

Abstract
Classical studies of vertebrate physiology have usually been confined to a given organ or cell type. The use of mouse genetics has changed this approach and has rejuvenated the concept of a whole-body study of physiology. One physiological system that has been profoundly influenced by mouse genetics is skeletal physiology. Indeed, genetic approaches have identified several unexpected organs that affect bone physiology. These new links have begun to provide a plausible explanation for the evolutionary involvement of hormones such as leptin with bone physiology. These genetic approaches have also revealed bone as a true endocrine organ capable of regulating energy metabolism and reproduction. Collectively, the body of work discussed below illustrates a new and unconventional role for bone in mammalian physiology.
Keywords: osteocalcin, serotonin, bone mass accrual, energy metabolism, reproduction
INTRODUCTION: THE OBVIOUS AND BEYOND
Physiology has been approached by two different methodologies. Molecular physiology is the most recent approach and deals with the function of one particular cell type or one protein (or group of proteins), usually with a focus on transcription factors, membrane surface receptors, or ion channels. This focused approach has provided a better understanding of molecular or cellular events and has contributed to novel rationales for effective treatments of human disease. However, the physiological approach that predates molecular methods is whole-organism physiology. Claude Bernard initially defined this aspect of physiology when he described the milieu interiéur. In the next century, W.C. Cannon forged the fundamental concept of homeostasis, and L.J. Henderson proposed that there is functional dependency between organs (1–4). Now, many decades later, the ability to spatially and temporally inactivate one gene in a particular cell type has provided critical experimental tools for studying and understanding whole-organism physiology. Below, we discuss how a whole-organism approach to physiology has influenced and transformed our understanding of bone physiology. This transformation was made possible by leveraging fully the concept of functional dependency and in all cases by providing a molecular basis for the novel functions that are described in detail in this review.
SPECIFIC FEATURES OF BONE AND THEIR IMPACT ON WHOLE-ORGANISM PHYSIOLOGY
To surmise which organs the skeleton and more precisely bones interact with, one needs to look at two characteristics of bone tissue. Bone is the only tissue in the body that contains a cell type whose function is to destroy (resorb) the host tissue, the osteoclast. Destruction of bone can be viewed as an autoimmune reaction and is required during bone modeling, which is responsible for linear growth in childhood, and during bone remodeling, which is responsible for maintenance of bone mass in adulthood (5–8).
Bone is also one of the largest tissues in the human body. This second important feature of bone implies that bone remodeling consumes a large amount of energy. For this reason, we hypothesized that bone remodeling must be coordinately regulated with energy metabolism (5, 9). Clinical evidence supports this hypothesis. For instance, anorexia nervosa in children leads to a complete arrest of skeletal growth. Likewise, adult anorectic patients develop osteoporosis, whereas adult obese patients display a higher bone mass that protects them from osteoporosis (10–14). Although these studies are subject to interpretation, they suggest a correlation between bone mass accrual and food intake.
Clinical experiences tell us one more thing. One of the most-established features of bone pathology is that osteoporosis, a low-bone-mass disease, appears after menopause (11, 15, 16). In other words, sex steroid hormones regulate bone mass. It is also possible that bone or bone-derived hormones reciprocate to regulate fertility. At a more global level, one wonders if bone mass accrual, energy metabolism, and reproduction are all coordinated by endocrine regulation (9). The possible cross talk between these three distinct physiological systems has several implications. First, such a coordinated regulation would begin with bony vertebrates because the energetic needs of bone modeling and remodeling justify its existence. Second, given the role of the brain in energy homeostasis, bone (re)modeling may be subject to central regulation. Finally, there are likely to be feedback loops that originate from bone and that affect energy metabolism. Current data supporting the hypothesis that bone mass, metabolism, and reproduction are linked are presented below.
BONE AS IT IS KNOWN TO BE: A TAKER
Coordinated Control of Bone Mass and Energy Metabolism: The Viewpoint of the Adipocyte (Part I)
Of all hormones known to regulate energy metabolism, leptin is the best candidate to test the hypothesis that bone mass, metabolism, and reproduction are linked. Leptin, an adipocyte-derived hormone, regulates appetite, energy expenditure, and fertility by signaling in the brain (9, 17–22). Thus, it fulfills many requirements of our hypothesis. A less obvious but equally important reason is that leptin appears during evolution with bone cells, not with appetite, reproduction, or adipocytes (23, 24). It is reasonable to assume that the appearance of a given gene during evolution coincides with the functional needs of a given organism. If we apply this general assumption to leptin, its appearance in bony vertebrates might suggest an endocrine link between the control of bone (re)modeling and the control of energy metabolism. This hypothesis has been addressed and was validated largely by using cell-specific loss-of-function leptin mouse models.
In addition to its role in energy metabolism, leptin is a powerful inhibitor of bone mass accrual. Hence, and in full agreement with the notion that there may be coregulation of bone (re)modeling and energy metabolism, leptin decreases both food intake and bone mass (5, 9, 22, 25, 26). Leptin is exceptionally powerful in inhibiting bone mass accrual. Indeed, mice or humans lacking leptin or its receptor develop a high-bone-mass phenotype even though they are hypogonadic, a condition that tends to greatly increase bone resorption. Only leptin signaling deficiency can achieve such a true biological tour de force. If this feature is taken at face value, it suggests that the inhibition of bone mass accrual may be a major function of leptin. This has been verified genetically through the use of a partial gain-of-function leptin signaling model that showed that leptin’s regulation of bone mass requires a lower threshold of leptin signaling than that needed for regulation of energy metabolism and reproduction (9). This latter observation resonates with the above-mentioned fact that leptin and bone appear simultaneously during evolution.
Leptin regulation of bone mass accrual revealed for the first time the existence of central control of bone mass. There are two known mediators linking leptin signaling in the brain to the osteoblasts, the ultimate target cell of leptin. The first one is the sympathetic nervous system, signaling through the β2-adrenergic receptor (Adrβ2) present in osteoblasts (Figure 1) (20, 22). In the osteoblast, sympathetic tone recruits several transcriptional components of the molecular clock, cMyc and cAMP response element binding (CREB) protein, to inhibit cell proliferation (Figure 1) (27–34). Sympathetic tone also increases expression in osteoblasts of RankL, the most powerful osteoclast differentiation factor (Figure 1) (20, 28, 33). Thus, the sympathetic tone inhibits bone formation and favors bone resorption, which in turn reduces bone mass accrual (Figure 1) (5, 35–38). As a result, β-blockers antagonizing Adrβ2 can cure osteoporosis in mice, rats, and humans (22, 39). The second mediator of leptin regulation of bone mass accrual is the cocaine amphetamine regulated transcript (CART), a peptide that is found in the brain and the general circulation and whose expression is regulated by leptin (20, 40–42). CART also acts on osteoblasts but inhibits RankL expression and bone resorption (Figure 1). This function of CART, the only one identified in CART-less or carpt−/− mice maintained on a normal diet, is important because the absence of CART increases bone resorption, as seen in leptin signaling–deficient mice (Figure 1) (20, 43). That the sympathetic tone through Adrβ2 and CART signaling is not involved in the control of appetite or energy expenditure in mice fed a normal diet or in fertility implies that if bone metabolism and energy metabolism are coregulated by the same molecules, such molecules must reside in the brain. The broader implication of these collective results is that the brain controls bone mass accrual (44, 45).
Figure 1.
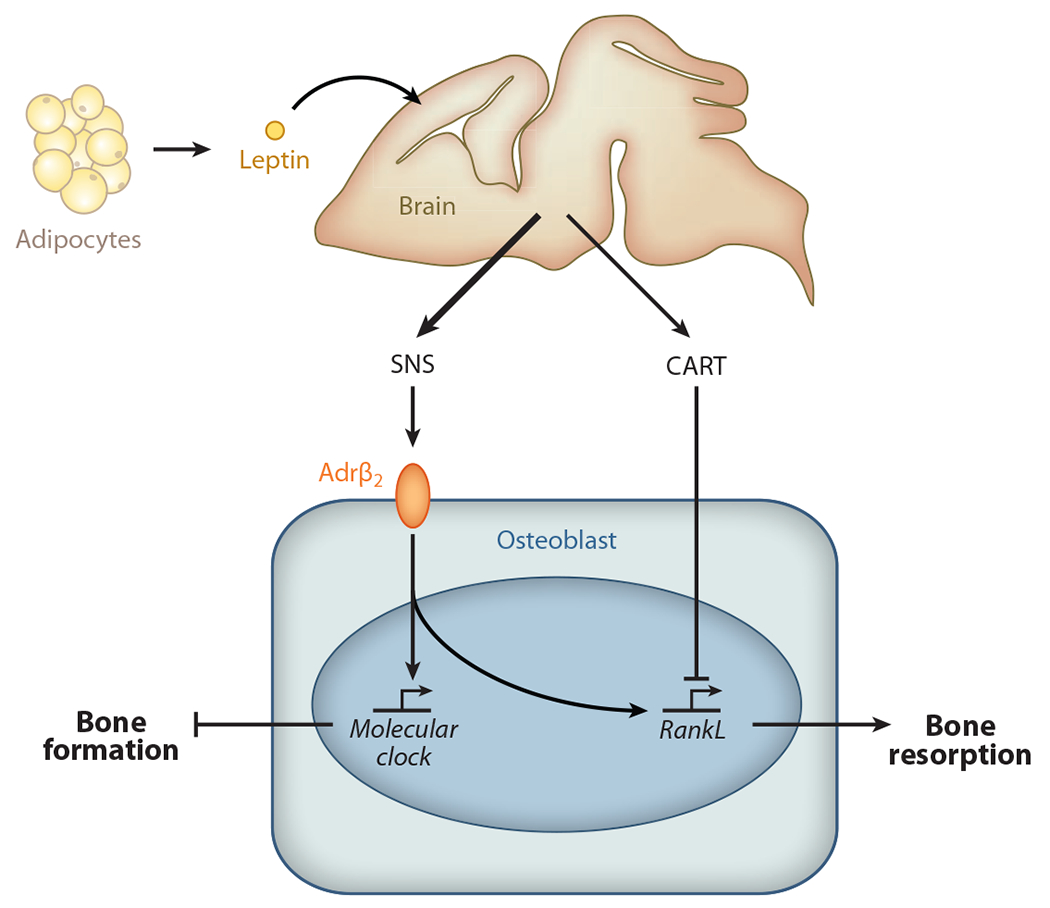
The sympathetic nervous system (SNS) and CART (cocaine amphetamine regulated transcript) mediate leptin signaling in the brain to the osteoblasts. The SNS inhibits bone formation and favors bone resorption. Following β2-adrenergic receptor (Adrβ2) activation in osteoblasts, the sympathetic tone favors expression of RankL, the most powerful osteoclast differentiation factor, and recruits several transcriptional components of the molecular clock, inhibiting bone formation. CART, the second mediator of the leptin regulation of bone mass accrual, also acts on osteoblasts, but by inhibiting RankL expression and bone resorption.
