

Abstract
BACKGROUND & AIMS:
Pancreatic ductal adenocarcinoma (PDAC) has a hypoxic, immunosuppressive stroma that contributes to its resistance to immune checkpoint blockade therapies. The hypoxia-inducible factors (HIFs) mediate the cellular response to hypoxia, but their role within the PDAC tumor microenvironment remains unknown.
METHODS:
We used a dual recombinase mouse model to delete Hif1α or Hif2α in α-smooth muscle actin–expressing cancer-associated fibroblasts (CAFs) arising within spontaneous pancreatic tumors. The effects of CAF HIF2α expression on tumor progression and composition of the tumor microenvironment were evaluated by Kaplan-Meier analysis, reverse transcription quantitative real-time polymerase chain reaction, histology, immunostaining, and by both bulk and single-cell RNA sequencing. CAF-macrophage crosstalk was modeled ex vivo using conditioned media from CAFs after treatment with hypoxia and PT2399, an HIF2 inhibitor currently in clinical trials. Syngeneic flank and orthotopic PDAC models were used to assess whether HIF2 inhibition improves response to immune checkpoint blockade.
RESULTS:
CAF-specific deletion of Hif2α, but not Hif1α, suppressed PDAC tumor progression and growth, and improved survival of mice by 50% (n ¼ 21–23 mice/group, Log-rank P ¼ .0009). Deletion of CAF-HIF2 modestly reduced tumor fibrosis and significantly decreased the intratumoral recruitment of immunosuppressive M2 macrophages and regulatory T cells. Treatment with the clinical HIF2 inhibitor PT2399 significantly reduced in vitro macrophage chemotaxis and M2 polarization, and improved tumor responses to immunotherapy in both syngeneic PDAC mouse models.
CONCLUSIONS:
Together, these data suggest that stromal HIF2 is an essential component of PDAC pathobiology and is a druggable therapeutic target that could relieve tumor microenvironment immunosuppression and enhance immune responses in this disease.
Keywords: Pancreatic Ductal Adenocarcinoma, Hypoxia, Cancer-Associated Fibroblasts, Tumor-Associated Macrophages
Graphical Abstract
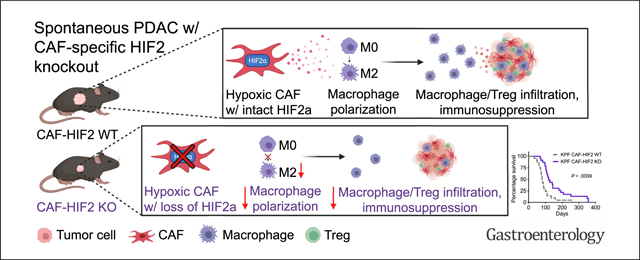
Pancreatic ductal adenocarcinoma (PDAC) responds poorly to most cancer treatments, including immunotherapy.1 This therapeutic recalcitrance may stem from the extensive desmoplastic stroma of PDAC, which suppresses antitumor immunity2 and increases intratumoral pressure,3 resulting in severe hypoxia4 and impaired drug delivery.5 Cancer-associated fibroblasts (CAFs) are the main components and producers of stroma in PDAC.6 Efforts to physically disrupt the hypoxic stromal component through Sonic hedgehog protein inhibition,7 selective fibroblast depletion,8 or recombinant human hyaluronidase9 have effectively lowered stromal content but paradoxically led to worse outcomes in both preclinical studies and clinical trials. These data argue that the initially promising strategy of physically ablating the PDAC stroma may be clinically counterproductive, warranting a different approach.
The hypoxia-inducible factors 1 (HIF1) and 2 (HIF2) are stabilized in low oxygen and have been hypothesized to mediate the therapeutic resistance10,11 and aggressive growth of PDAC.12,13 Deletion of HIF114 or HIF215 in the pancreatic epithelial compartment failed to change overall survival in mice with spontaneous PDAC; however, the function of HIFs in other prominent compartments of the pancreatic tumor microenvironment (TME) remains unclear. Given the importance of the tumor stroma in PDAC oncobiology, we investigated the role of HIF signaling in CAFs and its impact on the PDAC TME.
Here, we elucidated the function of the HIFs within the PDAC stroma using a dual recombinase model to spatiotemporally alter HIF1 or HIF2 signaling only in activated fibroblasts reprogrammed within spontaneous murine pancreatic tumors (also known as CAFs). We found that CAF-specific deletion of HIF2, but not HIF1, improved survival from pancreatic cancer by reducing the recruitment of immunosuppressive macrophages. We further showed that therapeutic HIF2 inhibition improved responses to immune checkpoint blockade, indicating this is a potential combinatorial therapeutic strategy for PDAC.
Materials and Methods
Mice
All experimental mouse work adhered to the standards articulated in the Animal Research: Reporting of In Vivo Experiments guidelines. In addition, all mouse work was approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center. Both female and male mice were used in this study. Mice were maintained on a 12-hour light/dark cycle and were provided with sterilized water and either standard rodent chow (Prolab Isopro RMH 3000, LabDiet, St. Louis, MO) or a tamoxifen diet (Teklad, TD.130855, Envigo, Madison, WI, 250 mg tamoxifen/ kg). Experiments were carried out during the light cycle.
FSF-KrasG12D/+;P53frt/frt mice were gifts from Dr David Kirsch (Duke University, Durham, NC).16,17 Pdx1Flp/+ mice were gifts from Dr Dieter Saur (Technical University, Munich, Germany).18 αSMACreERT2/+ mice were gifts from Dr Richard Premont (Case Western Reserve University, Cleveland, OH).19 Hif1αfl/fl (RRID:IMSR_JAX:007561), Hif2afl/fl (RRID: IMSR_JAX:008407), LSL-tdTomato (RRID:IMSR_JAX:007914), and C57BL/6 (RRID:IMSR_JAX:000664) mice were obtained from Jackson Laboratories (Bar Harbor, ME). FSF-KrasG12D/+; P53frt/frt mice were bred with Pdx1Flp mice to produce FSF-KrasG12D/+;P53frt/frt;Pdx1Flp/+ (KPF) mice. KPF mice were bred with αSMACreERT2/+ mice, and their progeny were bred with Hif1αfl/fl and Hif2αfl/fl mice to produce KPF CAF-HIF1 and KPF CAF-HIF2 mice, respectively. LSL-KrasG12D/+;Trp53fl/fl;Ptf1aCre/+ (KPC) mice and EGLN1/2/3fl/fl mice were previously bred and backcrossed to C57BL/6 mice in our laboratory.20,21 Genotyping was performed as described previously.22,23 Littermate controls were used in all experiments. Mice were screened for tumors by weekly ultrasounds as previously described.20
Isolation of Fibroblasts and CAFs
Mice harboring a tdTomato reporter were bred with αSMACreERT2/+;Hif2αfl/fl mice to produce αSMACreERT2/+; Hif2αfl/fl; tdTomatoLSL/LSL mice. Normal pancreata from the tdTomato progeny and from EGLN1/2/3fl/fl mice, and whole tumors from KPC and KPF CAF-HIF2 mice were minced and digested with 1 mg/mL Collagenase V (C9263–500MG; Sigma-Aldrich, St. Louis, MO) for 30 minutes at 37°C and 130 rpm/min followed by digestion with TrypLE (12605036; Thermo Fisher Scientific, Waltham, MA) for 10 minutes at 37°C. Cells were seeded in T175 flasks with Dulbecco’s modified Eagle’s medium (DMEM) (30–2002; ATCC, Manassas, VA) plus 10% (v/v) fetal bovine serum (FBS) (F4135; MilliporeSigma, Burlington, MA) and 1% Pen/Strep. On reaching 70% confluence, cells were passaged and incubated at 37°C for 30 minutes before the media was refreshed. The attached cells became enriched for fibroblasts or CAFs after 5 to 10 passages. Normal fibroblasts were immortalized with a pBABE-hygro-hTERT lentivirus24 (plasmid #1773; RRID: Addgene_1773; Addgene, Watertown, MA), which was a gift from Bob Weinberg.
