

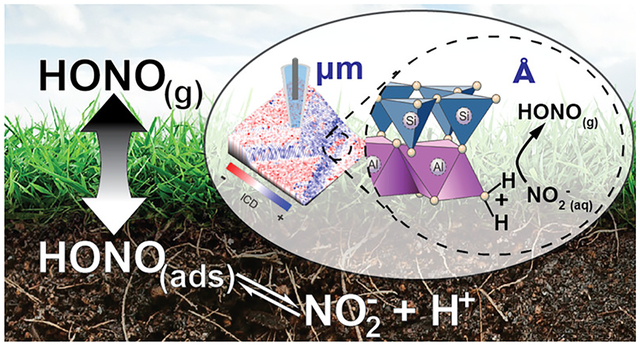
Abstract
Unique surface properties of aluminosilicate clay minerals arise from anisotropic distribution of surface charge across their layered structures. Yet, a molecular-level understanding of clay mineral surfaces has been hampered by the lack of analytical techniques capable of measuring surface charges at the nanoscale. This is important for understanding the reactivity, colloidal stability, and ion-exchange capacity properties of clay minerals, which constitute a major fraction of global soils. In this work, scanning ion conductance microscopy (SICM) is used for the first time to visualize the surface charge and topography of dickite, a well-ordered member of the kaolin subgroup of clay minerals. Dickite displayed a pH-independent negative charge on basal surfaces whereas the positive charge on edges increased from pH 6 to 3. Surface charges responded to malonate addition, which promoted dissolution/precipitation reactions. Results from SICM were used to interpret heterogeneous reactivity studies showing that gas-phase nitrous acid (HONO) is released from the protonation of nitrite at Al–OH2+ groups on dickite edges at pH well above the aqueous pKa of HONO. This study provides nanoscale insights into mineral surface processes that affect environmental processes on the local and global scale.
Keywords: microscopy, clay, surface charge, nitrous acid, atmospheric chemistry
Graphical Abstract
INTRODUCTION
Clay minerals belong to a group of natural aluminosilicates prevalent in soil. Due to their high abundance, small particle size, and unique surface charge characteristics, clay minerals contribute a significant fraction of the reactive mineral surface area present in many soils and sediments.1 Thus, clays are major contributors to a soil’s ion-exchange capacity and they facilitate organic matter–clay interactions that can stabilize soil organic matter and prevent their mineralization. In addition, the Brønsted and Lewis acid/base characteristics of clay mineral surfaces play an important role in controlling the soil–atmosphere exchange of small molecules such as nitrous acid (HONO), a weak acid that plays an important role in atmospheric photooxidation chemistry. HONO is formed on terrestrial surfaces2–14 and, once emitted to the atmosphere, is a major source of hydroxyl radical (OH) that controls the lifetime of greenhouse gases and leads to ozone and aerosol formation.15–18 Evidence suggests that proton transfer reactions occurring on clay mineral surfaces promote HONO emissions from soil having near-neutral bulk pH, despite the fact that the pKa of HONO is ~3.12,19 However, the precise identity of surface reaction sites has not been confirmed. Accurately parameterizing adsorbate–clay mineral interactions in predictive models is only possible if the surface charge characteristics of mineral surfaces are understood at the molecular level. Here, we use scanning ion conductance microscopy (SICM) to quantify the surface charge of the kaolin group clay mineral dickite and use the observations to gain unprecedented insights into the reactive sites responsible for the pH-dependent exchange of HONO from clay mineral surfaces.
The fundamental structural unit in kaolin minerals is comprised of siloxane (Si2O5)2− and gibbsite-like (OH)3Al2(OH)2+ sheets fused together via shared O2− atoms (Figure S4.1).20–22 Adjacent layers are held together by hydrogen bonding between oxygens of tetrahedral siloxane sheets and hydrogens on the adjacent gibbsite surface,20,21 giving kaolin minerals their characteristic planar crystal habit (Figures S4.1 and S4.2). This unique anisotropic structure yields three unique surfaces: (1) a silica-like siloxane basal surface that is negatively charged in water due to the presence of Si–O− groups (down to pH 2); (2) a gibbsite-like basal surface comprising Al–(OH2+)–Al groups (pKa = 4); and (3) edge surfaces consisting of both Al–OH and Si–OH groups with exchangeable protons that are involved in acid/base equilibria (pKa1 = 6.3 and pKa2 = 8.7 for Al–OH2+ and Al–OH, respectively; pKa = 6.9 for Si–OH). All three of these give rise to pH-dependent surface charges.23–26
Until recently, characterization of the charge properties of kaolin mineral surfaces at the nanoscale has not been possible. Conventional surface charge investigations of kaolin group minerals are limited to bulk methods such as potentiometric titrations27,28 and electrokinetic techniques.29–31 Point of zero charge (PZC) values determined for kaolinite using alkali-/acidimetric titration show significant variability from 3.8 to 7.5.32–36 Isoelectric points (IEPs) measured via electrokinetic techniques also show significant differences. For example, reported values include 2.2,30 3.7,31 and 4.29 Discrepancies have been explained by preparation methods and inherent differences in particle size, shape, or surface chemistry between different kaolinite samples. When samples are measured with macroscale measurement techniques, subtle differences accumulate and result in a significant variance in the PZCs or IEPs. Recently, the surface charge on kaolinite was studied at the nanoscale with atomic force microscopy (AFM), in which the surface charge at different surfaces of single crystals of kaolinite was measured by accurately positioning an AFM probe over an area of interest and measuring force–distance curves.37,38 Extensive studies of kaolinite samples with AFM techniques have provided evidence that both basal surfaces (siloxane and gibbsite-like) and edges of kaolinite exhibit a pH-dependent surface charge.38–44
SICM45,46 provides an alternative approach for investigating the surface charge distribution of a surface. SICM is a probe-based technique that collects high-resolution topography images by laterally scanning a nanoscale pipette across a sample surface.45,47,48 Recent studies have shown that ion current through the pipette is also affected by the surface charge of samples at small tip–sample separations.49,50 Several surface charge measurement methods have been developed51–58 and successfully applied to characterize the surface charge of ionogels,59 lipid membranes,55,56 DNA,60 live cells,54 bacteria,61 and human hair.57 Methods that utilize AFM to transduce surface charges have been shown as very useful, but also suffer from a possible convolution of the multiple types of forces interacting at the tip–sample gap, and relatively short interaction distances.62,63 Surface charge measurements and mapping surface charge can be more facile to implement with SICM, as benefits from the inherent noncontact nature of feedback and larger operational tip–sample separations (relative to AFM) are realized. Further, for measurements here, a self-referencing method is employed, which minimizes possible influence of changes in tip geometry during imaging.
The objective of this work is to apply SICM for the first time to study the nano/microscale surface charge characteristics of a kaolin group mineral and to use the insights provided to understand an environmentally relevant process that impacts atmospheric composition. Instead of the widely used kaolinite crystals, structurally similar dickite was selected due to the availability of large single crystals (typically 150 μm in length and 75 μm in width), presenting abundant basal planes and edge surfaces that are ideal for SICM measurements. The use of well-ordered dickite single crystals also avoids the complexities of using smaller and notoriously disordered kaolinite crystallites. Additionally, we examined the influence of pH on surface charge and interactions between dickite and exogenous dicarboxylic acid groups. Results were analyzed with finite-element methods (FEM) to quantify the surface charge on basal planes and edges of dickite crystals. As part of this work, we also carried out flow reactor studies to investigate protonation of adsorbed NO2− and emission of gaseous HONO from dickite and kaolinite surfaces. The combined SICM and reactivity studies provide molecular-level insights into the reactive sites responsible for the soil–atmosphere exchange of HONO—a process that ultimately influences atmospheric composition on a local to global scale.
EXPERIMENTAL SECTION
Chemicals and Solutions.
Potassium chloride from Sigma-Aldrich (St. Louis, MO) was used as received. Hydrochloric acid from EMD Millipore (Burlington, MA) was diluted with deionized water to create 100 mM and 10 mM stock solutions. Malonic acid (Sigma-Aldrich, St. Louis, MO) and potassium hydroxide pellets (KOH, Mallinckrodt, Paris, KY) were dissolved in deionized water to give a 300 mM potassium malonate stock solution. The pH of the potassium malonate solution was subsequently adjusted to 5.12 with 1 M HCl before use. All solutions were prepared with deionized water (resistivity ~ 18 MΩ cm at 25 °C, Millipore Corp., Danvers, MA) and filtered with sterile 0.20 μm nylon filters (DWK Life Sciences LLC, Millville, NJ) before use.
Polydimethyl siloxane (PDMS) substrates were created by mixing a SYLGARD 184 silicone elastomer and curing agent in a ratio of 10:1 (Dow Corning Corporation, Midland, MI). Dickite from St. Claire, PA (Ward’s Natural Science Establishment, Inc.) was used for all SICM measurements; kaolinite (KGa-1) from The Clay Minerals Society Source Clays Repository (Purdue University, West Lafayette, IN) was used for surface reactivity studies. Dickite samples used for surface charge measurements were transferred onto PDMS substrates and measured directly in a 100 mM KCl solution without purification. For the flow reactor study, mineral samples were washed three times with deionized water, centrifuged at 2000 rpm for 1 h between each wash, and then dried in an oven at 70 °C overnight. A slurry was created by mixing the minerals and deionized water in a ratio of 1:2 (w/w mineral/water).64 The pH was subsequently adjusted by adding H2SO4 or NaOH (Sigma-Aldrich). Finally, the pH-adjusted slurry was dried again in the oven. To investigate the influence of malonate adsorption on dickite reactivity, the washed and dried dickite crystals were immersed in 300 mM potassium malonate solution at pH 5.12 overnight prior to the flow tube reactor studies.
Pipette Fabrication.