Coordinated Regulation of Bone Mass and Energy Metabolism: The Viewpoint of the Brain
The mechanism whereby leptin signaling in the brain affects bone physiology provides a rare but frightening example of how a genetics-only approach to a biological problem could have misled scientists. Indeed, as explained below, a genetics-only approach would have misled scientists about the role of hypothalamic neurons by suggesting that such neurons are not involved in leptin signaling.
Chemical lesion experiments of rat hypothalamic neurons performed in the 1940s resulted in hyperphagia and obesity similar to those observed in leptin signaling–deficient mice (22, 46–51). The signaling form of the leptin receptor is highly expressed in neurons of the ventromedial hypothalamus (VMH) nuclei and arcuate hypothalamus nuclei, a part of the brain involved in the regulation of many homeostatic functions (19, 52–56). On the basis of these observations, we and other investigators in the field hypothesized that leptin signals directly in the hypothalamus to regulate bone mass accrual. This working hypothesis was initially supported by many experiments seeking to verify this model. For instance, chemical lesioning of VMH neurons resulted in a high-bone-mass phenotype similar to the one seen in ob/ob mice, which lack leptin, whereas lesioning in ob/ob mice, followed by leptin intracerebroventricular (ICV) infusion, failed to rescue the bone phenotype observed in these mice (22). Such evidence indicated that VMH neurons regulate bone mass accrual in a leptin-dependent manner, but direct proof that leptin actually binds these neurons was lacking.
A couple of years later, another group of investigators carried out a genetic experiment that involved inactivation of the leptin receptor selectively in VMH nuclei, in arcuate nuclei, or in both nuclei (57, 58). Surprisingly, all these mutant mice had normal bone mass. Even more remarkable, appetite, energy metabolism, and body weight were normal when these mutant animals were fed a normal diet, the diet on which leptin signaling–deficient mice display hyperphagia. How could one reconcile these contradictory sets of data, and were they really contradictory?
One possible way to explain this paradox is to consider differences between chemical lesions and genetic lesions. The genetic approach appears to have the advantage because it allows for more precise deletion. A second and possibly more constructive way to approach these data is to consider that both experimental approaches are valid and that each has a valid set of data. The chemical lesioning experiments indicate that leptin requires the integrity of the VMH and arcuate neurons to regulate bone mass and energy metabolism, respectively. The genetic data, in contrast, showed that leptin does not need to bind to VMH or to arcuate neurons to fulfill its functions. Thus, the two sets of data are complementary and suggest a novel hypothesis: Leptin may not signal in the hypothalamus but may signal elsewhere in the brain to regulate the synthesis and/or secretion of a neuromediator(s) that then acts in hypothalamic neurons.
As shown for other biomedical mysteries, clinical observations greatly helped in identifying this hypothetical mediator. Serotonin reuptake inhibitors (SSRIs), a class of drugs preventing serotonin reuptake in neurons, are widely used to treat depression and other mood disorders. Like most drugs, SSRIs have side effects that include bone loss, hyperphagia, and body weight gain (59–63). These clinical observations indicated that brain serotonin affects, in ways yet to be defined, bone mass accrual, appetite, and perhaps other aspects of energy metabolism.
These clinical observations provided true insight in the search for this hypothetical mediator. Serotonin is a neuromediator made by brainstem neurons and is also a hormone synthesized by the enterochromaffin cells of the duodenum (58, 64–68). However, serotonin does not cross the blood-brain barrier, and thus each pool of serotonin behaves as a totally independent entity with conceivably different functions (58, 68). Embryonic or postnatal inactivation of tryptophan hydroxylase 2 (Tph2),the initial enzyme necessary for the synthesis of serotonin, showed that brain serotonin is a powerful activator of bone mass accrual (Figure 2). Because serotonin does not cross the blood-brain barrier, this experiment identified it as the first neuromediator to truly affect bone mass (58–69). Brain serotonin is also an activator of appetite and a regulator of energy expenditure. Axon tracing and cell-specific and time-specific gene inactivation showed that serotonin signals in VMH and arcuate neurons through distinct receptors to postnatally regulate bone mass and appetite, respectively (Figure 2). Serotonin favors bone mass accrual by decreasing sympathetic tone in VMH neurons, and it also enhances appetite by favoring expression in arcuate neurons of pro-opiomelanocortin-α(Pomc), melanocortin receptor 4 (MC4R), and other genes regulating appetite (Figure 2) (58, 68). In-depth molecular studies showed that in both hypothalamic nuclei serotonin fulfills its function through the transcription factor CREB (Figure 2) (68).
Figure 2.
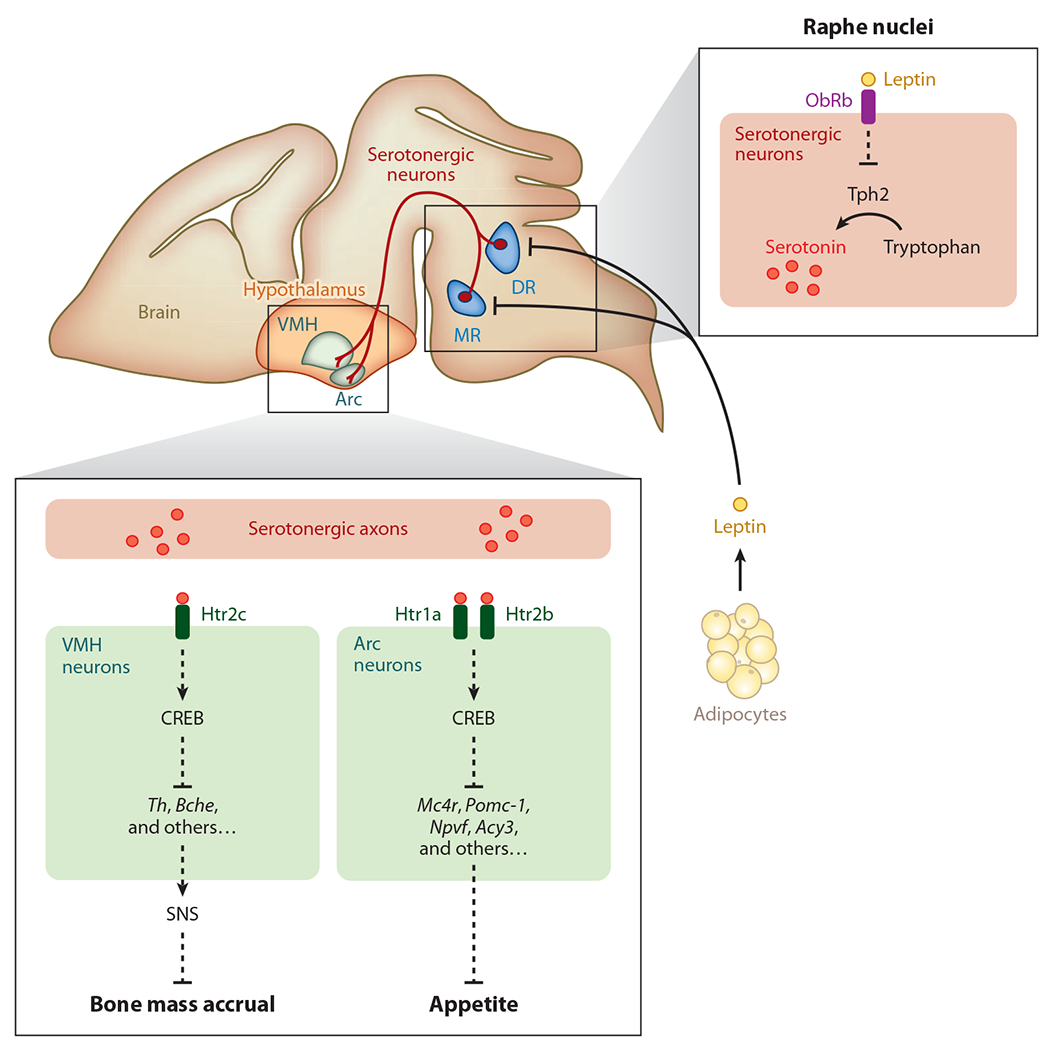
Brain-derived serotonin regulation of bone mass accrual and appetite. Brain-derived serotonin is synthesized by the hydroxylation of tryptophan, a rate-limiting reaction performed by the enzyme tryptophan hydroxylase 2 (Tph2) in the neurons of the dorsal raphe nuclei (DR) and median raphe nuclei (MR) in the brain stem. The axonal projections of serotonergic neurons reach ventromedial hypothalamus (VMH) and arcuate hypothalamus (Arc) neurons of the hypothalamus. Following its binding to the Htr2c receptor in neurons of the VMH nuclei, serotonin favors bone mass accrual, whereas following its binding to the Htr1a and Htr2b receptors in neurons of the Arc nuclei, serotonin favors appetite. The cAMP response element binding (CREB) protein is a crucial transcriptional effector of brain-derived serotonin in Arc neurons. CREB inhibits the expression of the genes encoding tyrosine hydroxylase (Th) and butyrylcholinesterase (Bche) in the VMH and the expression of several genes affecting appetite [melanocortin receptor 4 (Mc4r), pro-opiomelanocortin-α (Pomc-1), neuropeptide VF precursor (Npvf), aspartoacylase 3 (Acy3)] in Arc neurons. Leptin, an adipocyte-derived hormone, directly inhibits serotonin production and its release by the raphe nuclei neurons of the brain stem. The action of leptin is mediated by its receptor, ObRb, which is expressed on these neurons. SNS denotes sympathetic nervous system. Dashed lines indicate that regulation is not a primary signal (but direct); there may be other molecules in between.