Histopathology, Immunohistochemistry, and Immunofluorescence
For details regarding histopathology, immunohistochemistry (IHC), and immunofluorescence, please see the Supplementary Methods.
Bulk RNA Sequencing
Frozen tumors from KPF CAF-HIF2 wild-type (WT) and knockout (KO) mice were homogenized and RNA was purified using an RNeasy mini kit (74106; Qiagen, Hilden, Germany). Library preparation and sequencing were performed in the Sequencing and Microarray Facility at MD Anderson. The RSEM software package (RRID:SCR_013027) was used to quantitate transcript abundance from RNA sequencing (RNA-seq) data.25 Differential expression analysis was performed using the DESeq2 software package (RRID:SCR_015687). Gene Set Enrichment Analysis was performed to identify significantly enriched pathways (false discovery rate < 0.15). This dataset can be accessed in the National Center for Biotechnology Information’s Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/) using accession number GSE191474.
CAF Conditioned Media Harvest
CAFs isolated from KPC tumors and immortalized normal fibroblasts isolated from EGLN1/2/3fl/fl mice were seeded in DMEM with 10% FBS and 1% Pen/Strep at 5 × 105 density in 60-mm cell plates and cultured overnight. The media was replaced with DMEM containing 0.5% FBS, and cells were transferred to a hypoxia chamber (InvivO2; Baker Ruskinn, Pencoed, UK) set at 1% O2 and treated with increasing concentrations of PT2399 (Peloton Therapeutics/Merck, Kenilworth, NJ) for 48 hours.26 Cell media was collected and centrifuged at 1734 × g for 5 minutes, and the supernatant was filtered through a 40-μm strainer (352340; Corning, Corning, NY) and stored as conditioned media (CM) at −80°C. CM was diluted 1:1 with DMEM plus 10% FBS before experimental use to replenish nutrients depleted during media conditioning.
Macrophage Culture and Activation
Authenticated RAW 264.7 murine macrophages were purchased from ATCC (TIB-71, RRID:CVCL_0493) and maintained in DMEM with 10% FBS at 37°C with 5% CO2. For activation, macrophages were treated with 10 ng/ml IL-4 (214–14; PeproTech, Rocky Hill, NJ) and 10 ng/mL IL-13 (210–13; PeproTech) for 48 hours. Early passages were used for all experiments.
Macrophage Transwell Migration Assay
Migration was tested in 24-well transwell permeable plates with 8-μm-pore polyester membrane inserts (3464; Corning). Macrophages were resuspended in DMEM with 0.5% FBS and a total of 5 × 105 cells in 100 μL of cell suspension was added to the upper chamber, while 600 μL of CM from CAFs or normal fibroblasts was added to the lower chamber as a chemo-attractant. Cells were allowed to migrate through the membrane insert at 21% O2. After 22 hours, nonmigrating macrophages were gently removed from the top of the insert membrane with a cotton swab. Migrated macrophages were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 2 minutes, permeabilized with 100% methanol for 20 minutes, stained with 0.5% crystal violet solution in 70% ethanol for 15 minutes, and rinsed with deionized water, all at room temperature. Inserts were left drying overnight before imaging. At least 3 random nonoverlapping fields (×10) were imaged with a Leica (Wetzlar, Germany ) DMi1 or a BioTek (Winooski, VT) Cytation 5. ImageJ software (RRID:SCR_003070; NIH, Bethesda, MD; RRID:SCR_002285; Fiji, Tokyo, Japan) was used for analysis with a custom macro to identify, mask, and determine percent macrophage coverage. Macrophage migration was normalized to the control condition in each experiment and data from independent experiments were pooled.
Reverse Transcription Quantitative Real-Time Polymerase Chain Reaction
For details regarding primers and reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) analysis, please see the Supplementary Methods and Supplementary Table 1.
Single-Cell RNA-Seq
Single-cell suspensions were prepared by mincing KPF CAF-HIF2 WT and KO tumors, digesting them with 0.5 mg/mL Liberase (LIBTH-RO 5401135001; Sigma) for 30 minutes at 130 rpm, and passing them through a 100-μm cell strainer. Samples were then incubated with Accutase (A6964; Sigma) for 10 minutes at 37°C in a shaker, followed by treatment with ACK lysing buffer (A1049201; ThermoFisher) to eliminate erythrocytes. Samples were filtered through a 30-μm cell strainer and single cells were resuspended in PBS (SH30256.01; GE Healthcare Life Sciences, Piscataway, NJ) with 0.1% bovine serum albumin. Cell viability was measured using Trypan Blue (1450021; Bio-Rad, Hercules, CA). Single-cell suspensions were loaded into a 10x Genomics (Pleasanton, CA) Chromium instrument to generate gel beads in emulsion. Approximately 5000 cells were loaded per channel. Single-cell complementary DNA libraries were prepared using a Chromium Single-Cell 3’ Library and Gel Bead kit v2 (PN-120237; 10x Genomics) and sequenced using a NextSeq 500 (Illumina, San Diego, CA). The mean number of reads per cell was approximately 25,000 and the median number of genes detected per cell was approximately 2000.