Pipettes used in the experiments were pulled from quartz glass capillaries (Q100-70-7.5, Sutter Instrument, Novato, CA, USA) with a CO2-laser puller (P2000, Sutter Instrument, Novato, CA, USA). Typical pulling parameters used in the experiment were heat = 690, Fil = 4, Vel = 45, Del = 160, and Pul = 190. Pipettes were then characterized by scanning electron microscopy (SEM; FEI Quanta-FEG, Hillsboro, OR). Electron micrographs are shown in Section SI-1. The typical inner and outer radii of pipettes are 50 and 70 nm, respectively, with a half-cone angle of ~7.5°.
Instrument Setup and Scan Protocol for Surface Charge Measurement.
Briefly, an electrolyte-filled nanopipette was used as the probe. An Ag/AgCl electrode was back-inserted inside the pipette and functioned as the working electrode (WE). A second Ag/AgCl electrode was placed in the bulk electrolyte and functioned as the reference electrode (RE). A potential difference was applied between the electrodes to generate ion current for feedback control.
The surface charge at the sample was measured simultaneously with a self-referencing method. Details of the scan protocol are given elsewhere.58 Briefly, the pipette was advanced toward the surface until a predetermined setpoint was reached. A staircase potential waveform was then applied to the WE, and the current response at each step was recorded. The resulting current–voltage (I–V) response was then used for further analysis. The probe was subsequently retracted by a preset distance and a retracted I–V response was collected. The surface charge was then extracted based on relative current differences between the extended and retracted I–V curves. Typical pixel resolution used for experiments was 64 × 64. Scan areas used for coarse measurement and fine measurement were 16 × 16 and 8 × 8 μm to yield values of 250 and 125 nm2 per pixel, respectively. This resolution was found to be beneficial to optimize the imaging time and provide adequate resolution, with some sacrifice to resolution of transitions at sharp features (e.g., the area between basal planes and growth steps). Depending on the ion current magnitude at the scan potential, I–V curves were measured between ±0.6 or ±0.5 V with 19 even steps. The potential was maintained for 15 ms at each step, and the current values in the last 1 ms were averaged and recorded to generate an I–V curve.
To quantify the surface charge at the sample surface, the current differences at extreme potentials were averaged and the following equation was used to express the ion current difference (ICD)
| (1) |
in which Iext− and Iext+ represent current values at the maximum negative potential and maximum positive potential when the pipette was extended to the position near the surface. In contrast, Iret− and Iret+ denote current values when the pipette was retracted from the surface. With eq 1, ICD values for each point in the scan area can be calculated and plotted with the XY-coordinates to generate an ICD image, which reveals the surface charge distribution over the scan area. The surface charge image was then combined with the topography image via ImageJ65 to correlate topography and surface charge properties.
Flow Reactor Experiments to Study HONO Emissions.
A flow tube reactor coupled to a chemical ionization mass spectrometer (CIMS) was used to study HONO formation following the addition of nitrite to pH-adjusted minerals. The experimental setup is described elsewhere.66 Briefly, the system consists of a laminar horizontal flow reactor (100 cm length × 2 cm i.d.) into which a Teflon holder (13 mm inner diameter, 2 mm depth) containing the pH-adjusted mineral substrate is inserted (Figure S2.1). High-purity N2 humidified at 30% relative humidity was introduced at 1 L min−1 through the reactor to achieve laminar flow conditions (Reynolds number of 87) across a Teflon holder designed to minimize turbulence in the flow tube. A port on the flow tube mentioned above the sample holder permitted the addition of 5 μL of sodium nitrite solution at 200 μM (corresponding to 1 nmol of nitrite) onto ~100 mg of the substrate (Figure S2.1b,c). The port was sealed with a rubber septum that supported a 10 μL Hamilton syringe. The HONO mixing ratio was recorded at 0.75 Hz for 5 min before and after nitrite injection, and the integrated HONO emissions flux (over the 10 min following the injection) was estimated, with the 5 min before injection used to estimate the HONO signal background. At least three replicate experiments were performed at each pH. Additional details on the CIMS calibration, performance, experimental setup, and calculation are available in Section SI-2.
FEM Simulations.
FEM simulations were carried out with COMSOL Multiphysics (v5.4) with transport of diluted species and electrostatics modules. The pipette model in bulk electrolyte and near the charged substrate was simplified as a two-dimensional axisymmetric model. The pipette’s geometry was determined with SEM and used in the model to simulate the I–V response of the pipette and the surface charge of the substrate. For successive surface charge measurement experiments, the pipette radius was adjusted based on the change of the retracted I–V curve to compensate for subtle changes in the pipette during the scanning. Further details of the model and parameters used in the simulation are provided in Section SI-3.
RESULTS AND DISCUSSION
Surface Charge Distribution on Dickite Crystals.
Figure 1a,b shows optical and electron micrographs of dickite crystals examined here. Figure 1c illustrates the instrument setup for SICM. Dickite crystals with well-defined basal surfaces and steps were identified under an optical microscope and selected for SICM measurement. Figure 2a shows an optical micrograph of a typical dickite crystal selected for analysis. The area scanned by SICM is highlighted with a yellow square, and the shadow of the SICM pipette is indicated with an arrow. As shown in Figure 2b, the topography of the scan area comprises four basal surfaces and three growth steps with well-defined edges that are many layers thick. The angle between the edge and basal plane is ~70° and shows only minor differences between different steps. Figure 2c illustrates the surface charge of the scan area, represented as ICD (see Experimental Section). Compared with ΔICRR used in our previous report,58 the use of ICD simplifies comparison of the intensity of surface charge at positive and negative surfaces by providing an equivalent scale. To better illustrate the surface charge in the ICD image, the tricolor scale commonly used to display protein surface charge is adapted here, where negative charge, neutral, and positive charge are displayed with red, white, and blue color, respectively. As shown in Figure 2c, the surface charge image exhibits a negative charge with a magnitude of ~−100 pA at the basal surfaces. In contrast, the exposed edges show an unevenly distributed weak positive charge. The 3D topography of the scan area is plotted to correlate morphology and surface charge by loading the surface charge image as texture on the 3D topography image. Results shown in Figure 2d illustrate a negative surface charge at basal surfaces and a positive surface charge at edges.
Figure 1.
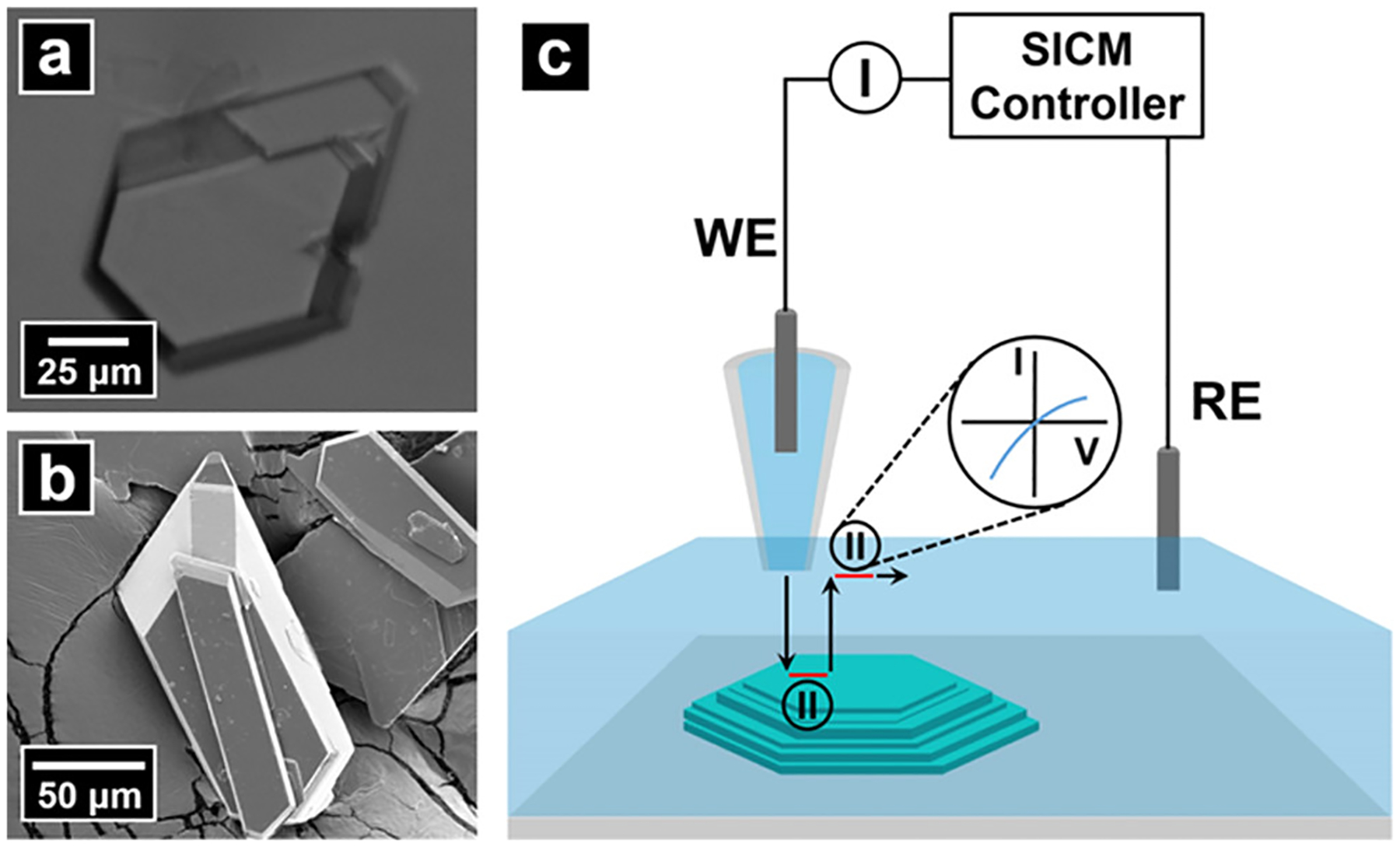
(a) Optical and (b) electron micrographs of dickite crystals examined. (c) Schematic for SICM measurements. An electrolyte-filled nanopipette was used as the probe and contained a back-inserted Ag/AgCl electrode (WE). A second Ag/AgCl electrode was placed in the bulk electrolyte and functioned as the RE. A potential difference was applied between the WE and RE to generate ion current for feedback control. Surface charge measurement was conducted by introducing an additional “pause state” when the pipette approached close to (extended) and retracted away from (retracted) the sample surface. At pause states, the feedback of SICM was disabled temporally, and the applied potential was swept to acquire a current–voltage response. Surface charge on the sample surface was then calculated at each pixel based on the difference of I–Vs at the extended position and retracted position.