That serotonin influences bone mass and energy metabolism in a manner opposite that of leptin suggested a model whereby leptin coordinates the inhibition of bone mass accrual and appetite by decreasing serotonin synthesis and/or release (Figure 2) (58). This model has now been verified in vivo. Classical neurophysiology, expression analyses, genetic epistasis studies, cell-specific gene inactivation experiments, and pharmacological interventions all demonstrated that, indeed, leptin binds to serotonergic neurons and inhibits serotonin synthesis and release from these neurons. Data gathered so far indicate that inhibition of serotonin synthesis and release by brainstem neurons are the main mechanisms whereby leptin postnatally coordinates the regulation of bone mass accrual and appetite (68). In addition, an inhibitor of serotonin signaling efficiently decreased appetite and body weight in leptin-deficient mice, further verifying that serotonin is a target of leptin signaling in the brain (68). This work has important therapeutic implications.
These and other data gathered in several laboratories paint a richer and perhaps more lucid picture of leptin biology. Leptin coevolved with bone tens of million years after fertility and appetite and at a time when food sources were limiting. This suggests that leptin arose to coordinate the regulation of appetite and bone mass accrual so that bone growth does not occur in the absence of food or with low energy intake. Thus, leptin is the first of a small group of hormones tightly linking bone physiology and energy metabolism.
Coordinated Regulation of Bone Mass and Energy Metabolism, a Detour Through the Gut: Surprising and Yet Predictable
The importance of brain-derived serotonin during bone (re)modeling, along with the fact that serotonin does not cross the blood-brain barrier, begged the question as to whether gut-derived serotonin exerts any influence on bone (re)modeling. This is the first question we began to address. Only later did we realize that two humans with skeletal dysplasia might help answer this research question. Initially, as a control for specificity to understand how brain serotonin functions in bone physiology, we inactivated tryptophan hydroxylase 1 (Tph1), the counterpart of Tph2 in the gut. Normally, Tph1 is expressed in enterochromaffin cells of the gut (Figure 3) (58, 64, 66, 70). This experiment revealed that gut serotonin influences bone formation in a manner opposite that of brain serotonin: Gut serotonin inhibits rather than enhances bone formation by osteoblasts (68). In essence, gut serotonin acts as a hormone, binding to receptors on osteoblasts that are distinct from those on VMH neurons. Moreover, in contrast to brain serotonin’s activity, gut serotonin inhibits the activity of the transcription factor CREB. As a result, it hampers osteoblast proliferation (Figure 3) (68). Thus, serotonin is a rare example of a single molecule that exerts totally opposite effects on the same physiological function, depending on its site of synthesis. Brain serotonin favors bone mass accrual, whereas gut serotonin inhibits bone formation and therefore bone mass accrual (68). That each pool of serotonin affects bone mass through CREB also identified serotonin as a major transcriptional regulator of bone (re)modeling by affecting transcriptional programs in both osteoblasts and hypothalamic neurons (Figure 3) (68, 69). One more surprise was the fact that removing serotonin on either side of the blood-brain barrier results in a bone phenotype identical to that observed after simply depleting the small amount of brain-derived serotonin (brain serotonin accounts for only 5% of total serotonin).
Figure 3.
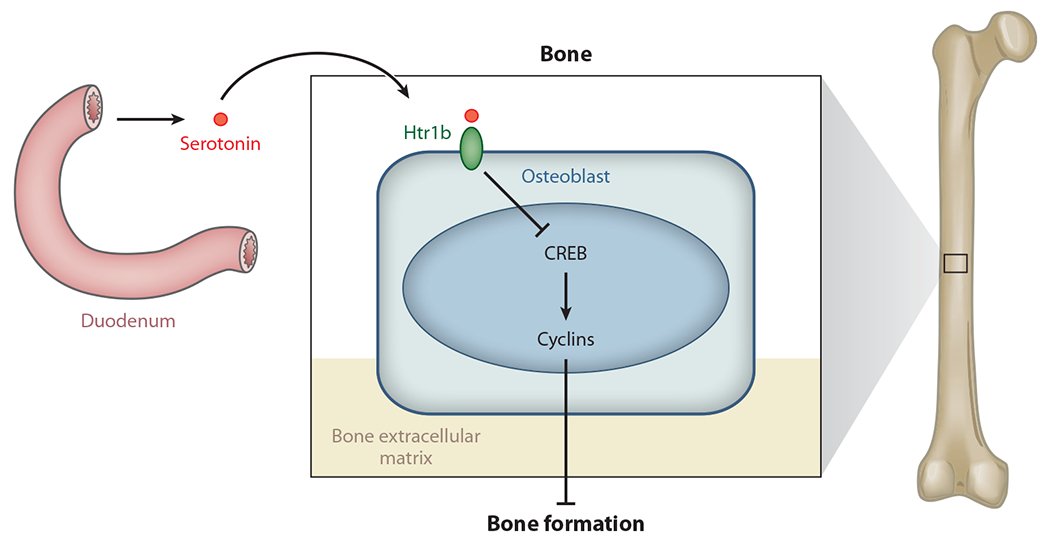
Gut-derived serotonin regulation of bone mass accrual. Gut-derived serotonin is synthesized in enterochromaffin cells of the duodenum and acts on osteoblasts through its receptor, Htr1b, and CREB to inhibit osteoblast proliferation.
At the time this review was submitted, published studies showed that circulating serotonin levels are high in patients suffering from osteoporosis pseudolioma disease and low in patients affected with high-bone-mass syndrome (70, 71). The medical relevance and therapeutic implication of these observations go well beyond these two tragic but rare diseases. Indeed, these findings imply that inhibiting Tph1 activity in enterochromaffin cells of the duodenum could become an anabolic treatment for osteoporosis. This therapeutic application has been validated in rodents, and its broader implications in humans are currently being tested. That an inhibitor of gut serotonin synthesis increases bone formation may be the most emphatic verification of the importance of serotonin in bone mass regulation (72).
BONE’S CHANGE OF IDENTITY: FROM A TAKER TO A GIVER
That both energy metabolism and the gastrointestinal tract influence bone (re)modeling is a novel notion; the same is true for the central control of bone mass. Yet these two novel modes of regulation of bone mass fit well with the well-established notion that bones are recipients of hormonal inputs, despite the broadly accepted view that bones are calcified tubes with only structural properties. To dispel this latter notion and to truly change this narrow concept of bones, one needs to show that bone is not only a recipient of external influences but an endocrine organ affecting functions that have nothing to do with its own integrity. The second part of this review article addresses this aspect of bone physiology.
Coordinated Regulation of Bone (Re)modeling and Energy Metabolism: The Viewpoint of the Osteoblasts
Although we had long suspected that bone must be an endocrine organ regulating energy metabolism, it took almost 10 years and a stroke of luck to demonstrate that this is the case. We began by elucidating the function of genes encoding secreted or signaling molecules that are expressed exclusively in osteoblasts. Esp (embryonic stem cell phosphatase), one such gene, eventually revealed the endocrine nature of bone.