The raw data were processed using cellranger count (Cell Ranger v2.1.1; 10x Genomics) based on the mm10 mouse reference genome. Subsequent data analysis was done in R using the Seurat package v3.1 (RRID:SCR_007322) with default parameters.27 Dead cells were excluded by retaining cells with less than 20% mitochondrial reads, leaving 20,802 cells for downstream analysis. We performed batch correction using the scMC algorithm with default parameters.28 Log normalization, variable feature identification (FindVariableFeatures), and z-scoring (ScaleData) were applied to the merged object of all cells, and principal component analysis (RunPCA, npcs = 30) with subsequent Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction and graph-based clustering of cells were performed. Markers for each cluster were identified using Seurat’s FindMarkers command and clusters were assigned to cell populations using published signature genes.29 Gene expression for genes of interest was then quantified across cell type groupings.
Immunotherapy Experiments
We obtained KPC cells from Dr Anirban Maitra that were authenticated by short tandem repeat profiling and were confirmed to be Mycoplasma free by real-time PCR (CellCheck Mouse 19 Plus; IDEXX Laboratories, Inc., Westbrook, ME). For the flank model, 1 × 106 KPC cells were resuspended in PBS and Matrigel (Corning) in a 1:1 ratio and subcutaneously implanted into the right flanks of syngeneic 10-week-old C57BL/6 female mice. Murine anti-CTLA4 antibody (αCTLA4) (BE0164; BioXCell, Lebanon, NH) or isotype control was administered intraperitoneally (IP) every 3 to 4 days at 250 μg/mouse beginning 13 days after implantation. PT2399 was resuspended in 10% ethanol, 30% PEG400, and 60% methylcellulose/water/Tween 80 and administered 5 days per week (Monday–Friday), twice daily, at 50 mg/kg via oral gavage. Treatments lasted 2 weeks, and tumor dimensions were measured with a caliper to calculate approximate volumes.
For the orthotopic model, 2 × 105 KPC cells were resuspended in PBS and Matrigel in a 1:1 ratio and injected into the tail of the pancreas of syngeneic 12-week-old C57BL/6 male mice. After 2 weeks of recovery, murine aCTLA4 (clone 9D9; Merck, Readington, NJ) and murine anti-PD1 antibody (αPD1) (muDX400; Merck) or isotype control were administered IP every 4 days at 20 μg/mouse, 200 μg/mouse, and 220 μg/ mouse, respectively, for 2 weeks. PT2399 was administered 5 days per week, twice daily for 3 weeks, at 50 mg/kg via oral gavage. Tumor burden was monitored by ultrasound, as previously described.20 Mice were age-matched, but group assignment was unblinded.
Statistical Methods
Survival was analyzed by the Kaplan-Meier method and log-rank test. Student t test and 1-way analysis of variance were used to analyze parametric data sets with 2 groups and more than 3 experimental groups, respectively, unless otherwise noted on the figure legend. The Mann-Whitney U test was used to analyze nonparametric data sets. All statistical analyses were performed using GraphPad (La Jolla, CA) Prism V.8 (RRID:SCR_002798), with a significance level of α = 0.05.
Results
Deletion of Stromal HIF2 Delays PDAC Progression and Enhances Survival
We used a dual recombinase system to constrain the deletion of Hif1α or Hif2α to CAFs within autochthonous PDAC tumors. Mice with FlpO-responsive alleles of both oncogenic Kras (FSF-KrasG12D/+)16 and homozygous Trp53 (Trp53frt/frt)17 were crossed with mice expressing FlpO in pancreatic tissue (Pdx1-FlpO)18 to generate KPF mice. These mice developed spontaneous PDAC over a timeframe and with a penetrance similar to those in KPC mice (Cre-driven model), and both models recapitulate human PDAC.18 KPF mice were subsequently bred with mice harboring conditional null alleles of Hif1α (Hif1αfl/fl)30 or Hif2α (Hif2afl/fl),23 driven by expression of the Cre-ERT2 transgene under the control of the α-smooth muscle actin (αSMA, also known as Acta2) promoter which marks CAFs (Figure 1A).19 We confirmed αSMA-Cre-ERT2– mediated deletion of Hif1α or Hif2α through ex vivo analyses of activated fibroblasts isolated from tdTomato reporter mice (Supplementary Figure 1A and B). Moreover, both immunofluorescence and immunohistochemical analyses of tumor sections showed a reduction of HIF2 expression in αSMA+ CAFs (Figure 1B and Supplementary Figure 1C). Once weaned, mice were fed normal chow or tamoxifen chow to generate KPF CAF-HIF1 and -HIF2 WT mice, or KPF CAF-HIF1 and -HIF2 KO mice, respectively (Figure 1C). Mice were screened for tumors weekly by ultrasound. The median age at tumor onset was 10.3 weeks (range: 7.1–21.1) in both the WT and KO groups.
Figure 1.

Deletion of stromal HIF2 delays PDAC progression and enhances survival. (A) Dual recombinase genetic strategy to develop a PDAC model with HIF1 or HIF2 KO in αSMA+ cells in a tamoxifen-induced manner using KPF mice. (B) Left: Representative immunofluorescence images of KPF CAF-HIF2 WT and KO tumors stained for αSMA and HIF2 (n = 3–5 fields of view/group). Scale bars, 50 μm and 10 μm. Right: Quantitative analysis of mean αSMA-specific HIF2 intensity per field of view. (C) Experimental design to generate KPF CAF-HIF2 WT control and KPF early CAF-HIF2 KO mice. (D) Left: Kaplan-Meier curves showing percentage survival for KPF CAF-HIF2 WT (n = 21) and KO (n = 23) mice. P, by log-rank test. Right: Tumor weights of KPF CAF-HIF2 WT (n = 21) and KO (n = 15) mice. Mean ± SEM; P, by Student t test. (E) Experimental design to generate postdiagnosis (post-Dx) KPF CAF-HIF2 WT and KO mice. (F) Kaplan-Meier curves showing percentage survival for post-Dx KPF CAF-HIF2 WT (n = 10) and KO (n = 7) mice. P, by log-rank test. See also Supplementary Figures 1–5.
Surprisingly, loss of stromal HIF1 had no effect on tumor growth or survival (median survival, 91 days for KO vs 100 days for WT; Supplementary Figure 1D and E). In contrast, HIF2 ablation in CAFs significantly decreased tumor growth and improved survival (median survival, 120 days for KO vs 80 days for WT; n = 21–23 mice/group, Log-rank P = .0009; Figure 1D). Histological analyses of the pancreata revealed well-differentiated PDAC foci in both CAF-HIF2 WT and CAF-HIF2 KO groups, yet remarkably, we found no gross or microscopic evidence of tumor tissue in the sections analyzed from 6 of the HIF2-depleted mice, suggesting that deletion of stromal HIF2 may also influence PDAC oncogenesis and/or progression (Supplementary Figure 2A–C)15. Importantly, there were no statistically significant differences in tumor fibrosis associated with KPF CAF-HIF2 KO (n = 8–12 tumors/group, P = .051; Supplementary Figure 2D), indicating that this approach does not physically disrupt the tumor stroma.31 Because stromal modulation, particularly of αSMA+ CAFs, impacts PDAC metastasis,8,32 we evaluated metastatic burden at the time of death and found no statistically significant differences in the proportion of KPF CAF-HIF2 WT and KO mice that presented with distant metastasis (0.45 for WT vs 0.32 for KO; n = 19–20 mice/group, P = .514; Supplementary Figure 2E). These results indicate that survival in KPF CAF-HIF2 KO mice was driven by lower primary tumor burden. Moreover, the survival advantage seen in mice with deletion of HIF2 in CAFs was not dependent on Trp53 gene dosage, as CAF-specific loss of HIF2 in KPF mice heterozygous for Trp53 also exhibited significantly improved survival compared with CAF-HIF2 WT littermates (median survival, 375 days for KO vs 237 days for WT; n = 5–10 mice/group, Log-rank P = .0103; Supplementary Figure 3A).