Figure 2.
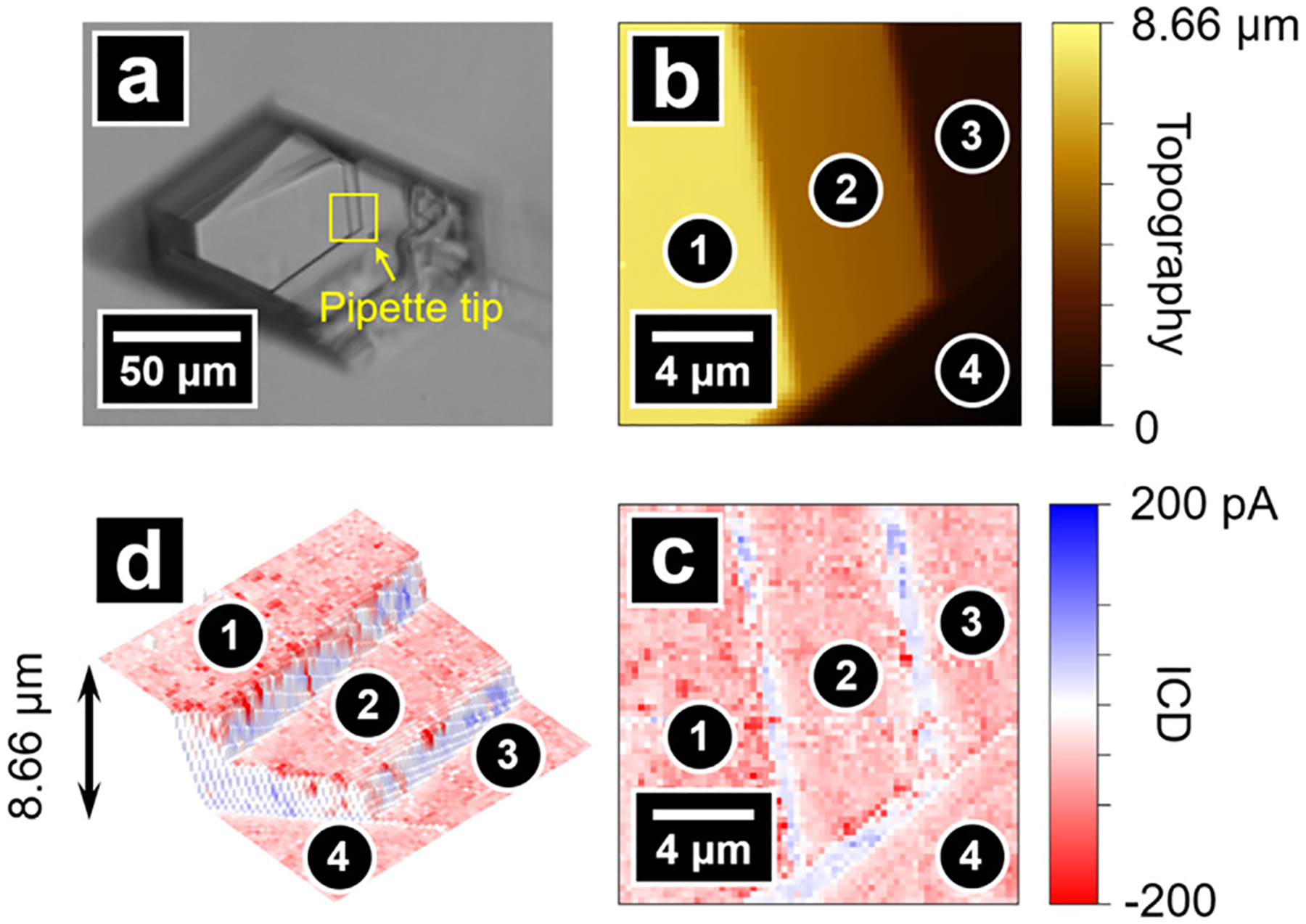
(a) Optical micrograph of a dickite crystal. The area scanned by SICM is highlighted with a yellow square, where the pipette tip can be seen in the bottom right corner of the square. Topography image (b) and the surface charge image (c) of the scan area. (d) Combined topography and surface charge image that illustrates negatively charged basal planes and positively charged edges.
Influence of pH on Surface Charge Density.
The pH dependence of surface charge at the dickite surface was measured on the same area shown in Figure 2 at pH 5, followed by pH 4 and 3. The combined 3D topography and surface charge images are shown in Figure 3. Over the pH range 3–5, the basal surfaces of the crystal show an invariant negative surface charge.67 Another basal surface displaying a pH-independent negative charge over the majority of the surface with minor positive charge at small defects on the surface (due to exposure of an underlaying gibbsite layer) (Figure S5.1) was used for FEM simulations (details of the FEM are included in Section SI-3 of the Supporting Information). The calculated value of the negative surface charge on dickite was estimated to be −133 mC/m2, which is similar to the charge density measured for aqueous suspensions of kaolinite (−110 to −150 mC/m2)68 and silica69 but higher in intensity than the value of −4 mC/m2 measured on the siloxane layer of kaolinite using AFM methods (pH 4, 1–10 mM electrolyte solutions).38,43 In contrast to the basal surfaces, edge surfaces shown in Figure 2 possess a positive charge that increases in intensity as pH is decreased from 5 to 3. The pH-dependent charge is attributed to Al–OH2+ groups on exposed edges of the octahedral gibbsite-like layers. Within the pH range of our experiments, Si–OH groups are also present at edges;26 however, they are only expected to contribute a significant negative charge to the edge at pH > 9.25
Figure 3.
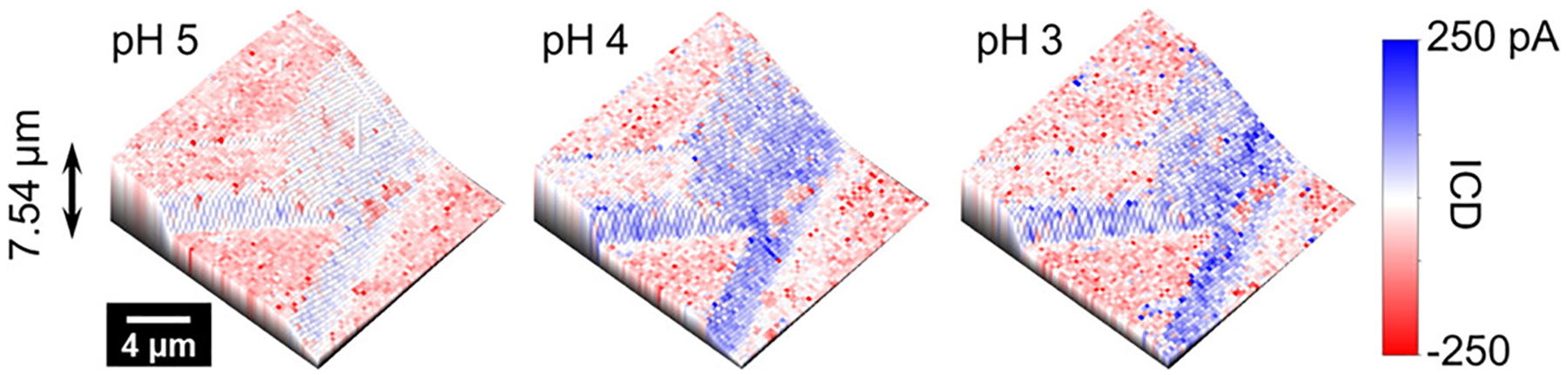
Combined topography and surface charge images of a dickite crystal at pH 5, 4, and 3. The addition of hydrochloric acid results in a greater positive surface charge on the edges. In contrast, the negative charge on basal surfaces is insensitive to change of solution pH.
The influence of sample slope on the tip–sample separation in SICM has been described previously by Thatenhorst et al.70 and is a relevant consideration for samples examined here (see Supporting Information). To obviate this consideration in images, dickite crystals with edge surfaces oriented perpendicular to the pipette probe were also measured to examine the possibility that the relatively large angle between the step edge surface and measurement plane influenced the SICM feedback and the I–V curves used for surface charge analysis. Figure 4 shows edge-on topography images of a dickite crystal (oriented similar to the case of Figure S4.2b), which is characterized by a positively charged gibbsite-like basal surface (receding into the distance) on the left half of Figure 4a,b and abundant edge surfaces on the right side of the images. Surface charge images reveal a predominantly positively charged surface over the scan area with faint negatively charged features between the positively charged lamellae, consistent with the topography image (composite topography/charge shown in Figure 4i). When the pH is adjusted from 5 to 3, the overall positive charge intensifies due to an increase in the surface density of edge Al–OH2+ sites. Simulations with FEM show an increase in charge at edge surfaces from 3 to 31 mC/m2 during the titration (see Table S3.2 for simulation parameters). The surface charge density calculated here is somewhat lower than the ~70 mC/m2 derived from potentiometric titrations on kaolinite over a similar pH range,24 and greater than the 3–6 mC/m2 determined for the gibbsite-like surface of kaolinite crystals derived from AFM studies (pH 4, 1–10 mM electrolyte solutions),38,43 with the added caveat that probe–sample distances are likely overestimated in models used here.70 In addition, randomly distributed islands of relatively strong negative charge are observed on the edge surfaces, which are likely small flakes of dickite that are electrostatically adsorbed to the positively charged edges. These flakes are also observed in electron micrographs of dickite samples (Figure S4.3) and in the topography images (correlated with the surface charge image) (Figure 4, examples highlighted with blue circles).