Esp encodes a large protein containing a long extracellular domain, a transmembrane domain, and an intracellular tyrosine phosphatase moiety (73–75). Remarkably, this gene is expressed in only two cell types, the osteoblast and the Sertoli cell of the testis. Its pattern of expression justified an in vivo functional analysis of this gene. This was done through two complementary strategies. The Smith laboratory knocked in a Lac Z allele in the Esp locus (73), whereas we removed the phosphatase domain of OST-PTP (osteoblast-testicular tyrosine phosphatase) in an osteoblast-specific manner (76). Both mutant mouse strains exhibited an identical metabolic phenotype, described below. This result shows that Esp influences insulin sensitivity through its expression in osteoblasts, as explained below.
The first phenotype noticed in both Esp−/− mice and Esposb−/− mice is that, although they were born at the expected Mendelian ratio, they had a strong tendency to die in the first 2 weeks of life. This pattern was so pronounced that at weaning we failed to obtain 25% of homozygous mutant mice when heterozygous mutant mice were intercrossed, despite a normal Mendelian ratio at birth. No obvious developmental defect of any kind could explain these postnatal deaths (74). In contrast, an extensive biochemical analysis showed that Esp−/− mice and Esposb−/− mice were hypoglycemic and hyperinsulinemic (74, 77). A more in-depth analysis showed that insulin secretion was increased, as was insulin sensitivity, in mice lacking Esp in osteoblasts, whereas mice overexpressing Esp in osteoblasts were glucose intolerant because of a decrease in insulin secretion and sensitivity. Thus, the analysis of Esp function showed unambiguously that the osteoblast influences insulin secretion from pancreatic β cells and alters insulin sensitivity in liver, muscle, and white adipose tissue (74). A simple yet powerful experiment confirmed this finding. In a coculture assay in which cells were separated by a filter, osteoblasts, but not a closely related cell type like fibroblasts, enhanced insulin secretion by islets or β cells (74). Therefore, the osteoblast is an endocrine cell favoring insulin secretion.
The protein encoded by Esp is not secreted and therefore cannot be a hormone. The search for the only known hormone that is made by osteoblasts and that regulates glucose metabolism was facilitated by what we thought a hormone should be and what we also knew about osteoblast biology. The requirement that hormones be cell-specific molecules narrowed the search dramatically because there is only one known secreted protein that is made only by osteoblasts: osteocalcin. Osteocalcin was an even more credible candidate to be a hormone regulating energy metabolism because Osteocalcin−/− mice exhibit an obvious increase in abdominal fat mass. Given the osteoblast-specific nature of osteocalcin and the fact that it is secreted, bone may affect energy metabolism (74).
Osteocalcin is extremely abundant in the bone extracellular matrix (ECM) and is a small protein (46 and 49 amino acids long in mice and in humans, respectively) that can be carboxylated on three glutamic acid residues (78). Carboxylation of glutamic acid residues is a posttranslational modification that increases a protein’s affinity for mineral ions. This feature of osteocalcin and the fact that it is so abundant in a mineralized ECM suggested that this protein is involved in bone ECM mineralization (79). Yet loss- and gain-of-function mutations in Osteocalcin have unambiguously established that this is not the case (79).
Besides being present in the bone ECM, osteocalcin is also found in the general circulation. So osteocalcin may be an osteoblast-derived hormone that regulates glucose metabolism and other aspects of energy metabolism. This hypothesis was verified by showing that, unlike wild-type osteoblasts, Osteocalcin−/− osteoblasts cannot induce insulin secretion by pancreatic β cells. Accordingly, Osteocalcin−/− mice have a metabolic phenotype that is the mirror image of the one observed in Esp−/− mice; they are hyperglycemic, hypoinsulinemic, and insulin resistant in liver, muscle, and white adipose tissue. That the glucose intolerance phenotype of Osteocalcin−/− mice was corrected by removing one allele of Esp from these mice established that Esp acts upstream of Osteocalcin. In other words, Esp−/− mice are a gain-of-function model for osteocalcin. Remarkably, Esp−/− mice or wild-type mice receiving exogenous osteocalcin do not develop an obesity phenotype or a glucose intolerance phenotype when fed a high-fat diet (79). These results raise the prospect that osteocalcin may become a treatment for type 2 diabetes, a hypothesis being tested currently. Using multiple methods, we established that the form of osteocalcin responsible for its metabolic function is not the carboxylated form but the undercarboxylated form, which is the least abundant form of circulating osteocalcin (Figure 4).
Figure 4.
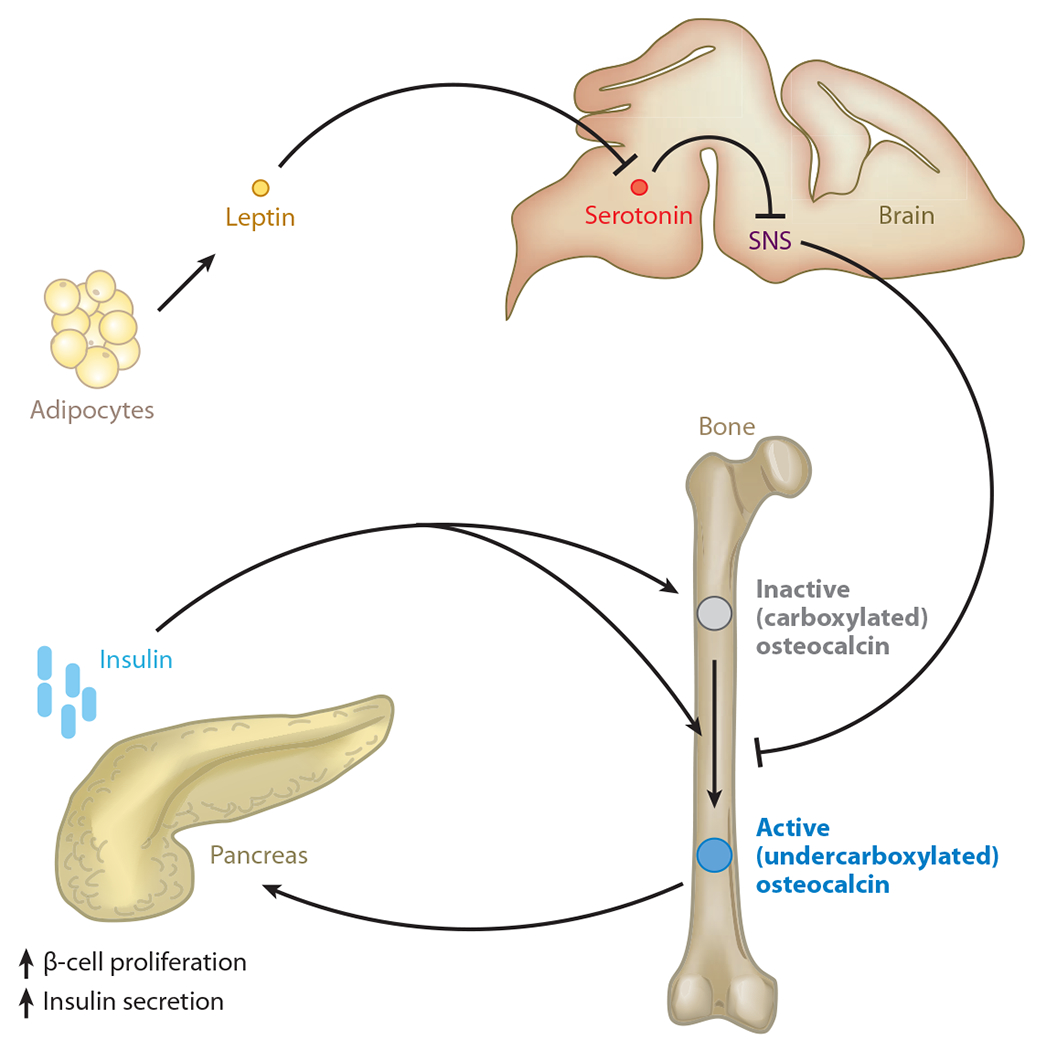
Endocrine regulation of energy metabolism by bone. Bone mediates such regulation by an osteoblast-specific secreted molecule, osteocalcin, that when undercarboxylated acts as a hormone favoring β-cell proliferation and insulin secretion in the pancreas. The mechanism by which osteocalcin may be activated is regulated in osteoblasts by insulin signaling, which favors osteocalcin bioavailability by promoting its undercarboxylation. In contrast, the sympathetic tone, which is regulated centrally by leptin, decreases osteocalcin bioactivation. SNS denotes sympathetic nervous system.
In summary, this work demonstrated that bone is an endocrine organ regulating energy metabolism, a function that is critical for bone (re)modeling. Further work also showed that Esp and Osteocalcin expression is regulated by activating transcription factor 4, an osteoblast-enriched transcription factor. These collective studies established the importance of the osteoblast as a cell type and more generally the importance of the skeleton as a determinant of whole-body glucose metabolism (80–83). Since the initial description of osteocalcin metabolic function was reported in the mouse, numerous studies have indicated that in humans, as in mice, serum total and/or undercarboxylated osteocalcin is a marker of glucose tolerance (84–93).
Coordinated Regulation of Bone Mass and Energy Metabolism: The Viewpoint of the Pancreas
It was quite unexpected to find two genes expressed in osteoblasts regulating glucose metabolism. However, that one of them encodes an intracellular phosphatase whereas the other one encodes a hormone that is not even the substrate of this phosphatase was puzzling. In addition, the regulation of insulin secretion by osteocalcin raised another question: Does insulin signaling in osteoblasts regulate the expression, secretion, or activation of osteocalcin? Such insulin signaling was found to regulate all three processes.