The tumor weights, histopathological quantification, and metastasis rate for Trp53het KPF CAF-HIF2 WT and KO mice are shown in Supplementary Figure 3B–E.
We next assessed how stromal HIF2 deletion after PDAC onset affected survival, as this would more closely reflect the timeline of therapeutic HIF2 targeting in patients. We generated another cohort of KPF αSMA-HIF2fl/fl mice and fed them normal chow or tamoxifen chow after tumors were diagnosed by ultrasound (Figure 1E). This late abrogation of HIF2 in CAFs still improved survival by 37.3% compared with the survival of control mice (median, 114 days vs 83 days; P = .002; Figure 1F), which was similar to the median survival of mice receiving tamoxifen chow at weaning. Moreover, we confirmed that CAF-HIF2 KO did not alter HIF1 messenger RNA or nuclear protein levels (Supplementary Figure 4). Together, these data suggest that stromal HIF2, but not HIF1, plays a critical role in PDAC development and progression.
To confirm that this survival advantage was mediated by HIF2 depletion in CAFs, and not in tumor cells, we isolated cancer cells from KPF tumors with Hif1αfl/fl or Hif2αfl/fl alleles and induced ex vivo recombination by infection with Cre or control GFP adenovirus. These KPF cells were orthotopically implanted into the pancreata of immunocompromised mice (Supplementary Figure 5A). We found that deletion of HIF1 or HIF2 in tumor cells had no impact on tumor growth (Supplementary Figure 5B and C), confirming cell nonautonomous functions of HIF in PDAC.
Stromal HIF2 Regulates Macrophage Recruitment to PDAC Tumors
We performed bulk RNA-seq to understand the mechanism by which loss of HIF2 in CAFs suppressed tumor growth. Transcriptomic analysis revealed a stromal HIF2dependent gene downregulation signature with the most conspicuous changes being in pathways related to myeloid/ macrophage biology (Figure 2A and Supplementary Figure 6). Deletion of HIF2 in CAFs led to downregulation of genes involved in macrophage migration, differentiation, and activation, including Mmp9, Cd74, Tgfb1, Itgam, and C3ar1; these results were validated by RT-qPCR (Figure 2B and C). Gene set enrichment analysis did not reveal any upregulated gene sets that achieved statistical significance. We next compared tumor-associated macrophage (TAM) infiltration by F4/80 IHC and observed significantly fewer TAMs in KPF CAF-HIF2 KO tumors than in controls (n = 5 tumors/group, P = .028; Figure 2D). These results suggest that HIF2 signaling in CAFs regulates macrophage recruitment to PDAC tumors.
Figure 2.

Stromal HIF2 regulates macrophage recruitment to PDAC tumors. (A) Heatmap of the top expressed genes using bulk RNA-seq data from KPF CAF-HIF2 tumors (n = 4/group). (B) Gene set enrichment analysis of tumors in (A) correlates CAF-HIF2 function with macrophage migration, differentiation, and activation. GO, gene ontology; NES, normalized enrichment score. (C) RT-qPCR confirmed the downregulation of genes involved in the pathways in (B). (D) Left: Representative IHC images of CAF-HIF2 tumors stained for F4/80 and counterstained with hematoxylin QS (n = 5/group). Scale bar, 50 μm. Right: Quantification of F4/80+ macrophages per field. All error bars represent mean ± SEM and each dot denotes a biological replicate. P, by Student t test. See also Supplementary Figure 6.
To evaluate this hypothesis, we established CAF and normal fibroblast lines from spontaneous pancreatic tumors and normal pancreata, respectively. Both cell lines were cultured in hypoxia to stabilize HIF2 and to approximate in vivo TME conditions, and were then treated with either vehicle or the clinical HIF2 inhibitor PT2399.26 We found that conditioned media from hypoxic CAFs stimulated macrophage migration in an HIF2-dependent fashion (Figure 3A and B). Stimulation of macrophage migration by CAFs appears to be specific to fibroblasts reprogrammed in the PDAC TME, as fibroblasts isolated from normal pancreata lacked the ability to stimulate macrophage migration (Supplementary Figure 7A and B). To confirm that the reduction in migration that occurred with HIF2-inhibited CAF conditioned media was not due to biological repercussions of carryover of residual PT2399, we directly added the drug to processed conditioned media made from untreated hypoxic CAFs and found no differences in macrophage migration (Supplementary Figure 7C and D). Moreover, direct treatment of macrophages with PT2399 had no effect on transwell migration (Figure 3C and D and Supplementary Figure 7E). These results strongly suggest that HIF2 coordinates CAF-TAM crosstalk and induces macrophage migration in a paracrine fashion.
Figure 3.

Hypoxic CAFs promote macrophage migration in a HIF2-dependent paracrine fashion. (A) CM was collected from hypoxic CAFs treated with vehicle (veh) or PT2399. This CM was subsequently placed in the bottom chamber of a transwell as a chemoattractant for macrophage migration. (B) Representative bright-field images (10×; left) and quantification of macrophage migration relative to macrophages treated with CM from hypoxic veh-treated CAFs (right) of the transwell assay depicted in (A) (n = 8/group pooled from 3 independent experiments). (C) Five percent FBS/DMEM with veh or PT2399 was used as chemoattractant for macrophage migration. (D) Quantification of macrophage migration relative to macrophages treated with veh of the transwell assay depicted in (C). All error bars represent mean ± SEM; P, by 1-way analysis of variance; ns, not significant. See also Supplementary Figure 7.
Hypoxic CAFs Promote Macrophage M2 Polarization in an HIF2-Dependent Paracrine Fashion
Macrophages are functionally classified as either M1, which are classically activated and proinflammatory, or M2, which are alternatively activated during the resolution phase of inflammation, and thus display an immunosuppressive phenotype.33 In many cancers, including PDAC, M2 macrophages are associated with worse outcomes because they promote metastasis and suppress antitumor immune responses by expressing checkpoint ligands and by inducing regulatory T cells (Tregs).33,34 CAFs have been linked to M2 repolarization of TAMs in PDAC,34 yet the roles of hypoxia and HIF2 in this context remain unclear.