Figure 4.
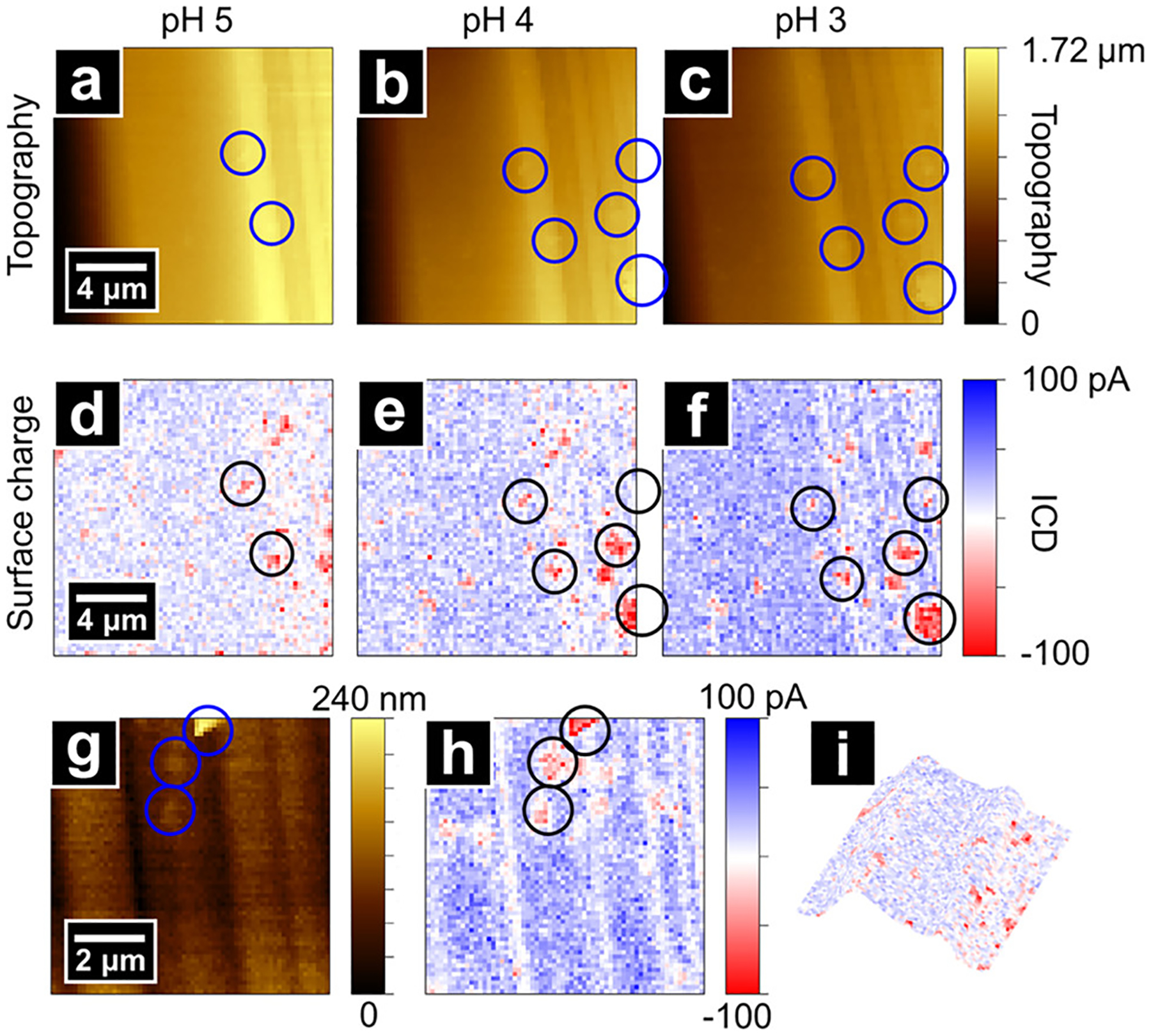
Topography and surface charge images of an area on edge surfaces of a dickite crystal at pH 5 (a,d), 4 (b,e), and 3 (c,f). The scan area is dominated by positively charged edges with neutral/negatively charged regions between lamellae. In addition, several negatively charged islands are observed due to small dickite flakes adhered to the surface. The addition of acid increases the positive charge intensity at edges whereas the negative surface charge remains constant. (g,h) High-resolution images within the area of (c,f). A 3D rendering of topography and surface charge from panels (a,d) is shown in panel (i).
The weak negative surface charge intensity measured between platelet edges is due to the probe geometry used for scanning and its size is relative to the surface structural features. As shown in Figure 4g, most negatively charged basal surfaces are in spaces between platelet edges and have widths similar to the outer diameter of the pipette tip used in the experiment (~100 nm). As a result, the tip–sample distance is larger over the groove compared with that on a flat surface, which reduces the influence of surface charge on the extended I–V response. Moreover, as the pipette moves closer to the groove, both the basal surface underneath the tip and the adjacent edge surfaces influence the ion current. As the basal surface and the edge carry opposite surface charge, the apparent negative surface charge measured by SICM is greatly diminished.
Interestingly, all dickite crystals studied (n > 15) with SICM exhibited negatively charged basal surfaces. This result was unexpected due to the anisotropic nature of the crystal structure and the expectation that for randomly oriented crystals there should be an equal probability of observing a positively charged gibbsite or negatively charged siloxane surface. This observation was confirmed for six additional crystals that were intentionally oriented on the PDMS substrate such that either the gibbsite or siloxane basal surface was presented to the SICM probe. For these experiments, each dickite crystal was individually placed on the PDMS substrate in a known orientation that was established from the face index and crystal structure determined by single-crystal X-ray diffraction measurements (see Section SI-4 and Table S4.1). Three crystals were oriented with siloxane basal surfaces facing the SICM probe, and another three had gibbsite basal surfaces exposed to the probe. Regardless of orientation, all basal surfaces probed by SICM were negatively charged.
Some studies have attributed a permanent negative charge on kaolinite basal surfaces to (Si–O–Al)− groups resulting from Al3+ substitution in the siloxane layer.24,68 Although isomorphous substitution is common in many phyllosilicate clay minerals, we are unaware of any direct evidence for such substitution in well-ordered euhedral members of the kaolin family such as dickite, which are known to have minimal substitution of variable-valence cations.20–22,25,71 Indeed, surface analysis using time-of-flight secondary ion mass spectrometry (SIMS) showed Si and Al close to the expected 1:1 ratio, with only trace amounts of a Mg2+ impurity (Figure S6.1). Another possibility is that Si and Al dissolve from the crystal and re-adsorb or precipitate on the surface.24 There is precedent for the precipitation of silica,72 aluminosilicate,73 and aluminum hydroxide74 phases during kaolinite dissolution, but such studies were carried out under higher temperatures, highly acidic or basic pH, and longer reaction times compared with our experimental conditions. Another possibility is that the basal planes are coated by an adsorbate layer stemming from prolonged exposure to contaminants in ambient air or in the subsurface environment in which the crystal formed. A freshly cleaved dickite surface would have a high surface free energy and would be prone to passivation by adsorbed impurities; in the bulk, the charges are balanced by strong interlayer interactions between tetrahedral and octahedral sheets. Resolving the origin of the negative surface charge will require elucidating the constitution of the basal surface layer, which should be possible using X-ray methods and synchrotron radiation in the future.
Interaction between Malonate and Dickite Crystals.
To further explore the positively charged edges and study the interactions between kaolin group minerals and natural carboxylic acids, potassium malonate was introduced into the bulk solution after measuring the topography and surface charge of dickite crystals. Malonate is known to undergo ligand–exchange reactions at Lewis acid Al sites, leading to the formation of coordination complexes on mineral surfaces.75,76 Dissolution of kaolinite is accelerated in the presence of dicarboxylic acids, leading to an increase in dissolved Al and Si.24,76 Malonate has the same effect on dickite crystals, as verified by the presence of dissolved Si and Al in malonate extracts that had been in contact with dickite crystals overnight (Tables S9.1 and S9.2).
As shown in Figure 5, addition of malonate induces significant changes in surface charge distribution. The most evident is the appearance of clusters of intense negative charge at edges and steps (indicated by black arrows). These clusters increased in size when the same area was scanned repeatedly under the same experimental conditions, suggesting that negatively charged material was depositing on surfaces over time. Deposits were also observed via SEM for dickite samples immersed overnight in KCl/potassium malonate (Figure S7.1). Again, clusters were found to be more concentrated at the boundaries and edges.
Figure 5.