An efficient way to regulate the activity of tyrosine kinase receptors is through the use of intracellular tyrosine protein phosphatases. The insulin receptor, a tyrosine kinase receptor, operates in this manner, and its activity is negatively regulated in many insulin target cells by a tyrosine phosphatase, PTP-1B. This observation suggests that, if expressed in osteoblasts, the insulin receptor may be a substrate of OST-PTP (94, 95). Therefore, insulin signaling in osteoblasts may be necessary for glucose homeostasis. This is a worthwhile question to address because inactivating the insulin receptor in classical target tissues such as muscle and white adipose tissue did not result in glucose intolerance when mice were fed a normal diet (96–100). Such experiments raise the prospect that insulin signals in additional cell types to fulfill its metabolic functions.
As hypothesized, the insulin receptor is expressed in osteoblasts and is a substrate of ESP. Moreover, selective inactivation of the insulin receptor in osteoblasts results in glucose intolerance and in a decrease in insulin secretion (77). Various biochemical and genetic evidence showed that insulin signaling in osteoblasts favors osteocalcin activation by decreasing its carboxylation through an increase in bone (re)modeling. Indeed, osteoblasts are multifunctional cells that are responsible for bone formation and that, through at least two genes, determine osteoclast differentiation. Those two genes are Rankl, a positive regulator, and Osteoprotegerin (Opg), a soluble receptor sequestering RANKL and thus a negative regulator of this process (33, 101). An analysis of mice lacking the insulin receptor in osteoblasts showed that insulin signaling in this cell type favors bone resorption by inhibiting the expression of Opg. One gene expressed in osteoclasts and regulated by OPG, Tcirg1, contributes to the acidification of the extracellular space around osteoclasts (102–105). Thus, insulin signaling in osteoblasts favors acidification of bone ECM, a necessary component of bone resorption.
Because the only known mechanism to decarboxylate a protein outside a cell is an acid pH, it was hypothesized that insulin signaling in osteoblasts increases osteocalcin decarboxylation by stimulating bone resorption by osteoclasts (104, 105). Biochemical and genetics approaches indeed suggested that insulin signaling in osteoblasts promotes decarboxylation, i.e., activation of osteocalcin, through the activation of osteoclastic function. This ultimately favors insulin secretion. Thus, in a feed-forward loop, insulin signals in osteoblasts to enhance bone resorption, which then activates osteocalcin and upregulates Insulin expression and secretion.
The elucidation of the role of insulin signaling in osteoblasts raised another question: Does the endocrine function of bone also exist in humans? Because bone is one of the youngest organs to appear during evolution, it seemed likely that critical functions and regulatory mechanisms of bone might differ greatly between mice and humans. However, the pathways used by leptin, serotonin, and insulin/phosphatase/osteocalcin have similar function in mice and humans. Indeed, an analysis of osteopetrotic patients showed that a decrease in osteoclast function results in a decrease in the active form of osteocalcin and hypoinsulinemia (104, 105). The only difference between mice and humans is that Esp, which is a pseudogene in humans, is replaced in human osteoblasts by PTP1B, which encodes a tyrosine phosphatase that dephosphorylates the insulin receptor (94, 95).
Coordinated Regulation of Bone Mass and Energy Metabolism: The Viewpoint of the Adipocyte (Part II)
That osteocalcin bioactivity is enhanced by insulin signaling in osteoblasts also implies that there must be a hormone(s) that, unlike insulin, will inhibit osteocalcin expression, secretion, or bioactivity to maintain blood glucose levels within a normal range. Leptin is the only known negative regulator of the osteocalcin endocrine function; it does so by favoring Esp expression.
Among the many metabolic functions of leptin is the inhibition of insulin secretion through a neuronal relay. Study of cell-specific mutant mouse strains lacking either the leptin receptor, Adrβ2, or Esp showed that leptin signaling in the brain relies on sympathetic signaling in osteoblasts to enhance Esp expression and to inhibit insulin secretion (106). This results in a decrease in osteocalcin bioactivity. That leptin regulates an aspect of energy metabolism through bone adds further credence to the notion that this hormone’s main function is to coordinate the regulation of energy metabolism and bone physiology (Figure 4).
That insulin and leptin act directly and indirectly, respectively, in osteoblasts to regulate energy metabolism underscores the importance of bone as an important determinant of energy metabolism. This notion is validated by the fact that the broadly expressed transcription factor Foxo1, which regulates glucose metabolism, does so in part through its osteoblast expression (107, 108). This body of work does not imply that the osteoblast is the most important cell involved in the regulation of energy metabolism. Instead, we suggest that it would be a mistake to ignore the importance of the osteoblast in this physiological process. Although we currently know of only one hormone that fulfills the metabolic functions of the osteoblast, other hormones that are made by osteoblasts and that regulate energy metabolism may exist.
Coordinated Regulation of Bone Mass and Fertility: The Viewpoint of the Osteoblasts
Although many important questions remain to be addressed regarding the regulation of energy metabolism by bone, there is a need to solidify our understanding of the endocrine role of bone, especially the link between bone remodeling and energy metabolism and the link between bone and reproduction.
Menopause favors bone loss (15, 16, 109, 110). This medical observation implies that gonads, mostly through sex steroid hormones, affect bone cell function (this aspect of bone physiology is not discussed here). The regulation of bone mass accrual by gonads also suggests that in turn bone, in its endocrine capacity, may affect reproductive function in one or both genders. Verifying this hypothesis would further enhance the emerging importance of bone as an endocrine organ.
Testing this hypothesis in vivo was greatly helped by a striking feature of mutant mice lacking osteocalcin: Whereas female Osteocalcin-deficient mice were normally fertile, the male mutant mice were rather poor breeders, whether their partners were wild type or Osteocalcin deficient. As was the case for energy metabolism, the demonstration that this phenotype betrays a true biological function of osteocalcin was made more complete and convincing because of the availability of gain-of-function (Esp−/−) and loss-of-function (Osteocalcin−/−) mutations for Osteocalcin (74). Osteocalcin-deficient mice showed decreased weights of the testes, epididymides, and seminal vesicles, whereas these organs’ weights were increased in Esp-deficient mice. Osteocalcin-deficient males showed a 50% decrease in sperm count with a corresponding impairment of Leydig cell maturation, whereas Esp-deficient male mice showed a 30% increase in sperm count (111). These phenotypes suggested that osteocalcin may enhance testosterone synthesis. Coculture assays and subsequent in vivo experiments confirmed this suggestion (111).
The supernatant of cultured wild-type osteoblasts was able to increased testosterone production by Leydig cells to far greater levels than those observed for other mesenchymal cells. In contrast, this same osteoblast culture supernatant did not affect estrogen production by ovarian explants. As predicted, the supernatant of Osteocalcin-deficient osteoblast cultures was ineffective in promoting testosterone production in Leydig cells. Again, further cell-based and in vivo assays showed that osteocalcin increases expression of all genes necessary for testosterone biosynthesis in Leydig cells (111). Accordingly, circulating testosterone levels are low in Osteocalcin−/− mice and are high in Esp−/− mice. In contrast, circulating estrogen levels, as well as the expression of Cyp19A1 (an aromatase enzyme needed to convert testosterone to estrogen), are not affected in Esp−/− and Osteocalcin−/− mice (111). That Osteocalcin−/− mice develop a peripheral testicular insufficiency in the face of high levels of pituitary hormones, including luteinizing hormone and follicle-stimulating hormone, underscores the regulatory role of osteocalcin in male reproduction and suggests that some male patients with gonadal insufficiency may be defective in components of this bone-testis axis.
To formally establish that osteocalcin regulates testosterone production as a bone-derived hormone and not as a testis-secreted growth factor, investigators generated mice that lacked Osteocalcin only in osteoblasts. Male Osteocalcinosb−/− mice exhibited the same defects in testosterone production as did Osteocalcin−/− mice; deletion of Osteocalcin in Leydig cells did not affect male fertility (111). Taken together, these experiments established that osteocalcin is a bone-derived hormone favoring fertility in male mice by promoting Leydig cell maturation and testosterone production (Figure 5). In other words, these experiments verified that for at least one gender the skeleton participates in endocrine regulation of reproduction. Such experiments also reveal that this novel aspect of reproductive endocrinology appears to be sexually dimorphic.
Figure 5.