To understand whether HIF2 signaling in CAFs drives macrophage M2 repolarization, we stimulated murine macrophages with conditioned CAF media and assessed expression of Arg1, an M2 polarization marker (Figure 4A). We found that hypoxia in CAFs, and therefore HIF2 expression, increased Arg1 levels in macrophages by 4-fold compared with normoxic CAFs (Supplementary Figure 8A). Moreover, HIF2 inhibition via PT2399 in hypoxic CAFs impaired the ability of conditioned media from these cells to induce M2 polarization, indicating that the paracrine CAF signal is HIF2-dependent (Figure 4B). Conditioned media from hypoxic normal pancreatic fibroblasts failed to induce M2 polarization (Supplementary Figure 8B and C), confirming that stimulation of macrophages by CAFs is specific to fibroblasts reprogrammed in the PDAC TME. Furthermore, direct treatment of hypoxic or normoxic macrophages with PT2399 did not affect their Arg1 levels (Figure 4C–F). To confirm that this polarization was specific to HIF2 function in CAFs and not an off-target effect of PT2399,35 we developed HIF2 WT and HIF2 KO CAFs from KPF tumors and deleted Hif2a ex vivo with Cre or control GFP adenovirus (Supplementary Figures 4A and B). We found that conditioned media from CAFs lacking Hif2α expression did not enhance Arg1 expression in macrophages, confirming that our phenotype is driven by intrinsic HIF2 signaling in CAFs (Supplementary Figure 8D and E). Taken together, these findings support the notion that hypoxic CAFs regulate TAMs in a HIF2-dependent paracrine fashion.
Figure 4.

Hypoxic CAFs promote macrophage M2 polarization in a HIF2-dependent paracrine fashion. (A) Macrophages were incubated under normoxic conditions with conditioned media collected from hypoxic CAFs treated with vehicle (veh) or PT2399 and Arg1 expression was measured by RT-qPCR. (B) Arg1 fold change over macrophages incubated with CM from veh-treated hypoxic CAFs is shown. Data are representative of 4 independent experiments. (C) Macrophages were incubated under hypoxic conditions and directly treated with veh or PT2399, and Arg1 expression was measured by RT-qPCR. (D) Arg1 fold change over veh-treated hypoxic macrophages is shown. (E) M2-polarized macrophages were incubated under normoxic conditions and directly treated with veh or PT2399 and Arg1 expression was measured by RT-qPCR. (F) Arg1 fold change over vehicle-treated normoxic M2 macrophages is shown. Data are representative of 3 independent experiments. All error bars represent mean ± SEM; P, by Student t test; *P ≤ .05; ns, not significant. See also Supplementary Figures 8 and 9.
Vascular endothelial growth factor (VEGF) is a potent immunosuppressive factor known to induce M2 repolarization in TAMs.36 Because Vegf is a hypoxia-inducible gene,37 we measured Vegf expression in hypoxic CAFs treated with PT2399 and found modest to no difference compared with vehicle-treated CAFs (Supplementary Figure 9A). Moreover, IHC staining of tumor sections for the endothelial marker Meca32 showed no differences in vessel density between CAF-HIF2 WT and KO mice (n = 5 tumors/group, P = .556; Supplementary Figure 9B and C). These data suggest that HIF2-regulated CAF-TAM crosstalk is independent of angiogenic signaling pathways.
Deletion of Stromal HIF2 Reduces PDAC Immunosuppression
We performed single-cell RNA-seq (scRNA-seq) to interrogate the impact of CAF-specific HIF2 signaling on other cells in the PDAC TME. We analyzed the transcriptomes from 20,802 single cells isolated from 3 KPF CAF-HIF2 WT tumors (9774 cells) and 3 KPF CAF-HIF2 KO tumors (11,028 cells). Graph-based clustering of cells after UMAP dimensionality reduction identified 15 clusters that were assigned to 6 major cell types using signature genes (Figure 5A and Supplementary Figure 10A and B). All the cell populations identified were represented in both experimental groups and in all 6 mice, with 48.7% of the cells analyzed being identified as epithelial/tumor cells, 25.4% as myeloid cells, 21.4% as fibroblasts, and the remaining cells as endothelial cells, B cells, and T cells, and neutrophils (Figure 5A and Supplementary Figure 10C).
Figure 5.

Deletion of HIF2 in CAFs reduces PDAC immunosuppression. (A) UMAP of scRNA-seq analysis of 20,802 cells isolated from KPF CAF-HIF2 WT tumors (9774 cells; n = 3 mice) and KPF CAF-HIF2 KO tumors (11,028 cells; n = 3 mice). Cell types were identified through graph-based clustering followed by manual annotation using known marker genes. (B) Average percentage of myeloid cells over immune cells sequenced per tumor. (C) M2-polarized TAMs were identified within the myeloid cell population via expression of Arg1 and Mrc1. Immunosuppressive TAMs were identified within the myeloid cell population via expression of Cd274 (Pdl1) and Cd86 (B7–2). (D) Violin plots showing findings on scRNA-seq analysis of Ctla4 in KPF CAF-HIF2 WT and KO tumors in all identified cell types. (E) Left: Representative IHC images of CAF-HIF2 WT and KO tumors stained for FoxP3 and counterstained with hematoxylin QS (n = 5–6/group); scale bars, 50 μm. Right: Quantification of FoxP3+ Tregs per field. All error bars represent mean ± SEM; P, by Student t test, unless otherwise noted. See also Supplementary Figure 10.
Quantification of the relative proportions of each cell type within tumors showed that HIF2 deletion in αSMA+ CAFs did not affect the total number of fibroblasts within tumors (Supplementary Figure 10C). Single-cell analyses were largely concordant with the bulk RNA-seq and IHC data, showing that CAF-HIF2 KO tumors had a significantly lower proportion of myeloid immune cells than CAF-HIF2 WT tumors (66.9% vs 83.24%; n = 3 tumors/group, P = .018; Figures 2B–D and 5B, and Supplementary Figure 10D). Further interrogation of the myeloid cell population revealed high expression of Arg1, Mrc1, Cd11b (Itgam), Cd68, and Adgre1 (F4/80), indicating a predominance of M2-polarized TAMs, which were reduced in CAF-HIF2 KO tumors (Figure 5C and Supplementary Figure 10E). A substantial proportion of these TAMs expressed the immunosuppressive checkpoint ligands Cd274 (Pdl1, ligand for PD-1), Cd80 and Cd86 (both ligands for CTLA-4; Figure 5C and Supplementary Figure 10E). Single-cell analysis also showed higher expression of Ctla4, Foxp3, and Pdcd1 (Pd1) in a subset of T cells (Figure 5D and Supplementary Figure 10E), indicating the presence of Tregs in KPF tumors. Moreover, IHC staining of tumor sections for the Treg marker FoxP3 showed that CAF-HIF2 KO tumors had significantly fewer Tregs than CAF-HIF2 WT tumors (n = 5–6 tumors/group, P = .014; Figure 5E). These data strongly suggest that deletion of HIF2 in CAFs reduces the PDAC immunosuppressive landscape.