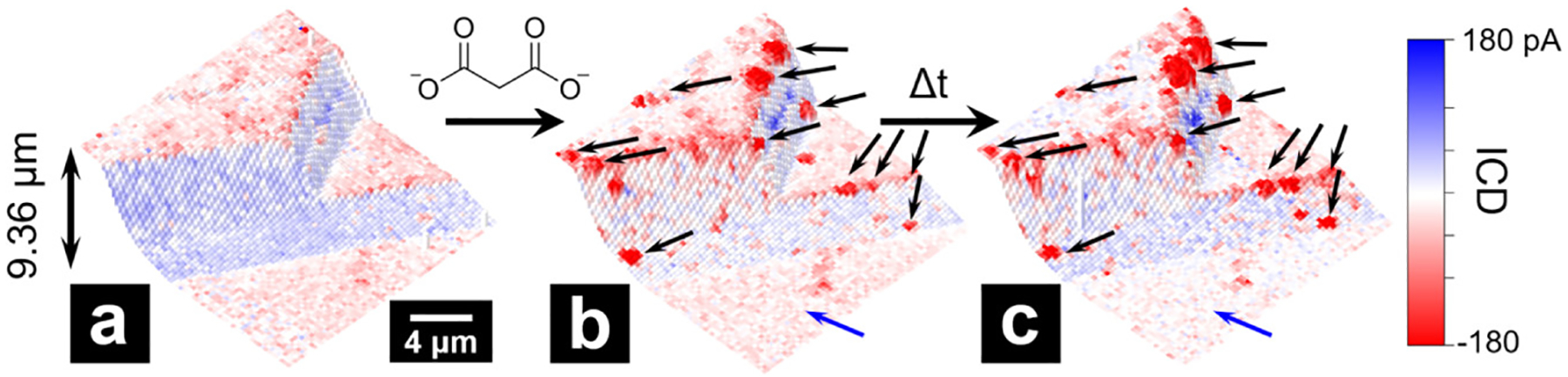
Combined topography and surface charge images on an area of a dickite crystal before addition of potassium malonate (a) and after the addition of malonate (b). The addition of malonate results in negatively charged clusters at edge surfaces and step edges and reduces the overall negative surface charge at the basal surfaces. The same area measured again under the same experimental conditions shows the growth of negatively charged clusters and further reduction of basal surface negative charge (c). Experiments carried out at pH = 5.
In addition to the appearance of negatively charged clusters on edge surfaces, the overall negative charge density on the basal surfaces also diminished after introducing potassium malonate (highlighted with blue arrows in Figure 5). A similar observation was made during a second experiment (Figure S7.2), where malonate addition resulted in a complete shift in the basal surface charge from negative to positive that coincided with the formation of small clusters of negative charge on edge surfaces. These changes are attributed to dissolution of the outermost (negatively charged) basal layer of dickite crystals in the presence of malonate, revealing portions of the positively charged gibbsite layer beneath. As mentioned above, there is precedent for the dissolution and subsequent precipitation of Al (hydr)oxide, mixed Al–Si phases, or silica onto kaolinite—a process likely enhanced by the presence of organic acids.24,77,78 These observations are consistent with a process in which malonate-induced dissolution of dickite surfaces is followed by precipitation of negatively charged material that forms islands that are likely electrostatically bound to positively charged aluminol sites.75 Over time, the entire surface becomes coated in the precipitate.
Repeating the dissolution experiments multiple times with different crystals appears to catch the dissolution process at different stages. For example, in a third experiment, addition of potassium malonate resulted in a negatively charged precipitate coating dickite. Combined topography and surface charge images (Figure S7.3) show an eventual intensification of negative charge on the basal surface and conversion of edge surface charge from positive to negative. The resulting negative surface charge did not change in intensity as the pH of the solution was decreased from 5 to 4 and finally 3, indicating the surface pKa of the newly deposited coating is less than 3. Although additional work is needed to explore ligand-promoted dissociation processes, these experiments demonstrate the usefulness of SICM to study surface reactions at the mineral–water interface on the nanoscale.
Effect of Surface pH on Nitrite-to-HONO(g) Conversion.
A flow reactor coupled to a CIMS was used to study the pH-dependent emission of gaseous HONO from bulk water, kaolinite, dickite, and dickite reacted overnight with malonate. Figure 6a shows the amount of HONO(g) released following titration of bulk water (in the absence of clay mineral) with 1 nmol of NO2−, as quantified by the integration of the HONO(g) signal following addition. The titration curve shows an inflection point at pH 3.3, which corresponds to the well-known pKa of HONO in bulk aqueous solutions.79–81 As expected, HONO(g) emissions are highest between pH 2–4, where HONO is in highest abundance and partitions to the gas phase. At pH > 3.3, NO2− is the predominant N(III) species and remains dissolved in bulk solution. This behavior is in stark contrast to the titration of kaolin mineral surfaces with aqueous NO2− (Figures 6b and S8.2). Although a maximum in HONO(g) emission is still observed at pH 3 for the clay minerals, HONO(g) emission persists out to pH 8 to 5 pH units above the pKa of HONO.
Figure 6.
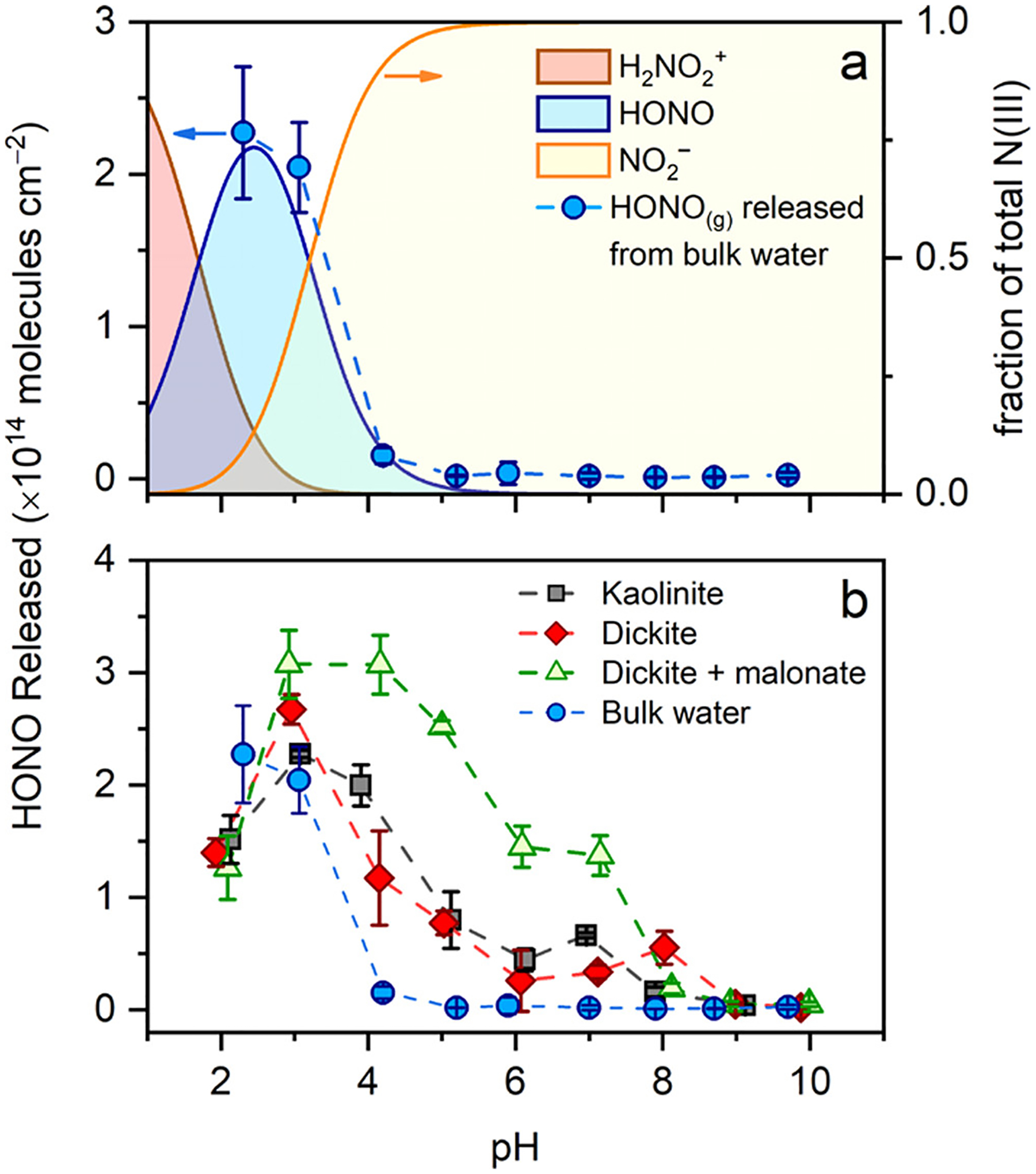
Amount of HONO(g) emitted following addition of 1 nmol of nitrite to pure water (a) or clay mineral surfaces (b) that were pH-adjusted. Data are normalized to the geometric surface area of the sample. Dotted lines are guides for the eye, and the error bars represent the 95% confidence interval of the mean of three replicate measurements. The equilibrium distribution of indicated N(III) species in aqueous solution as a function of pH is included for comparison in panel (a).
The amount of HONO(g) emitted from the substrates reflects the balance of HONO(g) that desorbs versus the amount of N(III) that remains adsorbed to the surface. In the case of the kaolin minerals, HONO(g) emissions are low at pH < 2 due to the presence of H2ONO+ as the major N(III) species (pKa = 1.8, Figure 6a).82 Between pH 4 and 8, a sufficient source of protons is clearly available to promote NO2− protonation and release HONO(g) from the surface. In this pH range, the SICM evidence (Figures 3 and 4) of persistent positively charged edges and steps on dickite surfaces at pH > 4 suggests that NO2− protonation occurs at Al–OH2+ sites and is likely facilitated by adsorbed water. At pH > 8, M–O− (where M = Al3+ and Si4+) is the dominant surface species and the lack of protons prevents NO2− to HONO conversion.
Differences in NO2− protonation mechanisms are evident in the kinetics traces collected during the titration (Figure S8.3). At pH < 4, titration of the clay mineral surface with NO2− results in an instantaneous pulse of HONO(g) emitted that decays exponentially to baseline levels by experiment’s end; this is interpreted as protonation of NO2− by abundant H3O+ within the multilayer water coating the mineral surface. In contrast, at pH > 4, HONO(g) emission is gradual and builds up over the course of the experiment, suggesting that a more complicated (slower) mechanism is operational. Based on the prevalence of positively charged M–OH2+ groups at pH > 4, we hypothesize that the slower process at higher pH stems from adsorption and protonation reactions involving Al–OH2+ on the kaolin steps and edges. The process is slower at higher pH because of competition between protonation of NO2− by Al–OH2+ and deprotonation of HONO(aq) by OH−(aq) before HONO(g) can escape the interface.