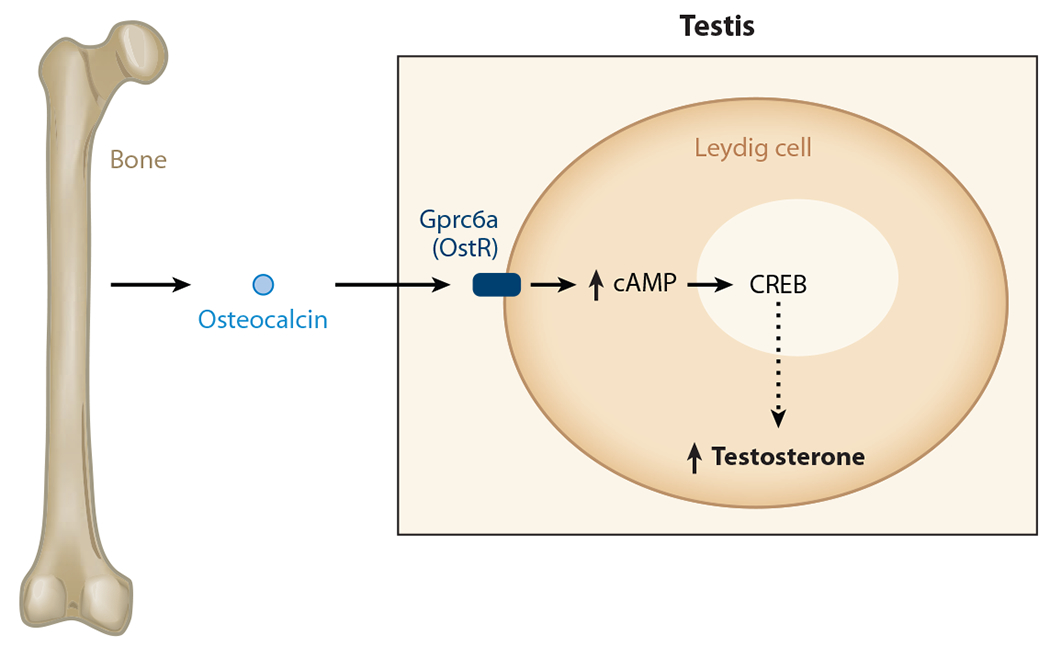
Endocrine regulation of male fertility by bone. Osteocalcin favors male fertility, increasing testosterone production by Leydig cells of the testes. By binding to a G protein–coupled receptor expressed in the Leydig cells of the testes, osteocalcin, an osteoblast-derived hormone, promotes testosterone production by the testes in a cAMP response element binding (CREB) protein–dependent manner. The dashed arrow indicates that regulation is not a primary signal (but direct); there may be other molecules in between.
Osteocalcin’s Molecular Mode of Action: Characterization of Its Receptor
In the molecular era, the identification of a novel hormone immediately begs the question of its mechanism of action. A prerequisite to answering this question is to characterize its cognate receptor on relevant target cells. In the case of osteocalcin, this was achieved through a two-step strategy that took advantage of the fact that osteocalcin regulates fertility in males but not in females (111).
The first step elucidated the signal transduction pathway affected by osteocalcin in two target cells, the β cell of the pancreas and the Leydig cell of the testis (74, 104, 105, 111). This approach identified cAMP production as the only intracellular signaling event triggered reproducibly by osteocalcin in these two cell types. We interpreted this result as suggesting that the osteocalcin receptor is probably a G protein–coupled receptor (GPCR) linked to adenylate cyclase. In the second step of this experimental strategy, we took advantage of the sexually dimorphic aspects of osteocalcin function by asking whether there were testis-specific orphan GPCRs. Out of more than 100 orphan GPCRs submitted to this test, 22 of them were expressed more highly in testes than in ovaries, and 4 were expressed predominantly or exclusively in Leydig cells (111). One of these 4 orphan GPCRs, Gprc6a, was a particularly good candidate to be an osteocalcin receptor because its inactivation in mice results in metabolic and reproduction phenotypes similar to those seen in Osteocalcin−/− mice (112). Furthermore, and although this was never tested through any binding assays, Gprc6a may be a calcium-serving receptor that functions better in the presence of osteocalcin (111).
Although the aforementioned result could not be reproduced, several criteria formally identified Gprc6a as an osteocalcin receptor present in Leydig cells (Figure 5) (111, 112). First, osteocalcin binds directly to wild-type cells, but not to Gprc6a-deficient Leydig cells. Second, osteocalcin increases cAMP production in wild-type cells, but not in Gprc6a-deficient Leydig cells. Third, and more convincingly, Leydig cell–specific deletion of Gprc6a revealed a reproduction phenotype caused by low testosterone production that was similar if not identical to the phenotype seen in the case of osteocalcin inactivation. Fourth, compound heterozygous mice lacking one copy of Osteocalcin and one copy of Gprc6a had a reproduction phenotype identical in all aspects to the one seen in Osteocalcin−/− or Gprc6a−/− mice. The identification of Gprc6a as an osteocalcin receptor led subsequently to the realization that CREB is a transcriptional effector of osteocalcin regulation of testosterone biosynthesis (Figure 5) (111). The identification of Gprc6a now allows us to specifically identify the functions of osteocalcin. It also enables us to perform a more sophisticated dissection of osteocalcin’s molecular mode of action in known and yet-to-be-identified target cells.
WHAT DID WE LEARN, AND WHERE DO WE STAND?
Although studies on the endocrine function of bone tissue are still ongoing, there are several lessons to be learned from this body of work. The first and most stimulating lesson is that because so much was discovered in a short amount of time about a single organ, namely the skeleton, there is probably much more to be learned about other organs. Second, this work demonstrates that genetics can uncover intimate connections between organs and can provide an ideal approach to link multiple physiologies and medical disciplines. For the foreseeable future, mouse genetics is the most powerful tool to map out and to study unidentified interorgan connections that exist in vertebrates.
The whole-organism molecular genetic approach to skeleton physiology may explain why a hormone like leptin co-appeared with bony vertebrates during evolution. Moreover, because the skeleton affects glucose homeostasis, energy expenditure, and fertility, bone may affect many more organs and physiological functions outside of the skeleton. As such, we suggest that vertebrate physiology is best studied in animal models containing a bony skeleton. Last, the demonstration that the osteoblast is an important endocrine cell suggests that further studies probing the molecular aspects of bone mass loss over time are needed.
If we look to a particular aspect of the skeleton, osteocalcin, we are struck by the fact that this hormone affects functions that go awry during aging. This observation is important because traditionally the skeleton is considered a victim of the aging process, as evidenced by age-dependent osteoporosis. Interestingly, two functions already ascribed to osteocalcin identify it as a fitness hormone affecting processes that deteriorate or disappear with aging. These findings therefore lead us to hypothesize that bone may both determine the aging process and be a victim of the aging process. Further knowledge about whole-organism physiology will be revealed from the study of this particular organ.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Bernard C 1865. An Introduction to the Study of Experimental Medicine. Paris: Flamarion [Google Scholar]
- 2.Cannon WB. 1929. Organization for physiological homeostasis. Physiol. Rev 9:399–431 [Google Scholar]
- 3.Cannon WB. 1932. The Wisdom of the Body. New York: Norton [Google Scholar]
- 4.Henderson LJ. 1913. The Fitness of the Environment: An Inquiry into the Biological Significance of the Properties of Matter. New York: Macmillan [Google Scholar]
- 5.Karsenty G 2006. Convergence between bone and energy homeostases: leptin regulation of bone mass. Cell Metab. 4:341–48 [DOI] [PubMed] [Google Scholar]
- 6.Rodan GA, Martin TJ. 2000. Therapeutic approaches to bone diseases. Science 289:1508–14 [DOI] [PubMed] [Google Scholar]
- 7.Teitelbaum SL. 2000. Osteoclasts, integrins, and osteoporosis. J. Bone Miner. Metab 18:344–49 [DOI] [PubMed] [Google Scholar]
- 8.Harada S, Rodan GA. 2003. Control of osteoblast function and regulation of bone mass. Nature 423:349–55 [DOI] [PubMed] [Google Scholar]
- 9.Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, et al. 2000. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100:197–207 [DOI] [PubMed] [Google Scholar]
- 10.Reid IR. 2002. Relationships among body mass, its components, and bone. Bone 31:547–55 [DOI] [PubMed] [Google Scholar]
- 11.Rigotti NA, Nussbaum SR, Herzog DB, Neer RM. 1984. Osteoporosis in women with anorexia nervosa. N. Engl. J. Med 311:1601–6 [DOI] [PubMed] [Google Scholar]
- 12.Wolfert A, Mehler PS. 2002. Osteoporosis: prevention and treatment in anorexia nervosa. Eat. Weight Disord 7:72–81 [DOI] [PubMed] [Google Scholar]
- 13.Zhao LJ, Liu YJ, Liu PY, Hamilton J, Recker RR, Deng HW. 2007. Relationship of obesity with osteoporosis. J. Clin. Endocrinol. Metab 92:1640–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zipfel S, Seibel MJ, Lowe B, Beumont PJ, Kasperk C, Herzog W. 2001. Osteoporosis in eating disorders: a follow-up study of patients with anorexia and bulimia nervosa. J. Clin. Endocrinol. Metab 86:5227–33 [DOI] [PubMed] [Google Scholar]
- 15.Khosla S 2010. Update on estrogens and the skeleton. J. Clin. Endocrinol. Metab 95:3569–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riggs BL, O’Fallon WM, Muhs J, O’Connor MK, Kumar R, Melton LJ 3rd. 1998. Long-term effects of calcium supplementation on serum parathyroid hormone level, bone turnover, and bone loss in elderly women. J. Bone Miner. Res 13:168–74 [DOI] [PubMed] [Google Scholar]
- 17.Ahima RS. 2004. Body fat, leptin, and hypothalamic amenorrhea. N. Engl. J. Med 351:959–62 [DOI] [PubMed] [Google Scholar]
- 18.Ahima RS, Saper CB, Flier JS, Elmquist JK. 2000. Leptin regulation of neuroendocrine systems. Front. Neuroendocrinol 21:263–307 [DOI] [PubMed] [Google Scholar]
- 19.Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, et al. 1998. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392:398–401 [DOI] [PubMed] [Google Scholar]
- 20.Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, et al. 2005. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434:514–20 [DOI] [PubMed] [Google Scholar]