Inhibition of HIF2 Signaling Enhances the Response of PDAC to Immunotherapy
PDAC is highly resistant to immunotherapy,1 but recent studies have suggested that targeting nonredundant pathways with a combined anti-CTLA4 and anti-PD1 approach may overcome inherent TME immunosuppression.38 Because HIF2 deletion in CAFs reduced the number of immunosuppressive M2-polarized TAMs and Tregs, we reasoned that PT2399 might improve response to checkpoint immunotherapy. To test this hypothesis, we implanted KPC cells subcutaneously into syngeneic C57BL/6 mice and assigned them to 1 of 4 treatments: vehicle plus immunoglobulin (Ig)G control, vehicle plus αCTLA4, PT2399 plus IgG, or PT2399 plus αCTLA4 (Figure 6A). We found that the combination of PT2399 with αCTLA4 significantly slowed tumor growth (n = 10 mice/group, P = .038), whereas treatment with either drug alone had no discernible effect (Figure 6B).
Figure 6.

Inhibition of HIF2 signaling enhances the response of PDAC to checkpoint immunotherapy. (A) Schematic for administration of PT2399 + αCTLA4 in a syngeneic flank KPC model. i.p., intraperitoneal; o.g., oral gavage; b.i.d., bid in die (twice a day). (B) Tumor growth curve from (A) (n = 10/group). Veh, vehicle; P, by Mann-Whitney U test. (C) Schematic for administration of PT2399 + αCTLA4/αPD1 in a syngeneic orthotopic KPC model. (D) Kaplan-Meier curves showing percentage survival for (C) (n = 10/group); P, by log-rank test. All error bars represent mean ± SEM; P, by Student t test unless otherwise noted. See also Supplementary Figures 11–13.
We next implanted KPC cells orthotopically into syngeneic C57BL/6 mice to test whether HIF2 inhibition enhanced response to dual checkpoint blockade (DCB) with αCTLA4 and αPD1. Mice were assigned to 1 of 4 treatments: vehicle plus IgG, vehicle plus DCB, PT2399 plus IgG, or PT2399 plus DCB, with the goal to assess 60-day survival (Figure 6C). The experiment was prematurely terminated because of institutional mandates related to COVID-19, yet the survival rate at 45 days in mice that received combined PT2399 and DCB was 100%, significantly higher than the survival rate in the groups treated with IgG control (n = 10 mice/group, P ≤ .0005), and trending toward improved survival compared with mice treated with DCB and vehicle (n = 10 mice/group, P = .067; Figure 6D). Both groups treated with DCB had slower growing tumors (Supplementary Figures 11 and 12). PT2399 induced modest anemia in a separate cohort of tumor-bearing mice, which is an expected on-target effect26 (Supplementary Figure 13). Taken together, these results suggest that HIF2 inhibition might enhance antitumor immune responses and improve survival.
Discussion
Our study addresses a long-standing knowledge gap about the relative roles of HIF signaling in the PDAC microenvironment. Here we show that CAF-specific expression of HIF2, but not HIF1, drives paracrine signals that increase the presence of immunosuppressive cells like TAMs and Tregs in a HIF2-dependent fashion. Furthermore, both genetic and pharmacologic inhibition of HIF2 improved survival in spontaneous and syngeneic mouse models.
Our study identifies HIF2 signaling in CAFs as a critical component of hypoxia-related immunosuppression in pancreatic cancer. We demonstrate that HIF2 signaling orchestrates immunosuppression within pancreatic tumors by shifting the immune cellular composition of the TME rather than by altering fibrosis, which was unchanged in our model. We observed more TAMs and Tregs in tumors from mice with intact CAF HIF2 function compared with mice with HIF2 deletion. These data contrast with findings from a previous study in which depletion of αSMA+ CAFs reduced fibrosis, increased Tregs, and drove cancer progression.8 These phenotypic differences are most likely explained by the different approaches of the 2 studies: we targeted CAF functionality under hypoxia, whereas the former study ablated CAFs altogether. Our dual recombinase system specifically interrogated HIF2 function in αSMA+ CAFs within autochthonous tumors, but did not address the potential role of HIF2 in other CAF subtypes that do not express αSMA.39 These other subtypes could be studied in future experiments using different Cre drivers, such as Fap or Fsp1, to address the dynamic relationships between CAF populations.
Hypoxia, and therefore HIF signaling, affects every cell type within a pancreatic tumor, but our data suggest that the detrimental effects of tumor hypoxia are mediated largely by HIF2-dependent crosstalk between CAFs and macrophages, which has not been previously reported. Using single-cell approaches, we found that M2-polarized TAMs were a major source of immunosuppressive CD86 and PD-L1, and this was reversed by the CAF-specific ablation of HIF2. Thus, we infer that these TAMs may be partially responsible for the subsequent reprogramming of effector T cells in the pancreatic TME. Like the findings in several clinical reports, reduced TAM density correlated with improved survival from pancreatic cancer33,34 in mice in the setting of CAF-HIF2 deletion. Furthermore, we demonstrate via ex vivo experiments that hypoxic CAFs stimulate the migration and M2 polarization of macrophages in a HIF2-dependent fashion. In contrast, normal pancreatic fibroblasts did not alter macrophage migration or polarization. It is not yet known if this novel crosstalk is mediated by a single soluble factor or by a combination of cytokines/ growth factors,34 metabolites,40 lipids,41 and/or exosomes.42
Although we focused on CAF-TAM crosstalk, we note that other HIF2-dependent pathways may be contributing to the overall phenotype. Hypoxia, and particularly HIF2, has also been shown to induce transforming growth factor-β (TGF-β) signaling,43,44 which could modulate the immune landscape in the TME. Notably, we found that Tgfb1 expression was reduced in CAF-HIF2 KO tumors, thus changes in TGF-β signaling could potentially have a role in HIF2-dependent CAF-mediated TME reprogramming. However, because HIF2 modulates multiple homeostatic pathways, our data do not rule out the contribution of HIF2dependent metabolic40 or epigenetic45 changes within CAFs that could mediate immune crosstalk.
