain G domain peptide, RKRLQVQLSIRT, inhibits epithelial branching morphogenesis of cultured embryonic mouse submandibular gland. Dev Dyn 1998, 212:394–402. [DOI] [PubMed] [Google Scholar]
- 122.Kadoya Y, Salmivirta K, Talts JF, Kadoya K, Mayer U, Timpl R, Ekblom P. Importance of nidogen binding to laminin γ1 for branching epithelial morphogenesis of the submandibular gland. Development 1997, 124:683–691. [DOI] [PubMed] [Google Scholar]
- 123.Menko AS, Kreidberg JA, Ryan TT, Van Bockstaele E, Kukuruzinska MA. Loss of α3β1 integrin function results in an altered differentiation program in the mouse submandibular gland. Dev Dyn 2001, 220:337–349. [DOI] [PubMed] [Google Scholar]