- 21.Spiegelman BM, Flier JS. 2001. Obesity and the regulation of energy balance. Cell 104:531–43 [DOI] [PubMed] [Google Scholar]
- 22.Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, et al. 2002. Leptin regulates bone formation via the sympathetic nervous system. Cell 111:305–17 [DOI] [PubMed] [Google Scholar]
- 23.Doyon C, Drouin G, Trudeau VL, Moon TW. 2001. Molecular evolution of leptin. Gen. Comp. Endocrinol 124:188–98 [DOI] [PubMed] [Google Scholar]
- 24.Huising MO, Geven EJ, Kruiswijk CP, Nabuurs SB, Stolte EH, et al. 2006. Increased leptin expression in common carp (Cyprinus carpio) after food intake but not after fasting or feeding to satiation. Endocrinology 147:5786–97 [DOI] [PubMed] [Google Scholar]
- 25.Elefteriou F, Takeda S, Ebihara K, Magre J, Patano N, et al. 2004. Serum leptin level is a regulator of bone mass. Proc. Natl. Acad. Sci. USA 101:3258–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karsenty G, Oury F. 2010. The central regulation of bone mass, the first link between bone remodeling and energy metabolism. J. Clin. Endocrinol. Metab 95:4795–801 [DOI] [PubMed] [Google Scholar]
- 27.Fu L, Patel MS, Bradley A, Wagner EF, Karsenty G. 2005. The molecular clock mediates leptin-regulated bone formation. Cell 122:803–15 [DOI] [PubMed] [Google Scholar]
- 28.Karsenty G, Kronenberg HM, Settembre C. 2009. Genetic control of bone formation. Annu. Rev. Cell Dev. Biol 25:629–48 [DOI] [PubMed] [Google Scholar]
- 29.Lowrey PL, Takahashi JS. 2004. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu. Rev. Genomics Hum. Genet 5:407–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morse D, Sassone-Corsi P. 2002. Time after time: inputs to and outputs from the mammalian circadian oscillators. Trends Neurosci. 25:632–37 [DOI] [PubMed] [Google Scholar]
- 31.Okamura H, Miyake S, Sumi Y, Yamaguchi S, Yasui A, et al. 1999. Photic induction of mPer1 and mPer2 in Cry-deficient mice lacking a biological clock. Science 286:2531–34 [DOI] [PubMed] [Google Scholar]
- 32.Perreau-Lenz S, Pevet P, Buijs RM, Kalsbeek A. 2004. The biological clock: the bodyguard of temporal homeostasis. Chronobiol. Int 21:1–25 [DOI] [PubMed] [Google Scholar]
- 33.Teitelbaum SL, Ross FP. 2003. Genetic regulation of osteoclast development and function. Nat. Rev. Genet 4:638–49 [DOI] [PubMed] [Google Scholar]
- 34.Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, et al. 2001. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell 105:683–94 [DOI] [PubMed] [Google Scholar]
- 35.Pasco JA, Henry MJ, Sanders KM, Kotowicz MA, Seeman E, Nicholson GC. 2004. β-Adrenergic blockers reduce the risk of fracture partly by increasing bone mineral density: Geelong Osteoporosis Study. J. Bone Miner. Res 19:19–24 [DOI] [PubMed] [Google Scholar]
- 36.Rejnmark L, Vestergaard P, Mosekilde L. 2006. Treatment with β-blockers, ACE inhibitors, and calcium-channel blockers is associated with a reduced fracture risk: a nationwide case-control study. J. Hypertens 24:581–89 [DOI] [PubMed] [Google Scholar]
- 37.Schlienger RG, Kraenzlin ME, Jick SS, Meier CR. 2004. Use of β-blockers and risk of fractures. JAMA 292:1326–32 [DOI] [PubMed] [Google Scholar]
- 38.Turker S, Karatosun V, Gunal I. 2006. β-Blockers increase bone mineral density. Clin. Orthop. Relat. Res 443:73–74 [DOI] [PubMed] [Google Scholar]
- 39.Bonnet N, Benhamou CL, Malaval L, Goncalves C, Vico L, et al. 2008. Low dose β-blocker prevents ovariectomy-induced bone loss in rats without affecting heart functions. J. Cell Physiol 217:819–27 [DOI] [PubMed] [Google Scholar]
- 40.Asnicar MA, Smith DP, Yang DD, Heiman ML, Fox N, et al. 2001. Absence of cocaine- and amphetamine-regulated transcript results in obesity in mice fed a high caloric diet. Endocrinology 142:4394–400 [DOI] [PubMed] [Google Scholar]
- 41.Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, et al. 1998. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron 21:1375–85 [DOI] [PubMed] [Google Scholar]
- 42.Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, et al. 1998. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature 393:72–76 [DOI] [PubMed] [Google Scholar]
- 43.Ahn JD, Dubern B, Lubrano-Berthelier C, Clement K, Karsenty G. 2006. Cart overexpression is the only identifiable cause of high bone mass in melanocortin 4 receptor deficiency. Endocrinology 147:3196–202 [DOI] [PubMed] [Google Scholar]
- 44.Abizaid A, Gao Q, Horvath TL. 2006. Thoughts for food: brain mechanisms and peripheral energy balance. Neuron 51(6):691–702 [DOI] [PubMed] [Google Scholar]
- 45.Sato S, Hanada R, Kimura A, Abe T, Matsumoto T, et al. 2007. Central control of bone remodeling by neuromedin U. Nat. Med 13(10):1234–40 [DOI] [PubMed] [Google Scholar]
- 46.Hetherington AW, Ranson SW. 1940. Hypothalamic lesions and adipocity in the rat. Anat. Rec 78:149–72 [Google Scholar]
- 47.Hetherington AW, Ranson SW. 1942. The relation of various hypothalamic lesions to adiposity in the rat. J. Comp. Neurol 76:475–99 [Google Scholar]
- 48.Brobeck JR. 1946. Mechanisms of the development of obesity in animals with hypothalamic lesions. Physiol. Rev 26:541–59 [DOI] [PubMed] [Google Scholar]
- 49.Anand BK, Brobeck JR. 1951. Localization of a feeding center in the hypothalamus of the rat. Proc. Soc. Exp. Biol. Med 77:323–24 [DOI] [PubMed] [Google Scholar]
- 50.Debons AF, Silver L, Cronkite EP, Johnson HA, Brecher G, et al. 1962. Localization of gold in mouse brain in relation to gold thioglucose obesity. Am. J. Physiol 202:743–50 [DOI] [PubMed] [Google Scholar]
- 51.Olney JW. 1969. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164:719–21 [DOI] [PubMed] [Google Scholar]
- 52.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, et al. 1996. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84:491–95 [DOI] [PubMed] [Google Scholar]
- 53.Fei H, Okano HJ, Li C, Lee GH, Zhao C, et al. 1997. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc. Natl. Acad. Sci. USA 94:7001–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, et al. 1996. Abnormal splicing of the leptin receptor in diabetic mice. Nature 379:632–35 [DOI] [PubMed] [Google Scholar]
- 55.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, et al. 1995. Identification and expression cloning of a leptin receptor, OB-R. Cell 83:1263–71 [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, et al. 1994. Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–32 [DOI] [PubMed] [Google Scholar]
- 57.Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, et al. 2004. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42:983–91 [DOI] [PubMed] [Google Scholar]
- 58.Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, et al. 2009. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell 138:976–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haney EM, Chan BK, Diem SJ, Ensrud KE, Cauley JA, et al. 2007. Association of low bone mineral density with selective serotonin reuptake inhibitor use by older men. Arch. Intern. Med 167:1246–51 [DOI] [PubMed] [Google Scholar]
- 60.Kaye W, Gendall K, Strober M. 1998. Serotonin neuronal function and selective serotonin reuptake inhibitor treatment in anorexia and bulimia nervosa. Biol. Psychiatry 44:825–38 [DOI] [PubMed] [Google Scholar]
- 61.Laekeman G, Zwaenepoel L, Reyntens J, de Vos M, Casteels M. 2008. Osteoporosis after combined use of a neuroleptic and antidepressants. Pharm. World Sci 30:613–16 [DOI] [PubMed] [Google Scholar]
- 62.Richards JB Papaioannou A, Adachi JD, Joseph L, Whitson HE, et al. 2007. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch. Intern. Med 167:188–94 [DOI] [PubMed] [Google Scholar]
- 63.Ziere G, Dieleman JP, van der Cammen TJ, Hofman A, Pols HA, Stricker BH. 2008. Selective serotonin reuptake inhibiting antidepressants are associated with an increased risk of nonvertebral fractures. J. Clin. Psychopharmacol 28:411–17 [DOI] [PubMed] [Google Scholar]
- 64.Gershon MD, Tack J. 2007. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology 132:397–414 [DOI] [PubMed] [Google Scholar]
- 65.Mann JJ, McBride PA, Brown RP, Linnoila M, Leon AC, et al. 1992. Relationship between central and peripheral serotonin indexes in depressed and suicidal psychiatric inpatients. Arch. Gen. Psychiatry 49:442–46 [DOI] [PubMed] [Google Scholar]
- 66.Walther DJ, Peter J-U, Bashammakh S, Hörtnagl H, Voits M, et al. 2003. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299:76. [DOI] [PubMed] [Google Scholar]
- 67.Yadav VK, Oury F, Tanaka K, Thomas T, Wang Y, et al. 2011. Leptin-dependent serotonin control of appetite: temporal specificity, transcriptional regulation, and therapeutic implications. J. Exp. Med 208:41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, et al. 2008. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 135:825–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oury F, Yadav VK, Wang Y, Zhou B, Liu XS, et al. 2010. CREB mediates brain serotonin regulation of bone mass through its expression in ventromedial hypothalamic neurons. Genes Dev. 24:2330–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, et al. 2002. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med 346:1513–21 [DOI] [PubMed] [Google Scholar]