We note that because our experimental system was designed to interrogate HIF2 function in CAFs, we cannot exclude the possibility that HIF2 signaling also may be critical in other cell types in the TME. Previous KO studies have shown that HIF2 has tumor supporting roles in macrophages46 and regulatory T cells47 in colorectal and hepatocellular cancer models, suggesting a potential therapeutic benefit with HIF2 inhibition. Pharmacologic HIF2 inhibition is global and affects all cells in the TME. Therefore, it was reassuring to demonstrate that HIF2 inhibition with PT2399 enhanced immune responses in our syngeneic pancreatic cancer models. It will be important to evaluate immune responses in autochthonous tumor models that contain higher stromal abundance,48 and we posit that with higher CAF content, the immune responses to HIF2 inhibition will be more robust. Belzutifan (PT2977, MK6487) is a second-generation HIF2 inhibitor that recently received approval from the Food and Drug Administration for use in von Hippel–Lindau–associated renal cell carcinoma.26,49 Thus, our approach of using HIF2 inhibition to prime immune responses could potentially be repurposed to treat pancreatic cancer.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
The desmoplastic stroma of pancreatic cancer promotes treatment resistance and immunosuppression, and there are currently no successful treatments to inhibit these oncogenic traits.
NEW FINDINGS
We found that loss of stromal hypoxia-inducible factor-2 decreased tumor growth and doubled the median survival of mice with pancreatic cancer, possibly through interfering with immunosuppressive cancer-associated fibroblast–macrophage crosstalk.
LIMITATIONS
This is a preclinical study and therefore it is not known if this approach will directly translate to human patients.
IMPACT
A hypoxia-inducible factor-2 inhibitor (Belzutifan) was recently approved by the Food and Drug Administration for use in the treatment of von Hippel–Lindau– associated renal cell carcinoma. Thus, it may be possible to repurpose this drug to prime immune responses in pancreatic cancer patients.
Acknowledgments
We are grateful to all the members and alumni of the Taniguchi Lab for their critical input throughout the course of this study. We also acknowledge Dr David Kirsch (Duke) and Dr Dieter Saur for their generous gift of KPF breeders for our colony and Dr Richard Premont (Case Western) for providing the aSMACreERT2 mice. Experimental design figures were made using BioRender.com.
Funding
Cullen M. Taniguchi was supported by funding from the National Institutes of Health (NIH) under award number R01CA227517-01A1 and from the Cancer Prevention & Research Institute of Texas (CPRIT) grant RR140012, the V Foundation (V2015-22), the Sidney Kimmel Foundation, a Sabin Family Foundation Fellowship, the Reaumond Family Foundation, the Mark Foundation, Childress Family Foundation, the McNair Family Foundation, and generous philanthropic contributions to The University of Texas MD Anderson Moon Shots Program. Carolina J. Garcia Garcia was supported by the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) of the NIH under award number F31DK121384 and by the NIH/National Cancer Institute (NCI) under award number U54CA096300/297. Natividad R. Fuentes was supported by a CPRIT Training Award (RP210028) and by an NIH Diversity Supplement under award number R01CA227517-01A1. This work was also supported by the NIH/NCI Cancer Center Support Grant (CCSG) P30CA016672, which supports MD Anderson Cancer Center’s Small Animal Imaging Facility, Sequencing and Microarray Facility, and Research Histology Core Laboratory, by the NIH/NCI GI SPORE Grant P50CA221707, and by the NIH/NIDDK grant DK056338, which supports the Texas Medical Center Digestive Diseases Center.
Writing Assistance: We thank the MD Anderson Research Library Editing Services for their input.
Data Transparency Statement: Data, materials, and reagents will be made available to other researchers on request. Bulk and single-cell RNA sequencing datasets have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/) and can be retrieved using accession numbers GSE191474 and GSE193416.
BioRxiv Preprint DOI: https://doi.org/10.1101/2021.05.21.445190
Abbreviations and Acronyms:
- αCTLA4
anti-CTLA4 antibody
- αPD1
anti-PD1 antibody
- αSMA
α-smooth muscle actin
- CAFs
cancer-associated fibroblasts
- CM
conditioned media
- DCB
dual checkpoint blockade
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- HIFs
hypoxia-inducible factors
- Ig
immunoglobulin
- IHC
immunohistochemistry
- KO
knockout
- KPC
LSL-KrasG12D/+;Trp53fl/fl;Ptf1aCre/+ mice
- KPF
FSF-KrasG12D/+;P53frt/frt;Pdx1Flp mice
- PBS
phosphate-buffered saline
- PDAC
pancreatic ductal adenocarcinoma
- RT-qPCR
reverse transcription quantitative real-time polymerase chain reaction
- RNA-seq
RNA sequencing
- scRNA-seq
single-cell RNA-seq
- TAM
tumor-associated macrophage
- TME
tumor microenvironment
- Tregs
regulatory T cells
- UMAP
Uniform Manifold Approximation and Projection
- veh
vehicle
- WT
wild-type
Footnotes
CRediT Authorship Contributions
Order of Authors (with Contributor Roles):
Carolina Jannet Garcia Garcia, BS (Conceptualization: Supporting; Data curation: Lead; Formal analysis: Equal; Investigation: Equal; Methodology: Equal; Project administration: Equal; Validation: Lead; Visualization: Equal; Writing – original draft: Lead; Writing – review & editing: Lead)
Yanqing Huang, PhD (Conceptualization: Supporting; Data curation: Equal; Formal analysis: Equal; Investigation: Lead; Methodology: Equal; Project administration: Equal; Software: Lead; Validation: Equal; Visualization: Equal)
Natividad R. Fuentes, PhD (Formal analysis: Equal; Investigation: Equal; Validation: Supporting; Visualization: Supporting; Writing – review & editing: Equal)
Madeleine C. Turner, BS (Formal analysis: Supporting; Investigation: Supporting; Methodology: Supporting; Visualization: Supporting; Writing – review & editing: Supporting)
Maria E. Monberg, BS (Data curation: Equal; Software: Equal; Validation: Supporting; Visualization: Supporting)
Daniel Lin, BS (Data curation: Supporting; Investigation: Supporting; Visualization: Supporting; Writing – original draft: Supporting; Writing – review & editing: Supporting)
Nicholas D. Nguyen, BS (Data curation: Supporting; Formal analysis: Supporting; Investigation: Supporting; Software: Supporting; Visualization: Supporting; Writing – original draft: Supporting)
Tara N. Fujimoto, BS (Investigation: Supporting)
Jun Zhao, MD (Investigation: Supporting)
Jaewon J. Lee, PhD (Formal analysis: Supporting; Software: Supporting; Visualization: Supporting)
Vincent Bernard, MD, PhD (Formal analysis: Supporting; Software: Supporting)
Meifang Yu, PhD (Investigation: Supporting)
Abagail M. Delahoussaye, BS (Investigation: Supporting)
Iancarlos Jimenez Sacarello, BS (Validation: Supporting)
Emily G. Caggiano, BS (Data curation: Supporting; Investigation: Supporting; Writing – review & editing: Supporting)
Jae L. Phan, BS (Investigation: Supporting)
Amit Deorukhkar, PhD (Investigation: Supporting)
Jessica M. Molkentine, BS (Investigation: Supporting)
Dieter Saur, MD, PhD (Resources: Supporting)
Anirban Maitra, MBBS (Resources: Supporting)
Cullen Mitsuo Taniguchi, MD, PhD (Conceptualization: Lead; Funding acquisition: Lead; Methodology: Equal; Project administration: Supporting; Resources: Lead; Supervision: Lead; Validation: Supporting; Visualization: Supporting; Writing – original draft: Supporting; Writing – review & editing: Equal)
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at http://www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2022.02.024.