Whereas data in Figure 6b display the amount of HONO(g) formed relative to geometric surface area, it is instructive to plot the amount of HONO emitted relative to the specific surface area of the mineral substrate (see Section SI-8), as doing so may reveal differences in reactivity between the substrates. As shown in Figure S8.2, kaolin minerals have similar reactivity profiles across the pH range. The exception is kaolinite, which was less reactive by a factor of 10 relative to the other minerals. This lower reactivity is related to the high specific surface area of kaolinite, which is the highest among the kaolin mineral substrates studied here (Table S9.3). The high surface area would also correspond to a high density of Lewis basic sites on gibbsite-like surface layers that are capable of chemisorbing NO2− and HONO and yielding covalently bonded nitrate, nitrito (–ONO−), and nitro species (–NO2−).83–87 As a result, kaolinite is expected to have an enhanced retention of adsorbed nitrite or promote reactive loss of N(III). This effect may be less pronounced in dickite due to its lower specific surface area and the negatively charged surface layer covering all basal surfaces. Dickite suspended in a malonate solution overnight exhibits lower HONO(g) emission than pure dickite at pH < 4 but higher reactivity at pH > 4 (Figure S8.2). Based on the SICM evidence discussed above, lower surface area (due to malonate-induced dissolution) and blocking of reactive edge sites by malonate deposits (e.g., Figure 5b) could be responsible for lower reactivity at lower pH, whereas dissolution of basal surface layers (Figure 5c) could increase reactivity by exposing additional reactive sites [e.g., Al–(OH2+)–Al or Al–OH2+]. Future work will investigate the impact of clay surface-organic matter interactions on the surface acidity of clay minerals.
Environmental Implications.
From a measurement science perspective, this work demonstrates the feasibility of using SICM for the simultaneous visualization of surface charge and topography of natural minerals. Dickite crystals were investigated and observed to carry a heterogeneous distribution of surface charges across different crystal faces. In the pH range of 3–5, basal surfaces exhibit a pH-independent negative charge. In contrast, edges carried a pH-dependent positive charge with increasing charge at more acidic conditions. The variable charge on the edges likely originates from acid/base equilibria involving Al–OH2+ groups. When malonate was introduced to the bulk solution, shifts in surface charge and clusters of negative charge suggest that malonate facilitates the dissolution of both siloxane and gibbsite-like layers, with subsequent precipitation of material at edge surfaces. These results clearly demonstrate the usefulness of SICM in studying dynamic surface reactions at mineral–water interfaces. In addition, the technique will be useful to other environmentally relevant research areas ranging from studying the chemical and physical properties of aerosols to elucidating mechanisms of pollutant degradation on catalyst surfaces.
From a larger, environmental perspective, this study also demonstrates the ability of SICM to provide molecular-level insights into mineral surface processes that ultimately can impact local and global atmospheric composition. Surface charge measurements on dickite suggest the release of HONO from kaolin mineral surfaces is due to protonation of NO2− by Al–OH2+ groups on edges and steps, which persist at near-neutral pH. This provides a mechanism with which to rationalize recent reports of atmospherically relevant emissions of HONO(g) from soil with pH values up to 8.8,13,14 a pH that is orders of magnitude higher than the pKa of HONO(g) in aqueous solution. Prior to these studies, it was assumed that atmosphere–soil exchange of HONO was solely dependent on bulk soil pH, Henry’s Law, and aqueous N(III) concentrations. This study clearly shows that not accounting for soil mineralogy and the presence of ubiquitous amphoteric clay minerals can lead to underestimation of the potential for soil to emit HONO(g). Although this work focused on an application of SICM to understand the acid/base chemistry and air-surface partitioning of HONO, microprobe techniques capable of measuring surface charge on the nanoscale have many potential applications in the field of biogeochemistry, especially in light of increasing evidence that surface charge determines the strength of mineral–organic interactions, which are key to retention and stabilization of soil organic matter against mineralization and subsequent conversion to greenhouse gases.88–91
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this study came from the U.S. Department of Energy, Office of Science, Early Career Research Program from Subsurface Biogeochemical Research Program (award DE-SC0014443 to J.D.R.), National Science Foundation (AGS-1352375 J.D.R.), and the National Science Foundation (CHE-1808133 L.A.B.). K.A. acknowledges support from a fellowship from the Graduate Training Program Quantitative and Chemical Biology (QCB) at Indiana University (grants T32 GM131994 and GM109825). The Nanoscale Characterization Facility at Indiana University is acknowledged for access to electron microscopy. SIMS data were obtained at the Materials Research Lab at the University of Illinois. We are grateful to Maren Pink at Indiana University’s Molecular Structure Center for carrying out single crystal X-ray diffraction analysis of dickite crystals and preparing the oriented dickite samples used for SICM analysis. Support for the acquisition of the Bruker Venture D8 diffractometer through the Major Scientific Research Equipment Fund from the President of Indiana University and the Office of the Vice President for Research is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.1c03455.
Electron micrographs of nanopipettes, experimental details of flow reactor studies, FEM simulations, additional characterization of dickite samples, X-ray crystallography details, SIMS, additional malonate interaction studies, HONO emission flux estimation, and dissolution and inductively coupled plasma–mass spectrometry (PDF)
Accession Codes
Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Inorganic Crystal Structure Database. Further details of the crystal structure investigations may be obtained from the joint CCDC/FIZ Karlsruhe online deposition service: https://www.ccdc.cam.ac.uk/structures/? by quoting the deposition number CSD-2083088-2083093.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.est.1c03455
The authors declare no competing financial interest.
Contributor Information
Cheng Zhu, Department of Chemistry, Indiana University, Bloomington, Indiana 47401, United States.
Gargi Jagdale, Department of Chemistry, Indiana University, Bloomington, Indiana 47401, United States.
Adrien Gandolfo, Paul H. O’Neill School of Public & Environmental Affairs, Indiana University, Bloomington, Indiana 47405, United States.
Kristen Alanis, Department of Chemistry, Indiana University, Bloomington, Indiana 47401, United States.
Rebecca Abney, Paul H. O’Neill School of Public & Environmental Affairs, Indiana University, Bloomington, Indiana 47405, United States; Warnell School of Forestry and Natural Resources, University of Georgia, Athens, Georgia 30602, United States.
Lushan Zhou, Department of Chemistry, Indiana University, Bloomington, Indiana 47401, United States.
David Bish, Department of Chemistry, Indiana University, Bloomington, Indiana 47401, United States.
Jonathan D. Raff, Department of Chemistry, Indiana University, Bloomington, Indiana 47401, United States; Paul H. O’Neill School of Public & Environmental Affairs, Indiana University, Bloomington, Indiana 47405, United States.
Lane A. Baker, Department of Chemistry, Indiana University, Bloomington, Indiana 47401, United States.
REFERENCES
- (1).Sparks DL Environmental Soil Chemistry, 2nd ed.; Academic Press: San Diego, CA, 2003; pp 1–368. [Google Scholar]
- (2).Kleffmann J; Kurtenbach R; Lörzer J; Wiesen P; Kalthoff N; Vogel B; Vogel H Measured and simulated vertical profiles of nitrous acid—Part I: Field measurements. Atmos. Environ 2003, 37, 2949–2955. [Google Scholar]
- (3).VandenBoer TC; Brown SS; Murphy JG; Keene WC; Young CJ; Pszenny AAP; Kim S; Warneke C; de Gouw JA; Maben JR; Wagner NL; Riedel TP; Thornton JA; Wolfe DE; Dubé WP;Öztörk F; Brock CA; Grossberg N; Lefer B; Lerner B; Middlebrook AM; Roberts JM Understanding the role of the ground surface in HONO vertical structure: High resolution vertical profiles during NACHTT-11. J. Geophys. Res. Atmos 2013, 118, 10155–10171. [Google Scholar]
- (4).Wong KW; Tsai C; Lefer B; Grossberg N; Stutz J Modeling of daytime HONO vertical gradients during SHARP 2009. Atmos. Chem. Phys 2013, 13, 3587–3601. [Google Scholar]