- 71.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, et al. 2001. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107:513–23 [DOI] [PubMed] [Google Scholar]
- 72.Yadav VK, Balaji S, Suresh PS, Liu XS, Lu X, et al. 2010. Pharmacological inhibition of gut-derived serotonin synthesis is a potential bone anabolic treatment for osteoporosis. Nat. Med 16:308–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee K, Nichols J, Smith A. 1996. Identification of a developmentally regulated protein tyrosine phosphatase in embryonic stem cells that is a marker of pluripotential epiblast and early mesoderm. Mech. Dev 59:153–64 [DOI] [PubMed] [Google Scholar]
- 74.Lee NK, Sowa H, Hinoi E, Ferron M, Ahn J, et al. 2007. Endocrine regulation of energy metabolism by the skeleton. Cell 130:456–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mauro LJ, Olmsted EA, Skrobacz BM, Mourey RJ, Davis AR, Dixon JE. 1994. Identification of a hormonally regulated protein tyrosine phosphatase associated with bone and testicular differentiation. J. Biol. Chem 269:30659–67 [PubMed] [Google Scholar]
- 76.Dacquin R, Mee PJ, Kawaguchi J, Olmsted-Davis EA, Gallagher JA, et al. 2004. Knock-in of nuclear localised β-galactosidase reveals that the tyrosine phosphatase Ptprv is specifically expressed in cells of the bone collar. Dev. Dyn 229:826–34 [DOI] [PubMed] [Google Scholar]
- 77.Ferron M, Hinoi E, Karsenty G, Ducy P. 2008. Osteocalcin differentially regulates β cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl. Acad. Sci. USA 105:5266–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hauschka PV, Lian JB, Cole DE, Gundberg CM. 1989. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol. Rev 69:990–1047 [DOI] [PubMed] [Google Scholar]
- 79.Ducy P, Desbois C, Boyce B, Pinero G, Story B, et al. 1996. Increased bone formation in osteocalcin-deficient mice. Nature 382:448–52 [DOI] [PubMed] [Google Scholar]
- 80.Kajimura D, Hinoi E, Ferron M, Kode A, Riley KJ, et al. 2011. Genetic determination of the cellular basis of the sympathetic regulation of bone mass accrual. J. Exp. Med 388:34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang X, Karsenty G. 2004. ATF4, the osteoblast accumulation of which is determined post-translationally, can induce osteoblast-specific gene expression in non-osteoblastic cells. J. Biol. Chem 279:47109–14 [DOI] [PubMed] [Google Scholar]
- 82.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, et al. 2004. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 117:387–98 [DOI] [PubMed] [Google Scholar]
- 83.Yoshizawa T, Hinoi E, Jung DY, Kajimura D, Ferron M, et al. 2009. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J. Clin. Investig 119:2807–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aonuma H, Miyakoshi N, Hongo M, Kasukawa Y, Shimada Y. 2009. Low serum levels of undercarboxylated osteocalcin in postmenopausal osteoporotic women receiving an inhibitor of bone resorption. Tohoku J. Exp. Med 218:201–5 [DOI] [PubMed] [Google Scholar]
- 85.Fernandez-Real JM, Izquierdo M, Ortega F, Gorostiaga E, omez-Ambrosi J, et al. 2009. The relationship of serum osteocalcin concentration to insulin secretion, sensitivity, and disposal with hypocaloric diet and resistance training. J. Clin. Endocrinol. Metab 94:237–45 [DOI] [PubMed] [Google Scholar]
- 86.Hwang YC, Jeong IK, Ahn KJ, Chung HY. 2009. The uncarboxylated form of osteocalcin is associated with improved glucose tolerance and enhanced β-cell function in middle-aged male subjects. Diabetes Metab. Res. Rev 25:768–72 [DOI] [PubMed] [Google Scholar]
- 87.Im JA, Yu BP, Jeon JY, Kim SH. 2008. Relationship between osteocalcin and glucose metabolism in postmenopausal women. Clin. Chim. Acta 396:66–69 [DOI] [PubMed] [Google Scholar]
- 88.Kanazawa I, Yamaguchi T, Yamamoto M, Yamauchi M, Kurioka S, et al. 2009. Serum osteocalcin level is associated with glucose metabolism and atherosclerosis parameters in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab 94:45–49 [DOI] [PubMed] [Google Scholar]
- 89.Kindblom JM, Ohlsson C, Ljunggren O, Karlsson MK, Tivesten A, et al. 2009. Plasma osteocalcin is inversely related to fat mass and plasma glucose in elderly Swedish men. J. Bone Miner. Res 24:785–91 [DOI] [PubMed] [Google Scholar]
- 90.Levinger I, Zebaze R, Jerums G, Hare DL, Selig S, Seeman E. 2011. The effect of acute exercise on undercarboxylated osteocalcin in obese men. Osteoporos. Int 2(5):1621–66 [DOI] [PubMed] [Google Scholar]
- 91.Pittas AG, Harris SS, Eliades M, Stark P, Dawson-Hughes B. 2009. Association between serum osteocalcin and markers of metabolic phenotype. J. Clin. Endocrinol. Metab 94:827–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Winhofer Y, Handisurya A, Tura A, Bittighofer C, Klein K, et al. 2010. Osteocalcin is related to enhanced insulin secretion in gestational diabetes mellitus. Diabetes Care 33:139–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yeap BB, Chubb SA, Flicker L, McCaul KA, Ebeling PR, et al. 2010. Reduced serum total osteocalcin is associated with metabolic syndrome in older men via waist circumference, hyperglycemia, and triglyceride levels. Eur. J. Endocrinol 163:265–72 [DOI] [PubMed] [Google Scholar]
- 94.Delibegovic M, Bence KK, Mody N, Hong EG, Ko HJ, et al. 2007. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol. Cell. Biol 27:7727–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Delibegovic M, Zimmer D, Kauffman C, Rak K, Hong EG, et al. 2009. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 58:590–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bluher M, Michael MD, Peroni OD, Ueki K, Carter N, et al. 2002. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell 3:25–38 [DOI] [PubMed] [Google Scholar]
- 97.Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, et al. 1998. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2:559–69 [DOI] [PubMed] [Google Scholar]
- 98.Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, et al. 2007. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 5:438–49 [DOI] [PubMed] [Google Scholar]
- 99.Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. 1999. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–39 [DOI] [PubMed] [Google Scholar]
- 100.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, et al. 2000. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 6:87–97 [PubMed] [Google Scholar]
- 101.Kong YY, Boyle WJ, Penninger JM. 1999. Osteoprotegerin ligand: a common link between osteoclastogenesis, lymph node formation and lymphocyte development. Immunol. Cell Biol 77:188–93 [DOI] [PubMed] [Google Scholar]
- 102.Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, et al. 2010. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142:296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, et al. 2010. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142:309–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, et al. 1998. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl. Acad. Sci. USA 95:13453–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Scimeca JC, Franchi A, Trojani C, Parrinello H, Grosgeorge J, et al. 2000. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone 26:207–13 [DOI] [PubMed] [Google Scholar]
- 106.Hinoi E, Gao N, Jung DY, Yadav V, Yoshizawa T, et al. 2008. The sympathetic tone mediates leptin’s inhibition of insulin secretion by modulating osteocalcin bioactivity. J. Cell Biol 183:1235–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rached MT, Kode A, Silva BC, Jung DY, Gray S, et al. 2010. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J. Clin. Investig 120:357–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rached MT, Kode A, Xu L, Yoshikawa Y, Paik JH, et al. 2010. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 11:147–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, et al. 2007. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell 130:811–23 [DOI] [PubMed] [Google Scholar]
- 110.Manolagas SC, Kousteni S, Jilka RL. 2002. Sex steroids and bone. RecentProg. Horm. Res 57:385–409 [DOI] [PubMed] [Google Scholar]
- 111.Oury F, Sumara G, Sumara O, Ferron M, Chang H, et al. 2011. Endocrine regulation of male fertility by the skeleton. Cell 144:796–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pi M, Chen L, Huang MZ, Zhu W, Ringhofer B, et al. 2008. GPRC6A null mice exhibit osteopenia, feminization and metabolic syndrome. PLoS ONE 3:e3858. [DOI] [PMC free article] [PubMed] [Google Scholar]