Conflict of interest
This author discloses the following: Cullen M. Taniguchi is on the medical advisory board of Accuray and is a paid consultant for Xerient Pharma and Phebra Pty, Ltd. The remaining authors disclose no conflicts.
References
- 1.Lee JS, Ruppin E. Multiomics prediction of response rates to therapies to inhibit programmed cell death 1 and programmed cell death 1 ligand 1. JAMA Oncol 2019; 5:1614–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tjomsland V, Niklasson L, Sandström P, et al. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin Devel Immunol 2011; 2011:212810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chauhan VP, Boucher Y, Ferrone CR, et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell 2014;26:14–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koong AC, Mehta VK, Le QT, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys 2000;48:919–922. [DOI] [PubMed] [Google Scholar]
- 5.Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer 2013;108:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whittle MC, Hingorani SR. Fibroblasts in pancreatic ductal adenocarcinoma: biological mechanisms and therapeutic targets. Gastroenterology 2019; 156:2085–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Jesus-Acosta A, Sugar EA, O’Dwyer PJ, et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br J Cancer 2020;122:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hakim N, Patel R, Devoe C, et al. Why HALO 301 failed and implications for treatment of pancreatic cancer. Pancreas (Fairfax) 2019;3:e1–e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res 1998;58:1408. [PubMed] [Google Scholar]
- 11.Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 2017;32:71–87.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colbert LE, Fisher SB, Balci S, et al. High nuclear hypoxia-inducible factor 1 alpha expression is a predictor of distant recurrence in patients with resected pancreatic adenocarcinoma. Int J Radiat Oncol Biol Phys 2015;91:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Lou Y, Zhang J, et al. Hypoxia-inducible factor-2a promotes tumor progression and has crosstalk with Wnt/b-catenin signaling in pancreatic cancer. Mol Cancer 2017;16:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KE, Spata M, Bayne LJ, et al. Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov 2016;6:256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Criscimanna A, Duan L-J, Rhodes JA, et al. PanIN-specific regulation of Wnt signaling by HIF2a during early pancreatic tumorigenesis. Cancer Res 2013;73:4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young NP, Crowley D, Jacks T. Uncoupling cancer mutations reveals critical timing of p53 loss in sarcomagenesis. Cancer Res 2011;71:4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee C-L, Moding EJ, Huang X, et al. Generation of primary tumors with Flp recombinase in FRT-flanked p53 mice. Dis Model Mech 2012;5:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schonhuber N, Seidler B, Schuck K, et al. A nextgeneration dual-recombinase system for time- and hostspecific targeting of pancreatic cancer. Nat Med 2014; 20:1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wendling O, Bornert JM, Chambon P, et al. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis 2009;47:14–18. [DOI] [PubMed] [Google Scholar]
- 20.Fujimoto TN, Colbert LE, Huang Y, et al. Selective EGLN inhibition enables ablative radiotherapy and improves survival in unresectable pancreatic cancer. Cancer Res 2019;79:2327–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taniguchi CM, Miao YR, Diep AN, et al. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci Transl Med 2014; 6:236ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bardeesy N, Aguirre AJ, Chu GC, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A 2006;103:5947–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruber M, Hu C-J, Johnson RS, et al. Acute postnatal ablation of Hif-2a results in anemia. Proc Natl Acad Sci U S A 2007;104:2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Counter CM, Hahn WC, Wei W, et al. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci U S A 1998;95:14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W, Hill H, Christie A, Kim MS, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016;539:112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butler A, Hoffman P, Smibert P, et al. Integrating singlecell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018; 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Nie Q. scMC learns biological variation through the alignment of multiple single-cell genomics datasets. Genome Biol 2021;22:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JJ, Bernard V, Semaan A, et al. Elucidation of tumor-stromal heterogeneity and the ligand-receptor interactome by single-cell transcriptomics in real-world pancreatic cancer biopsies. Clin Cancer Res 2021; 27:5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan HE, Poloni M, McNulty W, et al. Hypoxia-inducible Factor-1a is a positive factor in solid tumor growth. Cancer Res 2000;60:4010. [PubMed] [Google Scholar]
- 31.Spivak-Kroizman TR, Hostetter G, Posner R, et al. Hypoxia triggers hedgehog-mediated tumor–stromal interactions in pancreatic cancer. Cancer Res 2013; 73:3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity 2014;41:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang A, Qian Y, Ye Z, et al. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med 2017; 6:463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho H, Du X, Rizzi JP, et al. On-target efficacy of a HIF2alpha antagonist in preclinical kidney cancer models. Nature 2016;539:107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee WS, Yang H, Chon HJ, et al. Combination of antiangiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med 2020;52:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choueiri TK, Kaelin WG. Targeting the HIF2–VEGF axis in renal cell carcinoma. Nat Med 2020;26:1519–1530. [DOI] [PubMed] [Google Scholar]
- 38.Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate nonredundant immune mechanisms in cancer. Nature 2015;520:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elyada E, Bolisetty M, Laise P, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov 2019;9:1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016;536:479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auciello FR, Bulusu V, Oon C, et al. A stromal lysolipid– autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov 2019;9:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao H, Yang L, Baddour J, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016;5:e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim WY, Perera S, Zhou B, et al. HIF2a cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest 2009;119:2160–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wierenga ATJ, Vellenga E, Schuringa JJ. Convergence of hypoxia and TGFb pathways on cell cycle regulation in human hematopoietic stem/progenitor cells. PLoS One 2014;9:e93494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherman MH, Yu RT, Tseng TW, et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc Natl Acad Sci U S A 2017;114:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imtiyaz HZ, Williams EP, Hickey MM, et al. Hypoxiainducible factor 2a regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 2010;120:2699–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu T-S, Lin Y-L, Wang Y-A, et al. HIF-2a is indispensable for regulatory T cell function. Nat Commun 2020; 11:5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mallya K, Gautam SK, Aithal A, et al. Modeling pancreatic cancer in mice for experimental therapeutics. Biochim Biophys Acta 2021;1876:188554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel–Lindau disease. N Engl J Med 2021;385:2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.