- (5).Zhang N; Zhou X; Shepson PB; Gao H; Alaghmand M; Stirm B Aircraft measurement of HONO vertical profiles over a forested region. Geophys. Res. Lett 2009, 36, L15820. [Google Scholar]
- (6).Villena G; Kleffmann J; Kurtenbach R; Wiesen P; Lissi E; Rubio MA; Croxatto G; Rappenglück B Vertical gradients of HONO, NOx and O3 in Santiago de Chile. Atmos. Environ 2011, 45, 3867–3873. [Google Scholar]
- (7).Meng F; Qin M; Tang K; Duan J; Fang W; Liang S; Ye K; Xie P; Sun Y; Xie C; Ye C; Fu P; Liu J; Liu W High-resolution vertical distribution and sources of HONO and NO2 in the nocturnal boundary layer in urban Beijing, China. Atmos. Chem. Phys 2020, 20, 5071–5092. [Google Scholar]
- (8).Young CJ; Washenfelder RA; Roberts JM; Mielke LH; Osthoff HD; Tsai C; Pikelnaya O; Stutz J; Veres PR; Cochran AK; VandenBoer TC; Flynn J; Grossberg N; Haman CL; Lefer B; Stark H; Graus M; de Gouw J; Gilman JB; Kuster WC; Brown SS Vertically Resolved Measurements of Nighttime Radical Reservoirs in Los Angeles and Their Contribution to the Urban Radical Budget. Environ. Sci. Technol 2012, 46, 10965–10973. [DOI] [PubMed] [Google Scholar]
- (9).Kleffmann J Daytime Sources of Nitrous Acid (HONO) in the Atmospheric Boundary Layer. ChemPhysChem 2007, 8, 1137–1144. [DOI] [PubMed] [Google Scholar]
- (10).VandenBoer TC; Young CJ; Talukdar RK; Markovic MZ; Brown SS; Roberts JM; Murphy JG Nocturnal loss and daytime source of nitrous acid through reactive uptake and displacement. Nat. Geosci 2015, 8, 55–60. [Google Scholar]
- (11).Scharko NK; Martin ET; Losovyj Y; Peters DG; Raff JD Evidence for Quinone Redox Chemistry Mediating Daytime and Nighttime NO2-to-HONO Conversion on Soil Surfaces. Environ. Sci. Technol 2017, 51, 9633–9643. [DOI] [PubMed] [Google Scholar]
- (12).Donaldson MA; Bish DL; Raff JD Soil surface acidity plays a determining role in the atmospheric-terrestrial exchange of nitrous acid. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Oswald R; Behrendt T; Ermel M; Wu D; Su H; Cheng Y; Breuninger C; Moravek A; Mougin E; Delon C; Loubet B; Pommerening-Röser A; Sörgel M; Pöschl U; Hoffmann T; Andreae MO; Meixner FX; Trebs I HONO Emissions from Soil Bacteria as a Major Source of Atmospheric Reactive Nitrogen. Science 2013, 341, 1233–1235. [DOI] [PubMed] [Google Scholar]
- (14).Su H; Cheng Y; Oswald R; Behrendt T; Trebs I; Meixner FX; Andreae MO; Cheng P; Zhang Y; Pöschl U Soil Nitrite as a Source of Atmospheric HONO and OH Radicals. Science 2011, 333, 1616–1618. [DOI] [PubMed] [Google Scholar]
- (15).Zhang J; An J; Qu Y; Liu X; Chen Y Impacts of potential HONO sources on the concentrations of oxidants and secondary organic aerosols in the Beijing-Tianjin-Hebei region of China. Sci. Total Environ 2019, 647, 836–852. [DOI] [PubMed] [Google Scholar]
- (16).Elshorbany YF; Steil B; Brühl C; Lelieveld J Impact of HONO on global atmospheric chemistry calculated with an empirical parameterization in the EMAC model. Atmos. Chem. Phys 2012, 12, 9977–10000. [Google Scholar]
- (17).Elshorbany YF; Crutzen PJ; Steil B; Pozzer A; Tost H; Lelieveld J Global and regional impacts of HONO on the chemical composition of clouds and aerosols. Atmos. Chem. Phys 2014, 14, 1167–1184. [Google Scholar]
- (18).Gonçalves M; Dabdub D; Chang WL; Jorba O; Baldasano JM Impact of HONO sources on the performance of mesoscale air quality models. Atmos. Environ 2012, 54, 168–176. [Google Scholar]
- (19).Kebede MA; Bish DL; Losovyj Y; Engelhard MH; Raff JD The Role of Iron-Bearing Minerals in NO2 to HONO Conversion on Soil Surfaces. Environ. Sci. Technol 2016, 50, 8649–8660. [DOI] [PubMed] [Google Scholar]
- (20).Johnston CT; Agnew SF; Bish DL Polarized single-crystal Fourier-transform infrared microscopy of Ouray dickite and Keokuk kaolinite. Clays Clay Miner. 1990, 38, 573–583. [Google Scholar]
- (21).Bish DL; Johnston CT Rietveld refinement and Fourier-transform infrared spectroscopic study of the dickite structure at low temperature. Clays Clay Miner. 1993, 41, 297–304. [Google Scholar]
- (22).Johnston CT; Elzea Kogel J; L. Bish D; Kogure T; H. Murray H Low-Temperature FTIR Study of Kaolin-Group Minerals. Clays Clay Miner. 2008, 56, 470–485. [Google Scholar]
- (23).Van Olphen H An Introduction to Clay Colloid Chemistry, 2nd ed.; John Wiley and Sons: New York, 1977; pp 1–318. [Google Scholar]
- (24).Wieland E; Stumm W Dissolution kinetics of kaolinite in acidic aqueous solutions at 25°C. Geochim. Cosmochim. Acta 1992, 56, 3339–3355. [Google Scholar]
- (25).Brady PV; Cygan RT; Nagy KL Molecular Controls on Kaolinite Surface Charge. J. Colloid Interface Sci 1996, 183, 356–364. [DOI] [PubMed] [Google Scholar]
- (26).Liu X; Lu X; Sprik M; Cheng J; Meijer EJ; Wang R Acidity of edge surface sites of montmorillonite and kaolinite. Geochem. Cosmochim. Acta 2013, 117, 180–190. [Google Scholar]
- (27).Marcano-Martinez E; McBride MB Comparison of the Titration and Ion Adsorption Methods for Surface Charge Measurement in Oxisols. Soil Sci. Soc. Am. J 1989, 53, 1040–1045. [Google Scholar]
- (28).Van Raij B; Peech M Electrochemical Properties of Some Oxisols and Alfisols of the Tropics. Soil Sci. Soc. Am. J 1972, 36, 587–593. [Google Scholar]
- (29).Williams DJA; Williams KP Electrophoresis and zeta potential of kaolinite. J. Colloid Interface Sci 1978, 65, 79–87. [Google Scholar]
- (30).Smith RW; Narimatsu Y Electrokinetic behavior of kaolinite in surfactant solutions as measured by both the microelectrophoresis and streaming potential methods. Miner. Eng 1993, 6, 753–763. [Google Scholar]
- (31).Pierre AC; Ma K Sedimentation behaviour of kaolinite and montmorillonite mixed with iron additives, as a function of their zeta potential. J. Mater. Sci 1997, 32, 2937–2947. [Google Scholar]
- (32).Appel C; Ma LQ; Dean Rhue R; Kennelley E Point of zero charge determination in soils and minerals via traditional methods and detection of electroacoustic mobility. Geoderma 2003, 113, 77–93. [Google Scholar]
- (33).Zhou Z; Gunter WD The Nature of the Surface Charge of Kaolinite. Clays Clay Miner. 1992, 40, 365–368. [Google Scholar]
- (34).Huertas FJ; Chou L; Wollast R Mechanism of Kaolinite Dissolution at Room Temperature and Pressure: Part 1. Surface Speciation. Geochim. Cosmochim. Acta 1998, 62, 417–431. [Google Scholar]
- (35).Coppin F; Berger G; Bauer A; Castet S; Loubet M Sorption of lanthanides on smectite and kaolinite. Chem. Geol 2002, 182, 57–68. [Google Scholar]
- (36).Kretzschmar R; Holthoff H; Sticher H Influence of pH and Humic Acid on Coagulation Kinetics of Kaolinite: A Dynamic Light Scattering Study. J. Colloid Interface Sci 1998, 202, 95–103. [Google Scholar]
- (37).Gupta V; Hampton MA; Nguyen AV; Miller JD Crystal lattice imaging of the silica and alumina faces of kaolinite using atomic force microscopy. J. Colloid Interface Sci 2010, 352, 75–80. [DOI] [PubMed] [Google Scholar]
- (38).Gupta V; Miller JD Surface force measurements at the basal planes of ordered kaolinite particles. J. Colloid Interface Sci 2010, 344, 362–371. [DOI] [PubMed] [Google Scholar]
- (39).Gupta V; Hampton MA; Stokes JR; Nguyen AV; Miller JD Particle interactions in kaolinite suspensions and corresponding aggregate structures. J. Colloid Interface Sci 2011, 359, 95–103. [DOI] [PubMed] [Google Scholar]
- (40).Yin X; Miller JD Wettability of kaolinite basal planes based on surface force measurements using atomic force microscopy. Min., Metall., Explor 2012, 29, 13–19. [Google Scholar]
- (41).Liu J; Sandaklie-Nikolova L; Wang X; Miller JD Surface force measurements at kaolinite edge surfaces using atomic force microscopy. J. Colloid Interface Sci 2014, 420, 35–40. [DOI] [PubMed] [Google Scholar]
- (42).Kumar N; Zhao C; Klaassen A; van den Ende D; Mugele F; Siretanu I Characterization of the surface charge distribution on kaolinite particles using high resolution atomic force microscopy. Geochim. Cosmochim. Acta 2016, 175, 100–112. [Google Scholar]
- (43).Kumar N; Andersson MP; van den Ende D; Mugele F; Siretanu I Probing the Surface Charge on the Basal Planes of Kaolinite Particles with High-Resolution Atomic Force Microscopy. Langmuir 2017, 33, 14226–14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Guo Y; Xiong Y Characterizing the Surface Charge of Clay Minerals with Atomic Force Microscope (AFM). AIMS Mater. Sci 2017, 4, 582–593. [Google Scholar]
- (45).Hansma P; Drake B; Marti O; Gould S; Prater C The scanning ion-conductance microscope. Science 1989, 243, 641–643. [DOI] [PubMed] [Google Scholar]
- (46).Zhu C; Huang K; Siepser NP; Baker LA Scanning Ion Conductance Microscopy. Chem. Rev 2020, DOI: 10.1021/acs.chemrev.0c00962. In Press [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Korchev YE; Bashford CL; Milovanovic M; Vodyanoy I; Lab MJ Scanning ion conductance microscopy of living cells. Biophys. J 1997, 73, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chen C-C; Zhou Y; Baker LA Scanning Ion Conductance Microscopy. Annu. Rev. Anal. Chem 2012, 5, 207–228. [DOI] [PubMed] [Google Scholar]
- (49).Sa N; Baker LA Rectification of Nanopores at Surfaces. J. Am. Chem. Soc 2011, 133, 10398–10401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sa N; Lan W-J; Shi W; Baker LA Rectification of Ion Current in Nanopipettes by External Substrates. ACS Nano 2013, 7, 11272–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).McKelvey K; Kinnear SL; Perry D; Momotenko D; Unwin PR Surface Charge Mapping with a Nanopipette. J. Am. Chem. Soc 2014, 136, 13735–13744. [DOI] [PubMed] [Google Scholar]
- (52).Perry D; Al Botros R; Momotenko D; Kinnear SL; Unwin PR Simultaneous Nanoscale Surface Charge and Topographical Mapping. ACS Nano 2015, 9, 7266–7276. [DOI] [PubMed] [Google Scholar]
- (53).Page A; Perry D; Young P; Mitchell D; Frenguelli BG; Unwin PR Fast Nanoscale Surface Charge Mapping with Pulsed-Potential Scanning Ion Conductance Microscopy. Anal. Chem 2016, 88, 10854–10859. [DOI] [PubMed] [Google Scholar]
- (54).Perry D; Paulose Nadappuram B; Momotenko D; Voyias PD; Page A; Tripathi G; Frenguelli BG; Unwin PR Surface Charge Visualization at Viable Living Cells. J. Am. Chem. Soc 2016, 138, 3152–3160. [DOI] [PubMed] [Google Scholar]
- (55).Klausen LH; Fuhs T; Dong M Mapping surface charge density of lipid bilayers by quantitative surface conductivity microscopy. Nat. Commun 2016, 7, 12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Fuhs T; Klausen LH; Sønderskov SM; Han X; Dong M Direct measurement of surface charge distribution in phase separating supported lipid bilayers. Nanoscale 2018, 10, 4538–4544. [DOI] [PubMed] [Google Scholar]
- (57).Maddar FM; Perry D; Brooks R; Page A; Unwin PR Nanoscale Surface Charge Visualization of Human Hair. Anal. Chem 2019, 91, 4632–4639. [DOI] [PubMed] [Google Scholar]
- (58).Zhu C; Zhou L; Choi M; Baker LA Mapping Surface Charge of Individual Microdomains with Scanning Ion Conductance Microscopy. ChemElectroChem 2018, 5, 2986–2990. [Google Scholar]
- (59).Martinez J; Ashby D; Zhu C; Dunn B; Baker LA; Siwy ZS Probing ion current in solid-electrolytes at the meso- and nanoscale. Faraday Discuss. 2018, 210, 55–67. [DOI] [PubMed] [Google Scholar]
- (60).Møller Sønderskov S; Hyldgaard Klausen L; Amland Skaanvik S; Han X; Dong M In situ Surface Charge Density Visualization of Self-assembled DNA Nanostructures after Ion Exchange. ChemPhysChem 2020, 21, 1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Cremin K; Jones BA; Teahan J; Meloni GN; Perry D; Zerfass C; Asally M; Soyer OS; Unwin PR Scanning Ion Conductance Microscopy Reveals Differences in the Ionic Environments of Gram-Positive and Negative Bacteria. Anal. Chem 2020, 92, 16024–16032. [DOI] [PubMed] [Google Scholar]
- (62).Dorwling-Carter L; Aramesh M; Han H; Zambelli T; Momotenko D Combined Ion Conductance and Atomic Force Microscope for Fast Simultaneous Topographical and Surface Charge Imaging. Anal. Chem 2018, 90, 11453–11460. [DOI] [PubMed] [Google Scholar]
- (63).Drelich J; Long J; Yeung A Determining surface potential of the bitumen-water interface at nanoscale resolution using atomic force microscopy. Can. J. Chem. Eng 2007, 85, 625–634. [Google Scholar]
- (64).Guo M Soil Sampling and Methods of Analysis. J. Environ. Qual 2009, 38, 375. [Google Scholar]
- (65).Schneider CA; Rasband WS; Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Donaldson MA; Bish DL; Raff JD Soil surface acidity plays a determining role in the atmospheric-terrestrial exchange of nitrous acid. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Van Olphen H Rheological phenomena of clay sols in connection with the charge distribution on the micelles. Discuss. Faraday Soc 1951, 11, 82–84. [Google Scholar]
- (68).Bolland M; Posner A; Quirk J Surface charge on kaolinites in aqueous suspension. Soil Res. 1976, 14, 197–216. [Google Scholar]
- (69).Tadros Th. F.; Lyklema J Adsorption of potential-determining ions at the silica-aqueous electrolyte interface and the role of some cations. J. Electroanal. Chem. Interfacial Electrochem 1968, 17, 267–275. [Google Scholar]
- (70).Thatenhorst D; Rheinlaender J; Schäffer TE; Dietzel ID; Happel P Effect of Sample Slope on Image Formation in Scanning Ion Conductance Microscopy. Anal. Chem 2014, 86, 9838–9845. [DOI] [PubMed] [Google Scholar]
- (71).Newman ACD Chemistry of Clays and Clay Minerals; Longman Scientific & Technical: Essex, England, 1987; p 480. [Google Scholar]
- (72).Shaw SA; Peak D; Hendry MJ Investigation of acidic dissolution of mixed clays between pH 1.0 and −3.0 using Si and Al X-ray absorption near edge structure. Geochim. Cosmochim. Acta 2009, 73, 4151–4165. [Google Scholar]
- (73).Crosson GS; Choi S; Chorover J; Amistadi MK; O’Day PA; Mueller KT Solid-State NMR Identification and Quantification of Newly Formed Aluminosilicate Phases in Weathered Kaolinite Systems. J. Phys. Chem. B 2006, 110, 723–732. [DOI] [PubMed] [Google Scholar]
- (74).Nagy KL; Blum AE; Lasaga AC Dissolution and precipitation kinetics of kaolinite at 80 degrees C and pH 3; the dependence on solution saturation state. Am. J. Sci 1991, 291, 649–686. [Google Scholar]
- (75).Specht CH; Frimmel FH An in situ ATR-FTIR study on the adsorption of dicarboxylic acids onto kaolinite in aqueous suspensions. Phys. Chem. Chem. Phys 2001, 3, 5444–5449. [Google Scholar]
- (76).Chin P-KF; Mills GL Kinetics and mechanisms of kaolinite dissolution: effects of organic ligands. Chem. Geol 1991, 90, 307–317. [Google Scholar]
- (77).Nagy KL; Cygan RT; Hanchar JM; Sturchio NC Gibbsite growth kinetics on gibbsite, kaolinite, and muscovite substrates: Atomic force microscopy evidence for epitaxy and an assessment of reactive surface area. Geochim. Cosmochim. Acta 1999, 63, 2337–2351. [Google Scholar]
- (78).Yang L; Steefel CI Kaolinite dissolution and precipitation kinetics at 22 degrees C and pH 4. Geochem. Cosmochim. Acta 2008, 72, 99–116. [Google Scholar]
- (79).Park JY; Lee YN Solubility and decomposition kinetics of nitrous acid in aqueous solution. J. Phys. Chem 1988, 92, 6294–6302. [Google Scholar]
- (80).Becker KH; Kleffmann J; Kurtenbach R; Wiesen P Solubility of Nitrous Acid (HONO) in Sulfuric Acid Solutions. J. Phys. Chem 1996, 100, 14984–14990. [Google Scholar]
- (81).Anastasio C; Chu L Photochemistry of Nitrous Acid (HONO) and Nitrous Acidium Ion (H2ONO+) in Aqueous Solution and Ice. Environ. Sci. Technol 2009, 43, 1108–1114. [DOI] [PubMed] [Google Scholar]
- (82).Riordan E; Minogue N; Healy D; O’Driscol P; Sodeau JR Spectroscopic and Optimization Modeling Study of Nitrous Acid in Aqueous Solution. J. Phys. Chem. A 2005, 109, 779–786. [DOI] [PubMed] [Google Scholar]
- (83).Goodman AL; Bernard ET; Grassian VH Spectroscopic Study of Nitric Acid and Water Adsorption on Oxide Particles: Enhanced Nitric Acid Uptake Kinetics in the Presence of Adsorbed Water. J. Phys. Chem. A 2001, 105, 6443–6457. [Google Scholar]
- (84).Ozensoy E; Peden CHF; Szanyi J NO2 Adsorption on Ultrathin θ-Al2O3 Films: Formation of Nitrite and Nitrate Species. J. Phys. Chem. B 2005, 109, 15977–15984. [DOI] [PubMed] [Google Scholar]
- (85).Underwood GM; Miller TM; Grassian VH Transmission FT-IR and Knudsen Cell Study of the Heterogeneous Reactivity of Gaseous Nitrogen Dioxide on Mineral Oxide Particles. J. Phys. Chem. A 1999, 103, 6184–6190. [Google Scholar]
- (86).Hadjiivanov KI Identification of Neutral and Charged NxOy Surface Species by IR Spectroscopy. Catal. Rev 2000, 42, 71–144. [Google Scholar]
- (87).Donaldson MA; Berke AE; Raff JD Uptake of Gas Phase Nitrous Acid onto Boundary Layer Soil Surfaces. Environ. Sci. Technol 2014, 48, 375–383. [DOI] [PubMed] [Google Scholar]
- (88).Sollins P; Swanston C; Kleber M; Filley T; Kramer M; Crow S; Caldwell BA; Lajtha K; Bowden R Organic C and N stabilization in a forest soil: Evidence from sequential density fractionation. Soil Biol. Biochem 2006, 38, 3313–3324. [Google Scholar]
- (89).Hassink J The capacity of soils to preserve organic C and N by their association with clay and silt particles. Plant Soil 1997, 191, 77–87. [Google Scholar]
- (90).Kleber M; Eusterhues K; Keiluweit M; Mikutta C; Mikutta R; Nico PS Mineral–Organic Associations: Formation, Properties, and Relevance in Soil Environments. In Advances in Agronomy; Sparks DL, Ed.; Academic Press, 2015; Chapter 1, Vol. 130, pp 1–140. [Google Scholar]
- (91).Mikutta R; Turner S; Schippers A; Gentsch N; Meyer-Stüve S; Condron LM; Peltzer DA; Richardson SJ; Eger A; Hempel G; Kaiser K; Klotzbücher T; Guggenberger G Microbial and abiotic controls on mineral-associated organic matter in soil profiles along an ecosystem gradient. Sci. Rep 2019, 9, 10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.